CHAPTER 16 Transplantation Immunology
Transplantation is a widely used treatment for replacement of nonfunctioning organs and tissues with healthy organs or tissues. Technically, transplantation is the process of taking cells, tissues, or organs, called a graft, from one individual and placing them into a (usually) different individual. The individual who provides the graft is called the donor, and the individual who receives the graft is called either the recipient or the host. If the graft is placed into its normal anatomic location, the procedure is called orthotopic transplantation; if the graft is placed in a different site, the procedure is called heterotopic transplantation. Transfusion refers to the transfer of circulating blood cells or plasma from one individual to another. Clinical transplantation to treat human diseases has increased steadily during the past 45 years, and transplantation of kidneys, hearts, lungs, livers, pancreata, and bone marrow is widely used today (Fig. 16-1). More than 30,000 kidney, heart, lung, liver, and pancreas transplantations are currently performed in the United States each year. In addition, transplantation of many other organs or cells, including stem cells, is now being attempted.
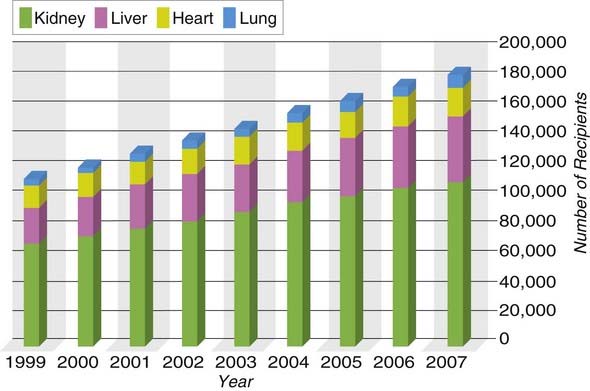
FIGURE 16–1 People in the United States living with functioning organ grafts, 1999-2007.
(Data from OPTN/SRTR Annual Report 2009. Available at: http://www.ustransplant.org/csr/current/fastfacts.aspx. Accessed April 2010.)
Transplantation of cells or tissues from one individual to a genetically nonidentical individual invariably leads to rejection of the transplant due to an adaptive immune response. Rejection has been a major barrier to successful transplantation of tissues. This problem was first appreciated when attempts to replace damaged skin on burn patients with skin from unrelated donors proved to be uniformly unsuccessful. During a matter of 1 to 2 weeks, the transplanted skin would undergo necrosis and fall off. The failure of the grafts led Peter Medawar and many other investigators to study skin transplantation in animal models. These experiments established that the failure of skin grafting was caused by an inflammatory reaction called rejection. The conclusion that graft rejection is the result of an adaptive immune response came from experiments demonstrating that the process had characteristics of memory and specificity and was mediated by lymphocytes (Fig. 16-2). For instance, rejection occurs in 7 to 14 days after the first transplant from a donor to a recipient (called first-set rejection) and more rapidly after the second transplant from the same donor to this recipient (called second-set rejection), implying that the recipient developed memory for the grafted tissue. Individuals who have rejected a graft from one donor show accelerated rejection of another graft from the same donor but not from a different donor, demonstrating that the rejection process is immunologically specific. These experimental results were recapitulated in clinical transplantation. Perhaps the most compelling evidence showing that allograft rejection is an adaptive immune response was the finding that the ability to rapidly reject a transplant can be transferred with lymphocytes from a sensitized to a naive host.
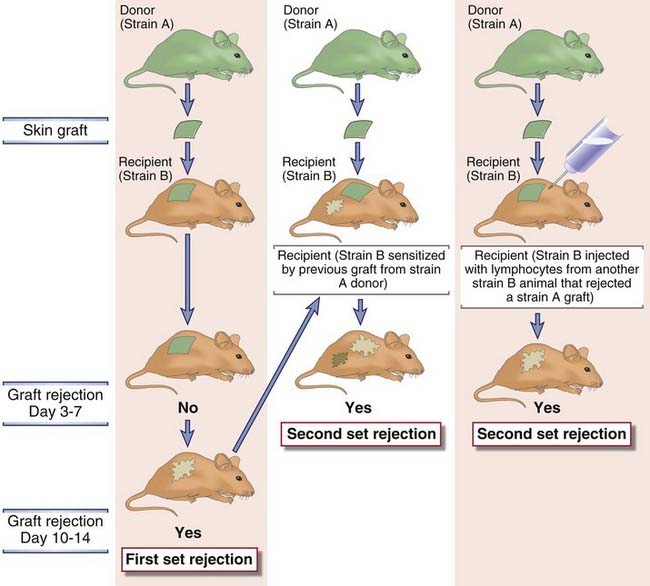
FIGURE 16–2 First- and second-set allograft rejection.
Results of the experiments shown indicate that graft rejection displays the features of adaptive immune responses, namely, memory and mediation by lymphocytes. An inbred strain B mouse will reject a graft from an inbred strain A mouse with first-set kinetics (left panel). An inbred strain B mouse sensitized by a previous graft from an inbred strain A mouse will reject a second graft from an inbred strain A mouse with second-set kinetics (middle panel), demonstrating memory. An inbred strain B mouse injected with lymphocytes from another strain B mouse that has rejected a graft from a strain A mouse will reject a graft from a strain A mouse with second-set kinetics (right panel), demonstrating the role of lymphocytes in mediating rejection and memory. An inbred strain B mouse sensitized by a previous graft from a strain A mouse will reject a graft from a third unrelated strain with first-set kinetics, thus demonstrating another feature of adaptive immunity, specificity (not shown). Syngeneic grafts are never rejected (not shown).
Transplant immunologists have developed a special vocabulary to describe the kinds of cells and tissues encountered in the transplant setting. A graft transplanted from one individual to the same individual is called an autologous graft. A graft transplanted between two genetically identical or syngeneic individuals is called a syngeneic graft. A graft transplanted between two genetically different individuals of the same species is called an allogeneic graft (or allograft). A graft transplanted between individuals of different species is called a xenogeneic graft (or xenograft). The molecules that are recognized as foreign on allografts are called alloantigens, and those on xenografts are called xenoantigens. The lymphocytes and antibodies that react with alloantigens or xenoantigens are described as being alloreactive or xenoreactive, respectively.
The immunology of transplantation is important for several reasons. First, immunologic rejection remains one of the major problems in clinical transplantation. Second, although transplantation of tissues is not a normal phenomenon, the immune response to allogeneic molecules has been a useful model for studying the mechanisms of lymphocyte activation. Third, many immunosuppressive therapies that have proved to be useful for a variety of immunologic and inflammatory diseases were first tested and shown to be effective for treatment of graft rejection, which is a clinically important immunologic reaction that can be measured rapidly and with precision. Most of this chapter focuses on allogeneic transplantation because it is far more commonly practiced and better understood than xenogeneic transplantation, which is discussed briefly at the end of the chapter. We consider both the basic immunology and some aspects of the clinical practice of transplantation. We conclude the chapter with a discussion of hematopoietic stem cell transplantation, which raises special issues not usually encountered with solid organ transplants.
Immune Responses to Allografts
Alloantigens elicit both cellular and humoral immune responses. In this section of the chapter, we discuss the molecular and cellular mechanisms of allorecognition, with an emphasis on the nature of graft antigens that stimulate allogeneic responses and the properties of the responding lymphocytes.
Recognition of Alloantigens
Recognition of transplanted cells as self or foreign is determined by polymorphic genes, called histocompatibility genes, which differ among different members of a species. This conclusion is based on the results of experimental transplantation between inbred strains of mice, and in some cases, the results have been confirmed in human transplantation. Remember that all the animals of an inbred strain are genetically identical, and they are homozygous for all genes (except the sex chromosomes in males). The basic rules of transplantation immunology, which are derived from such animal experiments, are the following (Fig. 16-3).
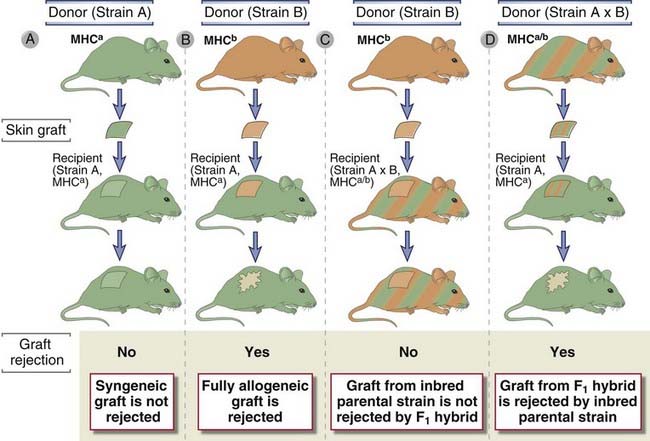
FIGURE 16–3 The genetics of graft rejection.
In the illustration, the two different mouse colors represent inbred strains with different MHC haplotypes. Inherited MHC alleles from both parents are codominantly expressed in the skin of an A × B offspring, and therefore these mice are represented by both colors. Syngeneic grafts are not rejected (A). Allografts are always rejected (B). Grafts from an A or B parent will not be rejected by an (A × B)F1 offspring (C), but grafts from the offspring will be rejected by either parent (D). These phenomena are due to the fact that MHC gene products are responsible for graft rejection; grafts are rejected only if they express an MHC type (represented by green or orange) that is not expressed by the recipient mouse.
Such results suggested that the molecules in the grafts that are responsible for eliciting rejection must be polymorphic and their expression is codominant. Polymorphic refers to the fact that these graft antigens differ among the individuals of a species (other than identical twins) or between different inbred strains of animals. Codominant expression means that every individual inherits genes encoding these molecules from both parents and both parental alleles are expressed. Therefore, (A × B)F1 animals express both A and B alleles and see both A and B tissues as self, whereas inbred A or B animals express only one allele and see (A × B)F1 tissues as partly foreign. This is why an (A × B)F1 animal does not reject either A or B strain grafts and why both A and B strain recipients reject an (A × B)F1 graft.
The molecules responsible for almost all strong (rapid) rejection reactions are called major histocompatibility complex (MHC) molecules. George Snell and colleagues used pairs of congenic strains of inbred mice, which were bred to be genetically identical to each other except for genes needed for graft rejection, to identify the polymorphic genes that encode the molecular targets of allograft rejection. This approach led to the identification of MHC genes as the underlying genetic basis of graft rejection.Transplants of most tissues between any pair of individuals, except identical twins, will be rejected because MHC molecules, the major polymorphic targets of graft rejection, are expressed on virtually all tissues. As discussed in Chapter 6, the normal function of MHC molecules is to present peptides derived from protein antigens in a form that can be recognized by T cells. The role of MHC molecules as the antigens that cause graft rejection is a consequence of the nature of T cell antigen recognition, as we will discuss later. Recall that human MHC molecules are called human leukocyte antigens (HLA), and in the context of human transplantation, the terms MHC and HLA are used interchangeably.
Allogeneic MHC molecules of a graft may be presented for recognition by the T cells of the recipient in two fundamentally different ways, called direct and indirect (Fig. 16-4). Initial studies showed that the T cells of a graft recipient recognize intact, unprocessed MHC molecules in the graft, and this is called direct presentation of alloantigens. Subsequent studies showed that sometimes, the recipient T cells recognize graft MHC molecules only in the context of the recipient’s MHC molecules, implying that the recipient’s MHC molecules must be presenting allogenic graft MHC proteins to recipient T cells. This process is called indirect presentation, and it is essentially the same as the presentation of any foreign (e.g., microbial) protein antigen. Not only MHC molecules but other alloantigens in a graft that are different between the donor and recipient can also be presented to host T cells by the indirect pathway. We discuss the mechanisms of direct and indirect presentation separately.
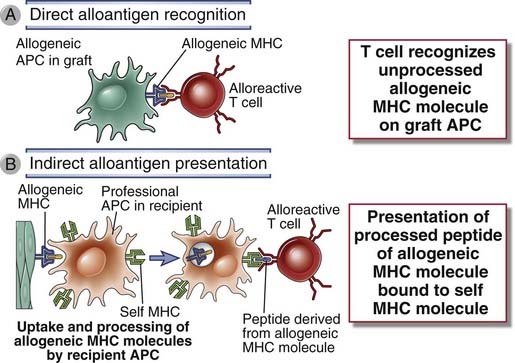
FIGURE 16–4 Direct and indirect alloantigen recognition.
A, Direct alloantigen recognition occurs when T cells bind directly to an intact allogeneic MHC molecule on a graft (donor) antigen-presenting cell (APC). B, Indirect alloantigen recognition occurs when allogeneic MHC molecules from graft cells are taken up and processed by recipient APCs and peptide fragments of the allogeneic MHC molecules containing polymorphic amino acid residues are bound and presented by recipient (self) MHC molecules.
Direct Presentation of MHC Alloantigens
In direct presentation, an intact MHC molecule is displayed by donor antigen-presenting cells (APCs) in the graft and recognized by recipient T cells without a need for host APCs. It may seem puzzling that T cells that are normally selected during their maturation to be self MHC restricted are capable of recognizing foreign (allogeneic or xenogeneic) MHC molecules. In fact, as we will discuss in more detail later, the frequency of T cells in a normal individual that recognize a single allogeneic MHC molecule is as high as 1% to 2% of all T cells, which is 100 to 1000 times greater than the frequency of T cells specific for any microbial peptide displayed by self MHC molecules. There are several likely explanations for this surprisingly strong recognition of foreign MHC molecules.
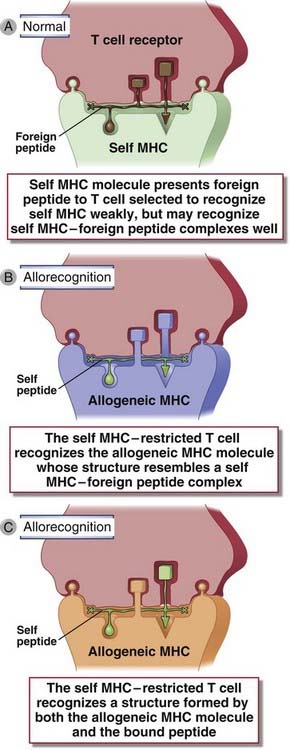
FIGURE 16–5 Molecular basis of direct recognition of allogeneic MHC molecules.
Direct recognition of allogeneic MHC molecules may be thought of as a cross-reaction in which a T cell specific for a self MHC molecule–foreign peptide complex (A) also recognizes an allogeneic MHC molecule (B, C). Nonpolymorphic donor peptides, labeled “self peptide,” may not contribute to allorecognition (B) or they may (C).
Direct allorecognition can generate both CD4+ and CD8+ T cells that recognize graft antigens and contribute to rejection. This aspect of the alloreactive T cell response is described later.
Indirect Presentation of Alloantigens
In the indirect pathway, donor (allogeneic) MHC molecules are captured and processed by recipient APCs that enter grafts, and peptides derived from the allogeneic MHC molecules are presented in association with self MHC molecules (see Fig. 16-4). Thus, peptides from the allogeneic MHC molecules are displayed by host APCs and recognized by T cells like conventional foreign protein antigens. Because allogeneic MHC molecules have amino acid sequences different from those of the host, they can generate foreign peptides associated with self MHC molecules on the surface of host APCs. In fact, MHC molecules are the most polymorphic proteins in the genome; therefore, each allogeneic MHC molecule may give rise to multiple foreign peptides, each recognized by different T cells. Indirect presentation may result in allorecognition by CD4+ T cells because alloantigen is acquired by host APCs primarily through the endosomal vesicular pathway (i.e., as a consequence of phagocytosis) and is therefore presented by class II MHC molecules. Some antigens of phagocytosed graft cells appear to enter the class I MHC pathway of antigen presentation and are indirectly recognized by CD8+ T cells. This phenomenon is an example of cross-presentation or cross-priming (see Chapter 6, Fig. 6-20), in which dendritic cells ingest antigens of another cell, from the graft, and present these antigens on class I MHC molecules to activate or “prime” CD8+ T lymphocytes.
Evidence that indirect presentation of allogeneic MHC molecules plays a significant role in graft rejection was obtained from studies with knockout mice lacking class II MHC expression. For example, skin grafts from donor mice lacking class II MHC are able to induce recipient CD4+ (i.e., class II restricted) T cell responses to the donor alloantigens, including peptides derived from donor class I MHC molecules. In these experiments, the donor class I MHC molecules are processed and presented by class II molecules on the recipient’s APCs and stimulate the recipient’s helper T cells. Evidence has also been obtained that indirect antigen presentation may contribute to late rejection of human allografts. For example, CD4+ T cells from heart and liver allograft recipients recognize and are activated by peptides derived from donor MHC when presented by the patient’s own APCs.
In the setting of any transplant between genetically nonidentical donor and recipient, there will be polymorphic antigens other than MHC molecules against which the recipient may mount an immune response. These antigens typically induce weak or slower (more gradual) rejection reactions than do MHC molecules and are therefore called minor histocompatibility antigens. Most minor histocompatibility antigens are proteins that are processed and presented to host T cells in association with self MHC molecules on host APCs (i.e., by the indirect pathway). The relevance of minor histocompatibility antigens in clinical solid organ transplantation is uncertain, mainly because there has been little success in identifying the relevant antigens. The male H-Y antigen appears to be a target of immune recognition by female recipients of male donor organs, and this correlates with a very slight increase in risk of rejection compared with sex-matched transplants. Antibodies specific for donor alleles of the class I MHC–like molecule MIC-A are detectable in some renal allograft recipients, and the presence of the antibodies correlates with reduced graft survival. This has led to speculation that these proteins are also minor histocompatibility antigens of relevance to graft rejection. Minor histocompatibility antigens play a more significant role in stimulating graft-versus-host responses after hematopoietic stem cell transplantation, discussed later, but the nature of the relevant antigens in that setting is also not defined.
Activation of Alloreactive Lymphocytes
Allografts stimulate T and B cell responses that are similar to immune responses to conventional protein antigens but also have some special features. Here we discuss these common and unique aspects of the immune responses to alloantigens.
T Cell Recognition of Alloantigens
The T cell response to an organ graft may be initiated in the lymph nodes that drain the graft (Fig. 16-6). Most organs contain resident APCs such as dendritic cells. Transplantation of these organs into an allogeneic recipient provides APCs that express donor MHC molecules as well as costimulators. It is believed that these donor APCs migrate to regional lymph nodes and present, on their surface, unprocessed allogeneic MHC molecules to the recipient’s T cells (the direct pathway of allorecognition). Host dendritic cells from the recipient may also migrate into the graft, pick up graft alloantigens, and transport these back to the draining lymph nodes, where they are displayed (the indirect pathway). Naive lymphocytes that normally traffic through the lymph node encounter these alloantigens and are induced to proliferate and differentiate into effector cells. This process is sometimes called sensitization to alloantigens. Effector T cells migrate back into the graft and mediate rejection.
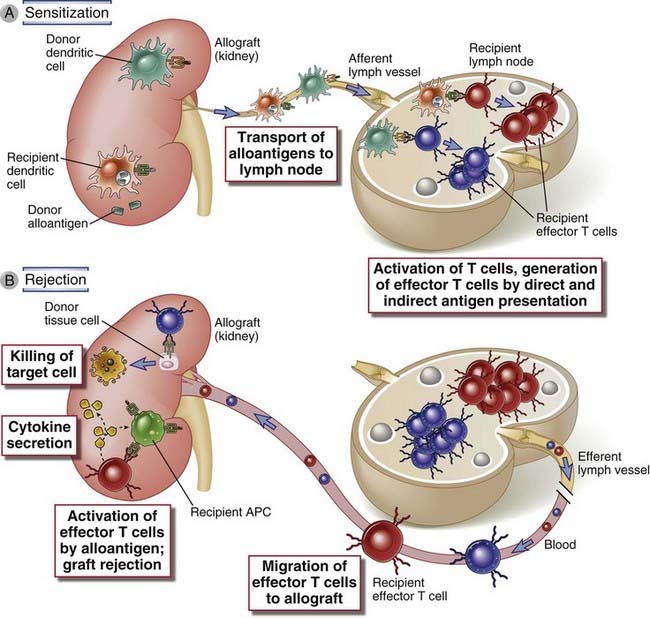
FIGURE 16–6 Activation of alloreactive T cells.
A, In the case of direct allorecognition, donor dendritic cells in the allograft migrate to secondary lymphoid tissues, where they present allogeneic MHC molecules to host T cells. B, In the case of indirect allorecognition, recipient dendritic cells that have entered the allograft transport donor MHC proteins to secondary lymphoid tissues and present peptides derived from these MHC proteins to alloreactive host T cells. In both cases, the T cells become activated and differentiate into effector cells. The alloreactive effector T cells migrate into the allograft, become reactivated by alloantigen, and mediate damage. Lymphatic drainage of grafted organs is not well described, and therefore the location of the relevant lymph nodes is uncertain.
As many as 1% to 2% of an individual’s T cells are capable of recognizing and responding to a single foreign MHC molecule, and this high frequency of T cells reactive with allogeneic MHC molecules is one reason that allografts elicit strong immune responses. Recall that the frequency of T cells reactive with any foreign (e.g., microbial) antigen is only 1 in 105 or 106. The likely reasons that each allogeneic MHC molecule is directly recognized by so many different TCRs were discussed earlier.
Many of the T cells that respond to an allogeneic MHC molecule, even on first exposure, are memory T cells. It is likely that these memory cells were generated during previous exposure to other foreign (e.g., microbial) antigens and cross-react with allogeneic MHC molecules. These memory cells are not only expanded populations of antigen-specific cells but also more rapid and powerful responders than naive lymphocytes, and thus contribute to the strength of the alloreactive T cell response. Memory cells are also thought to be more resistant to immunosuppression than are naive lymphocytes, and the presence of large numbers of memory cells may lead to poor outcomes of transplantation.
Role of Costimulation in T Cell Responses to Alloantigens
In addition to recognition of alloantigen, costimulation of T cells primarily by B7 molecules on APCs is important for activating alloreactive T cells. Rejection of allografts, and stimulation of alloreactive T cells in a mixed lymphocyte reaction (described later), can be inhibited by agents that block B7 molecules. Allografts survive for longer periods when they are transplanted into knockout mice lacking B7-1 (CD80) and B7-2 (CD86) compared with transplants into normal recipients. As we will discuss later, blocking of B7 costimulation is a therapeutic strategy to inhibit graft rejection in humans as well. There is experimental evidence, largely from rodents, that several other T cell costimulatory pathways, including ICOS ligand/ICOS and Ox40 ligand/Ox40, contribute to acute allograft rejection, but the relevance of these pathways to human transplantation has not yet been examined.
The requirement for costimulation leads to the interesting question of why these costimulators are expressed by graft APCs in the absence of infection, which we have previously discussed as the physiologic stimulus for the expression of costimulators (see Chapter 9). A likely possibility is that the process of organ transplantation is associated with ischemic damage and death of some cells in the graft, during the time the organ is removed from the donor and before it is surgically connected to the circulatory system of the recipient. Several molecules expressed by or released from ischemically damaged cells (so-called damage-associated molecular patterns) stimulate innate immune responses that result in increased expression of costimulators on APCs (see Chapter 4). In fact, the clinical experience is that the ischemia time of an organ is a determinant of the frequency and severity of acute rejection, and one reason for this may be that ischemic death of graft cells stimulates subsequent antigraft immune responses.
The Mixed Lymphocyte Reaction
The response of alloreactive T cells to foreign MHC molecules can be analyzed in an in vitro reaction called the mixed lymphocyte reaction (MLR). The MLR is used as a predictive test of T cell–mediated graft rejection. Studies of the MLR were among the first to establish the role of class I and class II MHC molecules in activating distinct populations of T cells (CD8+ and CD4+, respectively).
The MLR is induced by culturing mononuclear leukocytes (which include T cells, B cells, natural killer [NK] cells, mononuclear phagocytes, and dendritic cells) from one individual with mononuclear leukocytes derived from another individual. In clinical practice, these cells are typically isolated from peripheral blood; in mouse or rat experiments, mononuclear leukocytes are usually purified from the spleen or lymph nodes. If the two individuals have differences in the alleles of the MHC genes, a large proportion of the mononuclear cells will proliferate during a period of 4 to 7 days. This proliferative response is called the allogeneic MLR (Fig. 16-7). If cells from two MHC-disparate individuals are mixed, each can react against the other and both will proliferate, thus resulting in a two-way MLR. To simplify the analysis, one of the two leukocyte populations can be rendered incapable of proliferation before culture, either by γ-irradiation or by treatment with the antimitotic drug mitomycin C. In this one-way MLR, the treated cells serve exclusively as stimulators, and the untreated cells, still capable of proliferation, serve as the responders. Among the T cells that respond in an MLR, the CD4+ cells are specific for allogeneic class II MHC molecules and the CD8+ cells for class I molecules.
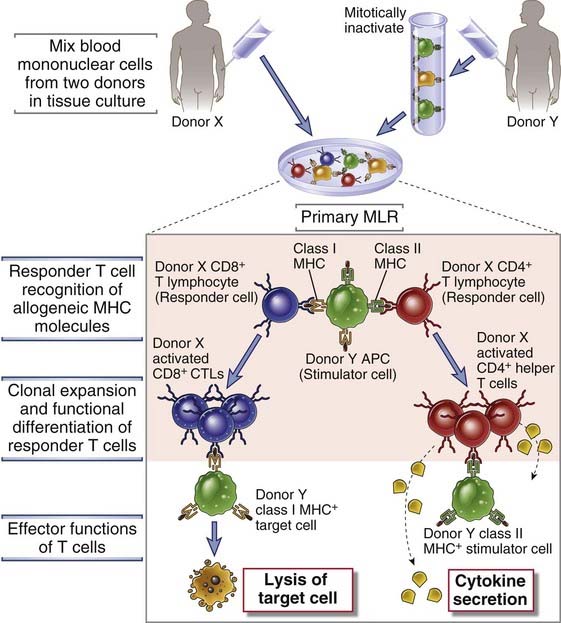
FIGURE 16–7 The mixed lymphocyte reaction (MLR).
In a one-way primary MLR, stimulator cells (from donor Y) activate and cause the expansion of two types of responder T cells (from donor X). CD4+ T cells from donor X react to donor Y class II molecules, and CD8+ T lymphocytes from donor X react to donor Y class I MHC molecules. The CD4+ T cells differentiate into cytokine-secreting helper T cells, and the CD8+ T cells differentiate into CTLs. APC, antigen-presenting cell.
Because of the high frequency of alloreactive T cells, primary responses to alloantigens are the only responses of naive T cells that can readily be detected in vitro. Responses of T cells to a protein antigen in vitro can be detected only if the T cells are from an individual who has previously been exposed to that antigen (e.g., by vaccination), because there are too few naive antigen–specific T cells to mount a detectable response. In contrast, naive T cells will proliferate vigorously when cultured with mononuclear cells from another individual in an MLR.
Effector Functions of Alloreactive T Cells
Alloreactive CD4+ and CD8+ T cells that are activated by graft alloantigens cause rejection by distinct mechanisms. The CD4+ helper T cells differentiate into cytokine-producing effector cells that damage grafts by cytokine-mediated inflammation, similar to a delayed-type hypersensitivity (DTH) reaction (see Chapters 10 and 18). Alloreactive CD8+ T cells differentiate into cytotoxic T lymphocytes (CTLs), which kill nucleated cells in the graft that express the allogeneic class I MHC molecules. CTLs also secrete inflammatory cytokines, which can contribute to graft damage.
Only CTLs that are generated by direct allogeneic MHC recognition can kill graft cells, whereas CTLs or helper T cells generated by either direct or indirect alloantigen recognition can cause cytokine-mediated damage to grafts. CD8+ CTLs that are generated by direct allorecognition recognize graft alloantigens and can, therefore, kill graft cells that express these same alloantigens. In contrast, any CD8+ CTLs that are generated by the indirect pathway are self MHC restricted, and they will not be able to kill the foreign graft cells because these cells do not express self MHC alleles displaying allogeneic peptides. Therefore, when alloreactive T cells are stimulated by the indirect pathway, the principal mechanism of rejection is not CTL-mediated killing of graft cells but inflammation caused by the cytokines produced by either CD8+ or CD4+ effector T cells. Presumably, these effector cells infiltrate the graft and recognize graft alloantigens being displayed by host APCs that have also entered the graft. The relative importance of the direct and indirect pathways in graft rejection is not definitively established. It may be that CD8+ CTLs induced by direct recognition of alloantigens are most important for acute cellular rejection of allografts, in which killing of graft cells is a prominent component, whereas CD4+ effector T cells stimulated by the indirect pathway play a greater role in chronic rejection. These differences may be of clinical significance because conventional immunosuppressive therapy for graft rejection seems to preferentially suppress CD8+ CTL responses induced by direct allorecognition and is less effective against CD4+ T cells activated by the indirect pathway.
Activation of Alloreactive B Cells and Production of Alloantibodies
Most high-affinity alloantibodies are produced by helper T cell–dependent activation of alloreactive B cells, much like antibodies against other protein antigens (see Chapter 11). The antigens most frequently recognized by alloantibodies in graft rejection are donor HLA molecules, including both class I and class II MHC proteins. The likely sequence of events leading to the generation of these alloantibody-producing cells is that naive B lymphocytes recognize foreign MHC molecules, internalize and process these proteins, and present peptides derived from them to helper T cells that were previously activated by the same peptides presented by dendritic cells. Thus, activation of alloreactive B cells is an example of indirect presentation of alloantigens. Anti-HLA antibodies contribute significantly to allograft rejection, as we will discuss below.
Patterns and Mechanisms of Allograft Rejection
Thus far, we have described the molecular basis of alloantigen recognition and the cells involved in the recognition of and responses to allografts. We now turn to a consideration of the effector mechanisms responsible for the immunologic rejection of allografts. In different experimental models and in clinical transplantation, alloreactive CD4+ and CD8+ T cells and alloantibodies have been shown to be capable of mediating allograft rejection. These different immune effectors cause graft rejection by different mechanisms (Fig. 16-8), and all three effectors may contribute to rejection concurrently.
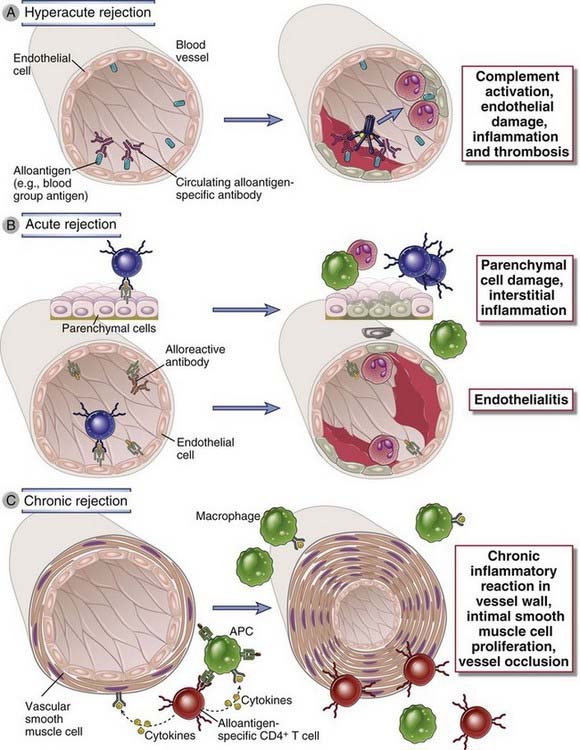
FIGURE 16–8 Immune mechanisms of graft rejection.
A, In hyperacute rejection, preformed antibodies reactive with vascular endothelium activate complement and trigger rapid intravascular thrombosis and necrosis of the vessel wall. B, In acute rejection, CD8+ T lymphocytes reactive with alloantigens on endothelial cells and parenchymal cells mediate damage to these cell types. Alloreactive antibodies formed after engraftment may also contribute to vascular injury. C, In chronic rejection with graft arteriosclerosis, injury to the vessel wall leads to intimal smooth muscle cell proliferation and luminal occlusion. This lesion may be caused by a chronic DTH reaction to alloantigens in the vessel wall.
For historical reasons, graft rejection is classified on the basis of histopathologic features or the time course of rejection after transplantation rather than on the basis of immune effector mechanisms. Based on the experience of renal transplantation, the histopathologic patterns are called hyperacute, acute, and chronic (see Fig. 16-8). These patterns are associated with different dominant immune effector mechanisms.
Hyperacute Rejection
Hyperacute rejection is characterized by thrombotic occlusion of the graft vasculature that begins within minutes to hours after host blood vessels are anastomosed to graft vessels and is mediated by preexisting antibodies in the host circulation that bind to donor endothelial antigens (Fig. 16-8A). Binding of antibody to endothelium activates complement, and antibody and complement products together induce a number of changes in the graft endothelium that promote intravascular thrombosis. Complement activation leads to endothelial cell injury and exposure of subendothelial basement membrane proteins that activate platelets. The endothelial cells are stimulated to secrete high-molecular-weight forms of von Willebrand factor that cause platelet adhesion and aggregation. Both endothelial cells and platelets undergo membrane vesiculation, leading to shedding of lipid particles that promote coagulation. Endothelial cells lose the cell surface heparan sulfate proteoglycans that normally interact with antithrombin III to inhibit coagulation. These processes contribute to thrombosis and vascular occlusion (Fig. 16-9A), and the grafted organ suffers irreversible ischemic damage.
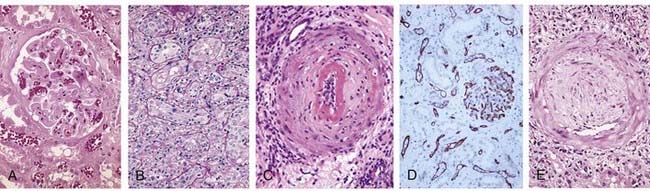
FIGURE 16–9 Histopathology of different forms of graft rejection.
A, Hyperacute rejection of a kidney allograft with endothelial damage, platelet and thrombin thrombi, and early neutrophil infiltration in a glomerulus. B, Acute rejection of a kidney with inflammatory cells in the connective tissue around the tubules and between epithelial cells of the tubules. C, Acute antibody-mediated rejection of a kidney allograft with destructive inflammatory reaction destroying the endothelial layer of an artery. D, Complement C4d deposition in vessels in acute antibody-mediated rejection. E, Chronic rejection in a kidney allograft with graft arteriosclerosis. The vascular lumen is replaced by an accumulation of smooth muscle cells and connective tissue in the vessel intima.
(Courtesy of Dr. Helmut Rennke, Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts.)
In the early days of transplantation, hyperacute rejection was often mediated by preexisting IgM alloantibodies, which are present at high titer before transplantation. Such “natural antibodies” are believed to arise in response to carbohydrate antigens expressed by bacteria that normally colonize the intestine. The best known examples of such alloantibodies are those directed against the ABO blood group antigens expressed on red blood cells, discussed later. ABO antigens are also expressed on vascular endothelial cells. Today, hyperacute rejection by anti-ABO antibodies is extremely rare because all donor and recipient pairs are selected so that they have the same ABO type. As we shall discuss later in this chapter, hyperacute rejection caused by natural antibodies is the major barrier to xenotransplantation and limits the use of animal organs for human transplantation.
Currently, hyperacute rejection of allografts, when it occurs, is usually mediated by IgG antibodies directed against protein alloantigens, such as donor MHC molecules, or against less well defined alloantigens expressed on vascular endothelial cells. Such antibodies generally arise as a result of previous exposure to alloantigens through blood transfusion, previous transplantation, or multiple pregnancies. If the titer of these alloreactive antibodies is low, hyperacute rejection may develop slowly, during several days. In this case, it is sometimes referred to as accelerated allograft rejection because the onset is still earlier than that typical for acute rejection. As we will discuss later in this chapter, patients in need of allografts are routinely screened before grafting for the presence of antibodies that bind to cells of a potential organ donor to avoid hyperacute rejection.
In rare cases in which grafts have to be done in ABO-incompatible recipients, survival may be improved by rigorous depletion of antibodies and B cells. Sometimes, if the graft is not rapidly rejected, it survives even in the presence of anti-graft antibody. One possible mechanism of this resistance to hyperacute rejection is increased expression of complement regulatory proteins on graft endothelial cells, a beneficial adaptation of the tissue that has been called accommodation.
Acute Rejection
Acute rejection is a process of injury to the graft parenchyma and blood vessels mediated by alloreactive T cells and antibodies. Before modern immunosuppression, acute rejection would often begin several days to a few weeks after transplantation. The delayed time of onset of acute rejection is because alloreactive effector T cells and antibodies take time to be generated from naive or resting memory T cells in response to the graft. In current clinical practice, episodes of acute rejection may occur at much later times, even years after transplantation, if immunosuppression is reduced for any number of reasons. Although the patterns of acute rejection are divided into cellular, mediated by T cells, and humoral, mediated by antibodies, both typically coexist in an acutely rejecting organ.
Acute Cellular Rejection
The principal mechanism of acute cellular rejection is CTL-mediated killing of cells in the graft (Fig. 16-8B). On histologic examination, this type of rejection is characterized by infiltrates of lymphocytes, which invade and destroy graft components (Fig. 16-9B). There are many lines of evidence that support the role of CTLs in acute cellular rejection. The cellular infiltrates present in grafts undergoing this type of rejection are markedly enriched for CD8+ CTLs specific for graft alloantigens. In fact, the presence of mRNAs encoding CTL-specific genes (e.g., perforin and granzyme B) is sometimes used as a specific and sensitive indicator of clinical acute rejection. Experimentally, alloreactive CD8+ CTLs can be used to adoptively transfer acute cellular graft rejection. The destruction of allogeneic cells in a graft is highly specific, a hallmark of CTL killing. The best evidence for this specificity has come from mouse skin graft experiments using chimeric grafts that contain two distinct cell populations, one syngeneic to the host and one allogeneic to the host. When these skin grafts are transplanted, the allogeneic cells are killed without injury to the “bystander” syngeneic cells.
In addition to direct killing of the graft cells by CTLs, activated CD4+ helper T cells and CTLs produce cytokines that recruit and activate inflammatory cells, which also injure the graft.
In vascularized grafts such as kidney grafts, endothelial cells are major targets of acute rejection. Microvascular endothelialitis is a frequent early finding in grafts undergoing acute rejection episodes. Endothelialitis or intimal arteritis in medium-sized arteries also occurs at an early stage of acute rejection and is indicative of severe rejection, which, if left untreated, will likely result in acute graft failure. Both CD8+ and CD4+ T cells may contribute to endothelial injury.
Acute Antibody-Mediated Rejection
Alloantibodies cause acute rejection by binding to alloantigens, mainly HLA molecules, on vascular endothelial cells, causing endothelial injury and intravascular thrombosis that results in graft destruction (see Fig. 16-8B). The binding of the alloantibodies to the endothelial cell surface triggers local complement activation, which leads to lysis of the cells, recruitment and activation of neutrophils, and thrombus formation. In addition, alloantibody binding to the endothelial surface may directly alter endothelial function by inducing intracellular signals that enhance surface expression of proinflammatory and procoagulant molecules.
The histologic hallmark of this form of acute rejection is transmural necrosis of graft vessel walls with acute inflammation (Fig. 16-9C), which is different from the thrombotic occlusion without vessel wall necrosis seen in hyperacute rejection. Immunohistochemical identification of the C4d complement fragment in capillaries of renal allografts is used clinically as an indicator of activation of the classical complement pathway and humoral rejection (Fig. 16-9D). In a significant fraction of cases of antibody-mediated rejection, there is no C4d deposition detectable, suggesting that damage is caused by the complement-independent effects of alloantibody binding to endothelial cells, mentioned before.
Chronic Rejection and Graft Vasculopathy
As therapy for acute rejection has improved, the major cause of the failure of vascularized organ allografts has become chronic rejection. Since 1990, 1-year survival of kidney allografts has been better than 90% but the 10-year survival has remained about 60% despite advances in immunosuppressive therapy. Chronic rejection develops insidiously during months or years and may or may not be preceded by episodes of acute rejection. Chronic rejection of different transplanted organs is associated with distinct pathologic changes. In the kidney and heart, chronic rejection results in vascular occlusion and interstitial fibrosis. Lung transplants undergoing chronic rejection show thickened small airways (bronchiolitis obliterans), and liver transplants show fibrotic and nonfunctional bile ducts (called the vanishing bile duct syndrome).
A dominant lesion of chronic rejection in vascularized grafts is arterial occlusion as a result of the proliferation of intimal smooth muscle cells, and the grafts eventually fail mainly because of the resulting ischemic damage (Fig. 16-8C). The arterial changes are called graft vasculopathy or accelerated graft arteriosclerosis (Fig. 16-9E). Graft vasculopathy is frequently seen in failed cardiac and renal allografts and can develop in any vascularized organ transplant within 6 months to a year after transplantation. The pathogenesis of the lesions remains poorly understood but likely involves a combination of immunologic and nonimmunologic processes. The likely mechanisms underlying the occlusive vascular lesions of chronic rejection are: activation of alloreactive T cells and secretion of cytokines that stimulate proliferation of vascular endothelial and smooth muscle cells; repair with fibrosis after repeated bouts of acute antibody-mediated or cellular rejection; and consequences of perioperative ischemia, toxic effects of immunosuppressive drugs, and even chronic viral infections. As the arterial lesions of graft arteriosclerosis progress, blood flow to the graft parenchyma is compromised, and the parenchyma is slowly replaced by nonfunctioning fibrous tissue. This process leads to congestive heart failure or arrhythmias in cardiac transplant patients or loss of function in glomeruli and ischemic renal failure in renal transplant patients.
Prevention and Treatment of Allograft Rejection
If the recipient of an allograft has a fully functional immune system, transplantation almost invariably results in some form of rejection. The strategies used in clinical practice and in experimental models to avoid or to delay rejection are general immunosuppression and minimizing the strength of the specific allogeneic reaction. An important goal in transplantation research is to find ways of inducing donor-specific tolerance, which would allow grafts to survive without nonspecific immunosuppression.
Immunosuppression to Prevent or to Treat Allograft Rejection
Immunosuppressive drugs that inhibit or kill T lymphocytes are the principal agents used to treat or prevent graft rejection. Several methods of immunosuppression are commonly used (Fig. 16-10).
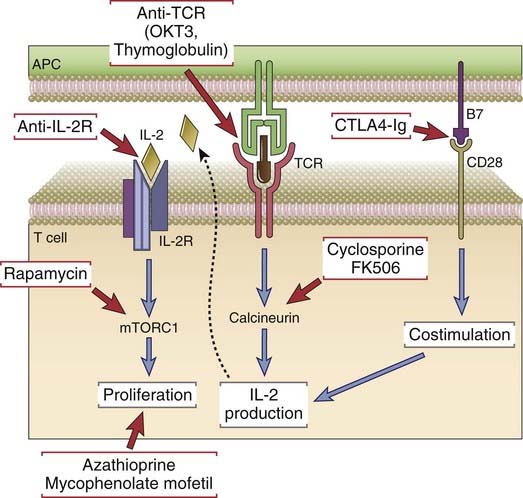
FIGURE 16–10 Mechanisms of action of immunosuppressive drugs.
Each major category of drugs used to prevent or to treat allograft rejection is shown along with the molecular targets of the drugs.
Inhibitors of T Cell Signaling Pathways
The calcineurin inhibitors cyclosporine and FK506 (tacrolimus) inhibit transcription of certain genes in T cells, most notably those encoding cytokines such as IL-2. Cyclosporine is a fungal peptide that binds with high affinity to a ubiquitous cellular protein called cyclophilin. The complex of cyclosporine and cyclophilin binds to and inhibits the enzymatic activity of the calcium/calmodulin-activated serine/threonine phosphatase calcineurin (see Chapter 7). Because calcineurin is required to activate the transcription factor NFAT (nuclear factor of activated T cells), cyclosporine inhibits NFAT activation and the transcription of IL-2 and other cytokine genes. The net result is that cyclosporine blocks the IL-2–dependent proliferation and differentiation of T cells. FK506 is a macrolide lactone made by a bacterium that functions like cyclosporine. FK506 and its binding protein (called FKBP) share with the cyclosporine-cyclophilin complex the ability to bind calcineurin and inhibit its activity.
The introduction of cyclosporine into clinical practice ushered in the modern era of transplantation. Before the use of cyclosporine, the majority of transplanted hearts and livers were rejected. Now as a result of the use of cyclosporine, FK506, and other more recently introduced drugs, the majority of these allografts survive for more than 5 years (Fig. 16-11). Nevertheless, these drugs have limitations. For example, at doses needed for optimal immunosuppression cyclosporine causes kidney damage, and some rejection episodes are refractory to cyclosporine treatment. FK506 was initially used for liver transplant recipients, but it is now used widely for immunosuppression of kidney allograft recipients, including those who are not adequately controlled by cyclosporine. FK506 is also used topically for some inflammatory skin diseases.
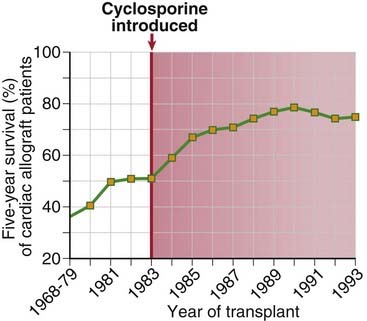
FIGURE 16–11 Influence of cyclosporine on graft survival.
Five-year survival rates for patients receiving cardiac allografts increased significantly beginning when cyclosporine was introduced in 1983.
(Data from Transplant Patient DataSource, United Network for Organ Sharing, Richmond, Virginia. Available at: http://207.239.150.13/tpd/. Accessed February 16, 2000.)
The immunosuppressive drug rapamycin (sirolimus) inhibits growth factor–mediated T cell proliferation. Like FK506, rapamycin binds to FKBP, but the rapamycin-FKBP complex does not inhibit calcineurin. Instead, this complex binds to and inhibits a cellular enzyme called mammalian target of rapamycin complex 1 (mTORC1), which is a serine/threonine protein kinase required for translation of proteins that promote cell survival and proliferation. mTORC1 is negatively regulated by a protein complex called tuberous sclerosis complex 1 (TSC1)–TSC2 complex. Phosphotidyinositol 3-kinase (PI3K)–Akt signaling results in phosphorylation of TSC2 and release of mTOR regulation. Several growth factor receptor signaling pathways, including the IL-2 receptor pathway in T cells, activate mTOR through PI3K-Akt, leading to translation of proteins needed for cell cycle progression. Thus, by inhibiting mTORC1 function, rapamycin blocks IL-2–driven T cell proliferation. Combinations of cyclosporine (which blocks IL-2 synthesis) and rapamycin (which blocks IL-2–driven proliferation) are potent inhibitors of T cell responses. Interestingly, rapamycin inhibits the generation of effector T cells but does not impair the survival and functions of regulatory T cells as much, which may promote immune suppression of allograft rejection. mTORC1 is involved in dendritic cell functions, and therefore rapamycin may suppress T cell responses by interfering with dendritic cell function as well. mTORC1 is also involved in B cell proliferation and antibody responses, and therefore rapamycin may also be effective in preventing or treating antibody-mediated rejection. In addition to rapamycin, other mTOR inhibitors have been developed for immunosuppression of allograft recipients and for cancer therapy.
Other molecules involved in cytokine and T cell receptor signaling are also targets of immunosuppressive drugs that are in early trials for treatment or prevention of allograft rejection. These target molecules include JAK3, a kinase linked to signaling of various cytokine receptors, including IL-2, and protein kinase C, an essential kinase in T cell receptor signaling.
Antimetabolites
Metabolic toxins that kill proliferating T cells are used in combination with other drugs to treat graft rejection. These agents inhibit the proliferation of lymphocyte precursors during their maturation and also kill proliferating mature T cells that have been stimulated by alloantigens. The first such drug to be developed for the prevention and treatment of rejection was azathioprine. This drug is still used, but it is toxic to precursors of leukocytes in the bone marrow and enterocytes in the gut. The most widely used drug in this class is mycophenolate mofetil (MMF). MMF is metabolized to mycophenolic acid, which blocks a lymphocyte-specific isoform of inosine monophosphate dehydrogenase, an enzyme required for de novo synthesis of guanine nucleotides. Because MMF selectively inhibits the lymphocyte-specific isoform of this enzyme, it has relatively few toxic effects on other cells. MMF is now routinely used, often in combination with cyclosporine or FK506, to prevent acute allograft rejection.
Function-Blocking or Depleting Antilymphocyte Antibodies
Antibodies that react with T cell surface structures and deplete or inhibit T cells are used to treat acute rejection episodes. One widely used antibody is a mouse monoclonal antibody called OKT3 that is specific for human CD3. Polyclonal rabbit or horse antibodies specific for a mixture of human T cell surface proteins, so-called antithymocyte globulin, have also been in clinical use for many years to treat acute allograft rejections. These anti–T cell antibodies deplete circulating T cells either by activating the complement system to eliminate T cells or by opsonizing them for phagocytosis. T cells that escape elimination by OKT3 probably do so by endocytosis (“modulation”) of CD3 off their surface, but such cells may be rendered nonfunctional.
Monoclonal antibodies are now in clinical use that are specific for CD25, the α subunit of the IL-2 receptor. These reagents presumably prevent T cell activation by blocking IL-2 binding to activated T cells and IL-2 signaling.
Another monoclonal antibody in use in clinical transplantation is a rat IgM specific for CD52, a cell surface protein expressed widely on most mature B and T cells whose function is not understood. Anti-CD52 was originally developed to treat B cell malignant neoplasms, and it was found to profoundly deplete most peripheral B and T cells for many weeks after injection into patients. In current trials, it has been administered just before and early after transplantation, with the hope that it may induce a prolonged state of graft tolerance as new lymphocytes develop in the presence of the allograft.
The major limitation to the use of monoclonal or polyclonal antibodies from other species is that humans given these agents produce anti-immunoglobulin (Ig) antibodies that eliminate the injected foreign Ig. For this reason, human-mouse chimeric (“humanized”) antibodies (e.g., against CD3 and CD25), which are less immunogenic, have been developed.
Costimulatory Blockade
Drugs that block T cell costimulatory pathways reduce acute allograft rejection. The rationale for the use of these types of drugs is to prevent the delivery of costimulatory signals required for activation of T cells (see Chapter 9). A soluble high-affinity form of CTLA-4 fused to an IgG Fc domain binds to B7 molecules on APCs and prevents them from interacting with T cell CD28 (see Chapter 9, Fig. 9-7) and is near approval for use in allograft recipients. Clinical studies have shown that CTLA-4–Ig can be as effective as cyclosporine in preventing acute rejection. An antibody that binds to T cell CD40 ligand and prevents its interactions with CD40 on APCs (see Chapter 9) has also proved beneficial for preventing graft rejection in experimental animals. In some experimental protocols, simultaneous blockade of both B7 and CD40 appears to be more effective than either alone in promoting graft survival. However, the anti-CD40L antibody has a serious side effect of thrombotic complications, apparently related to the expression of CD40L on platelets.
Drugs Targeting Alloantibodies and Alloreactive B Cells
As we have learned more about the importance of alloantibodies in mediating acute and perhaps chronic rejection, therapies targeting antibodies and B cells that were developed for other diseases are now being used in transplant patients. For example, plasmapheresis is sometimes used to treat acute antibody-mediated rejection. In this procedure, a patient’s blood is pumped through a machine that removes the plasma but returns the blood cells to the circulation. In this way, circulating antibodies, including pathogenic alloreactive antibodies, can be removed. Intravenous immune globulin (IVIG) therapy, which is used to treat several, often antibody-mediated, inflammatory diseases, is also being applied in the setting of acute antibody-mediated rejection. In IVIG therapy, pooled IgG from normal donors is injected intravenously into a patient. The mechanisms of action are not fully understood but likely involve binding of the injected IgG to the patient’s Fc receptors on various cell types, thereby reducing alloantibody production and blocking effector functions of the patient’s own antibodies. IVIG also enhances degradation of the patient’s antibodies by competitively inhibiting their binding to the neonatal Fc receptor (see Chapter 12). A monoclonal antibody specific for the B cell surface protein CD20 very effectively depletes mature B cells from the circulation and secondary lymphoid organs. Anti-CD20 has been used for treatment of B cell lymphomas and for autoimmune diseases and is now in clinical trials for treatment of antibody-mediated allograft rejection. These antibody and B cell targeted therapies have been used in combination to effectively treat antibody-mediated rejection.
Anti-inflammatory Drugs
Anti-inflammatory agents, specifically corticosteroids, are frequently used to reduce the inflammatory reaction to organ allografts. The proposed mechanism of action of these natural hormones and their synthetic analogues is to block the synthesis and secretion of cytokines, including tumor necrosis factor (TNF) and IL-1, and other inflammatory mediators, such as prostaglandins, reactive oxygen species, and nitric oxide, produced by macrophages and other inflammatory cells. The net result of this therapy is reduced leukocyte recruitment, inflammation, and graft damage. Very high doses of corticosteroids may inhibit T cell secretion of cytokines or even kill some T cells, but it is unlikely that the levels of corticosteroids achieved in vivo act in this way. Newer anti-inflammatory agents are in clinical trials, including soluble cytokine receptors and anticytokine antibodies.
Inhibitors of Leukocyte Migration
A new therapeutic agent, called fingolimod (FTY720), works by binding to and blocking sphingosine 1-phosphate (S1P) receptors on lymphocytes. S1P is required for the egress of lymphocytes from lymphoid organs (see Chapter 3), and blocking its action leads to the sequestration of lymphocytes in lymph nodes. Fingolimod inhibits allograft rejection in animal models. This drug is not yet used for clinical transplantation, but it is approved for treatment of multiple sclerosis, an autoimmune disease of the central nervous system.
Anti-integrin antibodies have proved to be effective treatments for some autoimmune diseases because they block leukocyte recruitment from the circulation into inflamed tissues (see Chapter 3). There are some early animal studies testing whether these drugs work to block allograft rejection, but so far there are too few data to predict if this approach will be useful.
Current immunosuppressive protocols have dramatically improved graft survival. Before the use of calcineurin inhibitors, the 1-year survival rate of unrelated cadaveric kidney grafts was between 50% and 60%, with a 90% rate for grafts from living related donors (which are better matched with the recipients). Since cyclosporine, FK506, rapamycin, and MMF have been introduced, the survival rate of unrelated cadaveric kidney grafts has increased to about 90% at 1 year. Heart transplantation, for which HLA matching is not practical, has also significantly benefited from the use of cyclosporine and now has a similar ~90% 1-year survival rate (see Fig. 16-11). Experience with other organs is more limited, but survival rates have also improved with modern immunosuppressive therapy, with 10-year patient survival rates of approximately 60% and 75% for pancreas and liver recipients, respectively, and 3-year patient survival rates of 70% to 80% for lung recipients.
Strong immunosuppression is usually started in allograft recipients at the time of transplantation with a combination of drugs, and after a few days, the drugs are changed for long-term maintenance of immunosuppression. For example, in the case of adult kidney transplantation, a patient may be initially induced with an anti–IL-2R or anti–T cell depleting antibody and a high-dose corticosteroid, and then maintained on a calcineurin inhibitor, an antimetabolite, and maybe low-dose steroids. Acute rejection, when it occurs, is managed by rapidly intensifying immunosuppressive therapy. In modern transplantation, chronic rejection has become a more common cause of allograft failure, especially in cardiac transplantation. Chronic rejection is more insidious than acute rejection, and it is much less reversible by immunosuppression.
Immunosuppressive therapy leads to increased susceptibility to various types of intracellular infections and virus-associated tumors. The major goal of immunosuppression to treat graft rejection is to reduce the generation and function of helper T cells and CTLs, which mediate acute cellular rejection. It is therefore not surprising that defense against viruses and other intracellular pathogens, the physiologic function of T cells, is also compromised in immunosuppressed transplant recipients. Reactivation of latent herpesviruses is a frequent problem in immunosuppressed patients, including cytomegalovirus, herpes simplex virus, varicella-zoster virus, and Epstein-Barr virus. For this reason, transplant recipients are now given prophylactic antiviral therapy for herpesvirus infections. Immunosuppressed allograft recipients are also at greater risk for a variety of so-called opportunistic infections, which normally do not occur in immunocompetent people, including fungal infections (Pneumocystis jiroveci pneumonia, histoplasmosis, coccidioidomycosis), protozoan infections (toxoplasmosis), and gastrointestinal parasitic infections (Cryptosporidium and Microsporidium). Immunosuppressed allograft recipients have a higher risk for development of neoplasias compared with the general population, including various forms of skin cancer. Some of the tumors that are more frequently found in allograft recipients are known to be caused by viruses, and therefore they may arise because of impaired antiviral immunity. These include uterine cervical carcinoma, which is related to human papillomavirus infection, and lymphomas caused by Epstein-Barr virus infection. The lymphomas found in allograft recipients as a group are called post-transplantation lymphoproliferative disorders, and most are derived from B lymphocytes.
Despite the risks of infections and neoplasias associated with the use of immunosuppressive drugs, the major limitation on the tolerated doses of most of these drugs, including calcineurin inhibitors, mTOR inhibitors, antimetabolites, and steroids, is direct toxicity to cells unrelated to immunosuppression. In some cases, the toxicities affect the same cells as rejection does, such as cyclosporine toxicity to renal tubular epithelial cells, which can complicate the interpretation of declining renal function in kidney allograft recipients.
Methods to Reduce the Immunogenicity of Allografts
In human transplantation, the major strategy to reduce graft immunogenicity has been to minimize alloantigenic differences between the donor and recipient. Several clinical laboratory tests are routinely performed to reduce the risk for immunologic rejection of allografts. These include ABO blood typing; the determination of HLA alleles expressed on donor and recipient cells, called tissue typing; the detection of preformed antibodies in the recipient that recognize HLA and other antigens representative of the donor population; and the detection of preformed antibodies in the recipient that bind to antigens of an identified donor’s leukocytes, called crossmatching. Not all of these tests are done in all types of transplantation. We will now summarize each of these tests and discuss their significance.
To avoid hyperacute rejection, the ABO blood group antigens of the graft donor are selected to be identical to those of the recipient. This test is uniformly used in renal transplantation because kidney grafts will typically not survive if there are ABO incompatibilities between the donor and recipient. Natural IgM antibodies specific for allogeneic ABO blood group antigens (discussed earlier) will cause hyperacute rejection. Blood typing is performed by mixing a patient’s red blood cells with standardized sera containing anti-A or anti-B antibodies. If the patient expresses either blood group antigen, the serum specific for that antigen will agglutinate the red cells. The biology of the ABO blood group system is discussed later in the chapter in the context of blood transfusion.
In kidney transplantation, the larger the number of MHC alleles that are matched between the donor and recipient, the better the graft survival (Fig. 16-12). HLA matching had a more profound influence on graft survival before modern immunosuppressive drugs were routinely used, but current data still show significantly greater survival of grafts when donor and recipient have fewer HLA allele mismatches. Past clinical experience with older typing methods had shown that of all the class I and class II loci, matching at HLA-A, HLA-B, and HLA-DR is most important for predicting survival of kidney allografts. (HLA-C is not as polymorphic as HLA-A or HLA-B, and HLA-DR and HLA-DQ are in strong linkage disequilibrium, so matching at the DR locus often also matches at the DQ locus.) Although current typing protocols in many centers include HLA-C, DQ, and DP loci, most of the available data in predicting graft outcome refer only to HLA-A, HLA-B, and HLA-DR mismatches. Because two codominantly expressed alleles are inherited for each of these HLA genes, it is possible to have zero to six HLA mismatches of these three loci between the donor and recipient. Zero-antigen mismatches predict the best survival of living related donor grafts, and grafts with one-antigen mismatches do slightly worse. The survival of grafts with two to six HLA mismatches is significantly worse than that of grafts with zero- and one-antigen mismatches. HLA matching has an even greater impact on nonliving (unrelated) donor renal allografts. Therefore, attempts are made to reduce the number of differences in HLA alleles expressed on donor and recipient cells, which will have a modest effect in reducing the chance of rejection.
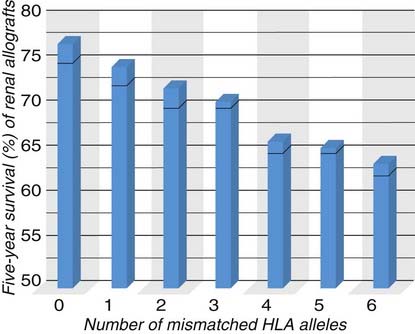
FIGURE 16–12 Influence of MHC matching on graft survival.
Matching of MHC alleles between the donor and recipient significantly improves renal allograft survival. The data shown are for deceased donor (cadaver) grafts. HLA matching has less of an impact on survival of renal allografts from live donors, and some MHC alleles are more important than others in determining outcome.
(Data from Organ Procurement and Transplantation Network/Scientific Registry annual report, 2010.)
HLA matching in renal transplantation is possible because donor kidneys can be stored in organ banks before transplantation until a well-matched recipient can be identified, and because patients needing a kidney allograft can be maintained on dialysis until a well-matched organ is available. In the case of heart and liver transplantation, organ preservation is more difficult, and potential recipients are often in critical condition. For these reasons, HLA typing is not considered in pairing of potential donors and recipients, and the choice of donor and recipient is based only on ABO blood group matching and anatomic compatibility. In heart transplantation, the paucity of donors, the emergent need for transplantation, and the success of immunosuppression override the possible benefit of reducing HLA mismatches between donor and recipient. As we will discuss later, in bone marrow transplantation, HLA matching is essential to reduce the risk of graft-versus-host disease.
Most HLA haplotype determinations are now performed by polymerase chain reaction (PCR), replacing older serologic methods. MHC genes can be amplified by PCR with use of primers that bind to conserved sequences within the 5′ and 3′ ends of exons encoding the polymorphic regions of class I and class II MHC molecules. The amplified segment of DNA can then be sequenced. Thus, the actual nucleotide sequence, and therefore the predicted amino acid sequence, can be directly determined for the MHC alleles of any cell, providing precise molecular tissue typing. On the basis of these DNA sequencing efforts, the nomenclature of HLA alleles has changed to reflect the identification of many alleles not distinguished by previous serologic methods. Each allele defined by sequence has at least a four-digit number, but some alleles require six or eight digits for precise definition. The first two digits usually correspond to the older serologically defined allotype, and the third and fourth digits indicate the subtypes. Alleles with differences in the first four digits encode proteins with different amino acids. For example, HLA-DRB1*1301 is the sequence-defined 01 allele of the 13-allele family of the gene encoding the HLA-DR β1 protein.
Patients in need of allografts are also tested for the presence of preformed antibodies against donor MHC molecules or other cell surface antigens. Two types of tests are done to detect these antibodies. In the panel reactive antibody test, patients waiting for organ transplants are screened for the presence of preformed antibodies reactive with allogeneic HLA molecules prevalent in the population. These antibodies, which may be produced as a result of previous pregnancies, transfusions, or transplantation, can identify risk for hyperacute or acute vascular rejection. Small amounts of the patient’s serum are mixed with multiple fluorescently labeled beads coated with defined MHC molecules, representative of the MHC alleles that may be present in an organ donor population. Each MHC allele is attached to a bead with a differently colored fluorescent label. Binding of the patient’s antibodies to beads is determined by flow cytometry. The results are reported as percent reactive antibody (PRA), which is the percentage of the MHC allele pool with which the patient’s serum reacts. The PRA is determined on multiple occasions while a patient waits for an organ allograft. This is because the PRA can vary, as each panel is chosen at random and the patient’s serum antibody titers may change over time.
If a potential donor is identified, the crossmatching test will determine whether the patient has antibodies that react specifically with that donor’s cells. The test is performed by mixing the recipient’s serum with the donor’s blood lymphocytes. Complement-mediated cytotoxicity tests or flow cytometric assays can then be used to determine if antibodies in the recipient serum have bound to the donor cells. For example, complement is added to the mixture of cells and serum, and if preformed antibodies, usually against donor MHC molecules, are present in the recipient’s serum, the donor cells are lysed. This would be a positive crossmatch, which indicates that the donor is not suitable for that recipient.
Methods to Induce Donor-Specific Tolerance
Allograft rejection may be prevented by making the host tolerant to the alloantigens of the graft. Tolerance in this setting means that the host does not injure the graft despite the absence or withdrawal of immunosuppressive and anti-inflammatory agents. It is presumed that tolerance to an allograft will involve the same mechanisms that are involved in peripheral tolerance to self antigens (see Chapter 14), namely, anergy, deletion, and active suppression of alloreactive T cells. Tolerance is desirable in transplantation because it is alloantigen specific and will therefore avoid the major problems associated with nonspecific immunosuppression, namely, immune deficiency leading to increased susceptibility to infection and development of tumors and drug toxicity. In addition, achieving graft tolerance may reduce chronic rejection, which has to date been unaffected by the commonly used immunosuppressive agents that prevent and reverse acute rejection episodes.
Various experimental approaches and clinical observations have shown that it should be possible to achieve tolerance to allografts. In experiments in mice, Medawar and colleagues found that if neonatal mice of one strain (the recipient) are given spleen cells of another strain (the donor), the recipients will subsequently accept skin grafts from the donor. Such tolerance is alloantigen specific because the recipients will reject grafts from mouse strains that express MHC alleles that differ from the donor’s. Renal transplant patients who have received blood transfusions containing allogeneic leukocytes have a lower incidence of acute rejection episodes than do those who have not been transfused. The postulated explanation for this effect is that the introduction of allogeneic leukocytes by transfusion produces tolerance to alloantigens. One underlying mechanism for tolerance induction may be that the transfused donor cells contain immature dendritic cells, which induce unresponsiveness to donor alloantigens. Indeed, pretreatment of potential recipients with blood transfusions is now used as prophylactic therapy to reduce rejection. Some recipients of liver allografts are able to retain healthy grafts even after withdrawal of immunosuppression. The mechanism underlying this apparent “spontaneous” tolerance is not known, and it seems to be unique to liver grafts.
Several strategies are being tested to induce donor-specific tolerance in allograft recipients.
Xenogeneic Transplantation
The use of solid organ transplantation as a clinical therapy is greatly limited by the lack of availability of donor organs. For this reason, the possibility of transplantation of organs from other mammals, such as pigs, into human recipients has kindled great interest.
A major immunologic barrier to xenogeneic transplantation is the presence of natural antibodies that cause hyperacute rejection. More than 95% of primates have natural IgM antibodies that are reactive with carbohydrate determinants expressed by cells of species that are evolutionarily distant, such as the pig. The majority of human anti-pig natural antibodies are directed at one particular carbohydrate determinant formed by the action of a pig α-galactosyltransferase enzyme. This enzyme places an α-linked galactose moiety on the same substrate that in human and other primate cells is fucosylated to form the blood group H antigen. Species combinations that give rise to natural antibodies against each other are said to be discordant. Natural antibodies are rarely produced against carbohydrate determinants of closely related, concordant species, such as humans and chimpanzees. Thus, organs from chimpanzees or other higher primates might theoretically be accepted in humans. However, ethical and logistic concerns have limited such procedures. For reasons of anatomic compatibility, pigs are the preferred xenogeneic species for organ donation to humans.
Natural antibodies against xenografts induce hyperacute rejection by the same mechanisms as those seen in hyperacute allograft rejection. These mechanisms include the generation of endothelial cell procoagulants and platelet-aggregating substances, coupled with the loss of endothelial anticoagulant mechanisms. However, the consequences of activation of human complement on pig cells are typically more severe than the consequences of activation of complement by natural antibodies on human allogeneic cells, possibly because some of the complement regulatory proteins made by pig cells, such as decay-accelerating factor, are not able to interact with human complement proteins and thus cannot limit the extent of complement-induced injury (see Chapter 12). A strategy for reducing hyperacute rejection in xenotransplantation is to breed transgenic pigs that cannot express enzymes that synthesize pig antigens or express human proteins that inhibit human complement activation. For example, α-galactosyltransferase knockout pigs and transgenic pigs expressing human complement regulatory proteins have been generated, and transplants of organs from these animals into primates are resistant to hyperacute rejection.
Even when hyperacute rejection is prevented, xenografts are often damaged by a form of acute vascular rejection that occurs within 2 to 3 days of transplantation. This form of rejection has been called delayed xenograft rejection, accelerated acute rejection, or acute vascular rejection and is characterized by intravascular thrombosis and necrosis of vessel walls. The mechanisms of delayed xenograft rejection are incompletely understood; recent findings indicate that there may be incompatibilities between primate platelets and porcine endothelial cells that promote thrombosis independent of antibody-mediated damage.
Xenografts can also be rejected by T cell–mediated immune responses to xenoantigens. The mechanisms of cell-mediated rejection of xenografts are believed to be similar to those that we have described for allograft rejection, and T cell responses to xenoantigens can be as strong as or even stronger than responses to alloantigens.
Blood Transfusion and the ABO and Rh Blood Group Antigens
Blood transfusion is a form of transplantation in which whole blood or blood cells from one or more individuals are transferred intravenously into the circulation of a host. Blood transfusions are most often performed to replace blood lost by hemorrhage or to correct defects caused by inadequate production of blood cells, which may occur in a variety of diseases. The major barrier to successful blood transfusions is the immune response to cell surface molecules that differ between individuals. The most important alloantigen system in blood transfusion is the ABO system, which we will discuss in detail below. ABO antigens are expressed on virtually all cells, including red blood cells. Individuals lacking a particular blood group antigen produce natural IgM antibodies against that antigen. If such individuals are given blood cells expressing the target antigen, the preexisting antibodies bind to the transfused cells, activate complement, and cause transfusion reactions, which can be life-threatening. Transfusion across an ABO barrier may trigger an immediate hemolytic reaction, resulting in both intravascular lysis of red blood cells, probably mediated by the complement system, and extensive phagocytosis of antibody- and complement-coated erythrocytes by macrophages of the liver and spleen. Hemoglobin is liberated from the lysed red cells in quantities that may be toxic for kidney cells, causing acute renal tubular cell necrosis and kidney failure. High fevers, shock, and disseminated intravascular coagulation may also develop, suggestive of massive cytokine release (e.g., of TNF or IL-1). The disseminated intravascular coagulation consumes clotting factors faster than they can be synthesized, and the patient may paradoxically die of bleeding in the presence of widespread clotting. More delayed hemolytic reactions may result from incompatibilities of minor blood group antigens. These result in progressive loss of the transfused red cells, leading to anemia, and jaundice, a consequence of overloading the liver with hemoglobin-derived pigments.
We will now discuss the ABO blood group antigens as well as other blood group antigens of clinical relevance.
ABO Blood Group Antigens
The ABO antigens are carbohydrates linked to cell surface proteins and lipids that are synthesized by polymorphic glycosyltransferase enzymes, which vary in activity depending on the inherited allele (Fig. 16-13). The ABO antigens were the first alloantigen system to be defined in mammals. All normal individuals synthesize a common core glycan, called the O antigen, that is mainly attached to plasma membrane proteins. Most individuals possess a fucosyltransferase that adds a fucose moiety to a nonterminal sugar residue of the O antigen, and the fucosylated glycan is called the H antigen. A single gene on chromosome 9 encodes a glycosyltransferase enzyme, which further modifies the H antigen. There are three allelic variants of this gene. The O allele gene product is devoid of enzymatic activity. The A allele–encoded enzyme transfers a terminal N-acetylgalactosamine moiety, and the B allele gene product transfers a terminal galactose moiety. Individuals who are homozygous for the O allele cannot attach terminal sugars to the H antigen and express only the H antigen. In contrast, individuals who possess an A allele (AA homozygotes, AO heterozygotes, or AB heterozygotes) form the A antigen by adding terminal N-acetylgalactosamine to some of their H antigens. Similarly, individuals who express a B allele (BB homozygotes, BO heterozygotes, or AB heterozygotes) form the B antigen by adding terminal galactose to some of their H antigens. AB heterozygotes form both A and B antigens from some of their H antigens. The terminology has been simplified so that OO individuals are said to be blood type O; AA and AO individuals are blood type A; BB and BO individuals are blood type B; and AB individuals are blood type AB. Mutations in the gene encoding the fucosyltransferase that produces the H antigen are rare; people who are homozygous for such a mutation are said to have the Bombay blood group and cannot produce H, A, or B antigens. These individuals make antibodies against H, A, and B antigens and cannot receive type O, A, B, or AB blood.
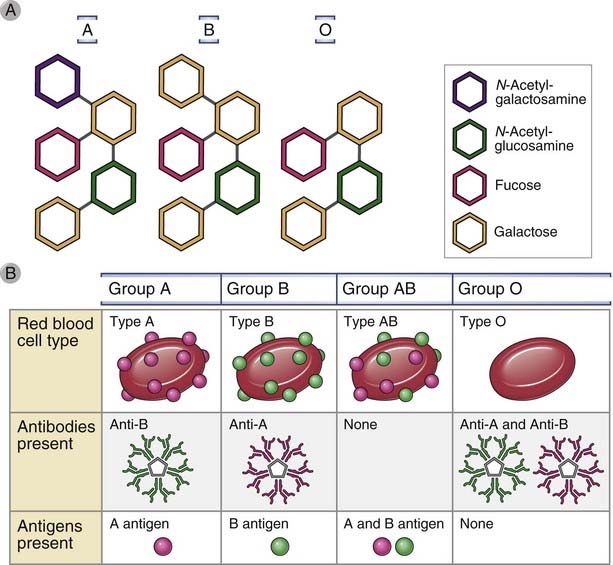
Figure 16–13 ABO blood group antigens.
A, Blood group antigens are carbohydrate structures added onto cell surface proteins by the action of glycosyltransferases. Most people inherit a gene that encodes l-fucosyltransferase, which produces the H antigen. Inheritance of a gene for N-acetyl-d-galactosaminyl transferase, which generates the A antigen, and the gene that encodes d-galactosyltransferase, which generates the B antigen, varies between people. A person who does not inherit genes for either of these enzymes will be type O; a person who inherits only one of these glycosal transferase genes will be type A or B; and a person who inherits genes for both enzymes will be type AB.
Individuals who express a particular ABO antigen are tolerant to that antigen, but individuals who do not express that antigen produce natural antibodies that recognize the antigen. Virtually all individuals express the H antigen, and therefore they are tolerant to this antigen and do not produce anti-H antibodies. Individuals who express A or B antigens are tolerant to these molecules and do not produce anti-A or anti-B antibodies, respectively. However, blood group O and A individuals produce anti-B IgM antibodies, and blood group O and B individuals produce anti-A IgM antibodies. On face value, it seems paradoxical that individuals who do not express a blood group antigen make antibodies against it. The likely explanation is that the antibodies are produced against glycolipids of intestinal bacteria that happen to cross-react with the ABO antigens, unless the individual is tolerant to one or more of these.
In clinical transfusion, the choice of blood donors for a particular recipient is based on the expression of blood group antigens and the antibody responses to them. If a patient receives a transfusion of red blood cells from a donor who expresses the antigen not expressed on self red blood cells, a transfusion reaction may result (described before). It follows that AB individuals can tolerate transfusions from all potential donors and are therefore called universal recipients; similarly, O individuals can tolerate transfusions only from O donors but can provide blood to all recipients and are therefore called universal donors. In general, differences in minor blood groups lead to red cell lysis only after repeated transfusions trigger a secondary antibody response.
ABO antigens are expressed on many other cell types in addition to blood cells, including endothelial cells. For this reason, ABO typing is critical to avoid hyperacute rejection of certain solid organ allografts, as discussed earlier in the chapter. ABO incompatibility between mother and fetus generally does not cause problems for the fetus because most of the anti-carbohydrate antibodies are IgM and do not cross the placenta.
Other Blood Group Antigens
Lewis Antigen
The same glycoproteins that carry the ABO determinants can be modified by other glycosyltransferases to generate minor blood group antigens. For example, addition of fucose moieties at other nonterminal positions can be catalyzed by different fucosyltransferases and results in epitopes of the Lewis antigen system. Lewis antigens have recently received much attention from immunologists because these carbohydrate groups serve as ligands for E-selectin and P-selectin.
Rhesus (Rh) Antigen
The Rhesus (Rh) antigens, named after the monkey species in which they were originally identified, are another clinically important group of blood group antigens. Rh antigens are nonglycosylated, hydrophobic cell surface proteins found in red blood cell membranes and are structurally related to other red cell membrane glycoproteins with transporter functions. Rh proteins are encoded by two tightly linked and highly homologous genes, but only one of them, called RhD, is commonly considered in clinical blood typing. This is because up to 15% of the population has a deletion or other alteration of the RhD allele. These people, called Rh negative, are not tolerant to the RhD antigen and will make antibodies to the antigen if they are exposed to Rh-positive blood cells.
The major clinical significance of anti-Rh antibodies is related to hemolytic reactions associated with pregnancy that are similar to transfusion reactions. Rh-negative mothers carrying an Rh-positive fetus can be sensitized by fetal red blood cells that enter the maternal circulation, usually during childbirth. Since the Rh antigen is a protein, as opposed to the carbohydrate ABO antigens, class-switched IgG antibodies are generated in Rh-negative mothers. Subsequent pregnancies in which the fetus is Rh positive are at risk because the maternal anti-Rh antibodies can cross the placenta and mediate the destruction of the fetal red blood cells. This causes erythroblastosis fetalis (hemolytic disease of the newborn) and can be lethal for the fetus. This disease can be prevented by administration of anti-RhD antibodies to the mother within 72 hours of birth of the first Rh-positive baby. The treatment prevents the baby’s Rh-positive red blood cells that entered the mother’s circulation from inducing the production of anti-Rh antibodies in the mother. The exact mechanisms of action of the administered antibodies are not clear but may include phagocytic clearance or complement-mediated lysis of the baby’s red cells or Fc receptor–dependent feedback inhibition of the mother’s RhD-specific B cells (see Chapter 11).
Hematopoietic Stem Cell Transplantation
The transplantation of allogeneic pluripotent hematopoietic stem cells is done commonly using an inoculum of bone marrow cells collected by aspiration, and the procedure is often called bone marrow transplantation. Hematopoietic stem cells can also be purified from the blood of donors after treatment with colony-stimulating factors, which mobilize stem cells from the bone marrow. The recipient is treated before transplantation to deplete bone marrow cells to free up niches for the transferred stem cells. After transplantation, stem cells repopulate the recipient’s bone marrow and differentiate into all the hematopoietic lineages. We consider bone marrow transplantation separately because this type of grafting has several unique features that are not encountered with solid organ transplantation.
Bone marrow transplantation is most often used clinically in the treatment of leukemias. In some forms of leukemia, the grafted cells are effective in destroying residual leukemia cells. In addition, the chemotherapeutic agents needed to destroy cancer cells also destroy normal marrow elements, and bone marrow transplantation is used to “rescue” the patient from the side effects of chemotherapy. Hematopoietic stem cell transplantation is also used clinically to treat diseases caused by inherited mutations in genes affecting only cells derived from hematopoietic stem cells, such as lymphocytes or red blood cells. Examples of such diseases that can be cured by hematopoietic stem cell transfer are adenosine deaminase (ADA) deficiency, X-linked severe combined immunodeficiency disease, and hemoglobin mutations such as beta-thalassemia major and sickle cell disease.
Allogeneic hematopoietic stem cells are rejected by even a minimally immunocompetent host, and therefore the donor and recipient must be carefully matched at all MHC loci. The mechanisms of rejection of bone marrow cells are not completely known, but in addition to adaptive immune mechanisms, hematopoietic stem cells may be rejected by NK cells. The role of NK cells in bone marrow rejection has been studied in experimental animals. Irradiated F1 hybrid mice reject bone marrow donated by either inbred parent. This phenomenon, called hybrid resistance, appears to violate the classical laws of solid tissue transplantation (see Fig. 16-2). Hybrid resistance is seen in T cell–deficient mice, and depletion of recipient NK cells with anti–NK cell antibodies prevents the rejection of parental bone marrow. Hybrid resistance is probably due to host NK cells reacting against bone marrow precursors that lack class I MHC molecules expressed by the host. Recall that normally, recognition of self class I MHC inhibits the activation of NK cells, and if these self MHC molecules are missing, the NK cells are released from inhibition (see Chapter 4, Fig. 4-6). Donor-versus-recipient NK cell alloreactivity has been used to reduce leukemia relapses after HLA haplotype–mismatched hematopoietic stem cell transplantation.
Even after successful engraftment, two additional problems are frequently associated with bone marrow transplantation, namely, graft-versus-host disease and immunodeficiency.
Graft-Versus-Host Disease
Graft-versus-host disease (GVHD) is caused by the reaction of grafted mature T cells in the marrow inoculum with alloantigens of the host. It occurs when the host is immunocompromised and therefore unable to reject the allogeneic cells in the graft. In most cases, the reaction is directed against minor histocompatibility antigens of the host because bone marrow transplantation is not performed when the donor and recipient have differences in MHC molecules. GVHD may also develop when solid organs that contain significant numbers of T cells are transplanted, such as the small bowel, lung, or liver.
GVHD is the principal limitation to the success of bone marrow transplantation. As in solid organ transplantation, GVHD may be classified on the basis of histologic patterns into acute and chronic forms.
Acute GVHD is characterized by epithelial cell death in the skin, liver (mainly the biliary epithelium), and gastrointestinal tract (Fig. 16-14). It is manifested clinically by rash, jaundice, diarrhea, and gastrointestinal hemorrhage. When the epithelial cell death is extensive, the skin or lining of the gut may slough off. In this circumstance, acute GVHD may be fatal.
FIGURE 16–14 Histopathology of acute GVHD in the skin.
A sparse lymphocytic infiltrate can be seen at the dermal-epidermal junction, and damage to the epithelial layer is indicated by spaces at the dermal-epidermal junction (vacuolization), cells with abnormal keratin staining (dyskeratosis), apoptotic keratinocytes, and disorganization of maturation of keratinocytes from the basal layer to the surface.
(Courtesy of Dr. Scott Grantor, Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts.)
Chronic GVHD is characterized by fibrosis and atrophy of one or more of the same organs, without evidence of acute cell death. Chronic GVHD may also involve the lungs and produce obliteration of small airways. When it is severe, chronic GVHD leads to complete dysfunction of the affected organ.
In animal models, acute GVHD is initiated by mature T cells present in the bone marrow inoculum, and elimination of mature donor T cells from the graft can prevent the development of GVHD. In clinical hematopoietic stem cell transplantation, efforts to eliminate T cells from the marrow inoculum have reduced the incidence of GVHD but also decrease the graft-versus-leukemia effect that is often critical in treating leukemias by this type of transplantation. T cell–depleted marrow also tends to engraft poorly, perhaps because mature T cells produce colony-stimulating factors that aid in stem cell repopulation. One approach that has been tried is to combine removal of T cells with supplemental colony-stimulating factor treatment to promote engraftment.
Although GVHD is initiated by grafted T cells recognizing host alloantigens, the effector cells that cause epithelial cell injury are less well defined. On histologic examination, NK cells are often attached to the dying epithelial cells, suggesting that NK cells are important effector cells of acute GVHD. CD8+ CTLs and cytokines also appear to be involved in tissue injury in acute GVHD.
The relationship of chronic GVHD to acute GVHD is not known and raises issues similar to those of relating chronic allograft rejection to acute allograft rejection. For example, chronic GVHD may represent the fibrosis of wound healing secondary to loss of epithelial cells. However, chronic GVHD can arise without evidence of prior acute GVHD. An alternative explanation is that chronic GVHD represents a response to ischemia caused by vascular injury.
Both acute and chronic GVHD are commonly treated with intense immunosuppression. It is not clear that either condition responds very well. A possible explanation for this therapeutic failure is that conventional immunosuppression is targeted against T lymphocytes, which may be only one of several mediators of GVHD. Cyclosporine and the metabolic toxin methotrexate are also used for prophylaxis against GVHD. Various new therapies are being studied in clinical trials, including rapamycin, anti-TNF antibodies, and regulatory T cell transfer.
Immunodeficiency After Bone Marrow Transplantation
Bone marrow transplantation is often accompanied by clinical immunodeficiency. Several factors may contribute to defective immune responses in recipients. Bone marrow transplant recipients may be unable to regenerate a complete new lymphocyte repertoire. Radiation therapy and chemotherapy used to prepare recipients for transplantation are likely to deplete the patient’s memory cells and long-lived plasma cells, and it can take a long time to regenerate these populations.
The consequence of immunodeficiency is that bone marrow transplant recipients are susceptible to viral infections, especially cytomegalovirus infection, and to many bacterial and fungal infections. They are also susceptible to Epstein-Barr virus–provoked B cell lymphomas. The immune deficiencies of bone marrow transplant recipients can be more severe than those of conventionally immunosuppressed patients. Therefore, bone marrow transplant recipients commonly receive prophylactic antibiotics and anti-cytomegalovirus therapy and are often actively immunized against capsular bacteria such as pneumococcus before transplantation.
There is great interest in the use of pluripotent stem cells to repair tissues with little natural regenerative capacity, such as cardiac muscle, brain, or spinal cord. One approach is to use embryonic stem cells, which are pluripotent stem cells derived from the blastocyst stage of human embryos. Although embryonic stem cells have not yet been used clinically, it is highly likely that a major barrier to their usefulness will be their alloantigenicity and rejection by the recipient’s immune system. A possible solution to this may be to use induced pluripotent stem (iPS) cells, which can be derived from adult somatic tissues by transduction of certain genes. The immunologic advantage of the iPS cell approach is that these cells can be derived from somatic cells harvested from the patient, and therefore they will be syngeneic to the patient.
Summary
Recognition and Rejection of Allogeneic Transplants
Baldwin WM, Valujskikh A, Fairchild RL. Antibody-mediated rejection: emergence of animal models to answer clinical questions. American Journal of Transplantation. 2010;10:1135-1142.
Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nature Review Immunology. 2005;5:807-817.
Heeger PS. T-cell allorecognition and transplant rejection: a summary and update. American Journal of Transplantation. 2003;3:525-533.
LaRosa DF, Rahman AH, Turka LA. The innate immune system in allograft rejection and tolerance. Journal of Immunology. 2007;178:7503-7509.
Li XC, Rothstein DM, Sayegh MH. Costimulatory pathways in transplantation: challenges and new developments. Immunological Reviews. 2009;229:271-293.
Chinen J, Buckley RH. Transplantation immunology: solid organ and bone marrow. Journal of Allergy and Clinical Immunology. 2010;125:S324-S335.
Lechler RI, Sykes M, Thomson AW, Turka LA. Organ transplantation—how much of the promise has been realized? Nature Medicine. 2005;11:605-613.
Ricordi C, Strom TB. Clinical islet transplantation: advances and immunological challenges. Nature Reviews Immunology. 2004;4:259-268.
Rogers NJ, Lechler RI. Allorecognition. American Journal of Transplantation. 2001;1:97-102.
Immunosuppression and Tolerance Induction to Allografts
Chidgey AP, Layton D, Trounson A, Boyd RL. Tolerance strategies for stem-cell-based therapies. Nature. 2008;453:330-377.
Gibbons C, Sykes M. Manipulating the immune system for anti-tumor responses and transplant tolerance via mixed hematopoietic chimerism. Immunological Reviews. 2008;223:334-360.
Halloran PF. Immunosuppressive drugs for kidney transplantation. New England Journal of Medicine. 2004;351:2715-2729.
Newell KA, Larsen CP, Kirk AD. Transplant tolerance: converging on a moving target. Transplantation. 2006;81:1-6.
Safinia N, Sagoo P, Lechler R, Lombardi G. Adoptive regulatory T cell therapy: challenges in clinical transplantation. Current Opinions in Organ Transplantation. 2010;15:427-434.
Turka LA, Lechler RI. Towards the identification of biomarkers of transplantation tolerance. Nature Reviews Immunology. 2009;9:521-526.