39 Neurodegenerative diseases
Overview
As a rule, dead neurons in the adult central nervous system (CNS) are not replaced,1 nor can their terminals regenerate when their axons are interrupted. Therefore any pathological process causing neuronal death generally has irreversible consequences. At first sight, this appears to be very unpromising territory for pharmacological intervention, and indeed drug therapy is currently very limited, except in the case of Parkinson’s disease (PD; see below). Nevertheless, the incidence and social impact of neurodegenerative brain disorders in ageing populations has resulted in a massive research effort in recent years.
In this chapter, we focus mainly on three common neurodegenerative conditions: Alzheimer’s disease (AD), PD and ischaemic brain damage (stroke). AD and PD are the commonest examples of a group of chronic, slowly developing conditions that include various prion diseases (e.g. Creutzfeldt–Jakob disease, CJD). They have a common aetiology in that they are caused by the aggregation of misfolded variants of normal physiological proteins. The high hopes that the new pathophysiological understanding that has emerged over the last two decades would lead to significant therapeutic progress in this important area remain largely unrealised, and to date the available therapeutic interventions are aimed at compensating for, rather than preventing or reversing, the neuronal loss.
Stroke, which is a common disorder of enormous socioeconomic importance, results from acute ischaemic brain damage, quite different from the aetiology of chronic neurodegenerative diseases, requiring different but equally challenging therapeutic approaches.
Protein Misfolding and Aggregation in Chronic Neurodegenerative Diseases
Protein misfolding and aggregation is the first step in many neurodegenerative diseases (see Stefani & Dobson, 2003; Forman et al., 2004; Selkoe, 2004). Misfolding means the adoption of abnormal conformations, by certain normally expressed proteins, such that they tend to form large insoluble aggregates (Fig. 39.1). The conversion of the linear amino acid chain produced by the ribosome into a functional protein requires it to be folded correctly into a compact conformation with specific amino acids correctly located on its surface. This complicated stepwise sequence can easily go wrong and lead to misfolded variants that are unable to find a way back to the correct ‘native’ conformation. The misfolded molecules are non-functional with respect to the normal function of the protein, but can nonetheless make mischief within the cell. The misfolding often means that hydrophobic residues that would normally be buried in the core of the protein are exposed on its surface, which gives the molecules a strong tendency to stick to cell membranes and aggregate, initially as oligomers and then as insoluble microscopic aggregates (Fig. 39.1), leading to the death of neurons. The tendency to adopt such conformations may be favoured by specific mutations of the protein in question, or by infection with prions (see below).
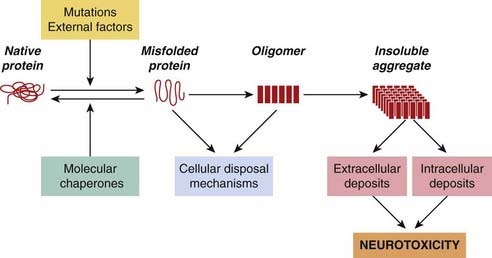
Misfolded conformations can be generated spontaneously at a low rate throughout life, so that aggregates accumulate gradually with age. In the nervous system, the aggregates often form distinct structures, generally known as amyloid deposits, that are visible under the microscope and are characteristic of neurodegenerative disease. Although the mechanisms are not clear, such aggregates, or the misfolded protein precursors, lead to neuronal death. Examples of neurodegenerative diseases that are caused by such protein misfolding and aggregation are shown in Table 39.1.
Table 39.1 Examples of neurodegenerative diseases associated with protein misfolding and aggregationa
The brain possesses a variety of protective mechanisms that limit the accumulation of such protein aggregates. The main ones involve the production of ‘chaperone’ proteins, which bind to newly synthesised or misfolded proteins and encourage them to fold correctly, and the ‘ubiquitination’ reaction, which prepares proteins for destruction within the cell. Accumulation of protein deposits occurs when these protective mechanisms are unable to cope.
Protein misfolding 
Mechanisms of Neuronal Death
Acute injury to cells causes them to undergo necrosis, recognised pathologically by cell swelling, vacuolisation and lysis, and associated with Ca2+ overload of the cells and membrane damage (see below). Necrotic cells typically spill their contents into the surrounding tissue, evoking an inflammatory response. Chronic inflammation is a feature of most neurodegenerative disorders (see Schwab & McGeer, 2008), and a possible target for therapeutic intervention.
Cells can also die by apoptosis or programmed cell death (see Ch. 5), a mechanism that is essential for many processes throughout life, including development, immune regulation and tissue remodelling. Apoptosis, as well as necrosis, occurs in both acute neurodegenerative disorders (such as stroke and head injury) and chronic ones (such as Alzheimer’s and Parkinson’s disease; see Okouchi et al., 2007). The distinction between necrosis and apoptosis as processes leading to neurodegeneration is not absolute, for challenges such as excitotoxicity and oxidative stress may be enough to kill cells directly by necrosis or, if less intense, may induce them to undergo apoptosis. Both processes therefore represent possible targets for putative neuroprotective drug therapy. Pharmacological interference with the apoptotic pathway may become possible in the future, but for the present most efforts are directed at the processes involved in cell necrosis, and at compensating pharmacologically for the neuronal loss.
Excitotoxicity
Despite its ubiquitous role as a neurotransmitter, glutamate is highly toxic to neurons, a phenomenon dubbed excitotoxicity (see Ch. 37). A low concentration of glutamate applied to neurons in culture kills the cells, and the finding in the 1970s that glutamate given orally produces neurodegeneration in vivo caused considerable alarm because of the widespread use of glutamate as a ‘taste-enhancing’ food additive. The ‘Chinese restaurant syndrome’—an acute attack of neck stiffness and chest pain—is well known, but so far the possibility of more serious neurotoxicity is only hypothetical.
Local injection of the glutamate receptor agonist kainic acid is used experimentally to produce neurotoxic lesions. It acts by excitation of local glutamate-releasing neurons, and the release of glutamate, acting on NMDA and also metabotropic receptors (Ch. 37), leads to neuronal death.
Calcium overload is the essential factor in excitotoxicity. The mechanisms by which this occurs and leads to cell death are as follows (Fig. 39.2):
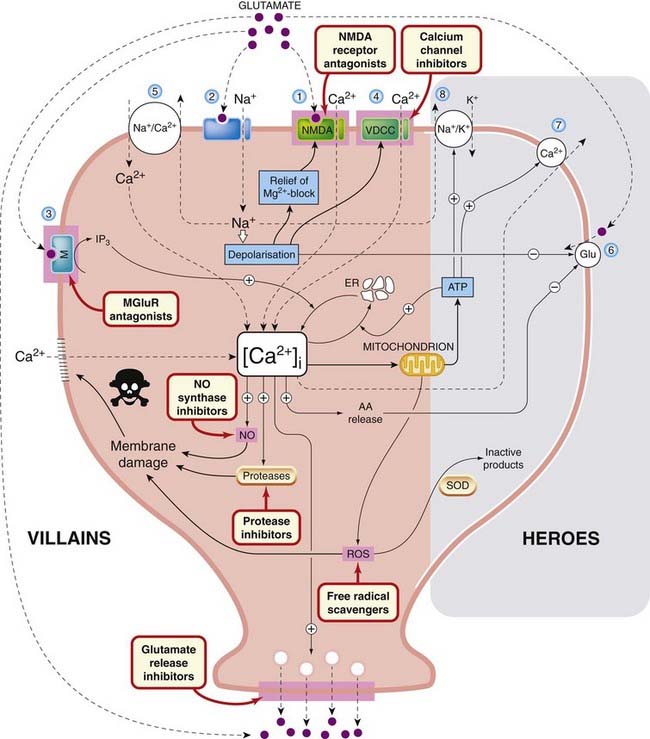
Fig. 39.2 Mechanisms of excitotoxicity.
Membrane receptors, ion channels and transporters, identified by numbers 1–8, are discussed in the text. Possible sites of action of neuroprotective drugs (not yet of proven clinical value) are highlighted. Mechanisms on the left (villains) are those that favour cell death, while those on the right (heroes) are protective. See text for details. AA, arachidonic acid; ER, endoplasmic reticulum; Glu, glutamate uptake; IP3, inositol trisphosphate; M, MGluR, metabotropic glutamate receptor; NO, nitric oxide; ROS, reactive oxygen species; SOD, superoxide dismutase; VDCC, voltage-dependent calcium channel.
Glutamate and Ca2+ are arguably the two most ubiquitous chemical signals, extracellular and intracellular, respectively, underlying brain function, so it is disconcerting that such cytotoxic mayhem can be unleashed when they get out of control. Both are stored in dangerous amounts in subcellular organelles, like hand grenades in an ammunition store. Defence against excitotoxicity is clearly essential if our brains are to have any chance of staying alive. Mitochondrial energy metabolism provides one line of defence (see above), and impaired mitochondrial function, by rendering neurons vulnerable to excitotoxic damage, may be a factor in various neurodegenerative conditions, including PD. Furthermore, impaired mitochondrial function can cause release of cytochrome c, which is an important initiator of apoptosis.
The role of excitotoxicity in ischaemic brain damage is well established (see below), and it is also believed to be a factor in other neurodegenerative diseases, such as those discussed below (see Lipton & Rosenberg, 1994).
 There are several examples of neurodegenerative conditions caused by environmental toxins acting as agonists on glutamate receptors. Domoic acid is a glutamate analogue produced by mussels, which was identified as the cause of an epidemic of severe mental and neurological deterioration in a group of Newfoundlanders in 1987. On the island of Guam, a syndrome combining the features of dementia, paralysis and PD was traced to an excitotoxic amino acid, β-methylamino-alanine, in the seeds of a local plant. Discouraging the consumption of these seeds has largely eliminated the disease.
There are several examples of neurodegenerative conditions caused by environmental toxins acting as agonists on glutamate receptors. Domoic acid is a glutamate analogue produced by mussels, which was identified as the cause of an epidemic of severe mental and neurological deterioration in a group of Newfoundlanders in 1987. On the island of Guam, a syndrome combining the features of dementia, paralysis and PD was traced to an excitotoxic amino acid, β-methylamino-alanine, in the seeds of a local plant. Discouraging the consumption of these seeds has largely eliminated the disease.
Disappointingly, intense effort, based on the mechanisms described above, to find effective drugs for a range of neurodegenerative disorders in which excitotoxicity is believed to play a part has had very limited success. Riluzole, a compound that inhibits both the release and the postsynaptic action of glutamate, retards to some degree the deterioration of patients with amyotrophic lateral sclerosis. Memantine, a compound first described 40 years ago, is a weak NMDA receptor antagonist that produces slight improvement in moderate-to-severe cases of AD, but is not recommended for routine clinical use.
Apoptosis
Apoptosis can be initiated by various cell surface signals (see Ch. 5). The cell is systematically dismantled, and the shrunken remnants are removed by macrophages without causing inflammation. Apoptotic cells can be identified by a staining technique that detects the characteristic DNA breaks. Many different signalling pathways can result in apoptosis, but in all cases the final pathway resulting in cell death is the activation of a family of proteases (caspases), which inactivate various intracellular proteins. Neural apoptosis is normally prevented by neuronal growth factors, including nerve growth factor and brain-derived neurotrophic factor, secreted proteins that are required for the survival of different populations of neurons in the CNS. These growth factors regulate the expression of the two gene products Bax and Bcl-2, Bax being proapoptotic and Bcl-2 being antiapoptotic (see Ch. 5). Blocking apoptosis by interfering at specific points on these pathways represents an attractive strategy for developing neuroprotective drugs, but one that has yet to bear fruit.
Oxidative Stress
The brain has high energy needs, which are met almost entirely by mitochondrial oxidative phosphorylation, generating ATP at the same time as reducing molecular O2 to H2O. Under certain conditions, highly reactive oxygen species (ROS), for example oxygen and hydroxyl free radicals and H2O2, may be generated as side products of this process (see Coyle & Puttfarken, 1993; Barnham et al., 2004). Oxidative stress is the result of excessive production of these reactive species. They can also be produced as a byproduct of other biochemical pathways, including nitric oxide synthesis and arachidonic acid metabolism (which are implicated in excitotoxicity; see above), as well as the P450 mono-oxygenase system (see Ch. 9). Unchecked, reactive oxygen radicals attack many key molecules, including enzymes, membrane lipids and DNA. Not surprisingly, defence mechanisms are provided, in the form of enzymes such as superoxide dismutase (SOD) and catalase, as well as antioxidants such as ascorbic acid, glutathione and α-tocopherol (vitamin E), which normally keep these reactive species in check. Some cytokines, especially tumour necrosis factor (TNF)-α, which is produced in conditions of brain ischaemia or inflammation (Ch.17), exert a protective effect, partly by increasing the expression of SOD. Transgenic animals lacking TNF receptors show enhanced susceptibility to brain ischaemia. Mutations of the gene encoding SOD (Fig. 39.2) are associated with amyotrophic lateral sclerosis (ALS, also known as motor neuron disease), a fatal paralytic disease resulting from progressive degeneration of motor neurons, and transgenic mice expressing mutated SOD develop a similar condition.2 Accumulation of aggregates of misfolded mutated SOD (see above) may also contribute to neurodegeneration.
Mitochondria play a central role in energy metabolism, failure of which leads to oxidative stress. Damage to mitochondria, leading to the release of cytochrome c into the cytosol, also initiates apoptosis. Mitochondrial integrity is therefore essential for neuronal survival, and mitochondrial dysfunction is seen as a major factor in many neurodegenerative disorders (see Petrozzi et al., 2007). It is possible that accumulated or inherited mutations in enzymes such as those of the mitochondrial respiratory chain lead to a congenital or age-related increase in susceptibility to oxidative stress, which is manifest in different kinds of inherited neurodegenerative disorders (such as Huntington’s disease), and in age-related neurodegeneration.
Oxidative stress is both a cause and consequence of inflammation (Ch. 6), which is a general feature of neurodegenerative disease and is thought to contribute to neuronal damage (see Schwab & McGeer, 2008).
Several possible targets for therapeutic intervention with neuroprotective drugs are shown in Figure 39.2.
Excitotoxicity and oxidative stress
Ischaemic Brain Damage
After heart disease and cancer, strokes are the commonest cause of death in Europe and North America, and the 70% that are non-fatal are the commonest cause of disability. Approximately 85% of strokes are ischaemic, usually due to thrombosis of a major cerebral artery. The remainder are haemorrhagic, due to rupture of a cerebral artery. Atherosclerosis is the usual underlying cause of both types.
Pathophysiology
Interruption of blood supply to the brain initiates the cascade of neuronal events shown in Figure 39.2, which lead in turn to later consequences, including cerebral oedema and inflammation, which can also contribute to brain damage (see Dirnagl et al., 1999). Further damage can occur following reperfusion,3 because of the production of reactive oxygen species when the oxygenation is restored. Reperfusion injury may be an important component in stroke patients. These secondary processes often take hours to develop, providing a window of opportunity for therapeutic intervention. The lesion produced by occlusion of a major cerebral artery consists of a central core in which the neurons quickly undergo irreversible necrosis, surrounded by a penumbra of compromised tissue in which inflammation and apoptotic cell death develop over a period of several hours. It is assumed that neuroprotective therapies, given within a few hours, might inhibit this secondary penumbral damage.
Glutamate excitotoxicity plays a critical role in brain ischaemia. Ischaemia causes depolarisation of neurons, and the release of large amounts of glutamate. Ca2+ accumulation occurs, partly as a result of glutamate acting on NMDA receptors, for both Ca2+ entry and cell death following cerebral ischaemia are inhibited by drugs that block NMDA receptors or channels (see Ch. 37). Nitric oxide is also produced in amounts much greater than result from normal neuronal activity (i.e. to levels that are toxic rather than modulatory).
Therapeutic Approaches
The only drug currently approved for treating strokes is recombinant tissue plasminogen activator, alteplase, given intravenously, which helps to restore blood flow by dispersing the thrombus (see Ch. 24). A controlled trial showed that it did not reduce mortality (about 8%), but gave significant functional benefit to patients who survive. To be effective, it must be given within about 3 h of the thrombotic episode. Also, it must not be given in the 15% of cases where the cause is haemorrhage rather than thrombosis, so preliminary computerised tomography (CT) scanning is essential. These stringent requirements seriously limit the use of fibrinolytic agents for treating stroke, except where specialised rapid response facilities are available.
A preferable approach would be to use neuroprotective agents aimed at rescuing cells in the penumbral region of the lesion, which are otherwise likely to die. In animal models involving cerebral artery occlusion, many drugs targeted at the mechanisms shown in Figure 39.2 (not to mention many others that have been tested on the basis of more far-flung theories) act in this way to reduce the size of the infarct. These include glutamate antagonists, calcium and sodium channel inhibitors, free radical scavengers, anti-inflammatory drugs, protease inhibitors and others (see Green, 2008). It seems that almost anything works. Altogether, Green et al. (2003) reported that more than 37 such agents had been tested in more than 114 clinical trials, and all had failed to show efficacy. The dispiriting list of failures includes calcium and sodium channel blockers (e.g. nimodipine, fosphenytoin), NMDA receptor antagonists (selfotel, eliprodil, dextromethorphan), drugs that inhibit glutamate release (adenosine analogues, lobeluzole), drugs that enhance GABA effects (e.g. chlormethiazole), 5-HT antagonists, metal chelators and various free radical scavengers (e.g. tirilazad). Green et al. (2003) argued, reasonably enough, that the animal models in use failed to replicate the clinical situation, and urged the use of more rigorous experimental protocols to make animal models more predictive, but 5 years later (see Green, 2008) the success rate was still zero, and the prospect for proven neuroprotective agents in clinical use remains bleak.4 Controlled clinical trials on stroke patients are problematic and very expensive, partly because of the large variability of outcome in terms of functional recovery, which means that large groups of patients (typically thousands) need to be observed for several months. The need to start therapy within hours of the attack is an additional problem.
Stroke treatment is certainly not—so far at least—one of pharmacology’s success stories, and medical hopes rest more on prevention (e.g. by controlling blood pressure, taking aspirin and preventing atherosclerosis) than on treatment.
Stroke
Alzheimer’s Disease
Loss of cognitive ability with age is considered to be a normal process whose rate and extent is very variable. AD was originally defined as presenile dementia, but it now appears that the same pathology underlies the dementia irrespective of the age of onset. AD refers to dementia that does not have an antecedent cause, such as stroke, brain trauma or alcohol. Its prevalence rises sharply with age, from about 5% at 65 to 90% or more at 95. Until recently, age-related dementia was considered to result from the steady loss of neurons that normally goes on throughout life, possibly accelerated by a failing blood supply associated with atherosclerosis. Studies over the past three decades have, however, revealed specific genetic and molecular mechanisms underlying AD (reviewed by Selkoe, 1997; Bossy-Wetzel et al., 2004). These advances have raised hopes of more effective treatments (see Yamada & Nabeshima, 2000), but success has proved elusive.
Pathogenesis of Alzheimer’s Disease
Alzheimer’s disease is associated with brain shrinkage and localised loss of neurons, mainly in the hippocampus and basal forebrain. The loss of cholinergic neurons in the hippocampus and frontal cortex is a feature of the disease, and is thought to underlie the cognitive deficit and loss of short-term memory that occur in AD. Two microscopic features are characteristic of the disease, namely extracellular amyloid plaques, consisting of amorphous extracellular deposits of β-amyloid protein (known as Aβ), and intraneuronal neurofibrillary tangles, comprising filaments of a phosphorylated form of a microtubule-associated protein (Tau). Both of these deposits are protein aggregates that result from misfolding of native proteins, as discussed above. They appear also in normal brains, although in smaller numbers. The early appearance of amyloid deposits presages the development of AD, although symptoms may not develop for many years. Altered processing of amyloid protein from its precursor (amyloid precursor protein, APP; see Bossy-Wetzel et al., 2004) is now recognised as the key to the pathogenesis of AD. This conclusion is based on several lines of evidence, particularly the genetic analysis of certain, relatively rare, types of familial AD, in which mutations of the APP gene, or of other genes that control amyloid processing, have been discovered. The APP gene resides on chromosome 21, which is duplicated in Down’s syndrome, in which early AD-like dementia occurs in association with overexpression of APP.
Amyloid deposits consist of aggregates of Aβ (Fig. 39.3), a 40 or 42 residue segment of APP, generated by the action of specific proteases (secretases). Aβ40 is produced normally in small amounts, whereas Aβ42 is overproduced as a result of the genetic mutations mentioned above. Both proteins aggregate to form amyloid plaques, but Aβ42 shows a stronger tendency than Aβ40 to do so, and appears to be the main culprit in amyloid formation. APP is a 770-amino acid membrane protein normally expressed by many cells, including CNS neurons. Cleavage by α-secretase releases the large extracellular domain as soluble APP, which is believed to serve a physiological trophic function. Formation of Aβ involves cleavage at two different points, including one in the intramembrane domain of APP, by β- and γ-secretases (Fig. 39.3). γ-Secretase is a clumsy enzyme—actually a large intramembrane complex of several proteins—that lacks precision and cuts APP at different points in the transmembrane domain, generating Aβ fragments of different lengths, including Aβ40 and 42. Mutations in this region of the APP gene affect the preferred cleavage point, tending to favour formation of Aβ42. Mutations of the unrelated presenilin genes result in increased activity of γ-secretase, because the presenilin proteins form part of the γ-secretase complex. These different AD-related mutations increase the ratio of Aβ42:Aβ40, which can be detected in plasma, serving as a marker for familial AD. Mutations in another gene, that for the lipid transport protein ApoE4 which facilitates the clearance of Aβ oligomers, also predispose to AD, probably because the mutant form of ApoE4 proteins are less effective in this function.
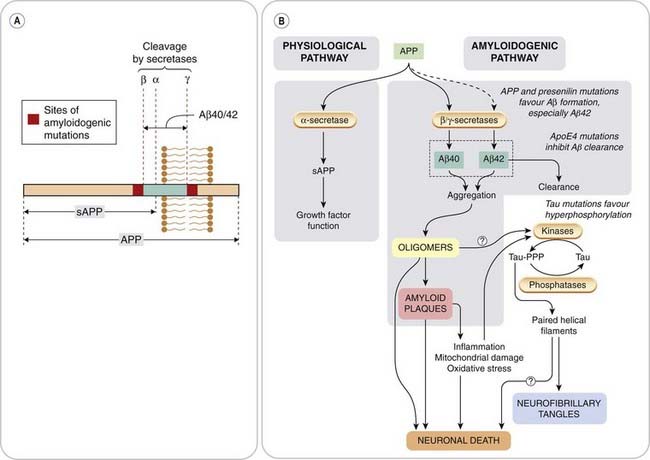
Fig. 39.3 Pathogenesis of Alzheimer’s disease.
[A] Structure of amyloid precursor protein (APP), showing origin of secreted APP (sAPP) and Aβ amyloid protein. The regions involved in amyloidogenic mutations discovered in some cases of familial Alzheimer’s disease are shown flanking the Aβ sequence. APP cleavage involves three proteases: secretases α, β and γ. α-Secretase produces soluble APP, whereas β- and γ-secretases generate Aβ amyloid protein. γ-Secretase can cut at different points, generating Aβ peptides of varying lengths, including Aβ40 and Aβ42, the latter having a high tendency to aggregate as amyloid plaques. [B] Processing of APP. The main ‘physiological’ pathway gives rise to sAPP, which exerts a number of trophic functions. Cleavage of APP at different sites gives rise to Aβ, the predominant form normally being Aβ40, which is weakly amyloidogenic. Mutations in APP or presenilins increase the proportion of APP, which is degraded via the amyloidogenic pathway, and also increase the proportion converted to the much more strongly amyloidogenic form Aβ42. Clearance of Aβ is impaired by mutations in the apoE4 gene. Hyperphosphorylated Tau results in dissociation of Tau from microtubules, misfolding and aggregation to form paired helical filaments, which enhance Aβ toxicity.
It is uncertain exactly how Aβ accumulation causes neurodegeneration, and whether the damage is done by soluble Aβ monomers or oligomers or by amyloid plaques. There is evidence that the cells die by apoptosis, although an inflammatory response is also evident. Expression of Alzheimer mutations in transgenic animals (see Götz & Ittner, 2008) causes plaque formation and neurodegeneration, and also increases the susceptibility of CNS neurons to other challenges, such as ischaemia, excitotoxicity and oxidative stress, and this increased vulnerability may be the cause of the progressive neurodegeneration in AD. These transgenic models are of great value in testing potential drug therapies aimed at retarding the neurodegenerative process.
The other main player on the biochemical stage is Tau, the protein of which the neurofibrillary tangles are composed (Fig. 39.3). Their role in neurodegeneration is unclear, although similar ‘tauopathies’ occur in many neurodegenerative conditions (see Brunden et al., 2009; Hanger et al., 2009). Tau is a normal constituent of neurons, being associated with the intracellular microtubules that serve as tracks for transporting materials along nerve axons. In AD and other tauopathies, Tau is abnormally phosphorylated by the action of various kinases, and dissociates from microtubules to be deposited intracellularly as paired helical filaments with a characteristic microscopic appearance. When the cells die, these filaments aggregate as extracellular neurofibrillary tangles. Tau phosphorylation is enhanced by the presence of Aβ, possibly by activation of kinases. Conversely, hyperphosphorylated Tau favours the formation of amyloid deposits. Whether hyperphosphorylation and intracellular deposition of Tau directly harms the cell is not certain, although it is known that it impairs fast axonal transport, a process that depends on microtubules.
Loss of cholinergic neurons
Although changes in many transmitter systems have been observed, mainly from measurements on postmortem AD brain tissue, a relatively selective loss of cholinergic neurons in the basal forebrain nuclei (Ch. 38) is characteristic. This discovery, made in 1976, implied that pharmacological approaches to restoring cholinergic function might be feasible, leading to the use of cholinesterase inhibitors to treat AD (see below).
Choline acetyl transferase activity, acetylcholine content, and acetylcholinesterase and choline transport in the cortex and hippocampus are all reduced considerably in AD but not in other disorders, such as depression or schizophrenia. Muscarinic receptor density, determined by binding studies, is not affected, but nicotinic receptors, particularly in the cortex, are reduced. The reason for the selective loss of cholinergic neurons resulting from Aβ formation is not known.
Alzheimer’s disease
Therapeutic Approaches
Unravelling the mechanism of neurodegeneration in AD has yet to result in therapies able to retard it. Currently, cholinesterase inhibitors (see Ch. 13) and memantine (see above) are the only drugs approved for treating AD. Many other approaches have been explored, based on the amyloid hypothesis as well as other ideas for neuroprotection (see Citron, 2004; Spencer et al., 2007), so far without success in clinical trials. The Web site http://www.alzforum.org keeps track of ongoing trials.5
Cholinesterase Inhibitors
Tacrine, the first drug approved for treating AD, was investigated on the basis that enhancement of cholinergic transmission might compensate for the cholinergic deficit. Trials showed modest improvements in tests of memory and cognition in about 40% of AD patients, but no improvement in other functional measures that affect quality of life. Tacrine has to be given four times daily and produces cholinergic side effects such as nausea and abdominal cramps, as well as hepatotoxicity in some patients, so it is far from an ideal drug. Later compounds, which also have limited efficacy but are more effective than tacrine in improving quality of life, include donepezil, rivastigmine and galantamine (Table 39.2). These drugs produce a measurable, although slight, improvement of cognitive function in AD patients, but this may be too small to be significant in terms of everyday life.
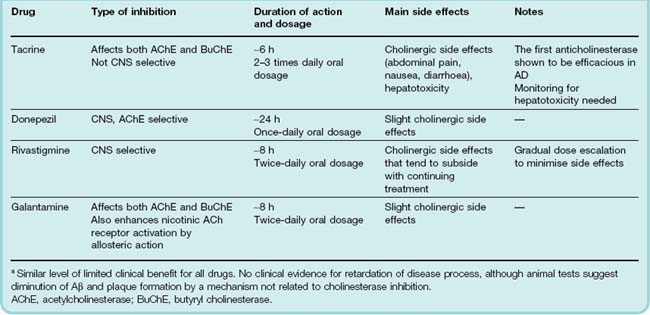
There is some evidence from laboratory studies that cholinesterase inhibitors may act somehow to reduce the formation or neurotoxicity of Aβ, and therefore retard the progression of AD as well as producing symptomatic benefit. Clinical trials, however, have shown only a small improvement in cognitive function, with no effect on disease progression.
Other drugs aimed at improving cholinergic function that are being investigated include other cholinesterase inhibitors and a variety of muscarinic and nicotinic receptor agonists, none of which looks promising on the basis of early clinical results.
Memantine
The other drug currently approved for the treatment of AD is memantine, an orally active antagonist at NMDA receptors, with weaker blocking actions on various other amine receptors. It was originally introduced as an antiviral drug, and resurrected as a potential inhibitor of excitotoxicity. It produces—surprisingly—a modest cognitive improvement in moderate or severe AD, but does not appear to be neuroprotective. It causes few side effects, and has a long plasma half-life.
Clinical use of drugs in dementia 
Acetylcholinesterase inhibitors
Inhibiting neurodegeneration
For most of the disorders discussed in this chapter, including AD, the Holy Grail, which so far eludes us, would be a drug that retards neurodegeneration. Now that we have several well-characterised targets, such as Aβ formation by the β- and γ-secretases, and Aβ neurotoxicity, together with a range of transgenic animal models of AD on which compounds can be tested, the prospects certainly look brighter than they did a decade ago. Particular developments are worth mentioning (see Selkoe & Schenk, 2003, and Citron, 2004, for more details).
Inhibitors of β- and γ-secretase have been identified and are undergoing clinical trials. Though they are effective in reducing Aβ formation, several have proved toxic to the immune system and gastrointestinal tract, and development has been halted.
Kinase inhibitors aimed at preventing Tau phosphorylation are also being investigated (see Brunden et al., 2009). The large number of phosphorylation sites and different kinases make this a difficult approach.
An ingenious new approach was taken by Schenk et al. (1999), who immunised AD transgenic mice with Aβ protein, and found that this not only prevented but also reversed plaque formation. Initial trials in humans had to be terminated because of neuroinflammatory complications, but monoclonal Aβ antibodies are undergoing clinical trials
Epidemiological studies reveal that some non-steroidal anti-inflammatory drugs (NSAIDs; see Ch. 26) used routinely to treat arthritis reduce the likelihood of developing AD. Ibuprofen and indometacin have this effect, although other NSAIDs, such as aspirin, do not, nor do anti-inflammatory steroids such as prednisolone. There is some evidence that certain NSAIDs may affect Aβ-induced neurotoxicity by mechanisms other than cyclo-oxygenase inhibition (see Weggen et al., 2007). Disappointingly, however, clinical trials with various NSAIDs have so far failed to show evidence of benefit.
Aβ plaques bind copper and zinc, and removal of these metal ions promotes dissolution of the plaques. The amoebicidal drug clioquinol is a metal-chelating agent that causes regression of amyloid deposits in animal models of AD, and showed some benefit in initial clinical trials. Clioquinol itself has known toxic effects in humans, which preclude its routine clinical use, but less toxic metal-chelating agents are under investigation.
Shortage of growth factors (particularly nerve growth factor) may contribute to the loss of forebrain cholinergic neurons in AD. Administering growth factors into the brain is not realistic for routine therapy, but alternative approaches, such as implanting cells engineered to secrete nerve growth factor, are under investigation.
Parkinson’s Disease
Features of Parkinson’s Disease
Parkinson’s disease (see review by Schapira, 2009) is a progressive disorder of movement that occurs mainly in the elderly. The chief symptoms are:
Parkinsonian patients walk with a characteristic shuffling gait. They find it hard to start, and once in progress they cannot quickly stop or change direction. PD is commonly associated with dementia, depression and autonomic dysfunction, probably because the degenerative process is not confined to the basal ganglia but also affects other parts of the brain. In the later stages of the disease, the non-motor symptoms often predominate.
Parkinson’s disease often occurs with no obvious underlying cause, but it may be the result of cerebral ischaemia, viral encephalitis or other types of pathological damage. The symptoms can also be drug induced, the main drugs involved being those that reduce the amount of dopamine in the brain (e.g. reserpine; see Ch. 14) or block dopamine receptors (e.g. antipsychotic drugs such as chlorpromazine; see Ch. 45). There are rare instances of familial early-onset PD, and several gene mutations have been identified, including those encoding synuclein and parkin. Study of these gene mutations has given some clues about the mechanism underlying the neurodegenerative process (see below).
Neurochemical changes
Parkinson’s disease affects the basal ganglia, and its neurochemical origin was discovered in 1960 by Hornykiewicz, who showed that the dopamine content of the substantia nigra and corpus striatum (see Ch. 38) in postmortem brains of PD patients was extremely low (usually less than 10% of normal), associated with a loss of dopaminergic neurons in the substantia nigra and degeneration of nerve terminals in the striatum. Other monoamines, such as noradrenaline and 5-hydroxytryptamine, were much less affected than dopamine. Gradual loss of dopamine occurs over several years, with symptoms of PD appearing only when the striatal dopamine content has fallen to 20–40% of normal. Lesions of the nigrostriatal tract or chemically induced depletion of dopamine in experimental animals also produce symptoms of PD. The symptom most clearly related to dopamine deficiency is hypokinesia, which occurs immediately and invariably in lesioned animals. Rigidity and tremor involve more complex neurochemical disturbances of other transmitters (particularly acetylcholine, noradrenaline, 5-hydroxytryptamine and GABA) as well as dopamine. In experimental lesions, two secondary consequences follow damage to the nigrostriatal tract, namely a hyperactivity of the remaining dopaminergic neurons, which show an increased rate of transmitter turnover, and an increase in the number of dopamine receptors, which produces a state of denervation hypersensitivity (see Ch. 12). The striatum expresses mainly D1 (excitatory) and D2 (inhibitory) receptors (see Ch. 38), but fewer D3 and D4 receptors. A simplified diagram of the neuronal circuitry involved, and the pathways primarily affected in PD and Huntington’s disease, is shown in Figure 39.4.
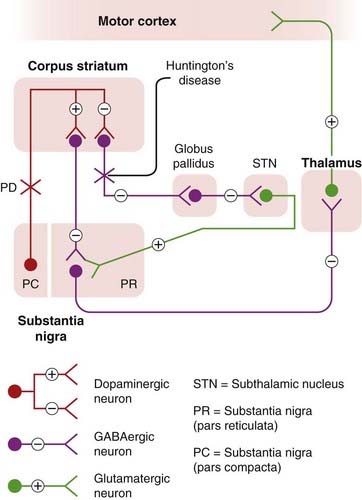
Fig. 39.4 Simplified diagram of the organisation of the extrapyramidal motor system and the defects that occur in Parkinson’s disease (PD) and Huntington’s disease.
Normally, activity in nigrostriatal dopamine neurons causes excitation of striatonigral neurons and inhibition of striatal neurons that project to the globus pallidus. In either case, because of the different pathways involved, the activity of GABAergic neurons in the substantia nigra is suppressed, releasing the restraint on the thalamus and cortex, causing motor stimulation. In PD, the dopaminergic pathway from the substantia nigra (pars compacta) to the striatum is impaired. In Huntington’s disease, the GABAergic striatopallidal pathway is impaired, producing effects opposite to the changes in PD.
Cholinergic interneurons of the corpus striatum (not shown in Fig. 39.4) are also involved in PD and Huntington’s disease. Acetylcholine release from the striatum is strongly inhibited by dopamine, and it is suggested that hyperactivity of these cholinergic neurons contributes to the symptoms of PD. The opposite happens in Huntington’s disease, and in both conditions therapies aimed at redressing the balance between the dopaminergic and cholinergic neurons are, up to a point, beneficial.
Pathogenesis of Parkinson’s Disease
Parkinson’s disease is believed to be caused mainly by environmental factors, although the rare types of hereditary PD have provided some valuable clues about the mechanism. As with other neurodegenerative disorders, the damage is caused by protein misfolding and aggregation, aided and abetted by other familiar villains, namely excitotoxicity, mitochondrial dysfunction, oxidative stress, inflammation and apoptosis (see Lotharius & Brundin, 2002; Schapira, 2009). Aspects of the pathogenesis and animal models of PD are described by Meredith et al. (2008).
Neurotoxins
New light was thrown on the possible aetiology of PD by a chance event. In 1982, a group of young drug addicts in California suddenly developed an exceptionally severe form of PD (known as the ‘frozen addict’ syndrome), and the cause was traced to the compound 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which was a contaminant in a preparation used as a heroin substitute (see Langston, 1985). MPTP causes irreversible destruction of nigrostriatal dopaminergic neurons in various species, and produces a PD-like state in primates. MPTP acts by being converted to a toxic metabolite, MPP+, by the enzyme monoamine oxidase (MAO, specifically by the MAO-B subtype; see Chs 14 and 46). MPP+ is taken up by the dopamine transport system, and thus acts selectively on dopaminergic neurons; it inhibits mitochondrial oxidation reactions, producing oxidative stress. MPTP appears to be selective in destroying nigrostriatal neurons and does not affect dopaminergic neurons elsewhere—the reason for this is unknown. Selegiline, a selective MAO-B inhibitor, prevents MPTP-induced neurotoxicity by blocking its conversion to MPP+. Selegiline is also used in treating PD (see below); as well as inhibiting dopamine breakdown, it might also work by blocking the metabolic activation of a putative endogenous, or environmental, MPTP-like substance, which is involved in the causation of PD. It is possible that dopamine itself could be the culprit, because oxidation of dopamine gives rise to potentially toxic metabolites. Whether or not the action of MPTP reflects the natural pathogenesis of PD, the MPTP model is a very useful experimental tool for testing possible therapies.
Impaired mitochondrial function is a feature of the disease in humans. Various herbicides, such as rotenone, that selectively inhibit mitochondrial function cause a PD-like syndrome in animals. PD in humans is more common in agricultural areas than in cities, suggesting that environmental toxins could be a factor in its causation.
Molecular aspects
Parkinson’s disease, as well as several other neurodegenerative disorders, is associated with the development of intracellular protein aggregates known as Lewy bodies in various parts of the brain. They consist largely of α-synuclein, a synaptic protein, present in large amounts in normal brains. Mutations occur in rare types of hereditary PD (see above), and it is believed that such mutations render the protein resistant to degradation within cells, causing it to pile up in Lewy bodies. It is possible (see Lotharius & Brundin, 2002) that the normal function of α-synuclein is related to synaptic vesicle recycling, and that the mutated form loses this functionality, with the result that vesicular storage of dopamine is impaired. This may lead to an increase in cytosolic dopamine, degradation of which produces reactive oxygen species and hence neurotoxicity. Consistent with the α-synuclein hypothesis, another mutation associated with PD (parkin) also involves a protein that participates in the intracellular degradation of rogue proteins. Other gene mutations that have been identified as risk factors for early-onset PD code for proteins involved in mitochondrial function, making cells more susceptible to oxidative stress.
Thus, a picture similar to AD pathogenesis is slowly emerging. Misfolded α-synuclein, facilitated by genetic mutations or possibly by environmental factors, builds up in the cell as a result of impaired protein degradation (resulting from defective parkin) in the form of Lewy bodies, which, by unknown mechanisms, compromise cell survival. If oxidative stress is increased, as a result of ischaemia, mitochondrial poisons or mutations of certain mitochondrial proteins, the result is cell death.
Parkinson’s disease
Drug Treatment of Parkinson’s Disease
Currently, the main drugs used (see Fig. 39.5) are:
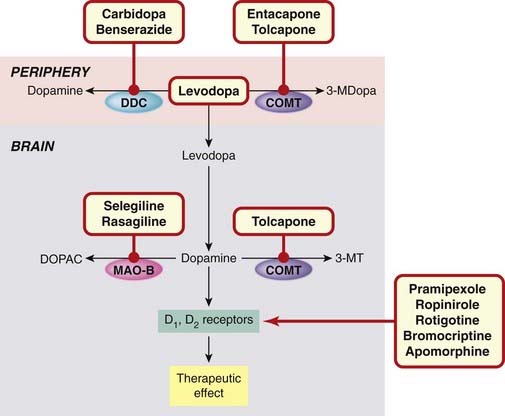
Fig. 39.5 Sites of action of drugs used to treat Parkinson’s disease.
Levodopa enters the brain and is converted to dopamine (the deficient neurotransmitter). Inactivation of levodopa in the periphery is prevented by inhibitors of DDC and COMT. Inactivation in the brain is prevented by inhibitors of COMT and MAO-B. Dopamine agonists act directly on striatal dopamine receptors. 3-MDopa, 3-methoxydopa; 3-MT, 3-methoxytyrosine; COMT, catechol-O-methyl transferase; DDC, DOPA decarboxylase; DOPAC, dihydroxyphenylacetic acid; MAO-B, monoamine oxidase B.
Amantadine (thought to act by releasing dopamine), and muscarinic ACh receptor antagonists (e.g. benztropine) are occasionally used.
Despite past optimism, none of the drugs used to treat PD affects the progression of the disease. For general reviews of current and future approaches, see Olanow (2004) and Schapira (2009).
Levodopa
Levodopa is the first-line treatment for PD and is combined with a dopa decarboxylase inhibitor, either carbidopa or benserazide, which reduces the dose needed by about 10-fold and diminishes the peripheral side effects. It is well absorbed from the small intestine, a process that relies on active transport, although much of it is inactivated by MAO in the wall of the intestine. The plasma half-life is short (about 2 h). Conversion to dopamine in the periphery, which would otherwise account for about 95% of the levodopa dose and cause troublesome side effects, is largely prevented by the decarboxylase inhitor. Decarboxylation occurs rapidly within the brain, because the decarboxylase inhibitors do not penetrate the blood–brain barrier. It is not certain whether the effect depends on an increased release of dopamine from the few surviving dopaminergic neurons or on a ‘flooding’ of the synapse with dopamine formed elsewhere. Because synthetic dopamine agonists (see below) are equally effective, the latter explanation is more likely, and animal studies suggest that levodopa can act even when no dopaminergic nerve terminals are present. On the other hand, the therapeutic effectiveness of levodopa decreases as the disease advances, so part of its action may rely on the presence of functional dopaminergic neurons. Combination of levodopa plus dopa decarboxylase inhibitor with entacapone, a catechol-O-methyl transferase (COMT) inhibitor (see Ch. 14) to inhibit its degradation, is used in patients troubled by ‘end of dose’ motor fluctuations.
Therapeutic effectiveness
About 80% of patients show initial improvement with levodopa, particularly of rigidity and hypokinesia, and about 20% are restored virtually to normal motor function. As time progresses, the effectiveness of levodopa gradually declines (Fig. 39.6). In a typical study of 100 patients treated with levodopa for 5 years, only 34 were better than they had been at the beginning of the trial, 32 patients having died and 21 having withdrawn from the trial. It is likely that the loss of effectiveness of levodopa mainly reflects the natural progression of the disease, but receptor downregulation and other compensatory mechanisms may also contribute. There is no evidence that levodopa can actually accelerate the neurodegenerative process through overproduction of dopamine, as was suspected on theoretical grounds (see above). Overall, levodopa increases the life expectancy of PD patients, probably as a result of improved motor function, although some symptoms (e.g. dysphagia, cognitive decline) are not improved.
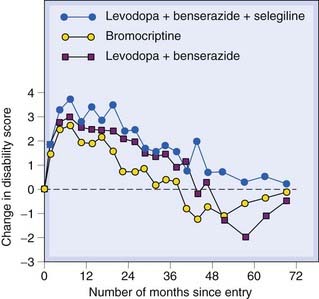
Fig. 39.6 Comparison of levodopa/benserazide, levodopa/benserazide/selegiline and bromocriptine on progression of Parkinson’s disease symptoms.
Patients (249–271 in each treatment group) were assessed on a standard disability rating score. Before treatment, the average rate of decline was 0.7 units/year. All three treatments produced improvement over the initial rating for 2–3 years, but the effect declined, either because of refractoriness to the drugs or disease progression. Bromocriptine appeared slightly less effective than levodopa regimens, and there was a higher drop-out rate due to side effects in this group.
(From Parkinson’s Disease Research Group 1993 Br Med J 307: 469–472.)
Unwanted effects
There are two main types of unwanted effect:
In addition to these slowly developing side effects, levodopa produces several acute effects, which are experienced by most patients at first but tend to disappear after a few weeks. The main ones are as follow:
Dopamine Agonists
Two older drugs, bromocriptine and pergolide, are orally active ergot derivatives that act mainly on D1 and D2 receptors (see Ch. 38). Bromocriptine, which inhibits the release of prolactin from the anterior pituitary gland, was first introduced for the treatment of galactorrhoea and gynaecomastia (Ch. 32). Though effective in controlling the symptoms of PD, their usefulness is limited by side effects, mainly nausea and vomiting and somnolence. Pergolide is also believed to cause heart valve disease. These disadvantages have led to the replacement of these drugs by the non-ergot compounds pramipexole and ropinirole, which are D2/3 selective and better tolerated, and do not show the fluctuations in efficacy associated with levodopa. They do, however, cause somnolence and sometimes hallucinations and recent evidence suggests that they may predispose to compulsive behaviours, such as excessive gambling,6 over-eating and sexual excess, related to the ‘reward’ functions of dopamine (see Ch. 48).
A disadvantage of current dopamine agonists is their short plasma half-life (6–8 h), requiring three-times daily dosage, though slow-release once-daily formulations are now available.
Rotigotine is a newer agent, delivered as a transdermal patch, with similar efficacy and side effects.
Apomorphine, given by injection, is sometimes used to control the ‘off effect’ with levodopa. Because of its powerful emetic action, it must be combined with an oral antiemetic drug. It has other serious adverse effects (mood and behavioural changes, cardiac dysrhythmias, hypotension) and is a last resort if other drugs fail.
MAO-B Inhibitors
Selegiline is a selective MAO-B7 inhibitor, which lacks the unwanted peripheral effects of non-selective MAO inhibitors used to treat depression (Ch. 46) and, in contrast to them, does not provoke the ‘cheese reaction’ or interact so frequently with other drugs. Inhibition of MAO-B protects dopamine from extraneuronal degradation and was initially used as an adjunct to levodopa. Long-term trials showed that the combination of selegiline and levodopa was more effective than levodopa alone in relieving symptoms and prolonging life. Recognition of the role of MAO-B in neurotoxicity (see above) suggested that selegiline might be neuroprotective rather than merely enhancing the action of levodopa, but clinical studies do not support this. A large-scale trial (Fig. 39.6) showed no difference when selegiline was added to levodopa/benserazide treatment. Selegiline is metabolised to amphetamine, and sometimes causes excitement, anxiety and insomnia. Rasagiline, a very similar drug, does not have this unwanted effect, and a recent trial (Olanow et al., 2009) suggests that it may somewhat retard disease progression, as well alleviating symptoms.
Other Drugs Used in Parkinson’s Disease
Amantadine
Amantadine was introduced as an antiviral drug and discovered by accident in 1969 to be beneficial in PD. Many possible mechanisms for its action have been suggested based on neurochemical evidence of increased dopamine release, inhibition of amine uptake or a direct action on dopamine receptors. Most authors now suggest, although not with much conviction, that increased dopamine release is primarily responsible for the clinical effects.
Amantadine is less effective than levodopa or bromocriptine, and its action declines with time. Its side effects are considerably less severe, although qualitatively similar to those of levodopa.
Acetylcholine antagonists
For more than a century, until levodopa was discovered, atropine and related drugs were the main form of treatment for PD. Muscarinic acetylcholine receptors exert an inhibitory effect on dopaminergic nerve terminals, suppression of which compensates for a lack of dopamine. The side effects of muscarinic antagonists—dry mouth, constipation, impaired vision, urinary retention—are troublesome, and they are now rarely used, except to treat parkinsonian symptoms in patients receiving antipsychotic drugs (which are dopamine antagonists and thus nullify the effect of L-dopa; see Ch. 45).
Drugs used in Parkinson’s disease
Neural Transplantation and Brain Stimulation
Parkinson’s disease is the first neurodegenerative disease for which neural transplantation was attempted in 1982, amid much publicity. Various transplantation approaches have been tried, based on the injection of dissociated fetal cells (neuroblasts) directly into the striatum. Trials in patients with PD (see Björklund & Lindvall, 2000; Barker & Rosser, 2001) have mainly involved injection of midbrain cells from aborted human fetuses. Such transplants have been shown to survive and establish functional dopaminergic connections, and to produce clinical benefit in many cases (see Lindvall & Kokaia, 2009). However, some patients have gone on to develop serious dyskinesias, possibly due to dopamine overproduction. It is not yet known whether the transplanted cells will prove vulnerable to the neurodegenerative process responsible for killing the resident dopaminergic neurons. The use of fetal material is, of course, fraught with difficulties (usually cells from five or more fetuses are needed for one transplant), and hopes for the future rest mainly on the possibility of developing stem cell transplants (see Lindvall & Kokaia, 2009).
Electrical stimulation of the subthalamic nuclei with implanted electrodes (which inhibits ongoing neural activity, equivalent to reversible ablation) is used in severe cases, and can improve motor dysfunction in PD, but does not improve cognitive and other symptoms (see Benabid et al., 2009).
Huntington’s Disease
Huntington’s disease (HD) is an inherited (autosomal dominant) disorder resulting in progressive brain degeneration, starting in adulthood and causing rapid deterioration and death. As well as dementia, it causes severe motor symptoms in the form of involuntary writhing movements, which are highly disabling. It is the commonest of a group of so-called trinucleotide repeat neurodegenerative diseases, associated with the expansion of the number of repeats of the CAG sequence in specific genes, and hence the number (50 or more) of consecutive glutamine residues at the N-terminal of the expressed protein (see Walker, 2007). The larger the number of repeats, the earlier the appearance of symptoms. The protein coded by the HD gene, huntingtin, which normally possesses a chain of fewer than 30 glutamine residues, is a soluble cytosolic protein of unknown function found in all cells. HD develops when the mutant protein contains 40 or more repeats. The long poly-Gln chains reduce the solubility of huntingtin, and favour the formation of aggregates, which are formed from proteolytic N-terminal fragments that include the poly-Gln region. As with AD and PD, aggregation is probably responsible for the neuronal loss, which affects mainly the cortex and the striatum, resulting in progressive dementia and severe involuntary jerky (choreiform) movements. Studies on postmortem brains showed that the dopamine content of the striatum was normal or slightly increased, while there was a 75% reduction in the activity of glutamic acid decarboxylase, the enzyme responsible for GABA synthesis (Ch. 37). It is believed that the loss of GABA-mediated inhibition in the basal ganglia produces a hyperactivity of dopaminergic synapses, so the syndrome is in some senses a mirror image of PD (Fig. 39.4). The effects of drugs that influence dopaminergic transmission are correspondingly the opposite of those that are observed in PD, dopamine antagonists being effective in reducing the involuntary movements, while drugs such as levodopa and bromocriptine make them worse. Drugs used to alleviate the motor symptoms include tetrabenazine (an inhibitor of the vesicular monoamine transporter (see Ch. 14) that reduces dopamine storage, dopamine antagonists such as chlorpromazine (Ch. 45) and the GABA agonist baclofen (Ch. 37). These do not affect dementia or retard the course of the disease, and it is possible that drugs that inhibit excitotoxicity, or possibly neural transplantation procedures when these become available (see above), may prove useful.
Neurodegenerative Prion Diseases
A group of human and animal diseases associated with a characteristic type of neurodegeneration, known as spongiform encephalopathy because of the vacuolated appearance of the affected brain, has recently been the focus of intense research activity (see Collinge, 2001; Prusiner, 2001). A key feature of these diseases is that they are transmissible through an infective agent, although not, in general, across species. The recent upsurge of interest has been spurred mainly by the discovery that the bovine form of the disease, bovine spongiform encephalopathy (BSE), is transmissible to humans. Different human forms of spongiform encephalopathy include Creutzfeld–Jacob disease (CJD) which is unrelated to BSE, and the new variant form (vCJD), which results from eating, or close contact with, infected beef or human tissue. Another human form is kuru, a neurodegenerative disease affecting cannibalistic tribes in Papua New Guinea. These diseases cause a progressive, and sometimes rapid, dementia and loss of motor coordination, for which no therapies currently exist. Scrapie, a common disease of domestic sheep, is another example, and it may have been the practice of feeding sheep offal to domestic cattle that initiated an epidemic of BSE in Britain during the 1980s, leading to the appearance of vCJD in humans in the mid-1990s. Although the BSE epidemic has been controlled, there is concern that more human cases may develop in its wake, because the incubation period—known to be long—is uncertain.
Prion diseases are examples of protein misfolding diseases (see above) in which the prion protein adopts a misfolded conformation that forms insoluble aggregates. The infectious agent responsible for transmissible spongiform encephalopathies such as vCJD is, unusually, a protein, known as a prion. The protein involved (PrPC) is a normal cytosolic constituent of the brain and other tissues, whose functions are not known. As a result of altered glycosylation, the protein can become misfolded, forming the insoluble PrPSc form, which has the ability to recruit normal PrPC molecules to the misfolded PrPSc, thus starting a chain reaction. PrPSc—the infective agent—accumulates and aggregates as insoluble fibrils, and is responsible for the progressive neurodegeneration. In support of this unusual form of infectivity, it has been shown that injection of PrPSc into normal mice causes spongiform encephalopathy, whereas PrP knockout mice, which are otherwise fairly normal, are resistant because they lack the substrate for the autocatalytic generation of PrPSc. Fortunately, the infection does not easily cross between species, because there are differences between the PrP genes of different species. It is possible that a mutation of the PrP gene in either sheep or cattle produced the variant form that became infective in humans.
This chain of events bears some similarity to that of AD, in that the brain accumulates an abnormal form of a normally expressed protein.
There is as yet no known treatment for this type of encephalopathy, but laboratory experiments suggest that two very familiar drugs, namely quinacrine (an antimalarial drug) and chlorpromazine (a widely used antipsychotic drug; Ch. 45), can inhibit PrPSc aggregation in mouse models. Both are under investigation for treating human CJD. Pentosan polyphosphate, a glycosidic polymer that binds PrP and inhibits disease progression when give by intracerebroventricular injection in animal models, is also being tested in humans. Other possible strategies, none yet tested in patients, are discussed by Mallucci & Collinge (2005).
References and Further Reading
General mechanisms of neurodegeneration
Barnham K.J., Masters C.L., Bush A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov.. 2004;3:205-214. (Update on the oxidative stress model of neurodegeneration, including evidence based on various transgenic animal models)
Bossy-Wetzel E., Schwarzenbacher R., Lipton S.A. Molecular pathways to neurodegeneration. Nat. Med.. 2004;10(Suppl.):S2-S9. (Review of molecular mechanisms underlying various chronic neurodegenerative diseases, including those discussed in the chapter)
Brunden K., Trojanowski J.O., Lee V.M.-Y. Advances in Tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat. Rev. Drug Discov.. 2009;8:783-793. (Good detailed review of the current status of Tau-directed drug discovery efforts, with a realistic assessment of the problems that have to be overcome)
Coyle J.T., Puttfarken P. Oxidative stress, glutamate and neurodegenerative disorders. Science. 1993;262:689-695. (Good review article)
Forman M.S., Trojanowski J.G., Lee V.M.-Y. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med.. 2004;10:1055-1063. (General review on pathogenesis of neurodegenerative diseases—not much on therapeutic approaches, despite the title)
Gross C.G. Neurogenesis in the adult brain: death of a dogma. Nat. Rev. Neurosci.. 2000;1:67-73.
Hanger D.P., Anderton B.H., Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med.. 2009;15:112-119.
Okouchi M., Ekshyyan O., Maracine M., Aw T.Y. Neuronal apoptosis in neurodegeneration. Antioxid. Redox. Signal. 2007;9:1059-1096. (Detailed review describing the role of apoptosis, the factors that induce it and possible therapeutic strategies aimed at preventing it, in various neurodegenerative disorders)
Petrozzi L., Ricci G., Giglioli N.J., et al. Mitochondria and neurodegeneration. Biosci. Rep.. 2007;27:87-104. (Summarises evidence for the involvement of mitochondrial dysfunction in several neurodegenerative diseases)
Stefani M., Dobson C.M. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med.. 2003;81:678-699. (Excellent review article on protein misfolding as the underlying cause of chronic neurodegenerative disease)
Yamada K., Nabeshima T. Animal models of Alzheimer’s disease and evaluation of anti-dementia drugs. Pharmacol. Ther.. 2000;88:93-113. (Describes pathology of AD, transgenic and other animal models, and therapeutic approaches)
Zhao C., Deng W., Gage F.H. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645-660. (Review by one of the pioneers in this controversial field. Neurogenesis probably contributes to learning, but evidence for involvement in neural repair is weak)
Citron M. Strategies for disease modification in Alzheimer’s disease. Nat. Rev. Neurosci.. 2004;5:677-685. (A review—generally optimistic—of the status of new therapeutic strategies for treating AD)
Götz J., Ittner L.M. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat. Rev. Neurosci.. 2008;9:532-544. (Detailed review focusing on transgenic models)
Rakic P. Neurogenesis in the primate cortex: an evaluation of the evidence. Nat. Rev. Neurosci.. 2002;3:65-71.
Schenk D., Barbour R., Dunn W., et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173-177. (Report of an ingenious experiment that could have implications for AD treatment in humans)
Schwab C., McGeer P.L. Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J. Alzheimer Dis.. 2008;13:359-369. (Discusses the role of inflammation in neurodegeneration and repair)
Selkoe D.J. Alzheimer’s disease: genotypes, phenotype and treatments. Science. 1997;275:630-631. (Short but informative summary of recent advances in Alzheimer genetics by one of the pioneers in identifying the amyloid hypothesis)
Selkoe D.J. Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol.. 2004;6:1054-1061. (Good review article)
Selkoe D.J., Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu. Rev. Pharmacol. Toxicol.. 2003;43:545-584. (Comprehensive review article)
Spencer B., Rockenstein E., Crews L., et al. Novel strategies for Alzheimer’s disease treatment. Exp. Opin. Biol. Ther.. 2007;7:1853-1867. (Focus on potential applications of gene therapy and other biological approaches)
Townsend K.P., Pratico D. Novel therapeutic opportunities for Alzheimer’s disease: focus on nonsteroidal antiinflammatory drugs. FASEB J.. 2005;19:1592-1601. (Discussion of possible drug targets for treatment of AD, including animal studies and clinical trials data—largely negative, so far)
Weggen S., Rogers M., Eriksen J. NSAIDs: small molecules for prevention of Alzheimer’s disease or precursors for future drug development. Trends Pharmacol. Sci.. 2007;28:536-543. (Summarises data relating to effects of NSAIDs on AD and concludes that mechanisms other than cyclo-oxygenase inhibition may be relevant in the search for new anti-AD drugs)
Barker R.A., Rosser A.E. Neural transplantation therapies for Parkinson’s and Huntington’s diseases. Drug Discov. Today. 2001;6:575-582. (Informative and balanced review article on a controversial topic)
Benabid A.L., Chabardes S., Mitrofanis J., Pollak P. Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson’s disease. Lancet Neurol.. 2009;8:67-81. (Review of current status of brain stimulation techniques, which are increasing in use)
Björklund A., Lindvall O. Cell replacement therapies for central nervous system disorders. Nat. Neurosci.. 2000;3:537-544. (Upbeat review by pioneers in the field of neural transplantation)
Langston W.J. MPTP and Parkinson’s disease. Trends Neurosci.. 1985;8:79-83. (Readable account of the MPTP story by its discoverer)
Lindvall O., Kokaia Z. Prospects of stem cell therapy for replacing dopamine neurons in Parkinson’s disease. Trends Pharmacol. Sci.. 2009;30:260-267. (Suggests the way ahead for neurotransplantation for treating PD)
Lipton S.A., Rosenberg P.A. Excitatory amino acids as a final common pathway for neurologic disorders. New Engl. J. Med.. 1994;330:613-622. (Review emphasising central role of glutamate in neurodegeneration)
Lotharius J., Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci.. 2002;3:833-842. (Review of PD pathogenesis, emphasising the possible role of dopamine itself as a likely source of neurotoxic metabolites)
Meredith G.E., Sonsalla P.K., Chesselet M.-F. Animal models of Parkinson’s disease progression. Acta. Neuropath. 2008;115:185-398. (Useful review of animal models of PD)
Olanow C.W. The scientific basis for the current treatment of Parkinson’s disease. Annu. Rev. Med.. 2004;55:41-60. (Detailed review of current PD therapeutics, based on knowledge of pathophysiology)
Olanow C.W., Rascol O., Hauser R., et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. New Engl. J. Med.. 2009;139:1268-1278. (Well-conducted trial showing that rasagiline can significantly retard disease progression in patients with early PD)
Schapira A.H.V. Neurobiology and treatment of Parkinson’s disease. Trends Pharmacol. Sci.. 2009;30:41-47. (Short review of pathophysiology and treatment of PD, including summary of recent trials)
Besancon E., Guo S., Lok J., Tymianski M., Lo E.H. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol. Sci.. 2008;29:268-275. (Further elaboration of the excitotoxicity model described in this chapter)
Dirnagl U., Iadecola C., Moskowitz M.A. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci.. 1999;22:391-397. (Useful review of mechanisms underlying neuronal damage in stroke)
Green A.R. Pharmacological approaches to acute ischaemic stroke: reperfusion certainly, neuroprotection possibly. Br. J. Pharmacol.. 2008;153(Suppl. 1):S325-S338. (Update on efforts—largely unsuccessful so far—to develop neuroprotective agents)
Green A.R., Odergren T., Ashwood T. Animal models of stroke: do they have value for discovering neuroprotective agents? Trends Pharmacol. Sci.. 2003;24:402-408. (Article suggesting reasons why drug efficacy in animal models does not predict success in the clinic)
Walker F.O. Huntington’s disease. Lancet. 2007;369:218-228. (General review of genetics, pathogenesis and treatment of HD)
Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci.. 2001;24:519-550. (Useful review article)
Mallucci G., Collinge J. Rational targeting for prion therapeutics. Nat. Rev. Neurosci.. 2005;6:23-34. (Realistic review of possible approaches to treating prion diseases; a very difficult problem with nothing really in sight yet)
Prusiner S.B. Neurodegenerative disease and prions. New Engl. J. Med.. 2001;344:1544-1551. (General review article by the discoverer of prions)
1It is recognised that new neurons are formed from progenitor cells (neurogenesis) in certain regions of the adult brain and can become functionally integrated, even in primates (see Rakic, 2002; Zhao et al., 2008). Neurogenesis in the hippocampus is thought to play a role in learning and memory, but plays little if any role in brain repair. However, learning how to harness the inherent ability of neuronal progenitors (stem cells) to form new neurons is seen as an obvious approach to treating neurodegenerative disorders.
2Surprisingly, some SOD mutations associated with ALS are more, rather than less, active than the normal enzyme. The mechanism responsible for neurodegeneration probably involves abnormal accumulation of the enzyme in mitochondria.
3Nevertheless, early reperfusion (within 3 h of the thrombosis) is clearly beneficial, based on clinical evidence with fibrinolytic drugs.
4Nevertheless, Besancon et al., 2008 retain their optimism that among the plethora of channels and transporters possessed by neurons and glia, there must be something that will prove to be a useful neuroprotective drug target.
5The authors admit to disappointment that, despite intense research efforts, no new drugs worthy of mention have emerged since the last edition of this book.
6In 2008, a plaintiff was awarded $8.2m damages by a US court, having become a compulsive gambler (and losing a lot of money) after taking pramipexole for PD—a side effect of which the pharmaceutical company had been aware.
7MAO-B in the brain is located mainly in glial cells, and also in 5-HT neurons (though, surprisingly, it does not appear to be expressed in dopamine neurons).