Chapter 13 Retinal Vascular Disease
Retinal circulation
Arterial system
Capillaries
Retinal capillaries supply the inner two-thirds of the retina, with the outer third being supplied by the choriocapillaris. The inner capillary network (plexus) is located in the ganglion cell layer, with an outer plexus in the inner nuclear layer. Capillary-free zones are present around arterioles (Fig. 13.1A) and at the fovea (foveal avascular zone – FAZ). Retinal capillaries are devoid of smooth muscle and elastic tissue and their walls consist of the following (Fig. 13.1B):
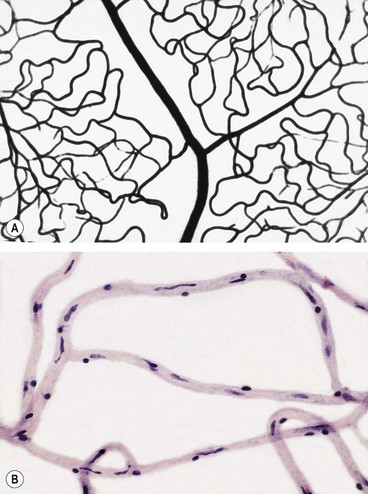
Fig. 13.1 Normal retinal capillary bed. (A) Periarteriolar capillary-free zone – flat preparation of Indian ink-injected retina; (B) endothelial cells with elongated nuclei and pericytes with rounded nuclei – trypsin digest preparation
(Courtesy of J Harry and G Misson, from Clinical Ophthalmic Pathology, Butterworth-Heinemann 2001)
Diabetic retinopathy
Introduction
Prevalence
The reported prevalence of diabetic retinopathy (DR) varies substantially between studies, even amongst contemporary diabetic populations in the same country, but is probably up to 40%. It is more common in type 1 diabetes than in type 2 and sight-threatening disease is present in up to 10%. Proliferative diabetic retinopathy (PDR) affects 5–10% of the diabetic population; type 1 diabetics are at particular risk with an incidence of about 60% after 30 years.
Risk factors
Pathogenesis
DR is predominantly a microangiopathy in which small blood vessels are particularly vulnerable to damage from hyperglycaemia. Direct hyperglycaemic effects on retinal cells are also likely to play a role.
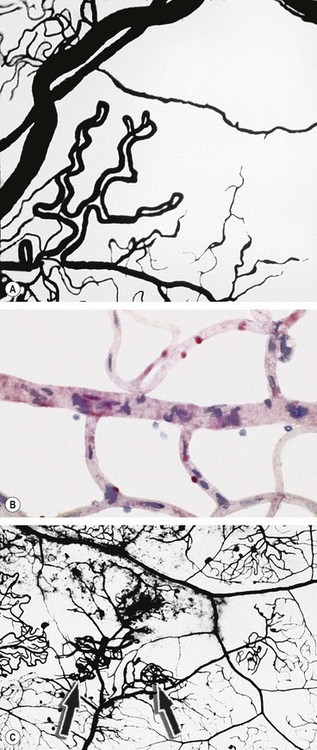
Fig. 13.2 The capillary bed in diabetic retinopathy. (A) Capillary closure with adjacent dilated and elongated capillaries – flat preparation of Indian ink-injected retina; (B) degenerate pericytes which are eosinophilic – trypsin digest preparation; (C) new capillaries (arrows) on the inner retinal surface growing from vessels in relation to non-perfused areas – flat preparation of Indian ink-injected retina
(Courtesy of J Harry and G Misson, from Clinical Ophthalmic Pathology, Butterworth-Heinemann 2001)
Many angiogenic stimulators have been identified; vascular endothelial growth factor (VEGF), especially VEGF-A, appears to be of particular importance. Others include platelet-derived growth factor and hepatocyte growth factor. Similarly, several endogenous inhibitors of angiogenesis have also been reported such as endostatin, angiostatin and pigment epithelium-derived factor. It has been hypothesized that a key determinant of the activity of retinopathy is the net balance between VEGF and endostatin.
Classification
The classification used in the Early Treatment Diabetic Retinopathy Study (the modified Airlie House classification) is widely used internationally. An abbreviated version is set out in Table 13.1, in conjunction with management guidelines. The following descriptive categories are also in widespread use in clinical practice:
Table 13.1 Abbreviated Early Treatment Diabetic Retinopathy Study classification of diabetic retinopathy
| Category/description | Management |
|---|---|
| Non-proliferative diabetic retinopathy (NPDR) | |
| No DR | Review in 12 months |
| Very mild | Review most patients in 12 months |
| Microaneurysms only | |
| Mild | Review range 6–12 months, depending on severity of signs, stability, systemic factors, and patient’s personal circumstances |
| Any or all of: microaneurysms, retinal haemorrhages, exudates, cotton wool spots, up to the level of moderate NPDR. No IRMA or significant beading | |
| Moderate | Review in approximately 6 months PDR in up to 26%, high-risk PDR in up to 8% within a year |
| Severe | Review in 4 months PDR in up to 50%, high-risk PDR in up to 15% within a year |
| The 4-2-1 rule; one or more of: |
|
| Very severe | Review in 2–3 months High-risk PDR in up to 45% within a year |
| Two or more of the criteria for severe | |
| Proliferative diabetic retinopathy (PDR) | |
| Mild-moderate | Treatment considered according to severity of signs, stability, systemic factors, and patient’s personal circumstances such as reliability of attendance for review. If not treated, review in up to 2 months |
| New vessels on the disc (NVD) or new vessels elsewhere (NVE), but extent insufficient to meet the high-risk criteria | |
| High-risk | Treatment advised – see text Should be performed immediately when possible, and certainly same day if symptomatic presentation with good retinal view |
| Advanced diabetic eye disease | See text |
| See text for description | |
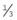 disc area)
disc area)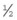 disc area with vitreous or preretinal haemorrhage (or haemorrhage with presumed obscured NVD/E)
disc area with vitreous or preretinal haemorrhage (or haemorrhage with presumed obscured NVD/E)Signs
Figure 13.3 shows the location of lesions in background diabetic retinopathy.
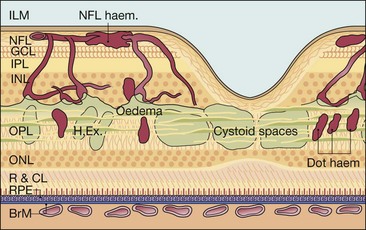
Microaneurysms
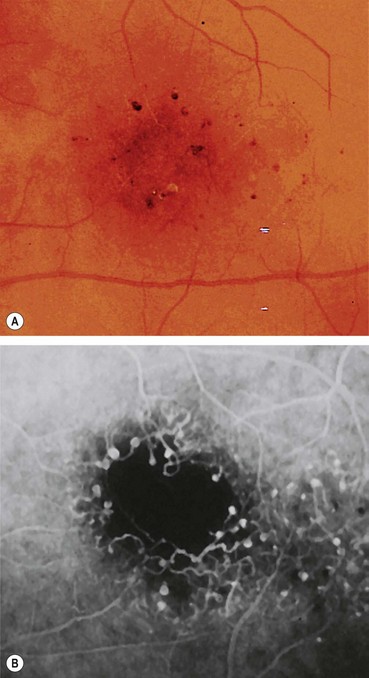
Microaneurysms are localized out-pouchings, mainly saccular, of the capillary wall that may form either by focal dilatation of the capillary wall where pericytes are absent, or by fusion of two arms of a capillary loop (Fig. 13.4A). Most develop in the inner capillary plexus (inner nuclear layer) frequently in relation to areas of capillary non-perfusion (Fig. 13.4B). Loss of pericytes may also lead to endothelial cell proliferation with the formation of ‘cellular’ microaneurysms (Fig. 13.4C). Microaneurysms may leak plasma constituents into the retina as a result of breakdown in the blood–retinal barrier, or become thrombosed (Fig. 13.4D).
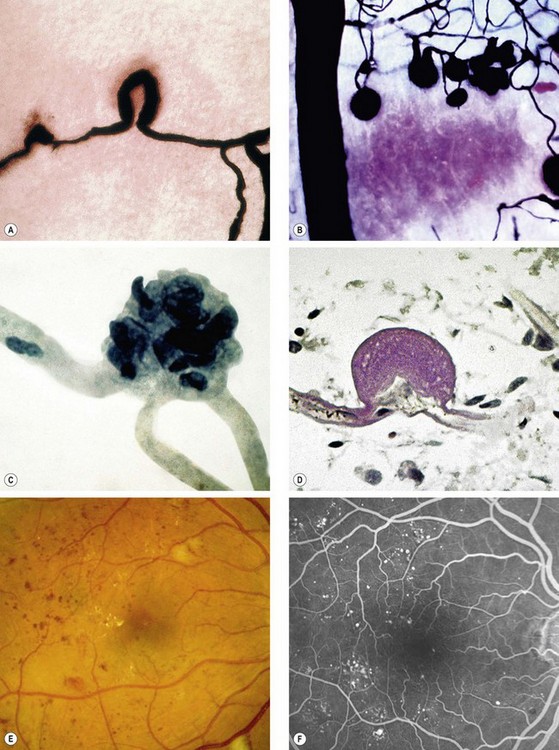
Fig. 13.4 Retinal microaneurysm. (A) Two arms of a capillary loop are not yet fused to become a microaneurysm – flat preparation of Indian ink-injected retina; (B) an area of capillary non-perfusion and adjacent microaneurysms – flat preparation of Indian ink-injected retina; (C) microaneurysm with endothelial cell proliferation (cellular microaneurysm) – trypsin digest preparation; (D) thrombosed microaneurysm – PAS and haematoxylin stain; (E) microaneurysms at the posterior pole; (F) FA shows scattered hyperfluorescent spots in the posterior fundus
(Courtesy of J Harry and G Misson, from Clinical Ophthalmic Pathology, Butterworth-Heinemann 2001 – fig. A; J Harry – figs B–D)
Retinal haemorrhages
Figure 13.5A is a histological section showing the location of blood.
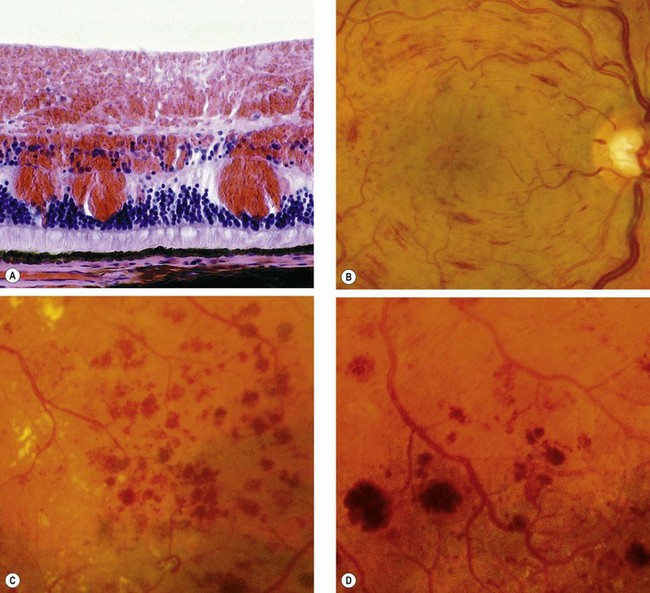
Fig. 13.5 Retinal haemorrhages. (A) Histology shows blood lying diffusely in the retinal nerve fibre and ganglion cell layers and as globules in the outer layers; (B) retinal nerve fibre layer haemorrhages; (C) deep dot and blot haemorrhages; (D) deep dark haemorrhages
(Courtesy of J Harry and G Misson, from Clinical Ophthalmic Pathology, Butterworth-Heinemann 2001 – fig. A; Moorfields Eye Hospital – fig. C)
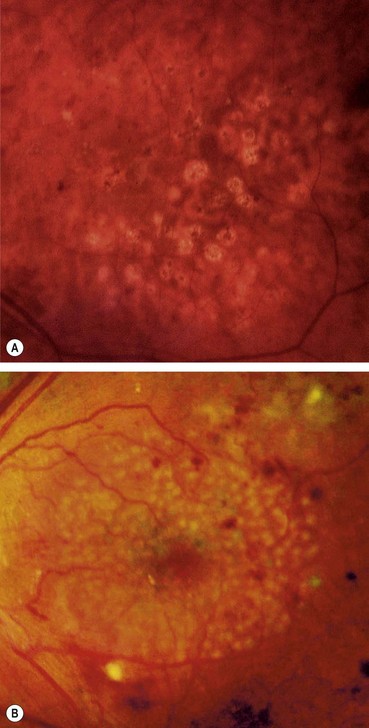
Exudates
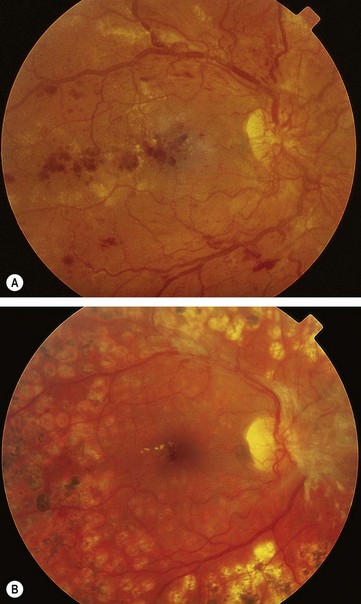
Exudates, sometimes termed ‘hard’ exudates to distinguish from the older term of ‘soft’ exudates for cotton wool spots, are caused by chronic localized retinal oedema and develop at the junction of normal and oedematous retina. They are composed of lipoprotein and lipid-filled macrophages located mainly within the outer plexiform layer (Fig. 13.6A). Hyperlipidaemia may increase the likelihood of exudate formation.
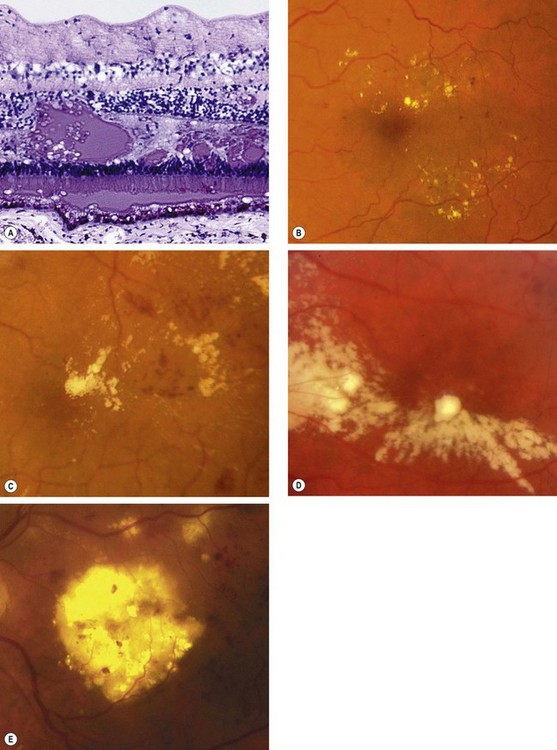
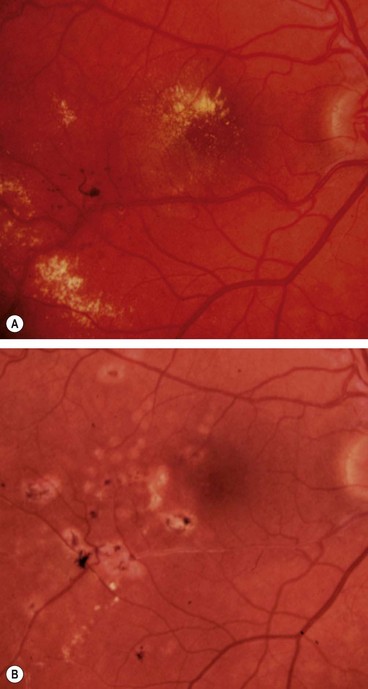
Fig. 13.6 Exudates. (A) Histology shows irregular eosinophilic deposits mainly in the outer plexiform layer; (B) small exudates and microaneurysms; (C) incomplete ring of exudates and a few small haemorrhages; (D) exudates involving the fovea; (E) plaque of exudates at the macula associated with cholesterol deposition
(Courtesy of J Harry – fig. A)
Diabetic macular oedema
Diabetic maculopathy (foveal oedema, exudates or ischaemia) is the most common cause of visual impairment in diabetic patients, particularly type 2. Diffuse retinal oedema is caused by extensive capillary leakage, and localized oedema by focal leakage from microaneurysms and dilated capillary segments. The fluid is initially located between the outer plexiform and inner nuclear layers; later it may also involve the inner plexiform and nerve fibre layers, until eventually the entire thickness of the retina becomes oedematous. With further accumulation of fluid the fovea assumes a cystoid appearance (cystoid macular oedema – CMO).
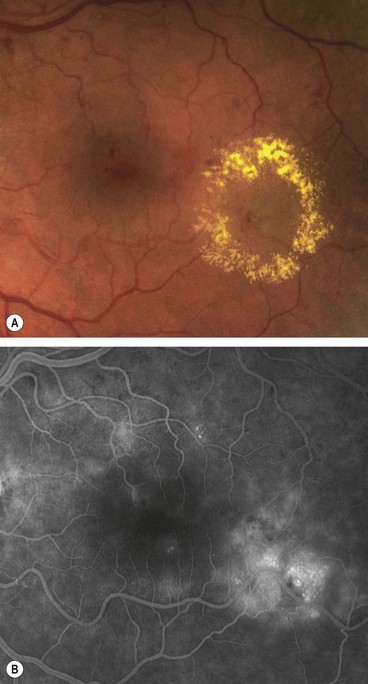
Focal maculopathy
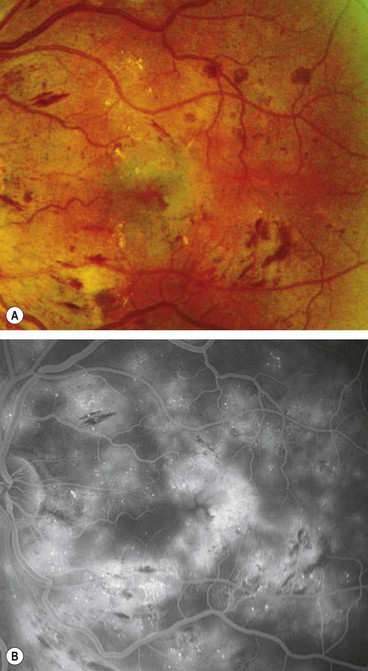
Diffuse maculopathy
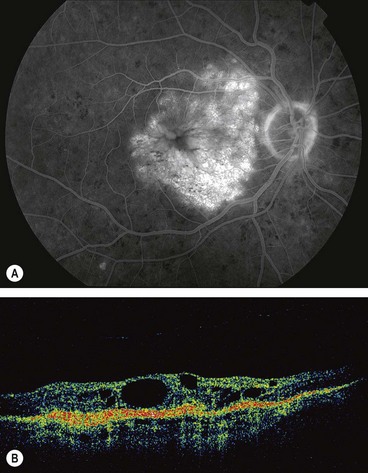
Ischaemic maculopathy
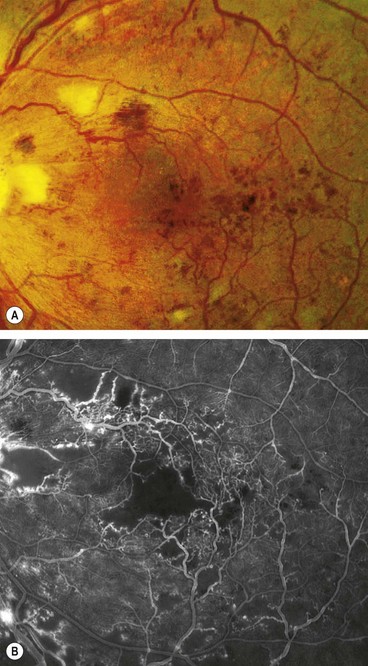
Clinically significant macular oedema
Clinically significant macular oedema (CSMO) was defined in the ETDRS (Fig. 13.11):
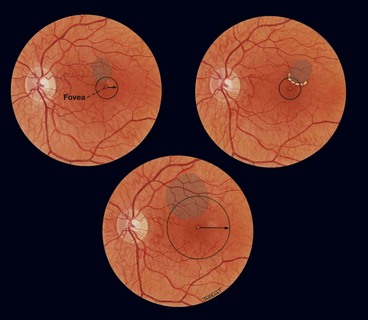
Cotton wool spots
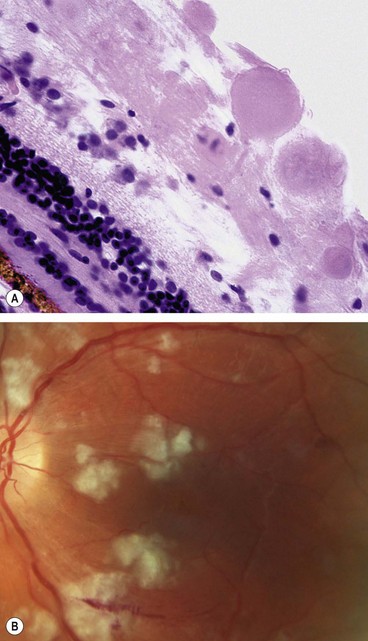
Cotton wool spots are composed of accumulations of neuronal debris within the nerve fibre layer. They result from disruption of nerve axons, the swollen ends of which are known as cytoid bodies, seen on light microscopy as globular structures in the nerve fibre layer (Fig. 13.12A). As cotton wool spots heal, debris is removed by autolysis and phagocytosis.
Venous changes
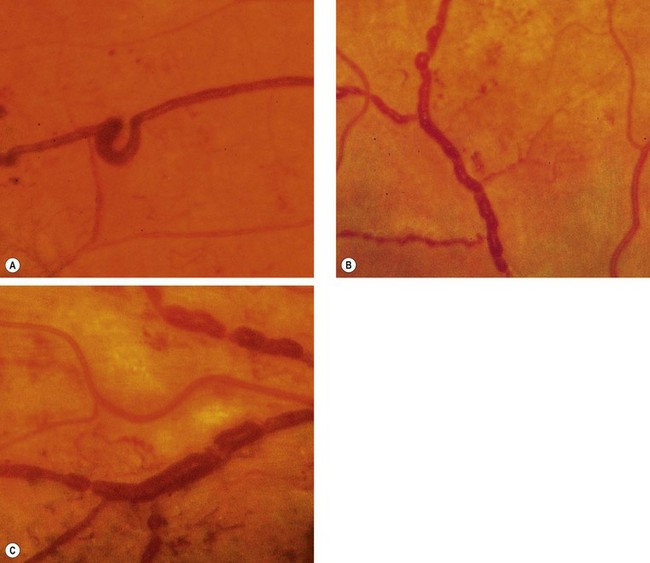
Venous anomalies seen in ischaemia consist of generalized dilatation and tortuosity, ‘looping’ (Fig. 13.13A), ‘beading’ (focal narrowing and dilatation – Fig. 13.13B) and ‘sausage-like’ segmentation (Fig. 13.13C). The extent of the retinal area exhibiting venous changes correlates well with the likelihood of developing proliferative disease.
Intraretinal microvascular abnormalities
Intraretinal microvascular abnormalities (IRMA) are arteriolar-venular shunts that run from retinal arterioles to venules, thus bypassing the capillary bed and are therefore often seen adjacent to areas of marked capillary hypoperfusion (Fig. 13.14A).
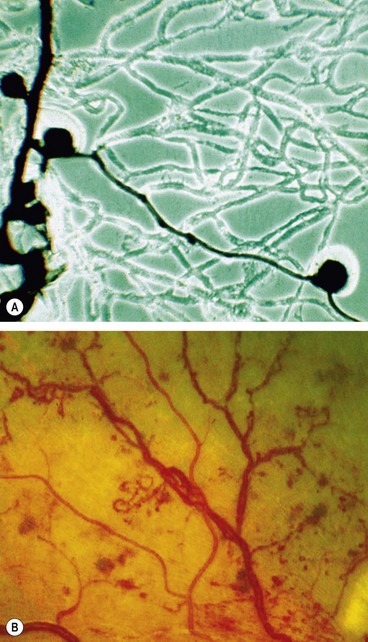
Fig. 13.14 Intraretinal microvascular abnormalities. (A) Histology shows arteriolar-venular shunt and a few microaneurysms within a poorly perfused capillary bed – flat preparation of Indian ink-injected retina; phase contrast microscopy; (B) clinical appearance
(Courtesy of J Harry – fig. A; Moorfields Eye Hospital – fig. B)
Arterial changes
Subtle retinal arteriolar dilatation may be an early marker of ischaemic dysfunction. When significant ischaemia is present these include peripheral narrowing, silver-wiring and obliteration (Fig. 13.15), similar to the late appearance following a branch retinal artery occlusion.
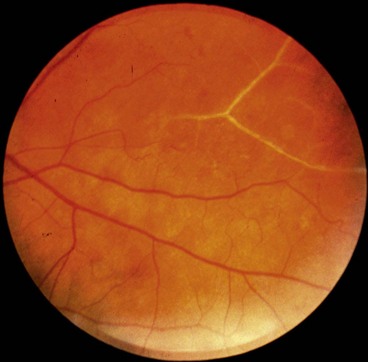
Proliferative retinopathy
It has been estimated that over one-quarter of the retina must be non-perfused before PDR develops. Although preretinal new vessels may arise anywhere in the retina, they are most commonly seen at the posterior pole. Fibrous tissue, initially fine, gradually develops in association as vessels increase in size.
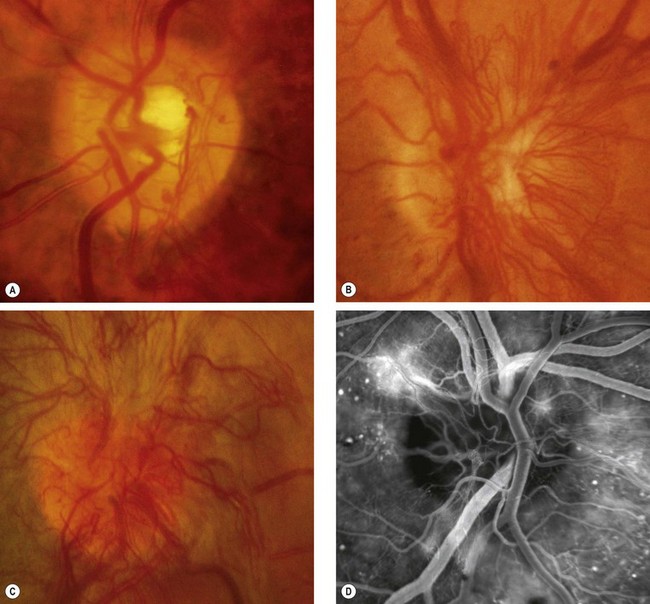
Treatment
Argon laser treatment of clinically significant macular oedema
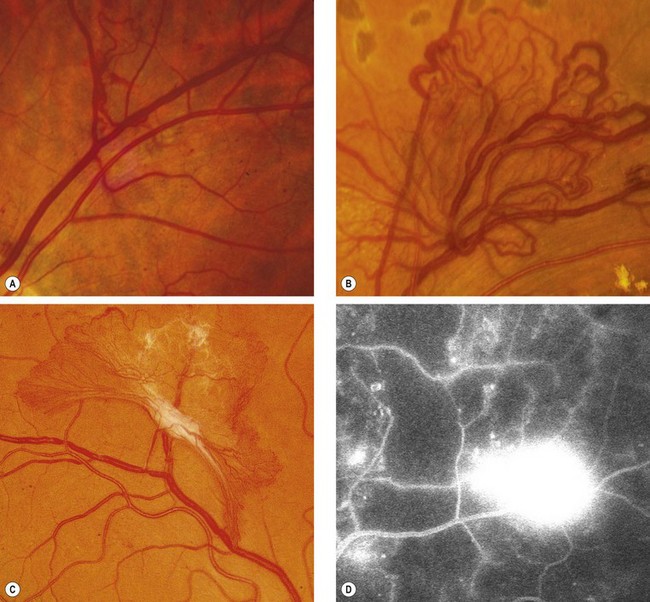
Other treatments for maculopathy
Argon laser remains the primary therapeutic modality, but numerous other forms of treatment have shown promising results.
Laser photocoagulation for proliferative retinopathy
The Diabetic Retinopathy Study (DRS) established the characteristics of high-risk proliferative disease and investigated the effect of panretinal photocoagulation (PRP). The benefits demonstrated included:
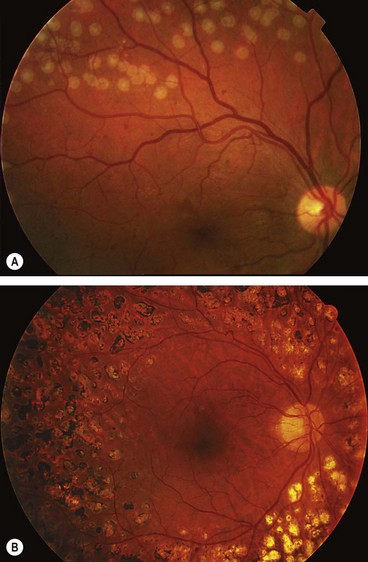
Fig. 13.19 (A) Appropriate laser burns; (B) appearance several weeks after completion of treatment
(Courtesy of C Barry – fig. B)
In very severe PDR it is advisable to treat the inferior fundus first, since any vitreous haemorrhage will gravitate inferiorly and obscure this area, precluding further treatment.
VEGF inhibition for proliferative retinopathy
Intravitreal anti-VEGF injection is likely to have an increasing role in the treatment of PDR, probably as an adjunct to laser. A particular indication may be to encourage the resolution of persistent vitreous haemorrhage, avoiding vitrectomy in some patients.
Advanced diabetic eye disease
Advanced diabetic eye disease is a serious vision-threatening complication of DR that occurs in patients in whom treatment has been inadequate or unsuccessful. Occasionally, advanced disease is evident at, or prompts, presentation.
Diagnosis
Indications for pars plana vitrectomy
Visual results of pars plana vitrectomy
Visual results depend on the specific indications for surgery and the complexity of pre-existing vitreoretinal abnormalities. In general, about 70% of cases achieve visual improvement, about 10% are made worse and the rest are unchanged. It appears that the first few postoperative months are vital. If an eye is doing well after 6 months, then the long-term outlook is good because the incidence of subsequent vision-threatening complications is low. Favourable prognostic factors include:
Retinal venous occlusive disease
Pathogenesis
Arteriolosclerosis is an important causative factor for branch retinal vein occlusion (BRVO). Because a retinal arteriole and its corresponding vein share a common adventitial sheath, thickening of the arteriole appears to compress the vein. This causes secondary changes, including venous endothelial cell loss, thrombus formation and potential occlusion. Similarly, the central retinal vein and artery share a common adventitial sheath at arteriovenous crossings posterior to the lamina cribrosa so that atherosclerotic changes of the artery may compress the vein and precipitate central retinal vein occlusion (CRVO). It therefore appears that both arterial and venous disease contribute to retinal vein occlusion. Venous occlusion causes elevation of venous and capillary pressure with stagnation of blood flow. Stagnation results in hypoxia of the retina drained by the obstructed vein, which, in turn, results in damage to the capillary endothelial cells and extravasation of blood constituents. The tissue pressure is increased, causing further stagnation of the circulation and hypoxia, so that a vicious cycle is established.
Predisposing factors
Common
Uncommon
Uncommon predispositions (listed below) may assume more importance in patients under the age of 50 years.
Factors that appear to decrease the risk of venous occlusion include increased physical activity and moderate alcohol consumption.
Systemic assessment
All patients
Selected patients according to clinical indication
Patients in whom these might be considered are those under the age of 50, those with bilateral RVO, a history of previous thromboses or a family history of thrombosis, and possibly in other patients in whom investigation for the common associations is negative.
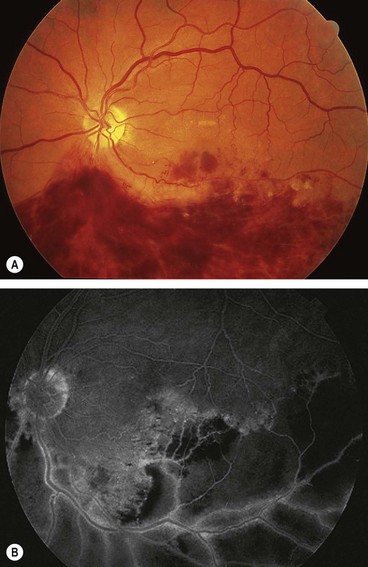
Branch retinal vein occlusion
Classification
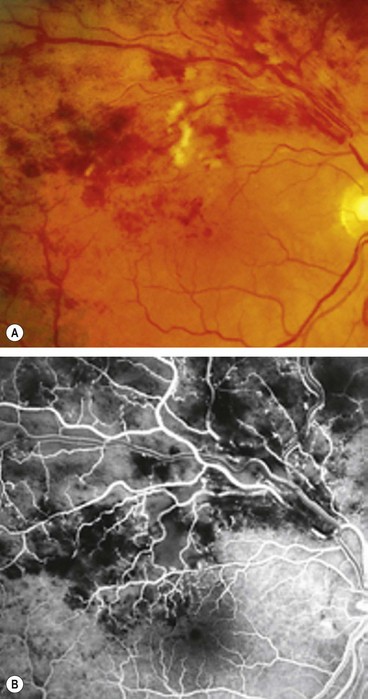
Diagnosis
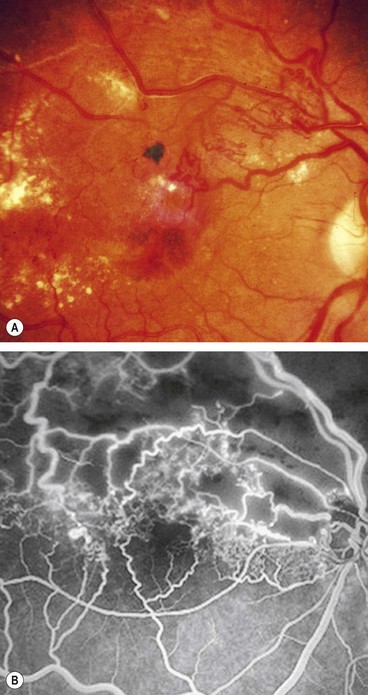
Prognosis
At 6 months about 50% of eyes achieve vision of 6/12 or better. Approximately 50% of untreated eyes with BRVO retain 6/12 or better whilst 25% will have vision of <6/60. The two main vision-threatening complications are:
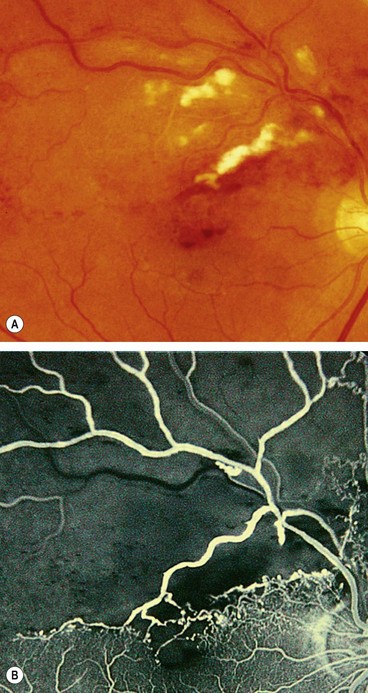
Further management
Follow-up should be at about 3 months with FA, if vision is compromised, provided retinal haemorrhages have cleared sufficiently. Further management depends on visual acuity and angiographic findings.
Treatment of macular oedema
Treatment of neovascularization
Neovascularization is not normally treated unless vitreous haemorrhage occurs because early treatment does not appear to affect the visual prognosis. If appropriate, scatter laser photocoagulation (200–500 µm size, 0.05–0.1 s duration and spaced one burn width apart) is performed with sufficient energy to achieve a medium reaction covering the entire involved sector (Fig. 13.31) as defined by the colour photograph and FA. A quadrant usually requires 400–500 burns. Follow-up should be after 4–6 weeks. If neovascularization persists re-treatment can be considered, and is usually effective in inducing regression.
Impending central retinal vein occlusion
Impending (partial) CRVO is a relatively poorly-defined condition which may resolve or progress to complete obstruction.
Non-ischaemic central retinal vein occlusion
Non-ischaemic CRVO is the most common type, accounting for about 75%.
Diagnosis
Follow-up
In a clearly non-ischaemic occlusion, initial follow-up should take place after 3 months. Defined arrangements for review of test results should be in place. The patient should be instructed to make contact if the vision deteriorates as this may indicate the development of significant ischaemia. Pain or redness (may indicate neovascular glaucoma and occasionally inflammation without rubeosis) should also be reported. Subsequent review is dependent on the clinical picture, with discharge from follow-up usually at 18–24 months.
Prognosis
In cases that do not subsequently become ischaemic, the prognosis is reasonably good with return of vision to normal or near normal in about 50%. The main cause for poor vision is chronic macular oedema (Fig. 13.34B), which may lead to secondary RPE changes. To a certain extent the prognosis is related to initial visual acuity as follows:
Treatment of macular oedema
Laser photocoagulation for macular oedema is not beneficial. Some of the following novel therapies have exhibited apparent significant benefit and may play an increasing role in management.
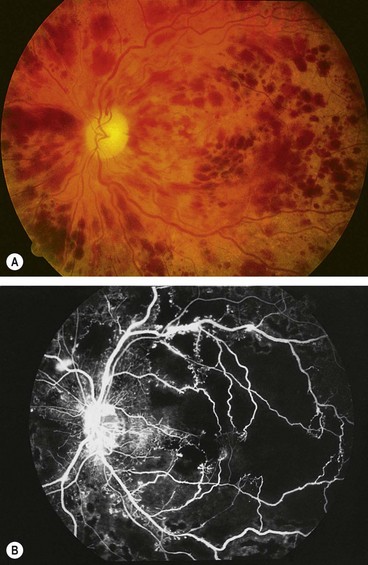
Ischaemic central retinal vein occlusion
Ischaemic CRVO is characterized by rapid onset venous obstruction resulting in decreased retinal perfusion, capillary closure and retinal hypoxia. This may lead to profound vascular leakage, rubeosis iridis and raised intraocular pressure. Neovascular glaucoma is one of the most common indications for enucleation in the Western world.
Diagnosis
Prognosis
The prognosis is extremely poor due to macular ischaemia. Rubeosis iridis develops in about 50% of eyes, usually between 2 and 4 months (100-day glaucoma), and there is a high risk of neovascular glaucoma. The development of opticociliary shunts (retinochoroidal collateral veins) may protect the eye from anterior segment neovascularization and probably indicates a dramatic reduction in risk. Retinal neovascularization occurs in about 5% of eyes and is therefore much less common than with BRVO.
Follow up
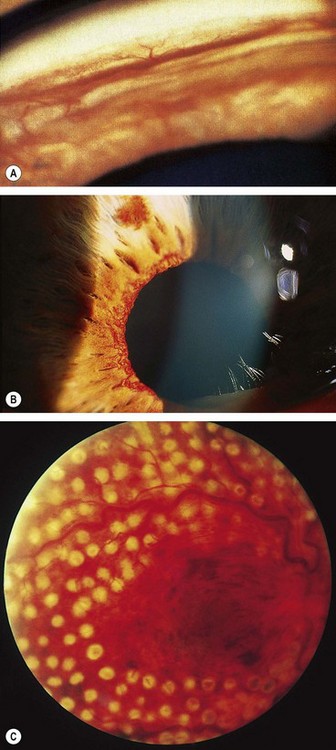
Where possible, patients with ischaemic CRVO should be seen monthly for 6 months to detect the onset of anterior segment neovascularization. Angle neovascularization (Fig. 13.36A), while not synonymous with progression to neovascular glaucoma, is the best clinical predictor of its development because it may occur in the absence of neovascularization at the pupillary margin (Fig. 13.36B). Routine gonioscopy of eyes at risk should therefore be performed and the pupillary margin should be examined prior to mydriasis. Prophylactic PRP is generally not recommended even with marked ischaemia unless iris new vessels develop, though may be considered in patients unlikely to attend scheduled review. Subsequent review should usually be for up to 2 years to detect significant ischaemia and macular oedema.
Treatment of neovascularization
Laser PRP should be performed without delay in eyes with angle neovascularization or rubeosis iridis. This involves the application of 1500–3000 burns (0.5–0.1 second, spaced one burn width apart), with sufficient energy to produce a moderate reaction in the periphery but avoiding areas of haemorrhage (Fig. 13.36C). Some cases require further treatment if rubeosis fails to regress or continues to progress. Intravitreal anti-VEGF injections may be used adjunctivally in selected cases.
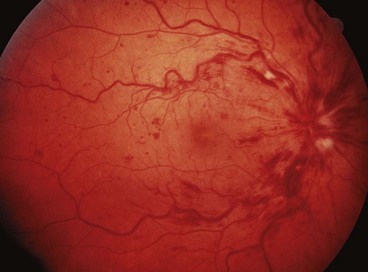
Papillophlebitis
Papillophlebitis (optic disc vasculitis) is an uncommon condition which typically affects otherwise healthy individuals under the age of 50 years. It is thought that the underlying lesion is optic disc swelling with resultant secondary venous congestion rather than venous thrombosis occurring at the level of the lamina cribrosa, as occurs in older patients.
Diagnosis
Hemiretinal vein occlusion
Hemiretinal vein occlusion is generally regarded as a variant of CRVO and may be ischaemic or non-ischaemic. It is less common than both BRVO and CRVO and involves occlusion of the superior or inferior branch of the CRV. A hemispheric occlusion blocks a major branch of the CRV at or near the optic disc. A hemicentral occlusion, which is less common, involves one trunk of a dual-trunked CRV, which persists in the anterior part of the optic nerve head as a congenital variant.
Systemic treatment in retinal vein occlusion
Retinal arterial occlusive disease
Aetiology
Atherosclerosis-related thrombosis
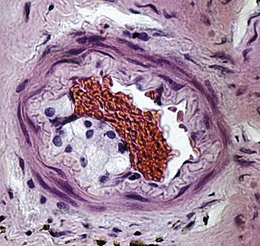
Atherosclerosis-related thrombosis at the level of the lamina cribrosa is by far the most common underlying cause of central retinal artery occlusion (CRAO), accounting for about 80% of cases. Atherosclerosis is characterized by focal intimal thickening comprising cells of smooth muscle origin, connective tissue and lipid-containing foam cells (Fig. 13.39). The incidence of atherosclerosis increases with age and is accelerated by hypertension, hyperlipidaemia, diabetes, oral contraceptives and hyperhomocysteinaemia. Other risk factors include obesity, tobacco smoking and a sedentary lifestyle.
Carotid embolism
Embolism is another key cause of retinal arterial compromise, including transient ischaemia. The origin of emboli is most commonly an atheromatous plaque at the carotid bifurcation, and less often the aortic arch and elsewhere. Since the ophthalmic artery is the first branch of the internal carotid artery, embolic material from the heart and carotid arteries has a fairly direct route to the eye. Carotid stenosis involves atheromatous narrowing, often associated with ulceration, at the bifurcation of the common carotid artery. The irregularity of the vessel wall may act as a source of cerebral and retinal emboli of the following types:
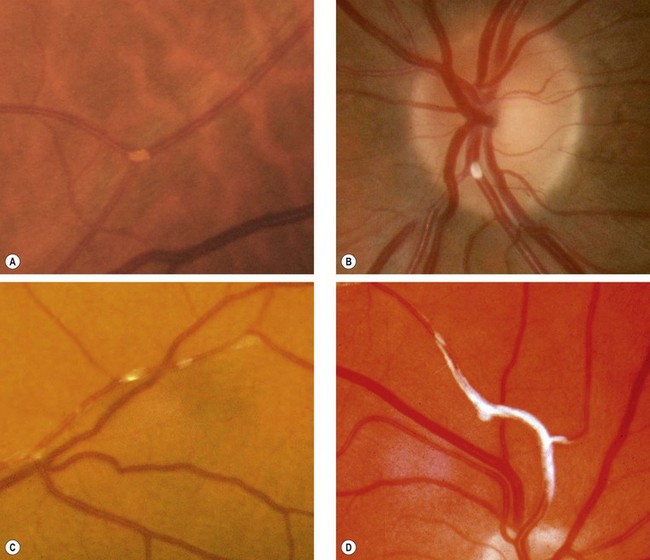
Uncommon causes
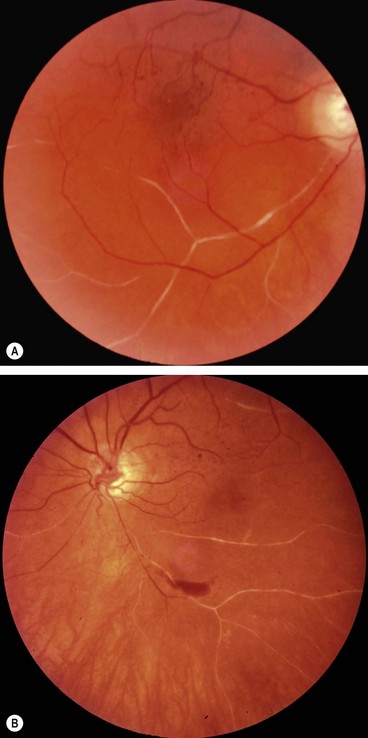
Systemic assessment
All patients
Many patients will have a history of vascular disease; enquiry should be made about smoking.
Selected patients
The following additional tests can be considered on a targeted basis in some patients, particularly if younger and with no known cardiovascular risk factors.
Amaurosis fugax
Amaurosis fugax is characterized by painless transient monocular loss of vision, often described as a curtain coming down over the eye, usually from top to bottom, but occasionally vice versa; it is common for patients to be unaware of whether transient unilateral visual loss affects one eye or the ipsilateral hemifield (cerebral ischaemia) of both. Visual loss, which may be complete, usually lasts a few minutes. Recovery is in the same pattern as the initial loss, although usually more gradual. Frequency of attacks may vary from several times a day to once every few months. The attacks may sometimes be accompanied by ipsilateral cerebral TIA, with contralateral neurological features. Investigation and systemic management is carried out as for persistent arterial occlusion.
Branch retinal artery occlusion
Diagnosis
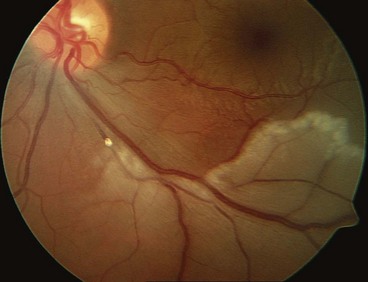
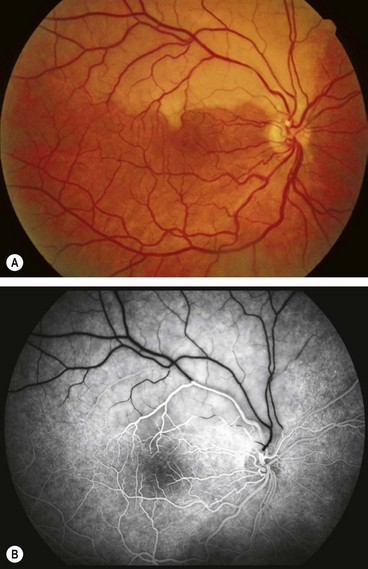
Follow-up
A single follow-up assessment in 3 months may be warranted to review the appearance of the fundus and provide advice on prognosis. This also provides an opportunity for a systemic management review, to ensure care by an appropriate specialist team was implemented.
Prognosis
In patients where central vision is severely compromised, the prognosis is commonly poor unless the obstruction is relieved within a few hours (see below). The visual field defect is likely to be permanent and the affected artery remains attenuated. Occasionally, recanalization of the obstructed artery may leave subtle or absent ophthalmoscopic signs.
Central retinal artery occlusion
Diagnosis
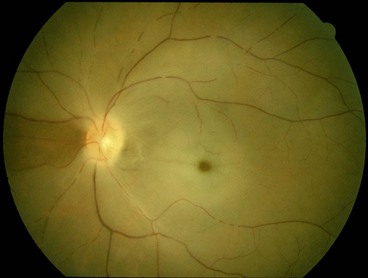
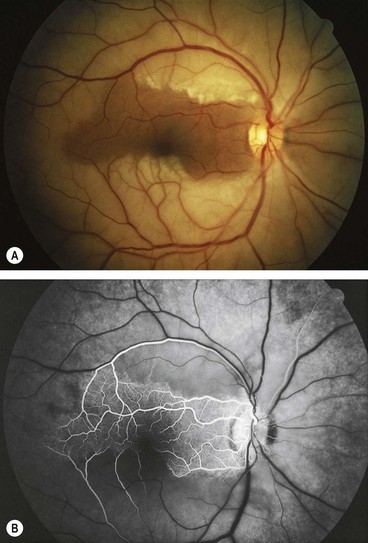
Follow-up
The patient should be seen by an ophthalmologist in 3–4 weeks and again a month later in order to detect incipient neovascularization, particularly of the anterior segment. In the minority of cases where referral to a specialist vascular team is not indicated, it should be ensured that the results of systemic investigations have been reviewed and necessary systemic treatment initiated.
Prognosis
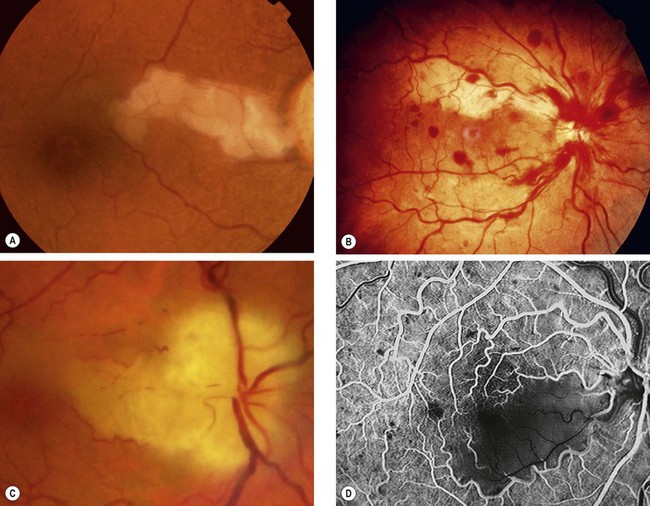
The prognosis is poor due to retinal infarction. Over a few days to weeks the retinal cloudiness and ‘cherry-red spot’ gradually disappear although the arteries remain attenuated. Later signs include optic atrophy (Fig. 13.46A), vessel sheathing, and patchy inner retinal atrophy and RPE changes. Histology shows atrophy of the inner retina and ganglion cells (Fig. 13.46B). Rubeosis iridis may occur in up to about 1 in 5, typically earlier than in CRVO (4–5 weeks compared with 3 months), and along with very poor vision may indicate ophthalmic artery occlusion. About 2% of eyes with CRAO develop retinal or disc neovascularization. PRP should be performed as for ischaemic CRVO, and intravitreal injection of vascular endothelial growth factor (VEGF) inhibitor might be considered.
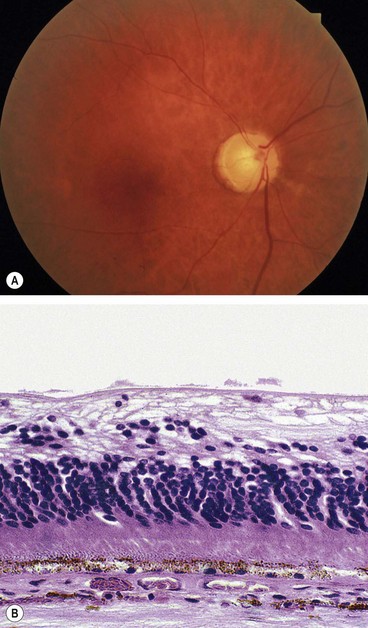
Cilioretinal artery occlusion
A cilioretinal artery, present in 20% of the population, arises from the posterior ciliary circulation but supplies the retina, commonly in the area of the macula and papillomacular bundle.
Treatment of acute retinal artery occlusion
Retinal artery occlusion is an emergency because it causes irreversible visual loss unless the retinal circulation is re-established prior to the development of retinal infarction. It appears that the prognosis for occlusions caused by calcific emboli is worse than those resulting from either cholesterol or platelet emboli. Theoretically, timely dislodgement of thrombus or emboli may ameliorate subsequent visual loss. The following treatments may be tried in patients with occlusions of less than 24–48 hours duration at presentation, though evidence of benefit is limited. The number of measures tried and the intensity of treatment should be tailored to the individual (more aggressive if lower duration of occlusion, good general health, monocularity; more aggressive systemic treatment may be avoided in the frail elderly); options, including the lack of evidence for clear benefit and the risks, should be discussed before use.
Systemic prophylaxis following retinal arterial occlusion
The risk of stroke is relatively high in the first few days following retinal artery occlusion or amaurosis fugax, and ‘fast-track’ referral to a specialist stroke clinic is advisable.
Asymptomatic retinal embolus
It is not uncommon to identify a retinal embolus on routine examination of an asymptomatic older patient. This indicates a substantially increased risk of stroke and ischaemic heart disease, and management should consist of evaluation and treatment of risk factors discussed above. A higher threshold for carotid surgery is appropriate.
Ocular ischaemic syndrome
Pathogenesis
Ocular ischaemic syndrome (OIS) is an uncommon condition which is the result of chronic ocular hypoperfusion secondary to severe ipsilateral atherosclerotic carotid stenosis of more than 90%, resulting in a 50% reduction of ipsilateral perfusion pressure. It typically affects patients during the 7th decade and may be associated with diabetes, hypertension, ischaemic heart disease and cerebrovascular disease. The male : female ratio is about 2 : 1. Five year mortality is in the order of 40%, most frequently from cardiac disease. Patients with ocular ischaemic syndrome may also give a history of amaurosis fugax due to retinal embolism.
Diagnosis
OIS is unilateral in 80% of cases. It affects both anterior and posterior segments. The signs are variable and may be subtle such that the condition is missed or misdiagnosed.
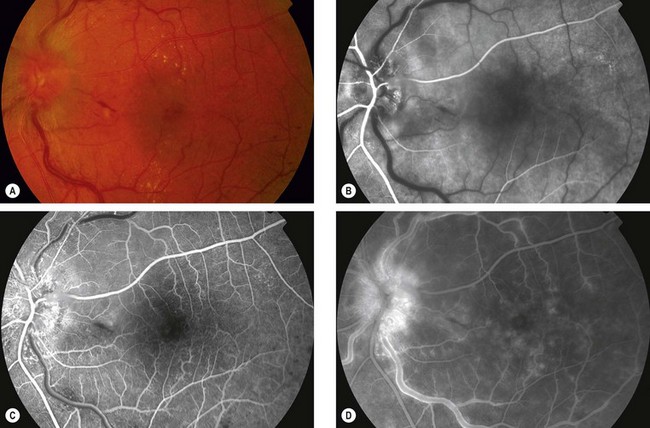
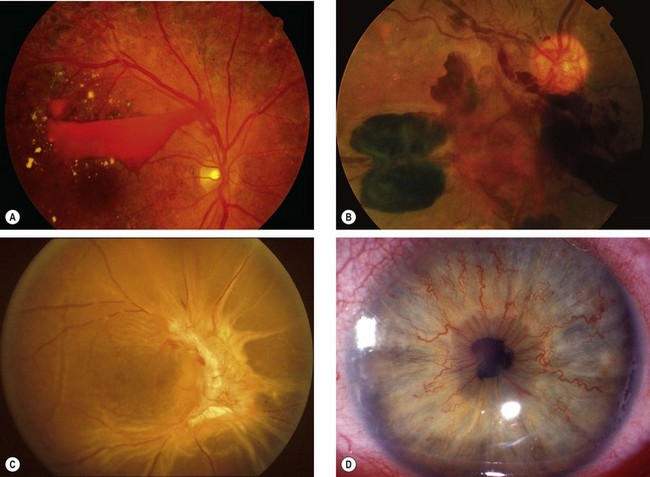
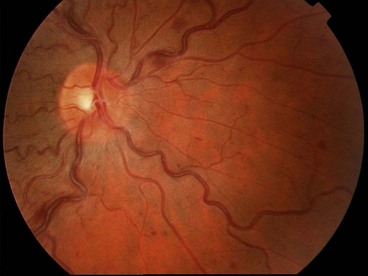
Fig. 13.48 Ocular ischaemic syndrome. (A) Venous dilatation, arteriolar narrowing, a few scattered flame-shaped haemorrhages and hard exudates, and disc oedema; (B and C) FA early phase shows delayed choroidal filling and prolonged arteriovenous transit; (D) FA late phase shows disc and perivascular hyperfluorescence, and spotty hyperfluorescence at the posterior pole due to leakage
(Courtesy of Moorfields Eye Hospital)
Management
Differential diagnosis
Hypertensive disease
Retinopathy
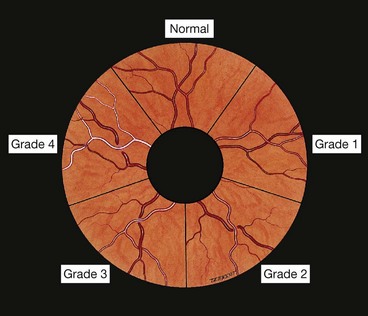
Retinopathy consists of a spectrum of retinal vascular changes that are pathologically related to both transient and persistent microvascular damage from elevated blood pressure. The primary response of the retinal arterioles to systemic hypertension is narrowing (vasoconstriction). However, the degree of narrowing is dependent on the amount of pre-existing replacement fibrosis (involutional sclerosis). For this reason, hypertensive narrowing is seen in its pure form only in young individuals. In older patients, rigidity of retinal arterioles due to involutional sclerosis prevents the same degree of narrowing seen in young individuals. In sustained hypertension the inner blood–retinal barrier is disrupted in localized areas, with increased vascular permeability.
Signs
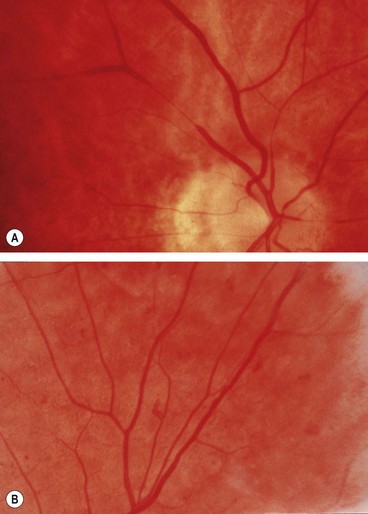
Fig. 13.49 Hypertensive retinopathy. (A) Focal arteriolar attenuation; (B) generalized arteriolar attenuation
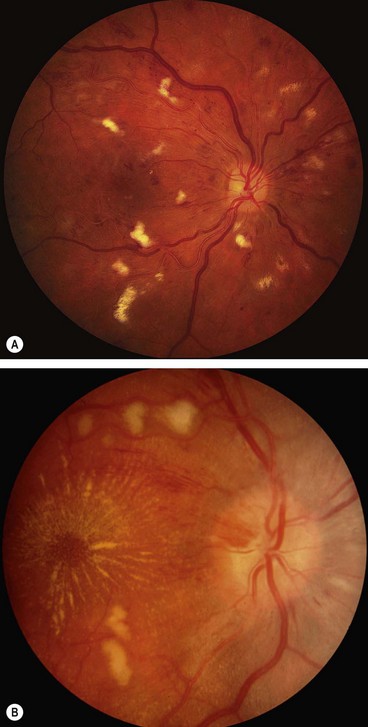
Fig. 13.50 Severe hypertensive retinopathy. (A) Cotton wool spots, a few flame-shaped haemorrhages and arteriolosclerosis; (B) cotton wool spots, macular star and mild disc swelling
(Courtesy of P Saine – fig. A)
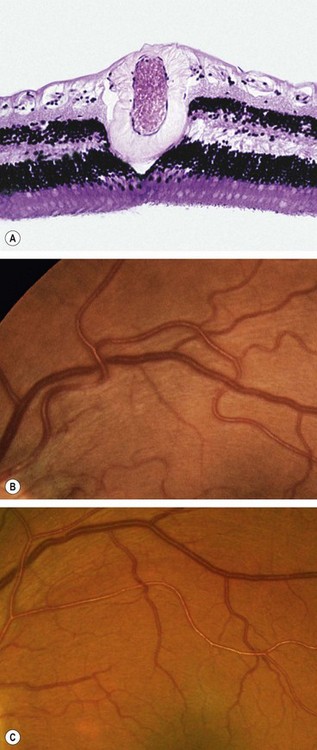
Choroidopathy
Choroidopathy is rare but may occur as the result of an acute hypertensive crisis (accelerated hypertension) in young adults.
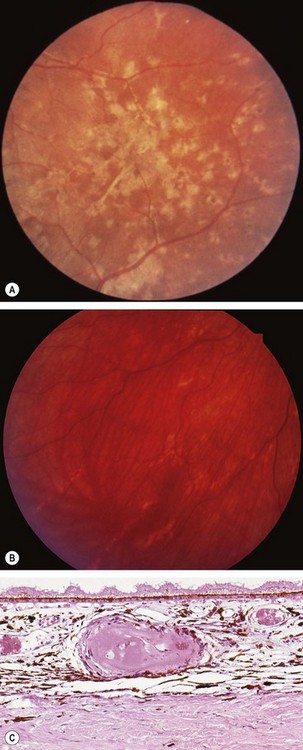
Sickle-cell retinopathy
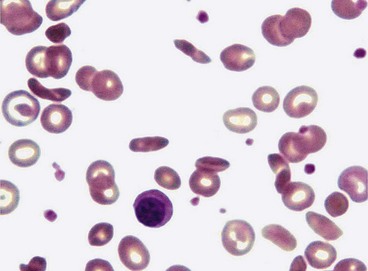
Sickling haemoglobinopathies
Sickling haemoglobinopathies are caused by one or more abnormal haemoglobins which induce the red blood cell to adopt an anomalous shape (Fig. 13.54) under conditions of hypoxia and acidosis. Because these deformed red blood cells are more rigid than healthy cells, they may become impacted in and obstruct small blood vessels. The sickling disorders in which the mutant haemoglobins S and C are inherited as alleles of normal haemoglobin A have important ocular manifestations. These abnormal haemoglobins may occur in combination with normal haemoglobin A or in association with each other as indicated below.
Proliferative retinopathy
Diagnosis
The development of proliferative retinopathy is usually insidious, and patients remain asymptomatic unless vitreous haemorrhage or retinal detachment occurs.
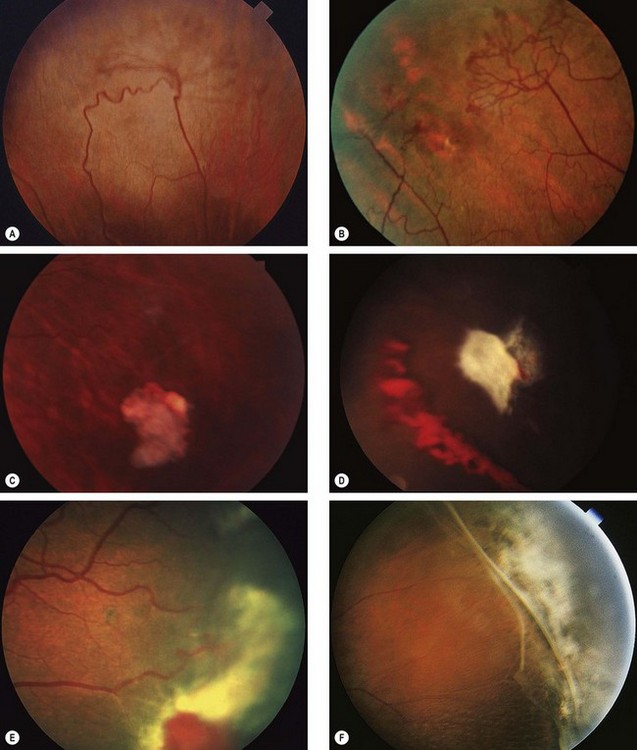
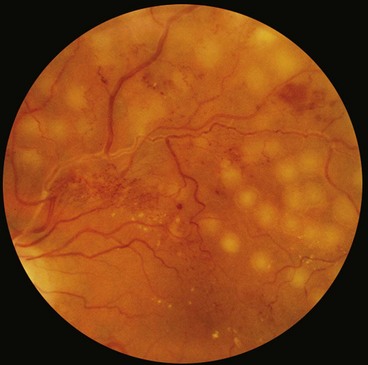
Fig. 13.55 Progression of proliferative sickle-cell retinopathy. (A) Peripheral arteriovenous anastomosis; (B) ‘sea-fan’ neovascularization; (C) spontaneous involution of a neovascular tuft; (D) haemorrhage due to traction; (E) extensive fibrovascular proliferation; (F) peripheral retinal detachment
(Courtesy of K Nischal – fig. A; R Marsh – figs B–F)
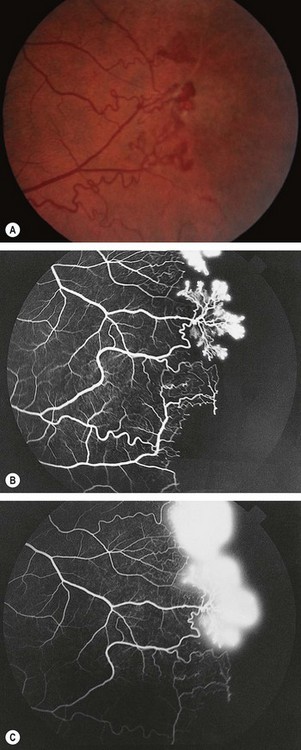
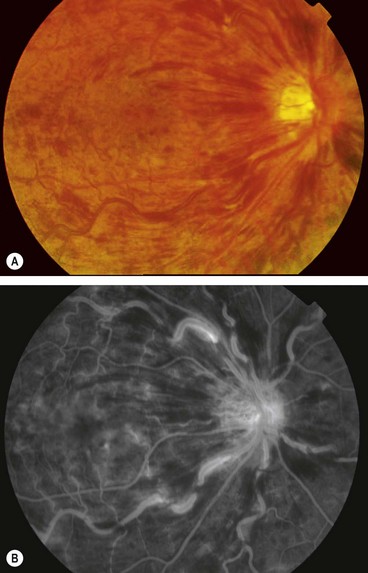
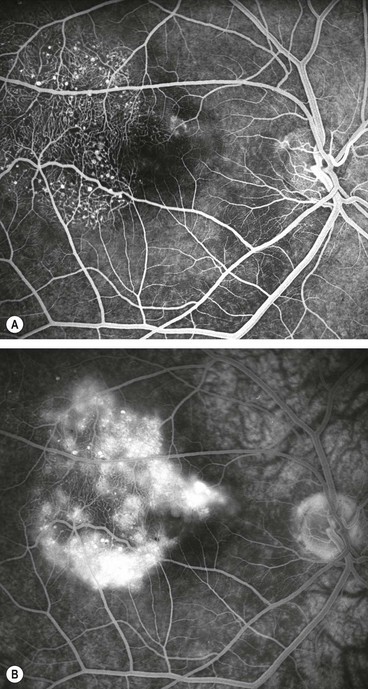
Fig. 13.56 (A) Proliferative sickle-cell retinopathy stage 3; (B) FA early phase shows filling of new vessels (sea-fans) and extensive peripheral retinal capillary non-perfusion; (C) late phase shows leakage from new vessels
FA in stage 3 shows filling of sea-fans and peripheral capillary non-perfusion (Fig. 13.56B) followed by extensive hyperfluorescence due to leakage from new vessels (Fig. 13.56C).
Treatment
Treatment is not required in most cases because new vessels tend to auto-infarct and involute spontaneously without treatment. PRP probably does not alter the natural history. Occasionally vitreoretinal surgery may be required for tractional retinal detachment and/or persistent vitreous haemorrhage.
Non-proliferative retinopathy
Asymptomatic lesions
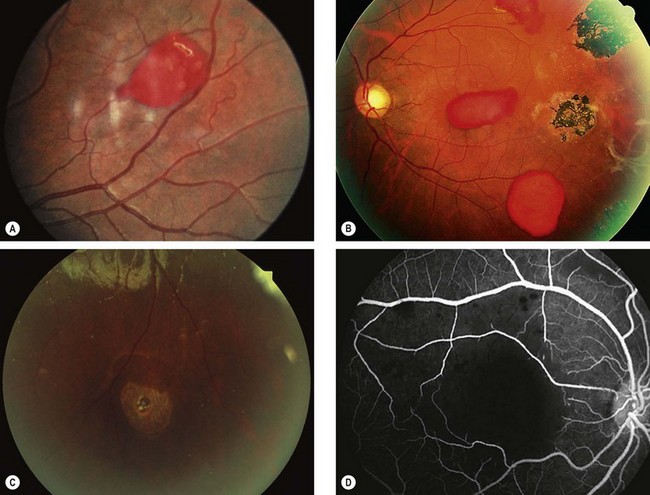
Fig. 13.57 Non-proliferative sickle-cell retinopathy. (A) Preretinal haemorrhage (‘salmon patch’); (B) RPE hyperplasia (‘black sunburst’) and preretinal haemorrhages; (C) retinal hole and an area of whitening superiorly; (D) FA shows macular ischaemia
(Courtesy of J Donald M Gass, from Stereoscopic Atlas of Macular Diseases, Mosby 1997 – fig. B)
Retinopathy of prematurity
Pathogenesis
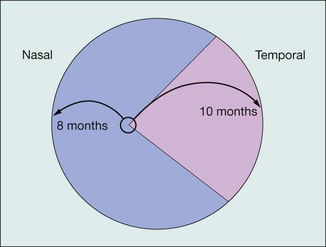
Retinopathy of prematurity (ROP) is a proliferative retinopathy affecting premature infants of very low birth weight, who have often been exposed to high ambient oxygen concentrations. The retina is unique among tissues in that it has no blood vessels until the fourth month of gestation, at which time vascular complexes emanating from the hyaloid vessels at the optic disc grow towards the periphery. These vessels reach the nasal periphery after 8 months of gestation, but do not reach the temporal periphery until about 1 month after delivery (Fig. 13.58). The incompletely vascularized retina is particularly susceptible to oxygen damage in the premature infant. A model of ROP suggests that the avascular retina produces VEGF (vascular endothelial growth factor) which in utero is the stimulus for vessel migration in the developing retina. With premature birth the production of VEGF is down-regulated by the relative hyperoxia and vessel migration is halted. Subsequently the increased metabolic demand of the growing eye allows excessive VEGF production which leads to the neovascular complications of ROP.
Active disease
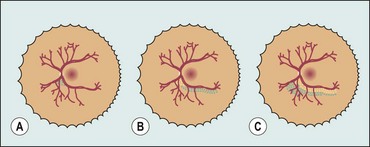
Location
For the purpose of defining the anteroposterior location of ROP, three concentric zones centred on the optic disc are described (Fig. 13.59).
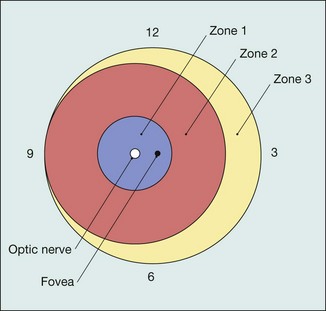
Extent
Extent of involvement is determined by the number of clock hours of retina involved (30° sectors).
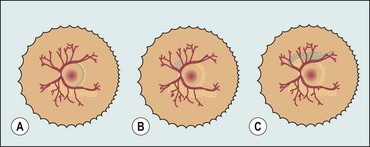
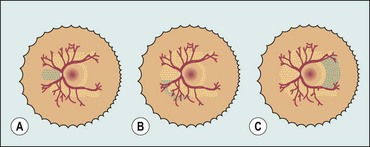
Staging
The following five stages are used to describe the abnormal vascular response at the junction of immature avascular peripheral retina and vascularized posterior retina. Because more than one ROP stage may be present in the same eye, staging for the eye as a whole is determined by the most severe manifestation.
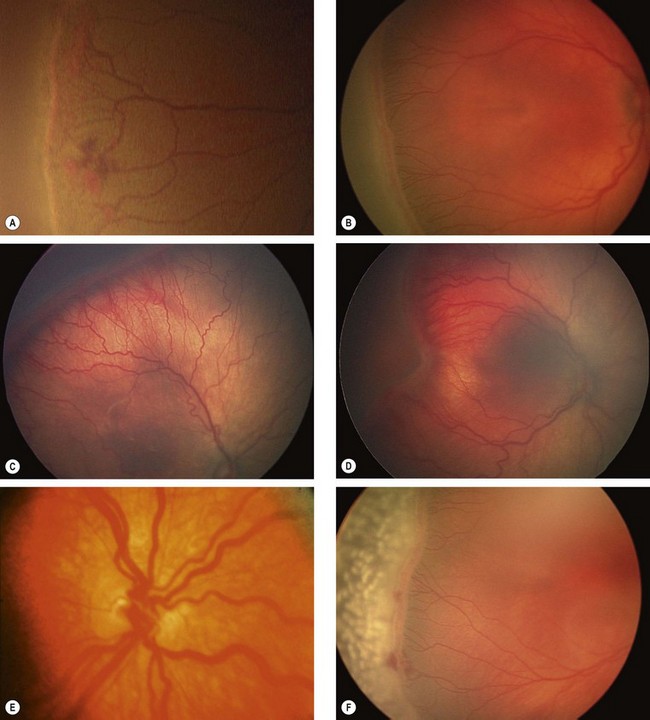
Fig. 13.60 Staging of active retinopathy of prematurity. (A) Stage 1 – demarcation line; (B) stage 2 – ridge; (C) stage 3 – ridge with extraretinal vascular proliferation; (D) stage 4a – partial extrafoveal retinal detachment; (E) ‘plus’ disease; (F) appearance immediately following laser photocoagulation for threshold disease
(Courtesy of L MacKeen – figs A, C and D; P Watts – figs B and F)
Other features
When these changes are present, a plus sign is added to the stage number.
Regression
In about 80% of cases ROP regresses spontaneously by a process of involution, or by evolution from a vasoproliferative to fibrotic phase leaving few if any residua. Spontaneous regression may even occur in eyes with partial retinal detachments.
Screening
Babies born at or before 31 weeks gestational age, or weighing 1500 g or less, should be screened for ROP. This may involve indirect ophthalmoscopy with a 28 D lens or a 2.2 panfunduscopic Volk lens, or a wide field retinal camera. Screening should begin 4–7 weeks postnatally to detect the onset of threshold disease. Subsequent review should be at 1–2-weekly intervals, depending on the severity of the disease and continue until retinal vascularization reaches zone 3. The pupils in a premature infant are dilated with 0.5% cyclopentolate and 2.5% phenylephrine. Only about 8% of babies screened actually require treatment.
Treatment
Cicatricial disease
About 20% of infants with active ROP develop cicatricial complications, which range from innocuous to extremely severe. In general, the more advanced or the more posterior the proliferative disease at the time of involution, the worse the cicatricial sequelae.
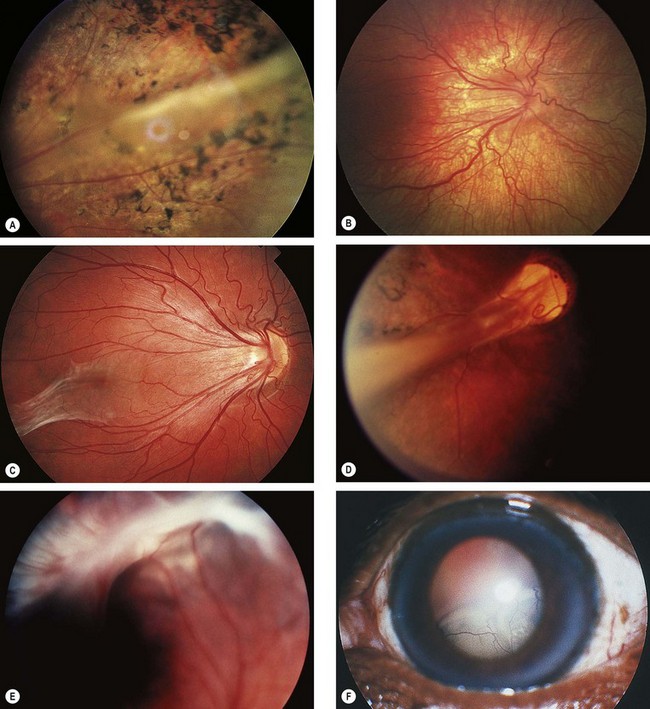
Fig. 13.61 Cicatricial retinopathy of prematurity. (A) Stage 1 – peripheral pigmentary disturbance; (B) early stage 2 – straightening of vascular arcades; (C) late stage 2 – ‘dragging’ of the disc and macula; (D) stage 3 – falciform fold; (E) stage 4 – retrolental fibrovascular tissue and partial retinal detachment; (F) stage 5 – total retinal detachment
Retinal artery macroaneurysm
A retinal artery macroaneurysm is a localized dilatation of a retinal arteriole; it usually occurs in the first three orders of the arterial tree. It has a predilection for elderly hypertensive women and involves only one eye in 90% of cases.
Diagnosis
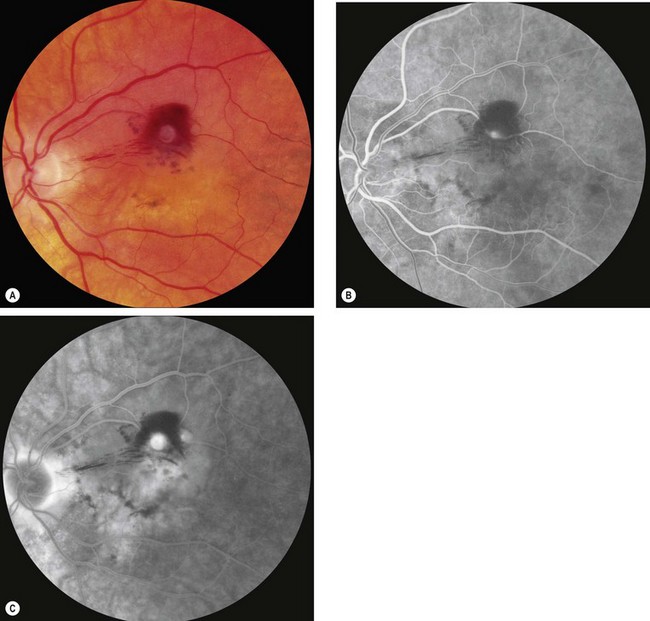
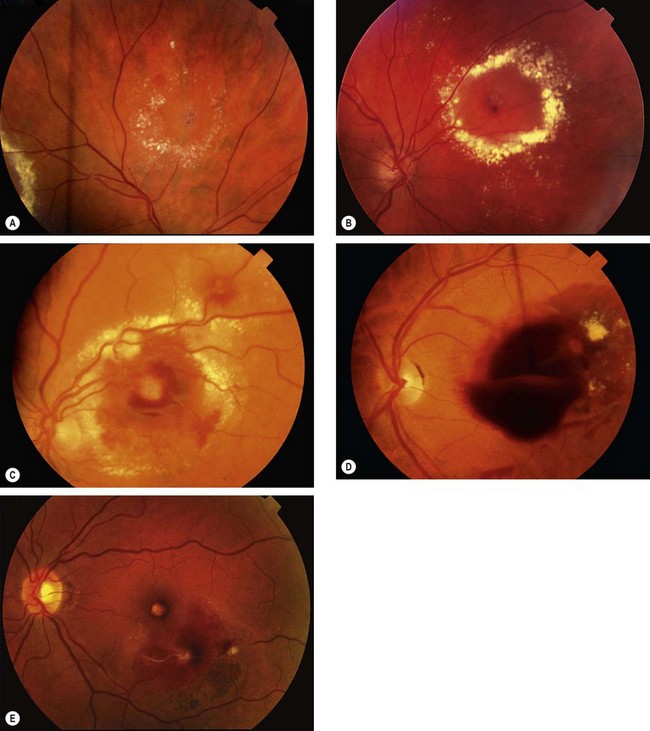
Prognosis
Eyes with vitreous or premacular haemorrhage tend to recover good vision, but central visual function in those with submacular haemorrhage generally remains poor.
Management
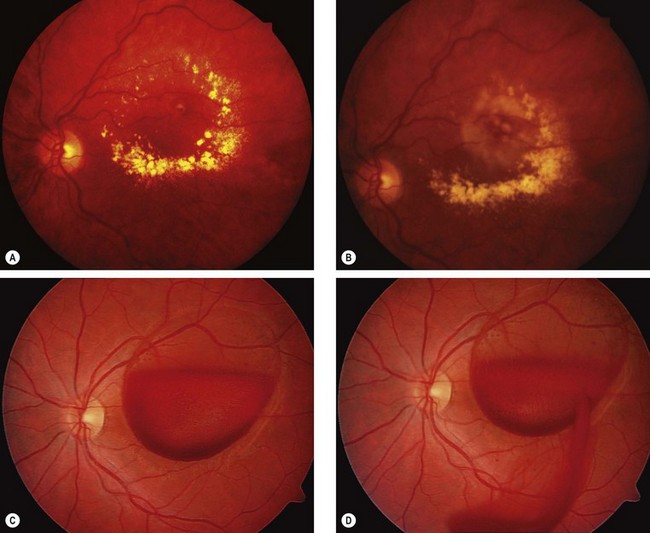
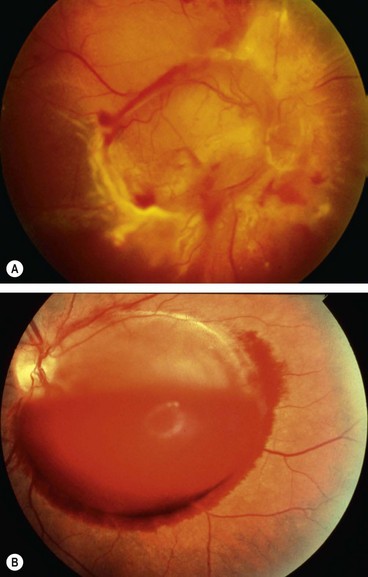
Fig. 13.64 Treatment of complications of retinal artery macroaneurysm. (A) Hard exudates at the macula due to chronic leakage; (B) following laser photocoagulation; (C) large preretinal haemorrhage overlying the macula; (D) following YAG laser hyaloidotomy the blood is dispersing into the vitreous
(Courtesy of P Gili – figs C and D)
Primary retinal telangiectasiA
Retinal capillary telangiectasia is relatively common. Most cases, however, are clearly secondary to another retinal condition, typically involving inflammation or vascular occlusion; examples include diabetic retinopathy and retinal venous occlusion. Primary retinal telangiectasis comprises a group of rare, idiopathic, congenital or acquired retinal vascular anomalies characterized by dilatation and tortuosity of retinal blood vessels, multiple aneurysms, vascular leakage and the deposition of hard exudates. Retinal telangiectasis involves the capillary bed, although the arterioles and venules may also be involved. The vascular malformations often progress and become symptomatic later in life.
Idiopathic macular telangiectasia
Idiopathic macular telangiectasia (IMT) is a rare condition of unknown pathogenesis. There are occasional reports of occurrence in close relatives, and although IMT is generally not considered familial it is suspected that a genetic element has a role. An updated and simplified classification has recently been suggested, reflecting increased knowledge of clinical and imaging features.
Type 1: aneurysmal telangiectasia
This may be closely related to Coats disease and may involve a variable area of the fundus including the periphery.
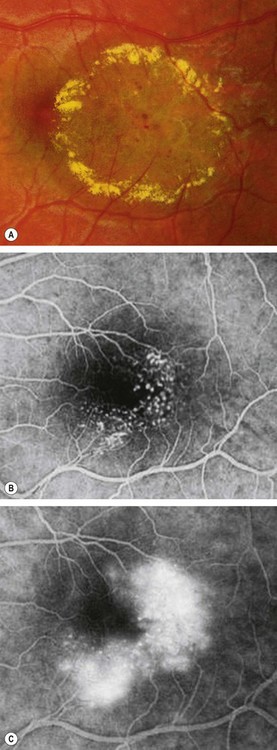
Type 2: perafoveal telangiectasia
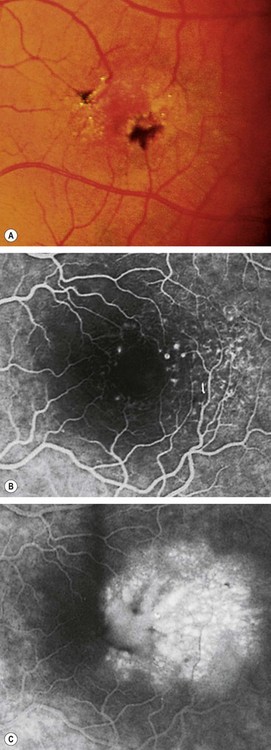
Occlusive telangiectasia
This is an extremely rare condition. The manifestations relate to capillary occlusion rather than telangiectasia, and probably result from a distinct pathogenesis. It has been omitted from the new classification system but was categorized as types 3A and B under the previous scheme. It carries a poor visual prognosis, and is frequently associated with systemic haematological or neurological disease.
Coats disease
Coats disease is an idiopathic retinal telangiectasia generally of onset in early childhood. It is associated with intraretinal and subretinal exudation, and frequently exudative retinal detachment, without signs of vitreoretinal traction. About 75% of patients are male and the great majority have involvement of only one eye. Although it is not clearly inherited, a genetic predisposition may be involved as at least some patients have a somatic mutation in the NDP gene, which is also mutated in Norrie disease. It is now considered that Leber miliary aneurysms, previously regarded as a distinct condition, represents a generally milder form of the same disease, presenting later, in a more localized pattern, and carrying a better visual prognosis.
Diagnosis
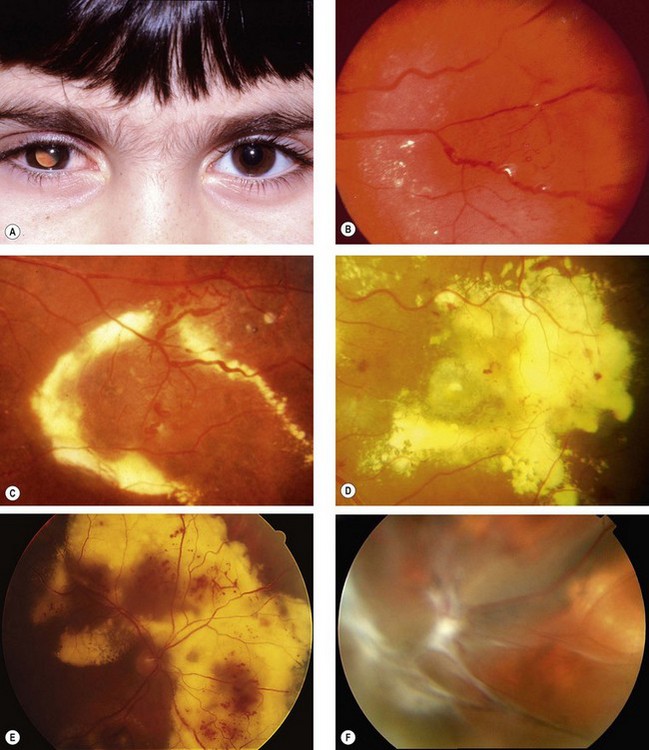
Treatment
Prognosis
The prognosis is variable and dependent on the severity of involvement at presentation. Young children, particularly those under 3 years of age, frequently have a more aggressive clinical course and often already have extensive retinal detachment at presentation. However, older children and young adults have a more benign disease with less likelihood of progressive exudation and retinal detachment and in some cases spontaneous regression may occur.
Eales disease
The eponym ‘Eales disease’ is used to describe patients with bilateral, idiopathic, occlusive, peripheral periphlebitis and neovascularization. The disease is rare in Caucasians but is an important cause of visual morbidity in young Asian males and is strongly associated with tuberculoprotein hypersensitivity.
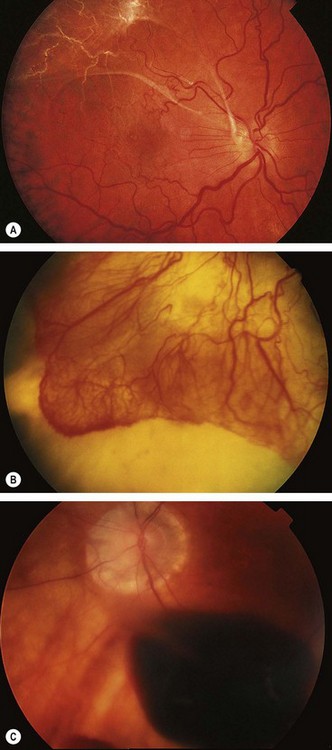
Radiation retinopathy
Radiation retinopathy may develop following treatment of intraocular tumours by plaque therapy (brachytherapy) or external beam irradiation of sinus, orbital or nasopharyngeal malignancies. It is characterized by delayed retinal microvascular changes with endothelial cell loss, capillary occlusion and microaneurysm formation. As with diabetic retinopathy its progress may be accelerated by pregnancy. Affected patients may also develop cataract and keratopathy. There is some evidence that it is more likely to occur in genetically predisposed patients.
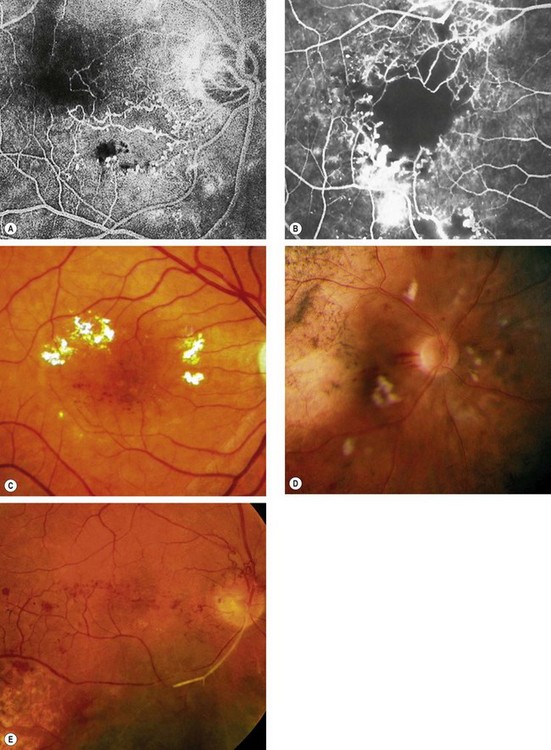
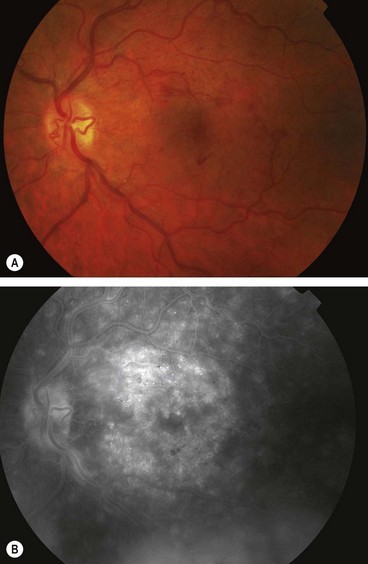
Fig. 13.72 Radiation retinopathy. (A) FA shows focal retinal capillary non-perfusion associated with microvascular abnormalities; (B) more severe retinal capillary non-perfusion and microvascular abnormalities; (C) microvascular abnormalities and hard exudates; (D) cotton wool spots and haemorrhages following brachytherapy for choroidal melanoma; (E) disc new vessels and arterial occlusion
(Courtesy of J Donald M Gass, from Stereoscopic Atlas of Macular Diseases, Mosby 1997 – fig. B; S Milenkovic – fig. C; B Damato – fig. D)
Purtscher retinopathy
Purtscher retinopathy is caused by microvascular damage with occlusion and ischaemia associated with severe trauma, especially to the head and in chest compressive injury. Other causes include embolism (fat, air or amniotic fluid) and systemic diseases (acute pancreatitis, pancreatic carcinoma, connective tissue diseases, lymphoma, thrombotic thrombocytopenic purpura and following bone marrow transplantation). Cases not associated with trauma are sometimes referred to as ‘Purtscher-like retinopathy’.
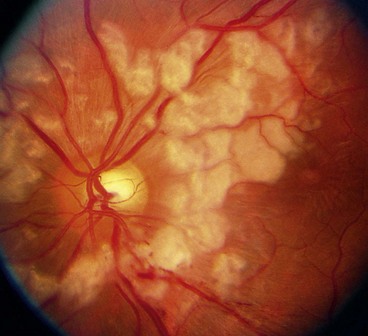
Benign idiopathic haemorrhagic retinopathy
Benign idiopathic haemorrhagic retinopathy is rare but important because it has a good prognosis without treatment.
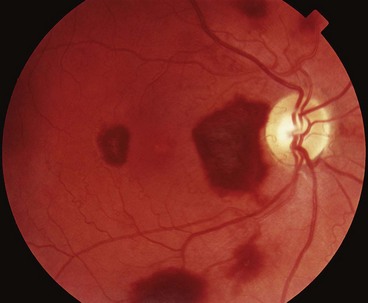
Valsalva retinopathy
The Valsalva manoeuvre comprises forcible exhalation against a closed glottis, thereby creating a sudden increase in intrathoracic and intra-abdominal pressure (e.g. weight-lifting, blowing up balloons). The associated sudden rise in venous pressure may result in rupture of perifoveal capillaries leading to unilateral or bilateral sub-internal limiting membrane haemorrhage at the macula of varying severity (Fig. 13.75).
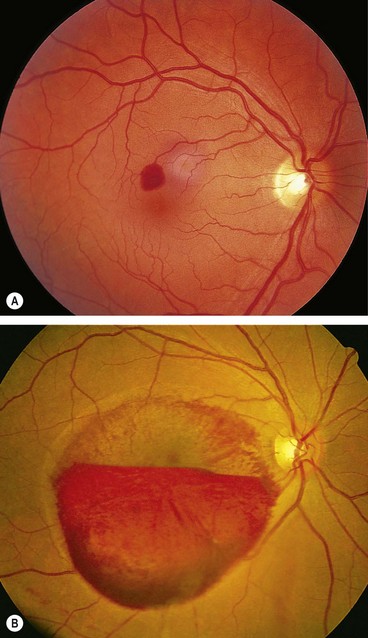
Lipaemia retinalis
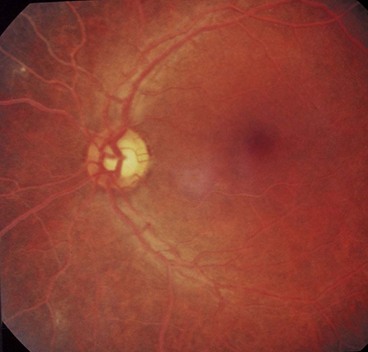
Lipaemia retinalis is a rare condition characterized by creamy-white coloured retinal blood vessels in patients with hypertriglyceridaemia (Fig. 13.76). The visualization of high levels of chylomycrons in blood vessels accounts for the fundus appearance. Visual acuity is usually normal but electroretinogram amplitude may be decreased.
Retinopathy in blood disorders
Leukaemia
Classification
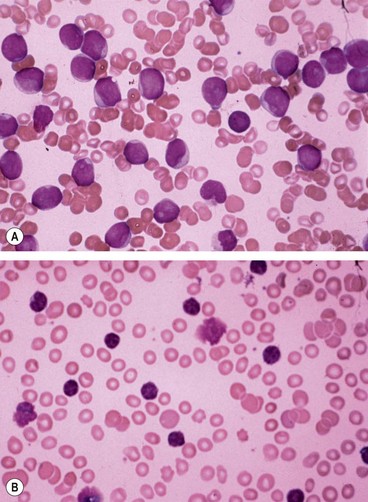
The leukaemias are malignancies of the haematopoietic stem cells characterized by abnormal proliferation of white blood cells. Acute leukaemias are characterized by replacement of bone marrow with very immature (blast) cells (Fig. 13.77A). Chronic leukaemias are associated, at least initially, with well-differentiated (mature) leucocytes (Fig. 13.77B) and occur almost exclusively in adults. The four major variants of leukaemia are:
Ocular features
Ocular involvement is more commonly seen in the acute than the chronic forms and virtually any ocular structure may be involved. It is, however, important to distinguish the fairly rare primary leukaemic infiltration from the more common secondary changes such as those associated with anaemia, thrombocytopenia, hyperviscosity and opportunistic infections; these may manifest as intraocular bleeding, infection and vascular occlusion.
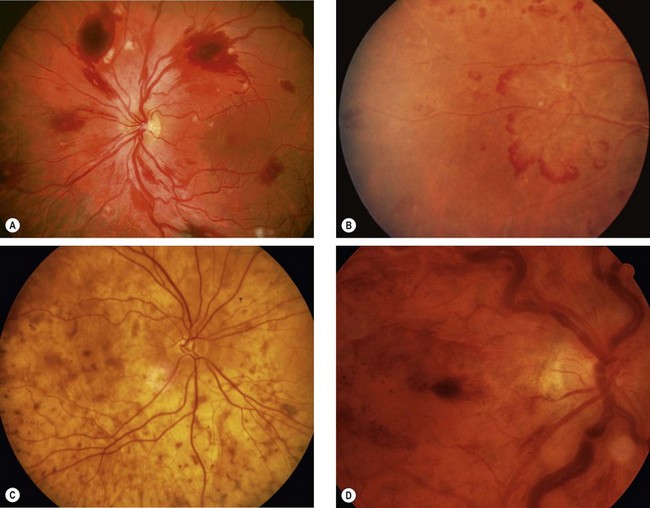
Fig. 13.78 Fundus changes in haematological disorders. (A) Retinal haemorrhages, cotton wool spots and Roth spots in acute leukaemia and anaemia associated with thrombocytopenia; (B) peripheral retinal neovascularization in chronic myeloid leukaemia; (C) ‘leopard-skin’ appearance due to choroidal infiltration in chronic leukaemia; (D) retinal haemorrhages, and gross venous dilatation and segmentation in hyperviscosity
(Courtesy of P Saine – fig. A; P Morse – fig. B)
Anaemia
The anaemias are a group of disorders characterized by a decrease in the number of circulating red blood cells, a decrease in the amount of haemoglobin in each cell, or both, that occurs when the equilibrium between blood loss and production are disturbed. Retinal changes in anaemia are usually innocuous and rarely of diagnostic importance.
Hyperviscosity
The hyperviscosity states are a diverse group of rare disorders characterized by increased blood viscosity due to polycythaemia or to abnormal plasma proteins (e.g. Waldenström macroglobulinaemia).
Congenital vascular anomalies
Retinal macrovessel
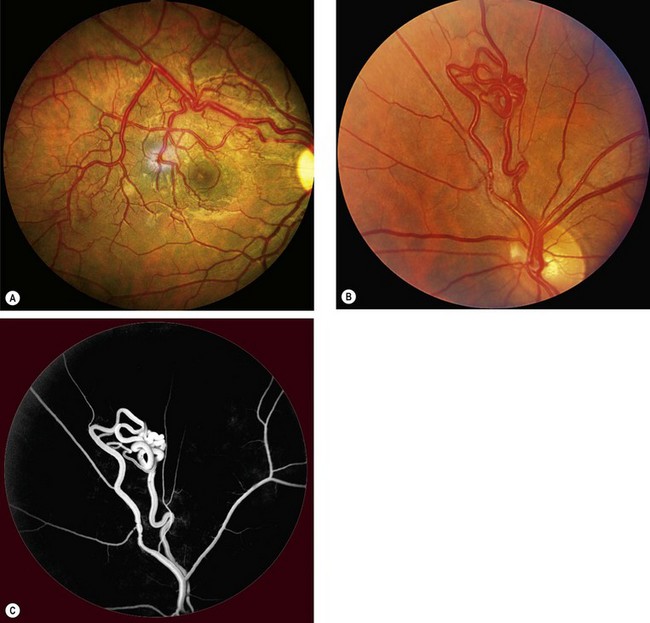
Arteriovenous communications
Congenital arteriovenous communications usually present on routine examination, with unilateral involvement in single or multiple sites of the same fundus. They have a predilection for the papillomacular bundle and the superotemporal quadrant. Occasionally reported complications include haemorrhage, exudation and vascular occlusion. Some patients may harbour similar systemic lesions. The malformations can be divided into the following three types on the basis of severity.