17 Local hormones
Cytokines, biologically active lipids, amines and peptides
Overview
In Chapter 6 we discussed the cellular players in host defence and alluded to the crucial role played by soluble chemical messengers in the inflammatory response. Here, we take a close look at these mediators as well as others which, while having a normal physiological role, are pressed into service by the host defence mechanism when necessary. The exceptions are the cytokines and chemokines which, as a general rule, are mainly of importance in inflammation and immunity. Many of the mediators described here are important targets for anti-inflammatory and other drug action.
Introduction
A ‘mediator’ is operationally defined as a substance that fulfils a set of criteria generally modelled on the original suggestions of Sir Henry Dale in 1933. A modified version, more applicable to the field today, was considered by Dale & Foreman (1994). They defined a ‘mediator’ as a substance that fulfils certain criteria, including the following:
The principal mediators of inflammation will be described below beginning with the cytokines.
Cytokines
‘Cytokine’ is an all-purpose functional term that is applied to protein or polypeptide mediators synthesised and released by cells of the immune system during inflammation. They are enormously important for the overall coordination of the inflammatory response. Cytokines act locally by autocrine or paracrine mechanisms. Their synthesis is massively upregulated during inflammatory episodes and they are usually active at very low (sub-nanomolar) concentrations.
On the target cell, they bind to and activate specific, high-affinity receptors that, in most cases, are also upregulated during inflammation. Except for chemokines (see below), which act on G-protein-coupled receptors, most cytokines act on kinase-linked receptors, regulating phosphorylation cascades that affect gene expression, such as the Jak/Stat pathway (Ch. 3).
In addition to their own direct actions on cells, some cytokines amplify inflammation by inducing the formation of other inflammatory mediators. Others can induce receptors for other cytokines on their target cell, or engage in synergistic or antagonistic interactions with other cytokines. Cytokines may be likened to a complex signalling language, with the final response of a particular cell involved being determined by the strength and number of different messages received concurrently at the cell surface.
Various systems for classifying cytokines can be found in the literature, as can a multitude of diagrams depicting complex networks of cytokines interacting with each other and with a range of target cells. No one system of classification does justice to the complexity of cytokine biology. The terminology and nomenclature are horrendous and a comprehensive coverage of this area is beyond the scope of this book. For the purposes of this chapter, however, Table 17.1 lists some significant species and their biological actions using a very simplified classification scheme. The cytokine aficionado can find further classification tables in Janeway et al. (2004), or by using the Web links listed at the end of the chapter.
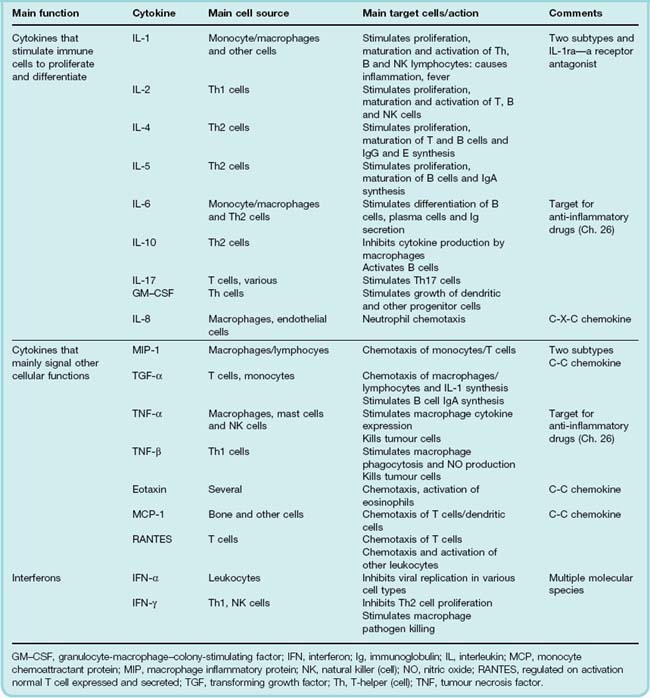
More than 100 cytokines have been identified, falling into four main groups, namely interleukins, chemokines, interferons and colony-stimulating factors (discussed separately in Ch. 25).
Interleukins
Originally so named as they signalled between leukocytes, the distinction has become less useful with time. The primary proinflammatory interleukins (IL) are usually considered to be tumour necrosis factor (TNF)-α and IL-1. The latter cytokine actually comprises a family of three cytokines consisting of two agonists, IL-1α, IL-1β and, surprisingly, an endogenous IL-1-receptor antagonist (IL-1ra).1 Mixtures of these are released from macrophages and many other cells during inflammation and can initiate the synthesis and release of a cascade of secondary cytokines, among which are the chemokines (see below). Some interleukins are anti-inflammatory. These include transforming growth factor (TGF)-β, IL-4, IL-10 and IL-13. They inhibit chemokine production, and the anti-inflammatory interleukins can inhibit responses driven by T-helper (Th)1 cells, whose inappropriate activation is involved in the pathogenesis of several diseases. Both TNF-α and IL-1 are important targets for anti-inflammatory biopharmaceuticals (Ch. 26).
Chemokines
Chemokines are defined as chemoattractant cytokines that control the migration of leukocytes, functioning as traffic coordinators during immune and inflammatory reactions. Again, the nomenclature (and the classification) is confusing, because some non-cytokine mediators also control leukocyte movement (C5a, LTB4, f-Met-Leu-Phe, etc; see Fig. 6.2). Furthermore, many chemokines have other actions, for example causing mast cell degranulation or promoting angiogenesis.
More than 40 chemokines have been identified, and for those of us who are not professional chemokinologists they can be conveniently distinguished by considering the configuration of key cysteine residues in their polypeptide chain. Chemokines with one cysteine are known as C-chemokines. If there are two adjacent residues they are called C-C chemokines. Other members have cysteines separated by one (C-X-C chemokines) or three other residues (C-XXX-C chemokines).
The C-X-C chemokines (main example IL-8; see Fig. 6.2) act on neutrophils and are predominantly involved in acute inflammatory responses. The C-C chemokines (main examples eotaxin, MCP-1 and RANTES2) act on monocytes, eosinophils and other cells, and are involved predominantly in chronic inflammatory responses.
 Chemokines generally act through G-protein-coupled receptors, and alteration or inappropriate expression of these is implicated in multiple sclerosis, cancer, rheumatoid arthritis and some cardiovascular diseases (Gerard & Rollins, 2001). Some types of virus (herpes virus, cytomegalovirus, pox virus and members of the retrovirus family) can exploit the chemokine system and subvert the host’s defences (Murphy, 2001). Some produce proteins that mimic host chemokines or chemokine receptors, some act as antagonists at chemokine receptors and some masquerade as growth or angiogenic factors. The AIDS-causing HIV virus is responsible for the most audacious exploitation of the host chemokine system. This virus has a protein (gp120) in its envelope that recognises and binds T-cell receptors for CD4 and a chemokine coreceptor that allows it to penetrate the T cell (see Ch. 51).
Chemokines generally act through G-protein-coupled receptors, and alteration or inappropriate expression of these is implicated in multiple sclerosis, cancer, rheumatoid arthritis and some cardiovascular diseases (Gerard & Rollins, 2001). Some types of virus (herpes virus, cytomegalovirus, pox virus and members of the retrovirus family) can exploit the chemokine system and subvert the host’s defences (Murphy, 2001). Some produce proteins that mimic host chemokines or chemokine receptors, some act as antagonists at chemokine receptors and some masquerade as growth or angiogenic factors. The AIDS-causing HIV virus is responsible for the most audacious exploitation of the host chemokine system. This virus has a protein (gp120) in its envelope that recognises and binds T-cell receptors for CD4 and a chemokine coreceptor that allows it to penetrate the T cell (see Ch. 51).
Interferons
There are three classes of interferon, termed IFN-α, IFN-β and IFN-γ. IFN-α is not a single substance but a family of approximately 20 proteins with similar activities. IFN-α and IFN-β have antiviral activity, and IFN-α also has some antitumour action. Both are released from virus-infected cells and activate antiviral mechanisms in neighbouring cells. IFN-γ has a role in induction of Th1 responses (Fig. 6.3; see also Abbas et al., 1996).
Clinical Use of Interferons
IFN-α is used in the treatment of chronic hepatitis B and C, and has some action against herpes zoster and in the prevention of the common cold. Antitumour action against some lymphomas and solid tumours has been reported. A variety of dose-related side effects may occur. IFN-β is used in some patients with multiple sclerosis, whereas IFN-γ is used in chronic granulomatous disease in conjunction with antibacterial drugs (see clinical box for more details).
Clinical uses of interferons 
Interfering with cytokine action using biopharmaceuticals has proved to be a particularly fertile area of drug development: several successful strategies have been adopted including direct antibody neutralisation or the use of ‘decoy’ receptor proteins that remove the biologically active pool from the circulation. These are explained in detail in Chapters 26 and 59.
Cytokines 
Histamine
In a classic study, Sir Henry Dale and his colleagues demonstrated that a local anaphylactic reaction (a type I or ‘immediate hypersensitivity reaction’; see below) was caused by antigen–antibody reactions in sensitised tissue, and found that histamine mimicked this effect both in vitro and in vivo. Later studies confirmed that histamine is present in tissues, and released (along with other mediators described below) during anaphylaxis.
Synthesis and Storage of Histamine
Histamine is a basic amine formed from histidine by histidine decarboxylase. It is found in most tissues but is present in high concentrations in the lungs and the skin, and in particularly high concentrations in the gastrointestinal tract. At the cellular level, it is found largely in mast cells (approximately 0.1–0.2 pmol/cell) and basophils (0.01 pmol/cell), but non-mast cell histamine occurs in ‘histaminocytes’ in the stomach and in histaminergic neurons in the brain (see Ch. 38). In mast cells and basophils, histamine is complexed in intracellular granules with an acidic protein and a high-molecular-weight heparin termed macroheparin.
Histamine Release
Histamine is released from mast cells by exocytosis during inflammatory or allergic reactions. Stimuli include C3a and C5a that interact with specific surface receptors, and the combination of antigen with cell-fixed immunoglobulin (Ig)E antibodies. In common with many secretory processes (Ch. 4), histamine release is initiated by a rise in cytosolic [Ca2+]. Various basic drugs, such as morphine and tubocurarine, release histamine through a non-receptor action. Agents that increase cAMP formation (e.g. β-adrenoceptor agonists; see Ch. 14) inhibit histamine secretion. Replenishment of secreted histamine by mast cells or basophils is a slow process, which may take days or weeks, whereas turnover of histamine in the gastric histaminocyte is very rapid. Histamine is metabolised by histaminase and/or by the methylating enzyme imidazole N-methyltransferase.
Histamine Receptors
Histamine acts on G-protein-coupled receptors, of which four main types have been identified; all four are implicated in the inflammatory response (see Gutzmer et al., 2005, for a review). Selective antagonists at H1, H2 and H3 receptors include mepyramine, cimetidine and thioperamide, respectively. Selective agonists for H2 and H3 receptors are, respectively, dimaprit and (R)-methylhistamine. Histamine H1 antagonists are the principal antihistamines used in the treatment of inflammation (notably rhinitis). Other clinical uses of subtype antagonists may be found in Chapters 27, 36 and 47. At the time of writing, the pharmacology of H4 receptors is less well developed.
Actions
Smooth muscle effects
Histamine, acting on H1 receptors, contracts the smooth muscle of the ileum, bronchi, bronchioles and uterus. The effect on the ileum is not as marked in humans as it is in the guinea pig (this tissue remains the de facto standard preparation for histamine bioassay). Histamine reduces air flow in the first phase of bronchial asthma (see Ch. 27 and Fig. 27.3).
Cardiovascular effects
Histamine dilates human blood vessels by an action on H1 receptors, the effect being partly endothelium dependent in some vascular beds. It also increases the rate and the output of the heart by action on cardiac H2 receptors.
Gastric secretion
Histamine stimulates the secretion of gastric acid by action on H2 receptors. In clinical terms, this is the most important action of histamine, because it is implicated in the pathogenesis of peptic ulcer. It is considered in detail in Chapter 29.
Itching
Itching occurs if histamine is injected into the skin or applied to a blister base, because it stimulates sensory nerve endings by an H1-dependent mechanism.
The ‘triple response’
When injected intradermally, histamine causes a reddening of the skin, accompanied by a weal with a surrounding flare. This is the triple response described by Sir Thomas Lewis over 80 years ago and is explained by the foregoing effects. The reddening reflects vasodilatation of the small arterioles and precapillary sphincters, and the weal the increased permeability of the postcapillary venules. These effects are mainly mediated through activation of H1 receptors. The flare is an axon reflex: stimulation of sensory nerve fibres evokes antidromic impulses through neighbouring branches of the same nerve, releasing vasodilators such as calcitonin gene-related peptide (CGRP; see Chs 19 and 26).
Despite the fact that histamine release is evidently capable of reproducing many of the inflammatory signs and symptoms, histamine H1 antagonists do not have much clinical utility in the acute inflammatory response per se, because other mediators are more important. Histamine is, however, significant in type I hypersensitivity reactions such as allergic rhinitis and urticaria. The use of H1 antagonists in these and other conditions is dealt with in Chapter 26.
Histamine
Eicosanoids
General Remarks
Unlike histamine, eicosanoids are not preformed in cells but are generated from phospholipid precursors on demand. They are implicated in the control of many physiological processes, and are among the most important mediators and modulators of the inflammatory reaction (Fig. 17.1) and are a very significant target for drug action.
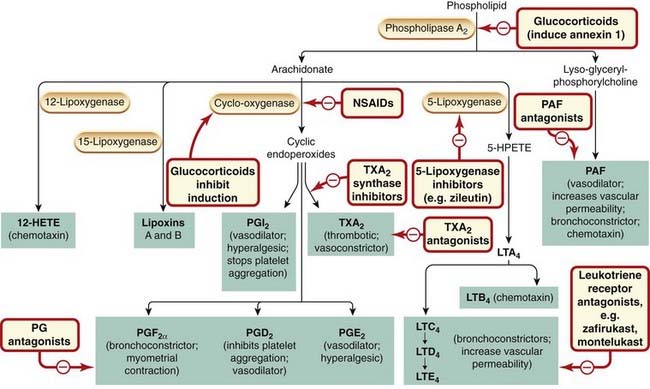
Fig. 17.1 Summary diagram of the inflammatory mediators derived from phospholipids, with an outline of their actions and the sites of action of anti-inflammatory drugs.
The arachidonate metabolites are eicosanoids. The glucocorticoids inhibit transcription of the gene for cyclo-oxygenase-2, induced in inflammatory cells by inflammatory mediators. The effects of prostaglandin (PG)E2 depend on which of the three receptors for this prostanoid are activated. HETE, hydroxyeicosatetraenoic acid; HPETE, hydroperoxyeicosatetraenoic acid; LT, leukotriene; NSAID, non-steroidal anti-inflammatory drug; PAF, platelet-activating factor; PGI2, prostacyclin; TX, thromboxane.
Interest in eicosanoids arose in the 1930s after reports that semen contained a lipid substance that contracted uterine smooth muscle. Later, it became clear that prostaglandin (as the factor was named3) was not a single substance but a whole family of compounds that could be generated from 20-carbon unsaturated fatty acids by virtually all cells.
Structure and Biosynthesis
In mammals, the main eicosanoid precursor is arachidonic acid (5,8,11,14-eicosatetraenoic acid), a 20-carbon unsaturated fatty acid containing four double bonds (hence eicosa, referring to the 20 carbon atoms, and tetraenoic, referring to the four double bonds). In most cell types, arachidonic acid is esterified in the phospholipid pool, and the concentration of the free acid is low. The principal eicosanoids are the prostaglandins, the thromboxanes and the leukotrienes, although other derivatives of arachidonate, for example the lipoxins, are also produced. (The term prostanoid will be used here to encompass both prostaglandins and thromboxanes.)
In most instances, the initial and rate-limiting step in eicosanoid synthesis is the liberation of arachidonate, usually in a one-step process catalysed by the enzyme phospholipase A2 (PLA2; Fig. 17.2), although a multi-step process involving phospholipases C or D in conjunction with diacylglycerol lipase is sometimes utilised. Several species of PLA2 exist, but the most important is probably the highly regulated cytosolic PLA2. This enzyme generates not only arachidonic acid (and thus eicosanoids) but also lysoglyceryl-phosphorylcholine (lyso-PAF), the precursor of platelet activating factor, another inflammatory mediator (see Fig. 17.2).
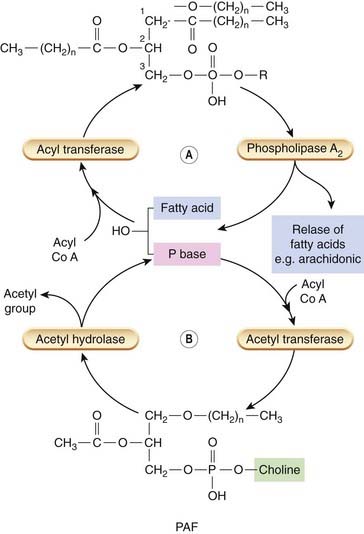
Fig. 17.2 The structure of phospholipids, the release of fatty acids and platelet-activating factor (PAF) precursors.
The structure of a ‘generic’ phospholipid is shown. Different bases are found at C3 yielding phosphatidyl-choline, -ethanolamine, -serine or -inositol species. Generally speaking, unsaturated fatty acids such as arachidonic acid are esterified at the C2 position and saturated fatty acids are linked to C1. Two bonds are possible: an ether linkage or an ester linkage. Arachidonic acid can be removed by phospholipase A2 and used for synthesis of eicosanoids. This yields a lyso-phospholipid that is normally rapidly reacylated and converted back to phospholipids (A). If the species is lysophosphatidyl choline and it contains an ether-linked hexadecyl or octadecyl fatty acid at C1, it can serve as a precursor for PAF. This is accomplished by a further acetylation step. PAF is inactivated by an acetylhydrolase that removes the acetyl group and converts it back to lyso-PAF, where it can be recycled (B).
Cytosolic PLA2 is activated (and hence arachidonic acid liberated) by phosphorylation. This occurs in response to signal transduction events triggered by many stimuli, such as thrombin action on platelets, C5a on neutrophils, bradykinin on fibroblasts and antigen–antibody reactions on mast cells. General cell damage also triggers the activation process. The free arachidonic acid is metabolised separately (or sometimes jointly) by several pathways, including the following.
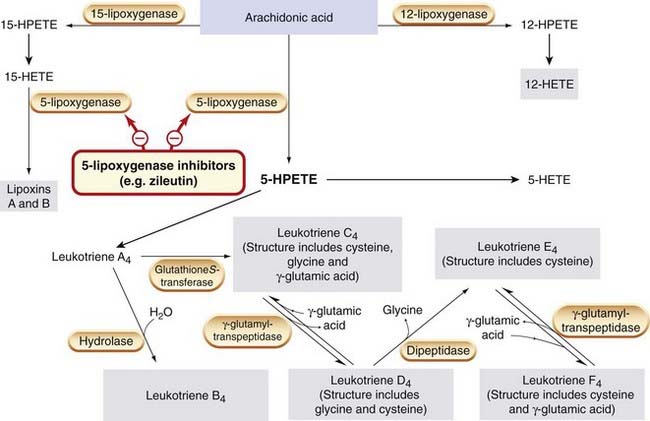
Fig. 17.3 The biosynthesis of leukotrienes from arachidonic acid.
Compounds with biological action are shown in grey boxes. HETE, hydroxyeicosatetraenoic acid; HPETE, hydroperoxyeicosatetraenoic acid.
Chapter 26 deals in detail with the way inhibitors of these pathways (including non-steroidal anti-inflammatory drugs [NSAIDs] and glucocorticoids) produce anti-inflammatory effects.
Mediators derived from phospholipids
Prostanoids
COX-1 is present in most cells as a constitutive enzyme that produces prostanoids that act as homeostatic regulators (e.g. modulating vascular responses), whereas COX-2 is not normally present (at least in most tissues) but it is strongly induced by inflammatory stimuli and therefore believed to be more relevant to inflammation therapy (see Ch. 26 for a full discussion of this point). Both enzymes catalyse the incorporation of two molecules of oxygen into each arachidonate molecule, forming the highly unstable endoperoxides PGG2 and PGH2. These are rapidly transformed by isomerase or synthase enzymes to PGE2, PGI2, PGD2, PGF2α and TXA2, which are the principal bioactive end products of this reaction. The mix of eicosanoids thus produced varies between cell types depending on the particular endoperoxide isomerases or synthases present. In platelets, for example, TXA2 predominates, whereas in vascular endothelium PGI2 is the main product. Macrophages, neutrophils and mast cells synthesise a mixture of products. If eicosatrienoic acid (three double bonds) rather than arachidonic acid is the substrate, the resulting prostanoids have only a single double bond, for example PGE1, while eicosapentaenoic acid, which contains five double bonds, yields PGE3. The latter substrate is significant because it is present in abundance in some fish oils and may, if present in sufficient amounts in the diet, come to represent a significant fraction of cellular fatty acids. When this occurs, the production of the proinflammatory PGE2 is diminished and, more significantly, the generation of TXA2 as well. This may partly underlie the beneficial anti-inflammatory and cardiovascular actions that are ascribed to diets rich in this type of marine product (see Resolvins below).
Catabolism of the Prostanoids
This is a multistep process. After carrier-mediated uptake, most prostaglandins are rapidly inactivated by ‘prostaglandin-specific’ enzymes, and the inactive products are further degraded by general fatty acid-oxidising enzymes. The prostaglandin-specific enzymes are present in high concentration in the lung, and 95% of infused PGE2, PGE1 or PGF2α is inactivated on first passage. The half-life of most prostaglandins in the circulation is less than 1 min.
PGI2 and TXA2 are slightly different. Both are inherently unstable and decay spontaneously and rapidly (within 5 min and 30 s, respectively) in biological fluids into inactive 6-keto-PGF1α and TXB2. Further metabolism occurs, but it is not really relevant to us here.
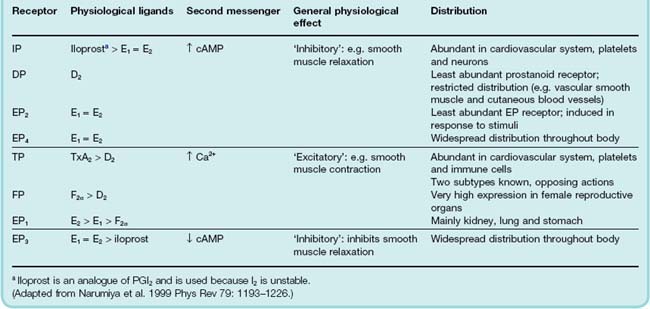
Prostanoid Receptors
There are five main classes of prostanoid receptors (Coleman & Humphrey, 1993), all of which are typical G-protein-coupled receptors (Table 17.2). They are termed DP, FP, IP, EP and TP receptors, respectively, depending on whether their ligands are PGD, PGF, PGI, PGE or TXA species. Some have further subtypes; for example, the EP receptors are subdivided into four subgroups.
Actions of the Prostanoids
The prostanoids affect most tissues and exert a bewildering variety of effects.
The Role of the Prostanoids in Inflammation
The inflammatory response is inevitably accompanied by the release of prostanoids. PGE2 predominates, although PGI2 is also important. In areas of acute inflammation, PGE2 and PGI2 are generated by the local tissues and blood vessels, while mast cells release mainly PGD2. In chronic inflammation, cells of the monocyte/macrophage series also release PGE2 and TXA2. Together, the prostanoids exert a sort of yin–yang effect in inflammation, stimulating some responses and decreasing others. The most striking effects are as follows.
In their own right, PGE2, PGI2 and PGD2 are powerful vasodilators and synergise with other inflammatory vasodilators such as histamine and bradykinin. It is this combined dilator action on precapillary arterioles that contributes to the redness and increased blood flow in areas of acute inflammation. Prostanoids do not directly increase the permeability of the postcapillary venules, but potentiate this effect of histamine and bradykinin. Similarly, they do not themselves produce pain, but potentiate the effect of bradykinin by sensitising afferent C fibres (see Ch. 41) to the effects of other noxious stimuli. The anti-inflammatory effects of NSAIDs stem largely from their ability to block these actions.
Prostaglandins of the E series are also pyrogenic (i.e. they induce fever). High concentrations are found in cerebrospinal fluid during infection, and there is evidence that the increase in temperature (attributed to cytokines) is actually finally mediated by the release of PGE2. NSAIDs exert antipyretic actions (Ch. 26) by inhibiting PGE2 synthesis in the hypothalamus.
However, some prostaglandins have anti-inflammatory effects under some circumstances. For example, PGE2 decreases lysosomal enzyme release and the generation of toxic oxygen metabolites from neutrophils, as well as the release of histamine from mast cells. Several prostanoids are available for clinical use (see clinical box).
Prostanoids
Clinical uses of prostanoids
Leukotrienes
Leukotrienes (leuko- because they are made by white cells, and -trienes because they contain a conjugated triene system of double bonds) are synthesised from arachidonic acid by lipoxygenase-catalysed pathways. These soluble cytosolic enzymes are mainly found in lung, platelets, mast cells and white blood cells. The main enzyme in this group is 5-lipoxygenase. On cell activation, this enzyme translocates to the nuclear membrane, where it associates with a crucial accessory protein affectionately termed FLAP (five-lipoxygenase activating protein). The 5-lipoxygenase incorporates a hydroperoxy group at C5 in arachidonic acid to form 5-hydroperoxytetraenoic acid (5-HPETE, Fig. 17.3), leading to the production of the unstable compound leukotriene (LT)A4. This may be converted enzymically to LTB4 and, utilising a separate pathway, is also the precursor of the cysteinyl-containing leukotrienes LTC4, LTD4, LTE4 and LTF4 (also referred to as the sulfidopeptide leukotrienes). Mixtures of these cysteinyl adducts constitute the slow-reacting substance of anaphylaxis (SRS-A), a substance shown many years ago to be generated in guinea pig lung during anaphylaxis, and believed to be important in asthma. LTB4 is produced mainly by neutrophils, and the cysteinyl-leukotrienes mainly by eosinophils, mast cells, basophils and macrophages. Lipoxins and other active products, some of which have anti-inflammatory properties, are also produced from arachidonate by this pathway (Fig. 17.3).
LTB4 is metabolised by a unique membrane-bound P450 enzyme in neutrophils, and then further oxidised to 20-carboxy-LTB4. LTC4 and LTD4 are metabolised to LTE4, which is excreted in the urine.
Leukotriene Receptors
Leukotriene receptors are termed BLT if the ligand is LTB4, and CysLT for the cysteinyl-leukotrienes. LTB4 acts on specific LTB4 receptors as defined by selective agonists and antagonists. The transduction mechanism utilises inositol trisphosphate and increased cytosolic Ca2+.
Leukotriene Actions
Cysteinyl-leukotrienes have important actions on the respiratory and cardiovascular systems, and specific receptors for LTD4 have been defined on the basis of numerous selective antagonists. The CysLT-receptor antagonists zafirlukast and montelukast are now in use in the treatment of asthma (see Ch. 27). Cysteinyl-leukotrienes may mediate the cardiovascular changes of acute anaphylaxis. Agents that inhibit 5-lipoxygenase are under development as antiasthmatic agents (see Ch. 27) and anti-inflammatory agents. One such drug, zileuton, is available in some parts of the world but has not won a definite place in therapy yet (see Larsson et al., 2006).
The respiratory system
Cysteinyl-leukotrienes are potent spasmogens, causing dose-related contraction of human bronchiolar muscle in vitro. LTE4 is less potent than LTC4 and LTD4, but its effect is much longer lasting. All cause an increase in mucus secretion. Given by aerosol to human volunteers, they reduce specific airway conductance and maximum expiratory flow rate, the effect being more protracted than that produced by histamine (Fig. 17.4).
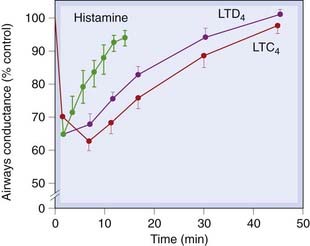
Fig. 17.4 The time course of action on specific airways conductance of the cysteinyl-leukotrienes and histamine, in six normal subjects.
Specific airways conductance was measured in a constant volume whole-body plethysmograph, and the drugs were given by inhalation.
(From Barnes P J, Piper P J, Costello J K 1984 Thorax 39: 500.)
The cardiovascular system
Small amounts of LTC4 or LTD4 given intravenously cause a rapid, short-lived fall in blood pressure, and significant constriction of small coronary resistance vessels. Given subcutaneously, they are equipotent with histamine in causing weal and flare. Given topically in the nose, LTD4 increases nasal blood flow and increases local vascular permeability.
The role of leukotrienes in inflammation
LTB4 is a potent chemotactic agent for neutrophils and macrophages (see Fig. 6.2). On neutrophils, it also upregulates membrane adhesion molecule expression, and increases the production of toxic oxygen products and the release of granule enzymes. On macrophages and lymphocytes, it stimulates proliferation and cytokine release. It is found in inflammatory exudates and tissues in many inflammatory conditions, including rheumatoid arthritis, psoriasis and ulcerative colitis.
The cysteinyl-leukotrienes are present in the sputum of chronic bronchitis patients in amounts that are biologically active. On antigen challenge, they are released from samples of human asthmatic lung in vitro, and into nasal lavage fluid in subjects with allergic rhinitis. There is evidence that they contribute to the underlying bronchial hyperreactivity in asthmatics, and it is thought that they are among the main mediators of both the early and late phases of asthma (Fig. 27.2).
Leukotrienes
Lipoxins and Resolvins
A recently identified group of trihydroxy arachidonate metabolites termed lipoxins (Fig. 17.3) are formed by the concerted action of the 5- and the 12- or 15-lipoxygenase enzymes during inflammation. They act on polymorphonuclear leukocytes to oppose the action of proinflammatory stimuli, supplying what might be called ‘stop signals’ to inflammation. Lipoxins utilise the same formyl peptide G-protein-coupled receptor system that recognises other endogenous anti-inflammatory factors such as annexin-A1. Oddly, aspirin (a COX inhibitor, see Ch. 26) stimulates the synthesis of these substances because COX-2 can still produce hydroxy fatty acids even when inhibited by aspirin, even though it cannot synthesise prostaglandins. The formation of lipoxins probably contributes to aspirin’s anti-inflammatory effects, some of which are not completely explained through inhibition of prostaglandin generation (see Gilroy & Perretti, 2005; Serhan, 2005). Resolvins, as the name implies, are a series of compounds that fulfil a similar function, but unlike lipoxins, their precursor fatty acid is eicosapentaenoic acid. Fish oils are rich in this fatty acid and it is likely that at least some of their anti-inflammatory benefit is produced through conversion to these highly active species (see Ariel & Serhan, 2007, for a review of this promising area). The leukocyte receptor for resolvins is called Chem 23.
Platelet-Activating Factor
Platelet-activating factor, also variously termed PAF-acether and AGEPC (acetyl-glyceryl-ether-phosphorylcholine), is a biologically active lipid that can produce effects at exceedingly low concentrations (less than 10−10 mol/l). The name is somewhat misleading, because PAF has actions on a variety of different target cells, and is believed to be an important mediator in both acute and chronic allergic and inflammatory phenomena. PAF is biosynthesised from acyl-PAF in a two-step process (Fig. 17.2). The action of PLA2 on acyl-PAF produces lyso-PAF, which is then acetylated to give PAF. PAF, in turn, can be deacetylated to the inactive lyso-PAF. It is produced by platelets in response to thrombin, and by activated inflammatory cells.
Actions and Role in Inflammation
By acting on specific receptors, PAF is capable of producing many of the signs and symptoms of inflammation. Injected locally, it produces vasodilatation (and thus erythema), increased vascular permeability and weal formation. Higher doses produce hyperalgesia. It is a potent chemotaxin for neutrophils and monocytes, and recruits eosinophils into the bronchial mucosa in the late phase of asthma (Fig. 27.3). It can activate PLA2 and initiates eicosanoid synthesis.
PAF stimulates arachidonate turnover and TXA2 generation by platelets, producing shape change and the release of the granule contents. This is important in haemostasis and thrombosis (see Ch. 24). PAF has spasmogenic effects on both bronchial and ileal smooth muscle.
The anti-inflammatory actions of the glucocorticoids may be caused, at least in part, by inhibition of PAF synthesis (Fig. 17.2). Competitive antagonists of PAF and/or specific inhibitors of lyso-PAF acetyltransferase could well be useful anti-inflammatory drugs and/or antiasthmatic agents. The PAF antagonist lexipafant is in clinical trial in the treatment of acute pancreatitis (see Leveau et al., 2005). Rupatidine is a combined H1 and PAF antagonist that is available in some parts of the world for treating allergic symptoms.
Platelet-activating factor
Bradykinin
Bradykinin and lysyl bradykinin (kallidin) are active peptides formed by proteolytic cleavage of circulating proteins termed kininogens through a protease cascade pathway (Fig. 6.1).
Source and Formation of Bradykinin
An outline of the formation of bradykinin from high-molecular-weight kininogen in plasma by the serine protease kallikrein is given in Figure 17.5. Kininogen is a plasma α-globulin that exists in both high- (Mr 110 000) and low- (Mr 70 000) molecular-weight forms. Kallikrein is derived from the inactive precursor prekallikrein by the action of Hageman factor (factor XII; see Ch. 24 and Fig. 6.1). Hageman factor is activated by contact with negatively charged surfaces such as collagen, basement membrane, bacterial lipopolysaccharides, urate crystals and so on. Hageman factor, prekallikrein and the kininogens leak out of the vessels during inflammation because of increased vascular permeability, and exposure to negatively charged surfaces promotes the interaction of Hageman factor with prekallikrein. The activated enzyme then ‘clips’ bradykinin from its kininogen precursor (Fig. 17.6). Kallikrein can also activate the complement system and can convert plasminogen to plasmin (see Fig. 6.1 and Ch. 24).
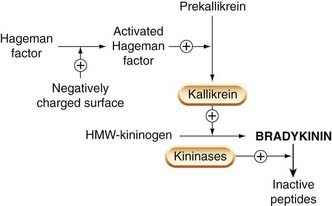
Fig. 17.5 The generation and breakdown of bradykinin.
High-molecular-weight kininogen (HMW-kininogen) probably acts both as a substrate for kallikrein and as a co-factor in the activation of prekallikrein.
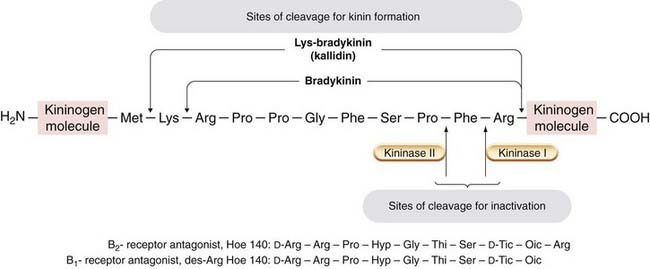
Fig. 17.6 Structure of bradykinin and some bradykinin antagonists.
The sites of proteolytic cleavage of high-molecular-weight kininogen by kallikrein kallidin involved in the formation of bradykinin are shown in the upper half of the figure; the sites of cleavage associated with bradykinin inactivation are shown in the lower half. The B2-receptor antagonist icatibant (Hoe 140) has a pA2 of 9, and the competitive B1-receptor antagonist des-Arg Hoe 140 has a pA2 of 8. The Hoe compounds contain unnatural amino acids: Thi, d-Tic and Oic, which are analogues of phenylalanine and proline.
In addition to plasma kallikrein, there are other kinin-generating isoenzymes found in pancreas, salivary glands, colon and skin. These tissue kallikreins act on both high- and low-molecular-weight kininogens and generate mainly kallidin, a peptide with actions similar to those of bradykinin.
Metabolism and Inactivation of Bradykinin
Specific enzymes that inactivate bradykinin and related kinins are called kininases (Fig. 17.5). One of these, kininase II, is a peptidyl dipeptidase that inactivates kinins by removing the two C-terminal amino acids. This enzyme, which is bound to the luminal surface of endothelial cells, is identical to angiotensin-converting enzyme (ACE; see Ch. 21), which cleaves the two C-terminal residues from the inactive peptide angiotensin I, converting it to the active vasoconstrictor peptide angiotensin II. Thus kininase II inactivates a vasodilator and activates a vasoconstrictor. Potentiation of bradykinin actions by ACE inhibitors may contribute to some side effects of these drugs (e.g. cough). Kinins are also metabolised by various less specific peptidases, including a serum carboxypeptidase that removes the C-terminal arginine, generating des-Arg9-bradykinin, a specific agonist at one of the two main classes of bradykinin receptor (see below).
Bradykinin Receptors
There are two bradykinin receptors, designated B1 and B2. Both are G-protein-coupled receptors and mediate very similar effects. B1 receptors are normally expressed at very low levels but are strongly induced in inflamed or damaged tissues by cytokines such as IL-1. B1 receptors respond to des-Arg9-bradykinin but not to bradykinin itself. A number of selective peptide and non-peptide antagonists are known. It is likely that B1 receptors play a significant role in inflammation and hyperalgesia (see Ch. 41), and antagonists could be developed for use in cough and neurological disorders (see Chung, 2005; Rodi et al., 2005).
B2 receptors are constitutively present in many normal cells and are activated by bradykinin and kallidin, but not by des-Arg9-bradykinin. Peptide and non-peptide antagonists have been developed, the best known being the bradykinin analogue icatibant, which has recently been approved by the European Medicines Agency for treating acute attacks in patients with hereditary angioedema (an uncommon disorder caused by deficiency of C1-esterase inhibitor that normally restrains complement activation).
Actions and Role in Inflammation
Bradykinin causes vasodilatation and increased vascular permeability. Its vasodilator action is partly a result of generation of PGI2 (Fig. 17.1) and release of nitric oxide (NO). It is a potent pain-producing agent at sensory neurons, and its action here is potentiated by prostaglandins (which are released by bradykinin). Bradykinin also has spasmogenic actions on intestinal, uterine and bronchial smooth muscle (in some species). The contraction is slow and sustained in comparison with that produced by histamine (hence brady, which means ‘slow’).
Although bradykinin reproduces many inflammatory signs and symptoms, its role in inflammation and allergy has not been clearly defined, partly because its effects are often part of a complex cascade of events triggered by other mediators. However, excessive bradykinin production contributes to the diarrhoea of gastrointestinal disorders, and in allergic rhinitis it stimulates nasopharyngeal secretion. Bradykinin also contributes to the clinical picture in pancreatitis. Physiologically, the release of bradykinin by tissue kallikrein may regulate blood flow to certain exocrine glands, and influence secretions. It also stimulates ion transport and fluid secretion by some epithelia, including intestine, airways and gall bladder.
Bradykinin
Nitric Oxide
Chapter 20 discusses NO in detail, and here we will consider only its role in inflammation. Inducible NO synthase (iNOS) is the chief isoform relevant to inflammation, and virtually all inflammatory cells express the enzyme in response to cytokine stimulation. iNOS is also present in the bronchial epithelium of asthmatic subjects, in the mucosa of the colon in patients with ulcerative colitis, and in synoviocytes in inflammatory joint disease. NO probably has a net proinflammatory effect: it increases vascular permeability and prostaglandin production, and is a potent vasodilator. Some other properties may be seen as anti-inflammatory; for example, endothelial NO inhibits adhesion of neutrophils and platelets, and platelet aggregation. NO, or compounds derived from it, also has cytotoxic actions, killing bacteria, fungi, viruses and metazoan parasites, so in this respect NO enhances local defence mechanisms. However, produced in excess, it may also harm host cells.
Inhibitors of iNOS are under investigation for treatment of inflammatory conditions. Patients with septic shock have benefited from inhibitors of iNOS, and in experimental arthritis iNOS inhibitors reduce disease activity. Laboratory studies on compounds consisting of NSAIDs coupled with NO-releasing groups suggest that these have fewer side effects than conventional NSAIDs and greater anti-inflammatory efficacy (see Ch. 26).
Neuropeptides
Neuropeptides released from sensory neurons cause neurogenic inflammation (Maggi, 1996). The main peptides involved are substance P, neurokinin A and CGRP (see Ch. 19). Substance P and neurokinin A (members of the tachykinin family) act on mast cells, releasing histamine and other mediators, and producing smooth muscle contraction and mucus secretion, whereas CGRP is a potent vasodilator. Neurogenic inflammation is implicated in the pathogenesis of several inflammatory conditions, including the delayed phase of asthma, allergic rhinitis, inflammatory bowel disease and some types of arthritis.
Concluding Remarks
Even from the, albeit superficial, sketch of the host defence response that we have presented here and in Chapter 6, it must be evident to the reader that this is among the most complicated of all physiological responses. Perhaps that is not surprising, given the central importance of its mission to the very survival of the organism. Perhaps it is also understandable that it can recruit so many different mediators that regulate and orchestrate the workings of the immune system that run into the hundreds.
What do come as a shock are experimental observations suggesting that the activity of many of these local hormones can apparently be blocked with little or no effect on the outcome of inflammation. This fact speaks to the redundancy of many of the component systems and goes at least some of the way to explaining why, until the advent of antibody-based therapies (see Ch. 26), our ability to curb the worst ravages of chronic inflammatory disease was very limited.
References and Further Reading
Abbas A.K., Murphy K.M., Sher A. Functional diversity of helper lymphocytes. Nature. 1996;383:787-793. (Excellent review, helpful diagrams; commendable coverage of Th1 and Th2 cells and their respective cytokine subsets)
Ariel A., Serhan C.N. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol.. 2007;28:176-183. (Very accessible review on these unusual lipid mediators which promote inflammatory resolution and the link with fish oils)
Coleman R.A., Humphrey P.A. Prostanoid receptors: their function and classification. In: Vane J., O’Grady J., editors. Therapeutic applications of prostaglandins. London: Edward Arnold; 1993:15-36. (Useful coverage; includes structures of prostanoids, their analogues and antagonists—a classification that brought forth order from chaos!)
Gerard C., Rollins B. Chemokines and disease. Nat. Immunol.. 2001;2:108-115. (Discusses diseases associated with inappropriate activation of the chemokine network, and discusses some therapeutic implications; describes how viruses evade the immune responses by mimicry of the chemokines or their receptors)
Gutzmer R., Diestel C., Mommert S., et al. Histamine H4 receptor stimulation suppresses IL-12p70 production and mediates chemotaxis in human monocyte-derived dendritic cells. J. Immunol.. 2005;174:5224-5232.
Horuk R. Chemokine receptors. Cytokine Growth Factor Rev.. 2001;12:313-335. (Comprehensive review focusing on recent findings in chemokine receptor research; describes the molecular, physiological and biochemical properties of each chemokine receptor)
Kim N., Luster A.D. Regulation of immune cells by eicosanoid receptors. ScientificWorldJournal. 2007;7:1307-1328. (Useful overview of eicosanoids, their biology and receptor family)
Luster A.D. Mechanisms of disease: chemokines—chemotactic cytokines that mediate inflammation. N. Engl. J. Med.. 1998;338:436-445. (Excellent review; outstanding diagrams)
Mackay C.R. Chemokines: immunology’s high impact factors. Nat. Immunol.. 2001;2:95-101. (Clear, elegant coverage of the role of chemokines in leukocyte–endothelial interaction, control of primary immune responses and T/B cell interaction, T cells in inflammatory diseases and viral subversion of immune responses)
Maggi C.A. Pharmacology of the efferent function of primary sensory neurones. In: Geppetti P., Holzer P., editors. Neurogenic inflammation. London: CRC Press, 1996. (Worthwhile. Covers neurogenic inflammation, the release of neuropeptides from sensory nerves and inflammatory mediators. Discusses agents that inhibit release and the pharmacological modulation of receptor-mediated release)
Mantovani A., Bussolino F., Introna M. Cytokine regulation of endothelial cell function: from molecular level to the bedside. Immunol. Today. 1997;5:231-239. (Pathophysiology of endothelial cell–cytokine interactions; detailed diagrams)
Murphy P.M. Viral exploitation and subversion of the immune system through chemokine mimicry. Nat. Immunol.. 2001;2:116-122. (Excellent description of viral/immune system interaction)
Pease J.E., Williams T.J. The attraction of chemokines as a target for specific anti-inflammatory therapy. Br. J. Pharmacol.. 2006;147(Suppl. 1):S212-S221. (Very good review of the history of chemokine research with particular emphasis on their potential role as drug targets)
Perretti M., D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol.. 2009;9:62-70. (Explores the role of the glucocorticoid-regulated protein annexin 1 in the control of inflammatory resolution. Easy to read and good diagrams)
Rodi D., Couture R., Ongali B., et al. Targeting kinin receptors for the treatment of neurological diseases. Curr. Pharm. Des.. 2005;11:1313-1326. (An overview of the potential role of kinin receptor antagonists in neurological diseases, dealing particularly with those of immunological origin)
Samuelsson B. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science. 1983;220:568-575. (Seminal article on leukotrienes)
Schulze-Topphoff U., Prat A. Roles of the kallikrein/kinin system in the adaptive immune system. Int. Immunopharmacol.. 2008;8:155-160. (Up-to-date overview of these mediators particularly with respect to their involvement in the adaptive response)
Szolcsànyi J. Neurogenic inflammation: reevaluation of the axon reflex theory. In: Geppetti P., Holzer P., editors. Neurogenic inflammation. London: CRC Press; 1996:33-42. (Good coverage of neurogenic inflammation)
Zhu Y., Michalovich D., Wu H., et al Cloning, expression, and pharmacological characterization of a novel human histamine receptor Mol. Pharmacol. 59:2001:434-441 (Describes the cloning of the fourth type of histamine receptor, H4)
Some relevant anti-inflammatory papers
Chung K.F. Drugs to suppress cough. Expert. Opin. Investig. Drugs. 2005;14:19-27. (Useful review of cough treatments, including a section on the role of neurokinin and bradykinin receptor antagonists)
Gilroy D.W., Perretti M. Aspirin and steroids: new mechanistic findings and avenues for drug discovery. Curr. Opin. Pharmacol.. 2005;5:405-411. (A very interesting review dealing with anti-inflammatory substances that are released during the inflammatory response and that bring about resolution; it also deals with a rather odd effect of aspirin—its ability to boost the production of anti-inflammatory lipoxins. Easy to read and informative)
Larsson B.M., Kumlin M., Sundblad B.M., et al. Effects of 5-lipoxygenase inhibitor zileuton on airway responses to inhaled swine house dust in healthy subjects. Respir. Med.. 2006;100:226-237. (A paper dealing with the effects of zileuton, a 5-lipoxygenase inhibitor, on the allergic response in humans; the results are not unequivocally positive, but the study is an interesting one)
Leveau P., Wang X., Sun Z., et al. Severity of pancreatitis-associated gut barrier dysfunction is reduced following treatment with the PAF inhibitor lexipafant. Biochem. Pharmacol.. 2005;69:1325-1331. (A paper dealing with the role of the PAF inhibitor lexipafant in pancreatitis; this is an experimental study using a rat model but provides a useful insight into the potential clinical role of such an antagonist)
Serhan C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fatty Acids. 2005;73:141-162. (A paper reviewing the lipoxins—anti-inflammatory substances formed by the 5-lipoxygenase enzyme; also discusses the action of aspirin in boosting the synthesis of these compounds and the receptors on which they act. A good review that summarises a lot of work)
Dale M.M., Foreman J.C., Fan T.-P., editors. Textbook of immunopharmacology, third ed, Oxford: Blackwell Scientific, 1994. (Excellent textbook written with second- and third-year medical and science students in mind; contains many sections relevant to this chapter)
Janeway C.A., Travers P., Nolan A., et al. Immunobiology: the Immune System in Health and Disease, sixth ed. Edinburgh: Churchill Livingstone; 2004. (Excellent textbook, good diagrams)
http://microvet.arizona.edu/Courses/MIC419/Tutorials/cytokines.html (This is a useful Web site with a series of immunological tutorials. The cytokines module is worth looking at, and it has a good [although not complete] list of the most important members of the family, their targets and function. Also contains other material that is likely to be useful in understanding this chapter)
http://www.copewithcytokines.de/ (A very comprehensive site dealing with practically all known cytokines. Also contains a list of terms, links to reviews and short pieces on individual cytokines. Worth a look if you are stuck for some information)
1One might have expected evolution to have generated more examples of endogenous receptor antagonists as physiological regulators, but apart from IL-1ra, they are only exploited as toxins directed against other species.
2MCP, monocyte chemoattractant protein; RANTES, regulated on activation normal T cell expressed and secreted.
3The name arose through an anatomical error. In some species it is difficult to differentiate the prostaglandin-rich seminal vesicles from the prostate gland which, ironically, contains virtually none. Nevertheless the name stuck, outlasting the term vesiglandin which, while being suggested later, would have been more appropriate.