6 Cellular mechanisms
Host defence
Overview
Everyone has experienced an inflammatory episode at some time or other and will be familiar with the characteristic redness, swelling, heat, pain and loss of function that this generally entails. In this chapter we list the cellular players involved in the host defence response and explain the bare bones of this crucial and sophisticated mechanism; inflammatory mediators are considered separately in Chapter 17. Understanding these cellular responses and their functions provides an essential basis for understanding the actions of anti-inflammatory and immunosuppressant drugs—a major class of therapeutic agents (see Ch. 26).
Introduction
All living creatures are born into a universe that poses a constant challenge to their physical well-being and survival. Evolution, which has equipped us with homeostatic systems that maintain a stable internal environment in the face of changing external temperatures and fluctuating supplies of food and water, has also provided us with mechanisms for combating the ever-present threat of infection and for promoting healing and restoration to normal function in the event of injury. In mammals, this function is subserved by the innate and acquired (or adaptive) immune systems, working together with a variety of mediators and mechanisms that collectively gives rise to what we term inflammation. Generally this response acts to protect us, but occasionally it goes awry, leading to a spectrum of inflammatory diseases, and it is under these circumstances that we need to resort to drug therapy to dampen or abolish the inflammatory response.
The main functions of this host inflammatory response then are defence and repair—in other words, nothing less than the security of the organism. This response is crucial to survival. If it is defective either through genetic causes (e.g. leukocyte adhesion deficiency), infection with organisms that subvert its function (e.g. HIV) or because of immunosuppressant drug therapy, then the outcome can be very serious or even fatal.
The inflammatory response 
Like border security systems in the mundane world, the body has the cellular and molecular equivalents of guards, identity checks, alarm systems and a communication network with which to summon back-up when required. It also has access to an astonishing data bank that memorises precise details of previous illegal immigrants and prevents them from returning. In this discussion it is convenient to divide the host response into two components, although it should be recognised at the outset that these two systems work hand-in-hand. The two principal components are:
The Innate Immune Response
The innate response is activated immediately following infection or injury.1 It is a system that is present in virtually all organisms and some of the mammalian gene families that control these responses were first identified in plants and insects.
The innate immune response
Pathogen Recognition
One of the most important functions of any security system is the ability to establish identity. How does an organism decide whether a cell is a bona fide citizen or an invading pathogen? In the case of the innate response this is achieved through a network of pattern recognition receptors (PRRs), found in virtually all organisms. They recognise pathogen-associated molecular patterns (PAMPs), common products produced by bacteria, fungi, viruses and so on that these organisms could not readily change to evade detection. These receptors include G-protein-coupled receptors such as the FPR (formyl peptide receptor) family that recognises N-formylated peptides characteristic of bacterial protein synthesis (although these are also liberated from mitochondria during host cell death as well) and cytoplasmic receptors such as the NOD-like receptors (nucleotide-binding oligomerization domain-like receptors)—a large family of intracellular proteins that recognise fragments of bacterial proteoglycan.
Among the best-studied of these PRRs are the Toll-like receptors (TLRs). The Toll2 gene was first identified in Drosophila in the mid-1990s. Analogous genes were soon found in vertebrates and it was quickly established that as a family, their main job was to detect highly conserved components in pathogens and to signal their presence to the different components of the immune system.
There are approximately 15 TLRs known but only some 10 occur in mammals. They belong to the class of receptor tyrosine kinases (see Ch. 3), and are phylogenetically highly conserved. Unlike the antigen receptors on T and B cells that are generated somatically as the cells develop, endowing each lymphocyte clone with a structurally unique receptor, TLRs are encoded in the host DNA. Table 6.1 lists these receptors and the pathogenic products that are recognised, where these are known. There are two types of TLR, located respectively on the cell surface and in endosomes. The latter type generally recognises pathogen RNA/DNA (presumably because they appear in phagosomes), while the former recognises other pathogen components such as cell wall material, endotoxin, etc. Some TLRs also recognise ligands released when host cells are damaged (e.g. heat shock proteins). Presumably this provides an additional way of monitoring damage.
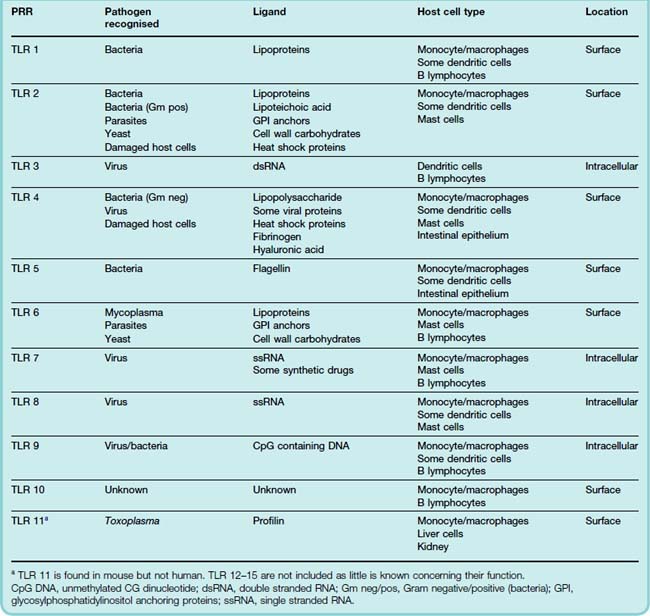
How a single family of receptors can recognise such a wide spectrum of different chemicals is a molecular mystery. Sometimes the problem is solved by recruiting additional ‘accessory’ binding proteins to assist this process. When activated, Toll receptors dimerise and initiate a complex signalling pathway that activates genes coding for proteins and factors crucial to the deployment of the inflammatory response, many of which we will discuss below. Interestingly from the pharmacological viewpoint, TLR 7 also recognises some synthetic antiviral compounds such as imidazoquinolones. The ability of these drugs to provoke TLR activation probably underlies their clinical effectiveness.
TLRs are strategically located on those ‘sentinel’ cells which are most likely to come into contact with pathogens in the first instance. These include mast cells, macrophages and dendritic cells, all of which are found in tissues throughout the body, as well as some intestinal epithelial cells (which are exposed to pathogens in the food that we eat) and other cells.
Having outlined how ‘non-self’ pathogens are detected by the innate immune system, we can now describe the events that follow the ‘raising of the alarm’.
Responses to Pattern Recognition
Vascular events
Interaction of a PAMP with TLRs triggers the sentinel cells to respond immediately by producing the main proinflammatory cytokines, tumour necrosis factor (TNF)-α and interleukin (IL)-1, as well as other mediators (such as prostaglandins and histamine) that act on the vascular endothelial cells of the postcapillary venules, causing expression of adhesion molecules on the intimal surface and an increase in vascular permeability.
White blood cells adhere to the endothelial cells through interactions between their cell surface integrins (see below) and adhesion molecules on endothelial cells. This enables them to migrate out of the vessels, attracted by chemotaxins generated by the microorganisms or as a result of their interaction with tissues (see below). Chemokines released during TLR activation play an important part in this. (Cytokines and chemokines are considered in Ch. 17.)
The initial vascular events include dilatation of the small arterioles, resulting in increased blood flow. This is followed by a slowing and eventually a stasis of blood, and an increase in the permeability of the postcapillary venules with exudation of fluid. The vasodilatation is brought about by mediators including histamine, prostaglandin (PG)E2 and PGI2 (prostacyclin) produced by the interaction of the microorganism with tissue, some of which act together with cytokines to increase vascular permeability.
The fluid exudate contains the components for four proteolytic enzyme cascades: the complement system, the coagulation system, the fibrinolytic system and the kinin system (see Fig. 6.1). The components of these cascades are proteases that are inactive in their native form but that are activated by proteolytic cleavage, each activated component then activating the next. The exudate is carried by lymphatics to local lymph nodes or lymphoid tissue, where the products of the invading microorganism trigger the adaptive phase of the response.
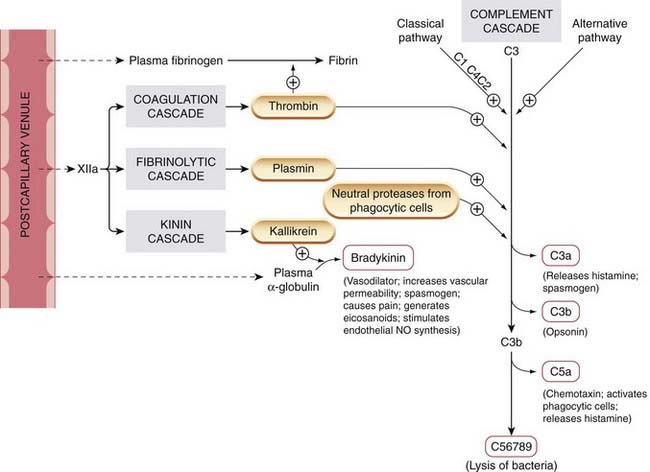
Fig. 6.1 Four enzyme cascades are activated when plasma leaks out into the tissues as a result of the increased vascular permeability of inflammation.
Factors causing exudation are depicted in Figure 6.2. Mediators generated are shown in red-bordered boxes. Complement components are indicated by C1, C2, etc. When plasmin is formed, it tends to increase kinin formation and decrease the coagulation cascade.
(Adapted from Dale M M, Foreman J C, Fan T-P (eds) 1994 Textbook of immunopharmacology, 3rd edn. Blackwell Scientific, Oxford.)
 The complement system comprises nine major components, designated C1 to C9. Activation of the cascade is initiated by substances derived from microorganisms, such as yeast cell walls or endotoxins. This pathway of activation is termed the alternative pathway (Fig. 6.1) as opposed to the classic pathway that is dealt with later. One of the main events is the enzymatic splitting of C3, giving rise to various peptides, one of which, C3a (termed an anaphylatoxin) stimulates mast cells to secrete further chemical mediators and can also directly stimulate smooth muscle, while C3b (termed an opsonin) attaches to the surface of a microorganism, facilitating ingestion by white blood cells. C5a, generated enzymatically from C5, also releases mediators from mast cells and is a powerful chemotactic attractant and activator of white blood cells.
The complement system comprises nine major components, designated C1 to C9. Activation of the cascade is initiated by substances derived from microorganisms, such as yeast cell walls or endotoxins. This pathway of activation is termed the alternative pathway (Fig. 6.1) as opposed to the classic pathway that is dealt with later. One of the main events is the enzymatic splitting of C3, giving rise to various peptides, one of which, C3a (termed an anaphylatoxin) stimulates mast cells to secrete further chemical mediators and can also directly stimulate smooth muscle, while C3b (termed an opsonin) attaches to the surface of a microorganism, facilitating ingestion by white blood cells. C5a, generated enzymatically from C5, also releases mediators from mast cells and is a powerful chemotactic attractant and activator of white blood cells.
The final components in the sequence, complement-derived mediators (C5 to C9) coalesce to form a ‘membrane attack complex’ that attaches to certain bacterial membranes, leading to lysis. Complement can therefore mediate the destruction of invading bacteria or damage multicellular parasites; however, it may sometimes cause injury to the host. The principal enzymes of the coagulation and fibrinolytic cascades, thrombin and plasmin, can also activate the cascade by hydrolysing C3, as can enzymes released from white blood cells.
The coagulation system and the fibrinolytic system are described in Chapter 24. Factor XII is activated to XIIa (e.g. by collagen), and the end product, fibrin, laid down during a host–pathogen interaction, may serve to limit the extent of the infection. Thrombin is additionally involved in the activation of the kinin (Fig. 6.1) and, indirectly, the fibrinolytic systems (see Ch. 24).
The kinin system is another enzyme cascade relevant to inflammation. It yields several mediators, in particular bradykinin (Fig. 6.1 and see below).
Cellular events
Of the cells involved in inflammation, some (e.g. vascular endothelial cells, mast cells, dendritic cells and tissue macrophages) are normally present in tissues, while other actively motile cells (e.g. leukocytes) gain access from the blood.
Polymorphonuclear leukocytes
Neutrophil polymorphs are the ‘shock troops’ of inflammation, and are the first of the blood leukocytes to enter an inflamed area (Fig. 6.2). The whole process is cleverly choreographed: under direct observation, the neutrophils may be seen first to roll along the activated endothelium, then to adhere and finally to migrate out of the blood vessel and into the extravascular space. This process is regulated by the successive activation of different families of adhesion molecules (selectins, intercellular adhesion molecule [ICAM] and integrins) on the inflamed endothelium that engage corresponding counter-ligands on the neutrophil, capturing it as it rolls along the surface, stabilising its interaction with the endothelial cells and enabling it to migrate out of the vessel (using a further adhesion molecule termed PECAM, platelet endothelium adhesion molecule). The neutrophil is attracted to the invading pathogen by chemicals termed chemotaxins, some of which (such as the tripeptide formyl-Met-Leu-Phe) are released by the microorganism, whereas others, such as C5a, are produced locally or released by nearby cells such as macrophages (e.g. chemokines such as IL-8).
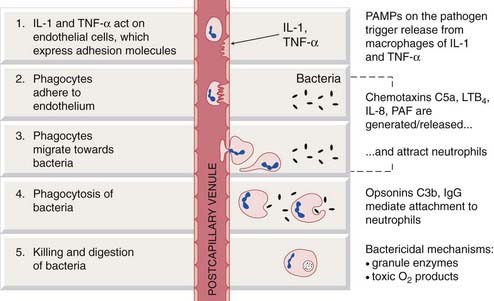
Fig. 6.2 Simplified diagram of the initial events in a local acute inflammatory reaction.
Recognition by tissue macrophages of pathogen-associated molecular patterns (PAMPs) on the pathogen triggers release, from tissue macrophages, of the proinflammatory cytokines interleukin (IL)-1 and tumour necrosis factor (TNF)-α. These act on the endothelial cells of postcapillary venules, causing exudation of fluid and expression of adhesion factors (e.g. selectins, integrins) to which counter-ligands on blood-borne neutrophils adhere. Subsequent steps are listed in the figure. C5a and C3b, complement components; IgG, immunoglobulin G; LTB4, leukotriene B4; PAF, platelet-activating factor.
Neutrophils can engulf, kill and digest microorganisms. Together with eosinophils, they have surface receptors for C3b, which acts as an opsonin that forms a link between neutrophil and invading bacterium. (An even more effective link may be made by antibody; see below.) Neutrophils kill microorganisms by generating toxic oxygen products and other mechanisms, and enzymatic digestion then follows. If the neutrophil is inappropriately activated, these weapons can cause damage to the host’s own tissues. When neutrophils have released their toxic chemicals, they undergo apoptosis and must be cleared by macrophages. It is this mass of live and apoptotic neutrophils that constitutes ‘pus’.
Mast cells
An important ‘sentinel’ cell that expresses TLRs, the mast cell also has surface receptors both for IgE and for the complement-derived anaphylotoxins C3a and C5a. Ligands acting at these receptors trigger mediator release, as does direct physical damage. One of the main substances released is histamine; others include heparin, leukotrienes, PGD2, platelet-activating factor (PAF), nerve growth factor and some interleukins. Unusually, mast cells have pre-formed packets of cytokines that they can release when stimulated. This makes them extremely effective triggers of the inflammatory response.
Monocytes/macrophages
Monocytes arrive in inflammatory lesions several hours after the polymorphs. Adhesion to endothelium and migration into the tissue follow a pattern similar to that of the neutrophils (see above), although monocyte chemotaxis utilises additional chemokines, such as MCP-13 (which, reasonably enough, stands for monocyte chemoattractant protein-1) and RANTES (which very unreasonably stands for regulated on activation normal T cell expressed and secreted: immunological nomenclature has excelled itself here!).
Once in tissues, blood monocytes differentiate into macrophages.4 The resultant ‘sentinel’ cell has a remarkable range of abilities, being not only a jack-of-all-trades but also master of many (see below). Activation of TLRs stimulates the generation and release of chemokines and other cytokines that act on vascular endothelial cells, attract other leukocytes to the area and give rise to systemic manifestations of the inflammatory response such as fever. Macrophages engulf tissue debris and dead cells, as well as phagocytosing and killing most (but unfortunately not all) microorganisms. They also play an important part in antigen presentation (see below). When stimulated by glucocorticoids, macrophages secrete annexin-1 (a potent anti-inflammatory polypeptide; see Ch. 32), which controls the extent of the local inflammatory reaction.
Dendritic cells
These are present in many tissues, especially when they subserve a barrier function (e.g. the skin, where they are sometimes referred to as Langerhans cells after their discoverer). As an important ‘sentinel cell’ they can recognise the presence of pathogens and when thus activated they can migrate into lymphoid tissue, where they play an important part in antigen presentation (see below).
Eosinophils
These cells have similar capacities to neutrophils but are also ‘armed’ with a battery of substances stored in their granules, which, when released, kill multicellular parasites (e.g. helminths). These include eosinophil cationic protein, a peroxidase enzyme, the eosinophil major basic protein and a neurotoxin. The eosinophil is considered by many to be of primary importance in the pathogenesis of the late phase of asthma where, it is suggested, granule proteins cause damage to bronchiolar epithelium (see Fig. 27.4).
Basophils
Basophils are very similar in many respects to mast cells. Except in certain parasitic infections and hypersensitivity reactions, the basophil content of the tissues is negligible and in health they form only 0.5% of circulating white blood cells.
Vascular endothelial cells
Vascular endothelial cells (see also Chs 22 and 23), originally considered as passive lining cells, are now known to play an active part in inflammation. Small arteriole endothelial cells secrete nitric oxide (NO), causing relaxation of the underlying smooth muscle (see Ch. 20), vasodilatation and increased delivery of plasma and blood cells to the inflamed area. The endothelial cells of the postcapillary venules regulate plasma exudation and thus the delivery of plasma-derived mediators (see Fig. 6.1). Vascular endothelial cells express several adhesion molecules (the ICAM and selectin families; see Fig. 6.2), as well as a variety of receptors including those for histamine, acetylcholine and IL-1. In addition to NO, the cells can synthesise and release the vasodilator agents PGI2 and PGE2, the vasoconstrictor agent endothelin, plasminogen activator, PAF and several cytokines. Endothelial cells also participate in the angiogenesis that occurs during inflammatory resolution, chronic inflammation and cancer (see Chs 5 and 55).
Platelets
Platelets are involved primarily in coagulation and thrombotic phenomena (see Ch. 24) but also play a part in inflammation. They have low-affinity receptors for IgE, and are believed to contribute to the first phase of asthma (Fig. 27.1). In addition to generating thromboxane (TX)A2 and PAF, they can generate free radicals and proinflammatory cationic proteins. Platelet-derived growth factor contributes to the repair processes that follow inflammatory responses or damage to blood vessels.
Natural killer cells
Natural killer (NK) cells are a specialised type of lymphocyte. In an unusual twist to the receptor concept, NK cells kill targets (e.g. virus-infected or tumour cells) that lack ligands for inhibitory receptors on the NK cells themselves. The ligands in question are the major histocompatibility complex (MHC) molecules, and any cells lacking these become a target for NK-cell attack, a strategy sometimes called the ‘mother turkey strategy’.5 MHC proteins are expressed on the surface of most host cells and, in simple terms, are specific for that individual, enabling the NK cells to avoid damaging host cells. NK cells have other functions: they are equipped with Fc receptors and, in the presence of antibodies directed against a target cell, they can kill the cell by antibody-dependent cellular cytotoxicity.
The Adaptive Immune Response
The adaptive response provides the physical basis for an ‘immunological memory’. It provides a more powerful defence than the innate response as well as being highly specific for the invading pathogen. Here we will provide only a simplified outline and stressing those aspects relevant for an understanding of drug action; for more detailed coverage, see Janeway et al. (2004).
The adaptive response
The key cells are the lymphocytes. These are long-lived cells derived from precursor cells within the bone marrow. Following release into the blood, they mature in the bone or thymus after which they enter the circulation and dwell in the lymphoid tissues such as the lymph nodes and spleen. Here, they are poised to detect, intercept and identify foreign proteins presented to them by antigen presenting cells (APCs) such as the macrophage or the dendritic cells. The three main groups of lymphocytes are:
Miraculously, T and B lymphocytes express antigen-specific receptors that recognise and react with virtually all foreign proteins and polysaccharides that we are likely to encounter during our lifetime. This receptor repertoire is generated randomly and so would recognise ‘self’ proteins as well as foreign antigens if it were not that tolerance to self antigens is acquired during fetal life by apoptotic deletion of T-cell clones that recognise the host’s own tissues. Dendritic cells and macrophages involved in the innate response also have a role in preventing harmful immune reactions against the host’s own cells (see below).
The specific immune response occurs in two phases termed the induction phase and the effector phase.
The Induction Phase
During the induction phase, antigen is ‘presented’ to T cells by macrophages or large dendritic cells, and this is followed by complex interactions of those T cells with B cells and other T cells (Fig. 6.3). The antigen may constitute part of an invading pathogen (e.g. the coat of a bacterium) or be released by such an organism (e.g. a bacterial toxin), or it may be a vaccine or a substance introduced experimentally in the laboratory to study the immune response (e.g. the injection of egg albumin into the guinea pig). APCs ingest and proteolytically ‘process’ the antigen and ‘present’ it on their surface to lymphocytes in combination with various MHC molecules once they reach local lymph nodes (Fig. 6.4). Two types of lymphocytes ‘attend’ APCs. They are generally distinguished by the presence, on their surface, of CD4 or CD8 receptors. These are co-receptors that cooperate with the main antigen-specific receptors in antigen recognition. Macrophages also carry surface CD4 proteins.
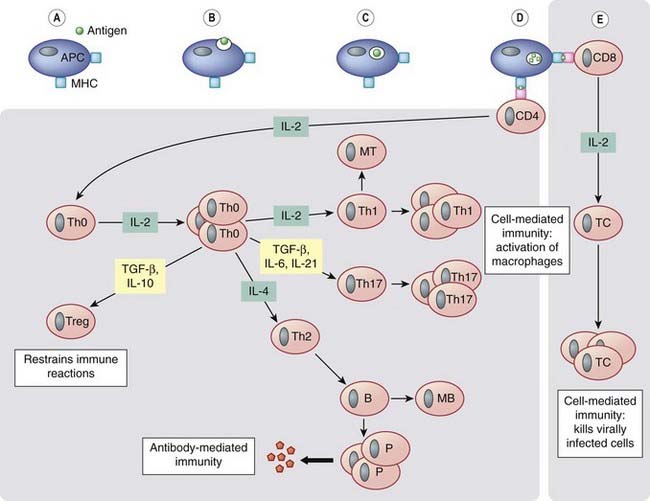
Fig. 6.3 Simplified diagram of the induction and effector phases of lymphocyte activation.
Antigen-presenting cells (APCs) ingest and process antigen (A–D) and present fragments to naive, uncommitted CD4 T cells in conjunction with major histocompatibility complex (MHC) class II molecules, or to naive CD8 T cells in conjunction with MHC class I molecules, thus ‘arming’ them. The armed CD4+ T cells synthesise and express interleukin (IL)-2 receptors and release this cytokine, which stimulates the cells by autocrine action, causing generation and proliferation of T-helper zero (Th0) cells. Autocrine cytokines (e.g. IL-4) cause proliferation of some Th0 cells to give Th2 cells, which are responsible for the development of antibody-mediated immune responses. These Th2 cells cooperate with and activate B cells to proliferate and give rise eventually to memory B cells (MB) and plasma cells (P), which secrete antibodies. Other autocrine cytokines (e.g. IL-2) cause proliferation of Th0 cells to give Th1, Th17 or Treg cells. Th1 and Th17 cells secrete cytokines that activate macrophages (responsible for some cell-mediated immune reactions). Treg cells restrain and inhibit the development of the immune response, thus preventing autoimmunity and excessive immune activation.
The armed CD8+ T cells (E) also synthesise and express IL-2 receptors and release IL-2, which stimulates the cells by autocrine action to proliferate and give rise to cytotoxic T cells (TC). These can kill virally infected cells. IL-2 secreted by CD4+ cells also plays a part in stimulating CD8+ cells to proliferate. Note that the ‘effector phase’ depicted above relates to the ‘protective’ action of the immune response. When the response is inappropriately deployed—as in chronic inflammatory conditions such as rheumatoid arthritis—the Th1/Th17 component of the immune response is dominant and the activated macrophages release IL-1 and tumour necrosis factor (TNF)-α, which in turn trigger the release of the chemokines and inflammatory cytokines that play a major role in the pathology of the disease. MT and MB, memory T and B cells, respectively.
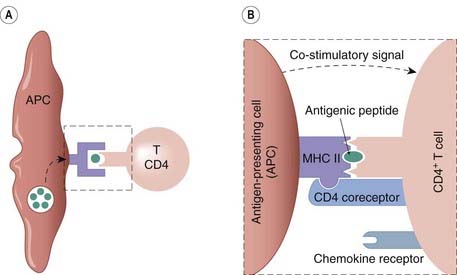
Fig. 6.4 The activation of a T cell by an antigen-presenting cell (APC).
[A] The APC encounters a foreign protein and this is proteolytically processed into peptide fragments. The activation process then involves three stages. (i) Interaction between the complex of pathogen-derived antigen peptide fragments with major histocompatibility complex (MHC) class II and the antigen-specific receptor on the T cell. [B] (ii) Interaction between the CD4 co-receptor on the T cell and an MHC molecule on the APC. (iii) A co-stimulatory signal from the APC to the T cell. The CD4 co-receptor, together with a T-cell chemokine receptor, constitute the main binding sites for the HIV virus (see Fig. 51.3).
The two types of lymphocyte involved in the adaptive response are:
Activation of a T cell by an APC requires that several signals pass between the two cells at this ‘immune synapse’ (Fig. 6.4; see Medzhitov & Janeway, 2000). After activation, the T cells both generate IL-2 and acquire IL-2 receptors. Some potent anti-inflammatory drugs block this receptor thus preventing lymphocyte proliferation (see Ch. 26). IL-2 has an autocrine7 action, stimulating proliferation and giving rise to a clone of T cells termed Th0 cells, which, depending on the prevailing cytokine milieu, give rise to different subsets of armed helper cells. There are four major types of these ‘helper cells’, each of which generate a characteristic cytokine profile, possess a unique surface marker profile and have different roles in disease. These characteristics are summarised in Table 6.2.
Table 6.2 Lymphocyte subsets and their role in host defence and relationship to inflammatory disease
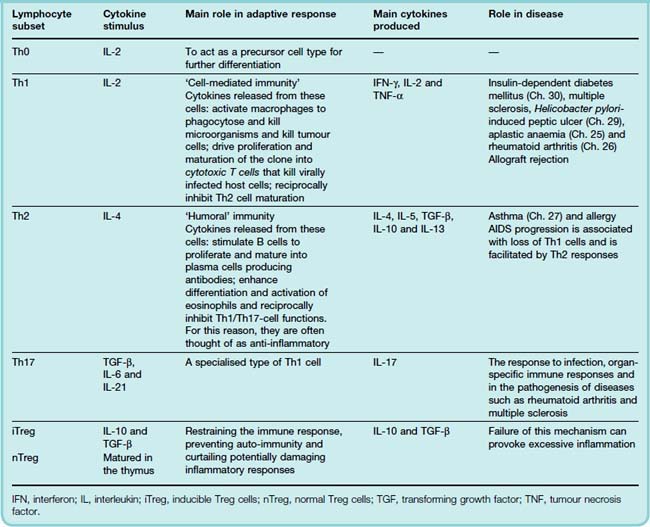
Understanding the relationship between T-cell subsets, their respective cytokine profiles and pathological conditions is expected to highlight ways to manipulate the immune responses for disease prevention and treatment. There are already many experimental models in which modulation of the Th1/Th2 balance with recombinant cytokines or cytokine antagonists alters the outcome of the disease.
The Effector Phase
During the effector phase, the activated B and T lymphocytes differentiate either into plasma cells or into memory cells. The B plasma cells produce antibodies, which are effective in the extracellular fluid, but which cannot neutralise pathogens within cells. T-cell-mediated immune mechanisms overcome this problem by activating macrophages or directly killing virus-infected host cells. Antigen-sensitive memory cells are formed when the clone of lymphocytes that are programmed to respond to an antigen is greatly expanded after the first contact with the organism. They allow a greatly accelerated and more effective response to subsequent antigen exposure. In some cases, the response is so rapid and efficient that, after one exposure, the pathogen can never gain a foothold again. Immunisation procedures make use of this fact.
The Antibody-Mediated (Humoral) Response
There are five main classes of antibody—IgG, IgM, IgE, IgA and IgD—which differ from each other in certain structural respects. All are γ-globulins (immunoglobulins), which both recognise and interact specifically with antigens (i.e. proteins or polysaccharides foreign to the host), as well as activating one or more further components of the host’s defence systems.
An antibody is a Y-shaped protein molecule (see Ch. 59) in which the arms of the Y (the Fab portions) are the recognition sites for specific antigens, and the stem of the Y (the Fc portion) activates host defences. The B cells that are responsible for antibody production recognise foreign molecules by means of surface receptors that are essentially the immunoglobulin which that B-cell clone will eventually produce. Mammals harbour a vast number of B-cell clones that produce different antibodies with recognition sites for different antigens.
The induction of antibody-mediated responses varies with the type of antigen. With most antigens, a cooperative process between Th2 cells and B cells is necessary to produce a response. B cells can also present antigen to T cells that then release cytokines that act further on the B cell. The anti-inflammatory glucocorticoids (see Chs 26 and 32) and the immunosuppressive drug ciclosporin (see Ch. 26) affect the events at the stage of induction. The cytotoxic immunosuppressive drugs (see Ch. 26) inhibit the proliferation of both B and T cells. Eicosanoids may play a part in controlling these processes as prostaglandins of the E series can inhibit lymphocyte proliferation, probably by inhibiting the release of IL-2.
As you might guess, the ability to make antibodies has huge survival value; children born without this ability suffer repeated infections such as pneumonia, skin infections and tonsillitis. Before the days of antibiotics, they died in early childhood, and even today they require regular replacement therapy with immunoglobulin. Apart from their ability to neutralise pathogens, antibodies can boost the effectiveness and specificity of the host’s defence reaction in several ways.
Antibodies and complement
Formation of the antigen–antibody complex exposes a binding site for complement on the Fc domain. This activates the complement sequence and sets in train its attendant biological effects (see Fig. 6.1). This route to C3 activation (the classic pathway) provides an especially selective way of activating complement in response to a particular pathogen, because the antigen–antibody reaction that initiates it is not only a highly specific recognition event, but also occurs in close association with the pathogen. The lytic property of complement can be used therapeutically: monoclonal antibodies (mAbs) and complement together can be used to rid bone marrow of cancer cells as an adjunct to chemotherapy or radiotherapy (see Ch. 55).
Antibodies and the phagocytosis of bacteria
When antibodies are attached to their antigens on microorganisms by their Fab portions, the Fc domain is exposed. Phagocytic cells (neutrophils and macrophages) express surface receptors for these projecting Fc portions, which serve as a very specific link between microorganism and phagocyte.
Antibodies and cellular cytotoxicity
In some cases, for example with parasitic worms, the invader may be too large to be ingested by phagocytes. Antibody molecules can form a link between parasite and the host’s white cells (in this case, eosinophils), which are then able to damage or kill the parasite by surface or extracellular actions. NK cells in conjunction with Fc receptors can also kill antibody-coated target cells (an example of antibody-dependent cell-mediated cytotoxicity).
Antibodies and mast cells or basophils
Mast cells and basophils have receptors for IgE, a particular form of antibody that can attach (‘fix’) to their cell membranes. When antigen reacts with this cell-fixed antibody, an entire panoply of pharmacologically active mediators is secreted. This very complex reaction is found widely throughout the animal kingdom and presumably offers clear survival value to the host. Having said that, its precise biological significance is not entirely clear, although it may be of importance in association with eosinophil activity as a defence against parasitic worms. When inappropriately triggered by substances not inherently damaging to the host, it is implicated in certain types of allergic reaction (see below) and apparently contributes more to illness than to survival in the modern world.
The Cell-Mediated Immune Response
Cytotoxic T cells (derived from CD8+ cells) and inflammatory (cytokine-releasing) Th1 cells are attracted to inflammatory sites in a similar manner to neutrophils and macrophages, and are involved in cell-mediated responses (see Fig. 6.3).
Cytotoxic T cells
Armed cytotoxic T cells kill intracellular microorganisms such as viruses. When a virus infects a mammalian cell, there are two aspects to the resulting defensive response. The first step is the expression on the cell surface of peptides derived from the pathogen in association with MHC molecules. The second step is the recognition of the peptide–MHC complex by specific receptors on cytotoxic (CD8+) T cells (Fig. 6.4 shows a similar process for a CD4+ T cell). The cytotoxic T cells then destroy virus-infected cells by programming them to undergo apoptosis. Cooperation with macrophages may be required for killing to occur.
Macrophage-activating CD4+ Th1 cells
Some pathogens (e.g. Mycobacteria, Listeria) survive and multiply within macrophages after ingestion. Armed CD4+ Th1 cells release cytokines that activate macrophages to kill these intracellular pathogens. Th1 cells also recruit macrophages by releasing cytokines that act on vascular endothelial cells (e.g. TNF-α) and chemokines (e.g. macrophage chemotactic factor-1; MCP-1) that attract the macrophages to the sites of infection.
A complex of microorganism-derived peptides plus MHC molecules is expressed on the macrophage surface and is recognised by cytokine-releasing Th1 cells, which then generate cytokines that enable the macrophage to deploy its killing mechanisms. Activated macrophages (with or without intracellular pathogens) are factories for the production of chemical mediators, and can generate and secrete not only many cytokines but also toxic oxygen metabolites and neutral proteases that kill extracellular organisms (e.g. Pneumocystis carinii and helminths), complement components, eicosanoids, NO, a fibroblast-stimulating factor, pyrogens and the ‘tissue factor’ that initiates the extrinsic pathway of the coagulation cascade (Ch. 24), as well as various other coagulation factors. It is primarily the cell-mediated reaction that is responsible for allograft rejection. Macrophages are also important in coordinating the repair processes that must occur for inflammation to ‘resolve’.
The specific cell-mediated or humoral immunological response is superimposed on the innate non-specific vascular and cellular reactions described previously, making them not only markedly more effective but much more selective for particular pathogens.
The general events of the inflammatory and hypersensitivity reactions specified above vary in some tissues. For example, in the airway inflammation of asthma, eosinophils and neuropeptides play a particularly significant role (see Ch. 27). In CNS inflammation, there is less neutrophil infiltration and monocyte influx is delayed, possibly because of lack of adhesion molecule expression on CNS vascular endothelium and deficient generation of chemotaxins. It has long been known that some tissues—the CNS parenchyma, the anterior chamber of the eye, and the testis—are immunologically privileged sites, in that a foreign antigen introduced directly does not provoke an immune reaction (which could be very disadvantageous to the host). However, introduction elsewhere of an antigen already in the CNS parenchyma will trigger the development of immune/inflammatory responses in the CNS.
Systemic Responses in Inflammation
In addition to the local changes in an inflammatory area, there are often general systemic manifestations of inflammatory disease, including fever, an increase in blood leukocytes termed leukocytosis (or neutrophilia if the increase is in the neutrophils only) and the release from the liver of acute-phase proteins. These include C-reactive protein, α2-macroglobulin, fibrinogen, α1-antitrypsin and some complement components. While the function of many of these components is still a matter of conjecture, they all seem to have antimicrobial actions. C-reactive protein, for example, binds to some microorganisms, and the resulting complex activates complement. Other proteins scavenge iron (an essential nutrient for invading organisms) or block proteases, perhaps protecting the host against the worst excesses of the inflammatory response.
The Role of the Nervous System in Inflammation
It has become clear in recent years that the central, autonomic and peripheral nervous systems all play an important part in the regulation of the inflammatory response. This occurs at various levels:
Unwanted Inflammatory and Immune Responses
The immune response has to strike a delicate balance. According to one school of thought, an infection-proof immune system would be a possibility but would come at a serious cost to the host. With approximately 1 trillion potential antigenic sites in the host, such a ‘superimmune’ system would be some 1000 times more likely to attack the host itself, triggering autoimmune disease. In addition, it is not uncommon to find that innocuous substances such as pollen or peanuts sometimes inadvertently activate the immune system. When this happens, the inflammation itself inflicts damage and may be responsible for the major symptoms of the disease—either acutely as in (for example) anaphylaxis, or chronically in (for example) asthma or rheumatoid arthritis. In either case, anti-inflammatory or immunosuppressive therapy may be required.
Unwanted immune responses, termed allergic or hypersensitivity reactions, have been classified into four types (Janeway et al., 2004).
Type I: immediate or anaphylactic hypersensitivity
Type I hypersensitivity (often known simply as ‘allergy’) occurs in individuals who predominantly exhibit a Th2 rather than a Th1 response to antigen. In these individuals, substances that are not inherently noxious (such as grass pollen, house dust mites, certain foodstuffs or drugs, animal fur and so on) provoke the production of antibodies of the IgE type.8 These fix on mast cells, in the lung, and also to eosinophils. Subsequent contact with the substance causes the release of histamine, PAF, eicosanoids and cytokines. The effects may be localised to the nose (hay fever), the bronchial tree (the initial phase of asthma), the skin (urticaria) or the gastrointestinal tract. In some cases, the reaction is more generalised and produces anaphylactic shock, which can be severe and life-threatening. Some important unwanted effects of drugs include anaphylactic hypersensitivity responses (see Ch. 57).
Type II: antibody-dependent cytotoxic hypersensitivity
Type II hypersensitivity occurs when the mechanisms outlined above are directed against cells within the host that are (or appear to be) foreign. For example, host cells altered by drugs are sometimes mistaken by the immune system for foreign proteins and evoke antibody formation. The antigen–antibody reaction triggers complement activation (and its sequelae) and may promote attack by NK cells. Examples include alteration by drugs of neutrophils, leading to agranulocytosis (see Ch. 56), or of platelets, leading to thrombocytopenic purpura (Ch. 24). These type II reactions are also implicated in some types of autoimmune thyroiditis (e.g. Hashimoto’s disease; see Ch. 33).
Type III: complex-mediated hypersensitivity
Type III hypersensitivity occurs when antibodies react with soluble antigens. The antigen–antibody complexes can activate complement or attach to mast cells and stimulate the release of mediators.
An experimental example of this is the Arthus reaction that occurs if a foreign protein is injected subcutaneously into a rabbit or guinea pig with high circulating concentrations of antibody. Within 3–8 h, the area becomes red and swollen because the antigen–antibody complexes precipitate in small blood vessels and activate complement. Neutrophils are attracted and activated (by C5a) to generate toxic oxygen species and to secrete enzymes.
Mast cells are also stimulated by C3a to release mediators. Damage caused by this process is involved in serum sickness, caused when antigen persists in the blood after sensitisation, causing a severe reaction, as in the response to mouldy hay (known as farmer’s lung), and in certain types of autoimmune kidney and arterial disease. Type III hypersensitivity is also implicated in lupus erythematosus (a chronic, autoimmune inflammatory disease).
Type IV: cell-mediated hypersensitivity
The prototype of type IV hypersensitivity (also known as delayed hypersensitivity) is the tuberculin reaction, a local inflammatory response seen when proteins derived from cultures of the tubercle bacillus are injected into the skin of a person who has been sensitised by a previous infection or immunisation. An ‘inappropriate’ cell-mediated immune response is stimulated, accompanied by infiltration of mononuclear cells and the release of various cytokines. Cell-mediated hypersensitivity is also the basis of the reaction seen in some other infections (e.g. mumps and measles), as well as with mosquito and tick bites. It is also important in the skin reactions to drugs or industrial chemicals (see Ch. 57), where the chemical (termed a hapten) combines with proteins in the skin to form the ‘foreign’ substance that evokes the cell-mediated immune response (Fig. 6.3).
In essence, inappropriately deployed T-cell activity underlies all types of hypersensitivity, initiating types I, II and III, and being involved in both the initiation and the effector phase in type IV. These reactions are the basis of the clinically important group of autoimmune diseases. Immunosuppressive drugs (Ch. 26) and/or glucocorticoids (Ch. 32) are routinely employed to treat such disorders.
The Outcome of the Inflammatory Response
It is important not to lose sight of the fact that the inflammatory response is a defence mechanism and not, ipso facto, a disease. Its role is to restore normal structure and function to the infected or damaged tissue and, in the vast majority of cases, this is what happens. The healing and resolution phase of the inflammatory response is an active process and does not simply ‘happen’ in the absence of further inflammation. This is an area that we are just beginning to understand, but it is clear that it utilises its own unique palette of mediators and cytokines (including various growth factors, annexin-A1, lipoxins and IL-10; see Ch. 17) to terminate residual inflammation and to promote remodelling and repair of damaged tissue.
In some cases, healing will be complete, but if there has been damage (death of cells, pus formation, ulceration) repair is usually necessary and may result in scarring. If the pathogen persists, the acute response is likely to transform into a chronic inflammatory response. This is a slow, smouldering reaction that can continue indefinitely, destroying tissue and promoting local proliferation of cells and connective tissue. The principal cell types found in areas of chronic inflammation are mononuclear cells and abnormal macrophage-derived cells. During healing or chronic inflammation, growth factors trigger angiogenesis and cause fibroblasts to lay down fibrous tissue. Infection by some microorganisms, such as syphilis, tuberculosis and leprosy, bears the characteristic hallmarks of chronic inflammation from the start. The cellular and mediator components of this type of inflammation are also seen in many, if not most, chronic autoimmune and hypersensitivity diseases, and are important targets for drug action.
References and Further Reading
The innate and adaptive responses
Abbas A.K., Murphy K.M., Sher A. Functional diversity of helper lymphocytes. Nature. 1996;383:787-793. (Excellent review, helpful diagrams; commendable coverage of Th1 and Th2 cells and their respective cytokine subsets)
Adams D.H., Lloyd A.R. Chemokines: leukocyte recruitment and activation cytokines. Lancet. 1997;349:490-495. (Commendable review)
Delves P.J., Roitt I.M. The immune system. N. Engl. J. Med.. 2000;343:37-49. 108–117. (A good overview of the immune system—a minitextbook of major areas in immunology; colourful three-dimensional figures)
Gabay C., Kushner I. Acute phase proteins and other systemic responses to inflammation. N. Engl. J. Med.. 1999;340:448-454. (Lists the acute-phase proteins and outlines the mechanisms controlling their synthesis and release)
Kärre K., Welsh R.M. Viral decoy vetoes killer cell. Nature. 1997;386:446-447.
Kay A.B. Allergic diseases and their treatment. N. Engl. J. Med.. 2001;344:30-37. 109–113. (Covers atopy and Th2 cells, the role of Th2 cytokines in allergies, IgE, the main types of allergy and new therapeutic approaches)
Mackay C.R., Lanzavecchia A., Sallusto F. Chemoattractant receptors and immune responses. Immunologist. 1999;7:112-118. (Masterly short review covering the role of chemoattractants in orchestrating immune responses—both the innate reaction and the Th1 and Th2 responses)
Medzhitov R. Toll-like receptors and innate immunity. Nat. Rev. Immunol.. 2001;1:135-145. (Excellent review of the role of Toll-like receptors in (a) the detection of microbial infection, and (b) the activation of innate non-adaptive responses, which in turn lead to antigen-specific adaptive responses)
Medzhitov R., Janeway C. Innate immunity. N. Engl. J. Med.. 2000;343:338-344. (Outstandingly clear coverage of the mechanisms involved in innate immunity and its significance for the adaptive immune response)
Mills K.H. Induction, function and regulation of IL-17-producing T cells. Eur. J. Immunol.. 2008;38:2636-2649. (This paper covers the biology of Th17 cells—a relatively recent addition to our understanding of Th biology. Accessible and has good diagrams)
Murphy P.M. Viral exploitation and subversion of the immune system through chemokine mimicry. Nat. Immunol.. 2001;2:116-122. (Excellent description of viral/immune system interaction)
Panes J., Perry M., Granger D.N. Leukocyte–endothelial cell adhesion: avenues for therapeutic intervention. Br. J. Pharmacol.. 1999;126:537-550. (Brief coverage of the principal cell adhesion molecules and factors affecting leukocyte–endothelial adhesion precedes consideration of potential therapeutic targets)
Parkin J., Cohen B. An overview of the immune system. Lancet. 2001;357:1777-1789. (A competent, straightforward review covering the role of the immune system in recognising, repelling and eradicating pathogens and in reacting against molecules foreign to the body)
Sternberg E.M. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat. Rev. Immunol.. 2006;6:318-328. (This paper and the next are both excellent and easy-to-read reviews covering the role of the CNS in inflammation. Some good diagrams)
Takeda K., Akira S. Toll receptors and pathogen resistance. Cell Microbiol.. 2003;5:143-153. (Useful review and easy to read. Also deals with Toll receptor signalling in some depth)
Tracey K.J. The inflammatory reflex. Nature. 2002;420:853-859.
Vasselon T., Detmers P.A. Toll receptors: a central element in innate immune responses. Infect. Immun.. 2002;70:1033-1041. (Another comprehensive review on this important topic)
Walker C., Zuany-Amorini C. New trends in immunotherapy to prevent atopic disease. Trends Pharmacol. Sci.. 2001;22:84-91. (Discusses potential therapies based on recent advances in the understanding of the immune mechanisms of atopy)
Wills-Karp M., Santeliz J., Karp C.L. The germless theory of allergic diseases. Nat. Rev. Immunol.. 2001;1:69-75. (Discusses the hypothesis that early childhood infections inhibit the tendency to develop allergic disease)
Textbook of immunopharmacology Dale M.M., Foreman J.C., Fan T.-P., editors third ed 1994 Blackwell Scientific Oxford (Excellent textbook written with second- and third-year medical and science students in mind; contains many sections relevant to this chapter and the next)
Janeway C.A., Travers P., Nolan A., et al. Immunobiology: the immune system in health and disease, sixth ed. Edinburgh: Churchill Livingstone; 2004. (Excellent textbook, good diagrams)
Roitt I., Brostoff J., Male D. Immunology, ninth ed. Oxford: Blackwell Science; 1998. (Excellent textbook; well illustrated)
http://www.biochemweb.org/fenteany/research/cell_migration/movement_movies.html (If you have never seen a neutrophil in hot pursuit of a bacterium, then you definitely need to look at this online movie. Great fun and highly instructive)
1Mucosal epithelial tissues constantly secrete antibacterial proteins and a type of ‘all purpose’ immunoglobulin (Ig)A as a sort of pre-emptive defensive strategy. One immunologist aptly referred to the innate response as the organism’s ‘knee jerk’ response to infection; it is an excellent description.
2The name, which loosely translates from German as ‘Great!’ or ‘Eureka!’, has remained firmly attached to the family.
3Human immunodeficiency virus-1 binds to the surface CD4 glycoprotein on monocytes/macrophages but is able to penetrate the cell only after binding also to MCP-1 and RANTES receptors.
4Literally ‘big eaters’, compared with neutrophils, originally called microphages or ‘little eaters’.
5Richard Dawkins in River out of Eden, citing the zoologist Schliedt, explains that the ‘rule of thumb a mother turkey uses to recognise nest robbers is a dismayingly brusque one; in the vicinity of the nest, attack anything that moves unless it makes a noise like a baby turkey’ (quoted by Kärre & Welsh, 1997).
6The main reason that it is difficult to transplant organs such as kidneys from one person to another is that their respective MHC molecules are different. Lymphocytes in the recipient will react to non-self (allogeneic) MHC molecules in the donor tissue, which is then likely to be rejected by a rapid and powerful immunological reaction.
7‘Autocrine’ signalling means that the mediator acts on the same cell that releases it. ‘Paracrine’ signalling means that the mediator acts on neighbouring cells.
8Such individuals are said to be ‘atopic’, from a Greek word meaning ‘out of place’.