Aldolase
Test Explanation
Serum aldolase is very similar to the enzymes aspartate aminotransferase (AST) (see p. 119) and creatine kinase (CK) (see p. 186). Aldolase is an enzyme used in the glycolytic breakdown of glucose. As with AST and CPK, aldolase is present in most tissues of the body. This test is most useful for identifying muscular or hepatic cellular injury or destruction. The serum aldolase level is very high in patients with muscular dystrophies, dermatomyositis, and polymyositis. Levels also are increased in patients with gangrenous processes, muscular trauma, and muscular infectious diseases (e.g., trichinosis). Elevated levels are also noted in chronic hepatitis, obstructive jaundice, and cirrhosis.
Neurologic diseases causing weakness can be differentiated from muscular causes of weakness with this test. Normal values are seen in patients with such neurologic diseases as poliomyelitis, myasthenia gravis, and multiple sclerosis. Elevated aldolase levels are seen in patients with primary muscular disorders.
Test Results and Clinical Significance
Related Test
Creatine Kinase (CK) (p. 186). CK is another muscular enzyme and is more frequently used for identifying cardiac and skeletal injury.
Aldosterone
Indications
This test is used to diagnose hyperaldosteronism. To differentiate primary aldosteronism (adrenal pathology) from secondary aldosteronism (extraadrenal pathology), a plasma renin assay must be performed simultaneously.
Test Explanation
Aldosterone, a hormone produced by the adrenal cortex, is a potent mineralocorticoid. Production of aldosterone is regulated primarily by the renin-angiotensin system. This system works as follows: a decreased effective renal blood flow triggers pressure-sensitive renal glomerular elements to release renin. The renin then stimulates the liver to secrete angiotensin I, which is converted to angiotensin II in the lung and kidney. Angiotensin II is a potent stimulator of aldosterone (see Figure 2-26, p. 448).
Secondarily, aldosterone is stimulated by adrenocorticotropic hormone (ACTH), low serum sodium levels, and high serum potassium levels. Aldosterone in turn stimulates the renal tubules to absorb sodium (water follows) and to secrete potassium in the urine. In this way, aldosterone regulates serum sodium and potassium levels. Because water follows sodium transport, aldosterone also partially regulates water absorption (and plasma volume).
Increased aldosterone levels are associated with primary aldosteronism in which a tumor (usually an adenoma) of the adrenal cortex (Conn syndrome) or bilateral adrenal nodular hyperplasia causes increased production of aldosterone. The typical pattern for primary aldosteronism is an increased aldosterone level and a decreased renin level. The renin level is low because the increased aldosterone level “turns off” the renin-angiotensin system. Patients with primary aldosteronism characteristically have hypertension, weakness, polyuria, and hypokalemia.
Increased aldosterone levels also occur with secondary aldosteronism caused by nonadrenal conditions. These include the following:
• Renal vascular stenosis or occlusion
• Hyponatremia (from diuretic or laxative abuse) or low salt intake
• Pregnancy or use of estrogens
• Poor perfusion states (e.g., congestive heart failure)
• Decreased intravascular volume (e.g., cirrhosis, nephrotic syndrome).
In secondary aldosteronism, aldosterone levels and renin levels are high.
The aldosterone assay can be performed on a 24-hour urine specimen or a plasma blood sample. The advantage of the 24-hour urine sample is that short-term fluctuations are eliminated. Plasma values are more convenient to sample, but they are affected by short-term fluctuations. Factors that can rapidly cause fluctuation in aldosterone levels include the following:
• Diurnal variation: Peak aldosterone levels occur in early morning. In late afternoon the levels are cut in half.
• Body position: In the upright position, plasma aldosterone levels are greatly increased.
• Diet: Levels of both urine and plasma aldosterone are increased by low-sodium diets and are decreased by high-sodium diets. (Diets high and low in potassium have the opposite effect.)
A 24-hour urine collection is therefore much more reliable because the effect of these interfering factors is dampened.
Primary aldosteronism can be diagnosed by demonstrating little or no increase in renin levels after aldosterone stimulation (using salt restriction as the stimulant). This is because aldosterone is already maximally secreted by the pathologic adrenal gland. Also, patients with primary aldosteronism fail to suppress aldosterone after saline infusion (1.5 to 2 L of normal saline solution infused between 8 AM and 10 AM). Aldosterone can be measured in blood obtained from adrenal venous sampling. In this situation, high levels from the right and left adrenal veins are diagnostic of bilateral adrenal hyperplasia. Unilateral high aldosterone levels are found in patients with aldosterone-producing tumors of the adrenal gland or renal artery stenosis. Renin levels are usually obtained at the same time. High unilateral renin levels with unilateral high aldosterone levels indicate renal artery stenosis. Aldosterone-producing tumors of the adrenal gland are characterized by unilateral high adrenal vein aldosterone and low renin levels.
Interfering Factors
• Strenuous exercise and stress can stimulate adrenocortical secretions and increase aldosterone levels.
• Excessive licorice ingestion can cause decreased levels, because it produces an aldosterone-like effect.
• Values are influenced by posture, diet, pregnancy, and diurnal variations.
• Patient position can significantly affect aldosterone levels.
 If the test is performed by radioimmunoassay, recently administered radioactive medications will affect test results.
If the test is performed by radioimmunoassay, recently administered radioactive medications will affect test results.
Drugs that may cause increased levels include diazoxide (Hyperstat), hydralazine (Apresoline), nitroprusside (Nipride), diuretics, laxatives, potassium, and spironolactone.
Drugs that may cause decreased levels include angiotensin-converting enzyme inhibitors (e.g., captopril), fludrocortisone (Florinef), licorice, and propranolol (Inderal).
 Clinical Priorities
Clinical Priorities
• Aldosterone levels exhibit a diurnal variation, with peak levels occurring early in the morning and lower levels in the late afternoon.
• Body position affects aldosterone levels. Levels are greatly increased in the upright position. Usually patients should be sitting up for at least 2 hours before blood is drawn.
• Levels of both urine and plasma aldosterone are increased by low-sodium diets and are decreased by high-sodium diets. Patients should maintain a normal-sodium diet (approximately 3 g/day) for at least 2 weeks before blood or urine collection.
Procedure and Patient Care
Before
 Explain the procedure for blood collection to the patient. Usually the patient is asked to be in the upright position (at least sitting) for a minimum of 2 hours before blood is drawn. Occasionally blood will be drawn again before the patient gets out of bed. Inform nonhospitalized patients when to arrive at the laboratory and to maintain the upright position for at least 2 hours.
Explain the procedure for blood collection to the patient. Usually the patient is asked to be in the upright position (at least sitting) for a minimum of 2 hours before blood is drawn. Occasionally blood will be drawn again before the patient gets out of bed. Inform nonhospitalized patients when to arrive at the laboratory and to maintain the upright position for at least 2 hours.
Tell the patient that no fasting is necessary.
Explain the procedure for collecting a 24-hour urine sample.
Give the patient verbal and written instructions regarding dietary and medication restrictions.
Instruct the patient to maintain a normal-sodium diet (approximately 3 g/day) for at least 2 weeks before blood or urine collection.
Have the patient ask the physician whether drugs that alter sodium, potassium, and fluid balance (e.g., diuretics, antihypertensives, steroids, oral contraceptives) should be withheld. Test results will be more accurate if these are suspended at least 2 weeks before either the blood or urine test.
Inform the patient that renin inhibitors (e.g., propranolol) should not be taken 1 week before the test if confirmed by the physician.
Tell the patient to avoid licorice for at least 2 weeks before the test because of its aldosterone-like effect.
During
Blood Collection
• Collect a venous blood sample in a gold-top (serum separator) tube. For hospitalized patients, the sample is occasionally drawn first with the patient in the supine position. A second specimen (upright sample) is collected 4 hours later after the patient has been up and moving.
• Obtain the specimen in the morning.
• Indicate on the laboratory slip if the patient was supine or standing during the venipuncture.
• Handle the blood specimen gently. Rough handling may cause hemolysis and alter the test results.
• Transport the specimen on ice to the laboratory.
• Indicate the source of the specimen (i.e., peripheral or adrenal vein).
Urine Collection
Instruct the patient to begin the 24-hour urine collection after discarding the first morning specimen. This is the start time of the 24-hour collection.
• Collect all urine passed over the next 24 hours into a container containing a boric acid preservative.
Instruct the patient to void before defecating so that the urine is not contaminated by feces.
Remind the patient not to put toilet paper in the collection container.
• Keep the urine specimen on ice or keep it refrigerated during the 24 hours.
• Collect the last specimen as close as possible to the end of the 24 hours.
Test Results and Clinical Significance
 Increased Levels
Increased Levels
Primary Aldosteronism
Secondary Aldosteronism
Diuretic ingestion resulting in hypovolemia and hyponatremia,
Poor perfusion states (e.g., congestive heart failure),
Decreased intravascular volume (e.g., cirrhosis, nephrotic syndrome),
Pregnancy and oral contraceptives,
The renin-angiotensin system is stimulated in these conditions. Renin levels are high, and aldosterone secretion is stimulated.
Cushing disease: Abnormally high ACTH levels secreted by a pituitary adenoma act as a direct stimulant to aldosterone.
 Decreased Levels
Decreased Levels
Renin deficiency: This is very rare and results in aldosterone deficiency.
Steroid therapy: ACTH is suppressed and therefore aldosterone is suppressed.
Addison disease: The adrenal cortex is not functional and therefore aldosterone cannot be secreted.
Patients on a high-sodium diet,
Antihypertensive therapy: Some antihypertensive medications inhibit aldosterone secretion.
Related Tests
Sodium, Blood (p. 466), Urine (p. 946), and Potassium, Blood (p. 409), Urine (p. 942). These are direct measurements of these electrolytes.
Adrenocorticotropic Hormone (ACTH) (p. 31). This is a test of anterior pituitary gland function.
Renin Assay (p. 447). This test is helpful in the differential diagnosis of primary versus secondary aldosteronism.
Alkaline Phosphatase (ALP)
Test Explanation
Although ALP is found in many tissues, the highest concentrations are found in the liver, biliary tract epithelium, and bone. The intestinal mucosa and placenta also contain ALP. This phosphatase enzyme is called alkaline because its function is increased in an alkaline (pH of 9 to 10) environment. This enzyme test is important for detecting liver and bone disorders. Within the liver, ALP is present in Kupffer cells. These cells line the biliary collecting system. This enzyme is excreted into the bile. Enzyme levels of ALP are greatly increased in both extrahepatic and intrahepatic obstructive biliary disease and cirrhosis. Other liver abnormalities, such as hepatic tumors, hepatotoxic drugs, and hepatitis, cause smaller elevations in ALP levels. Reports have indicated that the most sensitive test to indicate tumor metastasis to the liver is the ALP.
Bone is the most frequent extrahepatic source of ALP; new bone growth is associated with elevated ALP levels. Pathologic new bone growth occurs with osteoblastic metastatic (e.g., breast, prostate) tumors. Paget disease, healing fractures, rheumatoid arthritis, hyperparathyroidism, and normal-growing bones are sources of elevated ALP levels as well.
Isoenzymes of ALP are also used to distinguish between liver and bone diseases. These isoenzymes are most easily differentiated by the heat stability test and electrophoresis. The isoenzyme of liver origin (ALP1) is heat stable; the isoenzyme of bone origin (ALP2) is inactivated by heat. The detection of isoenzymes can help differentiate the source of the pathologic condition associated with the elevated total ALP. ALP1 would be expected to be high when liver disease is the source of the elevated total ALP. ALP2 would be expected to be high when bone disease is the source of the elevated total ALP. Another way to separate the source of elevated ALP is to simultaneously test for 5′-nucleotidase. This later enzyme is made predominantly in the liver. If total ALP and 5′-nucleotidase are concomitantly elevated, the disease is in the liver. If 5′-nucleotidase is normal, the bone is the most probable source.
 Age-Related Concerns
Age-Related ConcernsInterfering Factors
• Recent ingestion of a meal can increase the ALP level.
• Age: Young children with rapid bone growth have increased ALP levels. This is most magnified during the growth spurt. Females and males differ in age of growth spurt.
Drugs that may cause increased ALP levels include albumin made from placental tissue, allopurinol, antibiotics, azathioprine, colchicine, fluorides, indomethacin, isoniazid (INH), methotrexate, methyldopa, nicotinic acid, phenothiazine, probenecid, tetracycline, and verapamil.
Drugs that may cause decreased levels include arsenicals, cyanides, fluorides, nitrofurantoin, oxalates, and zinc salts.
Test Results and Clinical Significance
Related Tests
Alanine Aminotransferase (ALT) (p. 39). This liver enzyme can aid in the differential diagnosis of causes of ALP elevation. If the ALT is elevated along with the ALP, hepatocellular disease is suspected.
Aspartate Aminotransferase (AST) (p. 119). This liver enzyme can aid in the differential diagnosis of causes of ALP elevation. If the AST is elevated along with the ALP, hepatocellular disease is suspected.
Gamma-Glutamyl Transpeptidase (GGT) (p. 246). This liver enzyme can aid in the differential diagnosis of causes of ALP elevation. If the GGT is elevated along with the ALP, diseases affecting the biliary tree are suspected.
5′-Nucleotidase (p. 376). This liver enzyme can aid in the differential diagnosis of causes of ALP elevation. If the 5′-nucleotidase is elevated along with the ALP, diseases affecting the biliary tree are suspected.
Acid Phosphatase (p. 25). This bone enzyme can aid in the differential diagnosis of causes of ALP elevation. If the acid phosphatase is elevated along with the ALP, bone disease is suspected.
Creatine Kinase (CK) (p. 186). This enzyme exists predominantly in heart and skeletal muscle.
Lactic Dehydrogenase (LDH) (p. 329). This intracellular enzyme is used to support the diagnosis of injury or disease involving the heart, liver, red blood cells, kidneys, skeletal muscle, brain, and lungs.
Leucine Aminopeptidase (p. 337). This enzyme is specific to the hepatobiliary system. Diseases affecting that system will cause elevation of this enzyme.
Allergy Blood Testing (IgE Antibody Test, Radioallergosorbent test [RAST])
Normal Findings
| RAST Rating | IgE Level (KU/L) | Comment |
| 0 | <0.35 | Absent or undetectable allergen specific IgE |
| 1 | 0.35-0.69 | Low level of allergen specific IgE |
| 2 | 0.70-3.49 | Moderate level of allergen specific IgE |
| 3 | 3.50-17.49 | High level of allergen specific IgE |
| 4 | 17.50-49.99 | Very high level of allergen specific IgE |
| 5 | 50-100 | Very high level of allergen specific IgE |
| 6 | >100 | Extremely high level of allergen specific IgE |
Indications
Allergy blood testing is an alternative to allergy skin testing in diagnosing allergy as a cause of a particular symptom complex. It is also useful in identifying the specific allergen affecting a patient. It is particularly helpful when allergy skin testing is contraindicated.
Test Explanation
Measurement of serum IgE is an effective method to diagnose allergy and specifically identify the allergen (the substance to which the person is allergic). Serum IgE levels increase when allergic individuals are exposed to the allergen. Various classes of allergens can initiate the allergic response. They include animal dandruff, foods, pollens, dusts, molds, insect venoms, drugs, and agents in the occupational environment.
Although skin testing (see p. 1079) can also identify a specific allergen, measurement of serum levels of IgE is helpful when a skin test result is questionable, when the allergen is not available in a form for dermal injection, or when the allergen may incite an anaphylactic reaction if injected. IgE is particularly helpful in cases in which skin testing is difficult (e.g., in infants or in patients with dermatographism or widespread dermatitis), and it is not always necessary to remove the patient from antihistamines. The decision concerning which method to use to diagnose an allergy and to identify the allergen depends on the elapsed time between exposure to an allergen and testing, class of allergen, age of patient, the possibility of anaphylaxis, and the affected target organ (such as skin, lungs, or intestine). In general, allergy skin testing is the preferred method in comparison with various in vitro tests for assessing the presence of specific IgE antibodies because it is more sensitive and specific, simpler to use, and less expensive.
IgE levels, like provocative skin testing, are used not only to diagnose allergy, but also to identify the allergen so that an immunotherapeutic regimen can be developed. Increased levels of total IgE can be diagnostic of allergic disease in general. Specific IgE blood allergy testing, however, is an in vitro test for specific IgE directed to a specific allergen. Since the development of liquid allergen preparations, the use of in vitro blood allergy testing has increased considerably. It is more accurate and safer than skin testing.
Once the allergen has been identified, for most patients, the treatment would include avoidance of the allergen and use of bronchodilators, antihistamines, and possibly steroids. If aggressive antiallergy treatment is provided before testing, IgE levels may not rise despite the existence of an allergy.
Allergy to latex-containing products is an increasingly common allergy for which certain industrial and most medical personnel are at risk. It is an allergy that may develop in otherwise nonallergic patients because of overexposure. Furthermore, patients with latex exposure are at risk for allergic reaction if they undergo operative procedures or any procedure for which the health care personnel wear latex gloves. In these patients a latex-specific IgE can be easily identified with the use of an enzyme-labeled immunometric assay. This test is 94% accurate.
There are many methods of measuring IgE. One of the most commonly used methods is the radioallergosorbent test (RAST). In this method, the serum of a patient suspected of having a specific allergy is mixed with a specific allergen. The antibody-allergen complex is then incubated with one or more radiolabeled monoclonal anti-IgE antibodies. The total amount of IgE can be measured. Enzyme-conjugated, radioimmunometric, colorimetric, fluorometric, or chemiluminometric methods are used for allergen-specific IgE quantification. Accuracy varies between 45% and 95% depending on the allergen.
Allergy testing of IgG antibodies can also be performed and may provide a more accurate correlation between allergen and allergic symptoms. Like IgE antibody testing, IgG antibody testing is often performed in “panels.” For example, there are meat panels that might include IgE or IgG testing for chicken, duck, goose, and turkey. Testing a fruit panel might include IgE or IgG antibody testing for apples, bananas, peaches, and pears. Testing in panels diminishes the cost of testing. Specific allergen antibody testing can follow panel testing. Testing methods vary by laboratory. They may include ELISA/EIA or Quantitative Immunofluorescence Enzyme Assay.
Procedure
Before
Explain the procedure to the patient.
Remind the patient that the suspected allergen will be mixed with the patient's blood specimen in the laboratory. The patient will not experience any allergic reaction by this method of testing.
• Determine if the patient has recently been treated with a corticosteroid for allergies.
Related Tests
Allergy Skin Testing (p. 1079). Skin testing is the easiest and cheapest manner of determining specific allergic reactions. However, skin testing is not available for many allergens and may instigate an anaphylactic response.
Immunoglobulin Quantification (p. 312). This test is used to assist in the diagnosis of several different disease states. Elevated levels of IgE are occasionally detected, indicating that the disease is associated with an allergic response.
Alpha1-Antitrypsin (AAT, A1AT, AAT phenotyping)
Indications
Serum alpha1-antitrypsin (AAT) determinations are obtained in patients with a family history of emphysema, because there is a familial tendency for a deficiency of this anti-enzyme. Deficient or absent serum levels of this enzyme can cause the early onset of disabling emphysema. A similar deficiency in AAT is seen in children with cirrhosis and other liver diseases.
AAT is also an acute-phase reactant protein that is elevated in the presence of inflammation, infection, or malignancy. It is not specific regarding the source of the inflammatory process.
Test Explanation
AAT inactivates endoproteases (protein catabolic enzymes that are released in the body by degenerating and dying cells), such as trypsin and neutrophil elastase, that can break down elastic fibers and collagen, especially in the lung. Deficiencies of AAT can be genetic or acquired. Acquired deficiencies in AAT can occur in patients with protein-deficiency syndromes (e.g., malnutrition, liver disease, nephrotic syndrome, neonatal respiratory distress syndrome). People with AAT deficiency develop severe panacinar (although usually more severe in the lower third of the lungs) emphysema in the third or fourth decade of life. Their major clinical symptoms usually include progressive dyspnea with minimal coughing. Chronic bronchitis is prominent in those patients with deficient AAT levels who smoke. Bronchiectasis can also occur in these patients.
Inherited AAT deficiency is associated with symptoms earlier in life than acquired AAT disease. Inherited AAT is also commonly associated with liver and biliary disease. AAT Genetic Phenotyping (AAT phenotyping) has shown that most persons have two AAT “M” genes (designated as MM) and AAT levels over 250 mg/dL. “Z” and “S” gene mutations are typically associated with alterations in serum levels of AAT. Individuals who are ZZ or SS homozygous have serum levels below 50 mg/dL and often near zero.
Individuals who are MZ or MS heterozygous have diminished or low-normal serum levels of AAT. Approximately 5% to 14% of the adult population have this heterozygous state, which is considered to be a risk factor for emphysema. Homozygous individuals have severe pulmonary and liver disease very early in life. AAT Phenotyping is particularly helpful when blood AAT levels are suggestive but not definitive.
AAT is qualitatively measured using immunochemical methods. Quantification is possible but rarely useful with phenotyping. Routine serum protein electrophoresis (p. 424) is a good screening test for AAT deficiency, because AAT accounts for about 90% of the protein in the alpha1-globulin region on electrophoresis.
Procedure and Patient Care
After
• Apply pressure or a pressure dressing to the venipuncture site.
• Observe the venipuncture site for bleeding.
If the results show the patient is at risk for emphysema, begin patient teaching. Include such factors as avoidance of smoking, infection, and inhaled irritants; proper nutrition; adequate hydration; and education about the disease process of emphysema.
If the test is positive, genetic counseling is indicated. Other family members should be tested to determine their and their children's risks.
Test Results and Clinical Significance
Decreased Levels
Early onset of emphysema (adults),
Neonatal respiratory distress syndrome,
These diseases are a result of endoproteases working uninhibited (no AAT available) within the body. Collagen is broken down, setting up the destruction of lung and liver structures.
Low serum proteins: Diseases such as malnutrition, end-stage cancer, nephrotic syndrome, protein-losing enteropathy, and hepatic failure are associated with lack of protein synthesis. AAT is a protein and therefore will not be produced in adequate quantities in these diseases.
Alpha-Fetoprotein (AFP, Alpha1-Fetoprotein)
Indications
This test is used as a screening marker indicating increased risk for birth defects, such as fetal body wall defects, neural tube defects, and chromosomal abnormalities. It can also be used as a tumor marker to identify cancers.
Test Explanation
AFP is an oncofetal protein normally produced by the fetal liver and yolk sac. It is the dominant fetal serum protein in the first trimester of life and diminishes to very low levels by the age of 1 year. Normally it is found in very low levels in the adult.
AFP is an effective screening serum marker for fetal body wall defects. The most notable of these is neural tube defects, which can vary from a small myelomeningocele to anencephaly. If a fetus has an open body wall defect, fetal serum AFP leaks out into the amniotic fluid and is picked up by the maternal serum. Normally AFP from fetal sources can be detected in the amniotic fluid or the mother's blood after 10 weeks' gestation. Peak levels occur between 16 and 18 weeks. Maternal serum reflects that change in amniotic AFP levels. When elevated maternal serum AFP levels are identified, further evaluation with repeat serum AFP levels, amniotic fluid AFP levels, and ultrasound is warranted. Other examples of fetal body wall defects would include omphalocele and gastroschisis.
Elevated serum AFP levels in pregnancy may also indicate multiple pregnancy, fetal distress, fetal congenital abnormalities, or intrauterine death. Low AFP levels after correction for age of gestation, maternal weight, race, and presence of diabetes are found in mothers carrying a fetus with trisomy 21 (Down syndrome). There are other indicators of trisomy that are often performed simultaneously. See Maternal Screen Testing (p. 354) and Fetal Nuchal Translucency (p. 888).
AFP is also used as a tumor marker. Increased serum levels of AFP are found in as many as 90% of patients with hepatomas. The higher the AFP level, the greater the tumor burden. A decrease in AFP is seen if the patient is responding to antineoplastic therapy. AFP is not specific for hepatomas, although extremely high levels (above 500 ng/mL) are diagnostic for hepatoma. Other neoplastic conditions, such as nonseminomatous germ cell tumors and teratomas of the testes, yolk sac and germ cell tumors of the ovaries, and to a lesser extent Hodgkin disease, lymphoma, and renal cell carcinoma, are also associated with elevated AFP levels. Testing methods for AFP quantification include radioimmunoassay or enzyme-linked immunosorbent assay (ELISA) with a commercially available kit. Noncancerous causes of elevated AFP levels occur in patients with cirrhosis or chronic active hepatitis.
Procedure and Patient Care
If an AFP test is to be performed on amniotic fluid, follow “Procedure and Patient Care” for amniocentesis, p. 632.
Test Results and Clinical Significance
Increased Maternal Serum Levels
Neural tube defects (e.g., anencephaly, encephalocele, spina bifida, myelomeningocele),
Abdominal wall defects (e.g., gastroschisis, omphalocele):
If a fetus has an open body wall defect, fetal serum AFP leaks out into the amniotic fluid and is picked up by the maternal serum.
Multiple-fetus pregnancy: The multiple fetuses make large quantities.
Related Tests
Maternal Screen Testing (p. 354). This testing includes AFP and other serum markers that are accurate indicators of increased risk for birth defects.
Amniocentesis (p. 632). This is a procedure to obtain amniotic fluid for evaluation of fetal health.
Pelvic Ultrasonography (p. 887). Nuchal Translucency is an accurate indicator of trisomy chromosomal abnormalities.
Aluminum (Chromium and Other Heavy Metals)
Indications
This test is used to evaluate aluminum levels in patients with renal failure. Elevated concentrations of aluminum in a patient with an aluminum-based joint implant suggest significant prosthesis wear.
Test Explanation
Under normal physiologic conditions, the usual daily dietary intake of aluminum (5-10 mg) is completely excreted by the kidneys. Patients in renal failure (RF) lose the ability to clear aluminum and are at risk for aluminum toxicity. Aluminum-laden dialysis water and aluminum-based phosphate binder gels designed to decrease phosphate accumulation increase the incidence of aluminum toxicity in RF patients. Furthermore, the dialysis process is not highly effective at eliminating aluminum. If a significant load exceeds the body's excretory capacity, the excess is deposited in various tissues, including bone, brain, liver, heart, spleen, and muscle. This accumulation causes morbidity and mortality through various mechanisms. Brain deposition has been implicated as a cause of dialysis dementia. In bone, aluminum replaces calcium and disrupts normal osteoid formation.
Aluminum is absorbed from the GI tract in the form of oral phosphate-binding agents (aluminum hydroxide), parenterally via immunizations, via dialysate on patients on dialysis, via total parenteral nutrition (TPN) contamination, via the urinary mucosa through bladder irrigation, and transdermally in antiperspirants. Lactate, citrate, and ascorbate all facilitate GI absorption.
Serum aluminum concentrations are likely to be increased above the reference range in patients with metallic joint prosthesis. Serum concentrations >10 ng/mL in a patient with an aluminum-based implant suggest significant prosthesis wear. Chromium and other metals can be determined using similar laboratory techniques.
Interfering Factors
• Most of the common evacuated blood collection devices have rubber stoppers that are comprised of aluminum-silicate. Simple puncture of the rubber stopper for blood collection is sufficient to contaminate the specimen with aluminum; therefore special evacuated blood collection tubes are required for aluminum testing.
• Gadolinium- or iodine-containing contrast media that has been administered within 96 hours can alter test for heavy metals including aluminum.
Amino Acid Profiles (Amino Acid Screen)
Indications
Measurement of certain amino acids is performed to identify diseases associated with specific essential amino acid deficiencies.
Test Explanation
Amino acids are “building blocks” of proteins, hormones, nucleic acids, and pigments. They can act as neurotransmitters, enzymes, and coenzymes. There are eight essential amino acids that must be provided to the body by the diet. The body can make the others. The essential amino acids must be transported across the gut and renal tubular lining cells. The metabolism of the essential amino acids is critical to the production of other amino acids, proteins, carbohydrates, and lipids. Amino acid levels can thereby be affected by defects in renal tubule or gastrointestinal (GI) transport of amino acids.
When there is a defect in the metabolism or transport of any one of these amino acids, excesses of their precursors or deficiencies of their “end product” amino acid are evident in the blood and/or urine. There are more than 90 diseases described that are associated with abnormal amino acid function.
Clinical manifestations of these diseases may be precluded if diagnosis is early, and appropriate dietary replacement of missing amino acids is provided. Usually urine testing (see phenylketonuria [PKU] testing, p. 374) for specific amino acids is used to screen for some of these errors in amino acid metabolism and transport. Blood testing is very accurate. Federal law now requires hospitals to test all newborns for inborn errors in metabolism, including amino acids. Testing is required for errors in amino acid metabolism such as phenylketonuria (PKU), maple syrup urine disease (MSUD), and homocystinuria. Testing for more rare disorders may include testing for tyrosinemia and argininosuccinic aciduria.
A few drops of blood are obtained from the heel of a newborn baby to fill a few circles on filter paper (Guthrie card) labeled with names of infant, parent, hospital, and primary physician. The sample is usually obtained on the second or third day of life, after protein-containing feedings (i.e., breast milk or formula) have started,
Once a presumptive diagnosis is made, amino acid levels can be determined by chromatographic methods on blood or amniotic fluid. The genetic defects for many of these diseases are becoming more defined, allowing for even earlier diagnosis to be made in utero. Common examples of amino acid diseases include PKU, cystinosis, and cystic fibrosis.
Interfering Factors
• Amino acid levels are affected by the circadian rhythm. Levels are usually lowest in the morning and highest by mid-day.
• Levels of amino acids are generally higher in infants and children compared to adults.
• Pregnancy is associated with reduced levels of some amino acids.
• Normal values vary widely and only extremely abnormal results are diagnostic without genetic corroboratory evidence.
Drugs that may increase amino acid levels include bismuth, heparin, steroids, and sulfonamides.
Drugs that may decrease some amino acid levels include estrogens and oral contraceptives.
Procedure and Patient Care
Before
• Obtain a history of the patient symptoms.
• Obtain a pedigree highlighting family members with amino acid disorders.
Inform the patient that a 12-hour fast is generally required before blood collection. Occasionally, a particular protein or carbohydrate load is ordered to stimulate production of a particular amino acid metabolite.
Test Results and Clinical Significance
Increased Blood Levels
Specific aminoacidopathies (e.g., PKU, maple syrup disease): The parent amino acid is present at increased quantities because of a genetic defect that impairs catabolism of that particular amino acid. It is the excessive build up of that amino acid that causes disease.
Specific aminoacidemias (e.g., glutaric aciduria): Products in the catabolic pathway of a particular amino acid accumulate. Which particular product accumulates depends on which enzyme is deficient (usually as a result of a genetic defect).
Related Test
Phenylketonuria (PKU) (p. 374). This test is routinely performed on infants to exclude the diagnosis of PKU. It is part of the newborn screening panel.
Ammonia
Indications
Ammonia is used to support the diagnosis of severe liver diseases (fulminant hepatitis or cirrhosis), and for surveillance of these diseases. Ammonia levels are also used in the diagnosis and follow-up of hepatic encephalopathy.
Test Explanation
Ammonia is a by-product of protein catabolism. Most of it is made by bacteria acting on proteins present in the gut. By way of the portal vein, it goes to the liver, where it is normally converted into urea and then secreted by the kidneys. Ammonia cannot be catabolized in the presence of severe hepatocellular dysfunction. Furthermore, when portal blood flow to the liver is altered (e.g., in portal hypertension), ammonia cannot reach the liver to be catabolized. Ammonia blood levels rise. Plasma ammonia levels do not correlate well with the degree of hepatic encephalopathy. Inherited deficiencies of urea cycle enzymes, inherited metabolic disorders of organic acids, and the dibasic amino acids lysine and ornithine are a major cause of high ammonia levels in infants and adults. Finally, impaired renal function diminishes excretion of ammonia, and the blood levels rise. High levels of ammonia result in encephalopathy and coma. Arterial ammonia levels are more reliable than venous levels but more difficult to obtain and are therefore not routinely used.
Interfering Factors
• Hemolysis increases ammonia levels because the red blood cells (RBCs) have about three times the ammonia level content of plasma.
• Muscular exertion can increase ammonia levels.
• Cigarette smoking can produce significant increases in ammonia levels within 1 hour of inhalation.
• Ammonia levels may be factitiously increased if the tourniquet is too tight for a long period.
Drugs that may cause increased ammonia levels include acetazolamide, alcohol, ammonium chloride, barbiturates, diuretics (loop, thiazide), narcotics, and parenteral nutrition.
Drugs that may cause decreased levels include broad-spectrum antibiotics (e.g., neomycin), lactobacillus, lactulose, levodopa, and potassium salts.
Test Results and Clinical Significance
Increased Levels
Primary hepatocellular disease,
Severe heart failure with congestive hepatomegaly:
The portal blood flow from the gut to the liver is altered. The ammonia cannot get to the liver to be metabolized for excretion. Furthermore, the ammonia from the gut is rapidly shunted around the liver (by way of gastroesophageal varices) and into the systemic circulation.
Hemolytic disease of newborn (erythroblastosis fetalis): RBCs contain high amounts of ammonia. The newborn liver is not mature enough to metabolize all the ammonia presented to it by the hemolysis that occurs in this disease.
GI bleeding with mild liver disease,
GI obstruction with mild liver disease:
Ammonia production is increased because the bacteria have more protein (blood) to catabolize. An impaired liver may not be able to keep up with the increased load of ammonia presented to it.
Hepatic encephalopathy and hepatic coma: These neurologic states are a result of ammonia acting as false neurotransmitters. The brain cannot function properly.
Genetic metabolic disorder of urea cycle: Ammonia is catabolized by the urea cycle. Disruption of that cycle will inhibit excretion of ammonia and levels can be expected to rise.
Amylase, Blood
 Critical Values
Critical ValuesIndications
This test is used to detect and monitor the clinical course of pancreatitis. It is frequently ordered when a patient presents with acute abdominal pain.
Test Explanation
The serum amylase test, which is easy and rapidly performed, is most specific for pancreatitis. Amylase is normally secreted from pancreatic acinar cells into the pancreatic duct and then into the duodenum. Once in the intestine it aids in the catabolism of carbohydrates to their component simple sugars. Damage to pancreatic acinar cells (as in pancreatitis) or obstruction of the pancreatic duct flow (as in pancreatic carcinoma or common bile duct gallstones) causes an outpouring of this enzyme into the intrapancreatic lymph system and the free peritoneum. Blood vessels draining the free peritoneum and absorbing the lymph pick up the excess amylase. An abnormal rise in the serum level of amylase occurs within 12 hours of the onset of disease. Because amylase is rapidly cleared (2 hours) by the kidney, serum levels return to normal 48 to 72 hours after the initial insult. Persistent pancreatitis, duct obstruction, or pancreatic duct leak will cause persistent elevated amylase levels.
Although serum amylase is a sensitive test for pancreatic disorders, it is not specific. Other nonpancreatic diseases can cause elevated amylase levels in the serum. For example, during bowel perforation, intraluminal amylase leaks into the free peritoneum and is picked up by the peritoneal blood vessels. This results in an elevated serum amylase level. A penetrating peptic ulcer into the pancreas will also cause elevated amylase levels. Duodenal obstruction can be associated with less significant elevations in amylase. Because salivary glands contain amylase, elevations can be expected in patients with parotiditis (mumps). Amylase isoenzyme testing can differentiate pancreatic from salivary hyperamylasemia. Amylase is also found in low levels in the ovaries and skeletal muscles. Ectopic pregnancy and severe diabetic ketoacidosis are also associated with hyperamylasemia.
Patients with chronic pancreatic disorders that have resulted in pancreatic cell destruction or patients with massive hemorrhagic pancreatic necrosis often do not have high amylase levels, because there may be so few pancreatic cells left to make amylase.
Interfering Factors
• Serum lipemia may factitiously decrease amylase levels.
Intravenous dextrose solutions can lower amylase levels and cause a false-negative result.
Drugs that may cause increased serum amylase levels include aminosalicylic acid, aspirin, azathioprine, corticosteroids, dexamethasone, ethyl alcohol, glucocorticoids, iodine-containing contrast media, loop diuretics (e.g., furosemide), methyldopa, narcotic analgesics, oral contraceptives, and prednisone.
Drugs that may cause decreased levels include citrates, glucose, and oxalates.
Test Results and Clinical Significance
Increased Levels
Chronic relapsing pancreatitis:
Damage to pancreatic acinar cells, as in pancreatitis, causes an outpouring of amylase into the intrapancreatic lymph system and the free peritoneum. Blood vessels draining the free peritoneum and absorbing the lymph pick up the excess amylase.
Penetrating peptic ulcer into the pancreas: The peptic ulcer penetrates the posterior wall of the duodenum into the pancreas. This causes a localized pancreatitis with elevated amylase levels.
Gastrointestinal disease: In patients with perforated peptic ulcer, necrotic bowel, perforated bowel, or duodenal obstruction, amylase leaks out of the gut and into the free peritoneal cavity. The amylase is picked up by the blood and lymphatics of the peritoneum, where levels are demonstrated in excess.
Amylase is also present in the salivary glands, gallbladder, and fallopian tubes. Diseases affecting these organs will be associated with elevated levels of amylase.
Renal failure: Amylase is cleared by the kidney. Renal diseases will reduce excretion of amylase.
Related Tests
Urine Amylase (p. 909). Amylase can be detected in the urine long after serum amylase has cleared. If serum amylase levels are normal and pancreatitis is suspected, the period of peak amylase levels may have passed. Elevated amylase levels may still be found in the urine.
Lipase (p. 339). Lipase is similar to amylase except that it is more specific for the pancreas.
Angiotensin
Test Explanation
Renin (p. 447) is an enzyme that is released by the juxtaglomerular apparatus of the kidney. Its release is stimulated by hypokalemia, hyponatremia, decreased renal blood perfusion, or hypovolemia. Renin stimulates the release of angiotensinogen. Angiotensin-converting enzyme (ACE) (p. 64) metabolizes angiotensinogen to angiotensin I and subsequently to angiotensin II and III. Angiotensin then stimulates the release of catecholamines, antidiuretic hormone, ACTH, oxytocin, and aldosterone. Angiotensin is also a vasoconstrictor. Angiotensin is used to identify renovascular sources of hypertension. It can be measured as angiotensin I or angiotensin II. The test is performed by direct radioimmunoassay.
Procedure and Patient Care
Before
Explain the procedure to the patient.
Instruct the patient to maintain a normal diet with a restricted amount of sodium (approximately 3 g/day) for 3 days before the test.
Instruct the patient to check with a health care provider about discontinuing any medications that may interrupt in renin activity.
During
• Collect a venous blood sample and place it in a chilled lavender-top tube with ethylene diamine tetraacetic acid (EDTA) as an anticoagulant. Heparin can falsely decrease results.
• Gently invert the blood tube to allow adequate mixing of the blood sample and the anticoagulant.
• Record the patient's position, dietary status, and time of day on the laboratory slip.
• Place the tube of blood on ice, and immediately send it to the laboratory.
• In the laboratory, the blood will be centrifuged and the serum frozen.
Test Results and Clinical Significance
Increased Levels
Essential hypertension: A small percentage of these patients have renin hypertension and elevated angiotensin levels.
Malignant hypertension: A large percentage of these patients with aggressive hypertensive episodes have elevated angiotensin levels.
Renovascular hypertension: Renal artery stenosis or occlusion decreases the renal blood flow, which is a strong stimulant to angiotensin production.
Decreased Levels
Primary hyperaldosteronism: This is usually caused by an adrenal adenoma. Aldosterone levels are high and angiotensin levels are low.
Steroid therapy: Glucocorticosteroids also have an aldosterone effect, which acts to increase serum sodium levels, decrease potassium levels, and increase blood volume. These responses all tend to diminish angiotensin levels.
Congenital adrenal hyperplasia: An enzyme defect in cortisol synthesis causes an accumulation of cortisol precursors, some of which have strong aldosterone-like activity. These act to increase serum sodium levels, decrease potassium levels, and increase blood volume, all of which tend to diminish angiotensin levels.
Angiotensin-Converting Enzyme (ACE, Serum Angiotensin-Converting Enzyme [SACE])
Indications
ACE is used to detect and monitor the clinical course of sarcoidosis (a granulomatous disease that affects many organs, especially the lungs). Furthermore, it is used to differentiate between sarcoidosis and other granulomatous diseases. It is also used to differentiate active and dormant sarcoid disease.
Test Explanation
ACE is found in pulmonary epithelial cells and converts angiotensin I to angiotensin II (a potent vasoconstrictor). Angiotensin II is a significant stimulator of aldosterone. ACE is vital in the renin/aldosterone mechanism and therefore important in controlling blood pressure. Despite this, ACE is not very helpful in the evaluation of hypertension. Its value is in the detection of sarcoidosis.
Elevated ACE levels are found in a high percentage of patients with sarcoidosis. This test is primarily used in patients with sarcoidosis to evaluate the severity of disease and the response to therapy. Levels are especially high with active pulmonary sarcoidosis and can be normal with inactive (dormant) sarcoidosis. Elevated ACE levels also occur in conditions other than sarcoidosis, including Gaucher disease (a rare familial lysosomal disorder of fat metabolism), leprosy, alcoholic cirrhosis, active histoplasmosis, tuberculosis, Hodgkin disease, myeloma, scleroderma, pulmonary embolism, and idiopathic pulmonary fibrosis. ACE is elevated in the CSF of patients with neurosarcoidosis. An ACE assay can be performed using spectrophotometry or radioimmunoassay.
Test Results and Clinical Significance
Increased Levels
Sarcoidosis: This is the disease for which this test is primarily performed. The more severe the sarcoidosis, the greater the likelihood that ACE will be increased.
Other rare diseases that have been found to be associated with ACE elevations include Gaucher disease, tuberculosis, leprosy, alcoholic cirrhosis, active histoplasmosis, Hodgkin disease, myeloma, idiopathic pulmonary fibrosis, diabetes mellitus, primary biliary cirrhosis, amyloidosis, hyperthyroidism, scleroderma, and pulmonary embolism.
Anion Gap (AG, R factor)
Indications
Calculation of the anion gap (AG) assists in the evaluation of patients with acid-base disorders. It is used to attempt to identify the potential cause of the disorder and can also be used to monitor therapy for acid-base abnormalities.
Test Explanation
The anion gap (AG) is the difference between the cations and the anions in the extra-cellular space that are routinely calculated in the laboratory (i.e., AG = [sodium + potassium] − [chloride + bicarbonate]). In some laboratories, the potassium is not measured because the level of potassium in acid-base abnormalities varies. The normal value of the anion gap is adjusted downward if potassium is eliminated from the equation. The anion gap, although not real physiologically, is created by the small amounts of anions in the blood (such as lactate, phosphates, sulfates, organic anions, and proteins) that are not measured. Further, it is important to realize that the 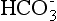 that is measured is actually the venous CO2, not the arterial .
that is measured is actually the venous CO2, not the arterial .
This calculation is most often helpful in identifying the cause of metabolic acidosis. As acids such as lactic acid or ketoacids accumulate in the bloodstream, bicarbonate neutralizes them to maintain a normal pH within the blood. Mathematically, when bicarbonate decreases, the AG increases. In general, most metabolic acidotic states (excluding some types of renal tubular acidosis) are associated with an increased anion gap. The higher the gap above normal, the more likely this will be the case. Proteins can have a significant effect on AG. As albumin (usually negatively charged) increases, AG will increase. In the face of normal albumin, a high AG is usually a result of an increase in non–chloride-containing acids or organic acids (such as lactic acid or ketoacids).
A decreased AG is very rare but can occur when there is an increase in unmeasured (calcium or magnesium) cations. A reduction in anionic proteins (nephrotic syndrome) will also decrease AG. For example, a 1 g/dL drop in serum protein is associated with a 2.5 mEq/L drop in AG. Because the anion proteins are lost, the increases to maintain electrical neutrality. Increase in cationic proteins (some immunoglobulins) will also decrease AG. Except for hypoproteinemia, conditions that cause a reduced or negative anion gap are relatively rare compared to those associated with an elevated anion gap.
AG measurement is also helpful in identifying the presence of a mixed acid-base situation. The arterial blood gases do not always tell the whole story, especially if there is a mixed metabolic acidosis and a concomitantly occurring alkalosis. An increased AG, despite a normal pH will indicate an acidotic component to the metabolic picture. When AG measurement is combined with the ABGs and electrolytes, complex metabolic clinical pictures can be more clearly elucidated. The AG calculation is indicated whenever an acid-base problem exists.
Interfering Factors
• Hyperlipidemia may cause under measurement of sodium and falsely decrease AG.
• Normal values of AG vary according to different normal values for electrolytes, depending on laboratory methods of measurement.
Drugs that increase AG are many. Examples include carbenicillin, carbonic anhydrase inhibitors (e.g., acetazolamide), diuretics, ethanol, methanol, penicillin, and salicylate.
Drugs that decrease AG are also many. Examples include acetazolamide, lithium, polymyxin B, spironolactone, and sulindac.
Test Results and Clinical Significance
Increased Levels
These diseases are associated with increased acid ions such as lactate, hydroxybutyrate, or acetoacetate. 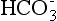 neutralizes these acids, levels fall and AG mathematically increases.
neutralizes these acids, levels fall and AG mathematically increases.
Renal failure: Uremic organic acid anions (phosphate, sulfates, etc.) accumulate in the blood as a result of poor excretion of these acids. The hydrogen combines with the bicarbonate to maintain a homeostatic pH. Bicarbonate levels diminish and AG mathematically rises.
Increased gastrointestinal (GI) losses of bicarbonate (e.g., diarrhea or fistulae): and other base losses can occur, thereby mathematically increasing AG. Not all GI losses result in AG differences, if mixed electrolyte imbalances occur.
Hypoaldosteronism: Aldosterone stimulates acid secretion in the distal renal tubule in exchange for sodium. With deficient quantities of aldosterone, acid builds up and is combined with bicarbonate. Bicarbonate levels diminish and AG rises.
Decreased Levels
Excess alkali ingestion: Increase in alkali products (antacids, boiled milk), especially in children, causes increased products and mathematically decreases AG.
Multiple myeloma: The M-chain component of the proteins produced by the neoplastic plasma cells are cationic, causing a compensatory decrease in measured cations and an increase in measured anions to maintain electrical neutrality.
Chronic vomiting or gastric suction: The loss of HCl causes a decrease in chloride and an increase in that mathematically decreases AG.
Hyperaldosteronism: These patients lose great amounts of potassium and hydrogen ions causing a metabolic alkalosis associated with a decreased AG.
Hypoproteinemia: Loss of anionic proteins directly causes a decrease in the AG.
Lithium toxicity: An increase in inorganic cations decreases the measured cations and thereby decreases AG.
Anticardiolipin Antibodies (aCL Antibodies, ACA, Antiphospholipid Antibodies, Lupus Anticoagulant)
Indications
This test is positive in some patients with systemic lupus erythematosus (SLE). The presence of this antibody places the patient at higher risk for “antiphospholipid syndrome” (i.e., venous or arterial thrombosis, thrombocytopenia, recurrent spontaneous abortions). This test is performed on patients with SLE to determine if the patient is at risk for developing the above-noted complications.
Test Explanation
Anticardiolipin antibodies (immunoglobulins G and M to cardiolipin) are antiphospholipid autoantibodies that attach to phospholipids of cell membranes and can interfere with the coagulation system. Antiphospholipid autoantibodies include anticardiolipin antibodies and the “lupus anticoagulant antibody.” Phospholipid antibodies occur in patients with a variety of clinical signs and symptoms, notably thrombosis (arterial or venous), pregnancy morbidity (unexplained fetal death, premature birth, severe preeclampsia, or placental insufficiency), unexplained cutaneous circulation disturbances (livido reticularis or pyoderma gangrenosum), thrombocytopenia or hemolytic anemia, and nonbacterial thrombotic endocarditis. Phospholipid antibodies and lupus anticoagulants are found with increased frequency in patients with systemic rheumatic diseases, especially lupus erythematosus. The term “antiphospholipid syndrome” (APS) or “Hughes syndrome” is used to describe the triad of thrombosis, recurrent fetal loss, and thrombocytopenia accompanied by phospholipid antibodies or a lupus anticoagulant. Both antibodies may be found in other autoimmune diseases, drug-induced lupus, and infection. These antibodies may be considered normal in the elderly person.
Antiphospholipid IgG and IgM antibodies are directed against a mixture of phosphatidylserine, phosphatidic acid, and beta-2 glycoprotein I antigens and are more specific than cardiolipin IgG and IgM antibodies in the diagnosis of antiphospholipid syndrome (APS). Testing is commonly performed by Semi-Quantitative Enzyme-Linked Immunosorbent Assay.
Interfering Factors
• Patients who have or had syphilis infections can have a cross reaction to the radiolabeled antibody used for radioimmune assay or the antibody used for enzyme-linked immunosorbent assay. These patients therefore will have a false-positive result.
• These transient antibodies can occur in patients with infections, acquired immunodeficiency syndrome (AIDS), inflammation, autoimmune diseases, or cancer.
False-positive results have been seen in patients who take medications such as chlorpromazine, hydralazine, penicillin, phenytoin, procainamide, and quinidine.
Anticentromere Antibody (Centromere Antibody)
Test Explanation
A centromere is the region of the chromosome referred to as the primary constriction that divides the chromosome into arms. During cell division the centromere exists in the pole of the mitotic spindle.
Anticentromere antibodies are a form of antinuclear antibodies. They are found in a very high percentage of patients with CREST syndrome, a variant of scleroderma. CREST is characterized by calcinosis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly (a hand deformity), and telangiectasia (permanent dilation of superficial capillaries and venules). Anticentromere antibodies, on the contrary, are present in only a small minority of patients with scleroderma, a disease that is difficult to differentiate from CREST. No correlation exists between antibody titer and severity of CREST disease.
Related Tests
Antinuclear Antibody (p. 88). This test is used to diagnose autoimmune-related diseases.
Antichromatin Antibody (Antinucleosome Antibodies [Anti-NCS], Antihistone Antibody [Anti-HST, AHA])
Test Explanation
There are several chromatin antinuclear antibodies associated with autoimmune diseases. Nucleosome (NCS) represents the main autoantigen-immunogen in systemic lupus erythematosus (SLE), and these antinucleosome antibodies are an important marker of the disease activity. Antinucleosome (antichromatin) antibodies play a key role in the pathogenesis of SLE. Nearly all patients with SLE have antinucleosome antibodies. Anti-NCS is one of the many antinuclear antibodies (see p. 88) that indicate autoimmune diseases. Anti-NCS has a sensitivity of 100% and specificity of 97% for SLE diagnosis. Anti-NCS antibodies show the highest correlation with disease activity. Anti-NCS antibodies also show strong association with renal damage (glomerulonephritis and proteinuria) associated with SLE. Anti-NCS autoantibodies are more prevalent than anti-DNA in patients with SLE.
Histone antibodies are present in 20% to 55% of idiopathic SLE cases and 80% to 95% of drug-induced lupus erythematosus cases. They occur in less than 20% of other types of connective tissue diseases. This antibody is particularly helpful in identifying patients with drug-induced lupus erythematosus caused by drugs such as procainamide, quinidine, penicillamine, hydralazine, methyldopa, isoniazid, and acebutolol. There are several subtypes of AHA. In drug-induced lupus erythematosus, a specific AHA (anti-[(H2A-H2B)-DNA] IgG) is produced. In most of the other associated diseases (rheumatoid arthritis, juvenile rheumatoid arthritis, primary biliary cirrhosis, autoimmune hepatitis, and dermatomyositis/polymyositis), the AHAs are of other varying specificities. A variety of immune testing methods are used to identify these antinuclear antibodies including ELISA and indirect immunofluorescence methods.
Test Results and Clinical Significance
Increased Levels
Systemic lupus erythematosus: This disease is most commonly associated with anti-NCS antibodies.
Drug-induced lupus erythematosus: This disease is most commonly associated with anti-HST antibodies.
Other autoimmune diseases: Diseases such as lupus hepatitis are occasionally associated with anti-NCS antibodies.
Anticyclic-Citrullinated Peptide Antibody (Cyclic Citrullinated Peptide Antibody, CCP IgG, Anti-CCP)
Indications
Anti-CCP is useful in the diagnosis of patients with unexplained joint inflammation, especially when the traditional blood test, rheumatoid factor (RF) (see p. 454), is negative or below 50 units/mL.
Test Explanation
Anti-CCP is known more formally as anticyclic-citrullinated peptide antibody and is formed by the intermediary conversion of the amino acid ornithine to arginine. Anti-CCP appears early in the course of rheumatoid arthritis (RA) and is present in the blood of most patients with the disease. When the citrulline antibody is detected in a patient's blood, there is a high likelihood that the patient has RA. Among patients with early RA, 30% to 40% may not have elevation of RF, making the diagnosis difficult in the initial stage. If the anti-CCP is elevated, the diagnosis of RA can be made even if RF is negative. This is particularly important because aggressive treatment in the early stages of RA prevents progression of joint damage. Anti-CCP may rise years before any clinical onset of arthritis or significant elevation of RF. At a cutoff of 5 units/mL, the sensitivity and specificity of anti-CCP for RA are 67.5% and 99.3%, respectively. RF has a sensitivity of 66.3% and a lower specificity (82.1%) than anti-CCP. When the two antibodies are used together, the specificity for diagnosing RA is 99.1%. Other autoimmune inflammatory diseases are rarely associated with elevated anti-CCP levels. It may also be useful in differentiating other entities that can resemble RA, and at times, cause RF positive test results (i.e., Polymyalgia Rheumatica and Parvoviral Arthropathy).
Anti-CCP is thought to be directly involved in the pathogenesis of RA. Citrullinated proteins are found in inflamed synovial tissue of patients with RA and may elicit a humoral mechanism for the joint inflammation that highlights RA. The presence of anti-CCP in RA indicates a more aggressive and destructive form of the disease. I–t is also a marker for disease progression. Some feel that anti–CCP-positive RA and anti–CCP-negative RA are clinically different disease entities—with the former having a far worse outcome. Anti–CCP-positive RA patients have more swollen joints and show more radiologic destruction than anti–CCP-negative patients with RA.
Anti-CCP ELISA testing is well correlated among the various commercially available assays with the same antigen specificity, but the numerical normal values for each assay differ widely. These methods include semi-quantitative enzyme-linked immunosorbent assay.
Related Tests
Rheumatoid Factor (RF) (p. 454). This is the most widely used test to assist in diagnosis and determining prognosis of RA.
Erythrocyte Sedimentation Rate (ESR) (p. 221). This test is used to assess RA disease activity.
C-Reactive Protein (CRP) (p. 184). This test is used to identify and assess treatment for most inflammatory diseases.
Antidiuretic Hormone (ADH, Vasopressin, Arginine Vasopressin [AVP])
Indications
ADH levels are tested in patients suspected of having diabetes insipidus or the syndrome of inappropriate ADH (SIADH). This test is often performed in patients who complain of polyuria or polydipsia and are found to have marked variations in blood and urine osmolarity or sodium levels.
Test Explanation
ADH, also known as vasopressin, is formed by the hypothalamus and stored in the posterior pituitary gland. It controls the amount of water reabsorbed by the kidney. ADH release is stimulated by an increase in serum osmolality or a decrease in intravascular blood volume. Physical stress, surgery, and even high levels of anxiety may also stimulate ADH release. On release of ADH more water is reabsorbed from the glomerular filtrate at the level of the distal convoluted renal tubule and collecting ducts. This increases the amount of free water within the bloodstream and causes highly concentrated urine.
With low ADH levels, water is excreted, thereby producing hemoconcentration and a more dilute urine. Diabetes insipidus (DI) occurs when ADH secretion is inadequate or when the kidney is unresponsive to ADH stimulation. Inadequate ADH secretion is usually associated with central neurologic abnormalities (neurogenic DI), such as trauma, tumor, or inflammation of the brain (hypothalamus). Surgical ablation of the pituitary gland will also result in the neurogenic form of DI; such patients excrete large volumes of free water in the dilute urine. Their blood is hemoconcentrated, producing a strong thirst.
Primary renal diseases may make the renal collecting system less sensitive to ADH stimulation (nephrogenic DI). Again, in this instance, dilute urine may be produced by excretion of high volumes of free water. To differentiate ADH deficiency (neurogenic DI) from renal resistance to ADH (nephrogenic DI), a water deprivation (ADH stimulation p. 979) test is performed. Water intake is restricted during this test. Urine osmolality is measured. Vasopressin is administered. In neurogenic DI, there is no rise in urine osmolality after water restriction, but there is a rise after vasopressin is given. In nephrogenic DI, there is no rise in urine osmolality after water deprivation or vasopressin administration. The diagnosis indicated by this test can be corroborated by a serum ADH level. ADH levels are low in neurogenic DI. ADH levels are high in nephrogenic DI.
High serum ADH levels are also associated with SIADH. In response to this inappropriately high level of ADH secretion, water is reabsorbed by the kidneys greatly in excess of normal amounts. Thus the patient's blood becomes very diluted and the urine is concentrated. Blood levels of important serum ions diminish, causing severe neurologic, cardiac, and metabolic alterations. SIADH can be associated with pulmonary diseases (e.g., tuberculosis, bacterial pneumonia), severe stress (e.g., surgery, trauma), CNS tumor, infection, or trauma. Ectopic secretion of ADH from neoplasm (paraneoplastic syndrome) can cause SIADH. The most common tumors associated with SIADH include carcinomas of the lung and thymus; lymphoma; leukemia; and carcinomas of the pancreas, urologic tract, and intestine. Patients with myxedema or Addison disease also can experience SIADH. Some drugs are know to cause SIADH (see Interfering Factors).
Interfering Factors
• Patients with dehydration, hypovolemia, or stress may have increased ADH levels.
• Patients with overhydration, decreased serum osmolality, and hypervolemia may have decreased ADH levels.
• Use of a glass syringe or collection tube causes degradation of ADH.
Drugs that increase ADH levels include acetaminophen, barbiturates, cholinergic agents, cyclophosphamide, diuretics (e.g., thiazides), estrogen, narcotics, nicotine, oral hypoglycemic agents (particularly sulfonylureas), and tricyclic or selective serotonin reuptake inhibitor (SSRI) antidepressants.
Drugs that decrease ADH levels include alcohol, beta-adrenergic agents, morphine antagonists, and phenytoin.
Test Results and Clinical Significance
Increased Levels
Central nervous system (CNS) tumors or infection,
Pneumonia or pulmonary tuberculosis,
Ectopic ADH secretion (usually from lung cancer),
Endocrinopathies, such as myxedema or Addison disease:
The above-noted diseases have been implicated to be associated with inappropriate high levels of ADH. Patients experience dilutional hyponatremia, hypoosmolality, and concentrated urine.
Nephrogenic DI caused by primary renal diseases: Because of primary renal disease, the kidneys cannot respond to ADH. The patient becomes hemoconcentrated and hyperosmolar. As a result, ADH is maximally stimulated, yet the kidneys cannot respond.
Severe physical stress (e.g., trauma, pain, prolonged mechanical ventilation):
Decreased Levels
Neurogenic (or central) DI caused by CNS trauma, tumor, or infection,
Surgical ablation of pituitary gland:
The hypothalamus or pituitary ADH-secreting cells are destroyed by the above-noted disease processes.
Hypervolemia: Increased blood volume is an inhibitor of ADH secretion.
Decreased serum osmolality caused by overhydration, nephrotic syndrome, psychogenic polydipsia, IV overinfusion of non–salt-containing fluid: Decreased serum osmolality is an inhibitor of ADH secretion.
Related Tests
Water Deprivation (p. 979). This test is helpful in differentiation of the causes of polyuria (neurogenic DI, nephrogenic DI, psychogenic polydipsia).
Osmolality, Blood (p. 378). This test is a measurement of solute load in the serum.
Osmolality, Urine (p. 938). This test is a measurement of solute load in the urine.
Sodium, Blood (p. 466). This is a direct measurement of sodium level in the blood.
Sodium, Urine (p. 946). This is a direct measurement of sodium level in the urine.
Antidiuretic Hormone Suppression (see following test). This test is used to differentiate SIADH from other causes of hyponatremia or edematous states.
Antidiuretic Hormone Suppression (ADH Suppression, Water Load)
Indications
This test is used to differentiate the syndrome of inappropriate ADH (SIADH) from other causes of hyponatremia or edematous states listed in Box 2-3.
Test Explanation
This test is used to evaluate the possibility of SIADH in patients with electrolyte abnormalities (such as hyponatremia) or edematous states. Usually this test is performed concomitantly with measurements of urine and serum osmolality. Patients with SIADH will excrete none or very little of the water load. Furthermore, their urine osmolality will never be less than 100, and the U/S ratio is greater than 100. Patients with hyponatremia, other edematous states, or chronic renal diseases will excrete up to 80% of the water load and will develop midrange osmolality results.
Interfering Factors
• Patients with dehydration, hypovolemia, hypotension, or stress may have increased ADH levels.
Drugs that increase ADH levels include acetaminophen, barbiturates, cholinergic agents, cyclophosphamide, estrogen, narcotics, nicotine, oral hypoglycemic agents, some diuretics (e.g., thiazides), and tricyclic antidepressants.
Procedure and Patient Care
Before
Explain the procedure to the patient.
Instruct the patient to fast after midnight before the test.
• The test is begun early in the morning.
Inform the patient of the early signs of water intoxication and instruct the patient to notify you if any occur.
• Place the patient in the recumbent position. The response to water loading in the upright position is reduced because this position is associated with increased ADH.
• One hour before the test administer 300 mL of water to replace fluids lost overnight. This is not part of the water load.
• Obtain a baseline serum sodium or a serum sodium level 24 hours before the test. If the sodium concentration is above a safe level (125 mmol/L), the test can proceed. If not, the test should be canceled until the sodium is brought to a safe level by water restriction or saline infusion. This precaution will minimize the risk of water intoxication.
During
• Administer water (approximately 20 mL/kg body weight up to 1500 mL) in 10 to 20 minutes.
• Collect urine every hour for 6 hours and send it for specific gravity and osmolality measurements. (Discard the first morning specimen.)
• Obtain blood for osmolality hourly or at specified times in serum (red-top) or heparinized (green-top) tube.
Related Tests
Antidiuretic Hormone (p. 73). This is a serum assay for direct measurement of ADH. This test is used in the differential diagnosis of neurogenic diabetes insipidus, nephrogenic diabetes insipidus, or psychogenic polydipsia.
Osmolality, Blood (p. 378). This test is a measurement of solute load in the serum.
Osmolality, Urine (p. 938). This test is a measurement of solute load in the urine.
Sodium, Blood (p. 466). This is a direct measurement of sodium level in the blood.
Sodium, Urine (p. 946). This is a direct measurement of sodium level in the urine.
Water Deprivation (p. 979). This is a test to assist in the differential diagnosis of diabetes insipidus.
Anti-DNA Antibody (Antideoxyribonucleic Acid Antibodies, Antibody to Double-Stranded DNA, Anti–Double-Stranded DNA [Anti–ds-DNA], DNA Antibody, Native Double-Stranded DNA)
Indications
The anti-DNA antibody test is useful for the diagnosis and follow-up of systemic lupus erythematosus (SLE).
Test Explanation
This antibody is found in approximately 65% to 80% of patients with active SLE and rarely in patients with other diseases. High titers are characteristic of SLE. Low to intermediate levels of this antibody may be found in patients with other rheumatic diseases and in those with chronic hepatitis, infectious mononucleosis, and biliary cirrhosis. The anti-DNA titer decreases with successful therapy and increases with exacerbation of SLE and especially with the onset of lupus glomerulonephritis. Near-negative values are seen in patients with dormant SLE. This test is semi-quantitative. Therefore small changes in antibody levels do not indicate disease activity.
The anti-DNA IgG antibody is a subtype of the antinuclear antibodies (ANAs) (p. 88). If the ANAs are negative, there is no reason to test for anti-DNA antibodies. There are two types of anti-DNA antibodies. The first and most commonly found is the antibody against double-stranded DNA (anti–ds-DNA). The second type is the antibody against single-stranded DNA (anti–ss-DNA). This is less sensitive and specific for SLE but is positive in other autoimmune diseases. These antibody-antigen complexes that occur with autoimmune disease are not only diagnostic but are major contributors to the disease process. These complexes induce the complement system, which then may cause local or systemic tissue injury.
There are several radioimmunoassay methods for measuring anti-DNA antibodies. The Farr method is the oldest and is more sensitive than the lupus erythematosus prep. It detects anti–ds-DNA and anti–ss-DNA antibodies. As a result, its specificity is not great.
Antiextractable Nuclear Antigen (Anti-ENA, Antibodies to Extractable Nuclear Antigens, Anti–Jo-1 [Antihistidyl Transfer Synthase], Antiribonucleoprotein [Anti-RNP], Anti-Smith [Anti-SM])
Indications
The anti-ENAs are used to assist in the diagnosis of systemic lupus erythematosus (SLE) and mixed connective tissue disease (MCTD) and to eliminate other rheumatoid diseases.
Test Explanation
Anti-ENAs are a type of antinuclear antibodies to certain nuclear antigens that consist of RNA and protein. The antigen is extracted from the thymus using phosphate-buffered saline solutions and therefore is sometimes referred to as saline-extracted antigen. The most common ENAs are Smith (SM) and ribonucleoprotein (RNP) types.
The anti-SM antibody is present in about 30% of patients with SLE and in about 8% of patients with MCTD diseases. However, it is not present in patients with most other rheumatoid-collagen diseases.
The anti-RNP antibody is reported in nearly 100% of patients with MCTD disease and in about 25% of patients with SLE, discoid lupus, and progressive systemic sclerosis (scleroderma). In high titers, anti-RNP is suggestive of MCTD.
The anti–Jo-1 (antihistidyl transfer synthase) antibody occurs in patients with autoimmune interstitial pulmonary fibrosis and in a minority of patients with aggressive autoimmune myositis. Two other antibodies to ENAs are anti–SS-A and anti–SS-B (see p. 98) and are used mainly in the diagnostic evaluation of Sjögren syndrome.
Antiglomerular Basement Membrane Antibody (Anti-GBM Antibody, AGBM, Glomerular Basement Antibody, Goodpasture's Antibody)
Indications
This test is used to detect the presence of circulating glomerular basement membrane antibodies commonly present in autoimmune-induced nephritis (Goodpasture syndrome).
Test Explanation
Goodpasture syndrome is an autoimmune disease characterized by the presence of circulating antibodies against an antigen in the renal glomerular basement membrane and the pulmonary alveolar basement membrane. These immune complexes activate the complement system and thereby cause tissue injury. Patients with this problem usually display a triad of glomerulonephritis (hematuria), pulmonary hemorrhage (hemoptysis), and antibodies to basement membrane antigens. This is a rare form of glomerular nephritis. About 60% to 75% of patients with immune-induced glomerular nephritis have pulmonary complications.
With the use of immunohistochemistry and now with radioimmunoassay, antibodies also can be demonstrated in the glomeruli, renal tubular basement membrane, and the pulmonary capillary basement membranes. Lung or renal biopsies are required to demonstrate these antibodies in tissue. Serum assays are a faster and more reliable method for diagnosing Goodpasture syndrome, especially in patients in whom renal or lung biopsy may be difficult or contraindicated. Furthermore, serum levels can be used in monitoring response to therapy (plasmapheresis or immunosuppression).
Anti-Glycan Antibodies (Crohn Disease Prognostic Panel, Multiple Sclerosis Antibody Panel)
Indications
This test is used to differentiate multiple sclerosis from other neurologic causes of weakness. It also is used to differentiate Crohn disease from other forms of inflammatory bowel diseases.
Test Explanation
Glycans (sugars or carbohydrates) exist on the surface of cells, such as erythrocytes. Anti-glycan antibodies are immunologically directed to these sugar-containing components. Antibodies to glycans can be instigated by bacterial, fungal, and parasitic infections. The use of glycan arrays for systematic screening of patients with multiple sclerosis (MS) and inflammatory bowel disease (particularly Crohn disease) has been helpful in enabling the diagnosis and prognosis in these patients.
Utilizing enzyme linked immunosorbent assay (ELISA), these antibodies can be identified and quantified. When used with other antibodies associated with Crohn disease (such as anti-Saccharomyces cerevisiae antibody [ASCA], anti-laminaribioside carbohydrate antibody [ALCA], anti-mannobioside carbohydrate antibody [AMCA], and anti-chitobiose carbohydrate antibody [ACCA]), anti-glycan antibodies are supportive of Crohn disease over ulcerative colitis or irritable bowel disease. Furthermore, higher levels of these antibodies are associated with a more complicated course of disease.
Other anti-glycan antibodies are specific for MS patients, enabling differentiation between MS patients and patients with other neurologic diseases.
Anti-Liver/Kidney Microsomal Type 1 Antibodies (Anti-LKM-1 Antibodies)
Indications
This test is used in the evaluation of patients suspected of having autoimmune hepatitis.
Test Explanation
Autoimmune liver disease (e.g., autoimmune hepatitis and primary biliary cirrhosis) is characterized by the presence of autoantibodies including smooth muscle antibodies (SMA), antimitochondrial antibodies (AMA), and anti-liver/kidney microsomal antibodies type 1 (anti-LKM-1). Subtypes of autoimmune hepatitis (AIH) are based on autoantibody reactivity patterns. For example, the presence of smooth muscle antibodies (SMA) is consistent with the diagnosis of chronic autoimmune hepatitis. The presence of anti-liver/kidney microsomal type 1 antibodies with or without SMA is consistent with autoimmune hepatitis, type 2. The presence of anti-mitochondrial antibodies is consistent with primary biliary cirrhosis.
Anti-LKM-1 antibodies serve as a serologic marker for AIH type 2 and typically occur in the absence of SMA and antinuclear antibodies. Children often have other autoantibodies (e.g., parietal cell antibodies and thyroid microsomal antibodies). These antibodies react with a short linear sequence of the recombinant antigen cytochrome monooxygenase P450 2D6. Patients with AIH type 2 more often tend to be young, female, and have severe disease that responds well to immunosuppressive therapy.
Patients with chronic hepatitis resulting from hepatitis C can also have elevated anti-LKM-1 antibodies. The diagnosis of autoimmune liver disease cannot be made on antibody testing alone. In many instances, autoimmune liver disease panel testing, including the antibodies discussed in the preceding paragraph, is performed. Testing is performed by semi-quantitative enzyme-linked immunosorbent assay/semi-quantitative indirect fluorescent antibody.
Related Tests
Aspartate Aminotransferase (p. 119). These hepatocellular enzymes are elevated in all forms of hepatitis.
Alanine Aminotransferase (p. 39). These hepatocellular enzymes are elevated in all forms of hepatitis.
Antinuclear Antibody (p. 88). These antibodies are associated with autoimmune liver disease.
Anti–Smooth Muscle Antibody (p. 95). These antibodies are associated with autoimmune liver disease.
Antimitochondrial Antibody (p. 84). These antibodies are associated with autoimmune liver disease.
Antimitochondrial Antibody (AMA, Mitochondrial Antibodies)
Test Explanation
AMA is an anticytoplasmic antibody directed against a lipoprotein in the mitochondrial membrane. Normally the serum does not contain AMA at a titer greater than 1:5. AMAs are found in 94% of patients with primary biliary cirrhosis or other autoimmune liver disease. This disease may be an autoimmune disease that occurs predominantly in young or middle-aged women. It has a slow progressive course marked by elevated liver enzymes, especially alkaline phosphatase and gamma-glutamyl transpeptidase, and a positive AMA test. Liver biopsy (see p. 734) is usually required to confirm the diagnosis because the AMA test can be positive in patients with chronic active hepatitis, drug-induced cholestasis, autoimmune hepatitis (e.g., scleroderma, systemic lupus erythematosus), extrahepatic obstruction, or acute infectious hepatitis. There are subgroups of AMA. The M-2 subgroup is highly specific for primary biliary cirrhosis. It is not useful in monitoring the course of the disease, however. For the AMA test, immunofluorescent assay (IFA) or enzyme-linked immunosorbent assay (ELISA) techniques can be used.
Test Results and Clinical Significance
Related Tests
Anti–Smooth Muscle Antibody (p. 95). This is another anticytoplasmic antibody that usually is present in patients with chronic active hepatitis and in 30% of patients with primary biliary cirrhosis.
Alkaline Phosphatase (p. 47). Although alkaline phosphatase (ALP) is found in many tissues, the highest concentrations are found in the liver, biliary tract epithelium, and bone.
Gamma-Glutamyl Transpeptidase (p. 246). This enzyme is found in the liver and is used to detect liver cell dysfunction and cholestasis.
Antimyocardial Antibody (AMA)
Indications
This test is used to detect an autoimmune source of myocardial injury and disease. AMAs may be detected in rheumatic heart disease, cardiomyopathy, postthoracotomy syndrome, and after myocardial infarction (MI). This test is not only used in the detection of an autoimmune cause for these conditions but also for monitoring response to treatment.
Test Explanation
A positive AMA test is associated with several forms of heart disease. AMAs may be detected before the development of clinical symptoms of heart disease. An immunologic basis has been suspected in rheumatic heart disease for a long time. Research has now documented the presence of serum antibodies against myocardial components and deposition of immunoglobulin and complement in lesional areas. Antibodies against heart muscle are also found in 20% to 40% of patients after cardiac surgery and in a smaller number of patients after MI. These antibodies are usually associated with pericarditis that follows the myocardial injury associated with cardiac surgery or MI (Dressler syndrome). AMAs have also been detected in patients with cardiomyopathy. Their role in this latter disease is unknown.
This test may be performed by indirect immunofluorescent technique. The patient's serum is added to a rat heart muscle extract. Antigen-antibody immune complexes are identified by immunofluorescent antihuman antibodies. Positive results (increased immunofluorescence) are reported in titers.
Test Results and Clinical Significance
Increased Levels
The myocardial antigen may be associated with streptococcus because the antibody may occur in patients with other streptococcal diseases.
Postthoracotomy (cardiac surgery) syndrome,
After MI (Dressler syndrome): Myocardial injury occurs and the antibody develops. The antibody-antigen complex may incite the pericarditis that follows the myocardial injury.
Cardiomyopathy: The association of cardiomyopathy and AMA is unknown. Whether the antibodies cause or contribute to the development of cardiomyopathy is being studied.
Antineutrophil Cytoplasmic Antibody (ANCA)
Normal Findings
| Components | Reference Interval |
| Anti-Neutrophil Cytoplasmic Antibody, IgG | <1:20: Not significant |
| Myeloperoxidase Antibody | Negative: ≤19 AU/mL Equivocal: 20-25 AU/mL Positive: ≥26 AU/mL |
| Serine Protease 3 Antibody | Negative: ≤19 AU/mL Equivocal: 20-25 AU/mL Positive: ≥26 AU/mL |
Indications
This blood test is used to assist in the diagnosis of Wegener granulomatosis (WG). It also is used to follow the course of the disease, monitor the response to therapy, and provide early detection of relapse.
Test Explanation
WG is a regional systemic vasculitis in which the small arteries of the kidneys, lungs, and upper respiratory tract (nasopharynx) are damaged by a granulomatous inflammation. Diagnosis can be made by biopsy of clinically affected tissue. Serologic testing plays a key role in the diagnosis of WG and other systemic vasculitis syndromes. Most patients with WG have circulating autoantibodies against neutrophil cytoplasm, which are useful in the diagnosis.
ANCAs are antibodies directed against cytoplasmic components of neutrophils. When ANCAs are detected with indirect immunofluorescence microscopy, two major patterns of staining are present: cytoplasmic ANCA (c-ANCA) and perinuclear ANCA (p-ANCA). Specific immunochemical assays demonstrate that c-ANCA consists mainly of antibodies to proteinase 3 (PR3) and p-ANCA consists of antibodies to myeloperoxidase (MPO). Using Semi-Quantitative Indirect Fluorescent Antibody/Semi-Quantitative Multi-Analyte Fluorescent Detection to characterize ANCA (rather than the pattern of immunofluorescence microscopy) is more specific and more clinically relevant; therefore, the terms proteinase 3-ANCA (PR3-ANCA) and myeloperoxidase-ANCA (MPO-ANCA) are used.
The PR3 autoantigen is highly specific (95% to 99%) for WG. When the disease is limited to the respiratory tract, the PR3 is positive in about 65% of patients. Nearly all patients with WG limited to the kidney do not have positive PR3. When WG is inactive, the percentage of positive drops to about 30%.
The MPO autoantigen is found in 50% of patients with WG centered in the kidney. It also occurs in patients with non-WG glomerulonephritis, such as microscopic polyangiitis (MPA).
P-ANCA antibodies can also differentiate various forms of inflammatory bowel disease. See also Anti-Glycan Antibodies (p. 82). P-ANCA are found in 50% to 70% of ulcerative colitis (UC) patients, but in only 20% of Crohn disease (CD) patients.
Antinuclear Antibody (ANA)
Indications
ANAs are used to diagnose systemic lupus erythematosus (SLE) and other autoimmune diseases. These antibodies are primarily used to screen for SLE. Because almost all patients with SLE develop autoantibodies, a negative ANA test excludes the diagnosis. If the ANA test is positive, other antibody studies must be done to corroborate the diagnosis.
Test Explanation
Autoantibodies are directed to nuclear material (ANAs) or to cytoplasmic material (anticytoplasmic antibodies) (Tables 2-3 and 2-4). Many abnormal antibodies are present in patients with autoimmune (also called rheumatic or connective tissue) diseases. ANA is a group of protein antibodies that react against cellular nuclear material. ANA is quite sensitive for detecting SLE. Positive results occur in approximately 95% of patients with this disease; however, many other rheumatic diseases are also associated with ANA (Table 2-5). ANA, therefore, is not a specific test for SLE (Table 2-6). ANA can be tested as a specific antibody or as a group with nonspecific antigens.
TABLE 2-3
Common Antinuclear Antibodies and Diseases They Cause
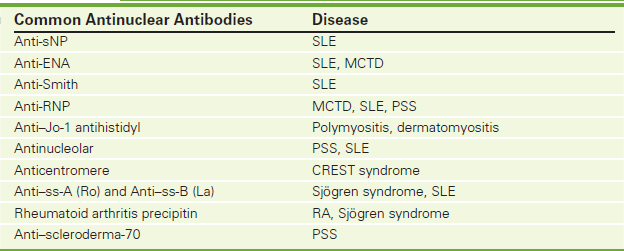
CREST, Calcinosis, Raynaud, esophageal dysfunction, sclerodactyly, telangiectasia; MCTD, mixed connective tissue disease; PSS, progressive systemic sclerosis (scleroderma); RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
TABLE 2-5
Autoimmune Disease and Positive Antibodies
| Autoimmune Disease | Positive Antibodies |
| SLE | ANA, SLE prep, dsDNA, ssDNA, anti-DNP, SS-A |
| Drug-induced SLE | ANA |
| Sjögren syndrome | RF, ANA, SS-A, SS-B |
| Scleroderma | ANA, Scl-70, RNA, dsDNA |
| Raynaud disease | ACA, Scl-70 |
| Mixed connective tissue disease | ANA, RNP, RF, ssDNA |
| Rheumatoid arthritis | RF, ANA, RANA, RAP |
| Primary biliary cirrhosis | AMA |
| Thyroiditis | Antimicrosomal, antithyroglobulin |
| Chronic active hepatitis | ASMA |

ANA tests are performed using different assays (indirect immunofluorescence microscopy or by enzyme-linked immunosorbent assay [ELISA]) and results are reported as a titer with a particular type of immunofluorescence pattern (when positive). Low-level titers are considered negative, while increased titers are positive and indicate an elevated concentration of antinuclear antibodies.
ANA shows up on indirect immunofluorescence as fluorescent patterns in cells that are fixed to a slide and are evaluated under an ultraviolet microscope. Different patterns are associated with a variety of autoimmune disorders. When combined with a more specific subtype of ANA (see Table 2-3), the pattern can increase specificity of the ANA subtypes for the various autoimmune diseases (Figure 2-5). An example of a positive result might be: “Positive at 1:320 dilution with a homogenous pattern.” This particular test is considered positive if ANA is found in a titer with a dilution of greater than 1:32. In general, the higher the titer of a certain ANA antibody known to be associated with a certain autoimmune disease, the more likely that disease exists and the more active the disease is. As the disease becomes less active because of therapy, the ANA titers can be expected to fall.
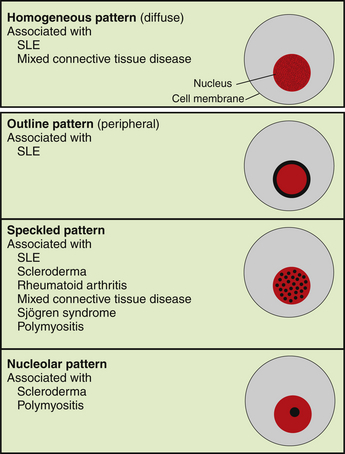
Often the ANA test is used to screen patients with suspected SLE. If the ANA test is negative, the patient probably does not have SLE. If positive, other corroborative serologic tests are performed (see Table 2-5). About 5% of patients with SLE have a negative ANA test. In this text, the more commonly clinically used ANA subtypes are separately discussed.
Interfering Factors
Drugs that may cause a false-positive ANA test include acetazolamide, aminosalicylic acid, chlorothiazides, chlorprothixene, griseofulvin, hydralazine, penicillin, phenylbutazone, phenytoin, procainamide, streptomycin, sulfonamides, and tetracyclines.
Drugs that may cause a false-negative test include steroids.
Procedure and Patient Care
After
• Apply pressure or a pressure dressing to the venipuncture site.
• Assess the venipuncture site for bleeding.
Because they are usually immunocompromised, patients with an autoimmune disease should be instructed to check for signs of infection at the venipuncture site. These patients often take steroids that further compromise their immune system.
Test Results and Clinical Significance
Increased Levels
Systemic lupus erythematosus (SLE): The signs and symptoms of this disease are vague and nonspecific. This disease is associated with a significant production of various autoimmune antibodies. Any organ in the patient's body can be the target of these autoantibodies. The immune complexes incite the complement system and thereby create tissue damage.
Rheumatoid arthritis (RA): The autoimmune response is targeted to the synovial tissues.
Periarteritis (polyarteritis) nodosa: The autoimmune response is targeted to the small vessels of various organs.
Scleroderma: The autoimmune response is targeted to the endothelium of blood vessels. Fibrosis occurs. This combined with deposit of collagen-related tissue creates the organ changes seen in the skin, gastrointestinal (GI) tract, and other internal organs.
Sjögren syndrome: The autoimmune response is targeted to the exocrine glands (lacrimal and salivary).
Raynaud phenomenon: This phenomenon is characterized by episodic digital ischemia as manifested by blanching and then cyanosis of the fingers in cold temperatures followed by rubor on rewarming. It is associated with many autoimmune diseases. The term “Raynaud disease” refers to this phenomenon without an associated autoimmune disease.
Related Tests
Anticentromere Antibody (p. 69). This test is used to diagnose CREST syndrome.
Anti-DNA Antibody (p. 78). This test is used to diagnose SLE.
Antiextractable Nuclear Antigen (ENA) (p. 79). This test is used to diagnose SLE and mixed connective tissue disease.
Antiscleroderma Antibody (p. 93). This test is used to diagnose scleroderma.
Anti–Parietal Cell Antibody (APCA)
Indications
Anti–parietal cell antibody (APCA) testing is used to diagnose an autoimmune cause of pernicious anemia.
Test Explanation
Parietal cells exist in the proximal stomach and produce hydrochloric acid and intrinsic factor. Intrinsic factor is necessary for the absorption of vitamin B12 (see p. 518). Anti–parietal cell antibodies are found in nearly 90% of patients with pernicious anemia. Nearly 60% of these patients also have anti–intrinsic factor antibodies. It is thought that these antibodies contribute to the destruction of the gastric mucosa in these patients. APCA is also found in patients with atrophic gastritis, gastric ulcers, and gastric cancer.
APCA is present in other autoimmune-mediated diseases such as thyroiditis, myxedema, juvenile diabetes, Addison disease, and iron-deficiency anemia. Nearly 10% to 15% of the normal population has APCA. As one ages, the incidence of having APCA increases (especially in relatives of patients with pernicious anemia).
Laboratory testing is usually done by indirect immunofluorescence using the Fluoro-Kit. With this test system, APCA can cross-react with other antibodies, especially anticellular and antithyroid antibodies. Titer levels greater than 1:240 are considered positive.
Test Results and Clinical Significance
Related Test
Vitamin B12 and Methylmalonic Acid Test (p. 518). This test measures the amount of vitamin B12 in the blood.
Antiscleroderma Antibody (Scl-70 Antibody, Scleroderma Antibody, RNA Polymerase III Antibody)
Indications
This antibody is diagnostic for scleroderma (progressive systemic sclerosis [PSS]) and is present in 45% of patients with that disease.
Test Explanation
Scl-70 antibody is an antinuclear antibody. On the fluorescent antinuclear antibody test, using the ultraviolet microscope, a specific pattern is created for the Scl-70 antibody. The pattern is a speckled group of dots throughout the nucleus (see Figure 2-5, p. 91). The test can be performed by fluorescent testing, enzyme-linked immunosorbent assay (ELISA), and enzyme immunoassay (EIA). Progressive serial dilutions are carried out.
PSS is a multisystem disorder characterized by inflammation with subsequent fibrosis of the small blood vessels in skin and visceral organs, including the heart, lungs, kidneys, and gastrointestinal (GI) tract. A collagen-like substance is also deposited into the tissue of these organs. In general, the higher the titer of Scl-70 antibody, the more likely it is that PSS exists and the more active the disease is. As the disease becomes less active because of therapy, the Scl-70 antibody titers can be expected to fall.
The absence of this antibody does not exclude the diagnosis of scleroderma. The antibody is rather specific for PSS but is occasionally seen in other autoimmune diseases, such as systemic lupus erythematosus (SLE), mixed connective tissue disease (MCTD), Sjögren syndrome, polymyositis, and rheumatoid arthritis.
RNA polymerase III antibodies are found in 11% to 23% of patients with PSS. Patients with PSS who are positive for RNA polymerase III antibodies form a distinct serologic subgroup and usually do not have any of the other antibodies typically found in PSS patients, such as anticentromere (p. 69) or anti–Sc1-70. PSS patients with anti-RNA polymerase III have an increased risk of the diffuse cutaneous form of scleroderma, with a high likelihood of skin involvement and hypertensive renal disease. A positive result supports a possible diagnosis of PSS. This autoantibody is strongly associated with diffuse cutaneous scleroderma and an increased risk of acute renal crisis. A negative result indicates no detectable IgG antibodies to RNA polymerase III, but does not rule out the possibility of PSS (11% to 33% sensitivity).
Related Test
Antinuclear Antibody (p. 88). This group of antibodies is used to diagnose systemic lupus erythematosus and other autoimmune diseases such as PSS.