Chapter 21 Fluid and Diuretic Therapy in Heart Failure
Congestive heart failure (CHF) is a clinical syndrome characterized by cardiac dysfunction, abnormal hemodynamics, neurohormonal activation, release of cytokines, and renal retention of sodium and water. A cardiac or vascular lesion that limits cardiac output and decreases arterial blood pressure (ABP) triggers heart failure, leading to a homeostatic state that is characterized by vasoconstriction and renal sodium retention. The stereotypical compensatory responses to heart failure support ABP but also promote a maladaptive state that leads to substantial morbidity and mortality.
Advanced CHF, as well as the therapy of this syndrome, often is associated with alterations in renal function and a variety of fluid, electrolyte, and serum biochemical abnormalities. Some of these disturbances are mild and seemingly well tolerated, but others, such as hyponatremia and acute renal failure, indicate severe circulatory dysfunction and a need for urgent therapy.75 There are circumstances in which cardiac patients actually require fluid therapy to maintain optimal ventricular filling and prevent deterioration of renal function. However, it is more common for fluid therapy to produce edema or effusions in a previously compensated cardiac patient. Safe restoration of fluid and electrolyte balance in the patient with cardiovascular disease is challenging. To orchestrate such treatment, the clinician must appreciate the pathophysiology of heart failure and the compensatory changes that develop. This chapter addresses some of the clinically relevant pathophysiologic and therapeutic aspects of heart failure.
Much of our understanding of hemodynamics, renal function, and neurohumoral activity in heart failure stems from many experimental studies in dogs and from a limited number of clinical investigations of dogs and cats with spontaneous heart disease. However, studies of fluid therapy in spontaneous CHF in dogs and cats are largely unavailable. Accordingly, the recommendations offered here represent our interpretation of relevant animal studies and personal experience with the treatment of dogs and cats with CHF.
The normal circulation
The central circulation is regulated largely by a need to maintain plasma volume, mean ABP, and tissue perfusion. Of prime importance is the maintenance of normal effective plasma volume and ABP in the central circulation.141,150 These two variables depend on cardiac output, systemic vascular impedance, and renal regulation of sodium and water excretion. The reflexes that control the circulation have evolved so that blood pressure and plasma volume are maintained within a narrow range even in the presence of sudden physiologic stresses, such as exercise, hypotension, or hemorrhage. Blood pressure and plasma volume are monitored by different mechanoreceptors and osmoreceptors located in the arteries, veins, heart, kidney, and central nervous system. Ultimately, two factors—cardiac output and systemic vascular resistance (more precisely, vascular resistance and arterial impedance)—determine ABP (Figure 21-1). A change in either one of these two variables causes a parallel change in blood pressure. Numerous physiologic variables can affect cardiac output and vascular impedance (Box 21-1), and many of these factors are perturbed in CHF. Of particular relevance in this chapter are determinants of plasma volume in health and disease (Box 21-2). Plasma volume is a major contributor to venous pressure and cardiac filling. The serum sodium concentration, as described more fully in Chapter 3, plays a central role in determining plasma volume. Renal tubular activity, vascular dynamics, hormones, and other vasoactive factors regulate sodium balance. Abnormalities of sodium excretion are pivotal to the development of CHF.
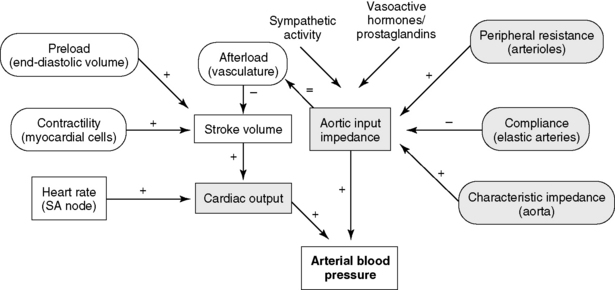
Figure 21-1 Control of arterial blood pressure. Arterial blood pressure is a function of the cardiac output and the arterial impedance. Contractility, preload, and afterload determine the stroke volume, which multiplied by the heart rate yields the cardiac output. Changes in arterioles, elastic arteries, and the aorta all influence the aortic input impedance (afterload). (+), Increases in the parameter increase aortic input impedance, stroke volume, cardiac output, or arterial blood pressure; (−), increases in the parameter decrease aortic input impedance or stroke volume.
(Modified from de Morais HSA. Pathophysiology of heart failure and clinical evaluation of heart function. In: Ettinger SJ, Feldman EC, editors. Textbook of veterinary internal medicine, 5th ed. Philadelphia: WB Saunders, 2000.)
Box 21-2 Factors Regulating Plasma Volume in Heart Failure
Plasma Protein (albumin)Drug Therapy
Diuretics
Loop diuretics (furosemide, bumetanide, torsemide)
Thiazide diuretics (hydrochlorothiazide)
Potassium-sparing diuretics (triamterene, amiloride)
Spironolactone (blocks renal effects of aldosterone)
Carbonic anhydrase inhibitors (acetazolamide)
Angiotensin-converting enzyme inhibitors (captopril, enalapril, benazepril, lisinopril, ramipril)Digitalis glycosides, pimobendan, and other cardiotonic drugsVasodilator drugs (hydralazine, nitrates, angiotensin-converting enzyme inhibitors)
Attention also must be directed to the microcirculation and factors controlling fluid movement across capillaries. Tissue perfusion is crucial for organ functions such as the formation of urine, muscle contraction, and exchange of oxygen and carbon dioxide. Assuming the maintenance of adequate mean ABP, regional vascular resistance largely governs tissue perfusion across the arterial side of the microcirculation. Vascular resistance for any regional circulation is the sum of structural, autonomic, hormonal, and local vasoactive factors (see Box 21-1). Conversely, plasma volume and venous pressure exert the greatest effect at the venous end of the capillary. The interplay of hydrostatic pressures, oncotic pressures, capillary permeability, and lymphatic function determines whether the interstitium and serous body cavities accumulate or remain free of excess solute and water.75,156,165 The effect of these so-called Starling forces on fluid dynamics is summarized in Figure 21-2.
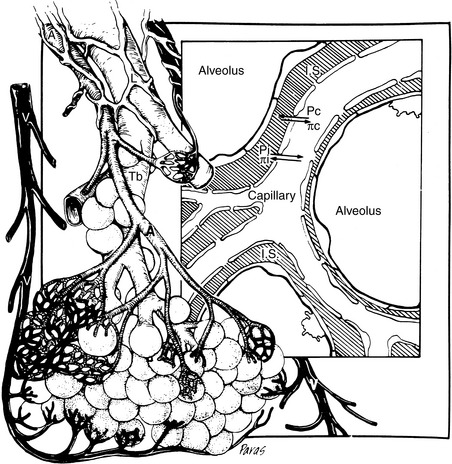
Figure 21-2 Microcirculation of the respiratory unit. The arteriole branches into the capillary plexus surrounding alveoli. Starling’s forces controlling fluid movement into or out of the capillary or interstitial space (IS) are indicated. Capillary hydrostatic pressure in the lung must generally exceed 20 to 25 mm Hg before edema develops. Chronically, even higher hydrostatic pressures can be tolerated before edema develops. This is explained by the increased lymphatic drainage of the interstitium that develops in chronic edematous states. Pc, Capillary hydrostatic pressure, which forces fluid into the interstitium; pc, capillary colloid osmotic pressure principally because of albumin, which causes fluid to be retained within the capillary; Pi, interstitial hydrostatic pressure, which is negative in the lung; pi, interstitial colloid osmotic pressure, which is controlled by pulmonary lymphatics and maintains the interstitium relatively free of albumin; A, arteriole; V, venule; L, lymphatic vessel; Tb, terminal bronchiole.
(From Ware WW, Bonagura JD. Pulmonary edema. In: Fox PR, editor. Canine and feline cardiology. Philadelphia: WB Saunders, 1999: 252. Medical illustration by Felicia Paras.)
The circulation in heart failure
Heart failure is characterized clinically by hemodynamic abnormalities triggered by cardiac dysfunction.81 The causes of heart failure include numerous structural and functional disorders of the cardiac valves, myocardium, pericardium, and blood vessels, as well as sustained cardiac arrhythmias (Box 21-3). In response to impaired cardiac output, potent homeostatic mechanisms are activated that preserve perfusion of the brain and heart but at the expense of less vital regional circulations. Preservation of blood pressure mandates dramatic alterations in neural, hormonal, and cardiovascular function and structure. These adaptations (summarized in Box 21-4) include (1) activation of the sympathetic nervous system23,49,178 and release of hormones,46,121 (2) increased systemic vascular resistance47,48 and impedance,39 (3) reduction of autonomic reflex activity,81,186 and (4) cardiac dilatation and hypertrophy that together with myocardial interstitial changes are collectively referred to as “cardiac remodeling.54,76,113” Heart failure also alters renal function122,186 and enhances reabsorption of sodium and water.16,179 In combination, these potent control systems are capable of maintaining normal ABP in all but the most severe cases of cardiac failure.
Box 21-3 Causes of Heart Failure
Myocardial Diseases
Box 21-4 Neurohormonal, Renal, and Cardiovascular Activities in Congestive Heart Failure
Hormonal or Autocrine
Hemodynamic abnormalities in the central circulation and microcirculation in CHF (Box 21-5) can be traced to both decreased cardiac performance and renal retention of sodium and water.122,141,152 Decreased cardiac output, valvular insufficiency, impaired myocardial relaxation, and reduced ventricular compliance increase ventricular end-diastolic pressure, which is transmitted back to the venous and capillary beds (“backward” failure). Higher venous and capillary pressures are augmented by renal fluid retention and expansion of the plasma volume. Renal sodium and water retention as a consequence of reduced cardiac output often is described in the medical literature as “forward” heart failure.141 Forward failure, in this regard, does not refer to clinical signs of low cardiac output but instead describes the renal responses triggered by low cardiac output. Forward failure is a critical factor in the development of edema and effusions in right-sided and biventricular heart failure. These concepts and some of the factors responsible for increased venous and capillary pressures are shown in Figure 21-3. The important role of the kidney in the pathogenesis of edema and effusions is discussed in the section on Renal Function in Heart Failure.
Box 21-5 Hemodynamic Consequences of Congestive Heart Failure
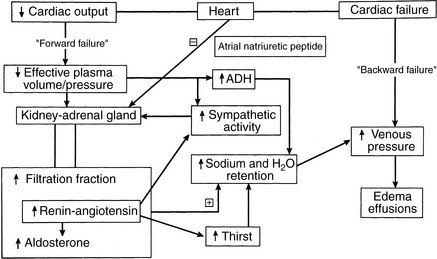
Figure 21-3 Prominent mechanisms responsible for fluid accumulation in heart failure. The combined effects of abnormally high venous pressure and renal retention of sodium and water can explain the development of pulmonary edema, subcutaneous edema, or the transudative effusions in body cavities. Ventricular systolic or diastolic failure increases venous pressure behind the failing ventricle (“backward” failure). This may be the predominant mechanism of edema formation in acute left-sided heart failure. In contrast, chronic heart failure, especially when right-sided or biventricular in nature, is characterized by avid sodium retention. Although atrial distention causes the release of atrial natriuretic peptide (ANP), the effects of sympathetic activity, angiotensin II, aldosterone, vasopressin (ADH), and local vasoconstrictor factors dominate, leading to vasoconstriction in systemic vessels and increased sodium and water reabsorption in the renal tubules. This is a simplified view because other local and systemic factors can be involved.
(Modified from Bonagura JD. Fluid management of the cardiac patient. Vet Clin North Am Small Anim Pract 1982;12:503.)
High venous pressures and increased ventricular end-diastolic pressure enhance cardiac filling and allow the ventricle to generate a greater contractile response. As shown in Figure 21-4, ventricular stroke volume is directly related to ventricular filling pressure.45,163 High venous pressure also maintains cardiac filling when ventricular compliance is decreased, as in hypertrophic cardiomyopathy, pericardial disease, or severe ventricular dilatation54 (see Figure 21-4, bottom). The clinical relevance of this relationship becomes obvious when the edematous patient is treated with diuretics and venous pressures, ventricular filling, and cardiac output decline, causing systemic hypotension or prerenal azotemia.
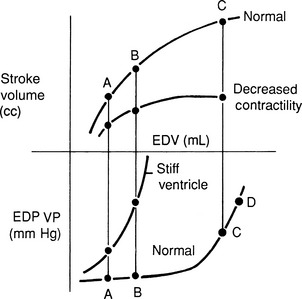
Figure 21-4 Ventricular function curves in heart failure. Ventricular function curves demonstrate the potential relationships between venous pressure (a determinant of ventricular filling and end-diastolic volume), ventricular compliance or distensibility (which determines the venous and atrial pressures required to fill the ventricle), and stroke volume (determined by ventricular end-diastolic volume, ventricular afterload, and myocardial contractility). The top of the graph demonstrates ventricular systolic function, and the lower curves demonstrate ventricular filling dynamics.
Top, When inotropic (“contractile”) state and afterload are held constant, the ventricular stroke volume depends on cardiac filling (preload), although this relationship is depressed in patients with myocardial failure. Patients treated with excessive dosages of diuretics may develop inadequate ventricular filling, leading to decreased stroke volume and cardiac output and causing prerenal azotemia. Reduction of diuretic dosage or fluid therapy is generally required to reestablish cardiac output.
Bottom, Ventricular distensibility—the tangent of any point on the diastolic pressure-volume curve—depends on the amount of ventricular hypertrophy, myocardial fibrosis, and the volume of the ventricle. Animals with stiff ventricles resulting from ventricular hypertrophy or myocardial fibrosis require high ventricular filling pressures and are poorly tolerant of fluid infusions. Note that increased cardiac filling can progress only at disproportionately higher venous pressures, a situation that predisposes to pulmonary edema. Recognize that even dilated ventricles can develop diastolic dysfunction (bottom right). Once the grossly dilated ventricle reaches a certain point, distensibility decreases. Compare the slope at the extreme right of this diastolic filling curve with that of the smaller, hypertrophied ventricle. The benefit of diuretic therapy in this setting can be appreciated because even small reductions in plasma volume and preload may permit the ventricle to fill at substantially lower venous pressures.
The term congestive heart failure implies a situation of increased venous and capillary hydrostatic pressures, increased transudation of fluid across capillary walls, and net accumulation of fluid in the interstitial compartment (i.e., edema) or serous body cavities (i.e., effusion). A safety margin normally prevents this accumulation of fluid, and venous pressures must increase substantially (usually to two or three times above the normal upper limit) before edema develops.60,157,165,180 Development of pulmonary edema in the dog usually requires left atrial pressure to increase acutely to more than 20 mm Hg.60 Substantial increases in lymphatic drainage permit much higher pressure to be tolerated chronically.16,34 In addition to increased venous pressures, hypoalbuminemia can contribute to edema formation.60,183 As a consequence of variable lymphatic drainage and other factors, such as capillary permeability and compartment compliance, edema is not uniformly distributed in the tissues.180 This nonuniform distribution is evident clinically inasmuch as acute cardiogenic pulmonary edema in the dog is most prominent in perihilar and in lung lobes on the rightside, although it can accumulate in cranial and ventral regions at the same time.
The edema of CHF develops predominantly in the capillary beds drained by the failing side of the heart. This finding is pertinent because CHF is classified clinically as left-sided, right-sided, or biventricular. Increased pulmonary venous and capillary hydrostatic pressures cause pulmonary edema (see Figure 21-2), the cardinal finding of left-sided CHF. Right-sided heart failure increases systemic venous pressures leading to jugular venous distention or pulsation, hepatic congestion, ascites, or (infrequently in small animals) subcutaneous edema. Increased systemic venous pressure even may contribute to pulmonary edema formation.103
Pleural effusions develop as a result of left-sided, right-sided, or, most often, biventricular failure. This finding can be explained by the dual venous drainage of the pleural surfaces (i.e., parietal drainage is systemic, whereas visceral drainage is pulmonary). Although veterinary textbooks usually attribute pleural effusion to isolated right-sided CHF, this is not common in human patients. Pleural effusion correlates better with pulmonary capillary wedge pressure than with right atrial pressure.183 Similarly, pleural effusions in small animals most often indicate biventricular CHF. Although pleural effusion does occur in some dogs and cats with predominantly right-sided cardiac disease (e.g., pulmonic stenosis, tricuspid malformation), ascites is more common in dogs. Clinically significant pleural effusions are rare in animals with isolated right ventricular failure caused by heartworm-induced pulmonary hypertension.15,169 Conversely, pleural effusions are common when end-stage CHF develops in dogs with severe mitral regurgitation, pulmonary hypertension, and secondary right ventricular dysfunction or in cats with any form of severe cardiomyopathy. Pleural effusion may become chylous in nature in those with advanced CHF.
The relative contribution of renal sodium retention in CHF probably depends on the type and acuteness of heart failure. The development of ascites, pleural effusion, or subcutaneous edema in right-sided or biventricular cardiac failure is accompanied by avid renal sodium retention (see Renal Function in Heart Failure section). Dramatic weight loss, sometimes exceeding 5 kg in giant-breed dogs, may be observed after successful diuresis. This degree of weight loss after diuretic therapy is uncommon in isolated left-sided failure. Thus successful therapy of right-sided CHF depends in the short term on initiation of a brisk diuresis or paracentesis. Long-term management hinges on improving cardiac function, reducing neurohormonal activation, and overcoming the potent sodium-retaining effects of forward cardiac failure.
In contrast to right-sided or biventricular CHF, severe left-sided heart failure can develop without substantial sodium retention or weight gain.69 Two common examples in veterinary medicine can be cited. The first example is rupture of a mitral chorda tendinea in an older dog with previously stable mitral regurgitation. The sudden increase in mitral regurgitant volume increases mean left atrial and pulmonary capillary pressures, leading to peracute pulmonary edema. The second example is a cat with hypertrophic cardiomyopathy and a noncompliant left ventricle (see Figure 21-4, bottom left curve). It is not uncommon for severe pulmonary edema to follow a bout of protracted tachycardia (e.g., stress). Development of pulmonary edema in these situations can be explained by acute deterioration of left ventricular systolic or diastolic performance that rapidly increases left atrial and pulmonary venous pressure. Although diuresis is a critical treatment in this situation, short-term success may hinge on therapy that reduces mitral regurgitant fraction (i.e., afterload reduction).
Another issue of relevance to CHF and fluid therapy of the cardiac patient is the relative size of the vascular compartments. The vascular compliance of the pulmonary circulation is much smaller than that of the systemic circulation, and sudden expansion of the plasma volume usually increases pulmonary venous pressure more than systemic venous pressure. This is particularly true in the patient with left-sided heart disease and explains why some dogs and cats develop pulmonary edema after intravenous administration of a so-called maintenance volume of crystalloid solution. Furthermore, central venous pressure (CVP) cannot be used to gauge the effect of intravenous fluid therapy on left-sided cardiac filling pressures, especially in the setting of isolated left-sided CHF.141 Owing to differences in vascular compliance and cardiac function, left-sided filling pressures may increase much more rapidly than CVP, though both increase simultaneously.
Renal function in heart failure
Remarkably, the kidney often is able to maintain glomerular filtration in the setting of decreased blood pressure or cardiac output. Decreases in renal perfusion are countered by dilatation of the afferent arteriole mediated by the release of prostaglandin E2, and constriction of the efferent arteriole primarily by angiotensin II. Efferent arteriolar constriction also is augmented by arginine vasopressin (antidiuretic hormone [ADH]) and norepinephrine.122 These microvascular responses increase glomerular filtration pressure, increase filtration fraction, and maintain glomerular filtration in the setting of reduced renal blood flow (see Chapter 2).116,138,159 However, considering normal renal function demands approximately 20% of a normal cardiac output, it is not surprising to identify azotemia in advanced CHF, especially during aggressive diuretic therapy. Progressive renal failure is common in dogs and cats with CHF, and the treatment of patients with both intrinsic renal disease and heart failure is especially difficult. This situation also occurs in human patients in whom worsening of renal function is associated with a poorer prognosis and higher mortality and often requires dialysis for control of both problems.57 Neurohormones and cytokines, angiotensin in particular, are considered central to the progression of renal disease in heart failure.80 Aggressive therapy of heart failure may slow progression of renal disease in humans.89
The renal response to decreased cardiac output is central to the pathogenesis of edema and effusions in heart failure. Studies of induced right-sided heart failure and spontaneous CHF in dogs have demonstrated avid retention of administered salt loads.9,84 Numerous mechanisms have been identified for persistent sodium retention in CHF (see Figure 21-3). These alterations include redistribution of renal blood flow,9,131 enhanced tubular sodium reabsorption,8,91,102 release of prostaglandins,36,38,111 greater renal sympathetic nerve activity,* increased renal interstitial pressure,58,101 and increased hormonal activity. The last includes increases in vasopressin (ADH),16,136 angiotensin II, and aldosterone† (see Box 21-4). Presumably, these mechanisms also operate in animals with spontaneous heart failure.134
Particular emphasis has been placed on the increased concentrations of renin, angiotensin II, and aldosterone found in patients with CHF.121 There are a number of triggers for the release of renin in the cardiac patient.113 One mechanism is the stimulation of renal ß-adrenergic receptors by sympathetic efferent traffic activated in response to hypotension. Renin also is released in response to reduced renal blood flow related to heart failure or volume depletion caused by diuretic therapy of CHF.182 Severe sodium restriction, especially in dogs with signs of heart disease but without overt CHF, can lead to renin release.127 Clinically, the effects of angiotensin II and aldosterone can be mitigated in part by drugs that inhibit formation of angiotensin II (angiotensin-converting enzyme [ACE] inhibitors such as enalapril and benazepril) or drugs that block the AT-1 receptor of angiotensin II, such as losartan and candesartan.
Other factors promote renal fluid retention in CHF. Changes in intrarenal blood flow can lead to redistribution of flow to the salt-conserving juxtamedullary nephrons.16,91,93 Increased filtration fraction maintains the glomerular filtration rate (GFR) but predisposes to renal tubular reabsorption of water (see Chapter 2). Arginine vasopressin (ADH) also plays a role. In CHF, increases in plasma ADH concentration probably represent nonosmotic release in response to low ABP.122 Increased thirst (mediated by angiotensin II), when combined with increases in ADH, can contribute to free-water retention and hyponatremia.35,99,120 Endothelin is another hormone released from endothelial cells in CHF.132,176 This hormone reduces renal blood flow, the GFR, and urinary sodium excretion.98,122 The sequence in which these mechanisms are activated varies with the type and severity of heart failure.46,141 However, it is clear that with deterioration in cardiac function, sodium- and water-retaining mechanisms are exacerbated, and further expansion of the plasma volume occurs. Blunting the renal response generally requires appropriate medical treatment of CHF, progressive restriction of dietary sodium, and administration of diuretics.
In CHF, the vasoconstrictive and sodium-retaining mechanisms overwhelm local and systemic vasodilator and natriuretic systems. Distention of the atria and ventricles signals release of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP). These peptides of cardiac origin stimulate formation of cyclic GMP, leading to diuresis, vasodilatation, and improved ventricular relaxation.107,113 Although increased circulating concentrations of ANP and BNP can be measured in dogs with experimentally induced and spontaneous CHF,* it also has been shown that the renal response to these hormones is blunted or antagonized. 21,104,135 If dogs or people with CHF are treated with pharmacologic doses of ANP, however, or if the degradation of ANP is reduced by administration of a neutral endopeptidase inhibitor, diuresis may follow.104,110 Other vascular-modulating factors, such as the vasodilator nitric oxide, are more difficult to assess in CHF, but metabolites of this endothelial-derived substance reportedly are decreased in some dogs with mitral regurgitation.126
Cardiovascular drugs and renal function
Effects of diuretics on renal function
Diuretics used in management of CHF prevent reabsorption of solute and water, leading to increased urine flow. Diuretics are essential to both the short- and long-term management of CHF. While there are no blinded clinical trials evaluating the efficacy of furosemide, experienced clinicians repeatedly observe the short-term benefit of furosemide diuresis in veterinary patients with life-threatening pulmonary edema. In a 3-week study of dogs with CHF, pulmonary wedge pressure, an estimate of left atrial pressure, declined within hours after initiating furosemide and remained below baseline after 21 days of therapy.71 Despite the lack of study in terms of chronic efficacy, diuretics are a mainstay for acute and chronic treatment of CHF in dogs, cats, and humans.70 They reduce preload by decreasing cardiac filling (venous) pressures and chronically prevent the excessive dietary sodium retention characteristic of CHF,9 allowing for a new steady state of sodium balance to develop.70 Diuretics may also reduce left ventricular afterload by reducing sodium loading and vascular resistance in arterioles.
The clinical pharmacology of these drugs and effects on renal function (Tables 21-1 and 21-2) are relevant to understanding their effectiveness and limitations. All of the commonly used diuretics, except spironolactone, are delivered by renal blood flow and secreted as organic acids into the proximal tubule. Circulatory failure, reduced renal blood flow, administration of nonsteroidal antiinflammatory drugs (NSAIDs), or primary renal failure may reduce the renal delivery of a diuretic. In the case of renal failure, endogenous organic acids can compete with furosemide for transport across the proximal nephron. Once secreted into the filtrate, a diuretic inhibits salt and water transport via a specific mechanism and at relatively specific sites along the nephron.72,112,142
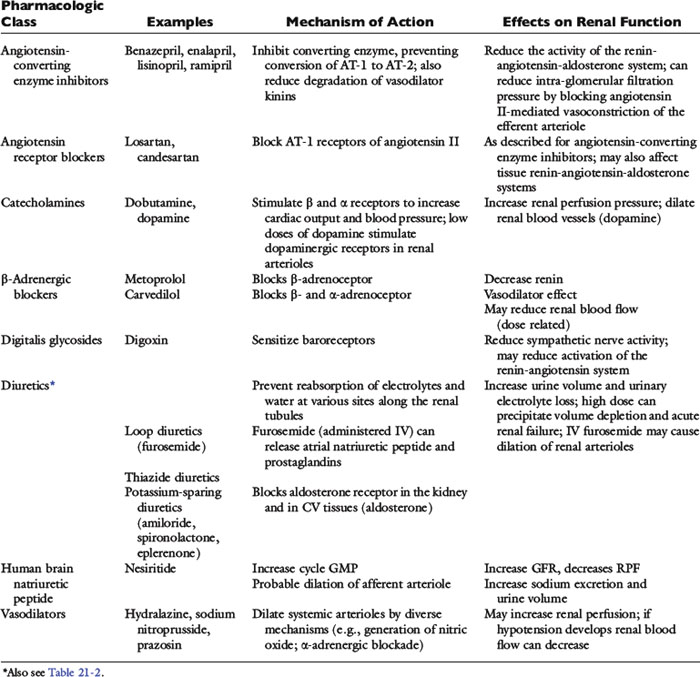
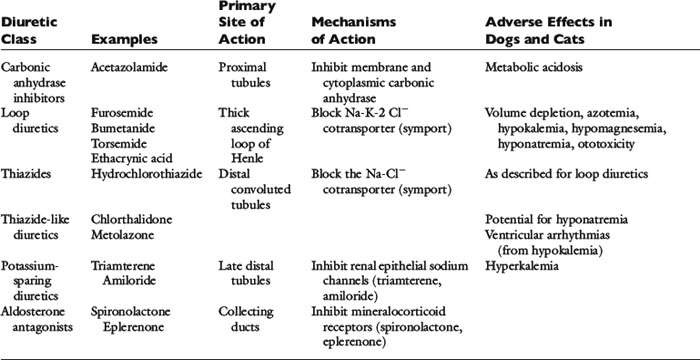
Figure 21-5 demonstrates the general sites of action of the commonly used diuretics. The importance of understanding these details can be illustrated by two examples. First, the effectiveness of a diuretic depends on the ability of cells distal to the site of diuretic action to reabsorb sodium and water. Initially in CHF, loop diuretics, which act on the thick portion of Henle’s loop, are highly effective. However, in severe chronic CHF, the more distal tubular cells can increase their reabsorption of sodium and water and overcome the effects of the diuretic.141 This problem can be counteracted with additional treatment such as the combination of hydrochlorothiazide and spironolactone, which act more in the distal nephron. This type of sequential nephron blockade can induce a marked diuresis in some dogs but not without risk of volume depletion and acute renal failure. A second example pertains to the adverse effects of diuretics. Loop diuretics increase the delivery of sodium to cells of the late distal convoluted tubules and collecting ducts. At those sites, sodium is reabsorbed in exchange for potassium (under the influence of aldosterone) or hydrogen ions that are secreted.72 These ion exchange mechanisms have the potential to cause hypokalemia and metabolic alkalosis, especially with high doses or chronic therapy.
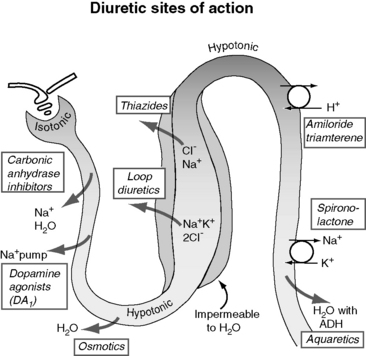
Figure 21-5 Renal effects of diuretics. Loop diuretics such as furosemide are most commonly used in treatment of congestive heart failure (CHF). Spironolactone works in the distal nephron and is therefore a relatively weak diuretic; however, it also demonstrates cardiac-protecting properties. In the concept of “sequential nephron blockade,” a loop diuretic would be combined with a thiazide and spironolactone or eplerenone to prevent solute and water reabsorption at multiple levels.
(From Opie LH, Gersch BJ. Drugs for the heart, 6th ed. Philadelphia: WB Saunders, 2005.)
The carbonic anhydrase inhibitors, such as acetazolamide, act on the proximal tubule by inhibiting bicarbonate reabsorption. These diuretics are limited in effectiveness because they induce metabolic acidosis, and the loop of Henle and distal nephron can reabsorb much of the increased salt and water that is delivered to these segments. Carbonic anhydrase inhibitors are not used in treating CHF.
Furosemide, ethacrynic acid, bumetanide, and torsemide exert their effects on the ascending limb of Henle’s loop.112,142,170 These so-called loop diuretics block the Na+-K+-2 Cl− cotransporter (symport) and prevent the active transport across the tubular lumen of two chloride ions, one sodium ion, and one potassium ion. Loop diuretics are potent with a good dose response, whereas higher dosages result in greater sodium loss (“high ceiling”). This is related to the high capacity for reabsorbing filtrate at this site (normally approximately 25% of the filtrate is reabsorbed there). Urinary concentration is impaired because blocking the Na+-K+-2 Cl− carrier impedes development of a hypertonic renal interstitium. Urinary dilution also is impaired because dilution of the filtrate is a normal function of this segment. After administration of a loop diuretic, there are substantial losses of chloride, sodium, water, and other electrolytes (including potassium, magnesium, and calcium) in the urine. In addition to tubular effects, some hemodynamic and extrarenal effects of loop diuretics may arise from vasodilatation or increased venous capacitance.32,70 For example, intravenous administration of furosemide releases vasodilator prostaglandins that increase venous capacity and renal blood flow.13,109,117,124 Dyspnea in human patients is relieved even before a reduction in lung water can be identified.70
The thiazide and thiazide-like diuretics act on the cortical distal convoluted tubule by competing with the luminal Na-Cl cotransporter and preventing movement of NaCl into the distal tubular cells. This effect impairs the ability to dilute urine (and excrete solute-free water) but does not necessarily affect urine concentration, which is a medullary function. Accordingly, in hyponatremia, when there is impaired free-water clearance, the thiazide diuretics are relatively contraindicated.75,142 The overall potency of thiazide diuretics is limited, in part because about 90% of the filtrate already has been reabsorbed before the distal nephron has been reached. For this reason these agents are low ceiling agents, and in human patients higher dosages do not lead to proportionate losses in sodium (but do predispose to more serious losses of potassium and magnesium).70 Nevertheless, when even low doses of a thiazide diuretic is combined with furosemide, dangerous volume depletion and electrolyte losses can result.
The late distal tubules and collecting ducts are sites of sodium reabsorption, aldosterone-controlled secretion of potassium ions, ADH-mediated water reabsorption, and urine concentration. Diuretics acting at these sites initiate diuresis by preventing movement of sodium through luminal channels, either by directly blocking the channels (e.g., amiloride, triamterene) or by antagonizing the effect of aldosterone (e.g., spironolactone, eplerenone). These drugs also exert a potassium-sparing effect and often are classified by that description. They act very distally in the nephron, and their quantitative potential to inhibit sodium reabsorption is low, resulting in a weak to nearly absent diuretic effect.74 The diuretic effect of spironolactone depends on prevailing aldosterone concentrations (which are low in animals with mild CHF or in those receiving appropriate dosages of ACE inhibitors). The main value of these drugs is for maintenance of normal serum potassium concentration or antagonism of aldosterone-induced cardiac injury.129,160,161,181
There are a number of clinically relevant issues regarding the dosage and administration of diuretics.30,142,153,158 Many of the commonly used diuretics are organic anions at physiologic pH and are highly bound to serum proteins. To be effective, the diuretic must be delivered to the urinary space by glomerular filtration or active secretion in the proximal renal tubule. Active secretion is the more important mechanism because the drug is concentrated in tubular fluid. Reduced renal perfusion associated with heart failure, as well as primary renal disease or NSAID administration, may limit the effectiveness of a diuretic unless a high dosage is used and the drug is sufficiently concentrated in renal tubular fluid. This concern about renal perfusion is one rationale for initial high-dose, intravenous administration of furosemide in patients with life-threatening CHF. It also explains in part why chronic diuresis may be associated with impaired response to diuretics. Drug delivery is also relevant in terms of oral dosing of diuretics. Gastrointestinal absorption may be impaired in CHF, especially with right-sided failure and intestinal edema. More importantly the time course from absorption to renal delivery is longer than for parenteral administration. This reduces the concentration of the drug acting at the tubules, decreasing the overall diuretic effect. Temporarily switching from oral to parenteral administration of furosemide at the same dose can have a dramatic diuretic benefit in some patients because of the more efficient delivery of the drug. Additional clinical situations in which diuretics may fail include treatment of pain with opiates (which stimulate ADH release), unanticipated high sodium intake, and acute worsening of heart failure. In these situations the diuretic dosage required to establish diuresis successfully may be substantially higher or an alternative route of administration may be required.
Diuretic therapy triggers neurohormonal responses,46,149,182 and diuretic monotherapy is not an appropriate management strategy for long-term treatment of CHF. Diuretic-induced volume depletion invariably leads to a rebound in renal retention of salt and water either at the previous or a new steady-state in terms of sodium balance. This concept, termed the braking phenomenon, is highly important for understanding the basis for multidrug therapy and why furosemide is typically given two or even three times daily. As an example, once-daily dosing of furosemide in human patients is associated with a brisk diuresis for about 6 hours. But over 24 hours there may be no net loss in total body sodium or edema because salt and water retention can occur for the balance of the day.70 This effect is mediated partly by decreased tubular flow rate, salt retention in segments of the nephron unaffected by the diuretic used, increased sympathetic activity, and activation of the renin-angiotensin-aldosterone system (RAAS).112,141 Thus control of edema in CHF requires a steady state of reduced sodium retention, and patients should receive a consistent dosage of furosemide along with an ACE inhibitor, spironolactone, and a sodium restricted diet.
The dosage of diuretics used must be effective but should be carefully controlled to minimize the common complications of dehydration, azotemia, electrolyte imbalance, and potentially deafness. The first dosage of furosemide chosen for a patient with life-threatening pulmonary edema often is high (2 to 5 mg/kg, intravenously every 1 to 3 hours) to ensure diuresis. The furosemide dosage is promptly reduced if symptomatic improvement and a brisk diuresis are observed. These effects can occur within 1 to 2 hours of administration of furosemide,71 but a lag period (12 to 24 hours) may be noted between obvious clinical improvement and clearing of radiographic pulmonary densities. Owing to the potential for overzealous diuresis and iatrogenic renal failure and electrolyte disturbances, the clinician should evaluate serum biochemistries every 24 to 48 hours until the patient is eating and drinking satisfactorily. After a stable diuretic course of 2 weeks, most dogs and cats maintain relatively stable renal function and serum potassium concentrations unless a decompensating factor (e.g., vomiting, anorexia) intervenes. This is especially true when ACE inhibitors and spironolactone are prescribed concurrently because they reduce aldosterone concentration or effect and decrease potassium and magnesium losses. Thus stable serum creatinine and potassium concentrations over two or three reevaluation periods are likely to be maintained for some time.141 The overall dosage of diuretics in dogs should be limited by using combination therapy for CHF, including progressive sodium restriction, ACE inhibitors, spironolactone, and pimobendan.10,61,62,78,95,123 Cats with chronic CHF typically receive furosemide, an ACE inhibitor, and sometimes pimobendan or spironolactone. Cats receiving furosemide are more prone to develop mild to moderate azotemia and hypokalemia than are dogs, even at dosages that are 50% lower than daily dosages typically used for dogs. Spironolactone is usually well tolerated in cats but may cause anorexia or ulcerative skin lesions.97
Effects of other cardiovascular drugs on renal function
Angiotensin II is one of the factors responsible for efferent arteriolar vasoconstriction and increased filtration fraction in CHF. The ACE inhibitors, such as enalapril, may antagonize efferent arteriolar constriction sufficiently in some patients to cause an abrupt decrease in glomerular perfusion pressure. This effect is especially likely in volume-depleted patients. The result is acute renal failure, with serum creatinine concentration often exceeding 5 mg/dL. Renal failure in this setting generally can be reversed by reducing diuretic dosage, decreasing the dosage of the ACE inhibitor, and providing judicious fluid therapy (see Therapy of Fluid and Electrolyte Imbalances in Congestive Heart Failure section). After volume repletion, the dosage of the ACE inhibitor is increased over 2 to 4 weeks, and the drug combination is adjusted while monitoring body weight, clinical signs of CHF, ABP, and serum creatinine concentration.
Normal autonomic responses to changes in blood pressure and normal heart rate variability are blunted in CHF.66,94 This is associated with dominant sympathetic activity in cardiac failure. Sympathetic nerve activity can increase renin release and affect renal blood flow.122 Digitalis glycosides such as digoxin appear to exert a neurotropic effect and restore baroreceptor sensitivity and parasympathetic tone, and this effect is independent of the inotropic action of the drug.81,166 By this or some other effect, digoxin also can blunt the RAAS in CHF. Although digoxin has been largely supplanted by the inodilator, pimobendan, due to this autonomic effect, digoxin therapy maintains a role in patients with atrial fibrillation and end-stage heart failure.
Cardiac patients sometimes are treated with aspirin and other antiprostaglandin drugs to prevent blood clots (cats) or to alleviate signs of osteoarthritis (dogs). These NSAIDs may be deleterious when used in CHF patients. By preventing prostaglandin-induced dilatation of the afferent arteriole, NSAIDs may decrease glomerular filtration pressure. They may be especially hazardous when used in combination with furosemide and ACE inhibitors. Practically speaking, the use of cyclooxygenase (COX) inhibitors can be tolerated in heart failure, but it is prudent to start at one third to one half of the normal dose and increase the dose while monitoring renal function (and for signs of gastrointestinal ulceration). Other drugs, including ß-adrenergic blockers, human BNP, neutral endopeptidase inhibitors, and direct vasodilators, also may affect renal function. Some of the major effects of these drugs are summarized in Table 21-1. The effects of BNP on renal hemodynamics are still under investigation, and these effects may differ in normal subjects from those in patients with CHF or systemic hypertension.73,172
Serum biochemical abnormalities in congestive heart failure
The majority of serum biochemical abnormalities in heart failure can be attributed to alterations in renal function, changes in dietary intake of water and electrolytes, diuretic and other drug therapy, and drug toxicosis. Most alterations are mild, and two surveys of serum biochemical concentrations of patients at our hospitals have failed to demonstrate severe changes in the majority of cardiac patients.10,12 Nevertheless, some animals with CHF develop substantial disorders of fluid and electrolyte balance that may require fluid therapy and adjustment of cardiac medications.
Sodium
Serum sodium concentration usually is normal in heart failure, but total body sodium and total body water are likely to be increased. Severe right-sided or biventricular CHF can be associated with hyponatremia. Salt wasting secondary to concurrent diuretic use may contribute to hyponatremia, but it is uncommon for low serum sodium concentration in an edematous patient to be caused solely by salt depletion. Multiple factors are probably involved.35,43,52,99,120 One likely cause of hyponatremia in CHF is dilution resulting from markedly reduced renal free-water clearance (see Chapter 3). This effect probably is mediated by the nonosmotic release of arginine vasopressin (ADH) and indicates insufficient cardiac output. Continued release of ADH and polydipsia are important factors to be considered in the pathogenesis of hyponatremia in the patient with CHF. In one study of dogs with CHF, dogs with severe heart failure caused by dilated cardiomyopathy were more likely to develop hyponatremia.178 Activation of the RAAS is predictable in the setting of severe CHF, and glomerular filtration pressure may depend largely on efferent arteriolar constriction mediated in part by angiotensin II.116,138 Consequently, ACE inhibitors must be used very carefully in such patients. However, such treatment often is effective in improving CHF and, despite a reduction in serum aldosterone concentration, increasing serum sodium concentration (see Therapy of Fluid and Electrolyte Imbalances section). Another reason for profound hyponatremia is concurrent use of several diuretics creating sequential nephron blockade from furosemide, a thiazide, and spironolactone. Although thiazides can reduce fluid retention in CHF of dogs and cats, when added to furosemide, azotemia, hyponatremia, hypokalemia, and hypochloremia can develop (sometimes in a matter of 1 or 2 days). Thiazides are more commonly associated with hyponatremia than are loop diuretics because they induce loss of effective solutes (sodium and potassium) in excess of water and do not interfere with the renal effects of ADH.140
Potassium
Serum potassium concentration may be normal, increased, or decreased in patients with heart failure. Mild hyperkalemia may be observed in acute low output heart failure because of an abrupt reduction in the GFR. Overzealous administration of potassium salts and potassium supplementation in the presence of potassium-sparing diuretics, β-blockers, or ACE inhibitors are causes of iatrogenic hyperkalemia.143 Profound hyperkalemia can occur in cats with CHF and concurrent aortic thromboembolism. This probably is related to multiple factors, such as muscle necrosis, reperfusion of infarcted tissues,126 metabolic acidosis, and renal failure with inadequate urinary excretion of potassium. Management of life-threatening hyperkalemia may be required as discussed in Chapter 5.
Hypokalemia is particularly injurious because it predisposes to premature complexes, digitalis intoxication, and muscular weakness along with rhabdomyolysis (with elevated serum creatine kinase concentration) in cats. Hypokalemia in the cat has been linked to abnormal taurine metabolism and taurine deficiency-associated myocardial failure.33 Numerous factors predispose to hypokalemia in the cardiac patient.141 Anorexia resulting from chronic disease or drug intoxication can lead to inadequate potassium intake. Cardiac cachexia and tissue wasting also lead to increased potassium loss. Activation of the RAAS may be important because potassium excretion is enhanced by aldosterone. Fortunately, aldosterone concentrations are readily reduced by administration of an ACE inhibitor. Reduced renal perfusion may influence potassium handling because inadequate delivery of sodium to the distal tubule causes potassium to be secreted with organic acids.75,142 Kaliuresis of variable magnitude occurs with diuretic therapy unless a potassium-sparing diuretic, such as spironolactone or triamterene, or an ACE inhibitor is prescribed. We have observed hypokalemia even when potassium-sparing diuretics have been administered. Cats seem particularly prone to diuretic-induced hypokalemia. The potent loop diuretics, such as furosemide, also promote kaliuresis by accelerating delivery of sodium to the distal nephron, leading to an overall increase in the rate of sodium-potassium exchange.20,75,142 Combination diuretic therapy with furosemide-hydrochlorothiazide is especially likely to lead to hypokalemia, even in the presence of ACE inhibitors or spironolactone. Lastly, metabolic alkalosis is a frequent complication of volume contraction, vomiting, or diuretic-induced chloriuresis.141,142 Alkalosis increases the concentration of potassium in the renal tubular cell and promotes its secretion into the tubular fluid.
Other electrolytes
Serum chloride concentration usually is normal in heart failure. However, it is common for an animal to develop mild hypochloremia after diuretic therapy. Mild hypochloremia is the most commonly observed diuretic-induced electrolyte disturbance in our practice. This observation probably is the result of the inhibitory effect of furosemide and other loop diuretics on chloride transport and may be associated with a small but commensurate increase in serum bicarbonate concentration as estimated by the total CO2. Serum calcium and phosphorus concentrations are normal in CHF unless renal failure or another unrelated disorder is present.
Hypomagnesemia has received little attention in veterinary medicine, but it is common in human patients undergoing diuresis induced by loop diuretics.28,146,162 In one veterinary hospital survey, cardiovascular disease was a prominent risk factor for development of hypomagnesemia.79 The potential importance of magnesium is emphasized by the association of hypomagnesemia with cardiac arrhythmias and the use of magnesium infusions to treat digitalis-induced cardiac arrhythmias in human patients. Serum magnesium concentration in dogs with CHF did not decrease significantly after furosemide therapy in one study of dogs40 but was 20% lower than that of a control population in another canine study.20 As with potassium, serum magnesium poorly reflects intracellular stores.70 Digitalis also has been shown to increase urinary magnesium excretion.
Acid-base disturbances
Blood pH in heart failure is the product of competing factors that alter acid-base balance. Complex acid-base disorders are common because of disturbances in tissue oxygenation and in pulmonary and renal function. As a result, simple determination of total CO2 without direct measurement of blood pH and calculation of bicarbonate may lead to erroneous conclusions (see Chapters 9 through 13). In our experience, respiratory alkalosis and metabolic acidosis are the most commonly encountered acid-base disorders in acute heart failure. Mild metabolic alkalosis is not uncommon in patients receiving chronic diuretic therapy.
Metabolic acidosis may be caused by a stagnant circulation with hypoxia and lactic acidemia,53 by prerenal azotemia, or by tissue ischemia as may occur with aortic thromboembolism. In uncomplicated cases, the venous pH and bicarbonate concentrations are mildly decreased and arteriovenous oxygen difference is increased. In severe CHF or cardiogenic shock, with avid vasoconstriction, mixed venous Po2 often is less than 30 mm Hg. Respiratory acidosis is a less common but more serious complication and indicates the presence of respiratory failure, pulmonary edema, compression atelectasis (from pleural effusion), or respiratory muscle fatigue. Respiratory acidosis is characterized by the development of arterial hypoxemia and hypercapnia and a decrease in blood pH unless a mixed disorder is present (see Chapter 12).
Metabolic alkalosis, with increased bicarbonate concentration and blood pH, is common and often is a complication of diuretic therapy with resultant volume contraction (contraction alkalosis) and renal loss of chloride and potassium.75,142 Vomiting, a complication of drug intoxication, also leads to chloride loss and metabolic alkalosis. Respiratory alkalosis with a low Pco2 may be detected in some patients because animals with moderate pulmonary edema tend to hyperventilate as a result of stimulation of stretch and nociceptive receptors in the lungs.141 Patients with low cardiac output without pulmonary edema have increased muscle fatigability that may be manifested as dyspnea and subsequent hypocapnia. Apparent dyspnea in this subset of patients may be related to skeletal muscle changes that occur during CHF. Abnormal muscle function during CHF has been linked to the decrease in muscle bulk, increased reliance on anaerobic metabolism, decreased muscle blood flow, and metaboreceptor activation.19
Serum proteins
Serum protein concentration frequently is decreased in severe heart failure, especially in dogs with right-sided or biventricular failure. In a survey of dogs with CHF and atrial fibrillation, about one fourth had low serum protein concentrations.12 Concurrent disorders (e.g., liver disease, renal disease, gastrointestinal disease) also may influence serum protein concentration.
The mechanisms responsible for decreased serum protein concentration in CHF are undetermined. Possible explanations include lymphatic loss of protein through a congested intestine, decreased hepatic synthesis, cardiac cachexia, and enhanced endothelial permeability caused by increased capillary pressure and hypoxia. Ascitic fluid is higher in protein concentration than is a transudate collecting in the pleural space because the hepatic sinusoid is more leaky than other capillary beds. Consequently, considerable protein can pool in the peritoneal cavity of a cardiac patient with ascites, and the protein concentration in ascitic fluid can exceed 3.5 g/dL. Repeated abdominal paracentesis also can contribute to total body depletion of protein. Plasma volume contraction after diuretic therapy usually increases serum protein concentration, but total serum protein concentration may remain subnormal or in the low-normal range. Hypoproteinemia in dogs with CHF caused by heartworm disease may be related to glomerular injury and renal protein loss. Dramatic proteinuria has been observed in heartworm-infected dogs with concurrent renal amyloidosis.
There are a number of clinical consequences of hypoproteinemia in CHF. Effective plasma volume is decreased further when moderate to severe hypoalbuminemia develops. As demonstrated in experimental studies of dogs with left atrial hypertension, edema is more likely to occur at lower venous pressures when there is hypoalbuminemia.60 Marked protein loss through the gut may indicate a need for additional nutritional support. Hypoalbuminemia also predisposes to metabolic alkalosis (see Chapter 10). Infusions of plasma may be required in the patient with severe hypoalbuminemia and may promote a substantial diuresis.
Renal function tests
The blood urea nitrogen (BUN) and serum creatinine concentrations may increase in CHF, indicating reduced glomerular filtration. There are several reasons for development of azotemia in heart failure, but the most common are preexisting renal disease, reduced cardiac output, and iatrogenic problems (i.e., overzealous use of diuretics and ACE inhibitors). Common causes of azotemia in dogs or cats with CHF are listed in Box 21-6.
Approximately 25% of dogs with CHF are azotemic at the time of admission.12 The magnitude of azotemia generally is mild to moderate. Renal function should be assessed both before and after initiation of therapy. Azotemia is common in patients with dilated cardiomyopathy and cardiogenic shock and may improve only after aggressive therapy with inotropic agents and reestablishment of hydration (see Therapy of Heart Failure section). The development of azotemia in a patient with previously normal renal function suggests overzealous diuresis, an adverse reaction to an ACE inhibitor, inappropriate water restriction, or a worsening of heart failure. Return of serum creatinine concentration to normal after intravenous or subcutaneous administration of a crystalloid solution or after reduction of the drug dosage indicates a prerenal or drug-induced cause of azotemia. Acute renal failure that responds promptly to intravenous administration of a crystalloid solution has been observed in some dogs treated with ACE inhibitors.
Therapy of heart failure
The initial goals of therapy in CHF include increasing arterial Po2, reducing oxygen demand, establishing a diuresis, and unloading the ventricles while supporting ABP, tissue perfusion, and renal function. Inotropic support is also beneficial in many patients with acute CHF. Long-term treatments are aimed at preventing fluid retention, load reduction, maintaining cardiac output to support exercise and organ perfusion, and blunting progressive neurohormonal injury to cardiac and vascular tissues.
Hospital therapy
The first goals are attained with supplemental oxygen therapy and sedation as needed to reduce distress or air hunger. Traditionally, dogs in heart failure have been sedated with morphine (initial dosage of 0.05 to 0.1 mg/kg intramuscularly), but vomiting after morphine injection occasionally precipitates cardiac arrest. For this reason, we prefer butorphanol (0.2 to 0.3 mg/kg, intramuscularly) as an effective and safer sedative for dogs in CHF. Stress in cats can be alleviated with an acepromazine-butorphanol combination (0.05 to 0.1 mg/kg acepromazine and 0.25 mg/kg butorphanol intramuscularly). In hypothermic or hypotensive cats, butorphanol is used but without acepromazine. In the presence of moderate to severe pleural effusion, thoracocentesis is performed to decrease pulmonary atelectasis. Tense ascites, sufficient to impair ventilation, is reduced by abdominocentesis. About one third to one half of the total ascitic volume is drained. The high protein content of hepatic lymph and the dynamic equilibrium between the third-space and plasma compartments argue against complete drainage of the peritoneal space.141 Pulmonary edema sufficient to cause respiratory failure and respiratory muscle fatigue is an indication for artificial ventilation.
Diuresis is initiated and maintained with parenterally administered furosemide. An initial intravenous bolus of 2 to 5 mg/kg can be followed by serial intravenous or intramuscular boluses of 1 to 4 mg/kg every 6 to 8 hours or more frequently when necessitated by insufficient clinical response. The use of constant rate infusion (CRI) of furosemide also may be used to treat dogs and cats with life-threatening pulmonary edema. In healthy dogs and in human patients with CHF, furosemide CRI increases urine output and minimizes electrolyte disturbances when compared with repeated bolus injections.2,31 Our approach for a CRI is to initially administer an intravenous bolus of furosemide, estimate the furosemide dosage required for the next 24 hours, and then infuse this volume by syringe pump. Supplemental boluses also can be given if required during the CRI. A novel approach for treatment of severe CHF in human patients has been advocated by Licata et al.92 They administered small-volume, hypertonic saline combined with furosemide and demonstrated enhanced diuresis in refractory CHF. This therapy has not been studied in animals with spontaneous disease but deserves consideration, especially in hyponatremic patients. Another approach that may be adopted in veterinary practice involves addition of intravenous synthetic human brain natriuretic factor (h-BNP) or nesiritide to the hospital treatment protocol.107 Although expensive, nesiritide is labeled for human use and appears to be effective in dogs. The h-BNP increases urine output and decreases the effects of aldosterone in furosemide-treated dogs,17 increases urine volume in normal dogs and those with experimental CHF,18,167 and has limited electrophysiologic effects on the canine heart.41
Both preload reduction and afterload reduction are beneficial to the failing left ventricle. The inotropic drug pimobendan (0.2 to 0.3 mg/kg PO q12h) also exerts vasodilator properties via phosphodiesterase-3 inhibition. Nitrates such as nitroglycerin ointment and sodium nitroprusside increase concentrations of the vasodilator nitric oxide in vascular smooth muscle, leading to relaxation of arterioles and systemic veins.113 Two percent nitroglycerin ointment ({1/4} to 1 inch of the 2% ointment, topically every 12 hours) acts primarily as a systemic venodilator, and this treatment is well tolerated by both dogs and cats although efficacy has not been demonstrated in a clinical study. The anticipated venodilation should work in concert with furosemide to decrease venous and capillary hydrostatic pressures. The need for arteriolar dilators in the hospital setting depends on the cause and severity of CHF, and in many cases furosemide, nitroglycerine, and pimobendan are the only drugs needed for initial control of CHF in dogs. In more advanced cases, systemic arterial dilation therapy may be beneficial. Although vasodilator therapy has the potential to induce systemic hypotension, such treatment generally is safe in dogs when baseline ABP is greater than 95 mm Hg. Sodium nitroprusside (1 to 5 µg/kg/min intravenously by CRI), enalapril (0.5 mg/kg orally every 12 hours), and hydralazine (1 to 2 mg/kg orally every 12 hours) all exert vasodilator effects in the hospital setting. Each drug can increase stroke volume and reduce pulmonary edema, especially in the setting of severe mitral regurgitation arising from valvular endocardiosis. Afterload reduction can also increase stroke volume when there is left ventricular dysfunction, as in dogs with dilated cardiomyopathy.11 The choice of vasodilator in dogs depends on the urgency of the situation. In florid pulmonary edema, nitroprusside can be infused to a specific endpoint, such as a systolic ABP of 85 to 90 mm Hg. In less urgent cases, or when intravenous therapy is impractical, enalapril or hydralazine can be administered orally to provide afterload reduction. After stabilization, enalapril or another ACE inhibitor is initiated (or continued) as part of the home treatment plan. Nitroprusside and hydralazine rarely are used in cats, and most cats with CHF are treated with furosemide, nitroglycerin, and eventually an ACE inhibitor. Pimobendan (1.25 mg/cat every 12 hours) can be used as an extralabel treatment in cats with severe CHF and hypotension.
Cardiac output, ABP, and tissue perfusion are supported when necessary by providing inotropic support. In dogs or cats with severe systemic hypotension (ABP <80 mm Hg), inotropic support with dobutamine (2.5 to 10 µg/kg/min) or dopamine (2 to 10 µg/kg/min) is indicated (in conjunction with pimobendan). Catecholamines most often are administered to dogs with CHF caused by dilated cardiomyopathy. Occasionally, this approach is used in patients with severe mitral regurgitation or pulmonary embolism. Cats with any form of cardiomyopathy may develop cardiogenic shock characterized by bradycardia, hypothermia, and hypotension. Treatment with dobutamine can be life saving in affected cats. Infusions should be titrated to a systolic ABP of 90 to 120 mm Hg and can be combined with slow external warming in an oxygen incubator. When treatment with catecholamines is impractical, oral administration of the calcium sensitizer pimobendan may be effective. Intravenous administration of a related compound, levosimendan, may become a treatment alternative in the future. Both pimobendan and levosimendan are considered “inodilators” owing to potent positive inotropic effects combined with vasodilatation, which unloads the left ventricle.95,164
Home therapy
Chronic therapy of CHF targets the kidney, heart, and vascular tree, while attempting to minimize neurohormonal injury to cardiac and vascular tissues. By combining diuretics (usually furosemide and spironolactone) with an ACE inhibitor and dietary sodium restriction, fluid retention is prevented. In dogs, cardiac performance is enhanced by administration of pimobendan.62 Further cardiac protection may be achieved by gradual up-titration of a ß-adrenergic blocker such as carvedilol or bucindolol, but definitive data supporting this therapy is unavailable. Digoxin may still be useful for dogs with atrial fibrillation or as a drug that will reduce sympathetic and heighten parasympathetic activity. Specific heart rhythm disturbances such as atrial fibrillation or ventricular tachycardia require additional antiarrhythmic drug treatments. Many of these treatments impact renal function and fluid and electrolyte balance in the cardiac patient. The rationale for medical therapy is considered below.
A fundamental feature of CHF is dominance of vasoconstrictive, sodium-retaining mechanisms over competing vasodilator-natriuretic systems.46,137,139 Chronic activation of the sympathetic nervous system, increased formation of endothelin, and progressive stimulation of the RAAS injures the myocardium, blood vessels, and the kidney.22,67,119,160 Neurohormonal activation clearly occurs in many dogs with spontaneous heart disease, especially those with advanced heart failure.85,125,155,178 Many laboratory and clinical investigations in humans have emphasized the beneficial effect that pharmacologic blockade of the RAAS has in limiting the progression of myocardial disease and reducing morbidity and mortality in CHF.26,113,154 Another therapeutic advance is aldosterone blockade at the tissue level by administration of an aldosterone antagonist (spironolactone or eplerenone).129,181 A tissue RAAS is present in the canine heart, and the local chymases that convert angiotensin to its active form may not be inhibited by ACE inhibitors. Aldosterone antagonism produces modest but measurable survival benefits in human patients with CHF and has been shown to reduce left ventricular remodeling in dogs with experimentally induced heart failure.161 Aldosterone also blunts baroreceptor reflexes in dogs, an effect that can be partially reversed by administration of digitalis glycosides.177
Two prospective North American studies of dogs with CHF have demonstrated the efficacy of enalapril at 0.5 mg/kg orally every 24 hours or every 12 hours,25,71 and another investigation demonstrated the relative renal safety of monotherapy with enalapril in dogs with asymptomatic valvular heart disease.5 That study also showed a modest trend for delay in onset of CHF in dogs with advanced chronic valvular disease,5b although no benefit was observed in another study of Cavalier King Charles spaniels.87 Other studies have shown efficacy for benazepril,82 including the relatively large BENCH study of dogs with CHF caused by mitral regurgitation or dilated cardiomyopathy.130 In the latter trial, benazepril dosages of 0.5 mg/kg every 24 hours or every 12 hours both were effective and well tolerated in terms of renal function. Other ACE inhibitors, such as ramipril, have been approved in countries outside of North America for management of canine heart failure. Based on clinical observations,3,145 cats with chronic CHF also can benefit from ACE inhibition, but a blinded, controlled, prospective study has not yet been published. The dosage of ACE inhibitors in cats with CHF is similar if slightly lower than that for dogs (enalapril, 0.25 mg/kg once or twice daily, increased after 1 or 2 weeks to 0.25 to 0.5 mg/kg every 12 hours orally). Renal function should be monitored before and after any dosage change.
Spironolactone is prescribed frequently for dogs and occasionally for cats with chronic heart failure. Although diuretic effects of this drug are minimal, ACE inhibition does not fully inhibit aldosterone formation in advanced CHF. One clinical study demonstrated the potential value of spironolactone in canine valvular heart disease in mixed populations of dogs with and without CHF10; however, in the author’s opinion, these survival benefits have not been demonstrated conclusively in well-defined patient populations. In cats spironolactone has been used for antifibrotic effects in hypertrophic cardiomyopathy, but at least one study failed to show clear benefit in terms of fibrosis or diastolic function.97
As with the ACE inhibitors and spironolactone, ß-adrenergic blockers also improve left ventricular ejection fraction and inhibit myocardial remodeling and fibrosis in humans and in animal models of myocardial failure.1,14,42,147,148,185 β-blockers are now standard therapy for human heart failure. However, this approach has not been widely accepted in veterinary medicine, with the possible exception of left ventricular systolic dysfunction leading to the “preclinical” (before CHF) phase of cardiac failure. One problem with use of β-blockers in veterinary patients relates to the advanced state of CHF so often observed. Furthermore, aside from canine model studies and a known benefit on reducing dynamic outflow obstruction in cardiomyopathy, there is no pivotal evidence supporting β-blockade in spontaneous animal diseases. An ongoing clinical trial with bucindolol may address some of the outstanding questions regarding efficacy and safety in dogs. Another issue relates to the specific ß-blocker because not every drug is equally cardioprotective. Metoprolol, carvedilol, and atenolol have been evaluated in canine models of heart failure,1,42,105,147,148,185 Currently, the author confines use of β-blockers in dogs with heart failure to those with echocardiographically demonstrated impaired systolic function but without overt CHF. We most often prescribe carvedilol (starting at 0.1 mg/kg q12h PO and up-titrating every 2 to 4 weeks to 0.6 mg/kg q12h PO). These dosages are well tolerated in “preclinical” disease. However, once CHF is evident, initiating ß-blocker therapy is contraindicated until failure is well controlled and dogs taking carvedilol may require a dosage reduction. Use of these drugs in established CHF is best monitored in consultation with a cardiologist. Finally, we do not routinely use ß-blockers in small breed dogs at any stage of valvular heart disease.
Thus the typical home therapy of CHF in dogs includes administration of an ACE inhibitor (generally enalapril or benazepril 0.25 to 0.5 mg/kg orally every 12 hours), furosemide (2 to 6 mg/kg orally every 12 to 8 hours), pimobendan (0.2 to 0.3 mg/kg orally every 12 hours), and spironolactone (2 mg/kg/day orally as one dose or in two divided doses).7 Digoxin (0.005 to 0.0075 mg/kg orally every 12 hours) is reserved for cases with atrial fibrillation or end-stage CHF), with contraindications including complex ventricular ectopy, moderate azotemia, or sinus node dysfunction. Once CHF is well controlled, an up-titration of carvedilol can be considered in selected cases as discussed above. Importantly, the negative inotropic effects of carvedilol and other β-blockers can worsen CHF and these drugs should not be given to “wet” patients. Additionally, the dosage may need to be reduced if fluid retention worsens despite diuretic and inotropic therapy. When CHF is complicated by atrial fibrillation, digoxin should be given along with diltiazem (starting at 1.5 mg/kg orally daily in two divided doses (for the long-acting drug) or three divided doses (for the standard drug). The total daily dosage can be increased to a maximum of about 6 mg/kg orally to gain heart rate control of 100 to 150 beats/min in the hospital. A β-blocker may also help in control of ventricular rate response. Both diltiazem and β-blockers are negative inotropes and must be used carefully in CHF. A serum digoxin concentration is measured after 1 week of treatment (trough target concentration of 0.9 to 1.2 ng/mL), and the dosage of diltiazem adjusted to control heart rate. When severe pulmonary hypertension leads to clinical signs such as exertional collapse, the phosphodiesterase-V inhibitor sildenafil is added to the treatment regimen (1 to 3 mg/kg q12h PO). Finally, amlodipine may be used for treatment of concurrent hypertension that is unresponsive to an ACE-inhibitor and diuretic. Vasodilator drugs such as amlodipine and hydralazine may activate the RAAS61 and can lead to additional fluid retention.
Dietary measures in treatment of CHF are often overlooked. Low sodium diets may be beneficial in dogs with CHF.144 Other dietary measures may be considered. The addition of omega-3 fatty acids found in fish oil may inhibit proinflammatory cytokines and reduce cardiac cachexia.50 Typical dosages are 30 to 40 mg/kg orally daily for eicosapentaenoic acid (EPA) and 20 to 25 mg/kg orally daily for docosahexaenoic acid (DHA). Nutraceuticals, such as taurine or L-carnitine, may be indicated for selected patients83 with dilated cardiomyopathy (consult a cardiologist).
Home management of cats with progressive CHF or recurrent pulmonary edema or pleural effusion secondary to cardiomyopathy generally includes furosemide (1 to 2 mg/kg orally every 24 hours or every 12 hours) and enalapril or benazepril (0.25 to 0.5 mg/kg every 24 hours to every 12 hours orally). Spironolactone (6.25 mg orally every 24 hours) also can be added to the treatment plan. Digoxin (one fourth of a 0.125-mg tablet orally every 48 hours) rarely is used in cats today. Once the cat with CHF is stabilized, a cardiologist should be consulted regarding other drug options, including inodilators such as pimobendan, which can provide clinical benefit in some cats with chronic cardiac failure.
Refractory edema and effusions
Some patients become refractory to diuretic therapy and continue to develop edema or effusions.142 Three commonly encountered examples of this problem are (1) progressive ascites and pleural effusion in dogs with biventricular heart failure, (2) progressive pleural effusion in cats with cardiomyopathy, and (3) recurrent pulmonary edema in dogs with left-sided heart failure. Successful therapy of some of these patients may be attained by skillful use of cardiac medications110 and by addressing the following points:
• Ensure medication compliance, and educate the client about medications, dosages, and methods of administration.
• Consistently enforce a restricted or low-sodium diet.
• Optimize current medication dosages (to full recommended dosages).
• Improve left-sided heart function with an additional afterload reducer such as amlodipine.
• Reduce the dosage of any negative inotropic drugs, such as β-blockers or diltiazem.
• Adjust the dosage or route of administration of furosemide.
• Consider using combination diuretic therapy.
• Identify and treat extracardiac complications such as hyperthyroidism, anemia, and hypertension.
The first three points are straightforward but by no means easy to achieve. With progressive CHF, the sodium intake should be progressively limited unless the patient is hyponatremic. Periods of enforced rest are useful in mobilizing edema and decreasing cardiac work. Rest alone can lead to considerable diuresis in patients with right-sided CHF. The remaining guidelines require some explanation.
Modifying the diuretic dosage may be necessary, especially in dogs with chronic renal failure, in those that develop severe polydipsia, and in those with apparent intestinal malabsorption of furosemide. Low dosages of furosemide (e.g., 1 to 2 mg/kg every 12 hours or every 24 hours), in combination with an ACE inhibitor and pimobendan, are quite effective in patients with mild heart failure. However, patients with renal failure or low cardiac output may require higher dosages to deliver sufficient amount of active drug to the renal tubules.55 In the case of furosemide, gradually increasing the dose and frequency from 2 mg/kg every 12 hours to 6 mg/kg every 8 hours may be sufficient to maximally inhibit renal tubular chloride and sodium reabsorption. Once this “ceiling” effect is achieved, no further diuresis develops with increasing the dosage.142 This “ceiling” effect is especially apparent with loop diuretics (e.g., furosemide, bumetanide, torsemide), which typically have a short duration of action (see previous Diuretics section). Furosemide may be poorly absorbed by a congested intestine,174 and subcutaneous administration of furosemide in patients with refractory ascites and pleural effusion should be considered. Frequently, the same dose, given subcutaneously instead of orally, leads to substantial diuresis. We have taught clients to administer one of the daily doses of furosemide subcutaneously to their animals every other day, and such therapy can be beneficial when used chronically. Combination diuretic therapy with sequential nephron blockade represents another option for the patient with refractory edema or effusion.65,112,115,142 The combination of three diuretics (furosemide, hydrochlorothiazide, and spironolactone) acting on different segments of the nephron (see Figure 21-5) may be effective in treating dogs with progressive ascites or pleural effusion. However, hyponatremia (<130 mEq/L) and hypokalemia are contraindications to thiazide diuretics, and thiazide diuretics often induce profound hyponatremia. When hydrochlorothiazide is prescribed, the initial dosage should be low, approximately 1 to 2 mg/kg orally every 48 hours. Renal function and serum electrolyte concentrations should be evaluated within 1 week of treatment before the dosage is increased.
Therapy of fluid and electrolyte imbalances in congestive heart failure
Indications
The cardiac patient, in contrast to many other sick animals, is not an ideal candidate for parenteral fluid therapy. Volume expansion poses substantial risks in terms of increasing venous pressures, sodium retention, and edema. In managing cardiac patients, we prefer to offer water (of low sodium content) ad libitum, provide a sodium-restricted but palatable diet, treat CHF medically, and allow the patient’s kidneys to correct any fluid and electrolyte disturbances. This approach may lack technical sophistication, but it often works well in the clinical setting. Aside from mild hypochloremia, dogs are especially resilient to the complications of diuretic therapy provided their intake of water and food is adequate. In fact, it is common to observe a dog or cat begin drinking shortly after receiving successful therapy for life-threatening pulmonary edema or pleural effusion.
Some patients with heart failure do develop problems that require fluid and electrolyte supplementation. Indications for fluid therapy in the patient with CHF include persistent anorexia, dehydration, renal failure, moderate to severe hypokalemia, digitalis intoxication, drug-induced hypotension, gastroenteritis, anemia, and serious metabolic (e.g., diabetes mellitus), neoplastic, or infectious diseases. Another indication is the need for intravenous infusion to deliver drugs such as dobutamine, sodium nitroprusside, or lidocaine. Ventricular filling is impaired in pericardial disease, and this abnormality may demand volume expansion with parenteral fluid therapy along with pericardiocentesis. When animals with heart disease undergo general anesthesia, a catheter should be placed and intravenous fluids administered although at a reduced rate. Hypertrophied ventricles may be more difficult to distend unless CVP is maintained at a normal to slightly increased level. However, overinfusion of fluids can lead to peracute pulmonary edema in dogs and in cats with marked left ventricular hypertrophy, and care must be taken.
Thus a number of situations may necessitate fluid therapy in the cardiac patient. What fluid should be infused? The following recommendations are based on our clinical experience and theoretical considerations for fluid, electrolyte, and diuretic therapy in patients with CHF. Controlled, prospective evaluations of such therapy in dogs and cats are unavailable. The following discussion considers basic principles of therapy; selection of fluids, additives, and rates of administration; monitoring of the patient (including Swan-Ganz catheterization); and our approach to some specific problems related to fluid therapy in the cardiac patient.
Parenteral solutions
Fluid Volume
The daily fluid volume is guided by the current state of edema, estimated maintenance needs (40 to 60 mL/ kg/day), hydration status, body weight, oral fluid intake, estimated urine output, total serum protein concentration, serum sodium concentration, serum creatinine concentration, and, when available, CVP and pulmonary capillary wedge pressure. It is prudent to consider a minimal fluid infusion initially (e.g., no more than 30 to 40 mL/kg/day) and to assess the effect of fluid therapy on the patient and serum biochemical values. The daily volume should be infused slowly and distributed evenly over 24 hours to reduce the risk of pulmonary edema and pleural effusion. The choice of fluid depends largely on concerns about sodium retention. The intravenous route of administration is preferred, but either 0.45% NaCl in 2.5% dextrose or lactated Ringer’s solution can be given subcutaneously if necessary. When the patient can drink, fluid therapy is tapered, low-sodium fresh water is supplied ad libitum, and dietary sodium intake is regulated while ensuring a palatable diet.
The CHF patient continues to retain sodium, and diuretics must be given concurrently to prevent untoward retention of sodium derived from the diet or crystalloid therapy. Although it may seem paradoxical to administer diuretics to a patient receiving fluid therapy, these drugs are important adjuncts to the overall fluid and electrolyte management in treatment of the edematous cardiac patient.64,75,142 Diuretic therapy also promotes redistribution of extracellular water from edematous sites to the venous system. Furosemide also acts initially to increase GFR (possibly by releasing vasodilating prostaglandins). After diuresis and contraction of the plasma volume, however, cardiac filling and GFR decrease unless the patient drinks adequately or receives supplemental fluid therapy. A fine balance is required, and the clinician must learn to control the risk of edema while preventing an increase in BUN or serum creatinine concentration. Human BNP (nesiritide) may represent another option for preventing fluid retention in cardiac patients receiving fluid therapy; however, this drug also increases the serum creatinine in some human patients.
Sodium
Dogs with cardiac failure do not respond normally to a sodium load, and after saline infusion, marked retention of sodium and water can occur.9 Healthy dogs can maintain normal serum sodium concentration with a diet containing sodium at only 0.5 mEq/kg/day (11.5 mg/kg/day).100,106 This amount is equivalent to approximately 175 mg of sodium or 435 mg of sodium chloride per day for a 15-kg dog. In Canine Prescription Diet H/d (Hill’s Pet Nutrition, Topeka, Kan.), there are approximately 23 mg of sodium and 542 kcal in a 418-g serving of canned food. The H/d dry product contains about 15 mg of sodium and 407 kcal in a 99-g serving. Another highly sodium-restricted diet, CV-Formula (Nestlé Purina PetCare Co., St. Louis), contains about 20 mg of sodium and 638 kcal in a 354-g serving. Early Cardiac Support Diet (Royal Canin) delivers 61 mg of sodium and 300 kcal in a 73-g, dry food serving. A 2.5-oz jar of chicken baby food contains approximately 40 to 60 mg of sodium. A number of over-the-counter dog foods also are relatively restricted in sodium (e.g., Cycle Senior [Del Monte, San Francisco], Alpo Senior [Nestlé Purina PetCare Co.]). The extent of dietary sodium restriction required in animals with CHF has not been determined, but it seems prudent to limit daily sodium intake to less than 12 mg/kg/day in dogs with end-stage cardiac failure. The average sodium content of Feline Prescription Diet H/d (Hill’s Pet Nutrition) is about 354 mg per 14.25-oz can (70 mg/100 kcal; 506 kcal/can), and a 5.5-oz can of CV-Formula contains about 112 mg of sodium (50 mg/100 kcal; 223 kcal/can). Dietary sodium requirements for cats with CHF are not available and their acceptance of sodium-restricted diets appears lower than for many dogs.
The clinician also must be mindful of the sodium content of crystalloid solutions. Normal saline solution (0.9% NaCl) contains 154 mEq of sodium per liter. Therefore 500 mL of 0.45% NaCl in 2.5% dextrose contains 37.5 mEq (862 mg) of sodium, an amount that conceivably represents the minimal daily requirement for a normal 75-kg dog. If severe metabolic acidosis in a cardiac patient must be treated with sodium bicarbonate, an additional sodium load is imposed because there are 23 mg of sodium per milliequivalent of sodium bicarbonate. Metabolic acidosis in those with CHF often is caused by lactic acidosis, a condition that may not be responsive to bicarbonate treatment, and is best treated by improving cardiac output (see Chapter 10).
Based on these concepts, either 5% dextrose or 0.45% NaCl in 2.5% dextrose, supplemented with potassium chloride, is recommended when routine fluid therapy is required for rehydration, maintenance of hydration, or drug infusions in patients with CHF. Unfortunately, therapy with 5% dextrose or 0.45% NaCl in 2.5% dextrose is sometimes associated with inadequate free-water excretion, weight gain, hyponatremia, and hypokalemia, especially when 5% dextrose is administered. These electrolyte disturbances are similar to those observed when some dogs and cats with severe CHF are treated with diuretics and given free access to water. Development of hyponatremia in this clinical setting is especially common in cats. Because of the potential for hyponatremia, either 0.45% NaCl in 2.5% dextrose or a balanced crystalloid, such as lactated Ringer’s solution or Plasmalyte, is used as a replacement fluid for cardiac patients with dehydration. The short-term use (<12 hours) of such sodium-replete fluids usually is well tolerated, provided the volume is small and the rate of infusion is slow (e.g., 2.5 to 5 mL/kg/hr). Therapy of hyponatremia is discussed later and in Chapter 3.
Potassium Supplementation
Potassium (as the chloride salt) is administered routinely to cardiac patients receiving fluid therapy. Administration of glucose-containing, salt-poor solutions, especially during diuretic therapy of anorexic patients, tends to decrease serum potassium concentration. Typical intravenous potassium dosages of 0.5 to 2.0 mEq/kg/day are given using accepted guidelines for intravenous administration of potassium (see Chapter 5). For hypokalemic animals, higher dosages of potassium chloride are used up to a rate not to exceed 0.5 mEq/ kg/hr intravenously. Oliguria, hyperkalemia, and concurrent administration of potassium-sparing diuretics, β-blockers, or ACE inhibitors are relative contraindications for parenteral potassium therapy, unless serum potassium concentration is known to be low. When providing the patient with oral potassium chloride supplementation, the clinician should consider that there is 1 mEq of potassium in each 89 mg of potassium chloride salt (or in 234 mg of potassium gluconate).
Blood Products
Moderate to severe anemia increases the demand for cardiac output and can precipitate CHF. In most cases, the packed cell volume must decrease to less than 22% or it must decrease rapidly for cardiac complications to occur. Although anemia alone can cause high output heart failure, the development of pulmonary edema or pleural effusion is even more common in the setting of a preexisting heart disease, such as cardiomyopathy or chronic valvular heart disease. Anemic patients often receive fluid therapy to maintain blood pressure and organ perfusion, and this poses another risk for the dog or cat with underlying cardiac dysfunction. Similarly, the hemoglobin solution Oxyglobin (Biopure Corp., Cambridge, Mass.) expands plasma volume and can cause CHF in susceptible patients (this product is at times unavailable). Management of these animals involves medical therapy of CHF, treatment of the underlying cause of anemia, and often a slow infusion of packed cells to reduce the demand for cardiac output.
Managing electrolyte disorders in CHF
Electrolyte disturbances, notably hypokalemia, hypochloremia, and metabolic alkalosis, are common complications of diuretic therapy. Digitalis intoxication with anorexia and vomiting can have similar effects. Mild reductions in serum chloride concentration are of limited concern, but hypokalemia should be avoided in cardiac patients because it predisposes them to cardiac arrhythmias, digitalis intoxication, muscle weakness (and necrosis), and renal fibrosis and may decrease serum taurine concentration in cats.33 Fortunately, most dogs develop only mild hypokalemia during the initial hospital therapy of CHF.11 With the widespread use of ACE inhibitors and spironolactone (which spare potassium loss), hypokalemia also is relatively uncommon during chronic management of CHF, except in the settings of digitalis intoxication, vomiting, or prolonged anorexia or when combination diuretic therapy is prescribed. Cats are more prone to hypokalemia. Even a 1-day course of parenteral furosemide can decrease the serum potassium concentration significantly in cats. Hypokalemia is also more common with chronic furosemide administration in cats unless prevented by an ACE inhibitor, spironolactone, or potassium supplementation.
Hypokalemia can be prevented in the hospital setting by encouraging food intake and supplementing parenteral fluids with KCl. Constant-rate infusion of furosemide also may be associated with less severe urinary potassium loss. Routine oral potassium supplementation is not needed in chronic CHF, 143 but a KCl “salt substitute” or solution (such as Renacare) can be administered if indicated by serum biochemical monitoring. In practice, mild hyperkalemia is not uncommon in patients receiving both an ACE inhibitor and spironolactone, but it usually is ignored. Use of oral potassium supplements and the hospital management of severe hypokalemia are described in detail in Chapter 5.
Serum sodium concentration generally is normal in cardiac patients, and the finding of hyponatremia is a serious sign. Low serum sodium concentration in the setting of excess extracellular fluid volume suggests decreased effective arterial blood volume with impaired renal water excretion related to persistent release of ADH. Diuretics also may contribute to hyponatremia (and hypochloremia) by causing hypokalemia, inducing plasma volume depletion and release of ADH, and impairing function in the diluting segments of the nephron.43,52,75 Thiazide diuretics are especially likely to cause hyponatremia because they favor excretion of relatively concentrated urine. These abnormalities are exacerbated by increased water intake associated with polydipsia, which can be prominent in dogs with CHF, or by infusion of a sodium-poor crystalloid. The general causes of and approach to hyponatremia are described in Chapter 3.
Therapy for hyponatremia in CHF is difficult. Mild hyponatremia (130 to 145 mEq/L in dogs) simply is an indication to adjust cardiac therapy. Moderate hyponatremia (<130 mEq/L), especially when associated with prerenal azotemia, is an indication for cage rest, mild water restriction, frequent determination of body weight and serum biochemistry, and vigorous therapy for CHF. Furosemide is continued because studies in human patients suggest that furosemide may promote the formation of more dilute urine, thereby increasing free-water clearance, whereas thiazides do not.142 Thiazide diuretics should be discontinued. If the patient is receiving fluids, either lactated Ringer’s solution or 0.9% NaCl, supplemented with KCl, should be used initially at conservative infusion rates (e.g., 20 to 30 mL/kg per 24 hours). Infusion for 48 to 72 hours of a catecholamine (dobutamine or dopamine) should be considered to increase cardiac output and the pimobendan dose in dogs may be administered (extralabel) at 0.2 mg/kg every 8 hours. Gradually increasing the dosage of the ACE inhibitor up to the maximal dosage tolerated (at least 0.5 mg/kg of enalapril or benazepril every 12 hours) is important to antagonize the RAAS.35,120 Despite the theoretical concern that an ACE inhibitor may reduce serum sodium concentration, clinical experience in this setting is just the opposite. Severe hyponatremia (<120 mEq/L) requires water restriction and cautious infusion of 0.9% saline or low-volume hypertonic saline to prevent the neurologic consequences of hyponatremia (see Chapter 3). Mannitol or low-volume hypertonic saline may increase delivery of filtrate to the distal diluting segments of the nephron and may increase free-water clearance. With few exceptions, patients with CHF and severe hyponatremia are unresponsive to therapy. Therapy with ADH (vasopressin) receptor antagonists that block the effects of ADH on the distal nephron are available for human use and may be a consideration. These antagonists have been effective in treatment of experimental canine CHF.108,150,184
Monitoring of patients
Cardiac patients require careful monitoring of clinical, hematologic, cardiac, radiographic, and hemodynamic variables. It is important to tabulate and establish the trend of important clinical signs: body temperature, respiratory rate and depth (in hospital and at home), breath sounds, heart rate, heart rhythm, mucous membrane color and refill time, pulse strength, attitude, and noninvasively determined arterial blood pressure (Box 21-7). Frequent determination of such simple variables as water and food intake, estimated urine output, body weight, and diuretic dosage provides the clinician with useful information about fluid dynamics and the need for fluid therapy. Home respiratory rates exceeding 35 to 40 in resting dogs correlates well with radiographic evidence of pulmonary edema. Serial determination of serum creatinine, BUN, sodium, and potassium concentrations is useful for monitoring fluid, diuretic, and cardiac therapy. Physical and radiographic signs of fluid accumulation may indicate a need to reduce fluid volume in hospitalized patients and to increase diuretic dosage or to consider additional treatments. For critically ill dogs, more accurate hemodynamic information can be obtained using a percutaneously placed pulmonary arterial catheter, as described in the following section. The effect of fluid therapy on CVP and pulmonary venous pressure is a prime concern in patients with heart failure and can be a major determinant of the rate of fluid administration. Insufficient venous pressure reduces cardiac output, whereas very high pressure promotes formation of edema. In heart failure, an optimal venous pressure is necessary to maintain cardiac output, but pulmonary venous pressure greater than 20 mm Hg and CVP greater than 10 to 12 cm H2O may be associated with formation of edema.
Box 21-7 Evaluation of the Cardiac Patient
Inspection and Examination
The CVP is simple to measure using an indwelling jugular venous catheter, and its determination quantifies and indicates the directional changes of right heart filling pressures. More practically, the inspection and estimation of jugular venous pressure provides similar qualitative information. A CVP line is useful in guiding fluid management of seriously ill patients without heart disease, but CVP is not an accurate reflection of pulmonary venous pressure in those with left-sided CHF. The ability of the left and right ventricles to accept and pump blood may be different in CHF. Accordingly, the effects of a volume infusion on the left ventricle and pulmonary circulation may not be accurately gauged by measuring the filling pressures of the right ventricle.44,45,163 It is common to observe animals with high pulmonary venous pressure but relatively low CVP. This is especially true after diuretic therapy. Even in animals with right-sided CHF, ascites may continue to develop despite a relatively low CVP, possibly as a result of avid sodium retention, hypoproteinemia, or the development of cardiac cirrhosis and portal hypertension secondary to chronic hepatic congestion. Noninvasive estimation of cardiac filling pressures can be accomplished using advanced Doppler echocardiographic techniques that record transmitral filling, pulmonary venous flow, and tissue ventricular movements during diastole, but these are not widely available and require advanced training to apply with any consistency. Experienced clinicians also recognize that a ventricular (S3) gallop sound typically corresponds to elevated filling pressures and as such will be diminished or eliminated with effective diuresis or management of heart failure.
To obtain direct measurements of pulmonary venous and left-sided cardiac filling pressures, a catheter must be advanced into a lobar pulmonary artery under fluoroscopic or pressure-monitored guidance (Figure 21-6). Special end-hole, balloon-tipped catheters (Swan-Ganz) can be used to occlude pulmonary arterial flow temporarily, permitting measurement of the damped left atrial pressure waveform, which is transmitted through the valveless pulmonary venous and capillary beds.44,45,78,163 The mean value of such a determination is called the pulmonary capillary wedge pressure and is equivalent to the mean left ventricular filling pressure (but not equivalent to the end-diastolic pressure in some patients) (Figure 21-7). Pulmonary edema generally is associated with pulmonary capillary wedge pressures greater than 20 to 25 mm Hg. These values are guidelines, and even higher values may not be associated with edema in chronic left-sided heart failure. The clinician can measure the pressure filling the left ventricle and estimate the tendency to form pulmonary edema by determining whether low (<7 mm Hg), optimal (12 to 18 mm Hg), or high (>20 mm Hg) venous pressures are present in the cardiac patient (see Figures 21-6 and 21-7).45 The rate of fluid administration, diuretic dosage, and cardiac therapy are guided by these measurements. Marked reductions in pulmonary capillary wedge pressure can be observed in some patients after administration of furosemide, hydralazine, or sodium nitroprusside. Noninvasive estimation of left atrial pressure may be possible in the future by application of advanced Doppler echocardiographic methods, in particular, the ratio of early ventricular filling velocity (E-wave) to the tissue Doppler velocity (Ea-wave).
Figure 21-6 Swan-Ganz pulmonary catheterization. A, Determination of central venous pressure (CVP) and pulmonary capillary wedge pressure. Determination of right and left ventricular filling pressures (left lateral view). A balloon-tipped, flow-directed catheter (Swan-Ganz) is inserted into the jugular vein and passed through the cranial vena cava (Cran VC), right atrium (RA), right ventricle (RV), and pulmonary artery (PA). Two independent catheter lumina permit pressure determinations in both the right atrium and the pulmonary artery. The proximal lumen in the RA measures the CVP, and the distal tip measures the PA pressure. When the balloon is inflated, blood flow is temporarily occluded, and the pulmonary capillary wedge pressure (PCWP) is measured (inset). Caud VC, Caudal vena cava. B, Cardiac output curve of a 21-kg dog in heart failure. The curve was obtained using a Swan-Ganz catheter equipped with a distal thermistor tip for measuring blood temperature. The recording demonstrates the change in blood temperature that developed after 3 mL of iced 5% dextrose was injected into the right atrial port of the catheter. Cardiac output is inversely related to the area under the curve. The calculated cardiac output in this case was 2.3 L/min.
(A, from Bonagura JD. Fluid management of the cardiac patient. Vet Clin North Am Small Anim Pract 1982;12:509.)
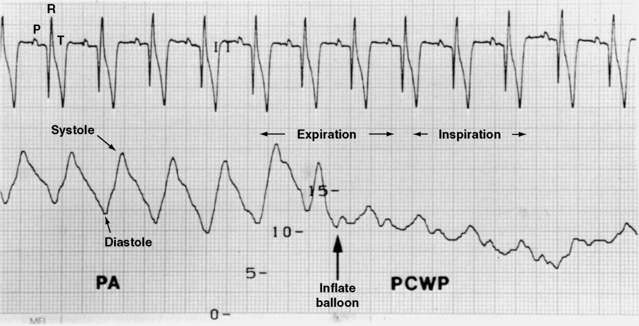
Figure 21-7 Pressure tracing recorded from the pulmonary artery of a dog undergoing diuretic treatment of congestive heart failure. The pulmonary artery pulsatile pressure and occlusion (wedge) pressure are indicated. Balloon inflation is complete at the arrow. Notice that the pulmonary artery diastolic pressure is closely related to the wedge pressure. The pulmonary artery diastolic pressure is often used to track changes in the wedge pressure (provided there is no pulmonary vascular disease). Variations in the pressure recording baseline are related to ventilation. Measurements are generally made during expiration to avoid the “dips” associated with negative intrapulmonary pressures.
Cardiac output also can be measured when the catheter is equipped with a thermistor near the catheter tip and a cardiac output computer is available (see Figure 21-6, B). With this information, four potential hemodynamic subsets may be encountered44:
• Normal cardiac output and normal pulmonary capillary wedge pressure (the normal situation)
• Normal cardiac output with high pulmonary capillary wedge pressure predisposing to edema (left-sided CHF with volume expansion)
• Low cardiac output and low pulmonary capillary wedge pressure (volume depletion, as with excessive diuresis)
• Low cardiac output and high pulmonary capillary wedge pressure (severe left-sided heart failure, cardiogenic shock)
The CVP also can be measured through dual-port Swan-Ganz catheters (see Figure 21-6, A). In biventricular CHF, both the wedge pressure and CVP are abnormally high (see Figures 21-7 and 21-8). With excessive diuresis of the patient, both pressures are reduced. A relatively common situation after diuresis in those with primary left-sided heart failure is persistently high wedge pressure with relatively low (<5 mm Hg) CVP and normal jugular venous pressure. This situation can lead to reduced right-sided filling and prerenal azotemia. Reducing the diuretic dosage improves cardiac output but may exacerbate pulmonary edema. Noninvasive determination of cardiac output is feasible with Doppler echocardiography, but requires specialized equipment and training. The greatest value of this technique is found in the following of trends, as the aortic velocity-time integral increases or decreases with changes in ventricular stroke volume.
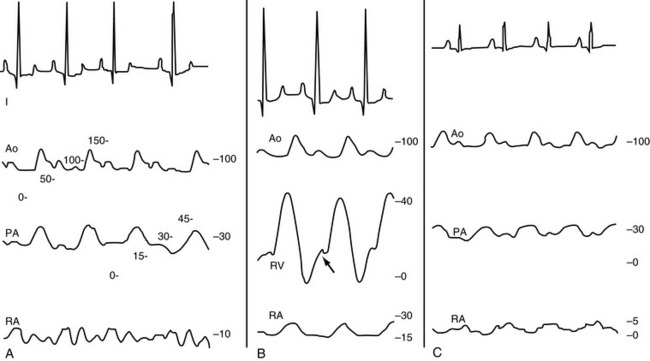
Figure 21-8 A, Electrocardiogram and pressure recordings from a standing dog with dilated cardiomyopathy and biventricular cardiac dysfunction. There is pulmonary hypertension secondary to abnormally high wedge pressure (not shown) and left-sided cardiac dysfunction. The pulmonary artery pressures are approximately 39/23 (systolic/diastolic) mm Hg. The right atrial pressure (central venous pressure) is also abnormally high, with a mean value of approximately 9 mm Hg. Scales indicate millimeters of mercury for the adjacent curves. Ao, Pressure in the descending aorta; PA, pressure in the pulmonary artery; RA, pressure in the right atrium. B, Electrocardiograph (ECG) and pressure recordings from a standing dog with dilated cardiomyopathy and mitral and tricuspid regurgitation during infusion of 0.9% saline solution. Volume expansion has led to a marked increase in the right atrial pressure (mean, 16 mm Hg). The right ventricular pressure (RV) tracing also indicates an increase in the end-diastolic ventricular pressure (arrow). Pulmonary hypertension from left-sided heart failure accounts for the increase in systolic RV pressure (42 mm Hg, normal is <25 mm Hg). Scales on the right indicate millimeters of mercury for the preceding curves. C, ECG and pressure recordings from a standing dog with mitral regurgitation and mild left-sided heart failure. Pulmonary hypertension secondary to abnormally high wedge pressure (not shown) and left-sided cardiac dysfunction are present. The pulmonary artery pressures are approximately 34/25 (systolic/diastolic) mm Hg. The wedge pressure in this case was approximately 23 mm Hg. Despite the high left-sided filling pressures, note that right atrial pressure is normal, with a mean (central venous pressure) value of approximately 3 mm Hg. Scales on the right indicate millimeters of mercury for the preceding curves.
1 Abbott J.A. Beta-blockade in the management of systolic dysfunction. Vet Clin North Am Small Anim Pract. 2004;34:1157-1170.
2 Adin D.B., Taylor A.W., Hill R.C., et al. Intermittent bolus injection versus continuous infusion of furosemide in normal adult greyhound dogs. J Vet Intern Med. 2003;17:632-636.
3 Amberger C.N., Glardon O., Glaus T., et al. Effects of benazepril in the treatment of feline hypertrophic cardiomyopathy: results of a prospective, open-label, multicenter clinical trial. J Vet Cardiol. 1999;1:19-26.
4 Asano K., Masuda K., Okumura M., et al. Plasma atrial and brain natriuretic peptide levels in dogs with congestive heart failure. J Vet Med Sci. 1999;61:523-529.
5 Atkins C.E., Brown W.A., Coats J.R., et al. Effects of long-term administration of enalapril on clinical indicators of renal function in dogs with compensated mitral regurgitation. J Am Vet Med Assoc. 2003;221:654-658.(Erratum in. J Am Vet Med Assoc. 2002;15:1149. 221
6 Atkins C.E., Keene B.W., Brown W.A., Coats J.R., Crawford M.A., DeFrancesco T.C., et al. Results of the veterinary enalapril trial to prove reduction in onset of heart failure in dogs chronically treated with enalapril alone for compensated, naturally occurring mitral valve insufficiency. J Am Vet Med Assoc. 2007;231(7):1061-1069.
7 Atkins C., Bonagura J., Ettinger S., Fox P., Gordon S., Haggstrom J., et al. Guidelines for the diagnosis and treatment of canine chronic valvular heart disease. J Vet Intern Med. 2009;23(6):1142-1150.
8 Auld R.B., Alexander E.A., Levinsky N.G. Proximal tubular function in dogs with thoracic caval constriction. J Clin Invest. 1971;50:2150-2158.
9 Barger A.C., Yates F.E., Rudolph A.M. Renal hemodynamics and sodium excretion in dogs with graded valvular damage, and in congestive failure. Am J Physiol. 1961;200:601-608.
10 Bernay F., Bland J.M., Häggström J., Baduel L., Combes B., Lopez A., et al. Efficacy of spironolactone on survival in dogs with naturally occurring mitral regurgitation caused by myxomatous mitral valve disease. J Vet Intern Med. 2010;24;2:331-341.(also see Letter to Editor:. J Vet Intern Med. 2010. in press
11 Bonagura J.D., Lehmkuhl L.B. Fluid therapy in heart failure. In: DiBartola S.J., editor. Fluid therapy in small animal practice. 1st ed. Philadelphia: WB Saunders; 1992:529-553.
12 Bonagura J.D., Ware W.A. Atrial fibrillation in the dog: clinical findings in 81 cases. J Am Anim Hosp Assoc. 1986;22:111-120.
13 Bourland W.A., Day D.K., Williams H.E. The role of the kidney in the early nondiuretic action of furosemide to reduce elevated left atrial pressure in the hypervolemic dog. J Pharmacol Exp Ther. 1977;202:221-229.
14 Bristow M.R. Mechanism of action of beta-blocking agents in heart failure. Am J Cardiol. 1997;80:26L-40L.
15 Calvert C.A., Rawlings C.A. Canine heartworm disease. In: Fox P.R., editor. Canine and feline cardiology. New York: Churchill Livingstone; 1988:519-549.
16 Cannon P.J. The kidney in heart failure. N Engl J Med. 1977;296:26-32.
17 Cataliotti A., Boerrigter G., Costello-Boerrigter L.C., et al. Brain natriuretic peptide enhances renal actions of furosemide and suppresses furosemide-induced aldosterone activation in experimental heart failure. Circulation. 2004;109:1680-1685.
18 Chen H.H., Grantham J.A., Schirger J.A., et al. Subcutaneous administration of brain natriuretic peptide in experimental heart failure. J Am Coll Cardiol. 2000;36:1706-1712.
19 Clark A.L., Poole-Wilson P.A., Coats A.J. Exercise limitation in chronic heart failure: central role of the periphery. J Am Coll Cardiol. 1996;28:1092-1102.
20 Cobb M., Michell A.R. Plasma electrolyte concentrations in dogs receiving diuretic therapy for cardiac failure. J Small Anim Pract. 1992;33:526-529.
21 Cogan M.G. Atrial natriuretic peptide. Kidney Int. 1990;37:1148-1160.
22 Cohn J.N., Levine T.B. Angiotensin-converting enzyme inhibition in congestive heart failure: the concept. Am J Cardiol. 1982;49:1480-1483.
23 Cohn J.N., Levine T.B., Olivar M.T., et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819-823.
24 Connolly D.J. Natriuretic peptides: the feline experience. Vet Clin North Am Small Anim Pract. 2010;40(4):559-570.
25 Co-operative Veterinary Enalapril (COVE) Study Group. Controlled clinical evaluation of enalapril in dogs with heart failure-results of the cooperative veterinary enalapril study group. J Vet Intern Med. 1995;9:243-252.
26 Creager M.A., Cusco J.A. Treatment of congestive heart failure with angiotensin-converting enzyme inhibitors. In: McCall D., Rahimtoola S.H., editors. Heart failure. New York: Chapman & Hall; 1995:316-344.
27 Creager M.A., Halperin J.L., Bernard D.B., et al. Acute regional circulatory and renal hemodynamic effects of converting-enzyme inhibition in patients with congestive heart failure. Circulation. 1981;64:483-489.
28 Decarli C., Spouse G., Larosa J.L. Serum magnesium levels in symptomatic atrial fibrillation and their relation to rhythm control by intravenous digoxin. Am J Cardiol. 1986;57:956-959.
29 DeFrancesco T.C., Rush J.E., Rozanski E.A., Hansen B.D., Keene B.W., Moore D.T., et al. Prospective clinical evaluation of an ELISA B-type natriuretic peptide assay in the diagnosis of congestive heart failure in dogs presenting with cough or dyspnea. J Vet Intern Med. 2007;21(2):243-250.
30 Dirks J.H., Cirksena W.J., Berliner R.W. Micropuncture study of the effect of various diuretics on sodium reabsorption by the proximal tubules of the dog. J Clin Invest. 1966;45:1875-1885.
31 Dormans T.P., Pickkers P., Russel F.G., et al. Vascular effects of loop diuretics. Cardiovasc Res. 1996;32:988-997.
32 Dormans T.P., van Meyel J.J., Gerlag P.G., et al. Diuretic efficacy of high dose furosemide in severe heart failure: bolus injection versus continuous infusion. J Am Coll Cardiol. 1996;28:376-382.
33 Dow S.J., Fettman M.J., Smith K.R., et al. Taurine depletion and cardiovascular disease in adult cats fed a potassium-depleted acidified diet. Am J Vet Res. 1992;53:402-405.
34 Dumont A.E., Clauss R.H., Reed G.E., et al. Lymph drainage in patients with congestive heart failure: comparison with findings in hepatic cirrhosis. N Engl J Med. 1963;269:949-952.
35 Dzau V.J., Hollenberg N.K. Renal response to captopril in severe heart failure: role of furosemide in natriuresis and reversal of hyponatremia. Ann Intern Med. 1984;100:777-781.
36 Dzau V.J., Swartz S.L. Dissociation of the prostaglandin and renin-angiotensin systems during captopril therapy for chronic congestive heart failure secondary to coronary artery disease. Am J Cardiol. 1987;60:1101-1105.
37 Dzau V.J., Colucci W.S., Hollenberg N.K., et al. Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation. 1981;63:645-651.
38 Dzau V.J., Packer M., Lilly L.S., et al. Prostaglandins in severe congestive heart failure: relation to activation of the renin-angiotensin system and hyponatremia. N Engl J Med. 1984;310:347-352.
39 Eaton G.E., Cody R.J., Brinkley P.F. Increase in aortic impedance precedes peripheral vasoconstriction at the early stage of ventricular failure in the paced canine model. Circulation. 1993;88:2714-2721.
40 Edwards J.N. Magnesium and congestive heart failure. Proceedings of the American College of Veterinary Internal Medicine. 1991:679-680. New Orleans
41 Fenelon G., Protter A.A., Stambler B.S. Examination of the in vivo cardiac electrophysiological effects of nesiritide (human brain natriuretic peptide) in conscious dogs. J Card Fail. 2002;8:320-325.
42 Feuerstein G.Z., Yue T.L., Cheng H.Y., et al. Myocardial protection by the novel vasodilating beta-blocker, carvedilol: potential relevance of anti-oxidant activity. J Hypertens Suppl. 1993;11:S41-S48.
43 Fichman M.P., Vorherr H., Kleeman C.R., et al. Diuretic-induced hyponatremia. Ann Intern Med. 1971;75:853-863.
44 Forrester J.S., Diamond G., Chatterjee K., et al. Medical therapy of acute myocardial infarction by application of hemodynamic subsets. N Engl J Med. 1976;295:1404-1413.
45 Franciosa J.A., Dunkman W.B., Wilen M., et al. Optimal” left ventricular filling pressure during nitroprusside infusion for congestive heart failure. Am J Med. 1983;74:457-464.
46 Francis G.S., McDonald K.M. Neurohumoral mechanisms in heart failure. In: McCall D., Rahimtoola S.H., editors. Heart failure. New York: Chapman & Hall; 1995:90-116.
47 Francis G.S., Goldsmith S.R., Levine T.B., et al. The neurohumoral axis in congestive heart failure. Ann Intern Med. 1984;101:370-377.
48 Francis G.S., Siegel R.M., Goldsmith S.R., et al. Acute vasoconstrictor response to intravenous furosemide in patients with chronic congestive heart failure. Activation of the neurohumoral axis. Ann Intern Med. 1985;103:1-6.
49 Francis G.S. Neurohumoral mechanisms involved in congestive heart failure. Am J Cardiol. 1985;55:15A-21A.
50 Freeman L.M., Rush J.E., Kehayias J.J., et al. Nutritional alterations and the effect of fish oil supplementation in dogs with heart failure. J Vet Intern Med. 1998;12:440-448.
51 Freeman R.H., Davis J.D., Williams G.M., et al. Effects of the oral angiotensin converting enzyme inhibitor, SQ 14225 in a model of low cardiac output in dogs. Circ Res. 1979;45:540-545.
52 Friedman E., Shadel M., Halkin H., et al. Thiazide-induced hyponatremia. Ann Intern Med. 1989;110:24-30.
53 Fulop M., Horowitz M., Aberman A., et al. Lactic acidosis in pulmonary edema due to left ventricular failure. Ann Intern Med. 1973;79:180-186.
54 Gaasch W.H., Freeman G.L. Functional properties of normal and failing hearts. In: McCall D., Rahimtoola S.H., editors. Heart failure. New York: Chapman & Hall; 1995:14-29.
55 Gerlag P.G.G., van Meijel J.J.M. High-dose furosemide in the treatment of refractory congestive heart failure. Arch Intern Med. 1988;148:286-291.
56 Gottlieb S.S., Weir M.R. Renal effects of angiotensin-converting enzyme inhibition in congestive heart failure. Am J Cardiol. 1990;66:14D-21D.
57 Gottlieb S.S., Abraham W., Butler J., et al. The prognostic importance of different definitions of worsening renal function in congestive heart failure. J Card Fail. 2002;8:136-141.
58 Granger J.P. Regulation of sodium excretion by renal interstitial hydrostatic pressure. Fed Proc. 1986;45:2892-2896.
59 Greco D.S., Biller B., Van Liew C.H. Measurement of plasma atrial natriuretic peptide as an indicator of prognosis in dogs with cardiac disease. Can Vet J. 2003;44:293-297.
60 Guyton A.C., Lindsey A.W. Effect of elevated left atrial pressure and decreased plasma protein concentration on the development of pulmonary edema. Circ Res. 1959;7:649-657.
61 Haggstrom J., Hansson K., Karlberg B.E., et al. Effects of long-term treatment with enalapril or hydralazine on the renin-angiotensin-aldosterone system and fluid balance in dogs with naturally acquired mitral valve regurgitation. Am J Vet Res. 1996;57:1645-1652.
62 Häggström J., Boswood A., O’Grady M., et al. Effect of pimobendan or benazepril hydrochloride on survival times in dogs with congestive heart failure caused by naturally occurring myxomatous mitral valve disease: the QUEST study. J Vet Intern Med. 2008;22(5):1124-1135.
63 Hall J.E., Guyton A.C., Jackson T.E., et al. Control of glomerular filtration rate by the renin-angiotensin system. Am J Physiol. 1977;233:F366-F372.
64 Hamlin R.L., Smith R.C., Powere T.E., et al. Efficacy of various diuretics in normal dogs. J Am Vet Med Assoc. 1965;146:1417-1420.
65 Heinemann H.O. Right-sided heart failure and the use of diuretics. Am J Med. 1978;64:367-369.
66 Higgins C.B., Vatner S.F., Eckberg D.L., et al. Alterations in the baroreceptor reflex in conscious dogs with heart failure. J Clin Invest. 1972:51715-51724.
67 Hirsch A.T., Muellerleile M., Dzau V.J. Cardiovascular tissue angiotensin systems: activation and actions in heart failure. In: McCall D., Rahimtoola S.H., editors. Heart failure. New York: Chapman & Hall; 1995:117-134.
68 Hirsch A.T., Pinto Y.M., Schunkert H., et al. Potential role of the tissue renin-angiotensin system in the pathophysiology of congestive heart failure. Am J Cardiol. 1990;66:22D-32D.
69 Hollander W., Judson W.E. The relationship of cardiovascular and renal hemodynamic function to sodium excretion in patients with severe heart disease but without edema. J Clin Invest. 1956;35:970-979.
70 Hollenberg N.K. Diuretics in congestive heart failure. In: Messerli F.N., editor. Cardiovascular drug therapy. 2nd ed. Philadelphia: WB Saunders; 1990:383-388.
70a Ichikawa I., pfeffer J.M., pfeffer M.A., et al. Role of Angiotensin II in the altered renal function of congestive heart failure, Circ Res. 1984;55:669-675.
71 IMPROVE Study Group. Acute and short-term hemodynamic, echocardiographic, and clinical effects of enalapril maleate in dogs with naturally acquired heart failure: results of the invasive multicenter prospective veterinary evaluation of enalapril study. J Vet Intern Med. 1995;9:234-242.
72 Jackson E.K. Diuretics. In: Hardman J.G., Limbird L.E., Molinoff P.B., et al, editors. Goodman & Gilman’s the pharmacological basis of therapeutics. 9th ed. New York: McGraw-Hill; 1996:685-714.
73 Jensen K.T., Carstens J., Pedersen E.B. Effect of BNP on renal hemodynamics, tubular function and vasoactive hormones in humans. Am J Physiol. 1998;274:F63-F72.
74 Jeunesse E., Woehrle F., Schneider M., Lefebvre H.P. Effect of spironolactone on diuresis and urine sodium and potassium excretion in healthy dogs. J Vet Cardiol. 2007;9(2):63-68.
75 Kaloyanides G.J. Pathogenesis and treatment of edema with special reference to the use of diuretics. In: Maxwell M.H., Kleeman C.R., editors. Clinical disorders of fluid and electrolyte metabolism. New York: McGraw-Hill; 1980:647-701.
76 Katz A.M. Cardiomyopathy of overload: a major determinant of prognosis in congestive heart failure. N Engl J Med. 1990;322:100-110.
77 Keane W.R., Shapiro B.E. Renal protective effects of angiotensin-converting enzyme inhibition. Am J Cardiol. 1990;65:491-531.
78 Keene B.W., Rush J.E. Therapy of heart failure. In: Ettinger S.J., editor. Textbook of veterinary internal medicine. 3rd ed. Philadelphia: WB Saunders; 1989:939-975.
79 Khanna C., Lund E.M., Raffe M., et al. Hypomagnesemia in 188 dogs: a hospital population-based prevalence study. J Vet Intern Med. 1998;12:304-309.
80 Kim S., Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev. 2001;52:11-34.
81 Kinugawa T., Thames M.C. Baroreflexes in heart failure. In: McCall D., Rahimtoola S.H., editors. Heart failure. New York: Chapman & Hall; 1995:30-45.
82 Kitagawa H., Wakamiya H., Kitoh K., et al. Efficacy of monotherapy with benazepril, an angiotensin converting enzyme inhibitor, in dogs with naturally acquired chronic mitral insufficiency. J Vet Med Sci. 1997;59:513-520.
83 Kittleson M., Keene B., Pion P.D., et al. Results of the multicenter spaniel trial (MUST): taurine and carnitine responsive dilated cardiomyopathy in American cocker spaniels with decreased plasma taurine concentration. J Vet Intern Med. 1997;11:204-211.
84 Knowlen G.G., Kittleson M.D., Nachreiner R.F., et al. Comparison of plasma aldosterone concentration among clinical status groups of dogs with chronic heart failure. J Am Vet Med Assoc. 1983;183:991-996.
85 Koch J., Pedersen H.D., Jensen A.L., et al. Activation of the renin-angiotensin system in dogs with asymptomatic and symptomatic dilated cardiomyopathy. Res Vet Sci. 1995;59:172-175.
86 Kubo S.H. Neurohormonal activation and the response to converting enzyme inhibitors in congestive heart failure. Circulation. 1990;81(Suppl. III):107-114.
87 Kvart C., Haggstrom J., Pedersen H.D., et al. Efficacy of enalapril for prevention of congestive heart failure in dogs with myxomatous valve disease and asymptomatic mitral regurgitation. J Vet Intern Med. 2002;16:80-88.
88 Levens N.R. Control of renal function by intrarenal angiotensin II in the dog. J Cardiovasc Pharmacol. 1990;16(Suppl. 4):565-569.
89 Levin A., Djurdjev O., Barret B., et al. Cardiovascular disease in patients with chronic kidney disease: getting to the heart of the matter. Am J Kidney Dis. 2001;38:1398-1407.
90 Levine T.B., Francis G.S., Goldsmith S.R., et al. Activity of the sympathetic nervous system and renin-angiotensin system assessed by plasma hormone levels and their relation to hemodynamic abnormalities in congestive heart failure. Am J Cardiol. 1982;49:1659-1666.
91 Levy M. Effects of acute volume expansion and altered hemodynamics on renal tubular function in chronic caval dogs. J Clin Invest. 1972;51:922-938.
92 Licata G., Di Pasquale P., Parrinello G., et al. Effects of high-dose furosemide and small-volume hypertonic saline solution infusion in comparison with a high dose of furosemide as bolus in refractory congestive heart failure: long-term effects. Am Heart J. 2003;145:459-466.
93 Lifschitz M.D., Schrier R.W. Alterations in cardiac output with chronic constriction of thoracic inferior vena cava. Am J Physiol. 1973;225:1364-1370.
94 Little C.J., Julu P.O., Hansen S., et al. Non-invasive real-time measurements of cardiac vagal tone in dogs with cardiac disease. Vet Rec. 2005;156:101-105.
95 Luis Fuentes V. Use of pimobendan in the management of heart failure. Vet Clin North Am Small Anim Pract. 2004;34:1145-1155.
96 MacDonald K.A., Kittleson M.D., Munro C., et al. Brain natriuretic peptide concentration in dogs with heart disease and congestive heart failure. J Vet Intern Med. 2003;17:172-177.
97 MacDonald K.A., Kittleson M.D., Kass P.H., White S.D. Effect of spironolactone on diastolic function and left ventricular mass in Maine coon cats with familial hypertrophic cardiomyopathy. J Vet Intern Med. 2008;22(2):335-341.
98 Margulies K.G., Hilderbrand F.L., Lerman A., et al. Increased endothelin in experimental heart failure. Circulation. 1990;82:2226-2232.
99 Mettauer B., Roulearu J.L., Bichet D., et al. Sodium and water excretion abnormalities in congestive heart failure. Ann Intern Med. 1986;105:161-167.
100 Michell A.R. Physiological aspects of the requirement for sodium in mammals. Nutr Res Rev. 1989;2:149-160.
101 Migdal S., Alexander E.A., Levinsky N.G. Evidence that decreased cardiac output is not the stimulus to sodium retention during acute constriction of the vena cava. J Lab Clin Med. 1977;89:809-816.
102 Millard R.W., Higgins C.B., Franklin D., et al. Regulation of the renal circulation during severe exercise in normal dogs and dogs with experimental heart failure. Circ Res. 1972;31:881-888.
103 Miller W.C., Simi W.W., Rice D.L. Contribution of systemic venous hypertension to the development of pulmonary edema in dogs. Circ Res. 1978;43:598-600.
104 Molina C.R., Fowler M.B., McCrory S., et al. Hemodynamic, renal, and endocrine effects of atrial natriuretic peptide infusion in severe heart failure. J Am Coll Cardiol. 1988;12:175-186.
105 Morita H., Suzuki G., Mishima T., et al. Effects of long-term monotherapy with metoprolol CR/XL on the progression of left ventricular dysfunction and remodeling in dogs with chronic heart failure. Cardiovasc Drugs Ther. 2002;16:443-449.
106 Morris M.L., Patton R.L., Teeter S.M. Low sodium diet in heart disease: how low is low? Vet Med Small Anim Clin. 1976;9:1225-1227.
107 Munagala V.K., Burnett J.C.Jr, Redfield M.M. The natriuretic peptides in cardiovascular medicine. Curr Probl Cardiol. 2004;29:707-769.
108 Naitoh M., Suzuki H., Murakami M., et al. Effects of oral AVP receptor antagonists OPC-21268 and OPC-31260 on congestive heart failure in conscious dogs. Am J Physiol. 1994;267:H2245-H2254.
109 Nomura A., Yasuda H., Minami M., et al. Effect of furosemide in congestive heart failure. Clin Pharmacol Ther. 1981;30:177-182.
110 O’Connor C.M., Gattis W.A., Swedberg K. Current and novel pharmacologic approaches in advanced heart failure. Am Heart J. 1998;135:S249-S263.
111 Oliver J.A., Sciacca R.R., Pinto J., et al. Participation of the prostaglandins in the control of renal blood flow during acute reduction of cardiac output in the dog. J Clin Invest. 1981;67:229-237.
112 Opie L.H., Kaplan N.M. Diuretics. In: Opie L.H., Gersh B.J., editors. Drugs for the Heart. 6th ed. Philadelphia: Elsevier/WB Saunders; 2005:80-103.
113 Opie L.H. Heart physiology: from cell to circulation, 4th ed. Baltimore: Lippincott Williams & Wilkins, 2003.
114 Osborn J.L., Holdaas H., Thames M.D., et al. Renal adrenoreceptor mediation of antinatriuretic and renin secretion responses to low frequency renal nerve stimulation in the dog. Circ Res. 1983;53:298-305.
115 Oster J.R., Epstein M., Smoller S. Combination therapy with thiazide-type and loop diuretic agents for resistant sodium retention. Ann Intern Med. 1983;99:405-406.
116 Packer M., Lee W.H., Kessler P.D. Preservation of glomerular filtration rate in human heart failure by activation of the renin-angiotensin system. Circulation. 1986;74:766-774.
117 Packer M., Lee W.H., Medina N., et al. Functional renal insufficiency during long-term therapy with captopril and enalapril in severe chronic heart failure. Ann Intern Med. 1987;106:346-354.
118 Packer M., Lee W.H., Medina N., et al. Influence of renal function on the hemodynamic and clinical responses to long-term captopril therapy in severe chronic heart failure. Ann Intern Med. 1986;104:147-154.
119 Packer M., McMurray J., Massie B.M., et al. Clinical effects of endothelin receptor antagonism with bosentan in patients with severe chronic heart failure: results of a pilot study. J Card Fail. 2005;11:12-20.
120 Packer M., Medina N., Yshak M. Correction of dilutional hyponatremia in severe chronic heart failure by converting-enzyme inhibition. Ann Intern Med. 1984;100:782-789.
121 Packer M. Neurohumoral interactions and adaptations in congestive heart failure. Am J Cardiol. 1985;55:1A-10A.
122 Paradiso G., Bakris G.L., Stein J.H. The kidney and heart failure. In: McCall D., Rahimtoola S.H., editors. Heart failure. New York: Chapman & Hall; 1995:135-158.
123 Parmley W.W. Pathophysiology of congestive heart failure. Am J Cardiol. 1985;55:9A-14A.
124 Patak R.V., Fadem S.Z., Rosenblatt S.G., et al. Diuretic-induced changes in renal blood flow and prostaglandin E excretion in the dog. Am J Physiol. 1979;236:F494-F500.
125 Pedersen H.D., Koch J., Poulsen K., et al. Activation of the renin-angiotensin system in dogs with asymptomatic and mildly symptomatic mitral valvular insufficiency. J Vet Intern Med. 1995;9:328-331.
126 Pedersen H.D., Schutt T., Sondergaard R., et al. Decreased plasma concentration of nitric oxide metabolites in dogs with untreated mitral regurgitation. J Vet Intern Med. 2003;17:178-184.
127 Pedersen H.D. Effects of mild mitral-valve insufficiency, sodium-intake, and place of blood-sampling on the renin-angiotensin system in dogs. Acta Vet Scand. 1996;37:109-118.
128 Pion P.D., Kittleson M.D. Therapy for feline aortic thromboembolism. In: Kirk R.W., editor. Current veterinary therapy X. Philadelphia: WB Saunders; 1989:295-301.
129 Pitt B., Zannad F., Remme W.J., et al. The randomized aldactone evaluation study investigators: the effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341:709-717.
130 Pouchelon J.L., (for the Benazepril in Canine Heart Disease Study Group (BENCH). The effect of benazepril on survival times and clinical signs of dogs with congestive heart failure: results of the multicenter, prospective, randomized, double-blind, placebo-controlled, long term clinical trial. J Vet Cardiol. 1999;1:7-18.
131 Priebe H., Heimann J.C., Hedley-Whyte J. Effects of renal and hepatic venous congestion on renal function in the presence of low and normal cardiac output in dogs. Circ Res. 1980;47:883-890.
132 Prosek R., Sisson D.D., Oyama M.A., et al. Plasma endothelin-1 immunoreactivity in normal dogs and dogs with acquired heart disease. J Vet Intern Med. 2004;18:840-844.
133 Prosek R., Sisson D.D., Oyama M.A., Solter P.F. Distinguishing cardiac and noncardiac dyspnea in 48 dogs using plasma atrial natriuretic factor, B-type natriuretic factor, endothelin, and cardiac troponin-I. J Vet Intern Med. 2007;21(2):238-242.
134 Reinhardt H.W., Kaczmarczy K.G., Eisele R., et al. Left atrial pressure and sodium balance in conscious dogs on a low sodium intake. Pflugers Arch. 1977;370:59-66.
135 Riegger G.A., Elsner D., Kroner E.P., et al. Atrial natriuretic peptide in congestive heart failure in the dog: plasma levels, cyclic guanidine monophosphate, ultrastructure of atrial myoendocrine cells, and hemodynamic, hormonal, and renal effects. Circulation. 1988;77:398-406.
136 Riegger G.A., Liebau G., Kochsiek K. Antidiuretic hormone in congestive heart failure. Am J Med. 1982;72:49-52.
137 Riegger G.A., Liebau G., Holzschuh M., et al. Role of the renin-angiotensin system in the development of congestive heart failure in the dog as assessed by chronic converting-enzyme blockade. Am J Cardiol. 1984;53:614-618.
138 Ritz E., Nowack R. Detrimental and beneficial effects of converting enzyme inhibitors on the kidney. J Cardiol Pharmacol. 1990;16(Suppl. 4):570-575.
139 Roche B.M., Schwartz D., Lehnhard R.A., et al. Changes in concentrations of neuroendocrine hormones and catecholamines in dogs with myocardial failure induced by rapid ventricular pacing. Am J Vet Res. 2003;63:1413-1417.
140 Rose B.D., Post T.W. Clinical physiology of acid base and electrolyte disorders, 5th ed, New York: McGraw Hill; 2001:702.
141 Rose B.D., Post T.W. Clinical physiology of acid base and electrolyte disorders, 5th ed, New York: McGraw Hill; 2001:677.
142 Rose B.D. Diuretics. Kidney Int. 1991;39:336-352.
143 Roudebush P., Allen T.A., Kuehn N.F., et al. The effect of combined therapy with captopril, furosemide, and a sodium-restricted diet on serum electrolyte concentrations and renal function in normal dogs and dogs with congestive heart failure. J Vet Intern Med. 1994;8:337-342.
144 Rush J.E., Freeman L.M., Brown D.J., et al. Clinical, echocardiographic, and neurohormonal effects of a sodium-restricted diet in dogs with heart failure. J Vet Intern Med. 2000;14:513-520.
145 Rush J.E., Freeman L.M., Brown D.J., et al. The use of enalapril in the treatment of feline hypertrophic cardiomyopathy. J Am Anim Hosp Assoc. 1998;34:38-41.
146 Ryan M.P. Diuretics and potassium/magnesium depletion: directions for treatment. Am J Med. 1987;82(Suppl. 3A):38-47.
147 Sabbah H.N., Sharov V.G., Gupta R.C., et al. Chronic therapy with metoprolol attenuates cardiomyocyte apoptosis in dogs with heart failure. J Am Coll Cardiol. 2000;36:1698-1705.
148 Sabbah H.N., Shimoyama H., Kono T., et al. Effects of long-term monotherapy with enalapril, metoprolol, and digoxin on the progression of left ventricular dysfunction and dilation in dogs with reduced ejection fraction. Circulation. 1994;89:2852-2859.
149 Sayer M.B., Atkins C.E., Fujii Y., Adams A.K., DeFrancesco T.C., Keene B.W. Acute effect of pimobendan and furosemide on the circulating renin-angiotensin-aldosterone system in healthy dogs. J Vet Intern Med. 2009;23(5):1003-1006.
150 Schrier R.W., Abraham W.T. Hormones and hemodynamics in heart failure. N Engl J Med. 1999;341:577-585.
151 Schrier R.W., Humphreys M.H., Ufferman R.C. Role of cardiac output and the autonomic nervous system in the antinatriuretic response to acute constriction of the thoracic superior vena cava. Circ Res. 1971;29:490-498.
152 Schrier R.W. Pathogenesis of sodium and water retention in high output and low output cardiac failure, nephrotic syndrome, cirrhosis and pregnancy. N Engl J Med. 1988;319:1065-1072. 1127–1134
153 Seely J.F., Dirks J.H. Site of action of diuretic drugs. Kidney Int. 1977;11:1-8.
154 Shimoyama H., Sabbah H.N., Rosman H., et al. Effects of long-term therapy with enalapril on severity of functional mitral regurgitation in dogs with moderate heart failure. J Am Coll Cardiol. 1995;25:768-772.
155 Sisson D.D. Neuroendocrine evaluation of cardiac disease. Vet Clin North Am Small Anim Pract. 2004;34:1105-1126.
156 Starling E.H. The influence of mechanized factors on lymph production. J Physiol (Lond). 1894;10:14-155.
157 Staub N.C. Pathophysiology of pulmonary edema. In: Staub N.C., Taylor A.E., editors. Edema. New York: Raven Press; 1984:719-746.
158 Suki W.N., Rector F.C.Jr, Seldin D.W. The site of action of furosemide and other sulfonamide diuretics in the dog. J Clin Invest. 1965;44:1458-1469.
159 Suki W.N. Renal hemodynamic consequences of angiotensin-converting enzyme inhibition in congestive heart failure. Arch Intern Med. 1989;149:669-673.
160 Sun Y., Weber K.T. Cardiac remodelling by fibrous tissue-Role of local factors and circulating hormones. Ann Med. 1998;30(Suppl. 1):S3-S8.
161 Suzuki G., Morita H., Mishima T., et al. Effects of long-term monotherapy with eplerenone, a novel aldosterone blocker, on progression of left ventricular dysfunction and remodeling in dogs with heart failure. Circulation. 2002;106:2967-2972.
162 Swales J.D. Magnesium deficiency and diuretics. BMJ. 1982;285:1377-1378.
163 Swan H.J.C., Ganz W., Forrester J., et al. Catheterization of the heart in man with use of a flow-directed balloon-tipped catheter. N Engl J Med. 1970;283:447-451.
164 Tachibana H., Cheng H.J., Ukai T., et al. Levosimendan improves LV systolic and diastolic performance at rest and during exercise after heart failure. Am J Physiol (Heart Circ Physiol). 2005;288:H914-H922.
165 Taylor A.E. Capillary fluid filtration: Starling forces and lymph flow. Circ Res. 1982;49:557-575.
166 Thames M.D. Acetylstrophanthidin-induced reflex inhibition of canine renal sympathetic nerve activity mediated by cardiac receptors with vagal afferents. Circ Res. 1979;44:8-15.
167 Thomas C.J., Woods R.L. Haemodynamic action of B-type natriuretic peptide substantially outlasts its plasma half-life in conscious dogs. Clin Exp Pharmacol Physiol. 2003;30:369-375.
168 Thomas J.A., Marks B.H. Plasma norepinephrine in congestive heart failure. Am J Cardiol. 1978;41:233-243.
169 Thrall D.E., Calvert C.A. Radiographic evaluation of canine heartworm disease coexisting with right heart failure. Vet Radiol. 1983;24:124-126.
170 Uechi M., Matsuoka M., Kuwajima E., et al. The effects of the loop diuretics furosemide and torsemide on diuresis in dogs and cats. J Vet Med Sci. 2003;65:1057-1061.
171 Uechi M., Sasaki T., Ueno K., et al. Cardiovascular and renal effects of carvedilol in dogs with heart failure. J Vet Med Sci. 2002;64:469-475.
172 van der Zander K., Houben A.J., Kroon A.A., et al. Does brain natriuretic peptide have a direct renal effect in human hypertensives? Hypertension. 2003;41:119-123.
173 Van Veldhuisen D.J., Genth-Zoth S., Brouwer J., et al. High-versus low-dose ACE inhibition in chronic heart failure: a double-blind, placebo-controlled study of imidapril. J Am Coll Cardiol. 1998;32:1811-1818.
174 Vasko M.R., Brown-Cartwright D., Knochel J.P., et al. Furosemide absorption altered in decompensated congestive heart failure. Ann Intern Med. 1985;102:314-318.
175 Vollmar A.M., Montag C., Preusser U., et al. Atrial natriuretic peptide and plasma volume of dogs suffering from heart failure or dehydration. Zentralbl Veterinarmed A. 1994;41:548-557.
176 Vollmar A.M., Preusser U., Gerbes A.L., et al. Endothelin concentration in plasma of healthy dogs and dogs with congestive heart failure, renal failure, diabetes mellitus, and hyperadrenocorticism. J Vet Intern Med. 1995;9:105-111.
177 Wang W. Chronic administration of aldosterone depresses baroreceptor reflex function in the dog. Hypertension. 1994;24:571-575.
178 Ware W.A., Lund D.D., Subieta A.R., et al. Sympathetic activation in dogs with congestive heart failure caused by chronic mitral valve disease and dilated cardiomyopathy. J Am Vet Med Assoc. 1990;197:1475-1481.
179 Watkins L.Jr, Burton J.A., Haber E., et al. The renin-angiotensin-aldosterone system in congestive failure in conscious dogs. J Clin Invest. 1976;57:1606-1617.
180 Weaver L.J., Carrico C.J. Congestive heart failure and edema. In: Staub N.C., Taylor A.E., editors. Edema. New York: Raven Press; 1984:543-562.
181 Weber K.T. Aldosterone and spironolactone in heart failure [editorial]. N Engl J Med. 1999;341:709-717.
182 Weber K.T. Furosemide in the long-term management of heart failure: the good, the bad, and the uncertain. J Am Coll Cardiol. 2004;44:1308-1310.
183 Wiener-Kronish J.P., Goldstein R., Matthay R.A., et al. Lack of association of pleural effusion with chronic pulmonary arterial and right atrial hypertension. Chest. 1987;92:967-970.
184 Yatsu T., Tomura Y., Tahara A., et al. Pharmacological profile of YM087, a novel nonpeptide dual vasopressin V1A and V2 receptor antagonist, in dogs. Eur J Pharmacol. 1997;321:225-230.
185 Zacà V., Rastogi S., Mishra S., Wang M., Sharov V.G., Gupta R.C., et al. Atenolol is inferior to metoprolol in improving left ventricular function and preventing ventricular remodeling in dogs with heart failure. Cardiology. 2009;112(4):294-302.
186 Zucker I.H., Share L., Gilmore J.P. Renal effects of left atrial distension in dogs with chronic congestive heart failure. Am J Physiol. 1979;236:H554-H560.