Adjuvant Analgesics for Persistent (Chronic) Neuropathic Pain
SOME of the adjuvant analgesic classes are conventionally used solely for persistent neuropathic pain. The drugs in these classes, combined with the drugs in classes subsumed under the category of multipurpose analgesics, offer a very large group of individual agents that might be useful for pains of this type. Antidepressants and anticonvulsants are the first-line adjuvant analgesics for a wide variety of neuropathic pain syndromes. The multipurpose antidepressants were discussed earlier in Chapter 22, and the anticonvulsants will be discussed in detail here. Refractory neuropathic pain, which has not responded to these first-line approaches, may be considered for trials of the other so-called multipurpose drugs, or other agents classified as drugs used conventionally for neuropathic pain. Other adjuvant agents used for refractory neuropathic pain include sodium channel blockers, several topical agents, gamma aminobutyric acid (GABA) agonists (baclofen [Lioresal]), N-methyl-d-aspartate (NMDA) receptor antagonists (e.g., dextromethorphan and ketamine), and the relatively new intrathecal drug, ziconotide (Prialt). In addition to persistent pain, certain adjuvant agents are used to manage the neuropathic component of some types of acute pain and for the purpose of preventing persistent neuropathic pain, such as persistent neuropathic postsurgical pain. These are discussed later in this section.
Recent systematic reviews, some with evidence-based guidelines for drug selection, provide information about a range of therapies used for neuropathic pain (Finnerup, Otto, McQuay, et al., 2005; Saarto, Wiffen, 2007; Kroenke, Krebs, Bair, 2009). Table 23-1 lists many of these drugs, and Table V-1 at the end of Section V provides dosing guidelines and other characteristics of many of the adjuvant analgesics for the treatment of neuropathic pain. See also Section II for assessment of neuropathic pain.
Guidelines
Table 23-1
Adjuvant Analgesics for Neuropathic Pain
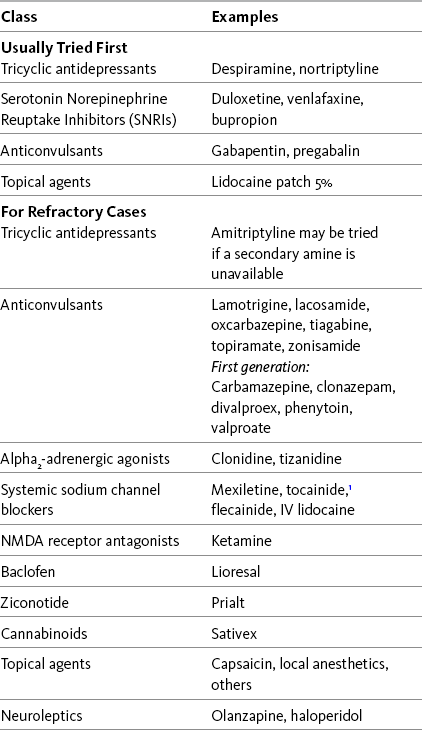
IV, Intravenous; NMDA, N-methyl-d-aspartate.
This table provides the classes of adjuvant analgesics for neuropathic pain and examples of those that should be used first and for refractory pain. See text for discussion and references.
1No longer marketed in the United States.
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 654, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Anticonvulsant Drugs
Several systematic reviews of randomized controlled trials of anticonvulsants for pain management have demonstrated strong evidence to support the efficacy of anticonvulsants in the management of both acute and persistent neuropathic pain (Backonja, Glanzman, 2003; Backonja, Serra, 2004; Goodyear-Smith, Halliwell, 2009; Serpell, Neuropathic Pain Study Group, 2002; Wiffen, Collins, McQuay, et al., 2005; Wiffen, McQuay, Rees, et al., 2005; Wiffen, McQuay, Moore, 2005). They are listed as first-line agents in several evidence-based neuropathic pain treatment guidelines (Argoff, Backonja, Belgrade, et al., 2006; Dworkin, Backonja, Rowbotham, et al., 2003; Dworkin, O’Connor, Backonja, et al., 2007; Moulin, Clark, Gilron, et al., 2007). There is good evidence to support the clinical impression that anticonvulsants may be effective for all qualities of neuropathic pain, including neuropathic pain that does not have a dysesthetic component, neuropathic pain described as continuous dysesthesia (e.g., burning), and neuropathic pain that is lancinating, sharp, shooting, stabbing, or knifelike (Backonja, Glanzman, 2003; Krafft, 2008; Serpell, Neuropathic Pain Study Group, 2002; Wiffen, McQuay, Rees, et al., 2005; Wiffen, McQuay, Moore, 2005).
Anticonvulsants are discussed today in terms of the length of time they have been available (Gilron, 2006). The “older” or “first-generation” anticonvulsants include carbamazepine (Tegretol), phenytoin (Dilantin), clonazepam (Klonopin), divalproex sodium (Depakote), and valproic acid or valproate (Depacon, Depakene). Anticonvulsants that are referred to as “newer” or “second-generation” include the alpha-2-delta-1 modulators gabapentin (Neurontin) and pregabalin (Lyrica), lamotrigine (Lamictal), oxcarbazepine (Trileptal), tiagabine (Gabitril), topiramate (Topamax), zonisamide (Zonegran), lacosamide (Vimpat), and felbamate (Felbatol). One group of researchers commented that the quality of research, and hence of the evidence, tends to be higher with the newer anticonvulsants (Goodyear-Smith, Halliwell, 2009). With the exception of felbamate, which has the potential for serious bone marrow toxicity, the newer anticonvulsants generally have better safety profiles and now are prescribed as the first-line drugs for epilepsy and neuropathic pain.
“Newer” Second-Generation Anticonvulsants
Following is a discussion of several of the “newer” second-generation anticonvulsants. See Chapter 26 for their use in acute pain treatment.
Gabapentin
Gabapentin (Neurontin) has been demonstrated to be analgesic in many types of neuropathic pain, some other types of persistent pain, and acute perioperative pain (Knotkova, Pappagallo, 2007; Kong, Irwin, 2007; Seib, Paul, 2006; Tiippana, Hamunen, Kontinen, et al., 2007; Wiffen, McQuay, Rees, et al., 2005). (See Chapter 26 for perioperative use of gabapentin.) For example, a large randomized, placebo-controlled study of 305 patients with diverse neuropathic pain syndromes, including postherpetic neuralgia, complex regional pain syndrome (CRPS), central pain, and persistent postsurgical pain, found that gabapentin in doses up to 2400 mg were well-tolerated and improved pain intensity by 21% compared with 14% with placebo (Serpell, Neuropathic Pain Study Group, 2002). Improvements were noted in patient-reported quality of life and functional indicators as well.
Other studies confirmed the potential utility of gabapentin in specific syndromes. A large, multicenter 7-week study randomized 334 patients with postherpetic neuralgia to receive 1800 or 2400 mg of gabapentin or placebo daily in three divided doses (Rice, Maton, Postherpetic Neuralgia Study Group, 2001). Within 1 week, pain scores were reduced, with a final improvement difference from baseline pain of approximately 34.5% for the 1800 mg dose and 34.4% for the 2400 mg dose. The drug was well-tolerated, with the worst adverse effects being dizziness and sedation, which were especially bothersome during the titration phase. Additional studies in the population with postherpetic neuralgia observed similar results (Backonja, Glanzman, 2003).
Guidelines recommend gabapentin as a first-line (Dworkin, O’Connor, Backonja, et al., 2007) or second-line (Argoff, Backonja, Belgrade, et al., 2006) analgesic for treatment of painful diabetic neuropathy. A 12-week open-label pilot study of 25 patients with type-II diabetes and neuropathy demonstrated greater pain reduction, decreased paresthesia, and less frequent adverse effects with gabapentin than with amitriptyline (Elavil) (Dallocchio, Buffa, Mazzarello, et al., 2000). Other studies in this condition have confirmed these positive results (Backonja, Glanzman 2003).
A randomized controlled trial evaluated gabapentin for the treatment of fibromyalgia (Arnold, Goldenberg, Stanford, et al., 2007). Patients received either gabapentin (1200 to 2400 mg/day) or placebo for 12 weeks. Those who took gabapentin experienced significant improvements in quality of life and functional outcomes and a 51% reduction in pain compared with 31% in the placebo group.
Central neuropathic pain such as from stroke or spinal cord injury can be particularly difficult to treat. An 18-week trial randomized 20 patients with traumatic spinal cord injury to receive gabapentin or placebo during a 4-week titration period followed by a 4-week stable dosing period. After a 2-week washout period, patients were crossed over to the alternative treatment for 4 weeks of titration followed by a 4-week stable dosing period (Levendoglu, Ogun, Ozerbil, et al., 2004). During the period of treatment with the active drug, an effort was made to titrate the dose to 900 mg/day by the end of the first week, 1800 mg/day by the second week, 2400 mg/day by the third, and 3600 mg/day by the fourth. Patients received the maximum tolerated dose during the 4 weeks of stable dosing. All patients completed the study, and gabapentin was shown to be efficacious for neuropathic pain, including all types of neuropathic phenomena except sensations described as itchy, sensitive, dull, and cold. Quality of life was also improved.
A retrospective study of 38 patients with central pain found similar dramatic results during gabapentin treatment, with improvements in sharp, burning pain and numerous quality of life and functional indicators such as ability to sleep and participate in domestic activities (To, Lim, Hill, et al., 2002). Some patients in this study reported that life would be unbearable without gabapentin. The range of dosing was 900 to 4800 mg/day. Follow-up interviews with 21 patients with traumatic spinal cord injury who were treated with gabapentin found that 67% reported a favorable response at 6-month interview, and 91% of these continued to report effective pain relief at 36-month interview (Putzke, Richards, Kezar, et al., 2002).
Gabapentin is also effective as an adjuvant to opioid analgesia for neuropathic pain treatment. A randomized controlled trial of 57 patients with postherpetic neuralgia or diabetic neuropathy administered placebo, gabapentin alone, morphine alone, or gabapentin plus morphine (Gilron, Bailey, Tu, et al., 2005). Pain relief was best with the combination of morphine plus gabapentin, and the maximum tolerated doses of gabapentin and morphine were lower with the combination than for each drug alone. Similar results have been found when gabapentin is added to opioids for neuropathic cancer pain, particularly if the patient has allodynia and burning, shooting pain (Caraceni, Zecca, Martini, et al., 1999; Keskinbora, Pekel, Aydinli, 2007). Its effectiveness for these symptoms was supported in a systematic review of randomized controlled trials that concluded that the drug was particularly effective in relieving the neuropathic symptoms of allodynia, burning pain, shooting pain, and hyperesthesia (Backonja, Glanzman, 2003).
Not all studies in neuropathic pain have yielded positive results. In an 8-week 3-phase crossover trial (N = 38) that compared gabapentin, amitriptyline, and diphenhydramine (Benadryl) for spinal cord injury–related pain, gabapentin was no more effective than diphenhydramine in patients who had the highest baseline pain scores; amitriptyline was more effective than diphenhydramine (Rintala, Holmes, Courtade, et al., 2007) (see Chapter 31 for more on antihistamines).
Although gabapentin frequently is given to patients with chemotherapy-induced peripheral neuropathy (Mao, Chen, 2000a), few controlled trials have been conducted, and investigations have shown conflicting results. A phase III multicenter, placebo-controlled, randomized trial (N = 115) using gabapentin for this type of pain failed to show any significant benefits (Rao, Michalak, Sloan, et al., 2007). However, another study grouped 75 patients with chemotherapy-induced neuropathy into three categories according to the severity of their pain (mild, moderate, or severe) and administered all of them a fixed dose of 800 mg/day of gabapentin (Tsavaris, Kopterides, Kosmas, et al., 2008). Results in these groups were compared with a control group that received naproxen and codeine plus acetaminophen. Of those who received gabapentin, approximately 25% had complete relief, 44% had partial relief, 25% had minor relief, and 5% had no relief. Of those in the control group, none had complete relief, approximately 5% had partial relief, 46% had minor relief, and 49% had no relief.
Small trials in phantom pain produced generally favorable results. A 14-week, randomized controlled trial that included 19 patients with phantom limb pain noted that gabapentin (titrated to 2400 mg) reduced pain but had no significant effect on mood, sleep interference, or activities of daily living compared with placebo (Bone, Critchley, Buggy, 2002). A report on 7 children and young adults with phantom limb pain revealed that 6 of the 7 had resolution of their phantom limb pain within 2 months of gabapentin treatment (Rusy, Troshynski, Weisman, 2001).
Some types of back pain may be responsive to anticonvulsants such as gabapentin (Backonja, Glanzman, 2003). One guideline suggests that gabapentin may provide short-term benefit for painful radiculopathy, but also concludes that evidence is limited and all but lacking for other anticonvulsants (Chou, Qaseem, Snow, et al., 2007).
The foregoing describes a small proportion of the large number of clinical trials, case reports, and reviews that have addressed the analgesic potential of gabapentin. A broader review of this literature reveals publications on the following conditions:
• Postherpetic neuralgia (Backonja, Glanzman, 2003; Chou, Carson, Chan, 2009; Dubinsky, Kabbani, El-Chami, et al., 2004; Mao, Chen, 2000a; Rosenberg, Harrell, Ristic, et al., 1997; Rice, Maton, Postherpetic Neuralgia Study Group, 2001; Rosner, Rubin, Kestenbaum, 1996; Rowbotham, Harden, Stacey, et al., 1998; Serpell, Neuropathic Pain Study Group, 2002)
• Painful diabetic neuropathy (Argoff, Backonja, Belgrade, et al., 2006; Backonja, Beydoun, Edwards, et al., 1998; Backonja, Glanzman, 2003; Boulton, Vinik, Arezzo, et al., 2005; Chou, Carson, Chan, 2009; Dallocchio, Buffa, Mazzarello, et al., 2000; Duby, Campbell, Setter, et al., 2004; Gilron, Bailey, Tu, et al., 2005; Hemstreet, Lapointe, 2001; Jensen, Larson, 2001; Mao, Chen, 2000a; Serpell, Neuropathic Pain Study Group, 2002; Veves, Backonja, Malik, 2008)
• Fibromyalgia (Arnold, Goldenberg, Stanford, et al., 2007) (see also Hauser, Thieme, Turk, 2009)
• Neuropathic cancer pain (Caraceni, Zecca, Martini, et al., 1999; Keskinbora, Pekel, Aydinli, 2007)
• Chemotherapy-induced pain (Mao, Chen, 2000a; Tsavaris, Kopterides, Kosmas, et al., 2008)
• Central pain from spinal cord injury (To, Lim, Hill, et al., 2002; (Levendoglu, Ogun, Ozerbil, et al., 2004)
• Central poststroke pain (Frese, Husstedt, Ringelstein, et al., 2006; Kumar, Kalita, Kumar, et al., 2009; Serpell, Neuropathic Pain Study Group, 2002)
• Multiple sclerosis (Mao, Chen, 2000a)
• Phantom limb pain (Bone, Critchley, Buggy, 2002; Serpell, Neuropathic Pain Study Group, 2002)
• CRPS (Backonja, Glanzman, 2003; Mao, Chen, 2000a; Mellick, Mellick, 1995; Serpell, Neuropathic Pain Study Group, 2002)
• Radiculopathy (Serpell, Neuropathic Pain Study Group, 2002)
• HIV-related neuropathy (Rosner, Rubin, Kestenbaum, 1996; Hahn, Arendt, Braun, et al., 2004)
• Spinal stenosis (Yaksi, Ozgonenel, Ozgonenel, 2007)
• Atypical facial pain and trigeminal neuralgia (Mao, Chen, 2000a; Serpell, Neuropathic Pain Study Group, 2002)
• Cluster and migraine headache (Kaniecki, 2008; Mathew, 2001; Tay, Ngan Kee, Chung, 2001)
• Neuroma of peripheral nerve (Serpell, Neuropathic Pain Study Group, 2002)
• Persistent neuropathic postsurgical pain syndromes (e.g., postmastectomy, postthoractomy, post inguinal hernia, cholecystectomy) (Backonja, Glanzman, 2003; Mao, Chen, 2000a; Pandey, Patra, Pant, et al., 2006; Serpell, Neuropathic Pain Study Group, 2002; Tiippana, Hamunen, Kontinen, et al., 2007)
• Persistent back pain (Backonja, Glanzman, 2003; Chou, Qaeem, Snow, et al., 2007; Serpell, Neuropathic Pain Study Group, 2002)
• Persistent masticatory muscle pain (Kimos, Biggs, Mah, et al., 2007)
• Guillain-Barré syndrome (Mao, Chen, 2000a; Pandey, Bose, Garg, et al., 2002)
Pregabalin
Pregabalin (Lyrica) is a newer gabapentinoid and a precursor to gabapentin. It has a similar mechanism of action and many of the same pharmacologic properties, but has different pharmacokinetics and can exert a different profile of effects than gabapentin in the individual patient.
The oral bioavailability of gabapentin is 27% to 60%, depending on dose (Lacy, Armstrong, Goldman, et al., 2008), and 90% for pregabalin (Gajraj, 2007). Similar to gabapentin, pregabalin is not metabolized and is essentially unchanged with renal elimination. Neither drug has known drug-drug interactions.
The onset of pregabalin analgesia is approximately 25 minutes (Hill, Balkenohl, Thomas, et al., 2001), compared with 1 to 3 hours for gabapentin (Twycross, Wilcock, Charlesworth, et al., 2003). This faster onset of analgesic action may be clinically relevant in some cases (Blommel, Blommel, 2007). Equally important, pregabalin can be more rapidly titrated to the typical effective dose range than gabapentin. The time to effective dose for pregabalin may be as brief as 1 to 2 days (Gajraj, 2007; Portenoy, Murphy, Young, et al., 2006), compared to approximately 9 days for gabapentin (Gajraj, 2007). (See Chapter 26 for the perioperative use of pregabalin.)
Gabapentin is not a controlled substance, but pregabalin is designated a Schedule V drug. The Drug Enforcement Administration (DEA) reportedly designated pregabalin as a Schedule V drug because it produces some pharmacologic effects similar to diazepam (Valium) and alprazolam (Xanax); however, the data to support this conclusion are limited, and the effects are not sustained over time (Blommel, Blommel, 2007).
Pregabalin is approved in the United States for treatment of postherpetic neuralgia, painful diabetic neuropathy, and fibromyalgia. In the latter condition, studies have shown that the drug improves several core symptoms including pain, fatigue, and overall health and function (Arnold, Russell, Diri, et al., 2008; Crofford, Rowbotham, Mease, et al., 2005; Lyseng-Williamson, Siddiqui, 2008).
Although pregabalin may have effects on co-morbid anxiety, as suggested in a positive trial in patients with central pain from spinal cord injury (Murphy, Siddall, Griesing, 2007), a controlled trial in fibromyalgia patients demonstrated that anxiolysis was not necessary for pain reduction (Arnold, Crofford, Martin, et al., 2007). Studies show that pregabalin also can reduce pain-related sleep interference (Freynhagen, Grond, Schupfer, et al., 2007; Lesser, Sharma, LaMoreaux, et al., 2004; Sabatowski, Galvez, Cherry, et al., 2004; van Seventer, Feister, Young, 2006). Compared with alprazolam (Xanax) and placebo in healthy volunteers without pain, pregabalin produced improvements in features of disturbed sleep that have been reported in patients with fibromyalgia and anxiety disorders (Hindmarch, Dawson, Stanley, 2005).
Recent evidence-based guidelines indicate that pregabalin (or gabapentin) should be considered the first-line drug for the treatment of postherpetic neuralgia, painful diabetic neuropathy, and other neuropathic pains, unless a co-morbid depression suggests that an analgesic antidepressant should be tried first (Argoff, Backonja, Belgrade, et al., 2006; Dworkin, O’Connor, Backonja, et al., 2007) (see Chapter 22). This conclusion gains support from the consistent results observed in randomized controlled trials.
A 4-week randomized, placebo-controlled trial (N = 269) showed that pregabalin produced significant reductions in the spontaneous pain and allodynia caused by postherpetic neuralgia (Stacey, Barrett, Whalen, et al., 2008). An interesting finding was that improvements in pain and allodynia were correlated, which led the researchers to suggest allodynia could serve as an outcome measure in future research of this type. This trial compared fixed (300 mg/day) and flexible (150 to 600 mg/day) dosing regimens, and the latter was recommended as a way to reduce discontinuations, facilitate higher final doses, and improve ultimate pain relief. Flexible dosing of pregabalin has been recommended by others as well (Baron, Brunnmuller, Brasser, et al., 2007; Freynhagen, Grond, Schupfer, et al., 2007; Freynhagen, Strojek, Griesing, et al., 2005; Rowbotham, Stacey, Phillips, et al., 2007; Vranken, Dijkgraaf, Kruis, et al., 2008) (see later discussion of dosing recommendations).
In a recent meta-analysis of placebo-controlled trials in populations with painful diabetic neuropathy, pregabalin treatment yielded pain reduction; higher quality of life scores; and increased risk of dizziness, sedation, and edema (Hurley, Lesley, Adams, et al., 2008). Others have found similar results (Richter, Portenoy, Sharma, et al., 2005; Rosenstock, Tuchman, LaMoreaux, et al., 2004). A systematic review of research conducted between 1966 and 2005 concluded that pregabalin had a lower number-needed-to-treat (NNT = 3.24) for achieving greater than 50% analgesia in patients with painful diabetic neuropathy than any other anticonvulsants studied (Gutierrez-Alvarez, Beltran-Rodriguez, Moreno, 2007).
Positive findings also have been demonstrated in the treatment of central pain caused by spinal cord injury or stroke. A 4-week randomized, placebo-controlled trial of a flexible dosing regimen of pregabalin in 40 patients with central pain from brain or spinal cord injury demonstrated significant decreases in mean pain score and improvements in health status, but no difference in Pain Disability Index scores on follow-up evaluation compared with placebo (Vranken, Dijkgraaf, Kruis, et al., 2008).
The safety and effectiveness of pregabalin was evaluated in several open-label trials. A study of 55 patients with diverse types of refractory pain in which each patient’s physician prescribed pregabalin with or without other analgesics according to their own preferences observed a reduction in the mean pain score from 6.5 at baseline to 5.5 on day 14 and to 4.9 on day 28; associated improvements in quality of life, sleep, and functional outcomes; and no serious adverse effects (Freynhagen, Grond, Schupfer, et al., 2007). A review of the open-label extension phases following 7 placebo-controlled trials reported that pain levels remained constant without clinically meaningful dose variations over a 2-year follow-up period (Portenoy, Murphy, Young, et al., 2006). A 15-month open-label trial that incorporated flexible dosing of pregabalin demonstrated persistent positive drug effects in a subset of patients (N = 81) with postherpetic neuralgia or painful diabetic neuropathy refractory to other adjuvant analgesics (e.g., gabapentin and antidepressants) (Stacey, Dworkin, Murphy, et al., 2008). Almost half of the patients had a greater than 30% reduction in pain, and the prevalence of severe pain declined from 63% on admission to the trial to only 23% after 15 months of pregabalin treatment; when pregabalin was stopped during the study drug holidays, pain rapidly returned. Because patients were allowed to continue to take their other analgesics during this study, pregabalin was seen as an add-on therapy, and the researchers cautioned that the results should be interpreted with this in mind.
The prior studies are representative of a larger literature documenting the clinical trials, case reports, and reviews that have evaluated the analgesic potential of pregabalin. Recent studies also suggest that the drug may have utility in several nonpainful conditions (Ehrchen, Stander, 2008; Porzio, Aielli, Verna, et al., 2006) and restless leg syndrome pain (Sommer, Bachmann, Liebetanz, et al., 2007). A broad review of the literature on pregabalin reveals publications on the following conditions:
• Painful diabetic neuropathy (Argoff, Backonja, Belgrade, et al., 2006; Baron, Brunnmuller, Brasser, et al., 2007; Boulton, Vinik, Arezzo, et al., 2005; Frampton, Scott, 2004; Frank, Cousins, 2008; Freeman, Durso-Decruz, Emir, 2008; Freynhagen, Strojek, Griesing, et al., 2005; Freynhagen, Grond, Schupfer, et al., 2007; Richter, Portenoy, Sharma, et al., 2005; Rosenstock, Tuchman, LaMoreaux, et al., 2004; Tolle, Freynhagen, Versavel et al., 2008; Veves, Backonja, Malik, 2008)
• Fibromyalgia (Arnold, Crofford, Martin, et al., 2007; Arnold, Russell, Diri, et al., 2008; Crofford, Rowbotham, Mease, et al., 2005) (see also Hauser, Thieme, Turk, 2009.)
• Postherpetic neuralgia (Baron, Brunnmuller, Brasser, et al., 2007; Dubinsky, Kabbani, El-Chami, et al., 2004; Dworkin, Corbin, Young, et al., 2003; Frampton, Foster, 2005; Freynhagen, Strojek, Griesing, et al., 2005; Rowbotham, Stacey, Phillips, et al., 2007; Stacey, Barrett, Whalen, et al., 2008; van Seventer, Feister, Young, et al., 2006)
• Central pain from brain or spinal cord injury (Siddall, Cousins, Otte, et al., 2006; Vranken, Dijkgraaf, Kruis, et al., 2008)
• Trigeminal neuralgia (Obermann, Yoon, Sensen, et al., 2008)
• Glossopharyngeal neuralgia (Guido, Specchio, 2006)
• Restless leg syndrome with or without neuropathic pain (Sommer, Bachmann, Liebetanz, et al., 2007).
Lamotrigine
A recent meta-analysis of clinical trials evaluating lamotrigine (Lamictal) for acute and persistent pain showed limited efficacy for neuropathic pain states and no studies on its use for acute pain (Wiffen, Rees, 2007). In practice, the drug may be tried in those with persistent neuropathic pain that has not responded to the gabapentinoids and one or more of the analgesic antidepressants. The limited data supporting the potential for analgesic efficacy includes randomized trials for trigeminal neuralgia (Zakrzewska, Chaudhry, Nurmikko, et al., 1997), HIV painful neuropathy (Simpson, Olney, McArthur, et al., 2000), and central poststroke pain (Frese, Husstedt, Ringelstein, et al., 2006; Kumar, Kalita, Kumar, et al., 2009; Vestergaard, Andersen, Gottrup, et al., 2001). Open-label trials suggest analgesic effects in trigeminal neuralgia (Canavero, Bonicalzi, 1997; Rosen, 2001), sciatic pain (Eisenberg, Damunni, Hoffer, et al., 2003), and pain associated with multiple sclerosis (Cianchetti, Zuddas, Randazzo, et al., 1999). In contrast, studies in populations with painful diabetic neuropathy have yielded conflicting results (Duby, Campbell, Setter, et al., 2004; Eisenberg, Alon, Ishay, et al., 1998; Jose, Bhansali, Hota, et al., 2007; Vinik, Tuchman, Safirstein, et al., 2007), and a controlled study that evaluated the addition of lamotrigine (up to 400 mg/day) to either a nonopioid analgesic, gabapentin, or a tricyclic antidepressant (TCA) for a variety of different types of neuropathic pains did not reveal an analgesic response (Silver, Blum, Grainger, et al., 2007).
Lamotrigine carries a relatively high risk of rash—up to 7% of patients in some studies—and serious cutaneous hypersensitivity (e.g., Stevens-Johnson syndrome or toxic epidermal necrolysis). This adverse effect occurs far more frequently with lamotrigine than during treatment with other adjuvant analgesics. The latter risk is increased in younger patients, and the drug should not be used in patients younger than 15 years old. Although the risk of serious rash in adults is low overall (less than 1%), the availability of other agents (e.g., antidepressants and other anticonvulsants) with less of this risk suggests that lamotrigine may best be relegated to a trial only after several other drugs have failed to provide benefit. If a lamotrigine trial is undertaken, dosing should follow the manufacturer’s recommendation for gradual titration from a low dose, which has been determined to reduce the risk of cutaneous toxicity.
Oxcarbazepine
Another relatively new anticonvulsant is oxcarbazepine (Trileptal), which structurally is a metabolite of carbamazepine (Tegretol). It is approved in the United States for the treatment of trigeminal neuralgia and has been described as the drug of choice for this condition (Carrazana, Mikoshiba, 2003; Jensen, 2002). Simple dose titration and dose adjustments and convenient twice-daily dosing are cited among the advantages of this drug. A meta-analysis supports effectiveness comparable to carbamazepine but with fewer adverse effects; 62% reported good to excellent tolerability with oxcarbazepine compared with 48% who took carbamazepine (Beydoun, Schmidt, D’Souza, et al., 2002). Open-label studies support these findings as well (Carrazana, Mikoshiba, 2003).
In contrast to the studies in trigeminal neuralgia, studies in diabetic painful neuropathy have yielded equivocal results. A 16-week randomized, placebo-controlled study (N = 146) titrated patients with painful diabetic neuropathy to a maximum dose of 1800 mg/day and reported a larger decrease in average visual analog scale (VAS) score (–24.3) compared with placebo (–14.7) (Dogra, Beydoun, Mazzola, et al., 2005). More patients had a greater than 50% reduction in pain with oxcarbazepine (35.2%) than with placebo (18.4%), and there were fewer arousals from sleep because of pain in those who took the active drug. Another large placebo-controlled trial (N = 347) evaluating the drug for painful diabetic neuropathy found a trend toward meaningful changes in pain scores from baseline to the last week of the study with oxcarbazepine 1200 mg/day and 1800 mg/day compared with placebo, but the changes did not reach statistical significance (Beydoun, Shaibani, Hopwood, et al., 2006). A 9-week open-label trial in 30 patients with painful diabetic neuropathy found that oxcarbazepine (highest dose of 1200 mg/day) produced significant improvements in pain relief, with a decrease in mean VAS score from 66.3 at baseline to 34.3 at the end of the trial (Beydoun, Kobetz, Carrazana, 2004). The most common adverse effects were sedation and dizziness, similar to other anticonvulsants.
The efficacy of oxcarbazepine has been studied in other conditions as well. A randomized controlled trial of 32 patients with colon cancer undergoing chemotherapy found a dramatically lower incidence of chemotherapy-induced neuropathy when oxcarbazepine was administered prior to chemotherapy (31.2%) compared with chemotherapy without oxcarbazepine (75%) (Argyriou, Chroni, Polychronopoulos, 2006). In contrast, a controlled study of patients with frequent migraine was negative, demonstrating no reduction in the number of attacks over 28 days of prophylactic treatment (Silberstein, Saper, Berenson, et al., 2008). Efficacy in postherpetic neuralgia and CRPS has been suggested in published case reports (Criscuolo, Auletta, Lippi, et al., 2004; Lalwani, Shoham, Koh, et al., 2005). Further research and clinical experience with this drug are needed to better evaluate its safety and what types of pain will benefit most (Guay, 2003).
Topiramate
Topiramate (Topamax) is approved in the United States for migraine prevention. A multicenter, double-blind study randomized 487 people with persistent migraine to receive placebo, or 50, 100, or 200 mg of topiramate daily for 26 weeks to determine efficacy and optimal dose (Silberstein, Neto, Schmitt et al., 2004). The percent of individuals who experienced 50% or greater reduction in monthly migraine were: 23% (placebo), 36% (50 mg), 54% (100 mg), and 52% (200 mg) (see dosing guidelines later in this chapter). Topiramate treatment also resulted in a reduction in the use of acute headache treatment medications in this study. The most frequent adverse effects were paresthesia, fatigue, nausea, anorexia, and abnormal taste. Weight loss, a common effect of topiramate, was experienced in those taking 100 and 200 mg. Other randomized controlled trials similarly support 100 mg/day as the most efficacious and best-tolerated dose of topiramate for migraine prevention (Brandes, Saper, Diamond, et al., 2004; Silberstein, 2005; Silberstein, Diener, Lipton, et al., 2008; Silberstein, Lipton, Dodick, et al., 2007).
Topiramate also has been shown to be effective for persistent cluster headache (Lainez, Pascual, Pascual, et al., 2003), idiopathic trigeminal neuralgia (Solaro, Uccelli, Brichetto, et al., 2001), and trigeminal autonomic cephalalgias, which is a grouping of headache syndromes that include paroxysmal hemicrania and short-lasting unilateral neuralgiform headache with conjunctival injection and tearing (SUNCT) syndrome in addition to cluster headache (Cohen, Matharu, Goadsby, 2007; May, Leone, Afra, et al., 2006).
Topiramate has been studied for other types of persistent pain, with equivocal outcomes. A 10-week randomized placebo-controlled trial (N = 96) demonstrated that topiramate titrated to a maximum dose of 300 mg/day was safe and significantly improved pain, quality of life, and functional outcomes in a group of patients with persistent low back pain (Muehlbacher, Nickel, Kettler, et al., 2006). Again, weight loss was notable in this study, which may be a significant benefit in some patients with persistent pain who are also overweight. A small pilot study of 4 patients with phantom limb pain showed that 3 of the 4 experienced significant decreases in pain with topiramate (Harden, Houle, Remble, et al., 2005); the peak effect was noted at 800 mg/day, but such high doses are not recommended (United States Food and Drug Administration [U.S. FDA], 2004). Topiramate has not been shown to be effective for painful diabetic neuropathy (Jensen, 2002), and guidelines list it as a drug with limited evidence for treatment of this condition (Argoff, Backonja, Belgrade, et al., 2006).
Metabolic acidosis caused by renal wasting of bicarbonate has been linked to the use of topiramate. Mild to moderate decreases in serum bicarbonate are most likely at doses of 400 mg/day and usually occur early in treatment; however, these effects have also been noted in doses as low as 50 mg/day (U.S. FDA, 2004). These effects are of particular concern when the drug is used on a long-term basis (Welch, Graybeal, Moe, et al., 2006). Nephrolithiasis also has been noted.
Other Second-Generation Anticonvulsants
Very few studies have evaluated the analgesic potential of zonisamide (Zonegran). A study that randomized 25 patients with painful diabetic neuropathy to receive placebo or zonisamide found that a mean dose of 540 mg/day over 6 weeks was associated with a nonsignificant improvement in pain (Atli, Dogra, 2005). A larger sample size would have likely demonstrated analgesic efficacy in this trial, but the dropout rate was high due to a variety of adverse effects (e.g., irritability, insomnia, metallic taste, and rash) in those taking zonisamide. Given the limited data, this drug is rarely tried for patients with neuropathic pain or migraine (Kaniecki, 2008), and further research is needed to evaluate its efficacy and safety for these conditions (Duby, Campbell, Setter, et al., 2004; Guay, 2003).
Lacosamide (Vimpat) was recently approved in the United States for the indication of seizures. There are data suggesting analgesic efficacy in painful diabetic neuropathy. A randomized controlled trial (N = 119) found lacosamide titrated to 100 to 400 mg/day or maximum tolerated dose produced significantly better pain relief and improvements in quality of life compared with placebo in patients with moderate to severe intensity painful diabetic neuropathy (Rauck, Shaibani, Bilton, et al., 2007). An open-label trial (N = 69) demonstrated both short-term and long-term (2.5 years) safety and efficacy at maximum titrated doses of 400 mg/day in patients with painful diabetic neuropathy (Shaibani, Biton, Rauck, et al., 2009). Other studies have produced similar findings (Hidvegi, Bretschneider, Thierfelder, et al., 2008; Kenney, Simpson, Koch, et al., 2006; Shaibani, Fares, Selam, et al., 2009). Long-term safety and efficacy was established for 400 mg/day of lacosamide for 18 weeks in a double-blind, randomized, placebo-controlled trial of 370 patients with painful diabetic neuropathy (Wymer, Simpson, Sen, et al., 2009). The lower dose of 200 mg/day reduced pain but failed to significantly dissociate from placebo for primary and secondary outcome measures.
No substantial metabolism, low or no inhibition of cytochrome P450 isoenzymes, and low protein binding contribute to a low incidence of drug-drug interactions with lacosamide (Kropeit, Scharfenecker, Schiltmeyer, et al., 2006). There is no interaction between lacosamide and the oral antidiabetic drug metformin, which is an advantage in patients who take that drug for diabetes and also have painful diabetic neuropathy (Schiltmeyer, Kropeit, Cawello, et al., 2006).
Tiagabine (Gabitril) is used for treatment of a wide variety of conditions including epilepsy and other seizure disorders, depression, anxiety, posttraumatic stress syndrome, and substance withdrawal symptoms. There has been little investigation of its analgesic potential. A 3-month, open-label study comparing tiagabine and gabapentin in 91 patients with persistent pain, such as back pain, musculoskeletal headache, and fibromyalgia, found that both drugs significantly reduced pain, but tiagabine showed greater improvements in sleep quality (Todorov, Kolchev, Todorov, 2005). The drug has been linked to new-onset seizures, which have occurred at doses as low as 4 mg/day and usually occur after dose increases; this adverse effect led the United States Food and Drug Administration (2005b) to recommend discontinuation of the drug in nonepileptic patients who develop seizures.
Felbamate (Felbatol) has been used anecdotally to treat hemifacial spasm (Mellick, 1995), a painless syndrome characterized by paroxysmal contraction of facial muscles. Although this observation and treatment of a small number of patients initially raised expectations, no follow-up studies have been done in populations with pain. The potential for lethal aplastic anemia from this drug has tempered enthusiasm for these trials (see adverse effects).
First-Generation Anticonvulsants
The older anticonvulsants have been used as analgesics for several decades. Although the newer drugs are now preferred, patients with refractory pain may be offered trials of these drugs.
Carbamazepine
Carbamazepine (Tegretol) has been reported to be effective for many pain syndromes, including diabetic neuropathy, postherpetic neuralgia, poststroke pain, and CRPS (Dobecki, Schocket, Wallace, 2006; Harke, Gretenkort, Ladleif, et al., 2001; Vurdelja, Budincevic, Prvan, 2008; Zin, Nissen, Smith, et al., 2008). It is approved for treatment of trigeminal neuralgia, and based on years of positive experience in this syndrome, often is selected as a first-line drug (Jensen, 2002; Jensen, Finnerup, 2007; Krafft, 2008; Sindrup, Jensen, 2002) (see also oxcarbazepine). Carbamazepine also has been used to treat cancer-related pruritus (Korfitis, Trafalis, 2008). A systematic review of controlled trials concluded that carbamazepine has analgesic properties for varied acute and persistent pains and can treat lancinating neuropathic pain, regardless of the specific pathologic condition contributing to the presence of this pain characteristic (Wiffen, McQuay, Moore, 2005). Although the numbers of patients in these clinical trials were small, clinicians wanting to use this agent as a second-line anticonvulsant can access important information as to how effective it is with certain types of pain from this review.
Clonazepam
Clonazepam (Klonopin) is a benzodiazepine and has been used primarily as an anticonvulsant. It has been suggested to be analgesic in the lancinating pain associated with phantom limb pain, neuropathic cancer-related pain, and myofascial pain (Bartusch, Sanders, D’Alessio, et al., 1996; Hugel, Ellershaw, Dickman, 2003; Fishbain, Cutler, Rosomoff, et al., 2000). The supporting evidence is very limited, however, and its use may be more related to its potential to help co-morbid anxiety than to its established analgesic efficacy. Given its long half-life, the potential for accumulation with repeated dosing must be recognized, and it must be used very cautiously in patients predisposed to adverse effects, including the cognitively impaired (particularly older adults) and patients with sleep apnea syndrome or advanced cardiopulmonary disease. (See clonazepam patient medication information, Form V-3 on pp. 763-764, at the end of Section V.)
Divalproex Sodium and Valproic Acid
Divalproex sodium (Depakote) is approved and widely used for migraine prevention (Freitag, Diamond, Diamond, et al., 2001; Silberstein, Collins, 1999). The drug is closely related to valproic acid and shares the same pharmacology, but differs in that it is available in gastro-resistant sprinkles and modified-release formulation. In a 12-month, open-label study of 241 adolescents with migraines who were given a titrated dose of modified-release divalproex to a maximum dose of 1000 mg/day, the median number of migraine attacks decreased 75% between the first and fourth month of the study and remained at or below this level for the remainder of the study (Apostol, Lewis, Laforet et al., 2009). The most common adverse effects were nausea, vomiting, and weight gain; 17% discontinued therapy because of an adverse event.
Divalproex sodium has not been studied in neuropathic pain, and the evidence for valproic acid is mixed. Whereas one study showed improvements in painful diabetic neuropathy (Kochar, Rawat, Agrawal, et al., 2004), another found no benefit in pain from polyneuropathy (Otto, Bach, Jensen, et al., 2004). Based on the limited data (Chong, Hester, 2007), divalproex sodium is rarely tried for neuropathic pain, and the IV formulation of valproic acid (Depacon) is only occasionally used in an effort to quickly reverse severe pain, typically in an inpatient setting.
Phenytoin
Similar to carbamazepine, phenytoin (Dilantin) has been used as an analgesic for decades, but has been largely supplanted by newer anticonvulsants (Jensen, 2002; Vanotti, Osio, Mailland, et al., 2007). In early controlled trials, phenytoin was shown to be an effective analgesic for neuropathic pain, particularly pains characterized by a prominent lancinating component (McCleane, 1999). Isolated case reports illustrate its use, and one in particular that shows it has benefits for treating crescendo pelvic cancer–related pain with lancinating quality (Chang, 1997). IV phenytoin, like IV valproate, is sometimes suggested as an option for treatment of acute attacks of neuropathic pain (Jensen, 2002; McCleane, 1999). An extensive review (Duby, Campbell, Setter, et al., 2004) and guidelines (Argoff, Backonja, Belgrade, et al., 2006) list phenytoin as a drug with limited evidence for treatment of painful diabetic neuropathy.
Summary
At present, gabapentin and pregabalin share with selected antidepressants first-line status for the treatment of neuropathic pain. Numerous other anticonvulsants have some evidence of efficacy, but in all cases, this evidence is limited or conflicting. Nonetheless, patients with neuropathic pain that has not responded to trials of the gabapentinoids and appropriate antidepressants should be considered candidates for trials of those anticonvulsants with some supporting data. Sequential trials are appropriate for patients with severe and intractable pain. Each anticonvulsant has a unique pharmacodynamic and pharmacokinetic profile (LaRoche, 2007), and their use as analgesics largely mirrors the prescribing guidelines employed for their primary indication.
Mechanism of Action
The specific mechanisms of the analgesia produced by anticonvulsant drugs are not known, but presumably relate to those actions underlying anticonvulsant effects (Lussier, Portenoy, 2004). Most of the anticonvulsants have multiple mechanisms of action (Gilron, 2006), and these may differ among the various anticonvulsants (Lussier, Portenoy, 2004).
One of the mechanisms is blockade of presynaptic voltage-gated ion channels, which prevents the generation of spontaneous ectopic discharges (Beydoun, Backonja, 2003; Taylor, 2009) (see Section I and Figure I-2, B on pp. 4-5). Some anticonvulsants (e.g., carbamazepine, felbamate, lamotrigine, oxcarbazepine, phenytoin, topiramate, and zonisamide) relieve pain, in part, by prolonging the recovery phase of sodium channels after their activation (Gilron, 2006; Soderpalm, 2002). Some (e.g., carbamazepine, felbamate, gabapentin, lamotrigine, pregabalin, valproic acid, and zonisamide) bind to presynaptic voltage-gated calcium channels and inhibit calcium influx and the release of excitatory neurotransmitters from primary afferent nerve fibers (Dickenson, Matthews, Suzuki, 2002; Gajraj, 2007; Gilron, 2006; Taylor, 2009; Tiippana, Hamunen, Kontinen, et al., 2007). Pregabalin has an even higher calcium-channel affinity than gabapentin and has no effect on sodium channels (Gilron, Watson, Cahill, et al., 2006; Mao, Chen, 2000a; Nicholson, 2000). Recent research confirmed that gabapentin had no effect on transient sodium currents but inhibited persistent sodium currents in a dose-dependent way (Yang, Wang, Chen, et al., 2009). The clinical significance of these findings is unknown.
Other mechanisms also may be important. Inhibition of glutamate, an excitatory neurotransmitter that promotes the transmission of pain through increased activity at the NMDA receptor and other receptors, may be relevant for some drugs (Jensen, 2002; Soderpalm, 2002) (see Section I and Figure I-2, B on pp. 4-5). Others may indirectly or directly augment inhibitory GABAergic neurotransmission (Gilron, 2006; Soderpalm, 2002). For example, gabapentin is a GABA analogue, and although it does not bind to GABA receptors or modulate GABA reuptake, it does enhance overall GABA-mediated inhibitory tone (Dickenson, Matthews, Suzuki, 2002; Jensen, 2002; Mao, Chen, 2000). Gabapentin and pregabalin share a specific high affinity drug binding site (calcium channel α2-δ ligands) localized at synapses and sufficient in amount to account for their analgesic action (Taylor, 2009).
Interestingly, there is evidence that some of the anticonvulsants have peripheral effects that may be involved in pain. For example, carbamazepine and phenytoin produce antiinflammatory effects, and local injection of gabapentin and lamotrigine in animals exerts an analgesic effect (Gilron, 2006).
The central effects of anticonvulsants have been evaluated in experimental studies. Animal and human research has shown that gabapentin selectively reduces nociception in a sensitized (damaged) nervous system but not in a normal nervous system (Gilron, 2002). Healthy volunteers in two separate randomized, placebo-controlled studies were subjected to experimental thermal injury (acute pain model [nociception]) (Werner, Perkins, Holte, et al., 2001) and heat-capsaicin sensitization (central hypersensitivity model [neuropathic]) (Dirks, Petersen, Rowbotham, et al., 2002). Gabapentin was found to have no effect on pain transmission on normal skin but significantly reduced hyperalgesia. Gilron (2002) pointed out that the clinical value of these findings is that gabapentin may reduce pathologic (neuropathic) pain while leaving other protective nociceptive mechanisms intact.
Adverse Effects
Anticonvulsants usually are well-tolerated (Collins, Moore, McQuay, et al., 2000). The most common adverse effects are dizziness and sedation. These are usually transient and most notable during the titration phase of treatment (Serpell, Neuropathic Pain Study Group, 2002). GI upset (e.g., nausea) can occur but usually decreases with time. Occasionally, patients report changes in mood, such as dysphoria or cognitive impairment, which may be subtle.
There is a long history of concern about the association between anticonvulsant use and loss of bone mineral density (Sheth, 2005), particularly during treatment with enzyme-inducing anticonvulsants, such as carbamazepine and phenytoin (Petty, Paton, O’Brien, et al., 2005). Risk factors for this adverse effect are ages 40 years and older and anticonvulsant use for more than 2 years. Gabapentin, pregabalin, lamotrigine, and topiramate are examples of non–enzyme-inducing anticonvulsants (Novy, Stupp, Rossetti, 2009). Valproate is a non–enzyme-inducing anticonvulsant but it is associated with reduced bone mineralization (Novy, Stupp, Rossetti, 2009).
Although there have been case reports of hepatotoxicity with gabapentin (Richardson, Williams, Kingham, 2002), they are rare. Neither gabapentin nor pregabalin undergo hepatic metabolism, a positive effect of which is a minimal risk of drug-drug interactions (Frank, Cousins, 2008). The new anticonvulsant, lacosamide, produces low or no inhibition of cytochrome P450 isoenzymes and also may have a reduced risk of interactions (Kropeit, Scharfenecker, Schiltmeyer, et al., 2006). Other anticonvulsants inhibit various isoenzymes of the cytochrome P450 enzyme system, which can result in drug-drug interactions (Virani, Mailis, Shapiro, et al., 1997) (see Chapter 11, for more on the cytochrome P450 enzyme system). Each anticonvulsant has a unique profile (LaRoche, 2007), and it is important for the clinician to become familiar with the potential for drug-drug interactions associated with the particular anticonvulsant being administered. Patients must be told to report adverse effects promptly, and in some cases serum drug concentrations should be closely monitored to prevent toxicity, such as in patients who take multiple other medications and those who take anticonvulsants during chemotherapy (Yap, Chui, Chan, 2008). Following is a more detailed discussion of adverse effects associated with selected anticonvulsants.
Gabapentin
Gabapentin has a relatively low adverse effect profile, and in clinical practice dizziness and sedation seem to be the most dose-limiting adverse effects. A meta-analysis of 36 randomized, placebo-controlled studies of several of the second-generation anticonvulsants used for seizure control confirmed that gabapentin was significantly associated with sedation and dizziness; however, comparisons between drugs were not possible (Zaccara, Gangemi, Cincotta, 2008). Gabapentin is reported to produce more sedation but less dry mouth than amitriptyline (Mao, Chen, 2000).
Confusion and weight gain are other less common effects of gabapentin (Mao, Chen, 2000). Ataxia and other movement disorders have been reported and appear to cease when the drug is discontinued (Buetefisch, Guiterrez, Gutmann, et al., 1996; Reeves, So, Sharbrough, et al., 1996). The most commonly reported adverse gabapentin effects in surgical patients are nausea, sedation, dizziness, and urinary retention (Tiippana, Hamunen, Kontinen, et al., 2007).
As mentioned, a major benefit of gabapentin is that it is not hepatically metabolized. This results in minimal drug-drug interactions and hepatic adverse effects. The drug is excreted entirely by the renal system. Gabapentin toxicity was reported in a case describing its use during an episode of acute renal failure (Miller, Price, 2009). The importance of recognizing the need to adjust the dose downward during acute illness, particularly when there is a decline in renal clearance, must be recognized. (See gabapentin patient medication information, Form V-7 on pp. 771-772, at the end of Section V.)
Pregabalin
Pregabalin also has a low adverse effect profile and is well tolerated (Gajraj, 2007). As with gabapentin, dizziness and somnolence are common, and these are typically dose related, transient, and mild to moderate in severity (Gajraj, 2007; Arnold, Russell, Diri, et al., 2008; Lyseng-Williamson, Siddiqui, 2008). Pregabalin has a documented low risk for blurred vision. Patients should be told to report this to their health care provider should it occur. Weight gain also has been noted.
Also, like gabapentin, pregabalin does not undergo hepatic metabolism and has no reported drug interactions of concern (Frank, Cousins, 2008). Additive pharmacodynamic effects of the type that may occur whenever two or more centrally-acting drugs are taken also occurs during pregabalin dosing; patients may report effects on cognitive and gross-motor function when the drug is co-administered with other drugs or alcohol (Blommel, Blommel, 2007). Being a relatively new drug, evaluation of the impact of its adverse effects in older patients requires further research and clinical experience (Guay, 2005). (See pregabalin patient medication information, Form V-11 on pp. 779-780, at the end of Section V.)
Oxcarbazepine
Oxcarbazepine has a better adverse effect profile and is better tolerated than the older anticonvulsants, such as carbamazepine (Jensen, 2002). Rare cases of anaphylactic reactions and angioedema resulted in postmarketing labeling changes for oxcarbazepine (U.S. FDA, 2007b). The drug should be discontinued permanently should these occur. Potentially serious skin reactions, including Stevens Johnson syndrome and toxic epidermal necrolysis, have been reported with the use of oxcarbazepine and also require prompt discontinuation of the drug (Lacy, Armstrong, Goldman, et al., 2008). Hyponatremia should be recognized as a rare adverse effect of this drug, and other effects, such as edema, diplopia, abnormal gait, cognitive slowing, and speech difficulties, have been noted (Guay, 2005; Lacy, Armstrong, Goldman, et al., 2008).
Topiramate
Topiramate is generally well tolerated. Dose-related paresthesia is the most common adverse effect (Adleman, Freitag, Lainez, et al., 2008). Fatigue, nausea, and difficulty concentrating are usually mild to moderate in severity, if they occur. Serious adverse effects are rare (2%) and include abdominal pain, vomiting, dehydration, anorexia, venous thrombosis, and renal calculi. Language disturbances (i.e., difficulty finding the right word) also have been associated with the drug (Coppola, Rossi, Mancini, et al., 2008). Weight loss has been observed during treatment with this drug; it is generally modest and yields clinically noticeable effects in a minority of patients (Adleman, Freitag, Lainez, et al., 2008).
Other Second-Generation Anticonvulsants
Other second-generation anticonvulsant drugs also are usually well tolerated but present a range of potential toxicities (Walia, Khan, Ko, et al., 2004). As noted, all of these drugs can produce the spectrum of adverse effects common to centrally-acting agents, including dizziness, cognitive slowing, mood change, and related experiences. Other adverse effects are more specific to one or another of the drugs. Felbamate, for example, has been associated with rare fatal aplastic anemia and liver failure, which has limited its use to patients with refractory epilepsy. It is generally not considered for neuropathic pain. Lamotrigine has been associated with a relatively high incidence of serious cutaneous hypersensitivity, both Stevens Johnson syndrome and toxic epidermal necrolysis (Lacy, Armstrong, Goldman, et al., 2008). The dropout rate in a clinical trial of zonisamide was high due to a variety of causes including irritability, insomnia, metallic taste, and rash (Atli, Dogra, 2005).
Carbamazepine
Carbamazepine commonly causes sedation, dizziness, nausea, and unsteadiness. These effects can be minimized by low initial doses and gradual dose titration. The intensity diminishes in most patients maintained on the drug for several weeks. Of much greater concern, carbamazepine may cause aplastic anemia, agranulocytosis, or thrombocytopenia in a very small percentage of patients (Hart, Easton, 1982). Therefore, it is critical to obtain a complete hematologic profile prior to therapy, after several weeks, and every 3 to 4 months thereafter. A leukocyte count less than 4000 is usually considered to be a contraindication to treatment, and a decline to less than 3000 (or an absolute neutrophil count of less than 1500) during therapy should lead to discontinuation of the drug. Patients need to be told to report any signs or symptoms of infection, easy bruising, or fatigue.
Other extremely rare adverse effects of carbamazepine include hepatic damage and hyponatremia caused by inappropriate secretion of antidiuretic hormone (Van Amelsvoort, Bakshi, Devaux, et al., 1994). Liver and renal function tests should be routinely evaluated at the start and during the course of therapy. Treatment with carbamazepine may be complicated in older patients who are at risk for cardiac disease, water retention, decreased osmolality, and hyponatremia (Jensen, 2002). Balancing adverse effects with optimal effects in this population can be challenging. Careful monitoring during therapy is recommended.
Clonazepam
Drowsiness is the most common and troubling adverse effect of clonazepam. Tolerance to the effect often develops within weeks after dosing is begun. Occasional patients develop ataxia, particularly at higher doses. Idiosyncratic reactions, including dermatitis, hepatotoxicity, and hematologic effects, appear to be very rare. Like other benzodiazepine drugs, a withdrawal syndrome may occur with abrupt discontinuation of relatively high doses, so tapering gradually for 2 weeks or more is recommended.
Phenytoin
Most of the common adverse effects of phenytoin are dose dependent and usually occur at plasma concentrations greater than the therapeutic range for seizure control. These include sedation or mental clouding, dizziness, unsteadiness, and diplopia. Occasional patients experience toxicity with lower concentrations of the drug in the blood. Of the idiosyncratic effects, the most serious are hepatotoxicity and exfoliative dermatitis. The occurrence of a maculopapular rash, which can be the harbinger of the more severe cutaneous reactions, should lead to discontinuation of the drug.
Valproic Acid
At therapeutic doses, the adverse effects of valproate are usually mild, and include sedation, nausea, tremor, and sometimes increased appetite. An enteric-coated tablet minimizes GI disturbances, and dose-dependent adverse effects are reduced by the use of low initial doses and gradual upward dose titration. Hepatotoxicity, encephalopathy, dermatitis, alopecia, and a rare hyperammonemia syndrome are among the reported idiosyncratic reactions. Because the idiosyncratic hyperammonemia syndrome can occur without abnormalities in other liver function tests, the occurrence of confusion during therapy should be evaluated with both liver function tests and serum ammonia level.
Indications
All types of neuropathic pain are generally considered the primary indication for trying an anticonvulsant drug. Neuropathic pain guidelines recommend gabapentin and pregabalin as first-line choices (Dubinsky, Kabbani, El-Chami, et al., 2004; Dworkin, O’Connor, Backonja, et al., 2007; Moulin, Clark, Gilron, et al., 2007).
Drug Selection
For patients with neuropathic pain, the widely accepted first-line drugs are either the anticonvulsants gabapentin or pregabalin, or antidepressants in the tricyclic antidepressant (TCA) or serotonin norepinephrine reuptake inhibitor (SNRI) classes (Dworkin, Backonja, Rowbotham, et al., 2003; Dworkin, O’Connor, Backonja, et al., 2007; Moulin, Clark, Gilron, et al., 2007). There are few comparative trials, and a recent meta-analysis could not find appreciable differences in the effectiveness of TCAs compared with the anticonvulsant gabapentin for the treatment of diabetic neuropathy and postherpetic neuralgia (Chou, Carson, Chan, 2009). A reasonable guideline would be to consider one of the gabapentinoids first, unless there is a concurrent indication for antidepressant therapy, such as depressed mood, anxiety, or insomnia. The variability in responses to anticonvulsant adjuvant analgesics is great, and sequential trials in patients with refractory pain are amply justified by clinical experience.
Various influences may bear on the selection of an anticonvulsant, particularly in the setting of neuropathic pain that has been poorly responsive to gabapentin and pregabalin and one or more of the analgesic antidepressants. These factors include the pain diagnosis, patient characteristics, prior response to a particular drug, issues with convenience and adherence to therapy, cost and availability of agents, and access to care and follow-up treatment.
Occasionally, a drug is selected based on the type of pain. This is most typical in the case of trigeminal neuralgia, for which treatment with carbamazepine or oxcarbazepine often is preferred, even before trials of the usual first-line drugs for neuropathic pain. Similarly, a trial of topiramate, valproate, or lamotrigine may be considered early if neuropathic pain is accompanied by frequent headache with migranous features.
Unique patient characteristics must be considered as well. For example, a trial of topiramate, which can lead to weight loss (Adleman, Freitag, Lainez, et al., 2008), may be preferred in the obese patient and discouraged in the very thin patient.
Dose Selection
Dosing guidelines used in the treatment of seizures are typically extrapolated for the management of pain. As with all of the adjuvant analgesics, doses of anticonvulsants must be tailored to meet the patient’s individual needs. Titration is almost always required. Dose escalation should continue until favorable effects occur, intolerable and unmanageable adverse effects supervene, or plasma drug concentration has reached some arbitrary level (customarily at the upper end of the therapeutic range for seizure control). Table V-1, pp. 748-756, at the end of Section V contains dosing recommendations for all of the commonly used anticonvulsants. Following is a discussion of dosing considerations for selected anticonvulsants.
Gabapentin
Although a comprehensive review of available data recommends treatment of persistent pain with a maintenance gabapentin dose of 900 mg/day (Backonja, Glanzman, 2003), clinical experience indicates that analgesic effectiveness often requires a higher dose, and some patients have good outcomes at lower amounts. A low starting dose gradually titrated upward is recommended, for example, starting with 300 mg/day for most patients and 100 mg/day for those with significant renal insufficiency advanced age, or serious medical co-morbidities. In most cases, the dose can be increased every 3 to 4 days, initially by an amount equal to the starting dose. The doses should be divided into twice-daily administration; some patients experience better outcomes with three divided doses and this should be explored if patients report adverse effects at the peak effect of a twice-daily dose. Dose titration usually continues with a target of 1800 mg/day to 3600 mg/day in two or three divided doses. The dose required to maintain analgesia varies widely, and one study reported good results with a dose range between 900 mg/day and 4800 mg/day (median dose 2400 mg/day) in patients with spinal cord injury (To, Lim, Hill, et al., 2002). Doses as high as 6000 mg/day have been taken for cancer pain (Farrar, Portenoy, 2001). Patients with renal insufficiency should be titrated to a lower level and observed more carefully. Table 23-2 provides recommendations for dosing adjustments in patients with renal impairment.
Guidelines
Table 23-2
Gabapentin and Pregabalin Dosing Adjustments in Renal Impairment
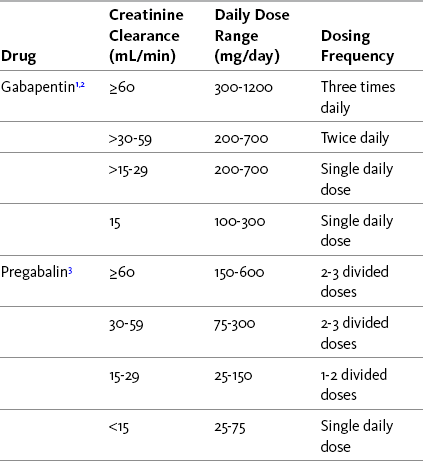
25 mg/day schedule: Single supplemental dose of 25 to 50 mg
25 to 50 mg/day schedule: Single supplemental dose of 50 to 75 mg
50 to 75 mg/day schedule: Single supplemental dose of 75 to 100 mg
75 mg/day schedule: Single supplemental dose of 100 to 150 mg
1Gabapentin posthemodialysis: Single supplemental dose of 125 to 300 mg administered after each 4 hours of hemodialysis.
2Reduce daily dose in proportion to creatinine clearance for creatinine clearance less than 15 mL/min.
3Pregabalin posthemodialysis:
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 664, St. Louis, Mosby. Data from Lacy, C. F., Armstrong, L. L., Goldman, M. P., et al (Eds.). (2008). Drug information handbook, ed. 17, Hudson, OH, Lexi-Comp Inc. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
The oral bioavailability of gabapentin is affected by a saturable absorption mechanism, such that doses above a certain level are characterized by a declining bioavailability. In one study, the drug was 60% bioavailable at 900 mg/day, 47% at 1200 mg/day, 34% at 2400 mg/day, 33% at 3600 mg/day, and 27% at 4800 mg/day (Lacy, Armstrong, Goldman, et al., 2008; Twycross, Wilcock, Charlesworth, et al., 2003). Dose escalation can be accompanied by a ceiling effect, which in the case of this drug, may be either pharmacokinetic or pharmacodynamic, or possibly both. If an increment in dose does not yield improved outcomes, titration should stop (the dose usually lowered) and a decision should be made about the value of the therapy overall. Dose escalation also can be limited by adverse effects, and should an increment produce intolerable effects, the same decision is required.
Gabapentin should be administered at least 2 hours after intake of antacids that contain aluminum, as concurrent administration decreases the bioavailability of gabapentin by 10% to 25% (Twycross, Wilcock, Charlesworth, et al., 2003). Capsules of gabapentin may be opened and contents mixed with juice or water for patients who have difficulty swallowing capsules or tablets.
The future may hold simpler dosing regimens for patients who take gabapentin. A modified-release formulation of gabapentin had been studied but not released at the time of this publication. The formulation was described as having a unique gastric-retentive characteristic made possible by polymer-based technology (Irving, Jensen, Cramer, et al., 2009). Administered with food, the modified-release tablet swells and is retained in the stomach for up to 8 hours, allowing gradual release of the drug over 10 hours. Patients (N = 158) with postherpetic neuralgia of at least 3 months duration were randomized to receive a placebo or the modified-release gabapentin once daily (1800 mg) or twice daily (600 mg in the morning and 1200 mg in the evening) (Irving, Jensen, Cramer, et al., 2009). A 50% or greater reduction in pain was reported in 25.5% (gabapentin once daily), 28.8% (gabapentin twice daily), and 11.8% (placebo) of the patients. The drug was well-tolerated, with dizziness and sedation being the most common adverse effects and experienced most often by those who received gabapentin once daily.
Pregabalin
Pregabalin is typically started at a dose of 75 mg/day or 150 mg/day. Older patients or those who are medically frail or have renal insufficiency typically are started at an even lower dose (e.g., 25 mg/day or 50 mg/day). The dose usually is titrated every 3 to 4 days by an amount equal to the starting dose (or higher as the dose goes up), and the total dose is divided. The usual effective dose is between 150 mg twice daily and 300 mg twice daily, but some patients have good outcomes at doses that are higher or lower than this range. A 15-month open-label trial of patients with refractory neuropathic pain showed that doses between 150 mg/day and 600 mg/day significantly improved pain (Stacey, Dworkin, Murphy, et al., 2008). The discontinuation rate for adverse effects was low (12.3%). Others have found similar results with doses of 150 to 600 mg/day (Frampton, Foster, 2005; Frampton, Scott, 2004; Freynhagen, Strojek, Griesing, et al., 2005). Pregabalin doses must be adjusted downward for patients with renal impairment, as the drug is eliminated almost entirely by renal excretion (Gajraj, 2007) (see Table 23-2 for dosing adjustments in patients with renal impairment).
Other Anticonvulsants
There is a general lack of research to guide dosing of other anticonvulsants. Following is information to consider related to specific agents.
• An initial dose of 100 mg/day of lacosamide followed by weekly increases of 100 mg/day to a maximum of 400 mg/day proved to be safe and effective in a study of patients with painful diabetic neuropathy (Shaibani, Biton, Rauck, et al., 2009). Clinical experience is limited with this drug, and it is prudent to initiate dosing at 50 mg/day in older or medically frail patients.
• To reduce the risk of cutaneous hypersensitivity, lamotrigine should be started at a relatively low dose for 1 month, after which the dose can be more rapidly increased (Jensen, 2002; Maizels, McCarberg, 2005). For example, one strategy administers the drug at 25 mg/day for the first week, 25 mg twice daily for the second, 25 mg in the morning and 50 mg at night for the third, and 50 mg twice daily for the fourth. Starting with the fifth week, the dose can be increased by 50% to 100% weekly, until the usual effective dose of 300 mg/day to 500 mg/day is reached. Another strategy for initiating treatment uses 25 mg twice daily for the first 2 weeks followed by 50 mg twice daily for the next 2 weeks, after which the dose is increased quickly.
• The recommended starting dose of oxcarbazepine for treatment of trigeminal neuralgia is 600 mg/day (300 mg twice daily) with increases of 150 to 300 mg every few days (Carrazana, Mikoshiba, 2003). Again, older or medically frail patients should be started at a dose as low as 150 mg/day. The drug has been shown to be efficacious at doses between 900 mg/day and 2100 mg/day, producing pain relief equivalent to carbamazepine at doses of 400 to 1200 mg/day. The effective dose range for most patients was 600 to 1200 mg/day (Carrazana, Mioshiba, 2003).
• Topiramate is often started at 25 mg twice daily followed by weekly increases of 25 mg as tolerated (Lacy, Armstrong, Goldman, et al., 2008). A 26-week trial of patients with migraine administered topiramate in daily doses of 50 mg, 100 mg, and 200 mg and found little difference between 50 mg and placebo; 100 mg and 200 mg doses resulted in significant reductions in migraine attacks (Silberstein, Neto, Schmitt, et al., 2004). Other research supports 100 mg/day as the most efficacious and best-tolerated dose of topiramate for migraine prevention (Brandes, Saper, Diamond, et al., 2004; Silberstein, 2005). Doses as high as 400 mg/day may be beneficial in some patients.
• Low initial doses are appropriate for carbamazepine, valproate, and clonazepam, but the administration of phenytoin often begins with the presumed therapeutic dose (e.g., 300 mg/day) or a prudent oral loading regimen (e.g., 400 mg followed by another 400 mg 2 to 3 hours later).
Sodium Channel Blockers
By far, the largest clinical use of traditional sodium channel blockers is as local anesthetics, drugs that are deposited adjacent to peripheral nerves and inhibit transduction or transmission of afferent input to the CNS, including information about noxious stimuli (see Section I and Figure I-2 on pp. 4-5). Local anesthetics at high concentration can block sensory and motor nerves and produce regional anesthesia; at lower concentrations, they may provide pain relief without blocking other nerve functions.
In pain management, local anesthetics may be delivered via direct injection or placement of a catheter into the tissues adjacent to major nerves or a nerve plexus (neural blockade), or by neuraxial (epidural or intrathecal) administration. In this way, they produce regional anesthesia or analgesia. Local anesthetics are effective by surgical site infiltration for postoperative pain associated with minor surgery, and an excellent systematic review provides data on this technique for control of pain following a variety of major surgical procedures as well (Dahl, Moiniche, 2009). Scalp infiltration of ropivacaine has been used to reduce both postoperative pain and the incidence of persistent postsurgical pain in patients following intracranial tumor resection (Batoz, Verdonck, Pellerin, et al., 2009). Local anesthetics have been administered by intraperitoneal instillation for postoperative pain treatment after minor surgery (Callesen, Hjort, Mogensen, et al., 1999; Hazinedaroglu, Kayaoglu, Ates, et al., 2006; Visalyaputra, Lertakyamanee, Pethpaisit, et al., 1999). These drugs also may be given by topical application, allowing absorption into the skin and underlying tissues (e.g., patches, gels, creams). Topical anesthetics produce analgesia or anesthesia depending on the amount absorbed.
Drugs that block sodium channels also have a long history as systemic analgesics, which may be administered orally or parenterally. The following text reviews this systemic use of traditional sodium channel blockers for the treatment of persistent neuropathic pain. The use of IV lidocaine for acute and postoperative pain and continuous neural blockade are discussed in Chapter 26. See also Chapter 24 for topical local anesthetics including the lidocaine patch 5%, and local anesthetics for procedural pain. Neuraxial (epidural and intrathecal) local anesthetics are discussed in Chapter 15. Table V-1, pp. 748-756, at the end of Section V presents the characteristics of the commonly used systemic local anesthetics.
Systemic (Oral and Parenteral) Sodium Channel Blockers
The systemic local anesthetics used most often for persistent neuropathic pain treatment are IV lidocaine and oral mexiletine (Mexitil). Mexiletine is described as an oral sodium channel antagonist that is structurally similar to lidocaine (Wallace, Magnuson, Ridgeway, 2000) and is classified as a cardiac antiarrhythmic. Other drugs in this class, such as flecainide (Tambocor) and tocainide (Tonocard), are similarly classified but rarely used for pain treatment (Wallace, Galer, Gammaitoni, 2006). Both IV lidocaine and mexiletine have efficacy for neuropathic pain, but the evidence of analgesic effects varies, and their respective roles in practice differ (Galer, Harle, Rowbotham, 1996). Both are considered third-line (Dworkin, O’Connor, Backonja, et al., 2007) or fourth-line (Moulin, Clark, Gilron, et al., 2007) options for neuropathic pain. A new drug in the United States, lacosamide (Vimpat), is a first-in-class sodium channel modulator that is now classified as an anticonvulsant (and is discussed earlier in the chapter).
There is relatively strong evidence of analgesic efficacy for IV lidocaine and little evidence of this effect for the oral compounds. Tremont-Lukats and colleagues (2005) conducted a systematic review of 9 mexiletine and 10 parenteral lidocaine trials and found that their therapeutic benefit was more consistent for neuropathic pain from trauma, diabetes, and cardiovascular (CV) disease and less for neuropathic pain from HIV, cancer, and infectious etiology. These researchers also conducted a more extensive Cochrane Collaboration Review of 32 controlled clinical trials (two of these were duplicate studies) (Challapalli, Tremont-Lukats, McNicol, et al., 2005), which included studies of IV lidocaine (16 trials), mexiletine (12 trials), IV lidocaine plus mexiletine sequentially (1 trial), and tocainide (1 trial). Lidocaine and mexiletine showed superior efficacy compared with placebo, and while data were limited for comparison, no differences in efficacy between these systemic sodium channel blockers and other more commonly used drugs for neuropathic pain (e.g., carbamazepine, gabapentin, morphine) were noted. The systemic sodium channel antagonists were found to be safe, and no deaths or life-threatening toxicities were associated with their use.
Some patients experience immediate analgesia from an IV lidocaine infusion, and this potential suggests that the utility of this approach is the management of “pain crises” or crescendo neuropathic pain. Some patients experience favorable effects that continue for a long enough time period after the infusion is completed that repeated IV infusions can be adopted as part of the long-term plan of care. Most patients, however, appear to experience relatively short-lasting relief after the infusion is discontinued, and this is a limitation of the therapy (Sharma, Rajagopal, Palat, et al., 2009). Other limitations are the incidence of adverse effects at optimal dose ranges and wide variations in response among patients and different pain conditions.
Information is lacking about the long-term safety and effectiveness of these drugs (see adverse effects). Given this limited experience, a trial of a systemic sodium channel blocker generally is reserved for pain that has not responded to the more typical first-line adjuvant analgesics. The exception, as noted, is the specific use of IV lidocaine in an effort to address unrelenting severe or progressive dysesthesias (a crescendo pain pattern).
Oral Sodium Channel Blockers
Of the oral sodium channel blockers, mexiletine has been used most often for pain treatment and should be tried before flecainide and tocainide (the latter is no longer marketed in the United States). Although there is some evidence of analgesic efficacy from mexiletine, data from controlled trials are very limited (Caroll, Kaplan, Mackey, 2008; Wallace, Magnuson, Ridgeway, 2000). For example, a small (N = 20) randomized controlled trial titrated oral mexiletine to a maximum dose of 900 mg/day or dose-limiting adverse effects in patients with allodynic neuropathic pain (Wallace, Magnuson, Ridgeway, 2000). The treatment had a significant effect on pain induced (evoked) by stroking but no effect on the area of allodynia or quality of life measurements. Peak lidocaine plasma levels (0.54 mcg/mL), reached on day 10, were below those associated with cardiac antiarrhythmic activity (1.5 mcg/mL) and toxicity (5 mcg/mL) (Lema, 1996).
Wallace and colleagues (2006) reported that although two double-blind, placebo-controlled trials of mexiletine for neuropathic pain showed positive results, four others showed no significant effect on painful diabetic neuropathy, spinal cord dysesthetic pain, and allodynic neuropathic pain. In contrast, a comprehensive critical review of research on analgesics for painful diabetic neuropathy listed mexiletine among those showing some benefit in alleviating this pain (Adriaensen, Plaghki, Mathieu et al., 2005). Mexiletine has been reported anecdotally to provide sustained pain relief for some patients with poststroke pain (Edmondson, Simpson, Stubler et al., 1993) and has been used to effectively treat alcoholic neuropathy, peripheral nerve injury pain, and thalamic pain (Wallace, Galer, Gammaitoni, 2006). Mexiletine was studied in 9 patients with refractory persistent headaches deriving some benefit (Marmura, Passero, Young, 2008). Of these, 7 had adverse effects; nevertheless, the investigators concluded that this agent may be useful for the management of daily headaches.
These studies of mexiletine have had small sample sizes and, in some cases, conflicting results. One review of four controlled trials suggested that there is no clear evidence of mexiletine superiority over placebo (Duby, Campbell, Setter, et al., 2004). Overall, it is more reasonable to conclude that there is evidence of analgesic effect, but compared to other groups of adjuvant analgesics, such as the antidepressants and the gabapentinoids, and compared to IV lidocaine, this evidence is sparse.
Some studies indicate that mexiletine has a relatively high adverse effect liability. The incidence of nausea and dizziness may prevent dose titration to levels that optimize positive clinical outcomes related to pain (Wallace, Magnuson, Ridgeway, 2000; Wallace, Galer, Gammaitoni, 2006).
Flecainide, another antiarrhythmic agent, has properties similar to mexiletine. Animal research demonstrated that the drug could suppress ectopic discharge from injured nerves (Ichimata, Kitano, Ikebe et al., 2001), and a phase II exploratory clinical trial of 14 patients with neuropathic pain demonstrated a 30% response rate after weeklong courses of flecainide (50 mg twice daily followed by 100 mg twice daily) (von Gunten, Eappen, Cleary, et al., 2007). In another study, significant improvement in pain was demonstrated in 15 patients with postherpetic neuralgia who, following a positive response to IV flecainide, were treated with the oral formulation (Ichimata, Ikebe, Yoshitake, et al., 2001). In contrast, flecainide has been shown to be ineffective in treating neuropathic cancer pain (Wallace, Galer, Gammaitoni, 2006), and paranoid psychosis from flecainide toxicity was reported in a patient with cancer-related neuropathic pain (Bennett, 1997).
Although tocainide, another antiarrhythmic agent, has analgesic activity, there is a lack of research supporting its use in humans (Wallace, Galer, Gammaitoni, 2006). In an early study, it showed efficacy similar to carbamazepine for trigeminal neuralgia (Lindstrom, Lindblom, 1987). The drug has also been found to reduce pain from postherpetic neuralgia (Ichimata, Ikebe, Yoshitake, et al., 2001). Withdrawal arrhythmias have been reported with sudden cessation of the drug (McCleane, 2009a)
On the basis of clinical experience and the limited data from studies, a trial of a systemic sodium channel blocker, specifically mexiletine, can be considered for refractory neuropathic pain. If administration yields meaningful partial analgesia, it should be continued. If a risk of drug interaction or additive toxicities exists, dosing must be very cautious and monitoring intensified.
Given the chemical similarities between IV lidocaine and the oral sodium channel blockers, there has been interest in determining whether the results of an IV infusion of lidocaine could be used to assess the potential benefit from long-term oral therapy (Carroll, 2007; Carroll, Kaplan, Mackey, 2008; Cohen, Kapoor, Rathmell, 2009; Galer, Harle, Rowbotham, 1996; McCleane, 2008). In an early study of 9 patients with persistent neuropathic pain of peripheral origin, a moderate or better response to IV lidocaine infusion correlated with a subsequent equal or better response to a 4-week trial of oral mexiletine (Galer, Harle, Rowbotham, 1996). In a case series of 37 patients with neuropathic pain, patients were given mexiletine after showing a benefit from IV lidocaine and were evaluated to determine predictors of a response (Carroll, Kaplan, Mackey, 2008). The median time to discontinuing mexiletine was 43 days, and fewer than 20% of the patients continued therapy for a year. The reasons for discontinuation were unknown; however, those who had a positive response to the IV lidocaine infusion prior to mexiletine therapy were more likely to accept and continue mexiletine after it was started. Younger age and male gender also predicted acceptance of mexiletine therapy. These data are too limited to conclude that the response to IV lidocaine adequately predicts the outcome of mexiletine therapy.
IV Lidocaine
Over the past several years, IV lidocaine has been widely studied and shown to be effective for a variety of painful conditions (McCleane, 2009a; Tremont-Lukats, Challapalli, McNicol, et al., 2005; Challapalli, Tremont-Lukats, McNicol, et al., 2005; Wallace, Galer, Gammaitoni, 2006). (See Chapter 26 for use of IV lidocaine for acute and postoperative pain.) McCleane (2009a) suggests that anecdotal reports of its use in over 7000 cases without untoward CV adverse effects should increase confidence in the use of IV lidocaine for pain treatment. The reader is referred again to the systematic reviews discussed above (Tremont-Lukats, Challapalli, McNicol, et al., 2005; Challapalli, Tremont-Lukats, McNicol, et al., 2005).
IV lidocaine appears to be more effective in relieving neuropathic pain arising from a lesion or damage of the peripheral nervous system than pain from CNS damage (Kawamata, Sugino, Narimatsu, et al., 2006; Kingery, 1997; McCleane, 2009a; Tremont-Lukats, Challapalli, McNicol, et al., 2005; Wallace, Galer, Gammaitoni, 2006; Wu, Tella, Staats, et al., 2002). A randomized, double-blind, active placebo-controlled, cross-over trial (N = 31) provided clues to a better understanding of the underlying mechanisms of stump and phantom limb pain and the effect of IV lidocaine on different types of neuropathic pain (Wu, Tella, Staats, et al., 2002). Individuals with both stump pain and phantom limb pain (N = 11) and those with either stump pain (N = 11) or phantom limb pain (N = 9) were titrated off all medications except acetaminophen and NSAIDs, and then admitted and randomly assigned to receive a series of three IV infusions: An initial bolus of lidocaine, morphine, or the active placebo diphenhydramine (Benadryl) was followed by an infusion of the same drug. The maximum infusion dose of lidocaine was 400 mg, and the maximum dose of morphine was 25 mg. There were 24-hour intervals between infusions, and doses were decreased for adverse effects. Compared with placebo, morphine significantly reduced both stump and phantom limb pain, and lidocaine reduced stump but not phantom limb pain. This suggests that the underlying mechanisms of stump and phantom pain differ and reinforces the suggestion that IV lidocaine is more effective in treating peripherally-mediated pain syndromes than centrally-mediated pain syndromes.
The relatively smaller effect of IV lidocaine in studies of central pain syndromes may reflect a need for a higher dose to address the underlying pathophysiology. A randomized study demonstrated that 5 mg/kg infused over 30 minutes resulted in reduction of spontaneous pain at and below the level of spinal cord injury, independent of the presence of evoked (induced) pain (Finnerup, Biering-Sorensen, Johannesen, et al., 2005), and an earlier study reported a similar reduction in spontaneous central poststroke pain at this dose (Attal, Gaude, Brasseur, et al., 2000). A small randomized controlled study (N = 10) found that lidocaine (2.5 mg/kg) had no significant effect on spinal cord injury pain (Kvarnstrom, Karlsten, Quiding, et al., 2004). These data suggest that the different responses between peripheral and central neuropathic pain observed in some studies may not reflect the inefficacy of sodium channel blockade in the latter syndromes, but instead, a need for higher serum concentration relative to peripherally-generated neuropathic pain. Evaluation of serum level to determine this may be helpful, and it has been suggested that serum levels of at least 4 mcg/mL may be required for reduction in central pain (Kingery, 1997).
Although it is possible that IV lidocaine is relatively less useful in the management of cancer-related neuropathic pain than other types of neuropathic pain, the data are conflicting. A previously discussed systematic review of mexiletine and parenteral lidocaine trials concluded this (Tremont-Lukats, Challapalli, McNicol, et al., 2005), but a phase II pilot study (N = 88) found that an IV lidocaine bolus (2 mg/kg) followed by infusion (2 mg/kg) produced significant improvements in pain relief for patients with opioid-refractory cancer pain (Sharma, Rajagopal, Palat, et al., 2009). Mean onset of analgesia was 40 minutes after initiation of infusion, and mean duration of analgesia was 9 days after infusion was completed.
As noted, there is extensive literature describing the response of varied populations with pain to IV lidocaine treatment. Following is a list of research, case reports, and reviews on the use of IV lidocaine for a variety of types of persistent pain and pain-related conditions.
• Painful diabetic neuropathy and other peripheral neuropathies (Kingery, 1997; McClean, 2009; Tremont-Lukats, Challapalli, McNicol, et al., 2005; Tremont-Lukats, Hutson, Backonja, 2006; Wallace, Galer, Gammaitoni, 2006)
• Postherpetic neuralgia (Baranowski, De Courcey, Bonello, 1999; Kingery, 1997; Tremont-Lukats, Challapalli, McNicol, et al., 2005; Wallace, Galer, Gammaitoni, 2006)
• Central pain (stroke, spinal cord injury) (Attal, Gaude, Brasseur, et al., 2000; Frese, Hustedt, Ringelstein, et al., 2006; Finnerup, Biering-Sorensen, Johannesen, et al., 2005; Kumar, Kalita, Kumar, et al., 2009; Kvarnstrom, Karlsten, Quiding, et al., 2004; Tremont-Lukats, Challapalli, McNicol, et al., 2005; Wallace, Galer, Gammaitoni, 2006)
• Phantom limb pain; stump pain (Wu, Tella, Staats, et al., 2002)
• CRPS (Kingery, 1997; Schwartzman, Patel, Grothusen, et al., 2009; Tremont-Lukats, Hutson, Backonja, 2006)
• Fibromyalgia (McClean, 2009; Wallace, Galer, Gammaitoni, 2006)
• Brachial plexopathy (Tremont-Lukats, Hutson, Backonja, 2006)
• Radicular pain (Tremont-Lukats, Hutson, Backonja, 2006)
• Sciatica (Medrik-Goldberg, Lifschitz, Pud, et al., 1999)
• Migraine and other headaches (Hand, Stark, 2000; Wallace, Galer, Gammaitoni, 2006)
• Cancer-related neuropathic pain (Brose, Cousins, 1991; Bruera, Ripamonti, Brenneis, et al., 1992; Ferrini, 2000; Galer, Harle, Rowbotham, 1996; Sharma, Rajagopal, Palat, et al., 2009; Tremont-Lukats, Challapalli, McNicol, et al., 2005)
Mechanism of Action
Peripheral nerve injury causes changes in the number and location of ion channels, particularly sodium channels. These channels, which abnormally accumulate in injured peripheral nociceptors, may lead to ectopic discharges and other phenomena that initiate and maintain neuropathic pain (Beydoun, Backonja, 2003; Bridges, Thomson, Rice, 2001). IV lidocaine, mexiletine, and the other drugs in this class relieve pain by blocking sodium channels, stabilizing nerve membranes, slowing depolarization, and reducing ectopic discharges (McCleane, 2009a) (see Section I and Figure I-2 on pp. 4-5).
Lidocaine also has significant antiinflammatory effects, which may account for its effectiveness in postoperative pain treatment (Martin, Cherif, Gentili, et al., 2008). Research in animals (Kawamata, Sugino, Narimatsu, et al., 2006) and humans (Kingery, 1997; McClean, 2009; Tremont-Lukats, Challapalli, McNicol, et al., 2005; Wallace, Galer, Gammaitoni, 2006; Wu, Tella, Staats, et al., 2002) as well as clinical experience support a peripheral analgesic action site for systemic local anesthetics.
Although peripherally-mediated effects appear to be very important in the analgesic effect of the sodium channel blockers, supraspinal and spinal mechanisms of action have been suggested for many years. IV lidocaine may activate the endogenous opioid system (Bach, Jensen, Kastrup, et al., 1990; Kastrup, Bach, Petersen, et al., 1989), and more recent research has demonstrated expression of sodium channels on dorsal horn neurons near spinal cord lesions, which were linked to central pain behaviors (Finnerup, Biering-Sorensen, Johannesen, et al., 2005; Hains, Klein, Saab, et al., 2003). This may help to explain the effectiveness of IV lidocaine for some types of central pain. Further research is needed, but the ability of IV lidocaine to reduce several features of pain caused by CNS injuries suggests a central action in the mechanisms underlying these types of pain (Attal, Gaude, Brasseur, et al., 2000; Finnerup, Biering-Sorensen, Johannesen, et al., 2005). Many questions remain, however, in light of other research that shows IV lidocaine was effective in relieving stump pain (peripherally mediated) but not phantom limb pain (centrally mediated) (Wu, Tella, Staats, et al., 2002).
Adverse Effects
The most common adverse effects of the systemic sodium channel blockers are nausea, vomiting, abdominal pain, diarrhea, dizziness, and perioral numbness (numbness and tingling inside and surrounding the mouth) (Tremont-Lukats, Challapalli, McNicol, et al., 2005). Although reviews are reassuring about the adverse effects associated with these drugs (Challapalli, Tremont-Lukats, McNicol, et al., 2005; Tremont-Lukats, Challapalli, McNicol, et al., 2005), they potentially can produce serious and even life-threatening effects involving the CNS and the CV system. These are directly related to serum concentration (Gordon, 2008). Lidocaine levels above 1.5 mcg/mL and 5 mcg/mL are associated with cardiac antiarrhythmic activity and toxicity, respectively (Lema, 1996). Observable CNS effects generally occur at a lower serum concentration than cardiac changes and are more common with IV lidocaine than with mexiletine (Tremont-Lukats, Challapalli, McNicol, et al., 2005). Dizziness, perioral numbness, other paresthesias (abnormal sensations), and tremor usually occur first (Catterall, Mackie, 2006). At higher plasma concentrations, progressive encephalopathy develops and seizures may occur. At concentrations above this, cardiac conduction disturbances and myocardial depression can occur.
All the sodium channel antagonists must be used cautiously in patients with pre-existing heart disease, and patients who have significant heart disease should undergo cardiologic evaluation before therapy is administered. It is prudent to avoid this therapy in patients with cardiac rhythm disturbances, those who are receiving alpha agonists or beta blockers, and those who have cardiac insufficiency (Gordon, 2008). Lidocaine infusion can aggravate and increase first- and second-degree heart conduction blocks. Periodic ECG evaluation is recommended during mexiletine therapy (Argoff, Backonja, Belgrade, et al., 2006). Withdrawal arrhythmias have been reported with sudden cessation of tocainide (McClean, 2009).
A bolus over 20 minutes and infusion over no less than 1 hour has been suggested to lower the risk of serious adverse effects with IV lidocaine (Sharma, Rajagopal, Palat, et al., 2009). Large-dose infusions have been safely administered via longer infusion times. A 5 mg/kg lidocaine infusion administered over 6 hours for persistent neuropathic pain was tolerated without serious adverse effects (Tremont-Lukats, Hutson, Backonja, 2006) (see the following paragraphs). Bolus doses and infusions should be administered via an infusion device that does not allow free flow to help ensure accurate dose delivery.
Should adverse effects occur during IV lidocaine therapy, the clinician should stop the infusion; monitor blood pressure closely; assess for the presence of associated symptoms including pain; obtain a stat blood sample for serum lidocaine level; and notify the primary prescriber, anesthesia provider, or pain service (Gordon, 2008). In the case of a life-threatening overdose, treatment may include (as indicated by patient condition) airway support, oxygenation and hyperventilation to raise seizure threshold, diazepam (Valium) for seizure, IV fluids and Trendelenburg position to increase fluid volume, and CV depression treatment including IV fat emulsion as an antidote (Gordon, 2008). Lipid emulsion has been used to successfully resuscitate several patients from cardiac arrest from local anesthetic-induced cardiotoxicity (Clark, 2008); however, this appears to be related to the type of local anesthetic administered. The effects appear to be positive for bupivacaine-induced but not ropivacaine- or mepivacaine-induced cardiac arrest (Espinet, Emmerton, 2009; Zausig, Zink, Keil, et al., 2009). Large doses of epinephrine are also reported to be required for reversal of bupivacaine-induced cardiotoxicity (Mulroy, 2002). These findings have implications for local anesthetics administered via neuraxial and regional techniques (see Chapters 19 and 26 for more information).
IV lidocaine therapy should not be administered to individuals who are allergic to amide local anesthetics such as bupivacaine or ropivacaine (Gordon, 2008). (Procaine [Novocaine] is not a contraindication, as procaine is an ester local anesthetic.) Patients with liver dysfunction, pulmonary disease with a prominent feature of carbon dioxide retention, or congestive heart failure are at higher risk for lidocaine-induced adverse effects (Gordon, 2008).
A less serious but common adverse effect of IV lidocaine is painful inflammation of the vein upon injection (McCleane, 2008). This is unrelated to the pH or presence of preservatives and is thought to be caused primarily by the concentration of the lidocaine infusion. The use of dilute solutions and a glyceryl trinitrate (GTN, nitroglycerine) patch above the infusion site are suggested to reduce the pain (McCleane, 2008). The nitrate patch is reported to have sufficient local antiinflammatory properties to negate this complication.
Adverse effects on postoperative wound healing during continuous IV lidocaine infusion have not been reported. One study described healing disturbances in one patient who received IV lidocaine and had minor skin wound irritation and one patient who received placebo and had a much more serious subphrenic abscess (Herroeder, Pecher, Schonherr, et al., 2007).
A few publications have reported the safety of long-term IV lidocaine therapy. Although there are no controlled studies, there are case reports of hospice patients tolerating in-home IV lidocaine infusions for as long as 240 days prior to death (Ferrini, 2000). Anecdotally, long-term subcutaneous administration of lidocaine was also reported to yield sustained relief of refractory neuropathic pain in cancer patients (Brose, Cousins, 1991).
Indications
Data from controlled trials and clinical experience suggest that any one of a variety of persistent pains can be considered a potential indication for systemic therapy with a sodium channel blocker. Although peripherally-generated neuropathic pain may be more likely to respond than central pain (Backonja, Arndt, Gombar, 1994; Galer, Miller, Rowbotham, 1993; Wallace, Galer, Gammaitoni, 2006; Kvarnstrom, Karlsten, Quiding, et al., 2004; Kingery, 1997; McClean, 2009; Tremont-Lukats, Challapalli, McNicol, et al., 2005; Wu, Tella, Staats, et al., 2002), the data are not clear-cut and patients with central pain may respond favorably (Galer, Miller, Rowbotham, 1993). It has also been suggested that IV lidocaine may reduce sympathetic activity, resulting in a subsequent reduction in sympathetically-mediated pain (Wallace, Galer, Gammaitoni, 2006). Further research is needed to confirm this.
On the basis of clinical experience, a trial of a brief lidocaine infusion is sometimes implemented in patients with severe neuropathic pain that has not responded promptly to other interventions and requires immediate relief. This technique, therefore, may be a useful approach to the uncommon circumstance of crescendo neuropathic pain.
As mentioned, some studies suggest that a brief IV infusion may predict response to oral therapy (Galer, Harle, Rowbotham, 1996), particularly in younger and male patients (Carroll, Kaplan, Mackey, 2008). However, if the IV infusion fails to provide pain relief, a trial of oral therapy is still appropriate. Older age and higher pain severity may be important predictors of a positive response to IV lidocaine for the treatment of neuropathic pain (Carroll, Gaeta, Mackey, 2007).
On the basis of the limited data available concerning long-term safety and efficacy, it is appropriate to position the oral sodium channel blockers as third-line (Dworkin, O’Connor, Backonja, et al., 2007) or fourth-line (Moulin, Clark, Gilron, et al., 2007) drugs for neuropathic pain. A trial is warranted in patients with neuropathic pain who do not respond adequately or who cannot tolerate first-line adjuvant analgesics (e.g., antidepressants and anticonvulsants). In the palliative care setting, therapy usually is considered only for the long-term management of opioid-refractory neuropathic pain. In all settings, a trial of IV lidocaine may be considered at any point in time if the indication of crescendo pain is present.
Drug Selection
On theoretical grounds (less potency at the sodium channel), mexiletine may be the oral local anesthetic least likely to produce serious toxicity. Although intraindividual variability in the response to different drugs in this class has not been systematically assessed, such variability has been observed commonly with other drug classes and is likely to exist with the oral sodium channel blockers as well. Thus if mexiletine does not provide relief to a patient with severe neuropathic pain that has already proved refractory to other drugs, a trial with an alternative oral sodium channel blocker may be justified. Lidocaine is currently the primary parenteral local anesthetic.
Dose Selection
IV lidocaine and mexiletine in varying doses for some types of persistent pain have been proven to be generally safe and efficacious in placebo-controlled trials (Tremont-Lukats, Challapalli, McNicol, et al., 2005; Challapalli, Tremont-Lukats, McNicol, et al., 2005). Low initial doses followed by slow titration reduce the likelihood of adverse effects.
Mexiletine
In the absence of contrary information, overall dosing should conform to that used in the treatment of cardiac arrhythmias. For example, mexiletine usually should be started at 150 to 200 mg once or twice per day (Guay, 2001). Doses are better tolerated when taken with food. If intolerable adverse effects do not occur, the dose can be increased by a like amount every few days until the usual dose of 300 mg three times per day is reached. A maximum daily dose of 1200 mg is recommended (Guay, 2001). (See the following paragraphs for a discussion of optimal serum levels.) (See mexiletine patient medication information, Form V-9 on pp. 775-776, at the end of Section V.)
IV Lidocaine
It is not possible to specify an exact dose of parenteral lidocaine that will be both safe and effective for treatment of neuropathic pain (Tremont-Lukats, Hutson, Backonja, 2006). Brief lidocaine infusions for persistent pain have been administered at varying doses, typically within a range of 1 to 5 mg/kg infused over 30 to 45 minutes (Guay, 2001; Tremont-Lukats, Hutson, Backonja, 2006). Slow bolus administration (over 20 minutes) and infusion rates (1 hour) have been suggested as a way to reduce the risk of serious adverse effects (Sharma, Rajagopal, Palat et al., 2009). One randomized controlled trial administered doses of 1, 3, and 5 mg/kg via IV infusion over a 6-hour period to patients with peripheral neuropathic pain and found the 5 mg/kg dose to be the most effective in relieving pain (Tremont-Lukats, Hutson, Backonja, 2006). This large dose given over an extended time produced no serious adverse effects, and pain relief lasted for 4 hours after administration. Although the speed of administration appears to affect the adverse effect profile of IV lidocaine, studies are needed to determine if the speed of administration also influences analgesic effectiveness (Baranowski, De Courcey, Bonello, 1999).
Dose must be individualized according to the patient’s condition and type of pain. In the medically frail patient, it may be prudent to start much lower than the usual starting dose, for example, 0.5 mg/kg of IV lidocaine infused over 45 to 60 minutes. Central neuropathic pain may require larger doses of IV lidocaine than the more responsive peripheral neuropathic pain (Galer, Miller, Rowbotham, 1993; Kingery, 1997; McClean, 2009; Tremont-Lukats, Challapalli, McNicol, et al., 2005). Whereas a small randomized controlled study conducted in patients with spinal cord injury pain found no significant relief with lidocaine at a dose of 2.5 mg/kg infused over 40 minutes (Kvarnstrom, Karlsten, Quiding, et al., 2004), randomized controlled studies demonstrated that a larger dose of 5 mg/kg of IV lidocaine resulted in significant reductions in spinal cord injury pain (Finnerup, Biering-Sorensen, Johannesen, et al., 2005) and poststroke pain (Attal, Gaude, Brasseur, et al., 2000) (see Table V-1 on pp. 748-756).
Dosing and Serum Lidocaine Levels
Evaluation of serum concentration to determine adequate levels may provide guidance in dosing for some types of pain or when pain relief is not apparent despite what is thought to be adequate dosing. For example, it has been suggested that relatively higher serum lidocaine levels (greater than 4 mcg/mL) may be required to relieve central pain, ischemic pain, or thermally-evoked pain, and that levels of 1.5 to 2 mcg/mL are sufficient to relieve allodynia and peripheral neuropathic pain (Kingery, 1997). Gradual dose escalation through repeated brief infusions should be considered if treatment initially fails (Ferrante, Paggioli, Cherukuri, et al., 1996). However, upward titration is always a balancing act, as increased doses bring increased adverse effects; cardiac antiarrhythmic activity and toxicity are associated with lidocaine serum levels of 1.5 mcg/mL and 5 mcg/mL, respectively (Lema, 1996; Groudine, Fisher, Kaufman, et al., 1998).
Serum lidocaine levels during IV lidocaine infusion vary widely among studies in large part because of different dosing protocols (dose administered, infusion with or without bolus, speed of bolus or infusion). One older study of patients with postherpetic neuralgia administered 1 and 5 mg/kg IV lidocaine infusions over a 2-hour period and observed that patients reported relief of pain and allodynia with the 1 mg/kg infusion and had a mean serum level of 1.7 mcg/mL (Baranowski, De Courcey, Bonello, 1999). The mean serum levels for those who received 5 mg/kg were significantly higher, with no appreciable benefits noted. Several patients reached toxic levels, but there were no CV complications, and only two patients in the higher-dose group reported symptoms (circumoral paresthesia). A later study reported a mean plasma concentration of 4.2 mcg/mL in patients with spinal cord injury who had significantly improved pain following a 5 mg/kg lidocaine 30-minute infusion; the majority of these patients rated adverse effects, such as sedation and dizziness, as mild (Finnerup, Biering-Sorensen, Johannessen, et al., 2005).
A summary of long-term, in-home lidocaine infusions in hospice patients reported effective pain control with a serum lidocaine level as high as 9.3 mcg/mL in a patient receiving a lidocaine infusion of 10 to 15 mg/h plus bolus doses (Ferrini, 2000). The patient experienced lightheadedness that resolved when the infusion rate was reduced. A letter to the editor described two cases of lidocaine toxicity during low-dose lidocaine infusion in medically ill patients (Tei, Morita, Shishido, et al., 2005). Both were older adults with advanced cancer and normal liver and renal function. One received a daily dose of 300 mg of IV lidocaine, and the other received 200 mg. Both developed somnolence within 1 week, which resolved within 24 to 48 hours after infusion was discontinued. Serum concentration in both cases was 8.4 mcg/mL, and the patients experienced acceptable pain control.
Higher doses and higher blood levels of mexiletine had no association with greater degrees of pain relief in an early study of patients with peripheral neuropathic pain and allodynia (Galer, Harle, Rowbotham, 1996). Dose-limiting adverse effects rather than a true lack of a dose-response relationship were proposed as an explanation for this finding. The highest tolerated mexiletine dose (878 mg/day) during the study produced a mean serum level of 0.76 mcg/mL. Following completion of the study, one patient eventually achieved complete pain relief with a very high mexiletine dose (1750 mg/day with a serum level of 1.3 mcg/mL). Another study reported a low peak plasma level of 0.54 mcg/mL but no effect on pain with a maximum mexiletine dose of 900 mg/day (Wallace, Magnuson, Ridgeway, 2000). The researchers concluded that adverse effects might preclude higher doses. Experts suggest that serum levels indicate a mexiletine dose of 1200 to 1500 mg daily are needed for optimal pain relief but are unlikely to be achieved without significant adverse effects (Wallace, Galer, Gammaitoni, 2006). This helps to explain mexiletine’s lack of efficacy for many neuropathic pain syndromes.
See Chapter 26 for a discussion of serum lidocaine levels during perioperative IV lidocaine infusions. Also see Table V-1, pp. 748-756, at the end of Section V for dosing recommendations for the commonly used systemic local anesthetics.
Gamma Aminobutyric Acid (GABA) Agonists
Among the GABA agonists used as adjuvant analgesics are the anticonvulsant tiagabine, the benzodiazepines (specifically clonazepam), and baclofen. Other GABA agonists, such as propofol (Diprivan) and fospropofol (Lusedra), are IV sedative hypnotics (anesthetics) used for anesthesia induction, sedation and analgesia in the critically ill, and procedural sedation. The latter two drugs are discussed in Chapter 27.
Baclofen
Baclofen (Lioresal) is an agonist at the GABA type B receptor and may be administered by the oral or intrathecal route. It is considered another alternative for the treatment of neuropathic pain that might be refractory to other adjuvant agents. GABA is the most abundant inhibitory neurotransmitter in the CNS (Dickenson, Matthews, Suzuki, 2002) and comprises a major inhibitory neurotransmitter system (Bridges, Thompson, Rice, 2001); when GABA inhibition is suppressed, abnormal pain processing occurs (Bridges, Thompson, Rice, 2001) (see Section I). As an agonist at GABA B receptors in the spinal cord, baclofen relieves pain by inhibiting excitatory neurotransmitters and enhancing the GABA inhibitory pathway.
Oral baclofen is an accepted second-line drug for trigeminal neuralgia (Lussier, Portenoy, 2004), and headache treatment guidelines list baclofen (15 to 30 mg orally) as a preventive drug for cluster headache (May, Leone, Afra, et al., 2006). Although studies are lacking in other persistent pain states (Siddall, 2005), oral baclofen also has been used empirically to treat other types of refractory neuropathic pain (Lussier, Portenoy, 2004). One early study showed that baclofen enhanced morphine analgesia for postoperative pain (Gordon, Gear, Heller et al., 1995), but its use is limited in this setting because of a lack of sufficient evidence of effectiveness and the potential for adverse effects, particularly sedation and confusion (Fitzgerald, Buggy, 2006).
Generally speaking, baclofen is reserved for intractable persistent neuropathic pain or muscle spasm–induced pain that is refractory to other adjuvant agents (Slonimski, Abram, Zuniga, 2004). Similarly, intrathecal baclofen usually is reserved for patients who have implanted infusion devices and are experiencing inadequate analgesia or intolerable and unmanageable adverse effects from opioids, local anesthetics, or clonidine (Slonimski, Abram, Zuniga, 2004; Zuniga, Schlicht, Abram, 2000). Intrathecal baclofen doses vary widely from 50 mcg/day to 460 mcg/day (Hassenbusch, Portenoy, Cousins, et al., 2004). Therapy must be individualized according to patient response.
Intrathecal baclofen may be administered alone (Harmer, Larson, 2002; Zuniga, Schlicht, Abram, 2000) or in combination with other drugs, including morphine (Zuniga, Schlicht, Abram, 2000), clonidine (Slonimski, Abram, Zuniga, 2004; Zuniga, Perera, Abram, 2002), and ziconotide (Saulino, Burton, Danyo, et al., 2009) (see later in this chapter for more on ziconotide). Compatibility and stability of baclofen with clonidine (Alvarez, Mazancourt, Chartier-Kastler, et al., 2004) and morphine are established (Slonomski, Abram, Zuniga, 2004) (see Chapter 15).
Baclofen has been reported to relieve sympathetically-mediated pain by inhibiting sympathetic nervous system activity (Slonimski, Abram, Zuniga, 2004). A double-blind, randomized, cross-over study found numerous dramatic improvements in pain, paresthesias, and dystonia following administration of intrathecal baclofen to 7 women with CRPS, in whom the distinguishing clinical feature was multifocal or generalized fixed dystonia (van Hilten, van de Beek, Hoff, et al., 2000). The effects were more prominent in the arms than in the legs. A study of 36 patients with CRPS demonstrated that intrathecal baclofen via implanted pump improved dystonia (abnormal involuntary muscle contractions that cause twisting or repetitive movements or sustained postures), pain, disability, and quality of life indices over a period of 1 year, but complications related to the drug or infusion pump or catheter were common (van Rijn, Munts, Marinus, et al., 2009). Case reports described the use of intrathecal baclofen alone and in combination with clonidine to improve pain, allodynia, and autonomic function in two patients with well-established and refractory lower-extremity CRPS (Zuniga, Perera, Abram, 2002).
An excellent case series described pain relief following intrathecal baclofen for a variety of neuropathic pain conditions including stump pain, low back pain, cerebral palsy–related back pain and radiculopathy, and persistent lower extremity pain (Zuniga, Schlicht, Abram, 2000). All involved patients had been previously unable to achieve pain relief with an exhaustive list of analgesics and interventions.
Intrathecal baclofen also has been used for many years for refractory cancer pain, alone or in combination with other analgesics, most often by implanted infusion technology (Newsome, Frawley, Argoff, 2008).
Two excellent references are recommended for clinicians and pharmacists who are involved in the care of patients receiving intrathecal baclofen: (1) A comprehensive review by Ghafoor and colleagues (2007) that includes pharmacology of the various medications, adverse effects and complications, and information about the implanted devices including information on compounding drugs for administration, and (2) nursing care guidelines by Bhimani (2008), which include patient monitoring and management of implantable devices used to administer baclofen intrathecally. Nurses are also referred to the American Society for Pain Management Nursing’s position paper, which describes the nurse’s responsibilities in administering analgesics by catheter techniques (Pasero, Eksterowicz, Primeau, et al., 2007).
Baclofen for Spasticity
A primary indication for baclofen is the treatment of spasticity of spinal origin, such as that associated with spinal cord injury or multiple sclerosis. As mentioned, it has also been given intrathecally for treatment of dystonia associated with CRPS (van Rijn, Munts, Marinus, et al., 2009). At least 50% of patients with these disorders experience muscle spasm, and oral baclofen is reported to improve spasticity in 70% to 87% of patients (Dario, Tomei, 2004; Slonomski, Abram, Zuniga, 2004). Intrathecal baclofen, which is approved in the United States for treatment of spasticity, is thought to be even more effective (Dario, Tomei, 2004). Oral baclofen reduced lower extremity but not upper extremity spasms in patients with acquired brain injury (Meythaler, Clayton, Davis, et al., 2004), and intrathecal baclofen significantly reduced spasms resulting from stroke (Meythaler, Guin-Renfroe, Brunner, et al., 2001) and cerebral palsy (Meythaler, Guin-Renfroe, Law, et al., 2001). A small (N = 11) prospective, placebo-controlled trial found intrathecal baclofen significantly improved function and comfort in patients with cerebral palsy (Van Schaeybroeck, Nuttin, Lagae, et al., 2000).
Oral Baclofen Dosing
The administration of oral baclofen for pain is undertaken in a manner similar to the use of the drug for its primary indication—spasticity (Lussier, Portenoy, 2004). A starting dose of 5 mg two to three times per day is gradually escalated to the range of 30 to 90 mg/day and sometimes higher if adverse effects do not occur. Occasionally patients require more than 200 mg/day to benefit maximally; however, doses greater than 60 mg are associated with a higher incidence of adverse effects (Dario, Tomei, 2004). It is appropriate to continue dose escalation until pain is relieved or limiting adverse effects occur. (See baclofen patient medication information, Form V-2 on pp. 761-762, at the the end of Section V.)
Adverse Effects
The common adverse effects of baclofen are dizziness, sedation, excessive weakness, GI distress, and cognitive disturbances (Dario, Tomei, 2004) that may range from mild confusion to rare transient global amnesia (Grande, Loeser, Samii, 2008). Others include headache, loss of deep-tendon reflexes, hypotension, slurred speech, urinary frequency, and sexual dysfunction (Slonomski, Abram, Zuniga, 2004). The incidence of adverse effects associated with oral baclofen ranges from 10% to 75%, and as many as 35% of patients experience adverse effects from intrathecal baclofen; an evaluation of risk and benefit prior to therapy and throughout the course of administration is recommended (Dario, Tomei, 2004). Although there are reports of the need for dose escalation during therapy, this appears to be rare (Slonomski, Abram, Zuniga, 2004).
Baclofen’s adverse effects are dose-related. Administration of the lowest effective dose is essential, and this dose is identified by using a low starting dose and gradual dose escalation (Dario, Tomei, 2004; Lussier, Portenoy, 2004).
The potential for a serious withdrawal syndrome, including delirium and seizures, exists with abrupt discontinuation of baclofen after prolonged use; therefore, doses should always be tapered before the drug is discontinued (Dario, Tomei, 2004; Hansen, Gooch, Such-Neibar, 2007; Lussier, Portenoy, 2004). Baclofen withdrawal syndrome following abrupt discontinuation of intrathecal baclofen is sometimes confused with malignant hyperthermia and neuroleptic malignant syndrome. If untreated, baclofen withdrawal syndrome can progress to rhabdomyolysis, hepatic and renal failure, and disseminated intravascular coagulation (DIC). Treatment is high-dose IV benzodiazepines and restoration of intrathecal baclofen delivery (Slonomski, Abram, Zuniga, 2004). (See Chapter 15 for intraspinal complications that can occur with intrathecal drug administration.)
N-Methyl-d-Aspartate (NMDA) Receptor Antagonists
The evidence of analgesic efficacy for the several oral NMDA receptor antagonists on the market is limited and conflicting, and these drugs—dextromethorphan, memantine and amantadine, and magnesium—play a limited role in pain management. The data supporting the analgesic efficacy of ketamine are more robust, and this parenteral drug is gaining increasing acceptance in the management of carefully selected patients. The NMDA receptor antagonists, particularly ketamine, may have a role in preventing neuropathic pain states, such as persistent postsurgical pain, by suppressing secondary hyperalgesia and central sensitization (De Kock, Lavand’homme, 2007; Manning, Jianren, Frenk, et al., 1996; Xuerong, Yuguang, Xia, et al., 2008) (see Section I).
Following is a discussion of the research and clinical use of the NMDA receptor antagonists for persistent noncancer pain, cancer pain, and a variety of neuropathic types of pain. Perioperative use of dextromethorphan and ketamine is presented in Chapter 26.
Ketamine
Ketamine is a so-called dissociative anesthetic with dose-dependent analgesic, sedative, and amnestic properties. Preclinical data support its analgesic efficacy (Kosson, Klinowiecka, Kosson, et al., 2008; Pelissier, Laurido, Kramer, et al., 2003). At the low doses typically used for analgesia, the psychomimetic effects of ketamine are minimized; analgesia occurs at lower doses than psychomimetic effects (Panzer, Moitra, Sladen, 2009). Case reports, open-label and randomized controlled trials, and systematic reviews have reported that ketamine has analgesic efficacy in a variety of acute and persistent pains. The data are mixed, however, and more work is needed to clarify the analgesic potential in varied clinical settings. The drug is typically given intravenously, but it is possible to treat using the oral, rectal, intranasal, or subcutaneous routes (see Chapter 24 for a discussion of topical and see following discussion or research on intranasal ketamine). A discussion of ketamine’s role in preventing persistent neuropathic pain syndromes by suppressing secondary hyperalgesia and central sensitization is found in Chapter 26. See Box 23-1 for guidelines on the administration of low-dose ketamine.
A systematic review of ketamine studies between 1966 and 2002 described the drug’s efficacy in the treatment of persistent pain as moderate to weak (Hocking, Cousins, 2003). The most frequent use was as a third-line analgesic for the management of acute episodes of pain superimposed on persistent neuropathic pain, particularly when opioid-induced hyperalgesia was believed to be present. A more recent review concluded that a short course of ketamine might benefit some patients with refractory neuropathic pain, but there is insufficient research to support longer use (Bell, 2009). The drug is not mentioned as an option in general neuropathic pain treatment guidelines (Dworkin, O’Connor, Backonja, et al., 2007; Moulin, Clark, Gilron, et al., 2007). Treatment guidelines for postherpetic neuralgia consider ketamine an unproven option (Dubinsky, Kabbani, El-Chami, et al., 2004).
Although a 2003 Cochrane Collaboration Review of randomized controlled trials concluded that further research is needed to establish ketamine’s efficacy in improving opioid analgesia for cancer pain (Bell, Eccleston, Kalso, 2003a, 2003b), some studies and case reports attest to benefits when ketamine is added to the treatment plan for some patients with intractable cancer-related neuropathic pain (Chung, Pharo, 2007; Jackson, Ashby, Martin, et al., 2001; Kannan, Saxena, Bhatnagar, et al., 2002; Kotlinska-Lemieszek, Luczak, 2004; Mercadante, Villari, Ferrara, et al., 2009; Sen, Aydin, Aydin, 2006; Tarumi, Watanabe, Bruera, et al., 2000). The administration of ketamine to improve cancer pain that is refractory to opioid treatment is called burst ketamine therapy and has been shown to be effective when other options have failed (Hocking, Viser, Schug, et al., 2007; Jackson, Ashby, Martin, et al., 2001). For example, a case report described the successful use of ketamine burst infusion (100 mg/day) for 2 consecutive days to relieve excruciating movement-related pain caused by bony metastases that was unresponsive to aggressive opioid administration in 2 patients (Mercadante, Villari, Ferrera et al., 2009). Both patients experienced excessive sedation and nausea during IV morphine titration and were switched to an IV methadone infusion with concomitant IV ketamine burst infusion. Within approximately 2 days of infusion, the patients were able to resume daily activities. The ketamine was discontinued, and both patients were switched to oral methadone and discharged with well-controlled baseline and movement-related pain.
Analgesic efficacy of ketamine in acute pain has been established in several contexts. It is useful as part of a multimodal plan for treatment of pain associated with major surgery, burns, trauma, and during painful procedures in the ICU (Panzer, Moitra, Sladen, 2009). A placebo-controlled trial of transdermal ketamine in gynecologic surgery revealed that patients who received ketamine experienced a longer time to first request for analgesia and consumed less supplemental analgesia compared with those who received a placebo (Azevedo, Lauretti, Pereira, et al., 2000). A controlled trial of intranasal ketamine for breakthrough pain demonstrated both safety and analgesic efficacy (Carr, Goudas, Denman, et al., 2004a). The latter study led to two excellent letters to the editor about misunderstandings related to the potential for abuse of ketamine (and other agents) by patients with pain (Carr, Goudas, Denman, et al., 2004b; Lynch, Clark, 2004), which were written in response to a critical editorial (Bell, Kalso, 2004).
The many studies and reviews and related literature about ketamine do not answer important questions about the analgesic potential of ketamine, but in aggregate, indicate that the drug should play a role as an adjuvant analgesic, particularly for refractory neuropathic pain of diverse types. The following is a list of case reports, reviews, and placebo-controlled research on the use of ketamine for cancer pain and other persistent pain states:
• Cancer-related pain (Bell, Eccleston, Kalso, 2003a, 2003b; Ben-Ari, Lewis, Davidson, 2007; Edmonds, Davis, 1996; Grande, O’Donnell, Fitzgibbon, et al., 2008; Jackson, Ashby, Martin, et al., 2001; Kannan, Saxena, Bhatnagar, et al., 2002; Lussier, Portenoy, 2004; Mercadante, Arcuri, Tirelli, et al., 2000; Mercadante, Villari, Ferrera, et al., 2009; Sen, Aydin, Aydin, 2006; Tarumi, Watanabe, Bruera, et al., 2000; Wood, 2006)
• Fibromyalgia (Graven-Nielsen, Aspegren, Henriksson, et al., 2000; Hocking, Cousins, 2003)
• Postherpetic neuralgia (Ben-Ari, Lewis, Davidson, 2007; Cvrcek, 2008; Eide, Stubhaug, Oye, et al., 1995; Hempenstall, Nurmikko, Johnson, et al., 2005; Hocking, Cousins, 2003)
• Painful diabetic neuropathy (Cvrcek, 2008)
• CRPS (Ben-Ari, Lewis, Davidson, 2007; Correll, Maleki, Gracely, et al., 2004; Hocking, Cousins, 2003; Kiefer, Rohr, Ploppa, et al., 2008; Schwartzman, Alexander, Grothusen, et al., 2009; Sigtermans, van Hilten, Bauer, et al., 2009)
• Ischemic pain (Capel, Jenkins, Jefferson, et al., 2008; Hocking, Cousins, 2003; Mitchell, Fallon, 2002)
• Phantom limb pain (prevention and treatment) (Ben-Ari, Lewis, Davidson, 2007; Eichenberger, Neff, Sveticic, et al., 2008; Hayes, Armstrong-Brown, Burstal, et al., 2004; Hocking, Cousins, 2003; Mitchell, 2001)
• Spinal cord injury pain (Hocking, Cousins, 2003; Kvarnstrom, Karlsten, Quiding, et al., 2004)
• Central poststroke pain (Kumar, Kalita, Kumar, et al., 2009)
• Multiple sclerosis (Sakai, Tomiyasu, Ono, et al., 2004)
• Burn pain (Gregoretti, Decaroli, Piacevoli, et al., 2008)
• Acute exacerbation of chronic pain; high-dose, opioid-related hyperalgesia (Hocking, Cousins, 2003)
• Restless leg syndrome (Kapur, Friedman, 2002)
• Various types of refractory pain of neuropathic origin (Bell, 2009; Ben-Ari, Lewis, Davidson, 2007; Chung, Pharo, 2007; Edmonds, Davis, 1996; Jackson, Ashby, Martin, et al., 2001; Kannan, Saxena, Bhatnagar, et al., 2002; Persson, Axelsson, Hallin, et al., 1995; Tarumi, Watanabe, Bruera, et al., 2000; Visser, Schug, 2006)
Other NMDA Receptor Antagonists
Although basic research conducted largely in the 1990s increased optimism that NMDA receptor antagonism could be a means by which many types of pain could be addressed, clinical trials of other commercially-available drugs with this mechanism yielded mixed results. Preclinical evidence suggested that dextromethorphan, the antitussive, is analgesic (Chow, Huang, Ho, et al., 2004). Clinical trials of oral dextromethorphan have not yielded impressive results, however. In one study, the drug was shown to produce dose-related analgesia for patients with painful diabetic neuropathy (N = 23) or postherpetic neuralgia (N = 21); median dose for both types of pain was 400 mg (Sang, Booher, Gilron, et al., 2002). Doses of 100 mg had no effect on the relief produced by IV morphine in another study (Heiskanen, Hartel, Dahl, et al., 2002), and others indicated no meaningful effect in trigeminal neuralgia or anesthesia dolorosa (Gilron, Booher, Rowan, et al., 2000). Although early research of a novel oral formulation that combined morphine with dextromethorphan (MorphiDex) suggested that the addition of dextromethorphan might inhibit the development of tolerance to morphine (Chevlen, 2000), three subsequent controlled studies evaluating 3-month use of the formulation or morphine alone failed to show enhanced analgesia or reduction in opioid tolerance in patients with persistent noncancer, nonneuropathic pain (Galer, Lee, Ma, et al., 2005).
Memantine (Ebixa, Namenda), a long-acting oral NMDA receptor antagonist, is approved for the treatment of Alzheimer’s disease. Randomized controlled trials have produced disappointing results as an analgesic (Buvanendran, Kroin, 2008). It was ineffective for phantom limb pain in one placebo-controlled, randomized trial (Wiech, Kiefer, Topfner, et al., 2004), and a later placebo-controlled trial that administered memantine immediately after traumatic upper limb amputation and for 4 weeks postoperatively demonstrated a four-fold decrease in the incidence of phantom limb pain at 4 weeks and 6 months but not at 12 months (Schley, Topfner, Wiech, et al., 2007). Case reports described very positive responses in patients with treatment-refractory phantom limb pain (Hackworth, Tokarz, Fowler, et al., 2008) and cancer pain (Grande, O’Donnell, Fitzgibbon, et al., 2008).
Amantadine (Symmetrel) is another NMDA receptor antagonist that has been used for treatment of Parkinson’s disease and spasticity but is rarely used for pain. A randomized controlled trial administered amantadine or placebo to women the day before mastectomy and axillary lymph node dissection and continued treatment for 14 days (Eisenberg, Pud, Koltun, et al., 2007). There were no differences between the groups in any of the neuropathic pain outcome measures. A preoperative dose of 200 mg of IV amantadine failed to enhance postoperative analgesia in women undergoing abdominal hysterectomy (Gottschalk, Schroeder, Ufer, et al., 2001).
Magnesium, another agent that blocks NMDA receptor activity, has been used for the treatment of pain but with disappointing or mixed results. A randomized controlled trial (N = 200) showed no analgesic or dose-sparing effects from a single 4 g IV bolus of magnesium after anesthesia induction in ambulatory surgery patients (Tramer, Glynn, 2007). A systematic review of research on the perioperative use of NMDA receptor antagonists showed promise with dextromethorphan and ketamine but no effect with magnesium (McCartney, Sinha, Katz, 2004). Another systematic review of 14 trials evaluated its effect on postoperative pain and concluded a lack of convincing evidence that the drug provides effective analgesia for this type of pain (Lysakowski, Dumont, Czarnetzki, et al., 2007). A placebo-controlled, cross-over study found improved pain scores in 7 patients with postherpetic neuralgia 20 to 30 minutes following IV infusion of magnesium (30 mg/kg) (Brill, Sedgwick, Hamann, et al., 2002), and a single IV dose of 500 mg or 1000 mg of magnesium was found to be effective in most of 12 patients with cancer pain that was refractory to opioid analgesics (Crosby, Wilcock, Corcoran, 2000). A pilot study reported improved pain, impairment level, and quality of life with 4-hour IV magnesium infusions (70 mg/kg; 25 mL/h/day) for 5 days in 8 patients with CRPS type 1 (Collins, Zurrmond, de Lange, et al., 2009). The treatment was well-tolerated with infusion site pain, flushing, burning eyes, and fatigue being the most common adverse effects.
Mechanism of Action
Research regarding the underlying mechanisms of action of the NMDA receptor antagonists is ongoing and believed to be complex (Hocking, Visser, Schug, et al., 2007). Afferent input caused by tissue injury causes a series of events, including changes in calcium channels, which facilitates the transmission of pain (De Kock, Lavand’homme, 2007) (see Section I and Figure I-2, B on pp. 4-5). This facilitation of transmission, or “wind up,” is thought to be associated with the development of neuropathic pain. NMDA antagonizes this facilitated response. The binding of antagonists to the NMDA receptor can produce analgesia in this way, and also can reduce the desensitization of the mu opioid receptor that follows binding to opioid compounds. In animal models, the latter effect is associated with the reversal of opioid tolerance. Other mechanisms also may be important. Ketamine, for example, inhibits serotonin and dopamine uptake and influences other ion channel activity (Hocking, Visser, Shug, et al., 2007), effects that could further explain its role in suppressing hyperalgesia and central sensitization (De Kock, Lavand’homme, 2007).
Adverse Effects
The most common adverse effects of the NMDA receptor antagonists are dose-related and include sedation, confusion that can progress to delirium, severe nightmares, hallucinations, and dysphoria (Pasero, McCaffery, 2005). Psychotomimetic effects and other adverse effects are rare with dextromethorphan and are minimized with ketamine by administration of low (subanesthetic) doses.
Even at low doses, sedation is common with ketamine, which is one reason it is sometimes used for procedural sedation. The drug increases muscle tone, and purposeless movements may occur. Other adverse effects are hypotension, tachycardia, and nausea and vomiting. Ketamine should not be used in patients with increased intracranial or intraocular pressure as it may worsen these pathologies. Slow IV administration of ketamine does not produce respiratory depression, which is a major advantage. Although hypotension can occur, CV function, hepatic blood flow, laryngeal protective reflexes, and bowel function are generally not depressed with low doses.
When administered intravenously (the usual approach), the risks associated with ketamine have led some experts to recommend that its use be limited to those skilled in pain management and anesthesiology (Bell, 2009; Cvrcek, 2008). Palliative care specialists are also among those knowledgeable in the safe administration of the drug. Co-administration of a benzodiazepine or a neuroleptic has been recommended to reduce the risk of psychotomimetic effects (Fitzgibbon, Hall, Schroder, et al., 2002; Mercadante, 1996). Some hospitals restrict the administration of ketamine to specific clinical areas, such as the intensive care unit (ICU), emergency department (ED), and palliative care. Nurses should be aware of their scope of practice, state board of nursing’s opinions, and institutional policies and procedures regarding the administration and monitoring of agents such as ketamine as these vary widely.
Ketamine’s adverse effect profile also makes it a less favorable therapy for long-term use (Cvrcek, 2008). A case series reported severe ulcerative cystitis associated with long-term recreational use of ketamine that has implications also for individuals using ketamine for persistent pain (Shahani, Streutker, Dickson, et al., 2007).
Other NMDA antagonists also can cause CNS toxicity, including dizziness, confusion, and dysphoria. The adverse effects are more pronounced in frail medically ill patients. Dextromethorphan inhibits the cytochrome P450 2D6 isoenzyme, an effect that has been associated with serious drug-drug interactions (Forget, le Polain de Waroux, Wallemacq, et al., 2008) (see Chapter 11 for more on the cytochrome P450 enzyme system). These drug-drug interactions, such as the potential for higher-than-expected TCA levels when co-administered with dextromethorphan, must be anticipated and monitored if the latter drug is added to an existing regimen.
Indications
Although two trials suggest that a brief IV infusion of ketamine may be a predictor of response to an oral agent, specifically dextromethorphan (Cohen, Verdolin, Chang, 2006; Cohen, Wang, Chen, et al., 2009), it remains uncertain that this novel use has sufficient predictive validity, or is widely enough available, to be useful. As an avenue of research leading to a more rational “mechanism-based” selection of analgesic therapy, it is interesting, and in the future, may be helpful in identifying patients with pain that is mediated by NMDA receptor activation.
At the present time, brief IV ketamine infusion is gaining increased acceptance as an approach to treat severe neuropathic pain that has been refractory to conventional oral therapies. Experience probably is greatest in the treatment of CRPS. Brief ketamine infusion also is a strategy considered for “crescendo” pain, and it may be selected to provide pain relief and sedation for intolerable distress at the end of life (in a treatment approach known as palliative sedation). Some patients are offered repeated brief infusions as a long-term strategy for persistent pain, and some pain specialists provide ongoing treatment by compounding an oral formulation (or having the patient take the injectable formulation by mouth). Finally, small numbers of patients with refractory CRPS have been treated with high-dose ketamine, with intriguing early observations (see list of case studies and research provided earlier in the chapter).
Trials of the oral NMDA receptor antagonists are occasionally considered for patients with refractory neuropathic pain. Memantine is most often tried.
Dose Selection
To date, there are no universally agreed-upon guidelines or protocols for ketamine, but there is available literature based on clinical experience that can be used to guide practice. See Box 23-1 for suggested dosing.
Ketamine
Several different dosing regimens have been used for ketamine treatment of refractory persistent pain, including IV boluses (0.25 to 0.5 mg/kg), continuous low- or high-dose IV infusion (up to 600 mg/24 h), IV burst infusion therapy (100 to 500 mg/24 h for 2 to 5 days [see the paragraphs that follow]), and oral doses (0.5 mg/kg three times daily) (Jackson, Ashby, Martin, et al., 2001; Lussier, Portenoy, 2004) (see Chapter 26 and Box 23-1 for dosing recommendations for acute pain). A titrated continuous infusion of ketamine is used to induce sedation in imminently dying patients. Ketamine infusions should be administered via an infusion device that does not allow free flow to insure accurate dose delivery.
A case report of patients with refractory pain recommended an initial low parenteral ketamine dose of 40 to 60 mg over 24 hours (Fitzgibbon, Hall, Schroder, et al., 2002). Conversion to oral ketamine was achieved without loss of pain control at doses 30% to 40% lower than those required by the parenteral route.
For oral therapy, an initial dose of 50 mg at bedtime increased as tolerated to 50 mg 3 times daily has been suggested for central types of refractory neuropathic pain (Hocking, Cousins, 2003). Another source suggests 0.5 mg/kg taken at bedtime (Reves, Glass, Lubarsky, et al., 2005). As with other adjuvant analgesics, low initial doses followed by titration upward to optimal pain relief with minimal adverse effects is recommended. The case reports of 21 patients with intractable persistent pain described a starting dose of oral ketamine of 100 mg/day (40 mg/day in sensitive individuals) and titration with 40 mg/day doses every 2 days until efficacy or adverse effects were encountered; the final median dose was 220 mg/day (range 40 to 500 mg/day) (Enarson, Hays, Woodruffe, 1999).
Outpatient treatment of persistent pain with ketamine followed by close monitoring during and after treatment with home health visits is possible (Webster, Walker, 2006). Experience with multi-day dosing for outpatients with CRPS has been described (Goldberg, Domsky, Scaringe, et al., 2005).
Oral and intranasal ketamine can produce a bitter taste and burning of the throat. When the injectable formulation is used orally, the bitter taste may be masked with orange juice or cola drinks.
Other NMDA Antagonists
In one study, the median dose of dextromethorphan for treatment of painful diabetic neuropathy or postherpetic neuralgia was 400 mg, much higher than the usual antitussive dose (Sang, Booher, Gilron, et al., 2002). Dextromethorphan has an extremely good safety profile and has been administered at doses higher than 1 g/day; however, sedation and confusion can occur with higher doses. The drug is available in cough suppressant products only in the United States, and at present, the most concentrated source is in a suspension of 30 mg/5 mL (e.g., Delsym 12-Hour). A trial of dextromethorphan may be initiated using a proprietary cough suppressant (ensuring that the product contains no alcohol or other active drugs). A prudent starting dose is 45 to 60 mg daily, which can be gradually escalated until favorable effects occur, adverse effects supervene, or a conventional maximum daily dose of 1 g is achieved (Lussier, Portenoy, 2004).
If memantine or amantadine is offered as a trial for neuropathic pain, dosing typically starts in a range similar to the primary indication for these drugs, such as memantine 10 mg/day. The dose is then gradually titrated upward until favorable effects occur or adverse effects develop. Dosing of IV magnesium usually takes the form of a bolus or brief infusion, usually at a 1 g dose. There is no experience with the use of oral magnesium for the longer-term management of neuropathic pain, but several products are available over the counter and provide guidelines for administration.
Ziconotide
Ziconotide (Prialt) is a nonopioid intrathecal analgesic currently approved for the treatment of intractable pain. The preparation is a synthetic peptide that is obtained from the marine snail Conus magus (Vitale, Battelli, Gasperoni, et al., 2008). Intrathecal ziconotide exerts its effects through blockade of the N-type calcium channel in the dorsal horn of the spinal cord (see Section I). It is administered via an implanted intrathecal infusion device, usually following a trial to assess its potential to relieve pain without toxicity. Ziconotide can be administered alone or with other analgesics, such as morphine (Wallace, Kosek, Staats, et al., 2008), hydromorphone (Deer, Krames, Hassenbusch, et al., 2007), and baclofen (Saulino, Burton, Danyo, et al., 2009).
Early studies of ziconotide revealed a high incidence of significant cognitive adverse effects, including hallucinations and psychosis. These studies evaluated a dose range higher than is now recommended (see the paragraphs that follow).
A phase II open-label study of 26 patients with primarily severe persistent low back pain demonstrated a 14.5% mean percentage improvement of baseline VAS scores (Wallace, Kosek, Staats, et al., 2008). The initial dose was 0.60 mcg/day, which was titrated to a median dose of 4.80 mcg/day (maximum 7.2 mcg/day) by week 5. Adverse effects were described as mild to moderate in most patients and included confusion, dizziness, abnormal gait, hallucinations, and anxiety. One patient experienced hallucinations for 57 days, and another had memory impairment for 163 days; these resolved in both patients after therapy was discontinued.
In a multicenter, open-label trial of 78 patients with persistent noncancer pain, the median ziconotide dose was 6.48 mcg/day at the start of the study and ranged from 5.52 to 7.20 mcg/day across all study visits (Webster, Fisher, Charapata, et al., 2009). With this dosing range and stable pain scores for the treatment duration of 3 years, the type and frequency of adverse events included memory impairment (11.3%); dizziness, nystagmus, and speech disorder (8.5% each); nervousness and somnolence (7.0% each); and abnormal gait (5.6%). Pain relief seemed to be relatively disappointing, with VAS improvements of just greater than 10% above baseline at most evaluation periods (see Section II for discussion of meaningful pain relief).
A 12-month study showed that ziconotide was effective for long-term treatment of a wide variety of persistent cancer and noncancer types of pain (Ellis, Dissanayake, McGuire, et al., 2008). Other research has also demonstrated efficacy for refractory cancer and AIDS-related pain (Staats, Yearwood, Charapata, et al., 2004) and refractory neuropathic pain and spasticity (Saulino, Burton, Danyo, et al., 2009).
Based on this research and clinical experience with ziconotide, the expert panel of the 2007 Polyanalgesic Consensus Conference suggested that ziconotide may be considered a first-line intrathecal agent, with indications beyond intractable pain in patients who had exhausted all other therapeutic options (Deer, Krames, Hassenbusch, et al., 2007). Given the cost and short history of this drug, its appropriate positioning within the armentarium of agents that may be explored for intrathecal use remains uncertain, however. It is likely that a larger proportion of trials of neuraxial analgesia will include a specific ziconotide trial in the future.
Ziconotide has a lag time before optimal pain relief (e.g., 9 to 14 days), which is achieved through slow dose titration to effect. One study reported a mean time to onset of analgesia as 15 weeks at a mean dose of 3.7 mcg/day (Saulino, Burton, Danyo et al., 2009). The therapeutic and safety dosing range is narrow. Further research is needed to determine ideal catheter tip location for optimal analgesic response (Deer, Krames, Hassenbusch, et al., 2007). The maximum recommended ziconotide dose is 19.2 mcg/day, although higher doses have been safely administered (Webster, Fisher, Charapata, et al., 2009). More research is needed with regard to the molecular stability of ziconotide. It appears to have stability at concentrations higher than 1 mcg/mL (Deer, Krames, Hassenbusch, et al, 2007). The rate of ziconotide degradation is accelerated when the agent is compounded with morphine and hydromorphone, so combinations of ziconotide with lower concentrations of the compounded opioid are expected to be more stable. Gradual dose escalation can reduce the occurrence and severity of CNS adverse effects, such as dizziness, nausea, and confusion (Klotz, 2006). Intolerable and unmanageable adverse effects are reported to resolve with dose reduction or termination of treatment (Webster, Fisher, Charapata, et al., 2009). Ziconotide does not cause respiratory depression or physical dependence, and tolerance has not been reported (Klotz, 2006).
Ziconotide must be administered by pain management specialists who are familiar with the drug, indications, dose ranges, adverse events, dose titration, and follow-up care. Patients receiving this agent must be closely followed for a treatment response and the occurrence of adverse effects. It is critical that patients have easy access to their care providers. They must be informed about the potential CNS adverse events and instructed to promptly report these if any are experienced. See Box 23-2 for guidelines on the administration of ziconotide.
Conclusion
There are several adjuvant analgesics that are used solely for persistent neuropathic pain. Along with the multipurpose adjuvant analgesics these drugs offer many options for the treatment of this type of pain. Among those discussed in this chapter are the anticonvulsants, which are first-line for neuropathic pain, and the sodium channel blockers, GABA agonists, NMDA receptor antagonists, and relatively new intrathecal drug ziconotide, which are options for neuropathic pain that is refractory to the recommended first-line options.