The Child with Endocrine Dysfunction

http://evolve.elsevier.com/wong/ncic

Abnormal Sexual Development, Ch. 11
Administration of Medication, Ch. 27
Family-Centered Home Care, Ch. 25
Genetic Evaluation and Counseling, Ch. 5
Hypocalcemia, Ch. 9
Hypoglycemia, Ch. 9
Single-Gene Disorders, Ch. 5
The Endocrine System
The endocrine system consists of three components: (1) the cell, which sends a chemical message by means of a hormone; (2) the target cells, or end organs, which receive the chemical message; and (3) the environment through which the chemical is transported (blood, lymph, extracellular fluids) from the site of synthesis to the sites of cellular action. The endocrine system controls or regulates metabolic processes governing energy production, growth, fluid and electrolyte balance, response to stress, and sexual reproduction (Baxter and Ribeiro, 2004).
The endocrine glands, which are distributed throughout the body, are listed in Box 38-1; also listed are several additional structures sometimes considered endocrine glands, although they are not usually included.
Hormones
A hormone is a complex chemical substance produced and secreted into body fluids by a cell or group of cells that exerts a physiologic controlling effect on other cells (Behrman, Kliegman, Jenson, et al, 2009). Some are local hormones, creating their effect near the point of secretion. For example, acetylcholine, released at the parasympathetic and skeletal nerve endings, mediates the synaptic activity of the nervous system; secretin, a digestive hormone secreted by certain cells lining the duodenum, stimulates the pancreas to release a watery secretion; and the prostaglandins, or tissue hormones, secreted by a wide variety of organs (including the seminal vesicles, kidneys, lungs, iris, brain, and thymus), usually diffuse only a short distance to integrate activities of neighboring cells.
General hormones are produced in one organ or part of the body and are carried through the bloodstream to a distant part, or parts, of the body where they initiate or regulate physiologic activity of an organ or group of cells. Some of these hormones (such as thyroid hormone [TH] and growth hormone [GH]) affect most cells of the body, whereas others (such as the tropic hormones) produce their effects on specific tissues, called target tissues. For example, the pituitary hormones stimulate the adrenal glands and the thyroid gland to secrete adrenocorticotropic hormone (ACTH) and thyroid-stimulating hormone (TSH), respectively.
Control of Hormone Secretion
Hormones are released by endocrine glands into the bloodstream, where they are carried to responsive tissues (Fig. 38-1). These responsive, or target, tissues may be another endocrine gland, an organ, or tissue (Baxter and Ribeiro, 2004). Regulation of hormonal secretion is based on negative feedback. As a rule, endocrine glands have a tendency to oversecrete their particular hormones. However, once the hormone’s physiologic effect has been achieved, this information is transmitted to the producing gland, either directly or indirectly, to inhibit further secretion. If the gland undersecretes, the inhibition is relieved, and the gland increases production of the hormone. As a result, the hormone is secreted according to the amount needed. This is the primary function of the tropic hormones.
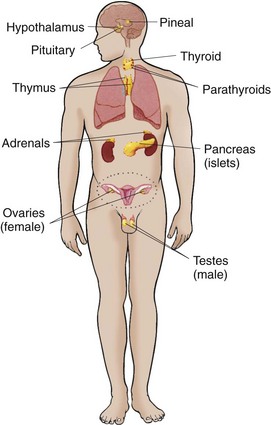
Fig. 38-1 Location of the endocrine glands and structures sometimes considered endocrine glands. (From Thibodeau GA, Patton KT: Structure and function of the body, ed 13, St Louis, 2008, Mosby.)
The endocrine gland primarily responsible for stimulation and inhibition of target glandular secretions is the anterior pituitary, or “master gland.” Tropic (which literally means “turning”) hormones secreted by the anterior pituitary regulate the secretion of hormones from various target organs (Fig. 38-2). As blood concentrations of the target hormones reach normal levels, a negative message is sent to the anterior pituitary to inhibit release of the tropic hormone. For example, TSH responds to low levels of circulating TH. As blood levels of TH reach normal concentrations, a negative feedback message is sent to the anterior pituitary, resulting in diminished release of TSH.
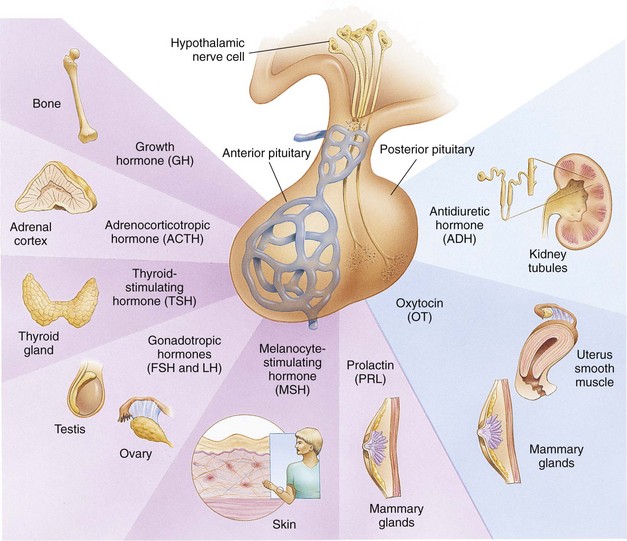
Fig. 38-2 Principal anterior and posterior pituitary hormones and their target organs. (From Thibodeau GA, Patton KT: Structure and function of the body, ed 13, St Louis, 2008, Mosby.)
The pituitary gland is, in turn, controlled by either hormonal or neuronal signals from the hypothalamus. Two types of substances are secreted from the hypothalamus: (1) releasing hormones and (2) inhibitory hormones, which are secreted within the hypothalamus and transported by way of the pituitary portal system to the anterior pituitary, where they stimulate the secretion of tropic hormones. An example of this is the secretion of corticotropin-releasing factor (CRF) by the hypothalamus, which stimulates the pituitary to secrete ACTH. In this instance the anterior pituitary is the target of the hypothalamus and secondarily affects a response from another target gland, the adrenals. The adrenals in turn secrete glucocorticoids, which have multiple target sites throughout the body. Pituitary hormones that lack feedback control from the product of a target tissue (GH, prolactin, and melanocyte-stimulating hormone) require hypothalamic inhibitors and stimulators for their control.
Not all hormones depend on other hormones for their release. For example, insulin is secreted in response to blood glucose concentrations. Other glandular hormones that are not under the control of the pituitary gland are glucagon, parathyroid hormone (parathormone, PTH), antidiuretic hormone (ADH), and aldosterone.
Neuroendocrine Interrelationships
Two regulatory systems maintain hemostasis: the endocrine and the autonomic nervous systems (collectively known as the neuroendocrine system) (Baxter and Ribeiro, 2004). The autonomic nervous system consists of the sympathetic and parasympathetic systems, which control nonvoluntary functions, specifically of smooth muscle, myocardium, and glands. The parasympathetic system is primarily involved in regulating digestive processes, whereas the sympathetic system functions to maintain homeostasis during stress.
The higher autonomic centers, located in the hypothalamus and limbic system, help control the functioning of both autonomic systems. Both sympathetic and parasympathetic nerve fibers secrete neurotransmitting substances: acetylcholine, released by cholinergic fibers, and norepinephrine, released by adrenergic fibers. Release of norepinephrine into the plasma produces the same effects as secretion of this substance by the adrenal medulla. Thus the interrelatedness between the two systems is demonstrated.
The neuroendocrine system acts by synthesizing and releasing various chemical substances that regulate body functions. Information is carried by means of neural impulses in the autonomic system and by the blood in the endocrine system. In general, neural responses are more rapid and localized; endocrine responses are more lasting and widespread. The two systems function synergistically because neural impulses transmitted to the central nervous system (CNS) stimulate the hypothalamus to manufacture and release several releasing or inhibiting factors.
Because of the interdependent relationship of these glands, a malfunction in one gland produces effects elsewhere. Endocrine dysfunction may result from an intrinsic defect in the target gland (primary) or from a diminished or elevated level of tropic hormones (secondary). Endocrine problems occur from hypofunction or hyperfunction of the glands. Primary hypofunction is usually associated with a more profound deficiency of the target gland hormone because little or no hormone is secreted. In secondary dysfunction the target glands secrete some of their hormones but in smaller amounts and less rapidly.
Hyperfunction or hypofunction may also result from an increase or decrease in secretion of the tropic hormones (primary) with a consequent increase in the target gland hormones (secondary) or an oversecretion or undersecretion of the target glands.
Disorders of Pituitary Function
The pituitary gland is divided into two lobes; the anterior pituitary and the posterior pituitary. Each lobe is responsible for the production, storage, and secretion of specific hormones (Hanberg, 2005). Deficiencies of the anterior pituitary hormones may be due to organic defects or have an idiopathic etiology and may occur as a single hormonal problem or in combination with other hormonal deficiencies. The clinical manifestations depend on the hormones involved and the age of onset. If the tropic hormones are involved, the resulting disorder reflects the altered stimulus to the target gland. For example, if TSH is deficient, TH is also deficient, and the child displays the manifestations of hypothyroidism.
An overproduction of the anterior pituitary hormones can result in gigantism (caused by excess GH production during childhood), hyperthyroidism, hypercortisolism (Cushing syndrome), and precocious puberty from excessive gonadotropins. Overproduction may be caused by hyperplasia of the pituitary cells—which may eventually progress to a tumor (adenoma)—or a primary hypothalamic defect that results in an excess of the hormone’s releasing factor. Although the initial clinical manifestations are a result of pituitary oversecretion, eventually pituitary insufficiency occurs, and the signs of panhypopituitarism become evident. Panhypopituitarism is often defined clinically as the loss of all anterior pituitary hormones, leaving only posterior pituitary function intact (Toogood and Stewart, 2008).
 NURSING ALERT
NURSING ALERT
Children with panhypopituitarism should wear medical identification, such as a bracelet.
Hypopituitarism
Hypopituitarism is diminished or deficient secretion of pituitary hormones. The consequences of the condition depend on the degree of dysfunction. It often leads to:
• Gonadotropin deficiency with absence or regression of secondary sexual characteristics
• GH deficiency, in which children display retarded somatic growth
• TSH deficiency, which produces hypothyroidism
• Corticotropin deficiency, which results in manifestations of adrenal hypofunction
Hypopituitarism can result from any of the conditions listed in Box 38-2. The most common organic cause of pituitary undersecretion is tumors in the pituitary or hypothalamic region, especially the craniopharyngiomas. These tumors usually invade the anterior and posterior pituitary lobes and the hypothalamus, causing panhypopituitarism (Box 38-3). The child may experience growth retardation for some time before developing any symptoms or signs of increased intracranial pressure, local compression, or the destructive effects of the tumor. Other potential causes of panhypopituitarism include encephalitis, radiation to the head or neck, head trauma, and congenital hypoplasia of the hypothalamic area (Hanberg, 2005; Darzy, 2009).
Congenital hypopituitarism, or congenital GH deficiency, can be seen in newborn infants, often as a result of birth trauma. Symptoms of hypoglycemia and seizure activity often manifest within the first 24 hours after birth (Toogood and Stewart, 2008).
Idiopathic hypopituitarism, or idiopathic pituitary growth failure, is usually related to GH deficiency, which inhibits somatic growth in all cells of the body (Miller and Zimmerman, 2004). Growth failure is defined as an absolute height of less than −2 SD for age, or a linear growth velocity consistently less than −1 SD for age. When this occurs without the presence of hypothyroidism, systemic disease, or malnutrition, then an abnormality of the GH–insulin-like growth factor (IGF) axis should be considered (Richmond and Rogol, 2008).
Although most children with hypopituitarism are normal at birth, they show growth patterns that progressively deviate from the normal growth rate, often beginning in infancy. The chief complaint in most instances is short stature. Of those who seek help, boys outnumber girls three to one. The extent of idiopathic GH deficiency may be complete or partial, but the cause is unknown. It is frequently associated with deficiencies of other pituitary hormone, such as TSH and ACTH; thus it is theorized that the disorder is probably secondary to hypothalamic deficiency. It has also been observed that there is a higher than average frequency in some families, which indicates a possible genetic origin in a number of instances.
Not all children with short stature have GH deficiency. In most instances the cause of short stature is either familial short stature or a simple constitutional growth delay. Familial short stature refers to otherwise healthy children who have ancestors with adult height in the lower percentiles, and whose height during childhood is appropriate for genetic background. Fig. 38-3 provides an overview of the possible causes of short stature in children.
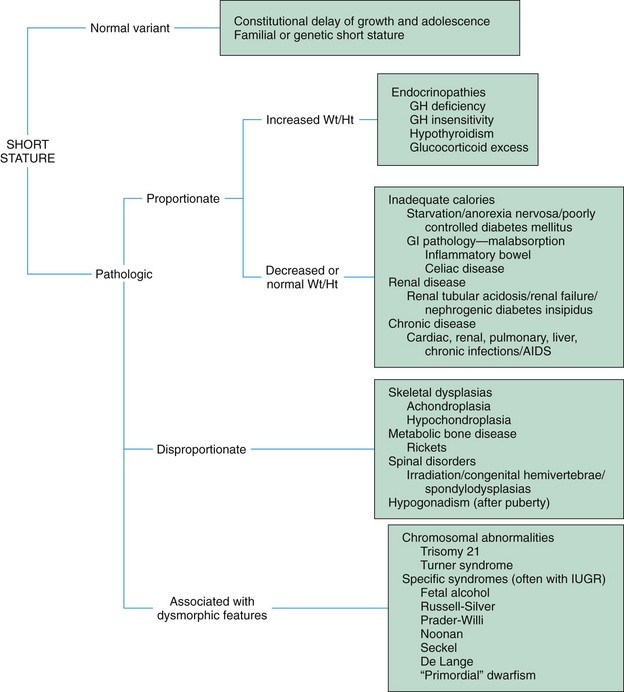
Fig. 38-3 Causes of short stature. AIDS, Acquired immunodeficiency syndrome; GI, gastrointestinal; GH, growth hormone; IUGR, intrauterine growth restriction; Wt/Ht, weight-to-height ratio. (From Vogiatzi MG, Copeland KC: The short child, Pediatr Rev 19(3):92-99, 1998.)
Constitutional growth delay refers to individuals (usually boys) with delayed linear growth, generally beginning as a toddler, and skeletal and sexual maturation that is behind that of age-mates (Miller and Zimmerman, 2004; Halac and Zimmerman, 2004). Typically these children will reach normal adult height. Often a history of a similar pattern of growth is found in one of the parents or other family members of children with constitutional growth delay. The untreated child proceeds through normal changes as expected on the basis of bone age. These changes, although occurring later than in the average child, appear in normal sequence and manner, and treatment with GH is not usually indicated. However, its use has become controversial, especially in relation to parental and child requests for treatment to accelerate growth.
Clinical Manifestations
Children with hypopituitarism generally grow normally during the first year and then follow a slowed growth curve that is below the 3rd percentile. In children with a partial GH deficiency, the growth retardation is less marked than in children with complete GH deficiency. Height may be retarded more than weight because, with good nutrition, these children can become overweight or even obese. Their well-nourished appearance is an important diagnostic clue to differentiation from other disorders such as failure to thrive.
Skeletal proportions are normal for the age, but these children appear younger than their chronologic age. However, later in life premature aging is common. The appearance of fine wrinkles about the eyes and mouth gives these children a peculiar impression of immaturity combined with presenility. They are no less active than other children if directed to size-appropriate sports, such as swimming, wrestling, gymnastics, soccer, or ballet. Bone age is nearly always retarded but is closely related to height age; the degree of retardation depends on the duration and extent of the hormonal deficiency. Children with diminished function of recent onset may show little retardation in skeletal age, whereas children with a longstanding deficiency may evidence a skeletal age only 40% to 50% of their chronologic age. It is difficult to predict their eventual height. Because the period of growth is prolonged past adolescence into the third or fourth decade, many of them reach a permanent height of 1.2 to 1.5 m (4 to 5 ft).
Usually, primary teeth appear at the expected age, but the eruption of the permanent teeth is delayed. Because of the underdeveloped jaw, the teeth are overcrowded and malpositioned. Sexual development is usually delayed but is otherwise normal. Even without GH replacement, adults with GH deficiency are able to reproduce normal offspring. However, if the gonadotropins are deficient, sexual maturation is absent.
Most of these children have normal intelligence. In fact, during early childhood they often appear precocious in their learning because their ability seems to exceed their small size. However, emotional problems are not uncommon, especially as they near puberty, when their smallness becomes increasingly apparent in comparison with their peers. Height discrepancy has been significantly correlated with emotional adjustment problems and may be a valuable predictor of the extent to which GH-delayed children will experience difficulty with anxiety, social skills, and positive self-esteem (Sandberg, Kranzler, Bukowski, et al, 1999). Academic problems are also common. A history often reveals repeated classes or enrollment in classes for children with learning disabilities. These children are usually not pushed to perform at their chronologic age but at their height age.
Diagnostic Evaluation
Only a small number of children with delayed growth or short stature have hypopituitary dwarfism. In the majority of instances the cause is constitutional delay. Diagnostic evaluation is aimed at isolating organic causes, which, in addition to GH deficiency, may include hypothyroidism, oversecretion of cortisol, gonadal aplasia, chronic illness, nutritional inadequacy, Russell-Silver dwarfism, or hypochondroplasia.
A complete diagnostic evaluation should include a family history, a history of the child’s growth patterns and previous health status, physical examination, psychosocial evaluation, radiographic surveys, and endocrine studies.
Family History: A family history is of utmost importance in relating short stature to genetic background. The midparental height is an important prognosticator of the child’s ultimate adult height. Normal adult height should fall within 5 cm (2 inches) of midparental height (Miller and Zimmerman, 2004). Children with constitutional delays frequently are the products of parents who experienced similar slow growth patterns and delayed sexual maturation. A small percentage of those with hypopituitarism demonstrate an autosomal recessive inheritance pattern. Height and weight of siblings should be compared with the child’s growth patterns at comparable age periods.
Child’s History: The child’s history should include a thorough prenatal history to rule out maternal disorders that may have influenced growth, such as malnutrition. Compare birth height and weight with gestational age. Children with hypopituitarism are usually of normal size and normal gestational age at birth.
Investigate the child’s health history for evidence of chronic illness that may have influenced growth patterns, although a chronic illness, such as congenital heart disease, malabsorptive disorders, severe anemia, or neurologic impairments, usually is identified long before the growth problem becomes a concern. Signs and symptoms suggesting a tumor, such as visual disturbances, headache, and signs of increasing intracranial pressure, are important. Such symptoms often precede retarded growth but may not have been regarded as significant. With lesions involving the hypothalamus, the history may also reveal characteristic manifestations of dysfunction such as somnolence, thermodysregulation, epilepsy, and polyphagia, resulting in obesity. Because a craniopharyngioma can affect the secretion of any of the pituitary hormones, assessment for hypothyroidism, hypoadrenalism, and hypoaldosteronism should also be included.
Whenever possible, evaluate the child’s growth patterns since birth, especially growth velocity, and compare them with standard measurements. The age of onset of short stature provides a significant diagnostic clue. When the clinician evaluates the results of plotting height and weight, upward or downward changes in height velocity in children older than 3 years may indicate a growth abnormality (Halac and Zimmerman, 2004). Progressive retardation in height and weight since early childhood suggests idiopathic hypopituitary dwarfism, whereas a recent change from normal growth is more characteristic of a tumor. In addition, these children are usually well nourished, ruling out other causes of growth failure.
Physical Examination: Accurate measurement of height (using a calibrated stadiometer) and weight and comparison with standard growth charts are essential (Box 38-4). Multiple height measures reflect a more accurate assessment of abnormal growth patterns (Hall, 2000). Other measurements may include crown-to-pubis and pubis-to-heel length to compare body proportions, and sexual development should be assessed and compared with age-appropriate development. Observation of general appearance yields valuable clues, especially signs of premature aging and infantile facial features. Perform a funduscopic examination and testing for visual acuity to detect evidence of ocular damage from a tumor.
Radiographic Surveys: A skeletal survey in children less than 3 years of age and radiographic examination of the hand-wrist for centers of ossification (bone age) (Box 38-5) in older children are important in evaluating growth. Epiphyseal maturation is retarded in hypopituitarism but consistent with retardation in height. This is in contrast to hypothyroidism, in which bone maturation is greatly retarded, or gonadal dysplasia, such as Turner syndrome, in which bone age is near normal. Radiographic studies should also include a skull series, which helps in identifying abnormalities such as an abnormally small sella turcica or evidence of a space-occupying lesion such as craniopharyngioma. Magnetic resonance imaging (MRI), computed tomography (CT), radionuclear scans, or carotid angiograms may be needed to establish diagnosis and localization of lesions (Stanhope, 2004).
Endocrine Studies: Definitive diagnosis of GH deficiency is based on absent or subnormal reserves of pituitary GH. Measuring a single GH level is inaccurate due to the pulsatile secretion of this hormone (Richmond and Rogol, 2008). Exercise is a natural and benign stimulus for GH release, and elevated levels can be detected after 20 minutes of strenuous exercise in normal children. Also, GH levels are elevated 45 to 90 minutes after the onset of sleep.
GH levels are normally so low in children that differentiation from abnormal concentrations is unreliable, GH secretion should be stimulated, followed by measurement of blood levels. Initial assessment of the serum IGF-I and IGF binding protein 3 (IGFBP3) indicates a need for further evaluation of GH dysfunction if levels are lower than −1 SD below the mean for age. It is recommended that GH stimulation tests be reserved for children with low serum IGF-I and IGFBP3 levels and poor growth who do not have other endocrine or nonendocrine causes for short stature (Aimaretti, Bellone, Baldelli, et al, 2004; Richmond and Rogol, 2008). However, although IGF-1 test is useful in detecting severe GH insensitivity, it may not be accurate in detecting less severe cases of idiopathic short stature (Cohen, Rogol, Deal, et al, 2008).
GH stimulation, or provocative testing, involves the use of pharmacologics to provoke the release of GH either directly or indirectly. Provocative testing involves the use of neuromodulators such as levodopa or agents such as clonidine, arginine, insulin, propranolol, or glucagon (Behrman, Kliegman, Jenson, et al, 2009; Richmond and Rogol, 2008). The GH stimulation test of choice is currently country dependent (Cohen, Rogol, Deal, et al, 2008). Studies have shown that traditional GH stimulation test results can be less reliable than previously accepted. Currently, findings in a child of poor linear growth, delayed bone age, and peak levels of GH at less than 10 ng/ml in two stimulation tests are consistent with GH deficiency (Behrman, Kliegman, Jenson, et al, 2009). New reference standards that decrease the lower limit of normal GH levels have been proposed (Cohen, Rogol, Deal, et al, 2008). GH-releasing hormone (GHRH), in combination with an agent that can stimulate GH response to GHRH, has also been used to detect GH deficiency (Aimaretti, Bellone, Baldelli, et al, 2004; Hilczer, Smyczynska, and Lewinski, 2006). The use of GHRH may be more accurate in detecting GH deficiency caused by hypothalamic disorders (Behrman, Kliegman, Jenson, et al, 2009). GH-dependent growth factors may be more sensitive indicators of GH deficiency than GH stimulation tests. Increasingly sensitive radioimmunoassays for GH levels have also been developed.
Therapeutic Management
Treatment of GH deficiency caused by organic lesions is directed toward correction of the underlying disease process (e.g., surgical removal or irradiation of a tumor). The definitive treatment of GH deficiency is replacement of GH, which is successful in 80% of affected children. For more than 20 years cadaver-derived human growth hormone (HGH) was used successfully to enhance linear growth in short children. In 1985 the U.S. Food and Drug Administration stopped use of the hormone in response to reported deaths resulting from Creutzfeldt-Jakob disease (CJD) in three former HGH recipients. Patients have been identified who both received HGH and became infected with CJD, a rare and fatal neurodegenerative condition iatrogenically transmitted through human tissue (Wetterau and Cohen, 2000). Donation of organs or tissues from HGH recipients for transplantation should be prohibited because of the inability to test for infection with CJD. Blood banks do not accept donations from former HGH recipients. Biosynthetic GH prepared by recombinant deoxyribonucleic acid (DNA) technology (and without the risk of CJD) is now available and is the therapy of choice (Miller and Zimmerman, 2004).
In the United States the recommended dosage range of recombinant GH is 0.037 to 0.18 mg/kg/week, divided into six or seven daily subcutaneous doses (Behrman, Kliegman, Jenson, et al, 2009; Cohen, Rogol, Deal, et al, 2008). Growth velocity increases in the first year of treatment and is often above the 95th percentile. With each consecutive year of treatment, growth rates decline (Behrman, Kliegman, Jenson, et al, 2009). A recent meta-analysis of the GH literature concluded that, although GH may improve growth velocity in children, individuals who receive treatment remain shorter than their peers (Bryant, Baxter, Cave, et al, 2007). For children to achieve their genetic growth potential, early diagnosis of and intervention for growth disorders are essential (Leschek, Rose, Yanovski, et al, 2004).
The child, family, and health care team make the decision jointly to stop GH therapy. Growth rates of less than 1 inch/yr and a bone age of more than 14 years in girls and more than 16 years in boys are often used as criteria to stop GH therapy (Behrman, Kliegman, Jenson, et al, 2009). Children with other hormone deficiencies require replacement therapy to correct the specific disorders. This may involve administration of thyroid extract, cortisone, testosterone, or estrogens and progesterone. The sex hormones are usually begun during adolescence to promote normal sexual maturation.
Nursing Care Management
 The principal nursing consideration is identifying children with growth problems. Despite the fact that the majority of growth problems are not a result of organic causes, any delay in normal growth and sexual development poses special emotional adjustments for these children.
The principal nursing consideration is identifying children with growth problems. Despite the fact that the majority of growth problems are not a result of organic causes, any delay in normal growth and sexual development poses special emotional adjustments for these children.
 Nursing Care Plan—The Child with Growth Failure
Nursing Care Plan—The Child with Growth Failure
The nurse may be a key person in helping establish a diagnosis. For example, if serial height and weight records are not available, the nurse can question parents about the child’s growth compared with that of siblings, peers, or relatives. Investigating clothing sizes is often helpful in determining growth at different ages. Parents may comment that the child wears out clothes before growing out of them or that, if the clothing fits the body, it often is too long in the sleeves or legs.
Because the behavioral or physical changes that suggest a tumor are insidious, they are frequently overlooked. It is important to correlate the onset of any positive findings with the initial evidence of growth retardation. For example, visual problems and headache are not uncommon in school-age children and can coincidentally occur after a growth problem is recognized. In fact, headache may represent the emotional trauma caused by short stature rather than be a symptom of a tumor. Pursue this line of questioning cautiously to avoid alarming parents unduly about the possibility of a brain tumor.
Part of a nurse’s role in helping establish a diagnosis is assisting with diagnostic tests. Preparation of the child and family is especially important if a number of tests are being performed, and the child requires particular attention during provocative testing. Blood samples are usually taken every 30 minutes for a 3-hour period. Children also have difficulty overcoming hypoglycemia generated by tests with insulin, so carefully observe them for signs of hypoglycemia. Those receiving glucagon are at risk of nausea and vomiting. Patients receiving clonidine require close blood pressure monitoring. Nursing administration of intravenous (IV) fluids may be required if hypotension is detected. The use of arginine is often well tolerated by children, but may cause hypoglycemia in some infants and toddlers. Therefore close monitoring for hypoglycemia is necessary.
Child and Family Support: Once an organic cause of the problem has been confirmed, the parents and child need an opportunity to express their thoughts and feelings. Frequently a growth problem that was present since birth is missed until adolescence, at which time the child’s difference in body development becomes dramatically evident in comparisons with peers. Family members may feel anger and resentment toward members of the health staff for not detecting the problem sooner. Parents may experience guilt for not seeking medical attention earlier, especially if the child has been miserable from the ridicule and criticism of peers. Appropriate emotional support from the nurse can include an affirmation of each person’s justified feelings, such as anger or guilt, and emphasis on the treatment plan and prospects for improvement in the future.
 DRUG ALERT
DRUG ALERT
Growth Hormone
GH is most effective when it is administered at bedtime. Physiologic release is more normally stimulated as a result of pituitary release of GH during the first 45 to 90 minutes after the onset of sleep.
Even when hormone replacement is successful, these children attain their eventual adult height at a slower rate than their peers; therefore they need assistance in setting realistic expectations regarding improvement. Both sexes need guidance toward appropriate vocational goals. Because these children appear younger than their chronologic age, others frequently relate to them in infantile or childish ways. Children having school problems need special counseling. Parents and teachers benefit from guidance directed toward setting realistic expectations for the child based on age and abilities. For example, in the home such children should have the same age-appropriate responsibilities as their siblings. As they approach adolescence, encourage them to participate in group activities with peers. They should wear styles that accentuate their actual age, not their size. If abilities and strengths are emphasized rather than physical size, such children are more likely to develop a positive self-image.
Professionals and families may find research, education, support, and advocacy from the Human Growth Foundation.* The treatment is expensive—up to $20,0000 to $30,0000 per year, depending on the dosage (Radetti, Buzi, Paganini, et al, 2003; Lee, Davis, Clark, et al, 2006). Usually the cost is partially covered by insurance if the child has a documented deficiency, and some pharmaceutical companies offer copay assistance.
Pituitary Hyperfunction
Excess GH before closure of the epiphyseal shafts results in proportional overgrowth of the long bones until the individual reaches a height of 2.4 m (8 feet) or more. Vertical growth is accompanied by rapid and increased development of muscles and viscera. Weight is increased but is usually in proportion to height. Proportional enlargement of head circumference also occurs and may result in delayed closure of the fontanels in young children. Children with a pituitary-secreting tumor may also demonstrate signs of increasing intracranial pressure, especially headache.
If oversecretion of GH occurs after epiphyseal closure, growth is in the transverse direction, producing a condition known as acromegaly. Typical facial features include:
• Overgrowth of the head, lips, nose, tongue, jaw, and paranasal and mastoid sinuses
• Separation and malocclusion of the teeth in the enlarged jaw
• Disproportion of the face to the cerebral division of the skull
• Increased facial hair; thickened, deeply creased skin
• Increased tendency toward hyperglycemia and diabetes mellitus (DM)
Excessive secretion of GH by a pituitary adenoma causes most cases of acromegaly. Acromegaly can develop slowly, with patients being diagnosed as much as 10 years after their symptoms first appear. Left untreated, these patients have a higher mortality rate due to potential cardiovascular, metabolic, and pulmonary complications (Katznelson, 2007; Natchtigall, Delgado, Swearingen, et al, 2008).
Diagnostic Evaluation
Diagnosis is based on a history of excessive growth during childhood and evidence of increased levels of GH. Radiographic studies may reveal a tumor in an enlarged sella turcica, normal bone age, enlargement of bones (such as the paranasal sinuses), and evidence of joint changes. Endocrine studies to confirm excess of other hormones, specifically thyroid, cortisol, and sex hormones, should also be included in the differential diagnosis.
Therapeutic Management
If a lesion is present, surgical treatment by cryosurgery or hypophysectomy is performed to remove the tumor when possible. External radiation or radioactive implants may be used to destroy GH-secreting tissue. New pharmacologic agents have evolved and may be used in combination with other therapies to treat this disease (Natchtigall, Delgado, Swearingen, et al, 2008). Depending on the extent of surgical extirpation and degree of pituitary insufficiency, hormone replacement with thyroid extract, cortisone, and sex hormones may be necessary.
Nursing Care Management
The primary nursing consideration is early identification of children with excessive growth rates. Although medical management is unable to reduce growth already attained, further growth can be retarded. The earlier the treatment, the greater the chances of attaining a normal adult height. Nurses in ambulatory settings who are frequently involved in growth screening should refer children who demonstrate excessive linear growth for a medical evaluation. They should also observe for signs of a tumor, especially headache, and evidence of concurrent hormonal excesses, particularly the gonadotropins, which cause sexual precocity.
Children with excessive growth rates require as much emotional support as those with short stature. However, girls may suffer from the effects of excessive height much more than boys, although, like boys, they may find the tallness an asset when pursuing sports such as basketball. Children and their parents need an opportunity to express their thoughts. A compassionate nurse can be supportive to these children, especially before adolescence when they are larger than their peers. The nurse can emphasize to a tall girl that as boys grow older, they become taller and she will not always be looking down at them. Because early adolescence is a time of idol worship, the nurse can point out marriages of celebrities in which the woman is taller than the man to help the girl realize that not all heterosexual relationships follow stereotypic models.
Precocious Puberty
Manifestations of sexual development before age 9 years in boys or age 8 years in girls have traditionally been considered precocious development, and these children were recommended for further evaluation (Kaplowitz, 2009), but guidelines on this have changed (see Research Focus box).
 RESEARCH FOCUS
RESEARCH FOCUS
Precocious Puberty
Recent examination of the age limit for defining when puberty is precocious reveals that the onset of puberty in girls is occurring earlier than previous studies have documented (Biro, Huang, Crawford, et al, 2006). Mean onset of puberty was 10.2 and 9.6 years in Caucasian and African-American girls, respectively. Based on these findings, precocious puberty evaluation for a pathologic etiology should be performed for Caucasian girls younger than 7 years of age or for African-American girls younger than 6 years of age. No change in the guidelines for evaluation of precocious puberty in boys is recommended. However, recent data suggest that boys may be beginning maturation earlier as well (Herman-Giddens, 2006).
Normally the hypothalamic-releasing factors stimulate secretion of the gonadotropic hormones from the anterior pituitary at the time of puberty. In the male, interstitial cell–stimulating hormone stimulates Leydig cells of the testes to secrete testosterone. In the female follicle-stimulating hormone and luteinizing hormone stimulate the ovarian follicles to secrete estrogens. This sequence of events is known as the hypothalamic-pituitary-gonadal axis. If for some reason the cycle undergoes premature activation, the child displays evidence of advanced or precocious puberty. Box 38-6 lists the causes of precocious puberty.
Isosexual precocious puberty is more common among girls than boys. Approximately 50% of children with precocious puberty have central precocious puberty (CPP), in which pubertal development is activated by the hypothalamic gonadotropin-releasing hormone (GnRH). This produces early maturation and development of the gonads with secretion of sex hormones, development of secondary sexual characteristics, and sometimes production of mature sperm and ova (Lee, 1999; Root, 2000). CPP occurs more frequently in girls and is usually idiopathic, with 95% demonstrating no causative factor (Nebesio and Eugster, 2007; Greiner and Kerrigan, 2006; Root 2000; Carel and Leger, 2008). A CNS insult or structural abnormality occurs in more than 90% of boys with CPP (Root, 2000).
Peripheral precocious puberty (PPP) includes early puberty resulting from hormone stimulation other than the hypothalamic GnRH–stimulated pituitary gonadotropin release. Isolated manifestations that are usually associated with puberty may be seen as variations in normal sexual development (Greiner and Kerrigan, 2006). They appear without other signs of pubescence and are probably caused by unusual end-organ sensitivity to prepubertal levels of estrogen or androgen. Included are premature thelarche (development of breasts in prepubertal girls), premature pubarche (premature adrenarche, early development of sexual hair), and premature menarche (isolated menses without other evidence of sexual development).
Therapeutic Management
Direct treatment of precocious puberty toward the specific cause when known. In 50% of cases, precocious pubertal development regresses or stops advancing without any treatment (Carel and Leger, 2008). If CPP progresses, it can be managed with monthly injections of a synthetic analog of luteinizing hormone–releasing hormone, which regulates pituitary secretions (Carel and Chaussain, 1999; Lee, 1999). A slow-release formulation of leuprolide acetate (Lupron Depot) is given in a dosage of 0.2 to 0.3 mg/kg intramuscularly q 4 wk. A longer lasting preparation may be given intramuscularly every 3 months and has also been successful in the treatment of CPP in a majority of patients. Although expensive, the GnRH analog (GnRHa) histrelin has been formulated as a subdermal implant and may be beneficial for some patients who would like to avoid injections (Kaplowitz, 2009).
After initiation of treatment, breast development regresses or does not advance, and growth returns to normal rates, enhancing predicted height. Some patients, however, do not attain adult targeted height during therapy. Researchers have taken different approaches to address this issue, proposing the use of GH and, more recently, conducting trials of a nonaromatizable anabolic steroid to improved adult height (Carel and Leger, 2008; Kaplowitz, 2009). Treatment is discontinued at a chronologically appropriate time, allowing pubertal changes to resume. Psychologic management of the patient and family is an important aspect of care. Both parents and the affected child should learn the injection procedure.
Nursing Care Management
Psychologic support and guidance of the child and family are the most important aspects of management. Parents and children need anticipatory guidance, support and information resources, and reassurance of the benign nature of the condition (Greiner and Kerrigan, 2006). Dress and activities for the physically precocious child should be appropriate to the chronologic age. Sexual interest is not usually advanced beyond the child’s chronologic age, and parents need to understand that the child’s mental age is congruent with the chronologic age and that the child’s normal, overt manifestations of affection are age appropriate and do not represent sexual advances.
Despite the early sexual development, maturation of the gonads and the appearance of secondary sexual characteristics proceed in the usual order. The most difficult time for the child is usually the school years before adolescence. After puberty, physical differences from peers are no longer present.
Although the child’s sexual behavior may be appropriate for the chronologic age, the nurse should emphasize to parents that the child may be fertile. Usually no form of contraception is necessary unless the child is sexually active. In this situation proper counseling is important because hormonal forms of birth control, such as estrogen pills, prematurely initiate epiphyseal closure, resulting in stunted linear growth.
Diabetes Insipidus
The principal disorder of posterior pituitary hypofunction is diabetes insipidus (DI), also known as neurogenic DI, resulting from undersecretion of ADH, or vasopressin, and producing a state of uncontrolled diuresis (Makaryus and McFarlane, 2006). This disorder is not to be confused with nephrogenic DI, a rare hereditary disorder affecting primarily males and caused by unresponsiveness of the renal tubules to the hormone. (See Chapter 30.)
Critical Thinking Case Study—Diabetes Insipidus
Neurogenic DI may result from a number of different causes. Primary causes are familial or idiopathic; of the total cases, approximately 45% to 50% are idiopathic. Secondary causes include trauma (accidental or surgical), tumors, granulomatous disease, infections (meningitis or encephalitis), and vascular anomalies (aneurysm). Certain drugs, such as alcohol or phenytoin (diphenylhydantoin), can cause a transient polyuria. DI may be an early sign of an evolving cerebral process (De Buyst, Massa, Christophe, et al, 2007).
Clinical Manifestations
The cardinal signs of DI are polyuria and polydipsia. In the older child, signs such as excessive urination accompanied by a compensatory insatiable thirst may be so intense that the child does little more than go to the toilet and drink fluids (Cheetham and Baylis, 2002). Frequently the first sign is enuresis. In the infant the initial symptom is irritability that is relieved with feedings of water but not milk. The infant is also prone to dehydration, electrolyte imbalance, hyperthermia, azotemia, and potential circulatory collapse.
Dehydration is usually not a serious problem in older children, who are able to drink larger quantities of water. However, any period of unconsciousness, such as after trauma or anesthesia, may be life threatening because the voluntary demand for fluid is absent. During such instances careful monitoring of urine volumes, blood concentration, and IV fluid replacement is essential to prevent dehydration.
Diagnostic Evaluation
The simplest test used to diagnose this condition is the water deprivation test, which restricts oral fluids and observes changes in urine volume and concentration. Normally, reducing fluids results in concentrated urine and diminished volume. In DI, fluid restriction has little or no effect on urine formation but causes weight loss from dehydration. Accurate results from this procedure require strict monitoring of fluid intake and urinary output, measurement of urine concentration (specific gravity or osmolality), and frequent weight checks. A weight loss between 3% and 5% indicates significant dehydration and requires termination of the fluid restriction.
NURSING ALERT
Small children require close observation during fluid deprivation to prevent them from drinking, even from toilet bowls, flower vases, or other unlikely sources of fluid.
If this test is positive, the child should be given a test dose of injected aqueous vasopressin (Pitressin), which should alleviate the polyuria and polydipsia. Unresponsiveness to exogenous vasopressin usually indicates nephrogenic DI. A rise by more than 50% in urine osmolality after vasopressin administration indicates central DI (Carr and Gill, 2007).
An important diagnostic consideration is differentiating DI from other causes of polyuria and polydipsia, especially DM. Other tests used in the diagnostic evaluation include CT of the brain or MRI to detect a tumor, kidney function tests, urine osmolality tests and blood electrolyte levels to assess renal failure, and specific endocrine studies to isolate associated problems (Carr and Gill, 2007). In rare instances a psychologic consultation may be warranted to confirm the possibility of compulsive water drinking related to psychogenic causes.
Therapeutic Management
The usual treatment is hormone replacement, either with an intramuscular or subcutaneous injection of vasopressin tannate in peanut oil or with a nasal spray of aqueous lysine vasopressin (Verbalis, 2003; Makaryus and McFarlane, 2006). The injectable form has the advantage of lasting for 48 to 72 hours, which affords the child a full night’s sleep. However, it has the disadvantage of requiring frequent injections and proper preparation of the drug.
DRUG ALERT
Vasopressin
To be effective, vasopressin must be thoroughly mixed in the oil by being held under warm running water for 10 to 15 minutes and shaken vigorously before being drawn into the syringe. If this is not done, the oil may be injected minus the ADH. Small brown particles, which indicate drug dispersion, must be seen in the suspension.
The nasal spray has the benefit of being a simple, painless route of administration. However, applications must be repeated every 8 to 12 hours to prevent recurrence of symptoms. To provide longer relief during the night, a cotton pledget moistened with the spray can be inserted into the nostril. However, mucous membrane irritation caused by a cold or allergy renders this route unreliable. Although the vaginal and buccal mucosae are substitute routes for the spray, this can be inconvenient. Desmopressin acetate (DDAVP), a long-acting analog of arginine vasopressin, which has fewer side effects, is administered intranasally by way of a flexible tube to achieve adequate control. The child’s response pattern is variable, with duration ranging from 6 to 24 hours (Verbalis, 2003; Makaryus and McFarlane, 2006). It is usually administered twice daily—at bedtime to allow the child to sleep through the night and in the morning to allow fewer interruptions in the school day. Some “breakthrough” urination is allowed during the evening hours as a precaution against overmedication. The signs of overmedication are similar to manifestations associated with syndrome of inappropriate ADH (SIADH). (See next section.)
Nursing Care Management
The initial objective is identification of the disorder. Because an early sign may be sudden enuresis in a child who is toilet trained, excessive thirst with bed-wetting is an indication for further investigation. Another clue is persistent irritability and crying in an infant that is relieved only by bottle-feedings of water. After head trauma or certain neurosurgical procedures, the development of DI can be anticipated; therefore closely monitor these patients.
Assessment includes measurement of body weight, serum electrolytes, blood urea nitrogen, hematocrit, and urine specific gravity taken before surgery and every other day after the procedure. Carefully measure and record fluid intake and output. Alert patients are able to adjust intake to urine losses, but unconscious or very young patients require closer fluid observation. In children who are not toilet trained, collection of urine specimens may require application of a urine-collecting device.
After confirmation of the diagnosis, parents need a thorough explanation regarding the condition with specific clarification that DI is a different condition from DM. They must realize that treatment is lifelong. If children are to receive the injectable vasopressin, ideally two caregivers should learn the correct procedure for preparation and administration of the drug. Once children are old enough, encourage them to assume full responsibility for their care.
For emergency purposes, these children should wear medical alert identification. Older children should carry the nasal spray with them for temporary relief of symptoms. School personnel need to be aware of the problem so they can grant children unrestricted use of the lavatory. Failure to permit this may result in embarrassing accidents that often result in a child’s unwillingness to attend school.
Syndrome of Inappropriate Antidiuretic Hormone
The disorder that results from oversecretion of the posterior pituitary hormone, or ADH, is known as SIADH. It occurs with increased frequency in a variety of conditions, especially those involving infections, tumors, or other CNS disease or trauma, and is the most common cause of hyponatremia in the pediatric population (Lin, Liu, and Lim, 2005; Rivkees, 2008).
The manifestations are directly related to fluid retention and hypotonicity. Excess ADH causes most of the filtered water to be reabsorbed from the kidneys back into central circulation. Serum osmolality is low, and urine osmolality is inappropriately elevated. When cells in the brain are exposed to too much water as opposed to sodium, swelling occurs (Rivkees, 2008). When serum sodium levels are diminished to 120 mEq/L, affected children display anorexia, nausea (and sometimes vomiting), stomach cramps, irritability, and personality changes. With progressive reduction in sodium, other neurologic signs, stupor, and convulsions may be evident. The symptoms usually disappear when the underlying disorder is corrected.
The immediate management consists of restricting fluids. Subsequent management depends on the cause and severity. Fluids continue to be restricted to one-fourth to one-half maintenance. When there are no fluid abnormalities but SIADH can be anticipated, fluids are often restricted expectantly at two-thirds to three-fourths maintenance.
Nursing Care Management
The first goal of nursing management is recognizing the presence of SIADH from symptoms described in patients at risk, especially those in the pediatric intensive care unit.
NURSING ALERT
Nausea, vomiting, and malaise may precede the onset of more severe stages such as disorientation, confusion, coma, and seizures (Majzoub and Muglia, 2003).
Accurately measuring intake and output, noting daily weight, and observing for signs of fluid overload are primary nursing functions, especially in the child receiving IV fluids. Seizure precautions are implemented, and the child and family need education regarding the rationale for fluid restrictions. The rare child with chronic SIADH is placed on long-term ADH-antagonizing medication, and the child and family require instructions for its administration.
Disorders of Thyroid Function
The thyroid gland secretes two types of hormones: TH, which consists of the hormones thyroxine (T4) and triiodothyronine (T3), and calcitonin. The secretion of thyroid hormones is controlled by TSH from the anterior pituitary, which in turn is regulated by thyrotropin-releasing factor (TRF) from the hypothalamus as a negative feedback response. Consequently, hypothyroidism or hyperthyroidism may result from a defect in the target gland or from a disturbance in the secretion of TSH or TRF. Because the functions of T3 and T4 are qualitatively the same, the term thyroid hormone (TH) is used throughout the discussion (Box 38-7).
The synthesis of TH depends on available sources of dietary iodine and tyrosine. The thyroid is the only endocrine gland capable of storing excess amounts of hormones for release as needed. During circulation in the bloodstream, T4 and T3 are bound to carrier proteins (thyroxine-binding globulin). They must be unbound before they are able to exert their metabolic effect.
The main physiologic action of TH is to regulate the basal metabolic rate and thereby control the processes of growth and tissue differentiation, as outlined in Box 38-7. Unlike GH, TH is involved in many more diverse activities that influence the growth and development of body tissues. Therefore a deficiency of TH exerts a more profound effect on growth than that seen in hypopituitarism.
Calcitonin helps maintain blood calcium levels by decreasing the calcium concentration. Its effect is the opposite that of PTH in that it inhibits skeletal demineralization and promotes calcium deposition in the bone.
Juvenile Hypothyroidism
Hypothyroidism is one of the most common endocrine problems of childhood. It may be either congenital (see Chapter 9) or acquired and represents a deficiency in secretion of TH (Foley, 2001). Hypothyroidism from dietary insufficiency of iodine is now rare in the United States, since iodized salt is a readily available source of the nutrient.
Critical Thinking Exercise—Hypothyroidism
Beyond infancy, a number of defects may cause primary hypothyroidism. For example, a congenital hypoplastic thyroid gland may provide sufficient amounts of TH during the first year or two but be inadequate when rapid body growth increases demands on the gland. A partial or complete thyroidectomy for cancer or thyrotoxicosis can leave insufficient thyroid tissue to furnish hormones for body requirements. Radiotherapy for Hodgkin disease or other malignancies may lead to hypothyroidism (Hudson, Krasin, Metzger, et al, 2011). Infectious processes may cause hypothyroidism. It can also occur when dietary iodine is deficient.
Clinical manifestations depend on the extent of dysfunction and the child’s age at onset. Primary congenital hypothyroidism is characterized by low levels of circulating thyroid hormones and raised levels of TSH at birth. If left untreated, congenital hypothyroidism causes decreased mental capacity. Improvements in newborn screening have led to earlier detection and prevention of cognitive dysfunction in many children (American Academy of Pediatrics, Rose, Section on Endocrinology and Committee on Genetics of the American Thyroid Association, et al, 2006). The presenting symptoms are decelerated growth from chronic deprivation of TH or thyromegaly. Growth and development are less impaired when hypothyroidism is acquired at a later age, and, because brain growth is nearly complete by 2 to 3 years of age, intellectual disability and neurologic sequelae are not associated with juvenile hypothyroidism. Some clinical manifestations of hypothyroidism in a child are myxedematous skin changes (dry skin, puffiness around the eyes, sparse hair), dry skin, constipation, sleepiness, lethargy, and mental decline. Growth failure, delayed puberty, and excessive weight gain can also be seen.
Therapy is TH replacement, the same as for hypothyroidism in the infant, although the prompt treatment needed in the infant is not required in the child. In children with severe symptoms, the restoration of euthyroidism is achieved more gradually with administration of increasing amounts of l-thyroxine over a period of 4 to 8 weeks to avoid symptoms of hyperthyroidism, which can occur with treatment of chronic hypothyroidism. Researchers have found that children treated early continue to have mild delays in reading, comprehension, and arithmetic but catch up by grade 6 (Rovet and Ehrlich, 2000). However, adolescents may demonstrate problems with memory, attention, and visuospatial processing.
Nursing Care Management
The importance of early recognition in the infant is discussed in Chapter 9. Growth cessation or retardation in a child whose growth has previously been normal should alert the observer to the possibility of hypothyroidism. After diagnosis and implementation of thyroxine therapy, the importance of compliance and periodic monitoring of response to therapy should be stressed to parents. Children should learn to take responsibility for their own health as soon as they are old enough, at about 9 or 10 years of age.
Goiter
A goiter is an enlargement or hypertrophy of the thyroid gland. It may occur with deficient (hypothyroid), excessive (hyperthyroid), or normal (euthyroid) TH secretion. It can be congenital or acquired. Congenital disease usually occurs as a result of maternal administration of antithyroid drugs or iodides during pregnancy. Acquired disease can result from increased secretion of pituitary TSH in response to decreased circulating levels of TH or from infiltrative neoplastic or inflammatory processes. In areas where dietary iodine (essential for TH production) is deficient, goiter can be endemic.
Enlargement of the thyroid gland may be mild and noticeable only when there is an increased demand for TH (e.g., during periods of rapid growth). Where iodine deficiency is severe, a large percentage of the population display goiters. Enlargement of the thyroid at birth can be sufficient to cause severe respiratory distress. Sporadic goiter is usually caused by lymphocytic thyroiditis, and intrinsic biochemical defects in synthesis of the hormones are associated with goiters. TH replacement is necessary to treat the hypothyroidism and reverse the TSH effect on the gland.
Nursing Care Management
Large goiters are identified by their obvious appearance. Smaller nodules may be evident only on palpation. Nurses in ambulatory settings need to be aware of the possibility of goiters and report such findings. Benign enlargement of the thyroid gland may occur during adolescence and should not be confused with pathologic states. Nodules rarely are caused by a cancerous tumor but always require evaluation. Include questions regarding exposure to radiation in the assessment.
NURSING ALERT
If an infant is born with a goiter, immediately begin precautions for emergency ventilation, such as having supplemental oxygen and a tracheostomy set nearby. Hyperextension of the neck often facilitates breathing.
Immediate surgery to remove part of the gland may be lifesaving in infants born with a goiter. When thyroid replacement is necessary, parents have the same needs regarding its administration as discussed for the parents of children who have hypothyroidism. (See Chapter 9.)
Lymphocytic Thyroiditis
Lymphocytic thyroiditis (Hashimoto disease, juvenile autoimmune thyroiditis) is the most common cause of thyroid disease in children and adolescents and accounts for the largest percentage of juvenile hypothyroidism (Szymborska and Staroszczyk, 2000). It accounts for many of the enlarged thyroid glands formerly designated as thyroid hyperplasia of adolescence or adolescent goiter. Although it can occur during the first 3 years of life, it occurs more frequently after age 6. It reaches a peak incidence during adolescence, and there is evidence that the disease is self-limiting. The presence of a goiter and elevated thyroglobulin antibody with progressive increase in both thyroid peroxidase antibody and TSH may be predictive factors for future development of hypothyroidism (Radetti, Gottardi, Bona, et al, 2006).
Pathophysiology
There is a strong genetic predisposition to the development of lymphocytic thyroiditis, although no mode of inheritance has been delineated and the basic stimulus or autoimmune defect is unknown. In families this disease is closely related to other thyroid disorders (Graves disease, idiopathic hypothyroidism, idiopathic myxedema) and autoimmune disorders (pernicious anemia, Addison disease, type 1 DM, and hypoparathyroidism).
The disease is characterized by lymphocytic infiltration of the gland, germinal center inflammation, and, in many patients, replacement with fibrous tissue. In the early stages there may be only hyperplasia. A defect in autoregulation allows the persistence of a T-cell clone, which induces a cell-mediated immune response. Several antithyroid antibodies have been recognized in patients with thyroiditis.
Clinical Manifestations
The practitioner usually detects the enlarged thyroid gland during a routine examination, although parents may notice it when the youngster swallows. In most children the entire gland is enlarged symmetrically (but may be asymmetric) and is firm, freely movable, and nontender. There may be manifestations of moderate tracheal compression (sense of fullness, hoarseness, and dysphagia), but it is extremely rare for a nontoxic diffuse goiter to enlarge to the extent that it causes mechanical obstruction. Most children are euthyroid, but some display symptoms of hypothyroidism. Others have signs suggestive of hyperthyroidism, such as nervousness, irritability, tachycardia, increased sweating, or hyperactivity.
Diagnostic Evaluation
Thyroid function tests are usually normal, although TSH levels may be slightly or moderately elevated. With progressive disease the T4 decreases, followed by a decrease in T3 levels and an increase in TSH. A variety of abnormalities in radioactive iodine uptake may be noted. The majority of children have serum antibody titers to thyroid antigens, but fewer children have a positive red blood cell hemagglutination test result. When both tests are used, almost all children with thyroid autoimmunity are detected. However, levels in children are lower than in adults; therefore repeated measurements may be needed in doubtful cases, since titers may increase later in the disease.
Therapeutic Management
In many cases the goiter is transient and asymptomatic and regresses spontaneously within a year or two. Therapy of a nontoxic diffuse goiter is usually simple, uncomplicated, and effective. Oral administration of TH decreases the size of the gland significantly and provides the feedback needed to suppress TSH stimulation, and the hyperplastic thyroid gland gradually regresses in size. Surgery is contraindicated in this disorder. Evaluate untreated patients periodically.
Hyperthyroidism
The largest percentage of hyperthyroidism in childhood is caused by Graves disease, which is usually associated with an enlarged thyroid gland and exophthalmos (Streetman and Khanderia, 2004; Thompson, 2002). Most cases of Graves disease in children occur between the ages 6 and 15 years, with a peak incidence at 12 to 14 years of age, but the disease may be present at birth in children of thyrotoxic mothers. The incidence is five times higher in girls than in boys.
The hyperthyroidism of Graves disease is apparently caused by an autoimmune response to TSH receptors, but no specific etiology has been identified. There is definitive evidence for familial association, with a high concordance incidence in twins. Patients with Graves disease possess the histocompatibility antigens A1, B8, and DR3 (Dallas and Foley, 2003; Simmonds, Howson, Heward, et al, 2005). Currently, there is no cure for Graves disease, but blocking the production of autoantibodies responsible for overstimulating the thyroid gland is being investigated (Siarkowski, 2005).
Clinical Manifestations
The development of manifestations is highly variable. Signs and symptoms develop gradually, with an interval between onset and diagnosis of approximately 6 to 12 months. The principal clinical features are excessive motion—irritability, hyperactivity, short attention span, tremors, insomnia, and emotional lability. Gradual weight loss despite a voracious appetite occurs in half the cases. Linear growth and bone age are usually accelerated. Muscle weakness often occurs. Hyperactivity of the gastrointestinal tract may cause vomiting and frequent stooling. Cardiac manifestations include a rapid, pounding pulse even during sleep; widened pulse pressure; systolic murmurs; and cardiomegaly. Dyspnea occurs during slight exertion, such as climbing stairs. The skin is warm, flushed, and moist. Heat intolerance may be severe and is accompanied by diaphoresis. The hair is unusually fine and unable to hold a wave.
Exophthalmos (protruding eyeballs), observed in many children, is accompanied by a wide-eyed staring expression, increased blinking, lid lag, lack of convergence, and absence of wrinkling of the forehead when looking upward. As protrusion of the eyeball increases, the child may not be able to completely cover the cornea with the lid. Visual disturbances may include blurred vision and loss of visual acuity. Ophthalmopathy can develop long before or after the onset of hyperthyroidism. A consistent pathogenic link between them has not been identified. It is now thought that Graves ophthalmopathy is a disorder of autoimmune origin caused by a complex interplay of endogenous and environmental factors (Bartalena, Tanda, Piantanida, et al, 2003).
Diagnostic Evaluation
The presence of a thyroid mass in a child requires a thorough history, including inquiry into prior irradiation to the head and neck and exposure to a goitrogen. The diagnosis is established on the basis of increased levels of T4 and T3. TSH is suppressed to unmeasurable levels. Other tests are rarely indicated.
Therapeutic Management
Therapy for hyperthyroidism is controversial, but all methods are directed toward retarding the rate of hormone secretion. The three acceptable modes available are the antithyroid drugs, which interfere with the biosynthesis of TH, including propylthiouracil (PTU) and methimazole (MTZ, Tapazole); subtotal thyroidectomy; and ablation with radioiodine (131I iodide) (Streetman and Khanderia, 2004; Rivkees and Cornelius, 2003). Each is effective, but each has advantages and disadvantages.
When affected children exhibit signs and symptoms of hyperthyroidism (e.g., increased weight loss, pulse, pulse pressure, and blood pressure), their activity should be limited to classwork only. Vigorous exercise is restricted until thyroid levels are decreased to normal or near-normal values.
The American Thyroid Association* has an extensive website with information relation to prevention, treatment, and cure of thyroid disease.
Drug Therapy: Most centers favor drugs as an initial therapy. An effective response to these drugs occurs after a latent period because they inhibit production of additional TH but do not retard secretion of stored supplies. Generally, some improvement is noted within the first 2 weeks, with evidence of decreased nervousness, less fatigue, increased strength, a lowered pulse, and weight gain. In many children an initial treatment course of 1 to 2 years is followed by a complete remission of the disorder. Those who relapse may benefit from a second course of therapy but may also be candidates for surgical intervention or radioiodine therapy (Siarkowski, 2005).
Disadvantages include toxic drug reactions requiring alternate therapy, chronic dependency on the drug, and failure to produce remission in a large number of patients. The most serious side effect of these antithyroid drugs is agranulocytosis (severe leukopenia), which generally occurs within the initial weeks or months of therapy. It is usually accompanied by a sore throat and fever. Treatment involves immediate discontinuation of the drug, social isolation of the child, and administration of antibiotics and glucocorticoids until symptoms resolve.
Thyroidectomy: Surgical treatment involves surgical ablation of the thyroid (thyroidectomy). Although this approach has the advantage of being a long-lasting form of therapy without the need for multiple-dose drug therapy, it has a number of serious disadvantages, including an increased incidence of hypothyroidism and the need for thyroxine therapy, infrequent recurrent laryngeal nerve palsy and permanent hypoparathyroidism, keloid formation of the anterior cervical scar, and (rarely) surgical mortality. Therefore surgery in most centers is reserved for children who do not respond to or comply with the use of antithyroid drugs or who are prone to recurrences.
Radioiodine Therapy: Radioiodine may be a therapy of choice in young patients with Graves disease who relapse after medical treatment (Cheetham, Hughes, Barnes, et al, 1998). Radioiodine therapy has become an even more acceptable option since it has become apparent that lifelong thyroxine replacement is required after either surgery or radioiodine therapy.
Thyrotoxicosis: Thyrotoxicosis (thyroid “crisis” or thyroid “storm”) may occur from sudden release of the hormone. Although thyrotoxicosis is unusual in children, a crisis can be life threatening. These “storms” are evidenced by the acute onset of severe irritability and restlessness, vomiting, diarrhea, hyperthermia, hypertension, severe tachycardia, and prostration. There may be rapid progression to delirium, coma, and even death. A crisis may be precipitated by acute infection, surgical emergencies, or discontinuation of antithyroid therapy. Treatment in addition to antithyroid drugs is administration of β-adrenergic blocking agents (propranolol), which provide relief from the adrenergic hyperresponsiveness that produces the disturbing side effects of the reaction. Therapy is usually required for 2 to 3 weeks.
Nursing Care Management
The initial nursing objective is identification of children with hyperthyroidism. Because the clinical manifestations often appear gradually, the goiter and ophthalmic changes may not be noticed, and the excessive activity may be attributed to behavioral problems. Nurses in ambulatory settings, particularly schools, need to be alert to signs that suggest this disorder, especially weight loss despite an excellent appetite, academic difficulties resulting from a short attention span and inability to sit still, unexplained fatigue and sleeplessness, and difficulty with fine motor skills such as writing. Exophthalmos may develop long before the signs and symptoms of hyperthyroidism and may be the only presenting sign (Thompson, 2002). Exophthalmos is less common in adults than children (Jospe, 2001).
Much of these children’s care is related to treating physical symptoms before a response to drug therapy is achieved. These children need a quiet, unstimulating environment that is conducive to rest. Sometimes hospitalization is necessary during the immediate treatment phase to remove a child from a troubled home. A regular routine is beneficial in providing frequent rest periods, minimizing the stress of coping with unexpected demands, and meeting the children’s needs promptly. Physical activity is restricted. For example, school physical education classes are discontinued.
Because the manifestations often interfere with schoolwork, a consultation with the child’s teachers is important to advise them of the medical reason for the problem and suggest ways of helping the child adjust. For example, the child may benefit from a shortened school day or at least study periods in a quiet area. Limiting demands on the child, such as reciting in class or participating in extracurricular activities, may help conserve strength for academic studies. Despite the excessive activity of these children, they tire easily, experience muscle weakness, and are unable to relax to recover their strength.
Emotional lability is often manifested by sudden episodes of crying or elation. Such behavior, coupled with irritability, disrupts interpersonal relationships, creating difficulties within and outside the home. Parents need help in understanding the uncontrollable nature of these outbursts and ways of minimizing them through decreased environmental stimulation, stress, and frustration. Encourage the child to express feelings about behavior and its effect on others. The nurse can encourage the child to concentrate on friendship with one special peer rather than a group until the condition is stabilized.
Heat intolerance may produce considerable family conflict. Preferring a cooler environment than others, the child is likely to open windows, complain about the heat, wear minimum clothing, and remove blankets while sleeping. Although the child should dress in accordance with climatic conditions, the use of light cotton clothing in the home, good ventilation, air conditioning or fans, frequent baths, and adequate hydration is helpful in providing comfort. Stress hygiene because of excessive sweating.
Adjust dietary requirements to meet the child’s increased metabolic rate. Although the need for calories is increased, these should be provided in wholesome foods rather than “junk” foods. The child may require vitamin supplements to meet daily requirement. Rather than three large meals, the child’s appetite may be better satisfied by five or six moderate meals throughout the day. Family members should refrain from making remarks about the child’s appetite because the child may voluntarily restrict his or her eating to avoid such attention.
Once therapy begins, the nurse explains the drug regimen, emphasizing the importance of observing for side effects of antithyroid drugs. Harmful effects of PTU and related compounds include urticarial rash, fever, arthritis, or arthralgia. There may be enlargement of the salivary and cervical lymph glands, a diminished sense of taste, hepatitis, and edema of the lower extremities. Parents should also be aware of the signs of hypothyroidism, which can occur from overdose of the drugs. The most common indications are lethargy and somnolence.
DRUG ALERT
Propylthiouracil and Methimazole
Children being treated with PTU or MTZ must be carefully monitored for side effects of the drug. Because sore throat and fever accompany the grave complication of leukopenia, these children should be seen by a practitioner if such symptoms occur. Parents and children should learn to recognize and report symptoms immediately.
Surgical Care: If surgery is anticipated, iodine is usually administered for a few weeks before the procedure. Because oral iodine preparations are unpalatable, they should be mixed with a strong-tasting fruit juice, such as grape or punch flavors, and be given through a straw. Compliance with iodine therapy is essential to avoid the danger of thyroid crisis after sudden discontinuation.
Psychologic preparation of children for thyroidectomy is similar to that for any other surgical procedure. (See Chapter 27.) However, of special consideration is the site of the incision. The fear of having the throat cut is very real and in older children is associated with death. The nurse should explain that the throat is not cut, only the skin, to remove the gland. Showing children a picture of the anatomic location of the thyroid around the trachea is often helpful. Children should be prepared for the dressing around the neck and the possibility of an endotracheal or “breathing” tube after surgery.
Postoperative care involves positioning with the neck slightly flexed to avoid strain on the sutures and observation for bleeding and complications. The children learn to support the neck in this position when they sit up. Damage to the laryngeal nerve is evidenced by severe stridor or hoarseness, although some hoarseness is expected. Laryngospasm, a spasmodic contraction of the larynx, can be a life-threatening complication of thyroidectomy. Signs of laryngospasm are stridor, hoarseness, and a feeling of tightness in the throat. Place a tracheostomy set near the bed for emergency use. The nurse should observe for signs of hypoparathyroidism, which causes hypocalcemia, in the immediate postoperative period.
NURSING ALERT
The earliest indication of hypoparathyroidism may be anxiety and mental depression, followed by paresthesia and evidence of heightened neuromuscular excitability, such as:
Chvostek sign—Facial muscle spasm elicited by tapping the facial nerve in the region of the parotid gland
Trousseau sign—Carpal spasm elicited by pressure applied to nerves of the upper arm
Tetany—Carpopedal spasm (sharp flexion of wrist and ankle joints), muscle twitching, cramps, seizures and stridor
Disorders of Parathyroid Function
The parathyroid glands secrete PTH, the main function of which, along with vitamin D and calcitonin, is homeostasis of serum calcium concentration (Perheentupa, 2003). The effect of PTH on calcium is opposite that of calcitonin. Box 38-8 lists the principal effects of PTH on its target sites.
The net result of the integrated action of PTH and vitamin D is maintenance of serum calcium levels within a narrow normal range and the mineralization of bone. Secretion of PTH is controlled by a negative feedback system involving the serum calcium ion concentration. Low ionized calcium levels stimulate PTH secretion, causing absorption of calcium by the target tissues; high ionized calcium concentrations suppress PTH.
Hypoparathyroidism
Hypoparathyroidism is a spectrum of disorders that result in deficient PTH. Congenital hypoparathyroidism may be caused by a specific defect in the synthesis or cellular processing of PTH or by aplasia or hypoplasia of the gland (Perheentupa, 2003).
Hypoparathyroidism can occur secondary to other causes. Postoperative hypoparathyroidism may follow thyroidectomy with acute or gradual onset and be transient or permanent. Two forms of transient hypoparathyroidism may be present in the newborn, both of which are the result of a relative PTH deficiency. One type is caused by maternal hyperparathyroidism or maternal DM. A more common, later form appears almost exclusively in infants fed a milk formula with a high phosphate-to-calcium ratio.
Clinical Manifestations
Symptoms vary from none to significant morbidity if treatment is not initiated. Mild deficiency may be identified through laboratory studies. Muscle cramps are an early symptom, progressing to numbness, stiffness, and tingling in the hands and feet. A positive Chvostek or Trousseau sign or laryngeal spasms may be present. Convulsions with loss of consciousness may occur. These episodes may be preceded by abdominal discomfort, tonic rigidity, head retraction, and cyanosis. Headaches and vomiting with increased intracranial pressure and papilledema may occur and may suggest a brain tumor (Behrman, Kliegman, Jenson, et al, 2009).
Children with longstanding deficiency may have dry, scaly, coarse skin with eruptions often caused by Candida organisms (Behrman, Kliegman, Jenson, et al, 2009). Dental and enamel hypoplasia often occurs. Cataracts develop in patients with untreated disease. Because hypoparathyroidism results in decreased bone resorption and inactive osteoclastic activity, skeletal growth is retarded.
Diagnostic Evaluation
The diagnosis of hypoparathyroidism is made on the basis of clinical manifestations associated with decreased serum calcium and increased serum phosphorus. Levels of plasma PTH are low in idiopathic hypoparathyroidism but high in pseudohypoparathyroidism. End-organ responsiveness is tested by the administration of PTH with measurement of urinary cyclic adenosine monophosphate. Kidney function tests are included in the differential diagnosis to rule out renal insufficiency. Although bone radiographs are usually normal, they may demonstrate increased bone density and suppressed growth.
Therapeutic Management
The objective of treatment is to maintain normal serum calcium and phosphate levels with minimum complications. Acute or severe tetany is corrected immediately by IV and oral administration of calcium gluconate and follow-up daily doses to achieve normal levels. Twice-daily serum calcium measurements are taken to monitor the efficacy of therapy and prevent hypercalcemia. When diagnosis is confirmed, vitamin D therapy is begun. Vitamin D therapy is somewhat difficult to regulate because the drug has a prolonged onset and a long half-life. Some authorities advocate beginning with a lower dose with stepwise increases and careful monitoring of serum calcium until stable levels are achieved. Others prefer rapid induction with higher doses and rapid reduction to lower maintenance levels.
Long-term management consists of administration of massive doses of vitamin D, and oral calcium supplementation may be useful in maintaining adequate serum calcium levels, although it is not essential. Blood calcium and phosphorus are monitored frequently until the levels have stabilized; they are then monitored monthly and less often until the child is seen at 6-month intervals. Renal function, blood pressure, and serum vitamin D levels are measured every 6 months. Serum magnesium levels are measured every 3 to 6 months to permit detection of hypomagnesemia, which may raise the requirement for vitamin D.
Nursing Care Management
The initial objective is recognition of hypocalcemia. Unexplained convulsions, irritability (especially to external stimuli), gastrointestinal symptoms (diarrhea, vomiting, cramping), and positive signs of tetany should lead the nurse to suspect this disorder. Much of the initial nursing care is related to the physical manifestations and includes institution of seizure and safety precautions; reduction of environmental stimuli (e.g., avoiding sudden or loud noise, bright lights, stimulating activities); and observation for signs of laryngospasm such as stridor, hoarseness, and a feeling of tightness in the throat. A tracheostomy set and injectable calcium gluconate should be located near the bedside for emergency use. The administration of calcium gluconate requires precautions against extravasation of the drug and tissue destruction.
After initiation of treatment, the nurse discusses with the parents the need for continuous daily administration of calcium salts and vitamin D. Because vitamin D toxicity can be a serious consequence of therapy, parents should watch for signs that include weakness, fatigue, lassitude, headache, nausea, vomiting, and diarrhea. Polyuria, polydipsia, and nocturia are signs of early renal impairment.
Hyperparathyroidism
Hyperparathyroidism is rare in childhood but can be primary or secondary. The most common cause of primary hyperparathyroidism is adenoma of the gland (Behrman, Kliegman, Jenson, et al, 2009). The most common causes of secondary hyperparathyroidism are chronic renal disease, renal osteodystrophy, and congenital anomalies of the urinary tract. The common factor is hypercalcemia. Box 38-9 lists the manifestations of hyperparathyroidism.
Diagnostic Evaluation
Blood studies to identify elevated calcium and decreased phosphorus levels are routinely performed. Measurement of PTH, as well as several tests to isolate the cause of the hypercalcemia, such as renal function studies, should be included. Other procedures used to substantiate the physiologic consequences of the disorder include electrocardiography and radiographic bone surveys.
Therapeutic Management
Treatment depends on the cause of hyperparathyroidism. The treatment of primary hyperparathyroidism is surgical removal of the tumor or hyperplastic tissue. Parathyroidectomy may cause recurrent laryngeal nerve damage, voice impairment, hypoparathyroidism, hypocalcemia, and tetany (Wang, Roman, and Sosa, 2008). Treatment of secondary hyperparathyroidism is directed at the underlying contributing cause, which subsequently restores the serum calcium balance. However, in some instances such as in chronic renal failure the underlying disorder is irreversible. In this case treatment is aimed at raising serum calcium levels to inhibit the stimulatory effect of low levels on the parathyroids. This includes oral administration of calcium salts, high doses of vitamin D to enhance calcium absorption, a low-phosphorus diet, and administration of a phosphorus-mobilizing aluminum hydroxide to reduce phosphate absorption.
Nursing Care Management
The initial nursing objective is recognition of the disorder. Because secondary hyperparathyroidism is a consequence of chronic renal failure, the nurse is always alert to signs that suggest this complication, especially bone pain and fractures. Because urinary symptoms are the earliest indication, assessment of other body systems for evidence of high calcium levels is indicated when polyuria and polydipsia coexist. Clues to the possibility of hyperparathyroidism include change in behavior, especially inactivity; unexplained gastrointestinal symptoms; and cardiac irregularities.
Much of the initial nursing care is related to the physical symptoms and prevention of complications. To minimize renal calculi formation, hydration is essential. Encourage the child to drink fruit juices that maintain a low urinary pH, such as cranberry or apple juice, since acidity of body fluids promotes calcium absorption. All urine should be strained for evidence of renal casts.
Safety precautions, such as keeping side rails in place at all times and assisting with ambulation, are instituted because of the tendency toward fractures and muscular weakness. Children with renal rickets (osteodystrophy) may wear braces to minimize skeletal deformities. These should be worn as prescribed. If the child is confined to bed, the nurse should consult with the physical therapist regarding proper use of orthopedic appliances.
Vital signs should be taken frequently, and the pulse should be counted for 1 full minute to detect irregularities. Report a decrease in pulse rate, since it may signal severe bradycardia and cardiac arrest. The diet needs supervision to ensure compliance with low-phosphate foods, particularly dairy products. The nurse should instruct parents regarding foods to avoid and the necessity of administering calcium and vitamin D.
If surgery is anticipated, care is similar to that discussed for the child with hyperthyroidism. Because hypocalcemia is a potential complication, observing for signs of tetany, instituting seizure precautions, and having calcium gluconate available for emergency use are part of the nursing care.
Disorders of Adrenal Function
The adrenal glands consist of two distinct portions: the cortex, or outer section, and the medulla, or inner core. The adrenal cortex secretes hormones, collectively called steroids, which are essential to life. The adrenal medulla produces the catecholamines, epinephrine and norepinephrine. Because these chemicals are also produced by the sympathetic nervous system, absence of the adrenal supply is compatible with life.
Adrenal Cortex
The cortex secretes three groups of hormones that are classified according to their biologic activity: (1) glucocorticoids (cortisol, corticosterone), (2) mineralocorticoids (aldosterone), and (3) sex steroids (androgens, estrogens, and progestins). The glucocorticoids and mineralocorticoids influence metabolic regulation and stress adaptation. The sex steroids influence sexual development but are not essential because the gonads secrete the major supply of these hormones.
Glucocorticoids: The most important glucocorticoids in humans are cortisol and corticosterone, the principal effects of which are listed in Box 38-10. Normally the hypothalamus secretes CRF, which causes the pituitary gland to produce ACTH, which stimulates the adrenal glands to synthesize glucocorticoids (primarily cortisol). The switch that controls this feedback is cortisol. When blood levels of cortisol are low, the system turns on. When blood levels of cortisol rise, the system turns off.
In times of stress the anterior pituitary is stimulated by CRF from the hypothalamus, which causes the release of increased amounts of ACTH. Stressful stimuli capable of provoking this response include trauma; anesthesia; surgical intervention; sepsis; acute anoxia; hypothermia; hypoglycemia; and emotional states, especially panic, anxiety, or anger.
Body rhythms also regulate secretion of the glucocorticoids. Blood levels of cortisol demonstrate a typical diurnal or circadian pattern. In individuals who follow a regular routine of nighttime sleeping, cortisol levels are highest in the early morning hours after arising and lowest in the evening hours before bedtime.
Mineralocorticoids: The most important mineralocorticoid is aldosterone. Like cortisol, it promotes sodium retention and potassium excretion in the renal tubules. The effect of aldosterone is many times more potent than that of the glucocorticoids in maintaining extracellular fluid volume, acid-base balance, and normal potassium levels.
Aldosterone synthesis is regulated primarily by the renin-angiotensin system of the kidney. A block in aldosterone synthesis causes high plasma renin activity levels (Huether, 2000). The juxtaglomerular cells of the kidney respond to decreased arterial pressure or blood volume and to decreased sodium concentrations by secreting the enzyme renin into the blood. Renin in turn converts angiotensinogen to angiotensin I and then to angiotensin II. Increased levels of angiotensin stimulate the adrenal cortex to secrete aldosterone, which preserves sodium, thereby retaining water. The renin-angiotensin mechanism also results in increased blood pressure.
Sex Steroids: Except for the first few days of life, the sex hormones are normally secreted in only minimum amounts until adolescence, at which time they play a role in pubertal changes. Their actions are the same as those of the gonadal hormones on internal and external sexual structures and skeletal growth.
Adrenal Medulla
The adrenal medulla secretes the catecholamines epinephrine and norepinephrine. Both hormones have essentially the same effects on different organs as those caused by direct sympathetic stimulation, except that the hormonal effects last several times longer. Their major actions are listed in Box 38-11.
Although the catecholamines evoke similar responses from target sites, there are some important differences. Epinephrine has a greater effect on cardiac activity than norepinephrine, but it causes only weak constriction of the blood vessels of muscles in comparison with the effect of norepinephrine. As a result, norepinephrine elevates blood pressure, whereas epinephrine increases cardiac output. Another important difference is their effect on metabolism. Epinephrine increases the metabolic rate to a much greater extent than norepinephrine. These differences in action have been attributed to the catecholamines’ effects on α- or β-adrenergic receptors. Supposedly norepinephrine can only affect those effector cells that contain α-receptors, which are mostly excitatory (constriction and contraction). Epinephrine, however, can affect both α- and β-receptors, and β-receptors are mostly inhibitory (dilation and relaxation). Control of secretion of catecholamines, primarily in response to physiologic or emotional stress, is through the hypothalamus. Also, stimulation of the sympathetic nervous system results in the release of epinephrine and norepinephrine from the sympathetic nerves and adrenal medulla. Both systems support each other, and one can be substituted for the other. For this reason no condition is attributable to hypofunction of the adrenal medulla. Even in bilateral adrenalectomy, catecholamine replacement is not necessary because the sympathetic release of these chemicals is sufficient to meet all the physiologic functions required to cope with stressful events.
Catecholamine-secreting tumors are the primary cause of adrenal medullary hyperfunction. In children the most common neoplasms of this type are pheochromocytoma (see p. 1593), neuroblastoma, and ganglioneuroma. Ganglioneuromas are neuroblastomas that have undergone maturation into a benign tumor composed of ganglion cells. These tumors are associated with less abnormal catecholamine secretion than the other two types, but persons with ganglioneuromas may have a clinical picture of chronic diarrhea, failure to thrive, skin rash, hypokalemia, persistent cough, and abdominal distention. The exact reason for these symptoms is unknown, although they are attributable to the tumor because they disappear after surgical removal of the mass.
Acute Adrenocortical Insufficiency
The acute form of adrenocortical insufficiency (adrenal crisis) may have a number of causes during childhood. Although a rare disorder, some of the more common etiologic factors include hemorrhage into the gland from trauma, which may be caused by a prolonged, difficult labor and rapidly progressing infections, such as meningococcemia, which result in hemorrhage and necrosis (Waterhouse-Friderichsen syndrome). Abrupt withdrawal of exogenous sources of cortisone or failure to increase endogenous supplies during stressor congenital adrenogenital hyperplasia of the salt-losing type can also cause adrenal crisis.
Clinical Manifestations
Early symptoms of adrenocortical insufficiency include increased irritability, headache, diffuse abdominal pain, weakness, nausea and vomiting, and diarrhea. Generalized hemorrhagic manifestations are present in Waterhouse-Friderichsen syndrome. Abnormal serum electrolyte levels include hyponatremia and hyperkalemia. Fever increases as the condition worsens and is accompanied by signs of CNS involvement, such as nuchal rigidity, convulsions, stupor, and coma. The child is in a shocklike state with a weak, rapid pulse; decreased blood pressure; shallow respirations; cold, clammy skin; and cyanosis. Circulatory collapse is the terminal event.
In the newborn, adrenal crisis is accompanied by extreme hyperpyrexia (high temperature), tachypnea, cyanosis, and seizures. Usually there is no evidence of infection or purpura. However, hemorrhage into the adrenal gland may be evident as a palpable retroperitoneal mass.
Diagnostic Evaluation
There is no rapid, definitive test for confirmation of acute adrenocortical insufficiency. Samples for measurement of plasma cortisol and ACTH levels should be sent for analysis, but this is too time-consuming to be practical for initial diagnosis. Therefore diagnosis is usually made based on clinical presentation, especially when a fulminating sepsis is accompanied by hemorrhagic manifestations and signs of circulatory collapse despite adequate antibiotic therapy. Serum electrolytes can be helpful in narrowing the diagnosis as well. Because there is no real danger in administering a cortisol preparation for a short period, institute treatment immediately. Improvement with cortisol therapy confirms the diagnosis.
Therapeutic Management
Treatment involves replacement of cortisol, replacement of body fluids to combat dehydration and hypovolemia, administration of glucose solutions to correct hypoglycemia, and specific antibiotic therapy in the presence of infection. Initially IV hydrocortisone (Solu-Cortef) is administered as a bolus, followed by a continuous infusion for 24 hours. Normal saline containing 5% glucose is given parenterally to replace lost fluid, electrolytes, and glucose. Persistent hyperkalemia that is associated with electrocardiographic changes may require the use of insulin and glucose or Kayexalate to reduce the potassium levels. If hemorrhage has been severe, whole blood may be replaced. In the event that these measures do not reverse the circulatory collapse, vasopressors are used for immediate vasoconstriction and elevation of blood pressure.
Once the child’s condition is stabilized, oral doses of cortisone, fluids, and salt are given, similar to the regimen used for chronic adrenal insufficiency. To maintain sodium retention, synthetic salt-retaining steroids replace aldosterone.
Nursing Care Management
Because of the abrupt onset and potentially fatal outcome of this condition, prompt recognition is essential. Vital signs and blood pressure are taken every 15 minutes to monitor the hyperpyrexia and shocklike state. Seizure precautions are instituted, since convulsions from the elevated temperature are not uncommon. As soon as therapy is instituted, the nurse should monitor the child’s response to fluid and cortisol replacement. Too rapid administration of fluids can precipitate cardiac failure, whereas overdosage with cortisol produces hypotension and a sudden fall in temperature.
The nurse should regulate IV infusions carefully to guard against too rapid administration of drugs. The nurse should also record intake and urinary output and monitor electrolytes frequently.
Once the acute phase is over and the hypovolemia is corrected, give the child oral fluids, such as small quantities of ginger ale, fruit juice, or ice pops. Too rapid ingestion of oral fluids may induce vomiting, which increases dehydration. Therefore the nurse should plan a gradual schedule for reintroducing liquids. For children who refuse to drink, the prospect of having the IV infusion removed once oral fluids are increased is often a motivating factor.
NURSING ALERT
Monitor serum electrolyte levels and observe for signs of hypokalemia or hyperkalemia (e.g., weakness, poor muscle control, paralysis, cardiac dysrhythmias, and apnea). The condition is rapidly corrected with IV or oral potassium replacement.
DRUG ALERT
Oral Potassium
When an oral potassium preparation is given, mix it with a small amount of strongly flavored fruit juice to disguise its bitter taste.
The sudden, severe nature of this disorder necessitates a great deal of emotional support for the child and family. The child may be placed in an intensive care unit where the surroundings are strange and frightening. Despite the need for emergency intervention, the nurse must be sensitive to the family’s psychologic needs and prepare them for each procedure, even if this is as brief as a statement such as “The IV infusion is necessary to replace fluid that the child is losing.” Because recovery within 24 hours is often dramatic, the nurse should keep the parents apprised of the child’s condition, emphasizing signs of improvement, such as a lowered temperature and normal blood pressure.
If treatment needs to be continued past the acute stage, parents require the same preparation as in the case of children with chronic adrenal insufficiency. Preparation for discharge should begin as soon as possible after the child’s condition has stabilized.
Chronic Adrenocortical Insufficiency (Addison Disease)
Chronic adrenocortical insufficiency is rare in children. Causes include infections (fungal, human immunodeficiency virus, tuberculosis), destructive lesions of the adrenal gland or neoplasms, and autoimmune processes, or the cause is idiopathic. At one time, generalized tuberculosis was the leading cause of adrenal gland destruction.
Evidence of this disorder is usually gradual in onset, since 90% of adrenal tissue must be nonfunctional before signs of insufficiency are manifested. However, during periods of stress, when demands for additional cortisol are increased, symptoms of acute insufficiency may appear in a previously well child (Table 38-1).
The initial step in the laboratory diagnosis of adrenal insufficiency is measurement of a fasting serum cortisol and ACTH in the early morning. An ACTH stimulation test can confirm the diagnosis. The cortisol and urinary 17-hydroxycorticosteroid levels are low and fail to rise, whereas plasma ACTH levels are elevated with corticotropin (ACTH) stimulation, the definitive test for the disease.
Therapeutic Management
Treatment involves replacement of glucocorticoids (cortisol) and mineralocorticoids (aldosterone). Some children are able to be maintained solely on oral supplements of cortisol (cortisone or hydrocortisone preparations) with a liberal intake of salt. During stressful situations, such as fever, infection, emotional upset, or surgery, the dosage must be tripled to accommodate the body’s increased need for glucocorticoids. Failure to meet this requirement will precipitate an acute crisis. Overdosage produces appearance of cushingoid signs (Fig. 38-4).
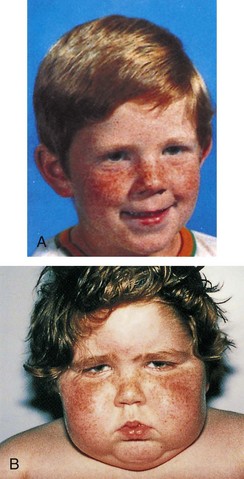
Fig. 38-4 A, Boy before development of Cushing syndrome. B, Same boy 4 months after onset of Cushing syndrome. (From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 5, St Louis, 2007, Mosby.)
Children with more severe states of chronic adrenal insufficiency require mineralocorticoid replacement to maintain fluid and electrolyte balance. Other forms of therapy include monthly injections of desoxycorticosterone acetate or implantation of desoxycorticosterone acetate pellets subcutaneously every 9 to 12 months.
Nursing Care Management
Once the disorder is diagnosed, parents need guidance concerning drug therapy. They must be aware of the continuous need for cortisol replacement. Sudden termination of the drug because of inadequate supplies or inability to ingest the oral form because of vomiting places the child in danger of an acute adrenal crisis. Therefore parents should always have a spare supply of the medication in the home. Ideally they will have a prefilled syringe of hydrocortisone and be instructed in proper technique for intramuscular administration of the drug in case of crisis. Unnecessary administration of cortisone will not harm the child, but if it is needed, it may be lifesaving. Report any evidence of acute insufficiency to the practitioner immediately.
Parents also need to be aware of side effects of the drugs. Undesirable side effects of cortisone include gastric irritation, which is minimized by ingestion with food or the use of an antacid; increased excitability and sleeplessness; weight gain that may require dietary management to prevent obesity; and, rarely, behavioral changes, including depression or euphoria. Parents should be aware of signs of overdose and report these to the practitioner. In addition, the drug has a bitter taste, which creates a challenge for nurses and parents in its administration.
DRUG ALERT
Mineralocorticoids
The side effects of mineralocorticoids are primarily caused by overdosage and include generalized edema, which is first noticed around the eyes; hypertension, which may cause headaches; cardiac arrhythmias; and signs of hypokalemia. Evaluate the child periodically for evidence of excessive medication. Emphasizing the importance of routine follow-up care is a significant nursing responsibility.
Because the body cannot supply endogenous sources of cortical hormones during times of stress, the home environment should be stable and relatively unstressful. Parents need to be aware that during periods of emotional or physical crisis the child requires additional hormone replacement. The child should wear medical identification, such as a bracelet, to permit medical personnel to adjust requirements during emergency care.