51 Antiviral drugs
Overview
This chapter deals with drugs used to treat infections caused by viruses. We give first some necessary information about viruses: a simple outline of virus structure, a list of the main pathogenic viruses and a brief summary of the life history of an infectious virus. We then continue with a consideration of the host–virus interaction: the defences deployed by the human host against viruses and the strategies employed by viruses to evade these measures. We then describe the various types of antiviral drugs and their mechanisms of action, with particular reference to the treatment of AIDS, an infection caused by the human immunodeficiency virus (HIV).
Background Information about Viruses
An Outline of Virus Structure
Viruses are small (usually in the range 20–30 nm) infective agents that are incapable of reproduction outside their host cells. The free-living (e.g. outside its host) virus particle is termed a virion, and consists of segments of nucleic acid (either RNA or DNA) enclosed in a protein coat comprised of symmetrical repeating structural units and called a capsid (Fig. 51.1). The viral coat, together with the nucleic acid core, is termed the nucleocapsid. Some viruses have, in addition, a further external lipoprotein envelope, which may be decorated with antigenic viral glycoproteins or phospholipids acquired from its host when the nucleocapsid buds through the membranes of the infected cell. Certain viruses also contain enzymes that initiate their replication in the host cell.
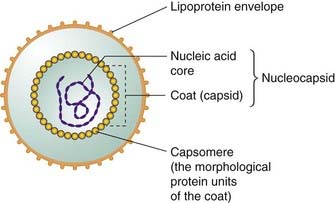
Viruses are generally characterised either as DNA or RNA viruses depending on the nature of their nucleic acid content. These two broad categories are conventionally subdivided into some six subgroups, which classify viruses according to whether they contain single- or double-stranded nucleic acids and how this functions during replication.
Examples of Pathogenic Viruses
Viruses can infect virtually all living organisms, and commonly cause disease in humans.
 Some important examples of the diseases they cause are as follow:
Some important examples of the diseases they cause are as follow:
Virus Function and Life History
As viruses have no metabolic machinery of their own, they have to attach to and penetrate a living host cell—animal, plant or bacterial—and hijack the victim’s own metabolic processes to replicate. The first step in this process is facilitated by polypeptide binding sites on the envelope or capsid, interacting with receptors on the host cell. These ‘receptors’ are normal membrane constituents, for example receptors for cytokines, neurotransmitters or hormones, ion channels, integral membrane glycoproteins, etc. Some examples of host cell receptors utilised by particular viruses are listed in Table 51.1.
Table 51.1 Some host cell structures that can function as receptors for viruses
| Host cell structurea | Virus(es) |
|---|---|
| Helper T lymphocytes CD4 glycoprotein | HIV (causing AIDS) |
| CCR5 receptor for chemokines MCP-1 and RANTES | HIV (causing AIDS) |
| CXCR4 chemokine receptor for cytokine SDF-1 | HIV (causing AIDS) |
| Acetylcholine receptor on skeletal muscle | Rabies virus |
| B-lymphocyte complement C3d receptor | Glandular fever virus |
| T-lymphocyte interleukin-2 receptor | T-cell leukaemia viruses |
| β-Adrenoceptors | Infantile diarrhoea virus |
| MHC molecules | Adenovirus (causing sore throat and conjunctivitis) T-cell leukaemia viruses |
MCP-1, monocyte chemoattractant protein-1; MHC, major histocompatibility complex; RANTES, regulated on activation normal T-cell expressed and secreted; SDF-1, stromal cell-derived factor-1.
a For more detail on complement, interleukin-2, the CD4 glycoprotein on helper T lymphocytes, MHC molecules, etc., see Chapter 6. For SDF-1, see Chapter 25.
Following attachment, the receptor–virus complex enters the cell (often by receptor-mediated endocytosis), during which time the virus coat may be removed by host cell enzymes (often lysosomal in nature). Some bypass this route. Once in the host cell, the nucleic acid of the virus then uses the host cell’s machinery to synthesise nucleic acids and proteins that are assembled into new virus particles. The actual way in which this occurs differs between DNA and RNA viruses.
Replication in DNA viruses
Viral DNA enters the host cell nucleus, where transcription into mRNA occurs catalysed by the host cell RNA polymerase. Translation of the mRNA into virus-specific proteins then takes place. Some of these proteins are enzymes that then synthesise more viral DNA, as well as structural proteins comprising the viral coat and envelope. After assembly of coat proteins around the viral DNA, complete virions are released by budding or after host cell lysis.
Replication in RNA viruses
Enzymes within the virion synthesise its mRNA from the viral RNA template, or sometimes the viral RNA serves as its own mRNA. This is translated by the host cell into various enzymes, including RNA polymerase (which directs the synthesis of more viral RNA), and also into structural proteins of the virion. Assembly and release of virions occurs as explained above. With these viruses, the host cell nucleus is usually not involved in viral replication, although some RNA viruses (e.g. orthomyxoviruses) replicate exclusively within the host nuclear compartment.
Replication in retroviruses
The virion in retroviruses1 contains a reverse transcriptase enzyme (virus RNA-dependent DNA polymerase), which makes a DNA copy of the viral RNA. This DNA copy is integrated into the genome of the host cell, and it is then termed a provirus. The provirus DNA is transcribed into both new viral genome RNA as well as mRNA for translation in the host into viral proteins, and the completed viruses are released by budding. Many retroviruses can replicate without killing the host cell.
The ability of several viruses to remain dormant within, and be replicated together with, the host genome is responsible for the periodic nature of some viral diseases, such as those caused by herpes labialis (cold sores) or the varicella zoster (chickenpox and shingles) virus, which recur when viral replication is reactivated by some factor (or when the immune system is compromised in some way). Some RNA retroviruses can transform normal cells into malignant cells.
The Host–Virus Interaction
Host Defences against Viruses
The first defence is the simple barrier function of intact skin, which most viruses are unable to penetrate. However, broken skin (e.g. at sites of wounds or insect bites) and mucous membranes are more vulnerable to viral attack. Should the virus gain entry to the body, then the host will deploy both the innate and subsequently the adaptive immune response (Ch. 6) to limit the incursion. The infected cell presents, on its surface, viral peptides complexed with major histocompatibility complex (MHC) class I molecules. This complex is recognised by T lymphocytes, which then kill the infected cell (Fig. 51.2). This may be accomplished by the release of lytic proteins (such as perforins, granzymes) or by triggering the apoptotic pathway in the infected cell by activation of its Fas receptor (‘death receptor’). The latter may also be triggered indirectly through the release of a cytokine such as tumour necrosis factor (TNF)-α. If the virus escapes immune detection by cytotoxic lymphocytes by modifying the expression of the peptide–MHC complex (see below), it may still fall victim to natural killer (NK) cells. This reaction to the absence of normal MHC molecules might be called the ‘mother turkey’ strategy (kill everything that does not sound exactly like a baby turkey). But some viruses also have a device for evading NK cells as well (see below).
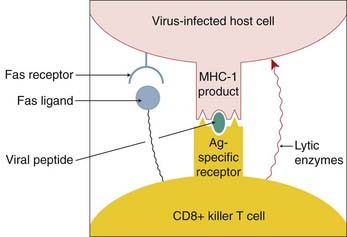
Fig. 51.2 The mechanisms whereby a CD8+ T cell kills a virus-infected host cell.
The virus-infected host cell expresses a complex of virus peptides plus major histocompatibility complex class I product (MHC-I) on its surface. This is recognised by the CD8+ T cell, which then releases lytic enzymes into the virus-infected cell and also expresses a Fas ligand. This triggers apoptosis in the infected cell by stimulating its Fas ‘death receptor’.
Within the cell itself, gene silencing may also provide a further level of protection (see Schutze, 2004). Short double-stranded fragments of RNA, such as those that could arise as a result of the virus’s attempts to recruit the host’s transcription/translational machinery, actually cause the gene coding for the RNA to be ‘silenced’—to be switched off, probably by DNA phosphorylation. This means that the gene is no longer able to direct further viral protein synthesis, thus interrupting the replication cycle. This mechanism can be exploited for experimental purposes in many areas of biology, and tailored siRNA (small- or short-interfering RNA) is a cheap and useful technique to suppress temporarily the expression of a particular gene of interest. Attempts to harness the technique for viricidal purposes have met with some success (see Barik, 2004).
Viral Ploys to Circumvent Host Defences
Viruses have evolved a variety of strategies to ensure successful infection, some entailing redirection of the host’s response for the advantage of the virus (discussed by Tortorella et al., 2000).
Subversion of the immune response
Viruses can inhibit the synthesis or action of the cytokines, such as interleukin-1, TNF-α and the antiviral interferons (IFNs), that normally coordinate the innate and adaptive immune responses. Following infection, for example, some poxviruses express proteins that mimic the extracellular ligand-binding domains of cytokine receptors. These pseudoreceptors bind cytokines, preventing them from reaching their natural receptors on cells of the immune system and thus moderating the normal immune response to virus-infected cells. Other viruses that can interfere with cytokine signalling include human cytomegalovirus, Epstein–Barr virus, herpesvirus and adenovirus.
Evasion of immune detection and attack by killer cells
Once within host cells, viruses may also escape immune detection and evade lethal attack by cytotoxic lymphocytes and NK cells in various ways, such as the following:
It is evident that evolution has equipped pathogenic viruses with many efficacious tactics for circumventing host defences, and understanding these in more detail is likely to suggest new types of antiviral therapy. Fortunately, the biological arms race is not one sided, and evolution has also equipped the host with sophisticated countermeasures. In most cases these prevail, and most viral infections eventually resolve spontaneously, except in an immunocompromised host. The situation does not always end happily though; some viral infections, such as Lassa fever and Ebola virus infection, have a high mortality, and we now discuss a further, grave example of this group: the HIV virus. This is appropriate because HIV exhibits many of the features common to other viral infections, and the sheer scale of the global AIDS problem has pushed HIV to the top of the list of antiviral targets.
Viruses 
HIV and AIDS
HIV is an RNA retrovirus. Two forms are known. HIV-1 is the organism responsible for human AIDS. The HIV-2 organism is similar to the HIV-1 virus in that it also causes immune suppression, but it is less virulent. HIV-1 is distributed around the world, whereas the HIV-2 virus is confined to parts of Africa.
The global situation is improving but even so, in 2007, the World Health Organization estimated that almost 33 million people were living with AIDS, and that women and children constituted approximately half that total number. During the same year, some 2 million people died of the disease (including 0.27 million children under 15 years), and there were a further 2.7 million new cases of AIDS infection reported. The epidemic is overwhelmingly centred on sub-Saharan Africa, which accounts for two-thirds of the total global number of infected persons, and where the adult prevalence is over 20 times greater than in Europe. For a review of the pathogenesis of AIDS, see Mindel & Tenant-Flowers (2001).
The interaction of HIV with the host’s immune system is complex, and although it involves mainly cytotoxic T lymphocytes (CTLs, CD8+ T cells) and CD4+ helper T lymphocytes (CD4+ cells), other immune cells, such as macrophages, dendritic cells and NK cells, also play a part. Antibodies are produced by the host to various HIV components, but it is the action of the CTLs and CD4+ cells that initially prevents the spread of HIV.
Cytotoxic T lymphocytes directly kill virally infected cells and produce and release antiviral cytokines (Fig. 51.2). The lethal event is lysis of the target cell, but induction of apoptosis by interaction of Fas ligand (see Fig. 5.5) on the CTL with Fas receptors on the virally infected cell can also play a part. CD4+ cells have an important role as helper cells, and it is the progressive loss of these cells that is the defining characteristic of HIV infection (see Fig. 51.4). Recent work suggests that CD4+ cells may themselves have a direct role (e.g. lysis of target cells) in the control of HIV replication (Norris et al., 2004).
The priming of naive T cells to become CTLs during the induction phase involves interaction of the T-cell receptor complex with antigenic HIV peptide in association with MHC class I molecules on the surface of antigen-presenting cells (APCs; see Figs 6.3 and 6.4). Priming also requires the presence and participation of CD4+ cells. It is thought that both types of cell need to recognise antigen on the surface of the same APC (Fig. 6.3).
The CTLs thus generated are effective during the initial stages of the infection but are not able to stop the progression of the disease. It is believed that this is because the CTLs have become ‘exhausted’ and dysfunctional, thus losing their protective function. Different mechanisms may be involved (see Jansen et al., 2004, and Barber et al., 2006, for further details).
The HIV virion cannily attaches to proteins on the host cell surface to gain entry to the cells. The main targets are CD4 (the glycoprotein marker of a particular group of helper T lymphocytes) and CCR5 (a co-receptor for certain chemokines, including monocyte chemoattractant protein-1 and RANTES [regulated on activation normal T-cell expressed and secreted]). CD4+ cells normally orchestrate the immune response to viruses, but by entering these cells and using them as virion factories, HIV virtually cripples this aspect of the immune response. Figure 51.3 shows an HIV virion infecting a CD4+ T cell. Infected activated CD4 T cells in lymphoid tissue form the major source of HIV production in HIV-infected individuals; infected macrophages are another source.
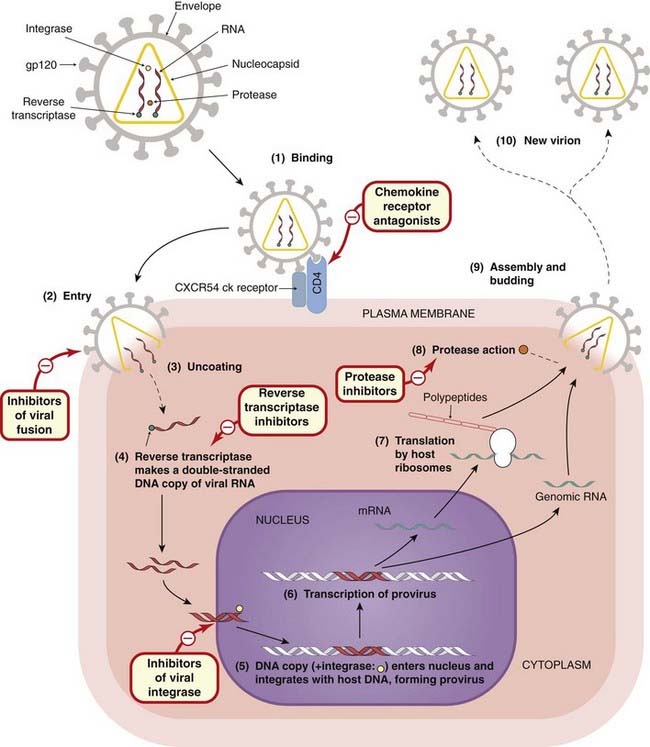
Fig. 51.3 Schematic diagram of infection of a CD4+ T cell by an HIV virion, with the sites of action of the two main classes of anti-HIV drugs.
The 10 steps of HIV infection, from attachment to the cell to release of new virions, are shown. The virus uses the CD4 co-receptor and the chemokine (ck) receptors CCR5/CXCR4 as binding sites to facilitate entry into the cell, where it becomes incorporated into host DNA (steps 1–5). When transcription occurs (step 6), the T cell itself is activated and the transcription factor nuclear factor κB initiates transcription of both host cell and provirus DNA. A viral protease cleaves the nascent viral polypeptides (steps 7 and 8) into structural proteins and enzymes (integrase, reverse transcriptase, protease) for the new virion. The new virions are assembled and released from the cells, initiating a fresh round of infection (steps 9 and 10). The sites of action of the currently used anti-HIV drugs are shown.
As for CCR5, evidence from exposed individuals who somehow evade infection indicates that this surface protein has a central role in HIV pathogenesis. Compounds that inhibit the entry of HIV into cells by blocking CCR5 are now available (see below).
When immune surveillance breaks down, other strains of HIV arise that recognise other host cell surface molecules such as CD4 and CXCR4. A surface glycoprotein, gp120, on the HIV envelope binds to CD4 and also to the T-cell chemokine co-receptor CXCR4. Another viral glycoprotein, gp41, then causes fusion of the viral envelope with the plasma membrane of the cell (Fig. 51.3).
Once within the cell, HIV is integrated with the host DNA (the provirus form), undergoing transcription and generating new virions when the cell is activated (Fig. 51.3). In an untreated subject, a staggering 1010 new virus particles may be produced each day. Intracellular HIV can remain silent (latent) for a long time.
Viral replication is error prone, and there are a large number of mutations daily at each site in the HIV genome, so HIV soon escapes recognition by the original cytotoxic lymphocytes. Although other cytotoxic lymphocytes arise that recognise the altered virus protein(s), further mutations, in turn, allow escape from surveillance by these cells too. It is suggested that wave after wave of cytotoxic lymphocytes act against new mutants as they arise, gradually depleting a T-cell repertoire already seriously compromised by the loss of CD4+ helper T cells, until eventually the immune response fails.
There is considerable variability in the progress of the disease, but the usual clinical course of an untreated HIV infection is shown in Figure 51.4. An initial acute influenza-like illness is associated with an increase in the number of virus particles in the blood, their widespread dissemination through the tissues and the seeding of lymphoid tissue with the virion particles. Within a few weeks, the viraemia is reduced by the action of cytotoxic lymphocytes as specified above.
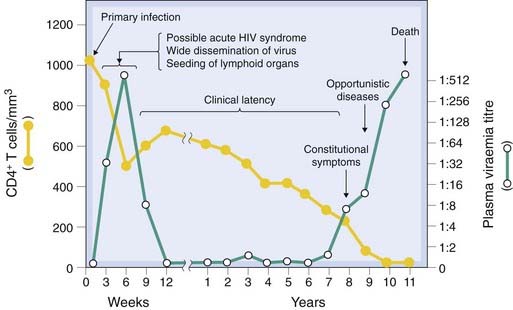
Fig. 51.4 Schematic outline of the course of HIV infection.
The CD4+ T-cell titre is often expressed as cells/mm3.
(Adapted from Pantaleo et al. 1993 N Engl J Med 328: 327–335.)
The acute initial illness is followed by a symptom-free period during which there is reduction in the viraemia accompanied by silent virus replication in the lymph nodes, associated with damage to lymph node architecture and the loss of CD4+ lymphocytes and dendritic cells. Clinical latency (median duration 10 years) comes to an end when the immune response finally fails and the signs and symptoms of AIDS appear—opportunistic infections (e.g. Pneumocystis pneumonia or tuberculosis), neurological disease (e.g. confusion, paralysis, dementia), bone marrow depression and cancers. Chronic gastrointestinal infections contribute to the severe weight loss. Cardiovascular damage and kidney damage can also occur. In an untreated patient, death usually follows within 2 years. The advent of effective drug regimens has greatly improved the prognosis in countries that are able to deploy them.
There is evidence that genetic factors play an important role in determining the susceptibility—or resistance—to HIV (see Flores-Villanueva et al., 2003).
Antiviral Drugs
Because viruses hijack many of the metabolic processes of the host cell itself, it is difficult to find drugs that are selective for the pathogen. However, there are some enzymes that are virus specific, and these have proved to be useful drug targets. Most currently available antiviral agents are effective only while the virus is replicating. Because the initial phases of viral infection are often asymptomatic, treatment is often delayed until the infection is well established. As is often the case with infectious diseases, an ounce of prevention is worth a pound of cure.
Antiviral drugs, of which many are available, fall into a few groups with similar mechanisms of action and side effects. Table 51.2 shows the commonest antiviral drugs, classified according to their mechanisms of action, some of the diseases they are used to treat and common side effects.
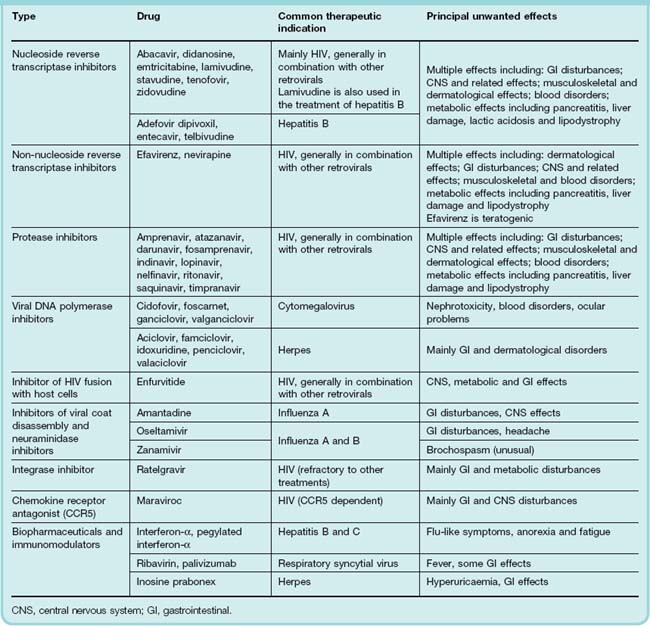
Reverse Transcriptase Inhibitors
The main group are nucleoside analogues, typified by zidovudine, all of which are phosphorylated by host cell enzymes to give the 5′-trisphosphate derivative. This moiety competes with the equivalent host cellular trisphosphate substrates for proviral DNA synthesis by viral reverse transcriptase (viral RNA-dependent DNA polymerase). Eventually, the incorporation of the 5′-trisphosphate moiety into the growing viral DNA chain results in chain termination. Mammalian α-DNA polymerase is relatively resistant to the effect. However, γ-DNA polymerase in the host cell mitochondria is more susceptible, and this may be the basis of some unwanted effects. The main utility of these drugs is the treatment of HIV, but a number of them have useful activity against other viruses also (e.g. hepatitis B).
Zidovudine
Zidovudine (AZT) was the first drug to be introduced for the treatment of HIV and retains an important place. It can prolong life in HIV-infected individuals and diminish HIV-associated dementia. Given to the parturient mother and then to the newborn infant, it can reduce mother-to-baby transmission by more than 20%. It is generally administered orally 2–3 times each day but can also be given by intravenous infusion. Its plasma half-life is 1 h, but the intracellular half-life of the active trisphosphate is 3 h. The concentration in cerebrospinal fluid (CSF) is 65% of the plasma level. Chemically, zidovudine is an analogue of thymidine. Most of the drug is metabolised to the inactive glucuronide in the liver, only 20% of the active form being excreted in the urine.
Because of rapid mutation, the virus is a constantly moving target, and resistance develops with long-term use of zidovudine, particularly in late-stage disease. Furthermore, resistant strains can be transferred between individuals. Other factors that underlie the loss of efficacy of the drug are decreased activation of zidovudine to the trisphosphate and increased virus load as the host immune response fails.
Unwanted effects include gastrointestinal disturbances (e.g. nausea, vomiting, abdominal pain), blood disorders (sometimes anaemia or neutropenia) and CNS effects (e.g. insomnia, dizziness, headache) as well as the risk of lactic acidosis in some patients, which are shared by this entire group of drugs to a greater or lesser extent.
Other, currently approved, drugs in this group include abacavir, adefovir dipivoxil, didanosine, emtricitabine, entecavir, lamivudine, stavudine, telbivudine and tenofovir.
Non-Nucleoside Reverse Transcriptase Inhibitors
Non-nucleoside reverse transcriptase inhibitors are chemically diverse compounds that bind to the reverse transcriptase enzyme near the catalytic site and inactivate it. Most non-nucleoside reverse transcriptase inhibitors are also inducers, substrates or inhibitors, to varying degrees, of the liver cytochrome P450 enzymes (Ch. 9). Currently available drugs are nevirapine and efavirenz.
Nevirapine has good oral bioavailability, and penetrates into the CSF. It is metabolised in the liver, and the metabolite is excreted in the urine. Nevirapine can prevent mother-to-baby transmission of HIV if given to the parturient mother and the neonate.
Efavirenz is given orally, once daily, because of its plasma half-life (~50 h). It is 99% bound to plasma albumin, and its CSF concentration is ~1% of that in the plasma. Nevertheless, its major adverse effects are insomnia, bad dreams and sometimes psychotic symptoms. It is also teratogenic if used in early pregnancy.
Unwanted effects common to both of these drugs include rash (common) as well as a cluster of other effects (see Table 51.2).
Protease Inhibitors
In HIV and many other viral infections, the mRNA transcribed from the provirus is translated into two biochemically inert polyproteins. A virus-specific protease then converts the polyproteins into various structural and functional proteins by cleavage at the appropriate positions (see Fig. 51.3). Because this protease does not occur in the host, it is a useful target for chemotherapeutic intervention. HIV-specific protease inhibitors bind to the site where cleavage occurs, and their use, in combination with reverse transcriptase inhibitors, has transformed the therapy of AIDS. Examples of current protease inhibitors are shown in Table 51.2 and are exemplified by drugs such as amprenavir, atazanavir, darunavir, fosamprenavir (prodrug of amprenavir), indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and timpranavir.
Ritonavir, a typical example, binds to and thus inactivates proteases from HIV-1 or HIV-2. It is often given in combination with other protease inhibitors (e.g. lopinavir) as it potentiates their action. Ritonavir is given orally, usually twice a day. It is usual to start at a low dose and increase gradually to a maximum over a period of a few days.
The plasma half-life of ritonavir is 3–5 h but oral absorption may be delayed in the presence of food. The drug is mainly (> 80%) excreted in the faeces with some 10% excreted in the urine. A major metabolite accounts for approximately one-third of all excreted drug.
Unwanted effects that are shared among this group include gastrointestinal disturbances (e.g. nausea, vomiting, abdominal pain), blood disorders (sometimes anaemia or neutropenia) and CNS effects (e.g. insomnia, dizziness, headache) as well as the risk of hyperglycaemia.
DNA Polymerase Inhibitors
Aciclovir
The era of effective selective antiviral therapy began with aciclovir, a guanosine derivative that is typical of drugs of this type.
Aciclovir is converted to the monophosphate by viral thymidine kinase, which is very much more effective in carrying out the phosphorylation than the enzyme of the host cell; it is therefore only activated adequately in infected cells. The host cell kinases then convert the monophosphate to the trisphosphate, the active form that inhibits viral DNA polymerase, terminating the nucleotide chain. It is 30 times more potent against the herpesvirus enzyme than the host enzyme. Aciclovir trisphosphate is fairly rapidly broken down within the host cells, presumably by cellular phosphatases. Resistance caused by changes in the viral genes coding for thymidine kinase or DNA polymerase has been reported, and aciclovir-resistant herpes simplex virus has been the cause of pneumonia, encephalitis and mucocutaneous infections in immunocompromised patients.
Aciclovir can be given orally, intravenously or topically. When it is given orally, only 20% of the dose is absorbed. The drug is widely distributed, and reaches effective concentrations in the CSF. It is excreted by the kidneys, partly by glomerular filtration and partly by tubular secretion.
Unwanted effects are minimal. Local inflammation can occur during intravenous injection if there is extravasation of the solution. Renal dysfunction has been reported when aciclovir is given intravenously; slow infusion reduces the risk. Nausea and headache can occur and, rarely, encephalopathy.
There are now many other drugs with a similar action to aciclovir including cidofovir, famciclovir (prodrug of penciclovir), ganciclovir, idoxuridine, penciclovir, valaciclovir (prodrug of aciclovir) and valganciclovir (prodrug of ganciclovir). Foscarnet achieves the same effect through a slightly different mechanism as does idoxuridine, which is sometimes used topically to treat herpes infections of the skin.

Neuraminidase Inhibitors and Inhibitors of Viral Coat Disassembly
Viral neuraminidase is one of three transmembrane proteins coded by the influenza genome. Infection with these RNA viruses begins with the attachment of the viral haemaglutinin to neuraminic (sialic) acid residues on host cells. The viral particle then enters the cell by an endocytic process. The endosome is acidified following influx of H+ through another viral protein, the M2 ion channel. This facilitates the disassembly of the viral structure, allowing the RNA to enter the host nucleus, thus initiating a round of viral replication. Newly replicated virions escape from the host cell by budding from the cell membrane. Viral neuraminidase promotes this by severing the bonds linking the particle coat and host sialic acid.
The neuraminidase inhibitors zanamivir and oseltamivir are active against both influenza A and B viruses, and are licensed for use at early stages in the infection or when use of the vaccine is impossible. Zanamivir is available as a powder for inhalation, and oseltamivir as an oral preparation. At the time of writing, governments around the world are stockpiling this latter drug in the expectation that it will mitigate the effects of the anticipated ‘swine’ (H1N1) flu pandemic.
Unwanted effects of both include gastrointestinal symptoms (nausea, vomiting, dyspepsia and diarrhoea), but these are less frequent and severe in the inhaled preparation.
Amantadine,2 quite an old drug (1966) and seldom recommended today, effectively blocks viral M2 ion channels, thus inhibiting disassembly. It is active against influenza A virus (an RNA virus) but has no action against influenza B virus. The closely related rimantadine is similar in its effects.
Given orally, amantadine is well absorbed, reaches high levels in secretions (e.g. saliva) and most is excreted unchanged via the kidney. Aerosol administration is feasible.
Unwanted effects are relatively infrequent, occurring in 5–10% of patients, and are not serious. Dizziness, insomnia and slurred speech are the most common adverse effects.
Drugs Acting By Other Mechanisms
Enfurvirtide inhibits the fusion of HIV with host cells. The drug is generally given by subcutaneous injection in combination with others to treat HIV when resistance becomes a problem or when the patient is intolerant of other antiretroviral drugs.
Unwanted effects include flu-like symptoms, central effects such as headache, dizziness, alterations in mood, gastrointestinal effects and sometimes hypersensitivity reactions.
Ratelgravir acts by inhibiting HIV DNA integrase, the enzyme that splices viral DNA into the host genome when forming the provirus. It is used for the treatment of HIV as part of combination therapy, and is generally reserved for cases that are resistant to other antiretroviral agents.
Maraviroc is a chemokine receptor antagonist—a novel concept in HIV therapy (see Dhami et al., 2009) and is the only such drug currently available.
CCR5, together with CXCR4, are cell surface chemokine receptors that have been hijacked by some strains of HIV to gain entry to the cell. In patients who are demonstrated to harbour ‘R5’ strains, maraviroc may be used, in combination with more conventional antiretroviral drugs. Its use in the UK is currently restricted. A similar compound, vicriviroc, is in clinical development.
Biopharmaceutical Antiviral Drugs
Biopharmaceuticals that have been recruited in the fight against virus infections include immunoglobulin preparations, interferons (IFNs) and monoclonal antibodies.
Immunoglobulin
Pooled immunoglobulin contains antibodies against various viruses present in the population. The antibodies are directed against the virus envelope and can ‘neutralise’ some viruses and prevent their attachment to host cells. If used before the onset of signs and symptoms, it may attenuate or prevent measles, German measles, infectious hepatitis, rabies or poliomyelitis. Hyperimmune globulin, specific against particular viruses, is used against hepatitis B, varicella zoster and rabies.
Palivisumab
Related in terms of its mechanism of action to immunoglobulins is palivisumab, a monoclonal antibody (see Chs 17 and 59) directed against a glycoprotein on the surface of respiratory syncytial virus. It is used (as an intramuscular injection) in infants to prevent infection by this organism.
Interferons
IFNs are a family of inducible proteins synthesised by mammalian cells and now generally produced commercially using recombinant DNA technology. There are at least three types, α, β, and γ, constituting a family of hormones involved in cell growth and regulation and the modulation of immune reactions. IFN-γ, termed immune interferon, is produced mainly by T lymphocytes as part of an immunological response to both viral and non-viral antigens, the latter including bacteria and their products, rickettsiae, protozoa, fungal polysaccharides and a range of polymeric chemicals and other cytokines. IFN-α and IFN-β are produced by B and T lymphocytes, macrophages and fibroblasts in response to the presence of viruses and cytokines. The general actions of the IFNs are described briefly in Chapter 17.
The IFNs bind to specific ganglioside receptors on host cell membranes. They induce, in host cell ribosomes, the production of enzymes that inhibit the translation of viral mRNA into viral proteins, thus halting viral replication. They have a broad spectrum of action and inhibit the replication of most viruses in vitro.
Given intravenously, IFNs have a half-life of 2–4 h. They do not cross the blood–brain barrier.
IFN-α-2a is used for treatment of hepatitis B infections and AIDS-related Kaposi sarcomas; IFN-α-2b is used for hepatitis C. There are reports that IFNs can prevent reactivation of herpes simplex after trigeminal root section in animals and can prevent spread of herpes zoster in cancer patients. Preparations of IFNs conjugated with polyethylene glycol (pegylated IFNs) have a longer lifetime in the circulation.
Unwanted effects are common and include fever, lassitude, headache and myalgia. Repeated injections cause chronic malaise. Bone marrow depression, rashes, alopecia and disturbances in cardiovascular, thyroid and hepatic function can also occur.
Other Agents
Immunomodulators are drugs that act by moderating the immune response to viruses or use an immune mechanism to target a virus or other organism. Inosine pranobex may interfere with viral nucleic acid synthesis but also has immunopotentiating actions on the host. It is sometimes used to treat herpes infections in mucosal tissues or on the skin.
Tribavirin is a synthetic nucleoside, similar in structure to guanosine. It is thought to act either by altering virus nucleotide pools or by interfering with the synthesis of viral mRNA. While it inhibits a wide range of DNA and RNA viruses, including many that affect the lower airways, it is mainly used in aerosol or tablet form to treat infections with respiratory syncytial virus (an RNA paramyxovirus). It has also been shown to be effective in hepatitis C as well as Lassa fever, an extremely serious arenavirus infection. When given promptly to victims of the latter disease, it has been shown to reduce to 9% a case fatality rate previously 76%.
Antiviral drugs
Most antiviral drugs generally fall into the following groups:
Combination Therapy for HIV
Two main classes of antiviral drugs are used to treat HIV: reverse transcriptase inhibitors and protease inhibitors. As they have different mechanisms of action (Fig. 51.3), they can usefully be deployed in combinations and this technique has dramatically improved the prognosis of the disease. The combination treatment is known as highly active antiretroviral therapy (HAART). A typical HAART 3- or 4-drug combination would involve two nucleoside reverse transcriptase inhibitors with either a non-nucleoside reverse transcriptase inhibitor or one or two protease inhibitors.
Using a HAART protocol, HIV replication is inhibited, the presence in the plasma of HIV RNA is reduced to undetectable levels and patient survival is greatly prolonged. But the regimen is complex and has many unwanted effects. Compliance is difficult and lifelong treatment is necessary. The virus is not eradicated but lies latent in the host genome of memory T cells, ready to reactivate if therapy is stopped.
Unwelcome interactions can occur between the component drugs of HAART combinations, and there may be interindividual variations in absorption. Some drugs penetrate poorly into the brain, and this could lead to local proliferation of the virus. So far, there is no cross-resistance between the three groups of drugs, but it needs to be borne in mind that the virus has a high mutation rate—so resistance could be a problem in the future. The AIDS virus has certainly not yet been outsmarted. Even with full compliance—which is often not achieved for long periods, given the complexity of the regimen and side effects—the virus can only be kept in check, not eliminated.
The choice of drugs to treat pregnant or breastfeeding women is difficult. The main aims are to avoid damage to the fetus and to prevent transmission of the disease to the neonate. Therapy with zidovudine alone is often used in these cases. Another area that requires special consideration is prophylaxis for individuals who may have been exposed to the virus accidentally. Specific guidelines have been developed for such cases, but they are beyond the scope of this chapter.
Other drugs such as enfurvitide, maraviroc and ratelgravir are used in combination therapy regimens and are seldom deployed alone.
Drugs for HIV infections
Prospects for New Antiviral Drugs
At the beginning of the 1990s, there were only five drugs available to treat viral infections; 20 years later, this number has increased some 10-fold. New strategies—based on the growing understanding of the biology of pathogenic viruses and their action on, and in, host cells—could well, if vigorously implemented, have the potential to target the viruses causing most viral diseases (see de Clercq, 2002). One such example has been the recent introduction of drugs that prevent CCR5 from serving as an entry portal for HIV. Work is underway to develop CXCR4 inhibitors for similar purposes, as are other approaches to disrupting this function of CCR5 (reviewed by Dhami et al., 2009).
However, the ultimate weapon in the fight against the virus is vaccination. This has proved to be highly effective in the past against diseases such as polio and smallpox, and more recently against influenza (both types) and hepatitis B. However, while there has been no shortage of candidate vaccines (some 40 have been trialled in thousands of volunteers), the prospect of a vaccine against HIV (and sadly many other viruses) still seems rather remote. Part of the problem is antigenic drift, a process whereby the virus mutates, thus presenting different antigenic structures and minimising the chance of an effective and long-lasting immune response or the production of a vaccine. The way forward is not totally clear, but the issue has stimulated research into the interface between the innate and adaptive immune systems in a quest to boost the effectiveness of vaccine design. The whole problem of HIV vaccines is the subject of numerous reviews (see Girard et al., 2006; Kaufman & Barouch, 2009; Rhee & Barouch, 2009).
References and Further Reading
Hanazaki K. Antiviral therapy for chronic hepatitis B: a review. Curr. Drug Targets Inflamm. Allergy. 2004;3:63-70. (Reviews the use of IFN and lamivudine, alone or in combination, in the treatment of this viral infection)
Lauer G.M., Walker B.D. Hepatitis C virus infection. N. Engl. J. Med.. 2001;345:41-52. (Comprehensive review of pathogenesis, clinical characteristics, natural history and treatment of hepatitis C infection)
Lee W.M. Hepatitis B virus infection. N. Engl. J. Med.. 1997;337:1733-1746. (Detailed coverage of the epidemiology and pathogenesis of hepatitis B, the life cycle of the virus in the human host and the treatment of the disease)
Schmidt A.C. Antiviral therapy for influenza: a clinical and economic comparative review. Drugs. 2004;64:2031-2046. (A useful review of influenza biology, together with a comprehensive evaluation of drug treatments, their mechanisms of action and relative economic costs)
Whitley R.J., Roizman B. Herpes simplex virus infections. Lancet. 2001;357:1513-1518. (A concise review of the viral replication cycle and the pathogenesis and treatment of herpes simplex virus infections)
Barber D.L., Wherry E.J., Masopust D., et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682-687. (Deals with a potential mechanism whereby the exhaustion of T cells may be reversed)
Jansen C.A., Piriou E., Bronke C., et al. Characterisation of virus-specific CD8(+) effector T cells in the course of HIV-1 infection: longitudinal analyses in slow and rapid progressors. Clin. Immunol.. 2004;11:299-309.
Mindel A., Tenant-Flowers M. Natural history and management of early HIV infection. Br. Med. J.. 2001;322:1290-1293. (A clinical review covering the current classification of HIV disease, clinical manifestations of primary HIV infection and general treatment of HIV patients)
Norris P.J., Moffett H.F., Brander C., et al. Fine specificity and cross-clade reactivity of HIV type 1 Gag-specific CD4+ T cells. AIDS Res. Hum. Retroviruses. 2004;20:315-325.
Mechanisms of immune evasion by viruses and drug resistance
Murphy P.M. Viral exploitation and subversion of the immune system through chemokine mimicry. Nat. Immunol.. 2001;2:116-122. (Excellent description of virus–immune system interaction)
Tortorella D., Gewurz B.E., Furman M.H., et al. Viral subversion of the immune system. Annu. Rev. Immunol.. 2000;18:861-926. (A comprehensive and clearly written review of the various mechanisms by which viruses elude detection and destruction by the host immune system)
Immune defences against viral attack
Levy J.A. The importance of the innate immune system in controlling HIV infection and disease Trends Immunol. 22:2001:312-316 (Stresses the role of innate immunity in the response to HIV; clear exposition of the various components of the innate and adaptive immune systems, as well as the role of non-cytotoxic CD8+ cell response to HIV)
Schutze N. siRNA technology. Mol. Cell. Endocrinol.. 2004;213:115-119. (An article explaining the siRNA concept)
Mechanisms of antiviral drug action
Balfour H.H. Antiviral drugs. N. Engl. J. Med.. 1999;340:1255-1268. (An excellent and comprehensive review of antiviral agents other than those used for HIV therapy; describes their mechanisms of action, adverse effects and clinical use)
de Clercq E. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov.. 2002;1:13-24. (Outstanding article describing the rationale behind current and future strategies for antiviral drug development)
Flexner C. HIV-protease inhibitors. N. Engl. J. Med.. 1998;338:1281-1292. (Excellent and comprehensive review covering mechanisms of action, clinical and pharmacokinetic properties, potential drug resistance and possible treatment failure)
Gubareva L., Kaiser L., Hayden F.G. Influenza virus neuraminidase inhibitors. Lancet. 2000;355:827-835. (Admirable coverage of this topic; lucid summary and clear diagrams of the influenza virus and its replication cycle; description of the structure and the action of, and resistance to, zanamivir and oseltamivir, and the relevant pharmacokinetic aspects and clinical efficacy)
Flexner C. Dual protease inhibitor therapy in HIV-infected patients: pharmacologic rationale and clinical benefits. Annu. Rev. Pharmacol. Toxicol.. 2000;40:649-674. (Review emphasising interactions between individual protease inhibitors and the potential benefits and disadvantages of dual therapy)
Richman D.D. HIV chemotherapy. Nature. 2001;410:995-1001. (Outstanding article; covers pathogenesis and natural history of HIV infection and the impact on viral dynamics and immune function of antiretroviral therapy; discusses the main antiretroviral drugs, drug resistance of HIV and targets for new drugs; excellent figures and comprehensive references)
Weller I.V.D., Williams I.G. Antiretroviral drugs. Br. Med. J.. 2001;322:1410-1412. (Part of a British Medical Journal series on the ABC of AIDS; clear, succinct coverage of antiretroviral regimens, and a list of potential targets for new drug development)
New leads in antiviral drug therapy
Barik S. Control of nonsegmented negative-strand RNA virus replication by siRNA. Virus Res.. 2004;102:27-35. (Interesting article explaining how siRNA technology might be used to inhibit viral replication)
Dhami H., Fritz C.E., Gankin B., et al. The chemokine system and CCR5 antagonists: potential in HIV treatment and other novel therapies. J. Clin. Pharm. Ther.. 2009;34:147-160. (Excellent and easy-to-read review of this area. Helpful diagrams)
Flores-Villanueva P.O., Hendel H., Caillat-Zucman S., et al. Associations of MHC ancestral haplotypes with resistance/susceptibility to AIDS disease development. J. Immunol.. 2003;170:1925-1929. (A paper that deals with the hereditary component of HIV susceptibility/resistance; a bit complex for the non-geneticist but worth the effort)
Girard M.P., Osmanov S.K., Kieny M.P. A review of vaccine research and development: the human immunodeficiency virus (HIV). Vaccine. 2006;24:4062-4081. (First-class review of HIV/AIDS, the difficulties of producing a vaccine and many other related issues. Easy to read)
Kaufman D.R., Barouch D.H. Translational mini-review series on vaccines for HIV: T lymphocyte trafficking and vaccine-elicited mucosal immunity. Clin. Exp. Immunol.. 2009;157:165-173. (This paper, together with the paper by Rhee et al. below, review new research that seeks to design better HIV vaccines through an increased understanding of the innate and adaptive immune systems. They are fairly advanced but worthwhile if you are interested in the topic)
Kilby J.M., Eron J.J. Novel therapies based on mechanisms of HIV-1 cell entry. N. Engl. J. Med.. 2003;348:2228-2238. (Excellent review on this innovative strategy)
Kitabwalla M., Ruprecht R.M. RNA interference: a new weapon against HIV and beyond. N. Engl. J. Med.. 2002;347:1364-1368. (An article in the series Clinical implications of basic research)
Moore J.P., Stevenson M. New targets for inhibitors of HIV-1 replication. Nat. Rev. Mol. Cell Biol.. 2000;1:40-49. (Excellent coverage of stages of the viral life cycle that might be susceptible to new drugs: attachment to host cell, membrane fusion, integration, accessory gene function and assembly. Introduces various potentially promising chemical compounds)
Rhee E.G., Barouch D.H. Translational mini-review series on vaccines for HIV: Harnessing innate immunity for HIV vaccine development. Clin. Exp. Immunol.. 2009;157:174-180. (See review of Kaufman & Barouch above)
Pisani E. The wisdom of whores. 2008 Granta Books London. (It is not enough to design and manufacture anti-HIV drugs—they have to be made available to those who need them. It is a sad fact that the most needy countries in this respect are those with the most poorly developed infrastructures for delivery of health care. This book is one of a kind: it is at once entertaining and informative. It chronicles the efforts made by an epidemiologist to pioneer HIV programmes in developing countries and the many bureaucratic and other obstacles that had to be overcome. Her portraits of the victims of the AIDS pandemic, many of whom work in the ‘sex industry’, are both amusing and poignant. There is also a Web site at http://www.wisdomofwhores.com/with an alternative view of much HIV/AIDS-related news. Highly recommended). http://www.wisdomofwhores.com/
http://www.aidsinfo.nih.gov/ (The official HIV/AIDS site of the US National Institutes of Health. This comprehensive Web site carries authoritative and completely up-to-date information on every aspect of this disease and its treatment, including data on drugs and drug action as well as the results of recent clinical trials and the latest progress in developing a vaccine. Superb)
http://www.unaids.org/en/default.asp (This is the official site of the United Nations Programme on HIV/AIDS. It deals with a wide range of issues but focuses on the demographics of the epidemic. It carries photographs, maps, slides, movies and statistics, as well as other resources that bring home the enormous problems faced by the international community in dealing with this disease. Prepare to be appalled)