CHAPTER 12 Effector Mechanisms of Humoral Immunity
Humoral immunity is mediated by secreted antibodies, and its physiologic function is defense against extracellular microbes and microbial toxins. This type of immunity contrasts with cell-mediated immunity, the other effector arm of the adaptive immune system, which is mediated by T lymphocytes and functions to eradicate microbes that infect and live within host cells (see Chapter 10). Humoral immunity against microbial toxins was discovered by von Behring and Kitasato in 1890 as a form of immunity that could be conveyed from immunized to naive individuals by the transfer of serum. The types of microorganisms that are combated by humoral immunity are extracellular bacteria, fungi, and even obligate intracellular microbes such as viruses, which are targets of antibodies before they infect cells or when they are released from infected cells. Defects in antibody production result in increased susceptibility to infection with many microbes, including bacteria, fungi, and viruses. Currently used vaccines induce protection primarily by stimulating the production of antibodies. Apart from their crucial protective roles, in allergic individuals and in certain autoimmune diseases, some specific antibodies can be harmful and mediate tissue injury. In this chapter, we discuss the effector mechanisms that are used by antibodies to eliminate antigens. The structure of antibodies is described in Chapter 5 and the process of antibody production in Chapter 11.
Overview of Humoral Immunity
Before we discuss the principal mechanisms by which antibodies provide protection against microbes, we will summarize some of the salient features of antibody-mediated host defense.
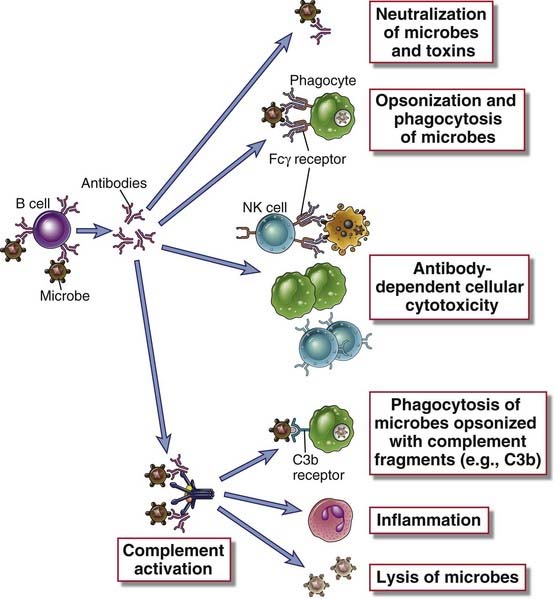
FIGURE 12–1 Effector functions of antibodies.
Antibodies against microbes (and their toxins, not shown here) neutralize these agents, opsonize them for phagocytosis, sensitize them for antibody-dependent cellular cytotoxicity, and activate the complement system. These various effector functions may be mediated by different antibody isotypes.
TABLE 12–1 Functions of Antibody Isotypes
| Antibody Isotype | Isotype-Specific Effector Functions |
|---|---|
| IgG | Opsonization of antigens for phagocytosis by macrophages and neutrophils |
| Activation of the classical pathway of complement | |
| Antibody-dependent cell-mediated cytotoxicity mediated by natural killer cells | |
| Neonatal immunity: transfer of maternal antibody across the placenta and gut | |
| Feedback inhibition of B cell activation | |
| IgM | Activation of the classical pathway of complement |
| Antigen receptor of naive B lymphocytes* | |
| IgA | Mucosal immunity: secretion of IgA into the lumens of the gastrointestinal and respiratory tracts |
| Activation of complement by the lectin pathway or by the alternative pathway | |
| IgE | Mast cell degranulation (immediate hypersensitivity reactions) |
| IgD | Antigen receptor of naive B lymphocytes* |
* These functions are mediated by membrane-bound and not secreted antibodies.
With this introduction to humoral immunity, we proceed to a discussion of the various functions of antibodies in host defense.
Neutralization of Microbes and Microbial Toxins
Antibodies against microbes and microbial toxins block the binding of these microbes and toxins to cellular receptors (Fig. 12-2). In this way, antibodies inhibit, or “neutralize,” the infectivity of microbes as well as the potential injurious effects of infection. Many microbes enter host cells by the binding of particular microbial surface molecules to membrane proteins or lipids on the surface of host cells. For example, influenza viruses use their envelope hemagglutinin to infect respiratory epithelial cells, and gram-negative bacteria use pili to attach to and infect a variety of host cells. Antibodies that bind to these microbial structures interfere with the ability of the microbes to interact with cellular receptors by means of steric hindrance and may thus prevent infection. In some cases, very few antibody molecules may bind to a microbe and induce conformational changes in surface molecules that prevent the microbe from interacting with cellular receptors; such interactions are examples of the allosteric effects of antibodies. Many microbial toxins mediate their pathologic effects also by binding to specific cellular receptors. For instance, tetanus toxin binds to receptors in the motor end plate of neuromuscular junctions and inhibits neuromuscular transmission, which leads to paralysis, and diphtheria toxin binds to cellular receptors and enters various cells, where it inhibits protein synthesis. Antibodies against such toxins sterically hinder the interactions of toxins with host cells and thus prevent the toxins from causing tissue injury and disease.
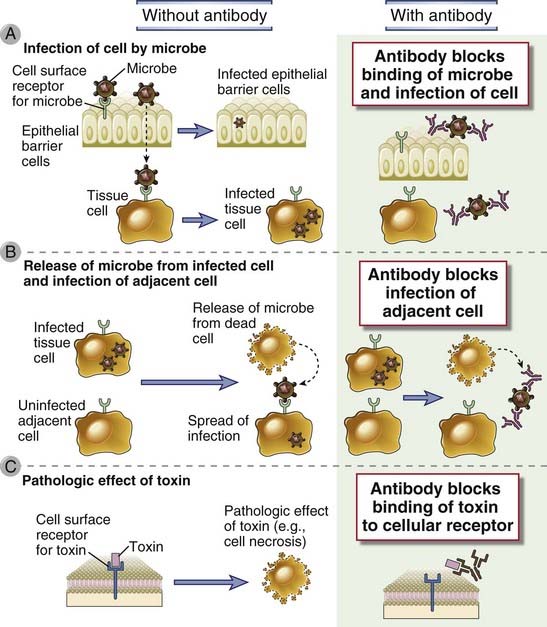
FIGURE 12–2 Neutralization of microbes and toxins by antibodies.
A, Antibodies prevent the binding of microbes to cells and thus block the ability of the microbes to infect host cells. B, Antibodies inhibit the spread of microbes from an infected cell to an adjacent uninfected cell. C, Antibodies block the binding of toxins to cells and thus inhibit the pathologic effects of the toxins.
Antibody-mediated neutralization of microbes and toxins requires only the antigen-binding regions of the antibodies. Therefore, such neutralization may be mediated by antibodies of any isotype in the circulation and in mucosal secretions and can experimentally also be mediated by Fab or F(ab′)2 fragments of specific antibodies, which lack the Fc regions of the heavy chains. Most neutralizing antibodies in the blood are of the IgG isotype; in mucosal organs, they are largely of the IgA isotype. The most effective neutralizing antibodies are those with high affinities for their antigens. High-affinity antibodies are produced by the process of affinity maturation (see Chapter 11). Many prophylactic vaccines work by stimulating the production of high-affinity neutralizing antibodies (Table 12-2). A mechanism that microbes have developed to evade host immunity is to mutate the genes encoding surface antigens that are the targets of neutralizing antibodies (see Chapter 15).
TABLE 12–2 Vaccine-Induced Humoral Immunity
| Infectious Disease | Vaccine | Mechanism of Protective Immunity |
|---|---|---|
| Polio | Oral attenuated poliovirus | Neutralization of virus by mucosal IgA antibody |
| Tetanus, diphtheria | Toxoids | Neutralization of toxin by systemic IgG antibody |
| Hepatitis, A or B | Recombinant viral envelope proteins | Neutralization of virus by systemic IgG antibody |
| Pneumococcal pneumonia, Haemophilus | Conjugate vaccines composed of bacterial capsular polysaccharide attached to a carrier protein | Opsonization and phagocytosis mediated by IgM and IgG antibodies, directly or secondary to complement activation |
Selected examples of vaccines that work by stimulating protective humoral immunity are listed.
Antibody-Mediated Opsonization and Phagocytosis
Antibodies of the IgG isotype coat (opsonize) microbes and promote their phagocytosis by binding to Fc receptors on phagocytes. Mononuclear phagocytes and neutrophils ingest microbes as a prelude to intracellular killing and degradation. These phagocytes express a variety of surface receptors that directly bind microbes and ingest them, even without antibodies, providing one mechanism of innate immunity (see Chapter 4). The efficiency of this process is markedly enhanced if the phagocyte can bind the particle with high affinity. Mononuclear phagocytes and neutrophils express receptors for the Fc portions of IgG antibodies that specifically bind antibody-coated (opsonized) particles. Microbes may also be opsonized by a product of complement activation called C3b and are phagocytosed by binding to a leukocyte receptor for C3b (described later in this chapter). The process of coating particles to promote phagocytosis is called opsonization, and substances that perform this function, including antibodies and complement proteins, are called opsonins.
Leukocyte Fc Receptors
Leukocytes express Fc receptors that bind to the constant regions of antibodies, and thereby promote the phagocytosis of Ig-coated particles and deliver signals that stimulate the microbicidal activities of the leukocytes and induce inflammation. Fc receptors for different Ig heavy chain isotypes are expressed on many leukocyte populations and serve diverse functions in immunity. Of these Fc receptors, the ones that are most important for phagocytosis of opsonized particles are receptors for the heavy chains of IgG antibodies, called Fcγ receptors, and these are the receptors that will primarily be considered in this chapter. The Fc receptors that bind to IgE are discussed in Chapter 19. We have already considered the neonatal Fc receptor (FcRn), which is expressed in the placenta, on intestinal epithelial cells, and on vascular endothelium, in Chapter 5. The poly-Ig receptor, which is involved in the transcytosis of IgA and IgM, is discussed in Chapter 13.
There are a number of different Fcγ receptors that have different affinities for the heavy chains of different IgG subclasses and are expressed on different cell types. Most Fc receptors result in cellular activation when triggered except for FcγRII, which is an inhibitory receptor. All Fcγ receptors contain a ligand-binding chain, called the α chain, that recognizes their IgG ligands. Differences in specificities or affinities of each FcγR for the various IgG isotypes are based on differences in the structure of these α chains. All Fc receptors are optimally activated by antibodies bound to their antigens and not by free, circulating antibodies. In all the FcRs except FcγRII, the α chain is associated with one or more additional polypeptide chains involved in signal transduction (Fig. 12-3). Signaling functions of FcγRII are mediated by the cytoplasmic tail of the α chain.
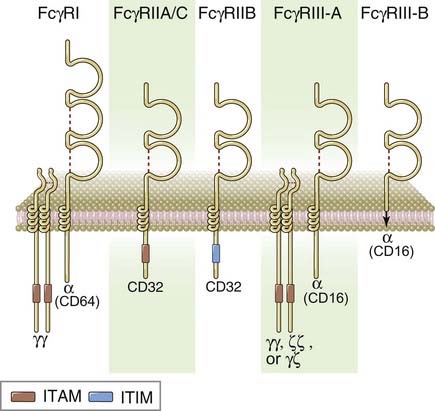
FIGURE 12–3 Subunit composition of Fcγ receptors.
Schematic models of the different human Fc receptors illustrate the Fc-binding α chains and the signaling subunits. FcγRIII-B is a glycophosphatidylinositol anchored membrane protein with no known signaling functions. FcγRIIA and IIC are structurally similar low-affinity activating receptors with slightly different patterns of expression.
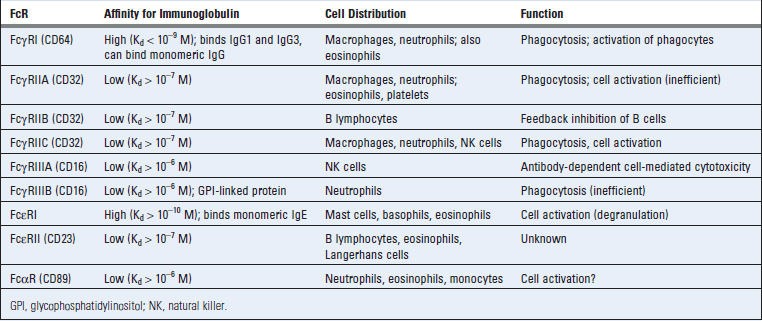
Fcγ receptors have been classified into three groups based on their affinities for heavy chains of different IgG subclasses (Table 12-3). Some of these Fc receptors have multiple isoforms that may differ in structure and function.
Transcription of the FcγRI gene and expression of FcγRI on macrophages is stimulated by interferon-γ (IFN-γ). The antibody isotypes that bind best to Fcγ receptors (such as IgG2a/2c in mice) are also produced in part as a result of IFN-γ–mediated isotype switching of B cells. In addition, IFN-γ directly stimulates the microbicidal activities of phagocytes (see Chapter 10).
Most of the FcRs serve to activate the cells on which they are expressed. FcεRI is described in Chapter 19, in the context of mast cell activation. The function of FcαR is not well established.
Role of Fcγ Receptors in Phagocytosis and Activation of Phagocytes
Phagocytosis of IgG-coated particles is mediated by binding of the Fc portions of opsonizing antibodies to Fcγ receptors on phagocytes. Therefore, the IgG subtypes that bind best to these receptors (IgG1 and IgG3) are the most efficient opsonins for promoting phagocytosis. As discussed before, FcγRI (CD64) is high-affinity Fcγ receptor on phagocytic cells, and it is the most important receptor for phagocytosis of opsonized particles.
Binding of Fc receptors on phagocytes to multivalent antibody-coated particles leads to engulfment of the particles and the activation of phagocytes (Fig. 12-4). The particles are internalized into vesicles known as phagosomes, which fuse with lysosomes, and the phagocytosed particles are destroyed in these phagolysosomes. Activation requires cross-linking of the FcRs by several adjacent Ig molecules (e.g., on antibody-coated microbes or in immune complexes). Cross-linking of the ligand-binding α chains of an FcR results in signal transduction events that are similar to those that occur after antigen receptor cross-linking in lymphocytes (see Chapter 7). These include Src kinase–mediated tyrosine phosphorylation of the ITAMs in the signaling chains of the FcRs; SH2 domain–mediated recruitment of Syk family kinases to the ITAMs; activation of phosphatidylinositol 3-kinase; recruitment of adaptor molecules, including SLP-76 and BLNK; and recruitment of enzymes such as phospholipase Cγ and Tec family kinases. These events lead to generation of inositol trisphosphate and diacylglycerol and sustained calcium mobilization. Responses to these mediators in leukocytes include transcription of genes encoding cytokines, inflammatory mediators, and microbicidal enzymes and mobilization of the cytoskeleton, leading to phagocytosis, granule exocytosis, and cell migration. One consequence of phagocyte activation is production of the enzyme phagocyte oxidase, which catalyzes the intracellular generation of reactive oxygen species that are cytotoxic for phagocytosed microbes. This process is called the respiratory burst. Another consequence of FcγRI activation is the activation of an enzyme called inducible nitric oxide synthase (iNOS), which triggers the production of nitric oxide that also contributes to the killing of pathogens. In addition, leukocytes that are activated by their Fc receptors secrete hydrolytic enzymes and reactive oxygen intermediates into the external milieu that are capable of killing extracellular microbes too large to be phagocytosed. The same toxic products may damage tissues; this mechanism of antibody-mediated tissue injury is important in hypersensitivity diseases (see Chapter 18). Knockout mice lacking the ligand-binding α chain of FcγRI or the signal-transducing FcR γ chain are defective in antibody-mediated defense against microbes and do not develop some forms of IgG antibody-mediated tissue injury, thus demonstrating the essential role of Fc receptors in these processes.
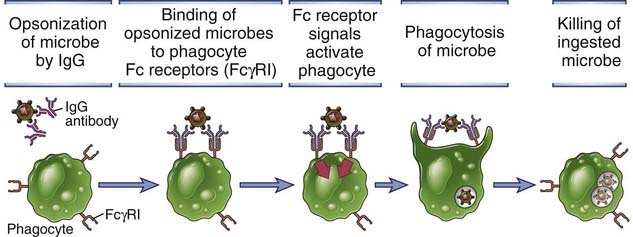
FIGURE 12–4 Antibody-mediated opsonization and phagocytosis of microbes.
Antibodies of certain IgG subclasses bind to microbes and are then recognized by Fc receptors on phagocytes. Signals from the Fc receptors promote the phagocytosis of the opsonized microbes and activate the phagocytes to destroy these microbes. The microbicidal mechanisms of phagocytes are described in Chapters 4 (see Fig. 4-13) and 10 (see Fig. 10-7).
Inhibitory Signaling by the FcγRIIB Receptor
The FcγRIIB receptor is an inhibitory Fc receptor that was described earlier in the context of inhibitory signaling in B cells and the phenomenon of antibody feedback (see Chapter 11). FcγRIIB is the only Fc receptor that has an ITIM motif in its cytoplasmic tail. When antibodies are produced during an immune response, these antibodies bind to remaining antigen, and the complex is simultaneously recognized by the antigen receptor and FcγRIIB on antigen-specific B cells. Immune complex–mediated cross-linking of the inhibitory FcγRIIB leads to tyrosine phosphorylation of the ITIM in the cytoplasmic tail, recruitment and activation of the SHIP inositol phosphatase, and subsequent inhibition of B cell receptor–mediated, ITAM-dependent activation pathways. FcγRIIB is also expressed on dendritic cells, neutrophils, macrophages, and mast cells and may play a role in regulating the responses of these cells to activating Fc receptors and other stimuli. One somewhat empirical but often useful treatment of many autoimmune diseases is the intravenous administration of pooled human IgG (IVIG). IVIG may engage FcγRIIB to deliver inhibitory signals to B lymphocytes and other cells, thus reducing antibody production and dampening inflammation. Another mechanism by which IVIG may ameliorate disease is by competing with circulating autoantibodies for the neonatal Fc receptor, which results in enhanced clearance of the antibodies (see Chapter 5).
Antibody-Dependent Cell-Mediated Cytotoxicity
Natural killer (NK) cells and other leukocytes bind to antibody-coated cells by Fc receptors and destroy these cells. This process is called antibody-dependent cellular cytotoxicity (ADCC) (Fig. 12-5). It was first described as a function of NK cells, which use their Fc receptor, FcγRIIIA, to bind to antibody-coated cells. FcγRIIIA (CD16) is a low-affinity receptor that binds clustered IgG molecules displayed on cell surfaces but does not bind circulating monomeric IgG. Therefore, ADCC occurs only when the target cell is coated with antibody molecules, and free IgG in plasma neither activates NK cells nor competes effectively with cell-bound IgG for binding to FcγRIII. Engagement of FcγRIII by antibody-coated target cells activates the NK cells to synthesize and secrete cytokines such as IFN-γ as well as to discharge the contents of their granules, which mediate the killing functions of this cell type (see Chapter 4). ADCC can be readily demonstrated in vitro, but its role in host defense against microbes is not definitively established. It is likely an important mechanism for the elimination of cells that are coated by specific therapeutic monoclonal antibodies, such as B cells and B cell–derived tumor cells that are targeted by anti-CD20 antibody.
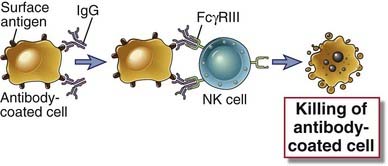
FIGURE 12–5 Antibody-dependent cell-mediated cytotoxicity.
Antibodies of certain IgG subclasses bind to cells (e.g., infected cells), and the Fc regions of the bound antibodies are recognized by an Fcγ receptor on NK cells. The NK cells are activated and kill the antibody-coated cells. Presumably, NK cells can lyse even class I MHC–expressing targets when these target cells are opsonized because the Fc receptor–mediated stimulation may overcome the inhibitory actions of class I MHC–recognizing NK cell inhibitory receptors (see Chapter 12).
Antibody-Mediated Clearance of Helminths
Antibodies, mast cells, and eosinophils function with antibodies to mediate the expulsion and killing of some helminthic parasites. Helminths (worms) are too large to be engulfed by phagocytes, and their integuments are relatively resistant to the microbicidal products of neutrophils and macrophages. They can, however, be killed by a toxic cationic protein, known as the major basic protein, present in the granules of eosinophils. IgE, IgG, and IgA antibodies that coat helminths can bind to Fc receptors on eosinophils and cause the degranulation of these cells, releasing the basic protein and other eosinophil granule contents that kill the parasites. The high-affinity Fcε receptor of eosinophils (FcεRI) lacks the signaling β chain and can only signal relatively weakly through the associated γ chain. In addition, IgE antibodies that recognize antigens on the surface of the helminths may initiate local mast cell degranulation through the high-affinity IgE receptor (see Chapter 19). Mast cell mediators may induce bronchoconstriction and increased local motility, contributing to the expulsion of worms from sites such as the airways and the lumen of the gastrointestinal tract. Chemokines and cytokines released by activated mast cells may attract eosinophils and cause their degranulation as well.
The Complement System
The complement system is one of the major effector mechanisms of humoral immunity and is also an important effector mechanism of innate immunity. We briefly discussed the role of complement in innate immunity in Chapter 4. Here we describe the activation and regulation of complement in more detail.
The name “complement” is derived from experiments performed by Jules Bordet shortly after the discovery of antibodies. He demonstrated that if fresh serum containing an antibacterial antibody is added to the bacteria at physiologic temperature (37°C), the bacteria are lysed. If, however, the serum is heated to 56°C or more, it loses its lytic capacity. This loss of lytic capacity is not due to decay of antibody activity because antibodies are relatively heat stable, and even heated serum is capable of agglutinating the bacteria. Bordet concluded that the serum must contain another heat-labile component that assists, or complements, the lytic function of antibodies, and this component was later given the name complement.
The complement system consists of serum and cell surface proteins that interact with one another and with other molecules of the immune system in a highly regulated manner to generate products that function to eliminate microbes. Complement proteins are plasma proteins that are normally inactive; they are activated only under particular conditions to generate products that mediate various effector functions of complement. Several features of complement activation are essential for its normal function.
Pathways of Complement Activation
There are three major pathways of complement activation: the classical pathway, which is activated by certain isotypes of antibodies bound to antigens; the alternative pathway, which is activated on microbial cell surfaces in the absence of antibody; and the lectin pathway, which is activated by a plasma lectin that binds to mannose residues on microbes (Fig. 12-6). The names classical and alternative arose because the classical pathway was discovered and characterized first, but the alternative pathway is phylogenetically older. Although the pathways of complement activation differ in how they are initiated, all of them result in the generation of enzyme complexes that are able to cleave the most abundant complement protein, C3. The alternative and lectin pathways are effector mechanisms of innate immunity, whereas the classical pathway is a major mechanism of adaptive humoral immunity.
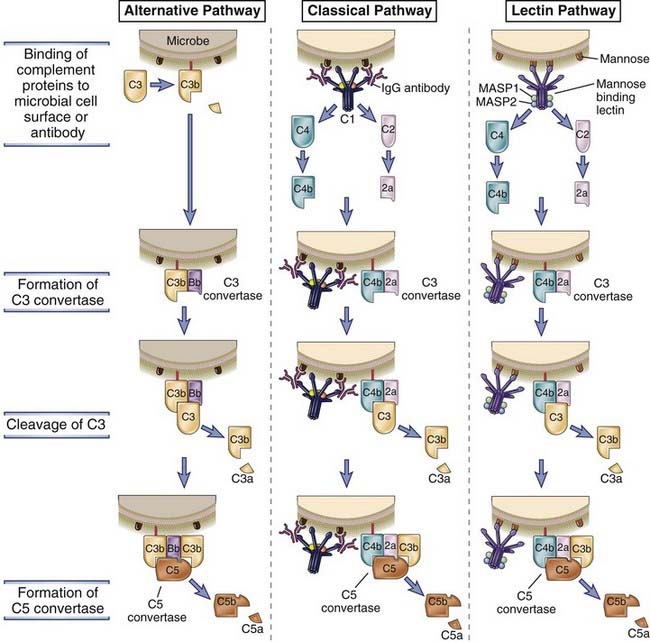
FIGURE 12–6 The early steps of complement activation by the alternative, classical, and lectin pathways.
The alternative pathway is activated by C3b binding to various activating surfaces, such as microbial cell walls; the classical pathway is initiated by C1 binding to antigen-antibody complexes; and the lectin pathway is activated by binding of a plasma lectin to microbes. The C3b that is generated by the action of the C3 convertase binds to the microbial cell surface or the antibody and becomes a component of the enzyme that cleaves C5 (C5 convertase) and initiates the late steps of complement activation. The late steps of all three pathways are the same (not shown here), and complement activated by all three pathways serves the same functions.
The central event in complement activation is proteolysis of the complement protein C3 to generate biologically active products and the subsequent covalent attachment of a product of C3, called C3b, to microbial cell surfaces or to antibody bound to antigen (see Fig. 12-6). Complement activation depends on the generation of two proteolytic complexes: the C3 convertase, which cleaves C3 into two proteolytic fragments called C3a and C3b; and the C5 convertase, which cleaves C5 into C5a and C5b. By convention, the proteolytic products of each complement protein are identified by lowercase letter suffixes, a referring to the smaller product and b to the larger one. C3b becomes covalently attached to the microbial cell surface or to the antibody molecules at the site of complement activation. All the biologic functions of complement are dependent on the proteolytic cleavage of C3. For example, complement activation promotes phagocytosis because phagocytes (neutrophils and macrophages) express receptors for C3b. Peptides produced by proteolysis of C3 (and other complement proteins) stimulate inflammation. The C5 convertase assembles after the prior generation of C3b, and this convertase contributes both to inflammation (by the generation of the C5a fragment) and to the formation of pores in the membranes of microbial targets. The pathways of complement activation differ in how C3b is produced but follow a common sequence of reactions after the cleavage of C5.
With this background, we proceed to more detailed descriptions of the alternative, classical, and lectin pathways.
The Alternative Pathway
The alternative pathway of complement activation results in the proteolysis of C3 and the stable attachment of its breakdown product C3b to microbial surfaces, without a role for antibody (Fig. 12-7 and Table 12-4). The C3 protein contains a reactive thioester bond that is buried in a region of the protein known as the thioester domain. When C3 is cleaved, the C3b molecule undergoes a dramatic conformational change and the thioester domain flips out (a massive shift of about 85 Å), exposing the previously hidden reactive thioester bond. Normally, C3 in plasma is being continuously cleaved at a low rate to generate C3b in a process that is called C3 tickover. A small amount of the C3b may become covalently attached to the surfaces of cells, including microbes, through the thioester domain, which reacts with the amino or hydroxyl groups of cell surface proteins or polysaccharides to form amide or ester bonds (Fig. 12-8). If these bonds are not formed, the C3b remains in the fluid phase, and the exposed and reactive thioester bond is quickly hydrolyzed, rendering the protein inactive. As a result, further complement activation cannot proceed.
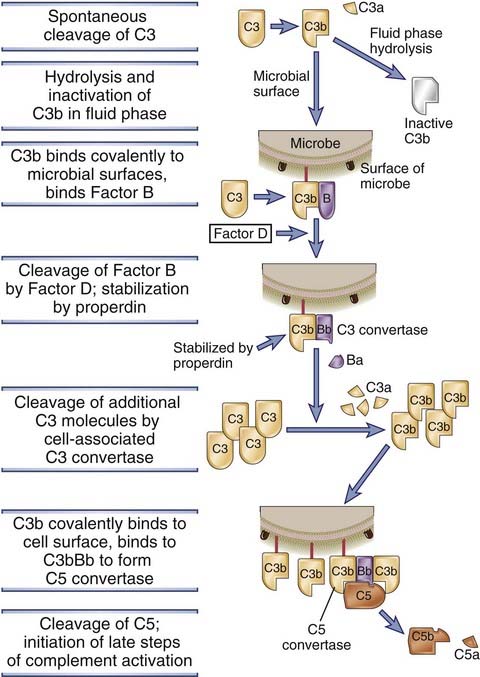
FIGURE 12–7 The alternative pathway of complement activation.
Soluble C3 in plasma undergoes slow spontaneous hydrolysis of its internal thioester bond, which leads to the formation of a fluid-phase C3 convertase (not shown) and the generation of C3b. If the C3b is deposited on the surfaces of microbes, it binds factor B and forms the alternative pathway C3 convertase. This convertase cleaves C3 to produce more C3b, which binds to the microbial surface and participates in the formation of a C5 convertase. The C5 convertase cleaves C5 to generate C5b, the initiating event in the late steps of complement activation.
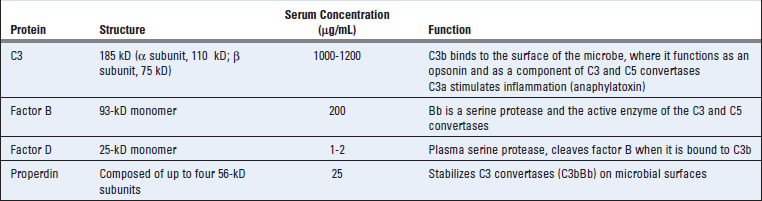
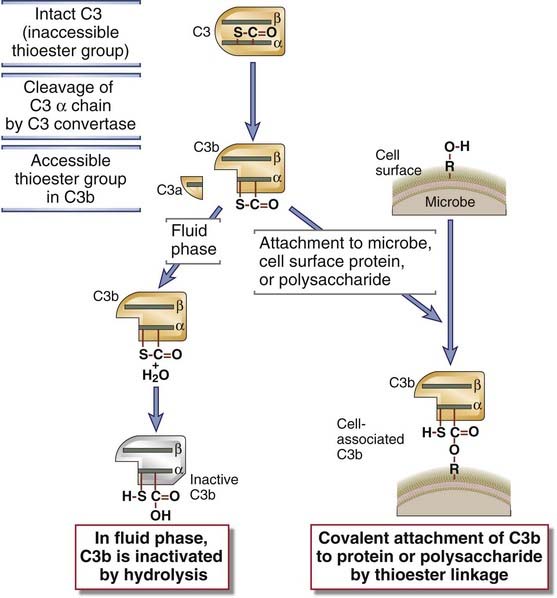
FIGURE 12–8 Internal thioester bonds of C3 molecules.
A schematic view is shown of the internal thioester groups in C3 and their role in forming covalent bonds with other molecules. Proteolytic cleavage of the α chain of C3 converts it into a metastable form in which the internal thioester bonds are exposed and susceptible to nucleophilic attack by oxygen (as shown) or nitrogen atoms. The result is the formation of covalent bonds with proteins or carbohydrates on the cell surfaces. C4 is structurally homologous to C3 and has an identical thioester group.
When C3b undergoes its post-cleavage conformational change, a binding site for a plasma protein called factor B is also exposed. Factor B then binds to the C3b protein that is now covalently tethered to the surface of a microbial or host cell. Bound factor B is in turn cleaved by a plasma serine protease called factor D, releasing a small fragment called Ba and generating a larger fragment called Bb that remains attached to C3b. The C3bBb complex is the alternative pathway C3 convertase, and it functions to cleave more C3 molecules, thus setting up an amplification sequence. Even when C3b is generated by the classical or lectin pathways, it can form a complex with Bb, and this complex is able to cleave more C3. Thus, the alternative pathway C3 convertase functions to amplify complement activation when it is initiated by the alternative, classical, or lectin pathways. When C3 is broken down, C3b remains attached to cells and C3a is released. This soluble fragment has several biologic activities that are discussed later.
Alternative pathway activation readily occurs on microbial cell surfaces and not on mammalian cells. If the C3bBb complex is formed on mammalian cells, it is rapidly degraded and the reaction is terminated by the action of several regulatory proteins present on these cells (discussed later). Lack of the regulatory proteins on microbial cells allows binding and activation of the alternative pathway C3 convertase. In addition, another protein of the alternative pathway, called properdin, can bind to and stabilize the C3bBb complex, and the attachment of properdin is favored on microbial as opposed to normal host cells. Properdin is the only known positive regulator of complement.
Some of the C3b molecules generated by the alternative pathway C3 convertase bind to the convertase itself. This results in the formation of a complex containing one Bb moiety and two molecules of C3b, which functions as the alternative pathway C5 convertase, which will cleave C5 and initiate the late steps of complement activation.
The Classical Pathway
The classical pathway is initiated by binding of the complement protein C1 to the CH2 domains of IgG or the CH3 domains of IgM molecules that have bound antigen (Fig. 12-9 and Table 12-5). Among IgG antibodies, IgG3 and IgG1 (in humans) are more efficient activators of complement than are other subclasses. C1 is a large, multimeric protein complex composed of C1q, C1r, and C1s subunits; C1q binds to the antibody, and C1r and C1s are proteases. The C1q subunit is made up of an umbrella-like radial array of six chains, each of which has a globular head connected by a collagen-like arm to a central stalk. This hexamer performs the recognition function of the molecule and binds specifically to the Fc regions of µ and some γ heavy chains (Fig. 12-10). Each Ig Fc region has a single C1q-binding site, and each C1q molecule must bind to at least two Ig heavy chains to be activated. This requirement explains why antibodies bound to antigens, and not free circulating antibodies, can initiate classical pathway activation (Fig. 12-11). Because each IgG molecule has only one Fc region, multiple IgG molecules must be brought close together before C1q can bind, and multiple IgG antibodies are brought together only when they bind to a multivalent antigen. Even though free (circulating) IgM is pentameric, it does not bind C1q because the Fc regions of free IgM are in a planar configuration that is inaccessible to C1q. Binding of the IgM to an antigen induces a conformational change into a “staple” form that exposes the C1q binding sites in the Fc regions and allows C1q to bind. Because of its pentameric structure, a single molecule of IgM can bind two C1q molecules, and this is one reason that IgM is a more efficient complement-binding (also called complement-fixing) antibody than IgG is.
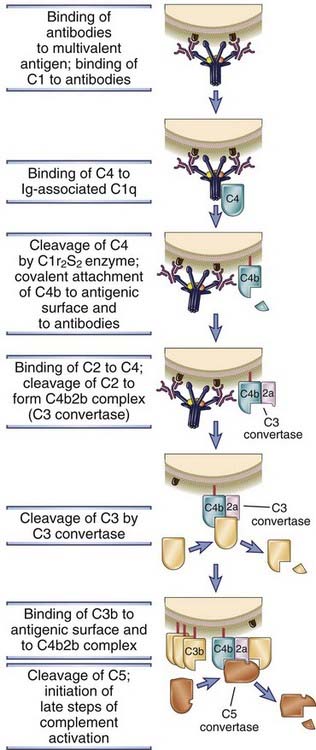
FIGURE 12–9 The classical pathway of complement activation.
Antigen-antibody complexes that activate the classical pathway may be soluble, fixed on the surface of cells (as shown), or deposited on extracellular matrices. The classical pathway is initiated by the binding of C1 to antigen-complexed antibody molecules, which leads to the production of C3 and C5 convertases attached to the surfaces where the antibody was deposited. The C5 convertase cleaves C5 to begin the late steps of complement activation.
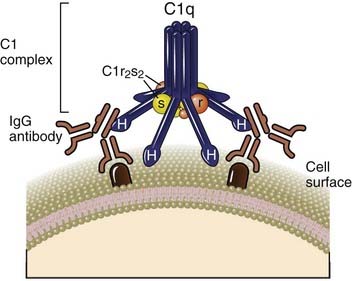
C1q consists of six identical subunits arranged to form a central core and symmetrically projecting radial arms. The globular heads at the end of each arm, designated H, are the contact regions for immunoglobulin. C1r and C1s form a tetramer composed of two C1r and two C1s molecules. The ends of C1r and C1s contain the catalytic domains of these proteins. One C1r2s2 tetramer wraps around the radial arms of the C1q complex in a manner that juxtaposes the catalytic domains of C1r and C1s.
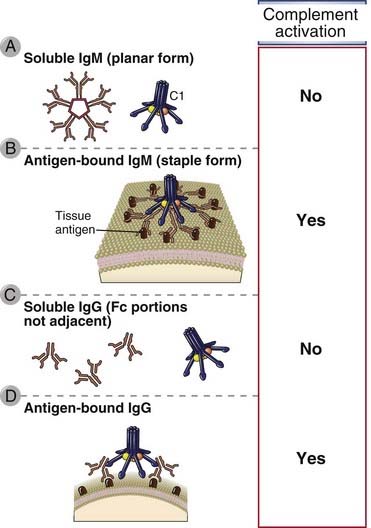
FIGURE 12–11 C1 binding to the Fc portions of IgM and IgG.
C1 must bind to two or more Fc portions to initiate the complement cascade. The Fc portions of soluble pentameric IgM are not accessible to C1 (A). After IgM binds to surface-bound antigens, it undergoes a shape change that permits C1 binding and activation (B). Soluble IgG molecules will also not activate C1 because each IgG has only one Fc region (C), but after binding to cell surface antigens, adjacent IgG Fc portions can bind and activate C1 (D).
C1r and C1s are serine proteases that form a tetramer containing two molecules of each protein. Binding of two or more of the globular heads of C1q to the Fc regions of IgG or IgM leads to enzymatic activation of the associated C1r, which cleaves and activates C1s (see Fig. 12-9). Activated C1s cleaves the next protein in the cascade, C4, to generate C4b. (The smaller C4a fragment is released and has biologic activities that are described later.) C4 is homologous to C3, and C4b contains an internal thioester bond, similar to that in C3b, that forms covalent amide or ester linkages with the antigen-antibody complex or with the adjacent surface of a cell to which the antibody is bound. This attachment of C4b ensures that classical pathway activation proceeds on a cell surface or immune complex. The next complement protein, C2, then complexes with the cell surface–bound C4b and is cleaved by a nearby C1s molecule to generate a soluble C2b fragment of unknown importance and a larger C2a fragment that remains physically associated with C4b on the cell surface. (Note that the nomenclature of C2 fragments that is now accepted is different from the other complement proteins because the attached fragment is called the a piece and the released part is the b fragment. This is because, for C2, the attached fragment is larger.) The resulting C4b2a complex is the classical pathway C3 convertase; it has the ability to bind to and proteolytically cleave C3. Binding of this enzyme complex to C3 is mediated by the C4b component, and proteolysis is catalyzed by the C2a component. Cleavage of C3 results in the removal of the small C3a fragment, and C3b can form covalent bonds with cell surfaces or with the antibody where complement activation was initiated. Once C3b is deposited, it can bind factor B and generate more C3 convertase by the alternative pathway, as discussed earlier. The net effect of the multiple enzymatic steps and amplification is that a single molecule of C3 convertase can lead to the deposition of hundreds or thousands of molecules of C3b on the cell surface where complement is activated. The key early steps of the alternative and classical pathways are analogous: C3 in the alternative pathway is homologous to C4 in the classical pathway, and factor B is homologous to C2.
Some of the C3b molecules generated by the classical pathway C3 convertase bind to the convertase (as in the alternative pathway) and form a C4b2a3b complex. This complex functions as the classical pathway C5 convertase; it cleaves C5 and initiates the late steps of complement activation.
An unusual antibody-independent variant form of the classical pathway, which is activated by carbohydrates binding to a cell surface lectin, occurs in pneumococcal infections. Splenic marginal zone macrophages express a cell surface C-type lectin called SIGN-R1 that can recognize the pneumococcal polysaccharide and can also bind C1q. Multivalent binding of whole bacteria or the polysaccharide to SIGN-R1 activates the classical pathway and permits the eventual coating of the pneumococcus with C3b. This is an example of a cell surface lectin that mediates activation of the classical pathway but without a requirement for antibody.
The Lectin Pathway
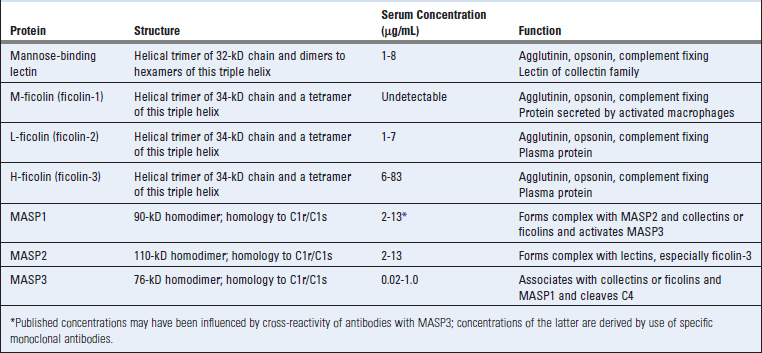
The lectin pathway of complement activation is triggered in the absence of antibody by the binding of microbial polysaccharides to circulating lectins, such as plasma mannose (or mannan)–binding lectin (MBL), or to ficolins (Table 12-6). These soluble lectins are collagen-like proteins that structurally resemble C1q (see Fig. 4-10, Chapter 4). MBL, L-ficolin, and H-ficolin are plasma proteins; M-ficolin is mainly secreted by activated macrophages in tissues. MBL is a member of the collectin family and has an N-terminal collagen-like domain and a C-terminal carbohydrate recognition domain. The ficolins have a similar structure with an N-terminal collagen-like domain and a C-terminal fibrinogen-like domain. The collagen-like domains help assemble basic triple-helical structures that can form higher order oligomers. MBL binds to mannose residues on polysaccharides; the ficolin fibrinogen-like domain binds N-acetylglucosamine–containing glycans. MBL and ficolins associate with MBL-associated serine proteases (MASPs) including MASP1, MASP2, and MASP3 (see Table 12-6). The MASP proteins are structurally homologous to the C1r and C1s proteases and serve a similar function, namely, the cleavage of C4 and C2 to activate the complement pathway. Higher order oligomers of MBL associate with MASP1 and MASP2, although MASP3/MASP2 complexes may also be found. MASP1 (or MASP3) can form a tetrameric complex with MASP2 similar to the one formed by C1r and C1s, and MASP2 is the protease that cleaves C4 and C2. Subsequent events in this pathway are identical to those that occur in the classical pathway.
Late Steps of Complement Activation
C5 convertases generated by the alternative, classical, or lectin pathway initiate activation of the late components of the complement system, which culminates in formation of the cytocidal membrane attack complex (MAC) (Table 12-7 and Fig. 12-12). C5 convertases cleave C5 into a small C5a fragment that is released and a two-chain C5b fragment that remains bound to the complement proteins deposited on the cell surface. C5a has potent biologic effects on several cells that are discussed later in this chapter. The remaining components of the complement cascade, C6, C7, C8, and C9, are structurally related proteins without enzymatic activity. C5b transiently maintains a conformation capable of binding the next proteins in the cascade, C6 and C7. The C7 component of the resulting C5b,6,7 complex is hydrophobic, and it inserts into the lipid bilayer of cell membranes, where it becomes a high-affinity receptor for the C8 molecule. The C8 protein is a trimer composed of three distinct chains, one of which binds to the C5b,6,7 complex and forms a covalent heterodimer with the second chain; the third chain inserts into the lipid bilayer of the membrane. This stably inserted C5b,6,7,8 complex (C5b-8) has a limited ability to lyse cells. The formation of a fully active MAC is accomplished by the binding of C9, the final component of the complement cascades, to the C5b-8 complex. C9 is a serum protein that polymerizes at the site of the bound C5b-8 to form pores in plasma membranes. These pores are about 100 Å in diameter, and they form channels that allow free movement of water and ions. The entry of water results in osmotic swelling and rupture of the cells on whose surface the MAC is deposited. The pores formed by polymerized C9 are similar to the membrane pores formed by perforin, the cytolytic granule protein found in cytotoxic T lymphocytes and NK cells (see Chapter 10), and C9 is structurally homologous to perforin.
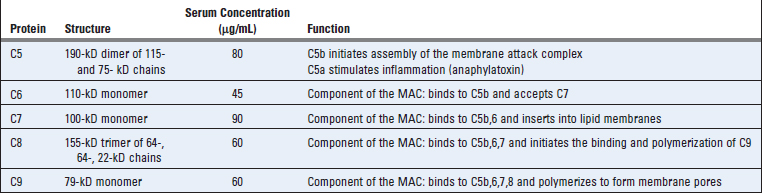
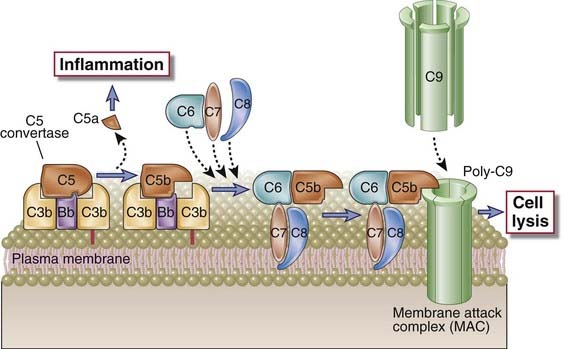
FIGURE 12–12 Late steps of complement activation and formation of the MAC.
A schematic view of the cell surface events leading to formation of the MAC is shown. Cell-associated C5 convertase cleaves C5 and generates C5b, which becomes bound to the convertase. C6 and C7 bind sequentially, and the C5b,6,7 complex becomes directly inserted into the lipid bilayer of the plasma membrane, followed by stable insertion of C8. Up to 15 C9 molecules may then polymerize around the complex to form the MAC, which creates pores in the membrane and induces cell lysis. C5a released on proteolysis of C5 stimulates inflammation.
Receptors for Complement Proteins
Many of the biologic activities of the complement system are mediated by the binding of complement fragments to membrane receptors expressed on various cell types. The best characterized of these receptors are specific for fragments of C3 and are described here (Table 12-8). Other receptors include those for C3a, C4a, and C5a, which stimulate inflammation, and some that regulate complement activation.
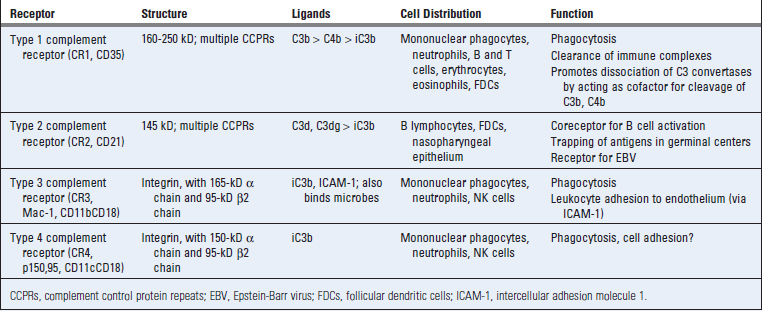
In humans, CR2 is the cell surface receptor for Epstein-Barr virus, a herpesvirus that causes infectious mononucleosis and is also linked to several malignant tumors. Epstein-Barr virus infects B cells and can remain latent in these cells for life.
Regulation of Complement Activation
Activation of the complement cascade and the stability of active complement proteins are tightly regulated to prevent complement activation on normal host cells and to limit the duration of complement activation even on microbial cells and antigen-antibody complexes. Regulation of complement is mediated by several circulating and cell membrane proteins (Table 12-9). Many of these proteins as well as several proteins of the classical and alternative pathways belong to a family called regulators of complement activity (RCA) and are encoded by homologous genes that are located adjacent to one another in the genome.
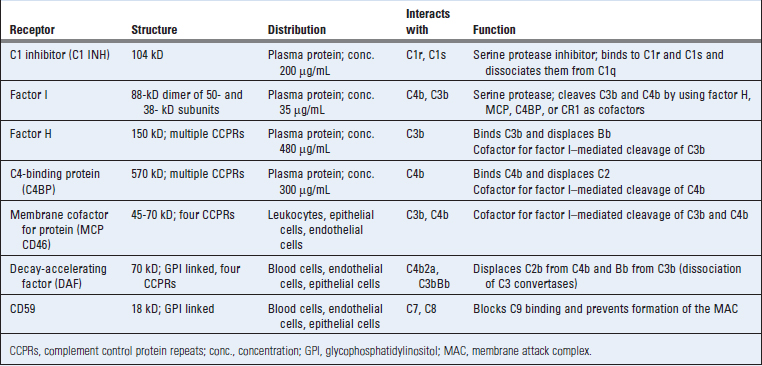
Complement activation needs to be regulated for two reasons. First, low-level complement activation goes on spontaneously, and if such activation is allowed to proceed, the result can be damage to normal cells and tissues. Second, even when complement is activated where needed, such as on microbial cells or antigen-antibody complexes, it needs to be controlled because degradation products of complement proteins can diffuse to adjacent cells and injure them. Different regulatory mechanisms inhibit the formation of C3 convertases in the early steps of complement activation, break down and inactivate C3 and C5 convertases, and inhibit formation of the MAC in the late steps of the complement pathway.
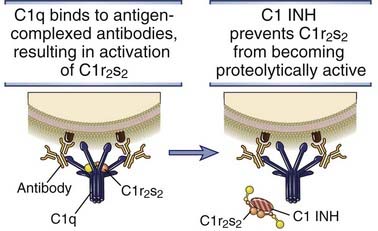
FIGURE 12–13 Regulation of C1 activity by C1 INH.
C1 INH displaces C1r2s2 from C1q and terminates classical pathway activation.
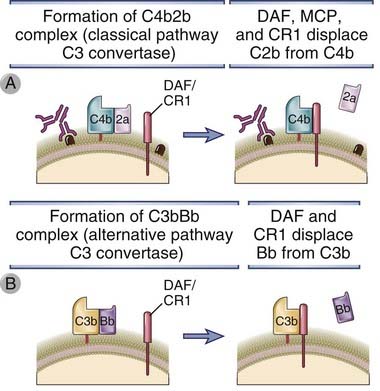
FIGURE 12–14 Inhibition of the formation of C3 convertases.
Several membrane proteins present on normal cells displace either C2a from the classical pathway C3 convertase (A) or Bb from the alternative pathway C3 convertase (B) and stop complement activation.
DAF is a glycophosphatidylinositol-linked membrane protein expressed on endothelial cells and erythrocytes. Genetic deficiency of an enzyme required to form such protein-lipid linkages results in failure to express many glycophosphatidylinositol-linked membrane proteins, including DAF and CD59 (see following), and causes a disease called paroxysmal nocturnal hemoglobinuria. This disease is characterized by recurrent bouts of intravascular hemolysis, at least partly attributable to unregulated complement activation on the surface of erythrocytes. Recurrent intravascular hemolysis in turn leads to chronic hemolytic anemia and venous thrombosis. An unusual feature of this disease is that the mutation in the defective gene is not inherited but represents an acquired mutation in hematopoietic stem cells.
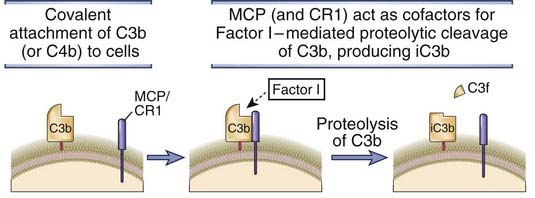
FIGURE 12–15 Factor I–mediated cleavage of C3b.
In the presence of cell membrane–bound cofactors (MCP or CR1), plasma factor I proteolytically cleaves C3b attached to cell surfaces, leaving an inactive form of C3b (iC3b). Factor H and C4-binding protein can also serve as cofactors for factor I–mediated cleavage of C3b. The same process is involved in the proteolysis of C4.
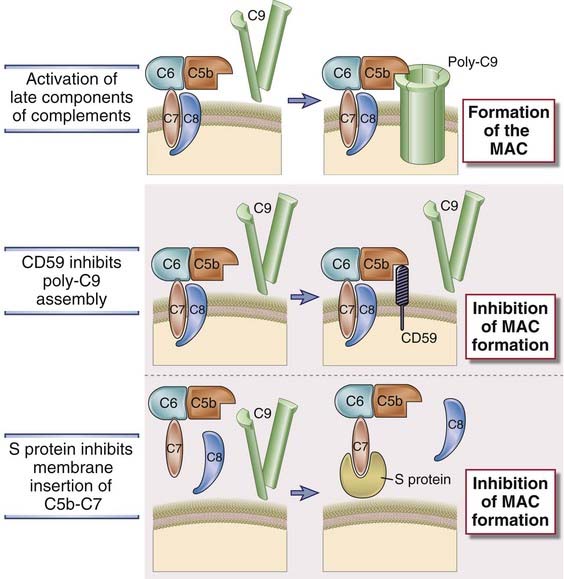
FIGURE 12–16 Regulation of formation of the MAC.
The MAC is formed on cell surfaces as an end result of complement activation. The membrane protein CD59 and S protein in the plasma inhibit formation of the MAC.
Much of the analysis of the function of complement regulatory proteins has relied on in vitro experiments, and most of these experiments have focused on assays that measure MAC-mediated cell lysis as an endpoint. On the basis of these studies, a hierarchy of importance for inhibiting complement activation is believed to be CD59 > DAF > MCP; this hierarchy may reflect the relative abundance of these proteins on cell surfaces.
The function of regulatory proteins may be overwhelmed by excessive activation of complement pathways. We have emphasized the importance of these regulatory proteins in preventing complement activation on normal cells. However, complement-mediated phagocytosis and damage to normal cells are important pathogenic mechanisms in many immunologic diseases (see Chapter 18). In these diseases, large amounts of antibodies may be deposited on host cells, generating enough active complement proteins that the regulatory molecules are unable to control complement activation.
Functions of Complement
The principal effector functions of the complement system in innate immunity and specific humoral immunity are to promote phagocytosis of microbes on which complement is activated, to stimulate inflammation, and to induce the lysis of these microbes. In addition, products of complement activation facilitate the activation of B lymphocytes and the production of antibodies. Phagocytosis, inflammation, and stimulation of humoral immunity are all mediated by the binding of proteolytic fragments of complement proteins to various cell surface receptors, whereas cell lysis is mediated by the MAC. In the following section, we describe each of these functions of the complement system and their role in host defense.
Opsonization and Phagocytosis
Microbes on which complement is activated by the alternative or classical pathway become coated with C3b, iC3b, or C4b and are phagocytosed by the binding of these proteins to specific receptors on macrophages and neutrophils (Fig. 12-17A). As discussed previously, activation of complement leads to the generation of C3b and iC3b covalently bound to cell surfaces. Both C3b and iC3b act as opsonins by virtue of the fact that they specifically bind to receptors on neutrophils and macrophages. C3b and C4b (the latter generated by the classical pathway only) bind to CR1, and iC3b binds to CR3 (Mac-1) and CR4. By itself, CR1 is inefficient at inducing the phagocytosis of C3b-coated microbes, but its ability to do so is enhanced if the microbes are coated with IgG antibodies that simultaneously bind to Fcγ receptors. Macrophage activation by the cytokine IFN-γ also enhances CR1-mediated phagocytosis. C3b- and iC3b-dependent phagocytosis of microorganisms is a major defense mechanism against infections in innate and adaptive immunity. One example of the importance of complement is host defense against bacteria with polysaccharide-rich capsules, such as pneumococci and meningococci, which is mediated primarily by humoral immunity. IgM antibodies against capsular polysaccharides bind to the bacteria, activate the classical pathway of complement, and cause phagocytic clearance of the bacteria in the spleen. In addition, SIGN-R1–expressing marginal zone macrophages may also bind to capsular polysaccharides and activate the classical pathway in the absence of antibody. This is why individuals lacking the spleen (e.g., as a result of surgical removal after traumatic rupture or in patients with autoimmune hemolytic anemia or thrombocytopenia) are susceptible to disseminated pneumococcal and meningococcal septicemia. C3-deficient humans and mice are extremely susceptible to lethal bacterial infections.
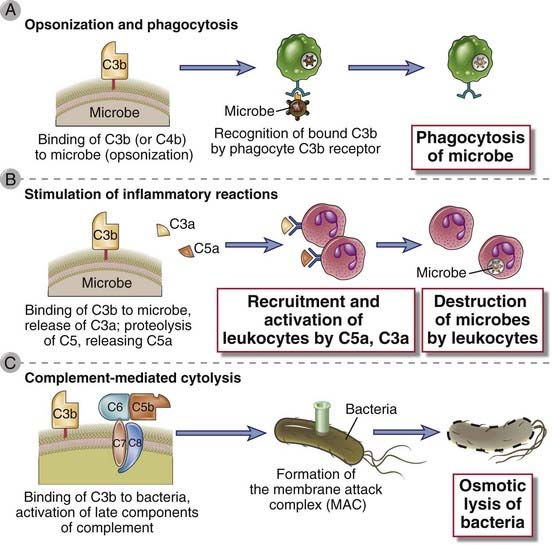
FIGURE 12–17 Functions of complement.
The major functions of the complement system in host defense are shown. Cell-bound C3b is an opsonin that promotes phagocytosis of coated cells (A); the proteolytic products C5a, C3a, and (to a lesser extent) C4a stimulate leukocyte recruitment and inflammation (B); and the MAC lyses cells (C).
Stimulation of Inflammatory Responses
The proteolytic complement fragments C5a, C4a, and C3a induce acute inflammation by activating mast cells and neutrophils (Fig. 12-17B). All three peptides bind to mast cells and induce degranulation, with the release of vasoactive mediators such as histamine. These peptides are also called anaphylatoxins because the mast cell reactions they trigger are characteristic of anaphylaxis (see Chapter 19). In neutrophils, C5a stimulates motility, firm adhesion to endothelial cells, and, at high doses, stimulation of the respiratory burst and production of reactive oxygen species. In addition, C5a may act directly on vascular endothelial cells and induce increased vascular permeability and the expression of P-selectin, which promotes neutrophil binding. This combination of C5a actions on mast cells, neutrophils, and endothelial cells contributes to inflammation at sites of complement activation. C5a is the most potent mediator of mast cell degranulation, C3a is about 20-fold less potent, and C4a is about 2500-fold less. The proinflammatory effects of C5a, C4a, and C3a are mediated by binding of the peptides to specific receptors on various cell types. The C5a receptor is the most thoroughly characterized. It is a member of the seven–α-helical transmembrane G protein–coupled receptor family. The C5a receptor is expressed on many cell types, including neutrophils, eosinophils, basophils, monocytes, macrophages, mast cells, endothelial cells, smooth muscle cells, epithelial cells, and astrocytes. The C3a receptor is also a member of the G protein–coupled receptor family.
Complement-Mediated Cytolysis
Complement-mediated lysis of foreign organisms is mediated by the MAC (Fig. 12-17C). Most pathogens have evolved thick cell walls or capsules that impede access of the MAC to their cell membranes. Complement-mediated lysis appears to be critical for defense against only a few pathogens that are unable to resist MAC insertion, such as infections by bacteria of the genus Neisseria that have very thin cell walls.
Other Functions of the Complement System
By binding to antigen-antibody complexes, complement proteins promote the solubilization of these complexes and their clearance by phagocytes. Small numbers of immune complexes are frequently formed in the circulation when an individual mounts a vigorous antibody response to a circulating antigen. If the immune complexes accumulate in the blood, they may be deposited in vessel walls and lead to inflammatory reactions that damage the vessels and surrounding tissue. The formation of immune complexes may require not only the multivalent binding of Ig Fab regions to antigens but also noncovalent interactions of Fc regions of juxtaposed Ig molecules. Complement activation on Ig molecules can sterically block these Fc-Fc interactions, thereby promoting dissolution of the immune complexes. In addition, as discussed earlier, immune complexes with attached C3b are bound to CR1 on erythrocytes, and the complexes are cleared by phagocytes in the liver.
The C3d protein generated from C3 binds to CR2 on B cells and facilitates B cell activation and the initiation of humoral immune responses. C3d is generated when complement is activated by an antigen, either directly (e.g., when the antigen is a microbial polysaccharide) or after the binding of antibody. Complement activation results in the covalent attachment of C3b and its cleavage product C3d to the antigen. B lymphocytes can bind the antigen through their Ig receptors and simultaneously bind the attached C3d through CR2, the coreceptor for the B cell antigen receptor, thus enhancing antigen-induced signaling in B cells (see Chapter 7). Opsonized antigens are also bound by follicular dendritic cells in the germinal centers of lymphoid organs. Follicular dendritic cells display antigens to B cells in the germinal centers, and this process is important for the selection of high-affinity B cells (see Chapter 11, Fig. 11-13). The importance of complement in humoral immune responses is illustrated by the severe impairment in antibody production and germinal center formation seen in knockout mice lacking C3 or C4 or the CR2 protein.
Although our discussion has emphasized the physiologic functions of complement as an effector mechanism of host defense, the complement system is also involved in several pathologic conditions. Some autoimmune diseases are associated with the production of autoantibodies specific for self proteins expressed on cell surfaces (see Chapter 18). Binding of these antibodies results in complement-dependent lysis and phagocytosis of the cells. In other diseases, immune complexes deposit in tissues and induce inflammation by complement-mediated recruitment and activation of leukocytes.
Complement Deficiencies
Genetic deficiencies of complement proteins and regulatory proteins are the causes of various human diseases. Inherited and spontaneous deficiencies in many of the complement proteins have been described in humans.
Pathologic Effects of a Normal Complement System
Even when it is properly regulated and appropriately activated, the complement system can cause significant tissue damage. Some of the pathologic effects associated with bacterial infections may be due to complement-mediated acute inflammatory responses to infectious organisms. In some situations, complement activation is associated with intravascular thrombosis and can lead to ischemic injury to tissues. For instance, antiendothelial antibodies against vascularized organ transplants and the immune complexes produced in autoimmune diseases may bind to vascular endothelium and activate complement, thereby leading to inflammation and generation of the MAC with damage to the endothelial surface, which favors coagulation. There is also evidence that some of the late complement proteins may activate prothrombinases in the circulation that initiate thrombosis independent of MAC-mediated damage to endothelium.
The clearest examples of complement-mediated pathology are immune complex–mediated diseases. Systemic vasculitis and immune complex glomerulonephritis result from the deposition of antigen-antibody complexes in the walls of vessels and kidney glomeruli (see Chapter 18). Complement activated by these deposited immune complexes initiates the acute inflammatory responses that destroy the vessel walls or glomeruli and lead to thrombosis, ischemic damage to tissues, and scarring. Studies with knockout mice lacking the complement proteins C3 or C4 or lacking Fcγ receptors suggest that Fc receptor–mediated leukocyte activation may also cause inflammation and tissue injury as a result of IgG deposition, even in the absence of complement activation.
Evasion of Complement by Microbes
Pathogens have evolved diverse mechanisms for evading the complement system. Some microbes express thick cell walls that prevent the binding of complement proteins, such as the MAC. Gram-positive bacteria and some fungi are examples of microbes that use this relatively nonspecific evasion strategy. A few of the more specific mechanisms employed by a small subset of pathogens will be considered here. These evasion mechanisms may be divided into three groups.
These examples illustrate how microbes have acquired the ability to evade the complement system, presumably contributing to their pathogenicity.
Neonatal Immunity
Neonatal mammals are protected from infection by maternally produced antibodies transported across the placenta into the fetal circulation and by antibodies in ingested milk transported across the gut epithelium of newborns by a specialized process known as transcytosis. Neonates lack the ability to mount effective immune responses against microbes, and for several months after birth, their major defense against infection is passive immunity provided by maternal antibodies. Maternal IgG is transported across the placenta, and maternal IgA and IgG in breast milk are ingested by the nursing infant. The transepithelial transport of maternal IgA into breast milk depends on the poly Ig receptor described in Chapter 13. Ingested IgA and IgG can neutralize pathogenic organisms that attempt to colonize the infant’s gut, and ingested IgG antibodies are also transported across the gut epithelium into the circulation of the newborn. Thus, a newborn contains essentially the same IgG antibodies as the mother.
Transport of maternal IgG across the placenta and across the neonatal intestinal epithelium is mediated by an IgG-specific Fc receptor called the neonatal Fc receptor (FcRn). The FcRn is unique among Fc receptors in that it resembles a class I major histocompatibility complex (MHC) molecule containing a transmembrane heavy chain that is noncovalently associated with β2-microglobulin. However, the interaction of IgG with FcRn does not involve the portion of the molecule analogous to the peptide-binding cleft used by class I MHC molecules to display peptides for T cell recognition.
Adults also express the FcRn in the endothelium and in many epithelial tissues. In the post-neonatal period, this receptor functions to protect plasma IgG antibodies from catabolism. This process was described in Chapter 5.
Summary
Carroll MC. Complement and humoral immunity. Vaccine. 2008;26:128-133.
Gros P, Milder FJ, Janssen BJ. Complement driven by conformational changes. Nature Reviews Immunology. 2008;8:48-58.
Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annual Review of Immunology. 2004;22:431-456.
Roozendaal R, Carroll MC. Emerging patterns in complement-mediated pathogen recognition. Cell. 2006;125:29-32.
Antibody Effector Functions and Fc Receptors
Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nature Reviews Immunology. 2008;8:205-217.
Lencer WI, Blumberg RS. A passionate kiss, then run: exocytosis and recycling of IgG by FcRn. Trends in Cell Biology. 2005;15:5-9.
Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nature Reviews Immunology. 2008;8:34-47.
Smith KG, Clatworthy MR. FcγRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nature Reviews Immunology. 2010;10:328-343.