CHAPTER 18 Hypersensitivity Disorders
Adaptive immunity serves the important function of host defense against microbial infections, but immune responses are also capable of causing tissue injury and disease. Disorders caused by immune responses are called hypersensitivity diseases. This term arose from the clinical definition of immunity as “sensitivity,” which is based on the observation that an individual who has been exposed to an antigen exhibits a detectable reaction or is “sensitive” to subsequent encounters with that antigen. Normally, immune responses eradicate infecting organisms without serious injury to host tissues. However, these responses are sometimes inadequately controlled, inappropriately targeted to host tissues, or triggered by commensal microorganisms or environmental antigens that are usually harmless. In these situations, the normally beneficial immune response is the cause of disease.
In this chapter, we describe the pathogenesis of different types of hypersensitivity diseases, with an emphasis on the effector mechanisms that cause tissue injury. We conclude with a brief consideration of the treatment of immunologic diseases and examples of diseases that illustrate important principles.
Causes of Hypersensitivity Diseases
Immune responses against antigens from different sources can be the underlying cause of hypersensitivity disorders.
In all these conditions, the mechanisms of tissue injury are the same as those that normally function to eliminate infectious pathogens. These mechanisms include innate immune responses, T lymphocytes, various other effector cells, and mediators of inflammation. The problem in hypersensitivity diseases is that the response is triggered and maintained inappropriately. Because the stimuli for these abnormal immune responses are difficult or impossible to eliminate (e.g., self antigens, commensal microbes, and environmental antigens) and the immune system has many built-in positive feedback loops (amplification mechanisms), once a pathologic immune response starts, it is difficult to control or to terminate it. Therefore, these hypersensitivity diseases tend to be chronic and progressive and are major therapeutic challenges in clinical medicine.
Mechanisms and Classification of Hypersensitivity Reactions
Hypersensitivity diseases are commonly classified according to the type of immune response and the effector mechanism responsible for cell and tissue injury (Table 18-1). This classification was originally developed by two British immunologists, Philip Gell and Robin Coombs.
TABLE 18–1 Classification of Immunologic Diseases
| Type of Hypersensitivity | Pathologic Immune Mechanisms | Mechanisms of Tissue Injury and Disease |
|---|---|---|
| Immediate hypersensitivity: type I | IgE antibody | Mast cells and their mediators (vasoactive amines, lipid mediators, cytokines) |
| Antibody mediated: type II | IgM, IgG antibodies against cell surface or extracellular matrix antigens | Opsonization and phagocytosis of cells Complement- and Fc receptor–mediated recruitment and activation of leukocytes (neutrophils, macrophages) Abnormalities in cellular functions, e.g., hormone receptor signaling |
| Immune complex mediated: type III | Immune complexes of circulating antigens and IgM or IgG antibodies | Complement- and Fc receptor–mediated recruitment and activation of leukocytes |
| T cell mediated: type IV | CD4+ T cells (cytokine-mediated inflammation) CD8+ CTLs (T cell–mediated cytolysis) |
Recruitment and activation of leukocytes Direct target cell killing, cytokine-mediated inflammation |
This classification is useful because distinct types of pathologic immune responses show different patterns of tissue injury and may vary in their tissue specificity. As a result, the different immunologic mechanisms cause disorders with distinct clinical and pathologic features. However, immunologic diseases in the clinical situation are often complex and caused by combinations of humoral and cell-mediated immune responses and multiple effector mechanisms. This complexity is not surprising given that a single antigen may normally stimulate both humoral and cell-mediated immune responses, in which several types of antibodies and effector T cells are produced. Because multiple mechanisms may be involved and inflammation, typically chronic inflammation, is a major component of the pathology and clinical manifestations of these disorders, they are sometimes grouped under the rubric immune-mediated inflammatory diseases. Considering these diseases together also has some clinical value because, as we shall discuss later in the chapter, many of them are treated with the same or related biologic agents. In the discussion that follows, we use descriptions that identify the pathogenic mechanisms rather than the less informative numerical designations for types of hypersensitivity.
With this background, we proceed to a discussion of antibody- and T cell-mediated diseases.
Diseases Caused by Antibodies
Antibody-mediated diseases are produced either by antibodies that bind to antigens on particular cells or in extracellular tissues or by antigen-antibody complexes that form in the circulation and are deposited in vessel walls (Fig. 18-1). To prove that a disease is caused by antibodies, one would need to demonstrate that the lesions can be induced in a normal animal by the adoptive transfer of immunoglobulin purified from the blood or affected tissues of individuals with the disease. An experiment of nature is occasionally seen in children of mothers suffering from antibody-mediated diseases. These infants may be born with transient manifestations of such diseases because of transplacental passage of antibodies. However, in clinical situations, the diagnosis of diseases caused by antibodies or immune complexes is usually based on the demonstration of antibodies or immune complexes in the circulation or deposited in tissues as well as clinicopathologic similarities with experimental diseases that are proved to be antibody mediated by adoptive transfer.
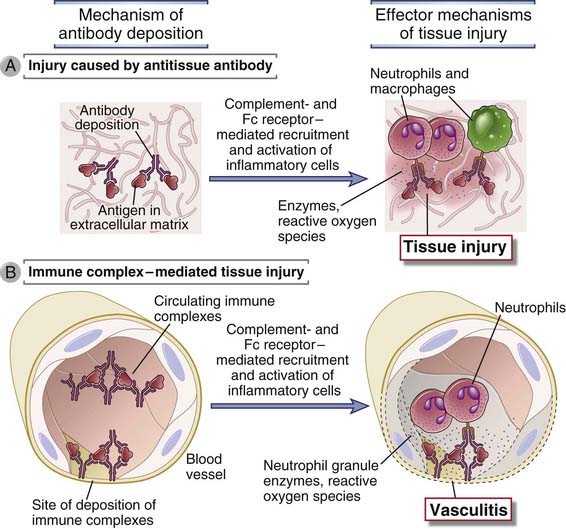
FIGURE 18–1 Types of antibody-mediated diseases.
Antibodies may bind specifically to tissue antigens (A), or they may be deposited as immune complexes that are formed in the circulation (B). In both cases, the deposited antibodies induce inflammation, leading to tissue injury.
Diseases Caused by Antibodies Against Fixed Cell and Tissue Antigens
Antibodies against cellular or matrix antigens cause diseases that specifically affect the cells or tissues where these antigens are present, and these diseases are often not systemic. Antibodies against tissue antigens cause disease by three main mechanisms (Fig. 18-2).
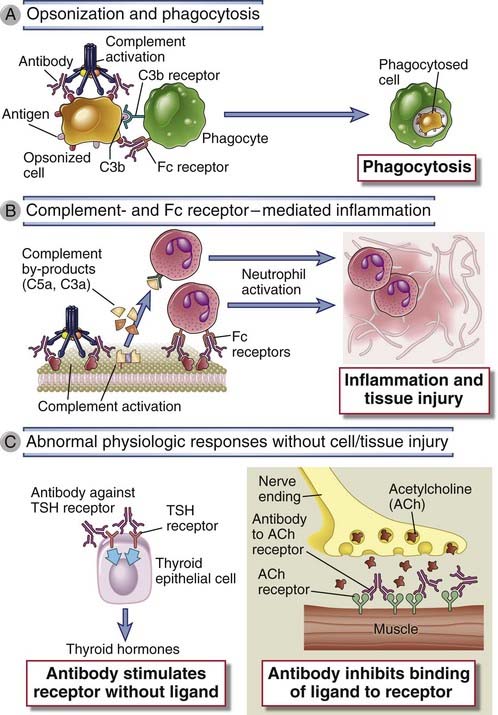
FIGURE 18–2 Effector mechanisms of antibody-mediated disease.
A, Antibodies opsonize cells and may activate complement, generating complement products that also opsonize cells, leading to phagocytosis of the cells through phagocyte Fc receptors or C3 receptors. B, Antibodies recruit leukocytes by binding to Fc receptors or by activating complement and thereby releasing byproducts that are chemotactic for leukocytes. C, Antibodies specific for cell surface receptors for hormones or neurotransmitters may stimulate the activity of the receptors even in the absence of the hormone (left panel) or may inhibit binding of the neurotransmitter to its receptor (right panel). TSH, thyroid-stimulating hormone.
Antibodies that cause cell- or tissue-specific diseases are usually autoantibodies produced as part of an autoimmune reaction against antigens in these cells or tissues. Examples of these autoantibodies are listed in Table 18-2. Less commonly, the antibodies may be produced against a foreign (e.g., microbial) antigen that is immunologically cross-reactive with a component of self tissues. In a rare sequel of streptococcal infection called rheumatic fever, antibodies produced against the bacteria cross-react with antigens in the heart, deposit in this organ, and cause inflammation and tissue damage. Tissue deposits of antibodies may be detected by morphologic examination in some of these diseases, and the deposition of antibody is often associated with local complement activation, inflammation, and tissue injury (Fig. 18-3A).
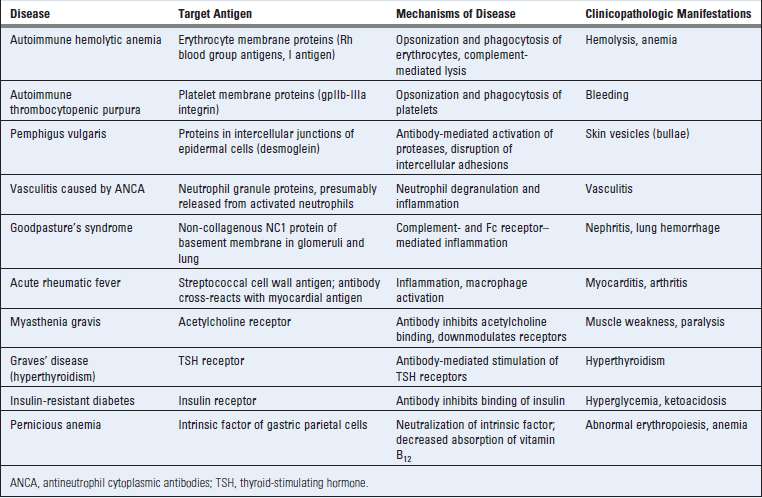
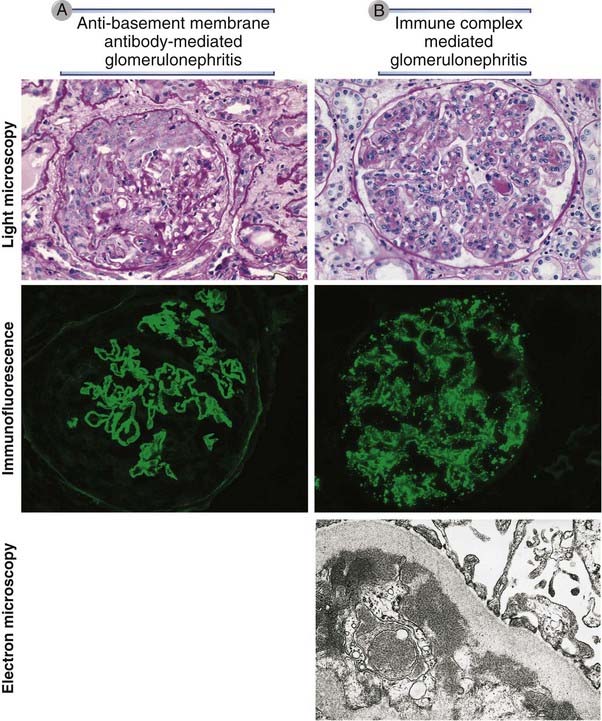
FIGURE 18–3 Pathologic features of antibody-mediated glomerulonephritis.
A, Glomerulonephritis induced by an antibody against the glomerular basement membrane (Goodpasture’s syndrome): the light micrograph shows glomerular inflammation and severe damage, and immunofluorescence shows smooth (linear) deposits of antibody along the basement membrane. B, Glomerulonephritis induced by the deposition of immune complexes (systemic lupus erythematosus): the light micrograph shows neutrophilic inflammation, and the immunofluorescence and electron micrograph show coarse (granular) deposits of antigen-antibody complexes along the basement membrane.
(Immunofluorescence micrographs are courtesy of Dr. Jean Olson, Department of Pathology, University of California, San Francisco, and the electron micrograph is courtesy of Dr. Helmut Rennke, Department of Pathology, Brigham and Women’s Hospital, Boston Massachusetts.)
Immune Complex–Mediated Diseases
Immune complexes that cause disease may be composed of antibodies bound to either self antigens or foreign antigens. The pathologic features of diseases caused by immune complexes reflect the site of immune complex deposition and are not determined by the cellular source of the antigen. Therefore, immune complex–mediated diseases tend to affect multiple tissues and organs, although some are particularly susceptible, such as kidneys and joints.
The occurrence of diseases caused by immune complexes was suspected as early as 1911 by an astute physician named Clemens von Pirquet. At that time, diphtheria infections were being treated with serum from horses immunized with the diphtheria toxin, which is an example of passive immunization against the toxin by the transfer of serum containing antitoxin antibodies. von Pirquet noted that joint inflammation (arthritis), rash, and fever developed in patients injected with the antitoxin-containing horse serum. Two clinical features of this reaction suggested that it was not due to the infection or a toxic component of the serum itself. First, these symptoms appeared even after the injection of horse serum not containing the antitoxin, so the lesions could not be attributed to the anti-diphtheria antibody. Second, the symptoms appeared at least a week after the first injection of horse serum and more rapidly with each repeated injection. von Pirquet concluded that this disease was due to a host response to some component of the serum. He suggested that the host made antibodies to horse serum proteins, these antibodies formed complexes with the injected proteins, and the disease was due to the antibodies or immune complexes. We now know that his conclusions were entirely accurate. He called this disease serum disease; it is now more commonly known as serum sickness and is the prototype for systemic immune complex–mediated disorders.
Experimental Models of Immune Complex–Mediated Diseases
Serum Sickness
Much of our current knowledge of immune complex diseases is based on analyses of experimental models of serum sickness. Immunization of an animal such as a rabbit with a large dose of a foreign protein antigen leads to the formation of antibodies against the antigen (Fig. 18-4). These antibodies bind to and form complexes with circulating antigen, which are initially cleared by macrophages in the liver and spleen. As more and more antigen-antibody complexes are formed, some of them are deposited in vascular beds. In these tissues, the antibodies in the complexes may activate complement, with a concomitant fall in serum complement levels. Complement activation leads to recruitment and activation of inflammatory cells, predominantly neutrophils, at the sites of immune complex deposition, and the neutrophils cause tissue injury. Neutrophils also bind to the immune complexes by their Fcγ receptors, and Fc receptor signaling activates the leukocytes to produce substances that damage tissues, as in diseases caused by antibodies against fixed tissues. Because the complexes are deposited mainly in small arteries, renal glomeruli, and the synovia of joints, the clinical and pathologic manifestations are vasculitis, nephritis, and arthritis. The clinical symptoms are usually short-lived, and the lesions heal unless the antigen is injected again. This type of disease is an example of acute serum sickness. A more indolent and prolonged disease, called chronic serum sickness, is produced by multiple injections of antigen, which lead to the formation of smaller complexes that are deposited most often in the kidneys, arteries, and lungs.
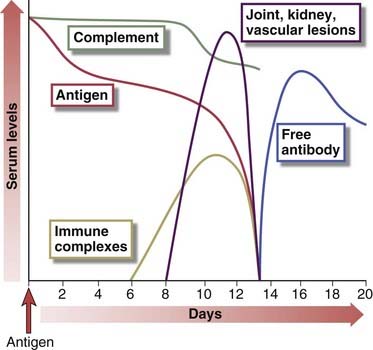
FIGURE 18–4 Sequence of immunologic responses in experimental acute serum sickness.
Injection of bovine serum albumin into a rabbit leads to the production of specific antibody and the formation of immune complexes. These complexes are deposited in multiple tissues, activate complement (leading to a fall in serum complement levels), and cause inflammatory lesions, which resolve as the complexes and the remaining antigen are removed and free antibody (not bound to antigen) appears in the circulation
(Adapted from Cochrane CG. Immune complex–mediated tissue injury. In Cohen S, PA Ward, and RT McCluskey [eds]. Mechanisms of Immunopathology. Werbel & Peck, New York, 1979, pp 29-48.
Arthus Reaction
A localized form of experimental immune complex–mediated vasculitis is called the Arthus reaction. It is induced by injection of an antigen subcutaneously into a previously immunized animal or an animal that has been given intravenous antibody specific for the antigen. Circulating antibodies rapidly bind to the injected antigen and form immune complexes that are deposited in the walls of small arteries at the injection site. This deposition gives rise to a local cutaneous vasculitis with tissue necrosis. This model has been used to study the cells and molecules involved in immune complex diseases.
Pathogenesis of Immune Complex–Mediated Diseases
Antigen-antibody complexes are produced during normal immune responses, but they cause disease only when they are produced in excessive amounts, are not efficiently cleared, and become deposited in tissues. The amount of immune complex deposition in tissues is determined by the nature of the complexes and the characteristics of the blood vessels. Small complexes are often not phagocytosed and tend to be deposited in vessels more than large complexes, which are usually cleared by phagocytes. Complexes containing cationic antigens bind avidly to negatively charged components of the basement membranes of blood vessels and kidney glomeruli. Such complexes typically produce severe and long-lasting tissue injury. Capillaries in the renal glomeruli and synovia are vessels in which plasma is ultrafiltered (to form urine and synovial fluid, respectively) by passing through the capillary wall at high hydrostatic pressure, and these locations are among the most common sites of immune complex deposition. However, immune complexes may be deposited in small vessels in virtually any tissue. Immune complexes may also bind to Fc receptors of mast cells and leukocytes and activate these cells to secrete cytokines and vasoactive mediators. These mediators may cause more immune complex deposition in vessel walls by increasing vascular permeability and blood flow.
The deposition of immune complexes in vessel walls leads to complement- and Fc receptor–mediated inflammation and injury to the vessels and adjacent tissues. Deposits of antibody and complement may be detected in the vessels, and if the antigen is known, it is possible to identify antigen molecules in the deposits as well (see Fig. 18-3B).
Many systemic immunologic diseases in humans are caused by the deposition of immune complexes in blood vessels. Some common examples of autoimmune immune complex diseases are systemic lupus erythematosus (SLE), in which the complexes consist of nuclear antigens and antibodies, and several forms of nephritis and vasculitis (Table 18-3). In almost 50% of cases of one type of immune complex–mediated vasculitis involving medium-size muscular arteries, called polyarteritis nodosa, the complexes are made up of viral antigen and antibodies, and the disease is a late complication of viral infection, most often with hepatitis B virus. This also is the mechanism of a disease called poststreptococcal glomerulonephritis that develops in rare cases after streptococcal infection and is caused by complexes of streptococcal antigen and antibodies depositing in the glomeruli of the kidney.
TABLE 18–3 Examples of Human Immune Complex–Mediated Diseases
| Disease | Antigen Involved | Clinicopathologic Manifestations |
|---|---|---|
| Systemic lupus erythematosus | DNA, nucleoproteins, others | Nephritis, arthritis, vasculitis |
| Polyarteritis nodosa | Hepatitis B virus surface antigen | Vasculitis |
| Poststreptococcal glomerulonephritis | Streptococcal cell wall antigens; may be “planted” in glomerular basement membrane | Nephritis |
| Serum sickness | Various proteins | Arthritis, vasculitis, nephritis |
Diseases Caused by T Lymphocytes
T lymphocytes injure tissues either by triggering inflammation or by directly killing target cells (Fig. 18-5). Inflammatory reactions are elicited mainly by CD4+ T cells of the TH1 and TH17 subsets, which secrete cytokines that recruit leukocytes. In some T cell–mediated disorders, CD8+ CTLs kill target cells bearing class I major histocompatibility complex (MHC)–associated antigens. The T cells that cause tissue injury may be autoreactive, or they may be specific for foreign protein antigens that are present in or bound to cells or tissues. T lymphocyte–mediated tissue injury may also accompany strong protective immune responses against persistent microbes, especially intracellular microbes that resist eradication by phagocytes and antibodies.
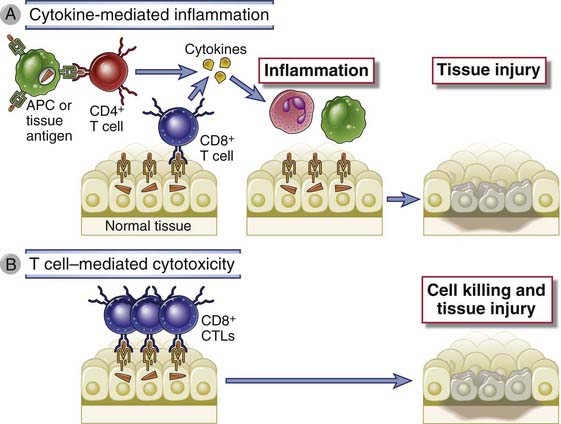
FIGURE 18–5 Mechanisms of T cell–mediated diseases.
A, In cytokine-mediated inflammatory reactions, CD4+ T cells (and sometimes CD8+ cells) respond to tissue antigens by secreting cytokines that stimulate inflammation and activate phagocytes, leading to tissue injury. APC, antigen-presenting cell. B, In some diseases, CD8+ CTLs directly kill tissue cells.
A role for T cells in causing a particular immunologic disease is suspected largely on the basis of the demonstration of T cells in lesions and the isolation of T cells specific for self or microbial antigens from the tissues or blood of patients. Animal models have been very useful for elucidating the pathogenesis of these disorders.
Diseases Caused by Cytokine-Mediated Inflammation
In immune-mediated inflammation, TH1 and TH17 cells secrete cytokines that recruit and activate leukocytes. IL-17, produced by TH17 cells, promotes neutrophil recruitment; interferon-γ (IFN-γ), produced by TH1 cells, activates macrophages; and tumor necrosis factor (TNF) and chemokines, produced by T lymphocytes and other cells, are involved in the recruitment and activation of many types of leukocytes. Tissue injury results from the products of activated neutrophils and macrophages, such as lysosomal enzymes, reactive oxygen species, nitric oxide, and proinflammatory cytokines (see Chapter 10). Vascular endothelial cells in the lesions may express increased levels of cytokine-regulated surface proteins such as adhesion molecules and class II MHC molecules. The inflammation associated with T cell–mediated diseases is typically chronic, but bouts of acute inflammation may be superimposed on a background of chronic inflammation. Delayed-type hypersensitivity (DTH) is an example of such inflammatory reactions and is described later. Chronic inflammatory reactions often produce fibrosis as a result of the secretion of cytokines and growth factors by the macrophages.
Many organ-specific autoimmune diseases are caused by interaction of autoreactive T cells with self antigens, leading to cytokine release and inflammation. This is believed to be the major mechanism underlying rheumatoid arthritis, multiple sclerosis, type 1 diabetes, psoriasis, and other autoimmune diseases (Table 18-4). Some of these are described in more detail at the end of the chapter.
TABLE 18–4 T Cell–Mediated Diseases
| Disease | Specificity of Pathogenic T Cells | Principal Mechanisms of Tissue Injury |
|---|---|---|
| Rheumatoid arthritis | Collagen? Citrullinated self proteins? |
Inflammation mediated by TH17 (and TH1?) cytokines Role of antibodies and immune complexes? |
| Multiple sclerosis | Protein antigens in myelin (e.g., myelin basic protein) | Inflammation mediated by TH1 and TH17 cytokines Myelin destruction by activated macrophages |
| Type 1 diabetes mellitus | Antigens of pancreatic islet β cells (insulin, glutamic acid decarboxylase, others) | T cell–mediated inflammation Destruction of islet cells by CTLs |
| Inflammatory bowel disease | Enteric bacteria Self antigens? |
Inflammation mediated by TH17 and TH1 cytokines |
| Autoimmune myocarditis | Myosin heavy chain protein | CTL-mediated killing of myocardial cells Inflammation mediated by TH1 cytokines |
Examples of human T cell–mediated diseases are listed. In many cases, the specificity of the T cells and the mechanisms of tissue injury are inferred on the basis of the similarity with experimental animal models of the diseases.
T cell reactions against microbes and other foreign antigens may also lead to inflammation and tissue injury at the sites of infection or antigen exposure. Intracellular bacteria such as Mycobacterium tuberculosis induce strong T cell and macrophage responses that result in granulomatous inflammation and fibrosis (described below); the inflammation and fibrosis may cause extensive tissue destruction and functional impairment, in this case in the lungs. Tuberculosis is a good example of an infectious disease in which tissue injury is mainly due to the host immune response (see Chapter 15). A variety of skin diseases that result from topical exposure to chemicals and environmental antigens, called contact sensitivity, are due to inflammatory reactions, presumably triggered by neoantigens formed by the binding of the chemicals to self proteins. Both CD4+ and CD8+ T cells may be the source of cytokines in contact sensitivity reactions. Examples of contact sensitivity include the rashes of poison ivy and poison oak (in which T cells react against chemicals made by the plants called urushiols) and rashes induced by contact with a variety of chemicals, such as thiuram, which is used in the manufacture of latex gloves. Some of these reactions become chronic and clinically are called eczema. T cell responses against intestinal bacteria are believed to underlie some forms of inflammatory bowel disease.
The classical T cell-mediated inflammatory reaction is called delayed type hypersensitivity, and is described next.
Delayed-Type Hypersensitivity
Delayed-type hypersensitivity (DTH) is an injurious cytokine-mediated inflammatory reaction resulting from the activation of T cells, particularly CD4+ T cells. The reaction is called hypersensitivity because it reflects excessive (i.e., injurious) immune responses (which are reflections of sensitivity to an antigen) and delayed because it typically develops during 24 to 48 hours after antigen challenge.
In the classic animal model of DTH, a guinea pig is first immunized by the administration of a protein antigen in adjuvant; this step is called sensitization. About 2 weeks later, the animal is challenged subcutaneously with the same antigen, and the subsequent reaction is analyzed; this step is called the elicitation phase. Humans may be sensitized for DTH reactions by microbial infection, by contact sensitization with chemicals and environmental antigens, or by intradermal or subcutaneous injection of protein antigens (Fig. 18-6). Subsequent exposure to the same antigen (called challenge) elicits the reaction. For example, purified protein derivative (PPD), a protein antigen of Mycobacterium tuberculosis, elicits a DTH reaction, called the tuberculin reaction, when it is injected into individuals who have been exposed to M. tuberculosis. A positive tuberculin skin test response is a widely used clinical indicator for evidence of previous or active tuberculosis infection.
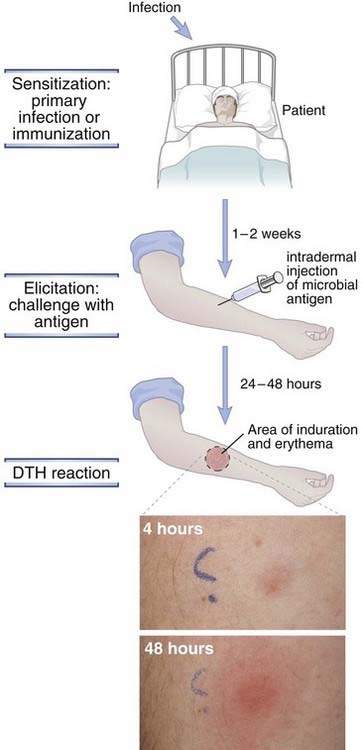
FIGURE 18–6 Delayed-type hypersensitivity reaction.
Infection or immunization (vaccination) sensitizes an individual, and subsequent challenge with an antigen from the infectious agent elicits a DTH reaction. The reaction is manifested by induration with redness and swelling at the site of the challenge, which is undetectable at ~4 hours and peaks at ~48 hours.
(Courtesy of Dr. J. Faix, Department of Pathology, Stanford University School of Medicine, Palo Alto, California.)
The characteristic response of DTH evolves during 24 to 48 hours. About 4 hours after the injection of antigen, neutrophils accumulate around the postcapillary venules at the injection site. By about 12 hours, the injection site becomes infiltrated by T cells and blood monocytes, also organized in a perivenular distribution (Fig. 18-7). The endothelial cells lining these venules become plump, show increased biosynthetic organelles, and become leaky to plasma macromolecules. Fibrinogen escapes from the blood vessels into the surrounding tissues, where it is converted into fibrin. The deposition of fibrin and, to a lesser extent, the accumulation of T cells and monocytes within the extravascular tissue space around the injection site cause the tissue to swell and become firm (indurated). Induration, a diagnostic feature of DTH, is detectable by about 18 hours after the injection of antigen and is maximal by 24 to 48 hours. In clinical practice, loss of DTH responses to universally encountered antigens (e.g., Candida antigens) is an indication of deficient T cell function, a condition known as anergy. (This general loss of immune responsiveness is different from lymphocyte anergy, a mechanism for maintaining tolerance to specific antigens, discussed in Chapter 14.)
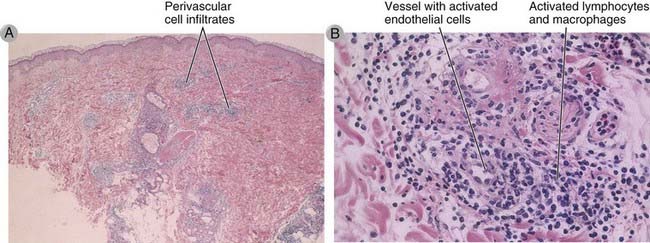
FIGURE 18–7 Morphology of a DTH reaction.
A, Histopathologic examination of the reaction in skin illustrated in Figure 18-6 shows perivascular mononuclear cell infiltrates in the dermis. B, At higher magnification, the infiltrate is seen to consist of activated lymphocytes and macrophages surrounding small blood vessels in which the endothelial cells are also activated.
(Courtesy of Dr. J. Faix, Department of Pathology, Stanford University School of Medicine, Palo Alto, California.)
Although DTH has traditionally been considered a TH1-mediated injurious reaction, other T cells may contribute to the inflammation. In some DTH lesions, neutrophils are prominent, suggesting the involvement of TH17 cells. In infections by some helminthic parasites, reactions against the parasite eggs elicit DTH with a strong component of eosinophils. In these cases, a role for TH2 cytokines has been demonstrated. CD8+ T cells also produce IFN-γ and may contribute to some DTH reactions, especially in the skin.
Chronic DTH reactions can develop if a TH1 response to an infection activates macrophages but fails to eliminate phagocytosed microbes. If the microbes are localized in a small area, the reaction produces nodules of inflammatory tissue called granulomas (Fig. 18-8A). Chronic DTH, as exemplified by granulomatous inflammation, is caused by prolonged cytokine signals (Fig. 18-8B). In such reactions, the activated T cells and macrophages continue to produce cytokines and growth factors, which amplify the reactions of both cell types and progressively modify the local tissue environment. The result is a cycle of tissue injury and chronic inflammation followed by replacement with connective tissue (fibrosis). In chronic DTH reactions, activated macrophages also undergo changes in response to persistent cytokine signals. These macrophages develop increased cytoplasm and cytoplasmic organelles and histologically may resemble skin epithelial cells, because of which they are sometimes called epithelioid cells. Activated macrophages may fuse to form multinucleate giant cells. Granulomatous inflammation is an attempt to contain the infection but is also the cause of significant tissue injury and functional impairment. This type of inflammation is a characteristic response to some persistent microbes, such as M. tuberculosis and some fungi, and represents a form of chronic DTH with fibrosis. Much of the respiratory difficulty associated with tuberculosis or chronic fungal infection of the lung is caused by replacement of normal lung with fibrotic tissue and not directly attributable to the microbes.
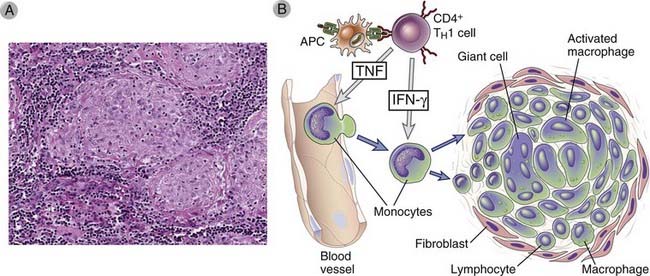
FIGURE 18–8 Granulomatous inflammation.
A, Lymph node from a patient with tuberculosis containing granulomas with activated macrophages, multinucleate giant cells, and lymphocytes. In some granulomas, there may be a central area of necrosis. Immunohistochemical studies would identify the lymphocytes as T cells. B, Mechanisms of granuloma formation. Cytokines are involved in the generation of TH1 cells, activation of macrophages, and recruitment of leukocytes. Prolonged reactions of this type lead to the formation of granulomas.
Diseases Caused by Cytotoxic T Lymphocytes
CTL responses to viral infection can lead to tissue injury by killing infected cells, even if the virus itself has no cytopathic effects. The principal physiologic function of CTLs is to eliminate intracellular microbes, primarily viruses, by killing infected cells. Some viruses directly injure infected cells and are said to be cytopathic, whereas others are not. Because CTLs may not be able to distinguish between cytopathic and noncytopathic viruses, they kill virally infected cells regardless of whether the infection itself is harmful to the host. Examples of viral infections in which the lesions are due to the host CTL response and not the virus itself include lymphocytic choriomeningitis in mice and certain forms of viral hepatitis in humans (see Chapter 15).
CTLs may contribute to tissue injury in autoimmune disorders that are caused primarily by CD4+ T cells, such as type 1 diabetes. Few examples of autoimmune diseases mediated only by CTLs have been documented. Myocarditis with infiltration of the heart by CD8+ T cells develops in mice and sometimes in humans after infection with coxsackievirus B, resulting in a form of dilated cardiomyopathy. Patients and experimental animals contain CTLs specific for myocyte proteins. It is postulated that the heart lesions are initiated by the virus infection and virus-specific CTLs, and myocardial injury leads to the exposure or alteration of self antigens and the subsequent development of autoreactive CTLs. However, in these forms of myocarditis, CD4+ T cells and antibodies are also likely involved in the pathogenesis.
Therapeutic Approaches for Immunologic Diseases
One of the most impressive accomplishments of immunology has been the development of novel therapies based on the understanding of basic science and its application to human disease. The therapies can be divided into several broad groups.
Anti-inflammatory Agents
The mainstay of therapy for hypersensitivity diseases for many years has been anti-inflammatory drugs, particularly corticosteroids. Such drugs are targeted at reducing tissue injury, specifically the inflammatory component of the pathologic immune responses.
Depletion of Cells and Antibodies
Monoclonal antibodies are given that deplete all lymphoid cells, only B cells, or only T cells. A recent and somewhat surprising development is the successful use of anti-CD20 antibody (rituximab), which depletes only B cells, to treat diseases that were thought to be caused primarily by T cell–mediated inflammation. This treatment has shown efficacy in some patients with rheumatoid arthritis and multiple sclerosis. Plasmapheresis has been used to eliminate circulating autoantibodies and immune complexes.
Anti-Cytokine Therapies
A large number of cytokines involved in inflammation are being targeted by specific antagonists for the treatment of chronic, T cell–mediated inflammatory diseases (Table 18-5). The first success with this class of biologic agents came with a soluble form of the TNF receptor and anti-TNF antibodies, which bind to and neutralize TNF. These agents are of great benefit in many patients with rheumatoid arthritis, Crohn’s disease, and the skin disease psoriasis. Antagonists of other proinflammatory cytokines, such as IL-1, the p40 chain that is present in both IL-12 and IL-23, IL-6, IL-17A, and many others, are in use or in clinical trials for inflammatory diseases.
TABLE 18–5 Examples of Cytokine Antagonists in Clinical Use or Trials
| Cytokine or Receptor Targeted | Predicted Biologic Effects Of Antagonist | Clinical Indications |
|---|---|---|
| TNF | Inhibits leukocyte migration into sites of inflammation | Rheumatoid arthritis, psoriasis, inflammatory bowel disease |
| IL-1 | Inhibits leukocyte migration into sites of inflammation | Rare autoinflammatory syndromes, severe gout, rheumatoid arthritis |
| IL-6 and IL-6 receptor | Inhibits synthesis of acute-phase proteins, antibody responses? | Juvenile idiopathic arthritis, rheumatoid arthritis |
| IL-17 | Inhibits leukocyte recruitment into sites of inflammation | Rheumatoid arthritis, psoriasis |
| p40 chain of IL-12 and IL-23 | Inhibits TH1 and TH17 responses | Inflammatory bowel disease, psoriasis |
| IL-2 receptor (CD25) | Inhibits IL-2–mediated T cell proliferation | Acute graft rejection |
| IFN-α | May be multiple effects on TH1 differentiation, antibody production | Systemic lupus erythematosus |
| IL-4 | Inhibits TH2 differentiation, IgE production | Asthma |
| IL-5 | Inhibits eosinophil activation | Asthma |
The table lists examples of antagonists against cytokines (antibodies or soluble receptors) that are approved for clinical use or in trials. IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
Agents That Inhibit Cell-Cell Interactions in Immune Responses
Agents that block B7 costimulators are approved for treatment of rheumatoid arthritis and psoriasis and are being tested in SLE and other diseases. Antibodies against CD40 ligand block T cell–mediated activation of B cells and macrophages and have been beneficial in patients with inflammatory bowel disease, but a small number of treated patients have developed thrombotic episodes, apparently because this molecule is expressed on human platelets (where its function is unknown). Antibodies against integrins have been used to inhibit leukocyte migration into tissues, particularly the central nervous system (CNS) in multiple sclerosis.
Intravenous IgG
Large doses of intravenous IgG (IVIG) have beneficial effects in some hypersensitivity diseases. It is not clear how this agent suppresses immune inflammation; one possibility is that the IgG binds to the inhibitory Fc receptor (FcγRIIB) on macrophages and B lymphocytes and thus attenuates inflammatory responses (see Chapter 12). IVIG may also compete with pathogenic antibodies for binding to the neonatal Fc receptor (FcRn), which functions in adults to protect antibodies from catabolism (see Chapter 5), resulting in reduced half-lives of the pathogenic antibodies.
There are ongoing attempts at more specific treatment, such as inducing tolerance in disease-producing T cells or inducing regulatory T cells specific for self antigens. Multiple sclerosis and type 1 diabetes are two immune diseases in which the target antigens have been defined; in both, clinical trials are starting in which the antigens (peptides of myelin basic protein and insulin, respectively) will be administered to patients in ways predicted to shut off specific immune responses. A risk of many treatments that block various components of the immune system is that these will interfere with the normal function of the immune system in combating microbes and thus make individuals susceptible to infections. Antigen-specific tolerance avoids this problem by selectively targeting the disease-causing lymphocytes. These general principles are similar to those on which treatment of transplant rejection is based (see Chapter 16).
Selected Immunologic Diseases: pathogenesis and therapeutic strategies
In the following section, we describe the pathogenesis of some selected diseases caused by antibodies and T cells and the application of novel therapies in these diseases to illustrate the principles that have been discussed before.
Systemic Lupus Erythematosus: The Prototypic Immune Complex–Mediated Disease
SLE is a chronic, remitting and relapsing, multisystem autoimmune disease that affects predominantly women, with an incidence of 1 in 700 among women between the ages of 20 and 60 years (about 1 in 250 among black women) and a female-to-male ratio of 10 : 1. The principal clinical manifestations are rashes, arthritis, and glomerulonephritis, but hemolytic anemia, thrombocytopenia, and CNS involvement are also common. Many different autoantibodies are found in patients with SLE. The most frequent are antinuclear, particularly anti-DNA, antibodies; others include antibodies against ribonucleoproteins, histones, and nucleolar antigens. Immune complexes formed from these autoantibodies and their specific antigens are responsible for glomerulonephritis, arthritis, and vasculitis involving small arteries throughout the body. Hemolytic anemia and thrombocytopenia are due to autoantibodies against erythrocytes and platelets, respectively. The principal diagnostic test for the disease is the presence of antinuclear antibodies; antibodies against double-stranded native DNA are specific for SLE.
Pathogenesis of Systemic Lupus Erythematosus
SLE is a complex disease in which genetic and environmental factors contribute to a breakdown of tolerance in self-reactive B and T lymphocytes. Among the genetic factors is the inheritance of particular HLA alleles. The odds ratio (relative risk) for individuals with HLA-DR2 or HLA-DR3 is 2 to 3, and if both haplotypes are present, the odds ratio is about 5. Genetic deficiencies of classical pathway complement proteins, especially C1q, C2, or C4, are seen in about 10% of patients with SLE. The complement deficiencies may result in defective clearance of immune complexes and apoptotic cells and the failure of B cell tolerance. A polymorphism in the inhibitory Fc receptor FcγRIIB has been described in some patients; this may contribute to inadequate control of B cell activation or a failure to attenuate inflammatory responses in innate immune cells. Many other genes have been detected by genome-wide association studies, but the role of each of these is not established, and their contribution to the development of the disease remains unclear. Environmental factors include exposure to ultraviolet (UV) light. It is postulated that this leads to apoptotic death of cells and release of nuclear antigens.
Two recent observations are leading to new hypotheses of the pathogenesis of SLE. First, studies in patients have revealed that blood cells show a striking molecular signature (pattern of gene expression) that indicates exposure to IFN-α, a type I interferon that is produced mainly by plasmacytoid dendritic cells. Some studies have shown that plasmacytoid dendritic cells from SLE patients also produce abnormally large amounts of IFN-α. Second, studies in animal models have shown that Toll-like receptors (TLRs) that recognize DNA and RNA, notably the DNA-recognizing TLR9, play an important role in the activation of B cells specific for self nuclear antigens. On the basis of these studies, a model for the pathogenesis of SLE has been proposed (Fig. 18-9). According to this model, UV irradiation and other environmental insults lead to the apoptosis of cells. Inadequate clearance of the nuclei of these cells, in part because of defects in clearance mechanisms such as complement proteins and receptors, results in a large burden of nuclear antigens. Polymorphisms in various susceptibility genes for lupus lead to defective ability to maintain self-tolerance in B and T lymphocytes, because of which self-reactive lymphocytes remain functional. Failure of B cell tolerance may be due to receptor editing or defective deletion of immature B cells in the bone marrow or defective peripheral tolerance. Self-reactive B cells that are not rendered tolerant are stimulated by the self nuclear antigens, and antibodies are produced against the antigens. Complexes of the antigens and antibodies bind to Fc receptors on dendritic cells and the antigen receptor on B cells and may be internalized. The nucleic acid components engage TLRs and stimulate B cells to produce autoantibodies and activate dendritic cells, particularly plasmacytoid dendritic cells, to produce IFN-α, which further enhance the immune response and cause more apoptosis. The net result is a cycle of antigen release and immune activation that leads to the production of high-affinity autoantibodies.
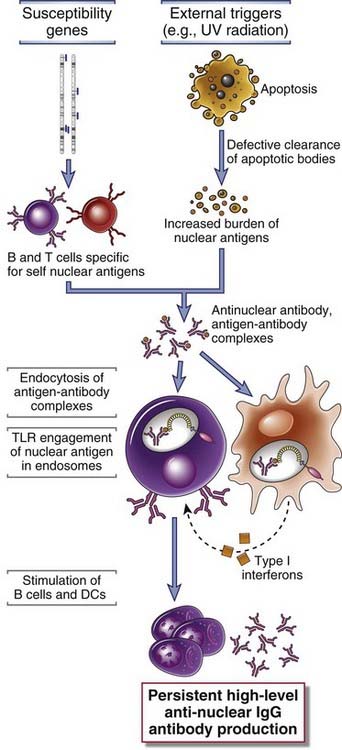
FIGURE 18–9 A model for the pathogenesis of systemic lupus erythematosus (SLE).
In this hypothetical model, various susceptibility genes interfere with the maintenance of self-tolerance, and external triggers lead to persistence of nuclear antigens. The result is an antibody response against self nuclear antigens, which is amplified by the TLR-dependent activation of dendritic cells and B cells by nucleic acids, and the production of type I interferons.
New Therapies for Systemic Lupus Erythematosus
The recent advances in our understanding of SLE are leading to novel therapeutic approaches. Clinical trials are under way to test the efficacy of anti–IFN-α antibodies in the disease, and attempts to inhibit TLR signals are being considered. There has been great interest in depleting B cells by use of an antibody against the B cell surface protein CD20. An antibody that blocks the B cell growth factor BAFF is now approved for the treatment of SLE.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is an inflammatory disease involving small and large joints of the extremities, including fingers, shoulders, elbows, knees, and ankles. The disease is characterized by inflammation of the synovium associated with destruction of the joint cartilage and bone, with a morphologic picture indicative of a local immune response. Both cell-mediated and humoral immune responses may contribute to development of synovitis. CD4+ TH1 and TH17 cells, activated B lymphocytes, plasma cells, and macrophages as well as other inflammatory cells are found in the inflamed synovium, and in severe cases, well-formed lymphoid follicles with germinal centers may be present. Numerous cytokines, including IL-1, IL-8, TNF, IL-6, IL-17, and IFN-γ, have been detected in the synovial (joint) fluid. Cytokines are believed to recruit leukocytes whose products cause tissue injury and also to activate resident synovial cells to produce proteolytic enzymes, such as collagenase, that mediate destruction of the cartilage, ligaments, and tendons of the joints. Increased osteoclast activity in the joints contributes to the bone destruction in RA, and this may be caused by the production of the TNF family cytokine RANK (receptor activator of nuclear factor κB) ligand by activated T cells. RANK ligand binds to RANK, a member of the TNF receptor family that is expressed on osteoclast precursors, and induces their differentiation and activation. Systemic complications of RA include vasculitis, presumably caused by immune complexes, and lung injury.
Although much of the emphasis in studies of RA has been on the role of T cells, antibodies may also contribute to the joint destruction. Activated B cells and plasma cells are often present in the synovia of affected joints. Patients frequently have circulating IgM or IgG antibodies that react with the Fc (and rarely Fab) portions of their own IgG molecules. These autoantibodies are called rheumatoid factors, and their presence is used as a diagnostic test for RA. Rheumatoid factors may participate in the formation of injurious immune complexes, but their pathogenic role is not established. Another type of antibody that has been detected in at least 70% of patients is specific for cyclic citrullinated peptides (CCP), which are derived from certain proteins that are modified in an inflammatory environment by the enzymatic conversion of arginine residues to citrulline. These so-called anti-CCP antibodies are a diagnostic marker for the disease and may be involved in tissue injury.
Pathogenesis of Rheumatoid Arthritis
Like other autoimmune diseases, RA is a complex disorder in which genetic and environmental factors contribute to the breakdown of tolerance to self antigens. The specificity of the pathogenic T and B cells remains unknown, because of which the understanding of the pathogenesis is incomplete. Susceptibility to RA is linked to the HLA-DR4 haplotype. Recent linkage and genome-wide association studies have revealed a large number of genes in which polymorphisms are associated with RA. There is an association with the gene encoding a tyrosine phosphatase, PTPN22, but the role of this enzyme in lymphocyte regulation is poorly understood (see Chapter 14).
The identification of anti-CCP immune responses has led to new ideas about the pathogenesis of RA (Fig. 18-10). According to one model, environmental insults, such as smoking and some infections, induce the citrullination of self proteins, leading to the creation of new antigenic epitopes. In genetically susceptible individuals, tolerance to these epitopes fails, resulting in T cell and antibody responses against the proteins. If these modified self proteins are also present in joints, the T cells and antibodies attack the joints. TH17 and perhaps TH1 cells secrete cytokines that recruit leukocytes into the joint and activate synovial cells to produce collagenases and other enzymes. The net result is the progressive destruction of cartilage and bone. The immune response in the joint may be strong enough that tertiary lymphoid tissues form in the synovium, and these may maintain and propagate the local inflammatory reaction.
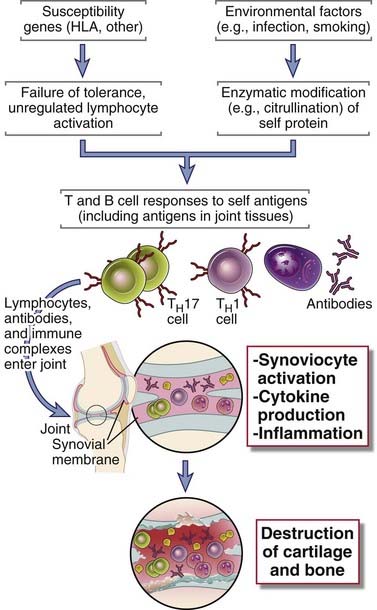
FIGURE 18–10 A model for the pathogenesis of rheumatoid arthritis.
According to this hypothesis, citrullinated proteins induced by environmental stimuli elicit T cell and antibody responses in genetically susceptible individuals. The T cells and antibodies enter joints, respond to the self proteins, and cause tissue injury mainly by cytokine secretion and perhaps also by antibody-dependent effector mechanisms. Protein modifications other than citrullination may lead to the same result.
New Therapies for Rheumatoid Arthritis
The realization of the central role of T cells and cytokines in the disease has led to a remarkable advance in treatment, in which specific molecules have been targeted on the basis of scientific understanding. Chief among these new therapies are antagonists against TNF, which have transformed the course of the disease in many patients from one of progressive and inexorable joint destruction to one of smoldering but manageable chronic inflammation. An IL-1 antagonist and an antibody against the IL-6 receptor are approved treatments, as is a fusion protein of the extracellular domain of CTLA-4 and the Fc portion of IgG, which binds to B7 molecules and blocks B7:CD28 interactions. Antibodies that block IL-17 are in clinical trials. The B cell–depleting anti-CD20 antibody is of benefit in some patients. The beneficial effect of B cell depletion does not seem to be attributable entirely to reduced production of autoantibodies, suggesting that B cells may play other roles in the disease, such as presenting antigens to pathogenic T cells.
Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis
Multiple sclerosis (MS) is an autoimmune disease of the CNS in which CD4+ T cells of the TH1 and TH17 subsets react against self myelin antigens, resulting in inflammation in the CNS with activation of macrophages around nerves in the brain and spinal cord, destruction of the myelin, abnormalities in nerve conduction, and neurologic deficits. It is the most common neurologic disease of young adults. On pathologic examination, there is inflammation in the CNS white matter with secondary demyelination. Multiple sclerosis is characterized clinically by weakness, paralysis, and ocular symptoms with exacerbations and remissions; CNS imaging suggests that in patients with active disease, there is frequent new lesion formation. The disease is modeled by experimental autoimmune encephalomyelitis (EAE) in mice, rats, guinea pigs, and nonhuman primates, and this is one of the best characterized experimental models of an organ-specific autoimmune disease mediated mainly by T lymphocytes. EAE is induced by immunizing animals with antigens normally present in CNS myelin, such as myelin basic protein, proteolipid protein, and myelin oligodendrocyte glycoprotein, with an adjuvant containing heat-killed mycobacterium, which is necessary to elicit a strong T cell response. About 1 to 2 weeks after immunization, animals develop encephalomyelitis, characterized by perivascular infiltrates composed of lymphocytes and macrophages in the CNS white matter, followed by demyelination. The neurologic lesions can be mild and self-limited or chronic and relapsing. These lesions result in progressive or remitting and relapsing paralysis. The disease can also be transferred to naive animals with T cells from diseased animals. Although antibodies against myelin antigens have been detected in patients as well as in the animal models, the pathogenic significance of these antibodies is not established.
Pathogenesis of Multiple Sclerosis
There is abundant evidence that in mice, EAE is caused by activated, CD4+ TH1 and TH17 cells specific for protein antigens in myelin. By analogy with the experimental disease, MS is also thought to be caused by myelin-specific TH1 and TH17 cells, and these cells have been detected in patients and isolated from the blood and CNS. How these cells are activated in patients remains an enigma. It has been suggested that an infection, most likely a viral infection, activates self myelin–reactive T cells by the phenomenon of molecular mimicry (see Chapter 14). Self-tolerance may fail because of the inheritance of susceptibility genes. Identical twins have a 25% to 40% concordance rate for development of MS, whereas nonidentical twins have a 1% concordance rate, implicating genetic factors in the development of the disease. Genetic polymorphisms associated with MS include the HLA locus, with HLA-DR2 being the strongest linkage. Genome-wide association studies have revealed an association with a polymorphism in the noncoding region of the gene encoding the IL-2 receptor α chain, CD25. Expression of CD25 on effector and memory T cells may be different in patients compared with healthy individuals, but how this results in disease is unclear. Some studies have suggested that regulatory T cells are defective in MS patients, but how this contributes to a failure of self-tolerance is also not known. Once myelin-specific T cells are activated, they migrate into the CNS, where they encounter myelin proteins and release cytokines that recruit and activate macrophages and more T cells, leading to myelin destruction. Studies of EAE suggest that the disease is propagated by the process known as epitope spreading (see Chapter 14). The tissue breakdown results in release of new protein antigens and expression of new, previously sequestered epitopes that activate more autoreactive T cells.
New Therapies for Multiple Sclerosis
Immunotherapy for MS has largely relied on approaches whose scientific basis is still not well understood. These include administration of β-interferon, which may alter cytokine responses, and treatment with a random polymer of four amino acids, which is postulated to bind to HLA molecules and block antigen presentation. More recently, an antibody against the VLA-4 integrin (see Chapter 3) has been used to block leukocyte migration into the CNS and has shown benefit in patients. However, in a small number of patients this treatment resulted in reactivation of a latent JC virus infection that causes a severe and sometime fatal CNS disease. Another recently approved drug to treat MS also interferes with leukocyte migration. The drug, called fingolimod (FTY720), blocks the sphingosine 1-phosphate–mediated pathway of T cell egress from lymphoid tissues (see Chapter 3). In a large subset of patients B cell depletion has been found to be therapeutically useful. An important role for B cells in the activation of pathogenic T cells is suggested by these results. Because myelin basic protein is known to be an important self antigen that is the target of the immune response in MS, attempts have been initiated to inject peptides derived from this antigen into patients, with the hope of inducing tolerance or generating regulatory T cells specific for the relevant antigen.
Type 1 Diabetes Mellitus
Type 1 diabetes mellitus, previously called insulin-dependent diabetes mellitus, is a multisystem metabolic disease resulting from impaired insulin production that affects about 0.2% of the U.S. population, with a peak age at onset of 11 to 12 years, and the incidence is increasing. The disease is characterized by hyperglycemia and ketoacidosis. Chronic complications of type 1 diabetes include progressive atherosclerosis of arteries, which can lead to ischemic necrosis of limbs and internal organs, and microvascular obstruction causing damage to the retina, renal glomeruli, and peripheral nerves. These patients have a deficiency of insulin resulting from immune-mediated destruction of the insulin-producing β cells of the islets of Langerhans in the pancreas, and continuous hormone replacement therapy is needed.
Pathogenesis of Type 1 Diabetes
Several mechanisms may contribute to β cell destruction, including inflammation mediated by CD4+ TH1 cells reactive with islet antigens (including insulin), CTL-mediated lysis of islet cells, local production of cytokines (TNF and IL-1) that damage islet cells, and autoantibodies against islet cells. In the rare cases in which the pancreatic lesions have been examined at the early active stages of the disease, the islets show cellular necrosis and lymphocytic infiltration consisting of both CD4+ and CD8+ T cells. This lesion is called insulitis. Autoantibodies against islet cells and insulin are also detected in the blood of these patients. In susceptible children who have not developed diabetes (such as relatives of patients), the presence of antibodies against islet cells is predictive of the development of type 1 diabetes, suggesting that the anti–islet cell antibodies are pathogenic. An informative animal model of the disease is the non-obese diabetic (NOD) mouse that develops spontaneous diabetes. In this model, there is evidence for defective survival and function of regulatory T cells as well as resistance of effector T cells to suppression.
Multiple genes are associated with type 1 diabetes. A great deal of attention has been devoted to the role of HLA genes. Between 90% and 95% of Caucasians with type 1 diabetes have HLA-DR3, or DR4, or both, in contrast to about 40% of normal subjects, and 40% to 50% of patients are DR3/DR4 heterozygotes, in contrast to 5% of normal subjects. Interestingly, susceptibility to type 1 diabetes is actually associated with alleles of DQ2 and DQ8 that are often in linkage disequilibrium with DR3 and DR4. Several non-HLA genes also contribute to the disease. The first of these to be identified is insulin, with tandem repeats in the promoter region being associated with disease susceptibility. The mechanism of this association is unknown; it may be related to the level of expression of insulin in the thymus, which determines whether insulin-specific T cells will be deleted (negatively selected) during maturation. Several other polymorphisms have been identified in patients and in NOD mice, including in the IL-2 and CD25 genes. The functional consequences of these polymorphisms are not known. Some studies have suggested that viral infections (e.g., with coxsackievirus B4) may precede the onset of type 1 diabetes, perhaps by initiating cell injury, inducing inflammation and the expression of costimulators, and triggering an autoimmune response. However, epidemiologic data suggest that repeated infections protect against type 1 diabetes, and this is similar to the NOD model. In fact, it has been postulated that one reason for the increased incidence of type 1 diabetes in developed countries is the control of infectious diseases.
New Therapies for Type 1 Diabetes
The most interesting new therapeutic strategies for type 1 diabetes are focused on inducing tolerance with diabetogenic peptides from islet antigens (such as insulin) or generating or giving regulatory T cells to patients. These clinical trials are in their infancy.
Inflammatory Bowel Disease
Inflammatory bowel disease consists of two disorders, Crohn’s disease and ulcerative colitis, in which T cell–mediated inflammation causes intestinal injury. Crohn’s disease is characterized by chronic inflammation and destruction of the intestinal wall, with frequent formation of fistulas. In ulcerative colitis, the lesions are largely confined to the mucosa and consist of ulcers with underlying foci of inflammation. The pathogenesis of inflammatory bowel disease was described in Chapter 13. New therapies for these diseases include antibodies against TNF, IL-17, and the p40 chain of IL-12 and IL-23.
Summary
Davidson A, Diamond B. Autoimmune diseases. New England Journal of Medicine. 2001;345:340-350.
Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25-35.
Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619-630.
Antibody and Immune Complex–Mediated Disorders
Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383-392.
D’Cruz DP, Khamashta MA, Hughes GR. Systemic lupus erythematosus. Lancet. 2007;369:587-596.
Fairhurst AM, Wandstrat AE, Wakeland EK. Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Advances in Immunology. 2006;92:1-69.
Jancar S, Sanchez M. Crespo. Immune complex–mediated tissue injury: a multistep paradigm. Trends in Immunology. 2005;26:48-55.
Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nature Reviews Rheumatology. 2010;6:280-289.
Plotz PH. The autoantibody repertoire: searching for order. Nature Reviews Immunology. 2003;3:73-78.
Frohman EM, Racke MK, Raine CS. Multiple sclerosis—the plague and its pathogenesis. New England Journal of Medicine. 2006;354:942-955.
Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. Journal of Clinical Investigation. 2007;117:1119-1127.
Imboden JB. The immunopathogenesis of rheumatoid arthritis. Annual Review of Pathology. 2009;4:417-434.
Palmer MT, Weaver CT. Autoimmunity: increasing suspects in the CD4+ T cell lineage. Nature Immunology. 2010;11:36-40.