Acyanotic defects: obstructive lesions
Pulmonary Stenosis
Pearls
• Pulmonary stenosis can present as critical stenosis in the newborn period, requiring urgent valvuloplasty.
• Pulmonary stenosis presenting in the older infant or child may be mild, moderate, or severe; mild stenosis is unlikely to progress in severity but moderate and severe stenosis typically will progress in severity.
• Diagnosis is made by echocardiography. The treatment of choice for most pulmonary stenosis is a valvuloplasty performed during cardiac catheterization.
• Surgical intervention is typically indicated when critical pulmonary stenosis is not responsive to valvuloplasty, when valve leaflets are dysplastic or when there are multiple areas of obstruction in the pulmonary outflow tract.
Etiology
Pulmonary Valve Stenosis
Pulmonary valve stenosis results from abnormal formation of pulmonary valve leaflets during fetal cardiac development. The normal pulmonary valve has three leaflets and is situated between the distal right ventricular outflow tract and the proximal main pulmonary artery. Pulmonary valve stenosis most commonly occurs when valve commissures fail to develop properly and the valve leaflets are thickened and fused. The result is an undersized valve orifice that provides increased resistance to blood flow. In a small number of patients the valve leaflets are dysplastic (abnormally shaped), and the pulmonary valve annulus is small. Either defect impedes flow from the right ventricle to the pulmonary artery.
The error in pulmonary valve formation likely occurs late in embryologic development. If the error occurred early in fetal life, the outcome would be complete underdevelopment of the pulmonary outflow tract. Familial cases of pulmonary valve stenosis have been reported, and siblings of patients with pulmonary stenosis have an increased incidence of congenital heart disease.711
Pulmonary stenosis is responsible for 8% to 12% of all congenital heart defects and may manifest with varying degrees of severity and at any age. Isolated pulmonary valve stenosis with an intact ventricular septum accounts for 80% to 90% of right ventricular outflow tract obstructive lesions and is one of the most common congenital heart defects.711 Isolated pulmonary valve stenosis is not commonly associated with other cardiac lesions.
The morphology of isolated pulmonary valve stenosis can be categorized into anatomic subgroups including descriptive terms such as domed, tricuspid, bicuspid, unicommissural, hypoplastic, and dysplastic. Thickened leaflets and commissural fusion are typically present in all subgroups except the dysplastic subgroup. Dysplastic stenosis accounts for approximately 10% to 20% of all cases of isolated pulmonary valve stenosis,620 and is the most common type of pulmonary stenosis associated with chromosomal anomalies and syndromes such as Noonan's syndrome.
Supravalvular and Infundibular Stenosis
Less common but additional forms of right ventricular outflow tract obstruction include supravalvular and infundibular pulmonary stenosis (also referred to as subvalvular stenosis). Supravalvular pulmonary stenosis is a rare lesion that occurs with Noonan's syndrome and occasionally with William's syndrome.451
Primary infundibular stenosis accounts for approximately 5% of all cases of right ventricular outflow tract obstruction. There are two types of isolated infundibular stenosis. The first type results from fibromuscular thickening in the wall of the right ventricular infundibulum. The other form is characterized by an obstructive muscle band at the junction of the right ventricle cavity and the proximal infundibulum.620 More commonly, infundibular pulmonary stenosis occurs in combination with three other lesions to create tetralogy of Fallot (see Tetralogy of Fallot later in this section of the chapter).
Pathophysiology
Pathophysiology and management strategies for all forms of isolated right ventricular outflow obstruction vary with the location and severity of the stenosis and the presentation of symptoms. Most patients have either critical pulmonary stenosis, which presents in the newborn period, or valvular pulmonary stenosis, which presents in older infants and children.620
With any pulmonary stenosis, the radius of the valve orifice is reduced and resistance to flow through the pulmonary valve is increased. To maintain normal flow across the small outflow tract, the right ventricle must generate a higher pressure proportional to the severity of the pulmonary stenosis. The resulting right ventricular hypertension and the obstruction of blood flow through the right ventricular outflow tract produces a quantifiable pressure gradient between the right ventricle and the main pulmonary artery that is directly related to the degree of stenosis. If the right ventricle cannot increase pressure proportionate to the severity of the pulmonary stenosis, blood flow (right ventricular output and pulmonary blood flow) will decrease.
The calculated gradient across the stenotic valve is categorized as mild (less than 40 mm Hg), moderate (40-80 mm Hg), or severe (greater than 80 mm Hg) based on the pressure gradient between the right ventricle and pulmonary artery. These severity classifications are shown in Box 8-26.
Box 8-26 Classification of Pulmonary Stenosis Based on Severity of Pressure Gradient Between Right Ventricle and Pulmonary Artery
Mild stenosis: <40 mm Hg gradient
Critical pulmonary stenosis of the newborn, if untreated, can lead to hemodynamic instability soon after birth. Severely limited right ventricular output and the presence of right ventricular hypertrophy at birth causes decreased diastolic compliance, elevated right ventricular pressure, and corresponding elevation in right atrial pressure. Typically newborns with critical pulmonary stenosis have an associated atrial shunt (through an atrial septal defect [ASD] or a patent foramen ovale [PFO]). The tricuspid valve and right ventricular size are usually normal, although some hypoplasia may be present. Because right atrial pressure substantially exceeds left atrial pressure, a right-to-left atrial level shunt develops, resulting in cyanosis. This shunt increases left ventricular output.
Myocardial oxygen supply may be compromised by hypoxemia and lower diastolic coronary perfusion pressure resulting from low-resistance runoff into the pulmonary circulation from the patent ductus arteriosus (PDA). If the defect is not detected early after birth, closure of the ductus arterious results in decreased pulmonary blood flow, worsening hypoxemia, acidosis, and cardiovascular collapse.620
When valvular pulmonary stenosis is present in older infants and children, the main hemodynamic effect is a rise in right ventricular pressure proportionate to the severity of valve obstruction. Chronic elevation of this pressure results in the development of right ventricular hypertrophy and decreased right ventricular compliance. Over time, this right ventricle may dilate and fail. Tricuspid regurgitation may develop and worsen right ventricular failure.451 Other associated right-heart findings include poststenotic dilation of the main pulmonary artery caused by the jet of blood flowing through the narrowed valve oriface.
When pulmonary stenosis is mild, it does not usually progress in severity. However, with moderate or severe forms of pulmonary stenosis, the severity typically increases with age because the size of stenotic orifice remains fixed as the patient grows.
Clinical Signs and Symptoms
This cardiac defect is often discovered because a murmur is detected during routine auscultation at birth. Ausculatory findings in valvular pulmonary stenosis reveal a normal first heart sound followed by a pulmonary ejection click in patients with mild or moderate stenosis. With severe stenosis the click occurs so early in systole that it merges with the first heart sound and becomes inaudible.711 A systolic ejection murmur is also present, loudest at the upper left sternal border with radiation over the entire precordium, axillae, and back. The intensity of the murmur generally increases with the degree of obstruction, and severe stenosis typically produces a grade IV murmur and a palpable thrill.
Neonates with critical pulmonary stenosis are cyanotic at birth for hemodynamic reasons discussed earlier. Initially cardiac output is maintained if an adequate atrial shunt is present, but cyanosis is apparent. With ductal closure, cyanosis worsens and hemodynamic deterioration is associated with tachycardia, tachypnea, and respiratory distress.620 Of note, patients with severe stenosis and right heart failure may have an unusually soft murmur because cardiac output is low, so blood flow through the stenotic pulmonary valve is limited.
Most infants and children with mild pulmonary valve stenosis are asymptomatic and have normal growth and development. Even with moderate pulmonary stenosis, symptoms often do not present until late infancy or childhood. Fatigue or exertional dyspnea may result because cardiac output is limited by the fixed obstruction. Severe, longstanding obstruction may cause symptoms of congestive heart failure. Less common findings include chest pain, syncope, or ventricular arrhythmias.620
Cardiac catheterization is no longer needed for the diagnosis of pulmonary stenosis but is indicated for therapeutic intervention. The ECG can be somewhat helpful in diagnosing pulmonary stenosis. About half of patients with mild pulmonary stenosis have a normal ECG and half demonstrate a right ventricular conduction delay.711 With moderate to severe pulmonary stenosis, the ECG is almost always abnormal with right axis deviation and right ventricular hypertrophy proportional to the severity of obstruction (see Table 8-26 as needed).
The definitive diagnostic technique for pulmonary stenosis is two-dimensional echocardiography with color Doppler. Echocardiography delineates the size of the pulmonary annulus, the level of obstruction, and valve leaflet mobility, shape, number, and thickness. Echocardiographic calculation of the pressure gradient across the pulmonary outflow tract has been shown to correlate well with direct measurements and is about 10% greater than peak-to-peak pressure gradients obtained in the cardiac catheterization laboratory.288 The degree of obstruction is classified as mild, moderate or severe based on pressure ranges detailed in Box 8-26.
Reports of the natural history for children with pulmonary valve stenosis are mixed. Several reports have documented that the degree of obstruction present at initial diagnosis correlates with progression of the obstruction over the child's lifetime751 and that children presenting at a younger age are more likely to have progression to severe obstruction.360 However, one study using serial echocardiograms documented a diminishing degree of stenosis in children with initial mild to moderate levels of obstruction.320 Therefore because it not currently possible to predict with certainty which subset of children will have significant progression of obstruction, all patients diagnosed with even mild stenosis require close followup with a pediatric cardiologist.
Management
Medical Management
Catheter intervention is the accepted first line treatment for symptomatic pulmonary valve stenosis. When relief of obstruction is indicated, balloon dilation has become the treatment of choice, and surgical intervention is rarely undertaken. A continuous infusion of prostaglandin E1 is required to maintain ductal patency in newborns with critical pulmonary stenosis as they await catheter intervention.
Pulmonary Valvuloplasty
Currently catheter intervention is the accepted initial treatment for pulmonary valve stenosis at any age and for all valve morphologies. Advances in equipment and technique allow even small neonates with critical pulmonary valve stenosis to undergo immediate valvuloplasty in the neonatal period.
Any symptomatic older infant or child requires catheter intervention as soon as the diagnosis of pulmonary stenosis is made. Even asymptomatic patients with severe obstruction should undergo semielective valvuloplasty.
The technique of percutaneous pulmonary balloon valvuloplasty was first described in early 1980s.430 Typically a balloon is chosen that is 20% to 40% larger than the pulmonary valve annulus measured angiographically. The balloon is positioned and inflated at the midpoint of the anatomic narrowing. Relief of obstruction results as the valve tears along fused commissures. In patients with a large pulmonary annulus, simultaneous inflation of two balloons may be necessary. The neonate or young infant with an extremely small pulmonary valve orifice may require initial dilation with a small coronary angioplasty balloon to enlarge the orifice enough to allow subsequent dilations with larger balloons.711 Any neonate with unsuccessful pulmonary balloon valvuloplasty should undergo immediate surgical valvotomy.
Results with percutaneous balloon valvuloplasty to treat isolated pulmonary valve stenosis have been very good.594 A successful outcome is defined as a postprocedure Doppler gradient less than 36 mm Hg without the need for repeat valvuloplasty or valvotomy. The Valvuloplasty and Angioplasty of Congenital Anomalies Registry reported followup data for 533 patients up to 8 years after initial catheter intervention. Eighty-four percent were noted to be free from further catheter or surgical procedures and 75% had gradients less than 36 mm Hg. Of those with an initial gradient greater than 36 mm Hg, just over half showed spontaneous regression of gradients to less than 36 mm Hg. Of the remaining 62 patients, 25 underwent subsequent surgery, 23 had repeat balloon valvuloplasty, and 14 remained with gradient greater than 36 mm Hg. Restenosis following initial intervention was also assessed. For patients with optimal immediate results only 12% had a significant gradient at followup or required reintervention for progressive stenosis.594
Patients with lower balloon valvuloplasty success rates are those with dysplastic pulmonary valves. Although balloon dilatation effectively splits fused commissures of typical stenotic valves, the thickened leaflets of a dysplastic valve do not respond as predictably to balloon dilation. Reported valvuloplasty associated complications (although uncommon) include vein tears, vein thrombosis, arrhythmia, perforation of the right ventricular outflow tract, tamponade, seizures, and stroke.828
Most patients treated with pulmonary valvuloplasty have some degree of pulmonary insufficiency following dilation. In a large follow-up study looking at degree of pulmonary regurgitation, 26% of patients had no regurgitation, 22% had trivial regurgitation, 45% had mild regurgitation, 7% had moderate regurgitation, and no patients demonstrated severe regurgitation.594 Some speculate that use of excessively large balloons corresponds with more severe insufficiency. Some residual postprocedure stenosis with little or no insufficiency is thought to be preferable to aggressive dilation that eliminates any gradient but is more likely to produce significant pulmonary regurgitation. Any degree of insufficiency must be monitored over time for possible progression and need for intervention. Overall, results of balloon dilation of pulmonary valve stenosis as long as 20 years ago are favorable.640
Despite successful relief of obstruction, a small percentage of infants with critical pulmonary stenosis may have persistent cyanosis after catheter intervention. Their right-to-left shunt probably results from low compliance of the right ventricle that is likely to be temporary. Typically the cyanosis resolves over a few weeks as right ventricular compliance improves, right atrial pressure decreases and the right-to-left shunt decreases. Temporary prostaglandin therapy can be used to augment insufficient forward flow across the pulmonary valve and into the pulmonary circulation.
If ductal dependency persists following valvuloplasty, one of two treatment strategies is undertaken.711 One available strategy is temporary placement of an aortopulmonary shunt that remains in place until the right heart grows sufficiently and right ventricular compliance improves. A second strategy is stenting (open) of the ductus in the interventional catheterization laboratory using an intravascular stent.640 At a later time when hemodynamically acceptable, these temporary shunts can be closed surgically or by interventional catheterization methods.
Patients who initially have mild hypoplasia of right heart structures at birth usually have sufficient growth of these underdeveloped areas over a period of months following relief of pulmonary outflow obstruction. However, in a small population of neonates with critical pulmonary stenosis, persistent right ventricular hypoplasia may necessitate staged single ventricle palliation (see section, Single Functioning Ventricle).
Surgical Management: Valvotomy
Isolated pulmonary stenosis is no longer a defect that is treated surgically. With the introduction of pulmonary balloon valvuloplasty, surgical pulmonary valvotomy is reserved for those patients with dysplastic valves resistant to dilation or when the pulmonary outflow tract has multiple areas of obstruction not conducive to valvuloplasty alone.451 As noted, any neonate with critical pulmonary valve stenosis not responsive to balloon dilation requires immediate surgical valvotomy.
When surgery is indicated to address isolated pulmonary valve stenosis, open commissurotomy is the most common surgical procedure performed. Via median sternotomy and using cardiopulmonary bypass, the main pulmonary artery is opened transversely or longitudinally to expose the valve. The valve commissures are incised to the annulus. If annular enlargement (enlargement of the valve orifice) is required, a transannular patch made of autologous pericardium or prosthetic material is inserted. In the case of dysplastic valves, a simple valvotomy may not be effective in relieving obstruction, and partial or even total removal of the valve may be required.
The degree of pulmonary insufficiency present following surgical intervention varies from patient to patient. Mild pulmonary insufficiency is usually well tolerated. However, patients requiring patch augmentation or valve removal are frequently left with moderate or severe pulmonary insufficiency. If the insufficiency is progressive and pulmonary regurgitation becomes severe, a valve replacement may be necessary.
Surgical Management of Supravalvular Pulmonary Artery Stenosis and Primary Infundibular Stenosis
Surgical management mirrors that for isolated pulmonary valve stenosis. However, unlike isolated pulmonary valve stenosis, the primary mode of management for these two lesions is surgical.620 In most cases of supravalvular stenosis, patch enlargement is all that is required. If infundibular obstruction is present, an extremely small incision is made in the pulmonary outflow tract to allow resection of the infundibular muscle under direct visualization. The infundibular stenosis may also be resected via transatrial route through the tricuspid valve. Recent surgical outcomes reported for this lesion are not available because balloon valvuloplasty has replaced surgery as the primary treatment strategy for this lesion.
Surgical Management: Pulmonary Valve Replacement
The presence of induced pulmonary insufficiency following surgical pulmonary valvotomy or pulmonary balloon valvuloplasty may necessitate the insertion of a competent valve in the pulmonary position. Controversy exists regarding the precise indications for intervention. At present, reported intervention criteria usually include echocardiographic evidence of progressive right ventricular dilation and associated enlargement of the pulmonary valve annulus leading to increasing tricuspid insufficiency. Clinical indications include progressive decrease in exercise tolerance, signs of right heart failure, or the presence of atrial or ventricular arrhythmias.250
A variety of prosthetic valves can be surgically placed in the pulmonary position, including homograft valves, xenograft valves, pericardial valves, and mechanical valves. Valve replacement requires open heart surgery using cardiopulmonary bypass. If a mechanical valve is placed, long-term anticoagulation is indicated (see Postoperative Care and Anticoagulation).
Ideally, pulmonary valve replacement should occur when the child is sufficiently grown to receive a valve that will last to adult years, but it should be performed before irreversible damage to the right ventricle and tricuspid valve has occurred. The concern is that implanted valves have limited longevity because calcifications and intimal thickening can develop over time, resulting in pulmonary stenosis or insufficiency. Most children requiring a prosthetic valve placement during infancy or early childhood will require subsequent valve replacement(s).
Since 2000, successful percutaneous pulmonary valve replacement has been performed using a bovine jugular venous valve sutured inside a stent.91 The possible benefit of eliminating the need for repeat thoracotomy and cardiopulmonary bypass makes this a potentially appealing option, and although long-term followup regarding effectiveness and safety of this intervention is not yet available, research in this area of interventional cardiac catheterization is ongoing.470
Antibiotic prophylaxis against endocarditis is no longer recommended postoperatively, with the exception of prosthetic pulmonary valves and for a 6-month period after placement of prosthetic material to augment the pulmonary outflow tract or repair the pulmonary valve.951
Advanced concepts regarding pulmonary stenosis are listed in Box 8-27.
Box 8-27 Advanced Concepts: Pulmonary Valve Stenosis
The intensity of a murmur of pulmonary stenosis often increases in proportion to the degree of obstruction of flow across the stenotic area. However, patients with severe pulmonary stenosis may have an unusually soft murmur because right heart failure and low cardiac output can limit blood flow across the right ventricular outflow tract.
Aortic Stenosis (AS)
Pearls
• Aortic stenosis may be valvular, subvalvular (subaortic), or supravalvular (supraaortic).
• The severity of aortic stenosis may be estimated by Doppler or during cardiac catheterization.
• Estimates of the severity of aortic stenosis will be falsely low if cardiac output is low.
• Percutaneous balloon aortic valvuloplasty has replaced surgical valvotomy as the therapy of choice for valvular aortic stenosis in infants and children requiring intervention.
• Subvalvular and supravalvular aortic stenosis require surgical intervention.
Etiology
The term aortic stenosis is used to indicate any obstruction of outflow from the left ventricle to the ascending aorta in the region of the aortic valve. Embryologically, the aortic valve develops from three ridges of subendocardial tissue that form when the aortopulmonary septum divides into the aortic and pulmonary trunks. A normal aortic valve is trileaflet and functions without obstruction. Abnormal development of either the number or morphology of the valve cusps and commissures results in varying forms and degrees of aortic stenosis.
The degree of abnormal development of the aortic outflow tract results in a clinical continuum of congenital aortic stenosis. This continuum can range from a normally functioning, but malformed bicuspid aortic valve, to severe aortic stenosis in the fetus resulting in hypoplastic left heart syndrome (HLHS; detailed elsewhere—see section, Single Functioning Ventricle). In general, obstructive lesions are categorized based on the location of the obstruction: valvular stenosis, subvalvular stenosis, or supravalvular stenosis.
Valvular aortic stenosis is the most common form, occurring in about 75% of all patients with aortic outflow obstruction. Stenosis of the valve is caused by decreased orifice size resulting from thickening and rigidity of valve leaflets. Males with valvular aortic stenosis outnumber females 4:1; however, subvalvular and supravalvular stenosis are only slightly more common in males that females. The most common abnormality of the aortic valve is a bicuspid aortic valve and occurs in about two thirds of patients with valvular aortic stenosis. An estimated 1% to 5% of all infants are born with a bicuspid aortic valve, although only a small percentage develops stenosis from fusion of the two abnormal leaflets.
Subvalvular aortic stenosis or subaortic stenosis is the second most common form of aortic obstruction and accounts for about 20% of patients with left ventricular outflow tract obstruction. Subaortic stenosis can occur as an isolated lesion or in association with other congenital heart defects. Three types of subaortic stenosis are described. The discrete membranous type is a fibromuscular ring with a central orifice that is located below the aortic valve. A second type is the hypertrophic type. This form results from hypertrophy of the interventricular septum and anterior leaflet of the mitral valve and leads to dynamic outflow tract obstruction that is often called hypertrophic cardiomyopathy (HCM). The least common type of subaortic stenosis is the fibromuscular tunnel type, which consists of a long segment of narrowing beneath the aortic valve.444
Supravalvular aortic stenosis, or supraaortic stenosis, the least common form of aortic obstruction, is caused by either localized or diffuse fibromembranous narrowing of the aorta above the aortic valve and coronary arteries. The aortic valve leaflets may also be thickened and abnormal and in some cases, the coronary artery ostia can be obstructed by membranous tissue.780
Aortic stenosis is believed to result from a complex interaction of genetic and environmental factors. Proposed causes include abnormal formation of the valve cusps during embryologic development and environmental influences such as prenatal infection or metabolic disturbances causing damage to the valve resulting in stenosis. Familial patterns of left ventricular outflow tract obstruction have been reported ranging from bicuspid aortic valve, to valvular AS, to coarctation of the aorta, to hypoplastic left heart syndrome (HLHS) and suggest a hereditary link as well. Recent genetic research has linked abnormalities in the chromosomal regions 5q, 13q, 18q,572 and the occurrence of aortic stenosis. Genetic defects associated with aortic stenosis include Turner syndrome and Jacobsen syndrome. Supravalvular aortic stenosis is associated with William's syndrome.780
Aortic stenosis is one of the more common forms of congenital heart disease (CHD), occurring in 4% to 8% of all infants born with CHD. It is now appreciated that congenital aortic stenosis is more common than previously recognized because many adults diagnosed with “acquired” aortic stenosis actually have congenitally bicuspid aortic valve. Additional or associated congenital heart defects occur in 20% of patients with congenital aortic valve stenosis. The most common are VSD, PDA, coarctation of the aorta, and mitral valve anomalies. Aortic insufficiency is often associated with aortic stenosis.
Pathophysiology
All forms of aortic stenosis produce obstruction of left ventricular outflow resulting in impedance to left ventricular ejection. Whenever there is obstruction to left ventricular outflow the left ventricle must generate higher pressure to maintain normal flow beyond the area of resistance. As a result, left ventricular hypertension develops that is proportional to the degree of aortic obstruction. This pressure overload leads to the development of left ventricular hypertrophy and can lead to left ventricular failure, resulting in elevated left ventricular end-diastolic and left atrial pressures and pulmonary edema.
Aortic stenosis can compromise blood flow to the coronary arteries, because the stenosis can reduce coronary perfusion pressure (difference between aortic end-diastolic pressure and right atrial pressure) and results in prolonged ejection (systole) and shortened diastole (the predominant time that the left coronary artery perfuses the left ventricle). The mismatch between coronary artery perfusion (oxygen delivery) and oxygen demand can be magnified during periods of stress or exercise when oxygen demand is increased; during these periods tachycardia further reduces ventricular diastolic filling and coronary perfusion time and the gradient across the aortic stenosis increases.531 If hypertrophy is severe, blood supply to the subendocardial tissue may be inadequate, resulting in subendocardial ischemia, arrhythmias, and even myocardial infarction.
When supravalvular aortic stenosis is present, left ventricular hypertrophy still occurs. However, coronary artery perfusion is usually adequate because the coronary artery ostia are located proximal to the aortic obstruction.
The degree of obstruction in AS is typically expressed in terms of catheter-derived peak-to-peak gradient, peak instantaneous echo Doppler gradient, mean systolic pressure gradient calculated from Doppler measurements, or mean pressure gradients derived from simultaneous catheter recordings. The pressure measurement necessary to quantify aortic stenosis as mild, moderate or severe is based on the measurement technique used and often varies from one institution to another.
Clinical Signs and Symptoms
Valvular Stenosis
Neonates, infants and children present with varying degrees of valvular aortic stenosis. Those with mild to even moderate stenosis are relatively asymptomatic. Some neonates have such left heart hypoplasia that they are incapable of sustaining systemic circulation; these infants require single ventricle palliation (see section, Single Functioning Ventricle).
Neonates with adequate left ventricular size but critical aortic valve obstruction may initially tolerate even moderate obstruction; however, gradients may increase substantially during the first days or weeks of life when left ventricular function becomes hyperdynamic, the ductus arteriosus closes, or associated defects such as muscular ventricular septal defects (VSD) close spontaneously. When the ductus arteriosus closes, pulmonary vascular resistance falls, and pulmonary blood flow and venous return to the left atrium increase. If the left ventricle cannot generate the high pressure required to maintain this blood flow through the stenotic outflow tract, signs and symptoms of heart failure or even cardiogenic shock develop and can be fatal without appropriate intervention.
Neonates with critical aortic stenosis are pale, dyspneic, and tachycardic. Physical examination reveals a hyperactive precordium, poor distal pulses, poor peripheral perfusion, and cyanosis. A cardiac murmur is frequently absent when low cardiac output is present. Although a gallop rhythm may be appreciated, an ejection click is rarely heard.780
Infants with severe but not critical AS typically present in infancy with signs and symptoms of heart failure, including poor feeding, tachypnea, and growth failure. Physical examination reveals a hyperactive precordium and a thrill is palpated in the suprasternal notch. A precordial thrill is often present with mild to moderate stenosis. On auscultation a characteristic ejection murmur is often heard along the left upper sternal border, radiating to the neck. A systolic ejection murmur may not be present if LV function is significantly reduced and blood flow across the aortic valve (cardiac output) is limited. The presence of a systolic ejection click points to valvular rather than supravalvular or subvalvular obstruction. In addition, patients may develop right ventricular failure and hepatomegaly.
Older children with valvular aortic stenosis can be relatively asymptomatic with appropriate growth and development. Some report easy fatigability, which appears to be unrelated to the severity of AS. Those who develop severe obstruction are at greatest risk for exercise intolerance, anginal pain, syncopal events, and even sudden death. Older children typically have normal vital signs, including blood pressure. Auscultation may reveal a systolic ejection murmur with intensity proportional to the extent of stenosis. Approximately one third of patients also have a diastolic murmur resulting from aortic regurgitation. More than half of children with valvular aortic stenosis have an ejection click heard best at the apex or lower left sternal border. Visible apical activity and an increased left ventricular impulse on palpation are found with severe aortic stenosis.
The diagnosis of aortic stenosis is almost always made by physical examination and echocardiography. Two-dimensional echocardiography with color Doppler analysis is used to estimate the level and severity of obstruction, evaluate valve and left ventricular outflow tract morphology, and estimate left ventricular function and the degree of aortic regurgitation. Doppler-derived peak instantaneous pressure gradients are calculated across the stenotic aortic valve. Traditionally, catheter-derived peak-to-peak pressure gradients recorded during cardiac catheterization were used to estimate the severity of stenosis and direct management. It is important to note that the Doppler-derived peak instantaneous pressure gradient represents a different physiologic parameter (i.e., calculated from blood flow velocities) than the catheter-derived peak-to-peak pressure gradient, and the two are not interchangeable. The Doppler peak instantaneous gradient is higher than peak-to-peak gradients obtained during catheterization.780 A mean systolic pressure gradient can be calculated from the Doppler measurements, and this echocardiographic assessment of severity of obstruction is shown to correlate well with mean pressure gradients derived from simultaneous catheter recordings in the catheterization laboratory.972 Many centers currently favor using mean pressure gradients to guide clinical management of aortic stenosis.
The ECG is of limited use in diagnosing children with AS and has limited utility in distinguishing mild from severe obstruction. A 24-hour ambulatory ECG (Holter) monitor in asymptomatic patients may detect ventricular arrhythmias. A strong relationship has been reported between arrhythmias and sudden death in patients with AS.780
The chest radiograph in newborns with critical aortic stenosis and CHF reveals cardiomegaly with venous congestion. A dilated ascending aorta may be seen in older patients with valvular aortic stenosis and results from post-stenotic dilatation. Exercise testing may be of value in evaluating children who want to participate in sports. The development of ST-segment or T-wave changes consistent with myocardial ischemia may indicate significant obstruction in an otherwise asymptomatic patient.
Cardiac catheterization is no longer used for routine diagnosis and is reserved for patients with associated defects that cannot be completely evaluated noninvasively, or if therapeutic intervention is indicated. Cardiac magnetic resonance imaging (MRI) is becoming a useful noninvasive mode for assessing ventricular mass and function as well as defining the morphology of the valve, annulus size, and coronary artery anatomy.
Subvalvular Aortic Stenosis
Subvalvular stenosis that occurs as an isolated lesion is rarely clinically significant in newborns and infants.544 Patients with mild or moderate obstruction are typically asymptomatic. The lesion is often discovered during evaluation of associated cardiac defects.
Frequently subvalvular aortic stenosis is identified when echocardiography is performed for evaluation of a murmur. More than one-half of affected patients have a characteristic harsh systolic ejection murmur and a high frequency diastolic murmur of AR is heard in some patients.
The physical examination can help distinguish fixed from dynamic subaortic stenosis as seen in hypertrophic cardiomyopathy. In patients able to perform a Valsalva maneuver, the intensity of the murmur typically decreases in fixed subvalvular AS and increases in hypertrophic cardiomyopathy (HCM). Subvalvular AS often progresses rapidly during infancy and early childhood; however, the disorder may remain stable for years as some adults have only mild obstruction.76 Diagnosis is confirmed by echocardiography, which enables determination of the location and extent of the obstruction and evaluation of left ventricular function and the integrity of the aortic and mitral valves.
Supravalvular Aortic Stenosis
Supravalvular AS is often suspected based on ausculatory findings. Affected children typically have a loud systolic ejection murmur. Unlike in valvular AS, there is no associated ejection click or diastolic murmur of aortic regurgitation. Other possible findings include a thrill in the suprasternal notch and a higher blood pressure in the right arm compared to the left arm caused by a jet of high-pressure flow in the ascending aorta (the Coanda effect). A blood pressure difference between the two arms of more than 10 mm Hg has been noted in approximately two thirds of patients.544
The diagnosis of supravalvular AS is confirmed by echocardiography and enables assessment of the severity of obstruction, left ventricular function, and the degree of left ventricular hypertrophy. Magnetic resonance imaging with angiography can be used when physical examination or other findings suggest associated defects of the vascular tree as MRI provides excellent anatomic detail of supravalvular aortic obstruction and associated aortic branch vessel disease.
Management
The American College of Cardiology and American Heart Association have developed joint ACC/AHA guidelines for the management of patients with aortic stenosis that consider age, clinical status and gradient across the stenosis.780 It is important to note that the gradient across the obstructed area will be falsely lowered when cardiac output is low. Conversely, conditions that increase cardiac output (e.g., anemia, fever, exercise) will result in an increase in the gradient across the obstructed area. Asymptomatic children with Doppler peak instantaneous gradients of greater than 70 mm Hg should be considered for cardiac catheterization with possible balloon valvuloplasty. If the catheter measured peak-to-peak gradient is greater than 60 mm Hg, balloon valvuloplasty is indicated.
Children with symptoms such as dyspnea on exertion, syncope, angina, or ischemic changes on a resting or exercise ECG should have valvuloplasty if the peak-to-peak gradient in greater than 50 mm Hg. If the gradient is less than 50 mm Hg other causes of these symptoms and ECG changes should be investigated.
Valvuloplasty is not recommended for asymptomatic children with peak-to-peak gradients less than 50 mm Hg unless there is a concern that low cardiac output is contributing to underestimation of the severity of obstruction. Even children with mild degrees of aortic stenosis are at risk for progression over time and require periodic echocardiographic assessment.62 If clinical findings do not appear to correlate with Doppler evaluation, cardiac catheterization may be indicated for direct measurement of peak-to-peak gradient.
Problematic aortic stenosis of all types requires treatment with either valvuloplasty or surgery. The goal of these interventions will be to relieve the aortic obstruction without creating significant aortic insufficiency. The management of each form of aortic stenosis, whether valvular, subvalvular, or supravalvular, is somewhat different and is summarized in the following pages.
Valvular Aortic Stenosis
The management approach is determined by the degree of obstruction present and is independent of the age of the patient. Medical therapy for neonates with critical AS includes intravenous administration of prostaglandin E1 to open or maintain the ductus arteriosus, thus providing a means of right-to-left shunting (from pulmonary artery to aorta) and adequate antegrade systemic perfusion and retrograde aortic and coronary artery perfusion.
Medical therapy in children and adolescents consist of periodic evaluation to monitor for potential progression of valve dysfunction and need for exercise restrictions. These children are managed according to ACC/AHA recommendations.780
Percutaneous balloon aortic valvuloplasty has replaced surgical valvotomy as the therapy of choice for valvular aortic stenosis in infants and children requiring intervention. The aortic valve leaflets in children are typically pliable and easy to dilate and/or tear. By comparison, adult AS is not as amenable to balloon dilation because calcification of valve leaflets in the adult makes them less amenable to successful dilation. The major risk of balloon valvuloplasty is the development of significant aortic regurgitation.
Valvuloplasty is accomplished through the retrograde insertion of a balloon catheter from the femoral artery to the aorta and then beyond the aortic valve. The balloon is inflated to tear and separate the valve leaflets. Excessive valve dilation is avoided to prevent the development of aortic insufficiency that may lead to left ventricular dilation and dysfunction. Repeat balloon dilation is often effective in patients who develop recurrent obstruction, with gradient reduction of at least 50% in most patients. Repeat intervention is required more often in newborns than in older children.771 Aortic wall injury has been reported as a procedure-related complication in neonates undergoing balloon aortic valvuloplasty is, but it has not resulted in significant mortality.117
The long-term outcome of balloon aortic valvuloplasty has been reported in two large series.593,629 In children undergoing intervention at ages 1 month to 20 years, the average reduction in peak transvalvular gradient was 56 to 60 mm Hg and procedural mortality was 0.7%. Moderate to severe aortic regurgitation was found in 13% of patients immediately after balloon dilatation and in 38% 3.5 years after intervention.593,629
Data regarding the outcome of neonatal balloon aortic valvuloplasty is available from retrospective reviews undertaken at two centers.346,599 In one study, early mortality fell significantly in recent years, from 22% (1985-1993) to 4% (1994-2002). The initial gradient reduction was 54% and significant AR developed in 15%.599 The second study evaluated neonates who underwent balloon aortic valvuloplasty between 1 and 30 days of age from 1994 to 2004.346 During the 3.5-year follow-up period, there were 31 reinterventions. Patients with a small aortic annulus were more likely to require aortic valve replacement. Catch-up growth of the left heart structures was reported but the size of the mitral valve remained below normal range for body surface area.599
In the treatment of valvular aortic stenosis, the alternative to balloon aortic valvuloplasty is surgical valvotomy performed via median sternotomy under cardiopulmonary bypass. An incision is made in the aorta, just above the coronary arteries, and the fused commissures are incised carefully under direct visualization. The goal of surgical intervention for valvular aortic stenosis is relief of the aortic obstruction without creation of significant aortic insufficiency. Incision is performed only for those commissures with adequate leaflet attachment; otherwise aortic insufficiency is likely to result. An alternative option is closed aortic valvotomy, performed without cardiopulmonary bypass, using calibrated dilators or balloon catheters. Currently this option is rarely used.
Children who develop severe aortic regurgitation following balloon dilation require surgical intervention. In such patients, valve repair can be effective and usually preferred to valve replacement. Surgical repair of residual aortic valve stenosis and aortic regurgitation is influenced by the size of the aortic annulus. If there is no significant annular hypoplasia, a surgical valve-sparing procedure can be performed. Repair techniques include commissurotomies, cusp extensions with pericardial patches, tightening of commissural edges, and shaving of fibrous material from valve cusps.
Aortic valve replacement is reserved for those patients with progressive aortic regurgitation not amenable to surgical intervention or those with recurrent stenosis refractory to balloon valvuloplasty. Options for aortic valve replacement during childhood include bioprosthetic or mechanical valves. Bioprosthetic valves, either homograft or heterograft, avoid the need for anticoagulation, but longevity of these valves and their lack of growth potential are limiting factors. In the early years of replacement, bioprosthetic valves had a failure rate in children as high as 20%. Failure resulted from progressive calcification in as little as 6 years.917 Replacement with a mechanical prosthesis is the most durable alternative; however, the valve has no growth potential and lifelong anticoagulation is required because of the risk of thromboembolic events. Anticoagulation introduces a risk of hemorrhagic complications. The child and parents require careful instruction about the importance of the anticoagulation, medical followup, and the signs of thromboembolic or hemorrhagic complications (see Postoperative Care and Anticoagulation).
An alternative to valve replacement in infants and small children with aortic stenosis is the Ross procedure (pulmonary autograft). The child's pulmonary valve (with a cuff of tissue) is transplanted to the aortic position, and a pulmonary homograft is placed in the pulmonary valve position. The coronary arteries are reimplanted into the cuff of tissue so they are located immediately above the transplanted pulmonary valve (Fig. 8-37). This procedure avoids the need for anticoagulation, and the neoaortic valve (pulmonary autograft) has potential for growth. The major disadvantage of the Ross procedure in infants and children is that pulmonary homograft dysfunction is inevitable and can occur soon after initial operation. Fortunately, recent modifications to the Ross procedure have reduced the frequency of pulmonary autograft dysfunction and in some reports the function of the neoaorta remains stable for many years. Long-term survival after the Ross procedure is greater than 95%.780
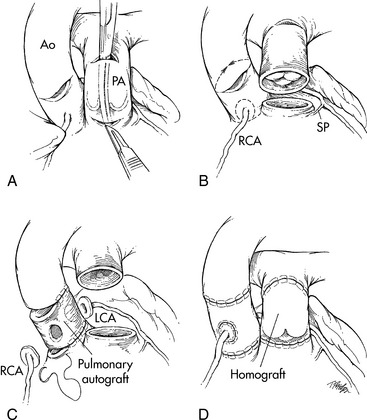
Fig. 8-37 Surgical correction of critical aortic stenosis with pulmonary autograft: Ross procedure. A, After the patient has been placed on cardiopulmonary bypass with cardioplegia solution, the pulmonary autograft procedure is performed by making a small incision in the aorta (Ao). If the aortic valve requires replacement, an incision is made in the main pulmonary artery (PA) adjacent to the origin of the right pulmonary artery, and a right-angle clamp is placed across the pulmonary artery and used to identify the right ventricular surface proximal to the pulmonary valve. An incision is then made in the right ventricle (RV); and B, the proximal pulmonary valve is removed with a cuff of muscle tissue. The posterior division of the valve from the right ventricle should follow a horizontal plane to prevent injury to the first septal perforator (SP) branch of the left anterior descending coronary artery. After the proximal portion of the pulmonary valve has been removed, the pulmonary artery is transected at the level of the bifurcation through the initial incision made in the pulmonary artery. C, With the autograft removed, the previously performed incision in the aorta is extended so that the aorta is transected above the level of the valve. The coronary arteries (right coronary artery [RCA] and left coronary artery [LCA] are then removed with large buttons of surrounding sinus tissue. The aortic valve and all remaining aortic tissue is removed from the base of the heart. The pulmonary autograft is then sewn to the base of the heart. The left coronary artery (LCA) and the right coronary artery (RCA) are implanted into the autograft. D, The procedure is completed by placing a cryopreserved homograft in the right ventricular outflow tract to reestablish right ventricle-to-pulmonary artery continuity.
(From Ungerleider RJ: Congenital aortic stenosis. In Nichols DG, et al, editors: Critical heart disease in infants and children. St Louis, 1995, Mosby.)
When aortic valve stenosis is complicated by obstruction from a hypoplastic aortic annulus or diffuse subaortic stenosis, a Konno procedure (aortoventriculoplasty) is performed to enlarge the annulus. Through a median sternotomy approach and under cardiopulmonary bypass, a longitudinal incision is made in the aorta. By way of a right ventriculotomy, the aortic annulus is entered and an incision is made into the ventricular septum. A prosthetic patch is used to enlarge the left ventricular outflow tract and a prosthetic aortic valve is placed. A pericardial patch is then used to close the right ventriculotomy (Fig. 8-38). Potential problems after the Konno procedure include complete heart block, right ventricular outflow tract obstruction and residual VSD.
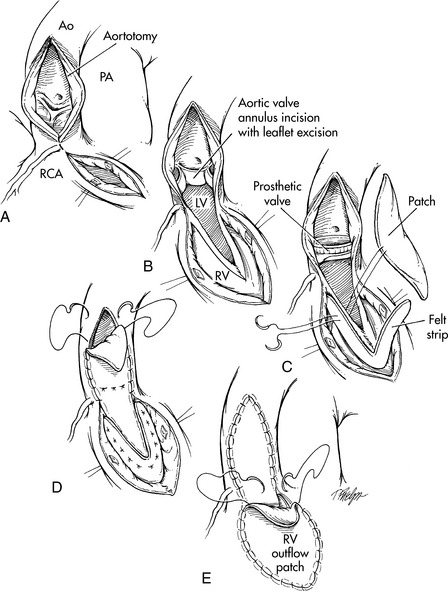
Fig. 8-38 Surgical correction of critical aortic stenosis: Konno procedure. An aortoventriculoplasty is performed with the patient on cardiopulmonary bypass, the aorta cross-clamped, and the heart protected with cardioplegia solution. A, A vertical aortotomy is extended just to the left side of the right coronary artery (RCA) and connected to a right ventriculotomy. The right ventriculotomy is performed below the pulmonary valve, and extended (see dotted line) toward the vertical aortotomy. Once these incisions are joined, another incision can be made across the aortic valve annulus B, and into the interventricular septum, so the incision is open from the right ventricle (RV) to the left ventricle (LV). The aortic valve leaflets are removed. C, A prosthetic valve is then sewn in the valve annulus, using interrupted (or continuous) sutures for the posterior portion of the valve ring. A prosthetic patch is placed and the valve ring sutures are brought through the patch, then through the interventricular septum, and then through a Teflon felt strip. This reconstructs and enlarges the left ventricular (LV) outflow tract and places the patch on the LV surface so the LV pressure will help to hold the patch in place. This patch is then secured. D, to the anterior portion of the prosthetic valve ring, and the suture line for the patch is continued around the aortotomy. E, Finally, the right ventricular (RV) outflow tract is repaired with an additional patch of either Gore-Tex or pericardium. AO, aorta; PA, pulmonary artery.
(From Ungerleider RM: Congenital aortic stenosis. In Nichols DG, et al, editors: Critical heart disease in infants and children. St Louis, 1995, Mosby.)
An evolving alternative to surgical valve placement is the percutaneous aortic valve implantation. The technique required for catheter delivery of a prosthetic aortic valve is progressing but not currently an option for children with aortic stenosis.780
There is limited comparative data between surgical valvotomy and balloon aortic valvuloplasty. In one series of 110 newborns with critical valvular aortic stenosis, surgical valvotomy was performed as the initial procedure in 28 and balloon aortic valvuloplasty was the initial procedure in 82, and survival and freedom from reintervention at 5 years were similar in both groups. Balloon aortic valvuloplasty resulted in a greater reduction in the systolic gradient (65% vs. 41%) and a lower mean residual postoperative gradient (20 vs. 36 mm Hg), but more frequent clinically significant aortic regurgitation (18% vs. 3%).595
Subvalvular Aortic Stenosis
Management of the asymptomatic infant or child with discrete subvalvular aortic stenosis is very similar to that of the child with valvular aortic stenosis. Children with a left ventricular outflow tract gradient less than 30 mm Hg and no significant left ventricular hypertrophy are monitored closely for progression, especially during the first several years of life. Echocardiographic evaluation is warranted for any change in symptoms or clinical examination.
Because obstruction is very likely to recur, recommendations for appropriate timing of surgical intervention range from soon after diagnosis to longer periods of observation. Surgery is typically deferred in the first decade of life if the obstruction is moderate or less and aortic regurgitation is trivial. The presence of significant aortic regurgitation is considered an indication for surgery even if the obstruction is moderate or less.
In contrast to valvular aortic stenosis, subvalvular aortic stenosis does not respond to balloon dilation, so surgical correction of the obstruction is the definitive therapy. The surgery is performed through a median sternotomy incision with use of cardiopulmonary bypass. An incision is made in the aorta above the aortic valve and resection of the subvalvular discrete membrane or fibromuscular ring is performed. If subaortic obstruction is the result of a tunnel-like narrowing of the left ventricular outflow tract and a small aortic valve annulus, a patch may be required to enlarge the entire left ventricular outflow tract and annulus also known as the Konno procedure (described earlier for aortic valvular stenosis). Modifications of this intervention are used if subvalvular obstruction is severe but the aortic annulus is adequate, including a modified patch enlargement of the left ventricular outflow tract alone.
Surgical outcomes have improved in recent years and operative mortality rates are very low. Reported postoperative complications include recurrent obstruction in addition to those previously associated with the Konno procedure. Recurrence of subaortic stenosis was evaluated in a recent report of 111 patients who underwent successful surgical resection of discrete subaortic stenosis between 1984 and 2001. Rate of re-operation was 14% at a median followup of 8.2 years. Factors associated with reoperation included a lesion in closer proximity to the aortic valve and a peak Doppler gradient of greater than or equal to 60 mm Hg. Age at initial operation was not a factor, but severity of disease at initial operation was associated with greater likelihood of reoperation.313
Supravalvular Aortic Stenosis
Infants and children with mild supravalvular aortic stenosis are followed at regular intervals to detect any increase in obstruction. Congestive heart failure and other signs of severe aortic outflow obstruction do not commonly occur in infants with supravalvular aortic stenosis. The risk of sudden death is approximately the same as that reported with valvular aortic stenosis.
Definitive therapy for supravalvular aortic stenosis, whether discrete or diffuse, consists of surgical correction of the obstruction. The indications for surgery vary based on type and degree of stenosis. Surgery for the discrete form of supravalvular AS is usually successful in alleviating the stenosis. Techniques range from single patch enlargement just above the aortic root to bifurcated patch placement extending into two sinuses, and even three sinus patch enlargements.144
Treatment of diffuse obstruction is more complex and surgical options include extensive endarterectomy with patch aortoplasty or resection of the stenotic segment with end-to-end anastomosis to the distal ascending aorta. Late complications of surgery include residual stenosis and valvular dysfunction requiring aortic valve replacement. Transcatheter stent placement has been undertaken at a few centers with varying results as an alternative therapy in children with associated involvement of aortic branch vessels.
Outcomes after surgical correction of supravalvular aortic stenosis include operative mortality rates ranging 1% to 9% with variability likely owing to nature of the stenosis and the presence of associated lesions. Main predictors of worse survival and more frequent reoperation were the presence of diffuse versus discrete stenosis and the presence of associated aortic valve disease.854-856 Operative risk is also higher in patients with diffuse arteriopathy as seen in William's syndrome.
The clinical course and natural history for patients with aortic stenosis is not completely known as diagnostic techniques and the evolution of treatment options have simultaneously progressed. It is generally accepted that aortic stenosis presenting in infancy is more severe and carries a higher mortality rate with or without treatment than cases presenting in childhood.
The long-term outcome of congenital aortic stenosis was evaluated in the Second Natural History Study of Congenital Heart Defects.443 This report included 371 patients with AS, mostly children, who had undergone diagnostic cardiac catheterization between 1958 and 1969. This report provides some useful information about the natural history of this defect, despite the fact that many of the patients did receive therapy. Patients with gradients less than 50 mm Hg were treated medically, those with intermediate gradients had either medical or surgical therapy, and a surgical valvotomy was performed for gradients across the aortic valve of greater than or equal to 80 mm Hg. A total of 92.3% of all participants were in New York Heart Association functional class I. The 25-year survival was 92.4% for patients with initial peak systolic ejection gradients less than 50 mm Hg and 81% for those greater than or equal to 50 mm Hg. The likelihood of requiring surgery in 25 years was 20%, 40%, and 60% for patients with initial gradients of less than 25, 25 to 49, and 50 to 79 mm Hg, respectively. Sudden death occurred in 5% of the patients and accounted for more than one-half of all cardiac deaths. The patients succumbing to sudden death were almost all older than 10 years of age and had significant obstruction and/or aortic regurgitation; 19 had prior surgery.
According to the 2007 American Heart Association endocarditis prophylaxis guidelines,951 antibiotic prophylaxis to prevent bacterial endocarditis is no longer recommended in patients with valvular aortic stenosis, supravalvular aortic stenosis, or subvalvular aortic stenosis except in those with a prior history of endocarditis or a repair that required prosthetic material or device. In the latter, antibiotic prophylaxis is recommended for the first 6 months after repair unless a residual defect is present in which case prophylactic antibiotics are continued beyond the 6-month period. In more severe cases of aortic obstruction, participation in competitive sports should probably be restricted along with avoidance of strenuous activity (see Bacterial Endocarditis later in this section of the chapter).
Please refer to Box 8-28 for advanced concepts in care of the patient with aortic stenosis
Box 8-28 Advanced Concepts: Severe Aortic Stenosis
• Patients with critical aortic stenosis may develop left ventricular ischemia during periods of stress, exercise, and tachycardia. These signs may include ST-segment elevation or depression and T-wave changes.
• Estimation of the gradient produced by the aortic obstruction is inaccurately low when cardiac output is low.
Coarctation of the Aorta
Etiology
Coarctation of the aorta is a constriction (narrowing) or stenosis of a portion of the aorta or aortic arch (Fig. 8-39). Most commonly there is a discrete congenital narrowing of the aortic arch occurring just distal to the left subclavian artery at the level of insertion of the ductus arteriosus. As a result, most coarctations are accurately described as juxtaductal in location. This description can be too simplistic, however, as coarctation anatomy can vary considerably. The coarctation may include stenosis of a long segment or it may be tortuous in presentation; it may be associated with transverse aortic arch hypoplasia or rarely the coarctation may be located in the abdominal aorta.
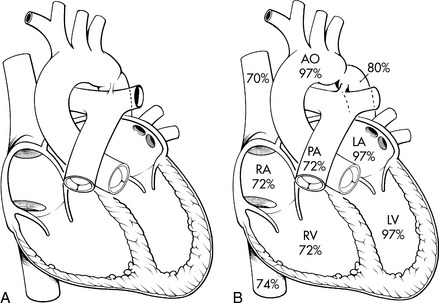
Fig. 8-39 Coarctation of the aorta. A, Postductal coarctation. This coarctation is in the region of the ductus arteriosus. B, Preductal coarctation. Typical oxyhemoglobin saturations in cardiac chambers and great vessels are depicted. When the ductus arteriosus is patent, the descending aorta is perfused largely with systemic venous blood from the right ventricle through the ductus. AO, aorta; LA, left atrium; LV, left ventricle; PA, pulmonary artery; RA, right atrium; RV, right ventricle.
Past classifications of simple coarctation included either preductal (infantile type) or postductal (adult type). This terminology, although accurate for some patients, is no longer used because the site is nearly always juxtaductal (i.e., adjacent to the insertion site of the ligamentum arteriosum or remnant of the ductus arteriosus) or located just distal to the origin of the left subclavian artery.421
Additional descriptive terms may be used to describe the coarctation or associated anomalies. With severe coarctation, underdevelopment or hypoplasia of the proximal descending aorta is referred to as long segment coarctation. The term hypoplastic aortic arch is used specifically when there is narrowing of the transverse and isthmus portions of the aortic arch. Some patients may develop enlargement of the descending aorta distal to the narrowed portion, referred to as poststenotic dilation.
Early theories speculated that coarctation resulted from decreased antegrade flow across the ascending aorta during fetal development.753 More recent evidence supports the role of ductal tissue in causing the most common juxtaductal coarctation.905 Thickening of the media of the aortic wall forms a ridge on the inner surface of the aorta, resulting in narrowing. Although the precise genetic and molecular abnormality of this defect is unknown, a well-documented genetic association has been noted in Turner syndrome and 45,XO karyotype. A reported 20% to 35% of patients with Turner syndrome are affected with coarctation of the aorta.68
Coarctation of the aorta constitutes approximately 8% of all congenital heart defects. The pathophysiology of coarctation varies with the severity of arch narrowing and presence of associated lesions. Bicuspid aortic valve is the most commonly associated cardiac congenital anomaly, present in as many as 50% to 80% of patients with coarctation. Other lesions frequently associated with coarctation include ventricular septal defect (VSD), truncus arteriosus, and transposition of the great vessels. Among coarctations requiring surgical intervention, the combination of coarctation with VSD is almost as common as coarctation alone.421 Coarctation is frequently associated with other left heart obstructive lesions, including aortic stenosis, subaortic narrowing, mitral stenosis, left ventricular and left ventricular outflow tract hypoplasia. Coarctation that occurs as one part of a constellation of left sided obstructive lesions is called Shone's complex (see section, Essential Anatomy and Physiology, Etiologies of CHD: Noninherited and Genetic Factors). The most severe form of this constellation of left heart lesions results in hypoplastic left heart syndrome (see later section, Single Functioning Ventricle).159
Pathophysiology
Following birth when the ductus arteriosus constricts, the medial layer of a portion of the aorta also constricts, creating a narrowing in the aorta. The area of stenosis can vary from a mild degree of narrowing producing only a small pressure gradient to severe obstruction causing near interruption of systemic blood flow to the lower body. Therefore, depending on the severity of coarctation and the presence of additional lesions, hemodynamic effects may range from mild upper extremity hypertension to congestive heart failure or even shock.
When there is obstruction to aortic flow, the left ventricle must generate higher pressure to maintain normal flow through the narrowed area, so hypertension will be present in the aorta and in the arteries branching from the aorta proximal to the obstruction. In addition, hypotension will be present in the aorta and arteries branching from the aorta distal to the stenotic area. The decreased flow and hypotension to the lower extremities and abdominal organs may result in ischemia of the organs perfused by the part of the aorta that is distal to the coarctation site. The head and neck vessels and coronary arteries are perfused from the part of the aorta that is proximal to the level of obstruction so they receive hypertensive blood flow.
Because the left ventricle must generate high pressure to maintain blood flow through the narrowed aorta, significant left ventricular hypertrophy may be present. Left ventricular failure may develop. If the coarctation is severe, ductal closure can result in acute development of left ventricular failure, including decreased stroke volume, elevated left ventricular end-diastolic and left atrial pressures, and pulmonary venous congestion. Low cardiac output can cause impaired myocardial perfusion, leading to cardiogenic shock.68 Inadequate systemic perfusion may produce severe acidosis and renal and gastrointestinal ischemia.
Some cases of significant coarctation result in development of arterial collateral vessels flowing from the aorta and arterial branches that are proximal to the level of obstruction to vessels that will carry blood flow back into the aorta distal to the level of obstruction. These vessels provide a source of low-pressure flow to the descending aorta and the tissues perfused by the descending aorta.
Clinical Signs and Symptoms
Clinical presentation for this cardiac defect can vary from cardiovascular collapse in neonates following ductal closure to asymptomatic hypertension in older infants and children. The pattern of clinical signs and symptoms can be divided into two categories: coarctation of the aorta presenting in the neonatal period and coarctation presenting in later infancy or during childhood.
Neonatal Coarctation of the Aorta
Neonatal coarctation of the aorta typically presents during the first week to 10 days of life and is typically associated with severe signs of shock. At birth, a newborn with severe coarctation may appear asymptomatic while the ductus arteriosus remains patent. Because pulmonary vascular resistance is high during the first hours of life, the presence of a patent ductus arteriosus (PDA) allows right-to-left shunting of blood from the main pulmonary artery to the descending aorta. Before spontaneous ductal closure the only symptom of coarctation may be mild cyanosis of the lower extremities (with a differential oxygen saturation noted with pulse oximetry). Following ductal closure, left ventricular failure and poor systemic perfusion develop and progressive signs and symptoms of deterioration include poor feeding or vomiting secondary to decreased bowel perfusion, tachypnea, pallor, listlessness and acidosis. Pulses in the lower extremities are weak or absent and severe hypotension is present in the lower extremities. Signs of multisystem organ failure can develop, including renal failure, seizures, necrotizing enterocolitis, and possible death.421
Coarctation of the Aorta Presenting During Infancy or Childhood
If coarctation of the aorta does not produce signs and symptoms during the first days and weeks of life, and if the degree of narrowing is mild or moderate in severity, the infant or child with coarctation often remains asymptomatic. Some patients may report headaches resulting from upper body hypertension or exercise intolerance and leg pain with activity, because it is impossible to increase cardiac output to tissues perfused by the distal aorta.421 Patients with severe coarctation can be asymptomatic if arterial collateral blood supply produces near normal blood flow into the descending aorta (beyond the coarctation) and into the femoral arteries, but this is uncommon.
Physical examination reveals diminished, delayed, or absent lower extremity pulses. In fact, the pathognomonic clinical findings of coarctation of the aorta include discrepant arterial pulses and systolic blood pressures between the upper and lower extremities.68 The upper extremity blood pressure is often elevated for age. Comparative upper and lower extremity measurements usually reveal a pressure gradient of 15 mm Hg or greater. In rare cases the presence of an aberrant right subclavian from the descending aorta (beyond the coarctation) will result in no blood pressure differential between upper and lower extremities.
To identify coarctation, the blood pressure in the right arm should be compared to the blood pressure in either lower extremity. The left arm blood pressure should not be compared to lower extremity blood pressures to identify a blood pressure gradient because the origin of the left subclavian artery is near the coarctation, and this location may result in lower blood pressure in the left arm than in the right arm.
Auscultation may reveal several different murmurs based on the location and severity of the coarctation and the presence of any arterial collateral vessels. A systolic ejection click may indicate the presence of a bicuspid aortic valve. A systolic ejection murmur produced by flow through the coarctation site is best heard at the upper left sternal border or over the left interscapular area of the back. A continuous murmur may also be produced from collateral vessels when present. Neonates with diminished cardiac output may have faint murmurs, and the presence of a gallop rhythm may be the most notable ausculatory finding.68
A plain chest radiograph in older patients presenting with coarctation of the aorta demonstrates a normal to mildly enlarged heart. Neonates or young infants with severe coarctation and congestive heart failure may demonstrate moderate to severe cardiomegaly and increased pulmonary vascular markings.
Erosion of the undersurface of the ribs (rib-notching) visible in an anterior-posterior (AP) chest radiograph results from dilation of intercostal vessels that provide arterial collateral blood supply. Rib-notching is not seen in infants because the collateral vessels must enlarge and it will take time to erode the lower surface of the ribs; typically rib-notching is found only in children older than 5 years with uncorrected coarctation.
A “figure 3 sign” may be identified on the AP chest radiograph, caused by the contour (silhouette) of the aortic arch. This contour includes a prominent aortic bulge proximal to the coarctation, a discrete indentation of the aorta at the coarctation site, and post-stenotic dilatation in the aorta immediately beyond the coarctation.68
The ECG is usually normal in neonates and infants with coarctation. The ECG of children and adolescents with coarctation may show increased left-sided voltages indicating left ventricular hypertrophy.
Magnetic resonance imaging (MRI) is a useful imaging tool for evaluating coarctation. It provides detailed imaging of head and neck vessels as well as the imaging of the immediate site of the coarctation, the entire aortic arch including the abdominal aorta, and it enables identification of collateral vessels if present. In addition, MRI studies can be used to estimate pressure gradients and aortic blood flow.
High-quality echocardiographic studies frequently provide sufficient physiologic and anatomic detail to accurately diagnose coarctation of the aorta. Images are readily obtainable in infants but can be more difficult to obtain in older children and adults because imaging windows are poor. Doppler imaging may be helpful: a high velocity signal in the area of the coarctation site and the presence of an altered waveform and diastolic runoff in the descending aorta is diagnostic of coarctation. Associated lesions can also be visualized via echocardiography, including bicuspid aortic valve and VSD. Left ventricular hypertrophy or dysfunction can be identified.
Diagnostic cardiac catheterization is only indicated if there are associated cardiac lesions. In the past, angiography was used to identify collateral blood flow or define arch anatomy poorly visualized by echocardiography; MRI is now diagnostic. Currently cardiac catheterization serves a therapeutic role in the management of some patients with coarctation of the aorta.
If left untreated, the natural history for patients with coarctation of the aorta is dismal. Severe coarctation in the neonate would likely be fatal, and mild or moderate coarctation and hypertension can eventually produce complications such as congestive heart failure, aortic rupture, and bacterial endocarditis. One large natural history study reported a mean age at death for patients with untreated coarctation of 34 years.134
Intervention is currently recommended for all patients diagnosed with coarctation of the aorta. The appropriate therapy is dictated by the presentation of the patient.
Management
Medical Management
Medical management is required to stabilize neonates and young infants with severe coarctation. Prostaglandin E1 (PGE1) infusion is provided to maintain patency of the ductus arteriosus or to reopen a closed ductus. The ductus provides a route of blood flow from the main pulmonary artery to the descending aorta, to maintain perfusion to the vital organs below the area of coarctation. Preoperative management with PGE1 improves organ perfusion, allowing correction of shock, acidosis and ischemia. After adequate medical stabilization, early surgical intervention is indicated.
Additional medical management during and beyond the neonatal period may include diuretic therapy for the infant with congestive heart failure. Support of organ function (e.g., renal replacement therapies) may be needed.
Surgical Therapy
Surgical therapy is the primary intervention for coarctation of the aorta in neonates, infants, and small children. Catheter intervention strategies such as balloon dilation and stent placement are alternative therapies for management of native coarctation in older patients. Surgical correction of coarctation of the aorta is usually a closed-heart procedure (i.e., without cardiopulmonary bypass) performed via a left thoracotomy. If other cardiac defects such as VSD require simultaneous repair, a sternotomy may be performed; cardiopulmonary bypass is used for the intracardiac repair.
A variety of surgical repairs have been used over the years to treat coarctation (Fig. 8-40). Presently the most frequently used approach is the end-to-end anastomosis and the second most common approach is the subclavian flap repair.
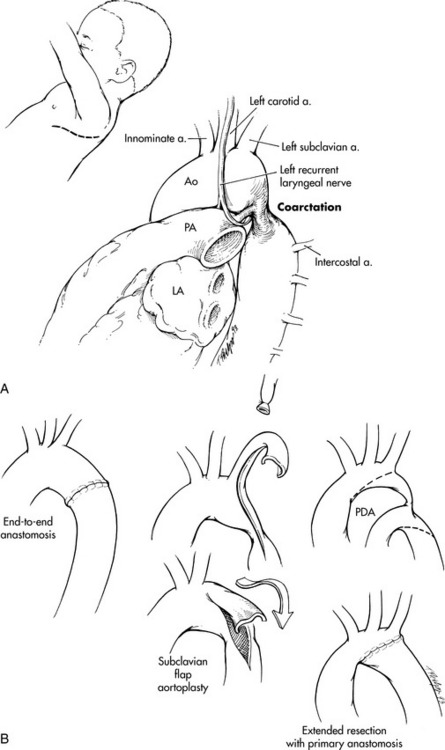
Fig. 8-40 Surgical approach to coarctation of the aorta (CoA). A, Typical surgical incision and surgical anatomy. B, Three operative procedures commonly used in repair of CoA: resection of the stenotic segment and end-to-end aortic anastomosis, subclavian flap aortoplasty, and extended resection with primary anastomosis. A, artery; Ao, aorta; LA, left atrium; PA, pulmonary artery; PDA, patent ductus arteriosus.
(From Schwengel DA, Nichols DG, Cameron DE: Coarctation of the aorta and interrupted aortic arch. In Nichols DG, et al, editors: Critical heart disease in infants and children, ed 2, St Louis, 2006, Mosby, Fig. 27-10.)
During the procedure, once the aorta is dissected, clamps are placed above and below the narrowed area. During dissection care is taken to avoid injury to lymphatic vessels as well as to the vagus, phrenic, and recurrent laryngeal nerves. If collateral vessels are present, they are generally ligated and divided to prevent intraoperative and postoperative bleeding.44
With the end-to-end approach the stenotic area of the aortic arch and ductal tissue are resected, and then the two ends of the aorta are anastomosed (sewn) together. Advantages to this technique include removal of ductal tissue and lack of prosthetic material (that can act as a site for infection). Disadvantages include a circumferential suture line and the resulting potential for recoarctation (narrowing) at that repair site.
A variation of this end-to-end anastomosis technique is used in cases of hypoplasia of the isthmus or transverse arch. The incision extends onto the transverse arch and under the left common carotid and the length of the incision is used to widen or extend the end-to-end anastomosis.
The subclavian flap procedure requires that the left subclavian artery be ligated (tied off) and divided (cut). A longitudinal incision is made through the proximal segment of the subclavian artery and down the aorta to the area just beyond the coarctation. The subclavian stump is then opened and turned down and stitched to the aorta, resulting in a patch of autologous tissue enlarging the area of coarctation. Potential advantages of this technique include avoidance of prosthetic material (that would act as a potential site for infection) and the potential for growth of the patch site as the child grows. In addition a circumferential suture line is avoided. The major disadvantage is the required sacrificing of the left subclavian artery.
More recently some centers have used a reverse left subclavian flap approach to repair discrete coarctation accompanied by distal arch hypoplasia. The left subclavian artery is mobilized as described. In this variation, the left subclavian flap is turned back in a reverse direction to augment the hypoplastic portion of the aortic arch. The segment of discrete narrowing is removed and an end-to-end anastomosis is performed, extending under the portion of the arch just enlarged by the reverse flap.421
Several techniques used in the past are not commonly used at present, but are mentioned here because they may be used for unique situations, and they may be present in a child who develops recoarctation several years after one of these procedures. Prosthetic patches were used many years ago for the aortoplasty, but they resulted in a higher rate of aneurysm formation than expected. A prosthetic patch is occasionally used in a small child with long segment of narrowing of the aortic arch. Another option for older patients with long-segment coarctation or recoarctation is placement of a prosthetic interposition graft. Rarely, a bypass graft or prosthetic tube is placed between the proximal and distal aorta, to bypass the stenotic area of the aorta.
Initially after repair, regardless of surgical technique used, most patients have a mild residual blood pressure gradient. Most gradients are less than 10 to 20 mm Hg.68
Optimal management of coarctation with a coexisting VSD remains controversial. A one stage repair includes VSD closure at the time of coarctation repair through a median sternotomy approach. Additional strategies include pulmonary artery banding at time of coarctation repair or coarctation repair alone with no initial VSD intervention. Often the coarctation repair alone will produce significant hemodynamic improvement, and the patient can be treated with diuretic therapy if the VSD remains symptomatic. In addition, the VSD may become relatively smaller or close completely during the next several months. If a pulmonary artery banding is performed, later debanding will be required, and it may be necessary to enlarge the pulmonary artery at the site of the band constriction.159
Surgical mortality for isolated coarctation of the aorta in infants and older children is near 0%. In the presence of associated lesions such as large VSD, mortality increases from 2% to 10% and is even higher when more complex cardiac defects are also present.68 In the Society of Thoracic Surgeons Congenital Heart Surgery Executive Summary, a hospital mortality rate of 1.5% is reported for all patients undergoing coarctation repair from 2006 to 2010.820
Potential early postoperative complications after repair of coarctation of the aorta include paradoxic hypertension, postcoarctectomy syndrome, paraplegia resulting from spinal cord ischemia, bleeding, residual coarctation, or injury to nearby structures such as lymphatics with resultant chylothorax.159 Late hypertension and recurrent coarctation may also develop.
Paradoxic hypertension is common postoperatively and appears to have multiple etiologies. In the first 24 to 48 hours after surgery, hypertension is likely related to elevated catecholamine levels from the stress of surgery and resetting of the baroreceptors in the aortic arch and carotid arteries. Medical management must include adequate sedation and analgesia (see Chapter 5). In addition, vasodilators, such as nitroprusside or hydralazine, used in conjunction with a beta-blocker such as esmolol or propranolol, are effective. Other strategies include treatment with labetalol, which produces both alpha- and beta-blocking effects.
A second phase of postoperative hypertension lasting several days to weeks is likely related to elevated angiotensin levels secondary to a fall in renal blood pressure relative to preoperative pressure. Effective management uses angiotensin converting enzyme inhibitors, such as intravenous enalapril, or oral preparations, such as captopril. Poorly controlled hypertension during either postoperative phase can result in additional complications, such as bleeding or postcoarctectomy syndrome.
Postcoarctectomy syndrome is a term to describe the development of mesenteric arteritis after coarctation repair. The cause is speculated to be either reflex vasoconstriction of mesenteric vessels or vessel injury after reintroduction of pulsatile (and possibly hypertensive) blood flow to the abdominal vessels. Postoperative mesenteric arteritis may produce abdominal pain and distension. With progression of the arteritis, bowel ischemia, gastrointestinal bleeding, and bowel necrosis can develop. Prevention and management include nasogastric decompression, administration of IV fluids, bowel rest, and cautious introduction of enteral feedings (see Chapter 14).
Spinal cord injury and subsequent paralysis is a rare but catastrophic complication of coarctation repair. Proposed theories for this injury include prolonged aortic cross-clamping time compromising flow to the descending aorta and spinal arteries, inadequate collateral arterial circulation during aortic clamping, or the inherent anatomy of the spinal artery. The anterior spinal artery receives collateral blood flow from intercostal arteries that branch from the descending aorta. The surgeon must ensure adequate distal aortic flow during aortic clamping and repair. Postoperatively a comprehensive lower body neurologic evaluation should be performed.
Suture line hemorrhage can occur following coarctation repair. A chest tube is left in place at the end of the procedure and should be monitored closely for excessive or sudden increase in blood output necessitating return to the operating room to control bleeding.
The term residual coarctation describes the presence of a significant gradient in the aortic arch immediately after repair. A residual coarctation is considered significant if the systolic blood pressure gradient between right arm and a lower extremity is greater than 20 mm Hg at rest.
Several key structures are located in the area near the aortic arch and injury during surgery can cause complications such as chylothorax if the thoracic duct is injured, paralysis of the hemi-diaphragm in the case of phrenic nerve damage, and vocal cord paralysis with recurrent laryngeal nerve injury.159 A chylothorax is likely present if milky chest tube drainage is observed; the volume of drainage will increase with resumption of enteral feeding postoperatively.
If a subclavian flap is utilized for the repair, the subclavian artery is sacrificed, so it will not be possible to obtain a cuff pressure or arterial sample from the left arm. In addition, venipunctures should not be performed on that arm. The arm may feel cool and appear mottled for several hours or even days after surgery until collateral vessels improve perfusion to the arm. Minor growth retardation of the left arm has been reported after use of the subclavian artery for arterioplasty.44
Careful and regular followup is required following repair of coarctation. Late complications after surgical repair of coarctation include the development of recurrent coarctation, hypertension not associated with residual coarctation, and aneurysm formation.
Recurrent coarctation implies the development of restenosis after an initially successful repair and has been reported after every type of coarctation repair.44 Restenosis is thought to be related to scar tissue formation at the site of aortic anastomosis or to inadequate resection of ductal tissue at the time of initial repair. Balloon angioplasty is currently the initial therapy of choice to address recoarctation.
Persistent systolic hypertension not caused by recurrent coarctation is more common when the initial coarctation repair is performed after 5 years of age. These patients require pharmacologic therapy, such as angiotensin converting enzyme (ACE) inhibitors (captopril or enalapril) to prevent secondary cardiovascular complications of hypertension.
Aneurysm formation in the aortic arch at the repair site can occur following any surgical approach although the highest incidence has been reported following prosthetic patch aortoplasty.111 If aneurysms are present, they must be monitored closely for signs of progression as rupture can cause sudden death.
Interventional Therapy
Interventional therapy for treatment of coarctation of the aorta includes percutaneous balloon angioplasty and intravascular stenting. These less invasive alternative therapies can be used to treat both native (never operated) and recurrent (after surgical repair) coarctation. Balloon dilation angioplasty was first undertaken to treat restenosis of surgically resected coarctation. Later the technique was expanded to include dilation of native coarctation. Balloon angioplasty has been successful in the treatment of discrete native coarctation,258 and some centers routinely manage coarctation in the interventional catheterization laboratory. This approach is not universal, however, chiefly because of concerns regarding residual stenosis and aneurysm formation. Documented significant (greater than 20 mm Hg) residual gradients have been reported in 14% to 27% of patients, and aneurysm formation in 5% to 10% of patients after angioplasty.611
The treatment of recurrent coarctation with balloon dilation has become the first-line intervention at most centers. The reported incidence of residual stenosis and of aneurysm formation following balloon dilation for recurrent coarctation are similar to those reported following treatment of native coarctation although the theoretical advantage of dilating a fibrous postsurgical scar is thought to reduce the likelihood of aneurysm development.
Complications from balloon dilation of recurrent and native coarctation are rare but can occur. Acute tears in the aortic wall are rare but serious complications. Aneurysms of the aortic wall can develop early following dilation or may appear years after the dilation. Arterial vessel injury was more frequently reported early in the balloon angioplasty experience; however, advances such as smaller balloon profiles and the use of indwelling sheaths has greatly reduced vessel injury.640
Coarctation stenting has been shown to be effective in treating discrete native and recurrent coarctation. The use of intravascular stents for primary treatment of coarctation in larger adolescents and adults has become the treatment of choice at some centers. General practice requires placement of a stent into the aorta only if it is expandable to the adult diameter of the aorta.640 Currently this prevents the use of stents in treatment of coarctation in infants and children because the large femoral sheath size necessary to deliver adult-size stents is too large for small patients. Balloon-expandable stents have the reported benefit of providing endovascular stability and may diminish the incidence of late aneurysm formation.417
Antibiotic prophylaxis against bacterial endocarditis in patients with coarctation of the aorta is recommended for 6 months after surgical repair using prosthetic material and indefinitely if a significant residual coarctation gradient is present. The presence of a bicuspid aortic valve alone no longer necessitates antibiotic prophylaxis; however, adherence to current American Heart Association guidelines for endocarditis prophylaxis is recommended (see Bacterial Endocarditis later in this section of the chapter).951
Advanced concepts in the management of the infant with severe coarctation of the aorta are listed in Box 8-29.
Box 8-29 Advanced Concepts: Coarctation of the Aorta
In cases of infant coarctation, a blood pressure gradient may not initially be detected if a patent ductus arteriosus is present. Because pulmonary vascular resistance may still be elevated in the first hours and days after birth, blood can flow to the descending aorta via the ductus arteriosus, resulting in adequate lower extremity perfusion and relatively equal upper and lower limb blood pressures. Blood flowing to the descending aorta is desaturated, so lower extremity oxyhemoglobin saturation is decreased.
Interrupted Aortic Arch
Etiology
Interrupted aortic arch (IAA) is defined as the congenital absence or atresia of a segment of the aortic arch resulting in complete separation between the ascending and descending aorta. Interrupted aortic arch is classified into three types is based on the site of interruption (Fig. 8-41). In Type A, the interruption occurs just distal to the left subclavian artery at the level of the isthmus. This subtype may be a variant of severe coarctation of the aorta and accounts for approximately 30% of patients with interrupted aortic arch. In Type B the aorta is interrupted between the left carotid and the left subclavian artery; the left subclavian artery arises from the descending aortic segment. This form of interrupted aortic arch is the most common, responsible for about 70% of all interrupted aortic arch. In Type C the interruption is between the right innominate artery and the left carotid artery. This type is very rare and accounts for only 1% of cases of interrupted aortic arch.
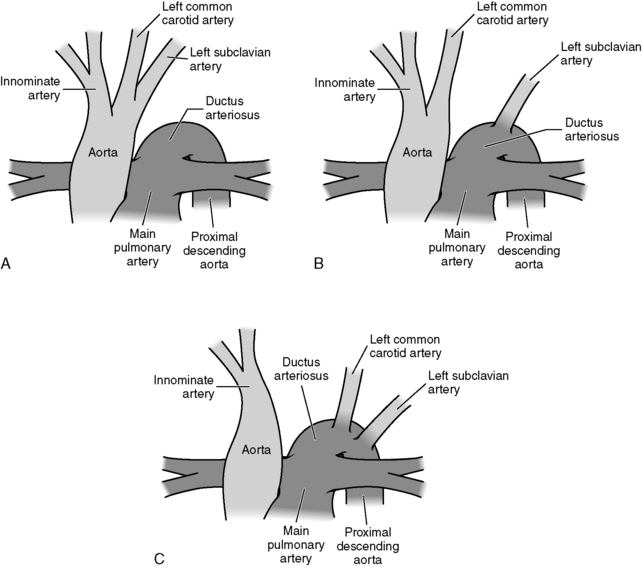
Fig. 8-41 Anatomic types of interrupted aortic arch. A, Type A, interruption just distal to the left subclavian artery. B, Type B, interruption between the left subclavian and left common carotid arteries. C, Type C, interruption between the left common carotid and innominate arteries.
(Redrawn from Mavroudis C, Backer CL, editors: Pediatric cardiac surgery, ed 3, Philadelphia, 2003, Mosby.)
This defect is thought to result from faulty formation of the aortic arch system during fetal development (see Evolve Fig. 8-1 in the Chapter 8 Supplement on the Evolve Website). Abnormalities in development of the left fourth arch in the embryo are felt to play a role in the occurrence of all three subtypes of IAA.930 There is also a strong link between chromosomal deletion of 22q11 (DiGeorge syndrome), and interrupted aortic arch, specifically Type B, so this syndrome should be ruled out in newborns with Type B IAA.
Interrupted aortic arch accounts for approximately 1.5% of all congenital heart defects. Interrupted aortic arch is rarely present as an isolated defect; most infants have additional congenital heart defects. The most commonly associated lesion is an isolated VSD. Other accompanying defects include ASD, truncus arteriosus, aortopulmonary window, transposition of the great arteries, double outlet right ventricle, and single ventricle.421 Type A interruption has been noted in a markedly large subgroup of patients with transposition of the great arteries, whereas Type B interruptions are more commonly associated conotruncal anomalies, malalignment-type VSD, and subaortic obstruction.930
Pathophysiology
Presentation of these patients typically is similar to presentation for other ductal-dependent left heart and aortic obstructive lesions, such as critical coarctation of the aorta. The neonate becomes acutely ill with signs of heart failure in the first days of life. After spontaneous closure of the ductus arteriosus, acute cardiovascular collapse ensues, because perfusion of the descending aorta and lower body occurs only through arterial collateral flow. If untreated, ischemic injury to the gut, liver, and kidneys will occur. Soon severe acidosis will develop resulting in injury to all body systems, including the heart and brain.420
Clinical Signs and Symptoms
Clinical signs and symptoms of interrupted aortic arch are the same as those described for critical coarctation of the aorta in the neonate. The neonate is tachypneic with evidence of decreased systemic perfusion, severe metabolic acidosis, and oliguria or anuria. The discrepancy in peripheral pulses will be affected by the anatomic subtype of interrupted aortic arch and the presence of an open or closed ductus arteriosus. For example, a neonate with Type B interruption and a closed ductus should have a palpable pulse in the right arm but absent left arm and femoral pulses.
Cyanosis is not a common clinical finding, particularly when the ductus closes, because the head and right arm are perfused with oxygenated blood from the left ventricle, and the left arm and lower body are perfused only with blood flow delivered to the descending aorta via collateral circulation.
In rare instances, patients with interrupted aortic arch have survived to adulthood without symptoms. These patients usually have extensive collateral circulation that maintains adequate descending aortic blood flow.
Prenatal diagnosis of many cardiac defects, including interrupted aortic arch, allows early intervention to maintain patency of the ductus arteriosus, preventing ductal constriction and the resulting period of low cardiac output. For those patients not identified prenatally, the diagnosis of interrupted aortic arch is suspected by the clinical presentation. It must be differentiated from coarctation of the aorta, aortic atresia, and hypoplastic left heart syndrome.
Two-dimensional (2D) echocardiography is the most important diagnostic tool in identifying this defect. Images pinpoint the site of interruption, the length of discontinuity, the diameter of the aortic annulus, and the diameter of the ascending aorta. All of these anatomic details influence the surgical management of this cardiac lesion. Angiography is still used in some centers to confirm the diagnosis and further delineate arch anatomy. However, accurate diagnosis can be made using echocardiography alone, eliminating the need for invasive cardiac catheterization in a neonate who is often in shock. Recently three-dimensional cardiac magnetic resonance imaging (3D cardiac MRI) has been used to identify the arterial branching pattern and area of separation between the proximal and distal aorta.
Management
Initial medical management includes maintenance or restoration of ductal patency with infusion of prostaglandin E1, fluid resuscitation, correction of acid-base and electrolyte imbalances, and inotropic support as necessary to optimize systemic perfusion (see Shock, Chapter 6). Once the neonate is stabilized, surgical correction is undertaken as soon as possible.
The surgical approach is via a median sternotomy. Typically all types of interrupted aortic arch can be reconstructed by direct anastomosis of the upper and lower segments of the arch after liberal dissection of each component. If indicated, homograft augmentation of the resected area can be performed to achieve an adequate arch diameter. Prosthetic tube grafts are generally not used for initial neonatal repair because they are rapidly outgrown and may complicate reintervention at a later date.
Whenever possible, simultaneous correction of coexisting cardiac defects such as VSD, transposition of the great arteries, and aortic stenosis is performed. Palliative operations used in the past are not often employed because primary one-stage repair in the neonatal period is considered optimal management.
Postoperative complications include those discussed after repair of severe coarctation of the aorta during infancy. In addition, pulmonary hypertension may be present in the early postoperative period (see section, Common Clinical Conditions, Pulmonary Hypertension) and residual obstruction within the arch or subaortic area may require surgical reintervention.158
The natural history of unrepaired interrupted aortic arch is death within days of birth, and surgical mortality as recently as the 1990s was high.673 With advances in surgical intervention and postoperative care, survival for interrupted aortic arch with VSD is 92% at 1 month.820
Antibiotic prophylaxis against bacterial endocarditis is recommended for a 6-month period after surgical repair if prosthetic material was used. Also, if a significant residual aortic arch gradient is present following repair, adherence to current American Heart Association guidelines for endocarditis prophylaxis is recommended (for further information, see Bacterial Endocarditis later in this section of the chapter).951
Mitral Valve Dysfunction
Etiology
A normal mitral valve is made up of five parts: the valve leaflets, valve annulus, papillary muscles, chordae tendineae, and left ventricular wall (Fig. 8-42). The valve has a smaller anterior or aortic leaflet and a larger posterior or mural leaflet. Congenital mitral valve abnormalities are relatively rare but include isolated mitral stenosis and mitral regurgitation.
Fig. 8-42 Mitral valve anatomy, shown in relationship to tricuspid valve (TV), pulmonary valve (PV) and aortic valve. Ao/RCC, aortic valve, right coronary cusp; Ao/NCC, aortic valve, non-coronary cusp; Ao/LCC, aortic valve, left coronary cusp, RCA, right coronary artery; LCA, left coronary artery; LAD, left anterior descending branch of LCA; LCir, left circumflex branch of LCA.
(From Nichols DG, et al, editors: Critical heart disease in infants and children, ed 2, St Louis, 2006, Mosby, Fig. 28-1.)
Congenital mitral valve abnormalities are commonly associated with additional congenital heart defects. Congenital mitral stenosis frequently occurs in association with other left heart obstructive lesions, such as coarctation of the aorta or aortic stenosis. Congenital mitral regurgitation is more commonly associated with atrioventricular septal defect (particularly complete AV canal), isolated mitral valve cleft, or mitral valve prolapse.
Most mitral valve disease in the pediatric population is acquired. Causes of acquired mitral stenosis and mitral regurgitation are listed in Box 8-30 and detailed in the sections immediately following.
Mitral Valve Stenosis
Mitral stenosis is defined as a narrowing of the mitral valve orifice that can involve the leaflets, the annulus, or both. Isolated mitral stenosis is a rare form of congenital heart disease and accounts for less than 0.5% of congenital heart defects.
Stenosis of the mitral valve results in obstruction of flow to the left ventricle from the left atria. It can be caused by pathology in the supravalvular, valvular, or subvalvular regions. Congenitally stenotic valves frequently produce obstruction at more than one level.
A variety of terms are used to describe abnormal stenotic mitral valves. The term mitral arcade valve implies the presence of thickened leaflets, absent or abnormal chordal insertions, and fused commissures. A “parachute” mitral valve takes the form of a funnel or the dome of a parachute with multiple small holes in the sides of the tissue and no true central orifice; it is considered the most severe form of congenital mitral stenosis.309 Lastly, the mitral valve can be entirely normal from a structural perspective but hypoplastic. This small size makes the valve functionally stenotic as is often the case in hypoplastic left heart syndrome (see section, Single Functioning Ventricle).
Mitral Valve Regurgitation
Mitral regurgitation, also referred to as mitral insufficiency, is far more common than mitral stenosis. Causes include dilation of the valve annulus to a point where leaflets no longer appose each other, resulting in central regurgitation. Mitral regurgitation can also result from structural abnormalities similar to those causing mitral stenosis such as dysplastic leaflets, isolated leaflet cleft, leaflet prolapse, or abnormal chords and papillary muscles.
Mitral Valve Prolapse
Mitral valve prolapse is present when the mitral valve leaflets move back across or prolapse across the mitral valve annulus into the left atrium during ventricular systole. In the past mitral valve prolapse was often over diagnosed in children using echocardiographic criteria. However, more recent studies using consistent diagnostic criteria found rates of occurrence in the general population between 0.6% and 2.4%.271,279
Mitral valve prolapse occurs both as a primary lesion and in association with a variety of other disorders. It is strongly associated with connective tissue disorders such as Ehlers-Danlos syndrome and is almost always present in patients with Marfan syndrome. Mitral valve prolapse is also associated with congenital heart defects such as ASD, Ebstein anomaly, and corrected transposition.
Pathophysiology
Mitral Valve Stenosis
When there is obstruction to blood flow from the left atrium to the left ventricle, the left atrium dilates and left atrial pressure increases. This left atrial hypertension causes elevation in pulmonary venous pressure and pulmonary congestion. The increased pulmonary venous pressure will cause elevated pulmonary arterial pressure, requiring increased right ventricular systolic pressure. In addition, severe obstruction of blood flow from the left atrium to the left ventricle can produce decreased cardiac output, although left ventricular function is usually maintained. Extreme mitral stenosis, as seen in Shone syndrome or hypoplastic left heart syndrome may prevent the left heart from supporting systemic circulation and the child may be dependent on flow from the right ventricle through the pulmonary artery and ductus arteriosus to maintain adequate systemic blood flow.421
Mitral Valve Regurgitation
As with mitral stenosis, mitral regurgitation increases left atrial and pulmonary venous pressures and ultimately increases pulmonary artery pressure. However, mitral regurgitation also results in left ventricular volume overload and may produce left ventricular dysfunction. Once mitral regurgitation of any origin is present, it is likely to progress because the regurgitation itself results in left ventricular dilation and further mitral valve annulus dilation and insufficiency.
Mitral Valve Prolapse
The consequences of mitral valve prolapse are determined by the degree of mitral regurgitation, which in turn is directly proportional to the amount of prolapse and the extent of annular dilation. The hemodynamic consequences of mitral regurgitation apply to infants and children with mitral valve prolapse.
Clinical Signs and Symptoms
The clinical presentation of mitral stenosis and mitral regurgitation is multifactorial and depends on the severity of obstruction or regurgitation and potential compounding effects if both lesions are present. Mild mitral stenosis or regurgitation is usually not associated with symptoms. In the setting of moderate mitral stenosis, infants present with symptoms typical of congestive heart failure, including tachypnea, diaphoresis, poor feeding, and failure to thrive. The presence of pulmonary venous congestion increases risk of respiratory infections. Older children may present with orthopnea, dyspnea on exertion, and exercise intolerance. Symptoms of moderate to severe mitral insufficiency are often identical to those of mitral stenosis.
Arrhythmias, including atrial flutter and atrial fibrillation can develop in patients with significant atrial enlargement caused by either mitral stenosis or insufficiency. These arrhythmias can be detected by Holter or event monitor. Echocardiography is the diagnostic tool of choice in patients with mitral stenosis and mitral regurgitation.
Mitral Valve Stenosis
Ausculatory findings in mitral stenosis include an opening snap, which is a sharp high-pitched sound occurring after the second heart sound (S2), corresponding to the mitral valve opening. The murmur associated with mitral stenosis is heard at the apex and is a low-frequency, mid-to-late diastolic murmur. The pulmonary component of the second heart sound is increased if pulmonary hypertension is present.
Increased right heart forces and right-axis deviation are present on the ECG for both mitral stenosis and regurgitation. The chest x-ray of patients with moderate to severe mitral stenosis demonstrates prominent pulmonary vasculature and cardiomegaly with left atrial enlargement.
In assessing mitral stenosis, 2D echocardiographic images can identify structural abnormalities at the valvular, subvalvular, and supravalvular levels. Doppler flow studies across the mitral valve enable calculation of the flow gradient from the left atrium to the left ventricle. Mild stenosis results in a mean gradient of 4 to 5 mm Hg, a moderate gradient is 6 to 12 mm Hg, and a gradient greater than 13 mm Hg indicates severe stenosis. Severe stenosis is often associated with systemic right heart pressures. It is important to remember, however, that the estimated gradient is affected by flow through the mitral valve and the presence of a left-to-right atrial level shunt can decrease the estimated gradient (because flow across the valve is decreased) and a left-to-right ventricular level shunt may increase the gradient (because pulmonary blood flow and the flow of pulmonary venous return across the mitral valve is increased).
A second echocardiographic method of evaluating the severity of mitral stenosis is to measure functional size of the mitral valve orifice. Normal values have been identified for children and relate to body surface area, allowing calculation of Z scores. A Z score of −2.5 to −3.0 indicates a low likelihood that the mitral valve is functional.421
Cardiac catheterization may be helpful in some cases of mitral stenosis if echocardiography alone is unable to determine Doppler gradients or right heart pressures. Ideally, left atrial and left ventricular pressures are measured simultaneously to definitively calculate the pressure gradient. The left atrial pressure can be estimated via pulmonary artery wedge pressure: after a balloon-tipped catheter is inserted through the right heart and into the pulmonary artery, inflation of the balloon occludes the antegrade flow through the pulmonary artery so the pressure distal to the balloon can reflect left atrial pressure. However there are several caveats to this estimation of left atrial pressure (see Pulmonary Artery Catheters in Chapter 21).
Mitral Regurgitation
Physical examination findings with mitral regurgitation include a hyperactive precordium and a high frequency holosystolic blowing murmur heard at the apex with radiation to the left axillae. If mitral insufficiency is severe, a diastolic rumble can be heard secondary to increased blood flow crossing the mitral valve. The pulmonary component of the second heart sound may be increased if pulmonary hypertension has developed.
Patients with mitral regurgitation have an enlarged left ventricle. Increased right heart forces and right-axis deviation are seen on the ECG for both mitral stenosis and regurgitation, but patients with mitral regurgitation also demonstrate increased left heart forces.
Echocardiography can very effectively diagnose mitral regurgitation, often identifying even very trivial regurgitation. Mitral regurgitation is usually graded as mild, moderate, or severe. Echocardiography is also able to localize the regurgitation as either central or through a cleft and can identify the presence or progression of left atrial and left ventricular enlargement. Cardiac catheterization is not necessary or specifically useful in identifying the source or severity of insufficiency.
Mitral Valve Prolapse
Current diagnostic criteria for mitral valve prolapse include both ausculatory and echocardiographic findings. A mid-to-late systolic click with or without a murmur of mitral regurgitation is present on physical examination. Two-dimensional echocardiography to confirm the diagnosis of mitral valve prolapse requires visualization of the mitral valve apparatus falling back into the left atrium across the plane of the mitral valve in systole.
Patients with mitral valve prolapse can be asymptomatic. Presenting complaints may include palpitations, atypical or nonspecific chest pain, decreased exercise tolerance or fatigue, and syncopal or near syncopal episodes. The palpitations are attributed to premature atrial contractions or premature ventricular contractions. Supraventricular tachycardia and ventricular arrhythmias are common in those patients with moderate to severe mitral regurgitation resulting in left atrial and left ventricular enlargement. The type of chest pain reported is of variable severity and not reproducible during an exercise stress test.
Management
Mitral Valve Stenosis
Management of mitral stenosis is determined by the severity of the obstruction. Patients with mild mitral stenosis are often followed clinically with serial echocardiograms performed to evaluate any left atrial enlargement and the progression of the gradient across the mitral valve. These patients are often asymptomatic and the stenosis may or may not progress.
Moderate degrees of mitral stenosis produce pulmonary venous obstruction and pulmonary venous congestion that can often be successfully managed medically while closely monitoring for signs of deteriorating left or right ventricular function or worsening pulmonary hypertension. Patients with severe mitral stenosis require early intervention.
Medical Therapy
Treatment targets relief of symptoms of congestive heart failure. Pulmonary edema is treated with diuretic therapy. Patients with poor weight gain, frequent respiratory infections, or echocardiographic signs of worsening ventricular function require further intervention.
Pulmonary venous congestion from mitral stenosis increases the risk of severe respiratory illnesses including respiratory syncytial virus (RSV). The administration of palivizumab (Synagis), a humanized monoclonal antibody against the RSV is recommended in infants and children younger than 24 months with hemodynamically significant heart disease such as mitral stenosis. Palivizumab is administered monthly during RSV season and timing varies by geographic region.
Interventional Therapy
The low incidence of congenital mitral stenosis has limited the experience with balloon mitral valvuloplasty in smaller children. As a result, mitral valve balloon valvuloplasty is primarily performed in patients with rheumatic mitral stenosis. Balloon dilation should not be attempted for treatment of mitral stenosis if the valve leaflets are poorly defined or there is parachute-like anatomy.
Successful balloon dilation reduces the degree of stenosis and delays the need for surgical intervention until the child is larger. Unfortunately, the unwanted tradeoff for relief of stenosis is mitral insufficiency. Currently percutaneous balloon valvuloplasty of the mitral valve should be contemplated only in select patients at centers with established experience and skill.92
Surgical Therapy
Surgical intervention for mitral stenosis involves repair or replacement of the mitral valve. Typically much effort is made to avoid mitral valve surgery in infants and children because results are variable, morbidity can be high, and often there is need for valve replacement. The surgical approach for each patient must consider valve anatomy and the presence of any associated lesions. Closure of an associated PDA or VSD may sufficiently reduce the pulmonary venous return and blood flow through the mitral valve that the mitral valve gradient is reduced and need for mitral valve surgery is eliminated or postponed. Approaches to repairing a stenotic mitral valve include resection of a supravalvular mitral ring, commissurotomy, splitting of fused cords or papillary muscles, muscle resection, or enlargement of the mitral annulus.
When mitral valve repair is not possible, replacement becomes necessary. Most commonly the valve is replaced with a prosthetic (mechanical) valve although placement of a bioprosthetic valve (homograft or heterograft) in the mitral position is an option. Unfortunately, bioprosthetic valves exposed to left ventricular (systemic) pressures deteriorate quickly, requiring early reintervention.164
Mitral valve replacement in small patients requires careful consideration of valve size because a prosthetic or bioprosthetic mitral valve placed during childhood is eventually outgrown and will require another valve replacement. Alternately, placing too large a prosthesis into the native valve annulus can result in complete heart block.
Placement of a mechanical mitral valve results in the need for long-term anticoagulation and its associated risks in infants and children (see Postoperative Care and Anticoagulation). In adolescent females the use of bioprosthetic valves is preferred because the warfarin required for anticoagulation of mechanical valves has teratogenic potential.65 The preferred valve for smaller children is a low-profile bileaflet valve. For infants with a valve annulus smaller than the smallest prosthetic valve available, supra-annular mitral valve replacement is a surgical option.421
Mitral Valve Regurgitation
Therapy to treat mitral regurgitation varies with the severity of the insufficiency. Mild to moderate regurgitation can be palliated temporarily with medical anticongestive therapy (diuretics) and close monitoring of left ventricular function. Progression to moderate insufficiency or the acute development of severe regurgitation necessitates surgical intervention.
Medical Therapy
Medical management of mild to moderate mitral regurgitation includes standard pharmacologic therapy for congestive heart failure. Specifically, afterload reduction with use of angiotensin converting enzyme inhibitors is beneficial in treating mitral insufficiency and should be maximized.
Interventional Therapy
Currently there are no interventional catheter techniques that are effective in the management of mitral regurgitation.
Surgical therapy
The criteria for surgical intervention to treat mitral regurgitation are less stringent than for repair of mitral stenosis because surgical intervention generally substantially improves the degree of insufficiency regardless of the anatomic cause. Delay in repair can worsen the chance of successful repair if left ventricular dysfunction develops or secondary changes to the valve occur.
The initial surgical attempt is almost always valve repair with valve replacement reserved as a final option. Surgical intervention for a regurgitant valve consists of assessment of annulus size, ensuring that leaflets are mobilized, leaflet repair if necessary, and closure of any clefts.509
Operative techniques for addressing the mitral regurgitation include cleft closure, chordal shortening, and annuloplasty for central regurgitation.421 The placement of an annuloplasty ring to aid in reduction of annulus size is typically avoided in infants and children because it may restrict the growth of the annulus. The procedure for mitral valve replacement for mitral regurgitation is the same as for mitral stenosis; however, the presence of annulus dilation eliminates the need for supra-annular valve placement.
Postoperative Care
Postoperative care after mitral valve surgery requires close assessment for changes in left ventricular compliance, and monitoring for elevated left atrial pressure and pulmonary hypertension. The placement of a left atrial pressure catheter intraoperatively facilitates this postoperative management.155
In addition to monitoring for possible development of complete heart block mentioned earlier, other potential postoperative complications following valve implantation include thrombus formation on the surface of the mechanical valve and hemolysis. The absence of a mechanical click raises concern regarding possible thrombus formation on the mechanical valve leaflets. The development of a perivalvular leak can result in a shearing force that lyses red blood cells.
Results for surgical management of mitral valve disease vary by complexity of the repair needed and the presence of associated cardiac defects. Straightforward repair of mitral regurgitation carries the lowest surgical risk with documented hospital mortality of 1.1% to 3.3%. However, for surgical repair of complex mitral anomalies with concurrent intracardiac disease, mortality prior to discharge as high as 26.6% has been reported.337,955 Unfortunately, patients undergoing surgical repair of the mitral valve often require reoperation and possible valve replacement.
In general, mitral valve replacement carries a higher mortality rate and less favorable prognosis than mitral valve repair. For children requiring valve replacement, early series reported operative mortality as high as 33%; however, recent advances in perioperative management have decreased hospital mortality to 5.8% for isolated mitral valve replacement.337
Mitral Valve Prolapse
The natural history of mild mitral valve prolapse with mild mitral regurgitation is generally excellent. Surgical repair or replacement of the mitral valve is needed for those patients with moderate to severe mitral regurgitation, and should be undertaken before the development of significant left atrial or left ventricular enlargement. Operative techniques include those reviewed in the discussion of surgical management of mitral stenosis and mitral regurgitation.
Patients with mitral valve prolapse may require endocarditis prophylaxis. For those with mitral valve prolapse without valvular regurgitation, endocarditis prophylaxis is not recommended. If there is valvular dysfunction and associated insufficiency, the patient falls into the moderate risk category and endocarditis prophylaxis is recommended. And finally, those with mitral valve prolapse resulting in a bioprosthetic or prosthetic cardiac valve placement are considered to be high-risk for endocarditis and endocarditis prophylaxis is recommended (see Bacterial Endocarditis later in this section of the chapter).952
Advanced concepts for the care of the child with mitral valve dysfunction are listed in Box 8-31.
Box 8-31 Advanced Concepts: Mitral Valve Dysfunction
• The ausculatory exam findings alone can often diagnose mitral valve prolapse. Patients with mitral valve prolapse have a mid-systolic click and late systolic murmur. The most important feature of the murmur and click in mitral valve prolapse is its variability with maneuvers designed to increase or decrease the left ventricular systolic volume.
 With maneuvers such as moving from a squat to a standing position, which decrease left ventricular systolic function and thus decrease venous return, the click and murmur occur earlier in systole. With maneuvers that increase left ventricular systolic volume, such as moving from a standing to a squatting position, the click and murmur occur later in systole.
With maneuvers such as moving from a squat to a standing position, which decrease left ventricular systolic function and thus decrease venous return, the click and murmur occur earlier in systole. With maneuvers that increase left ventricular systolic volume, such as moving from a standing to a squatting position, the click and murmur occur later in systole.• The timing of the click can also vary with the severity of the prolapse and regurgitation. A later click and murmur are usually associated with milder prolapse, whereas more significant prolapse and regurgitation cause the click to move earlier into systole.
Cyanotic defects
General principles in the care of the child with cyanotic heart disease are summarized in the section, Common Clinical Conditions, Hypoxemia. When the newborn demonstrates cyanosis it is important to determine if the cyanosis is cardiac (i.e., caused by congenital heart disease) or pulmonary in origin. The hyperoxia test is often performed (Box 8-32).364
Box 8-32 Hyperoxia Test for Newborns with Suspected Cyanotic Heart Disease (to distinguish intrapulmonary from intracardiac shunt)364
1. Administer room air to the newborn (10-15 min, if tolerated)
2. Obtain baseline arterial PO2 from right radial artery (via arterial puncture or transcutaneous oxygen monitor—pulse oximetry oxyhemoglobin saturation cannot be used)
3. Administer 100% oxygen (via mask, oxygen hood, or endotracheal tube, if intubated) for 10–15 min
4. Obtain arterial PO2 from right radial artery (via arterial puncture or transcutaneous oxygen monitor—pulse oximetry oxyhemoglobin saturation cannot be used)
If cyanotic heart disease is likely or possible and pulmonary or systemic blood flow is dependent on the ductus arteriosus, prostaglandin E1 is administered to reopen and keep the ductus patent. The administration of PGE1 is summarized in Box 8-33. Further evaluation and management is described for each congenital defect.
Tetralogy of Fallot and Pulmonary Atresia with Ventricular Septal Defect
Pearls
• The degree of cyanosis is determined by the severity of right ventricular outflow tract obstruction.
• Neonates with tetralogy of Fallot (TOF) with severe pulmonary stenosis have ductal-dependent pulmonary blood flow, requiring PGE1 and surgical intervention.
• Hypoxemic spells can occur during activities that increase oxygen demand such as crying and feeding. Note that blood flow to the lungs is limited by the right ventricular outflow tract obstruction. Conditions that may contribute to hypoxemic spells:
Etiology
Tetralogy of Fallot refers to the association of four cardiac abnormalities described in detail in 1888 by French physician Etienne Fallot. The four cardiac anomalies include a ventricular septal defect, right ventricular outflow tract obstruction, overriding aorta (it arises above the ventricular septal defect) and right ventricular hypertrophy (Fig. 8-43). If an atrial septal defect is also present, the defect is known as pentalogy of Fallot. In the classic form of pentalogy of Fallot, the left ventricle is small, because much of pulmonary venous return is diverted to the right side of the heart through the ASD.
Fig. 8-43 Tetralogy of Fallot. This defect is defined as the association of four anomalies: a ventricular septal defect, pulmonary infundibular stenosis (often the pulmonary valve and main pulmonary artery are small), dextroposition of the aorta (the aorta shifts to the right and overrides the VSD), and right ventricular hypertrophy. Pentalogy of Fallot includes these four defects plus an atrial septal defect; this association of anomalies often occurs with a small left ventricle. The arrows show direction of the blood flow from the right ventricle into the pulmonary artery and aorta. The more severe the obstruction, the smaller the pulmonary blood flow and the larger the shunt from the right ventricle into the aorta.
Tetralogy of Fallot is thought to result from inadequate development of the subpulmonary conus during fetal life. This not only produces pulmonary infundibular stenosis, but also causes malalignment of the conal septum during fetal cardiac development, resulting in a large unrestrictive ventricular septal defect that is approximately equal to the size of the aorta. The bundle of His is generally located at the posteroinferior edge of the defect, placing the infant at risk for arrhythmias during the postoperative phase.
In addition to infundibular stenosis, valvular and supravalvular pulmonary stenosis may be present in varying degrees. The pulmonary valve is almost always involved in the obstruction. A bicuspid pulmonary valve is present in nearly two-thirds of patients with TOF and the pulmonary annulus is typically smaller than normal for age.886
The aorta is displaced to the right (toward the right ventricle) because the subpulmonary conus and pulmonary outflow tract have not developed normally. As a result, the aorta sits directly over the ventricular septal defect; this also may be referred to as an overriding aorta. Right ventricular hypertrophy is merely a compensatory response to the obstruction to right ventricular outflow.
Tetralogy of Fallot is the most common cyanotic congenital heart lesion and is responsible for approximately 9% of all congenital heart defects.886 It may be associated with additional intracardiac anomalies that are likely to alter the clinical presentation or management. Common combinations include pulmonary atresia with ventricular septal defect, tetralogy of Fallot with absent pulmonary valve, and tetralogy of Fallot with atrioventricular canal defect. Patients with tetralogy of Fallot often have a right aortic arch, multiple ventricular septal defects, or persistent left superior vena cava.
When the pulmonary stenosis is extreme, there is no anatomic connection between the right ventricle and the pulmonary artery. This severe form of tetralogy of Fallot may be referred to as pulmonary atresia with ventricular septal defect, or pseudotruncus arteriosus. It is discussed briefly here and again in the Truncus Arteriosus section.
Rarely, tetralogy of Fallot is associated with a rudimentary or absent pulmonary valve. This occurs in 5% of children with tetralogy of Fallot, and produces valvular insufficiency during the neonatal period.886 As a result, aneurysmal dilation of the main pulmonary artery as well as the right and left branch pulmonary arteries develops. The dilated pulmonary artery and branch arteries compress the trachea and right and left main bronchii and may result in significant respiratory compromise.
Tetralogy of Fallot may be associated with an atrioventricular septal defect (see Atrioventricular Septal Defect earlier in this chapter). This combination occurs in less than 2% of infants with tetralogy of Fallot and is associated with trisomy 21. As part of the atrioventricular canal portion of the defect, an atrial septal defect is present in addition to an anterior valve leaflet that is common to both the tricuspid and mitral valves. Moderate or severe atrioventricular valve insufficiency is present.
Pathophysiology
Tetralogy of Fallot
The hemodynamic changes that develop with uncomplicated tetralogy of Fallot are determined by the severity of obstruction to pulmonary blood flow. When pulmonary infundibular and valvular stenosis is mild, the right ventricular pressure is only mildly increased; there is minimal shunting of blood through the ventricular septal defect because pulmonary vascular resistance is approximately equal to systemic vascular resistance. The result is a balanced shunt and balanced pulmonary and systemic circulations. The pulmonary stenosis is protective; it prevents development of a significant pulmonary shunt once pulmonary vascular resistance falls. This form of tetralogy is often referred to as “pink” (acyanotic) tetralogy of Fallot. It does not require treatment in the newborn period and patients are typically discharged home from the newborn nursery to feed and grow, on no medications. Hypoxemia and cyanosis will typically develop with progression of right ventricular outflow tract obstruction. As right ventricular obstruction increases, more blood shunts from the right ventricle across the ventricular septal defect into the aorta, causing systemic arterial oxygen desaturation. Initially, oxygen desaturation is only noted during exertion (e.g., during vigorous cry); however, when significant pulmonary stenosis is present, cyanosis is present even at rest. The increased right ventricular obstruction results in right ventricular hypertrophy.
Suprasystemic right ventricular hypertension does not develop in patients with tetralogy of Fallot because the ventricular septal defect serves to “vent” the right ventricle. The greater the resistance to pulmonary blood flow, the greater is the volume of the right-to-left shunt through the ventricular septal defect into the aorta.886
Approximately 8% of children with tetralogy of Fallot have abnormalities in coronary artery anatomy.886 The most common abnormalities include a single coronary artery arising from the aorta (with later branching into the right and left coronary arteries), or a left anterior descending coronary artery arising from the right coronary artery. One result of these anomalies is that the left anterior descending coronary artery may cross over the right ventricular outflow tract. It is extremely important that this anomalous coronary artery distribution be identified preoperatively so that surgical repair can be planned to avoid coronary artery injury.886 This may necessitate cardiac catheterization to delineate coronary anatomy if the coronary arteries are not clearly visualized with echocardiography. In addition, it is important to identify the presence of multiple ventricular septal defects, aortopulmonary collaterals, and the extent of right ventricular outflow tract obstruction before surgical repair.
Pulmonary Atresia with VSD
When pulmonary blood flow is significantly compromised such as in the neonate with tetralogy of Fallot with severe pulmonary stenosis or pulmonary atresia with VSD, pulmonary blood flow is dependent on the ductus arteriosus. As a result, severe hypoxemia develops when the ductus begins to close. Rarely, older children with unrepaired tetralogy of Fallot are seen for evaluation. These children generally develop collateral vessels from the descending aorta to the bronchial arteries to supply pulmonary arteries with additional blood flow.
Tetralogy of Fallot with Absent Pulmonary Valve
If an infant has tetralogy with absent pulmonary valve, pulmonary insufficiency is present from birth and produces right ventricular dysfunction. There is aneurysmal dilation of the main pulmonary artery that may involve the branch pulmonary arteries. The dilated pulmonary artery compresses the tracheobronchial tree, producing airway obstruction and air trapping with possible emphysema. These infants also have right-to-left shunting of blood through the ventricular septal defect, with resultant arterial oxygen desaturation.
Tetralogy of Fallot with Atrioventricular Septal Defect
The combination of tetralogy of Fallot with atrioventricular septal defect produces a left-to-right shunt at the atrial level. Mitral insufficiency is present and if significant, will increase left atrial pressure and increase the magnitude of left-to-right shunting through the atrial septal defect. If a ventricular component is present, right-to-left shunting will occur through the AVSD, with resulting hypoxemia.
Clinical Signs and Symptoms
Tetralogy of Fallot
The hallmark symptom of tetralogy of Fallot is cyanosis that is directly proportional to the degree of pulmonary stenosis. The infant with mild pulmonary stenosis may not demonstrate any symptoms because there is minimal shunt through the ventricular septal defect. If the pulmonary stenosis is severe or pulmonary atresia is present, the right-to left shunt through the ventricular septal defect will be significant and the infant will demonstrate severe cyanosis even at rest. (See Hypoxemia in the second section of this chapter for a discussion of the potential systemic consequences of polycythemia.)
Neonates with tetralogy of Fallot and mild pulmonary stenosis generally are monitored in the hospital until the patent ductus arteriosus closes to ensure there is a sufficient amount of antegrade blood flow across pulmonary valve. After the ductus constricts, the neonate may demonstrate cyanosis with exertion, but should generally maintain an oxyhemoglobin saturation greater than 75%. As the infant grows and becomes more active, the right ventricular outflow tract obstruction usually becomes more severe because it does not grow proportionate to the child's growth. The fixed obstruction caused by the pulmonary stenosis prevents an increase in pulmonary blood flow and oxygen delivery during periods of increased oxygen requirement. The infant usually begins to demonstrate progressive cyanosis and decreased exercise tolerance at 4 months of age.
If a newborn presents with cyanosis of unknown origin at birth, a hyperoxia test is performed. One hundred percent oxygen is administered to the infant in an attempt to determine whether the etiology of the cyanosis is cardiac or pulmonary related (see Box 8-32). If the arterial oxygen tension (PaO2) remains less than 50 mm Hg after administration of 100% oxygen, the cyanosis is likely caused by cyanotic congenital heart disease.
The neonate with tetralogy of Fallot and severe pulmonary stenosis or small branch pulmonary arteries generally presents with cyanosis at birth. In these infants, most pulmonary blood flow is provided through the ductus arteriosus. If the infant has been diagnosed in utero, a prostaglandin E1 infusion is initiated at birth to prevent constriction of the ductus arteriosus (see Box 8-33). If prostaglandin E1 is not administered, the neonate will likely become profoundly hypoxemic and acidotic as the duct closes. These neonates will also require surgical correction or a palliative intervention to improve pulmonary blood flow.
Unrepaired infants with tetralogy of Fallot may begin to develop hypoxemic spells as early as the first months of life depending on the severity and progression of pulmonary stenosis. Hypoxemic spells are thought to be caused by a transient increase in right ventricular outflow tract obstruction and a fall in systemic vascular resistance; these changes decrease pulmonary blood flow and promote right-to-left shunting across the ventricular septal defect, resulting in progressive hypoxemia. The spells typically occur in the morning, particularly during activities that increase oxygen demand such as during crying, defecation, or feeding. Infants who are dehydrated, anemic, acidotic, or have increased circulating catecholamines are at particular risk for developing hypoxemic spells.
With the onset of hypoxemic spells (also called “tet” spells or hypercyanotic spells), the infant becomes acutely cyanotic, hyperpneic, irritable, and diaphoretic and arterial oxyhemoglobin saturation falls. Late in the spell the infant may become limp and lose consciousness. If an arterial blood gas is obtained during the spell, hypercapnia, hypoxemia, and acidosis will be noted. Hypercyanotic spells can result in stroke, seizures, or death, so the development of hypoxemic spells is typically considered an indication for surgery. Administration of a beta-blocker such as propranolol may be indicated if a delay in surgery is warranted. Beta-blockade acts to relax the right ventricular infundibulum, decrease right ventricular response to agitation, and decrease the incidence and severity of hypoxemic spells.
Children with tetralogy of Fallot who are not repaired in infancy develop polycythemia caused by chronic hypoxemia. When the hematocrit approaches 60%, the infant may demonstrate a more rapid respiratory rate and increased work of breathing because polycythemia increases blood viscosity, which decreases the velocity of pulmonary blood flow. Infants with unrepaired tetralogy and chronic hypoxemia demonstrate clubbing of the tips of the fingers and toes.
If the iron intake of the infant with chronic hypoxemia is inadequate, a microcytic anemia will develop that will not only decrease arterial oxygen content but will increase risk of cerebrovascular accident (stroke). The infant may have a normal hemoglobin concentration for age but may demonstrate a relative anemia because polycythemia is present. The mean corpuscular hemoglobin concentration (MCHC) and mean corpuscular volume (MCV) should be followed to identify and treat microcytic anemia.
On physical examination, a systolic ejection murmur can be heard best at the second intercostal space along the left sternal border. This murmur is caused by flow through the narrowed pulmonary outflow tract. The murmur disappears during hypoxemic episodes because blood flow across the narrowed pulmonary outflow tract decreases substantially. Bruits may be heard over the child's back if the child has developed extensive collateral circulation to the lungs.
If moderate or severe pulmonary stenosis is present, the presence of a sternal lift indicates right ventricular hypertrophy. Right ventricular hypertrophy in addition to right axis deviation will be evident on the electrocardiogram. Echocardiography can fully delineate the intracardiac and extracardiac anatomy (including size and position of the ventricular septal defect, appearance of the right ventricular outflow tract, aortic position, and size of main and branch pulmonary arteries) and can enable estimation of right ventricular pressure and the gradient across the pulmonary valve.
Cardiac catheterization is rarely performed because of the advanced imaging capabilities of echocardiography. Indications for cardiac catheterization include the presence of multiple ventricular septal defects or the need to further delineate the pulmonary vascular and coronary artery anatomy. These children can develop a hypoxemic episode during the procedure. As a result, noninvasive diagnostic modalities such as computed tomography-angiography or magnetic resonance-angiography provide safer diagnostic options.
On chest radiograph, a narrow mediastinum is observed because the main pulmonary artery segment is small. The classic radiographic cardiac contour in the infant with tetralogy of Fallot resembles the shape of a boot. The apex of the heart is elevated because right ventricular hypertrophy is present; as a result, the apex resembles the upturned toe of a boot. Pulmonary vascular markings are decreased when pulmonary stenosis is severe, unless collateral vessels to the lungs have developed. Approximately one-fourth of patients with tetralogy of Fallot have a right aortic arch (see Table 8-25).
Pulmonary Atresia with VSD and Collateral Pulmonary Blood Flow
Prenatal diagnosis assists in the management of this group of patients. Postnatal diagnosis may be delayed if aorta to pulmonary artery collateral flow provides sufficient pulmonary blood flow. In many instances, the collaterals become stenotic, at which time the child will develop cyanosis. If collateral flow is significant, signs and symptoms of congestive heart failure and failure to thrive develop by about 3 to 6 months of age. Other significant findings include a continuous murmur due to collateral flow, and a single S2.
The chest radiograph reveals mild to moderate cardiomegaly with no main pulmonary artery segment and increased pulmonary vascular markings. Cardiac catheterization is routine to assist with delineation of the branch pulmonary artery anatomy, which may be small and nonconfluent.
Tetralogy of Fallot with Absent Pulmonary Valve
Infants with tetralogy of Fallot and absent pulmonary valve often have mild cyanosis, congestive heart failure, and significant respiratory distress. These infants have a muffled, single second heart sound because only the aortic valve closure is heard. A harsh, systolic ejection murmur (caused by pulmonary infundibular stenosis) and a prominent, low-frequency diastolic murmur (resulting from pulmonary insufficiency) may be present and accompanied by a thrill.
Right ventricular hypertrophy is evident on clinical examination and on the ECG. The chest radiograph typically reveals an enlarged heart, increased pulmonary vascular markings and a large pulmonary artery silhouette. Echocardiography reveals right ventricular dilation as well as dilation of main, right and left pulmonary arteries. The pulmonary valve is absent. Cardiac catheterization is rarely needed for diagnosis.
Tetralogy of Fallot with Atrioventricular Canal Defect
This combination of lesions often tempers the symptoms that normally would be produced by either lesion alone. The pulmonary stenosis associated with tetralogy of Fallot prevents excessive pulmonary blood flow that normally would result from the atrioventricular septal defect, so that signs of severe congestive heart failure are not observed. If pulmonary stenosis is mild or moderate, pulmonary blood flow is decreased, and cyanosis may be readily apparent. Atrioventricular valve regurgitation is usually present.
The child with tetralogy of Fallot and atrioventricular septal defect demonstrates a pulmonary systolic murmur as well as a systolic murmur heard over the left lower sternal border (VSD murmur). An apical systolic murmur produced by mitral insufficiency is also present.
The ECG confirms the presence of right ventricular hypertrophy, but a superior axis deviation consistent with atrioventricular septal defect is also noted. Findings on chest radiograph are dependent on the severity of the combined lesions. Echocardiography reveals features consistent with tetralogy of Fallot as well as the presence of a common atrioventricular valve leaflet. Cardiac catheterization is rarely performed in these children because echocardiography provides adequate preoperative information.
Management
Tetralogy of Fallot
With the expanded use of fetal ultrasound, a diagnosis of congenital heart disease is often established before birth. This allows the medical team to prepare for the immediate initiation of PGE1 and transfer to a cardiac center for medical and surgical management at birth. Many of these neonates have umbilical lines placed and they may be intubated as a precautionary measure for transport to a tertiary care center.
Medical Management of Tetralogy of Fallot
If the neonate is diagnosed postnatally, the infant will present with cyanosis shortly after birth. A hyperoxia test is typically administered to assist in differentiating the cause of the hypoxia. Failure to respond to 100% oxygen (i.e., PaO2 less than 50 mm Hg) is most likely caused by a cyanotic cardiac lesion, and initiation of prostaglandin E1, and an echocardiogram is warranted.
Initial medical management in the newborn period is directed at maintaining adequate oxygenation and preventing hypoxemic spells until surgical correction is performed. If mild pulmonary stenosis is present, the infant will generally be monitored in the hospital while the patent ductus closes and then follow-up care is provided to prevent complications until surgical care is performed. Clinicians should consider chromosomal analysis and fluorescent in situ hybridization (FISH) testing as part of routine newborn care before discharge. Ten percent of infants with conotruncal defects have an associated chromosomal abnormality. The most common genetic disorder is 22q11 microdeletion, which places the infant at risk for vascular anomalies in addition to the cardiac lesion (see Section, Essential Anatomy and Physiology, Etiologies of CHD: Noninherited and Genetic Factors).
The infant should be kept well hydrated to prevent hemoconcentration, and microcytic anemia should be avoided because it decreases oxygen content and increases the child's risk of cerebral thromboembolic events. Parents should be taught when to notify a physician or nurse practitioner if the infant develops diarrhea, nausea, vomiting, or fever, so that dehydration can be prevented or promptly treated and antibiotic prophylaxis can be prescribed if needed. The parents should be taught to monitor for signs of hypoxemic episodes. Instruction should include potential triggers and should include alleviating maneuvers such as calming the child and placement in the knee-chest position. If a hypoxemic spell does develop, surgery should be scheduled.
Whenever the infant or child with uncorrected tetralogy of Fallot is admitted to the hospital, it is essential that no air be allowed to enter any IV line because systemic venous blood may shunt directly into the aorta and any IV air may cause a cerebral air embolus. All staff members should be aware that infants with tetralogy of Fallot may develop hypoxemic spells.
Hypoxemic spells are treated by calming the infant, placing the child in the knee-chest position and administering oxygen, a potent pulmonary vasodilator. The on-call physician or nurse practitioner should be notified immediately if any spells develop, and the bedside nurse should anticipate administering a weight-appropriate dose of intravenous morphine sulfate (0.1 mg/kg). If these maneuvers do not alleviate the spell, propranolol, phenylephrine or ketamine may be given. Sodium bicarbonate may be administered to correct acidosis. In addition, volume in the form of packed red blood cells may be ordered to maintain the hematocrit greater than or equal to 45% and alpha-agonists may be administered to increase systemic vascular resistance (and reduce the right-to-left shunt, thereby increasing pulmonary blood flow). A beta-blocking agent may be given to relax the RV infundibulum and decrease RV outflow tract obstruction (Box 8-34).
Box 8-34 Management of Hypercyanotic Spells
• Place child in knee chest position
• Administer morphine (0.1 mg/kg IV or IM) to improve pulmonary blood flow
• Administer intravenous fluids (bolus isotonic crystalloids or packed red blood cells if needed to maintain adequate hemoglobin and hematocrit)
• Administer propranolol (beta-adrenergic blocking agent)
• Administer IV phenylephrine (alpha-agonist)
produces arterial and venous constriction. This should increase venous return to the right ventricle, and reduce right-to-left shunt and hypoxemia.• Ketamine (anesthetic) may be considered because it increases systemic vascular resistance (and therefore reduces the right-to-left shunt and hypoxemia and should improve pulmonary blood flow). A sedative IV dose is 0.25-0.5 mg/kg.
Complete corrective surgery is typically performed for the symptomatic full-term neonate with tetralogy of Fallot. The optimal age for elective correction of tetralogy of Fallot remains controversial. Surgery is generally recommended when the child's oxyhemoglobin saturation decreases to less than 70% to 80%.
Palliation of Tetralogy of Fallot
Many centers use a staged approach for premature or small-for-gestational-age infants. A staged surgical approach involves the placement of a Blalock-Taussig or modified Blalock-Taussig systemic to pulmonary artery shunt. The modified Blalock-Taussig shunt procedure involves placement of a 3.5- to 4.0-mm Gore-Tex tube between the innominate artery and ipsilateral pulmonary artery (Fig. 8-44). Additional palliative shunts for cyanotic heart disease are illustrated in the Evolve Fig. 8-5 and described in Evolve Table 8-2 in the Chapter 8 Supplement on the Evolve Website. Creation of a systemic to pulmonary artery shunt can be performed through a thoracotomy; however, a median sternotomy approach may be considered to enable initiation of cardiopulmonary bypass to support oxygenation in infants considered to be at high risk for hypoxemic spells during anesthesia induction. Placement of the Blalock-Taussig shunt can result in scarring and branch pulmonary artery distortion at the distal anastomosis; such scarring and distortion must be addressed at the time of complete repair.
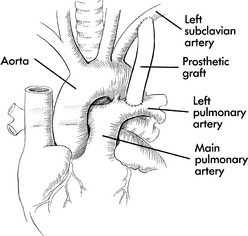
Fig. 8-44 Modified (Prosthetic) Blalock-Taussig shunt. A prosthetic shunt, usually made of polytetrafluoroethylene (Gore-Tex or Impra) is sewn between the patient's subclavian artery and the pulmonary artery to provide a systemic-to-pulmonary artery shunt.
There are technical challenges to the placement of an optimal shunt: too large a shunt can result in excessive pulmonary blood flow with symptoms of congestive heart failure, a wide pulse pressure and bounding pulses resulting from aortic runoff. Too small a shunt results in continued hypoxemia. Rarely, reoperation is required to revise the size of the shunt.
The expected range of oxyhemoglobin saturation after the placement of a systemic to pulmonary artery shunt is 75% to 85%. During the immediate postoperative period, the nurse should monitor for signs of hypovolemia caused by osmotic diuresis, especially if the infant was placed on cardiopulmonary bypass for the procedure. This places the infant at risk for hemoconcentration, clot formation, and shunt occlusion. Arterial punctures and cuff blood pressure measurement should be avoided on the arm and artery used for the shunt.
Immediate postoperative complications also include bleeding and shunt occlusion. When a polytetrafluoroethylene shunt is used for the creation of the systemic-to-pulmonary artery shunt, the infant's platelet count will likely fall in the first day after surgery because platelets adhere to the shunt material until it endothelializes. The infant's platelet count should be monitored and it should gradually return to normal. To prevent clot formation, a heparin infusion may be ordered until the infant is tolerating enteral feedings and aspirin therapy (10 mg/kg every other day, or per protocol) can be started (see Antiplatelet Therapy in section, Postoperative Care and Anticoagulation for the Child with Heart Disease).
Signs of inadequate pulmonary blood flow or possible shunt occlusion include a change in the intensity of the continuous shunt murmur, a drop in baseline oxyhemoglobin saturation, increased cyanosis or acidosis. If shunt occlusion is suspected based on physical exam and echocardiographic findings, the nurse should prepare the infant for cardiac catheterization and/or surgery. In some cases, the nurse may administer a bolus of heparin and begin a heparin drip (if one is not present) in the hopes of maintaining shunt patency.
Occasionally, injury to the phrenic nerve, thoracic duct, or recurrent laryngeal nerve may occur when the surgical approach is via left thoracotomy. Phrenic nerve injury causes diaphragm paralysis, which may not be apparent while the child receives positive pressure ventilation, but should be suspected if the infant has difficulty weaning from ventilator support. A chest radiograph obtained while the infant is removed briefly from positive pressure ventilator support will reveal elevation of the hemidiaphragm on the involved side. If the diagnosis still remains unclear, observation of spontaneous breathing under fluoroscopy can confirm the diagnosis. Diaphragm paralysis is generally temporary and function returns within several weeks. If the condition prevents effective spontaneous ventilation, the team may consider diaphragm plication.
Injury to the thoracic duct can result in chylothorax that is not apparent until the infant begins taking oral or enteral nutrition containing long chain triglycerides. If the thoracic duct has been injured, the chest tube drainage will turn to a milky white. If a chest tube in not in place, the infant may develop a pleural effusion on the left side. (See Postoperative section, Chylothorax, for management strategies.)
Injury to the recurrent laryngeal nerve can lead to vocal cord paralysis. This condition is usually temporary and unilateral. Diagnosis of this condition typically begins when a weak or absent cry is noted after extubation. If persistent, the infant may be at risk for aspiration; therefore, a speech evaluation should take place to assess whether the infant can safely take oral feedings. If vocal cord paralysis is suspected, an otolaryngology specialist should be consulted to confirm the diagnosis.
Nonsurgical palliation has been reported for a select subgroup of infants with tetralogy of Fallot. These procedures include a balloon valvuloplasty to open the stenotic pulmonary valve and improve antegrade flow through the native pulmonary artery. In addition, a stent may be inserted to open the narrow right ventricular outflow tract. Both procedures are performed in the cardiac catheterization laboratory in high-risk infants and can only be done if the branch pulmonary arteries are contiguous. Both procedures attempt to provide a secure source of pulmonary blood flow until corrective surgery is performed; they may prove to be a viable alternative in the staging process.
Surgical Correction of Tetralogy of Fallot
Corrective surgery is electively performed at approximately 3 to 6 months of age; however, some centers delay elective repair until 1 year of age. The goals for surgical repair include closure of the ventricular septal defect and reconstruction of the right ventricular outflow tract.
Whenever possible a ventriculotomy is avoided and the repair is usually accomplished via a transatrial and transpulmonary approach. An incision is made in the right atrium and the surgeon gains access to the right ventricle through the tricuspid valve and closes the VSD from this approach (Fig. 8-45). Through this approach, the VSD is closed using a Dacron patch, directing LV flow to the aorta. Resection of the pulmonary stenosis is accomplished through the pulmonary artery. The surgeon makes an incision in the pulmonary artery, opens the pulmonary valve, and resects the subvalvular stenosis. The hypertrophic right ventricular infundibular muscle is resected and a pulmonary valvotomy is performed if needed. A right ventriculotomy is occasionally needed. Any existing systemic to pulmonary artery shunt is taken down at the time of repair.
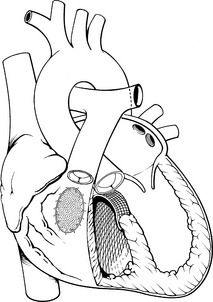
Fig. 8-45 Surgical correction of tetralogy of Fallot. Pulmonary infundibular stenosis is resected, and the ventricular septal defect (VSD) is closed with a patch. The pulmonary outflow tract is enlarged with one or two patches if necessary (one is depicted here). When possible, to avoid a right ventriculotomy incision, an atriotomy incision is performed, and the surgeon enters the right ventricle through the open tricuspid valve. The VSD is identified and closed from this approach. An incision is also made in the main pulmonary artery above the pulmonary valve, the pulmonary valve is opened and subpulmonary stenosis is resected.
If the pulmonary outflow tract and main pulmonary artery are small, a patch may be placed across the pulmonary outflow tract and if necessary, in the main pulmonary artery with extension onto the branch pulmonary arteries. Use of a transannular patch to enlarge the pulmonary valve annulus can reduce resistance to right ventricular ejection but can create valvular insufficiency. If pulmonary insufficiency is severe, the volume load produced by the insufficiency may cause severe postoperative right ventricular dysfunction.
Reoperation late in adolescence may be required to correct the valvular insufficiency. Reoperation may also be required if residual pulmonary branch stenosis is present because this increases resistance to pulmonary flow and magnifies pulmonary insufficiency.
Immediate postoperative complications include bleeding, low cardiac output, right ventricular dysfunction, and arrhythmias, especially junctional ectopic tachycardia. Additional complications include heart block, residual pulmonary stenosis, right ventricular outflow tract obstruction, and/or ventricular septal defect, pulmonary insufficiency, or damage to the aortic valve. Right heart failure may result from right ventricular dysfunction and is more likely to occur in neonates with pulmonary hypertension or those with residual pulmonary stenosis or significant pulmonary insufficiency. Left ventricular dysfunction can result from the sudden increase in pulmonary blood flow and subsequent pulmonary venous return, especially if the left ventricle is borderline in size.
If low cardiac output develops during the postoperative period, the healthcare team should assess for the presence of right ventricular dysfunction or residual lesions such as a residual ventricular septal defect. Treatment of low cardiac output during the postoperative period consists of judicious fluid administration, diuresis, inotropic support and afterload reduction (see Postoperative Care, Low Cardiac Output).
Arrhythmias are common after surgical correction of tetralogy of Fallot. Right bundle branch block is inevitable if a right ventriculotomy was performed during the procedure. Junctional ectopic tachycardia is also common in newborns after complete repair of tetralogy of Fallot. Management includes mild hypothermia, digoxin, and avoiding inotropic drugs. If the arrhythmia is compromising hemodynamics, sedation and paralysis as well as the use of a beta-blocker or amiodarone may be used. If the junctional rate is slowed with beta-blockade and the patient has temporary pacing wires in place, overdrive pacing may allow for capture and conversion to sinus rhythm. Other arrhythmias seen during the postoperative period include heart block, supraventricular tachycardia, and premature ventricular contractions.
Abnormal neurodevelopmental outcomes have been found on follow-up and may be related to the exposure of the neonatal brain to cardiopulmonary bypass and prolonged hospitalizations. Studies comparing neurodevelopmental outcomes and the timing of the complete repair using cardiopulmonary bypass have found lower intelligence scores in the children who underwent complete repair as a neonate.257,538,618
Early mortality after complete repair has been reported at 5%. The incidence of sudden death late after tetralogy of Fallot repair is reported to be approximately 2% to 7%.886 This risk of sudden death is extremely low among those patients with an excellent operative result, but is significant among patients with persistent severe right ventricular hypertension and a history of ventricular tachycardia. Life-long follow-up is required by this population for the management of residual pulmonary stenosis, chronic pulmonary regurgitation, and ventricular arrhythmias. Late reoperation may be necessary to preserve right ventricular function and exercise tolerance.49 Endocarditis is a lifelong risk because there is typically some residual defect (e.g., mild pulmonary stenosis or insufficiency) adjacent to the VSD patch (and any other patches placed during the repair).
Pulmonary Atresia with VSD
The neonate will require administration of prostaglandin E1 to maintain ductal patency. Surgical intervention will be required.
Surgical Correction of Pulmonary Atresia with VSD
Eligible children generally present in congestive heart failure with adequate arterial oxygen saturations and adequate pulmonary artery segments with a left-to-right (aorta to pulmonary) shunt. Surgical repair often consists of several steps: first, the pulmonary arteries are unifocalized; second, a right ventricular to pulmonary artery conduit is placed to promote growth of the proximal and distal pulmonary arteries. The ventricular septal defect is completely closed in children who have forward flow through the right ventricular outflow tract and adequately sized pulmonary arteries.
A postoperative right ventricular-to-left ventricular pressure ratio of less than two- thirds to three-fourths to one on direct measurement in the operating room is associated with good long-term results. Postoperative complications include right ventricular dysfunction, residual ventricular septal defect, pulmonary artery stenosis, right ventricular hypertension, aortic regurgitation, and arrhythmias, including heart block. Subsequent cardiac catheterizations may be necessary to assess the pulmonary arteries and need for balloon angioplasty or stenting. Reoperation will be required for conduit replacement if significant conduit stenosis or regurgitation develop. Endocarditis is a lifelong risk (see Bacterial Endocarditis later in this section).
Tetralogy of Fallot with Absent Pulmonary Valve
If the neonate with tetralogy of Fallot with absent pulmonary valve is symptomatic, generally with respiratory compromise, then severe dilation of the pulmonary artery and compression of the tracheobronchial tree is probably present. This condition produces signs of airway obstruction and air trapping necessitating the use of mechanical ventilation with positive end-expiratory pressure. Surgical repair requires cardiopulmonary bypass. The aneurysmal main and branch pulmonary arteries are plicated to relieve compression on the distal trachea and bronchus; the ventricular septal defect is closed and a right ventricle to pulmonary conduit is inserted.
Early mortality is significant, especially among symptomatic neonates with airway compromise. Postoperative complications include persistent respiratory failure, congestive heart failure, shock, and arrhythmias. Aggressive pulmonary toilet must be provided to these neonates to prevent further complications. Some infants require a tracheostomy.
Tetralogy of Fallot with Atrioventricular Canal
Definitive repair for this complex heart defect is typically performed between 6 and 12 months of age when the infant becomes symptomatic and is refractory to medical therapy. Cardiopulmonary bypass is instituted to perform a two-patch repair of the atrioventricular canal. An atriotomy is used to place a Dacron patch to close the ventricular septal defect and a pericardial patch is used to close the atrial septal defect. The common leaflet is sandwiched between the two patches, and this prevents the need for the surgeon to incise the common leaflet. The cleft in the mitral valve is closed with sutures and an incision is made in the pulmonary artery to facilitate resection of the pulmonary infundibular stenosis. Patch enlargement of the pulmonary outflow tract or insertion of a valved conduit is occasionally necessary. Postoperative complications include atrioventricular valve regurgitation, low cardiac output, congestive heart failure, arrhythmias, heart block, and bleeding. Right ventricular outflow tract obstruction may result from suboptimal placement of the intraventricular patch.
Advanced Concepts
Because many patients with tetralogy of Fallot have associated defects that affect the clinical presentation and medical and surgical management, it can be challenging to anticipate the clinical course and management (see Box 8-35). Skilled nursing assessment and care are required.
Box 8-35 Advanced Concepts: Tetralogy of Fallot (TOF)
• TOF with mild PS produces a balanced shunt: the pulmonary stenosis prevents a large left-to-right shunt through the VSD, but it is not so severe as to produce a right-to-left shunt.
• TOF with severe PS presents with cyanosis and pulmonary blood flow is decreased substantially.
• Hypoxemic spells can be triggered by events that increase myocardial oxygen consumption. However, because of the obstruction to pulmonary blood flow is fixed, pulmonary blood flow cannot increase. This causes tissue hypoxia, acidosis, and further decrease in pulmonary blood flow. If the cycle is not broken, the infant will become severely cyanotic and acidotic. Shock can develop.
Ebstein Malformation
Etiology
Ebstein malformation is a congenital anomaly of the tricuspid valve that was first described by Wilhelm Ebstein in 1866. In this rare defect the tricuspid valve leaflets do not attach normally to the tricuspid valve annulus. The valve leaflets are dysplastic, and two of the leaflets (the medial or septal leaflet and the posterior leaflet) are displaced inferiorly, adhering to the right ventricular wall (Fig. 8-46).61
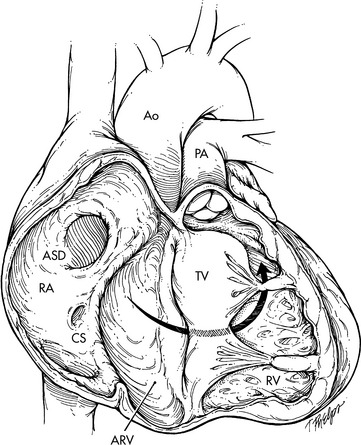
Fig. 8-46 Native anatomy of Ebstein anomaly. The tricuspid valve (TV) is displaced into the right ventricle (RV), leaving an atrialized portion of the RV (ARV) above the level of the displaced TV leaflets but below the level of what should be the tricuspid annulus. The anterior leaflet of the TV is large and sail-like and may obstruct the RV outflow tract. The posterior and septal TV leaflets are small, and a large ostium secundum defect (ASD) is seen. Ao, aorta; CS, coronary sinus; PA, pulmonary artery; RA, right atrium.
(From Davidson DL, Bando K, Haelnaer M, Cameron DE: Ebstein anomaly. In Nichols DG, editor: Critical heart disease in infants and children, ed 2, Philadelphia, 2005, Elsevier, p. 908, Fig. 37-1.)
The cause of Ebstein malformation is unknown. The defect results when the tricuspid valve leaflets fail to develop normally from the interior aspect of the embryonic right ventricular myocardium during the fifth week of fetal cardiac development.
The anatomy of this defect varies widely, and additional associated intracardiac abnormalities are common. Atrial or ventricular septal defects and L-transposition (“corrected transposition” or ventricular inversion) are often present. When L-transposition is present, the anomalous tricuspid valve is on the left side, receiving pulmonary venous return.
Pathophysiology
Hemodynamic alterations resulting from Ebstein anomaly are related to the effects of this anomaly on right atrial size and pressure, tricuspid valve function, and right ventricular size and function. The inferior displacement of the tricuspid valve leaflets effectively incorporates a variable portion of the right ventricle into the right atrium, so a portion of the ventricle is “atrialized.” As a result, the right atrium is dilated and right ventricular size is compromised. The atrialized portion of the right ventricle may appear to be relatively normal, or may be extremely thin with little ability to contract.61
The function of the tricuspid valve varies widely in Ebstein. The function is affected by the chordal attachments of the valve leaflets. Frequently the leaflets are attached abnormally (tethered) to the right ventricular wall; this restricts leaflet motion and results in valvular insufficiency and stenosis. If the valve is minimally displaced and minimally tethered and is relatively competent, hemodynamic effects may be minimal.
Significant displacement and tethering of the tricuspid valve leaflets results in tricuspid insufficiency and stenosis. Right atrial pressure rises and right-to-left shunting of blood occurs through a foramen ovale or (less commonly) through a true atrial septal defect, resulting in hypoxemia.
During atrial systole, blood is propelled from the right atrium into the atrialized portion of the right ventricle, as well as into the true remaining right ventricle. During ventricular systole any contraction of the atrialized right ventricle results in regurgitation of blood into the true right atrium.
Right-to-left shunting of blood at the atrial level is typically greatest during the neonatal period. Until pulmonary vascular resistance falls, right ventricular systolic and end-diastolic pressures will be high; this increases tricuspid insufficiency. In addition, the foramen ovale opens, allowing an atrial shunt even in the absence of a true atrial septal defect.
If the right-to-left atrial shunt is large, flow into the right ventricle will be compromised and pulmonary blood flow will be reduced. This will further increase systemic hypoxemia. If a significant portion of the right ventricle is atrialized, severe right ventricular dysplasia (decreased wall thickness) and dysfunction probably will be present.
Severe tricuspid insufficiency and right atrial hypertension are associated with signs of systemic venous congestion. Progressive right atrial dilation results in the development of atrial tachyarrhythmias, including atrial flutter or fibrillation. Supraventricular tachyarrhythmias also may result from the presence of accessory intraatrial conduction pathways. Progressive right heart dilation will compress the left ventricle, obstructing the left ventricular outflow tract.
Additional associated intracardiac defects will modify the pathophysiology. If L-transposition is present, insufficiency of the (left-sided) tricuspid valve will result in left atrial hypertension and pulmonary edema. Bidirectional shunting of blood at the atrial level is usually present.
Clinical Signs and Symptoms
Because the severity of the tricuspid valve dysfunction varies widely, the clinical spectrum of this defect also varies widely. If the valve is not significantly stenotic or insufficient and displacement is minimal, the patient may be asymptomatic during infancy and early childhood. Cyanosis may be present during the neonatal period, but disappears when pulmonary vascular resistance falls. Older infants may demonstrate cyanosis only during exercise. Late development of atrial arrhythmias is also common.61
Although Ebstein anomaly is a relatively rare congenital heart defect, it is one of the most common congenital heart defects diagnosed in utero. If the tricuspid valve is severely dysfunctional, significant right atrial dilation and systemic edema will be present in utero, producing hydrops, fetal pleural and pericardial effusions, and cardiomegaly, which are detected readily by fetal echocardiogram. Those defects diagnosed in utero are usually of the most severe form and carry the worst prognosis.
After birth, tricuspid valve dysfunction and right atrial hypertension result in a large right-to-left atrial shunt (through a foramen ovale), so that severe cyanosis is observed during the first days of life. In addition, signs of congestive heart failure also are observed. Cyanosis and congestive heart failure may be severe until pulmonary vascular resistance falls (when the neonate is several weeks old); at that point the infant's condition often improves.
Right ventricular dysplasia and dysfunction contribute further to the signs of tricuspid insufficiency, congestive heart failure, and cyanosis. Severe right heart dilation produces bulging of the ventricular septum toward the left ventricle, with resultant obstruction of the left ventricular outflow tract. Right atrial dilation also may be associated with stasis of systemic venous blood and paradoxic emboli to the left atrium; these may embolize to the systemic arterial circulation.
The hypoxemic child with Ebstein anomaly will develop compensatory polycythemia and is at risk for the development of systemic complications of this polycythemia. For further information, see Common Clinical Conditions, Hypoxemia in the second section of this chapter.
A systolic murmur of tricuspid insufficiency often is heard best at the left lower sternal border. Note that this location is unusual for tricuspid valve sounds but occurs because the valve leaflets are displaced inferiorly. The first heart sound may be normal or diminished in intensity, and tricuspid closure may produce a click. A diastolic murmur may be present, although its origins are unclear.899
Radiographic appearance of the heart and pulmonary vasculature vary widely among patients with Ebstein anomaly. The heart size and pulmonary vascular markings may be normal if the tricuspid valve is affected mildly; these findings most commonly are observed in older children. In the symptomatic infant the heart often is massively enlarged, with decreased pulmonary vascular markings (this distinguishes Ebstein anomaly from many other cyanotic heart lesions). Marked convexity of the right heart shadow (indicative of right atrial enlargement) is usually apparent. Massive right atrial enlargement produces a cardiac silhouette that resembles an inverted funnel; the mediastinum with small pulmonary artery silhouette creates a narrow top of the funnel, and the widened cardiac silhouette produced by right atrial enlargement creates the widened bottom of the funnel.
The electrocardiogram is always abnormal; right bundle branch block and right atrial enlargement are the most consistent features. Wolff-Parkinson-White syndrome (supraventricular tachycardia resulting from accelerated intraatrial conduction pathways) or other supraventricular atrial tachyarrhythmias are also common. The P-R interval usually is prolonged, and right axis deviation is common (see Table 8-26 and Common Clinical Conditions, Arrhythmias).
The echocardiogram enables thorough evaluation of the location and chordal attachments of the tricuspid valve leaflets, the size and wall thickness of the right ventricle, and the function of the heart in general. Those echocardiographic features associated with a severe form of Ebstein malformation and poor prognosis include: tethered distal attachments of the anterosuperior tricuspid leaflet, right ventricular dysplasia, left ventricular outflow tract obstruction (resulting from right heart dilation and septal deviation), and total combined area of the right atrium and atrialized right ventricle that is greater than the total combined area of the functional right ventricle, left atrium, and left ventricle. These risk factors are similar for patients of all ages with Ebstein malformation.
Cardiac catheterization is rarely necessary because the anatomy of Ebstein anomaly can be documented clearly by echocardiography. In fact the risk of fatal arrhythmias is so high that catheterization is avoided in many institutions. If catheterization and angiocardiography are performed, the child's heart rate and rhythm must be monitored closely and antiarrhythmic drugs must be prepared at the bedside.
The child with Ebstein anomaly is at risk for all of the systemic consequences of hypoxemia and polycythemia. Intracardiac conduction defects, including the presence of intraatrial conduction pathways, may also be present, and supraventricular tachycardia often develops. The PR interval is usually prolonged.
Management
Nonsurgical support of the patient with Ebstein anomaly requires treatment of congestive heart failure and management of arrhythmias. Diuresis cannot be too aggressive because hemoconcentration increases the risk of thromboembolic phenomena. Throughout therapy, until final surgical correction is performed, it is imperative that absolutely no air be allowed to enter any intravenous line because it may be shunted into the systemic arterial circulation, producing a cerebral air embolus (stroke). These children should not be allowed to become dehydrated; that may result in hemoconcentration and increased risk of thromboembolic events (see section, Common Clinical Conditions, Hypoxemia).
If pulmonary blood flow is severely compromised and hypoxemia is severe, the neonate should receive prostaglandin E1 to maintain ductal patency (see Box 8-33). Indications for surgical intervention in any patient with Ebstein malformation include severe cyanosis and increasing polycythemia (including neonates dependent on ductal pulmonary blood flow), congestive heart failure refractory to medical management, tachyarrhythmias secondary to an accessory intraatrial conduction pathway, paradoxic emboli, or progressive disability.
Any surgical intervention for Ebstein anomaly will be palliative in nature. Classical palliative procedures for cyanotic heart disease, including systemic to pulmonary shunts, are generally not associated with relief of symptoms and clinical improvement. “Corrective” surgical procedures for Ebstein anomaly are still being refined, but if two ventricles are to be maintained, either tricuspid valve replacement or reconstruction is performed.
Although valve replacement can be performed, complications are higher than for other valve replacements. In addition, the child will outgrow the valve and subsequent re-replacement will be needed.
Reconstruction of the tricuspid valve requires plication of the atrialized portion of the right ventricle and repair of the tricuspid valve, freeing it from abnormal right ventricular attachments. Plication of the atrialized right ventricle is performed through insertion of mattress sutures passed through pledgets of Teflon cloth from the normal valve ring through folds created in the atrialized portion of the right ventricle to the rim of the displaced valve leaflets; the sutures are pulled together to pull the valve leaflets toward their normal position (Fig. 8-47). Sutures through the atrialized wall must be carefully placed to avoid injuring coronary artery branches and the surgeon assesses the effects of suture placement. An annuloplasty is performed to reduce the size of the tricuspid valve orifice and ensure satisfactory tricuspid valve function. The associated atrial septal defect or patent foramen ovale is closed with a patch.163 Successful tricuspid valvuloplasty is dependent on an intact anterior valve leaflet. If the anterior leaflet is abnormal or the surgeon is unable to fashion a functioning valve from existing tissue, a prosthetic valve is inserted. If accessory intraatrial conduction pathways are producing supraventricular arrhythmias, these are interrupted at the time of surgery.
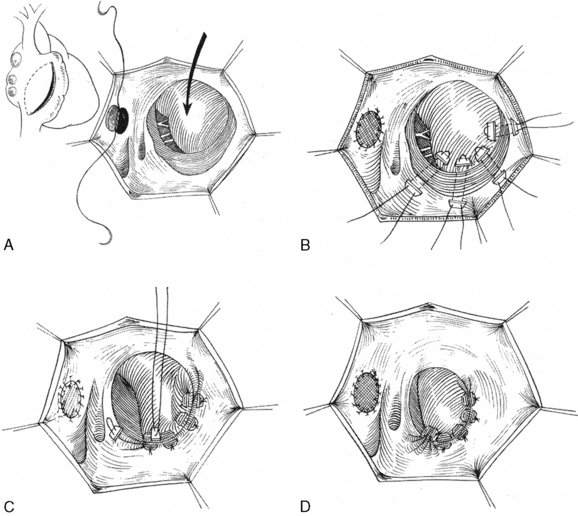
Fig. 8-47 Danielson repair of Ebstein anomaly, view from right atrial incision (atriotomy). A, Atrial septal defect is closed with a patch. Displaced tricuspid valve is indicated by arrow. B and C, Mattress sutures are passed through pledgets of woven material; they appear as rectangles above the valve annulus (in this figure they are depicted near the lower edge of the atrial incision at the bottom of each figure). The mattress sutures are used to pull folds of the atrialized ventricle and the displaced valve ring into the normal tricuspid valve position. D, A tricuspid annuloplasty is performed to reduce the size of the annulus.
(Reproduced with permission from Perloff M: Congenital heart disease in adults, ed 3, Philadelphia, 2008, Saunders, Fig. 15-17.)
The child may be managed as a patient with tricuspid atresia, in effect ignoring the right ventricle. Palliation and correction will be as for tricuspid atresia (see Tricuspid Atresia later in this section).
Children with Ebstein anomaly may be referred for cardiac transplantation rather than corrective surgery. Long-term results of transplantation for children with complex heart disease have not yet been determined, although it is hoped that these children will have better functional outcome immediately after transplantation than has been reported after surgical correction of the Ebstein anomaly.693
Perioperative mortality related to correction of Ebstein anomaly varies widely, ranging from 7% to 20%.36 Mortality rates appear to be highest in patients with severe congestive heart failure preoperatively. Postoperative complications after traditional correction of Ebstein anomaly include low cardiac output and sudden malignant arrhythmias (including complete heart block and ventricular fibrillation). Late deaths have been reported from sudden arrhythmias, progressive heart failure and low cardiac output.36,693 Transplantation may be recommended if clinical deterioration develops postoperatively.
Nonsurgical management of the child with Ebstein anomaly requires treatment of CHF and management of atrial arrhythmias. Aggressive diuresis must be avoided, because it may lead to hemoconcentration. Because a right-to-left shunt is often present, no air can be allowed to enter any IV system. Antibiotic prophylaxis will be required throughout the child's life during periods of increased risk of bacteremia.