Multipurpose Adjuvant Analgesics
DATA supporting the analgesic efficacy of some adjuvant drug classes are derived from numerous studies of very diverse syndromes. These drugs may be termed multipurpose and can be considered for any type of pain, fundamentally similar in this way to the opioids and nonopioid analgesics (Portenoy, 2000). The multipurpose adjuvant analgesics that are currently considered to be among the more useful in clinical practice include antidepressants, corticosteroids, and alpha2-adrenergic agonists (e.g., clonidine). Many of the multipurpose adjuvant analgesics are appropriate for both acute pain and persistent pain. Antidepressants have a delayed onset of analgesia, making them inappropriate for acute pain. Other drug classes, such as the sodium channel blockers and cannabinoids, have evidence to suggest broad applicability, but conventional use continues to position them solely for neuropathic pain (Lussier, Portenoy, 2004). Although the use of topical drugs is restricted by the location of pain, there are many types, and, combined, they too may be considered to have multiple purposes (see Chapter 24). See Table V-1, pp. 748-756, at the end of Section V for the characteristics and dosing guidelines for many of the multipurpose adjuvant analgesics.
Antidepressant Drugs
Antidepressant adjuvant analgesics are usually divided into two major groups: the tricyclic antidepressants (TCAs) and the newer biogenic amine reuptake inhibitors (Table 22-1). Of the latter group, the serotonin and norepinephrine reuptake inhibitors (SNRIs) are clearly analgesics, whereas research is lacking or inconsistent regarding the analgesic potential of the selective serotonin reuptake inhibitors (SSRIs) (Arnold, 2007; Collins, Moore, McQuay, et al., 2000; Kroenke, Krebs, Bair, 2009; Saarto, Wiffen, 2007; Veves, Backonja, Malik, 2008).
Table 22-1
Antidepressant Adjuvant Analgesics: Classes with Examples of Drugs
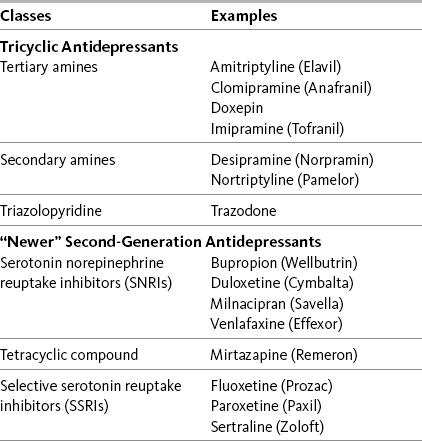
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 637, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
The multipurpose nature of antidepressants is demonstrated in their efficacy as analgesics in neuropathic pain states, such as painful diabetic neuropathy and postherpetic neuralgia, and other types of persistent pain states, such as headache, fibromyalgia, and some types of back pain. A meta-analysis of 18 randomized controlled trials concluded that there is strong evidence for the use of antidepressants in the treatment of fibromyalgia, with impressive improvements in pain, depression, fatigue, sleep disturbances, and health-related quality of life (Hauser, Bernardy, Uceyler, et al., 2009). These data have influenced treatment guidelines for fibromyalgia, which now include the early use of antidepressants (Hauser, Thieme, Turk, 2009). Guidelines also suggest that antidepressants should be considered an option for persistent low back pain, notwithstanding the lack of research focused on the newer drugs, specifically the SNRIs (Chou, Qaseem, Snow, et al., 2007).
An early systematic review of randomized controlled trials summarized the compelling evidence that antidepressants are efficacious in varied types of neuropathic pain (McQuay, Tramer, Nye, et al., 1996). Antidepressants are now identified as first-line analgesics in neuropathic pain guidelines (Dworkin, Backonja, Rowbotham, et al., 2003; Dworkin, O’Connor, Backonja, et al., 2007; Moulin, Clark, Gilron, et al., 2007). Anticonvulsants are another first-line choice for neuropathic pain and are discussed later in this section (see Chapter 23). Several excellent systematic reviews, some with related evidence-based guidelines for drug selection, provide additional support for the use of antidepressants in the management of neuropathic pain (Finnerup, Otto, McQuay, et al., 2005; Saarto, Wiffen, 2007; Kroenke, Krebs, Bair, et al., 2009). Data from another systematic review showed that there is no difference between antidepressants and anticonvulsants in the likelihood of achieving pain control (Chou, Carson, Chan, 2009).
Tricyclic Antidepressants (TCAs)
Evidence is compelling that the TCAs produce analgesia for a variety of chronic (persistent) pain syndromes including both neuropathic and other types of persistent pain (Argoff, Backonja, Belgrade, et al., 2006; Arnold, 2007; Hauser, Bernardy, Uceyler, et al., 2009; Dworkin, Backonja, Rowbotham, et al., 2003; Dworkin, O’Connor, Backonja, et al., 2007; Moulin, Clark, Gilron, et al., 2007; Perrot, Javier, Marty, et al., 2008; Tomkins, Jackson, O’Malley, et al., 2001; Verdu, Decosterd, Buclin, et al., 2008). The efficacy of the tertiary amine compounds has been demonstrated in a large number of controlled and uncontrolled trials (see each TCA listed below). Amitriptyline (Elavil), a tertiary amine, has an abundance of research showing efficacy and has been used for many years to treat a variety of types of persistent pain. Adverse effects are common, however, and the risk of more serious adverse effects, such as orthostatic hypotension, is relatively high (Curtis, Ostbye, Sendersky, et al., 2004; Dworkin, O’Connor, Backonja, et al., 2007).
Although research is more limited with the secondary amines, such as desipramine (Norpramin) and nortriptyline (Aventyl, Pamelor), they have also been shown to be efficacious in a variety of painful conditions and usually are better tolerated (American Geriatrics Society, 2002; Dworkin, Backonja, Rowbotham, et al., 2003; Dworkin, O’Connor, Backonja, et al., 2007; Gore, Dukes, Rowbotham, et al., 2006; Simon, Lipman, Caudill-Slosberg, et al., 2002). Some neuropathic guidelines recommend the use of a tertiary amine TCA only if a secondary amine TCA is unavailable (Dworkin, Backonja, Rowbotham, et al., 2003; Dworkin, O’Connor, Backonja, et al., 2007). This recommendation is particularly appropriate in high-risk populations, such as older adults who are at greater risk for antidepressant adverse effects, especially when they are administered in high doses and in combination with other CNS-acting medications (Wright, Roumani, Boudreau, et al., 2009). An increasingly popular alternative to the secondary amine TCAs is the SNRI antidepressants, all of which have a better toxicity profile than the TCAs (see adverse effects).
The best evidence for the use of TCAs for the treatment of neuropathic pain is summarized in several clinical reviews (Collins, Moore, McQuay, et al., 2000; Dobecki, Schocket, Wallace, 2006; Jensen, Finnerup, 2007; Dworkin, Backonja, Rowbotham, et al., 2003; Verdu, Decosterd, Buclin, et al., 2008). Agents in this class are useful for managing all types of dysesthesias (abnormal unpleasant sensations), such as burning, electrical-like, shock-like, shooting, and lancinating (stabbing). A Cochrane Collaboration Review of 61 randomized controlled trials documented the efficacy of TCAs and reported the following additional findings (Saarto, Wiffen, 2007):
• The number-needed-to-treat (NNT) when TCAs are studied as analgesics for varied types of neuropathic pain averages 3.6 (95% CI 3 to 4.5), which means that it is necessary to treat 3 to 4 patients to find one who gets at least a 50% reduction in pain; in other words, one-third of patients with neuropathic pain who take TCAs achieve moderate pain relief.
• Amitriptyline has been the best studied TCA, and in a range of doses up to 150 mg/day (10 studies, 588 patients), this drug has an NNT of 3.1 (95% CI 2.5 to 4.2).
• Studies of painful HIV-related neuropathy have not confirmed the efficacy of the TCAs.
• Analgesic doses for TCAs are typically less than antidepressant doses, and the effect on pain can be independent of any effect on depression.
• Although several small studies have suggested that analgesic efficacy correlates with dose, dose-response data have not been established; nonetheless, TCAs must be titrated in individual patients to identify responders and, within the group of responders, achieve the most effective dose (gradual titration also reduces the risk of adverse effects and is especially important in older patients).
• Across studies (N = 453), 13% of participants dropped out of active groups for a variety of reasons including intolerable adverse effects; discontinuation of dosing due to adverse effects is likely to be more prevalent in clinical populations, and prescribers usually attempt to select specific drugs based on relatively more favorable adverse effect profiles.
Amitriptyline
Amitriptyline (Elavil) is a tertiary amine TCA and has been extensively studied as an analgesic. The following are among the pain syndromes for which this drug has established analgesic efficacy:
• Postherpetic neuralgia (Argoff, Backonja, Belgrade, et al., 2006; Bowsher, 2003; Collins, Moore, McQuay, et al., 2000; Dubinsky, Kabbani, El-Chami, et al., 2004; Rowbotham, Reisner, Davies, et al., 2005)
• Painful diabetic neuropathy (Boulton, Vinik, Arezzo, et al., 2005; Collins, Moore, McQuay, et al., 2000; Duby, Campbell, Setter, et al., 2004; Max, Culnane, Schafer, et al., 1987; Max, Lynch, Muir, et al., 1992)
• Fibromyalgia (Arnold, 2007; Nishishinya, Urrutia, Walitt, et al., 2008; Hauser, Bernardy, Uceyler, et al., 2009; Heymann, Helfenstein, Feldman, 2001; Uceyler, Hauser, Sommer, 2008) (see also Hauser, Thieme, Turk, 2009)
• Migraine and other types of headache (Ashina, Bendtsen, Jensen, 2004; Descombes, Brefel-Courbon, Thalamas, et al., 2001; Keskinbora, Aydinli, 2008; Krymchantowski, Silva, Barbosa, et al., 2002)
• Arthritis (Frank, Kashini, Parker, et al., 1988; Katz, Rothenberg, 2005; Simon, Lipman, Caudill-Slosberg, et al., 2002)
• Central spinal cord injury pain (Cardenas, Warms, Turner, et al., 2002)
• Central post-stroke pain (Frese, Husstedt, Ringelstein, et al., 2006; Kumar, Kalita, Kumar, et al., 2009)
• Persistent facial pain (List, Axelsson, Leijon, 2003)
• Cancer-related neuropathic pain (Miaskowski, Cleary, Burney, et al., 2005; Ventafridda, Bonezzi, Caraceni, et al., 1987)
• Chemotherapy-induced neuropathy (Note: 50 mg/day was thought to be too low to produce positive effects) (Kautio, Haanpaa, Saarto, et al., 2008)
• Interstitial cystitis (van Ophoven, Pokupic, Heinecke, et al., 2004)
• As mentioned, antidepressants are not appropriate for treatment of acute pain because of the delay in time before appreciable analgesia. However, although not approved for use in the United States at the time of publication, a phase I trial of IV amitriptyline established the safety of a 25 mg to 50 mg preoperative infusion (Fridrich, Colvin, Zizza, et al., 2007). (See amitriptyline patient medication information, Form V-1 on pp. 759-760, at the end of Section V).
Desipramine
Desipramine (Norpramin) is a secondary amine TCA. It has relatively more effect on norepinephrine reuptake than amitriptyline and usually causes fewer adverse effects. (See desipramine patient medication information, Form V-5 on pp. 767-768, at the end of Section V). Clinical reviews and single studies have shown its efficacy in the following:
• Postherpetic neuralgia (Collins, Moore, McQuay, et al., 2000; Dubinsky, Kabbani, El-Chami, et al., 2004; Max, Lynch, Muir, et al., 1992; O’Connor, Noyes, Holloway, 2007; Zin, Nissen, Smith, et al., 2008)
• Painful diabetic neuropathy (Argoff, Backonja, Belgrade, et al., 2006; Boulton, Vinik, Arezzo, et al., 2005; Collins, Moore, McQuay, et al., 2000; Duby, Campbell, Setter, et al., 2004; Zin, Nissen, Smith, et al., 2008)
• Cancer-related neuropathic pain (Miaskowski, Cleary, Burney, et al., 2005)
Nortriptyline
Nortriptyline (Aventyl, Pamelor) also is a secondary amine compound. (See nortriptyline patient medication information, Form V-10 on pp. 777-778, at the end of Section V). It has been researched for the following types of pain:
• Fibromyalgia (Heyman, Helfenstein, Feldman, 2001) (See also Hauser, Thieme, Turk, 2009.)
• Postherpetic neuralgia (Dubinsky, Kabbani, El-Chami, et al., 2004)
• Painful diabetic neuropathy (Boulton, Vinik, Arezzo, et al., 2005)
• Persistent lumbar radiculopathy (Khoromi, Cui, Nackers, et al., 2007)
• Cancer-related neuropathic pain (Miaskowski, Cleary, Burney, et al., 2005)
• Chemotherapy-induced neuropathy (Hammack, Michalak, Loprinzi, et al., 2002)
“Newer” Antidepressants
Evidence of analgesic efficacy is compelling for the drugs that are synaptic reuptake blockers of both serotonin and norepinephrine. The SNRIs have a better adverse effect profile than TCAs (Zin, Nissen, Smith, et al., 2008). Duloxetine (Cymbalta) has been most studied, and milnacipran (Savella), an SNRI recently released in the United States, also has been approved for the treatment of fibromyalgia pain. There is limited evidence that venlafaxine (Effexor) is analgesic and, as yet, no evidence in support of the newer desvenlafaxine (Pristiq).
Bupropion (Aplenzin, Budeprion, Wellbutrin) and mirtazapine (Remeron) are relatively older drugs and have structures and actions distinct from the TCAs and SNRIs. Both increase activity in the pathways that use biogenic amines, including serotonin, norepinephrine, and dopamine. Evidence of analgesia from these drugs is very limited, but they are sometimes considered for patients with persistent pain, particularly when a concurrent symptom, such as fatigue or insomnia, would benefit from targeted treatment.
Guidelines for the treatment of neuropathic pain that recommend the analgesic antidepressants as possible first-line or second-line drugs suggest that consideration be given to the use of the SNRIs over the TCAs because of a more favorable adverse effect profile (Dworkin, Backonja, Rowbotham, et al., 2003; Moulin, Clark, Gilron, et al., 2007). There have been no comparative effectiveness trials, and the decision to select one or another is based on risk assessment and best clinical judgment. The newer antidepressants are discussed in alphabetical order in the following paragraphs.
Bupropion
Bupropion (Aplenzin, Budeprion, Wellbutrin) is distinguished from the TCAs and SNRIs because it inhibits neuronal norepinephrine reuptake and, less potently, dopamine reuptake (Katz, Penella-Vaughn, Hetzel, et al., 2005). The drug can be useful in the management of neuropathic pain (Semenchuk, Davis, 2000; Semenchuk, Sherma, Davis, 2001), but was found ineffective for relief of non-neuropathic persistent low back pain (Katz, Pennella-Vaughan, Hetzel, et al., 2005). An extensive review suggested bupropion as an option for painful diabetic neuropathy (Duby, Campbell, Setter, et al., 2004), but guidelines list it as a drug with limited evidence for this condition (Argoff, Backonja, Belgrade, et al., 2006). Reported case series indicate that this agent can help relieve pain associated with persistent headache (Pinsker, 1998).
Bupropion has a low risk of somnolence and sexual dysfunction, adverse effects that may be limiting with other antidepressants. Some patients report increased energy that appears to be unrelated to mood effects. This has led to empirical use of this drug for fatigue.
Duloxetine
Duloxetine (Cymbalta) is an established analgesic based on the results of numerous controlled trials (Dworkin, Backonja, Rowbotham, et al., 2003; Kroenke, Krebs, Bair, 2009). In the United States, it is approved for the treatment of fibromyalgia, and randomized controlled studies have shown its effectiveness for this type of pain (Arnold, Lu, Crofford, et al., 2004; Arnold, Rosen, Pritchett, et al., 2005; Hauser, Bernardy, Ucelyler, et al., 2009). A large (N = 520) randomized controlled 6-month trial of patients with fibromyalgia with or without major depression demonstrated improvements in both pain severity and Patient Global Impressions of Improvement (PGI-I) scores regardless of whether or not the patients were depressed (Russell, Mease, Smith, et al., 2008). Others have observed similar findings (Arnold, Hudson, Wang, et al., 2009). A 1-year trial established the long-term safety and efficacy of duloxetine in 350 women with fibromyalgia (Chappell, Littlejohn, Kajdasz, et al., 2009).
A number of randomized controlled studies also report that duloxetine is efficacious for depression-associated pain (Brecht, Courtecuisse, Debieuvre, et al., 2007; Perahia, Pritchett, Raskin, 2006; Raskin, Wiltse, Siegal, et al., 2007). In older depressed patients, duloxetine can improve mood, cognition, and pain (Raskin, Wiltse, Siegal, et al., 2007). However, a meta-analysis of five randomized controlled trials observed a lack of consistency in research findings and suggested that reports of pain relief in patients with depression may be overestimated (Spielmans, 2008).
Duloxetine also is approved for treatment of painful diabetic neuropathy. Numerous randomized controlled trials have shown its efficacy for this type of pain (Goldstein, Lu, Detke, et al., 2005; Kajdasz, Iyengar, Desaiah, et al., 2007; Pritchett, McCarberg, Watkin, et al., 2007; Raskin, Pritchett, Wang, et al., 2005), and some guidelines recommend it as a first-line analgesic for this condition (Argoff, Backonja, Belgrade, et al., 2006). One study suggested that relatively high pain intensity, but no other variable (e.g., age, type and duration of diabetes, or severity of neuropathy), predicted the efficacy of duloxetine (Ziegler, Pritchett, Wang, et al., 2007). A meta-analysis concluded that duloxetine had efficacy and tolerability comparable to gabapentin and pregabalin in diabetic peripheral neuropathic pain (Quilici, Chancellor, Lothgren, et al., 2009).
A study conducted in the United Kingdom concluded that duloxetine is a cost-effective and efficacious agent for the pain of diabetic peripheral neuropathy (Beard, McCrink, Le, et al., 2008). Similar results were found in a study in the United States, in which patients who participated in a previous randomized controlled study of painful diabetic neuropathy were re-randomized for a 52-week trial of 60 mg of duloxetine compared with routine treatment, which most often included gabapentin, venlafaxine, or amitriptyline (Wu, Birnbaum, Mareva, et al., 2006). Duloxetine was found to be more cost-effective than routine treatment from both employer and societal perspectives, which included patients’ out-of-pocket expenses.
Other studies have shown that the benefits produced by duloxetine can be sustained over time (Mease, Russell, Kajdasz, et al., 2009; Russell, Mease, Smith, et al., 2008; Wernicke, Wang, Pritchett, et al., 2007) and that improved functional outcomes accompany analgesia (Armstrong, Chappell, Le, et al., 2007; Arnold, Lu, Crofford, et al., 2004; Arnold, Rosen, Pritchett, et al., 2005; Russell, Mease, Smith, et al., 2008; Sullivan, Benlety, Fan, et al., 2009; Wernicke, Wang, Pritchett, et al., 2007). In summary, these favorable data support recent guidelines for the treatment of neuropathic pain, which designate the SNRIs as either first-line analgesics (Dworkin, O’Connnor, Backonja, et al., 2007) or second-line analgesics (Moulin, Clark, Gilron, et al., 2007).
Duloxetine may be effective for central pain as well. An open-label study revealed beneficial effects of duloxetine for the treatment of pain associated with Parkinson disease (Djaldetti, Yust-Katz, Kolianov, et al., 2007).
Although an early review concluded that research does not provide convincing support for the use of antidepressants for musculoskeletal pain (Curatolo, Bogduk, 2001), later research calls for further evaluation of duloxetine for osteoarthritis (OA) pain (Sullivan, Bentley, Fan, et al., 2009). Patients in the latter study were given two weeks of placebo followed by 10 weeks of duloxetine. Self-reported function improved, and pain intensity was reduced 30% as measured on the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) between 2 and 12 weeks of treatment. Similarly, improvements in pain (30%) and physical function were found in patients who received duloxetine in another randomized, placebo-controlled trial (Chappell, 2009). More and larger studies on the use of duloxetine for this type of pain are needed. (See duloxetine patient medication information, Form V-6 on pp. 769-770, at the end of Section V).
Milnacipran
Milnacipran (Savella) is another SNRI that has been used for many years in Europe and Japan and was approved for use in the United States in 2009 for the treatment of fibromyalgia (12.5 mg to 100 mg/day). A 15-week multicenter randomized, placebo-controlled trial (N = 1196) reported significant improvements in pain, physical functioning, and fatigue associated with fibromyalgia (Clauw, Mease, Palmer, et al., 2008). A 27-week randomized, placebo-controlled trial (N = 888) found similar improvements in pain, fatigue, cognition, and other functional domains in patients with fibromyalgia (Mease, Clauw, Gendreau, et al., 2009). The drug is well tolerated even at high doses (Vitton Gendreau, Gendreau, et al., 2004), and response to treatment is reported to have a durability of up to one year (Owen, 2008). An interesting case report described the complete resolution of phantom limb pain with the use of milnacipran (50 mg/day) (Sato, Higuchi, Hishikawa, 2008). Further research on the use of this drug for this and other types of pain is needed.
Mirtazapine
Mirtazapine (Remeron) is distinguished from the other antidepressants by its tetracyclic chemical structure. Its antidepressant efficacy is similar to the SSRIs, such as fluoxetine (Prozac) (Howland, 2008). A meta-analysis of 12 new-generation antidepressants concluded that mirtazapine is similar to venlafaxine and more efficacious than duloxetine, fluoxetine, and paroxetine (Paxil) for the treatment of depression (Cipriani, Furukawa, Salanti, et al., 2009). It may be better tolerated than TCAs with fewer anticholinergic effects.
Well-controlled trials that fully evaluate the analgesic effects of the drug are lacking at this time; however, a randomized, placebo-controlled trial in 10 healthy adults found that a single dose of mirtazapine (30 mg) significantly increased pain tolerance during electrical stimulation (Arnold, Vuadens, Kuntzer, et al., 2008). A 4-week prospective, open-label study of cancer patients reported rapid improvements in pain, nausea, sleep disturbance, depression, and quality of life (Kim, Shin, Kim, et al., 2008). However, a study of cancer patients with major depression, anxiety, or adjustment disorders found that mirtazapine was effective in resolving insomnia, anxiety, and depressive symptoms, but not pain (Cankurtaran, Ozalap, Soygur, et al., 2008). Mirtazapine has been suggested as an option for treatment of tension-type headaches (Bigal, Rapoport, Hargreaves, 2008). It was also reported to resolve postdural puncture headache, but the authors of this paper noted that the drug has unknown fetal effects and cannot be routinely recommended for the headache that follows inadvertent dural puncture in the laboring patient (Sheen, Ho, 2008). Preoperative administration of mirtazapine has been shown to reduce preoperative anxiety and postoperative nausea and vomiting (Chen, Lin, Ko, et al., 2008).
Venlafaxine
In a study of experimental pain, venlafaxine (Effexor) increased pain tolerance (Enggaard, Klitgaard, Gram, et al., 2001), and in a study of patients with neuropathic pain, the drug reduced hyperalgesia and temporal summation (repeated neuronal stimulation and action potentials) but not intensity and pain detection thresholds (Yucel, Ozyalcin, Talu, et al., 2005). Electrocardiogram (ECG) changes have been associated with venlafaxine, so cautious use in patients with high cardiovascular (CV) risk is recommended (Dworkin, Backonja, Rowbotham, et al., 2003). (See venlafaxine patient medication information, Form V-12 on pp. 781-782, at the end of Section V). Among the studies of venlafaxine for the treatment of pain are the following:
• Persistent pain and associated depression (Bradley, Barkin, Jerome, et al., 2003)
• Painful diabetic neuropathy (Davis, Smith, 1999; Duby, Campbell, Setter, et al., 2004; Dworkin, Backonja, Rowbotham, et al., 2003)
• Postherpetic neuralgia (Dworkin, Backonja, Rowbotham, et al., 2003)
• Chemotherapy-induced peripheral neuropathy (Durand, Alexandre, Guillevin, et al., 2005)
• Neuropathic back pain (Pernia, Mico, Calderon, et al., 2000; Sumpton, Moulin, 2001)
• Persistent pelvic pain (Karp, 2004)
• Tension headaches (Zissis, Harmoussi, Vlaikidis, et al., 2007)
Selective Serotonin Reuptake Inhibitors (SSRIs)
The evidence of analgesic efficacy for both the TCAs and the SNRIs far exceeds the SSRIs. Favorable anecdotal reports of fluoxetine (Prozac) analgesia (Diamond, Frietag, 1989; Geller, 1989) were refuted in a controlled trial, which showed no benefit in patients with painful diabetic neuropathy (Max, Lynch, Muir, et al., 1992). Other studies of fluoxetine yielded mixed results. A double-blind comparison of fluoxetine, desipramine, and amitriptyline in patients with postherpetic neuralgia revealed that all three were analgesic; although fluoxetine produced the fewest adverse effects, the dropout rate during fluoxetine treatment was highest (Davies, Reisner-Keller, Rowbotham, 1996). Another controlled study of 59 patients with rheumatic pain revealed that both amitriptyline 25 mg and fluoxetine 20 mg produced significant pain relief compared with placebo during 4 weeks of treatment; fluoxetine was considered superior to amitriptyline because of fewer adverse effects (Rani, Naidu, Prasad, et al., 1996).
Paroxetine (Paxil), another SSRI, has some benefit in the control of pain from diabetic neuropathy (Sindrup, Gram, Brosen, et al., 1990), and while one study reported that the drug was effective in improving mood and reducing anxiety in 116 patients with fibromyalgia, its pain relieving properties were far less apparent (Patkar, Masand, Krulewicz, et al., 2007). In contrast, a later meta-analysis found strong evidence for both paroxetine and fluoxetine in reducing pain but just small effects for mood and no effects on fatigue and sleep (Hauser, Bernardy, Uceyler, et al., 2009).
Fewer clinical trials have specifically compared the efficacy of antidepressants as analgesics for cancer pain. Nonetheless, there are general recommendations to advocate for the use of TCAs and SRNIs rather than SSRIs for treatment of neuropathic pain syndromes caused by progressive cancer and cancer treatments (Dworkin, O’Connor, Backonja, et al., 2007).
Summary
Substantial evidence exists that antidepressant drugs (both TCAs and SNRIs) have analgesic effects for diverse types of persistent pain. Given the range of pain syndromes that are potentially responsive, it is appropriate to classify these drugs as nonspecific, multipurpose analgesics. These agents are especially useful for the treatment of neuropathic pain states. The strongest evidence of analgesic efficacy is found in the numerous controlled trials of the tertiary amine drugs, of which amitriptyline is the best studied. There is less abundant data to support efficacy for the secondary amine TCAs nortriptyline and desipramine. Nevertheless, guidelines recommend the use of a tertiary amine TCA only if a secondary amine is unavailable because of a more favorable adverse effect profile (Argoff, Backonja, Belgrade, et al., 2006; Dworkin, O’Connor, Backonja, et al., 2007).
Antidepressants with more norepinephrine selective actions are also analgesic, and very impressive data have been generated for the SNRIs, with evidence mounting for their role in the treatment of numerous pain syndromes. Clinical interest is less for SSRIs despite their relatively good adverse effect profile. Among the SSRIs, limited evidence supports the analgesic efficacy of paroxetine and fluoxetine, but data are equivocal or absent for the others.
Mechanism of Action
The antidepressants presumably produce analgesic effects through increased activity in endogenous monoaminergic pain modulating pathways. Specific pathways originate from neuronal pools in the brainstem and descend to the spinal cord, where they release substances, such as the amines serotonin (5HT), and norepinephrine (NE), that inhibit the transmission of nociceptive impulses (see Section I and Figure I-2, D on pp. 4-5). By blocking the reuptake (resorption) of serotonin and norepinephrine at the synapse, the antidepressants presumably increase activity in these pathways (Maizels, McCarberg, 2005; Veves, Backonja, Malik, 2008). Norepinephrine is thought to play a more significant role than other amines in the endogenous analgesia pathways, which helps to explain the greater analgesic effectiveness of the SNRIs compared with the SSRIs (Veves, Backonja, Malik, 2008). Conversely, serotonin may be the more powerful mediator of depression, and low serotonin levels are associated with depression.
Other mechanisms also may be involved in antidepressant-mediated analgesia. Some of these drugs block peripheral sodium channels, but the extent to which this effect contributes to analgesia is uncertain (Gerner, 2004; Strumper, Durieux, 2004). The SSRIs are less potent sodium channel blockers than the TCAs and SNRIs (Dick, Brochu, Purohit, et al., 2007). Some of the antidepressants also block histamine (H1) receptors, and this, too, could contribute to analgesic effects (Mays, 2006).
Although effective treatment of concurrent depression can contribute to a favorable outcome from antidepressant therapy in patients with persistent pain, the analgesic effect of these drugs is not dependent on their antidepressant activity. Many controlled studies of TCAs, for example, have demonstrated that they are effective in relieving pain often at lower doses than those required to treat depression, and that the onset of analgesia typically occurs much sooner than the antidepressant effect, usually within one week. Moreover, patients who are not depressed can experience analgesia, and depressed patients can report pain relief without a change in mood (Kishore-Kumar, Max, Schafer, et al., 1990; Max, Lynch, Muir, et al., 1992).
Adverse Effects
Although serious adverse effects associated with the TCAs are uncommon at the doses usually administered for pain, less serious adverse effects are frequent and include dry mouth, sedation, dizziness, mental clouding, weight gain, and constipation, especially with the more anticholinergic TCAs such as amitriptyline. Even at low doses, patients with major organ dysfunction or those who might be taking multiple other drugs may experience troublesome adverse effects (Kurella, Bennett, Chertow, 2003). Moreover, some patients who receive low doses of the TCAs actually attain relatively high plasma drug concentrations. Rarely, serious anticholinergic toxicity can occur, including precipitation of acute angle closure glaucoma, tachycardia, severe constipation, or urinary retention. Amitriptyline is not recommended in older patients as they have increased sensitivity to its anticholinergic and sedative effects (see Table 13-4 on p. 336). Nevertheless the drug continues to be used in older adults. An extensive review of a health insurance claims database in the United States revealed that 20% of patients ages 65 years and older with a diagnosis of painful diabetic neuropathy were prescribed TCAs, primarily amitriptyline, and over half of these had co-morbidities or were taking other medications that could render the prescribing of a TCA potentially inappropriate and dangerous (Berger, Dukes, Edelsberg, et al., 2007).
When relatively mild, the anticholinergic adverse effects, such as dry mouth, blurred vision, or constipation, can usually be managed or tolerated. In a double-blind study of 26 older patients receiving nortriptyline, adverse effects of dry mouth and blurred vision were safely reduced with bethanechol (Urecholine) 10 mg three times a day (Rosen, Pollock, Altieri, et al., 1993). The drug was well tolerated except for causing an increase in orthostatic hypotension. The decrease in salivation caused by TCAs is especially troublesome for patients with dentures. Precautions should be taken to ensure that all patients practice good oral hygiene, keep well hydrated, and have regular dental examinations.
Of the TCAs, the secondary amines, desipramine and nortriptyline, are less anticholinergic than the tertiary amines, so are preferred particularly in populations who are at high risk for anticholinergic adverse effects, such as older adults, individuals who are otherwise predisposed to these symptoms, and those who have experienced distressing adverse effects during a trial of a tertiary amine drug.
Orthostatic hypotension is a potentially serious TCA adverse effect. Of the TCAs, nortriptyline and desipramine are the least likely to cause this. Orthostatic hypotension appears to be much more likely in older adults and, combined with the sedative effects of these drugs, probably accounts for an increased risk of hip fracture in this population (Ray, Griffin, Schaffner, et al., 1987). It is prudent to consider an SNRI before a TCA for patients who are predisposed to orthostasis. Care should always focus on preventing adverse events, and patients receiving TCAs should be advised to get up slowly from a supine position and rely on assistance if they are feeling dizzy or lightheaded.
The most serious adverse effect of TCAs is cardiotoxicity, which was recognized decades ago (Glassman, Bigger, 1981). While extremely rare, the potential for cardiotoxicity should always be considered when selecting an antidepressant (Dworkin, O’Connor, Backonja, et al., 2007). Patients at risk are those who have significant heart disease, including conduction disorders, dysrhythmias, or heart failure. Conduction abnormalities and a recent cardiac event are contraindications for TCAs (Maizels, McCarberg, 2005). Doses higher than 100 mg/day have been associated with sudden cardiac death (Ray, Meredith, Thapa, et al., 2004). If patients with known heart problems are treated with TCAs, initial doses should be low, dose escalation should be gradual, and the ECG should be regularly assessed as doses reach relatively high levels. SNRIs and SSRIs do not carry the same risk for cardiotoxic effects, and therefore, are acceptable alternatives for antidepressant analgesia in high-risk patients.
Both SNRIs and SSRIs have a more favorable adverse effect profile than the TCAs. They are less likely to cause mental clouding, confusion, or somnolence, and overall, appear to be better tolerated. The most common adverse effects of the SNRIs and SSRIs are nausea, headache, sedation, insomnia, weight gain, impaired memory, sweating, tremor, and sexual dysfunction. Of the SNRIs, duloxetine and milnacipran appear better tolerated than venlafaxine (Stahl, Grady, Moret, et al., 2005). Venlafaxine can cause dose-related hypertension, and, if appropriate, blood pressure (BP) monitoring should be done during initiation of treatment. A meta-analysis of four randomized controlled trials concluded that SSRIs taken alone and in combination with NSAIDs substantially increases upper gastrointestinal (GI) hemorrhage (Loke, Trivedi, Singh, 2008).
A troubling adverse effect for many patients taking any of the antidepressants is sexual dysfunction. Haberfellner (2007) provides an extensive review of 79 clinical trials describing this adverse effect and can be referred to for guidance related to specific antidepressants. Of interest is a suggestion to add bupropion to SSRI treatment for major depression as a means of reversing sexual adverse effects and bolstering antidepressant effects (Zisook, Rush, Haight, et al., 2006). Other strategies include: (1) switching to another antidepressant, (2) selecting an antidepressant with reduced likelihood for sexual dysfunction, (3) selecting an antidepressant with a short half-life, and (4) watchful waiting to see if this adverse effect may improve over time (Taylor, 2006). The use of a phosphodiesterase (PDE) inhibitor (e.g., sildenafil [Viagra], tadalafil [Cialis]) for men with erectile dysfunction can be considered as well (Rudkin, Taylor, Hawton, 2004; Taylor, 2006). Low-dose psychostimulant therapy, such as dextroamphetamine or methylphenidate 5 mg sublingually, one or more hours before intercourse, has been anecdotally reported to enhance sexual performance (Bartlik, Kaplan, Kaplan, 1995).
Drug-drug interactions are relatively common with antidepressants because they inhibit the various isoenzymes in the cytochrome P450 enzyme system (Virani, Mailis, Shapiro, et al., 1997) (see Chapter 11). Each antidepressant has a unique profile, and it is important for the clinician to become familiar with the potential for drug-drug interactions and toxicity associated with the particular antidepressant being administered. Patients must be told to report adverse effects promptly, and in some cases, serum drug concentrations should be monitored when adding a new drug or changing the dose of a drug in an existing regimen.
Caution is warranted when drugs that increase CNS levels of serotonin, such as antidepressants, monoamine oxidase inhibitors (MAOIs), and tramadol or tapentadol, are combined. This can precipitate serotonin syndrome (serotonin toxicity), which is characterized by nausea, agitation, confusion, tremulousness, hyperreflexia, and hyperthermia. Diagnosis can be challenging, particularly when medications in the inpatient setting are added to preconsumed outpatient prescriptions (Altman, Jahangiri, 2010). Treatment of serotonin syndrome depends on the severity of symptoms but can include reducing or discontinuing the causative medications, symptom management, and administering antiserotonergic drugs such as chlorpromazine (Thorazine) (Skinner, Epstein, Pappagallo, 2009). Rarely, patients are treated for respiratory failure, renal failure, and coagulopathy (Altman, Jahangiri, 2010).
All of the TCAs are metabolized in the liver so must be used with caution in individuals with hepatic impairment (Mays, 2006). An increase in hepatotoxicity has been linked to the use of duloxetine in individuals with pre-existing liver disease, suggesting that the drug may aggravate the disease (United States Food and Drug Administration [U.S. FDA], 2005a). All patients taking antidepressants should be told to report pruritus, dark urine, jaundice, right upper-quadrant tenderness, or unexplained flu-like symptoms, and prescribers should investigate these symptoms promptly. It is also important to recommend smoking cessation in patients taking duloxetine, as the drug’s bioavailability is reduced by as much as one-third in smokers (U.S. FDA, 2007a). Antidepressants are generally considered safe in patients with renal disease; however, their metabolites rely on renal clearance, and patients should be watched for signs of metabolite accumulation, such as increased sedation and anticholinergic effects (Leo, 2008; Mays, 2006).
Rapid discontinuation of the SNRIs or SSRIs after an extended period of dosing can cause a discontinuation syndrome characterized by both somatic (dizziness, lightheadedness, nausea, fatigue, lethargy, sleep disturbances) and psychologic (anxiety, agitation, crying, irritability) symptoms (Rosenbaum, Zajecka, 1997). If discontinued, all antidepressants that have been taken for more than 6 weeks should be tapered over 2 weeks to reduce the risk of any withdrawal phenomena.
Indications
As multipurpose analgesics, antidepressant drugs could potentially be considered for the treatment of any persistent pain syndrome. Opioids are first-line therapy for moderate or severe persistent pain in populations with active cancer or other serious advanced illness, and in these populations, analgesic antidepressants usually are considered for opioid-refractory neuropathic pain. In other types of persistent neuropathic pain, antidepressants are widely considered first-line or second-line treatment (e.g., after a trial of gabapentin or pregabalin) (Dworkin, O’Connor, Backonja, et al., 2007; Moulin, Clark, Gilron, et al., 2007). Similar guidelines can be applied to fibromyalgia. The positioning of the analgesic antidepressants in other types of persistent pain is a matter of clinical judgment.
Great variability exists in the range of symptoms presented by patients with neuropathic pains. Conceivably, specific symptoms may indicate the existence of mechanisms that respond differently to drugs with varying modes of action (Jensen, Baron, 2003). Statements in older literature suggest that antidepressants are more useful for neuropathic pains characterized by continuous dysesthesias (e.g., “burning” pain or hypersensitivity to stimuli) than pains described as lancinating (“stabbing” pain). This impression is not supported by individual controlled trials (Kishore-Kuman, Max, Schafer, et al., 1990; Mishra, Bhatnagar, Gupta, et al., 2009) or a systematic review (McQuay, Tramer, Nye, et al., 1996). At this time, symptom-specific drug selection remains a future goal.
In a recent meta-analysis of randomized clinical trials, it was not possible to find appreciable differences in the effectiveness of TCAs compared with the anticonvulsant gabapentin for the treatment of diabetic neuropathy and postherpetic neuralgia (Chou, Carson, Chan, 2009). However, TCAs are often preferred for initial treatment of neuropathic pain if the patient has coexisting insomnia, anxiety, or depression, or if cost is a consideration. In the treatment of postherpetic neuralgia in older adults, desipramine was shown to be more effective and less expensive compared with the gabapentenoids (gabapentin and pregabalin) (O’Connor, Noyes, Holloway, 2007). However, duloxetine has been shown to be more cost-effective than some of the other drugs used for neuropathic pain, including amitriptyline, in cost comparison studies (Beard, McCrink, Le, et al., 2008; Wu, Birnbaum, Mareva et al., 2006). As mentioned, multiple factors must be considered when determining the most cost-effective drug for a given patient.
In terminally ill patients, early use of an adjuvant analgesic is also considered when pain is accompanied by other symptoms that may respond to a nonanalgesic effect of the drug. For example, antidepressants are commonly used when pain is complicated by depression, and TCAs are especially useful when pain is accompanied by the inability to sleep or insomnia.
Drug Selection
A systematic review could not identify significant differences in analgesic efficacy across TCAs (McQuay, Tramer, Nye, et al., 1996). This conclusion is tentative, however, because very few studies have directly compared these drugs (Kroenke, Krebs, Bair, 2009). Although the extensive data from controlled clinical trials suggests that amitriptyline might be considered first when an antidepressant is indicated, despiramine and nortriptyline usually are better tolerated and may be more appropriate first-line options (Dworkin, O’Connor, Backonja, et al., 2007). Doxepin is sometimes used for intractable itching (pruritus), and an oral doxepin rinse has been shown to be effective in reducing the pain associated with oral mucositis due to cancer therapy (Epstein, Epstein, Epstein, et al., 2006).
There is sufficient evidence of analgesic efficacy for some of the SNRIs that a recent guideline on the treatment of neuropathic pain considers these drugs as first-line options (Dworkin, O’Connor, Backonja, et al., 2007). Other guidelines have recommended them as second-line treatment, to be tried if a TCA is ineffective, poorly tolerated, or relatively contraindicated (Moulin, Clark, Gilron, et al., 2007). Of the SNRIs, duloxetine has the most supporting data from randomized trials.
Substantial variability exists in the analgesic response to the different antidepressants. Failure of one drug might reasonably be followed by a trial of an alternative. No guidelines exist for drug selection during these sequential trials, and the process usually proceeds by trial and error.
Dose Selection
The starting dose of the TCAs should be low, 10 mg in older adults and 25 mg in younger adults (Dworkin, O’Connor, Backonja, et al., 2007). Beginning with a low initial dose and titrating relatively slowly thereafter, with intervals between dose changes as long as every 1 to 2 weeks, has been recommended for patients with organ failure (Kurella, Bennett, Chertow, 2003). Based on clinical experience, the interval between dose changes can be shorter—several days to one week—in those without significant medical illness.
The usual early dose increases are the same size as the starting dose; as the baseline dose increases, the increments can be greater, but usually no more than one-third of the baseline dose. For example, an older adult patient may begin with 10 mg of desipramine. Every 3 to 5 days, the dose may be increased by 10 mg until 50 mg is reached. In the absence of any adverse effects, the next increment may be 25 mg, and these 25 mg increments every few days may continue until the dose is 125 mg to 150 mg. Above this, the increments may be 50 mg.
The usual effective analgesic dose range for all of the commonly used TCAs is 50 to 150 mg, and the maximum recommended dose usually is 150 mg daily (Dworkin, O’Connor, Backonja, et al., 2007). Some patients will benefit from doses less than or greater than these parameters, however, and at doses above 100 mg it is prudent to obtain an ECG and to measure the plasma drug concentration, if possible. If the concentration of drug and metabolites is below the reported upper range for antidepressant effects, cautious titration may be continued (Dworkin, O’Connor, Backonja, et al., 2007). This evaluation of plasma concentration is not to imply that antidepressant blood levels are needed for analgesia; in fact, the doses of TCAs used for analgesia are usually lower than those used for depression (Kroenke, Krebs, Bair, 2009). A check of the concentration can be reassuring, however, and allow dose escalation to explore the analgesic potential of the drug.
Duloxetine can be started at 20 mg to 30 mg once daily and increased after 1 week to 40 mg to 60 mg once daily. The effective dose in clinical trials was 60 mg daily (Dworkin, O’Connor, Backonja, et al., 2007). Although doses of 60 mg twice daily do not appear to produce significantly better pain relief and are associated with increased adverse effects, individual patients benefit and dose escalation to this limit is appropriate if outcomes are not favorable and adverse effects are not a problem.
Milnacipran is started at a dose of 12.5 mg daily, rapidly titrated to 12.5 mg twice daily, and then 25 mg twice daily. The dose can then be gradually increased to a dose as high at 100 mg twice daily. Venlafaxine can be initiated using the long-acting formulation at a dose of 37.5 mg daily (Dworkin, O’Connor, Backonja, et al., 2007). The dose can be increased as tolerated each week, up to a maximum dose of 225 mg to 375 mg/day. Most patients who obtain pain relief achieve it at a dose lower than this maximum. Reductions in doses of venlafaxine, duloxetine, and milnacipran by 25% for mild-to-moderate renal or hepatic function impairment and 50% for those with moderate-to-severe renal or hepatic insufficiency are needed to minimize adverse effects (Leo, 2008). The manufacturer of duloxetine does not recommend its use in dialysis-dependent patients.
In the use of all the analgesic antidepressants, a dose lower than the usual antidepressant dose is expected to yield analgesic effects in responding patients, but it is impossible to know whether those who initially fail to respond at a lower dose than the antidepressant dose would in fact attain benefit if the dose were increased. Accordingly, a patient who does not benefit from the usual analgesic dose and has no adverse effects should have the dose titrated upward until the antidepressant dose is reached. This course is clearly justified in patients with a coexistent depression but should be considered even in patients without depression. As noted previously, laboratory measurement of plasma drug concentration is possible for most of the TCAs, and monitoring of these levels may reveal that typical doses are yielding low levels, indicating rapid metabolism of the drug or the possibility that the patient is not taking all doses. If adherence is not an issue, rapid metabolizers can undergo dose titration beyond the usual dose range, with repeated monitoring of the plasma concentration.
Most patients take a single nighttime dose of the TCAs to prevent excessive daytime sedation. The less sedating SNRIs and SSRIs are usually taken in a single daytime dose, or twice daily. If pain relief is adequate throughout the day during treatment with a TCA but the patient experiences “hangover” or double vision in the morning, the total daily dose may be divided into two doses given in the early and late evening. Similarly, if the patient has increased pain in the afternoon, the total daily dose may be divided into two doses, one given in the morning and one in the evening.
As noted, dose tapering is important when planning to discontinue an analgesic, particularly if treatment has continued for more than 6 weeks. In the absence of serious toxicity, slow tapering over 2 weeks reduces the risk of any withdrawal phenomena. Table V-1, pp. 748-756, at the end of Section V contains dosing recommendations for all of the commonly used antidepressants.
Monitoring Therapy
Changes in pain, mood, cognitive status, sleep pattern, and other effects must be carefully monitored during antidepressant analgesic dose escalation. Although favorable analgesic effects are usually observed within 1 week after achieving an effective dosing level, some patients accrue benefit over a longer period, and antidepressant effects may be further delayed in onset. The potential delay to realize the full benefit from these drugs, combined with the period required for dose titration, may result in a relatively lengthy period during which patients experience unsatisfactory effects from the therapy. Unless the patient is well-informed about this potential, the patient is likely to discontinue the drug.
Corticosteroids
Corticosteroids are considered multipurpose analgesics. Their use in this capacity is largely limited to the cancer population. (See Chapter 30 for a discussion of their use in the treatment of malignant bowel obstruction.) In the heterogeneous population with persistent noncancer pain, corticosteroids often are used as analgesics for a brief trial, based on the goal of “breaking the cycle” of pain, or they are used on a prolonged basis in inflammatory conditions as disease-modifying agents.
In the cancer population, corticosteroids are used to treat many pain syndromes, including bone pain and pain from stretching of the liver capsule due to metastasis, neuropathic pain from infiltration or compression of neural structures, headache caused by increased intracranial pressure, arthralgia, and pain caused by obstruction of hollow viscera (e.g., bowel or ureter) (Knotkova, Pappagallo, 2007; Lussier, Portenoy, 2004; Lussier, Huskey, Portenoy, 2004; Miaskowski, Cleary, Burney, et al., 2005). An evidence-based review of analgesic options for cancer pain evaluated the available data and also found support for the administration of steroids for spinal cord compression (Dy, Asch, Naeim, et al., 2008).
A limited course of corticosteroids has been recommended for pain associated with complex regional pain syndrome (CRPS) (Quisel, Gill, Witherell, 2005). Short-term administration for analgesic and disease-modifying purposes often is used as well in patients with inflammatory arthritis (Simon, Lipman, Caudill-Slosberg, et al., 2002). Intraarticular corticosteroids are used to alleviate pain associated with intense flares of OA and rheumatoid arthritis (RA) (Furtado, Oliveira, Natour, 2005; Lavelle, Lavelle, Lavelle, 2007; Simon, Lipman, Caudill-Slosberg, et al., 2002). Long-term corticosteroid use, which is conventional practice in some populations with chronic autoimmune disease, is not an accepted therapy for the pain of OA.
In the cancer population, dexamethasone (Decadron and others) 8 mg given before radiation therapy can effectively reduce the incidence of pain flare (Chow, Loblaw, Harris et al., 2007), as well as improve functional outcomes (Loblaw, Perry, Chambers, et al., 2005). Pain flare occurs in as many as 25% of patients following palliative radiation therapy and can persist for several days (Loblaw, Wu, Kirkbride, et al., 2007).
Multiple symptoms occur among those with advanced illnesses, and corticosteroid therapy is favored because of the potential benefit of these drugs on anorexia, nausea, malaise, and overall quality of life (Lussier, Portenoy, 2004; Lussier, Huskey, Portenoy, 2004; Mercadante, Fulfaro, Casuccio, 2001; Mercadante, Berchovich, Casuccio, et al., 2007; Shih, Jackson, 2007). These drugs are also particularly useful in alleviating symptoms associated with primary brain tumors and metastases to the brain (Newton, 2007).
Corticosteroids are seldom used in the setting of acute, self-limited pain, but a randomized controlled trial that compared intraarticular dexamethasone and intraarticular morphine found that both reduced pain for 5 days following knee surgery in patients with chronic OA (Stein, Yassouridis, Szopko, et al., 1999). Numerous guidelines and systematic reviews also recommend that corticosteroids be considered for the prevention and treatment of postoperative nausea and vomiting (PONV) (Gan, Meyer, Apfel, et al., 2003, 2007; Habib, Gan, 2004; Habib, El-Moalem, Gan, 2004), and they are often included in multimodal postoperative treatment plans that identify prevention of PONV as a primary goal (Kehlet, Wilmore, 2008) (see Chapter 19). Coating endotracheal tubes with topical betamethasone may help to decrease edema and inflammation with a subsequent reduction in sore throat, hoarseness, and cough after tracheal intubation (Ayoub, Ghobashy, Koch, et al., 1998).
Mechanism of Action
Steroids bind to glucocorticoid receptors in cells to produce antiinflammatory and immunosuppressive effects (Buvanendran, Kroin, 2007). The relationship between these effects and analgesia is poorly understood. Any of several processes may be involved. Compression of pain-sensitive structures may be relieved by reduction of inflammation and edema or, in the case of steroid-responsive neoplasms, by shrinkage of a tumor mass (Lussier, Portenoy, 2004). Activation of nociceptors may be lessened by reduced tissue concentrations of some inflammatory mediators, specifically prostaglandins and leukotrienes. Aberrant electrical activity in damaged nerves may also be tempered by these agents (Lussier, Portenoy, 2004).
Adverse Effects
Well-recognized adverse effects are associated with short-term and long-term administration of corticosteroids and with withdrawal of these drugs after long-term use (Lussier, Portenoy, 2004). The risk of serious toxicity increases with the dose, duration of therapy, and predisposing factors associated with the medical condition of the patient (Buvanendran, Kroin, 2007). Concern about toxicity is the reason that long-term use of these drugs generally is considered only when they are required as disease-modifying agents or when the target population has advanced disease and short life expectancies.
Acute, short-term use of corticosteroids is generally well tolerated. Potential adverse effects include hyperglycemia, fluid retention, and GI disturbances ranging from dyspepsia to ulceration (Lussier, Portenoy, 2004). Similar to short-term use, long-term use in low doses is usually well-tolerated; however, long-term use can cause cushingoid habitus, skin changes, weight gain, hypertension, osteoporosis, myopathy, increased risk of serious infection, and GI toxicity (Lussier, Portenoy, 2004). Dexamethasone (16 mg to 24 mg/day) produces the least mineralcorticoid effect, which may reduce the risk of fluid overload and hypertension (Miaskowski, Cleary, Burney, et al., 2005). Treatment with a bisphosphonate, calcium, and vitamin D should be considered to lower the risk of corticosteroid-induced osteoporosis. Long-term co-administration of an NSAID is not recommended, if it can be avoided, given the increased risk of peptic ulcer disease (Lussier, Portenoy, 2004). If combined therapy is used, concomitant administration of a proton pump inhibitor is recommended (see Chapter 6).
Neuropsychologic toxicity can occur with short- or long-term corticosteroid administration and ranges from changes in mood and cognitive functioning to delirium. Mood changes may be at either end of the spectrum, that is, depression to euphoria (Jenkins, Bruera, 2000; Lussier, Portenoy, 2004). Those at highest risk are cognitively impaired patients and older patients (Jenkins, Bruera, 2000). Several mechanisms are thought to be responsible for neuropsychologic changes including their interaction at the neurotransmitter level at multiple receptor sites and enhanced catecholamine synthesis (Jenkins, Bruera, 2000). Neuropsychologic toxicity usually occurs early during treatment and when relatively high doses are administered. No specific steroid has been identified as more likely than another to produce the effect. This adverse effect can be difficult to evaluate and diagnose in patients with multisystem disease, such as those with advanced cancer (Jenkins, Bruera, 2000). Patients must be monitored for changes in mood and cognition during corticosteroid therapy, and the occurrence of these adverse effects typically requires a change in therapy. In some settings, the problem is best treated by the addition of an antidepressant, a mood-stabilizing drug, or a neuroleptic (Jenkins, Bruera, 2000). In others, the steroid should be tapered, and ultimately discontinued, or treatment should be switched to another corticosteroid to determine whether toxicities are different.
Systematic assessment of response to steroid therapy should include neurologic examination of muscle strength, in an effort to identify a steroid-induced myopathy at an early point in its course. Changes in mood, appetite, fluid status, and blood glucose levels associated with high-dose steroids also should be evaluated regularly during therapy. Depending on the context, any of these toxicities may necessitate tapering and discontinuation of the drug.
Withdrawal after long-term corticosteroid use can produce myalgia and arthralgia (“pseudorheumatism”), headache, and mood disturbance. Flares of symptoms for which steroid treatment was initiated may be experienced as well (Lussier, Portenoy, 2004). The rate at which to taper corticosteroids to prevent severe withdrawal phenomena is not known (Jenkins, Bruera, 2000). Slow dose reduction from long-term therapy is prudent. Some corticosteroids have a long half-life (e.g., dexamethasone), and elimination of the drug and resolution of adverse effects may take weeks (Jenkins, Bruera, 2000).
Indications
In the cancer population, corticosteroid administration is strongly indicated in the management of spinal cord compression, superior vena cava syndrome, and increased intracranial pressure (Dy, Asch, Naeim, 2008; Jenkins, Bruera, 2000; Lussier, Portenoy, 2004). The purpose in these conditions is to modify the disease process by reducing peritumoral edema, as well as providing pain relief. In an early survey of 83 patients given high doses of dexamethasone (96 mg/day for 2 weeks) for malignant epidural spinal cord compression, pain relief was observed in 64% of patients within hours of the initial dose (Greenberg, Kim, Posner, 1980). A relatively high-dose regimen of this type usually is considered in the setting of emerging spinal cord or cauda equina signs; a lower dose regimen (e.g., 8 mg dexamethasone twice daily) often is used to reduce symptoms when epidural disease is present or suspected, but emerging cord or cauda equina signs do not exist (Rolles, 2005). Radiation therapy for appropriately selected patients is included in evidence-based guidelines for the treatment of spinal cord compression and associated pain (Abrahm, 2004; McQuay, Collins, Caroll, et al., 1999; Miaskowski, Cleary, Burney, et al., 2005), and as mentioned earlier, prophylactic dexamethasone may help to prevent pain flares following radiotherapy (Chow, Loblaw, Harris, et al., 2007).
On the basis of anecdotal experience, corticosteroids are also administered for many other cancer pain syndromes, including metastatic bone pain, neuropathic pain caused by compression or infiltration of peripheral nerves or nerve plexus, painful lymphedema, pain caused by obstruction of hollow viscera, and pain caused by organ capsule dissention (Lussier, Portenoy, 2004). The usual approach is a relatively low-dose regimen, typically equivalent to 1 mg to 4 mg of dexamethasone 2 to 4 times per day.
In patients with advanced illness, corticosteroids are usually added to an opioid regimen after the opioid dose has been increased to dose-limiting adverse effects. Patients with these pain syndromes commonly have other symptoms that could potentially be improved by steroid therapy such as nausea or malaise, and corticosteroid therapy may be considered earlier if primarily indicated by these other symptoms (Lussier, Portenoy, 2004). The low-dose regimen also is preferred in this setting.
Drug Selection
The relative risks and benefits of the various corticosteroids are unknown. In the United States, dexamethasone usually is a first choice and one that gains theoretical support from the relatively low mineralocorticoid effects (less fluid and electrolyte retention) (Lussier, Portenoy, 2004). Prednisone (Sterapred) and methylprednisolone (Depo-Medrol, Medrol, Solu-Medrol) have also been used. For convenience in prescribing, many clinicians consider these equivalencies: methylprednisolone 8 mg = prednisone 10 mg = dexamethasone 2 mg (see Table 22-2 for equivalent doses of corticosteroids).
Table 22-2
Equivalent Doses of Corticosteroids
| Corticosteroid | Equivalent Dose (mg) |
| Cortisone | 25 |
| Hydrocortisone | 20 |
| Prednisolone | 5 |
| Prednisone | 5 |
| Methylprednisolone | 4 |
| Triamcinolone | 4 |
| Dexamethasone | 0.75-1 |
| Betamethasone | 0.6 |
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 647, St. Louis, Mosby. Data from Clinical Pharmacology Online. Gold Standard, Inc. Available at http://clinicalpharmacology.com. Accessed August 1, 2009. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Dose Selection
As noted, corticosteroids are usually administered either in a high-dose regimen or a low-dose regimen. These recommendations derive from very few data and mostly reflect clinical experience. Table V-1, pp. 748-756, at the end of Section V contains dosing recommendations for corticosteroids.
High-Dose Regimen
A high-dose regimen (e.g., dexamethasone, 100 mg, followed initially by 96 mg/day in divided doses, usually 24 mg every 6 hours) has been used for oncologic emergencies, such as emerging spinal cord or cauda equina compression or worsening superior vena cava obstruction, and also has been advocated for patients who experience an acute episode of very severe pain that cannot be promptly reduced with opioids, such as that associated with a rapidly worsening malignant plexopathy (Lussier, Portenoy, 2004). The dose can be tapered over weeks, concurrent with the initiation of other analgesic approaches, such as radiotherapy.
Low-Dose Regimen
A low-dose corticosteroid regimen (e.g., usually dexamethasone, 1 to 2 mg twice daily, with a range of 1 mg twice daily to 4 mg every 6 hours) has been used for patients with advanced medical illness who continue to have pain despite optimal dosing of opioid drugs. In most cases, long-term therapy is planned. Although the risks associated with prolonged steroid use in this setting are more than balanced by the need for enhanced comfort, repeated assessments are required to ensure that benefits are sustained (Lussier, Portenoy, 2004). Ineffective regimens should be tapered and discontinued and, in all cases, the lowest dose that yields the desired results should be sought.
As mentioned, short-term and long-term corticosteroid therapy also is administered for other types of pain. A brief “pulse” of an oral drug, e.g., for 1 to 2 weeks, may be used to treat an acute flare of an inflammatory disorder, such as RA, or acute neuropathic pains of various types (e.g., acute herpes zoster). The dose selected is variable and may range from prednisone 15 mg/day to 80 mg/day, or the equivalent dose of another steroid. Long-term treatment usually is undertaken as a disease-modifying therapy appropriate for chronic inflammatory conditions. The dose usually is equivalent to prednisone 10 mg/day, or less (Simon, Lipman, Caudill-Slosberg, et al., 2002).
Alpha2-Adrenergic Agonists
Animal and human studies have led to the classification of alpha2-adrenergic agonists as nonspecific, multipurpose analgesics for a variety of types of acute and persistent pain (Lussier, Portenoy, 2004). The most commonly used alpha2-adrenergic agonists are clonidine (Catapres, Duraclon), dexmedetomidine (Precedex), and tizanidine (Zanaflex). Each has unique properties and indications, adverse effect profiles, and preferable routes of administration. Following is a discussion of clonidine and tizanidine for persistent pain treatment. See Chapter 24 for more on transdermal clonidine use for persistent pain, Chapter 26 for perioperative clonidine use, and Chapter 27 for the use of dexmedetomidine for intravenous (IV) procedural sedation and goal-directed sedation in the critically ill.
Clonidine
Clonidine (Catapres, Duraclon) is a centrally-acting alpha2-adrenergic agonist that has been used for years to treat hypertension and more recently has been used as an aid in smoking cessation. Clonidine is approved in the United States for epidural administration, and both epidural and intrathecal administration are commonplace when it is used in neuraxial analgesia (Ackerman, Follett, Rosenquist, 2003; Christo, Mazloomdoost, 2008; Eisenach, Hood, Curry, 2000; Nielsen, Sjogren, 2008). Its use as a systemic analgesic (administered as a tablet or transdermal patch) is limited (see the paragraphs that follow).
An early meta-analysis reported that successful outcomes with systemic clonidine can only be expected in pain states associated with increased sympathetic nervous system activity (Motsch, Kamler, 1997). Indeed, clonidine has been specifically suggested as an option for complex regional pain syndrome (CRPS) (Berthelot, 2006). Although some have reported less benefit with intrathecal clonidine for this condition (Ackerman, Follett, Rosenquist, 2003), methodologic problems and the need for improvements in the research of this very complex pain syndrome have been cited as a challenge in interpreting observations of this type (Perez, Kwakkel, Zuurmond, et al., 2001).
Despite the suggestion that systemic clonidine may have particular usefulness in syndromes characterized by sympathetically-maintained pain, it has been explored for other types of persistent pain, including persistent headaches, various neuropathic pains, and some cancer pain syndromes (Ackerman, Follett, Rosenquist, 2003; Christo, Mazloomdoost, 2008; Eisenach, Du Pen, Dubois, et al., 1995; Lussier, Portenoy, 2004; Tumber, Fitzgibbon, 1998). Some recommend a trial of clonidine for intractable central pain, such as following spinal cord injury (Devulder, Crombez, Mortier, 2002).
Systemic therapy using the commercially-available transdermal clonidine patch was studied in a randomized controlled trial that included 41 patients with painful diabetic neuropathy; there was little effect overall, but a secondary analysis suggested that patients with sharp and shooting pain may benefit (Byas-Smith, Max, Muir, et al., 1995). Others have found benefit with clonidine cream for orofacial neuropathic pain described as lancinating and sharp more than burning and aching (Epstein, Grushka, Le, 1997).
The transdermal clonidine patch also was tested in postoperative pain. The clonidine patch or a placebo patch was applied to patients the evening before undergoing abdominal hysterectomy followed by an IV clonidine or placebo infusion before anesthesia induction (Dimou, Paraskeva, Papilas, et al., 2003). Although those in the clonidine group required less analgesia during the first 24 hours postoperatively, there were no differences in pain relief or analgesic requirements thereafter.
Oral or transdermal clonidine is started at 0.1 mg/day (tablet) or TTS-1 patch (equivalent to 0.1 mg twice daily) and then is titrated upward until benefit occurs or adverse effects supervene (Elliott, 2009). If the patch is used, it is changed weekly. If doses lower than those commercially available are desirable, the transdermal system can be cut into pieces without changing its delivery properties.
Tizanidine
Tizanidine (Zanaflex), another centrally-acting alpha2-adrenergic agonist, is approved in the United States for the treatment of spasticity (Lussier, Portenoy, 2004). A Cochrane Collaboration Review concluded that there is not enough evidence to adequately compare the effectiveness of the various antispasmodics for treatment of multiple sclerosis spasticity (Shakespeare, Boggild, Young, 2003). However, a systematic review of various skeletal muscle relaxants used to treat spasticity related to multiple sclerosis found that tizanidine was more effective than placebo and had efficacy and toxicity comparable to baclofen (Lioresal) (Chou, Peterson, Helfand, 2004) (see Chapter 24 for a discussion of baclofen). Other systematic reviews have reported similar limited evidence for the effectiveness of tizanidine and many of the other commonly-used agents for spasticity associated with multiple sclerosis (Beard, Hunn, Wight, 2003; Paisley, Beard, Hunn, et al., 2002).
Conclusions were similar for a Cochrane Collaboration Review of the use of the various antispasmodics used to treat spasticity associated with spinal cord injury (Taricco, Adone, Pagliacci, et al., 2000). A cross-over, randomized controlled trial (N = 19) found tizanidine, titrated to a maximum tolerated dose over a 6-week period, also reduced spasticity associated with acquired brain injury, such as from stroke or trauma (Meythaler, Guin-Renfroe, Johnson, 2001).
Tizanidine has been reported to relieve neuropathic pain (Knotkova, Pappagallo, 2007; Semenchuk, Sherman, 2000), persistent neck and lower back pain that has a myofascial component (Malanga, Reiter, Garay, 2008), and myofascial pain syndrome (Malanga, Gwynn, Smith, et al., 2002). A combination of tizanidine and amitriptyline was found to be more effective for prevention of persistent tension headache than amitriptyline alone (Bettuci, Testa, Calzoni, et al., 2006).
Mechanism of Action
Alpha2 receptors are located in presynaptic sympathetic nerve endings and noradrenergic nerve endings in the dorsal horn of the spinal cord (Pandharipande, Ely, Maze, et al., 2006). When alpha2-adrenergic agonists bind to the latter receptor sites, the endogenous inhibitory pathway is activated and pain is diminished (Phillips, Gadiraju, Dickey, et al., 2007) (see Section I).
The ability of the alpha2-adrenergic agonists to block the sympathetic nervous system, which regulates CV functions, explains the efficacy of these drugs for the treatment of hypertension, although the introduction of angiotensin-converting enzyme (ACE) inhibitors and β-adrenergic antagonists has relegated clonidine to a third-line treatment option today (Jones, Maze, 2001). This also explains the adverse effects of bradycardia and hypotension that can occur with administration of alpha2-adrenergic agonists (see the discussion on adverse effects in the following paragraphs). Research is beginning to uncover the potential ability of some of the alpha2-adrenergic agonists to provide renal, cardiac, and neurocognitive protection (Sanders, Maze, 2007; Kamibayashi, Mace, 2000).
The capacity of alpha2-adrenergic agonists to inhibit nociceptive input to the CNS may indicate a role in the prevention of different types of pain syndromes. For example, by reducing sympathetic tone, clonidine may produce analgesia for pain that is sustained, at least in part, by circulating catecholamines or efferent activity in the sympathetic nervous system (so-called sympathetically-maintained pain) (Lussier, Portenoy, 2004). Although exact mechanisms are unclear, antihyperalgesia properties have been noted with both clonidine and dexmedetomidine (Kamibayashi, Mace, 2000) (see Section I for more on hyperalgesia).
The alpha2 receptors are thought to influence spatial memory (recording information about the environment). Dexmedetomidine is the most highly selective in this class for the alpha2 receptors, possessing a receptor affinity eight times that of clonidine. This strong affinity for this receptor subtype underlies its unique characteristic of enhancing, rather than diminishing, cognitive performance during sedation (Kamibayashi, Mace, 2000). This intense binding also has implications for its administration, i.e., the loading dose must be infused very slowly (e.g., over 20 minutes) to minimize bradycardia and hypotension (Phillips, Gadiraju, Dickey, et al., 2007). Dexmedetomidine is also shorter acting than clonidine, making it more appropriate for short-procedure sedation (see Chapter 27 for more on dexmedetomidine).
Adverse Effects
The most common adverse effects of the alpha2-adrenergic agonists are excessive sedation, hypotension, bradycardia, and dry mouth (Lussier, Portenoy, 2004). However, sedation is not accompanied by respiratory depression, and delayed sedation has not been observed. The occurrence of sedation can be more common and pronounced in patients who are debilitated or at the end of life. Cautious use of clonidine is recommended in older adults as orthostatic hypotension and CNS adverse effects can be more pronounced in this population.
Alpha2-adrenergic agonists can produce either hypotension or hypertension (Kamibayashi, Mace, 2000). With high doses or rapid bolus dosing of dexmedetomidine, there is an initial peripheral vasoconstriction and hypertension caused by activation of the alpha2 receptors located in the smooth muscle cells in vessels (Golembiewski, 2005; Kamibayashi, Mace, 2000). Reflex bradycardia can occur as well. These effects are transient and can be reduced or managed by decreasing the rate of administration or stopping infusion if they persist. An initial low heart rate that returns to baseline after administration is discontinued has been noted (Venn, Grounds, 2001). An early controlled trial of epidural clonidine in cancer pain demonstrated that this drug produced sustained hypotensive effects in almost 50% of patients but was considered serious in only 2 patients (Eisenach, Du Pen, Dubois, et al., 1995). Although most patients experience no hypotension, or mild and transient changes in BP, alpha2-adrenergic agonists generally should be avoided in patients who are hemodynamically unstable or predisposed to serious hypotension (e.g., autonomic neuropathy, intravascular volume depletion, or concurrent therapy with potent hypotensive agents).
The most frequent adverse effects of tizanidine are dry mouth and excessive sedation. The drug is often compared to baclofen and possesses similar adverse effects except that tizanidine produces more dry mouth and baclofen produces more muscle weakness (Chou, Peterson, Helfand, 2004). Liver function should be checked during administration of tizanidine as elevated liver function tests have been reported (Semenchuk, Sherman, 2000), and the drug should be stopped if hepatotoxicity occurs. Tizanidine is not recommended in patients with severe hepatic dysfunction, and the dose should be reduced with renal impairment, especially if creatinine clearance is less than 25 mL/min (Elliott, 2009). A number of drugs, including oral contraceptives, some antibiotics, some histamine receptor type-2 (H2) antagonists, and several antiarrhythmic drugs affect clearance of the drug (Elliott, 2009).
Indications
Clonidine and tizanidine can be considered for the treatment of all types of intractable persistent pain (Lussier, Portenoy, 2004). Both systemic administration (by the oral or transdermal route) and intraspinal administration can yield favorable effects. A trial of a systemic formulation (oral or transdermal clonidine or oral tizanidine) typically is considered only after therapeutic trials of many other adjuvant analgesics have proved ineffective. This is appropriate given the limited supporting data, adverse effect liability, and observations suggesting that a minority of those with intractable pain (less than 25%) are likely to respond. The limited experience with the use of alpha2-adrenergic agonists in advanced illness suggests that these drugs are appropriate only after titrated opioids and other adjuvant analgesics have failed to provide acceptable pain relief (Lussier, Portenoy, 2004).
Although clonidine has more evidence of analgesic efficacy than tizanidine, the latter drug may be preferable if hypotension is a concern (Lussier, Portenoy, 2004). Tizanidine also is preferred for the treatment of pain associated with spasm caused by multiple sclerosis or spinal cord or brain injury (Chou, Peterson, Helfand, 2004; Meythaler, Guin-Renfroe, Johnson, 2001).
Alpha2-adrenergic agonists blunt the signs of drug withdrawal, which include sympathetic hyperactivity (e.g., hypertension and tachycardia), anxiety, agitation, and generalized pain. Withdrawal may accompany cessation or dose reduction of diverse drug classes, including opioids, benzodiazepines, nicotine, and alcohol. Reports of the successful use of clonidine and dexmedetomidine to reduce these symptoms can be found in the literature (Bamgdade, 2006; Multz, 2003; Jones, Maze, 2001; Kamibayashi, Maze, 2000; Moss, Glick, 2005; Pandharipande, Ely, Maze, 2006; Westfall, Westfall 2006). If the clinical scenario suggests that pain could be related to substance withdrawal, an earlier trial of clonidine would be justified.
Routes of Administration
Clonidine can be administered via the oral, transdermal, IV, and intraspinal routes. Tizanidine is available in oral capsules and tablets.
Although evidence is strong that neuraxial clonidine administration is an effective therapy for some patients with refractory pain, this route of administration may not be possible in clinical settings that might lack pain specialists who can insert catheters and implantable devices or nursing support to manage therapy. An alternative may be the oral or transdermal route (see Chapter 24 for more on transdermal clonidine). As mentioned, the drug is well-absorbed by the oral route and has 100% bioavailability. Tizanidine, on the other hand, undergoes significant first-pass effect and has an oral bioavailability of 40% (Elliott, 2009) (see Chapter 11 for discussion of first-pass effect).
Because clonidine is lipophilic, it is rapidly absorbed into the systemic circulation after epidural administration. However, it is approximately twice as effective given epidurally as intravenously, a reflection of its spinal site of action for analgesia. The relative potency of epidural to intrathecal clonidine has been shown to differ depending on type of pain. Twenty-four healthy volunteers were randomized to receive either intrathecal or epidural clonidine in various doses and were subjected to thermal noxious stimuli (acute pain model [nociception]) or intradermal capsaicin (central hypersensitivity model [neuropathic]) (Eisenach, Hood, Curry, 2000). Intrathecal clonidine was found to be at least 10 times more potent than epidural clonidine to relieve acute thermal pain but two or less times more potent than epidural clonidine for capsaicin-induced mechanical hyperalgesia and allodynia. For acute thermal pain, intrathecal clonidine produced a dose-dependent analgesia with a lumbar > thoracic > cervical gradient; however, only one dose of epidural clonidine reduced thermal pain and only at the thoracic testing site.
Intraspinal clonidine is administered via implanted and external infusion devices and is usually combined with other drugs. Research has established the stability and compatibility of admixtures containing clonidine and other agents, including baclofen (Alvarez, de Mazancourt, Chartier-Kastler et al., 2004), bupivacaine (Classen, Wimbish, Kupiec, 2004; Wulf, Gleim, Mignat, 1994), hydromorphone (Rudich, Peng, Dunn, et al., 2004), meperidine (Vranken, van Kan, van der Vegt, 2006), and morphine (Classen, Wimbish, Kupiec, 2004; Hildebrand, Elsberry, Hassenbusch, 2003) (see Chapter 15).
Dose Selection
To limit the risk of adverse effects, doses of the alpha2-adrenergic agonists should be started low and increased slowly. Monitoring of both pain and adverse effects, particularly BP, is necessary during gradual dose escalation. An analgesic ceiling dose has not been determined for systemic clonidine therapy, but there appears to be a ceiling on intrathecal clonidine at 75 mcg (Eisenach, Hood, Curry, 2000). Table V-1, pp. 748-756, at the end of Section V contains dosing recommendations for the commonly used alpha2-adrenergic agonists.
Cannabinoids
Although the use of cannabinoids for pain is still very limited, emerging preclinical and clinical data suggest that they are best categorized as multipurpose drugs. Historically, these agents sparked considerable controversy, prejudice, and confusion concerning their appropriateness as medicinal agents, mostly because cannabinoids have some relation to the cannabis plant—also known as marijuana.
A synthesized form of one of marijuana’s active ingredients is delta-9-tetrahydrocannabinol (THC), which is marketed as dronabinol (Marinol), available in 2.5, 5, and 10 mg capsules, and approved for medical use in the United States, Canada, and the United Kingdom. Nabilone, another derivative, also is available in the United States for the indication of nausea. In the United Kingdom and Canada, a cannabis extract (Sativex), a spray that contains THC and cannabidiol and is delivered under the tongue, has been approved for the treatment of neuropathic pain from multiple sclerosis and advanced cancer pain. In the United States, some states already have legislation or ballot initiatives for crude herbal cannabis for medical use with specific indications (McCarberg, 2007).
Cannabinoids exert effects on the endogenous cannabinoid system to produce a wide variety of effects, including analgesia, psychologic effects, sedation, and appetite stimulation (Martin, Wiley, 2004). The analgesic effects of cannabinoids are mediated through the cannabinoid-1 (CBI) and cannabinoid-2 (CB2) receptors in the periphery and CNS (McCarberg, 2007; Hosking, Zajicek, 2008; Huskey, 2006). CBI stimulation modulates central neurotransmission in the serotonergic, dopaminergic, and glutamatergic systems as well as other systems (see Section 1). It is also believed that cannabinoids enhance the endogenous opioid system. The CB2 receptor is involved in the inflammatory response.
Smoking marijuana or cannabis rapidly increases plasma levels of THC, but the amount is highly variably, primarily dependent on the composition of the marijuana cigarette and inhalation technique. Thus, this form of use is both illegal and associated with unpredictable and less reliable results in relieving pain and symptoms (Hosking, Zajicek, 2008); it cannot be recommended. It also carries risks associated with any smoked substance.
Commercially available oral preparations offer better absorption and bioavailability, and are much more reliable and predictable. In an early single-dose study of patients with cancer pain, THC, 10 mg orally, was well tolerated and produced analgesic effects similar to 60 mg codeine, but a higher dose yielded severe adverse effects in many patients (Noyes, Brunk, Avery, et al., 1976). Thus, the therapeutic window for this drug appears to be narrow, and maximal efficacy at tolerable doses is limited (Martin, Wiley, 2004). A more recent crossover study randomized 32 patients with fibromyalgia to receive 0.5 mg of nabilone or 10 mg of amitriptyline before sleep; after 1 week, the dose was doubled based on whether or not the physician thought the patient might benefit from an increase in dose (Ware, Fitzcharles, Joseph, et al., 2010). More patients taking amitriptyline (26 of 28) required a dose increase at 1 week than patients taking nabilone (21 or 29). Both drugs improved the quality of sleep, but nabilone was superior. There were no effects on pain, mood, or quality of life. Nabilone adverse effects were mild to moderate and included dizziness, nausea, and dry mouth.
Recent studies of nabilone also suggest limited utility from this drug. Although an observational study in 112 cancer patients yielded promising results (Maida, Ennis, Irani, et al., 2008), a randomized, cross-over trial (N = 96) indicated that the drug was no more effective than dihydrocodeine and was less well-tolerated (Frank, Serpell, Hughes, et al., 2008).
In contrast, recent large and well-controlled studies of the THC-cannabidiol mixture (Sativex) yielded positive outcomes. A randomized controlled study of 125 patients with neuropathic pain of peripheral origin showed a significant reduction in pain and allodynia, favorable effects that persisted during an open-label extension study for 52 weeks (Nurmikko, Serpell, Hoggart, et al., 2007). Another controlled study of 66 patients with central pain from multiple sclerosis (Rog, Nurmikko, Friede, et al., 2005) revealed that the drug was well-tolerated and was effective in reducing pain and sleep disturbance. Other studies in patients with refractory cancer pain led to the approval of the drug for this indication in two countries, with development proceeding in the United States and elsewhere.
The reason that a mixture of cannabinoid alkaloids is more effective than single compounds is not known. The studies with the extract confirm that cannabinoids are analgesic and can benefit patients with diverse types of pain. The utility of these drugs will increase with the availability of the THC-cannabidiol formulation and other formulations on the market. At this time, trials of THC or nabilone should be considered only in the situation of pain that has not responded to many other, more established drug therapies.
Dizziness, somnolence, and dry mouth are the most common adverse effects associated with the cannabinoids. With pharmaceutical preparations, the incidence of adverse effects with short-term use involving euphoria, disorientation, confusion, dissociation, or depressed mood is relatively low. Studies of the THC-cannabidiol mixture suggest that better outcomes occur with drugs that include a combination of cannabinoid compounds.
There are many other beneficial effects for cannabinoids, such as alleviation of nausea and increased appetite (Martin, Wiley, 2004). In addition to some pain relief, they may also have opioid dose-sparing effects, possibly reduce opioid tolerance, and even ameliorate symptoms of opioid withdrawal (Svendsen, Jensen, Bach, 2004; McCarberg, 2007). Additional studies may provide new agents for clinical use and hopefully clarify the relative benefits and burdens.
Some authors have explored the controversial elements inherent in the use of cannabis substances for medical purposes (Degenhardt, Hall, 2008). First, the approval of cannabis for treatment of medical conditions in certain states and countries has occurred without strong evidence for its safety and efficacy. Second, the review of evidence from clinical trials involving cannabinoids and cannabis extracts for medical indications shows that the short-term benefits of this therapy and adverse effects have been limited for the drugs that have been available, although this is likely to change with new formulations. Last, there is a compelling need for more rigorous and scientific research to evaluate adverse effects, especially with long-term use for conditions such as multiple sclerosis and persistent pain. A systematic review and meta-analysis of cannabis treatment for persistent pain concluded that beneficial effects of the treatment may be partially or completely offset by potentially serious harms and urged more research to clarify the true balance of benefits to harms (Martin-Sanchez, Furukawa, Taylor et al., 2009).
Conclusion
Multipurpose adjuvant analgesic drugs have been shown to be effective for a variety of diverse pain syndromes and can be considered for the treatment of any type of pain. The multipurpose agents that are currently thought to be among the more useful in clinical practice include antidepressants, corticosteroids, and alpha2-adrenergic agonists. Except for antidepressants, the multipurpose adjuvant analgesics are appropriate for both acute and persistent pain. Antidepressants are not appropriate for acute pain because of their delayed onset of action.