5 Cell proliferation, apoptosis, repair and regeneration
Overview
This chapter deals with cell proliferation, apoptosis, repair and regeneration and how these relate to the actions of drugs dealt with in this book. About 10 billion new cells are manufactured in the body daily through cell division—an output that must be counterbalanced by the elimination of a similar number of cells. We deal first with the changes that occur within an individual cell when, after stimulation by growth factors, it gears up to divide into two daughter cells. We then consider the interaction of cells, growth factors and the extracellular matrix in cell proliferation. We describe the phenomenon of apoptosis (the programmed series of events that lead to cell death), outlining the changes that occur in a cell that is preparing to die, and the intracellular pathways that lead to its demise. We consider how these processes relate to the repair of damaged tissue and the possibility of its regeneration. Lastly, we consider the pathophysiological significance of these events, and implications for the potential development of clinically useful drugs.
Cell Proliferation
Cell proliferation is involved in many physiological and pathological processes including growth, healing, repair, hypertrophy, hyperplasia and the development of tumours. Angiogenesis (the development of new blood vessels) necessarily occurs during many of these processes.
Proliferating cells go through what is termed the cell cycle, during which the cell replicates all its components and then bisects itself into two identical daughter cells. Important components of the signalling pathways in proliferating cells are receptor tyrosine kinases or receptor-linked kinases, and the mitogen-activated protein kinase (MAP kinase) cascade (see Ch. 3). In all cases, the pathways eventually lead to transcription of the genes that control the cell cycle.
The Cell Cycle
The cell cycle is an ordered series of events consisting of several sequential phases (Fig. 5.1). These are:
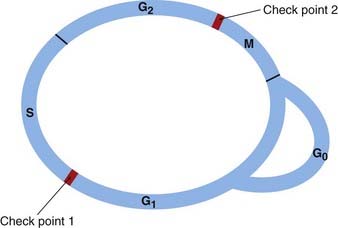
In cells that are dividing continuously, G1, S and G2 comprise interphase—the phase between one mitosis and the next.
Cell division requires the controlled timing of two critical events of the cell cycle: S phase (DNA replication) and M phase (mitosis). Entry into each of these phases is closely regulated, and there are two ‘check points’ (restriction points) in the cycle at the start of S and M, respectively. DNA damage results in the cycle being stopped at one or other of these. The integrity of the check points is critical for the maintenance of genetic stability and failure of the check points to stop the cycle when it is appropriate to do so is a hallmark of cancer.
In the adult, most cells do not constantly divide; most spend a varying amount of time in a quiescent phase outside the cycle in the phase termed G0 (Fig. 5.1). Neurons and skeletal muscle cells spend all their lifetime in G0; bone marrow cells and the lining cells of the gastrointestinal tract divide daily.
Quiescent cells can be activated into G1 by chemical stimuli associated with damage; for example, a quiescent skin cell can be stimulated by a wound into dividing and repairing the lesion. The impetus for a cell to start off on the cell cycle (i.e. to move from G0 into G1) can be provided by several stimuli, the most important being growth factors acting on growth factor receptors, though the action of ligands on G-protein-coupled receptors (see Ch. 3) can also stimulate the cell to embark on the cell cycle.
Growth factors stimulate the production of signals of two types:
The maintenance of normal cell numbers in tissues and organs requires that there be a balance between the positive regulatory forces and the negative regulatory forces. Apoptosis also has a role in the control of cell numbers (see below).
Positive Regulators of the Cell Cycle
The cycle starts when a growth factor acts on a quiescent cell, provoking it to divide. Growth factors stimulate production of the cell cycle regulators, which are coded for by the delayed response genes.
Two families of proteins, cyclins and cyclin-dependent kinases (cdks), control progress through the cycle. The cdks, functioning sequentially, phosphorylate various proteins (e.g. enzymes)—activating some and inhibiting others—to coordinate their activities.
Each cdk is inactive until it binds to a cyclin, the binding enabling the cdk to phosphorylate the protein(s) necessary for a particular step in the cycle. It is the cyclin that determines which protein(s) is phosphorylated. After the phosphorylation event has taken place, the cyclin is degraded (Fig. 5.2) by the ubiquitin/protease system. This involves several enzymes acting sequentially to add small molecules of ubiquitin to the cyclin, with the resulting ubiquitin polymer acting as an ‘address label’ that directs the cyclin to the proteasome where it is degraded.
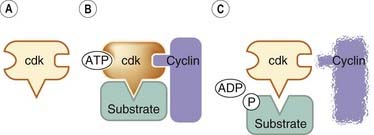
Fig. 5.2 Schematic representation of the activation of a cyclin-dependent kinase (cdk).
[A] An inactive cdk. [B] The inactive cdk is activated by being bound to a cyclin; it can now phosphorylate a protein substrate (e.g. an enzyme). [C] After the phosphorylating event, the cyclin is degraded.
There are eight main groups of cyclins. Those important in the control of the cell cycle are cyclins A, B, D and E. Each cyclin is associated with and activates a particular cdk. Cyclin A activates cdks 1 and 2; cyclin B, cdk 1; cyclin D, cdks 4 and 6; and cyclin E, cdk 2. Precise timing of each activity is essential, and many cycle proteins are degraded after they have carried out their functions. The actions of the cyclin/cdk complexes in the cell cycle are depicted in Figure 5.3.
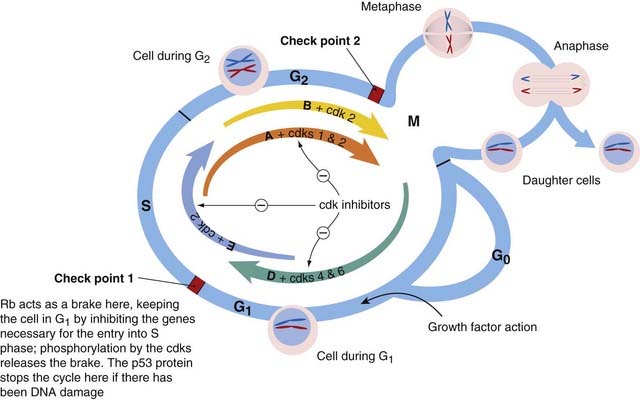
Fig. 5.3 Schematic diagram of the cell cycle, showing the role of the cyclin/cyclin-dependent kinase (cdk) complexes.
The processes outlined in the cycle occur inside a cell such as the one shown in Figure 5.4. A quiescent cell (in G0 phase), when stimulated to divide by growth factors, is propelled into G1 phase and prepares for DNA synthesis. Progress through the cycle is determined by sequential action of the cyclin/cdk complexes—depicted here by coloured arrows, the arrows being given the names of the relevant cyclins: D, E, A and B. The cdks are given next to the relevant cyclins. The thickness of each arrow represents the intensity of action of the cdk at that point in the cycle. The activity of the cdks is regulated by cdk inhibitors. If there is DNA damage, the products of the tumour suppressor gene p53 stop the cycle at check point 1, allowing for repair. If repair fails, apoptosis (see Fig. 5.5) is initiated. The state of the chromosomes is shown schematically in each G phase—as a single pair in G1, and each duplicated and forming two daughter chromatids in G2. Some changes that occur during mitosis (metaphase, anaphase) are shown in a subsidiary circle. After the mitotic division, the daughter cells may enter G1 or G0 phase. Rb, retinoblastoma gene.
The activity of these cyclin/cdk complexes is modulated by various negative regulatory forces (considered below), most of which act at one or other of the two check points.
In quiescent G0 cells, cyclin D is present in low concentration, and an important regulatory protein—the Rb protein1—is hypophosphorylated. Hypophosphorylated Rb holds the cell cycle in check at check point 1 by inhibiting the expression of several proteins critical for cell cycle progression. The Rb protein accomplishes this by binding to transcription factors, which control the expression of the genes that code for cyclins E and A, for DNA polymerase, for thymidine kinase, for dihydrofolate reductase, etc.—all essential for DNA replication during S phase.
Growth factor action on a cell in G0 propels it into G1, the phase in which the cell is preparing for S phase by synthesising the messenger RNAs and proteins needed for DNA replication.
During G1, the concentration of cyclin D increases and the cyclin D/cdk complex phosphorylates and activates the necessary proteins.
In mid-G1, the cyclin D/cdk complex phosphorylates the Rb protein, releasing a transcription factor that activates the genes for the components essential for the next phase—DNA synthesis.The action of the cyclin E/cdk complex is necessary for transition from G1 to S phase, i.e. past check point 1.
Once past check point 1, into the S-phase, the processes that have been set in motion cannot be reversed, and the cell is committed to continue with DNA replication and mitosis. Cyclin E/cdk and cyclin A/cdk regulate progress through S phase, phosphorylating and thus activating proteins/enzymes involved in DNA synthesis.
In G2 phase, the cell, which now has double the number of chromosomes, must duplicate all other cellular components for allocation to the two daughter cells. Synthesis of the necessary messenger RNAs and proteins occurs.
Cyclin A/cdk and cyclin B/cdk complexes are active during G2 phase and are necessary for entry into M phase, i.e. for passing check point 2. The presence of cyclin B/cdk complexes in the nucleus is required for mitosis to commence.
Mitosis occurs in four stages:
During metaphase, the cyclin A and B complexes phosphorylate cytoskeletal proteins, histones and possibly components of the spindle (the microtubules along which the chromatids are pulled during metaphase).
Negative Regulators of the Cell Cycle
One of the main negative regulators is the Rb protein (see above) that—while it is hypophosphorylated—holds the cycle in check. Inhibitors of the cdks also serve as negative regulators, their main action being at check point 1.
There are two families of inhibitors:
The action of p21 serves as an example of the role of a cyclin/cdk inhibitor. Protein p21 is under the control of the p53 gene—a particularly important negative regulator which is relevant in carcinogenesis—that operates at check point 1.
Inhibition of the cycle at check point 1
The p53 gene has been called the ‘guardian of the genome’. It codes for a transcription factor—the p53 protein. In normal healthy cells, the steady-state concentration of the p53 protein is low. But when there is DNA damage, the protein accumulates and activates the transcription of several genes, one of which codes for p21. Protein p21 inactivates cyclin/cdk complexes, thus preventing Rb phosphorylation, which means that the cycle is arrested at check point 1. This allows for DNA repair. If the repair is successful, the cycle proceeds past check point 1 into S phase. If the repair is unsuccessful, the p53 gene triggers apoptosis—cell suicide (see below).
Inhibition of the cycle at check point 2
DNA damage can result in the cycle being stopped at check point 2, but the mechanisms involved are poorly understood. Inhibition of the accumulation of cyclin B/cdk complex in the nucleus seems to be a factor.
For more detail on the control of the cell cycle, see under MicroRNAs (below) and Swanton (2004).
The cell cycle 
Interactions between Cells, Growth Factors and the Extracellular Matrix
During cell proliferation, there is integrated interplay between growth factors, cells, the extracellular matrix (ECM), and the matrix metalloproteinases (MMPs, see below). The ECM supplies the supporting framework for the cells and is secreted by the cells themselves. It also profoundly influences cell behaviour through the cell’s integrins (see below). Matrix expression is regulated by the action on the cell of growth factors and cytokines (see Verrecchia & Mauviel, 2007; Järveläinen et al., 2009). The activation status of some growth factors is, in turn, determined by the matrix, because they are sequestered by interaction with matrix components and released by enzymes (e.g. MMPs) secreted by the cells.
The action of growth factors—which act through receptor tyrosine kinases or receptor-coupled kinases (see Ch. 3) initiating the cell cycle—is a fundamental part of these processes. There are numerous growth factors, important examples being fibroblast growth factor (FGF), epidermal growth factor (EGF), platelet-dependent growth factor (PDGF), vascular endothelial growth factor (VEGF) and transforming growth factor (TGF)-β.
The main components of the extracellular matrix are:
Other proteins in the ECM are thrombospondin (Ch. 24) and osteopontin (Ch. 35) which are not structural elements but modulate cell–matrix interactions and repair processes. The production of the ECM components is regulated by growth factors, particularly transforming growth factor-β (TGF-β).
 Until recently, the importance of the ECM in drug action has been overlooked. Both beneficial and adverse effects of some drugs are due to effects on the ECM. Thus glucocorticoids decrease collagen synthesis in chronic inflammation, cyclo-oxygenase (COX)-2 inhibitors can modify fibrotic processes through a proposed action on TGF-β and statins can decrease fibrosis by inhibiting angiotensin-induced connective tissue growth factor production (Rupérez et al., 2007). The action of statins (see Ch. 23) in reducing circulating MMPs and decreasing MMP expression may contribute to their effects in cardiovascular diseases (Tousoulis et al., 2009). The adverse actions of some drugs attributable to an effect on the ECM include the osteoporosis and skin thinning caused by glucocortoicoids (discussed in Järveläinen et al., 2009). The ECM is also an important target in the search for new drugs.
Until recently, the importance of the ECM in drug action has been overlooked. Both beneficial and adverse effects of some drugs are due to effects on the ECM. Thus glucocorticoids decrease collagen synthesis in chronic inflammation, cyclo-oxygenase (COX)-2 inhibitors can modify fibrotic processes through a proposed action on TGF-β and statins can decrease fibrosis by inhibiting angiotensin-induced connective tissue growth factor production (Rupérez et al., 2007). The action of statins (see Ch. 23) in reducing circulating MMPs and decreasing MMP expression may contribute to their effects in cardiovascular diseases (Tousoulis et al., 2009). The adverse actions of some drugs attributable to an effect on the ECM include the osteoporosis and skin thinning caused by glucocortoicoids (discussed in Järveläinen et al., 2009). The ECM is also an important target in the search for new drugs.
The Role of Integrins
Integrins are transmembrane kinase-linked receptors (see Ch. 3), with α and β subunits that on interaction with the ECM elements outside the cell (e.g. fibronectin) mediate various cell responses, such as cytoskeletal rearrangement (not considered here) and co-regulation of growth factor function. Intracellular signalling by both growth factor receptors and integrins is important for optimal cell proliferation (Fig. 5.4). Integrin stimulation activates an intracellular transduction pathway, which, through an adapter protein and an enzyme (focal adhesion kinase), can activate the kinase cascade that forms part of the growth factor signalling pathway. Cross-talk between the integrin and growth factor pathways occurs by several other means as well (Streuli & Akhtar, 2009). Autophosphorylation of growth factor receptors (Ch. 3) is enhanced by integrin activation, and integrin-mediated adhesion to the extracellular matrix (Fig. 5.4) not only suppresses the concentrations of cdk inhibitors, but is required for the expression of cyclins A and D, and therefore for the progression of the cell cycle. Furthermore, integrin action stimulates apoptosis-inhibiting signals (see below), further facilitating growth factor action. See reviews by Gamberg et al., 2009 and Barczyk et al., 2010.
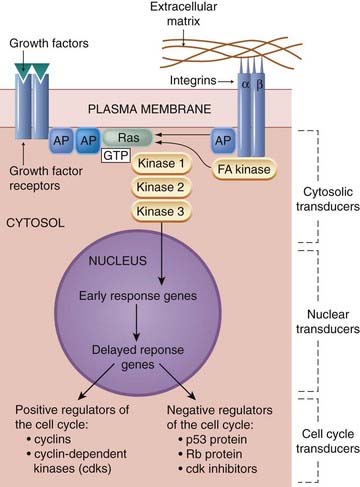
Fig. 5.4 Simplified diagram of the effect of growth factors on a cell in G0.
The overall effect of growth factor action is the generation of the cell cycle transducers. A cell such as the one depicted will then embark on G1 phase of the cell cycle. Most growth factor receptors have integral tyrosine kinase (see Fig. 3.15). These receptors dimerise (form pairs), then phosphorylate each other’s tyrosine residues. The early cytosolic transducers include proteins that bind to the phosphorylated tyrosine residues. Optimum effect requires cooperation with integrin action. Integrins (which have α and β subunits) connect the extracellular matrix with intracellular signalling pathways and also with the cell’s cytoskeleton (not shown here). G-protein-coupled receptors can also stimulate cell proliferation, because their intracellular pathways can connect with the Ras/kinase cascade (not shown). AP, adapter protein; FA kinase, focal adhesion kinase; Rb, retinoblastoma.
Several recently-introduced monoclonal antibodies are targeted at integrins, including natalizumab, used for multiple sclerosis (Baker & Hagg, 2007) and abciximab, an antithrombotic (Ch. 24).
The Role of Matrix Metalloproteinases
Degradation of the extracellular matrix by metalloproteinases is necessary during the growth, repair and remodelling of tissues. These enzymes are secreted as inactive precursors by local cells. When growth factors stimulate a cell to enter the cell cycle, they also stimulate the secretion of metalloproteinases, which then sculpt the matrix—producing the local changes necessary for the resulting increase in cell numbers. Metalloproteinases in turn play a part in releasing growth factors from the matrix as described above and, in some cases (e.g. interleukin [IL]-1β), in processing them from precursor to active form.
The action of these enzymes is regulated by TIMPS (tissue inhibitors of metalloproteinases), which are also secreted by local cells.
In addition to the physiological function outlined above, metalloproteinases are involved in the tissue destruction that occurs in various diseases, such as rheumatoid arthritis, osteoarthritis, periodontitis, macular degeneration and myocardial restenosis. They also have a critical role in the growth, invasion and metastasis of tumours, etc. See reviews by Clark et al. (2008), Skiles et al. (2004) and Marastoni et al. (2008). Much effort has gone into developing synthetic MMP inhibitors for treating cancers and inflammatory disorders, but clinical trials so far have shown limited efficacy and significant adverse effects (see Fingleton, 2008). Doxycycline, an antibiotic, also inhibits MMPs, and is used experimentally for this purpose.
Interactions between cells, growth factors and the matrix
Angiogenesis
Angiogenesis, which normally accompanies cell proliferation, is the formation of new capillaries from existing small blood vessels, without which new tissues, including tumours, cannot grow. Angiogenic stimuli, in the context of cell proliferation, include the action of various growth factors and cytokines, in particular vascular endothelial growth factor (VEGF). The sequence of events is as follows:
A monoclonal antibody, bevacizumab, directed against VEGF, is used as adjunct treatment for various cancers (see Ch. 55), and also, by injection into the eye, to treat age-related macular degeneration, a condition in which retinal blood vessels proliferate, causing blindness.
Apoptosis and Cell Removal
Apoptosis is cell suicide by a built-in self-destruct mechanism consisting of a genetically programmed sequence of biochemical events. It is thus unlike necrosis, which is disorganised disintegration of damaged cells resulting in products that trigger the inflammatory response. For a detailed review see Aslan & Thomas (2009).
Apoptosis plays an essential role in embryogenesis, helping to shape organs during development by eliminating cells that have become redundant. It is the mechanism that each day unobtrusively removes 10 billion cells from the human body. It is involved in numerous physiological events: the shedding of the intestinal lining, the death of time-expired neutrophils and the turnover of tissues as the newborn infant grows to maturity. It is the basis for the development of self-tolerance in the immune system (Ch. 6) and acts as a first-line defence against carcinogenic mutations by purging cells with abnormal DNA that could become malignant.
Disturbed apoptosis is also implicated in the pathophysiology of many conditions. Conditions associated with excessive apoptosis include:
Examples of defective apoptosis include:
Apoptosis is particularly important in the regulation of the immune response and in the many conditions in which it is an underlying component. There is recent evidence that T cells have a negative regulatory pathway controlled by surface programmed cell death receptors (e.g. the PD-1 receptor), and that there is normally a balance between the stimulatory pathways triggered by antigens and this negative regulatory apoptosis-inducing pathway. The balance is important in the maintenance of peripheral tolerance. A disturbance of this balance is seen in autoimmune disease, in the ‘exhaustion’ of T cells in chronic viral diseases such as HIV, and possibly in tumour escape from immune destruction (Zha et al., 2004).
Apoptosis is a default response, i.e. continuous active signalling by tissue-specific trophic factors, cytokines and hormones, and cell-to-cell contact factors (adhesion molecules, integrins, etc.) may be required for cell survival and viability, and the self-destruct mechanism is automatically triggered unless it is actively and continuously inhibited by these anti-apoptotic factors. Different cell types require differing sets of survival factors, which function only locally. If a cell strays or is dislodged from the area where its paracrine survival signals operate, it will die.
Withdrawal of these cell survival factors—which has been termed ‘death by neglect’—is not the only pathway to apoptosis (see Fig. 5.5). The death machinery can be activated by ligands that stimulate death receptors (‘death by design’) and by DNA damage. But it is generally accepted that cell proliferation processes and apoptosis are tightly connected (see below).
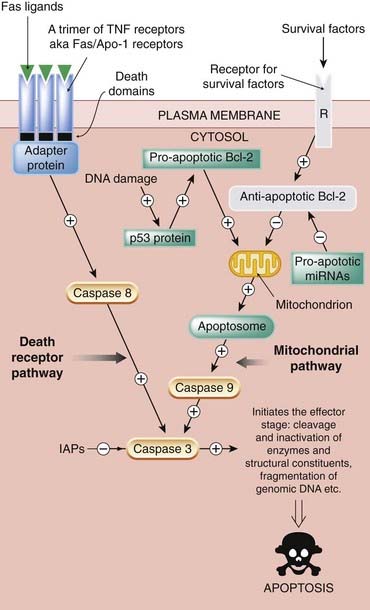
Fig. 5.5 Simplified diagram of the two main signalling pathways in apoptosis.
The death receptor pathway is activated when death receptors such as members of the tumour necrosis factor (TNF) family are stimulated by specific death ligands. This recruits adapter proteins that activate initiator caspases (e.g. caspase 8), which in turn activate effector caspases such as caspase 3. The mitochondrial pathway is activated by diverse signals, one being DNA damage. In the presence of DNA damage that cannot be repaired, the p53 protein (see text and Figs 5.3 and 5.4) activates a subpathway that results in release of cytochrome c from the mitochondrion, with subsequent involvement of the apoptosome and activation of an initiator caspase, caspase 9. The apoptosome is a complex of procaspase 9, cytochrome c and apoptotic-activating protease factor-1 (Apaf-1). Both these pathways converge on the effector caspase (e.g. caspase 3), which brings about the demise of the cell. The survival factor subpathway normally holds apoptosis at bay by inhibiting the mitochondrion pathway through activation of the antiapoptotic factor Bcl-2. The receptor labelled ‘R’ represents the respective receptors for trophic factors, growth factors, cell-to-cell contact factors (adhesion molecules, integrins), etc. Continuous stimulation of these receptors is necessary for cell survival/proliferation. If this pathway is non-functional (as depicted here by being shown in grey), this antiapoptotic drive is withdrawn. IAP, inhibitor of apoptosis.
Morphological Changes in Apoptosis
As the cell dies it rounds up, the chromatin condenses into dense masses, the cytoplasm shrinks, there is blebbing of the plasma membrane, and finally, by the action of a family of proteolytic enzymes known as caspases (see below), there is transformation of the cell into a cluster of membrane-bound entities, the corpse of the cell, which display ‘eat me’ signals—surface exposure of phosphatidylserine, etc. Macrophages recognise these signals and phagocytose the remains. The fact that the remains are membrane bound is important because release of the internal cell constituents could trigger an unwanted inflammatory reaction. An additional safeguard against this is that macrophages engaged in the clearance of the cell corpses release anti-inflammatory mediators such as TGF-β and IL-10.
The Major Players in Apoptosis
The repertoire of reactions in apoptosis is extremely complex and can vary not only between species but between cell types. Yet it could be that the pivotal reaction(s) that lead to either cell survival or cell death are controlled by a single gene or combination of genes. If so, these genes could be attainable targets in the development of drugs for many proliferative diseases. The use of gene silencing by RNA interference (RNAi) technology permits very efficient and precise block of gene expression (see Ch. 59) and is being used to identify antiapoptotic genes.
Only a simple outline of the complex apoptotic repertoire of reactions can be given here. The major players are the caspases—a family of cysteine proteases present in the cell in inactive form. These undertake delicate protein surgery, selectively cleaving a specific set of target proteins (enzymes, structural components), inactivating some and activating others. A cascade of about nine different caspases takes part in bringing about apoptosis, some functioning as initiators that transmit the initial apoptotic signals, and some being responsible for the final phase of cell death (Fig. 5.5).
The executioner caspases (e.g. caspase 3) cleave and inactivate cell constituents such as the DNA repair enzymes, protein kinase C, and cytoskeletal components. A DNAase is activated and cuts genomic DNA between the nucleosomes, generating DNA fragments of approximately 180 base pairs.
Besides the caspases, another pathway involves a protein termed apoptotic initiating factor (AIF) that is released from the mitochondria, enters the nucleus and triggers cell suicide.
Not all caspases are death-mediating enzymes; some have a role in the processing and activating of cytokines (e.g. caspase 8 is active in processing the inflammatory cytokines IL-1 and IL-18).
Pathways to Apoptosis
There are two main routes to cell death, one involving stimulation of death receptors by external ligands, and one arising within the cell and involving the mitochondria. Both these routes activate initiator caspases and both converge on a final common effector caspase pathway.
The Death Receptor Pathway
Lurking in the plasma membrane of most cell types are members of the tumour necrosis factor receptor (TNFR) superfamily (also known as Fas receptors), which function as death receptors (Fig. 5.5). Important family members are TNFR-1 and CD95 (also known as Fas ligands or Apo-1), but there are many others (e.g . PD-1, a death receptor that can be induced on activated T cells, as discussed above).
Each receptor has a ‘death domain’ in its cytoplasmic tail. Stimulation of the receptors by an external ligand such as tumour necrosis factor (TNF) itself or TRAIL2 causes them to get together in threes (trimerise), and recruit an adapter protein that complexes with the trimer by associating with the death domains. The resulting complex activates caspase 8, an initiator caspase that in turn activates the effector caspases (Fig. 5.5).
The Mitochondrial Pathway
This pathway can be called into action in two principal ways: by DNA damage and by withdrawal of the action of cell survival factors.
In the presence of DNA damage that cannot be repaired, the p53 protein activates a subpathway involving the p21 protein (see above) and proapoptotic members of the Bcl-2 protein family—Bid, Bax and Bak. In addition to these proapoptotic individuals, this family has antiapoptotic members (e.g. Bcl-2 itself, the first of these regulators to be discovered).3 They meet at the surface of mitochondria and compete with each other. The proapoptotic branch of the family (e.g. Bax) promotes release of cytochrome c from the mitochondria; the antiapoptotic branch inhibits this. The released cytochrome c complexes with a protein termed Apaf-1 (apoptotic protease-activating factor-1), and the two then combine with procaspase 9 and activate it. This latter enzyme orchestrates the effector caspase pathway. The three-party composite of cytochrome c, Apaf-1 and procaspase 9 is termed the apoptosome (Fig. 5.5). See Riedl & Salvesen (2007).
Nitric oxide (see Ch. 20) is another mediator that can have proapoptotic and antiapoptotic actions.
In normal cells, survival factors (specified above) continuously activate antiapoptotic mechanisms, and the withdrawal of survival factors can cause death in several different ways depending on the cell type. But a common mechanism is a tipping of the balance between Bcl-2 family members leading to loss of the stimulation of antiapoptotic protein action, with resultant unopposed action of the proapoptotic Bcl-2 proteins (see Fig. 5.5).
The two main pathways to cell death are connected to each other, in that caspase 8 in the death receptor pathway can activate the proapoptotic Bcl-2 and thus activate the mitochondrial pathway.
MicroRNAs, the cell cycle and apoptosis
MicroRNAs (miRNAs), discovered only in the past decade, are a family of small non-coding RNAs present in the genomes of plants and animals and now known to inhibit the expression of genes coding for cell cycle regulation, apoptosis (Fig 5.5), cell differentiation and development (Carleton et al., 2007; Lynam-Lennon et al., 2009). About 3% of human genes encode for miRNA and it is proposed that up to 30% of human genes coding for proteins are regulated by miRNAs. Altered miRNA expression is now believed to be linked to a variety of diseases including diabetes, obesity, Alzheimer’s, cardiovascular system diseases, inflammatory conditions, neurodegenerative diseases (Barbato et al., 2009) and various cancers (Wurdinger & Costa, 2007). Dysregulation of miRNA is believed to be involved in carcinogenesis, metastasis and resitance to cancer therapies (Garzon et al., 2009). There is in fact evidence that miRNAs are also believed to function as oncogenes and/or tumour suppressor genes and to regulate T cells (Zhou et al., 2009). Not surprisingly, miRNAs are being regarded as targets for new drug development for a variety of disease states (Liu et al., 2008; Stenvang et al., 2008; Tsai & Yu, 2010).
Apoptosis
Pathophysiological Implications
As mentioned above, cell proliferation and apoptosis are involved in many physiological and pathological processes. These are:
The role of cell proliferation and apoptosis in the first two processes listed is self evident and needs no further comment, and their involvement in immune tolerance is discussed briefly above. But the other processes need further comment.
Repair and Healing
Repair occurs when there has been damage or loss of tissue; it is also implicated in the resolution of the local inflammatory reaction to a pathogen or chemical irritant. In some instances, damage or tissue loss can lead to regeneration, which is quite different to repair and is considered separately below.
In repair and healing, there is an ordered series of events involving cell migration, angiogenesis, proliferation of connective tissue cells, synthesis of extracellular matrix and finally remodelling—all coordinated by the growth factors and cytokines that are relevant for the particular tissue involved. TGF-β is a key cytokine in several of these processes.
There is considerable overlap between the inflammatory reaction and repair in terms of the cells and mechanisms activated.
Repair, healing and regeneration
Hyperplasia
Hyperplasia (cell proliferation and matrix expansion) are hallmarks of chronic inflammatory, hypersensitivity and autoimmune diseases such as rheumatoid arthritis (Chs. 6, 17 & 26), psoriasis, chronic ulcers, chronic obstructive lung disease, the processes underlying the bronchial hyperreactivity of chronic asthma (Ch. 27) and glomerular nephritis. The cells that take part and the events themselves are described in more detail in Chapter 6.
Cell proliferation and apoptotic events are also implicated in atherosclerosis (Ch. 23), restenosis and myocardial repair after infarction (Ch. 21).
The Growth, Invasion and Metastasis of Tumours
Perturbations in the growth factor signalling pathways, the antiapoptotic pathways and the function of the cell cycle controllers have an important role in the pathogenesis of malignancy. New understanding of this is leading to novel approaches to the treatment of cancer. See below and Chapter 55.
Stem Cells and Regeneration
Regeneration after damage or tissue loss implies restitution or replacement of the area so that it is identical to what was there before.
Many animals (e.g. amphibians and other lower orders) have an impressive power to regenerate their tissues, even to regrow an organ such as a limb. The essential process is the activation of stem cells—undifferentiated cells that have the potential to develop into any or most of the specialised cells in the body. Amphibians have a plentiful supply of these primitive cells in their organs and, furthermore, many of their specialised cells can dedifferentiate to become stem cells. These stem cells then multiply and retrace the pathways that generated the organ (e.g. a limb) during fetal life, proliferating again and again and eventually differentiating into the various cell types needed to replace the missing part.
However, during evolution, mammals have lost this ability and now have regenerative capacity in only a few tissues. Blood cells, intestinal epithelium and the outer layers of the skin are replaced continuously throughout life. Of the more discrete organs, there is a low degree of turnover and replacement of cells in such organs as liver, kidney and bone. This is in essence physiological renewal and is effected by local tissue-specific stem cells.
Almost alone, the liver has significant ability to replace itself if much of it is removed. It can regenerate to its original size in a remarkably short time, provided that at least 25% has been left intact.4 And the mature parenchymal liver cells participate in this process as well as all the other cellular components of the liver.
Although stem cells are known to exist in most tissues in adult mammals, they are very few in number, the vast majority of cells in most tissues being irreversibly differentiated. If a mammal is injured or its tissue is removed, repair processes—often with subsequent scarring—usually make good the damage. It seems that rapid closure of the defect after tissue loss (which is much more speedily accomplished by repair mechanisms) takes priority over regeneration.
Until recently, it was assumed that this was an unalterable situation, except for a few examples, some mentioned above. But recent work has suggested that it might be possible to activate in mammals the original regenerative pathways—at least to some extent and in some organs. Regeneration of a lost limb as happens in amphibians is manifestly not possible in humans, but regeneration of limited areas of a tissue or of a small part of an organ may well be feasible. For this to happen, it would be necessary to encourage some stem cells to proliferate, develop and differentiate at the relevant sites. Or—and this is a rather more remote prospect in humans—to persuade some local specialised cells to dedifferentiate. This can occur in some mammals under special circumstances (see below). However, it may be that repair is the Janus face of regeneration, repair being an evolutionary trade-off in mammals for the lost power of regeneration.
Where are the relevant stem cells that could be coaxed into regenerative service? Various possibilities are being vigorously investigated and in some cases tested clinically. These include:
For a tissue such as the liver to regenerate, local tissue-specific stem cells must be stimulated by growth factors to enter the cell cycle and continue to proliferate. Other essential processes are:
Concomitant replacement of components of the lost connective tissue (fibroblasts, macrophages, etc.) would also be necessary.
Because most tissues do not regenerate spontaneously, mechanisms that could awaken the lost regenerative ability could be of immense value in numerous diseases. Two areas where recent progress has been reported include the regeneration of heart muscle after an infarction (Ch. 21) and replacement of insulin-secreting cells for the treatment of type I diabetes mellitus (Ch. 30).
Heart Muscle
Until recently it was assumed that cardiac muscle had no power to regenerate. But in a particular strain of mouse, when part of the heart is damaged by freezing, repair processes do not start up; instead, the area is replaced by regeneration within a few months. Regeneration of heart tissue also occurs in dogs after acute heart failure. Mitosis of myocytes is seen in the normal human heart, and proliferation of myocytes immediately after infarction has been reported. Indeed, the sequence of events described above has been shown to occur during the process of remodelling after myocardial infarction in rodents (Nian et al., 2004).
More recently, stem cell therapy has been shown to improve ventricular function in the failing heart (Gaetani et al., 2009) and to reduce infarct size and end systolic function in patients with myocardial infarction (Piepoli & Capucci, 2009).
Insulin-Secreting Cells
The results of ongoing clinical trials in patients with type I diabetes suggest that haemopoietic stem cell transplantation can remove the need for daily insulin injections (Voltarelli et al., 2007).
Therapeutic Prospects
Considerable effort is being expended on finding compounds that will inhibit or modify the processes described in this chapter. So far there are few in clinical use, the main examples being those mentioned earlier, but it is likely that such agents will figure strongly in the pharmacology of the next decade, much work being aimed at developing new drugs for cancer therapy. Theoretically, all the processes could constitute targets for new drug development. Here we concentrate on those approaches that are proving or are likely to prove fruitful.
Apoptotic Mechanisms
Compounds that could modify apoptosis are being intensively investigated (Melnikova & Golden, 2004; MacFarlane, 2009). Here we can only outline some of the more important approaches.
Drugs that promote apoptosis by various mechanisms were heralded as a potential new approach to cancer treatment, and are actively being studied, though none has yet been approved for clinical use. Potential proapoptotic therapeutic approaches need to be targeted precisely to the diseased tissue to avoid the obvious risks of damaging other tissues. Examples include the following:
Despite the appeal of inhibiting apoptosis as a means of preventing or treating a wide range of common degenerative disorders, success in developing inhibitors for clinical use has so far proved elusive, and a number of such compounds have been found to lack efficacy in clinical trials:
Angiogenesis and Metalloproteinases
Metalloproteinases and angiogenesis have critical roles in physiological (e.g. growth, repair) and pathological processes (e.g. tumour growth, chronic inflammatory conditions). The search for clinically useful MMP inhibitors is continuing, but has not so far been successful. At present, only one new drug has been approved for use in cancer treatment: the antiangiogenesis compound bevacizumab, a monoclonal antibody that acts against VEGF (see above) which is also used to treat age-related macular degeneration, a disease of the retina associated with excessive proliferation of retinal blood vessels.
Cell Cycle Regulation
The main endogenous positive regulators of the cell cycle are the cdks. Several small molecules that inhibit cdks by targeting the ATP-binding sites of these kinases have been developed; an example is flavopiridol, currently in clinical trials, which inhibits all the cdks, causing arrest of the cell cycle; it also promotes apoptosis, has antiangiogenic ability and can induce differentiation (Dickson & Schwartz, 2009).
Some compounds affect upstream pathways for cdk activation and may find uses in cancer treatment. Examples are perifosine (currently in development for cancer treatment) and lovastatin (a cholesterol-lowering drug, see Ch. 23, which may also have anticancer properties).
Bortezomib, a boronate compound, covalently binds the proteasome, inhibiting the degradation of proapoptotic proteins. It is used in treating multiple myeloma (see Ch. 55).
Of the various components of the growth factor signalling pathway, receptor tyrosine kinases, the Ras protein and cytoplasmic kinases have been the subjects of most interest. Kinase inhibitors recently introduced for cancer treatment include imatinib, gefitinib and erlotinib (see Ch. 55).
References and Further Reading
Cell cycle and apoptosis (general)
Ashkenasi A. Targeting death and decoy receptors of the tumour necrosis receptor superfamily. Nat. Rev. Cancer. 2002;2:420-429. (Exemplary review, comprehensive; good diagrams)
Aslan J.E., Thomas G. Death by committee: organellar trafficking and communication in apoptosis. Traffic. 2009;10:1390-1404.
Barbato C., Ruberti F., Cogoni C. Searching for MIND: microRNAs in neurodegenerative diseases. J. Biomed Biotechnol.. 2009. 2009, 871313
Barber D.L., Wherry E.J., Masopust D., et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682-687. (Antibody that blocks the interaction between the PD-1 receptor and its ligand reverses T-cell exhaustion in a chronic viral infection)
Carleton M., Cleary M.C., Linsley P.S. MicroRNAs and cell cycle regulation. Cell Cycle. 2007;6:2127-2132. (Describes how specific microRNAs act to control cell cycle check points)
Cummings J., Ward T., Ranson M., Dive C. Apoptosis pathway-targeted drugs—from the bench to the clinic. Biochim. Biophys Acta.. 2004;1705:53-66. (Good review discussing—in the context of anticancer drug development—Bcl-2 proteins, IAPs, growth factors, tyrosine kinase inhibitors and assays for apoptosis-inducing drugs)
Danial N.N., Korsmeyer S.J. Cell death: critical control points Cell 116:2004:205-219 (Definitive review of the biology and control of apoptosis; includes evidence from C. elegans, Drosophila and mammals)
Dickson M.A., Schwartz G.K. Development of cell-cycle inhibitors for cancer therapy. Curr. Oncol.. 2009;16:36-43. (Discusses drugs which target the cell cycle that have entered clinical trials)
Elmore S. Apoptosis: a review of programmed cell death. Toxicol. Pathol.. 2007;35:495-516. (A general overview of apoptosis including structural changes, biochemistry and the role of apoptosis in health and disease)
Garzon R., Calin G.A., Croce C.M. MicroRNAs in Cancer. Annu. Rev. Med.. 2009;60:167-179.
Giaccone G., Rajan A. Met amplification and HSP90 inhibitors. Cell Cycle. 2009;8:2682.
Hipfner D.R., Cohen S.M. Connecting proliferation and apoptosis in development and disease. Nat. Rev. Mol. Cell Biol.. 2004;5:805-811. (Extensive, detailed review)
Jannsen E.M., Droin N.M., Lemmens E.E. CD4+ T-cell-help controls CD4+ T cell memory via TRAIL-mediated activation-induced cell death Nature 434:2005:88-92(Control of TRAIL expression could explain the role of CD4+ T cells in CD8+ T-cell help)
Liu Z., Sall A., Yang D. MicroRNA: an emerging therapeutic target and intervention tool. Int. J. Mol. Sci.. 2008;9:978-999.
Lynam-Lennon N., Maher S.M., Reynolds J.V. The roles of microRNAs in cancer and apoptosis. Biol. Rev.. 2009;84:55-71. (Detailed review of the role of microRNAs in cell proliferation and cell death and their potential roles as oncogenes and tumour suppressor genes)
MacFarlane M. Cell death pathways—potential therapeutic targets. Xenobiotica. 2009;39:616-624. (Excellent up-to-date review with table of agents in early clinical trial)
Melnikova A., Golden J. Apoptosis-targeting therapies. Nat. Rev. Drug Discov.. 2004;3:905-906. (Crisp overview)
Riedl S.J., Salvesen G.S. The apoptosome: signalling platform of cell death. Nat. Rev. Mol. Cell Biol.. 2007;8:405-413. (Discusses the formation of the apoptosome and the activation of its effector, caspase-9)
Riedl S.J., Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol.. 2004;5:897-905. (Systematic review)
Rupérez M., Rodrigues-Díez R., Blanco-Colio L.M., et al. HMG-CoA reductase inhibitors decrease angiotensin II-induced vascular fibrosis: role of RhoA/ROCK and MAPK pathways. Hypertension. 2007;50:377-383.
Stenvang J., Lindow M., Kauppinen S. Targeting of microRNAs for therapeutics. Biochem. Soc. Trans.. 2008;36:1197-1200.
Swanton C. Cell-cycle targeted therapies. Lancet. 2004;5:27-36. (Definitive review of the protein families controlling the cell cycle, their alterations in malignancy and their potential as targets for new drugs)
Tousoulis D., Andreou I., Tentolouris C., et al. Comparative effects of rosuvastatin and allopurinol on circulating levels of matrix mettalloproteinases in patients with chronic heart failure. Int. J. Cardiol.. 2010;145:438-443.
Tsai L.M., Yu D. MicroRNAs in common diseases and potential therapeutic applications. Clin. Exp. Pharmacol. Physiol.. 2010;37:102-107.
Verrecchia F., Mauviel A. Transforming growth factor-beta and fibrosis. World J. Gastroenterol.. 2007;13:3056-3062.
Williams M.A., Bevan M.J. Exhausted T cells perk up. Nature. 2006;439:669-670. (Succinct article assesses the work on reversing T-cell exhaustion)
Wurdinger T., Costa F.F. Molecular therapy in the microRNA era. Pharmacogenomics J.. 2007;7:297-304.
Yang B.F., Lu Y.J., Wang Z.G. MicroRNAs and apoptosis: implications in molecular therapy of human disease. Clin. Exp. Pharmacol. Physiol.. 2009;36:951-960. (Comprehensive review of the apotosis-regulating miRNAs and apoptotic cell death)
Zha Y., Blank C., Gajewski T.F. Negative regulation of T-cell function by PD-1. Crit. Rev. Immunol.. 2004;24:229-237. (Article on the balance between stimulatory and inhibitory signalling and its relevance to self-tolerance and the pathogenesis of autoimmune diseases)
Zhou L., Seo K.H., Wong H.K., Mi Q.S. MicroRNAs and immune regulatory T cells. Int. Immunopharmacol. 2009;9:524-527.
Integrins, extracellular matrix, metalloproteinases and angiogenesis
Baker K.A., Hagg T. Developmental and injury-induced expression of alpha1beta1 and alpha6beta1 integrins in the rat spinal cord. Brain Res.. 2007;1130:54-66.
Barczyk M., Carracedo S., Gullberg D. Integrins. Cell Tissue Res.. 2010;339:269-280.
Clark I.M., Swingler T.E., Sampieri C.L., Edwards D.R. The regulation of matrix metalloproteinases and their inhibitors. Int. J. Biochem. Cell Biol.. 2008;40:1362-1378.
Fingleton B. MMPs as therapeutic targets—still a viable option? Semin. Cell Biol. Dev.. 2008;19:61-68. (Somewhat discouraging review of clinical trials data with MMP inhibitors)
Gahmberg C.G., Fagerholm S.C., Nurmi S.M., et al. 2009. Regulation of integrin activity and signalling. Biochim. Biophys Acta.. 1790:431-444. (Crisp review of the control of cell signalling by integrins)
Järveläinen H., Sainio A., Koulu M., Wight T.N., Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol. Rev.. 2009;61:198-223. (Comprehensive review of the role of the extracellular matrix [ECM] in the cellular events involved in proliferation and differentiation with discussion of the ECM as a potential target for new drug development)
Marastoni S., Ligresti G., Lorenzon E., Colombatti A., Mongiat M. Extracellular matrix: a matter of life and death. Connect. Tissue Res.. 2008;49:203-206. (Crisp analysis of the ECM and its role in cell survival, growth and proliferation)
Skiles J.W., Gonnella N.C., Jeng A.Y. The design, structure and clinical update of small molecular weight matrix metalloproteinase inhibitors. Curr. Med. Chem.. 2004;11:2911-2977. (Results of trials with early matrix metalloproteinases were disappointing; the authors discuss the proposed usefulness of matrix metalloproteinase inhibitors and review patented drugs)
Sternlicht M.D., Werb Z. How matrix metalloproteinases regulate cell behaviour. Annu. Rev. Cell Dev. Biol.. 2001;17:463-516. (Comprehensive review of the regulation of metalloproteinases, their regulation of cell signalling and their role in development and disease)
Streuli C.H., Akhtar N. Signal co-operation between integrins and other receptor systems. Biochem. J.. 2009;418:491-506. (Deals with integrin interaction with growth factors to regulate angiogenesis, their interplay with tyrosine kinases and with cytokine receptors)
Von Adrian U.H., Engelhardt B. a4 Integrins as therapeutic targets in autoimmune disease N. Engl. J. Med. 348:2003:68-72 (Describes physiological and pathological functions of a4 integrins [with good diagram], and a recombinant antibody against a4 integrins in clinical trial for multiple sclerosis and Crohn’s disease)
Stem cells, regeneration and repair
Aldhous P. How stem cell advances will transform medicine. New Scientist. 2008;2654:40-43. (Clear, simple article)
Gaetani R., Barile L., Forte E., et al. New perspectives to repair a broken heart. Cardiovasc Hematol. Agents Med. Chem.. 2009;7:91-107. (Discusses sources of cardiomyogenic cells and their potential for diseased or injured myocardium)
Huang N.F., Lam A., Fang Q., et al. Bone marrow-derived mesenchymal stem cells in fibrin augment angiogenesis in the chronically infarcted myocardium. Regen. Med.. 2009;4:527-538.
Nat. Rev. Drug Discov. August 2006. Vol. 5 has a series of articles on nerve regeneration. (The articles ‘highlight recent progress in knowledge of the molecular, cellular and circuitry level responses to injuries to the adult mammalian CNS, with a view to understanding the underlying mechanism that will enable the development of appropriate therapeutic strategies’)
Nian M., Lee P., Khaper N., Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ. Res.. 2004;94:1543-1553. (Excellent, forward-looking article on the possibility of using cytokines to improve healing and cardiac regeneration)
Nishikawa S., Goldstein R.A., Nierras C.R. The promise of human induced pluripotent stem cells for research and therapy. Nat. Rev. Mol. Cell Biol.. 2008;9:725-729. (Induced pluripotent stem cells [iPS] are human somatic cells that have reprogrammed to be pluripotent. Discusses the reprogramming of human somatic cells to become plutipotent. The reprogramming process is straightforward and has been replicated in several laboratories)
Piepoli M.F., Capucci A. Cardiac regeneration by progenitor cells: what is it known as and what is it still to be known as? Cardiovasc. Hematol. Agents Med. Chem.. 2009;7:127-136. (Intracoronary infusion of bone marrow mononuclear cells reduced infarct size and end systolic volumes in patients with acute myocardial infarction. The article discusses this therapeutic approach)
Rosenthal N. Prometheus’s vulture and the stem-cell promise. N. Engl. J. Med.. 2003;349:267-286. (Excellent article, entrancingly written; discusses the problem of regeneration of tissues and organs)
Sinanan A.C., Buxton P.G., Lewis M.P. Muscling in on stem cells. Biol. Cell. 2006;98:203-214.
Stapenbeck T.S., Miyoshi H. The role of stromal cells in tissue regeneration and wound repair. Science. 2009;26:1666-1669. (Succinct article on the possibility of mammalian stromal cells performing the same function as blastema cells in lower organisms)
Sylvester K.G., Longmaker M.T. Stem cells: review and update. Arch. Surg.. 2004;139:93-99. (Concise review)
Voltarelli J.C., Couri C.E., Stracieri A.B., et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA. 2007;297:1568-1576. (Successful early trial of stem cell transplantation)
Wilson C. The regeneration game. New Scientist. 2003;179:2414-2427. (Very readable article on the possibility of regeneration of mammalian tissues and organs)
1So named because mutations of the Rb gene are associated with retinoblastoma tumours.
2TRAIL is tumour necrosis factor-α–related apoptosis-inducing ligand, of course; what else? See Janssen et al. (2005) for discussion of a role of TRAIL. PD-L1, a ligand for the PD-1 receptor, is found on all haemopoietic cells and many other tissues.
3Another brake on the cell death mechanisms is a family of caspase-inhibiting proteins called IAPs (inhibitors of apoptosis proteins).
4There is an account of liver regeneration in Greek myths. Prometheus stole the secret of fire from Zeus and gave it to mankind. To punish him, Zeus had him shackled to a crag in the Caucasus, and every day an eagle tore at his flesh and devoured much of his liver. But during the night, it regenerated and in the morning was whole again. The legend doesn’t say whether the requisite 25% was left after the eagle had had its fill, and the regeneration described is unphysiologically speedy—rat liver takes 2 weeks or more to get back to the original size after 66% hepatectomy.