25 Haemopoietic system and treatment of anaemia
Overview
This chapter summarises the different kinds of anaemia, caused by nutrient deficiencies, bone marrow depression or increased red cell destruction, and covers the main haematinic agents used to treat them. We describe haemopoietic growth factors for red and white blood cells, and conclude by mentioning two drugs (hydroxycarbamide and eculizumab) used in treating, respectively, sickle cell anaemia and paroxysmal nocturnal haemoglobinuria.
Introduction
In this chapter we briefly review the haemopoietic system and different types of anaemia due to deficiency of nutrients, depression of the bone marrow or increased destruction of red cells (haemolytic anaemias). Nutritional deficiencies of iron, vitamin B12 or folic acid are common and important and most of the chapter is devoted to these haematinic agents (i.e. nutrients needed for healthy haemopoiesis and related drugs). Treatment of many forms of bone marrow depression is mainly supportive, but haemopoietic growth factors (especially epoetins—preparations of the natural hormone erythropoietin) have a place, especially in patients with chronic renal failure, and are covered briefly, as are other haemopoietic factors, known as colony-stimulating factors (CSFs), which are used to increase numbers of circulating white blood cells. Treatment of haemolytic anaemias is again mainly supportive, but we mention two drugs (hydroxycarbamide and eculizumab) that provide mechanistic insights as well as clinical benefit in two specific haemolytic disorders.
The Haemopoietic System
The main components of the haemopoietic system are the blood, bone marrow, lymph nodes and thymus, with the spleen, liver and kidneys as important accessory organs. Blood consists of formed elements (red and white blood cells and platelets) and plasma. This chapter deals mainly with red cells, which have the principal function of carrying oxygen. Their oxygen-carrying power depends on their haemoglobin content. The most important site of formation of red blood cells in adults is the bone marrow, whereas the spleen acts as their graveyard. Red cell loss in healthy adults is precisely balanced by production of new cells. The liver stores vitamin B12 and is involved in the process of breakdown of the haemoglobin liberated when red blood cells are destroyed. The kidney manufactures erythropoietin, a hormone that stimulates red cell production. Cells from various organs synthesise and release CSFs, which regulate the production of leukocytes and platelets. Drugs used in the chemotherapy of leukemias are described in Chapter 55.
Types of Anaemia
Anaemia is characterised by a reduced concentration of haemoglobin in the blood. It may cause fatigue but, especially if it is chronic, is often surprisingly asymptomatic. The commonest cause is blood loss resulting from menstruation, drug treatment (e.g. with aspirin or other non-steroidal anti-inflammatory drugs; Ch. 26) or pathological processes such as colonic carcinoma or (especially in developing countries) parasitic infestation (Ch. 54). Pregnancy and child bearing are other important physiological drains on iron reserves. There are several different types of anaemia and several different diagnostic levels. Determining indices of red cell size and haemoglobin content and microscopical examination of a stained blood smear allow characterisation into:
Further evaluation may include determination of concentrations of ferritin, iron, vitamin B12 and folic acid in serum, and microscopic examination of smears of bone marrow. This leads to more precise diagnostic groupings of anaemias into:
Haematinic Agents
It is important to note that the use of haematinic agents is often only an adjunct to treatment of the underlying cause of the anaemia—for example, surgery for colon cancer (a common cause of iron deficiency) or anthelminthic drugs for patients with hookworm (a frequent cause of anaemia in parts of Africa and Asia; Ch. 54). Sometimes treatment consists of stopping an offending drug, for example a non-steroidal anti-inflammatory drug that causes blood loss from the stomach (Ch. 26).
Iron
Iron is a transition metal with two important properties relevant to its biological role:
The body of a 70 kg man contains about 4 g of iron, 65% of which circulates in the blood as haemoglobin. About one-half of the remainder is stored in the liver, spleen and bone marrow, chiefly as ferritin and haemosiderin. The iron in these molecules is available for haemoglobin synthesis. The rest, which is not available for haemoglobin synthesis, is present in myoglobin, cytochromes and various enzymes.
The distribution and turnover of iron in an average adult man are shown in Table 25.1 and Figure 25.1. The corresponding values in a woman would be about 55% of the values in Table 25.1. Because most of the iron in the body is either part of—or destined to be part of—haemoglobin, the most obvious clinical result of iron deficiency is anaemia, and the only indication for therapy with iron is for treatment or prophylaxis of iron deficiency anaemia.
Table 25.1 The distribution of iron in the body of a healthy 70 kg man
| Protein | Tissue | Iron content (mg) |
|---|---|---|
| Haemoglobin | Erythrocytes | 2600 |
| Myoglobin | Muscle | 400 |
| Enzymes (cytochromes, catalase, guanylyl cyclase, etc.) | Liver and other tissues | 25 |
| Transferrin | Plasma and extracellular fluid | 8 |
| Ferritin and haemosiderin | Liver | 410 |
| Spleen | 48 | |
| Bone marrow | 300 |
Data from Jacobs A, Worwood M 1982 Chapter 5. In: Hardisty R M, Weatherall D J [eds] Blood and its disorders. Blackwell Scientific, Oxford.
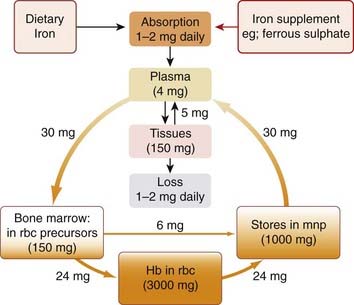
Fig. 25.1 Distribution and turnover of iron in the body.
The quantities by the arrows indicate the usual amounts transferred each day. The transfer of 6 mg from red cell precursors to phagocytes represents aborted cells that fail to develop into functional red blood cells. Hb, haemoglobin; mnp, mononuclear phagocytes (mainly in liver, spleen and bone marrow); rbc, red blood cells.
Haemoglobin is made up of four protein chain subunits (globins), each of which contains one haem moiety. Haem consists of a tetrapyrrole porphyrin ring containing ferrous (Fe2+) iron. Each haem group can carry one oxygen molecule, which is bound reversibly to Fe2+ and to a histidine residue in the globin chain. This reversible binding is the basis of oxygen transport.
Iron Turnover and Balance
Both the normal physiological turnover of iron and pharmacokinetic factors affecting iron when it is given therapeutically will be dealt with here. The normal daily requirement for iron is approximately 5 mg for men, and 15 mg for growing children and for menstruating women. A pregnant woman needs between 2 and 10 times this amount because of the demands of the fetus and increased requirements of the mother.1 The average diet in Western Europe provides 15–20 mg of iron daily, mostly in meat. Iron in meat is generally present as haem, and about 20–40% of haem iron is available for absorption.
 Humans are adapted to absorb haem iron. It is thought that one reason why modern humans have problems in maintaining iron balance (there are an estimated 500 million people with iron deficiency in the world) is that the change from hunting to grain cultivation 10 000 years ago led to cereals, which have a relatively small amount of utilisable iron, replacing meat in the diet. Non-haem iron in food is mainly in the ferric state, and this needs to be converted to ferrous iron for absorption. Ferric iron, and to a lesser extent ferrous iron, has low solubility at the neutral pH of the intestine; however, in the stomach iron dissolves and binds to a mucoprotein carrier. In the presence of ascorbic acid, fructose and various amino acids, iron is detached from the carrier, forming soluble low-molecular-weight complexes that enable it to remain in soluble form in the intestine. Ascorbic acid stimulates iron absorption partly by forming soluble iron–ascorbate chelates and partly by reducing ferric iron to the more soluble ferrous form. Tetracycline forms an insoluble iron chelate, impairing absorption of both substances.
Humans are adapted to absorb haem iron. It is thought that one reason why modern humans have problems in maintaining iron balance (there are an estimated 500 million people with iron deficiency in the world) is that the change from hunting to grain cultivation 10 000 years ago led to cereals, which have a relatively small amount of utilisable iron, replacing meat in the diet. Non-haem iron in food is mainly in the ferric state, and this needs to be converted to ferrous iron for absorption. Ferric iron, and to a lesser extent ferrous iron, has low solubility at the neutral pH of the intestine; however, in the stomach iron dissolves and binds to a mucoprotein carrier. In the presence of ascorbic acid, fructose and various amino acids, iron is detached from the carrier, forming soluble low-molecular-weight complexes that enable it to remain in soluble form in the intestine. Ascorbic acid stimulates iron absorption partly by forming soluble iron–ascorbate chelates and partly by reducing ferric iron to the more soluble ferrous form. Tetracycline forms an insoluble iron chelate, impairing absorption of both substances.
The amount of iron in the diet and the various factors affecting its availability are thus important determinants in absorption, but the regulation of iron absorption is a function of the intestinal mucosa, influenced by the body’s iron stores. Because there is no mechanism whereby iron excretion is regulated, the absorptive mechanism has a central role in iron balance as it is the sole mechanism by which body iron is controlled.
The site of iron absorption is the duodenum and upper jejunum, and absorption is a two-stage process involving first a rapid uptake across the brush border and then transfer into the plasma from the interior of the epithelial cells. The second stage, which is rate limiting, is energy dependent. Haem iron in the diet is absorbed as intact haem, and the iron is released in the mucosal cell by the action of haem oxidase. Non-haem iron is absorbed in the ferrous state. Within the cell, ferrous iron is oxidised to ferric iron, which is bound to an intracellular carrier, a transferrin-like protein; the iron is then either held in storage in the mucosal cell as ferritin (if body stores of iron are high) or passed on to the plasma (if iron stores are low).
Iron is carried in the plasma bound to transferrin, a β-globulin with two binding sites for ferric iron, which is normally only 30% saturated. Plasma contains 4 mg of iron at any one time, but the daily turnover is about 30 mg (Fig. 25.1). Most of the iron that enters the plasma is derived from mononuclear phagocytes, following the degradation of time-expired erythrocytes. Intestinal absorption and mobilisation of iron from storage depots contribute only small amounts. Most of the iron that leaves the plasma each day is used for haemoglobin synthesis by red cell precursors (erythroblasts). These have receptors that bind transferrin, releasing it again when its cargo of iron has been captured.
Iron is stored in two forms: soluble ferritin and insoluble haemosiderin. Ferritin is present in all cells, the mononuclear phagocytes of liver, spleen and bone marrow containing especially high concentrations. It is also present in plasma. The precursor of ferritin, apoferritin, is a protein of molecular weight 450 000, composed of 24 identical polypeptide subunits that enclose a cavity in which up to 4500 iron atoms can be stored. Apoferritin takes up ferrous iron, oxidises it and deposits the ferric iron in its core. In this form, it constitutes ferritin, the primary storage form of iron, from which the iron is most readily available. The lifespan of this iron-laden protein is only a few days. Haemosiderin is a degraded form of ferritin in which the iron cores of several ferritin molecules have aggregated, following partial disintegration of the outer protein shells.
The ferritin in plasma has virtually no iron associated with it. It is in equilibrium with the storage ferritin in cells, and its concentration in plasma provides an estimate of total body iron stores.
The body has no means of actively excreting iron. Small amounts leave the body through desquamation (peeling off) of mucosal cells containing ferritin, and even smaller amounts leave in the bile, sweat and urine. A total of about 1 mg is lost daily. Iron balance is therefore critically dependent on the active absorption mechanism in the intestinal mucosa. This absorption is influenced by the iron stores in the body, but the precise mechanism of this control is still a matter of debate; the amount of ferritin in the intestinal mucosa may be important, as may the balance between ferritin and the transferrin-like carrier molecule in these cells. The daily movement of iron in the body is illustrated in Figure 25.1. Since red cells contain approximately 0.6 mg iron per ml of blood, loss of only a few ml of blood per day substantially increases dietary iron requirement.
Administration of Iron
Iron is usually given orally, e.g. as ferrous sulfate. Other salts for oral administration are ferrous succinate, gluconate or fumarate.
Parenteral iron (e.g. iron dextran, iron sucrose) may be necessary in individuals who are not able to absorb oral iron because of malabsorption syndromes, or as a result of surgical procedures or inflammatory conditions involving the gastrointestinal tract. It is also used for patients who do not tolerate oral preparations, and patients with chronic renal failure or with chemotherapy-induced anaemia who are receiving treatment with erythropoietin (see below). Iron-dextran can be given by deep intramuscular injection or slow intravenous infusion; iron-sucrose is given by slow intravenous infusion. A small initial dose is given because of the risk of anaphylactoid reaction.
Unwanted effects
The unwanted effects of oral iron administration are dose related and include nausea, abdominal cramps and diarrhoea. Parenteral iron can cause anaphylactoid reactions (Ch. 57). Iron is an important nutrient for several pathogens and there is concern that excessive iron could worsen the clinical course of infection. Iron treatment is usually avoided during infection for this reason.
Acute iron toxicity, usually seen in young children who have swallowed attractively coloured iron tablets in mistake for sweets, can result in severe necrotising gastritis with vomiting, haemorrhage and diarrhoea, followed by circulatory collapse.
Iron overload
Chronic iron toxicity or iron overload occurs in chronic haemolytic anaemias requiring frequent blood transfusions, such as the thalassaemias (a large group of genetic disorders of globin chain synthesis) and haemochromatosis (a genetic iron storage disease with increased iron absorption, resulting in damage to liver, islets of Langerhans, joints and skin2).
The treatment of acute and chronic iron toxicity involves the use of iron chelators such as desferrioxamine. This is not absorbed from the gut but is nonetheless given intragastrically following acute overdose (to bind iron in the bowel lumen and prevent its absorption) as well as intramuscularly and, if necessary, intravenously. In severe poisoning, it is given by slow intravenous infusion. Desferrioxamine forms a complex with ferric iron and, unlike unbound iron, this is excreted in the urine. Deferiprone, an orally absorbed iron chelator, is an alternative treatment for iron overload in patients with thalassaemia major who are unable to take desferrioxamine. Agranulocytosis and other blood dyscrasias are serious potential adverse effects. Defasirox, an oral iron chelator, is used for selected patients with thalassaemia.
Iron 
Folic Acid and Vitamin B12
Vitamin B12 and folic acid are essential constituents of the human diet, being necessary for DNA synthesis and consequently for cell proliferation. Their biochemical actions are interdependent (see below), and treatment of vitamin B12 deficiency with folic acid corrects some, but not all, of the features of vitamin B12 deficiency. Deficiency of either vitamin B12 or folic acid affects tissues with a rapid cell turnover, particularly bone marrow, but vitamin B12 deficiency also causes important neuronal disorders, which are not corrected (or may even be made worse) by treatment with folic acid. Deficiency of either vitamin causes megaloblastic haemopoiesis, in which there is disordered erythroblast differentiation and defective erythropoiesis in the bone marrow. Large abnormal erythrocyte precursors appear in the marrow, each with a high RNA : DNA ratio as a result of decreased DNA synthesis. The circulating erythrocytes (macrocytes) are large fragile cells, often distorted in shape. Mild leukopenia and thrombocytopenia usually accompany the anaemia, and the nuclei of polymorphonuclear leukocytes are abnormal (hypersegmented). Neurological disorders caused by deficiency of vitamin B12 include peripheral neuropathy and dementia, as well as subacute combined3 degeneration of the spinal cord. Folic acid deficiency is caused by dietary deficiency, especially in settings of increased demand (e.g. during pregnancy—especially important because of the link between folate deficiency and neural tube defects in the baby [see Ch. 57] or because of chronic haemolysis in patients with haemoglobinopathies such as sickle cell anaemia—see below). Vitamin B12 deficiency, however, is usually due to decreased absorption (see below).
Folic Acid
Some aspects of folate structure and metabolism are dealt with in Chapters 50 and 55, because several important antibacterial and anticancer drugs are antimetabolites that interfere with folate synthesis in microorganisms or tumour cells. Liver and green vegetables are rich sources of folate. In healthy non-pregnant adults, the daily requirement is about 0.2 mg daily, but this is increased during pregnancy.
Mechanism of action
Reduction of folic acid, catalysed by dihydrofolate reductase in two stages yields dihydrofolate (FH2) and tetrahydrofolate (FH4), co-factors which transfer methyl groups (1-carbon transfers) in several important metabolic pathways. FH4 is essential for DNA synthesis because of its role as co-factor in the synthesis of purines and pyrimidines. It is also necessary for reactions involved in amino acid metabolism.
FH4 is especially important for the conversion of deoxyuridylate monophosphate (DUMP) to deoxythymidylate monophosphate (DTMP). This reaction is rate limiting in mammalian DNA synthesis and is catalysed by thymidylate synthetase, with FH4 acting as methyl donor.
Pharmacokinetic aspects
Therapeutically, folic acid is given orally and is absorbed in the ileum. Methyl-FH4 is the form in which folate is usually carried in blood and which enters cells. It is functionally inactive until it is demethylated in a vitamin B12-dependent reaction (see below). Folate is taken up into hepatocytes and bone marrow cells by active transport. Within the cells, folic acid is reduced and formylated before being converted to the active polyglutamate form. Folinic acid, a synthetic FH4, is converted much more rapidly to the polyglutamate form.
Unwanted effects
Unwanted effects do not occur even with large doses of folic acid—except possibly in the presence of vitamin B12 deficiency, when administration of folic acid may improve the anaemia while exacerbating the neurological lesion. It is therefore important to determine whether a megaloblastic anaemia is caused by folate or vitamin B12 deficiency and treat accordingly.
Vitamin B12
Vitamin B12 is a complex cobalamin. The vitamin B12 preparation used therapeutically is hydroxocobalamin. The principal dietary sources are meat (particularly liver, where it is stored), eggs and dairy products. For activity, cobalamins must be converted to methylcobalamin (methyl-B12) or 5′-deoxyadenosylcobalamin (ado-B12). The average European diet contains 5–25 µg of vitamin B12 per day, and the daily requirement is 2–3 µg. Absorption requires intrinsic factor (a glycoprotein secreted by gastric parietal cells). Vitamin B12, complexed with intrinsic factor, is absorbed by active transport in the terminal ileum. Healthy stomach secretes a large excess of intrinsic factor, but in patients with pernicious anaemia (an autoimmune disorder where the lining of the stomach atrophies), or following total gastrectomy, the supply of intrinsic factor is inadequate to maintain vitamin B12 absorption in the long term. Surgical removal of the terminal ileum, for example to treat Crohn’s disease (see Ch. 29), can also impair B12 absorption.
Vitamin B12 is carried in the plasma by binding proteins called transcobalamins. It is stored in the liver, the total amount in the body being about 4 mg. This store is so large compared with the daily requirement, that if vitamin B12 absorption stops suddenly—as after a total gastrectomy—it takes 2–4 years for evidence of deficiency to become manifest.
Mechanism of action
Vitamin B12 is required for two main biochemical reactions in humans.
The conversion of methyl-FH4 to FH4
The role of vitamin B12 in folate coenzyme synthesis is illustrated in Figure 25.2. It is through these mechanisms that the metabolic activities of vitamin B12 and folic acid are linked and implicated in the synthesis of DNA. It is also through this pathway that folate/vitamin B12 treatment can lower plasma homocysteine concentration. Because increased homocysteine concentrations may have undesirable vascular effects (Ch. 23, Table 23.1), this has potential therapeutic and public health implications. The reaction involves conversion of both methyl-FH4 to FH4 and homocysteine to methionine. The enzyme that accomplishes this (homocysteine–methionine methyltransferase) requires vitamin B12 as co-factor and methyl-FH4 as methyl donor. The methyl group from methyl-FH4 is transferred first to B12, and then to homocysteine to form methionine (Fig. 25.2). Vitamin B12 deficiency thus traps folate in the inactive methyl-FH4 form, thereby depleting the folate polyglutamate coenzymes needed for DNA synthesis (see above). Vitamin B12-dependent methionine synthesis also affects the synthesis of folate polyglutamate coenzymes by an additional mechanism. The preferred substrate for polyglutamate synthesis is formyl-FH4, and the conversion of FH4 to formyl-FH4 requires a formate donor such as methionine.
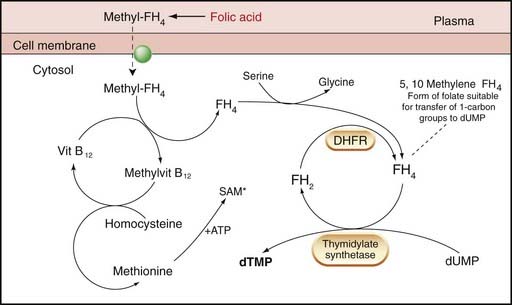
Fig. 25.2 Simplified diagram of the role of folate and vitamin B12 in the reactions necessary for the eventual synthesis of thymidylate.
Methyl-FH4 enters cells from the plasma by carrier. The methyl group is transferred to homocysteine to form methionine via vitamin B12, which is bound to a methyltransferase (not shown). Methionine reacts with ATP to form S-adenosyl methionine (SAM*) which is a universal methyl donor for several reactions including methylation of cytosine in DNA molecules. FH4 functions as a carrier of a one-carbon unit, providing the methyl group necessary for the conversion of 2′deoxyuridylate monophosphate (DUMP) to 2′ deoxythymidylate (DTMP) by thymidylate synthetase. During the transfer of the one-carbon unit, FH4 is oxidised to FH2, which must be reduced by dihydrofolate reductase (DHFR) to FH4 (before it can act again). The thymidylate synthetase action is rate-limiting in DNA synthesis. Note that in all the actions of folates it is the polyglutamate form that is most active. DHFR, dihydrofolate reductase; DTMP, thymidylate; DUMP, deoxyuridylate monophosphate.
Isomerisation of methylmalonyl-CoA to succinyl-CoA
This isomerisation reaction is part of a route by which propionate is converted to succinate. Through this pathway, cholesterol, odd-chain fatty acids, some amino acids and thymine can be used for gluconeogenesis or for energy production via the tricarboxylic acid cycle. Coenzyme B12 (ado-B12) is an essential co-factor, so methylmalonyl-CoA accumulates in vitamin B12 deficiency. This distorts the pattern of fatty acid synthesis in neural tissue and may be the basis of neuropathy in vitamin B12 deficiency.
Administration of vitamin B12
When vitamin B12 is used therapeutically (as hydroxocobalamin), it is usually given by injection4 because, as explained above, vitamin B12 deficiency commonly results from malabsorption. Patients with pernicious anaemia require life-long therapy. Hydroxocobalamin does not cause unwanted effects.
Vitamin B12 and folic acid
Both vitamin B12 and folic acid are needed for DNA synthesis. Deficiencies particularly affect erythropoiesis, causing macrocytic megaloblastic anaemia.
Folic acid
Vitamin B12 (hydroxocobalamin)
Haemopoietic Growth Factors
Every 60 seconds, a human being must generate about 120 million granulocytes and 150 million erythrocytes, as well as numerous mononuclear cells and platelets. The cells responsible for this remarkable productivity are derived from a relatively small number of self-renewing, pluripotent stem cells laid down during embryogenesis. Maintenance of haemopoiesis necessitates a balance between self-renewal of the stem cells on the one hand, and differentiation into the various types of blood cell on the other. The factors involved in controlling this balance are the haemopoietic growth factors, which direct the division and maturation of the progeny of these cells down eight possible lines of development (Fig. 25.3). These cytokine growth factors are highly potent glycoproteins, acting at concentrations of 10−12 to 10−10 mol/l. They are present in plasma at very low concentrations under basal conditions, but on stimulation their concentrations can increase within hours by 1000-fold or more. Erythropoietin regulates the red cell line, and the signal for its production is blood loss and/or low tissue oxygen tension. Colony-stimulating factors (CSFs) regulate the myeloid divisions of the white cell line, and the main stimulus for their production is infection (see also Ch. 6).
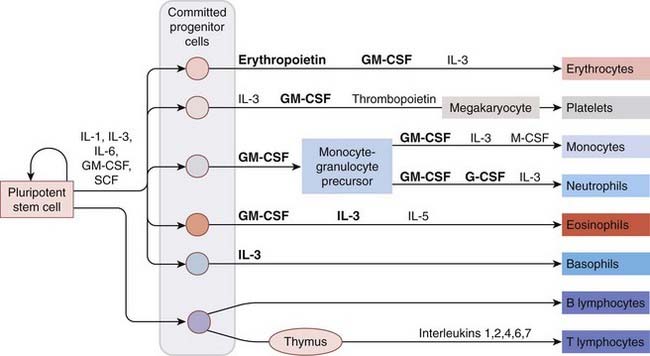
Fig. 25.3 Haemopoietic growth factors in blood cell differentiation.
Various preparations of the factors shown in bold are in clinical use (see text). Most T cells generated in the thymus die by apoptosis; those that emerge are either CD4 or CD8 T cells. The colours used for the mature blood cells reflect how they appear in common staining preparations (and after which some are named). CSF, colony-stimulating factor; G-CSF, granulocyte CSF; GM-CSF, granulocyte–macrophage CSF; IL-1, interleukin-1; IL-3, interleukin-3 or multi-CSF; M-CSF, macrophage CSF; SCF, stem cell factor. (See also Ch. 6.)
Recombinant erythropoietin (epoetin),5 and recombinant granulocyte CSF (filgrastim, lenograstim, pegfilgrastim) are used clinically (see below); thrombopoietin is available in recombinant form but there are concerns about effects on tumour progression (it activates a cell surface protein that is an oncogene product). Some of the other haemopoietic growth factors (e.g. interleukin-3, interleukin-5 and various other cytokines) are covered in Chapter 6.
Erythropoietin
Erythropoietin is produced in juxtatubular cells in the kidney and also in macrophages; it stimulates committed erythroid progenitor cells to proliferate and generate erythrocytes (Fig. 25.3). Recombinant human erythropoietins are used to treat symptomatic anaemia caused by erythropoietin deficiency. Darbepoetin, a hyperglycosylated form of epoetin, has a longer half-life and can be administered less frequently; methoxy polyethylene glycol-epoetin beta is another preparation with long half-life. Epoetin and darbopoietin are given intravenously or subcutaneously, the response being greatest after subcutaneous injection and fastest after intravenous injection.
Epoetins are reaching the end of their periods of patent protection and the first ‘biosimilar’ products have recently been licensed; unlicensed uses include its use in sport (e.g. ‘blood-doping’ in cyclists)—see Chapter 58.
Unwanted effects
Transient influenza-like symptoms are common. Hypertension is also common and can cause encephalopathy with headache, disorientation and sometimes convulsions. Iron deficiency can be induced because more iron is required for the enhanced erythropoiesis. Blood viscosity increases as the haematocrit (i.e. the fraction of the blood that is occupied by red blood cells) rises, increasing the risk of thrombosis, especially during dialysis. There have been rare reports of a devastating chronic condition known as pure red cell aplasia, connected with development of antibodies directed against erythropoietin.
Clinical use
Iron or folate deficiency must be corrected before starting treatment. Parenteral iron preparations are often needed (see above). Haemoglobin must be monitored and maintained within the range 10–12 g/dl to avoid the unwanted effects described above. The clinical use of epoetin is given in the box below.
Colony-Stimulating Factors
The CSFs are cytokines that stimulate the formation of maturing colonies of leukocytes, observable in tissue culture. They not only stimulate particular committed progenitor cells to proliferate (Fig. 25.3) but also cause irreversible differentiation. The responding precursor cells have membrane receptors for specific CSFs and may express receptors for more than one factor, thus permitting collaborative interactions between factors.
Granulocyte CSF is produced mainly by monocytes, fibroblasts and endothelial cells, and controls primarily the development of neutrophils, increasing their proliferation and maturation, stimulating their release from bone marrow storage pools and enhancing their function. Recombinant forms (filgrastim, which is not glycosylated, and glycosylated lenograstim) are used therapeutically. Pegfilgrastim is a derivative of filgrastim conjugated with polyethylene glycol (‘pegylated’), which has the effect of increasing its duration of action.
Thrombopoietin, made in liver and kidney, stimulates proliferation and maturation of megakaryocytes to form platelets. Recombinant thrombopoietin is not used clinically.
Administration and unwanted effects
Filgrastim and lenograstim are given either subcutaneously or by intravenous infusion. Pegfilgrastim is administered subcutaneously. Gastrointestinal effects, fever, bone pain, myalgia and rash are recognised adverse effects; less common effects include pulmonary infiltrates and enlargement of liver or spleen.
Haemopoietic growth factors
Clinical uses of epoetin 
Haemolytic Anaemia
Anaemia associated with increased red cell destruction can arise from genetic causes (e.g. sickle cell disease, thalassaemia) or a variety of non-genetic causes such as autoimmunity, infections and adverse drug reactions.
Adult haemoglobin (haemoglobin A) contains two α- and two β-globin chains. The cause of sickle cell anaemia is a mutation in the gene that codes the β-globin chain, resulting in a single amino acid substitution. The abnormal haemoglobin (haemoglobin S) can polymerise when deoxygenated, changing the physical properties of the red cells (which deform to a sickle shape, hence the name) and damaging the cell membranes. This can block the microcirculation, causing painful crises, and haemolysis can reduce the availability of nitric oxide (Ch. 20). Polymerisation is markedly reduced when other forms of haemoglobin are present. Almost 100% of the haemoglobin is in the form haemoglobin A in healthy adults of African (or European) origin, but in some ethnic groups (from Saudi Arabia), fetal haemoglobin (haemoglobin F, which contains two α- and two γ-globin chains) persists into adulthood. Such individuals, if they inherit the sickle cell gene, suffer a milder form of the illness. Since all adults possess the gene to make γ-globin, a means to turn it back on again might ameliorate the course of sickle cell disease.
Paroxysmal nocturnal haemoglobinuria (PNH) is a rare and previously untreatable form of haemolytic anaemia caused by clonal expansion of haemopoietic stem cells with somatic mutations in an X-linked gene PIG-A. The mutation prevents formation of glycophosphatidylinositol (GPI) which anchors many proteins to the cell surface. Haemolysis is a feature of PNH because of the absence of a GPI-linked protein, CD59, which blocks the formation of the terminal complement complex (the membrane attack complex) on the cell surface. In addition to anaemia, patients with PNH suffer from other features including thrombosis, attacks of abdominal pain and pulmonary hypertension (Ch. 22).
Eculizumab, now licensed for clinical use, is a humanised monoclonal antibody which blocks the terminal complement protein C5 (Ch. 17). In a double-blind, randomised, controlled trial in 87 patients, treatment with eculizumab dramatically reduced haemolysis and transfusion requirement during 6 months of treatment (Fig. 25.4). Patients must be innoculated against meningococcal infection before treatment. It is administered by intravenous infusion weekly for 4 weeks and then approximately every 2 weeks. Serious adverse effects include infection, notably meningococcal infection, but are uncommon. The commonest adverse effects are headache and back pain.
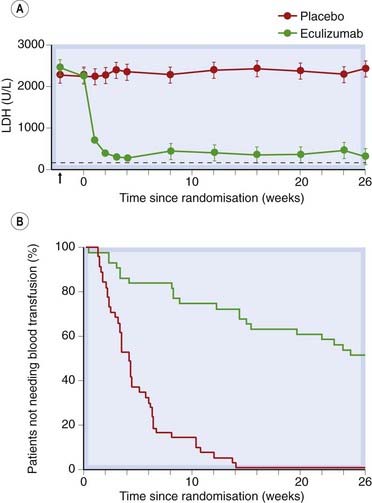
Fig 25.4 Effect of eculizumab in patients with paroxysmal nocturnal haemoglobinuria (PNH).
[A] Effect on plasma lactate dehydrogenase (LDH) activity, a measure of haemolyisis. The horizontal dotted line shows the upper limit of normal. The arrow shows the baseline level at screening (n = 44 in placebo group, n = 43 in eculizumab group, P < 0.001). [B] Kaplan–Meier curves for the time to first transfusion during treatment in the same patients shown in [A] (P < 0.001).
(Redrawn from Hillmen et al. 2006 NEJM 355: 1233–43.)
In most forms of haemolytic anaemia, treatment is symptomatic (e.g. analgesia for painful crises in patients with sickle cell disease) and supportive (e.g. attention to fluid balance, oxygen therapy, blood transfusion when essential, treatment of iron overload, provision of adequate folate to support increased red cell turnover and, in some cases, antibiotics and immunisation). Acute haemolytic anaemia associated with autoantibodies may respond to treatment with glucocorticoids (Ch. 32).
Hydroxycarbamide
Hydroxycarbamide (previously known as hydroxyurea) is a cytotoxic drug that has been used for decades to lower the red cell and platelet counts in patients with polycythaemia rubra vera (a myeloproliferative disorder affecting especially the red cell lineage) or to treat chronic myeloid leukemia. It shifts haemoglobin production from haemoglobin S to haemoglobin F (for further details see Platt, 2008). In one randomised, controlled trial in 499 adults with sickle cell anaemia, hydroxycarbamide ameliorated the clinical course with fewer painful crises and less need for transfusion. There were no serious adverse effects, but long-term safety is uncertain (Charache et al., 1995).
Mechanism of action
Hydroxycarbamide inhibits DNA synthesis by inhibiting ribonucleotide reductase and is S-phase specific (Ch. 5). Consequently, it is relatively selective for rapidly dividing red cells and reduces the production of red cells containing haemoglobin S while favouring production of red cells containing a high concentration of haemoglobin F (rapidly dividing F cells). Hydroxycarbamide metabolism gives rise to nitric oxide, which may contribute to its beneficial effect in sickle cell disease. Some of its beneficial effect in reducing painful crises could relate to anti-inflammatory effects secondary to its cytotoxic action.
Administration and unwanted effects
Hydroxycarbamide is administered by mouth once daily in rather lower starting dose than is used for treating malignant disease; reduced doses are used in patients with impaired renal function. The blood count and haemoglobin F are monitored and the dose adjusted accordingly. Once stabilised, treatment may be continued indefinitely.
Myelosuppression, nausea and rashes are the commonest adverse effects. Secondary leukaemias have occurred during treatment of myeloproliferative disorders, but it is unknown if this is drug related rather than part of the natural history of these disorders. Animal studies demonstrated teratogenicity, and potential adverse effects on spermatogenesis.
References and Further Reading
Fishbane S. Erythropoiesis-stimulating agent treatment with full anemia correction: a new perspective. Kidney Int.. 2009;75:358-365.
Fishman S.M., Christian P., West K.P. The role of vitamins in the prevention and control of anaemia. Public Health Nutr.. 2000;3:125-150.
Hoelzer D. Haemopoietic growth factors—not whether, but when and where. N. Engl. J. Med.. 1997;336:1822-1824. (Edifying editorial comment)
Kurzrock R. Thrombopoietic factors in chronic bone marrow failure states: the platelet problem revisited. Clin. Cancer Res.. 2005;11:1361-1367. (Slow progress)
Andrews N.C. Disorders of iron metabolism. N. Engl. J. Med.. 1999;341:1986-1995.
Provan D., Weatherall D. Red cells, II: acquired anaemias and polycythaemia. Lancet. 2000;355:1260-1268.
Toh B.-H., van Driel I.R., Gleeson P.A. Pernicious anaemia. N. Engl. J. Med.. 1997;337:1441-1448. (Immunopathogenesis of pernicious anaemia; excellent figures)
Folic acid and vitamin, B12 and their deficiencies
Botto L.D., Lisi A., Robert-Gnansia E., et al. International retrospective cohort study of neural tube defects in relation to folic acid recommendations: are the recommendations working? Br. Med. J.. 2005;330:571-573. (Not as well as might be hoped)
Refsum H. Folate, vitamin, B12 and homocysteine in relation to birth defects and pregnancy outcome. Br. J. Nutr.. 2001;85(Suppl. 2):S109-S113.
Lieschke G.J., Burges A.W. Granulocyte colony-stimulating factor and granulocyte–macrophage colony-stimulating factor. N. Engl. J. Med.. 1992;327:1-35. 99–106
Mohle R., Kanz L. Hematopoietic growth factors for hematopoietic stem cell mobilization and expansion. Semin. Hematol.. 2007;44:193-202.
Charache S., Terrin M.L., Moore R.D., et al. Effect of hydroxyurea on the frequency of painful crises in sickle-cell-anemia. N. Engl. J. Med.. 1995;332:1317-1322. (Important randomised, controlled trial evidence of efficacy and safety over mean follow-up of 21 months)
Hillmen P., Young N.S., Schubert J., et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med.. 2006;355:1233-1243. (Eculizumab is an effective therapy for PNH)
Platt O.S. Hydroxyurea for the treatment of sickle cell anaemia. N. Engl. J. Med.. 2008;358:1362-1369. (Clinical vignette and discussion of this form of treatment)
1Each pregnancy ‘costs’ the mother 680 mg of iron, equivalent to 1300 ml of blood, owing to the demands of the fetus, plus requirements of the expanded blood volume and blood loss at delivery.
2‘Bronze diabetes’—where chronic iron overload is treated by repeated venesection, one of the few modern uses of this once near-universal ‘remedy’.
3‘Combined’ because the lateral as well as the dorsal columns are involved, giving rise to motor as well as sensory symptoms.
4At least in Anglo-Saxon countries; in France, very large doses of vitamin B12 are given by mouth to achieve sufficient absorption for therapeutic efficacy despite the absence of intrinsic factor. Either method is a great improvement on eating the prodigious quantities of raw liver required by Minot and Murphy’s ‘liver diet’ of 1925!
5The first therapeutic agent to be produced by recombinant technology, by Amgen in 1989—a huge commercial success, heralding the emergence of the new biotechnology industry.