CHAPTER 13 Regional Immunity
Specialized Immune Responses in Epithelial and Immune Privileged Tissues
Most of our discussion of innate and adaptive immunity so far in this book has covered features and mechanisms of immune responses in any anatomic location in the mammalian body. However, the immune system has evolved specialized properties in different parts of the body, especially at epithelial surfaces. These features are essential for protection against the types of microbial challenges that are most often encountered at these locations, and they also ensure tolerance to nonpathogenic commensal organisms that live on epithelia and in the lumens of mucosal organs (Table 13-1). The collection of components of the immune system serving specialized functions at a particular anatomic location is called a regional immune system. Most of this chapter is devoted to a discussion of these specialized immune systems. We end with a consideration of some tissues that do not normally support immune responses and are said to be immune privileged.
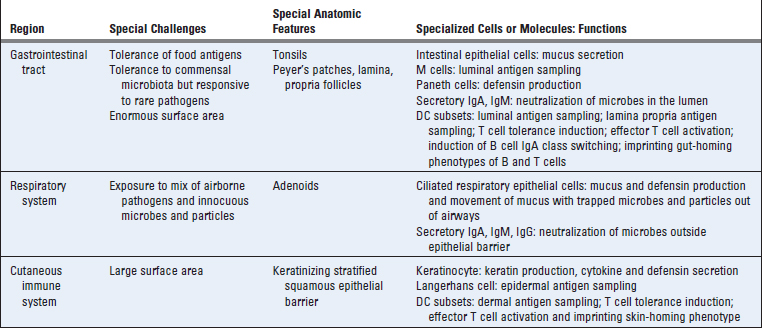
General Features of Immunity at Epithelial Barriers
Regional immune systems include the mucosal immune system, which protects the gastrointestinal, bronchopulmonary, and genitourinary mucosal barriers, and the cutaneous (skin) immune system. The gastrointestinal immune system is the largest and most complex. By two simple metrics, including the number of lymphocytes located in the tissue and the amount of antibodies made there, the gastrointestinal system dwarfs all other parts of the immune system combined. The human intestinal mucosa is estimated to contain approximately 50 × 109 lymphocytes (Table 13-2). The dedication of so many immune system resources to the gut reflects the large surface area of the intestinal mucosa, which has evolved to maximize the primary absorptive function of the tissue but must also resist invasion by trillions of bacteria in the lumen. The skin is also a barrier tissue with vast surface area that must be protected from the environmental microbes that have ready access to the external lining. The total number of lymphocytes in the skin is estimated to be about 20 × 109, about twice the total number of circulating lymphocytes (see Table 13-2). The different physical features of the mucosa (soft, wet, and warm) and the skin (tough, dry, and cool) favor colonization and invasion by different types of microbes. Therefore, it is not surprising that the immune system is specialized in different ways in these two types of tissues.
TABLE 13–2 Numbers of Lymphocytes in Different Tissues
| Spleen | 72 × 109 |
| Bone marrow | 50 × 109 |
| Blood | 10 × 109 |
| Skin | 20 × 109 |
| Gastrointestinal tract | 50 × 109 |
Data from Clark RA, B Chong, N Mirchandani, NK Brinster, K Yamanaka, RK Dowkiert, and TS Kupper. The vast majority of CLA+ T cells are resident in normal skin. Journal of Immunology 176:4431-4439, 2006; Ganusov AA and De Boer RJ. Do most lymphocytes in humans really reside in the gut? Trends in Immunology 28:514-518, 2007.
The major regional immune systems share a basic anatomic organization in which there is an outer epithelial barrier that serves essential functions to prevent microbial invasion, underlying connective tissue containing diffusely distributed cells of various types that are important for innate and adaptive immune responses to local microbes, and more distant draining lymph nodes where adaptive immune responses to invading microbes are initiated and amplified. The epithelial barrier may be several layers thick, as in the skin, or a single layer sitting on a basement membrane, as in the intestines. The underlying connective tissue, such as the dermis in the skin or the lamina propria in the gut, contains numerous scattered lymphocytes, dendritic cells (DCs), macrophages, and mast cells that mediate innate immune responses and the effector arm of adaptive immune responses. Mucosal tissues also contain unencapsulated but organized secondary lymphoid tissues just under the epithelial barrier, which include B and T lymphocytes, DCs, and macrophages. These collections of immune cells, often called mucosa-associated lymphoid tissue (MALT), are sites where adaptive immune responses specialized for the particular mucosa are initiated. Adaptive immune responses in regional immune systems are also induced in draining lymph nodes that are located outside the barrier tissues. In skin and mucosal tissues, antigens outside the epithelial barrier are sampled by specialized cells within the epithelium and are delivered to draining lymph nodes or MALT. Remarkably, the effector lymphocytes that are generated in the draining lymph nodes or MALT of a particular regional immune system (e.g., skin, small bowel) will enter the blood and preferentially home back into the subepithelial connective tissue of the same organ (e.g., dermis, lamina propria).
Each regional immune system is defined, in part, by the unique anatomy of the tissues in that region, including secondary lymphoid tissues. For example, the sampling of antigens in the gut and their transport to secondary lymphoid tissues rely on cell types and routes of lymphatic drainage that are fundamentally different from what takes place in the skin or internal organs. Furthermore, the MALT structures in different regions of the gut and in other mucosal organs have distinct features.
Regional immune systems each contain specialized cell types and molecules that may not be abundant in other sites. The cell types that are restricted to one or more regional immune systems but are not present throughout the immune system include subsets of DCs (e.g., Langerhans cells in the skin), antigen transport cells (e.g., M cells in the gut), T lymphocytes (e.g., γδ T cells in epithelia), and subsets of B lymphocytes (e.g., B cells and plasma cells in mucosal tissues that produce IgA). The localization of subsets of lymphocytes to different tissues is in part due to tissue-specific homing mechanisms that direct these subsets from the blood into particular secondary lymphoid organs or peripheral tissues, which we will discuss in detail later in the chapter.
Regional immune systems have important regulatory functions that serve to prevent unwanted responses to nonpathogenic microbes and foreign substances that are present at different barriers. The clearest example is the gut-associated immune system, which must suppress responses to commensal bacteria that colonize the intestinal mucosa as well as to foreign food substances but must respond to less frequent pathogenic bacteria. The suppression of immune responses to nonpathogenic organisms and harmless foreign substances is also important in other sites of the body, including the skin, lung, and genitourinary tract, that are not sterile and are constantly exposed to the environment.
With this introduction, we will now discuss the details of these various features in different regional immune systems, beginning with the largest.
Immunity in the Gastrointestinal System
The gastrointestinal system, like other mucosal tissues, is composed of a tube-like structure lined by a continuous epithelial cell layer sitting on a basement membrane that serves as physical barrier to the external environment. Underlying the epithelium is a layer of loose connective tissue, called the lamina propria in the gut, that contains blood vessels, lymphatic vessels, and mucosa-associated lymphoid tissues (Fig. 13-1). The submucosa is a dense connective tissue layer that connects the mucosa with layers of smooth muscle.
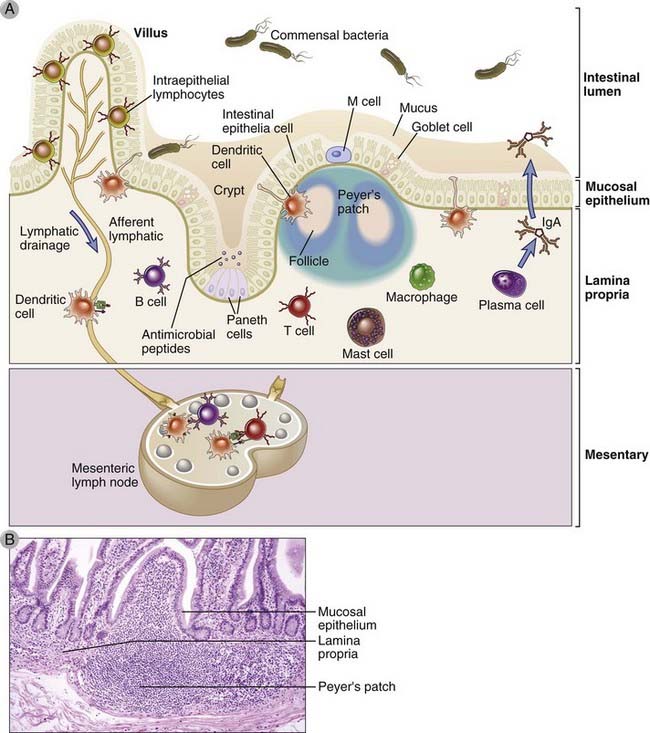
FIGURE 13–1 The gastrointestinal immune system.
A, Schematic diagram of the cellular components of the mucosal immune system in the intestine. B, Photomicrograph of mucosal lymphoid tissue in the human intestine. Similar aggregates of lymphoid tissue are found throughout the gastrointestinal tract.
From the perspective of the immunologist, the gastrointestinal tract has two remarkable properties. First, the combined mucosa of the small and large bowel has a total surface area of more than 200 m2 (the size of a tennis court), made up mostly of small intestinal villi and microvilli. Second, the lumen of the gut is teeming with microbes, many of which are ingested along with food and most of which are continuously growing on the mucosal surface in healthy individuals as commensals. It is estimated that more than 500 different species of bacteria, amounting to approximately 1014 cells, live in the mammalian gut. This is 10 times more than the number of all the cells in the body, prompting some microbiologists to point out that we humans are actually only 10% “human” and 90% bacterial! We have evolved to depend on these commensals for several functions, including the degradation of components of our diet that our own cells cannot digest. Although the commensal organisms are beneficial when they are contained on the outside of the gut mucosal barrier, they are potentially lethal if they cross the mucosal barrier and enter the circulation or traverse the bowel wall, especially in immune-compromised individuals. The huge surface area and the high density of mucosal commensals in the gut therefore represent a constant potential danger that must be guarded against. Furthermore, noncommensal pathogenic organisms may become part of the diverse mixture of organisms that make up the gut flora at any time if they are ingested in contaminated food or water. These pathogenic organisms, including bacteria, viruses, protozoa, and helminthic parasites, can cause significant disease, often without invading the epithelial lining and even if they represent a tiny fraction of the microbes in the gut lumen. For health to be maintained, the mucosal immune system must be able to recognize and eliminate these numerically rare pathogens in the presence of overwhelming numbers of nonpathogenic microbes. These challenges have been met by the evolution of a complex set of innate and adaptive immune recognition strategies and effector mechanisms, which we will now describe. Some of these we understand well, and others remain incompletely characterized. Many of the features of the gastrointestinal immune system are shared by other mucosal tissues, and we will point out these common features of mucosal immunity. Unfortunately, intestinal infections by pathogenic organisms are frequently not controlled by mucosal immunity and account for millions of deaths each year throughout the world.
Innate Immunity in the Gastrointestinal Tract
Intestinal epithelial cells lining the small and large bowel are an integral part of the gastrointestinal innate immune system, involved in responses to pathogens, tolerance to commensal organisms, and antigen sampling for delivery to the adaptive immune system in the gut. There are several different types of intestinal epithelial cells, all derived from a common precursor found in the crypts of intestinal glands. Among these are the mucus-secreting goblet cells, which reside at the top of the intestinal villi; cytokine-secreting absorptive epithelial cells; antigen-sampling M cells, found in specialized dome structures overlying lymphoid tissues; and antibacterial peptide-secreting Paneth cells, found at the bottom of the crypts. All these cell types contribute in different ways to the barrier function of the mucosa, as we will discuss later.
Innate immune protection in the gut is mediated in part by the nonspecific physical and chemical barrier provided by the mucosal epithelial cells and their mucus secretions. Adjacent intestinal epithelial cells are held together by proteins that form tight junctions, including zonula occludens 1 and claudins, and these block the movement of bacteria and pathogen-associated molecular patterns (PAMPs) between the cells into the lamina propria. In addition, mucosal epithelial cells produce antimicrobial substances, and several cell types located in the mucosa, including epithelial cells, DCs, and macrophages, are capable of mounting inflammatory and antiviral responses. Most of these responses are induced by pattern recognition receptor engagement of PAMPs, which we discussed in Chapter 4. Interestingly, some innate immune receptors that promote inflammation in other parts of the body have anti-inflammatory actions in the gut. In this section, we will describe features of innate immunity that are unique to the intestines.
Several different extensively glycosylated proteins, called mucins, form a viscous physical barrier that prevents microbes from contacting the cells of the gastrointestinal tract. Mucins contain many different O-linked oligosaccharides and include secreted and cell surface glycoproteins. The secreted mucins, including MUC2, MUC5, and MUC6, form a hydrated gel ranging from 300 to 700 µm in thickness that can prevent microbial contact with the epithelial lining cells and also serves as a matrix for display of antimicrobial substances produced by the epithelial cells. Some mucins act as decoy molecules, which can be shed from the epithelial cells and bind to the adhesin proteins that pathogenic bacteria use to attach to host cell membranes. In addition to the secreted mucus, the apical surface of gastrointestinal epithelial cells is coated with membrane-bound mucin proteins, including MUC1, MUC3A/b, MUC12, MUC13, MUC17. These membrane-bound mucins combine with various glycolipids to form a dense macromolecular layer at the epithelial cell surface called the glycocalyx, which ranges from 30 to 500 nm in thickness in different locations in the gut. The glycocalyx, like the secreted mucus, serves as a physical barrier to prevent microbial contact.
A remarkable property of the mucous barrier of the intestine is its rapid turnover and responsiveness to various environmental and immune signals, which allows rapid increases in mucosal barrier function. Mucins are constitutively produced both by the surface epithelial cells in the gastrointestinal tracts and by submucosal glands and are replaced by newly synthesized molecules every 6 to 12 hours. Several different environmental and immune stimuli can induce dramatic increases in mucin production. These stimuli include cytokines (IL-1, IL-4, IL-6, IL-9, IL-13, tumor necrosis factor [TNF], and type I interferons), neutrophil products (such as elastase), and microbial adhesive proteins. These stimuli not only increase mucin gene expression but also alter the glycosylation of the mucins because of induced changes in the expression of glycosyltransferase enzymes. The changes in quantity and glycosylation of mucins are thought to increase barrier function against pathogens.
Defensins produced by intestinal epithelial cells provide innate immune protection against luminal bacteria, and defects in their production are associated with bacterial invasion and inflammatory bowel disease. Defensins are peptides produced by various cell types in the body that exert lethal toxic effects on microbes by inserting into and causing loss of integrity of their outer phospholipid membranes (see Chapter 4). In the small bowel, the major defensins are the α-defensins, including human defensin 5 (HD5) and HD6, produced constitutively as inactive precursor proteins by Paneth cells located at the base of crypts between microvilli. Active HD5 and HD6 peptides are generated by proteolytic cleavage mediated by trypsin, also produced by Paneth cells. In the colon, β-defensins are produced by absorptive epithelial cells in the intestinal crypts, some constitutively and others in response to IL-1 or invasive bacteria. In addition, neutrophil granules are rich in α-defensins, which likely contributes to their antimicrobial functions in the setting of infections of the bowel wall. Several studies have identified defects in defensin production by epithelial cells in affected regions of bowel in Crohn’s disease, a chronic inflammatory disease that can involve the entire gastrointestinal tract. Because there is a significant inheritable risk for development of Crohn’s disease, it is possible that a genetically determined defect in production of defensins may be a predisposing factor for the disease, and reduced expression of defensin genes has been associated with a subset of Crohn’s disease.
Toll-like receptors (TLRs) and cytoplasmic Nod-like receptors (NLRs) expressed by intestinal epithelial cells promote immune responses to invasive pathogens but are also regulated to limit inflammatory responses to commensal bacteria. In Chapter 4, we defined TLRs and NLRs as cellular receptors that recognize PAMPs produced by microbes and generate signals that promote inflammatory and antiviral responses by the cells. Most luminal bacteria of the gut are nonpathogenic if they are retained outside the epithelial barrier, yet they may express the same array of PAMPs that pathogenic bacteria express, such as lipopolysaccharide, peptidoglycans, CpG DNA, and flagellin. Because inflammatory responses that involve the intestinal epithelial cells can impair barrier function and can lead to bacterial invasion and pathologic inflammation, it is not surprising that stringent control mechanisms have evolved to limit TLR-induced proinflammatory responses to commensal bacteria. Intestinal epithelial cells express a wide range of TLRs, including TLRs 2, 4, 5, 6, 7, and 9, with different receptors expressed in different regions of the gut. Ligation of some TLRs results in the phosphorylation and reorganization of zona occludens 1 and increased strength of the tight junctions between epithelial cells, and TLR signaling also increases intestinal epithelial motility and proliferation. These functional responses to TLR signaling increase barrier function but not inflammation. TLR responses in the gut appear also to be regulated by levels of expression or compartmentalized expression in only certain sites (Fig. 13-2). For example, TLR5, which recognizes bacterial flagellins, is exclusively expressed on the basolateral surface of intestinal epithelial cells, where it will be accessible only to bacteria that have invaded through the barrier. Similarly, NLR family receptors for flagellins (e.g., NAIP and IPAF-1) are expressed in the cytoplasm of intestinal epithelial cells and will activate inflammatory responses only when pathogenic bacteria or their products gain access to the cytosol. There is also evidence that regulators of TLR signaling inside intestinal epithelial cells maintain a higher threshold for activation of inflammatory responses compared with epithelial cells and DCs in other tissues (see Fig. 13-2).
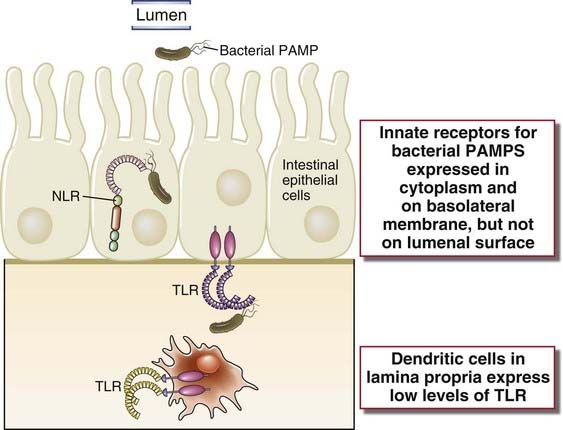
FIGURE 13–2 Mechanism of regulation of innate immune responses in the intestinal mucosa.
Pattern recognition receptor expression and function in intestinal epithelial cells and lamina propria DCs minimize inflammatory responses to commensal bacteria in the lumen but promote responses to microbes that traverse the barrier and enter the lamina propria. Top, Pattern recognition receptors that recognize bacterial flagellin are compartmentalized in the cytosol (NLR) or basal membrane (TLR5) of intestinal epithelial cells but not on the apical/lumen membrane. Bottom,TLR4, which recognizes bacterial lipopolysaccharides, is expressed at low levels on intestinal epithelial cells and lamina propria DCs. TLR signaling does not induce inflammatory gene expression in lamina propria DCs because of more dominant effect of intracellular regulators of TLR signal transduction, such as TOLLIP and IRAK-M, compared with conventional DCs in other tissues.
In healthy individuals, lamina propria DCs and macrophages in the gut inhibit inflammation and serve to maintain homeostasis. Overall, intestinal macrophages have a unique phenotype that enables them to phagocytose and kill microbes but at the same time to secrete anti-inflammatory cytokines, such as IL-10. This phenotype is apparently induced in the local mucosal environment by transforming growth factor-β (TGF-β). TLR4 expression on both macrophages and DCs in the lamina propria is lower than in other tissues, and inflammatory gene expression in these cells is often inhibited by microbial products. This may be an evolved mechanism to prevent damaging inflammation in response to commensal bacteria and bacterial products that may traverse the epithelial barrier.
Adaptive Immunity in the Gastrointestinal Tract
The adaptive immune system in the gastrointestinal tract has features that are distinct from adaptive immune functions in other organ systems.
We will now discuss the special features of adaptive immunity in the gastrointestinal system, including anatomic organization, antigen sampling, lymphocyte homing and differentiation, and antibody delivery to the lumen.
The Functional Anatomy of the Adaptive Immune System in the Gastrointestinal Tract
In this section, we will discuss the anatomic organization of cells within the intestines and the relationship of this organization to how adaptive immune responses are initiated, carried out, and regulated. In general terms, the functional anatomy of the adaptive immune system in the gut has evolved to effectively deal with the conditions we emphasized earlier of abundant commensal microbes and rare pathogens just outside an epithelial barrier of enormous surface area.
Adaptive immune responses in the gut are initiated in discretely organized collections of lymphocytes and antigen-presenting cells closely associated with the mucosal epithelial lining of the bowel and in mesenteric lymph nodes (see Fig. 13-1). Naive lymphocytes are exposed to antigens in these sites and differentiate into effector cells. These gut-associated lymphoid tissues adjacent to the mucosal epithelium are sometimes referred to as GALT, which is the gastrointestinal version of MALT, although the terms are often used interchangeably. Up to 30% of the lymphocytes in the body are found in the GALT. The most prominent GALT structures are Peyer’s patches, found mainly in the distal ileum, and smaller aggregates of lymphoid follicles or isolated follicles in the appendix and colon. Peyer’s patches have the structure of lymphoid follicles, with germinal centers containing B lymphocytes, follicular helper T cells, follicular dendritic cells (FDCs), and macrophages. The germinal centers in the follicles are surrounded by IgM- and IgD-expressing naive follicular B cells. A region called the dome is located between the follicles and the overlying epithelium and contains B and T lymphocytes, DCs, and macrophages. Between the follicles are T cell–rich parafollicular areas, similar to lymph nodes, but overall, the ratio of B cells to T cells in GALT is about five times higher than in lymph nodes. Also in distinction from lymph nodes, GALT structures are not encapsulated, and there are routes of antigen delivery to these structures that are independent of lymphatics. Development of both aggregates of follicles, such as Peyer’s patches, and isolated follicles in the gut lamina propria requires lymphoid tissue inducer cells, which express the RORγT transcription factor and produce the cytokine lymphotoxin-β (LTβ).
A major pathway of antigen delivery from the lumen to the GALT is through specialized cells within the gut epithelium called microfold (M) cells (Fig. 13-3). M cells are located in regions of the gut epithelium called follicle-associated or dome epithelium that overlie the domes of Peyer’s patches and other GALT structures. Although M cells and the more numerous epithelial cells with absorptive function likely arise from a common epithelial precursor, the M cells are distinguishable by a thin glycocalyx, their relatively short, irregular microvilli (referred to as microfolds), and large fenestrations in their membranes, all features that enhance the uptake of antigens from the gut lumen. The main function of M cells is transcellular transport of various substances from the lumen of the intestine across the epithelial barrier to underlying antigen-presenting cells. M cells take up luminal contents efficiently and in various ways, including phagocytosis in a manner similar to macrophages, and either clathrin-coated vesicular or fluid-phase endocytosis. These pathways enable uptake of whole bacteria, viruses, and soluble microbial products. Unlike macrophages or DCs, M cells do not engage in extensive processing of the substances they take up, but rather they move the particles and molecules through endocytic vesicles across the cytosol and deliver them by exocytosis at the basolateral membrane to DCs in the dome regions of underlying GALT structures. Although M cells play an important role in protective immunity to luminal microbes, some microbes have evolved to take advantage of M cells as a route of invasion through the mucosal barrier. The best described example of this is Salmonella typhimurium, similar to the human pathogen S. typhi that causes typhoid fever. M cells express specific lectins that allow these bacteria to specifically bind and be internalized. The bacteria are cytotoxic to the M cells, leading to gaps in the epithelium that promote invasion of more organisms. M cell lectins may also promote infection by certain enteric viruses.
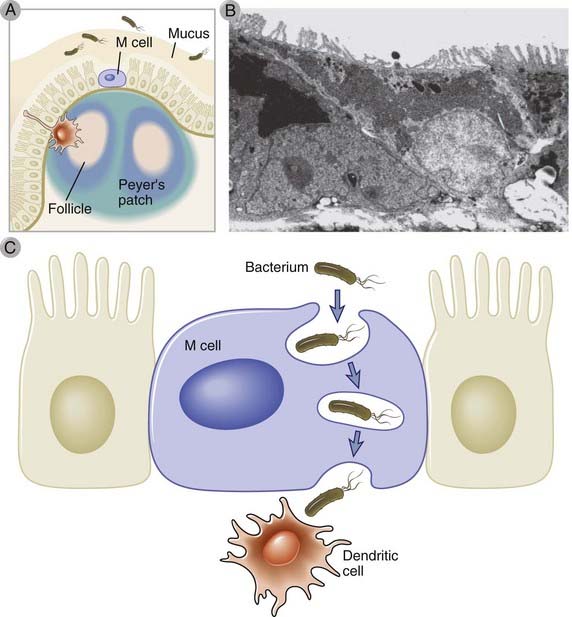
FIGURE 13–3 M cells in the small intestine.
M cells are specialized intestinal epithelial cells found in the small bowel epithelium overlying Peyer’s patches and lamina propria lymphoid follicles (A). Unlike neighboring epithelial cells with tall microvillous borders and primary absorptive functions, M cells have shorter villi (B) and engage in transport of intact microbes or molecules across the mucosal barrier into gut-associated lymphoid tissues, where they are handed off to DCs (C).
(Electron micrograph from Corr SC, CC Gahan, and C Hill. M-cells: origin, morphology and role in mucosal immunity and microbial pathogenesis. FEMS Immunology and Medical Microbiology 52:2-12, 2008.)
Microbial antigens in the gut lumen can be sampled by lamina propria DCs that extend cytoplasmic processes between the intestinal epithelial cells and by Fc receptor–dependent uptake of IgG opsonized antigens by the epithelial cells (Fig. 13-4). Antigen-sampling DCs are numerous in certain regions of the intestine, especially the terminal ileum, where they extend dendrites through the junctions between adjacent epithelial cells, apparently without disrupting the tight junctions. These antigen-sampling DCs belong to a subset of mucosal DCs that promote effector T cell responses, which we will discuss later in the chapter. Unlike M cells, these DCs are capable of processing and presenting protein antigens to T cells within the GALT. Antigens in the lumen that have been opsonized by antibodies can be delivered to the GALT by Fc receptor–mediated pathways. There is evidence from mouse studies that IgG-opsonized antigens, such as bacterial flagellin, can be transported across the gut epithelium through the neonatal Fcγ receptor (FcRn, see Chapter 12) and passed on to DCs in the GALT, leading to T cell responses to the antigens.
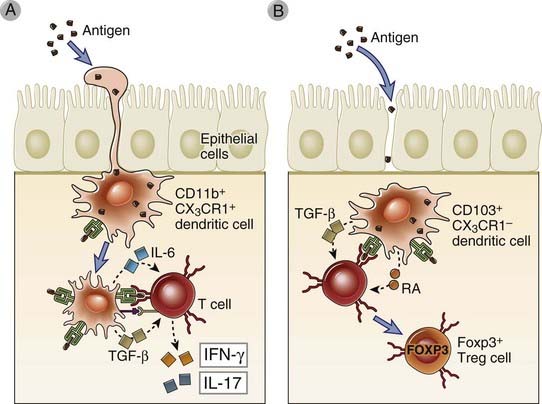
FIGURE 13–4 DCs in the intestinal mucosa.
There are several different subsets of DCs constitutively present in the intestinal mucosa that are defined by cell surface molecules and function. Two such subsets are shown that are also present in other mucosal tissues. A, Antigen-sampling DCs extend dendritic processes between intestinal epithelial cells into the lumen to sample antigens and then migrate to mesenteric lymph nodes, where they initiate activation and differentiation of proinflammatory effector T cells. These DCs express the CD11b integrin chain and the CX3CR1 chemokine receptor. B, Other DCs present in the lamina propria, which express the integrin CD103, present antigens to naive T cells and induce their differentiation of regulatory T cells, in part by secreting TGF-β and retinoic acid (RA). The regulatory function of these DCs depends on factors secreted by intestinal epithelial cells.
Mesenteric lymph nodes collect lymph-borne antigens from the small and large bowel and are sites of differentiation of effector and regulatory lymphocytes that home back to the lamina propria. There are 100 to 150 of these lymph nodes located between the membranous layers of the mesentery. Mesenteric lymph nodes serve some of the same functions as GALT, including differentiation of B cells into dimeric IgA–secreting plasma cells and the development of effector T cells as well as regulatory T cells. The cells that differentiate in the mesenteric lymph nodes in response to bowel wall invasion by pathogens or commensals often home to the lamina propria. We will discuss imprinting of homing properties of lymphocytes activated in mesenteric lymph nodes later.
Lingual and palatine tonsils are nonencapsulated lymphoid structures located beneath stratified squamous epithelial mucosa in the base of the tongue and oropharynx, respectively, and are sites of immune responses to microbes in the oral cavity. These tonsils, together with nasopharyngeal tonsils, form a ring of lymphoid tissues called Waldeyer’s ring. The bulk of the tonsillar tissue is composed of lymphoid follicles, usually with prominent germinal centers. There are multiple narrow and deep invaginations of the surface squamous epithelium, called crypts, that grow into the follicular tissue. Although these tonsils are often considered part of the GALT, they are distinct in that they are separated from the microbe-rich oral cavity by multiple layers of squamous epithelial cells rather than the single epithelial cell layer of the gut. The mechanism of antigen sampling from oral cavity microbes is not well described; the crypts are possible sites where this may happen. Nonetheless, the lingual and palatine tonsils respond to infections of the epithelial mucosa by significant enlargement and vigorous, mainly IgA, antibody responses. Typical infections that are associated with tonsillar enlargement, usually in children, are caused by streptococci and the Epstein-Barr virus.
Effector lymphocytes that are generated in the GALT and mesenteric lymph nodes are imprinted with selective integrin- and chemokine receptor–dependent gut-homing properties, and they circulate from the blood back into the lamina propria of the gut (Fig. 13-5). Both IgA-secreting B cells and effector T cells acquire the gut-homing phenotype. Thus, the effector arm of the gastrointestinal immune system depends on a large number of antibody-secreting cells and T cells that recirculate back into the lamina propria and respond rapidly to pathogens. The major integrin on gut-homing B and T lymphocytes is α4β7, which binds to the MadCAM-1 protein expressed on postcapillary venular endothelial cells in the gut lamina propria. Gut homing also requires the chemokine receptor CCR9 on the B and T lymphocytes and its chemokine ligand CCL25, which is produced by intestinal epithelial cells. The combined expression of MadCAM-1 and CCL25 is restricted to the gut. Homing of IgA-producing cells to the colon also requires CCR10 expression and the chemokine CCL28, but this is not a gut-specific pathway because CCL28 is expressed by epithelial cells in other mucosal tissues, such as the lung and genitourinary tract. Blocking monoclonal antibodies specific for the α4 chain of α4β7 have been used to treat patients with inflammatory bowel disease on the basis of the knowledge that effector T cells use this integrin to enter gut tissues in this disease. (We will discuss inflammatory bowel disease later in the chapter.)
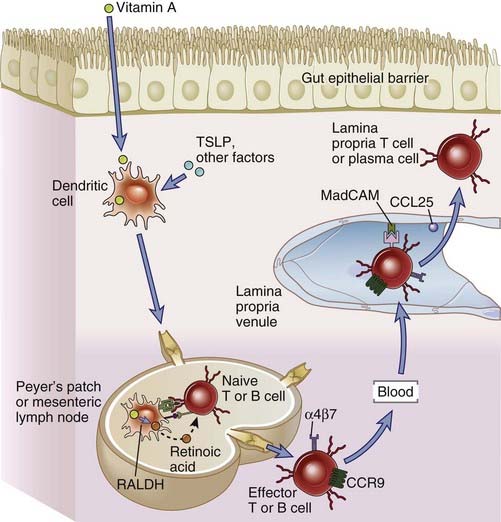
FIGURE 13–5 Homing properties of intestinal lymphocytes.
The gut-homing properties of effector lymphocytes are imprinted in the lymphoid tissues where they have undergone differentiation from naive precursors. DCs in gut-associated lymphoid tissues, including Peyer’s patches and mesenteric lymph nodes, are induced by thymic stromal lymphopoietin (TSLP) and other factors to express retinaldehyde dehydrogenase (RALDH), which converts dietary vitamin A into retinoic acid. When naive B or T cells are activated by antigen in GALT, they are exposed to retinoic acid produced by the DCs, and this induces the expression of the chemokine receptor CCR9 and the integrin α4β7 on the plasma cells and effector T cells that arise from the naive lymphocytes. The effector lymphocytes enter the circulation and home back into the gut lamina propria because the chemokine CCL25 (the ligand for CCR9) and the adhesion molecule MadCAM (the ligand for α4β7) are displayed on lamina propria venular endothelial cells.
The gut-homing phenotype of IgA-producing cells and effector T cells is imprinted by DCs and the action of retinoic acid during the process of T cell activation (see Fig. 13-5). In addition to promoting naive T cell differentiation into effector T cells and naive B cell differentiation into IgA antibody–secreting cells, discussed later in the chapter, DCs in GALT and mesenteric lymph nodes also provide signals that lead to the expression of α4β7 and CCR9 on these effector cells. The induction of these homing molecules depends on secretion of retinoic acid by the DCs, although the mechanisms are not well understood. The selective induction of gut-homing cells in the gut lymphoid tissues is explained by the fact that gut lymphoid tissues are exposed to dietary vitamin A, and DCs in GALT and mesenteric lymph nodes express retinal dehydrogenases (RALDH), the enzyme needed for retinoic acid synthesis from vitamin A, whereas DCs in other tissues do not. In addition, intestinal epithelial cells also express RALDH and can synthesize retinoic acid. Consistent with these properties of the intestinal humoral immune system, it is known that oral vaccination not only favors the expansion of IgA-producing B cells, compared with intradermal immunization, but that oral vaccines also induce higher levels of α4β7 on B cells.
The lamina propria contains diffusely distributed effector lymphocytes, DCs, and macrophages and is the site of the effector phase of gastrointestinal adaptive immune responses. As discussed before, effector lymphocytes generated in Peyer’s patches, other GALT structures, and mesenteric lymph nodes home back into the lamina propria. In this location, T cells can respond to invading pathogens, and B cells can secrete antibodies that are transported into the lumen and neutralize pathogens before they invade.
Humoral Immunity in the Gastrointestinal Tract
Humoral immunity in the gut is dominated by production of secretory IgA in the GALT and transport of the antibody across the mucosal epithelium into the lumen. Smaller but significant quantities of IgG and IgM are also secreted into the gut lumen. Within the lumen, IgA, IgG, and IgM antibodies bind to microbes and toxins and neutralize them by preventing their binding to receptors on host cells. This form of humoral immunity is sometimes called secretory immunity and has evolved to be particularly prominent in mammals. Antibody responses to antigens encountered by ingestion are typically dominated by IgA, and secretory immunity is the mechanism of protection induced by oral vaccines such as the polio vaccine. Several unique properties of the gut environment result in selective development of IgA-secreting cells that either stay in the gastrointestinal tract or, if they enter the circulation, home back to the lamina propria of the intestines. The result is that IgA-secreting cells efficiently accumulate next to the epithelium that will take up the secreted IgA and transport it into the lumen.
IgA is produced in larger amounts than any other antibody isotype. It is estimated that a normal 70-kg adult secretes about 2 g of IgA per day, which accounts for 60% to 70% of the total production of antibodies. This tremendous output of IgA is because of the large number of IgA-producing plasma cells in the GALT, which by some estimates amount to about 1010 cells per meter of bowel (Fig. 13-6). Because IgA synthesis occurs mainly in mucosal lymphoid tissue and transport into the mucosal lumen is efficient, this isotype constitutes less than one quarter of the antibody in plasma and is a minor component of systemic humoral immunity compared with IgG and IgM.
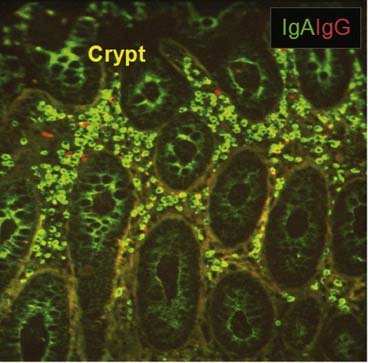
FIGURE 13–6 IgA-secreting plasma cells in the intestine.
The abundance of IgA-producing plasma cells (green) in colon mucosa compared with IgG-secreting cells (red) is shown by immunofluorescence staining. IgA that is being secreted can be seen as green cytoplasm in the crypt epithelial cells.
(From Brandtzaeg P. The mucosal immune system and its integration with the mammary glands. The Journal of Pediatrics 156[Suppl 1]:S8-S16, 2010.)
The dominance of IgA production by intestinal plasma cells is due in part to selective induction of IgA isotype switching in B cells in GALT and mesenteric lymph nodes. IgA class switching in the gut can occur by T-dependent and T-independent mechanisms (Fig. 13-7). In both cases, the molecules that drive IgA switching include both soluble cytokines and membrane proteins on other cell types that bind to signaling receptors on B cells (see Chapter 11). TGF-β is required for IgA isotype switching in the gut as well as in other mucosal compartments, and this cytokine is produced by intestinal epithelial cells and DCs in GALT. Furthermore, GALT DCs express the αvβ8 integrin, which is required for activation of TGF-β. Several molecules that promote IgA class switching are expressed by intestinal epithelial cells or GALT DCs in response to TLR signaling, and the commensal bacteria in the gut lumen produce the ligands that bind to the relevant TLRs. For example, T-independent IgA and IgG switching requires binding of the TNF family cytokine APRIL to the TACI receptor on B cells, and intestinal epithelial cells produce APRIL in response to TLR ligands made by commensal bacteria. Intestinal epithelial cells also produce thymic stromal lymphopoietin (TSLP) in response to TLR signals, and TSLP stimulates additional APRIL production by GALT DCs. TLR ligands made by commensal bacteria in the gut also increase expression of inducible nitric oxide synthase in DCs, leading to nitric oxide production. Nitric oxide is thought to promote both T-dependent and T-independent IgA class switching, in part because nitric oxide enhances TGF-β signaling in B cells and also synthesis of APRIL by GALT DCs. Finally, intestinal B cell IgA production is at least partly dependent on the vitamin A metabolite all-trans retinoic acid, which is made by intestinal epithelial cells and GALT DCs, although the mechanisms by which retinoic acid promotes IgA production are not known. Retinoic acid is also important in B cell homing to the gut, as we discussed earlier. There is an abundance of many of these molecules within the GALT and mesenteric lymph nodes compared with nonmucosal lymphoid tissues such as spleen and skin-draining lymph nodes, largely accounting for the propensity of B cells in the GALT to switch to IgA production.
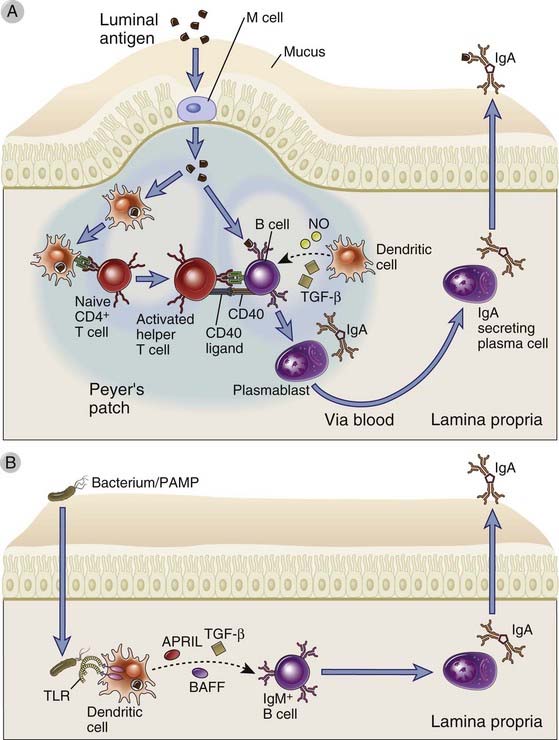
FIGURE 13–7 IgA class switching in the gut.
IgA class switching in the gut occurs by both T-dependent and T-independent mechanisms. A, In T-dependent IgA class switching, DCs in the subepithelial dome of Peyer’s patches capture bacterial antigens delivered by M cells and migrate to the interfollicular zone, where they present antigen to naive CD4+ T cells. The activated T cells differentiate into helper T cells and engage in cognate interactions with antigen-presenting IgM+IgD+ B cells that have also taken up and processed the bacterial antigen. B cell class switching to IgA is stimulated through T cell CD40L binding to B cell CD40, together with the action of TGF-β. IgA class switching may be enhanced by NO production by DCs, which upregulates TGF-β receptor on B cells. This T cell–dependent pathway yields high-affinity IgA antibodies that preferentially target pathogens and toxins. B, T-independent IgA class switching involves dendritic cell activation of IgM+IgD+ B cells, including B-1 B cells. TLR ligand–activated DCs secrete factors that induce IgA class switch, including BAFF, APRIL, and TGF-β. The DCs also produce IL-6 and retinoic acid. This T cell–independent pathway yields relatively low-affinity IgA antibodies to intestinal bacteria. The molecular mechanisms of class switching are described in Chapter 11.
The dominance of IgA production by intestinal plasma cells is enhanced by selective gut-homing properties of IgA-producing cells that arise in GALT and mesenteric lymph nodes (see Fig. 13-5). Some of the IgA that is transported across the intestinal epithelium may be produced by plasma cells that differentiated and remained within underlying GALT follicles. However, IgA-secreting plasma cells are widely dispersed in the lamina propria of the gastrointestinal tract, not just in lymphoid follicles. As we discussed earlier, activated B cells that undergo isotype switching into IgA-producing cells in the GALT and mesenteric lymph nodes may enter into the systemic circulation and then selectively home back to the intestinal lamina propria, where they may reside as plasma cells.
Secreted IgA is transported through epithelial cells into the intestinal lumen by an IgA/IgM-specific Fc receptor called the poly-Ig receptor (Fig. 13-8). The IgA produced by plasma cells in the lamina propria is in the form of a dimer that is held together by the coordinately produced J chain, which is covalently bound by disulfide bonds to the Fc regions of the α heavy chains of two IgA molecules. Mucosal plasma cells produce abundant J chain, even more than plasma cells in nonmucosal tissues, and serum IgA is usually a monomer lacking the J chain. From the lamina propria, the IgA must be transported across the epithelium into the lumen by a process known as transcytosis, and this function is mediated by the poly-Ig receptor. This receptor is synthesized by mucosal epithelial cells and expressed on their basal and lateral surfaces. It is an integral membrane glycoprotein with five extracellular domains homologous to Ig domains and is thus a member of the Ig superfamily. The J chain of secreted dimeric IgA and pentameric IgM contains a domain required for poly-Ig receptor binding, and therefore dimeric IgA (and multimeric IgM) binds to the poly-Ig receptor on mucosal epithelial cells (see Fig. 13-8). This complex is endocytosed into the epithelial cell and actively transported in vesicles to the luminal surface. Here the poly-Ig receptor is proteolytically cleaved, its transmembrane and cytoplasmic domains are left attached to the epithelial cell, and the extracellular domain of the receptor, which carries the IgA molecule, is released into the intestinal lumen. The soluble IgA–associated component of the receptor is called the secretory component. IgM produced by lamina propria plasma cells is also a polymer (pentamer) associated noncovalently with the J chain, and the poly-Ig receptor also transports IgM into intestinal secretions. This is why this receptor is called the poly-Ig receptor. It is believed that the bound secretory component protects polymeric IgA and IgM from proteolysis by enzymes present in the intestinal lumen and these antibodies are therefore able to serve their function of neutralizing microbes and toxins in the lumen. The poly-Ig receptor is also responsible for the secretion of IgA into bile, milk, sputum, saliva, and sweat.
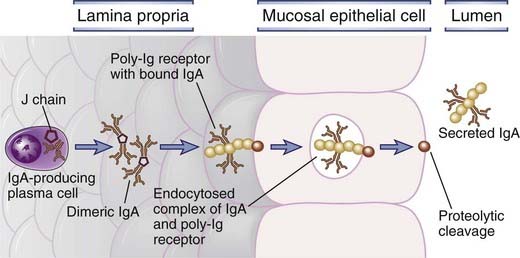
FIGURE 13–8 Transport of IgA across epithelial cells.
IgA is produced by plasma cells in the lamina propria of mucosal tissue and binds to the poly-Ig receptor at the base of an epithelial cell. The complex is transported across the epithelial cell, and the bound IgA is released into the lumen by proteolytic cleavage. The process of transport across the cell, from the basolateral to the luminal surface in this case, is called transcytosis.
IgG is present in intestinal secretions at levels equal to IgM but lower than IgA. In some mucosal secretions (i.e., in the rectum, genitourinary tract, and airways), IgG levels are high and often exceed IgA. The transport of IgG into mucosal secretions is due to another transcytosing receptor, the neonatal Fc receptor (FcRn), which we discussed in Chapter 12. In contrast to the poly-Ig receptor, which transports IgA unidirectionally (from the basal side to the apical/lumen side), FcRN can mediate bidirectional transport of IgG. Therefore, FcRn-mediated IgG transport likely contributes to humoral immunity against luminal intestinal pathogens and may also contribute to uptake of antibody-coated microbes and other antigens from the lumen into the GALT.
IgA produced in lymphoid tissues in the mammary gland is secreted into colostrum and mature breast milk through poly-Ig receptor–mediated transcytosis and mediates passive mucosal immunity in breast-fed children. The human lactating mammary gland contains a large number of IgA-secreting plasma cells, and the mammary gland epithelium can store large quantities of secretory IgA. The plasma cells in the breast originate from various mucosa-associated lymphoid tissues. They home to the breast because most IgA plasmablasts express CCR10, no matter which lymphoid tissues they were generated in, and the breast tissues express CCL28, the chemokine that binds CCR10. Therefore, during breast-feeding, a child ingests a significant quantity of maternal IgA, which provides broad polymicrobial protection in the infant’s gut. Moderate amounts of IgG and IgM are also secreted into breast milk and contribute to the passive immunity of breast-fed children. Many epidemiologic studies have shown that breast-feeding significantly reduces the risk of diarrheal disease and sepsis, especially in developing countries, and this correlates with the presence of secretory IgA in breast milk specific for enterotoxic species of bacteria including Escherichia coli and Campylobacter.
T Cell–Mediated Immunity in the Gastrointestinal Tract
T cells play important roles in protection against microbial pathogens in the gastrointestinal system and in regulating responses to food and commensal antigens. Furthermore, T cells contribute to inflammatory diseases in the gastrointestinal tract. As in other parts of the body, T cell immunity in the gut involves different subsets of T cells and is influenced in various ways by antigen-presenting DCs, which also belong to different subsets. In this section, we will discuss important features of T cell and DC functions in the intestines.
T cells are found within the gut epithelial layer, scattered throughout the lamina propria and submucosa, and within Peyer’s patches and other organized collections of follicles. In humans, most of the intraepithelial T cells are CD8+ cells. In mice, about 50% of intraepithelial lymphocytes express the γδ form of the TCR, similar to intraepidermal lymphocytes in the skin. In humans, only about 10% of intraepithelial lymphocytes are γδ cells, but this proportion is still higher than the proportions of γδ cells found among T cells in other tissues. Both the αβ and the γδ TCR-expressing intraepithelial lymphocytes show limited diversity of antigen receptors. These findings support the idea that mucosal intraepithelial lymphocytes have a limited range of specificity, distinct from that of most T cells, and this restricted repertoire may have evolved to recognize microbes that are commonly encountered at the epithelial surface. Lamina propria T cells are mostly CD4+, and most have the phenotype of activated effector or memory T cells, the latter with an effector memory phenotype (see Chapter 9). Recall that these lamina propria effector and memory T cells are generated from naive precursors in the GALT and mesenteric lymph nodes, enter the circulation, and preferentially home back into the lamina propria (see Fig. 13-5). T cells within Peyer’s patches and in other follicles adjacent to the intestinal epithelium include CD4+ helper T cells and regulatory T cells.
DCs are abundant in the gastrointestinal immune system and can be broadly divided into two functional subgroups, which participate in stimulating protective effector T cell responses or inducing regulatory T cell responses that suppress immunity to ingested antigens and commensal organisms (see Fig. 13-4). These subsets, which are also present in mucosal tissues other than the gut, are sometimes called effector DCs and regulatory DCs. They can be distinguished by their differential expression of integrins and chemokine receptors: effector DCs are CD11b+CX3CR1+, and regulatory DCs are CD103+CX3CR1−. Effector DCs are the antigen-sampling DCs we discussed earlier in the chapter that project dendrites between epithelial cells and sample luminal contents. They can interact with naive T cells (and B cells) in the GALT, or they can migrate, through lymphatic drainage, into mesenteric lymph nodes, where they present processed protein antigens to naive T cells and induce the differentiation of these T cells into IFN-γ– or IL-17–producing effector cells. Regulatory DCs, which do not directly sample luminal contents, induce the differentiation of naive T cells into FoxP3+ regulatory T cells. Regulatory DCs are thought to be conditioned by mucosal epithelial cells to secrete TGF-β and retinoic acid at the time of antigen presentation to naive T cells; these molecules favor Treg differentiation. DC heterogeneity in the gut as well as in other tissues is actually more complex than we have discussed, with 5 to 10 different subsets having been identified by patterns of expression of multiple surface molecules. The functional relevance of these subsets remains incompletely understood.
In the gastrointestinal tract, different subsets of effector CD4+ T cells are induced by and protect against different microbial species. In Chapters 9 and 10, we introduced the concept that helper T cell subsets that secrete different cytokines are specialized for particular types of antimicrobial responses. This fundamental concept is highly relevant to the mucosal immune system. The commensal bacterial microflora of the gut lumen exerts profound influences on T cell phenotypes even during homeostasis.
Regulation of Immunity in the Gastrointestinal Tract by Regulatory T Cells and Cytokines
Regulatory T cells are abundant in GALT and prevent inflammatory reactions against intestinal commensal microbes. It is estimated that there are about twice as many FoxP3+ Treg among CD4+ cells in the lamina propria as in other peripheral lymphoid tissues. Many of these Treg are likely induced in the gut in response to antigens encountered locally and thus belong to the category of adaptive Treg (see Chapter 14). The factors that contribute to the generation of these Treg include CD103+ DCs, local production of retinoic acid (which promotes FoxP3 expression), and local production of TGF-β (which also promotes FoxP3 expression and inhibits the generation of TH1 and TH2 cells). As we discuss in Chapter 14, Treg are thought to suppress immune responses by several mechanisms. Of these, the dominant mechanism in the gut seems to be production of the immunosuppressive cytokine IL-10, as discussed later.
Several cytokines, including TGF-β, IL-10, and IL-2, appear to play crucial roles in maintaining homeostasis in the gut immune system, and deficiencies in these cytokines or their receptors result in pathologic bowel inflammation. Much of our knowledge of cytokine-mediated regulation in the gut comes from studies with cytokine or cytokine receptor gene knockout mice. A major feature of the phenotype of mice with engineered deficiencies in TGF-β, IL-10, IL-10 receptor, IL-2, and IL-2 receptor is uncontrolled inflammation in the bowel. Mutations in the IL-10 and IL-10 receptor genes are also associated with severe inflammatory bowel disease in children, confirming the importance of IL-10 in preventing pathologic gut inflammation in humans. The uncontrolled inflammation observed in the gut in the absence of these cytokines or their receptors is most likely caused by innate and adaptive immune responses to commensal gut flora because the inflammation does not occur in mice raised in germ-free conditions. The cellular sources of the cytokines and the relevant receptor-expressing target cells that are critical for prevention of bowel inflammation are not completely understood. Mouse models in which cytokines, cytokine receptors, and cytokine receptor signaling are genetically ablated only in specific cell types have been used to address the question of which cell types are important. In the case of TGF-β– and IL-10–dependent regulation of gut inflammation, evidence indicates that Treg and macrophages are both important sources of these cytokines. For example, selective deletion of the IL10 gene in FoxP3+ cells rapidly leads to severe colitis but no other manifestations of inflammatory disease, consistent with the critical role of Treg-produced IL-10 in maintaining homeostasis in the gastrointestinal tract. The target cells that express receptors for and are regulated by TGF-β and IL-10 likely include DCs, effector T cells, innate effector cells such as macrophages, and epithelial cells. Inflammatory bowel disease in mice lacking IL-2 or its receptor is a consequence of the defects in the development and function of Treg, which require IL-2, (see Chapter 14).
Oral Tolerance and Oral Vaccines
Oral tolerance is systemic adaptive immune tolerance to antigens that are ingested or otherwise administered orally and is a potential way of treating diseases in which unwanted immune responses occur, such as autoimmunity. Oral tolerance has been most clearly demonstrated in experimental rodent models. Mice fed high doses of a protein antigen may subsequently have impaired humoral and T cell–mediated responses to the same antigen administered by other routes, such as through the skin. A similar phenomenon can be demonstrated when antigens are administered through the nasal passages into the respiratory mucosa, and the more general term mucosal tolerance is used to describe tolerance induced by either oral or nasal antigen administration. The physiologic role of oral tolerance is speculated to be the prevention of potentially harmful immune responses to food proteins and commensal bacteria. The underlying mechanisms of oral tolerance are not well understood but likely include the same mechanisms of peripheral tolerance discussed in Chapter 14, such as anergy, deletion, and Treg-mediated suppression. The propensity of the immune system in the gut to suppress local immune responses to antigens in the intestinal lumen could be manifested in other parts of the body because of circulation of Treg to other tissues and deletion or anergy of effector T cells in the gut, which are no longer available to respond to antigens at other sites. Attempts to treat autoimmune disease or allergies by oral or nasal administration of relevant self antigens or allergens have so far been unsuccessful.
Oral administration of antigen in the setting of concomitant stimulation of innate immunity can lead to productive adaptive immune responses, as in the use of oral viral vaccines to induce protective antibody responses to viruses. These vaccines are live attenuated viruses that may infect DCs in the intestine and stimulate strong innate responses that then promote T and B cell activation.
Diseases Related to Immune Responses in the Gut
Given the abundance of immune cells and their constant activity in the intestinal mucosa, it is not surprising that there are many intestinal diseases related to abnormal immune responses. These diseases are generally caused by unregulated responses to commensal organisms or to antigens in the food. We will now discuss selected examples of these diseases; they are more thoroughly described in medical textbooks.
Inflammatory bowel disease is a heterogeneous group of disorders characterized by chronic remitting inflammation in the small or large bowel, likely due to poorly regulated responses to commensal bacteria. The two main types of inflammatory bowel disease are Crohn’s disease, which can affect the entire thickness of the bowel wall tissue in any part of the gastrointestinal tract but most frequently involves the terminal ileum, and ulcerative colitis, which is restricted to the colonic mucosa. Symptoms include abdominal pain, vomiting, diarrhea, and weight loss. Treatments include various anti-inflammatory drugs, such as sulfasalazine, corticosteroids, TNF antagonists, and antimetabolites. Although the etiology of Crohn’s disease and ulcerative colitis is poorly understood, several types of evidence suggest that these disorders are a result of defects in the regulation of immune responses to commensal organisms in the gut in a genetically susceptible host. A number of immunologic abnormalities may contribute to the development of inflammatory bowel disease.
Celiac disease (gluten-sensitive enteropathy or nontropical sprue) is an inflammatory disease of the small bowel mucosa caused by immune responses against ingested gluten proteins present in wheat. Celiac disease is characterized by chronic inflammation in the small bowel mucosa, leading to atrophy of villi, malabsorption, and various nutritional deficiencies that lead to extraintestinal manifestations. The disease is treated by dietary restriction to gluten-free foods. Patients produce IgA and IgG antibodies specific for gluten as well as IgA and IgG autoantibodies specific for transglutaminase 2A, an enzyme that modifies the gluten protein gliadin. These autoantibodies are thought to arise when transglutaminase-specific B cells endocytose host transglutaminase covalently associated with gliadin and then present gliadin peptides to helper T cells, which then provide help for the anti-transglutaminase antibody response. Whether these antibodies contribute to disease development is not known, but they are a sensitive diagnostic marker for the disease. There is strong evidence that CD4+ T cell responses to gliadin are involved in disease pathogenesis. T cells specific for gliadin peptides are found in celiac patients, and the inflammatory process in the bowel includes T cells and T cell cytokines. Gliadin peptides bind strongly to two class II MHC alleles found in most patients, namely, HLA-DQ2 and HLA-DQ8, and there is a high relative risk for development of the disease among people with these two alleles. We will further discuss the association of autoimmune diseases with MHC alleles in Chapter 14. In addition to CD4+ T cell responses, CD8+ cytotoxic T lymphocyte (CTL) killing of intestinal epithelial cells may also contribute to celiac disease, although not by recognition of gliadin peptides. Instead, gliadin stimulates epithelial cells to secrete IL-15, which induces expression of the activating receptor NKG2D on CTLs, and gliadin induces the expression of the ligands for NKG2D (MICA, MICB) on intestinal epithelial cells. The net result is a lowering of the threshold for CTL killing of the epithelial cells, although the source of the actual peptides recognized by the CTLs is not clear.
Food allergies are caused by TH2 responses to many different food proteins and cause acute inflammatory responses locally in the gut and systemically on ingestion of these proteins. Allergies result from TH2-dependent IgE responses to environmental antigens (allergens), which are either proteins or chemicals that modify (haptenate) self proteins. In the case of food allergies, the environmental antigens are ingested, and this is another example of a failure of adaptive immune tolerance to food antigens. The anti-allergen antibodies bind to Fc receptors on mast cells, and subsequent exposure to the allergen will cause cross-linking of the Fc receptors, activation of the mast cells, and release of potent proinflammatory amine and lipid mediators and cytokines. There are abundant mast cells in the lamina propria of the bowel. Therefore, reingestion of a food allergen by a person who has previously mounted a TH2 and IgE response to the allergen will trigger mast cell activation, with its pathologic consequences. Cytokines produced by TH2 cells also directly stimulate peristalsis and may trigger symptoms of food allergies even without the participation of IgE. These reactions may cause gastrointestinal symptoms like nausea, vomiting, diarrhea, and abdominal pain, but the allergen can be absorbed into the blood and end up activating mast cells in many different tissues, producing systemic manifestations. We will discuss allergic reactions in more detail in Chapter 19.
Prolonged immune responses to gastrointestinal microbes can lead to tumors arising in the gastrointestinal tract. The best documented example of this is the so-called MALT lymphomas in the stomach of people with chronic Helicobacter pylori infection. These lymphomas are tumors arising from malignantly transformed follicular B cells in lymphoid follicles of the gastric lamina propria. It is believed that H. pylori sets up an inflammatory reaction that promotes the development and growth of tumors induced by B cell intrinsic oncogenic events. Remarkably, if gastric MALT lymphomas are diagnosed before they spread beyond the stomach wall, patients can be cured by antibiotic treatment of the H. pylori infection.
Immunity in other Mucosal Tissues
Like the gastrointestinal mucosa, the mucosae of the respiratory system, the genitourinary system, and the conjunctiva must maintain a barrier against invasion of diverse microbes in the environment and balance effective protective responses to invading microbes and suppression of responses to numerous commensal organisms. Many of the features we described for gastrointestinal immunity are shared by mucosal immunity in these different locations. These shared features include relatively impermeable mucus- and defensin-secreting epithelial barriers; localized collections of lymphoid tissues just beneath the epithelium; the constant sampling of antigens located outside the barriers by immune cells within the barrier; the constant integration of proinflammatory and regulatory signals generated by microbial products binding to epithelial and DC TLRs; the strong reliance on secretory IgA–mediated humoral immunity to prevent microbial invasion; and the presence of effector and regulatory DC populations that stimulate particular types of effector and regulatory T cell responses. In addition to these shared features, each different mucosal tissue has unique features that reflect the distinct functions and anatomy of the organs it is part of and the distinct range of environmental antigens and microbes that are present at each site. We will now discuss some of the major features of mucosal immunity in these organs, focusing mainly on the respiratory system.
Mucosal Immunity in the Respiratory System
The mucosa of the respiratory system lines the nasal passages, nasopharynx, trachea, and bronchial tree. Alveoli, the epithelium-lined sac-like termini of the bronchial airways, may also be considered part of the respiratory mucosa. Inhalation of air exposes the respiratory mucosa to a wide variety of foreign substances, including airborne infectious organisms, plant pollens, dust particles, and various other environmental antigens. The microbial flora of the airways is far less dense and less diverse than that in the gut, and the deep airways and alveoli are usually sterile. Nonetheless, similar mechanisms have evolved in the respiratory mucosal immune system to achieve a fine balance between immune activation to protect against pathogens and immune regulation to avoid unnecessary or excessive responses that might impair the physiologic functions. Failure of the immune system to control bronchopulmonary infections and excessive immune or inflammatory responses to infections are major causes of morbidity and mortality worldwide.
Innate Immunity in the Respiratory System
The pseudostratified, ciliated columnar epithelium that lines most of the respiratory mucosa, including nasal passages, nasopharynx, and bronchial tree, performs similar physical and chemical barrier functions as gut epithelium, by virtue of tight junctions between cells and secretion of mucus, defensins, and cathelicidins. The mucus in the airways traps foreign substances including microbes, and the cilia move the mucus and trapped microbes up and out of the lungs. The importance of mucus and cilia in innate immune protection in the lung is illustrated by the greatly increased frequency of serious bronchopulmonary infections in people with decreased cilia function, such as heavy smokers, or impaired mucus production, such as patients with cystic fibrosis.
Innate responses in the alveolus serve antimicrobial functions but are tightly controlled to prevent inflammation, which would impair gas exchange. The alveoli are normally sterile but are susceptible to infection spreading from bronchopneumonia, and alveolar lining cells can be directly infected by viruses. Surfactant proteins A (SP-A) and D (SP-D), which are secreted into the alveolar spaces, are members of the collectin family (see Chapter 4) and bind to carbohydrate PAMPs on the surface of many pathogens. These surfactants are involved in viral neutralization and clearance of microbes from the airspaces, but they also suppress inflammatory and allergic responses in the lung. For example, SP-A inhibits TLR2 and TLR4 signaling and inflammatory cytokine expression in alveolar macrophages, and SP-A also binds to TLR4 and inhibits lipopolysaccharide binding. SP-A and SP-D reduce the phagocytic activity of alveolar macrophages.
Alveolar macrophages represent the majority of free cells within the alveolar spaces. These cells are functionally distinct from macrophages in most other tissues in that they maintain an anti-inflammatory phenotype. They express IL-10, nitric oxide, and TGF-β and are poorly phagocytic compared with resident macrophages in other tissues, such as spleen and liver. Alveolar macrophages inhibit T cell responses as well as the antigen presentation function of CD103+ airway DCs.
Adaptive Immunity in the Respiratory System
Protective humoral immunity in the airways is dominated by secretory IgA, as in other mucosal tissues, although the amount of IgA secreted is much less than in the gastrointestinal tract. Secretory IgG plays an important role in the upper airway. The anatomic sites of naive B cell activation, differentiation, and IgA isotype class switching may vary but include tonsils and adenoids in the nasopharynx and lymph nodes in the mediastinum and adjacent to bronchi in the lungs. There are relatively few aggregated or isolated lymphoid follicles in the lamina propria in the lower airways compared with the gut and likely less initiation of humoral immune responses in these locations. The homing of IgA-secreting plasma cells back into the airway tissue in proximity to respiratory mucosal epithelium depends on the chemokine CCL28 secreted by respiratory epithelium and its receptor CCR10 on the plasma cells. IgA and IgG are transported into the airway lumen by the same poly-Ig receptor and FcRn mechanism of transcellular transport as in the gut. IgE responses to airway antigens occur frequently and are involved in allergic diseases of the respiratory system, including hay fever and asthma. IgE performs its inflammatory effector functions when bound to mast cells, which are abundant in the airways.
T cell responses in the lung are initiated by DC sampling of airway antigens and presentation of these antigens to naive T cells in peribronchial and mediastinal lymph nodes. A network of DCs is present in the mucosa of the airways, and these DCs can be subdivided into subsets based on surface markers and location. The CD103+CD11b− DCs extend dendrites between the bronchial epithelial cells into the airway lumen. These DCs sample airway antigens, migrate to draining lymph nodes, present the processed antigens to naive T cells, and have a propensity to drive differentiation of these T cells to the TH2 subset. The TH2 cells home back into the bronchial mucosa, where they may be reactivated by allergens presented by DCs in lamina propria. This pathway is considered central to the development of allergic asthma (see Chapter 19). Other DCs are found in the lamina propria beneath the epithelial cells, and these are mainly CD103−CD11b+.
Mucosal Immunity in the Genitourinary System
Innate immune defense against microbial invasion and infection in the genitourinary mucosa relies mainly on the epithelial lining, as in other mucosal barriers. Stratified squamous epithelium lines the vaginal mucosa and terminal male urethra, and a single layer of mucus-secreting columnar epithelium lines the upper female genital tract. The vaginal epithelium contains Langerhans cells, and a variety of DCs and macrophages have been described beneath the epithelium in vagina, endocervix, and urethra. There are also resident B and T cells in the genital mucosa. Differences in the phenotype of the DCs and macrophages in the female genital mucosa from those in the gastrointestinal tract may underlie the greater susceptibility of the latter to HIV infection. There is little regional specialization of the adaptive immune system in the genitourinary mucosa, which lacks prominent mucosa-associated lymphoid tissues. Unlike other mucosa, in which IgA is the dominant antibody isotype, most of the antibodies in genital secretions are IgGs, about half of which are produced by plasma cells in genital tract mucosa; the rest are from the circulation.
The Cutaneous Immune System
The skin includes two main layers, the outer epidermis composed mainly of epithelial cells and, separated by a thin basement membrane, the underlying dermis composed of connective tissue and specialized adnexal structures such as hair follicles and sweat glands. Within both of these layers, a variety of different cell types and their products, composing the cutaneous immune system (Fig. 13-9), provide physical barrier and active immune defense functions against microbes. The skin of an adult is about 2 m2 in area and is the second largest barrier of the body against environmental microbes and other foreign materials. Nonetheless, given its outermost location, the skin is normally colonized by many microbes and is frequently breached by trauma and burns. Therefore, the skin is a common portal of entry of a wide variety of microbes and other foreign substances and is the site of many immune responses.
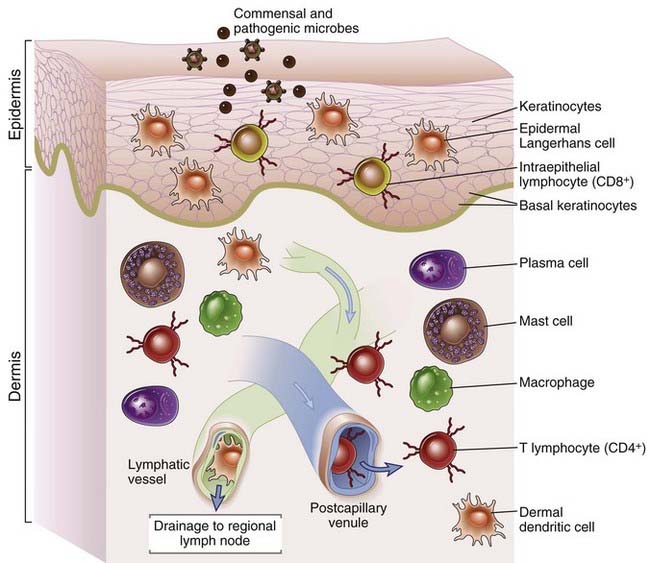
FIGURE 13–9 Cellular components of the cutaneous immune system.
The major components of the cutaneous immune system shown in this schematic diagram include keratinocytes, Langerhans cells, and intraepidermal lymphocytes, all located in the epidermis, and T lymphocytes, DCs, and macrophages, located in the dermis.
Innate and Adaptive Immune Reponses in the Skin
The epidermis provides a physical barrier to microbial invasion. The epidermis consists of multiple layers of stratified squamous epithelium, made up almost entirely of specialized epithelial cells called keratinocytes. The basal layer of keratinocytes, anchored onto the basement membrane, are continuously proliferating, and their maturing progeny cells are displaced upward and differentiate to form several different layers. In the top layer, called the stratum corneum, the cells undergo programmed death, thereby forming a keratin- and lipid-rich permeability barrier that is important for protection against microbes as well as harmful physical and chemical agents.
In addition to their role in forming a physical barrier, keratinocytes respond to pathogens and injury by producing antimicrobial peptides, which kill microbes, and various cytokines, which promote and regulate immune responses. The antimicrobial peptides that keratinocytes produce include defensins, S100, and cathelicidins (see Chapter 4). The cytokines made by keratinocytes include TNF, IL-1, IL-6, and IL-18, which promote inflammation; GM-CSF, which induces differentiation and activation of DCs in the epidermis, discussed later; and IL-10, which controls immune responses. Keratinocytes produce the chemokines CCL17, CCL20, and CCL27, which participate in recruitment of lymphocytes expressing CCR4, CCR6, and CCR27. The induced expression of defensins, cytokines, and chemokines by keratinocytes depends on innate immune receptors including TLRs and NLRs. Keratinocytes express most of the TLRs and NLRP3, which forms the IL-1–processing inflammasome (see Chapter 4). Keratinocytes in normal skin constitutively synthesize pro–IL-1 and pro–IL-18. Stimuli such as UV irradiation activate the inflammasome to process these pro-cytokines to the active forms, which explains the inflammatory response to sunburn. When signal transduction pathways linked to inflammatory responses, such as the NF-κB and STAT3 pathways, are genetically activated only in keratinocytes, mice develop inflammatory skin diseases, showing the potential of keratinocytes to act as central players of cutaneous immune responses.
Several DC populations are normally present in the skin and contribute both to innate immune responses and to initiation of T cell responses to microbial and environmental antigens that enter the body through the skin. In the epidermis, the most abundant DCs are the Langerhans cells, which express a C-type lectin receptor called langerin (CD207) and have numerous Birbeck granules in the cytoplasm (see Fig. 6-4, Chapter 6). The dendrites of Langerhans cells form a dense meshwork between the keratinocytes of the epidermis. In the dermis, there are relatively sparse langerin-expressing CD103+ DCs, which are a distinct lineage from Langerhans cells, and langerin-negative DCs, such as plasmacytoid DCs. Each of these dendritic cell populations express innate pattern recognition receptors for PAMPs expressed on microbes and damage-associated molecular patterns (DAMPs) expressed on injured cells, and the DCs respond by secreting inflammatory cytokines.
Skin DCs take up foreign proteins, transport them to draining lymph nodes, and present processed peptides from these proteins to T cells or pass the protein antigens to other lymph node–resident DCs. When Langerhans cells encounter microbes, they are activated by engagement of Toll-like receptors (see Chapter 6). The cells lose their adhesiveness for the epidermis, enter lymphatic vessels, begin to express the CCR7 chemokine receptor, and migrate to the T cell zones of draining lymph nodes in response to chemokines produced in that location. The Langerhans cells also mature into efficient antigen-presenting cells. What remains unclear is the relative contribution of the different skin DC subsets to the initiation of T cell responses. Mouse models have been developed in which langerin-expressing DCs can be selectively eliminated, and under the proper conditions, the mice lack Langerhans cells but have dermal DCs. Using these models, investigators have shown that some T cell responses to chemically modified self proteins, a model for contact hypersensitivity, occur in the absence of Langerhans cells. Furthermore, T cell responses to certain viruses, including herpes viruses, depend on dermal langerin+ CD103+ DCs but not Langerhans cells. Langerhans cells do appear to be required for TH2 responses that cause atopic dermatitis (contact hypersensitivity and atopic dermatitis will be discussed later). The role of the different skin DC populations could vary with the antigen dose and type and likely will differ between mice and humans.
Normal human skin contains many T cells, 95% of which have a memory phenotype. Human skin contains about 1 million T cells/cm2, which is about 2 × 1010 total T cells in the skin. About 98% of these T cells are present in the dermis, and 2% are intraepidermal lymphocytes. Dermal T lymphocytes (both CD4+ and CD8+ cells) are predominantly in a perivascular location and usually express phenotypic markers typical of activated or memory cells. It is not clear whether these cells reside permanently within the dermis or are merely in transit between blood and lymphatic capillaries as part of memory T cell recirculation. CD4+ T cells of each major subset, TH1, TH2, TH17, and Treg, are found in the skin. TH1 and TH17 cells are important for microbial defense, against intracellular and extracellular microbes, respectively, as in other tissues. The two signature TH17 cytokines, IL-17 and IL-22, are known to induce expression of defensins by keratinocytes. Intraepidermal T cells, most of which are CD8+ cells, may express a more restricted set of antigen receptors than do T lymphocytes in most extracutaneous tissues. In mice (and some other species), many intraepidermal lymphocytes are T cells that express the γδ T cell antigen receptor.
T cells in the skin express homing molecules that direct their migration out of dermal microvessels (Fig. 13-10). Migration of effector or memory T cells into the skin depends on T cell expression of cutaneous lymphocyte antigen (CLA), which is an E-selectin–binding carbohydrate moiety displayed on various glycoproteins on the endothelial cell plasma membrane. In addition, T cell expression of CCR4 and CCR10, which bind the chemokines CCL17 and CCL27, respectively, is also required for T cell trafficking to skin. The skin-homing properties of T cells are imprinted during activation in skin-draining lymph nodes, by a process analogous to imprinting of gut-homing properties of T cells in mesenteric lymph nodes, discussed earlier in the chapter. When naive T cells recognize antigens presented by DCs in skin-draining lymph nodes, they receive signals from the DCs that not only induce proliferation and differentiation into effector cells but also induce expression of the skin-homing molecules CLA, CCR4, and CCR10. Interestingly, sunlight and vitamin D appear to play an important role in T cell migration to the skin, analogous to the role of vitamin A and its metabolite retinoic acid in lymphocyte migration to the gut. UVB rays in sunlight act on 7-dehydrocholesterol made in the basal layer of the epidermis, converting it to previtamin D3. Dermal DCs express vitamin D3 hydroxylases that convert previtamin D3 to the active form, 1,25(OH)2D3. When the DCs present antigen to T cells, the active 1,25(OH)2D3 is released, enters the T cells, translocates to the nucleus, and activates transcription of CCR10. 1,25(OH)2D3 produced in dermal DCs may be transported in free form or within migrating DCs to skin-draining lymph nodes. Within the node, 1,25(OH)2D3 induces CCR10 expression on naive T cells activated by antigen-presenting DCs. IL-12 made by the DCs participates in induction of CLA. CCR4 is also upregulated, and the gut-homing integrin α4β7 is downregulated, both by unknown signals, during T cell activation in skin-draining lymph nodes. Thus, naive T cells activated in skin-draining lymph nodes will differentiate into effector T cells that preferentially home back into the skin. 1,25(OH)2D3 may also act locally within the dermis on effector and memory T cells to upregulate CCR10 and promote migration of the T cells into the epidermis because the CCR10 ligand CCL27 is made by keratinocytes.
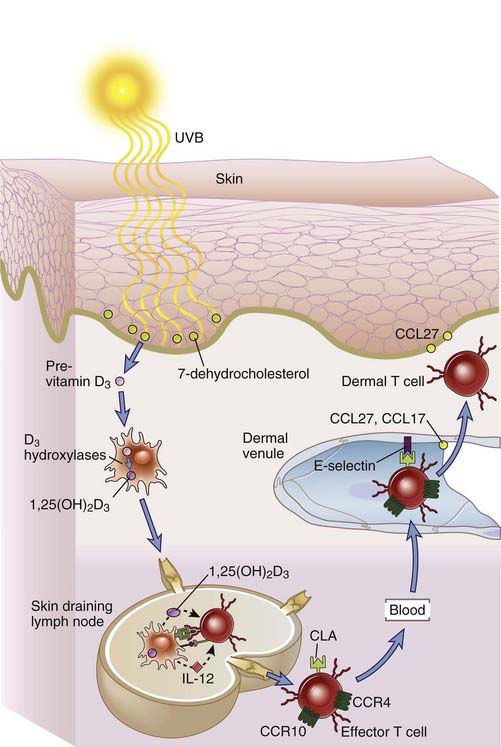
FIGURE 13–10 Homing properties of skin lymphocytes.
The skin-homing properties of effector lymphocytes are imprinted in skin-draining lymph nodes where they have undergone differentiation from naive precursors. The vitamin D precursor 7-dehydrocholesterol made in the basal layer of the epidermis is converted into 1,25(OH)2D3 (active vitamin D) by sequential actions of ultraviolet rays in sunlight (UVB) and hydroxylases in dermal dendritic cells. During naive T cell activation by dendritic cells in skin-draining lymph nodes, 1,25(OH)2D3 induces expression of CCR10, IL-12 induces expression of the E-selectin ligand cutaneous lymphocyte antigen (CLA), and other signals induce CCR4 expression. The differentiated effector T cells enter the circulation, and the homing molecules they now express direct migration out of dermal venules into the skin because of the expression of E-selectin on the endothelium of the venules and production of CCL27 and CCL17 (the ligands of CCR10 and CCR4) in the skin.
Diseases Related to Immune Responses in the Skin
There are many different inflammatory diseases that are caused by dysregulated or inappropriately targeted immune responses in the skin. We will now discuss two selected examples of these diseases. In addition to these inflammatory diseases, there are several malignant lymphomas that primarily affect the skin. Most of these are derived from skin-homing T cells.
Psoriasis, a chronic inflammatory disorder of the skin characterized by red scaly plaques, is caused by dysregulated innate and T cell–mediated immune responses triggered by various environmental stimuli. DCs are central to the pathogenesis, and reduction in numbers of skin DCs by treatment with the ultraviolet A (UVA) absorbing chemical psoralen plus UVA light (PUVA therapy) reduces disease manifestations. There is evidence that psoriasis is initiated when trauma or infection induces production of the cathelicidin LL-37 by keratinocytes, which forms complexes with host DNA and then activates plasmacytoid DCs in the skin through TLR9. Activated plasmacytoid DCs produce abundant IFN-α, and psoriatic skin has a strong type I interferon signature (i.e., expression of many interferon-inducible genes). One of the effects of IFN-α is activation of other DCs that are induced to migrate to lymph nodes, activate helper T cells of unknown antigen specificity, and induce their differentiation into skin-homing effector cells. These T cells circulate to the dermis and further promote an inflammatory cascade and persistent keratinocyte proliferation. Both TH1 and TH17 cells have been implicated in this phase of the disease. Various therapies have been approved or are in clinical trials targeting T cell migration and cytokines that drive TH1 and TH17 differentiation (IL-12, IL-23) or are produced by these T cells (IL-17, IL-22, TNF). A central unanswered question about this disease is the identity of the antigens recognized by the T cells.
Atopic dermatitis is a chronic inflammatory disease of the skin characterized by itchy rashes, in which IgE specific for environmental antigens and cells expressing high-affinity Fc receptors for IgE (FcεRI) play a central role. Antigen-mediated cross-linking of FcεRI on mast cells is important in many allergic diseases, as we will discuss in detail in Chapter 19, and mast cells are implicated in the pathogenesis of atopic dermatitis. In addition, FcεRI is expressed on skin DCs, including Langerhans cells and plasmacytoid DCs, and DC activation also plays a central role in atopic dermatitis, as in psoriasis. However, in atopic dermatitis, the response of DCs to TLR signaling is believed to be influenced by FcεRI signaling and by the cytokine thymic stromal lymphopoietin (TSLP), made by keratinocytes, leading to an initial TH2 response. Underlying the inflammatory cascade of atopic dermatitis may be genetically determined sensitivity to environmental antigens, perhaps because of impaired epithelial barrier function.
Immune Privileged Tissues
Immune responses and associated inflammation in certain parts of the body, including brain, eye, testes, placenta, and fetus, carry a high risk of lethal organ dysfunction or reproductive failure. These tissues, which have evolved to be protected, to a variable degree, from immune responses, are called immune privileged sites. The Nobel laureate immunologist Sir Peter Medawar coined the term immune privilege in the 1940s to describe the lack of immune responses to tissue transplanted into the brain or the anterior chamber of the eye of experimental animals. Foreign antigens that would evoke an immune response in most tissues are often tolerated in these immune privileged sites. The mechanisms underlying immune privilege vary between these tissues and are not fully understood. Some of the mechanisms are similar to mechanisms of regulation in gut and skin discussed earlier in this chapter and mechanisms of self tolerance discussed in Chapter 14. In this section of the chapter, we will discuss some of the distinguishing features of immune privilege in different tissues.
Immune Privilege in the Eye, Brain, and Testis
The Eye
Vision, which is essential to survival to most mammals, can be easily impaired by inflammation within the eye. Evolved mechanisms that minimize the likelihood of immune responses and inflammation in the eye have been most thoroughly described in the anterior chamber, a fluid-filled space between the transparent cornea in front and the iris and lens behind. Inflammation in this chamber could lead to opacification of the transparent cornea and lens, with loss of sight. At least some of the properties of immune privilege studied in the anterior chamber also apply to other ocular sites, such as the vitreous cavity and the subretinal space. Anatomic features of the anterior chamber that contribute to immune privilege include the tight junctions and resistance to leakiness of blood vessels in the tissues adjacent to the anterior chamber (the so-called blood-eye barrier), the avascular nature of the cornea, and the absence of lymphatics draining the anterior chamber, which limits access of the adaptive immune system to antigens in the eye. There are several soluble factors with immunosuppressive/anti-inflammatory properties in the aqueous humor that fills the anterior chamber, including neuropeptides (α-melanocyte–stimulating hormone, vasointestinal peptide, somatostatin), TGF-β, and indolamine 2,3-dioxygenase. Cells lining the anterior chamber, including the epithelium of the iris and the endothelium, constitutively express Fas ligand and PD-L1, which can induce death or inactivation of T cells, respectively.
Anterior chamber–associated immune deviation is a phenomenon in which introduction of foreign protein antigen into the anterior of the eye actively induces systemic tolerance to that antigen. This phenomenon presumably reduces the chance that adaptive immune responses will be mounted to foreign antigens that may be located in the eye. The tolerance is detectable as a diminished inflammatory T cell or antibody response to the same antigen when it is introduced at extraocular sites compared with the response in individuals who were not given intraocular antigen. Anterior chamber–associated immune deviation may be mediated by Treg. Studies in mice show that the antigen introduced in the anterior chamber is transported by macrophages or DCs, through the blood, to the spleen, and presented by splenic B cells to naive T cells, inducing the generation of regulatory T cells specific for the antigen.
In contrast to induced tolerance to foreign antigens introduced into the anterior chamber, self antigens in the eye are isolated from the immune system, and systemic tolerance to these antigens is not induced. This lack of tolerance becomes a problem only when eye trauma exposes the eye antigens to the immune system. A striking example of this is sympathetic ophthalmia, in which trauma to one eye causes release of eye antigens leading to autoimmune disease in both the injured eye and the uninjured eye. Presumably, although self antigens in the normal eye are inaccessible to the extraocular immune system to induce tolerance, activated immune effector cells and antibodies that are generated in the periphery when one eye is injured have access to and cause injury to the normal eye.
The Brain
Inflammation in the brain can lead to functional derangement and death of neurons, with disastrous consequences. Anatomic features of the brain that impair initiation of adaptive immunity to antigens include an absence of conventional lymphatic drainage and a scarcity of DCs. Delivery of immune cells and inflammatory mediators into the brain is impaired by the nature of the tight junctions between brain microvascular endothelial cells (the so-called blood-brain barrier). Some of the mechanisms operative in the eye may also apply to the brain, including the action of neuropeptides. The brain is rich in resident macrophages, called microglia, which become activated in response to tissue damage or infections in the brain. The threshold for their activation, however, may be higher than that of macrophages in other tissues. One putative mechanism for maintaining this high threshold is inhibitory signaling by the CD200 receptor, which is expressed by microglia. The ligand for this receptor, CD200, is highly expressed in the brain on neurons and other cell types.
Contrary to previously common assumptions based on classic experiments, there is evidence that immune surveillance against microbes does occur in the central nervous system. For example, the frequency of some opportunistic infections within the brain increases significantly in immunosuppressed patients. Patients treated with certain monoclonal antibodies that block lymphocyte and monocyte adhesion to endothelial cells have a significantly increased although still small risk for activation of latent JC virus, leading to a uniformly fatal central nervous system disease called progressive multifocal leukoencephalopathy. This finding suggests that T cell or monocyte trafficking into the brain is necessary to keep latent viruses in check and argues that the brain is not a stringently immune privileged site.
The Testis
Immune privilege in the testis serves to limit inflammation that may impair male fertility. Many self antigens in the adult testis are first expressed at the time of puberty, well after the development of a competent immune system, which may include testis antigen–specific precursor T and B cells. Therefore, immune privilege in the testis may also serve to prevent autoimmunity. The testis, like the eye and brain, has a blood-tissue barrier with specialized endothelial tight junctions that limit delivery of cells and molecules to the sites of spermatogenesis. The hormonal milieu of the testis, which is rich in androgens, has an anti-inflammatory influence on macrophages. TGF-β is produced by Leydig, Sertoli, and peritubular cells and likely contributes to local immune suppression.
Immune Privilege of the Mammalian Fetus
The mammalian fetus expresses paternally inherited genes that are allogeneic to the mother, but fetuses are not normally rejected by the mother. In essence, the fetus is a naturally occurring allograft, but one that is protected from graft rejection. It is clear that the mother is exposed to fetal antigens during pregnancy because maternal antibodies against paternal MHC molecules are easily detectable. Obviously, there has been very strong selective pressure that has led to the evolution of mechanisms that protect the fetus from the maternal immune system, yet these mechanisms remain poorly understood. Probably several different special molecular and barrier features of the placenta and local immunosuppression contribute.
Several experimental observations indicate that the anatomic location of the fetus is a critical factor in the absence of rejection. For example, pregnant animals are able to recognize and reject allografts syngeneic to the fetus placed at extrauterine sites without compromising fetal survival. Wholly allogeneic fetal blastocysts that lack maternal genes can successfully develop in a pregnant or pseudopregnant mother. Thus, neither specific maternal nor paternal genes are necessary for survival of the fetus. Hyperimmunization of the mother with cells bearing paternal antigens does not compromise placental and fetal growth.
The failure to reject the fetus has focused attention on the region of physical contact between the mother and fetus. The fetal tissues of the placenta that most intimately contact the mother are composed of either vascular trophoblast, which is exposed to maternal blood for purposes of mediating nutrient exchange, or implantation site trophoblast, which diffusely infiltrates the uterine lining (decidua) for purposes of anchoring the placenta to the mother.
One simple explanation for fetal survival is that trophoblast cells fail to express paternal MHC molecules. Class II molecules have not been detected on trophoblast. In mice, cells of implantation trophoblast, but not of vascular trophoblast, do express paternal class I MHC molecules. In humans, the situation may be more complex in that trophoblast cells express only a nonpolymorphic class IB molecule called HLA-G. This molecule may be involved in protecting trophoblast cells from maternal NK cell–mediated lysis. A specialized subset of NK cells called uterine NK cells are the major type of lymphocyte present at implantation sites, and IFN-γ production by these cells is essential for decidual development. The way in which uterine NK cells are stimulated and their role in maternal responses to fetal alloantigens are not known. Even if trophoblast cells do express classical MHC molecules, they may lack costimulator molecules and fail to act as antigen-presenting cells.
The uterine decidua may be a site where immune responses are functionally inhibited. In support of the idea is the observation that mouse decidua is highly susceptible to infection by Listeria monocytogenes and cannot support a delayed-type hypersensitivity response. The basis of immunologic privilege is clearly not a simple anatomic barrier because maternal blood is in extensive contact with trophoblast. Rather, the barrier is likely to be created by functional inhibition. Cultured decidual cells directly inhibit macrophage and T cell functions, perhaps by producing inhibitory cytokines, such as TGF-β. Some of these inhibitory decidual cells may be resident regulatory T cells, although the evidence for this is limited. Some experiments have led to the suggestion that TH2 cytokines are produced at the maternal-fetal interface and are responsible for local suppression of TH1 responses to fetal antigens. However, this idea is not supported by the finding that IL-4 and IL-10 knockout mice have normal pregnancies. There is expansion of systemic and decidual Treg in mothers during pregnancy, and the fetus contains abundant regulatory T cells.
Immune responses to the fetus may be regulated by local concentrations of tryptophan and its metabolites in the decidua. The enzyme indolamine 2,3-dioxygenase (IDO) catabolizes tryptophan, and the IDO-inhibiting drug 1-methyl-tryptophan induces abortions in mice in a T cell–dependent manner. These observations led to the hypothesis that T cell responses to the fetus are normally blocked because decidual tryptophan levels are kept low or the levels of toxic metabolites are high.
Several other mechanisms may also dampen maternal immune response of the fetus, including FasL expression by fetal trophoblast cells that promote apoptosis of activated Fas-expressing maternal lymphocytes, generation of tolerogenic DCs in response to galectin-1 expressed in the decidua, and impaired DC migration from the uterus to lymph nodes.
Trophoblast and decidua may also be resistant to complement-mediated damage activation. In mice, these tissues express a C3 and C4 inhibitor called Crry. Crry-deficient embryos die before birth and show evidence of complement activation on trophoblast cells. Thus, this inhibitor may block maternal alloantibody- and complement-mediated damage. However, Crry or equivalent molecules have not been found in humans.
Summary
Brandtzaeg P. Mucosal immunity: induction, dissemination, and effector functions. Scandinavian Journal of Immunology. 2009;70:505-515.
Doss M, White MR, Tecle T, Hartshorn KL. Human defensins and LL-37 in mucosal immunity. Journal of Leukocyte Biology. 2010;87:79-92.
Dubin PJ, Kolls JK. Th17 cytokines and mucosal immunity. Immunological Reviews. 2008;226:160-171.
Gastrointestinal Immune System, General
Duerkop BA, Vaishnava S, Hooper LV. Immune responses to the microbiota at the intestinal mucosal surface. Immunity. 2009;31:368-376.
Eberl G, Lochner M. The development of intestinal lymphoid tissues at the interface of self and microbiota. Mucosal Immunology. 2009;2:478-485.
Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell. 2010;140:859-870.
Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nature Reviews Immunology. 2010;10:159-169.
Innate Immune Responses in the Gastrointestinal Immune System
Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nature Reviews Immunology. 2010;10:131-144.
Corr SC, Gahan CC, Hill C. M-cells: origin, morphology and role in mucosal immunity and microbial pathogenesis. FEMS Immunology and Medical Microbiology. 2008;52:2-12.
Dommett R, Zilbauer M, George JT, Bajaj-Elliott M. Innate immune defence in the human gastrointestinal tract. Molecular Immunology. 2005;42:903-912.
Antigen-Presenting Cells and T Cell Responses in the Gastrointestinal Immune System
Barnes MJ, Powrie F. Regulatory T cells reinforce intestinal homeostasis. Immunity. 2009;31:401-411.
Johansson-Lindbom B, Agace WW. Generation of gut-homing T cells and their localization to the small intestinal mucosa. Immunological Reviews. 2007;215:226-242.
Maynard CL, Weaver CT. Intestinal effector T cells in health and disease. Immunity. 2009;31:389-400.
Rescigno M, Di Sabatino A. Dendritic cells in intestinal homeostasis and disease. The Journal of Clinical Investigation. 2009;119:2441-2450.
Varol C, Zigmond E, Jung S. Securing the immune tightrope: mononuclear phagocytes in the intestinal lamina propria. Nature Reviews Immunology. 2010;10:415-426.
Antibody Production in the Gastrointestinal Immune System
Cerutti A, Rescigno M. The biology of intestinal immunoglobulin A responses. Immunity. 2008;28:740-750.
Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell–dependent and T cell–independent IgA synthesis. Annual Review of Immunology. 2010;28:243-273.
Macpherson AJ, McCoy KD, Johansen FE, Brandtzaeg P. The immune geography of IgA induction and function. Mucosal Immunology. 2008;1:11-22.
Mora JR, von Andrian UH. Differentiation and homing of IgA-secreting cells. Mucosal Immunology. 2008;1:96-109.
Diseases of the Gastrointestinal Immune System
Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nature Reviews Immunology. 2009;9:858-870.
Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annual Review of Immunology. 2010;28:573-621.
Respiratory Mucosal Immune System
Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nature Reviews Immunology. 2008;8:142-152.
Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412-424.
Wissinger E, Goulding J, Hussell T. Immune homeostasis in the respiratory tract and its impact on heterologous infection. Seminars in Immunology. 2009;21:147-155.
Clark RA. Skin-resident T cells: the ups and downs of on site immunity. Journal of Investigative Dermatology. 2010;130:362-370.
Kupper TS, Fuhlbrigge RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nature Reviews Immunology. 2004;4:211-222.
Metz M, Maurer M. Innate immunity and allergy in the skin. Current Opinion in Immunology. 2009;21:687-693.
Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nature Reviews Immunology. 2009;9:679-691.
Romani N, Clausen BE, Stoitzner P. Langerhans cells and more: langerin-expressing dendritic cell subsets in the skin. Immunological Reviews. 2010;234:120-141.
Other Specialized Immune Systems
Erlebacher A. Why isn’t the fetus rejected? Current Opinion in Immunology. 2001;13:590-593.
Streilein JW. Ocular immune privilege: the eye takes a dim but practical view of immunity and inflammation. Journal of Leukocyte Biology. 2003;74:179-185.
Trowsdale J, Betz AG. Mother’s little helpers: mechanisms of maternal-fetal tolerance. Nature Immunology. 2006;7:241-246.
von Rango U. Fetal tolerance in human pregnancy—a crucial balance between acceptance and limitation of trophoblast invasion. Immunology Letters. 2008;115:21-32.