CHAPTER 10 Effector Mechanisms of Cell-Mediated Immunity
Cell-mediated immunity is the type of host defense that is mediated by T lymphocytes, and it serves as the defense mechanism against microbes that survive and replicate inside phagocytes and nonphagocytic cells. Historically, immunologists have divided adaptive immunity into humoral immunity, which can be adoptively transferred from an immunized donor to a naive host by antibodies in the absence of cells, and cell-mediated immunity, which can be adoptively transferred only by viable T lymphocytes. The effector phase of humoral immunity is triggered by the recognition of antigen by secreted antibodies; therefore, humoral immunity neutralizes and eliminates extracellular microbes and toxins that are accessible to antibodies, but it is not effective against microbes inside cells. In contrast, in cell-mediated immunity, the effector phase is initiated by the recognition of antigens by T cells. T lymphocytes recognize protein antigens of microbes that are displayed on the surfaces of infected cells as peptides bound to self major histocompatibility complex (MHC) molecules. Therefore, cell-mediated immunity is specific for cell-associated microbes. Defects in cell-mediated immunity result in increased susceptibility to infection by viruses and intracellular bacteria as well as some extracellular bacteria and fungi that are ingested by phagocytes. T cell–mediated reactions are also important in allograft rejection (see Chapter 16), anti-tumor immunity (see Chapter 17), and immune-mediated inflammatory diseases (see Chapter 18). In Chapter 9, we described the activation of T lymphocytes and the differentiation of naive T cells into effector and memory cells. In this chapter, we discuss the reactions of effector T cells in cell-mediated immunity.
Types of Cell-Mediated Immune Reactions
Different populations of T cells have evolved to combat different types of infectious pathogens. Thus, cell-mediated immunity provides excellent examples of the specialization of adaptive immunity. There are several important general principles of cell-mediated immune reactions.
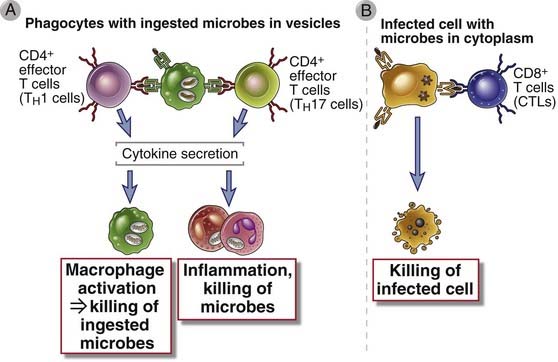
FIGURE 10–1 Types of T cell–mediated immune reactions.
A, CD4+ T cells recognize antigens of phagocytosed and extracellular microbes and produce cytokines that activate the phagocytes to kill the microbes and stimulate inflammation. CD8+ T cells can also secrete cytokines and participate in similar reactions. B, CD8+ cytotoxic T lymphocytes (CTLs) recognize antigens of microbes residing in the cytoplasm of infected cells and kill the cells.
Cell-mediated immune responses consist of the development of effector T cells from naive cells in peripheral lymphoid organs, migration of these effector T cells and other leukocytes to sites of infection, and either cytokine-mediated activation of leukocytes to destroy microbes or direct killing of infected cells (Fig. 10-2). The development of effector T cells involves the sequence of antigen recognition, clonal expansion, and differentiation that is characteristic of all adaptive immune responses; these processes were described in Chapter 9. In this chapter, we describe the reactions of effector T cells and their roles in host defense and tissue injury. We start with the process that brings effector T cells to the site of infection or tissue damage.
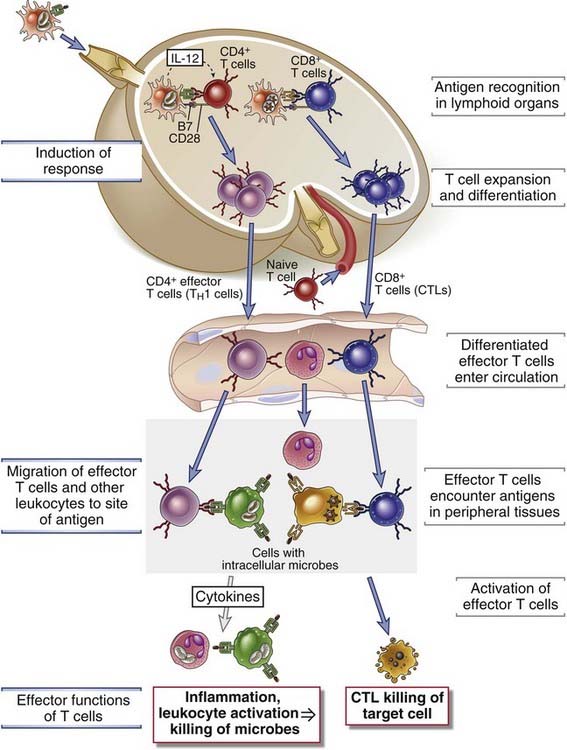
FIGURE 10–2 The induction and effector phases of cell-mediated immunity.
Induction of response: CD4+ T cells and CD8+ T cells recognize peptides that are derived from protein antigens and presented by dendritic cells in peripheral lymphoid organs. The T lymphocytes are stimulated to proliferate and differentiate into effector (and memory) cells, which enter the circulation.
Migration of effector T cells and other leukocytes to the site of antigen: Effector T cells and other leukocytes migrate through blood vessels in peripheral tissues by binding to endothelial cells that have been activated by cytokines produced in response to infection in these tissues.
Effector functions of T cells: Effector T cells recognize the antigen in the tissues and respond by secreting cytokines that recruit more leukocytes and activate phagocytes to eradicate the infection. CTLs also migrate to tissues and kill infected cells.
Migration of Effector T Lymphocytes to Sites of Infection
Some effector T cells exit the lymphoid organs where they were generated and preferentially home to sites of infection in peripheral tissues, where they are needed to eliminate microbes during the effector phase of adaptive immune responses (Fig. 10-3). In Chapter 3, we described naive T cell recirculation through lymphoid tissues and effector T cell migration into nonlymphoid tissues and the roles of adhesion molecules and chemokines in these processes. The differentiation of naive T cells into effector cells, which occurs in the peripheral lymphoid organs, is associated with a change in expression of the chemokine receptors and adhesion molecules that determine the migratory behavior of these cells. The expression of molecules involved in naive T cell homing into lymph nodes, including L-selectin and CCR7, decreases shortly after antigen-induced activation of the naive T cells, and the cell surface expression of the sphingosine 1-phosphate receptor S1PR1 increases. As a result, the effector cells that develop are no longer constrained to stay in the node and are attracted to enter the blood or efferent lymphatics, which ultimately drain into the blood through the thoracic duct. Furthermore, unlike naive T cells, effector T cells express chemokine receptors that bind chemokines produced at sites of infection and adhesion molecules that bind to endothelial adhesion molecules that are induced on postcapillary venules by cytokines, including IL-1 and tumor necrosis factor (TNF), that are produced at infection sites. Therefore, after they enter the circulation, effector T cells home preferentially to sites of infection.
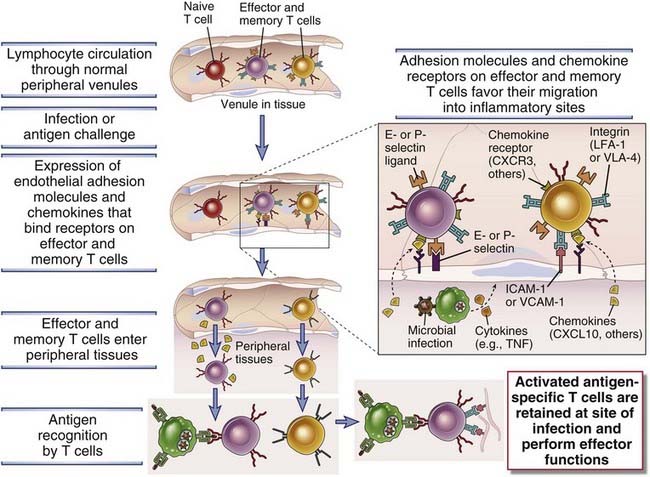
FIGURE 10–3 Migration and retention of effector and memory T cells at sites of infection.
Previously activated effector and memory T cells, but not naive cells, migrate through endothelium that is activated by cytokines (e.g., TNF) produced at a site of infection. Activated endothelial cells express E- and P-selectins and ligands for integrins, and effector T cells express selectin ligands and integrins. These adhesion molecules mediate the interaction between the T cells and endothelium. Chemokines play additional critical roles in the migration of the T cells through the vessel wall. In extravascular tissue, the T cells that specifically recognize antigen are activated and retained, whereas T cells that do not encounter the antigen for which they are specific either die or return to the circulation, largely through lymphatic vessels (not shown).
TH1, TH2, and TH17 subsets of CD4+ T cells each have distinct homing phenotypes that direct them to migrate into different sites of infections. For example, during differentiation from naive precursors, TH1 cells gain the capacity to produce functional ligands that bind E-selectin and P-selectin, whereas TH2 cells express lower levels of selectin ligands. This aspect of TH1 differentiation involves T-bet–dependent expression of glycosyltransferases that are required for synthesis of the carbohydrate moieties to which the selectins bind. Furthermore, the chemokine receptors CXCR3 and CCR5, which bind to chemokines elaborated in tissues during innate immune responses, are expressed at high levels by TH1 cells but not by TH2 cells. Therefore, TH1 cells tend to be abundant at sites of infection where the infectious agents trigger strong innate immune reactions; these agents include many bacteria and viruses. In general, CTLs migrate in similar ways as TH1 cells. In contrast, TH2 cells express the chemokine receptors CCR3, CCR4, and CCR8, which recognize chemokines that are highly expressed at sites of helminth infection or allergic reactions, particularly in mucosal tissues, and so TH2 cells tend to migrate to these tissues. TH17 cells express CCR6, which binds the chemokine CCL20, and migration of TH17 cells into inflammatory sites is dependent on CCR6. CCL20 is produced by various tissue cells and macrophages in many bacterial and fungal infections.
After effector T cells enter the site of infection and are activated once again by antigen, they produce more cytokines and chemokines and stimulate much greater leukocyte migration. This later, enhanced inflammation is sometimes called immune inflammation to indicate the role of T lymphocytes (immune cells) in the process.
The migration of effector T cells from the circulation to peripheral sites of infection is largely independent of antigen, but cells that recognize antigen in extravascular tissues may be preferentially retained there (see Fig. 10-3). T cell migration through blood and lymphatic vessels is controlled mainly by adhesion molecules and chemokines, which will engage T cells of any antigen specificity. Therefore, this process of migration results in the recruitment of circulating effector T cells to inflammatory sites, regardless of antigen specificity, ensuring that the maximum possible number of previously activated T cells have the opportunity to locate infectious microbes and to eradicate the infection. Some memory T cells also migrate into peripheral nonlymphoid tissues regardless of antigen specificity. Once in the tissues, the T cells encounter microbial antigens presented by macrophages and other antigen-presenting cells (APCs). T cells that specifically recognize antigens receive signals through their antigen receptors that increase the affinity of integrins for their ligands. Two of these integrins, VLA-4 and VLA-5, bind to fibronectin in extracellular matrices, and a third adhesion molecule, CD44, which is also highly expressed on effector and memory T cells, binds to hyaluronan. As a result, antigen-specific effector and memory T cells that encounter the antigen are preferentially retained at the extravascular site where the antigen is present. T cells not specific for the antigen that migrate into a site of inflammation may die in the tissue or return through lymphatic vessels to the circulation.
Effector Functions of CD4+ Helper T Cells
Effector T cells of the CD4+ lineage function by secreted cytokines and cell surface molecules to activate other cells to eliminate microbes. CD4+ T cells also participate indirectly in host defense by helping B lymphocytes to produce high-affinity antibodies against extracellular microbes and by promoting the development of fully functional CTLs that combat intracellular microbes such as viruses. The roles of helper T cells in antibody responses are described in Chapter 11 and in CTL responses in Chapter 9. Here our focus is on the roles of CD4+ T cells as the effector cells of cell-mediated immunity.
The functions of CD4+ effector cells in cell-mediated immunity can be divided into several steps (Fig. 10-4):
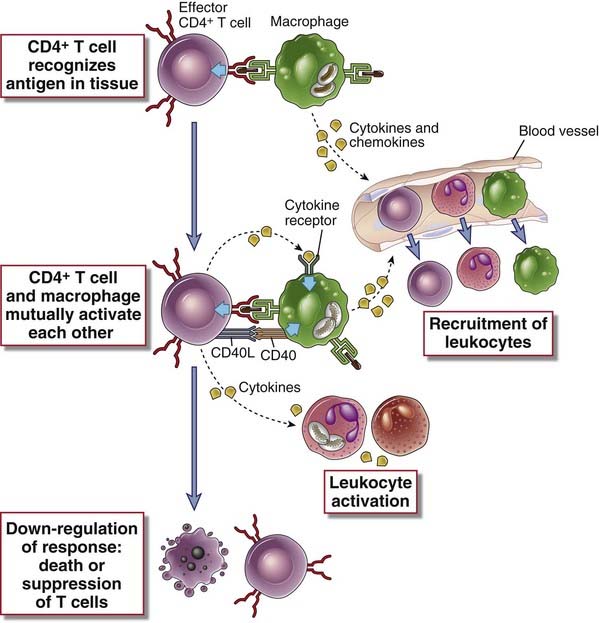
FIGURE 10–4 Sequence of events in the reactions of effector CD4+ T cells.
The sequence of events in the functional responses of effector CD4+ T cells is shown. Effector cells are recruited from the circulation, activated by antigens displayed by macrophages that have phagocytosed microbes, and secrete cytokines that amplify and control the reaction. The T cells use CD40L and cytokines to activate phagocytes and cytokines to recruit more leukocytes.
With this overview, we proceed to a discussion of the functions of the three major subsets of effector CD4+ T cells in cell-mediated immunity.
Functions of TH1 Cells
The principal function of TH1 cells is to activate macrophages to ingest and destroy microbes (Fig. 10-5). Recall that phagocytosed intracellular microbes are powerful stimuli for the generation of TH1 cells (see Chapter 9). Thus, TH1 effector cells develop in response to the pathogens that these cells are designed to eradicate, an excellent example of the specialization of adaptive immunity. The same reaction of TH1-mediated macrophage activation is involved in injurious delayed-type hypersensitivity, which is a component of many inflammatory diseases, and in granulomatous inflammation, which is typical of tuberculosis and is also seen in some other infectious and inflammatory disorders. These pathologic reactions are described in Chapter 18.
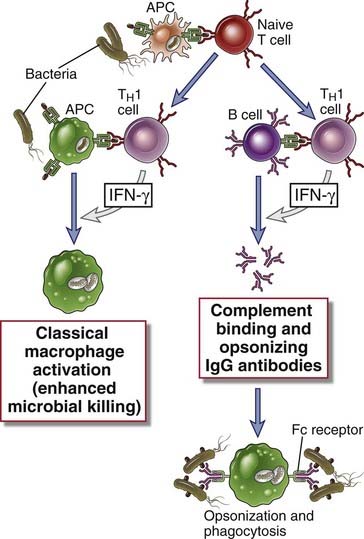
FIGURE 10–5 Functions of TH1 cells.
CD4+ T cells that differentiate into TH1 cells secrete IFN-γ, which acts on macrophages to increase phagocytosis and killing of microbes in phagolysosomes and on B lymphocytes to stimulate production of IgG antibodies that opsonize microbes for phagocytosis. The cells also produce TNF, which activates neutrophils and promotes inflammation (not shown).
Before discussing the activation of macrophages and how they destroy microbes, we describe the properties of interferon-γ (IFN-γ), the cytokine responsible for most of the specialized functions of TH1 cells.
Cytokines Produced by TH1 Cells
The signature cytokine of TH1 cells is IFN-γ. TH1 cells also produce TNF, some chemokines, and other cytokines.
Interferon-γ
IFN-γ is the principal macrophage-activating cytokine and serves critical functions in immunity against intracellular microbes. IFN-γ is also called immune or type II interferon. Although it has some antiviral activity, it is not a potent antiviral cytokine, and it functions mainly as an activator of effector cells of the immune system.
IFN-γ is a homodimeric protein belonging to the type II cytokine family (see Chapter 7). In addition to CD4+ TH1 cells, IFN-γ is also produced by NK cells and CD8+ T cells. NK cells secrete IFN-γ in response to activating ligands on the surface of infected or stressed host cells (see Chapter 2) or in response to IL-12; in this setting, IFN-γ functions as a mediator of innate immunity. In adaptive immunity, T cells produce IFN-γ in response to antigen recognition, and production is enhanced by IL-12 and IL-18.
The receptor for IFN-γ is composed of two structurally homologous polypeptides belonging to the type II cytokine receptor family, called IFNγR1 and IFNγR2. IFN-γ binds to and induces the dimerization of the two receptor chains. This leads to activation of the associated JAK1 and JAK2 kinases and ultimately to phosphorylation and dimerization of STAT1, which stimulates transcription of several genes (see Chapter 7). IFN-γ–induced genes encode many different molecules that mediate the biologic activities of this cytokine, described next.
The functions of IFN-γ are important in cell-mediated immunity against intracellular microbes (see Fig. 10-5).
The actions of IFN-γ together result in increased ingestion of microbes and the destruction of the ingested pathogens. Individuals with rare inherited inactivating mutations in the IFN-γ receptor and knockout mice lacking IFN-γ or the IFN-γ receptor or molecules required for TH1 differentiation or IFN-γ signaling (IL-12, T-bet, STAT1) are susceptible to infections with intracellular microbes, such as mycobacteria, because of defective macrophage-mediated killing of the microbes.
Other TH1 Cytokines
In addition to IFN-γ, TH1 cells produce TNF and various chemokines, which contribute to the recruitment of leukocytes and enhanced inflammation. Somewhat surprisingly, TH1 cells are also important sources of IL-10, which functions mainly to inhibit dendritic cells and macrophages and thus to suppress TH1 activation. This is an example of a negative feedback loop in T cell responses.
TH1-Mediated Classical Macrophage Activation and Killing of Phagocytosed Microbes
In cell-mediated immune responses against phagocytosed microbes, T cells specifically recognize microbial antigens but phagocytes actually destroy the pathogens. This fundamental concept was first appreciated from studies of cell-mediated immunity to the intracellular bacterium Listeria monocytogenes (Fig. 10-6). It was shown in the 1950s that mice infected with a low dose of Listeria were protected from challenge with higher, lethal doses. Protection could be transferred to naive animals with lymphocytes (later shown to be T lymphocytes) from the infected mice but not with serum. In vitro, the bacteria were killed not by T cells from immune animals but by activated macrophages, emphasizing the central role of macrophages in the execution of effector function.
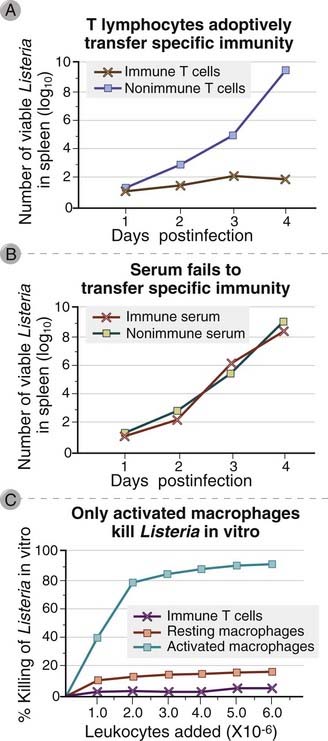
FIGURE 10–6 Cell-mediated immunity to Listeria monocytogenes.
Immunity to L. monocytogenes is measured by inhibition of bacterial growth in the spleens of animals inoculated with a known dose of viable bacteria. Such immunity can be transferred to normal mice by T lymphocytes (A) but not by serum (B) from syngeneic mice previously immunized with killed or low doses of L. monocytogenes. In an in vitro assay of cell-mediated immunity, the bacteria are actually killed by activated macrophages and not by T cells (C).
At any site of infection, as part of the innate immune response, monocytes are recruited from blood into tissues by chemokines produced by macrophages and other cells resident at the site (see Chapter 4). These monocytes mature into tissue macrophages and first attempt to phagocytose and destroy the pathogen. If the microbe has evolved to resist elimination by the macrophages, it survives within the phagosomes. In these infected cells, microbial peptides are processed and presented as peptides associated with class II MHC molecules. At the same time, TH1 effector cells are generated in an adaptive immune response in secondary lymphoid tissues, by processes described in Chapter 9. These T cells are recruited to the site of infection, where they recognize antigenic peptides (the same as those that initiated the response) displayed by the microbe-bearing macrophages. The macrophages are exposed to signals from the TH1 effector cells, which activate the macrophages to kill the ingested microbes. Activation consists of increased expression of various proteins that endow activated macrophages with the capacity to perform specialized functions, such as microbial killing. In the following sections, we describe the T cell signals that activate macrophages in cell-mediated immune reactions and the functions of these macrophages.
CD4+ TH1 cells activate macrophages by contact-mediated signals delivered by CD40L-CD40 interactions and by IFN-γ (Fig. 10-7). When the TH1 cells are stimulated by antigen, the cells express CD40L on their surface and secrete IFN-γ. The actions of IFN-γ on macrophages, described earlier, synergize with the actions of CD40 ligand, and together they are potent stimuli for macrophage activation. The importance of the CD40 pathway in cell-mediated immunity is illustrated by the immunologic defects in humans with inherited mutations in CD40L (X-linked hyper-IgM syndrome) and in mice in which the gene for CD40 or CD40L is knocked out (see Chapter 20). All these disorders are characterized by severe deficiencies in cell-mediated immunity to intracellular microbes, and children with the X-linked hyper-IgM syndrome often succumb to infection by the intracellular pathogen Pneumocystis jiroveci. As expected, these patients and knockout mice also have defects in helper T cell–dependent antibody production. The requirement for interactions between the surface molecules CD40 on the macrophages and CD40L on the T cells ensures that macrophages that are presenting antigens to the T cells (i.e., the macrophages that are harboring intracellular microbes) are also the macrophages that are most efficiently activated by the T cells. The role of IFN-γ as the major macrophage-activating cytokine was discussed before. The same principles are applicable to the T cell–dependent activation of B lymphocytes—helper T cells stimulate B lymphocyte proliferation and differentiation by CD40-mediated signals and cytokines (see Chapter 11).
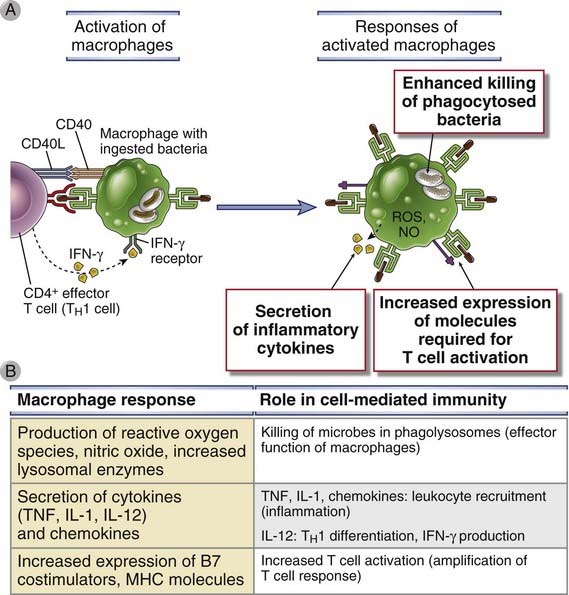
FIGURE 10–7 Macrophage activation by TH1 cells.
A, Macrophages are activated by CD40L-CD40 interactions and by IFN-γ expressed by TH1 cells and perform several functions that kill microbes, stimulate inflammation, and enhance the antigen-presenting capacity of the cells. B, The principal molecules that mediate the functions of macrophages are listed. Macrophages are also activated during innate immune reactions and perform the same functions (see Chapter 4).
Activated macrophages kill phagocytosed microbes mainly by the actions of reactive oxygen species, nitric oxide, and lysosomal enzymes. All these potent microbicidal agents are produced within the lysosomes of macrophages and kill ingested microbes after phagosomes fuse with lysosomes (see Chapter 4, Figure 4-12). These toxic substances may also be released into adjacent tissues, where they kill extracellular microbes and may cause damage to normal tissues. This pathway of macrophage activation is called classical (Fig. 10-8) to distinguish it from alternative activation, described later.
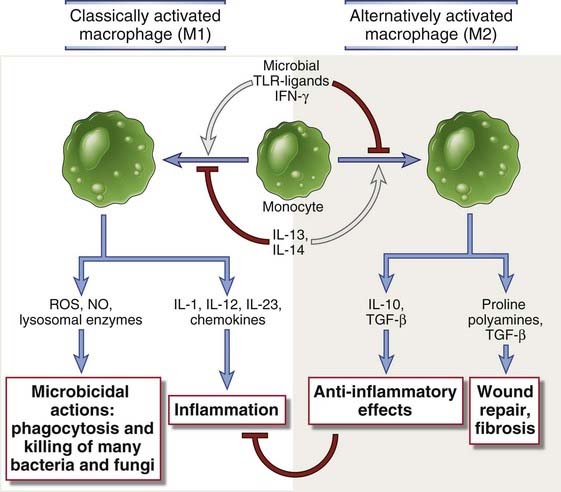
FIGURE 10–8 Classical and alternative macrophage activation.
Subsets of activated macrophages. Different stimuli activate monocytes-macrophages to develop into functionally distinct populations. Classically activated macrophages are induced by microbial products and cytokines, particularly IFN-γ, and are microbicidal and involved in potentially harmful inflammation. Alternatively activated macrophages are induced by IL-4 and IL-13 produced by TH2 cells and other leukocytes and are important in tissue repair and fibrosis.
Activated macrophages are involved in several other reactions of host defense (see Fig. 10-7). They stimulate inflammation through the secretion of cytokines, mainly TNF, IL-1, and chemokines, and short-lived lipid mediators such as prostaglandins, leukotrienes, and platelet-activating factor. The collective action of these macrophage-derived cytokines and lipid mediators is to recruit more leukocytes, which improves the ability to destroy infectious organisms. Activated macrophages amplify cell-mediated immune responses by becoming more efficient APCs because of increased levels of molecules involved in antigen processing and increased surface expression of class II MHC molecules and costimulators and by producing cytokines (such as IL-12) that stimulate T lymphocyte differentiation into effector cells.
Some tissue injury may normally accompany TH1 cell–mediated immune reactions to microbes because the microbicidal products released by activated macrophages and neutrophils are capable of injuring normal tissue and do not discriminate between microbes and host tissue. However, this tissue injury is usually limited in extent and duration, and it resolves as the infection is cleared. As mentioned earlier, delayed hypersensitivity is an example of a TH1-mediation reaction that can cause significant tissue injury (see Chapter 18).
Functions of TH2 Cells
TH2 cells stimulate IgE- and eosinophil-mediated reactions that serve to eradicate helminthic infections (Fig. 10-9). Helminths are too large to be phagocytosed by neutrophils and macrophages and may be more resistant to the microbicidal activities of these phagocytes than are most bacteria and viruses. Therefore, special mechanisms are needed for defense against helminthic infections. TH2 cells secrete IL-4, IL-5, and IL-13, which work cooperatively to eradicate these infections. We first describe the properties of these cytokines and then their roles in host defense.
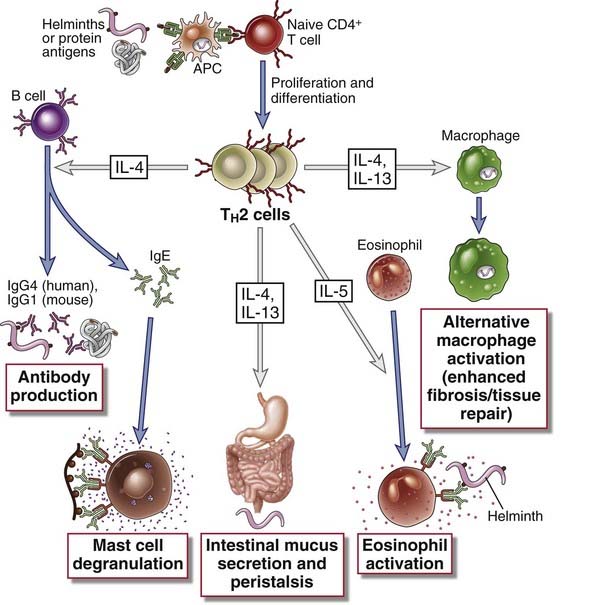
FIGURE 10–9 Functions of TH2 cells.
CD4+ T cells that differentiate into TH2 cells secrete IL-4, IL-5, and IL-13. IL-4 (and IL-13) act on B cells to stimulate production of antibodies that bind to mast cells, such as IgE. IL-4 is also an autocrine growth and differentiation cytokine for TH2 cells. IL-5 activates eosinophils, a response that is important for defense against helminthic infections. IL-4 and IL-13 are involved in barrier immunity, induce an alternative pathway of macrophage activation, and inhibit classical, TH1-mediated macrophage activation.
Cytokines Produced by TH2 Cells
The functions of TH2 cells are mediated by IL-4, which induces IgE antibody responses; IL-5, which activates eosinophils; and IL-13, which has diverse actions.
Interleukin-4
IL-4 is the major stimulus for the production of IgE antibodies and for the development of TH2 cells from naive CD4+ helper T cells. IL-4 is the signature cytokine of the TH2 subset and functions as both an inducer and an effector cytokine of these cells.
IL-4 is a member of the four–α-helical cytokine family. The principal cellular sources of IL-4 are CD4+ T lymphocytes of the TH2 subset and activated mast cells. The IL-4 receptor of lymphoid cells consists of a cytokine-binding α chain that is a member of the type I cytokine receptor family, associated with the γc chain shared by other cytokine receptors. This IL-4Rαγc receptor signals by the JAK-STAT pathway (JAK3 or JAK4 and STAT6) and by a pathway that involves the insulin response substrate (IRS) called IRS-2. IL-4 and IL-13 activate the STAT6 protein, which induces transcription of genes that account for many of the actions of these cytokines. IL-4 also binds to the IL-13 receptor (described below).
IL-4 has important actions on several cell types.
Interleukin-13
IL-13 is structurally and functionally similar to IL-4 and also plays a key role in defense against helminths (see Chapter 15) and in allergic diseases (see Chapter 19). IL-13 is a member of the four–α-helical cytokine family, with limited sequence homology but significant structural similarity to IL-4. IL-13 is produced mainly by the TH2 subset, but basophils, eosinophils, and NKT cells may also produce the cytokine. The functional IL-13 receptor is a heterodimer of the IL-4Rα chain and the IL-13Rα1 chain. This complex can bind both IL-4 and IL-13 with high affinity and accounts for the fact that most of the biologic effects of IL-13 are shared with IL-4. The receptor is expressed on a wide variety of cells, including B cells, mononuclear phagocytes, dendritic cells, eosinophils, basophils, fibroblasts, endothelial cells, and bronchial epithelial cells. T cells do not express the IL-13 receptor. IL-13R signaling is similar to IL-4R signaling.
IL-13 works together with IL-4 in producing biologic effects associated with allergic inflammation, discussed in detail in Chapter 19, and in defense against helminths. Some of the actions of IL-13 overlap those of IL-4, and others are distinct. IL-13 functions with IL-4 to induce alternative macrophage activation, which contributes to tissue repair and fibrosis. IL-13 stimulates mucus production by airway epithelial cells, an important component of allergic reactions such as asthma. As mentioned before, both IL-13 and IL-4 can activate B cells to switch to IgE and some IgG isotypes and recruit leukocytes. Unlike IL-4, IL-13 is not involved in TH2 differentiation.
Interleukin-5
IL-5 is an activator of eosinophils and serves as the principal link between T cell activation and eosinophilic inflammation. It is a homodimer of a polypeptide containing a four–α-helical domain and is a member of the type I cytokine family. It is produced by TH2 cells and by activated mast cells. The IL-5 receptor is a heterodimer composed of a unique α chain and a common β chain (βc), which is also part of the IL-3 and granulocyte-macrophage colony-stimulating factor (GM-CSF) receptors (see Fig. 7-23, Chapter 7). The major IL-5–induced signaling pathway involves JAK2 and STAT3.
The principal actions of IL-5 are to activate mature eosinophils and to stimulate the growth and differentiation of eosinophils. Activated eosinophils are able to kill helminths. Eosinophils express Fc receptors specific for IgE and some IgG antibodies and are thereby able to bind to microbes, such as helminths, that are opsonized by these antibodies. IL-5 also stimulates the production of IgA antibodies.
Roles of TH2 Cells in Host Defense
TH2 cells function in defense against helminth infections by several mechanisms (see Fig. 10-9).
Functions of TH17 Cells
TH17 cells secrete cytokines that recruit leukocytes, mainly neutrophils, to sites of infection (Fig. 10-10). Because neutrophils are a major defense mechanism against extracellular bacteria and fungi, TH17 cells play an especially important role in defense against these infections.
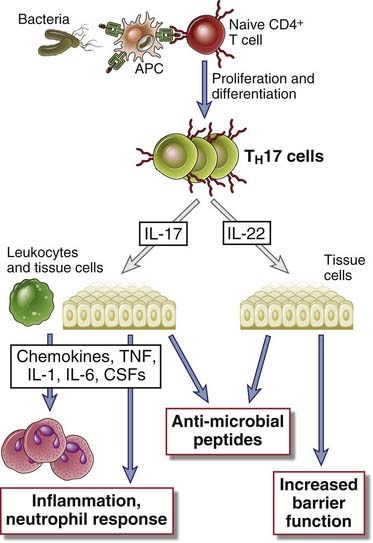
FIGURE 10–10 Functions of TH17 cells.
Cytokines produced by TH17 cells stimulate local production of chemokines and inflammation and the production of antimicrobial peptides (defensins) and promote epithelial barrier functions.
Cytokines Produced by TH17 Cells
TH17 cells produce several cytokines. Most of the inflammatory actions of these cells are mediated by IL-17, but other cytokines produced by this subset may also contribute.
Interleukin-17
IL-17 is an unusual cytokine because neither it nor its receptor is homologous to any other known cytokine-receptor pair. The IL-17 family includes six structurally related proteins, of which IL-17A and IL-17F are the most similar, and the immunologic activities seem to be mediated primarily by IL-17A. IL-17A and IL-17F are produced mainly by TH17 cells, whereas the other members of the family are produced by diverse cell types. IL-17 receptors are multimeric and expressed on a wide range of cells. Their structure and signaling mechanisms are not well defined.
IL-17 is an important link between T cell–mediated adaptive immunity and the innate immune system, especially the inflammatory component of innate responses.
Other TH17 Cytokines
IL-22 is a member of the IL-10 cytokine family. It is produced by activated T cells, particularly TH17 cells, and by NK cells. The actions of IL-22 appear contradictory. Some studies indicate that it contributes to inflammation and tissue injury, but the bulk of the available data suggests that it is produced in epithelial tissues, especially of the skin and gastrointestinal tract, and serves to maintain epithelial integrity, mainly by promoting the barrier function of epithelia and by stimulating repair reactions.
IL-21 is produced by activated CD4+ T cells, including TH17 cells, which has a wide variety of effects on B and T cells and NK cells. The IL-21 receptor belongs to the type I cytokine receptor family, consists of a ligand-binding chain and the γc subunit, and activates a JAK-STAT signaling pathway in which STAT3 is especially prominent. An important function of IL-21 is in antibody responses, especially the reactions that occur in germinal centers (see Chapter 11). IL-21 is required for the generation of follicular helper T cells and is also produced by follicular helper cells and stimulates B cells in germinal centers. IL-21 has also been shown to promote the differentiation of TH17 cells, especially in humans, providing an autocrine pathway for amplifying TH17 responses. Some of the other reported actions of IL-21 include increasing the proliferation and effector function of CD8+ T cells and NK cells.
Roles of TH17 Cells in Host Defense
The principal effector function of TH17 cells is to induce neutrophilic inflammation, which serves to destroy extracellular bacteria and fungi (see Fig. 10-10). The ability of IL-17 to recruit neutrophils accounts for the central role of TH17 cells in adaptive immune reactions in which neutrophilic inflammation is prominent. The recruited neutrophils ingest and kill extracellular microbes, including fungi and bacteria. The importance of this role of TH17 cells is illustrated by the inherited disease called the hyper-IgE syndrome (or Job’s syndrome), which is characterized by increased susceptibility to cutaneous fungal and bacterial infections, and is caused by mutations in the transcription factor STAT3, which is essential for the development of TH17 cells (see Chapter 9).
TH17 cells are also important in the pathogenesis of to many inflammatory diseases, such as psoriasis, inflammatory bowel disease, rheumatoid arthritis, and multiple sclerosis. Antibodies that block the development or functions of TH17 cells are in clinical trials for several of these diseases. TH1 and TH17 cells may both be present in the lesions in these diseases, and their relative contribution to the development and propagation of the disorders is an area of active research. TH17 cells may also serve to maintain normal epithelial function in the gut and skin, mainly by virtue of the actions of IL-22.
Effector Functions of CD8+ Cytotoxic T Lymphocytes
CD8+ CTLs eliminate intracellular microbes mainly by killing infected cells (see Fig. 10-1B). The development of a CD8+ CTL response to infection proceeds through similar steps as those described for CD4+ T cell responses, including antigen-mediated stimulation of naive CD8+ T cells in lymphoid organs, clonal expansion, differentiation, and migration of differentiated CTLs into tissues. These events were described in Chapter 9. In addition to direct cell killing, CD8+ T cells secrete IFN-γ and thus contribute to macrophage activation in host defense and in hypersensitivity reactions. Here we discuss the mechanisms by which differentiated CTLs kill cells harboring microbes.
Mechanisms of CTL-Mediated Cytotoxicity
CTL-mediated killing involves specific recognition of target cells and delivery of proteins that induce cell death. CTLs kill targets that express the same class I–associated antigen that triggered the proliferation and differentiation of naive CD8+ T cells from which they are derived and do not kill adjacent uninfected cells that do not express this antigen. In fact, even the CTLs themselves are not injured during the killing of antigen-expressing targets. This specificity of CTL effector function ensures that normal cells are not killed by CTLs reacting against infected cells. The killing is highly specific because an “immunologic synapse” (see Chapter 6) is formed at the site of contact of the CTL and the antigen-expressing target, and the molecules that actually perform the killing are secreted into the synapse and cannot diffuse to other nearby cells.
The process of CTL-mediated killing of targets consists of antigen recognition, activation of the CTLs, delivery of the “lethal hit” that kills the target cells, and release of the CTLs (Fig. 10-11). Each of these steps is controlled by specific molecular interactions.
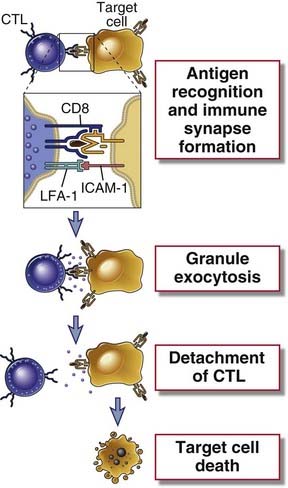
FIGURE 10–11 Steps in CTL-mediated lysis of target cells.
A CTL recognizes the antigen-expressing target cell and is activated. Activation results in the release of granule contents from the CTL into the target cell through the area of contact (the immunologic synapse). Granule contents deliver a lethal hit to the target. The CTL may detach and kill other target cells. The formation of conjugates between a CTL and its target and activation of the CTL also require interactions between accessory molecules (LFA-1, CD8) on the CTL and their specific ligands on the target cell; these are not shown.
Recognition of Antigen and Activation of CTLs
The CTL binds and reacts to the target cell by using its antigen receptor, coreceptor (CD8), and adhesion molecules. To be efficiently recognized by CTLs, target cells must express class I MHC molecules complexed to a peptide (the complex serving as the ligand for the T cell receptor (TCR) and the CD8 coreceptor) and intercellular adhesion molecule 1 (ICAM-1, the principal ligand for the LFA-1 integrin). The CTLs and their target cells form tight conjugates (Fig. 10-12). This immunologic synapse (see Chapter 7) formed between the two cells is characterized by a ring of close apposition between the CTL and target cell membranes, mediated by LFA-1–ICAM-1 binding, and an enclosed gap or space inside the ring. Distinct regions of the CTL membrane can be observed by immunofluorescence microscopy within the ring, including a signaling patch, which includes the TCR, protein kinase C-θ, and Lck, and a secretory domain, which appears as a gap to one side of the signaling patch. This interaction results in the initiation of biochemical signals that activate the CTL, which are essentially the same as the signals involved in the activation of helper T cells (see Chapter 7). Cytokines and costimulators provided by dendritic cells, which are required for the differentiation of naive CD8+ T cells into CTLs, are not necessary for triggering the effector function of CTLs (i.e., target cell killing). Therefore, once CD8+ T cells specific for an antigen have differentiated into fully functional CTLs, they can kill any nucleated cell that displays that antigen.
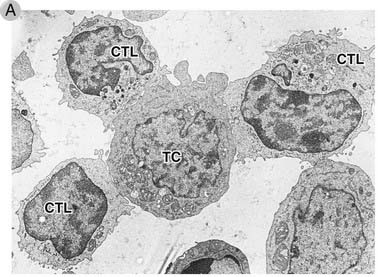
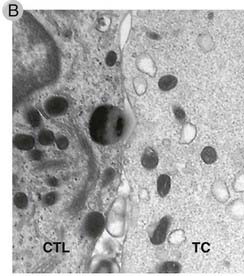
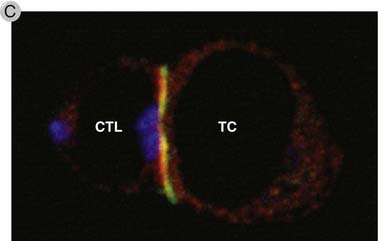
FIGURE 10–12 Formation of conjugates between CTLs and a target cell.
A, Electron micrograph of three CTLs from a cloned cell line specific for the human MHC molecule HLA-A2 binding to an HLA-A2–expressing target cell (TC) within 1 minute after the CTLs and targets are mixed. Note that in the CTL on the upper left, the granules have been redistributed toward the target cell.
(Courtesy of Dr. P. Peters, Netherlands Cancer Institute, Amsterdam.)
B, Electron micrograph of the point of membrane contact between a CTL (left) and target cell (right). Two CTL granules are near the synapse. Several mitochondria are also visible.
(Reprinted from Stinchcombe JC, G Bossi, S Booth, and GM Griffiths. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 8:751-761, 2001, © Cell Press, with permission from Elsevier).
C, Confocal fluorescence micrograph of an immune synapse between a CTL (left) and target cell (right) stained with antibodies against cathepsins in a secretory granule (blue), LFA-1 (green), and the cytoskeletal protein talin (red). The image demonstrates the central location of the secretory granule and the peripheral location of the adhesion molecule LFA-1 and associated cytoskeletal protein talin.
(Reprinted from Stinchcombe JC, and GM Griffiths. The role of the secretory immunological synapse in killing by CD8+ CTL. Seminars in Immunology 15:301-305,
In addition to the T cell receptor, CD8+ CTLs express receptors that are also expressed by NK cells, which contribute to both regulation and activation of CTLs. Some of these receptors belong to the killer immunoglobulin receptor (KIR) family, discussed in Chapter 4, and recognize class I MHC molecules on target cells but are not specific for a particular peptide-MHC complex. These KIRs transduce inhibitory signals that may serve to prevent CTLs from killing normal cells. In addition, CTLs express the NKG2D receptor, described in Chapter 4, that recognizes class I MHC–like molecules MIC-A, MIC-B, and ULBP, expressed on infected or neoplastic cells. NKG2D may serve to deliver signals that act together with TCR recognition of antigen to enhance killing activity.
Killing of Target Cells by CTLs
Within a few minutes of a CTL’s antigen receptor recognizing its antigen on a target cell, the target cell undergoes changes that induce it to die by apoptosis. Target cell death occurs during the next 2 to 6 hours and proceeds even if the CTL detaches. Thus, the CTL is said to deliver a lethal hit to the target cell. The principal mechanism of CTL-mediated target cell killing is the delivery of cytotoxic proteins stored within cytoplasmic granules (also called secretory lysosomes) to the target cell, thereby triggering apoptosis of the target cell (Fig. 10-13). As discussed earlier, CTL recognition of the target leads to activation of the CTL, one consequence of which is cytoskeleton reorganization such that the microtubule organizing center of the CTL moves to the area of the cytoplasm near the contact with the target cell. The cytoplasmic granules of the CTL are transported along microtubules and become concentrated in the region of the synapse, and the granule membrane fuses with the plasma membrane at the secretory domain. Membrane fusion results in exocytosis of the CTL’s granule contents into the confined space within the synaptic ring, between the plasma membranes of the CTL and target cell.
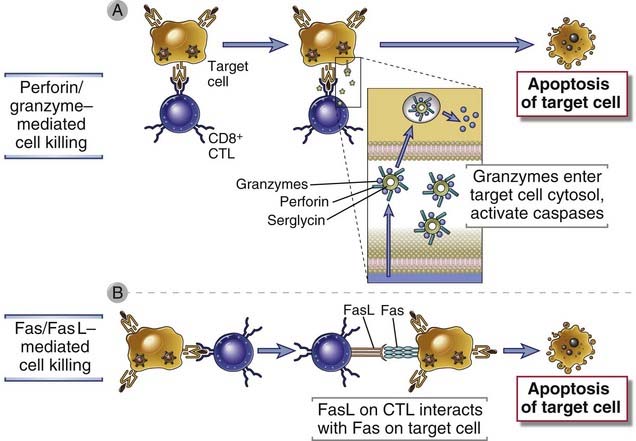
FIGURE 10–13 Mechanisms of CTL-mediated killing of target cells.
CTLs kill target cells by two main mechanisms. A, Complexes of perforin and granzymes are released from the CTL by granule exocytosis and enter target cells. The granzymes are delivered into the cytoplasm of the target cells by a perforin-dependent mechanism, and they induce apoptosis. B, FasL is expressed on activated CTLs, engages Fas on the surface of target cells, and induces apoptosis.
The cytotoxic proteins in the granules of CTLs (and NK cells) include granzymes and perforin. Granzymes A, B, and C are serine proteases that cleave proteins after aspartate residues. Perforin is a membrane-perturbing molecule homologous to the C9 complement protein. The granules also contain and a sulfated proteoglycan, serglycin, which serves to assemble a complex containing granzymes and perforin. The main function of perforin is to facilitate delivery of the granzymes into the cytosol of the target cell. How this is accomplished is still not well understood. Perforin may polymerize and form aqueous pores in the target cell membrane through which granzymes enter, but there is no proof that this is critical for CTL-mediated cell killing. According to another current model, complexes of granzyme B, perforin, and serglycin are discharged from the CTL onto the target cell and are internalized into endosomes by receptor-mediated endocytosis. Perforin may act on the endosomal membrane to facilitate the release of the granzymes into the target cell cytoplasm. Once in the cytoplasm, the granzymes cleave various substrates, including caspases, and initiate apoptotic death of the cell. For example, granzyme B activates caspase-3 as well as the Bcl-2 family member Bid, which triggers the mitochondrial pathway of apoptosis (see Fig. 14-7, Chapter 14). Another protein found in human CTL (and NK cell) granules, called granulysin, can alter the permeability of target cell and microbial membranes, but its importance in cell killing by CTLs is not established.
CTLs also use a granule-independent mechanism of killing that is mediated by interactions of membrane molecules on the CTLs and target cells. On activation, CTLs express a membrane protein, called Fas ligand (FasL), that binds to the death receptor Fas, which is expressed on many cell types. This interaction also results in activation of caspases and apoptosis of Fas-expressing targets (see Fig. 14-7, Chapter 14). Studies with knockout mice lacking perforin, granzyme B, or FasL indicate that granule proteins are the principal mediators of killing by CD8+ CTLs. Some CD4+ T cells are also capable of killing target cells (which, of course, must express class II MHC–associated peptides to be recognized by the CD4+ cells). CD4+ T cells are deficient in perforin and granzymes, and FasL may be more important for their killing activity.
After delivering the lethal hit, the CTL is released from its target cell, which usually occurs even before the target cell goes on to die. CTLs themselves are not injured during target cell killing, probably because the directed granule exocytosis process during CTL-mediated killing preferentially delivers granule contents into the target cell and away from the CTL. In addition, CTL granules contain a proteolytic enzyme called cathepsin B, which is delivered to the CTL surface on granule exocytosis, where it degrades errant perforin molecules that come into the vicinity of the CTL membrane.
Roles of CD8+ CTLs in Host Defense
In infections by intracellular microbes, the killing activity of CTLs is important for eradication of the reservoir of infection (see Fig. 10-1B). There are two types of situations in which cells cannot destroy microbes that infect them. First, some viruses live and replicate in cells that are incapable of destroying microbes (such as hepatitis viruses in liver cells). Second, even in phagocytes, some microbes escape from vesicles and live in the cytoplasm, where the microbicidal mechanisms of phagocytes are ineffective because these mechanisms are largely restricted to vesicles (to protect the cells from damage). Such infections can be eliminated only by destroying the infected cells, and in adaptive immune responses, CD8+ CTLs are the principal mechanism for killing infected cells. In addition, the caspases that are activated in target cells by granzymes and FasL cleave many substrates and activate enzymes that degrade DNA, but they do not distinguish between host and microbial proteins. Therefore, by activating nucleases in target cells, CTLs can initiate the destruction of microbial DNA as well as the target cell genome, thereby eliminating potentially infectious DNA. The massive expansion of CD8+ T cells that follows infections (see Fig. 9-12, Chapter 9) provides a large pool of CTLs to combat these infections. Defects in the development and activity of CTLs result in increased susceptibility to viral and some bacterial infections and reactivation of latent virus infections (such as infection by the Epstein-Barr virus), which are normally kept in check by virus-specific CTLs.
Destruction of infected cells by CTLs is a cause of tissue injury in some diseases. For instance, in infection by hepatitis B and C viruses, the infected liver cells are killed by the host CTL (and NK cell) response and not by the viruses. These viruses are not cytopathic, but the host senses and reacts against the infectious microbe and is not able to distinguish microbes that are intrinsically harmful or relatively harmless (see Chapter 18).