CHAPTER 4 Innate Immunity
Innate immunity is the first line of defense against infections. The cells and soluble molecules of innate immunity either exist in a fully functional state before encounter with microbes or are rapidly activated by microbes, faster than the development of adaptive immune responses (see Chapter 1, Fig. 1-1). Innate immunity coevolved with microbes to protect all multicellular organisms from infections. Some components of the mammalian innate immune system are remarkably similar to components in plants and insects, suggesting that these appeared in common ancestors long ago in evolution. For example, peptides that are toxic to bacteria and fungi, called defensins, are found in plants and mammals and have essentially the same tertiary structure in both life forms. A family of receptors that we will discuss in significant detail later in this chapter, called Toll-like receptors, are proteins that respond to the presence of pathogenic microbes by activating antimicrobial defense mechanisms in the cells in which they are expressed. Toll-like receptors are found in every life form in the evolutionary tree from insects up to mammals. The major signal transduction pathway that Toll-like receptors engage to activate cells, called the NF-κB pathway in mammals, also shows remarkable evolutionary conservation. In fact, most of the mechanisms of innate immune defense that we will discuss in this chapter appeared very early in evolution, after the development of complex multicultural organisms, about 750 million years ago. An adaptive immune system, in contrast, is clearly recognizable only in vertebrates about 500 million years ago. Adaptive immunity improves on some of the antimicrobial mechanisms of innate immunity by making them more powerful. In addition, adaptive immunity can recognize a much broader range of substances and, unlike innate immunity, displays memory of antigen encounter and specialization of effector mechanisms.
In this chapter, we describe the components, specificity, and anti-microbial mechanisms of the innate immune system. The remainder of this book is largely devoted to the role of the adaptive immune response in host defense and disease.
Innate immunity serves three important functions.
Different innate immune mechanisms work at different stages of infections. Epithelial barriers impair microbial entry into the host. Resident and recruited phagocytes in subepithelial and other tissues provide protection if the barriers are breached, and plasma proteins and circulating phagocytes provide protection if microbes reach the blood stream.
The two major types of responses of the innate immune system that protect against microbes are inflammation and antiviral defense. Inflammation is the process by which leukocytes and circulating plasma proteins are brought into sites of infection and activated to destroy and eliminate the offending agents. Inflammation is also the major reaction to damaged or dead cells and to accumulations of abnormal substances in cells and tissues. Antiviral defense consists of changes in cells that prevent virus replication and increase susceptibility to killing by lymphocytes, thus eliminating reservoirs of viral infection. In addition to these reactions, innate immune mechanisms include physical and chemical defense at epithelial barriers and activation of several circulating cells and proteins that can eliminate microbes in the blood independent of inflammation. The mechanisms by which the innate immune system works to protect against infections are described later in the chapter.
Many cells and tissues in higher organisms are endowed with the ability to contribute to innate immune reactions. Some components of innate immunity function at all times, even before infection; these components include barriers to microbial entry provided by epithelial surfaces, such as the skin and lining of the gastrointestinal and respiratory tracts. Other components of innate immunity are normally inactive but poised to respond rapidly to the presence of microbes and damaged cells; these components include phagocytes and the complement system. We begin our discussion of innate immunity by describing how the innate immune system recognizes microbes and host cells that are damaged by microbial infection. We will then proceed to the individual components of innate immunity and their functions in host defense.
Recognition of Microbes and Damaged Self by the Innate Immune System
The specificities of innate immune recognition have evolved to combat microbes and are different from the specificities of the adaptive immune system in several respects (Table 4-1).
TABLE 4–1 Specificity of Innate and Adaptive Immunity
| Innate Immunity | Adaptive Immunity | |
|---|---|---|
| Specificity | For structures shared by classes of microbes (pathogen-associated molecular patterns)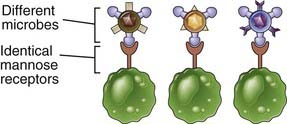 |
For structural detail of microbial molecules (antigens); may recognize nonmicrobial antigens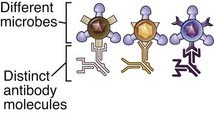 |
| Receptors | Encoded in germline; limited diversity (pattern recognition receptors)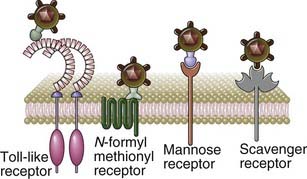 |
Encoded by genes produced by somatic recombination of gene segments; greater diversity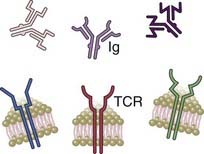 |
| Distribution of receptors | Nonclonal: identical receptors on all cells of the same lineage | Clonal: clones of lymphocytes with distinct specificities express different receptors |
| Discrimination of self and non-self | Yes; healthy host cells are not recognized or they may express molecules that prevent innate immune reactions | Yes; based on elimination or inactivation of self-reactive lymphocytes; may be imperfect (giving rise to autoimmunity) |
The innate immune system recognizes molecular structures that are characteristic of microbial pathogens but not mammalian cells. The microbial substances that stimulate innate immunity are called pathogen-associated molecular patterns (PAMPs). Different classes of microbes (e.g., viruses, gram-negative bacteria, gram-positive bacteria, fungi) express different PAMPs. These structures include nucleic acids that are unique to microbes, such as double-stranded RNA found in replicating viruses and unmethylated CpG DNA sequences found in bacteria; features of proteins that are found in microbes, such as initiation by N-formylmethionine, which is typical of bacterial proteins; and complex lipids and carbohydrates that are synthesized by microbes but not by mammalian cells, such as lipopolysaccharide (LPS) in gram-negative bacteria, lipoteichoic acid in gram-positive bacteria, and mannose-rich oligosaccharides found in microbial but not in mammalian glycoproteins (Table 4-2). In actuality, there are only a limited number of fundamental differences between microbial molecules and the molecules that higher organisms produce. Thus, the innate immune system has evolved to recognize only a limited number of molecules, most of which are unique to microbes, whereas the adaptive immune system is capable of recognizing a much wider array of foreign substances whether or not they are products of microbes.
TABLE 4–2 Examples of PAMPs and DAMPs
| Pathogen-Associated Molecular Patterns | Microbe Type | |
|---|---|---|
| Nucleic acids | ssRNA | Virus |
| dsRNA | Virus | |
| CpG | Virus, bacteria | |
| Proteins | Pilin | Bacteria |
| Flagellin | Bacteria | |
| Cell wall lipids | LPS | Gram-negative bacteria |
| Lipoteichoic acid | Gram-positive bacteria | |
| Carbohydrates | Mannan | Fungi, bacteria |
| Dectin glucans | Fungi | |
| Damage-Associated Molecular Patterns | ||
| Stress-induced proteins | HSPs | |
| Crystals | Monosodium urate | |
| Nuclear proteins | HMGB1 | |
CpG, cytidine-guanine dinucleotide; dsRNA, double-stranded RNA; HMGB1, high-mobility group box 1; HSPs, heat shock proteins; LPS, lipopolysaccharide; ssRNA, single-stranded RNA.
The innate immune system recognizes microbial products that are often essential for survival of the microbes. This feature of innate immune recognition is important because it ensures that the targets of innate immunity cannot be discarded by microbes in an effort to evade recognition by the host. An example of a target of innate immunity that is essential for microbes is double-stranded viral RNA, which plays a critical role in the replication of certain viruses. Similarly, LPS and lipoteichoic acid are structural components of bacterial cell walls that are recognized by innate immune receptors; both are required for bacterial survival and cannot be discarded. In contrast, as we shall see in Chapter 15, microbes may mutate or lose many of the antigens that are recognized by the adaptive immune system, thereby enabling the microbes to evade host defense without compromising their own survival.
The innate immune system also recognizes endogenous molecules that are produced by or released from damaged and dying cells. These substances are called damage-associated molecular patterns (DAMPs) (see Table 4-2). DAMPs may be produced as a result of cell damage caused by infections, but they may also indicate sterile injury to cells caused by any of myriad reasons, such as chemical toxins, burns, trauma, or decreased blood supply. DAMPs are generally not released from cells dying by apoptosis. In some cases, healthy cells of the immune system are stimulated to produce and release DAMPs, which enhances an innate immune response to infections.
The innate immune system uses several types of cellular receptors, present in different locations in cells, and soluble molecules in the blood and mucosal secretions to recognize PAMPs and DAMPs (Table 4-3). Cell-associated recognition molecules of the innate immune system are expressed by phagocytes (primarily macrophages and neutrophils), dendritic cells, epithelial cells that compose the barrier interface between the body and the external environment, and many other types of cells that occupy tissues and organs. These cellular receptors for pathogens and damage-associated molecules are often called pattern recognition receptors. They are expressed on the plasma membrane or endosomal membranes of various cell types and also in the cytoplasm of these cells. These various locations of the receptors ensure that the innate immune system can respond to microbes that may be present outside cells or within different cellular compartments (Fig. 4-1). When these cell-associated pattern recognition molecules bind to PAMPs and DAMPs, they activate signal transduction events that promote the antimicrobial and proinflammatory functions of the cells in which they are expressed. In addition, there are many proteins present in the blood and extracellular fluids (see Table 4-3) that recognize PAMPs. These soluble molecules are responsible for facilitating the clearance of microbes from blood and extracellular fluids by enhancing uptake into cells or by activating extracellular killing mechanisms.
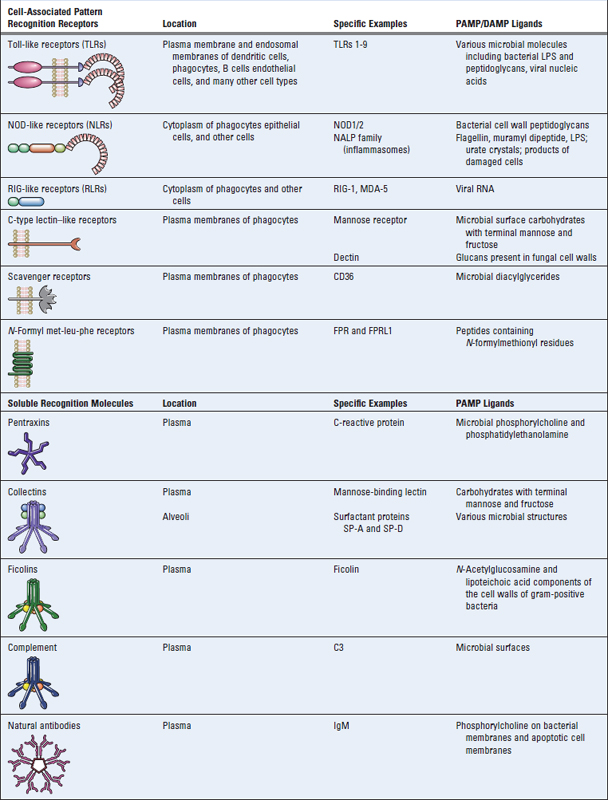
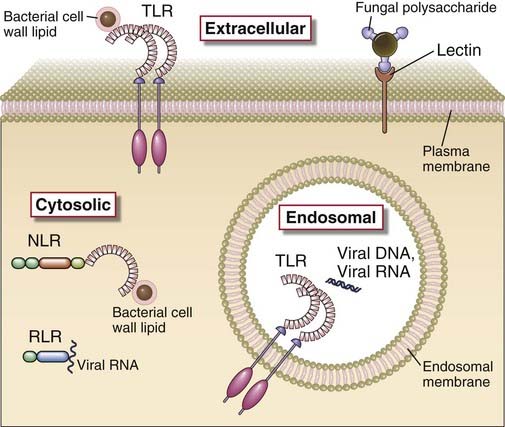
FIGURE 4–1 Cellular locations of pattern recognition molecules of the innate immune system.
Some pattern recognition molecules of the TLR family (see Fig. 4-2) are expressed on the cell surface, where they may bind extracellular pathogen-associated molecular patterns. Other TLRs are expressed on endosomal membranes and recognize nucleic acids of microbes that have been phagocytosed by cells. Cells also contain cytoplasmic sensors of microbial infection (discussed later in the chapter), including the NLR family of proteins, which recognize bacterial peptidoglycans, RIG-like receptors, which bind viral RNA, and plasma membrane lectin-like receptors that recognize fungal glycans. Cytoplasmic receptors that recognize products of damaged cells as well as some microbes are shown in Fig. 4-4.
The receptors of the innate immune system are encoded in the germline, whereas the receptors of adaptive immunity are generated by somatic recombination of receptor genes in the precursors of mature lymphocytes. As a result, the repertoire of specificities of innate immune system receptors is small compared with that of B and T cells of the adaptive immune system. It is estimated that the innate immune system can recognize about 103 molecular patterns. In contrast, the adaptive immune system is capable of recognizing 107 or more distinct antigens. Furthermore, whereas the adaptive immune system can distinguish between antigens of different microbes of the same class and even different antigens of one microbe, innate immunity can distinguish only classes of microbes, or only damaged cells from healthy cells, but not particular species of microbes or cell types.
The innate immune system does not react against normal, healthy cells and tissues. This characteristic is, of course, essential for the health of the organism. It is determined in part by the specificity of innate immune mechanisms for PAMPs and DAMPs and in part by regulatory proteins expressed by normal cells that prevent activation of various components of innate immunity. We will discuss examples of such regulation later in the chapter.
Cell-Associated Pattern Recognition Receptors of Innate Immunity
With this introduction, we can proceed to a discussion of the large variety of molecules in the body that are capable of recognizing PAMPs and DAMPs, focusing on their specificity, location, and functions. We will begin with cell-associated molecules expressed on membranes or in the cytoplasm of cells. The soluble recognition and effector molecules of innate immunity, found in the blood and extracellular fluids, are described later.
Most cell types express pattern recognition receptors and therefore are capable of participating in innate immune responses. Phagocytes, including neutrophils and macrophages, and dendritic cells express the widest variety and greatest amount of these receptors, which is in keeping with their fundamental role in detecting microbes and damaged cells and ingesting them for destruction (as the neutrophils and macrophages do) or reacting in ways that elicit inflammation and subsequent adaptive immunity (which is an important function of dendritic cells). Pattern recognition receptors are linked to intracellular signal transduction pathways that activate various cellular responses, including the production of molecules that promote inflammation and defend against microbes.
We will organize our discussion around several distinct classes of cellular pattern recognition receptors, which differ in their structure and specificity for various types of microbes.
Toll-like Receptors
The Toll-like receptors (TLRs), an evolutionarily conserved family of pattern recognition receptors expressed on many cell types, recognize products of a wide variety of microbes. Toll was originally identified as a Drosophila gene involved in establishing the dorsal-ventral axis during embryogenesis of the fruit fly, but subsequently it was discovered that the Toll protein also mediated antimicrobial responses in these organisms. This discovery led to the identification of mammalian homologues of Toll, which were named Toll-like receptors. There are 9 different functional TLRs in humans, named TLR1 to TLR9 (Fig. 4-2). The TLRs are type I integral membrane glycoproteins that contain leucine-rich repeats flanked by characteristic cysteine-rich motifs in their extracellular regions, which are involved in ligand binding, and a Toll/IL-1 receptor (TIR) homology domain in their cytoplasmic tails, which is essential for signaling. TIR domains are also found in the cytoplasmic tails of the receptors for the cytokines IL-1 and IL-18, and similar signaling pathways are engaged by TLRs, IL-1, and IL-18.
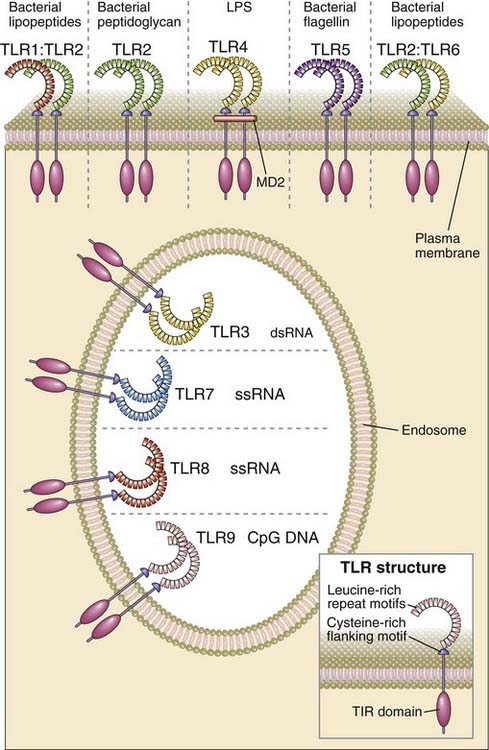
FIGURE 4–2 Structure, location, and specificities of mammalian TLRs.
Note that some TLRs are expressed in endosomes and some on the cell surface.
Mammalian TLRs are involved in responses to a wide variety of molecules that are expressed by microbial but not by healthy mammalian cells. The ligands that the different TLRs recognize are structurally diverse and include products of all classes of microorganism (see Fig. 4-2). Examples of bacterial products that bind to TLRs are LPS and lipoteichoic acid, constituents of the cell walls of gram-negative bacteria and gram-positive bacteria, respectively, and flagellin, the protein subunit component of the flagella of motile bacteria. Examples of TLR ligands produced by viruses are double-stranded RNAs, which compose the genomes of some viruses and are generated during the life cycle of most RNA viruses but are not produced by eukaryotic cells, and single-stranded RNAs, which are distinguished from cellular cytoplasmic single-stranded RNA transcripts by their location within endosomes and by their high guanosine and uridine content. Fungal mannose polysaccharides (mannans) are also TLR ligands.
TLRs are also involved in response to endogenous molecules whose expression or location indicates cell damage. Examples of host molecules that engage TLRs include heat shock proteins (HSPs), which are chaperones induced in response to various cell stresses, and high-mobility group box 1 (HMGB1), an abundant DNA-binding protein involved in transcription and DNA repair. Both HSPs and HMGB1 are normally intracellular but may become extracellular when released from injured or dying cells. From their extracellular location, they activate TLR2 and TLR4 signaling in dendritic cells, macrophages, and other cell types.
The structural basis of TLR specificities resides in the multiple extracellular leucine-rich modules of these receptors, which bind directly to PAMPs or to adaptor molecules that bind the PAMPs. There are between 16 and 28 leucine-rich repeats in TLRs, and each of these modules is composed of 20 to 30 amino acids that include conserved LxxLxLxxN motifs (where L is leucine, x is any amino acid, and N is asparagine) and amino acid residues that vary between different TLRs. The ligand-binding variable residues of the modules are on the convex surface formed by α helices and β turns or loops. These repeats contribute to the ability of some TLRs to bind hydrophobic molecules such as bacterial LPS. Ligand binding to the leucine-rich domains causes physical interactions between TLR molecules and the formation of TLR dimers. The repertoire of specificities of the TLR system is extended by the ability of TLRs to heterodimerize with one another. For example, dimers of TLR2 and TLR6 are required for responses to peptidoglycan.
Specificities of the TLRs are also influenced by various non-TLR accessory molecules. This is best defined for the TLR4 response to LPS. LPS first binds to soluble LPS-binding protein in the blood or extracellular fluid, and this complex serves to facilitate delivery of the LPS to the surface of the responding cell. An extracellular protein called MD2 (myeloid differentiation protein 2) binds to the lipid A component of LPS, forming a complex that then interacts with TLR4 and initiates signaling. Another protein called CD14 is also required for efficient LPS-induced signaling. CD14 is expressed by most cells (except endothelial cells) as a soluble protein or as a glycophosphatidylinositol-linked membrane protein. Both CD14 and MD2 can also associate with other TLRs. Thus, different combinations of accessory molecules in TLR complexes may serve to broaden the range of microbial products that can induce innate immune responses.
TLRs are found on the cell surface and on intracellular membranes and are thus able to recognize microbes in different cellular locations (see Fig 4-2). TLRs 1, 2, 4, 5, and 6 are expressed on the plasma membrane, where they recognize various PAMPs in the extracellular environment. Some of the most potent microbial stimuli for innate immune responses bind to these plasma membrane TLRs, such as bacterial LPS and lipoteichoic acid, which are recognized by TLRs 2 and 4, respectively. In contrast, TLRs 3, 7, 8, and 9 are mainly expressed inside cells on endoplasmic reticulum and endosomal membranes, where they detect several different nucleic acid ligands (see Fig. 4-2). Some of these nucleic acids are much more abundantly expressed by microbes than by mammals, such as double-stranded RNA, which is made by RNA viruses and binds to TLR3, and unmethylated CpG motifs common in prokaryotic DNA, which bind to TLR9. Single-stranded RNA, which binds to TLR8, and single- or double-stranded DNA, which binds to TLR9, are not uniquely expressed by microbes, but the relative specificity of these TLRs for microbial products is linked to their endosomal location. Host cell RNA and DNA are not normally present in endosomes, but microbial RNA and DNA may end up in endosomes of neutrophils, macrophages, or dendritic cells when the microbes are phagocytosed by these cells. Furthermore, host DNA from cells that have died because of infection or other causes may end up in the endosomes of the phagocytes. In other words, TLRs 3, 7, 8, and 9 may distinguish healthy self from foreign or unhealthy self on the basis, in part, of the cellular location of the nucleic acids they bind. A protein in the endoplasmic reticulum called UNC-93B is required for the endosomal localization and proper function of TLR3, 7, 8 and 9.
TLR recognition of microbial ligands results in the activation of several signaling pathways and ultimately transcription factors, which induce the expression of genes whose products are important for inflammatory and antiviral responses (Fig. 4-3). The signaling pathways are initiated by ligand binding to the TLR at the cell surface or in the endoplasmic reticulum or endosomes, leading to dimerization of the TLR proteins. Ligand-induced TLR dimerization is predicted to bring the TIR domains of the cytoplasmic tails of each protein close to one another. This is followed by recruitment of TIR domain–containing adaptor proteins, which facilitate the recruitment and activation of various protein kinases, leading to the activation of different transcription factors. The major transcription factors that are activated by TLR signaling pathways are nuclear factor κB (NF-κB), activation protein 1 (AP-1), interferon response factor 3 (IRF3), and IRF7. NF-κB and AP-1 stimulate the expression of genes encoding many of the molecules required for inflammatory responses including inflammatory cytokines (e.g., TNF and IL-1), chemokines (e.g., CCL2 and CXCL8), and endothelial adhesion molecules (e.g., E-selectin) (discussed later). IRF3 and IRF7 promote production of type I interferons (IFN-α and IFN-β), important for anti-viral innate immune responses.
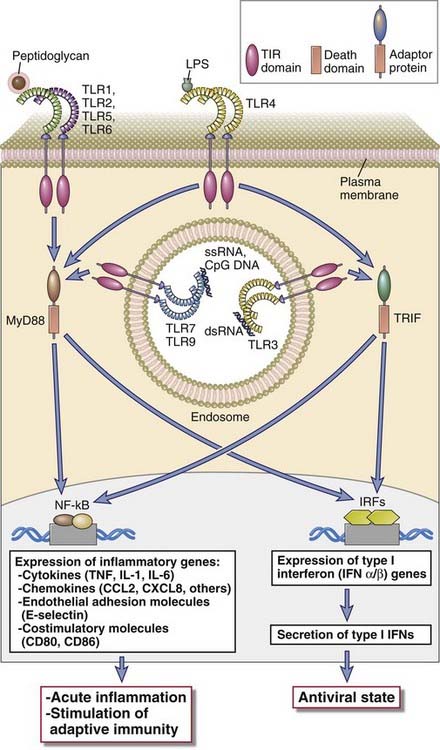
FIGURE 4–3 Signaling functions of TLRs.
TLRs 1, 2, 5, and 6 use the adaptor protein MyD88 and activate the transcription factors NF-κB and AP-1. TLR3 uses the adaptor protein TRIF and activates the IRF3 and IRF7 transcription factors. TLR4 can activate both pathways. TLRs 7 and 9 in the endosome use MyD88 and activate both NF-κB and IRF7 (not shown).
Different combinations of adaptors and signaling intermediates are used by different TLRs, which is the basis for common and unique downstream effects of the TLRs. For example, cell surface TLRs that engage the adaptor MyD88 lead to NF-κB activation, and TLR signaling that uses the adaptor called TRIF (TIR domain–containing adaptor inducing IFN-β) leads to IRF3 activation. All TLRs except TLR3 signal through MyD88 and are therefore capable of activating NF-κB and inducing an inflammatory response. TLR3 signals through TRIF and therefore activates IRF3 and induces expression of type I interferons. TLR4 signals through both MyD88 and TRIF and is able to induce both types of responses. Endosomal TLRs 7 and 9, which are most highly expressed in plasmacytoid dendritic cells, signal through a MyD88-dependent, TRIF-independent pathway that activates both NF-κB and IRF4. Therefore, TLR7 and TLR9, like TLR4, induce both inflammatory and antiviral responses. Details of NF-κB activation are discussed in Chapter 7.
Cytosolic Receptors for PAMPs and DAMPs
In addition to the membrane-bound TLRs, which sense pathogens outside cells or in endosomes, the innate immune system has evolved to equip cells with pattern recognition receptors that detect infection or cell damage in the cytoplasm (see Fig. 4-1 and Table 4-3). The two major classes of these cytoplasmic receptors are NOD-like receptors and RIG-like receptors. These cytoplasmic receptors, like TLRs, are linked to signal transduction pathways that promote inflammation or type I interferon production. The ability of the innate immune system to detect infection in the cytoplasm is important because parts of the normal life cycles of some microbes, such as viral gene translation and viral particle assembly, take place in the cytoplasm. Some bacteria and parasites have mechanisms to escape from phagocytic vesicles into the cytoplasm. Microbes can produce toxins that create pores in host cell plasma membranes, including endosomal membranes, through which microbial molecules can enter the cytoplasm. These pores can also result in changes in the concentration of endogenous molecules in the cytoplasm, which are reliable signs of infection and damage and are detected by the cytoplasmic receptors.
NOD-like Receptors
NOD-like receptors (NLRs) are a family of more than 20 different cytosolic proteins, some of which sense cytoplasmic PAMPs and DAMPs and recruit other proteins to form signaling complexes that promote inflammation. This family of proteins is named after NOD (nucleotide oligomerization domain–containing protein). Typical NLR proteins contain at least three different domains with distinct structures and functions. These include a leucine-rich repeat domain that senses the presence of ligand, similar to the leucine-rich repeats of TLRs; a NACHT (neuronal apoptosis inhibitory protein [NAIP], CIITA, HET-E, and TP1) domain, which allows NLRs to bind to one another and form oligomers; and an effector domain, which recruits other proteins to form signaling complexes. There are three NLR subfamilies, the members of which use different effector domains to initiate signaling, called CARD, Pyrin, and BIR domains. NLRs are found in a wide variety of cell types, although some NLRs have restricted tissue distributions. Some of the best studied NLRs are found in immune and inflammatory and epithelial barrier cells.
NOD1 and NOD2, members of the CARD domain–containing NOD subfamily of NLRs, are expressed in the cytoplasm of several cell types including mucosal epithelial cells and phagocytes, and they respond to bacterial cell wall peptidoglycans. NOD2 is particularly highly expressed in intestinal Paneth cells, where it stimulates expression of antimicrobial substances called defensins in response to pathogens. NOD1 recognizes substances derived mainly from gram-negative bacteria, whereas NOD2 recognizes a distinct molecule called muramyl dipeptide from both gram-negative and gram-positive organisms. These peptides are released from intracellular or extracellular bacteria; in the latter case, their presence in the cytoplasm requires specialized mechanisms of delivery of the peptides into host cells. These mechanisms include type III and type IV secretion systems, which have evolved in pathogenic bacteria as a means of delivering toxins into host cells. When oligomers of NODs recognize their peptide ligands, including bacterial toxins, a conformational change occurs that allows the CARD effector domains of the NOD proteins to recruit multiple copies of the kinase RIP2, forming a signaling complex that has been called the NOD signalosome. The RIP2 kinases in these complexes activate NF-κB, which promotes inflammatory gene expression, similar to TLRs that signal through MyD88, discussed earlier. Both NOD1 and NOD2 appear to be important in innate immune responses to bacterial pathogens in the gastrointestinal tract, such as Helicobacter pylori and Listeria monocytogenes. There is great interest in the finding that certain NOD2 polymorphisms increase the risk for an inflammatory disease of the bowel called Crohn’s disease, probably because of a defective innate response to commensal and pathogenic organisms in the intestine. Also, mutations of NOD2 that cause increased NOD signaling lead to a systemic inflammatory disease called Blau’s syndrome.
The NLRP subfamily of NLRs respond to cytoplasmic PAMPs and DAMPs by forming signaling complexes called inflammasomes, which generate active forms of the inflammatory cytokine IL-1 (Fig. 4-4). There are 14 NLRPs (NLR family, pyrin-domain-containing proteins), most of which share a Pyrin effector domain, named after the Greek root pyro, meaning heat, because it was first identified in a mutated gene that is associated with an inherited febrile illness. Inflammasomes containing only three of these NLRPs have been well studied, notably IPAF/NLRC4, NLRP3, and NLRP1. When these NLRPs are activated by the presence of microbial products or changes in the amount of endogenous molecules or ions in the cytoplasm, they bind other proteins through homotypic interactions between shared structural domains, thereby forming the inflammasome complex. For example, after binding of a ligand, multiple identical NLRP3 proteins interact to form an oligomer, and individual NLRP3 proteins in the oligomer each bind an adaptor protein called ASC. The adaptors then bind an inactive precursor form of the enzyme caspase-1 through interactions of caspase-recruitment domains on both proteins. Caspases are proteases with cysteine residues in their active site that cleave substrate proteins at aspartate residues. Caspase-1 becomes active only after recruitment to the inflammasome complex. Although several other caspases participate in a form of cell death called apoptosis (see Chapter 14), the main function of caspase-1 is to cleave the inactive cytoplasmic precursor forms of two homologous cytokines called IL-1β and IL-18. Caspase-1 cleavage generates active forms of these cytokines, which then leave the cell and perform various proinflammatory functions. We will describe the action of these cytokines and the inflammatory response in detail later in the chapter. Suffice it to say here that the inflammation induced by IL-1 serves a protective function against the microbes that incite the formation of the inflammasome. When inflammasome activity is abnormally stimulated, the abundant IL-1 that is produced can cause tissue damage. For example, some of the hereditary periodic fevers (also called autoinflammatory syndromes), which are rare diseases characterized by repeated bouts of fever, inflammation, and tissue destruction, are caused by gain-of-function mutations of the NLRP3 gene, and IL-1 antagonists are very effective in treating these diseases.
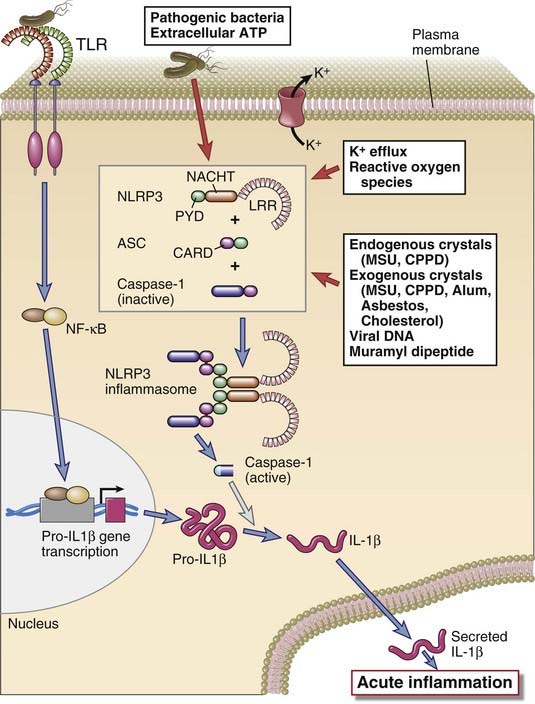
The activation of the NLRP3 inflammasome, which processes pro–IL-1β to active IL-1, is shown. Inflammasomes with other NLRP proteins function in a similar way. Pro–IL-1β expression is induced by various PAMPs or DAMPs through pattern recognition receptor signaling, such as a TLR, as shown. CPPD, calcium pyrophosphate dihydrate; MSU, monosodium urate.
NLRP-inflammasome responses are induced by a wide variety of cytoplasmic stimuli, including microbial products, environmentally or endogenously derived crystals, and reduction in cytoplasmic potassium ion (K+) concentrations, that are often associated with infections and cell stress (see Fig. 4-4). Microbial products that activate NLRP-inflammasomes include bacterial molecules such as flagellin, muramyl dipeptide, LPS, and pore-forming toxins as well as bacterial and viral RNA. Crystalline substances are also potent activators of inflammasomes, and these crystals can be derived from the environment, such as asbestos and silica, or they can be endogenously derived from dead cells, such as monosodium urate and calcium pyrophosphate dehydrate. Another endogenous stimulus of inflammasome activation is extracellular ATP, perhaps released from dead cells and transported into the cytoplasm of the responding cell. The structural diversity of the agents that activate the inflammasome suggests that they do not directly bind to NLRP proteins but may act by inducing a smaller set of changes in endogenous cytoplasmic conditions that activate the NLRPs. Reduced cytoplasm potassium ion concentrations may be one such common mechanism because reductions in cellular K+ induced by some bacterial pore-forming toxins can activate inflammasomes, and many of the other known inflammasome activators cause increased K+ efflux from cells. Another common mechanism implicated in inflammasome activation is the generation of reactive oxygen species, which are toxic free radicals of oxygen that are often produced during cell injury. A type of inflammasome that uses a protein called AIM2 (absent in melanoma-2) rather than an NLRP-family protein, recognizes cytosolic dsDNA.
The discovery that some crystalline substances are potent inflammasome activators has changed our understanding of certain inflammatory diseases. Gout is a painful inflammatory condition of the joints that has long been known to be caused by deposition of monosodium urate crystals in joints. Based on the understanding that urate crystals activate the inflammasome, there is interest in using IL-1 antagonists to treat cases of severe gout that are resistant to conventional anti-inflammatory drugs. Similarly, pseudogout is caused by deposition of calcium pyrophosphate crystals and inflammasome activation. Occupational inhalation of silica and asbestos can cause chronic inflammatory and fibrotic disease of the lung, and there is also interest in the potential of blocking the inflammasome or IL-1 to treat these diseases.
RIG-like Receptors
RIG-like receptors (RLRs) are cytosolic sensors of viral RNA that respond to viral nucleic acids by inducing the production of the antiviral type I interferons. RLRs can recognize double-stranded and single-stranded RNA, which includes the genomes of RNA viruses and RNA transcripts of RNA and DNA viruses. The two best characterized RLRs are RIG-I (retinoic acid–inducible gene I) and MDA5 (melanoma differentiation-associated gene 5). Both of these proteins contain two N-terminal caspase recruitment domains, which interact with other signaling proteins, and an RNA-helicase domain of unknown function. RIG-I and MDA5 display different specificities for viral RNA, partly based on length of double-stranded RNA genome, which may enhance sensitivity in detecting a wide range of viral double-stranded RNAs with heterogeneous lengths. RLRs also can discriminate viral single-stranded RNA from normal cellular single-stranded RNA transcripts. For example, RIG-I will only recognize RNA with a 5′ triphosphate moiety, which is not present in mammalian host cell cytoplasmic RNA because of addition of a 7-methylguanosine cap or removal of the 5′ triphosphate. RLRs are expressed in a wide variety of cell types, including bone marrow–derived leukocytes and various tissue cells. Therefore, these receptors enable the many cell types susceptible to infection by RNA viruses to participate in innate immune responses to these viruses.
On binding RNA, the RLRs initiate signaling events that lead to IRF3 and IRF7 activation, and these transcription factors induce production of type I interferons. In addition, RLR signaling can also activate NF-κB. Signaling of both RIG-I and MDA5 depends on their binding to adaptor proteins and activation of signaling cascades that lead to either IRF3/7 or NF-κB activation.
Other Cell-Associated Pattern Recognition Receptors
Several types of plasma membrane and cytoplasmic receptors other than the classes described before are expressed on the plasma membranes of various cell types and recognize microbial molecules (see Table 4-3). Some of these receptors transmit activating signals, similar to TLRs, that promote inflammatory responses and enhance killing of microbes. Other receptors mainly participate in the uptake of microbes into phagocytes.
Receptors for Carbohydrates
Receptors that recognize carbohydrates on the surface of microbes facilitate the phagocytosis of the microbes and stimulate subsequent adaptive immune responses. These receptors belong to the C-type lectin family, so called because they bind carbohydrates (hence, lectins) in a Ca++-dependent manner (hence, C-type). Some of these are soluble proteins found in the blood and extracellular fluids (discussed later); others are integral membrane proteins found on the surfaces of macrophages, dendritic cells, and some tissue cells. All these molecules contain a conserved carbohydrate recognition domain. There are several types of plasma membrane C-type lectins with specificities for different carbohydrates, including mannose, glucose, N-acetylglucosamine, and β-glucans. In general, these cell surface lectins recognize carbohydrate structures found on the cell walls of microorganisms but not mammalian cells. Some of these C-type lectin receptors function in the phagocytosis of microbes, and others have signaling functions that induce protective responses of host cells to microbes.
Scavenger Receptors
Scavenger receptors comprise a structurally and functionally diverse collection of cell surface proteins that were originally grouped on the basis of the common characteristic of mediating the uptake of oxidized lipoproteins into cells. Some of these scavenger receptors, including SR-A and CD36, are expressed on macrophages and mediate the phagocytosis of microorganisms. In addition, CD36 functions as a coreceptor in TLR2/6 recognition and response to bacterially derived lipoteichoic acid and diacylated lipopeptides. There is a wide range of molecular structures that bind to each scavenger receptor, including LPS, lipoteichoic acid, nucleic acids, β-glucan, and proteins. The significance of scavenger receptors in innate immunity is highlighted by increased susceptibility to infection in gene knockout mice lacking the receptors and by the observations that several microbial pathogens express virulence factors that block scavenger receptor–mediated recognition and phagocytosis.
N-Formyl Met-Leu-Phe Receptors
N-Formyl met-leu-phe receptors, including FPR and FPRL1 expressed by neutrophils and macrophages, respectively, recognize bacterial peptides containing N-formylmethionyl residues and stimulate directed movement of the cells. Because all bacterial proteins and few mammalian proteins (only those synthesized within mitochondria) are initiated by N-formylmethionine, FPR and FPRL1 allow phagocytes to detect and respond preferentially to bacterial proteins. The bacterial peptide ligands that bind these receptors are some of the first identified and most potent chemoattractants for leukocytes. Chemoattractants include several types of diffusible molecules, often produced at sites of infection, that bind to specific receptors on cells and direct their movement toward the source of the chemoattractant. Other chemoattractants, such as the chemokines discussed in Chapter 3, are made by host cells. FPR and FPRL1, along with all other chemoattractant receptors, belong to the seven-transmembrane, guanosine triphosphate (GTP)–binding (G) protein–coupled receptor (GPCR) superfamily. These receptors initiate intracellular responses through associated trimeric G proteins (see Chapter 7). The G proteins stimulate many types of cellular responses, including cytoskeletal changes, resulting in increased cell motility.
Cellular Components of the Innate Immune System
The cells of the innate immune system perform several functions that are essential for defense against microorganisms. Some cells form physical barriers that impede infections. Several cell types express the various pattern recognition receptors we have just discussed, and after recognizing PAMPs and DAMPs, they respond by producing inflammatory cytokines and antiviral proteins and by killing microbes or infected cells. In addition, some of the cells of innate immunity are critical for stimulating subsequent adaptive immune responses. We will now discuss the cell types that perform these functions.
Epithelial Barriers
Intact epithelial surfaces form physical barriers between microbes in the external environment and host tissue, and epithelial cells produce antimicrobial chemicals that further impede the entry of microbes (Fig. 4-5). The main interfaces between the environment and the mammalian host are the skin and the mucosal surfaces of the gastrointestinal, respiratory, and genitourinary tracts. These interfaces are lined by continuous layers of specialized epithelial cells that serve many physiologic functions, including preventing the entry of microbes. Loss of the integrity of these epithelial layers by trauma or other reasons predisposes an individual to infections. The protective barrier function is in large part physical. The epithelial cells form tight junctions with one another, blocking passage of microbes between the cells. The outer layer of keratin, which accumulates as surface skin keratinocytes die, serves to block microbial penetration into deeper layers of the epidermis. Mucus, a viscous secretion containing glycoproteins called mucins, is produced by respiratory, gastrointestinal, and urogenital epithelial cells. Mucus physically impairs microbial invasion and facilitates microbe removal by ciliary action in the bronchial tree and peristalsis in the gut. Although these physical barrier properties alone are very important in host defense, other epithelial defense mechanisms have evolved to complement the physical barrier.
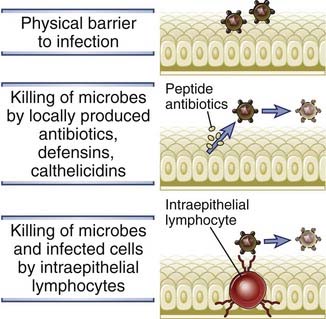
FIGURE 4–5 Epithelial barriers.
Epithelia at the portals of entry of microbes provide physical barriers, produce antimicrobial substances, and harbor intraepithelial lymphocytes that are believed to kill microbes and infected cells.
Epithelial cells as well as some leukocytes produce peptides that have antimicrobial properties. Two structurally distinct families of antimicrobial peptides are the defensins and the cathelicidins.
Barrier epithelia contain certain types of lymphocytes, including intraepithelial T lymphocytes, that recognize and respond to commonly encountered microbes. Intraepithelial T lymphocytes are present in the epidermis of the skin and in mucosal epithelia. Various subsets of intraepithelial lymphocytes are present in different proportions, depending on species and tissue location. These subsets are distinguished mainly by the type of T cell antigen receptors (TCRs) they express. Some intraepithelial T lymphocytes express the conventional αβ form of TCR, which is present on most T cells in lymphoid tissues. Other T cells in epithelia express a form of antigen receptor called the γδ receptor that may recognize peptide and nonpeptide antigens. A common characteristic of these T cells is the limited diversity of their antigen receptors compared with most T cells in the adaptive immune system. The intraepithelial T lymphocytes are believed to recognize a limited number of commonly encountered microbial structures (e.g., PAMPs). Intraepithelial lymphocytes may function in host defense by secreting cytokines, activating phagocytes, and killing infected cells.
Phagocytes
Cells that have specialized phagocytic functions, primarily macrophages and neutrophils, are the first line of defense against microbes that breach epithelial barriers. We have introduced these cell types in Chapter 2, and we will discuss many other details of their functions later in this chapter and in other chapters. For now, it is important to know that these phagocytic cells perform two general types of functions in defense against microbes. First, they are able to internalize and kill microbes. Neutrophils and macrophages are particularly good at this function. Second, phagocytes respond to microbes by producing various cytokines that promote inflammation and also enhance the antimicrobial function of host cells at the site of infection. Among the “professional phagocytes,” macrophages are particularly good at this second function. Macrophages are also involved in the repair of damaged tissues, which is another function important in host defense. The essential role that phagocytes play in innate immune defense against microbes is demonstrated by the high rate of lethal bacterial and fungal infections in patients with low blood neutrophil counts caused by bone marrow cancers or cancer therapy and in patients with inherited deficiencies in the functions of phagocytes.
Dendritic Cells
Dendritic cells perform essential recognition and effector roles in innate immunity. We introduced dendritic cells in Chapter 2, and their role in antigen presentation to T cells is discussed in Chapter 6. Recall that this heterogeneous family of bone marrow–derived cells with long dendrite-like cytoplasmic processes are constitutively present in epithelia and most tissues of the body. Because of their placement and morphology, these cells are poised to detect invading microbes. Furthermore, dendritic cells express more different types of TLRs and cytoplasmic pattern recognition receptors than any other cell type, making them the most versatile sensors of PAMPs and DAMPs among all cell types in the body. One particular subset of dendritic cells, called plasmacytoid dendritic cells because their morphology is similar to antibody-producing plasma cells, is the major source of antiviral cytokines, type I interferons, produced in response to viral infections. This feature of plasmacytoid dendritic cells is due in part to the fact that these cells, more than other cell types, abundantly express the endosomal TLRs (TLRs 3, 7, 8, 9) that recognize nucleic acids of viruses that have been internalized into the cell. We will discuss the antiviral actions of type I interferons in more detail later in the chapter.
Dendritic cells are uniquely capable of triggering and directing adaptive T cell–mediated immune responses, and this is dependent on their innate immune responses to microbes. This capability reflects the ability of dendritic cells to take up microbial protein antigens, to transport them to lymph nodes where naive T cells home, and to alter and display the protein antigens in a way that the T cells can recognize. These functions will be discussed in great detail in Chapter 6. Importantly, the innate response of dendritic cells to PAMPs is essential for these functions, which are enhanced by TLR signaling. Furthermore, TLR signaling induces dendritic cell expression of molecules, including costimulatory molecules and cytokines, that are needed, in addition to antigen, for the activation of the naive T cells and their differentiation into effector T cells. Depending on the nature of the microbe that induces the innate response, a dendritic cell will direct naive T cell differentiation into distinct types of effector cells, such as IFN-γ–producing TH1 cells or IL-17–producing TH17 cells. The influence of dendritic cells on T cell activation and differentiation will be discussed further in Chapter 9.
Natural Killer Cells
Natural killer (NK) cells are lymphocytes distinct from T and B cells that play important roles in innate immune responses mainly against intracellular viruses and bacteria. The term natural killer derives from the fact that these cells are capable of performing their killing function without a need for clonal expansion and differentiation, which is required for effector responses of the immune system’s other killer cells, the cytotoxic T lymphocytes (CTLs). NK cells constitute 5% to 15% of the mononuclear cells in the blood and spleen. They are rare in other lymphoid organs but are concentrated in certain organs such as the liver and gravid uterus. NK cells arise from bone marrow precursors and appear as large lymphocytes with numerous cytoplasmic granules. NK cells do not express highly diverse, clonally distributed antigen receptors typical of B and T cells. Rather, they use germline DNA-encoded receptors, discussed later, to distinguish pathogen-infected from healthy cells. They can be identified in the blood by expression of CD56 and the absence of CD3, two membrane proteins often found together on activated CTLs.
Recognition of Infected and Stressed Cells by NK Cells
NK cells distinguish infected and stressed cells from healthy cells, and NK cell activation is regulated by a balance between signals that are generated from activating receptors and inhibitory receptors. There are several families of these receptors (Fig. 4-6), some members of which we will discuss later. These receptors recognize molecules on the surface of other cells and generate activating or inhibitory signals that promote or inhibit NK responses. In general, the activating receptors recognize ligands on infected and injured cells, and the inhibitory receptors recognize healthy normal cells. When an NK cell interacts with another cell, the outcome is determined by the integration of signals generated from the array of inhibitory and activating receptors that are expressed by the NK cell and that interact with ligands on the other cell. Because of the stochastic nature of their expression, there is significant diversity in the array of activating and inhibitory receptors that different NK cells express in any one individual. The result of this is that an individual’s NK cells will respond to different types of microbes or infected cells. Furthermore, the genes encoding many of these receptors are polymorphic, meaning that there are several variants of the genes in the population, so that one person will express a slightly different form of the receptors than another person.
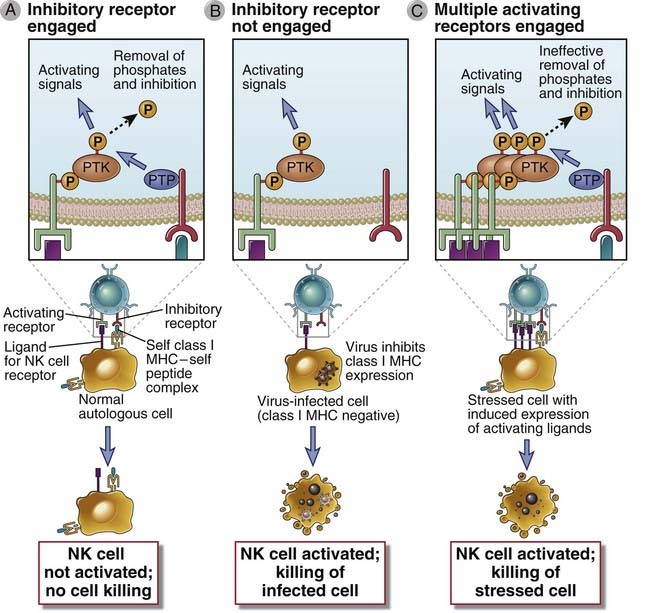
FIGURE 4–6 Functions of activating and inhibitory receptors of NK cells.
A, Activating receptors of NK cells recognize ligands on target cells and activate protein tyrosine kinase (PTK), whose activity is inhibited by inhibitory receptors that recognize class I MHC molecules and activate protein tyrosine phosphatases (PTP). NK cells do not efficiently kill class I MHC–expressing healthy cells. B, If a virus infection or other stress inhibits class I MHC expression on infected cells and induces expression of additional activating ligands, the NK cell inhibitory receptor is not engaged and the activating receptor functions unopposed to trigger responses of NK cells, such as killing of target cells and cytokine secretion. C. Cells stressed by infection or neoplastic transformation may express increased amounts of activating ligands, which bind NK cell activating receptors and induce more tyrosine phosphorylation than can be removed by inhibitory receptor associated phosphatases, resulting in killing of the stressed cells. Structural details and ligands of inhibitory and activating NK cell receptors are shown in Figure 4-7.
Most NK cells express inhibitory receptors that recognize class I major histocompatibility complex (MHC) molecules, which are cell surface proteins normally expressed on almost all healthy cells in the body (Fig. 4-7). A major function of class I MHC molecules, distinct from their role in regulating NK cell activation, is to display peptides derived from cytoplasmic proteins, including microbial proteins, on the cell surface for recognition by CD8+ T lymphocytes. We will describe the structure and function of MHC molecules in relation to CD8+ T cell antigen recognition in Chapter 6. For now, it is important to understand that NK cells use fundamentally different types of receptors than do T cells to recognize class I MHC molecules. Unlike T cells, many of the NK receptors for class I MHC respond by inhibiting NK activation. This is useful because normal cells express class I MHC molecules, and many viruses and other causes of cell stress lead to a loss of cell surface expression of class I MHC. Thus, NK cells interpret the presence of class I MHC molecules as markers of normal, healthy self, and their absence is an indication of infection or damage. Conversely, NK cells will not receive inhibitory signals from infected or stressed cells. At the same time, the NK cells are likely to receive activating signals from the same infected cells through activating receptors. The net result will be activation of the NK cell to secrete cytokines and to kill the infected or stressed cell. This ability of NK cells to become activated by host cells that lack class I MHC has been called recognition of missing self.
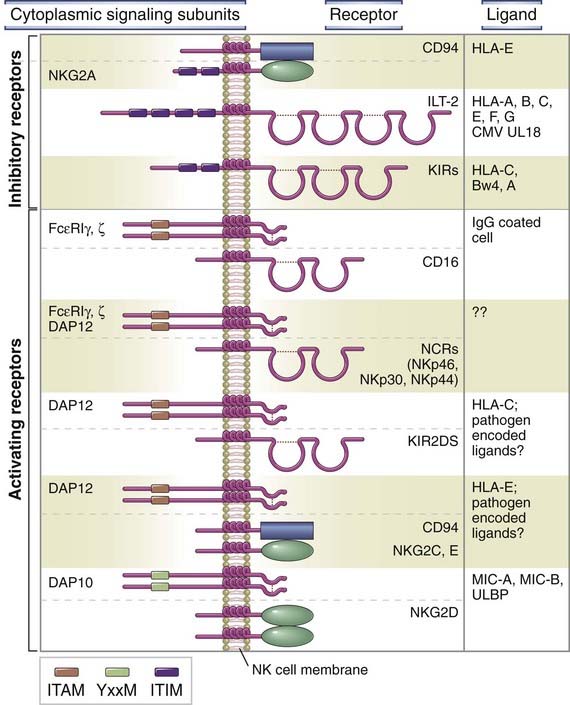
FIGURE 4–7 Structure and ligands of activating and inhibitory receptors of NK cells.
Examples of inhibitory and activating NK cell receptors and their ligands. CD16 and the natural cytotoxic receptors (NCRs) associate with ζ chain homodimers, FcεRIγ homodimers, or ζ-FcεRIγ heterodimers. There are multiple different KIRs, with varying ligand specificities.
Inhibitory receptors of NK cells share the common feature of a structural motif in their cytoplasmic tails, called an immunoreceptor tyrosine-based inhibition motif (ITIM), which engages molecules that block the signaling pathways of activating receptors (see Figs. 4-6 and 4-7). ITIMs contain tyrosine residues that are phosphorylated on ligand binding to the inhibitory receptor. This leads to the recruitment and activation of phosphatases, which remove phosphates from several signaling proteins or lipids generated by the signaling pathways downstream of NK activating receptors. The end result is blocking of the signaling functions of activating receptors. ITIMs are found in cytoplasmic tails of other receptors besides NK inhibitory receptors, and their structure and signaling functions are discussed in more detail in Chapter 7.
The largest group of NK inhibitory receptors are the killer cell immunoglobulin-like receptors (KIRs), which are members of the immunoglobulin (Ig) superfamily. Members of this family all contain a structural domain called an Ig fold, first identified in antibody (also known as Ig) molecules, discussed in Chapter 5. KIRs bind a variety of class I MHC molecules. A second important group of NK inhibitory receptors belong to the C-type lectin family, which includes proteins with carbohydrate-binding properties, as discussed earlier. One of these receptors is a heterodimer called CD94/NKG2A, which recognizes a class I MHC molecule called HLA-E. Interestingly, HLA-E displays peptides derived from other class I MHC molecules, so in essence, CD94/NKG2A is a surveillance receptor for several different class I MHC molecules. A third family of NK inhibitory receptors, called the leukocyte Ig-like receptors (LIRs), are also Ig superfamily members that bind class I MHC molecules, albeit with lower affinity than the KIRs, and are more highly expressed on B cells than on NK cells.
Activating receptors on NK cells recognize a heterogeneous group of ligands, some of which may be expressed on normal cells and others of which are expressed mainly on cells that have undergone stress, are infected with microbes, or are transformed. The molecular features of the ligands for many of these receptors are not well characterized. The induced expression of ligands on unhealthy cells that bind to activating receptors on NK cells may lead to signals that overwhelm the signals from inhibitory receptors, especially if class I MHC is also reduced or lost on the unhealthy cell (see Fig. 4-6).
Most activating NK receptors share the common feature of a structural motif in their cytoplasmic tails, called an immunoreceptor tyrosine-based activation motif (ITAM), which engages in signaling events that promote target cell killing and cytokine secretion (see Fig. 4-7). In some of these receptors, a single polypeptide chain contains the ITAM as well as the extracellular ligand-binding portion. In other receptors, the ITAMs are in separate polypeptide chains, such as FcεRIγ, ζ, and DAP12, that do not bind ligand but are noncovalently associated with the ligand-binding chain. ITAMs are also found in cytoplasmic tails of other multichain signaling receptors in the immune system, including the antigen receptors on T and B cells. After ligand binding to the NK cell activating receptors, tyrosine residues within the ITAMs become phosphorylated by cytoplasmic kinases, other protein kinases are recruited to the modified ITAMs and become activated, and these kinases contribute to further signaling by phosphorylating additional proteins. The structure and signaling functions of ITAMs are discussed in more detail in Chapter 7.
Many of the NK cell activating receptors are members of the C-type lectin or KIR families, which also include inhibitory receptors, as discussed before. Some of the activating receptors appear to bind class I MHC molecules, like the inhibitory receptors, but it is not known how these receptors are preferentially activated by infected or damaged cells. It is also clear that the activating receptors recognize ligands other than classical MHC molecules. One well-studied NK cell activating receptor in the C-type lectin family is NKG2D, which binds class I MHC–like proteins, including MIC-A and MIC-B, that are found on virally infected cells and tumor cells but not normal cells. The NKG2D receptor associates with a signaling subunit named DAP10, which has a signaling motif different from the ITAMs in other activating receptors but also enhances NK cell cytotoxicity against target cells.
Another important activating receptor on NK cells is CD16 (FcγRIIIa), which is a low-affinity receptor for IgG antibodies. Antibody molecules have highly variable antigen-binding ends, and on the opposite end, they have an invariant structure, called the Fc region, that interacts with various other molecules in the immune system. We will describe the structure of antibodies in detail in Chapter 5 but, for now, it is sufficient to know that CD16 binds to the Fc regions of certain types of antibodies called IgG1 or IgG3. CD16 associates with one of three different ITAM-containing signaling proteins (e.g., FcεRIγ, ζ, and DAP12 proteins). During an infection, the adaptive immune system produces IgG1 and IgG3 antibodies that specifically bind to the infecting microbes and their antigens on infected cells, and CD16 on NK cells can bind to the Fc parts of these antibodies. As a result, CD16 generates activating signals, through the associated signaling partners, and the NK cells may kill the infected cells that have been coated with antibody molecules. This process is called antibody-dependent cell-mediated cytotoxicity; it is an effector function of adaptive immunity and will be discussed in Chapter 12 when we consider humoral immunity.
The ability of activating receptors to induce functional responses in NK cells is enhanced by cytokines. The major cytokines of the innate immune system that stimulate NK function are IL-12, IL-15, IL-18, and type I interferons (discussed later). Each of these cytokines enhances the cytotoxic activity of NK cells and the amount of the cytokine IFN-γ the NK cells secrete. IFN-γ has various antimicrobial effects and will be discussed in detail in Chapter 10. In addition, IL-12 and IL-15 are important growth factors for NK cells.
KIR genes are polymorphic, meaning that there are several allelic variants in the human population, and groups of KIR alleles are often inherited together from a single parent. These groups of linked genes are called KIR haplotypes. There are two major KIR haplotypes and some rarer ones. Haplotypes differ in the number of receptors encoded, and some have more or fewer activating receptors than others. Some haplotypes are associated with increased susceptibility to some diseases, including spontaneous abortion and uveitis.
Effector Functions of NK Cells
The effector functions of NK cells are to kill infected cells and to activate macrophages to destroy phagocytosed microbes (Fig. 4-8). The mechanism of NK cell–mediated cytotoxicity is essentially the same as that of CD8+ CTLs, which we will describe in detail in Chapter 10. NK cells, like CTLs, have granules containing proteins that mediate killing of target cells. When NK cells are activated, granule exocytosis releases these proteins adjacent to the target cells. One NK cell granule protein, called perforin, facilitates the entry of other granule proteins, called granzymes, into the cytoplasm of target cells. The granzymes are enzymes that initiate a sequence of signaling events that cause death of the target cells by apoptosis. The signaling pathways that cause apoptosis are discussed in Chapter 14. By killing cells infected by viruses and intracellular bacteria, NK cells eliminate reservoirs of infection. Some tumors, especially those of hematopoietic origin, are targets of NK cells, perhaps because the tumor cells do not express normal levels or types of class I MHC molecules.
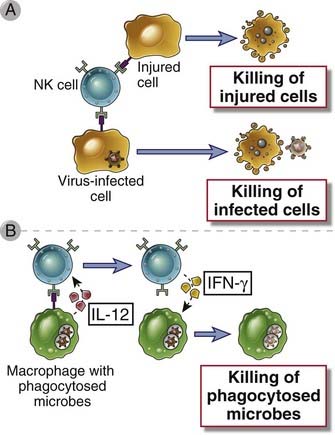
FIGURE 4–8 Functions of NK cells.
A, NK cells recognize ligands on infected cells or cells undergoing other types of stress and kill the host cells. In this way, NK cells eliminate reservoirs of infection as well as dysfunctional cells. B, NK cells respond to IL-12 produced by macrophages and secrete IFN-γ, which activates the macrophages to kill phagocytosed microbes.
NK cell–derived IFN-γ serves to activate macrophages, like IFN-γ produced by T cells, and increases the capacity of macrophages to kill phagocytosed bacteria (see Chapter 10). IFN-γ produced by NK cells in lymph nodes can also direct the differentiation of naive T cells into TH1 cells (see Chapter 9).
NK cells play several important roles in defense against intracellular microbes. They kill virally infected cells before antigen-specific CTLs can become fully active, that is, during the first few days after viral infection. Early in the course of a viral infection, NK cells are expanded and activated by IL-12 and IL-15, and they kill infected cells, especially those that display reduced levels of class I MHC molecules. In addition, IFN-γ secreted by NK cells activates macrophages to destroy phagocytosed microbes. This IFN-γ–dependent NK cell–macrophage reaction can control an infection with intracellular bacteria such as Listeria monocytogenes for several days or weeks and thus allow time for T cell–mediated immunity to develop and eradicate the infection. Depletion of NK cells leads to increased susceptibility to infection by some viruses and intracellular bacteria. In mice lacking T cells, the NK cell response may be adequate to keep infection with such microbes in check for some time, but the animals eventually succumb in the absence of T cell–mediated immunity. NK cells may also be important later in the body’s response to infection by killing infected cells that have escaped CTL-mediated immune attack by reducing expression of class I MHC molecules. Because NK cells can kill certain tumor cells in vitro, it has also been proposed that NK cells serve to kill malignant clones in vivo.
T and B Lymphocytes with Limited Antigen Receptor Specificities
As we will discuss in greater detail in later chapters, most T and B lymphocytes are components of the adaptive immune system and are characterized by a highly diverse repertoire of specificities for different antigens. The diversity of antigen receptors is generated by random somatic recombination of a large set of germline DNA segments as well as modification of nucleotide sequences at the junctions between the recombined segments, yielding unique antigen receptor genes in each lymphocyte clone (see Chapter 8). However, certain subsets of T and B lymphocytes have very little diversity because the same antigen receptor gene DNA segments are recombined in each clone and there is little or no modification of junctional sequences. It appears that these T and B cell subsets recognize structures expressed by many different or commonly encountered microbial species; in other words, they recognize PAMPs. T cell subsets with limited antigen receptor diversity include invariant natural killer T cells (iNKT), γδ T cells, and intraepithelial T cells with αβ TCRs (mentioned earlier). B cell subsets that produce antibodies with a limited set of specificities include B-1 B cells and marginal zone B cells. Although these T and B cells perform similar effector functions as do their more clonally diverse counterparts, the nature of their specificities places them in a special category of lymphocytes that is akin more to effector cells of innate immunity than to cells of adaptive immunity. These special T and B cell subsets are described in Chapters 10 and 11, respectively.
Mast Cells
Mast cells are present in the skin and mucosal epithelium and rapidly secrete proinflammatory cytokines and lipid mediators in response to infections and other stimuli. We introduced mast cells in Chapter 2. Recall that these cells contain abundant cytoplasmic granules containing various inflammatory mediators that are released when the cells are activated, either by microbial products or by a special antibody-dependent mechanism. The granule contents include vasoactive amines (such as histamine) that cause vasodilation and increased capillary permeability, and proteolytic enzymes that can kill bacteria or inactivate microbial toxins. Mast cells also synthesize and secrete lipid mediators (such as prostaglandins) and cytokines (such as TNF). Because mast cells are usually located adjacent to blood vessels (see Fig. 2-1), their released granule contents rapidly induce changes in the blood vessels that promote acute inflammation. Mast cells express TLRs, and TLR ligands can induce mast cell degranulation. Mast cell–deficient mice are impaired in controlling bacterial infections, probably because of impaired innate immune responses. Mast cell products also provide defense against helminths and are responsible for symptoms of allergic diseases. We will return to a detailed discussion of mast cells in relation to allergic diseases in Chapter 19.
Soluble Recognition and Effector Molecules of Innate Immunity
Several different kinds of molecules that recognize microbes and promote innate responses exist in soluble form in the blood and extracellular fluids. These molecules provide early defense against pathogens that are present outside host cells at some part of their life cycle. The soluble effector molecules function in two major ways.
The soluble effector molecules are sometimes called the humoral branch of innate immunity, analogous to the humoral branch of adaptive immunity mediated by antibodies. The major components of the humoral innate immune system are natural antibodies, the complement system, collectins, pentraxins, and ficolins. We will next describe the major features and functions of these components of innate immunity.
Natural Antibodies
Many antibodies with millions of different fine specificities are produced in humoral immune responses by B lymphocytes and their progeny, as part of the adaptive immune system, and we will describe antibodies and B cell responses in detail in later chapters. However, there are subsets of B cells that produce antibodies with only a limited number of specificities without overt exposure to foreign antigens, and these are called natural antibodies. As is typical for other components of innate immunity, natural antibodies are already present before infections, and they recognize common molecular patterns on microbes or stressed and dying cells. Natural antibodies are usually specific for carbohydrate or lipid molecules but not proteins, and most are IgM antibodies, one of several structural classes of Ig molecules (see Chapter 5). A remarkably large proportion of the natural antibodies in humans and mice are specific for oxidized lipids, including phospholipid head groups such as lysophosphatidylcholine and phosphorylcholine, which are found on bacterial membranes and on apoptotic cells but are not exposed on the surface of healthy host cells. Some experimental evidence indicates that the natural antibodies specific for these phospholipids provide protection against bacterial infections and facilitate the phagocytosis of apoptotic cells. The anti-ABO blood group antibodies, another example of natural antibodies, recognize certain glycolipids (blood group antigens) expressed on the surface of many cell types, including blood cells. Blood group antigens and antibodies are important for transplantation but not for host defense and are discussed in Chapter 16.
The Complement System
The complement system consists of several plasma proteins that work together to opsonize microbes, to promote the recruitment of phagocytes to the site of infection, and in some cases to directly kill the microbes (Fig. 4-9). Complement activation involves proteolytic cascades, in which an inactive precursor enzyme, called a zymogen, is altered to become an active protease that cleaves and thereby induces the proteolytic activity of the next complement protein in the cascade. As the cascade proceeds, the enzymatic activities result in tremendous amplification of the amount of proteolytic products that are generated. These products perform the effector functions of the complement system. Other proteolytic cascades include the blood coagulation pathways and the kinin-kallikrein system that regulates vascular permeability.
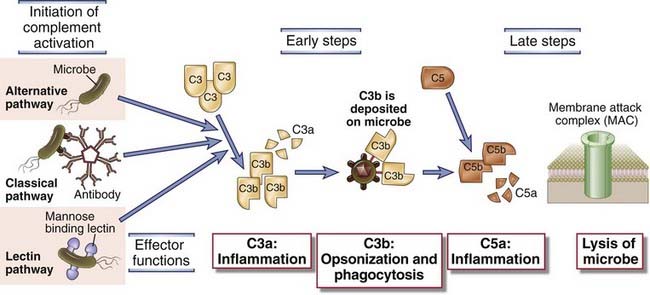
FIGURE 4–9 Pathways of complement activation.
The activation of the complement system may be initiated by three distinct pathways, all of which lead to the production of C3b (the early steps). C3b initiates the late steps of complement activation, culminating in the production of peptides that stimulate inflammation (C5a) and polymerized C9, which forms the membrane attack complex, so called because it creates holes in plasma membranes. The principal functions of major proteins produced at different steps are shown. The activation, functions, and regulation of the complement system are discussed in much more detail in Chapter 12.
The first step in activation of the complement system is recognition of molecules on microbial surfaces but not host cells, and this occurs in three ways, each referred to as a distinct pathway of complement activation.
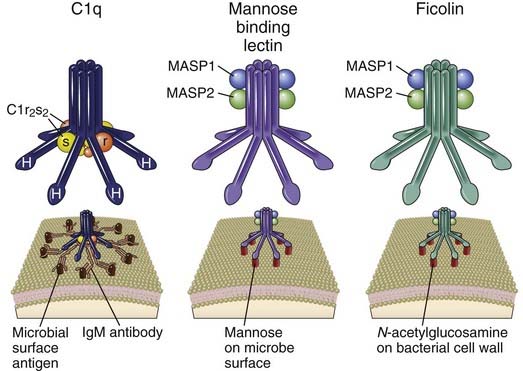
FIGURE 4–10 C1, mannose-binding lectin, and ficolin.
These three homologous pentameric proteins can all initiate complement activation on binding to their ligands on cell surfaces. C-type lectin–like globular heads at the end of collagenous-like stalks in the C1q and mannose-binding lectin proteins bind the Fc regions of IgM or mannose on the surface of microbes, respectively. Fibrinogen-like globular heads on ficolin bind N-acetylglucosamine on the surface of microbes. Binding results in conformational changes that activate the serine protease activity of C1r and C1s, associated with C1q, or MASP1 and MASP2, associated with mannose-binding lectin and ficolin.
Recognition of microbes by any of the three complement pathways results in sequential recruitment and assembly of additional complement proteins into protease complexes (see Fig. 4-9). One of these complexes, called C3 convertase, cleaves the central protein of the complement system, C3, producing C3a and C3b. The larger C3b fragment becomes covalently attached to the microbial surface where the complement pathway was activated. C3b serves as an opsonin to promote phagocytosis of the microbes. A smaller fragment, C3a, is released and stimulates inflammation by acting as a chemoattractant for neutrophils. C3b binds other complement proteins to form a protease called C5 convertase that cleaves C5, generating a secreted peptide (C5a) and a larger fragment (C5b) that remains attached to the microbial cell membranes. C5a is also a chemoattractant; in addition, it induces changes in blood vessels that make them leak plasma proteins and fluid into sites of infections. C5b initiates the formation of a complex of the complement proteins C6, C7, C8, and C9, which are assembled into a membrane pore, called the membrane attack complex (MAC), that causes lysis of the cells where complement is activated.
The complement system is an essential component of innate immunity, and patients with deficiencies in C3 are highly susceptible to recurrent, often lethal, bacterial infections. However, genetic deficiencies in MAC formation (the terminal product of the classical pathway) increase susceptibility to only a limited number of microbes, notably Neisseria bacteria, which have thin cell walls that make them especially susceptible to the lytic action of the MAC. The complement system will be discussed in more detail in Chapter 12.
Pentraxins
Several plasma proteins that recognize microbial structures and participate in innate immunity belong to the pentraxin family, which is a phylogenetically old group of structurally homologous pentameric proteins. Prominent members of this family include the short pentraxins C-reactive protein (CRP) and serum amyloid P (SAP) and the long pentraxin PTX3. Both CRP and SAP bind to several different species of bacteria and fungi. The molecular ligands recognized by CRP and SAP include phosphorylcholine and phosphatidylethanolamine, respectively, which are found on bacterial membranes and on apoptotic cells, as discussed earlier. PTX3 recognizes various molecules on fungi, selected gram-positive and gram-negative bacteria, and viruses. CRP, SAP, and PTX3 all activate complement by binding C1q and initiating the classical pathway.
Plasma concentrations of CRP are very low in healthy individuals but can increase up to 1000-fold during infections and in response to other inflammatory stimuli. The increased levels of CRP are a result of increased synthesis by the liver induced by the cytokines IL-6 and IL-1, which are produced by phagocytes as part of the innate immune response. Liver synthesis and plasma levels of several other proteins, including SAP and others unrelated to the pentraxins, also increase in response to IL-1 and IL-6, and as a group these plasma proteins are called acute-phase reactants.
PTX3 is produced by several cell types, including dendritic cells, macrophages, and endothelial cells, in response to TLR ligands and inflammatory cytokines, such as TNF, but it is not an acute-phase reactant. PTX3 is also stored in neutrophil granules and released as neutrophils die. PTX3 recognizes apoptotic cells and certain microorganisms. Studies with knockout mice reveal that PTX3 provides protection against some microbes, including the fungus Aspergillus fumigatus.
Collectins and Ficolins
The collectins are a family of trimeric or hexameric proteins, each subunit of which contains a collagen-like tail connected by a neck region to a calcium-dependent (C-type) lectin head. Three members of this family serve as soluble effector molecules in the innate immune system; these are mannose-binding lectin (MBL) and pulmonary surfactant proteins SP-A and SP-D.
MBL, which is a soluble pattern recognition receptor that binds carbohydrates with terminal mannose and fucose, was discussed earlier in relation to the lectin pathway of complement activation (see Fig. 4-10). MBL can also function as an opsonin by binding to and enhancing phagocytosis of microbes. Recall that opsonins simultaneously bind microbes and a surface receptor on phagocyte membranes, and in the case of MBL, the surface receptor is called the C1q receptor because it also binds C1q. This receptor mediates the internalization of microbes that are opsonized by MBL. The gene encoding MBL is polymorphic, and certain alleles are associated with impaired hexamer formation and reduced blood levels. Low MBL levels are associated with increased susceptibility to a variety of infections, especially in combination with other immunodeficiency states.
Surfactant protein A (SP-A) and surfactant protein D (SP-D) are collectins with lipophilic surfactant properties shared by other surfactants. They are found in the alveoli of the lungs, and their major functions appear to be as mediators of innate immune responses in the lung. They bind to various microorganisms and act as opsonins, facilitating ingestion by alveolar macrophages. SP-A and SP-D can also directly inhibit bacterial growth, and they may activate macrophages. SP-A– and SP-D–deficient mice have impaired abilities to resist a variety of pulmonary infections.
Ficolins are plasma proteins that are structurally similar to collectins, possessing a collagen-like domain, but instead of a C-type lectin domain, they have a fibrinogen-type carbohydrate recognition domain (see Fig. 4-10). Ficolins have been shown to bind several species of bacteria, opsonizing them and activating complement in a manner similar to that of MBL. The molecular ligands of the ficolins include N-acetylglucosamine and the lipoteichoic acid component of the cell walls of gram-positive bacteria.
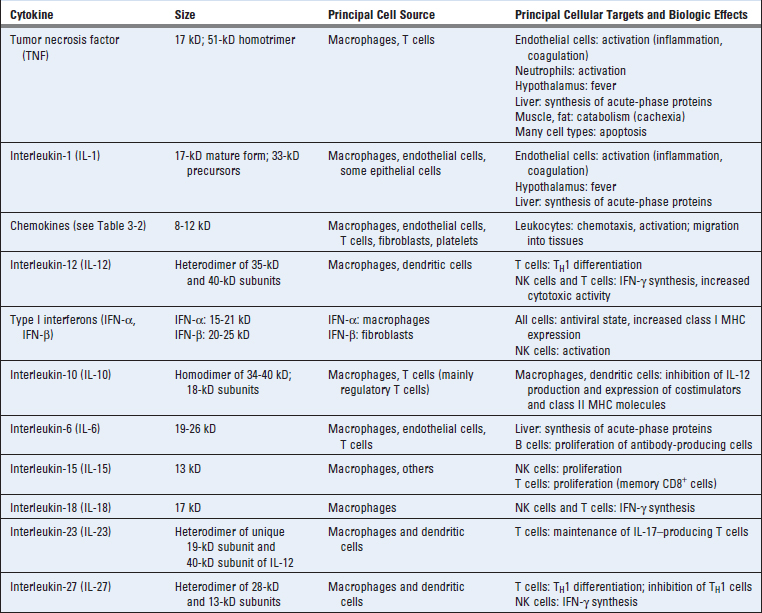
Now that we have discussed the general properties and various components of the innate immune system, including the cells, cellular pathogen recognition receptors, and soluble recognition and effector molecules, we can consider how these various components work to protect against pathogens. The three major ways in which the innate immune system protects against infections is by inducing inflammation, inducing antiviral defense, and stimulating adaptive immunity. Many of these reactions are mediated by cytokines, which serve diverse and important roles in innate immunity (Table 4-4). As we discuss below, these cytokines act mainly close to their site of production (paracrine actions), but some of them can also have distant effects (endocrine actions).
The Inflammatory Response
The major way by which the innate immune system deals with infections and tissue injury is to stimulate acute inflammation, which is the accumulation of leukocytes, plasma proteins, and fluid derived from the blood at an extravascular tissue site of infection or injury. The leukocytes and plasma proteins normally circulate in the blood and are recruited to sites of infection and injury, where they perform various effector functions that serve to kill microbes and begin to repair tissue damage. Typically, the most abundant leukocyte that is recruited from the blood into acute inflammatory sites is the neutrophil, but blood monocytes, which become macrophages in the tissue, become increasingly prominent over time and may be the dominant population in some reactions. Among the important plasma proteins that enter inflammatory sites are complement proteins, antibodies, and acute-phase reactants. The delivery of these blood-derived components to the inflammatory site is dependent on reversible changes in blood vessels in the infected or damaged tissue. These changes include increased blood flow into the tissue due to arteriolar dilation, increased adhesiveness of circulating leukocytes to the endothelial lining of venules, and increased permeability of the capillaries and venules to plasma proteins and fluid. All these changes are induced by cytokines and small-molecule mediators initially derived from resident cells in the tissue, such as mast cells, macrophages, and endothelial cells, in response to PAMP or DAMP stimulation. As the inflammatory process develops, the mediators may be derived from newly arrived and activated leukocytes and complement proteins.
Acute inflammation can develop in minutes to hours and last for days. Chronic inflammation is a process that takes over from acute inflammation if the infection is not eliminated or the tissue injury is prolonged. It usually involves recruitment and activation of monocytes and lymphocytes. Chronic inflammatory sites also often undergo tissue remodeling, with angiogenesis and fibrosis. Although innate immune stimuli may contribute to chronic inflammation, the adaptive immune system may also be involved because cytokines produced by T cells are powerful inducers of inflammation (see Chapter 10). Detailed descriptions of the various mediators and pathologic manifestations of acute and chronic inflammation can be found in pathology textbooks. We will focus our discussion on particular aspects of the acute inflammatory process that have broad relevance to both innate and adaptive immunity and immune-mediated inflammatory diseases.
The Major Proinflammatory Cytokines TNF, IL-1, and IL-6
One of the earliest responses of the innate immune system to infection and tissue damage is the secretion of cytokines by tissue cells, which is critical for the acute inflammatory response. Three of the most important proinflammatory cytokines of the innate immune system are TNF, IL-1 (which we have mentioned already several times), and IL-6 (see Table 4-4). Tissue macrophages and mast cells are the major source of these cytokines, although other cell types, including endothelial and epithelial cells, can also produce IL-1 and IL-6. We will discuss the major features of these cytokines, focusing mainly on TNF and IL-1, before describing their role in acute inflammation.
Tumor Necrosis Factor
Tumor necrosis factor (TNF) is a mediator of the acute inflammatory response to bacteria and other infectious microbes. The name of this cytokine derives from its original identification as a serum substance (factor) that caused necrosis of tumors, now known to be the result of local inflammation and thrombosis of tumor blood vessels. TNF is also called TNF-α to distinguish it from the closely related TNF-β, also called lymphotoxin. TNF is produced by macrophages, dendritic cells, and other cell types. In macrophages, it is synthesized as a nonglycosylated type II membrane protein and is expressed as a homotrimer, which is able to bind to one form of TNF receptor. The membrane form of TNF is cleaved by a membrane-associated metalloproteinase, releasing a polypeptide fragment, and three of these polypeptide chains polymerize to form a triangular pyramid-shaped circulating TNF protein (Fig. 4-11). The receptor-binding sites are at the base of the pyramid, allowing simultaneous binding of the cytokine to three receptor molecules.
FIGURE 4–11 Structure of the TNF receptor with bound lymphotoxin.
The ribbon structure depicts a top view of a complex of three TNF receptors (TNF-RI) and one molecule of the bound cytokine, revealed by x-ray crystallography. Lymphotoxin is a homotrimer in which the three subunits are colored dark blue. The lymphotoxin homotrimer forms an inverted three-sided pyramid with its base at the top and its apex at the bottom. Three TNF-RI molecules, colored magenta, cyan, and red, bind one homotrimer of lymphotoxin, with each receptor molecule interacting with two different lymphotoxin monomers in the homotrimer complex. Disulfide bonds in the receptor are colored yellow. TNF is homologous to lymphotoxin and presumably binds to its receptors in the same way.
(From Banner DW, et al., Cell: Crystal structure of the soluble human 55 kd TNF receptor–human TNFβ complex: 73:431-445. © Cell Press, 1993).
There are two distinct TNF receptors called type I (TNF-RI) and type II (TNF-RII). The affinities of TNF for its receptors are unusually low for a cytokine, the Kd being only ~1 × 10−9 M for binding to TNF-RI and approximately 5 × 10−10 M for binding to TNF-RII. Both TNF receptors are present on most cell types. The TNF receptors are members of a large family of proteins called the TNF receptor superfamily, many of which are involved in immune and inflammatory responses. These receptors exist as trimers in the plasma membrane. Cytokine binding to some TNF receptor family members, such as TNF-RI, TNF-RII, and CD40, leads to the recruitment of proteins, called TNF receptor–associated factors (TRAFs), to the cytoplasmic domains of the receptors. The TRAFs activate transcription factors, notably NF-κB and AP-1. Cytokine binding to other family members, such as TNF-RI, leads to recruitment of an adaptor protein that activates caspases and triggers apoptosis. Thus, different members of the TNF receptor family can induce gene expression or cell death, and some can do both (see Chapter 7).
TNF production by macrophages is stimulated by PAMPs and DAMPs. TLRs, NLRs, and RLRs can all induce TNF gene expression, in part by activation of the NF-κB transcription factor. Many different microbial products can therefore induce TNF production. Large amounts of this cytokine may be produced during infections with gram-negative and gram-positive bacteria, which release the TLR ligands LPS and lipoteichoic acid, respectively, from their cell walls. Septic shock, a life-threatening condition caused when bacteria enter the blood stream, is mediated in large part by TNF. We will discuss septic shock later in this chapter.
Interleukin-1
Interleukin-1 (IL-1) is also a mediator of the acute inflammatory response and has many similar actions as TNF. The major cellular source of IL-1, like that of TNF, is activated mononuclear phagocytes. Unlike TNF, IL-1 is also produced by many cell types other than macrophages, such as neutrophils, epithelial cells (e.g., keratinocytes), and endothelial cells. There are two forms of IL-1, called IL-1α and IL-1β, that are less than 30% homologous to each other, but they bind to the same cell surface receptors and have the same biologic activities. The main biologically active secreted form is IL-1β.
IL-1 production usually requires two distinct signals, one that activates new gene transcription and production of a 33-kD precursor pro–IL-1β polypeptide, and a second signal that activates the inflammasome to proteolytically cleave the precursor to generate the 17-kD mature IL-1β protein (see Fig. 4-4). As discussed earlier in the chapter, IL-1β gene transcription is induced by TLR and NOD signaling pathways that activate NF-κB, whereas pro–IL-1β cleavage is mediated by the NLRP3 inflammasome. IL-1 is secreted by a nonclassical pathway because, unlike most secreted proteins, neither IL-1α nor IL-1β has hydrophobic signal sequences to target the nascent polypeptide to the endoplasmic reticulum membrane. One possibility is that mature IL-1 is released mainly when infected cells or activated macrophages die. Some pathogenic bacteria induce both inflammasome-mediated processing of IL-1β and IL-18 in macrophages and caspase-1–dependent cell death, which leads to the release of the inflammatory cytokines. TNF can also stimulate phagocytes and other cell types to produce IL-1. This is an example of a cascade of cytokines that have similar biologic activities.
IL-1 mediates its biologic effects through a membrane receptor called the type I IL-1 receptor, which is expressed on many cell types, including endothelial cells, epithelial cells, and leukocytes. This receptor is an integral membrane protein that contains an extracellular ligand-binding Ig domain and a Toll/IL-1 receptor (TIR) signaling domain in the cytoplasmic region, described earlier in reference to TLRs. The signaling events that occur when IL-1 binds to the type I IL-1 receptor are similar to those triggered by TLRs and result in the activation of NF-κB and AP-1 transcription factors (see Chapter 7). The type II IL-1 receptor appears incapable of activating downstream signals.
Interleukin-6
IL-6 is another important cytokine in acute inflammatory responses that has both local and systemic effects, including the induction of liver synthesis of a variety of other inflammatory mediators, the stimulation of neutrophil production in the bone marrow, and the differentiation of IL-17–producing helper T cells. IL-6 is synthesized by mononuclear phagocytes, vascular endothelial cells, fibroblasts, and other cells in response to PAMPs and in response to IL-1 and TNF. IL-6 is a homodimer of type I cytokine family polypeptides. The receptor for IL-6 consists of a cytokine-binding polypeptide chain and a signal-transducing subunit (called gp130) that is also the signaling component of receptors for other cytokines. The IL-6 receptor engages a signaling pathway that activates the transcription factor STAT3.
Recruitment of Leukocytes to Sites of Infection
Recruitment of large numbers of neutrophils, followed by monocytes, from blood into tissues typically occurs as part of the acute inflammatory response to infections and tissue injury. The cytokines TNF, IL-1, and IL-6 and chemokines, which are secreted in the local sites of infection or tissue injury, have multiple effects on vascular endothelial cells, leukocytes, and bone marrow, which together increase the local delivery of cells that can fight infections and repair tissues (see Fig. 3-3, Chapter 3). Leukocyte recruitment was described in Chapter 3 and will be only briefly considered here.
Both TNF and IL-1 induce postcapillary venule endothelial cells to express E-selectin and to increase their expression of ICAM-1 and VCAM-1, the ligands for leukocyte integrins. These changes in endothelial adhesion molecule expression are the result of TNF and IL-1 activation of transcription factors, including NF-κB, leading to new adhesion molecule gene transcription. P-selectin expression is also induced on venular endothelial cells at sites of infection and tissue injury, but in large part, this is due to the effects of histamine and thrombin, which stimulate the rapid mobilization of P-selectin stored in granules in the endothelial cell to the cell surface.
TNF and IL-1 also stimulate various cells to secrete chemokines, such as CXCL1 and CCL2, that bind to receptors on neutrophils and monocytes, respectively, increase the affinity of leukocyte integrins for their ligands, and stimulate directional movement of leukocytes. The result of increased selectin, integrin, and chemokine expression is an increase in neutrophil and monocyte adhesion to endothelial cells and transmigration through the vessel wall. The leukocytes that accumulate in the tissues compose an inflammatory infiltrate. The actions of TNF on endothelium and leukocytes are critical for local inflammatory responses to microbes. If inadequate quantities of TNF are present (e.g., in patients treated with drugs that block TNF or in TNF gene knockout mice), a consequence may be failure to contain infections.
In addition, TNF, IL-1, and IL-6 produced at inflammatory sites may enter the blood and be delivered to the bone marrow, where they enhance the production of neutrophils from bone marrow progenitors, usually acting in concert with colony-stimulating factors. In this way, these cytokines increase the supply of cells that can be recruited to the sites of infection.
Phagocytosis and Killing of Microbes by Activated Phagocytes
Neutrophils and macrophages that are recruited into sites of infections ingest microbes into vesicles by the process of phagocytosis and destroy these microbes (Fig. 4-12). Phagocytosis is an active, energy-dependent process of engulfment of large particles (>0.5 µm in diameter) into vesicles. Phagocytic vesicles fuse with lysosomes, where the ingested particles are destroyed, and in this way, the mechanisms of killing, which could potentially injure the phagocyte, are isolated from the rest of the cell.
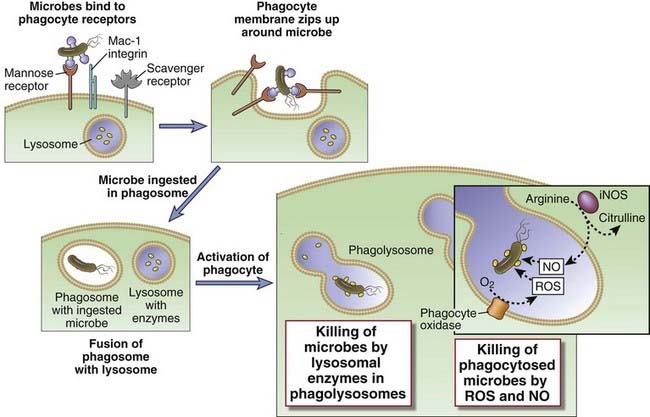
FIGURE 4–12 Phagocytosis and intracellular destruction of microbes.
Microbes may be ingested by different membrane receptors of phagocytes; some directly bind microbes, and others bind opsonized microbes. (Note that the Mac-1 integrin binds microbes opsonized with complement proteins, not shown.) The microbes are internalized into phagosomes, which fuse with lysosomes to form phagolysosomes, where the microbes are killed by reactive oxygen and nitrogen species and proteolytic enzymes. iNOS, inducible nitric oxide synthase; NO, nitric oxide; ROS, reactive oxygen species.
Neutrophils and macrophages express receptors that specifically recognize microbes, and binding of microbes to these receptors is the first step in phagocytosis. Some of these receptors are pattern recognition receptors, including C-type lectins and scavenger receptors, which we discussed previously. Pattern recognition receptors can contribute to phagocytosis only of organisms that express particular molecular patterns, such as mannose for the mannose receptor. Phagocytes also have high-affinity receptors for certain opsonins, including antibody molecules, complement proteins, and plasma lectins; these receptors are critical for phagocytosis of many different microbes that are coated with the opsonins. One of the most efficient systems for opsonizing microbes is coating them with antibodies. Recall that antibody molecules have antigen-binding sites at one end, and the other end, the Fc region, interacts with effector cells and molecules of the innate immune system. There are several types of antibodies, which we will discuss in detail in Chapters 5 and 12. Phagocytes express high-affinity Fc receptors called FcγRI specific for one type of antibody called IgG (see Chapter 12). Thus, if an individual responds to an infection by making IgG antibodies against microbial antigens, the IgG molecules bind to these antigens, the Fc ends of the bound antibodies can interact with FcγRI on phagocytes, and the end result is efficient phagocytosis of the microbes. Because many different antibodies may be produced that bind to many different microbial products, antibody-mediated opsonization contributes to the phagocytosis of a broader range of microbes than do pattern recognition receptors. Antibody-dependent phagocytosis illustrates a link between innate and adaptive immunity—antibodies are a product of the adaptive immune system (B lymphocytes) that engage innate immune system effector cells (phagocytes) to perform their protective functions.
Once a microbe or particle binds to receptors on a phagocyte, the plasma membrane in the region of the receptors begins to redistribute and extends a cup-shaped projection around the microbe. When the protruding membrane cup extends beyond the diameter of the particle, the top of the cup closes over, or “zips up,” and pinches off the interior of the cup to form an inside-out intracellular vesicle (see Fig. 4-12). This vesicle, called a phagosome, contains the ingested foreign particle, and it breaks away from the plasma membrane. The cell surface receptors also deliver activating signals that stimulate the microbicidal activities of phagocytes. Phagocytosed microbes are destroyed, as described next; at the same time, peptides are generated from microbial proteins and presented to T lymphocytes to initiate adaptive immune responses (see Chapter 6).
Activated neutrophils and macrophages kill phagocytosed microbes by the action of microbicidal molecules in phagolysosomes (see Fig. 4-12). Several receptors that recognize microbes, including TLRs, G protein–coupled receptors, antibody Fc and complement C3 receptors, and receptors for cytokines, mainly IFN-γ, function cooperatively to activate phagocytes to kill ingested microbes. Fusion of phagocytic vacuoles (phagosomes) with lysosomes results in the formation of phagolysosomes, where most of the microbicidal mechanisms are concentrated. Three types of microbicidal mechanisms are known to be the most important.
When neutrophils and macrophages are strongly activated, they can injure normal host tissues by release of lysosomal enzymes, ROS, and nitric oxide. The microbicidal products of these cells do not distinguish between self tissues and microbes. As a result, if these products enter the extracellular environment, they are capable of causing tissue injury.
Other Functions of Activated Macrophages
In addition to killing phagocytosed microbes, macrophages serve many other functions in defense against infections (Fig. 4-13). Several of these functions are mediated by the cytokines the macrophages produce. We have already described how TNF, IL-1, and chemokines made by phagocytes enhance the inflammatory reactions to microbes and bring in more leukocytes and plasma proteins. Activated macrophages also produce growth factors for fibroblasts and endothelial cells that participate in the remodeling of tissues after infections and injury. The role of macrophages in cell-mediated immunity is described in Chapter 10.
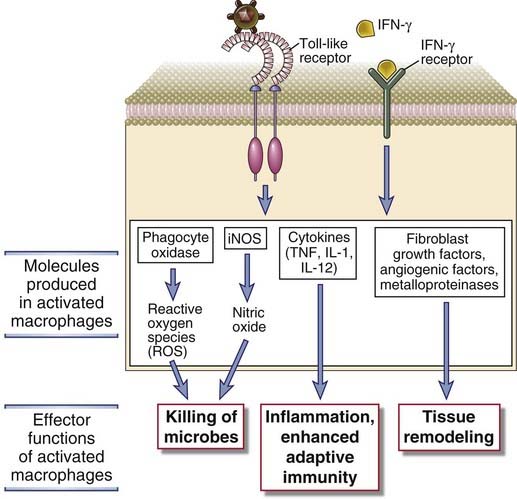
FIGURE 4–13 Effector functions of macrophages.
Macrophages are activated by microbial products such as LPS and by NK cell–derived IFN-γ (described earlier in the chapter). The process of macrophage activation leads to the activation of transcription factors, the transcription of various genes, and the synthesis of proteins that mediate the functions of these cells. In adaptive cell-mediated immunity, macrophages are activated by stimuli from T lymphocytes (CD40 ligand and IFN-γ) and respond in essentially the same way (see Chapter 10, Fig. 10-7).
Other Cytokines Produced During Innate Immune Responses
In addition to TNF, IL-1, and IL-6, dendritic cells and macrophages activated by PAMPs and DAMPs produce other cytokines that have important roles in innate immune responses (see Table 4-4). Some of the main features of these cytokines and their roles in innate immunity are discussed in this section. These cytokines also have important effects in stimulating adaptive immunity, as we will discuss later in this chapter and in more detail in Chapters 9 and 10.
IL-12 is secreted by dendritic cells and macrophages and stimulates IFN-γ production by NK cells and T cells enhances NK cell and CTL-mediated cytotoxicity, and promotes differentiation of Th1 cells. IL-12 exists as a disulfide-linked heterodimer of 35-kD (p35) and 40-kD (p40) subunits. The p35 subunit is a member of the type I cytokine family. In addition to IL-12, there are other heterodimeric cytokines whose subunits are homologous to either or both of the IL-12 p35 and p40 chains, including IL-23, IL-27, and IL-35. This is of significance because therapeutic antibodies specific for shared subunits are in development for treatment of inflammatory diseases, and some of these antibodies may block the function of more than one cytokine. The principal sources of IL-12 are activated dendritic cells and macrophages. Many cells appear to synthesize the p35 subunit, but macrophages and dendritic cells are the main cell types that produce the p40 component and therefore the biologically active cytokine. During innate immune reactions to microbes, IL-12 is produced in response to TLR and other pattern recognition receptor signaling induced by many microbial stimuli, including bacterial LPS or lipoteichoic acid and virus infections. IFN-γ produced by NK cells or T cells also stimulates IL-12 production, contributing to a positive feedback loop.
The receptor for IL-12 (IL-12R) is a heterodimer composed of β1 and β2 subunits, both of which are members of the type I cytokine receptor family. Both chains are required for high-affinity binding of IL-12 and for signaling, which activates the transcription factor STAT4. Expression of the β2 chain of the IL-12 receptor is itself enhanced by IFN-γ, whose production is stimulated by IL-12, and this is another example of a positive amplification loop in immune responses. Studies with gene knockout mice and the phenotype of rare patients with mutations in the IL-12 receptor support the conclusion that IL-12 is important for IFN-γ production by NK cells and T cells and for host resistance to intracellular bacteria and some viruses. For example, patients with mutations in the IL-12 receptor β1 subunit have been described, and they are highly susceptible to infections with intracellular bacteria, notably Salmonella and atypical mycobacteria. IL-12 secreted by DCs during antigen presentation to naive CD4+ T cells promotes their differentiation into the TH1 subset of helper T cells, which are important for defense against intracellular infections (see Chapter 9). This is a key way in which innate immunity shapes adaptive immune responses.
IL-18 enhances the functions of NK cells, similar to IL-12. Recall that the production of IL-18, like that of IL-1, is dependent on the inflammasome. Also like IL-1, IL-18 binds to a receptor that signals through a TIR domain.
IL-15 is cytokine that serves important growth-stimulating and survival functions for both NK cells and T cells. IL-15 is structurally homologous to the T cell growth factor IL-2, and the heterotrimeric IL-15 receptor shares two subunits with the IL-2 receptor. An interesting feature of IL-15 is that it can be expressed on the cell surface bound to the α chain of its receptor and in this form can be presented to and stimulate nearby cells that express a receptor composed of the other two chains (β and γ). IL-15 presented this way by dendritic cells to NK cells in lymph nodes activates signaling pathways that promote NK cell IFN-γ production. IL-15 also serves as a survival factor for NK and memory CD8+ T cells.
Systemic and Pathologic Consequences of the Acute Inflammatory Responses
TNF, IL-1, and IL-6 produced during the innate immune response to infection or tissue damage have systemic effects that contribute to host defense and are responsible for many of the clinical signs of infection and inflammatory disease (Fig. 4-14).
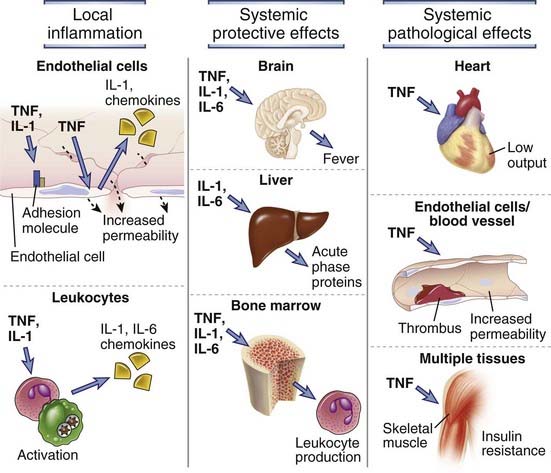
FIGURE 4–14 Local and systemic actions of cytokines in inflammation.
TNF, IL-1, and IL-6 have multiple local and systemic inflammatory effects. TNF and IL-1 act on leukocytes and endothelium to induce acute inflammation, and both cytokines induce the expression of IL-6 from leukocytes and other cell types. TNF, IL-1, and IL-6 mediate protective systemic effects of inflammation, including induction of fever, acute-phase protein synthesis by the liver, and increased production of leukocytes by the bone marrow. Systemic TNF can cause the pathologic abnormalities that lead to septic shock, including decreased cardiac function, thrombosis, capillary leak, and metabolic abnormalities due to insulin resistance.
In severe infections, TNF may be produced in large amounts and causes systemic clinical and pathologic abnormalities. If the stimulus for cytokine production is sufficiently strong, the quantity of TNF may be so large that it enters the blood stream and acts at distant sites as an endocrine hormone (see Fig. 4-14). The principal systemic actions of TNF are the following:
A complication of severe bacterial sepsis is a syndrome called septic shock, which may be caused by LPS released from gram-negative bacteria (in which case it is called endotoxin shock) or lipoteichoic acid from gram-positive bacteria. Septic shock is characterized by vascular collapse, disseminated intravascular coagulation, and metabolic disturbances. This syndrome is due to LPS- or lipoteichoic acid–induced TLR signaling leading to the production of TNF and other cytokines, including IL-12, IFN-γ, and IL-1. The concentration of serum TNF may be predictive of the outcome of severe bacterial infections. Septic shock can be reproduced in experimental animals by administration of LPS, lipoteichoic acid, or TNF. Antagonists of TNF can prevent mortality in the experimental models, but clinical trials with anti-TNF antibodies or with soluble TNF receptors have not shown benefit in patients with sepsis. The cause of this therapeutic failure is not known, but it may be because other cytokines elicit the same responses as TNF, an example of redundancy.
Acute inflammation may cause tissue injury because the effector mechanisms that phagocytes use to kill microbes are also highly toxic to host tissues. The proteolytic enzymes and reactive oxygen species produced by phagocytes that accumulate at a site of infection can injure host cells and degrade extracellular matrix if they are generated in large quantities, especially if the microbes resist being killed and continue to stimulate the innate immune responses. In fact, much of the pathology associated with infections is due to the inflammatory responses and not direct toxic effects of the microbes. Acute inflammation also causes tissue damage in the setting of autoimmune diseases, in which case neutrophils and macrophages accumulate and become activated secondarily to stimulation of the adaptive immune system by self antigens (see Chapter 14). As in inflammation induced by infections, TNF, IL-1, IL-6, and IL-12 are the key inducers of inflammation in autoimmune disease. Antagonists against TNF, IL-1, and IL-12 and antibodies against IL-6 receptors are in clinical use or in trials to reduce inflammation in patients with some of these diseases, such as rheumatoid arthritis, inflammatory bowel disease, and psoriasis.
The Antiviral Response
The major way by which the innate immune system deals with viral infections is to induce the expression of type I interferons, whose most important action is to inhibit viral replication. Earlier in the chapter, we have discussed how several of the pattern recognition receptors, including some TLRs, NLRs, and RLRs, generate signals that stimulate IFN-α and IFN-β gene expression in many different cell types. The type I interferons are secreted from these cells and act on other cells to prevent spread of viral infection. In this section, we will describe the major properties of type I interferons and the antiviral effects of these cytokines.
Type I interferons are a large family of structurally related cytokines that mediate the early innate immune response to viral infections. The term interferon derives from the ability of these cytokines to interfere with viral infection. There are many type I interferons, all of which have considerable structural homology and are encoded by genes in a single cluster on chromosome 9. The most important type I interferons in viral defense are IFN-α (which actually includes 13 different closely related proteins) and IFN-β, which is a single protein. Plasmacytoid dendritic cells are the major sources of IFN-α but it may also be produced by mononuclear phagocytes. IFN-β is produced by many cells. The most potent stimuli for type I interferon synthesis are viral nucleic acids. Recall that RIG-like receptors in the cytosol and TLRs 3, 7, 8, and 9, all in endosomal vesicles, recognize viral nucleic acids and initiate signaling pathways that activate the interferon regulatory factor (IRF) family of transcription factors, which induce type I interferon gene expression. In adaptive immunity, antigen-activated T cells stimulate mononuclear phagocytes to synthesize type I interferons. The receptor for type I interferons, which binds both IFN-α and IFN-β, is a heterodimer of two structurally related polypeptides, IFNAR1 and IFNAR2, expressed on all nucleated cells. This receptor signals to activate STAT1, STAT2, and IRF9 transcription factors, which induce expression of several different genes that have the following effects in antiviral defense (Fig. 4-15):
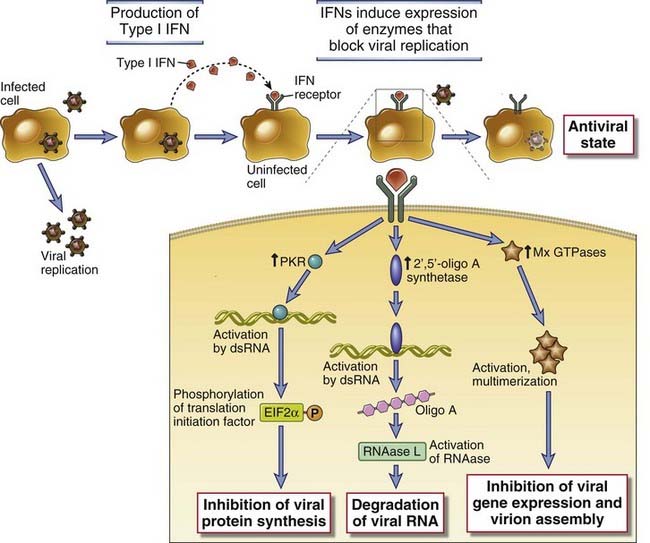
FIGURE 4–15 Biologic actions of type I interferons.
Type I interferons (IFN-α, IFN-β) are produced by virus-infected cells in response to intracellular TLR signaling and other sensors of viral RNA. Type I interferons bind to receptors on neighboring uninfected cells and activate JAK-STAT signaling pathways, which induce expression of genes whose products interfere with viral replication. Type I interferons also bind to receptors on infected cells and induce expression of genes whose products enhance the cell’s susceptibility to CTL-mediated killing.
Thus, the principal activities of type I interferon work in concert to combat viral infections. Knockout mice lacking the receptor for type I interferons are susceptible to viral infections. IFN-α is in clinical use as an antiviral agent in certain forms of viral hepatitis. IFN-α is also used for the treatment of some tumors, perhaps because it boosts CTL activity or interferes with cell growth. IFN-β is used as a therapy for multiple sclerosis, but the mechanism of its beneficial effect in this disease is not known.
Protection against viruses is due, in part, to the activation of intrinsic apoptotic death pathways in infected cells and enhanced sensitivity to extrinsic inducers of apoptosis. For example, virally infected cells can sense abnormal DNA replication and abnormal glycoprotein synthesis, leading to initiation of p53-dependent or endoplasmic reticulum–dependent apoptotic pathways, respectively. In addition, virally infected cells are sensitized to TNF-induced apoptosis. Abundant TNF is made by plasmacytoid dendritic cells and macrophages in response to viral infections, in addition to type I interferons. The type I TNF receptor engages both proinflammatory and proapoptosis death pathways (see Chapter 7). The dominant pathway that is activated upon TNF binding depends on the state of protein synthesis in the responding cells, and viral infection can shift this balance toward apoptosis.
Stimulation of Adaptive Immunity
The innate immune response provides signals that function in concert with antigen to stimulate the proliferation and differentiation of antigen-specific T and B lymphocytes. As the innate immune response is providing the initial defense against microbes, it also sets in motion the adaptive immune response. The activation of lymphocytes requires two distinct signals, the first being antigen and the second being molecules that are produced during innate immune responses to microbes or injured cells (Fig. 4-16). This idea is called the two-signal hypothesis for lymphocyte activation. The requirement for antigen (so-called signal 1) ensures that the ensuing immune response is specific. The requirement for additional stimuli triggered by innate immune reactions to microbes (signal 2) ensures that adaptive immune responses are induced when there is a dangerous infection and not when lymphocytes recognize harmless antigens, including self antigens. The molecules produced during innate immune reactions that function as second signals for lymphocyte activation include costimulators (for T cells), cytokines (for both T and B cells), and complement breakdown products (for B cells). We will return to the nature of second signals for lymphocyte activation in Chapters 9 and 11.
FIGURE 4–16 Stimulation of adaptive immunity by innate immune responses.
Antigen recognition by lymphocytes provides signal 1 for the activation of the lymphocytes, and molecules induced on host cells during innate immune responses to microbes provide signal 2. In this illustration, the lymphocytes are B cells, but the same principles apply to T lymphocytes. The nature of second signals differs for B and T cells and is described in later chapters.
The second signals generated during innate immune responses to different microbes not only enhance the magnitude of the subsequent adaptive immune response but also influence the nature of the adaptive response. A major function of T cell–mediated immunity is to activate macrophages to kill intracellular microbes and to induce robust acute inflammatory responses, beyond those directly induced by the innate immune system, so that a sufficiently large army of phagocytes is called into a site of infection. Infectious agents that engage TLRs and other pattern recognition receptors will tend to stimulate T cell–mediated immune responses. This is because the signaling from these pattern recognition receptors enhances the ability of antigen-presenting cells to induce the differentiation of naive CD4+ T cells into effector cells called TH1 and TH17 cells. TH1 cells produce the cytokine IFN-γ, which can activate macrophages to kill microbes that might otherwise survive within phagocytic vesicles. TH17 cells produce the cytokine IL-17, which can induce neutrophil-rich inflammation. TH1 and TH17 cell-mediated immunity is discussed in detail in Chapters 9 and 10. Many extracellular microbes that enter the blood activate the alternative complement pathway, which in turn enhances the production of antibodies by B lymphocytes. Some of these antibodies opsonize the bacteria and thereby promote their phagocytosis by neutrophils and macrophages. Therefore, humoral immune response serves to eliminate extracellular microbes. The role of complement in enhancing B cell activation is discussed in Chapter 11.
Cytokines produced by cells during innate immune responses to microbes stimulate the proliferation and differentiation of lymphocytes in adaptive immune responses. Examples of cytokines secreted by PAMP-stimulated cells acting on B cells, CD4+ T cells, and CD8+ T cells are given here. The details of the lymphocyte responses to these cytokines will be discussed in more detail in later chapters.
Adjuvants, which are substances that need to be administered together with purified protein antigens to elicit maximal T cell–dependent immune responses (see Chapter 6), work by stimulating innate immune responses at the site of antigen exposure. Adjuvants are useful in experimental immunology and in clinical vaccines. Many adjuvants in experimental use are microbial products, such as killed mycobacteria and LPS, that engage TLRs. The only routinely used adjuvant in human vaccines is alum, composed of either aluminum hydroxide or aluminum phosphate. Among their important effects, adjuvants activate dendritic cells to express more major histocompatibility molecules that are part of the antigen (signal 1) that T cells recognize, increase the expression of costimulators (signal 2) and cytokines needed for T cell activation, and stimulate migration of the dendritic cells to lymph nodes where T cells are located.
Feedback Mechanisms that Regulate Innate Immunity
The magnitude and duration of innate immune responses are regulated by a variety of feedback inhibition mechanisms that limit potential damage to tissues. Whereas the inflammatory response is critically important for protection against microbes, it has the potential to cause tissue injury and disease. Several mechanisms have evolved to provide a break on inflammation, and these mechanisms come into play at the same time as or shortly after the initiation of inflammation. Furthermore, the stimuli for the initiation of many of these control mechanisms include the same PAMPs and DAMPs that induce inflammation. We will describe a selected group of these regulatory mechanisms.
IL-10 is a cytokine that is produced by and inhibits activation of macrophages and dendritic cells. IL-10 inhibits the production of various inflammatory cytokines by activated macrophages and dendritic cells, including IL-1, TNF, and IL-12. Because it is both produced by macrophages and inhibits macrophage functions, IL-10 is an excellent example of a negative feedback regulator. It is not clear whether different stimuli may act on macrophages to induce the production of a regulatory cytokine like IL-10 or effector cytokines like TNF and IL-12, or whether the same stimuli elicit production of all these cytokines but with different kinetics. IL-10 is also produced by some nonlymphoid cell types (e.g., keratinocytes). The Epstein-Barr virus contains a gene homologous to human IL-10, and viral IL-10 has the same activities as the natural cytokine. This raises the intriguing possibility that acquisition of the IL-10 gene during the evolution of the virus has given the virus the ability to inhibit host immunity and thus a survival advantage in the infected host. IL-10 is also produced by regulatory T cells, and we discuss IL-10 in more detail in Chapter 14 in this context.
Mononuclear phagocytes produce a natural antagonist of IL-1 that is structurally homologous to the cytokine and binds to the same receptors but is biologically inactive, so that it functions as a competitive inhibitor of IL-1. It is therefore called IL-1 receptor antagonist (IL-1RA). Synthesis of IL-1RA is induced by many of the same stimuli that induce IL-1 production, and some studies in IL-1RA–deficient mice suggest that this inhibitory cytokine is required to prevent inflammatory diseases of joints and other tissues. Recombinant IL-1RA has been developed as a drug that is effective in the treatment of systemic juvenile rheumatoid arthritis and familial fever syndromes in which IL-1 production is dysregulated. Regulation of IL-1–mediated inflammation may also occur by expression of the type II receptor, which binds IL-1 but does not transduce an activating signal. The major function of this receptor may be to act as a “decoy” that competitively inhibits IL-1 binding to the type I signaling receptor.
Secretion of inflammatory cytokines from a variety of cell types appears to be regulated by the products of autophagy genes. Autophagy is a mechanism by which cells degrade their own organelles, such as mitochondria, by sequestering them within membrane-bound vesicles and fusing the vesicles with lysosomes. This process requires the coordinated actions of many different proteins that are encoded by autophagy (Atg) genes. Targeted mutations in different Atg genes result in enhanced secretion of type I interferons, IL-1, and IL-18 by various cell types and the development of inflammatory bowel disease. The mechanisms by which Atg proteins impair cytokine synthesis are not well understood, but evidence exists for their binding to and inhibition of RLRs and regulation of inflammasome formation. A role for Atg proteins in regulating innate immune responses is further supported by the discovery that polymorphisms in a human Atg are associated with inflammatory bowel disease.
There are numerous negative regulatory signaling pathways that block the activating signals generated by pattern recognition receptors and inflammatory cytokines. Suppressors of cytokine signaling (SOCS) proteins are inhibitors of JAK-STAT signaling pathways linked to cytokine receptors. TLR signaling in macrophages and dendritic cells induces the expression of SOCS proteins, which limit responses of these cells to exogenous cytokines such as type I interferons. Proinflammatory responses of cells to TLR signaling are negatively regulated by SHP-1, an intracellular protein phosphatase that negatively regulates numerous tyrosine kinase–dependent signaling pathways in lymphocytes. There are many other examples of kinases and phosphatases that inhibit TLR, NLR, and RLR signaling.
Summary
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783-801.
Areschoug T, Gordon S. Scavenger receptors: role in innate immunity and microbial pathogenesis. Cellular Microbiology. 2009;11:1160-1169.
Blasius AL, Beutler B. Intracellular Toll-like receptors. Immunity. 2010;32:305-315.
Chen G, Shaw MH, Kim YG, Nuñez G. Nod-like receptors: role in innate immunity and inflammatory disease. Annual Review of Pathology. 2009;4:365-398.
Hornung V, Latz E. Intracellular DNA recognition. Nature Reviews Immunology. 2010;10:123-130.
Ip WK, Takahashi K, Ezekowitz RA, Stuart LM. Mannose-binding lectin and innate immunity. Immunological Reviews. 2009;230:9-21.
Janeway CA, Medzhitov R. Innate immune recognition. Annual Review of Immunology. 2002;20:197-216.
Jeannin P, Jaillon S, Delneste Y. Pattern recognition receptors in the immune response against dying cells. Current Opinions in Immunology. 2008;20:530-537.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature Immunology. 2010;11:373-384.
Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39-44.
Pichlmair A, Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370-383.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805-820.
Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nature Reviews Immunology. 2007;7:179-190.
Cells of the Innate Immune System
Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. 2008;112:935-945.
Lanier LL. NK cell recognition. Annual Review of Immunology. 2005;23:225-274.
Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunological Reviews. 2007;219:88-102.
Segal AW. How neutrophils kill microbes. Annual Review of Immunology. 2005;23:197-223.
Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annual Review of Immunology. 2008;26:421-452.
Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annual Review of Immunology. 2002;20:825-852.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nature Immunology. 2008;9:503-510.
Effector Molecules of Innate Immunity
Bottazzi B, Doni A, Garlanda C, Mantovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annual Review of Immunology. 2010;28:157-183.
Klotman ME, Chang TL. Defensins in innate antiviral immunity. Nature Reviews Immunology. 2006;6:447-456.
Linden SK, Sutton P, Karlsson NG, Korolik V, McGuckin MA. Mucins in the mucosal barrier to infection. Mucosal Immunology. 2008;1:183-197.
Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annual Review of Immunology. 2010;28:321-342.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821-832.
Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nature Immunology. 2005;6:551-557.
Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nature Reviews Immunology. 2010;10:89-102.
Van de Wetering JK, van Golde LMG, Batenburg JJ. Collectins: players of the innate immune system. European Journal of Biochemistry. 2004;271:229-249.
Diseases Caused by Innate Immunity
Cinel I, Opal SM. Molecular biology of inflammation and sepsis: a primer. Critical Care Medicine. 2009;37:291-304.
Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. New England Journal of Medicine. 2003;348:138-150.
Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annual Review of Immunology. 2009;27:621-668.
Weighardt H, Holzmann B. Role of Toll-like receptor responses for sepsis pathogenesis. Immunobiology. 2007;212:715-722.