9 Drug metabolism and elimination
Overview
We describe phases I and II of drug metabolism, emphasising the importance of the cytochrome P450 monooxygenase system. We then cover the processes of biliary excretion and enterohepatic recirculation of drugs, and of drug and drug metabolite elimination by the kidney.
Introduction
Drug elimination is the irreversible loss of drug from the body. It occurs by two processes: metabolism and excretion. Metabolism consists of anabolism and catabolism, i.e. respectively the build-up and breakdown of substances by enzymic conversion of one chemical entity to another within the body, whereas excretion consists of elimination from the body of chemically unchanged drug or its metabolites. The main routes by which drugs and their metabolites leave the body are:
Most drugs leave the body in the urine, either unchanged or as polar metabolites. Some drugs are secreted into bile via the liver, but most of these are then reabsorbed from the intestine. There are, however, instances (e.g. rifampicin; Ch. 50) where faecal loss accounts for the elimination of a substantial fraction of unchanged drug in healthy individuals, and faecal elimination of drugs such as digoxin that are normally excreted in urine (Ch. 21) becomes progressively more important in patients with advancing renal failure. Excretion via the lungs occurs only with highly volatile or gaseous agents (e.g. general anaesthetics; Ch. 40). Small amounts of some drugs are also excreted in secretions such as milk or sweat. Elimination by these routes is quantitatively negligible compared with renal excretion, although excretion into milk can sometimes be important because of effects on the baby (e.g. see McNamara & Abbassi, 2004; Ito, 2000).
Lipophilic substances are not eliminated efficiently by the kidney. Consequently, most lipophilic drugs are metabolised to more polar products, which are then excreted in urine. Drug metabolism occurs predominantly in the liver, especially by the cytochrome P450 (CYP) system. Some P450 enzymes are extrahepatic and play an important part in the biosynthesis of steroid hormones (Ch. 32) and eicosanoids (Ch. 17), but here we are concerned with catabolism of drugs by the hepatic P450 system.
Drug Metabolism
Animals have evolved complex systems that detoxify foreign chemicals (‘xenobiotics’), including carcinogens and toxins present in poisonous plants. Drugs are a special case of such xenobiotics and, like plant alkaloids, they often exhibit chirality (i.e. there is more than one stereoisomer), which affects their overall metabolism. Drug metabolism involves two kinds of reaction, known as phase 1 and phase 2. These often, although not invariably, occur sequentially. Both phases decrease lipid solubility, thus increasing renal elimination.
Phase 1 Reactions
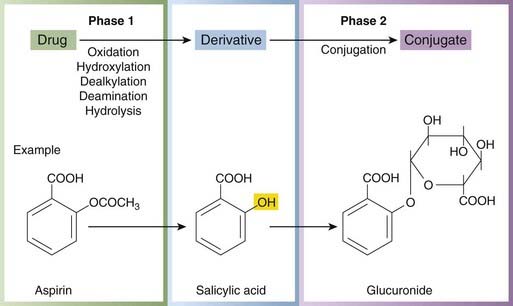
Phase 1 reactions are catabolic (e.g. oxidation, reduction or hydrolysis), and the products are often more chemically reactive and hence, paradoxically, sometimes more toxic or carcinogenic than the parent drug. Phase 1 reactions often introduce a reactive group, such as hydroxyl, into the molecule, a process known as ‘functionalisation’. This group then serves as the point of attack for the conjugating system to attach a substituent such as glucuronide (Fig. 9.1), explaining why phase 1 reactions so often precede phase 2 reactions (see below). Phase 1 reactions take place mainly in the liver. Many hepatic drug-metabolising enzymes, including CYP enzymes, are embedded in the smooth endoplasmic reticulum. They are often called ‘microsomal’ enzymes because, on homogenisation and differential centrifugation, the endoplasmic reticulum is broken into very small fragments that sediment only after prolonged high-speed centrifugation in the microsomal fraction. To reach these metabolising enzymes in life, a drug must cross the plasma membrane. Polar molecules do this less readily than non-polar molecules except where there are specific transport mechanisms (Ch. 8), so intracellular metabolism is important for lipid-soluble drugs, while polar drugs are at least partly excreted unchanged in the urine.
The P450 Monooxygenase System
Nature, classification and mechanism of P450 enzymes
Cytochrome P450 enzymes are haem proteins, comprising a large family (’superfamily’) of related but distinct enzymes, each referred to as CYP followed by a defining set of numbers and a letter. These enzymes differ from one another in amino acid sequence, in sensitivity to inhibitors and inducing agents (see below), and in the specificity of the reactions that they catalyse (see Anzenbacher, 2007 for reviews). Different members of the family have distinct, but often overlapping, substrate specificities, and may act on the same substrates but at different rates. Purification of P450 enzymes and complementary DNA cloning form the basis of the current classification, which is based on amino acid sequence similarities. Seventy-four CYP gene families have been described, of which three main ones (CYP1, CYP2 and CYP3) are involved in drug metabolism in human liver. Examples of therapeutic drugs that are substrates for some important P450 isoenzymes are shown in Table 9.1. Drug oxidation by the monooxygenase P450 system requires drug (substrate, ‘DH’), P450 enzyme, molecular oxygen, NADPH and a flavoprotein (NADPH–P450 reductase). The mechanism involves a complex cycle (Fig. 9.2), but the overall net effect of the reaction is quite simple, namely the addition of one atom of oxygen (from molecular oxygen) to the drug to form a hydroxyl group (product, ‘DOH’), the other atom of oxygen being converted to water.
Table 9.1 Examples of drugs that are substrates of P450 isoenzymes
| Isoenzyme P450 | Drug(s) |
|---|---|
| CYP1A2 | Caffeine, paracetamol (→NAPQI), tacrine, theophylline |
| CYP2B6 | Cyclophosphamide, methadone |
| CYP2C8 | Paclitaxel, repaglinide |
| CYP2C19 | Omeprazole, phenytoin |
| CYP2C9 | Ibuprofen, tolbutamide, warfarin |
| CYP2D6 | Codeine, debrisoquine, S-metoprolol |
| CYP2E1 | Alcohol, paracetamol |
| CYP3A4, 5, 7 | Ciclosporin, nifedipine, indinavir, simvastatin |
(Adapted from http://medicine.iupui.edu/flockhart/table.htm.)
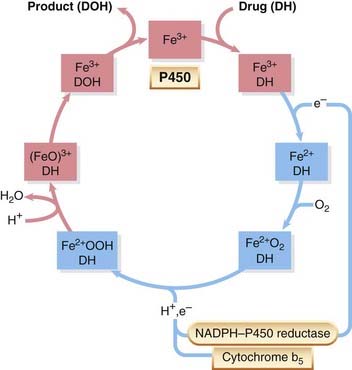
Fig. 9.2 The monooxygenase P450 cycle.
Each of the pink or blue rectangles represents one single molecule of cytochrome P450 (P450) undergoing a catalytic cycle. Iron in P450 is in either the ferric (pink rectangles) or ferrous (blue rectangles) state. P450 containing ferric iron (Fe3+) combines with a molecule of drug (‘DH’); receives an electron from NADPH–P450 reductase, which reduces the iron to Fe2+; combines with molecular oxygen, a proton and a second electron (either from NADPH–P450 reductase or from cytochrome b5) to form an Fe2+OOH–DH complex. This combines with another proton to yield water and a ferric oxene (FeO)3+–DH complex. (FeO)3+ extracts a hydrogen atom from DH, with the formation of a pair of short-lived free radicals (see text), liberation from the complex of oxidised drug (‘DOH’), and regeneration of P450 enzyme.
 P450 enzymes have unique spectral properties, and the reduced forms combine with carbon monoxide to form a pink compound (hence ‘P’) with absorption peaks near 450 nm (range 447–452 nm). The first clue that there is more than one form of CYP came from the observation that treatment of rats with 3-methylcholanthrene (3-MC), an inducing agent (see below), causes a shift in the absorption maximum from 450 to 448 nm—the 3-MC-induced isoform of the enzyme absorbs light maximally at a slightly shorter wavelength than the un-induced enzyme.
P450 enzymes have unique spectral properties, and the reduced forms combine with carbon monoxide to form a pink compound (hence ‘P’) with absorption peaks near 450 nm (range 447–452 nm). The first clue that there is more than one form of CYP came from the observation that treatment of rats with 3-methylcholanthrene (3-MC), an inducing agent (see below), causes a shift in the absorption maximum from 450 to 448 nm—the 3-MC-induced isoform of the enzyme absorbs light maximally at a slightly shorter wavelength than the un-induced enzyme.
P450 and biological variation
There are important variations in the expression and regulation of P450 enzymes between species. For instance, the pathways by which certain dietary heterocyclic amines (formed when meat is cooked) generate genotoxic products involves one member of the P450 superfamily (CYP1A2) that is constitutively present in humans and rats (which develop colon tumours after treatment with such amines) but not in cynomolgus monkeys (which do not). Such species differences have crucial implications for the choice of species to be used for toxicity and carcinogenicity testing during the development of new drugs for use in humans.
Within human populations, there are major sources of interindividual variation in P450 enzymes that are of great importance in therapeutics. These include genetic polymorphisms (alternative sequences at a locus within the DNA strand—alleles—that persist in a population through several generations; Ch. 11). Environmental factors (Ch. 56) are also important, since enzyme inhibitors and inducers are present in the diet and environment. For example, a component of grapefruit juice inhibits drug metabolism (leading to potentially disastrous consequences, including cardiac dysrhythmias; Ch. 56), whereas Brussels sprouts and cigarette smoke induce P450 enzymes. Components of St John’s wort (used to treat depression in ‘alternative’ medicine; Ch. 46) induce CYP450 isoenzymes as well as P-glycoprotein (P-gp) (see Ch. 8 and below, and Henderson et al., 2002).
Not all drug oxidation reactions involve the P450 system: some drugs are metabolised in plasma (e.g. hydrolysis of suxamethonium by plasma cholinesterase; Ch. 13), lung (e.g. various prostanoids; Ch. 17) or gut (e.g. tyramine, salbutamol; Chs 14 and 27). Ethanol (Ch. 48) is metabolised by a soluble cytoplasmic enzyme, alcohol dehydrogenase, in addition to CYP2E1. Other P450-independent enzymes involved in drug oxidation include xanthine oxidase, which inactivates 6-mercaptopurine (Ch. 55), and monoamine oxidase, which inactivates many biologically active amines (e.g. noradrenaline [norepinephrine], tyramine, 5-hydroxytryptamine; Chs 14 and 15).
Hydrolytic reactions (e.g. of aspirin; Fig. 9.1) do not involve hepatic microsomal enzymes but occur in plasma and in many tissues. Both ester and (less readily) amide bonds are susceptible to hydrolysis. Reductive reactions are much less common than oxidations, but some are important. For example, warfarin (Ch. 24) is inactivated by conversion of a ketone to a hydroxyl group by CYP2A6.
Phase 2 Reactions
Phase 2 reactions are synthetic (‘anabolic’) and involve conjugation (i.e. attachment of a substituent group), which usually results in inactive products, although there are exceptions (e.g. the active sulfate metabolite of minoxidil, a potassium channel activator used to treat severe hypertension, Ch. 22; morphine-6-glucuronide is an active metabolite of morphine that is being developed as an analgesic agent [Ch. 41]—on acute administration it induces less nausea and vomiting than the parent drug perhaps because, being more polar, it fails to access the vomiting centres). Phase 2 reactions also take place mainly in the liver. If a drug molecule has a suitable ‘handle’ (e.g. a hydroxyl, thiol or amino group), either in the parent molecule or in a product resulting from phase 1 metabolism, it is susceptible to conjugation. The groups most often involved are glucuronyl (Fig. 9.3), sulfate, methyl and acetyl. The tripeptide glutathione can conjugate drugs or their phase 1 metabolites via its sulfhydryl group, as in the detoxification of paracetamol (see Fig. 57.1, p. 701). Glucuronide formation involves the formation of a high-energy phosphate compound, uridine diphosphate glucuronic acid (UDPGA), from which glucuronic acid is transferred to an electron-rich atom (N, O or S) on the substrate, forming an amide, ester or thiol bond. UDP-glucuronyl transferase, which catalyses these reactions, has very broad substrate specificity embracing many drugs and other foreign molecules. Several important endogenous substances, including bilirubin and adrenal corticosteroids, are conjugated by the same system.
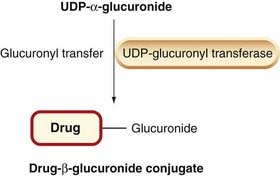
Fig. 9.3 The glucuronide conjugation reaction.
A glucuronyl group is transferred from uridine diphosphate glucuronic acid (UDPGA) to a drug molecule.
Acetylation and methylation reactions occur with acetyl-CoA and S-adenosyl methionine, respectively, acting as the donor compounds. Many of these conjugation reactions occur in the liver, but other tissues, such as lung and kidney, are also involved.
Stereoselectivity
Many clinically important drugs, such as sotalol (Ch. 21), warfarin (Ch. 24) and cyclophosphamide (Ch. 55), are mixtures of stereoisomers, the components of which differ not only in their pharmacological effects but also in their metabolism, which may follow completely distinct pathways (see Campo et al., 2009 for a recent review). Several clinically important drug interactions involve stereospecific inhibition of metabolism of one drug by another (Ch. 56). In some cases, drug toxicity is mainly linked to one of the stereoisomers, not necessarily the pharmacologically active one. Where practicable, regulatory authorities urge that new drugs should consist of single isomers to avoid these complications.1
Inhibition of P450
Inhibitors of P450 differ in their selectivity towards different isoforms of the enzyme, and are classified by their mechanism of action. Some drugs compete for the active site but are not themselves substrates (e.g. quinidine is a potent competitive inhibitor of CYP2D6 but is not a substrate for it). Non-competitive inhibitors include drugs such as ketoconazole, which forms a tight complex with the Fe3+ form of the haem iron of CYP3A4, causing reversible non-competitive inhibition. So-called mechanism-based inhibitors require oxidation by a P450 enzyme. Examples include the oral contraceptive gestodene (CYP3A4) and the anthelminthic drug diethylcarbamazine (CYP2E1). An oxidation product (e.g. a postulated epoxide intermediate of gestodene) binds covalently to the enzyme, which then destroys itself (’suicide inhibition’; see Pelkonen et al., 2008 for a fuller review). Many clinically important interactions between drugs are the result of inhibition of P450 enzymes (see Ch. 56).
Induction of Microsomal Enzymes
A number of drugs, such as rifampicin (Ch. 50), ethanol (Ch. 48) and carbamazepine (Ch. 44), increase the activity of microsomal oxidase and conjugating systems when administered repeatedly. Many carcinogenic chemicals (e.g. benzpyrene, 3-MC) also have this effect, which can be substantial; Figure 9.4 shows a nearly 10-fold increase in the rate of benzpyrene metabolism 2 days after a single dose. The effect is referred to as induction, and is the result of increased synthesis and/or reduced breakdown of microsomal enzymes—see Park et al. (1996), Dickins (2004) and Pelkonen et al. (2008) for more detail.
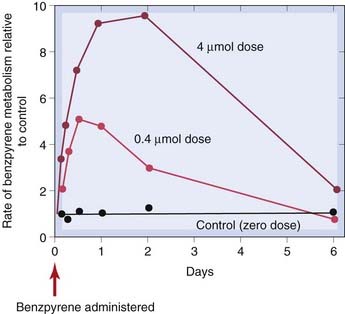
Fig. 9.4 Stimulation of hepatic metabolism of benzpyrene.
Young rats were given benzpyrene (intraperitoneally) in the doses shown, and the benzpyrene-metabolising activity of liver homogenates was measured at times up to 6 days.
(From Conney A H et al. 1957 J Biol Chem 228: 753.)
Enzyme induction can increase drug toxicity and carcinogenicity (Park et al., 2005), because several phase 1 metabolites are toxic or carcinogenic: paracetamol is an important example of a drug with a highly toxic metabolite (see Ch. 57).
The mechanism of induction is incompletely understood but is similar to that involved in the action of steroid and other hormones that bind to nuclear receptors (see Ch. 3). The most thoroughly studied inducing agents are polycyclic aromatic hydrocarbons (e.g. 3-MC). These bind to the ligand-binding domain of a soluble protein, termed the aromatic hydrocarbon (Ah) receptor. This complex is transported to the nucleus by an Ah receptor nuclear translocator and binds Ah receptor response elements in the DNA, thereby promoting transcription of the gene CYP1A1. In addition to enhanced transcription, some inducing agents (e.g. ethanol, which induces CYP2E1 in humans) also stabilise mRNA or P450 protein.
First-Pass (Presystemic) Metabolism
Some drugs are extracted so efficiently by the liver or gut wall that the amount reaching the systemic circulation is considerably less than the amount absorbed. This is known as first-pass or presystemic metabolism and reduces bioavailability (Ch. 8) even when a drug is well absorbed. Presystemic metabolism is important for many therapeutic drugs (Table 9.2 shows some examples), and is a problem because:
Table 9.2 Examples of drugs that undergo substantial first-pass elimination
| Aspirin | Metoprolol |
| Glyceryl trinitrate | Morphine |
| Isosorbide dinitrate | Propranolol |
| Levodopa | Salbutamol |
| Lidocaine | Verapamil |
Pharmacologically Active Drug Metabolites
In some cases (see Table 9.3), a drug becomes pharmacologically active only after it has been metabolised. For example, azathioprine, an immunosuppressant drug (Ch. 26), is metabolised to mercaptopurine; and enalapril, an angiotensin-converting enzyme inhibitor (Ch. 22), is hydrolysed to its active form enalaprilat. Such drugs, in which the parent compound lacks activity of its own, are known as prodrugs. These are sometimes designed deliberately to overcome problems of drug delivery (Ch. 8). Metabolism can alter the pharmacological actions of a drug qualitatively. Aspirin inhibits some platelet functions and has anti-inflammatory activity (Chs 24 and 26). It is hydrolysed to salicylic acid (Fig. 9.1), which has anti-inflammatory but not antiplatelet activity. In other instances, metabolites have pharmacological actions similar to those of the parent compound (e.g. benzodiazepines, many of which form long-lived active metabolites that cause sedation to persist after the parent drug has disappeared; Ch. 43). There are also cases in which metabolites are responsible for toxicity. Hepatotoxicity of paracetamol is one example (see Ch. 57), and bladder toxicity of cyclophosphamide, which is caused by its toxic metabolite acrolein (Ch. 55), is another. Methanol and ethylene glycol both exert their toxic effects via metabolites formed by alcohol dehydrogenase. Poisoning with these agents is treated with ethanol (or with a more potent inhibitor), which competes for the active site of the enzyme. Disulfiram inhibits CYP2E1 and reduces substantially the formation of trifluoroacetic acid during halothane anaesthesia, raising the intriguing possibility that it could prevent halothane hepatitis (see Kharasch, 2008).
Table 9.3 Some drugs that produce active or toxic metabolites
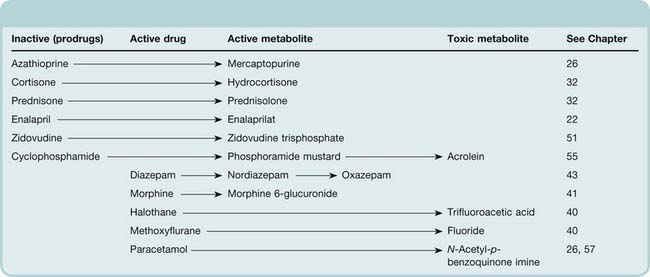
Drug metabolism 
Drug and Metabolite Excretion
Biliary Excretion and Enterohepatic Circulation
Liver cells transfer various substances, including drugs, from plasma to bile by means of transport systems similar to those of the renal tubule including organic cation transporters (OCTs), organic anion transporters (OATs) and P-glycoproteins (P-gp) (see Ch. 8). Various hydrophilic drug conjugates (particularly glucuronides) are concentrated in bile and delivered to the intestine, where the glucuronide is usually hydrolysed, releasing active drug once more; free drug can then be reabsorbed and the cycle repeated (enterohepatic circulation). The effect of this is to create a ‘reservoir’ of recirculating drug that can amount to about 20% of total drug in the body and prolongs drug action. Examples where this is important include morphine (Ch. 41) and ethinylestradiol (Ch. 34). Several drugs are excreted to an appreciable extent in bile. Vecuronium (a non-depolarising muscle relaxant; Ch. 13) is an example of a drug that is excreted mainly unchanged in bile. Rifampicin (Ch. 50) is absorbed from the gut and slowly deacetylated, retaining its biological activity. Both forms are secreted in the bile, but the deacetylated form is not reabsorbed, so eventually most of the drug leaves the body in this form in the faeces.
Renal Excretion of Drugs and Metabolites
Drugs differ greatly in the rate at which they are excreted by the kidney, ranging from penicillin (Ch. 50), which is cleared from the blood almost completely on a single transit through the kidney, to diazepam (Ch. 43), which is cleared extremely slowly. Most drugs fall between these extremes, and metabolites are nearly always cleared more quickly than the parent drug. Three fundamental processes account for renal drug excretion:
Glomerular Filtration
Glomerular capillaries allow drug molecules of molecular weight below about 20 000 to pass into the glomerular filtrate. Plasma albumin (molecular weight approximately 68 000) is almost completely impermeant, but most drugs—with the exception of macromolecules such as heparin (Ch. 24) or biological products (Ch. 59)—cross the barrier freely. If a drug binds to plasma albumin, only free drug is filtered. If, like warfarin (Ch. 24), a drug is approximately 98% bound to albumin, the concentration in the filtrate is only 2% of that in plasma, and clearance by filtration is correspondingly reduced.
Tubular Secretion
Up to 20% of renal plasma flow is filtered through the glomerulus, leaving at least 80% of delivered drug to pass on to the peritubular capillaries of the proximal tubule. Here, drug molecules are transferred to the tubular lumen by two independent and relatively non-selective carrier systems (see Ch. 8). One of these, the OAT, transports acidic drugs (as well as various endogenous acids, such as uric acid), while an OCT handles organic bases. Some important drugs that are transported by these two carrier systems are shown in Table 9.4. The OAT carrier can transport drug molecules against an electrochemical gradient, and can therefore reduce the plasma concentration nearly to zero, whereas OCT facilitates transport down an electrochemical gradient. Because at least 80% of the drug delivered to the kidney is presented to the carrier, tubular secretion is potentially the most effective mechanism of renal drug elimination. Unlike glomerular filtration, carrier-mediated transport can achieve maximal drug clearance even when most of the drug is bound to plasma protein.2 Penicillin (Ch. 50), for example, although about 80% protein bound and therefore cleared only slowly by filtration, is almost completely removed by proximal tubular secretion, and is therefore rapidly eliminated.
Table 9.4 Important drugs and related substances secreted into the proximal renal tubule by OAT or OCT transporters
| OAT | OCT |
|---|---|
| p-Aminohippuric acid | Amiloride |
| Furosemide | Dopamine |
| Glucuronic acid conjugates | Histamine |
| Glycine conjugates | Mepacrine |
| Indometacin | Morphine |
| Methotrexate | Pethidine |
| Penicillin Probenecid Sulfate conjugates Thiazide diuretics Uric acid |
Quaternary ammonium compounds |
| Quinine | |
| 5-Hydroxytryptamine (serotonin) | |
| Triamterene |
Many drugs compete for the same transport system (Table 9.4), leading to drug interactions. For example, probenecid was developed originally to prolong the action of penicillin by retarding its tubular secretion.
Diffusion Across the Renal Tubule
Water is reabsorbed as fluid traverses the tubule, the volume of urine emerging being only about 1% of that of the glomerular filtrate. Consequently, if the tubule is freely permeable to drug molecules, some 99% of the filtered drug will be reabsorbed passively down the resulting concentration gradient. Lipid-soluble drugs are therefore excreted poorly, whereas polar drugs of low tubular permeability remain in the lumen and become progressively concentrated as water is reabsorbed. Polar drugs handled in this way include digoxin and aminoglycoside antibiotics. These exemplify a relatively small but important group of drugs (Table 9.5) that are not inactivated by metabolism, the rate of renal elimination being the main factor that determines their duration of action. These drugs have to be used with special care in individuals whose renal function may be impaired, including the elderly and patients with renal disease or any severe acute illness (Ch. 56).
Table 9.5 Examples of drugs that are excreted largely unchanged in the urine
| Percentage | Drugs excreted |
|---|---|
| 100–75 | Furosemide, gentamicin, methotrexate, atenolol, digoxin |
| 75–50 | Benzylpenicillin, cimetidine, oxytetracycline, neostigmine |
| ∼50 | Propantheline, tubocurarine |
The degree of ionization of many drugs—weak acids or weak bases—is pH dependent, and this markedly influences their renal excretion. The ion-trapping effect means that a basic drug is more rapidly excreted in an acid urine which favours the charged form and thus inhibits reabsorption. Conversely, acidic drugs are most rapidly excreted if the urine is alkaline (Fig. 9.5). Urinary alkalinisation is used to accelerate the excretion of salicylate in treating selected cases of aspirin overdose.
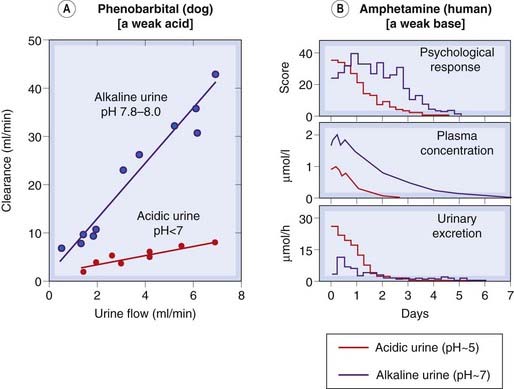
Fig. 9.5 The effect of urinary pH on drug excretion.
[A] Phenobarbital clearance in the dog as a function of urine flow. Because phenobarbital is acidic, alkalinising the urine increases clearance about five-fold. [B] Amphetamine excretion in humans. Acidifying the urine increases the rate of renal elimination of amphetamine, reducing its plasma concentration and its effect on the subject’s mental state.
(Data from Gunne & Anggard 1974. In: Torrell T et al. (eds) Pharmacology and pharmacokinetics. Plenum, New York.)
Renal Clearance
Elimination of drugs by the kidneys is best quantified by the renal clearance (CLr). This is defined as the volume of plasma containing the amount of substance that is removed from the body by the kidneys in unit time. It is calculated from the plasma concentration, Cp, the urinary concentration, Cu, and the rate of flow of urine, Vu, by the equation:
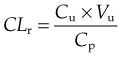
CLr varies greatly for different drugs, from less than 1 ml/min to the theoretical maximum set by the renal plasma flow, which is approximately 700 ml/min, measured by p-aminohippuric acid (PAH) clearance (renal extraction of PAH approaches 100%).
References and Further Reading
Nassar A.F. Drug Metabolism Handbook: Concepts and Applications. Hoboken, NJ: Wiley-Blackwell; 2009. (Multi-authored handbook aimed at bench scientists; will be invaluable for pharmaceutical industry scientists)
Testa B., Krämer S.D. The biochemistry of drug metabolism. Weinheim: Wiley-VCH; 2009. (Two-volume reference work)
Anzenbacher P. Special issue: cytochrome P450. BBA general subjects. 2007;1770(3):313-494. (Contains sections on mechanisms and principles; structural insights and biophysics; substrates, tissue specificities and regulation; clinical implications)
Campo V.L., Bernardes L.S.C., Carvalho I. Stereoselectivity in drug metabolism: molecular mechansisms and analytical methods. Curr. Drug Metab.. 2009;10:188-205.
Coon M.J. Cytochrome P450: nature’s most versatile biological catalyst. Annu. Rev. Pharmacol. Toxicol.. 2005;45:1-25. (Summarises the individual steps in the P450 and reductase reaction cycles)
Kharasch E.D. Adverse drugs reactions with halogenated anesthetics. Clin. Pharmacol. Ther.. 2008;84:158-162. (This review focuses on adverse effects that are attributable to anesthetic metabolism)
Kinirons M.T., O’Mahony M.S. Drug metabolism and ageing. Br. J. Clin. Pharmacol.. 2004;57:540-544. (Reviews age-related changes in drug metabolism)
P450 enzyme induction and inhibition
Dickins M. Induction of cytochromes P450. Curr. Top. Med. Chem.. 2004;4:1745-1766. (Recent advances)
Henderson L., Yue Q.Y., Bergquist C., et al. St John’s wort (Hypericum perforatum): drug interactions and clinical outcomes. Br. J. Clin. Pharmacol.. 2002;54:349-356. (Reviews the induction of CYP450 isoenzymes and of P-glycoprotein by constituents in this herbal remedy)
Pelkonen O., Turpeinen M., Hakkola J., et al. Inhibition and induction of human cytochrome P450 enzymes: current status. Arch. Toxicol.. 2008;82:667-715. (Review)
Ito S. Drug therapy: drug therapy for breast-feeding women. N. Engl. J. Med.. 2000;343:118-126.
Keppler D., Konig J. Hepatic secretion of conjugated drugs and endogenous substances. Semin. Liver Dis.. 2000;20:265-272. (‘Conjugate export pumps of the multidrug resistance protein-MRP-family mediate ATP-dependent secretion of anionic conjugates across the canalicular and the basolateral hepatocyte membrane into bile and sinusoidal blood, respectively. Xenobiotic and endogenous lipophilic substances may be conjugated with glutathione, glucuronate, sulfate, or other negatively charged groups and thus become substrates for export pumps of the MRP family’)
Kusuhara H., Sugiyama Y. In vitro–in vivo extrapolation of transporter-mediated clearance in the liver and kidney. Drug Metab. Pharmacokinet.. 2009;24:37-52. (Review)
1No doubt a good idea though the usefulness of effort directed towards developing ‘novel’ entities that are actually just the active isomers of well-established and safe racemates has been questioned.
2Because filtration involves isosmotic movement of both water and solutes, it does not affect the free concentration of drug in the plasma. Thus the equilibrium between free and bound drug is not disturbed, and there is no tendency for bound drug to dissociate as blood traverses the glomerular capillary. The rate of clearance of a drug by filtration is therefore reduced directly in proportion to the fraction that is bound. In the case of active tubular secretion, this is not so; secretion may be retarded very little even though the drug is mostly bound. This is because the carrier transports drug molecules unaccompanied by water. As free drug molecules are taken from the plasma, therefore, the free plasma concentration falls, causing dissociation of bound drug from plasma albumin. Consequently, effectively 100% of the drug, bound and free, is available to the carrier.