Questions about the effects of interventions
examples of appraisals from different health professions
Tammy Hoffmann, John W Bennett, Mal Boyle, Jeff Coombes, Mark R Elkins, Edzard Ernst, Angela Morgan, Lisa Nissen, Claire Rickard, Sharon Sanders, Alan Spencer and Caroline Wright
This chapter is an accompaniment to the previous chapter (Chapter 4) in which you learnt how to critically appraise evidence about the effects of interventions. In order to further illustrate the key points from Chapter 4, this chapter contains a number of worked examples of questions about the effects of interventions. As we mentioned in the preface of the book, we believe that it can be easier to learn the process of critical appraisal when you see some worked examples of how it is done, and it is even better when the examples are from your own health profession. Therefore, this chapter (and likewise Chapters 7, 9 and 11) contains examples from a range of health professions. Some of the clinical examples are relevant to more than one health profession. Each example is formatted in a similar manner and contains the following elements:
• a clinical scenario that explains the origins of the clinical question
• the search terms and databases used to find evidence to answer the clinical question
• a brief description of the article chosen and the reason for its selection
• a structured abstract of the chosen article
• an appraisal of the risk of bias of the evidence
• a summary of the main results of the article that are relevant to the clinical question
• a brief discussion about how the evidence can be used to inform practice.
You will notice that in most of the examples the type of article chosen to be appraised is a randomised controlled trial. You may wonder why this is the case when Chapters 2 and 4 explained that systematic reviews of randomised controlled trials should be the first choice of study design to answer questions about the effects of an intervention. There is a good reason behind this. The authors of the examples contained in this chapter were asked to not choose (or indeed, specifically search for) systematic reviews if these were available to answer their question. Why? Because it is easier to learn how to appraise a systematic review of randomised controlled trials if you have first learnt how to appraise a randomised controlled trial. This chapter and Chapter 4 are designed to help you learn how to appraise a randomised controlled trial. Once you know how to do this, Chapter 12 will help you learn how to appraise a systematic review.
As you read through these examples, keep in mind that the suggestions that the authors of these worked examples have provided in the ‘how might we use this evidence to inform practice’ section have been drawn from only one individual study. In reality, additional studies would need to be located and appraised prior to drawing clear conclusions about what should be done in clinical practice.
When appraising an article, you need to obtain and carefully read the full text of the article. We have not included the full text of the articles that are appraised. However, for each of the examples in this chapter (and Chapters 7, 9 and 11), the authors of the examples have prepared a structured abstract that summarises the article. This has been done so that you have some basic information about each article. As we mentioned in Chapter 1, the more you practise doing the steps of evidence-based practice, the easier it will become. This is particularly true of the critical appraisal step. You may find it useful if you approach these worked examples as a self-assessment activity and try to obtain a copy of the article that is appraised in each of the examples (or just the ones that are relevant to your health profession if you feel more comfortable with that). You can then critically appraise the articles for yourself and check your answers with those that are presented in the worked examples.
One other thing to note about the examples in this chapter (and Chapters 7, 9 and 11) is that the appraisal of articles is not an exact science and sometimes there are no definite right or wrong answers. As with evidence-based practice in general, the health professional's clinical experience has an important role to play, particularly in deciding about issues such as baseline similarity (as we saw in Chapter 4) and clinical significance (as we saw in Chapters 2 and 4). Some of the examples may contain statements that you do not completely agree with and that you, as a health professional, would interpret a little differently. Also, the examples are provided to give you an overall sense of the general process of evidence-based practice. The content that is presented in the examples is not exhaustive (particularly in the ‘how do we use this evidence to inform practice’ section), and there may be other factors or issues that you, as a health professional, would suggest or consider if you were in that situation. That is OK.
Occupational therapy example
Clinical scenario
You are an occupational therapist who works in a recently opened community adult health centre. In the last few weeks, you have received referrals for a number of patients who have been diagnosed with rheumatoid arthritis in the last 1 or 2 years. Most of these patients are female and middle-aged, and their arthritis is causing them considerable pain (particularly in the hand) and affecting their ability to perform self-care activities. You are considering running an education group about joint protection techniques and wonder if this will be effective in reducing your patients' pain and improving their function.
In adults with rheumatoid arthritis, is group education about joint protection techniques effective in reducing hand pain and improving function?
Search terms and databases used to find the evidence
Database: OTseeker.
Search terms: ‘rheumatoid arthritis’ AND ‘joint protection’
This search retrieved 14 randomised controlled trials. Only four of these articles specifically evaluated the effectiveness of a joint protection program and the two most relevant articles were both based on the same study, with one describing the 6- and 12-month results and the other reporting the long-term (4-year) effects of the intervention. You choose to appraise the article that presents the long-term results to see if there is a sustained effect of joint protection education. As you discover that the full methodology of the trial is described in the earlier article (2001), you also obtain the full text of this article.
Hammond A, Freeman K. The long-term outcomes from a randomized controlled trial of an educational–behavioural joint protection programme for people with rheumatoid arthritis. Clin Rehabil 2004;18:520–8.
Hammond A, Freeman K. One-year outcomes from a randomized controlled trial of an educational–behavioural joint protection programme for people with rheumatoid arthritis. Rheumatology 2001; 40:1044–51.
Structured abstract (adapted from the above)
Study design: Randomised controlled trial.
Setting: Outpatients from the occupational therapy departments of two hospitals in the UK.
Participants: 127 patients with rheumatoid arthritis; aged between 18 and 65 years (mean age 50.5 years, 76% female); diagnosed with rheumatoid arthritis within the last 5 years, a history of wrist or metacarpophalangeal joint pain and inflammation and experiencing hand pain during activity. Exclusion criteria included no other medical condition that affected hand function.
Intervention: Two small-group education interventions, both of 8 hours duration, were compared. One group attended a standard arthritis education program, which included 2.5 hours of joint protection education. Approximately 15–45 minutes was spent practising joint protection techniques. The other group attended a joint protection education program that used educational–behavioural teaching methods and aimed to enhance self-efficacy and motor learning. Participants in this program spent about two-thirds of their time practising and receiving feedback about hand joint protection methods.
Outcomes: Primary measures: hand pain experienced during a moderate activity (such as cooking or housework) within the last week (measured using a 100 mm visual analogue scale); adherence with joint protection. Secondary measures: functional status (measured using the Arthritis Impact Measurement Scales [AIMS2] with a range of 0–10 where 0 indicated better function), indicators of disease severity, hand status and psychological status.
Follow-up period: 4 years. Assessments were performed at baseline, 6 months, 12 months and 48 months.
Main results: At 4 years, compared with participants in the standard group, participants in the joint protection group had significantly better adherence to joint protection, less early-morning stiffness, higher AIMS2 activities of daily living (ADL) scores and fewer deformities in metacarpophalangeal and wrist joints.
Conclusion: Among those who attended the joint protection program, some of the significant improvements (in adherence and maintenance of functional ability) that were found at 1 year were maintained at 4 years, suggesting that an educational–behavioural joint protection program should be used in clinical practice.
Is the evidence likely to be biased?
• Was the assignment of participants to groups randomised?
Yes. Participants were randomly allocated, using a four-block sequence.
• Was the allocation sequence concealed?
Yes. Allocation occurred using sealed envelopes that had been prepared in advance.
• Were the groups similar at the baseline or start of the trial?
Yes. The baseline characteristics were similar between the two study groups. There is a small difference between the two groups in terms of steroid use (6% of participants in the standard group and 20% of participants in the joint protection group), but this difference is probably not large enough to have affected the results. The baseline scores of the outcome measures were also similar between the two groups.
• Were participants blind to which study group they were in?
No. For this trial, it was not possible for participants to be blinded to group allocation.
• Were the health professionals who provided the intervention blind to participants' study group?
No. For this trial, it was not possible for the health professionals who provided the intervention to be blinded to group allocation.
• Were the assessors blind to participants' study group?
Yes, for some measures. Assessments were conducted by independent assessors who were not informed of group allocation. The assessor was also asked to avoid discussing the education programs with the participants. However, for the outcomes that were measured by participant self-report (such as hand pain), the assessment of these outcomes was not blind as participants were not blinded to group allocation.
• Were all participants who entered the trial properly accounted for at its conclusion, and how complete was follow-up?
Yes. The follow-up rate was 95.3% at 6 months and 96.8% at 12 months. By 4 years, the follow-up rate was 77%. Reasons are given as to why some participants were not able to be followed up, as well as which group they were in.
• Was intention-to-treat analysis used?
Yes. The authors state that an intention-to-treat analysis was conducted.
• Did the study have enough participants to minimise the play of chance?
Yes. Based on data from a previous study, the authors conducted a power analysis and determined that, with a power of 80% and a significance level of 0.05, 63 participants would be needed in each group.
These are given in Table 5.1. For hand pain at 1 year, the effect size is 12.98, which is a statistically significant result. Both the p value (0.02) and confidence interval (CI), which does not include the no effect value of zero, show this. As hand pain was measured using a 100 mm visual analogue scale, an effect size (between groups) of 13 is probably large enough to also be considered clinically significant. However, the confidence interval is very wide, indicating some imprecision in the result. Thus, for some people the intervention effect may not be large enough to be considered clinically significant, whereas for other people the effect could be quite large and deemed clinically significant.
TABLE 5.1:
Hand pain and functional outcome for intervention and control groups at 1 year and 4 years
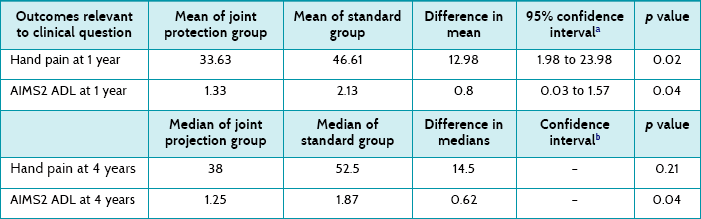
ADL = activities of daily living; AIMS2 = Arthritis Impact Measurement Scales.
aNot provided by authors—calculated using data provided in the article. Confidence intervals may vary slightly depending on the formula used and the extent of the rounding.
bNot able to be calculated based on the information provided by authors in the article.
However, by 4 years, the between-group difference in hand pain is no longer statistically significant. Unfortunately, in the 4-year follow-up article the authors present results differently than they did in the 1-year article. Two of the problems with this are that: (1) as medians are used to present 4-year results, confidence intervals cannot easily be calculated; and (2) it is difficult to compare the results of the 1- and 4-year follow-up papers as the results are presented differently.
With respect to function, the ADL subscale of the AIMS2 was used to measure participants' ability to perform self-care and household activities. The effect size for the ADL subscale at 12 months was 0.8, which is statistically significant (confirmed by both the p value and the confidence interval). However, as the AIMS2 is scored on a scale of 0–10, an effect size of 0.8 is small and may not be considered by many to be a clinically significant result. However, if we consider that the upper end of the confidence interval is 1.57, it is possible that the effect of this intervention could be clinically significant for some people. As explained in Chapter 4, the decision about clinical significance is a subjective one and depends on a number of factors such as the costs involved and preferences of the individual concerned.
At 4 years, the between-group difference remains statistically significant, but because the results are now presented in medians, it is difficult to compare the change from 1 year, not possible to calculate confidence intervals and difficult to consider clinical significance.
How might we use this evidence to inform practice?
As you are reasonably confident about the validity of the study's results and some of the results were of clinical importance, you proceed to assessing the applicability of this evidence to your clinical scenario. The patients with rheumatoid arthritis that you see are similar to the study participants in a number of ways such as age, gender (predominantly female) and disease duration. The majority of the participants in the study were assessed as having mild or moderate rheumatoid arthritis. You have yet to assess the disease severity in your recently referred patients. Once you do, you will check whether their disease severity is comparable to that of the study participants before making a decision about whether to implement a joint protection program.
Before making the decision, you will also need to consider whether you and the health centre where you work have the resources available to offer a joint protection program such as the one that was evaluated in the study. You will also need to obtain further, more detailed, information about the content of the program and the teaching strategies that were used. The article mentions that further details may be obtained in another published article, so you will start by obtaining that article and, if you have questions after reading that, you will contact the authors of the article for more information.
Although the study found a statistically and clinically significant effect of the intervention on hand pain at 1 year, this was not maintained by 4 years. The effect on the other outcome (self-care) that you were particularly interested in may not be conclusively clinically significant at either 1 year or 4 years. Although it is unlikely that the program would cause any harm and it appears to offer some benefits to participants, these mainly appear to be in the short term and there are considerable staffing resources associated with providing the program. Before making a decision about whether to implement the program at your workplace, you decide to search for other approaches to providing joint protection education and training to people with rheumatoid arthritis (and not necessarily education that is group-based or uses an educational–behavioural approach).
Physiotherapy example
Clinical scenario
As a physiotherapist on the cardiac surgery ward, you attend a conference and hear that some of your peers at another hospital are providing inspiratory muscle training to patients who are undergoing coronary artery bypass graft (CABG) surgery. The training is performed preoperatively in an attempt to reduce the likelihood of pulmonary complications occurring postoperatively. You are unaware of any research that has investigated the use of this intervention in patients undergoing cardiac surgery, and decide to search for evidence to decide whether this is something you should institute at the hospital where you work.
Does preoperative inspiratory muscle training in addition to standard care prevent postoperative pulmonary complications in patients who are undergoing CABG surgery?
Search terms and databases used to find the evidence
Database: PEDro (using the Advanced Search option).
Search terms: You decide that inspiratory muscle training is likely to be mentioned in the title of the article, although it may be described as respiratory muscle training. You therefore enter *spiratory muscle training in the ‘Title’ field. You consider that the patient population might be broadly defined in the title using a phrase like cardiac surgery, but you expect that it would be defined using coronary artery bypass graft or coronary artery bypass surgery in the abstract. Therefore, you enter coronary artery bypass in the ‘Abstract & Title’ field. You want both of these terms to be present, so you select the option ‘Match all search terms’ when searching.
The search returns eight records, but this represents only three separate trials as some of these trials had multiple publications that were retrieved by the search. For the first trial, by Hulzebos et al, there is the main publication which is accompanied by a report of pilot data, an English translation of the primary Dutch publication, a consumer summary of the trial and an analysis of the feasibility of the intervention using data from the trial. For the second trial, there is the main publication and a Hebrew-to-English translation. The third trial has only one publication, but it is about postoperative respiratory muscle training, so it is not relevant to your question.
Although the first and second trials examine inspiratory muscle training for patients who are undergoing CABG surgery, several features of the first trial show that it probably provides less-biased evidence to answer your question. Unlike the second trial, it used concealed allocation, blinded outcome assessment and intention-to-treat analysis and fewer participants were lost to follow-up. In addition, the sample size of the trial was larger (n = 279 vs n = 84) and the trial was conducted more recently, so the surgical procedure and standard care in the postoperative period are more consistent with that offered at your hospital. Therefore, you decide on the first article.
Hulzebos E, Helders P, Favie N, et al. Preoperative intensive inspiratory muscle training to prevent postoperative pulmonary complications in high-risk patients undergoing CABG surgery: a randomized clinical trial. JAMA 2006;296:1851–7.
Structured abstract (adapted from the above)
Study design: Randomised controlled trial.
Setting: University medical centre in Utrecht, the Netherlands.
Participants: 279 patients (mean age 66.9 years; 77.9% male) undergoing CABG surgery at high risk of developing postoperative pulmonary complications, indicated by the presence of two or more of these criteria: age >70 years, productive cough, diabetes mellitus, smoking, chronic obstructive pulmonary disease and body mass index >27. Exclusion criteria included: surgery within 2 weeks of initial contact; a history of stroke; use of immunosuppressive medication for 30 days before surgery; and presence of a neuromuscular disorder, cardiovascular instability or an aneurysm.
Intervention: Participants were randomly assigned to receive either preoperative inspiratory muscle training (n = 140) or usual care (n = 139). The intervention group trained daily (20 minutes), 7 times a week (6 times without supervision), for at least 2 weeks before the surgery. Both groups received the same postoperative physical therapy and other standard care.
Outcomes: Primary outcome: the incidence of postoperative pulmonary complications, defined according to recognised criteria. Secondary outcome: duration of postoperative hospitalisation, in days.
Main results: Postoperative pulmonary complications occurred in 25 (18%) of the participants in the intervention group and 48 (35%) of the control group, odds ratio 0.52 (95% CI 0.30 to 0.92). Median duration of hospitalisation was 7 days (range 5–41) in the intervention group and 8 days (range 6–70) in the control group (p = 0.02).
Conclusion: Preoperative inspiratory muscle training reduced the incidence of pulmonary complications and the duration of postoperative hospitalisation in patients who were at high risk of developing a pulmonary complication after CABG surgery.
Is the evidence likely to be biased?
• Was the assignment of participants to groups randomised?
Yes. Participants were appropriately randomised, using a computer-generated number list.
• Was the allocation sequence concealed?
Yes. The number list was sealed in envelopes which were held by an external investigator.
• Were the groups similar at the baseline or start of the trial?
Yes. The baseline characteristics were similar between the two study groups. Although in the table of baseline characteristics the authors of the article have reported a difference between the two groups in terms of median duration of mechanical ventilation (4 hours in the intervention group vs 5 hours in the control group), this is technically not a baseline characteristic as it was measured after the intervention had been provided. Additionally, it is unlikely that a difference of this size would be of clinical importance and have affected the results.
• Were participants blind to which study group they were in?
No. For this trial, it was not possible for participants to be blinded to group allocation.
• Were the health professionals who provided the intervention blind to participants' study group?
No. For this trial, it was not possible for the therapists who provided the intervention to be blinded to group allocation.
• Were the assessors blind to participants' study group?
Yes. The investigators who assessed outcomes were blinded to participants' treatment group.
• Were all participants who entered the trial properly accounted for at its conclusion, and how complete was follow-up?
Yes. Apart from three participants who died before surgery, all participants were followed up. This is a 98.9% follow-up rate.
• Was intention-to-treat analysis used?
Yes. It is stated that an intention-to-treat analysis was conducted.
• Did the study have enough participants to minimise the play of chance?
Yes. A power calculation was performed, but the trial was terminated before this number was reached because safety monitoring determined that statistically and clinically significant results had been achieved at the interim analysis.
Pulmonary complication: The risk of pulmonary complications is presented appropriately, using an odds ratio with a 95% confidence interval (refer to Table 5.2). The reduction in risk is both statistically and clinically significant. The 95% confidence interval includes only clinically worthwhile reductions in risk. Therefore, the results are sufficiently precise to make clinical recommendations.

The data for the first outcome, pulmonary complications, can be used to estimate two useful statistics. First, let us calculate the absolute risk reduction (ARR), which is simply the risk in the control group minus the risk in the intervention group: 35% – 18% = 17%. Using the formula (95% confidence interval ≈ difference in risk ± 1 √–nav ) that was provided in Box 4.3 in Chapter 4, you also calculate the 95% confidence interval of the ARR to be 9% to 25%. The ARR statistic is useful in clinical practice because, if you decide to implement the intervention, you can use it to explain to patients the value that they can expect from undertaking the inspiratory muscle training regimen. After explaining what a pulmonary complication is and how it can delay recovery, many patients would consider the training program worthwhile to reduce their risk of such a complication from 35% to 18%.
An alternative statistic is the relative risk reduction (RRR). This is calculated by dividing the difference in risk between the two treatment groups by the risk in the control group:
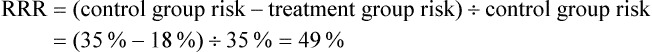
When an adverse outcome occurs fairly frequently in a study, the RRR is a useful statistic; but when the outcome is rare, a large RRR may still be found even though the ARR is very small, which can be misleading. Therefore you decide to use ARR instead of RRR.
You then use the ARR to calculate the number of people that need to be treated in order to prevent one pulmonary complication:

The 95% confidence interval of this number needed to treat is 4 to 12. This was calculated by using the inverse of the numbers in the confidence interval of the ARR that you calculated earlier (so 1/25% and 1/9%). Therefore, for every 6 high-risk patients that we treat, 1 pulmonary complication will be prevented that would have otherwise occurred with usual care. However, this number needed to treat could be as low as 4 or as high as 12.
Duration of hospitalisation: The median duration of postoperative hospitalisation was 7 days (range 5 to 41) in the treatment group and 8 days (range 6 to 70) in the control group, and this difference was statistically significant.
How might we use this evidence to inform practice?
You decide that the trial is valid and that the results are important. Pulmonary complications are dangerous and uncomfortable for the patient and they are expensive to treat. Therefore, the effect of this intervention on this outcome alone is clinically worthwhile, and the number needed to treat of 6 is useful in justifying the introduction of the service. Further justification of the clinical worth of this intervention comes from the other significant outcome, which was a reduction in the duration of hospitalisation. You plan to discuss the results of this study with your head of department as there are resource implications associated with introducing the service, particularly providing an intervention of the same intensity as was provided in the study. However, it is your recommendation that this intervention should be introduced.
Podiatry example
Clinical scenario
You are a podiatrist who works in a private practice. In recent days, you have seen several patients with heel pain which you believe is plantar fasciitis. Your management of this condition often includes prescription of customised orthoses (devices made to a cast of the patient's foot with an individualised prescription). Because it takes several weeks for the orthotics to be manufactured, you wonder whether prefabricated orthotics (mass-produced devices made to fit a generic foot shape) which can be bought over the counter and started immediately are as effective. You decide to search for the evidence.
In people with plantar fasciitis, are prefabricated orthotic devices as effective as customised orthoses in reducing pain and improving foot function?
Search terms and databases used to find the evidence
Database: The Cochrane Library. A systematic review of randomised controlled trials or a randomised controlled trial would be the ideal study design to answer this question. Ideally the randomised controlled trial would have three arms, allowing comparison of the two types of orthotic devices with each other and a control group.
Search terms: You start by checking for MeSH terms related to the population and intervention of interest, and combine these with textword terms using the Boolean operator OR in the ‘Search History’ and ‘Advanced Search’ features of The Cochrane Library:
(Fasciitis, plantar (MeSH) OR plantar fasciitis OR (heel OR calcan* NEAR pain)) AND (orthotic devices (MeSH) OR orthotic* OR orthoses)
This search produced 30 hits, comprising 7 Cochrane Reviews, 3 other non-Cochrane systematic reviews, 19 trials and 1 economic evaluation. Two Cochrane reviews look as though they may answer your question. Unfortunately, the first one you look at, titled ‘Interventions for the treatment of heel pain’, has been withdrawn from the Library because it is out of date. The second review, titled ‘Custom-made orthoses for the treatment of foot pain’, examines the effect of customised orthoses on foot pain of any type or aetiology. As you read through the review, you find that it included six trials which assessed the effect of customised orthoses for plantar fasciitis, and in three of these, customised orthoses were compared with prefabricated devices. The review is very helpful as it not only describes, assesses the risk of bias and synthesises the results of studies which address your clinical question, but it also provides information on the comparative effectiveness of other interventions such as night splinting and stretching.
Of the three relevant trials, one is a three-arm trial which compares the effect of customised, prefabricated and sham orthoses in participants with plantar fasciitis. Measures of pain and function at 3 and 12 months are reported. The other trials also compare customised orthoses with a range of other interventions, including prefabricated orthoses and in-shoe devices; however, participants in these studies were only followed up for a maximum of 3 months. You decide that assessment of pain and function at 12 months is preferable, so you select the trial titled ‘Effectiveness of foot orthoses to treat plantar fasciitis’ for appraisal.
Landorf KB, Keenan A, Herbert RD. Effectiveness of foot orthoses to treat plantar fasciitis. Arch Intern Med 2006;6:1305–10.
Structured abstract (adapted from the above)
Study design: Randomised controlled trial.
Setting: A university podiatry clinic, Melbourne, Australia.
Participants: 136 participants, mean age 48 years, 67% female, with a clinical diagnosis of plantar fasciitis who had experienced symptoms for at least 4 weeks. People with a major orthopaedic or medical condition that may have influenced the condition were excluded.
Intervention: Participants were randomised to receive one of: (a) ‘sham’ orthoses which were made of soft foam moulded over an unmodified cast of the foot; (b) prefabricated orthoses made from a thicker, firmer-density foam moulded over the cast; or (c) customised foot orthoses made from semi-rigid polypropylene moulded over neutral-position plaster casts with a firm foam heel post.
Outcomes: The primary outcome of the trial was self-reported pain and function at 3 and 12 months, which were measured using the pain and function domains of the Foot Health Status Questionnaire (on a 0–100 measurement scale).
Main results: This trial found that pain and function improved in all three groups over time. Participants receiving prefabricated and customised orthoses showed greater improvement in function than the sham group at 3 months (mean difference of 8.4 points on the function domain between prefabricated and sham orthoses, and mean difference of 7.5 points between customised and sham orthoses). Prefabricated and customised orthoses also reduced pain compared with sham orthoses at 3 months (8.7 points and 7.4 points, respectively), although these differences were not statistically significant. At 12 months, there were no significant differences in pain and function between any of the three groups.
Conclusion: Both prefabricated and customised orthoses have similar small short-term benefits for people with plantar fasciitis and negligible long-term effects.
Is the evidence likely to be biased?
• Was the assignment of participants to groups randomised?
Yes. The randomisation sequence was generated using an appropriate method (computer-generated randomisation sequence).
• Was the allocation sequence concealed?
Yes. The allocation was concealed from participants and from the investigator enrolling participants in the trial. The allocation sequence appears to be held off-site and, once a participant was enrolled and baseline assessments were completed, the allocation sequence was obtained by telephone or email.
• Were the groups similar at the baseline or start of the trial?
Table 1 in the article shows that study groups were balanced in terms of most of the baseline variables that were measured. One important exception is participant's weight, where the mean weight of participants in the prefabricated orthoses group was approximately 10 kg more than in the other two study groups. This is a clinically important difference that may influence the outcome. You notice that the results are not adjusted for this difference and keep this in mind. You also notice a considerable difference between the sham and prefabricated groups in the foot function score at baseline, but are satisfied that this has been adjusted for in the analysis.
• Were participants blind to which study group they were in?
Cannot tell. Although the article states that it was a ‘participant blinded’ trial, you cannot tell this for sure. The investigators attempted to ‘blind’ participants to the orthoses they received by making the devices as similar as possible in terms of colour and shape. Participants were told they would receive soft, medium or hard orthoses. All participants had a cast of the foot taken. However, because the material used for the three devices was different (foam, polyethylene, polypropylene), some participants may have been able to work out which device they had received. Because some participants may not have been blind to which study group they were in and because the study outcomes were measured by participant self-report, you cannot be sure that any treatment effect (or lack of effect) that is reported in this study is not biased. Evaluation of the success of participant blinding was not reported.
• Were the health professionals who provided the intervention blind to participants' study group?
No. Due to the nature of the intervention, the podiatrist who assessed the participants and provided them with their orthoses was not blind to the intervention allocation.
• Were the assessors blind to participants' study group?
Cannot tell. For the same reason that participants may not have been blind to study group allocation, the investigators who were in contact with participants during outcome measurement may also not have been blind to study group allocation. To prevent study investigators from biasing measures of outcome (and potentially influencing participants' responses), participants completed the Foot Health Status Questionnaire at the beginning of each appointment before they had any interaction with the investigator. However, as the Foot Health Status Questionnaire is a self-report outcome, if participants are not blind to their allocation then the assessment cannot be considered blind either.
• Were all participants who entered the trial properly accounted for at its conclusion, and how complete was follow-up?
Yes. The flow of participants through this study is clearly reported in a flow diagram. Five of the 136 participants (4%) were lost to follow-up, three from the sham orthoses group (though one of these withdrew before any baseline measures were conducted or treatment was received) and one each from the prefabricated and customised orthoses groups. The article also reports the number of participants who crossed over to alternative orthoses. This was highest in the sham orthoses group, with two of the 44 participants using alternative orthoses at 3 months and seven (of the 43) at 12 months.
• Was intention-to-treat analysis used?
Yes. The article states that an intention-to-treat analysis was conducted.
• Did the study have enough participants to minimise the play of chance?
Yes. The authors report conducting a power calculation prior to commencing participant recruitment. They determined the number of participants required based on the significance level (risk of type 1 error 5%), statistical power (90%) and an estimated difference in effect between any of the study groups. According to the power calculation, 136 participants were required, which is how many were randomised.
As you are interested in the comparison between prefabricated orthoses and customised orthoses, you look for these results first. The results are presented in the article as the mean difference (and 95% confidence intervals) in pain and function scores between the groups at 3 and 12 months (adjusted for baseline scores). See Table 5.3 for a summary of these results.

Pain: At 3 months, the mean difference in pain between prefabricated and customised orthoses was 1.3 (95% CI –7.6 to 10.2) points on the Foot Health Status Questionnaire pain domain, and at 12 months, it was 2.3 points (95% CI –5.6 to 10.1).
Function: On the function domain, the mean difference between prefabricated and customised orthoses at 3 months was 0.9 points (95% CI –6.3 to 8.1), and at 12 months, it was 1.2 points (95% CI –6.1 to 8.5).
None of these results are statistically significant (as seen by the confidence intervals which all contain the no effect value of zero). You therefore do not need to consider the clinical significance of these results. The results show that the effects of prefabricated and customised orthoses on pain and function are similar.
The study also presents the effects on foot function as a number needed to treat by dichotomising the data. In the dichotomisation, an improvement in function was considered to have occurred when function increased by more than one-third of the baseline value. The prefabricated foot orthoses produced one additional improved outcome for every six people treated for 3 months, and the customised foot orthoses produced one additional beneficial outcome for every four people treated for 3 months.
Other results show that pain and function improved in all study groups over time and that, at 12 months, differences in pain and function between all the groups were small and not statistically significant. However, at 3 months, the prefabricated and customised orthoses group showed greater improvement in pain and function than the sham group. The mean difference in function was statistically significant—a difference of 8.4 (95% CI 1.0 to 15.8) points for prefabricated orthoses vs sham orthoses, and a difference of 7.5 (95% CI 0.3 to 14.7) points for customised orthoses vs sham orthoses. Although the authors do not provide any guidance about the amount of difference on the 0–100 scale that would be needed for this to be considered a clinically significant difference, the upper ends of the confidence intervals indicate that these intervention effects may be considered clinically significant to some people, depending on their preferences. However, the lower ends of the confidence intervals suggest that it is also possible that some people may not find much benefit from this intervention.
How might we use this evidence to inform practice?
You decide that the trial is valid and that the results are important. Given the similarity in the effects of the prefabricated and the customised orthoses, it would be reasonable to prescribe a prefabricated device rather than a custom-made devices for your patients with plantar fasciitis. A major benefit you see in this approach is that the patient can start the treatment almost straight away, as opposed to the several weeks it takes for customised devices to be manufactured. However, you think there are other factors to consider. First, several of your patients have had symptoms for only a few weeks. You wonder if they would respond the same way as participants in the trial where the condition was more chronic (symptoms were experienced for a median of 12 months). You also wonder how the results might vary if you use different prefabricated orthoses or customised orthoses manufacturers to those that were used in the study.
The prefabricated orthoses used in the study are easily obtained for a cost of around AUD $40 to $70. This is considerably cheaper than customised orthoses, which can cost anywhere from AUD $250 to $400. However, prefabricated devices do not last as long as customised devices, which are usually made from more-durable polypropylene, and may need to be replaced a number of times depending on wear. If, as the results of this study suggest, most people recover from plantar fasciitis by 1 year, then two or three pairs of prefabricated devices over a 12-month period would still be cheaper than customised devices. You know from experience, though, that once people start to wear orthoses and feel they are beneficial, they often continue to use them long-term and, if this is the case, customised orthoses which last for several years may be more economical. Your experience also tells you that often patients also need to purchase footwear that can accommodate orthotic devices. This, together with the cost of the actual orthoses, can be an issue for some patients. You decide that it is important to discuss the results of this study with your patients, working with them to determine the best management option for each situation.
Midwifery example
Clinical scenario
You are a midwife who is newly employed in the postnatal unit of a tertiary hospital. You have noticed diversity of practice surrounding the provision of perineal ice-packs for pain relief in patients after spontaneous vaginal delivery. In addition to providing oral anti-inflammatories, some of your colleagues provide perineal ice-packs to these women in order to relieve perineal postpartum pain, while other nurses and midwives do not. You decide to find out about the effectiveness of an ice-pack applied during the immediate postpartum period in reducing perineal pain.
Following spontaneous vaginal delivery, does the application of an ice-pack to the perineal area reduce postpartum perineal pain?
Search terms and databases used to find the evidence
Database: PubMed—Clinical Queries (with ‘therapy category’ and ‘narrow scope’ selected).
Search terms: (perineal pain) AND ice
This search results in 5 articles. One matches your clinical question and you retrieve its full text.
Leventhal L, de Oliveira S, Nobre M, et al. Perineal analgesia with an ice pack after spontaneous vaginal birth: a randomised controlled trial. J Midwifery Women's Health 2011;56:141–6.
Structured abstract (adapted from the above)
Study design: Randomised controlled trial.
Setting: In-hospital birth centre averaging 900 births per month in São Paulo, Brazil.
Participants: 114 nulliparous women recovering from spontaneous vaginal deliveries, who experienced perineal pain ≥ 3 (0 = no pain to 10 = worst imaginable pain) within 2 and 48 hours following childbirth, regardless of perineal condition. Exclusion criteria included maternal, obstetric or clinical complications such as haemorrhoids, haematomas, newborn complications or instrumental births, multiple births, non-cephalic fetal presentation, postpartum fever treatment with antibiotics or had received a postpartum analgesic within 6 hours prior to inclusion into the study.
Intervention: The intervention group applied an ice-pack for a single instance for a 20-minute period in the perineal region between 2 and 48 hours postpartum. The placebo group applied a water-pack at room temperature for a single instance for a 20-minute period in the perineal region between 2 and 48 hours postpartum. The control group received no treatment in addition to routine care. All participants received routine care, consisting of 500 mg of metamizole, an anti-inflammatory agent which was administered orally every 8 hours. No other non-pharmacological treatment to alleviate perineal pain was provided.
Outcomes: The primary outcome measure was perineal pain, measured using a 0 (no pain) to 10 (worst imaginable pain) scale.
Follow-up period: Perineal pain was assessed prior to the intervention (baseline), immediately after the application of the pack (20 minutes), then 20 minutes later (40 minutes) and again 20 minutes later (60 minutes).
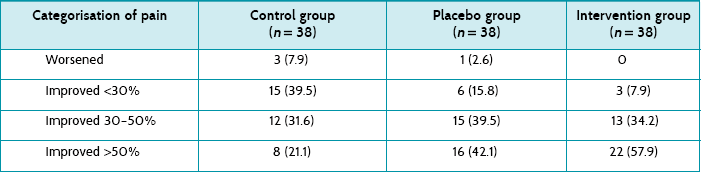
Main results: A comparison of the mean pain score at baseline and at 20 minutes showed a significant reduction of pain (p < 0.001) in all three groups. However, the intervention group had a lower average pain score at 20 minutes compared with the control group (1.6 versus 3.3, p = 0.032).
Conclusion: The use of ice-packs for 20 minutes was effective for the relief of perineal pain for women after spontaneous vaginal delivery.
Is this evidence likely to be biased?
• Was the assignment of participants to groups randomised?
Yes. Participants were randomly allocated to intervention, placebo or control group in a 1 : 1 : 1 ratio by computer-generated randomisation. Blocks of six were used.
• Was the allocation sequence concealed?
Yes. Allocation was concealed in an opaque, numbered sealed envelope which was opened by an individual not involved in the research.
• Were the groups similar at the baseline or start of the trial?
Yes. The three groups had similar baseline mean pain scores and were similar on other characteristics such as socio-demographic characteristics, ambient, axillary and perineal temperatures and birth-related characteristics (for example, degree of perineal trauma).
• Were participants blind to which study group they were in?
No. For this type of intervention it was not possible to blind participants.
• Were the health professionals who provided the intervention blind to participants' study group?
No. Due to the nature of the intervention the research nurse who applied the intervention could not be blinded.
• Were the assessors blind to participants' study group?
No. Even though the article states that there was an outcome assessor who was blinded and that participants were instructed not to inform the researcher which intervention they received, as the outcome was pain that was measured by participant self-report, the assessment of this was not blind as participants were not blinded to group allocation.
• Were all participants who entered the trial properly accounted for at its conclusion, and how complete was the follow-up?
Yes. Although there is no figure showing participant flow through the trial, the article reports that no participants withdrew from the study (most likely to due to the short duration of the intervention and follow-up period) and that there was no migration of women among the groups.
• Was intention-to-treat analysis used?
Cannot tell. The article does not specifically mention using intention-to-treat analysis, although it is likely that this is what occurred as there were no participants lost to follow-up or migration among groups.
• Did the study have enough participants to minimise the play of chance?
Yes. The authors had undertaken a pilot study to inform sample size calculations and concluded that for a significance level of 5% and power level of 80%, a sample size of 38 patients per group was required. They adequately recruited to this sample size.
The study reports the mean pain scores (and 95% confidence intervals) for all three groups at all time points (see Table 5.4). Although the article reports that all three groups had a statistically significant reduction in the mean pain score at 20 minutes compared with baseline, it is the between-group comparisons that you are interested in. The only statistically significant between-group difference was between the intervention (ice) group and control group at 20 minutes (a difference of 1.7). As standard deviations are not presented, a confidence interval for this effect size cannot be calculated.
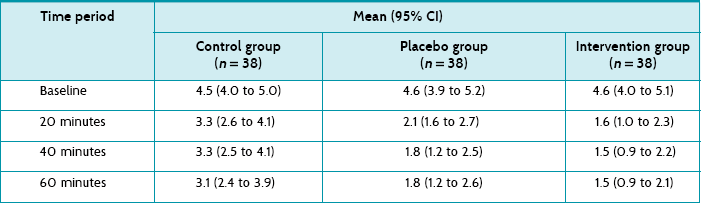
The article also presents the data transformed into categories (pain worsened, improved <30%, improved 30–50%, improved >50%) at the end of the intervention (presumably 60 minutes); see Table 5.5. The authors nominate a decrease in pain of ≥30% as clinically significant. It is unclear whether the analysis by categorisation was planned a priori or an attempt to find significant results. If you use the authors' cut-off of ≥30% improvement and calculate the number of participants in each group who reported ≥30% improvement in pain, using the data in Table 5.5, this gives 20/38 (53%) in the control group, 31/38 (82%) in the placebo group, and 35/38 (92%) in the intervention group. You then calculate the following (using the formula for calculating 95% CI for absolute differences given in Box 4.3 in Chapter 4):
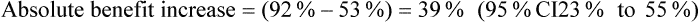
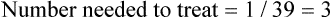
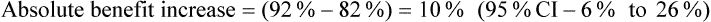
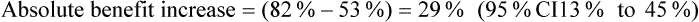
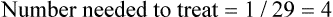
How might we use this evidence to inform practice?
You are reasonably confident of the results of this study. While there was no difference in pain outcomes between the intervention (ice-pack) and placebo (water-pack) groups, there was a difference between any pack (that is, either water-pack or ice-pack) and no pack. This difference may have been due to the placebo effect. In addition, all participants had a researcher remain at their side for 20 minutes providing guidance about postpartum care such as breastfeeding and postpartum bleeding, and this may have confounded the results. That is, the reduction in pain in all groups may be partially explained by the psychosocial support and distraction of having an experienced midwife's presence and support.
As there is unlikely to be any harm from using ice-packs (although it is advised that they should be removed after 20 minutes' use) and they are inexpensive, you decide that it may be worthwhile having ice-packs available to women as an option. Some women may not like using an ice-pack (in this study, 3 out of 117 declined to participate), so asking women if they wish to use this intervention will be important. Your ward already has the logistical issues associated with using ice-packs (such as a suitably-sized freezer that is accessible to staff, and appropriate infection control processes) in place.
Nutrition therapy example
Clinical scenario
As a clinical dietitian who works in the orthopaedic ward of a hospital, you are asked to review a 72-year-old woman admitted to the accident and emergency department 2 days ago with a fractured neck of femur. She went to theatre yesterday morning. Today, the orthopaedic team have commenced her on a full ward diet, but due to her thin and malnourished appearance they are concerned that she may develop a pressure ulcer. They have requested a dietitian consult for advice about whether supplementary nutritional support may reduce the risk of this occurring.
Does nutritional support prevent pressure ulcer development in people who have undergone surgery for a fractured neck of femur?
Search terms and databases used to find the evidence
Database: PubMed—Clinical Queries (with ‘therapy category’ and ‘broad scope’ selected).
Search terms: ((hip fracture*) OR (fractured neck of femur)) AND (nutritional support) AND (pressure ulcer*)
This search yielded three clinical studies and one systematic review. The systematic review relates to enteral nutrition and is therefore not relevant to the clinical question. Of the three clinical studies, only two relate to the clinical question; and as only one is a randomised controlled trial, this study is chosen.
Houwing R, Rozendaal M, Wouters-Wesseling W, et al. A randomised, double-blind assessment of the effect of nutritional supplementation on the prevention of pressure ulcers in hip-fracture patients. Clin Nutr 2003; 22:401–5.
Structured abstract (adapted from the above)
Study design: Randomised controlled trial.
Setting: Three medical centres in the Netherlands.
Participants: 103 participants who were post-surgery for a hip fracture and had a pressure ulcer risk score >8 according to the CBO risk-assessment tool. Exclusion criteria included: terminal care, metastatic hip fracture, insulin-dependent diabetes, renal disease (creatinine >176 mmol/L), hepatic disease, morbid obesity, need for a therapeutic diet incompatible with supplementation, and pregnancy or lactation.
Intervention: Participants in the intervention group (n = 51) received 400 mL daily of a nutritional supplement enriched with protein, arginine, zinc and antioxidants. Participants in the control group (n = 52) received a water-based placebo. The supplement and placebo were commenced postoperatively and continued for 4 weeks or until discharge. Participants in both groups also received a regular diet.
Outcomes: Pressure ulcer development (assessed using a four-stage classification); time of onset, size and location of any pressure ulcer(s) were also recorded.
Follow-up period: 4 weeks or until discharge. Participants were assessed daily by nursing staff.
Main results: There was no difference in the incidence of developing a pressure ulcer between the intervention (55%) and placebo (59%) groups; however, the supplemented group had a lower incidence of stage II ulcers and showed a trend towards later development of a pressure ulcer.
Conclusion: People with hip fracture are prone to developing pressure ulcers at an early postoperative stage. Initiating nutritional support at this stage may delay ulcer development and progression, but is unlikely to prevent this process occurring. The authors surmise that nutritional supplementation may be more effective if it is started earlier.
Is the evidence likely to be biased?
• Was the assignment of participants to groups randomised?
Yes, participants were randomised to receive the study or placebo supplement in addition to their regular diet; however, the method of randomisation is not specified.
• Was the allocation sequence concealed?
Cannot tell. The article does not describe how participants were allocated to groups.
• Were the groups similar at the baseline or start of the trial?
The baseline characteristics of participants appear similar between the two study groups. There was also no difference in the median time between the start of supplementation and surgery or admission to hospital between the two study groups.
• Were participants blind to which study group they were in?
Yes. Although participants may have been aware of what they were receiving due to the different taste and viscosity of the two drinks, it is unlikely that this was the case as there was no crossover of drinks between the study groups and participants would have been unlikely to know what each drink tasted like prior to the study.
• Were the health professionals who provided the intervention blind to participants' study group?
Cannot tell. It is stated that nursing staff recorded each participant's daily intake of the supplement, but it is not clear who gave the supplements to the participants in between their regular meals each day.
• Were the assessors blind to participants' study group?
Yes. The authors of the study acknowledge that, although each of the supplements (intervention supplement and placebo supplement) had a different look and taste, both supplements were provided in similar blinded packages to mask this difference. It is therefore unlikely that nursing staff would have been able to tell the difference between the two supplements. The presence of pressure ulcers was assessed daily by nursing staff according to a four-stage classification system based on the European Pressure Ulcer Advisory Panel Guidelines.
• Were all participants who entered the trial properly accounted for at its conclusion, and how complete was follow-up?
No. The article does not state whether any participants were lost to follow-up or, if so, the reasons for this. However, based on the data provided in Table 4 of the article, it appears that two participants were lost from the intervention group and one from the control group. This would give a 97% follow-up rate.
• Was intention-to-treat analysis used?
Cannot tell. The authors do not explicitly state that an intention-to-treat analysis was conducted; therefore, it is assumed that it was not. There was no mention of percentage error in drinks given, which may mean that there was no error; however, this should also have been stated in the article.
• Did the study have enough participants to minimise the play of chance?
No. This study did not have enough participants to identify a significant difference in outcome between the two groups. A power calculation is described indicating that 350 participants were required in each group to detect a 25% decrease in pressure ulcer incidence. The study only recruited 14% of this required amount.
As indicated by the confidence intervals and the p values, there were no statistically significant differences in any of the outcomes shown in Table 5.6. Based on the data that are provided in the article, you were able to calculate the absolute risk reduction (ARR), the 95% confidence interval associated with the ARR, and the number needed to treat. The development of one pressure ulcer would be prevented for every 27 people who receive the nutritional supplement and the development of one stage II pressure ulcer would be prevented for every 11 people who receive the nutritional supplement.
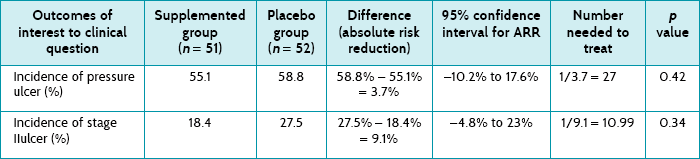
How might we use this evidence to inform practice?
There are a number of pieces of information which are relevant to this study but are not discussed in the article. For example, were participants using a pressure-ulcer relieving mattress prior to and/or after the surgery? What was the immediacy of surgery and had participants been fasted a number of times preoperatively?
It may be difficult to generalise the results of this study to other populations as the sample in the study had specific characteristics, such as a pressure ulcer risk score of around 11, average age of 81 years, 81% of the group being female, a well-nourished population (average body mass index 23.5–24.5 kg/m2) and a haemoglobin level of 7.1 g/L. These specific characteristics may partly explain the low number of participants who were able to be recruited into this study. The consequence of such specific characteristics is that the results can only be translated to patients who have similar characteristics. The patient in your clinical scenario differs from the study sample in a number of ways, and you do not feel confident that you can generalise the results to her. Additionally, the intervention was with a very specific supplement which was enriched with arginine, zinc and antioxidants (Cubitan®, from N.V. Nutricia in the Netherlands), and this product is not readily available in your region.
In conclusion, although this study had reasonably good internal validity, it does little to inform the dietitian about changing practices because it was severely underpowered and the sample population had a restricted profile. As such, nutritional practitioners should continue to provide nutritional support based on stronger data in the literature from different, yet related, patient populations. You are aware of good quality research studies that have demonstrated the positive effects of nutritional supplementation on pressure ulcer development in different clinical populations, for example malnourished patients or elderly people recovering from the acute phase of a critical illness, and so wonder if the lack of statistically significant results in this study was due to the fact that it was underpowered. Until a larger, sufficiently powered study is conducted with people with hip fractures, your practice will be guided by the research that has been conducted with other clinical populations.
Radiation therapy example
Clinical scenario
You are a radiation therapist who is currently assisting at a radiation therapy clinic in preparation for your new advanced practice role in radiation therapy side-effect review. One of the clinic patients is Mr Jones who is 58 years old and has a glioblastoma (a brain tumour). He is being treated with palliative high-dose radiation therapy to 60 Gy in 30 fractions, over 6 weeks. Prior to starting his treatment, Mr Jones had surgery to de-bulk as much of the tumour as possible. When he attends the weekly radiation oncology review at the clinic with his wife, Mrs Jones says that they have been looking at the internet for other possible treatment options for her husband's terminal brain tumour. She mentions that they have seen some information which suggests that a drug named temozolomide is being used in conjunction with radiation therapy in other hospitals to improve survival and control of the disease. They ask why it is not being used in their case. You decide to look for evidence about the use of this drug, particularly its success with respect to survival post-treatment.
Does the use of concomitant and adjuvant temozolomide with radiation therapy improve survival for people with glioblastoma compared with high-dose palliative radiation therapy alone?
Search terms and databases used to find the evidence
Database: PubMed—Clinical Queries (with ‘therapy category’ and ‘narrow scope’ selected).
Search terms: (glioblastoma) AND (temozolomide) AND (radiotherapy OR radiation therapy) AND (survival)
The search resulted in 23 articles. Of these, three articles are relevant to your question and compare radiation therapy combined with temozolomide to radiation therapy alone. One of these is a Phase II randomised controlled trial with 130 participants. In this study, overall survival, progression-free survival and toxicity are the primary outcomes. The other publication (a 2005 article) is a Phase III international multi-centre randomised controlled trial with 573 participants. The primary outcome of this study is overall survival, with secondary outcomes being progression-free survival, safety and quality of life. You choose the second article to fully appraise, as it is the most relevant to your question and because it is a Phase III trial and had a larger sample size. The third article is a more recent (2009) article that reports the 5-year follow up results of the Phase III trial, and you decide to review both articles related to this study.
Stupp R, Mason W, van den Bent M, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96.
Stupp R, Hegi M, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009;10:459–66.
Structured abstract (adapted from the above)
Study design: Phase III, multi-centre, randomised controlled trial.
Setting: Participants were recruited from 85 radiation oncology institutions in 15 countries throughout Europe and Canada.
Participants: 573 participants aged between 18 and 70 years (median age 56 years) with newly diagnosed histologically confirmed glioblastoma. Participants needed a World Health Organization performance status of 2 or less (indicating that they were relatively fit and self-caring). Additional specific inclusion criteria related to haematological, renal and hepatic function; serum creatinine and bilirubin levels.
Intervention: Participants in the control group (n = 286) received only radiation therapy (60 Gy, 30 fractions, 5 days a week for 6 weeks). Participants in the intervention group (n = 287) received radiation therapy (same dose and fractionation as the control group) and concomitant temozolomide (75 mg per square metre of body surface area per day, given 7 days a week from the first to the last day of radiation therapy, but for no longer than 49 days). Intervention group participants then had a 4-week break before receiving up to 6 cycles of adjuvant temozolomide (5-day schedule every 28 days) as well as prophylactic antibiotics due to increased chance of infection and prophylactic anti-emetics due to increased chance of nausea/vomiting.
Outcomes: The primary outcome was survival. Secondary outcomes were progression-free survival, safety (toxicity) and quality of life.
Follow-up period: Median follow-up was 51 months (in 2009 article). During radiation therapy, participants were seen every week. After the radiation therapy was completed, participants were seen 21–28 days after this, and every 3 months thereafter. Participants in the intervention group received a monthly clinical evaluation while receiving the adjuvant temozolomide therapy and a comprehensive assessment at the end of cycles 3 and 6.
Main results: During the 5 years of follow-up, 254 (89%) of the intervention group participants and 278 (97%) of the control group participants died. Overall survival was 27.2% (95% CI 22.2 to 32.5) at 2 years, 16.0% (12.0 to 20.6) at 3 years, 12.1% (8.5 to 16.4) at 4 years and 9.8% (6.4 to 14.0) at 5 years for the intervention group, compared with 10.9% (7.6 to 14.8), 4.4% (2.4 to 7.2), 3.0% (1.4 to 5.7) and 1.9% (0.6 to 4.4) in the control group (hazard ratio 0.6, 95% CI 0.5 to 0.7, p < 0.0001).
Conclusion: Benefits of temozolomide in addition to radiation therapy for people with newly diagnosed glioblastoma lasted throughout 5 years of follow-up.
Is the evidence likely to be biased?
• Was the assignment of participants to groups randomised?
Yes. Participants were randomly allocated to either the intervention or the control group, although details of the exact method of randomisation are not provided. Participants were stratified according to performance status, previous surgery and treatment centre.
• Was the allocation sequence concealed?
Yes. The article states that participants were centrally randomised over the phone or internet at the headquarters of a European research centre.
• Were the groups similar at the baseline or start of the trial?
Yes. The article provides details about a large range of participant characteristics and the two groups appear to have been well balanced at baseline.
• Were participants blind to which study group they were in?
No. For this trial, it was not possible for participants to be blinded to group allocation.
• Were the health professionals who provided the intervention blind to participants' study group?
No. For this trial, it was not possible for the health professionals who provided the intervention to be blinded to group allocation.
• Were the assessors blind to participants' study group?
No. Because of the nature and toxic effects of both treatments it was not possible to blind the assessors to the study group. The assessments were performed by qualified radiation oncologists.
• Were all participants who entered the trial properly accounted for at its conclusion, and how complete was follow-up?
Yes. The article includes a flowchart showing participant flow throughout the trial. Of the 287 participants randomised to the intervention group, 3 did not start treatment, 14 discontinued the radiotherapy and 37 discontinued the temozolomide; 287 were included in the intention-to-treat analysis and 284 in the safety analysis. Of the 286 control group participants, 7 did not start treatment and 19 discontinued treatment; 286 were included in the intention-to-treat analysis and 279 in the safety analysis. Because of the population (palliative patients) and primary outcome of this study (survival), participants who died would not have been considered as lost to follow-up.
• Was intention-to-treat analysis used?
Yes. The article stated that all analyses were conducted on an intention-to-treat basis, which is important as there were a number of reported deviations from the treatment allocation and treatment protocol.
• Did the study have enough participants to minimise the play of chance?
Yes. This study had 80% power at a significance level of 0.05 to detect a 33% increase in median survival (hazard ratio for death, 0.75), assuming that 382 deaths occurred.
Survival and disease progression: A total of 254 (89%) of the intervention group participants and 278 (97%) of the control group participants had died by the 5-year follow-up. The article reports that the hazard ratio for survival among participants in the intervention group compared with those in the control group was 0.6 (95% CI 0.5 to 0.7), which is statistically significant. Table 5.7 shows the overall survival according to group.
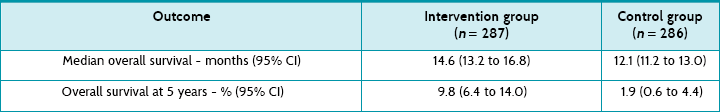
The median difference between the two groups in length of survival (that is, the median survival benefit) was 14.6 – 12.1 = 2.5 months. In the 2005 article it is reported that during the concomitant temozolomide therapy, 7% of participants had a grade 3 (severe adverse effect) or 4 (life-threatening effect) haematological effect. At the 5-year follow up, severe late toxicity was observed in 2 patients in the intervention group (visual disturbance and seizure) and in 1 patient in the control group (fatigue).
How do we use this evidence to inform practice?
You are satisfied with the validity of the study's results and consider the results to be important and, when you compare the demographic and clinical details of Mr Jones to the study participants, they compare well. Therefore, you decide to consider the side-effect profile of temozolomide (which includes fatigue and immunosuppression) before discussing this study with the radiation oncologist for consideration about the potential use of temozolomide in the clinic where you work.
The article does not provide data about the effects of the interventions on participants' quality of life. The article describes collecting data about quality of life as a secondary endpoint but does not report the data in this main article. As this is an important issue to consider, you will search for an article that contains the quality-of-life data.
There are also a number of resource issues related to the use of temozolomide which have to be evaluated in order to ensure that, if the decision is made to use this treatment, there is adequate funding for staff and equipment to support its use. If the clinic decides to implement the use of temozolomide, together with the radiation oncologist, you will make a time to explain the results of the study to Mr and Mrs Jones.
Human movement example
Clinical scenario
As a clinical exercise physiologist working in a health and fitness centre you have received a referral from a general practitioner for a 53-year-old male who underwent a prostatectomy 14 months ago and has been on androgen suppression therapy (AST) since the operation. You undertake a fitness appraisal and determine that the man has an average exercise capacity but has very low strength. He complains of feeling tired and weak and is now not strong enough to complete tasks around the home and activities such as lifting up his grandchildren. You wonder whether there are any benefits of resistance training in the context of AST.
Is resistance training effective at improving muscle strength in a man taking androgen suppression therapy?
Search terms and databases used to find the evidence
Database: PubMed—Clinical Queries (with ‘therapy category’ and ‘narrow scope’ selected).
Search terms: You are aware that the terms ‘androgen suppression therapy’ and ‘androgen deprivation therapy’ are used synonymously, therefore your search terms are: (androgen (suppression OR deprivation) therapy) AND (resistance training)
This search retrieved 5 results. A quick read of the abstracts showed that two were protocol papers, and one used resistance training only and looked at adherence. Another compared the effects of aerobic training, resistance training and a control group (three groups). The other trial (Galvão et al 2010) included 57 patients in a two-group design where the effects of a combined aerobic and resistance training program was compared with a control group. Given that you are considering giving your patient an exercise program that contains aerobic training, you choose the Galvão et al (2010) study to determine whether resistance training is able to improve strength alongside aerobic exercise.
Galvão D, Taaffe DR, Spry N, et al. Combined resistance and aerobic exercise program reverses muscle loss in men undergoing androgen suppression therapy for prostate cancer without bone metastases: a randomised controlled trial. J Clin Oncol 2010;28:340–7.
Structured abstract (adapted from the above)
Study design: Randomised controlled trial.
Setting: Participants were recruited from a hospital in Perth, Western Australia. The exercise training site locations were not supplied.
Participants: 57 participants directly referred by oncologists. Inclusion criteria: histologically documented prostate cancer, minimum prior exposure to AST longer than 2 months, without prostate-specific antigen evidence of disease activity, and anticipated to remain hypogonadal for the subsequent 6 months. Exclusion criteria: bone metastatic disease, musculoskeletal, cardiovascular or neurological disorders that could inhibit them from exercising, inability to walk 400 metres or undertake upper and lower limb exercise, and resistance training in the previous 3 months.
Intervention: Participants were randomly assigned to a program of resistance and aerobic exercise (n = 29) or usual care (n = 28) for 12 weeks. The resistance exercises included chest press, seated row, shoulder press, triceps extension, leg press, leg extension and leg curl, with abdominal crunches also performed. Sessions commenced and concluded with general flexibility exercises. The resistance exercise program was designed to progress from 12- to 6-repetition maximum (RM) for two to four sets per exercise. The aerobic component of the training program included 15 to 20 minutes of cardiovascular exercises (cycling and walking/jogging) at 65–80% maximum heart rate and perceived exertion at 11 to 13 (6 to 20 point, Borg scale). Sessions were conducted in small groups of one to five participants under direct supervision of an exercise physiologist.
Outcomes: Primary outcomes: whole-body and regional lean mass, fat mass and percentage fat. Secondary outcomes: muscle strength and endurance, functional performance, cardiorespiratory capacity balance, falls self-efficacy, blood biomarkers and quality of life.
Follow-up period: 12 weeks. Assessments were administered at baseline and at the end of the 12-week intervention.
Main results: Compared with usual care, participants in the intervention group had significant increases in lean mass for total body, upper limb and lower limb, increased muscle strength (chest press, seated row, leg press and leg extension) and 6-metre walk time, improvement in some aspects of quality of life (general health and less fatigue), and decreased levels of C-reactive protein.
Conclusion: Resistance exercise in combination with aerobic training improved muscle mass, strength, physical function and balance in hypogonadal men.
Is the evidence likely to be biased?
• Was the assignment of participants to groups randomised?
Yes, Participants were randomly allocated using a computer random-assignment program.
• Was the allocation sequence concealed?
Yes. The allocation sequence was concealed from the project coordinator and exercise physiologist involved in assigning participants to groups.
• Were the groups similar at the baseline or start of the trial?
Yes. There were no significant differences between groups at baseline in regard to demographics, clinical characteristics or primary and secondary outcome measures.
• Were participants blind to which study group they were in?
No. For this trial it was not possible for participants to be blinded to group allocation.
• Were the health professionals who provided the intervention blind to participants' study group?
No. For this trial it was not possible for the exercise physiologists who provided the intervention to be blinded to group allocation.
• Were the assessors blind to participants' study group?
Cannot tell. There is no mention in the article of whether the assessors were blinded to group allocation, so it is assumed that they were not.
• Were all participants who entered the trial properly accounted for at its conclusion, and how complete was follow-up?
Yes. The study had a follow-up rate of 96.5%. One participant in the usual care group was lost to follow-up and one in the intervention group discontinued the intervention.
• Was intention-to-treat analysis used?
Yes. The authors explicitly state that intention-to-treat analysis was used.
• Did the study have enough participants to minimise the play of chance?
Yes. A power calculation was performed with change in whole-body lean mass used as the primary outcome measure. This found that 25 participants per group were needed to detect a mean difference in change of 1 kg (SD = 1.25) at the end of the 12 weeks (80% power and alpha = 0.05). The study recruited a total of 57 participants.
The outcome measures relevant to the clinical question are shown in Table 5.8. In the article, the authors provide the mean scores (and their standard deviation) at baseline and at 12 weeks for both groups, along with the adjusted group difference in mean change over the 12 weeks and the 95% confidence interval for the mean change. For all of the strength outcome measures shown in the table, the between-group differences were statistically significant (note that none of the confidence intervals include the no effect value of 0).
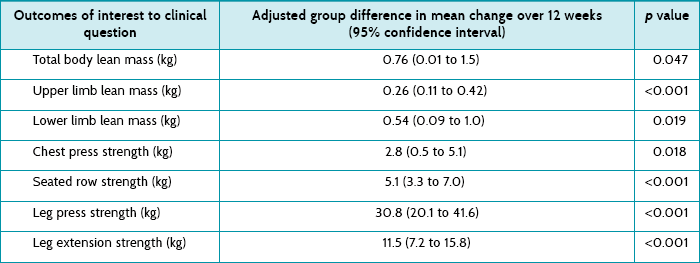
How might we use this evidence to inform practice?
After critically appraising the internal validity of this trial, you believe that there is only a low likelihood that these results are biased. There was a statistically significant between-group difference, in favour of the intervention group, for the strength outcome measures. You discuss the results of the trial with your patient, the benefits and likely increase that he may experience in lean mass and strength and explain what the exercise program would involve. He decides that he would like to try it.
Pharmacy example
Clinical scenario
You are the professional services programs manager for a large group of community pharmacies. Your role is to oversee the programs offered by the pharmacies, including the development and implementation of new professional programs and services for delivery across the group. There are 20 pharmacies in the group, including medical centre, strip and shopping centre pharmacies in metropolitan and regional locations. Your pharmacies are very actively engaged with the wider community, currently offering professional services in the area of asthma, sleep, healthy eating (weight), wound care and baby care. Many of these services involve the integration of other health professionals within and outside of the pharmacy.
Although these current services are working well, patients with diabetes are becoming a key focus of the pharmacies you work with. Many of them already offer a variety of diabetes products, and links with National Diabetes Services and other local health professionals. Diabetes is a significant health issue in the wider community and the majority of the patients seen in the pharmacies have type 2 diabetes. The pharmacists within the group would like to provide a more structured and focused pharmacy-based service for patients with type 2 diabetes. They think this service should be centred on the monthly visits that patients make to collect their regular prescriptions, and strongly believe that this service would improve patients' clinical care. They have approached you as the professional services programs manager to investigate whether this type of program would be feasible for the group. You search the literature for evidence to support their assertions.
In adults with type 2 diabetes, can a focused, community-pharmacy-delivered education and management service improve clinical outcomes?
Search terms and databases used to find the evidence
Database: PubMed—Clinical Queries (with ‘therapy category’ and ‘narrow scope’ selected).
Search terms: community AND pharmac* AND education AND diabetes
This search resulted in 15 articles. Only two are relevant to your question, and you choose the one that most closely matches it.
Krass I, Armour C, Mitchell B, et al. The Pharmacy Diabetes Care Program: assessment of a community pharmacy diabetes service model in Australia. Diab Med 2007;24:677–83.
Structured abstract (adapted from the above)
Study design: Randomised controlled trial.
Setting: 56 randomly selected urban and rural Australian community pharmacies (28 intervention, 28 control).
Participants: 289 patients with type 2 diabetes (149 in intervention group and 140 in control group). Inclusion criteria were patients who had (1) HbA1c ≥7.5% and were taking at least one oral glucose-lowering medication or insulin; or (2) HbA1c ≥7.0% and were taking at least one oral glucose-lowering medication or insulin and who were on at least one anti-hypertensive, angina or lipid-lowering drug.
Intervention: Pharmacy-delivered diabetes service which comprised an ongoing cycle of assessment, management and review, provided at regular intervals (5 times) over 6 months. Services included support for self-monitoring of blood glucose, education, adherence support and reminders of checks for diabetes complications. Control pharmacists provided usual care (that is, no specialised diabetes service).
Outcomes: Primary outcome was change in HbA1c (%); secondary outcomes were: blood pressure, lipid measures, body mass index and a quality-of-life measure (EQ-5D assessment tool).
Main results: There was a significant improvement in glycaemic control in the intervention group compared with the control group: a reduction in HbA1c in intervention group participants of –0.97% (95% CI –0.8 to –1.14) compared with –0.27% (95% CI –0.15 to –0.39) in control group participants. Improvements in blood pressure control and quality-of-life indicators were also seen in the intervention group.
Conclusion: The pharmacy-delivered diabetes service resulted in significant improvements in clinical and humanistic outcomes for type 2 diabetes patients.
Is the evidence likely to be biased?
• Was the assignment of participants to groups randomised?
Yes. Pharmacies were randomly allocated to either the intervention or the control group (that is, this was a cluster trial), but no further details are given about the process.
• Was the allocation sequence concealed?
Cannot tell. There is no mention in the article of concealed allocation.
• Were the groups similar at the baseline or start of the trial?
Yes, for some characteristics. The article reports that the two groups were similar in terms of only very basic pharmacy (cluster) characteristics (number of staff, location), pharmacists (age, gender, position in pharmacy) and most participant characteristics (demographics, diabetes history and most clinical characteristics). However, there was a baseline difference in the primary outcome of HbA1c (8.9% in intervention vs 8.3% in control group), which the authors adjusted for in the analyses.
• Were participants blind to which study group they were in?
No. For this intervention, it was not possible to blind participants to group allocation.
• Were the health professionals who provided the intervention blind to participants' study group?
No. For this intervention, it was not possible to blind the pharmacies to group allocation.
• Were the assessors blind to participants' study group?
No for some measures, and cannot tell for other measures. Data for the outcomes of HbA1c, blood pressure and lipids were obtained from participants' GPs. It is not described whether participants' GPs knew which group participants were in, but it is possible that at least some of them would have been aware of group allocation (if for no other reason than that they would have been involved in increased management of participants in the intervention group). For the primary outcome measure of HbA1c, the uncertainty regarding blinding is not of concern as it is an objective outcome measure. However, for other measures such as quality of life, assessment was not blinded.
• Were all participants who entered the trial properly accounted for at its conclusion, and how complete was follow-up?
Yes and no. The article provides a participant flowchart showing that 19 (12%) participants withdrew from the control group (n = 159 at baseline) and 27 (15%) from the intervention group (n = 176 at baseline). In addition, the article reports that final data from participants' GPs were unable to be obtained for 33 intervention and 39 control group participants (some who withdrew and some who did not), giving loss to follow-up of approximately 22%. However, as this is a cluster trial it would also have been useful to have information about whether any clusters withdrew, but this information is not provided.
• Was intention-to-treat analysis used?
No. The article does not mention whether intention-to-treat analysis was conducted. There is no mention of any attempt to handle the data that were missing for some participants, and therefore it is assumed that intention-to-treat analysis was not done.
• Did the study have enough participants to minimise the play of chance?
No. The article stated that to detect a 0.5% reduction in HbA1c in the intervention group compared with the control group, with a power of 90% at the 5% significance level, at least 180 patients were required in each group. The study collected baseline data for 159 control group participants and 176 intervention group participants, which is less than the required amount.
Table 5.9 shows the outcomes for which there was a statistically significant between-group difference in the change scores at follow-up. The main benefit of the study results are the improvement of HbA1c scores by a mean change of 0.7%, which was statistically significant (unfortunately, confidence intervals for this cannot be calculated from the data the authors provide). We must now ask ourselves if this is large enough to be considered clinically significant and therefore enough to change practice. Questions about clinical significance are subjective. Ideally a ‘minimally important clinical difference’ in HbA1c would be set before the study was conducted (in fact, as a hypothesis). Since this did not happen, we must use our judgment. A reduction of a patient's HbA1c by between 0.5% and 1% is associated with a better outcome from diabetes, and so this does appear to be an important enough improvement. There were no significant between-group differences for body mass index, blood pressure, total cholesterol, triglycerides or the utility score of the quality-of-life measure.
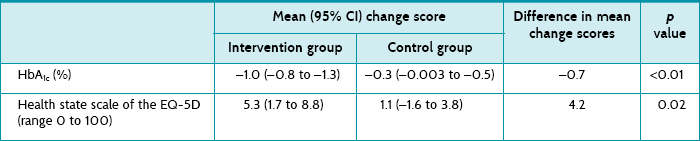
How might we use this evidence to inform practice?
First of all, do we believe the results of the study? Our critical appraisal suggests that there are some threats to the internal validity of the study. Again, we have to exercise judgment to decide whether the study was robust enough to change clinical practice according to its results. Added to this concern is the issue of whether the right outcome measure was measured. We must remember that HbA1c is a ‘surrogate measure’; it is not a direct measure of the patient's health status. Instead we are reliant on a series of assumptions about a lower HbA1c leading to better health, something that is actually somewhat controversial. Do we have sufficient well-established evidence that high HbA1c is a cause of diabetes complications, such that reducing it means that complications will be inevitably reduced? Not everyone believes that this is the case, and you are aware that some recent trials have found this may not be the case.
The intervention that was evaluated is a complex intervention that had lots of components. It is not clear which part of the intervention was effective. Was it the self-monitoring, the education, adherence support or reminders for diabetes checks, and in what combination? If we decided to implement this intervention, we would have to try to replicate the exact same intervention as in the study (which is not clearly outlined in the article). You decide that there is sufficient concern about the surrogate nature of the outcome measure (HbA1c), which of the intervention components was effective and the risk of bias in the results that you will await further research before asking for additional funds to provide this extra service in your group of pharmacies.