Conditions Caused by Defects in Physical Development
Defects in Physical Development
Malformations of the Central Nervous System

http://evolve.elsevier.com/wong/ncic

Alternative Feeding Techniques, Ch. 27
Anaphylaxis, Ch. 29
Assessment (Newborn), Ch. 8
Autosomal Inheritance Patterns, Ch. 5
Birth Injuries, Ch. 9
Birthmarks, Ch. 9
Cerebral Palsy, Ch. 40
Family-Centered Home Care, Ch. 25
Health Promotion of the Newborn and Family, Ch. 8
The High-Risk Newborn and Family, Ch. 10
Hypertrophic Pyloric Stenosis, Ch. 33
Multifactorial Disorders, Ch. 5
Neonatal Pain, Ch. 7
Osteogenesis Imperfecta, Ch. 39
Pain Assessment; Pain Management, Ch. 7
Preparation for Diagnostic and Therapeutic Procedures; Surgical Procedures, Ch. 27
Promotion of Parent-Infant Bonding (Attachment), Ch. 8
Defects in Physical Development
Congenital malformations, also called congenital anomalies or birth defects, may be caused by genetic or environmental factors, but not all congenital defects are malformations (e.g., inborn errors of metabolism that cause neurocognitive impairment). However, this chapter is primarily concerned with structural abnormalities and with the impact on the family of the birth of a child with a physical defect. The genetic basis of physical defects is discussed in Chapter 5, and other specific disorders are presented as appropriate throughout the book.
Prenatal Development
Fetal Growth and Differentiation
Development consists of two distinct but interrelated processes: growth and differentiation. Growth results when cells divide and synthesize new proteins and is reflected in increased size and weight. It is accomplished by two mechanisms: (1) hyperplasia (increase in cell number) and (2) hypertrophy (increase in cell size). Hyperplasia is the predominant form of growth during the embryonic period. Although the rate slows during later stages of gestation, cell division continues in variable degrees throughout childhood. Hypertrophy is more prominent during later periods of growth.
Each organ and tissue has a typical growth pattern, and all organs progress from a stage characterized by an increase in cell number to one of growth by increase in cell size. Any interference with this pattern of growth results in a reduction in the size and weight of that organ. However, the consequences of the inhibiting factor depend on whether the insult is inflicted during a period of hyperplasia or during a period of hypertrophy. Interference with growth during a period of cell proliferation is likely to cause irreversible growth restriction of that organ with a permanent deficit in overall cell numbers. Interruption of growth during cell enlargement is usually only temporary and can be overcome with proper intervention.
Differentiation is the process by which early cells are systematically modified and specialized to form all the tissues necessary to ensure an organized, coordinated individual. Each step in this process depends on successful completion of a previous step. Anything that interferes with one of these steps, such as a mutant gene or environmental agent, will cause an arrest in the development of that particular tissue or organ. Divergence from the normal course of development will result in maldevelopment of a part or, if it occurs at an early age, a sequence of distortions causing more severe or multiple malformations.
A relationship appears to exist between the incidence of one congenital anomaly and the presence of additional anomalies in an affected child. For example, malformed ears and kidney abnormalities have an association that reflects an event occurring at a common developmental stage. Knowledge of the stage of development for a variety of organs and systems provides a valuable clue for the examiner. When one defect is observed, closer scrutiny may reveal defects in another organ or system related to the same stage of development.
Extremely rapid development and change take place during the first 8 to 12 weeks of fetal life, and the beginnings of all major organ systems are formed (organogenesis). The embryo begins to acquire the specific functions needed to integrate these organs and organ systems into an organized, coordinated whole. It is also the period during which the organism is most vulnerable to structural disturbance from environmental hazards.
Sensitive Periods in Prenatal Development
Every organ, system, and body part goes through a period during which it experiences the most rapid cell division and differentiation. During this time the organism displays a marked susceptibility to injurious influences. These specific stages of crucial developmental advancement are termed sensitive or critical periods, and the major impact of environmental factors on development always coincides with these periods. The origin or method by which prenatal growth processes are disturbed to produce a structural or functional defect is termed teratogenesis (from the Greek teratos, “monster,” and genesis, “production”). An agent capable of producing such an effect is a teratogen. (See Problems Caused by Perinatal Environmental Factors, Chapter 9.)
The sensitive periods for all organs or parts do not occur simultaneously. A part that is susceptible to adverse influences at one particular time may be resistant to the same influences at other periods of development, while another part may be highly sensitive at that moment. Susceptibility to environmental influences decreases as organ formation advances—the younger the organism and the fewer the cells, the greater is the extent of involvement when an adverse influence is applied.
During the period of intensive differentiation, most teratogenic agents are highly effective and may produce a variety of deformities. The type of defect produced depends on which organ is most susceptible at the time of application. The susceptibility of most tissues to teratogenic influences decreases rapidly in the later periods of development, which are characterized by growth and elaboration of established organs. However, some tissues, particularly those of the central nervous system (CNS), are sensitive to varying degrees throughout fetal life and even beyond. Fig. 11-1 illustrates the approximate times of critical differentiation for some of the major organs and systems.
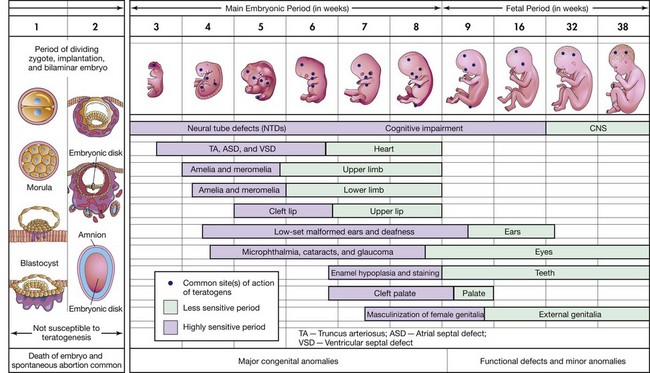
Fig. 11-1 Critical periods in human prenatal development. The mauve areas denote highly sensitive periods, when major defects may be produced. The green sections indicate stages that are less sensitive to teratogens, when minor defects may be induced. CNS, Central nervous system. (Modified from Moore KL, Persaud TVN: Before we are born: essentials of embryology and birth defects, ed 7, Philadelphia, 2008, Saunders.)
Birth of a Child with a Physical Defect
Part of the preparation for childbirth involves fantasies and images of the expected infant. Normally, parents hope for a perfect child, but at the same time they fear that the infant will be abnormal. Parents often express this fear when they state that their concern is not whether the child is a girl or a boy, but just that the infant is healthy. One of the first things the mother wants confirmed at the time of birth is: “Is my baby all right?” In many instances some discrepancy exists between the parents’ idealized child and the infant the mother delivers, as, for example, the birth of a boy when they had hoped for a girl. Resolution of this discrepancy is a developmental task of parenthood and is essential to the establishment of a healthy parent-child relationship. If this discrepancy is major, as when the infant has a birth defect or the wishes of the parents are unrealistic, the resulting emotional stress may be overwhelming.
The more severe the defect, the greater the impact of the experience, especially for the mother. The birth of a child with a physical imperfection abruptly ends the psychologic attachment the mother has formed during pregnancy with the idealized child. She and the father must now deal with loss of the anticipated healthy child while they face meeting the demands of the affected child for care and affection. The birth of an infant with a defect evokes the same psychologic reaction as the death of a child. The parents’ need to grieve for the loss of the expected child while adapting to the care of the child with a disability places overwhelming demands on them at a time when their own psychologic and physiologic resources have been depleted by the birth experience. The impact of this new and unexpected burden inhibits the accomplishment of the grief work that normally follows a loss.
The grief reaction experienced by parents at the birth of a child with a physical disability is the same as the response that follows the loss of any valued or significant object. The parents experience shock, frustration, and anger at what has happened to them, and they ask themselves, “Why? Why me?” Parents may feel shame and embarrassment, often with feelings of personal failure and guilt. Frequently the mother believes that she might have caused harm to the unborn child, and she may associate the condition with wrongdoing or evil thoughts, especially if the pregnancy was unwanted initially. She may believe the defect to be a result of passive or active attempts to terminate the pregnancy, such as deliberate attempts to induce abortion or failure to obtain prenatal care or comply with the practitioner’s instructions. The father may react to the situation by becoming withdrawn from the newborn and the mother. Anger is common, and parents may direct it at health care workers involved in the child’s care. Inwardly the father may blame himself for the child’s “imperfection” yet project that blame to others. The mother may not understand the father’s withdrawal, and this may compound her distress.
The phase of overwhelming shock is accompanied by weeping and feelings of helplessness. To deal with stress and anxiety, parents use defense mechanisms that have provided protection in the past. A common response is disbelief and denial, which may be short lived or may last for many months. They do not appear to “hear” what is told to them about their child, and they behave as though nothing is wrong with the child. Denial during the shock phase of the grief process can serve as a constructive means for parents to deal with the sudden and profound impact of the initial stress until they are better able to cope with the situation.
When parents are unable to face the reality of the infant’s condition, they may withdraw from the situation either physically or emotionally. They frequently become incapacitated and unable to function in their usual manner. They may avoid interpersonal contacts. Unable to face relatives and friends for fear of the reactions they may encounter, parents choose the protection of isolation. They feel as though they are alone in a world all their own. Avoidance behaviors on the part of others, including health workers, contribute to this withdrawal and compound the loneliness that is so common in parents of an affected infant.
Parents may extend this avoidance behavior to include each other or the infant. They may seem unable to face the infant, and visits may become sporadic or nonexistent. It may take time for the parents to master their own feelings before they are able to deal constructively with the situation. A more subtle form of isolation occurs in parents who are objective in their behavior toward the infant and the defect. They are intellectually concerned with the infant’s medical care but display no emotional involvement. Their attention is focused on the abnormality, not on the infant.
Parental reactions vary and include guilt, anger, anxiety, and sadness, which often last for years and depend to a large extent on the type and severity of the defect. A visible anomaly, especially one involving the face, usually elicits a more intense emotional response than one that is less apparent, such as a heart defect. The extent of the impairment does not determine the degree of parental reactions. Because of their limited contact with congenital defects, parents’ perception of the abnormality and its implications may be distorted, and much depends on previous feelings they may have experienced with a similar abnormality. Therefore their reactions may seem out of proportion to the actual extent and severity of the impairment as viewed by health professionals.
Nursing Care Management
The attitudes and behaviors of nurses and other health care providers at the birth of a child with a birth defect significantly influence the effect of the situation on the parents. During this time parents are particularly sensitive and responsive to the behaviors of those with whom they are in contact. Therefore the reactions of health professionals toward the infant and the parents provide cues to the parents that can affect their feelings toward the infant and themselves. Parents exert the greatest influence on the child’s growth and development, and their initial relationship with the child significantly affects the subsequent course of interaction.
Initial Contact: The first indication that all is not well often occurs at the time of delivery. The atmosphere of happy anticipation suddenly changes to one laden with anxiety. Even when the mother is unable to see the infant, she may be terrifyingly aware of the heightened tension in the room, which conveys to her that something is seriously wrong. Health professionals, unprepared for this disturbing experience, find it difficult to cope with their own feelings and react with frustration and resentment toward a situation that they are powerless to change. As a result, they may forget about or retreat from the parents, who at this moment are suffering the most.
Most practitioners believe it is their responsibility to inform the parents of a congenital anomaly. At the time of delivery, unless a pediatrician or nurse practitioner is in attendance, there is a delay while the practitioner is involved with the mother’s care. During this period the mother, unable to see her child and feeling the tense atmosphere, will believe either that the child is normal but that others do not share her enthusiasm or that the child has a defect that is so terrible the professional people in the room are unable to talk about it. A nurse, the person who is most likely to be free to support the mother and who is familiar with most common congenital anomalies, can make truthful statements about the defect.
The manner in which nurses present the infant to the parents may well set the tone for the early parent-child relationship. It is probably best to explain briefly, in simple language, the nature of the defect and to reinforce and help clarify information given by the practitioner before the infant is shown to them. At this time they are more likely to “hear” what is said. Parents attach a great deal of meaning to the behavior of others during this critical period and will watch the facial expressions of others closely for signs of revulsion or rejection. Presenting the infant as something precious and emphasizing the well-formed aspects of the infant’s body provide some reassurance to parents during this crisis period.
It is important to allow time and opportunity for the parents to express their initial response to the situation. Many issues may surface, such as the importance placed on this particular infant or the cultural significance of one sex over the other. Encourage parents to ask questions and to receive honest, straightforward answers without undue optimism or pessimism.
Family Support: Parents need time to grieve for the loss of the expected child before they are able to form an emotional attachment to the child they have. It is a nursing responsibility to help parents with their grief work and to facilitate the formation of a satisfactory adjustment to the child with a defect. They need help to see their infant as a person, support in coping with their situation, and guidance in physical care of the child.
Nurses who understand the grief response will be prepared to support the parents through this necessary process. This is particularly important with the birth of a child with a defect, since the parents may not begin to invest any feeling for the child until they are able to talk about and work through their feelings of disappointment, resentment, guilt, and helplessness. The supportive nurse creates and maintains an atmosphere that encourages expression of feelings. Open expression is difficult for many people, and the parent(s) may hesitate to display intense feelings. Containing those feelings expends considerable energy that would be better used later on to develop a relationship with the infant. Nurses therefore need to listen closely for cues that indicate areas of discomfort or readiness to talk.
Parents may not be ready to talk about their feelings during the first few days after the birth. Their dream has vanished, and when others avoid them, they often interpret it as another abandonment. Staying near and available tells them that they are not alone and that someone cares about them and their feelings. What is said to them is also important. Clichés such as “You will be able to have more children” or “It could be a lot worse” are not a comfort to the parents. Such behavior implies that this infant is not important, and this behavior may destroy the parents’ trust. Parents may also attach inordinate significance to statements made by a health care worker about the prognosis of the infant; such statements may be recalled by the parent years later.
Initiating a discussion about matters that were of concern to others in a similar situation may help the parents to know that their feelings are natural. Parents need to be allowed silence and solitude if this is their wish. The parents are likely to be angry and often direct this anger at anyone nearby—physicians, nurses, friends, and families who have normal children. Directing their frustrations at a nonjudgmental target helps parents relieve some of their distress. Nurses must be prepared to accept any or all of the parental reactions and defenses—anger, hostility, rejection, dependency—without showing anger or withdrawing from the situation. If nurses make themselves available to the parents for support, they can often find nonthreatening ways to help and comfort. Most important, nurses need to promote communication and understanding within the family and help strengthen family interpersonal relationships.
Care of the Infant: Many parents are uneasy about handling their infant and require support and encouragement in their caregiving tasks. These parents need a longer period of dependency to muster their resources for coping. Although they should not feel forced to care for the infant until they are ready for the responsibility, they should be given opportunities to assume care as soon as possible to help them deal with the reality of the infant’s condition. Parents’ responses are highly individual and must be evaluated on this premise. However, all parents need sympathetic, patient, and understanding help to gain feelings of adequacy in the care of their child and to facilitate development of a positive relationship with the infant later on. As anxiety and the intensity of emotional responses decrease, parents begin to feel more comfortable with the infant and more confident in their ability to provide needed care.
Supplying Information: Parents need accurate, up-to-date information given to them early and in language they can understand. Because they do not hear everything the first time it is said, they need careful, repeated explanations about the child’s defect, the treatments outlined, and what will be expected of them. Parents often misinterpret information, another reason for repeated explanations. Often the nurse’s responsibility is to explain, interpret, and clarify and to answer questions about information that the practitioner has given. Following the basic concepts of informational needs assessment, the nurse determines what the parents know and proceeds from that point. One cannot assume that the parents’ failure to ask questions means they understand. Most parents have little or no knowledge of basic anatomy or physiology; therefore use pictures and other tangible visual aids to explain both normal and deviant structures.
Teaching the parents to provide the special care that is frequently required for an infant with a physical defect is an important nursing responsibility. Nurses need to explain and demonstrate special feeding, holding, and positioning techniques. Anticipatory guidance regarding problems that are unique to each condition reduces apprehension and stimulates the parents to institute preventive measures and to make alert observations.
Numerous agencies and organizations offer services to families of children with congenital defects. Some provide services for a variety of conditions; others are devoted to specific disorders. They help families with ongoing problems and with anticipating problems, including financial burdens, they will encounter in raising a child with a defect. Many have local support groups. All have unique and specialized services to support the family and aid parents in problem solving. Among those that include most types of defects and conditions are the Easter Seals,* the March of Dimes,† and Birth Defect Research for Children, Inc.,‡ most of which have branches in all major cities and communities. The Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities has a website with information on birth defects.§
Nursing Care of the Surgical Neonate
Advances in early detection of defects (including prenatal diagnosis), surgical techniques, and anesthesia have made it possible for correction or amelioration of many physical defects in the newborn period. Fortunately, most malformations, even those with a dramatic presentation, are correctable with a high degree of success.
Preoperative Care
Most of the problems encountered with the infant undergoing surgery are discussed in relation to the high-risk infant (e.g., airway maintenance, cardiovascular support, thermoregulation, fluid and electrolyte balance, and nutritional needs). Electronic monitoring of cardiovascular and respiratory status is implemented and maintained, as are regular comprehensive assessments. (See Assessment, Chapter 10.) Monitoring and assessments are continued in the postoperative period. Some congenital defects are often associated with other anomalies; therefore assessment should include careful observation for evidence of complications related to these.
Before surgery the infant usually requires peripheral intravenous (IV) access for fluids and glucose. Any electrolyte problems, acid-base imbalance, and anemia are corrected. In some instances a blood product such as packed red blood cells or whole blood is placed on reserve in case blood loss is anticipated. Prophylactic antibiotic administration may begin before surgery, and the infant is observed and monitored for any evidence of infection. In addition to routine care, special attention is directed to specific defects, such as abdominal decompression, protection and management of open wounds, and specific measurements (e.g., abdominal girth, head dimensions). (See also discussion of specific defects.) A preoperative assessment of the infant’s behavior is essential because postoperative deviations may be a manifestation of pain or unstable condition.
Compounding the initial shock of having an infant born with a physical defect, the parents are often further traumatized by the prospect of surgery, sometimes shortly after birth. Health care personnel provide parents with accurate information regarding the type of surgical procedure anticipated, method of anesthesia, and, most important, what to expect postoperatively. (Parents are sometimes mentally unprepared for the infant’s appearance postoperatively; some may have false hopes or expectations that the infant will be perfect in appearance after surgery.) The nurse also assures parents that the infant’s pain management needs will be evaluated and met postoperatively.
When an infant is transported to a tertiary center for surgery shortly after birth, it is helpful for the nurse to stay in contact with the parents, especially the mother, regarding the infant’s condition. Photographs and even videos, when possible, are helpful tools to relieve the mother’s anxiety; without seeing her infant and without adequate communication, the mother’s anxiety and fears about her infant’s condition may be far worse than the reality. During this time the father may serve as the vital link of information between the mother, siblings, and the tertiary center where the infant is undergoing surgery.
Postoperative Care
Surgery imposes significant stresses on the neonate, especially the preterm or ill infant. The assessment and observations remain much the same as for preoperative care, with the additional problems related to surgery, such as anesthesia and pain. It is essential to maintain physiologic stability to avoid undesirable consequences. Because the neonate is subject to many adverse effects of stress in all physiologic parameters, continual vigilance is mandatory.
Many of the physiologic problems to which the neonate is vulnerable are discussed in relation to assessment and nursing care of the normal newborn (Chapter 8) and the high-risk infant (Chapter 10). Optimum ventilation, cardiac function, thermoregulation, fluid regulation, care of the operative site, and pain management are primary concerns (Table 11-1). Table 11-2 further outlines some of the possible reactions, their probable cause, and the nursing responsibilities.
TABLE 11-1
CRITICAL GUIDELINES FOR NEONATAL POSTOPERATIVE CARE
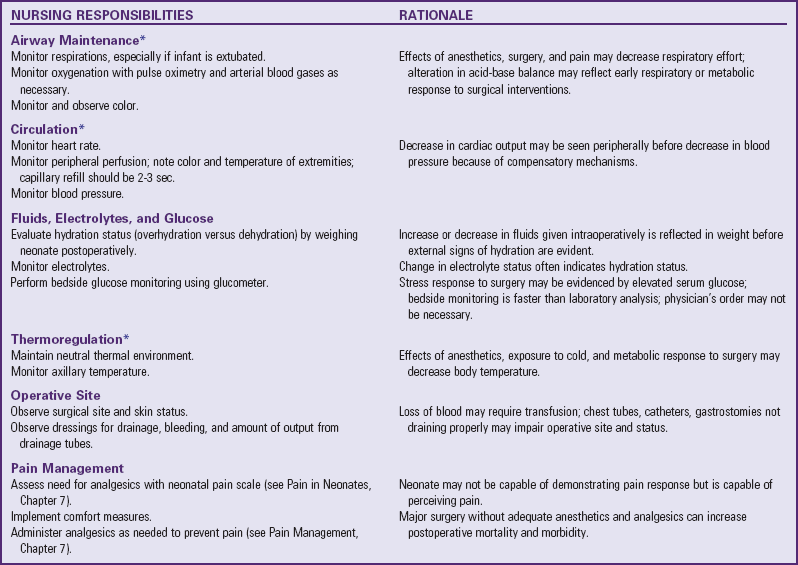
*Suggested interval for monitoring vital signs postoperatively in neonate: every 15 min for 4 hr; every 30 min for 2 hr; every 1 hr for 6 hr; then every 2 hr for 24 hr. More frequent monitoring may be needed based on nurse’s judgment of infant’s status.
TABLE 11-2
POSSIBLE EFFECTS OF SURGERY ON SELECTED SYSTEMS
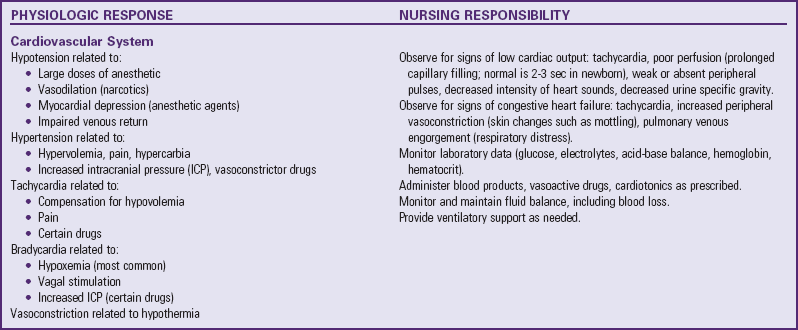




Data from Rushton CH: The surgical neonate: principles of nursing management, Pediatr Nurs 14:141-151, 1988.
Because of the respiratory characteristics of newborns, some compromising responses may occur. The newborn’s poor chest wall stability, smaller and more reactive airways, fewer and smaller alveoli, and poorly developed accessory muscles contribute to respiratory dysfunction. Compression by intrapleural fluid, air, blood, or a distended abdomen can further compromise pulmonary efforts. Respiratory distress is a common problem in preterm infants. Many postoperative neonates require mechanical ventilation, which may be further influenced by the type, duration, and urgency of the surgery. Neonates are highly subject to acidosis and hypoxia and require continuous monitoring of oxygen and acid-base status. Preterm infants require close monitoring for respiratory complications from general anesthesia.
Cardiovascular support is of particular importance because the immature sympathetic innervation of the myocardium makes the neonate sensitive to vagal stimulation induced by many postoperative procedures, such as nasogastric (NG) tubes, endotracheal (ET) tubes, and tracheal suctioning. The nurse notes any evidence of early compensation for diminished cardiac output and implements interventions before decompensation occurs.
Careful management of fluid and electrolyte status is vital to neonatal surgical care. The natural tendency for rapid fluid shifts related to characteristics of the neonate (see Chapter 28) may be aggravated by stress and any abnormal losses associated with some surgical procedures. (See Hydration, Chapter 10.)
Pain Management: During the postoperative period it is essential to assess and manage neonatal pain. This task is complicated by the variability with which neonates respond to painful stimuli and the lack of physiologic responses that may occur as a result of anesthesia. The use of muscle-paralyzing agents may further mask physiologic manifestations of pain in the postoperative period. It is often noted that the more preterm or physiologically immature the infant, the more difficult it becomes to measure pain responses, particularly when major surgery is involved. Because infants of any gestational age are capable of experiencing pain and being adversely affected by it during and after operative procedures, it is important to advocate for appropriate pharmacologic therapy to improve neonatal pain. Both pharmacologic and nonpharmacologic pain management therapies may be used in the postoperative period to effectively reduce neonatal pain. (See Pain in Neonates, Chapter 7.)
Malformations of the Central Nervous System
Defects of Neural Tube Closure
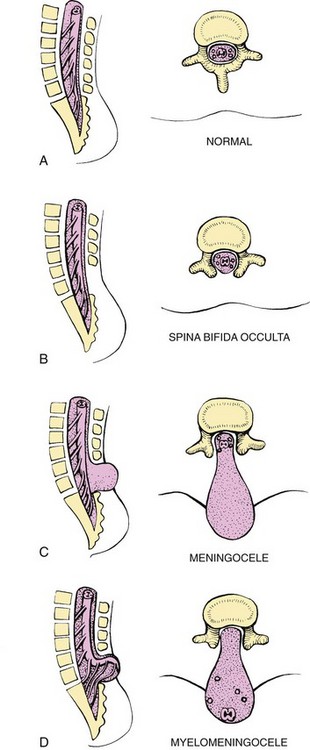
Abnormalities that come from the embryonic neural tube (neural tube defects [NTDs]) constitute the largest group of congenital anomalies with multifactorial inheritance. Normally the spinal cord and cauda equina are encased in a protective sheath of bone and meninges (Fig. 11-2, A). Failure of neural tube closure produces defects of varying degrees (Box 11-1). They may involve the entire length of the neural tube or may be restricted to a small area.`
Etiology
Two of the defects, anencephaly and spina bifida (SB), occur in association with one another more often than would be expected by chance, suggesting a common origin. The CNS defects may alternate in siblings, which also tends to support the theory of a common origin. The incidence of SB is higher in girls than in boys, and it is three times more likely to occur in Caucasians than in African-Americans. In the United States, rates of NTDs declined by as much as 23% between 1995-1996 and 2000. NTD rates decreased an additional 6.9% between 2000 and 2005, primarily among African-American mothers. One concern is that NTD rates have not decreased among Hispanic and non-Hispanic Caucasian mothers since 1999 (Centers for Disease Control and Prevention, 2009). The decline in NTDs in the late 1990s has been attributed in large part to the addition of folic acid to cereal grain products (Honein, 2001). In 2005 the rates for SB were estimated by the Centers for Disease Control and Prevention to be 17.96 per 100,000 live births, thus making this one of the most common birth defects in the United States (Matthews, 2009; Wolff, Witkop, Miller, et al, 2009). Increased use of prenatal diagnostic techniques and termination of pregnancies have also affected the overall incidence of NTDs.
Most authorities believe that the primary defect in NTDs is a failure of neural tube closure during the embryo’s early development (between the third and fourth week). However, evidence also implicates a multifactorial origin, including drugs, radiation, maternal malnutrition, chemicals, and possibly a genetic mutation in folate pathways in some cases, which may result in abnormal development (Kinsman and Johnston, 2007). Additional factors predisposing the infant to NTDs include prepregnancy maternal obesity, previous NTD pregnancy, and the use of antiepileptic drugs (e.g., valproic acid) in pregnancy (Frey and Hauser, 2003; Finnell, Gould, and Spiegelstein, 2003; Stothard, Tennant, Bell, et al, 2009). The degree of neurologic dysfunction depends on where the sac protrudes through the vertebrae, the anatomic level of the defect, and the amount of nerve tissue involved. Most myelomeningoceles involve the lumbar or lumbosacral area.
The American Academy of Pediatrics (2007) recommends daily intake of folic acid for all women of childbearing age. The recommended 0.4-mg daily dose is supplied safely in many multivitamin preparations. Because the greatest risk factor is a previous pregnancy affected by NTDs, women in this category should increase their daily folic acid dose to 4 mg, under a practitioner’s supervision, beginning at least 1 month before they plan a pregnancy and through the first trimester, since the neural tube closes about 1 month after conception. In 2009 the U.S. Preventive Services Task Force published a statement indicating there is ample evidence to support the recommendations for folic acid supplementation to decrease the incidence of NTDs (Wolff, Witkop, Miller, et al, 2009). In 1998 the U.S. Food and Drug Administration (FDA) authorized the fortification of cereal grains (including corn meal, grits, and wheat flour) with folic acid. It remains important for all women of childbearing age to take a multivitamin with 0.4 mg folic acid daily (American Academy of Pediatrics, 2007).
The following discussion of NTDs is limited to the two most common types: anencephaly, a defect incompatible with life; and SB, in particular, myelomeningocele, an abnormality that causes significant disability.
Anencephaly
Anencephaly, the most serious NTD, is a congenital malformation in which both cerebral hemispheres are absent. The condition is incompatible with life, and many affected infants are stillborn. For those who survive, no specific treatment is available. The infants have a portion of the brainstem and are able to maintain vital functions (such as temperature regulation and cardiac and respiratory function) for a few hours to several weeks but eventually die of respiratory failure.
Traditionally these infants have been provided comfort measures, but with no effort at resuscitation. Ethical and moral questions are encountered regarding treatment and withdrawal of support systems (e.g., feedings) if the newborn survives the first few days of life, as well as use of the organs for donor transplants. During this time the family requires emotional support and counseling to cope with the birth of an infant with a fatal defect.
Spina Bifida and Myelodysplasia
Myelodysplasia refers broadly to any malformation of the spinal canal and cord. Midline defects involving failure of the osseous (bony) spine to close are called spina bifida, the most common defect of the CNS. SB is categorized into two types: SB occulta and SB cystica.
SB occulta refers to a defect that is not visible externally. It occurs most commonly in the lumbosacral area (L5 and S1) (Fig. 11-2, B). Routine radiographic examinations indicate that the disorder may occur in as many as 10% to 30% of the general population. However, it may not be apparent unless there are associated cutaneous manifestations or neuromuscular disturbances. Superficial cutaneous indications include a skin depression or dimple (which may also mark the outlet of a dermal sinus tract that extends to the subarachnoid space); port-wine angiomatous nevi; dark tufts of hair; and soft, subcutaneous lipomas. These signs may be absent, appear singly, or be present in combination.
If associated neurologic involvement is present, the defect is known as occult spinal dysraphism. Fibrous bands and adhesions, an intraspinal lipoma (fatty tumor) or subcutaneous lipoma (lipomyelomeningocele), a dermoid or epidermoid cyst, diastematomyelia (spinal cord split in two), or a tethered cord can distort the spinal cord or roots. The usual cause is abnormal adhesion, or tethering, to a bony or fixed structure, resulting in traction on the spinal cord and cauda equina. (See Figs. 40-5 and 40-7 for areas innervated by specific spinal nerves.)
Neuromuscular disturbances usually consist of progressive or static changes in gait with foot weakness, foot deformity, or bowel and bladder sphincter disturbances. Some manifestations may not be evident until the child walks or is toilet trained.
Plain radiography is employed to disclose the precise bony defect in the symptomatic lesion and to establish the diagnosis in the suspected, nonsymptomatic occult variety. Magnetic resonance imaging (MRI) is the most sensitive tool for evaluating the defect. Computed tomography (CT), ultrasonography, and myelography are also used to differentiate between SB occulta and other spinal disorders.
SB cystica refers to a visible defect with an external saclike protrusion. The two major forms of SB cystica are meningocele, which encases meninges and spinal fluid, but no neural elements (Fig. 11-2, C), and myelomeningocele (or meningomyelocele), which contains meninges, spinal fluid, and nerves (Fig. 11-2, D). Neurologic deficit is not associated with meningocele but occurs in varying, often serious, degrees in myelomeningocele.
Myelomeningocele (Meningomyelocele)
 Myelomeningocele develops during the first 28 days of pregnancy when the neural tube fails to close and fuse at some point along its length. It may be detected prenatally or at birth, accounts for 90% of spinal cord lesions, and may be located at any point along the spinal column. Usually the sac is encased in a fine membrane that is prone to tears through which cerebrospinal fluid (CSF) leaks. In other instances the sac may be covered by dura, meninges, or skin, in which case there is rapid and spontaneous epithelialization. The largest number (75%) of myelomeningoceles occur in the lumbar or lumbosacral area (Fig. 11-3). The location and magnitude of the defect determine the nature and extent of neurologic impairment. When the defect is below the second lumbar vertebra, the nerves of the cauda equina are involved, giving rise to symptoms such as flaccid, areflexic partial paralysis of the lower extremities and varying degrees of sensory deficit. Unlike a spinal cord injury, the degree of deficit is not necessarily uniform on both sides but may vary between extremities, depending on the compromise to specific nerves from malformation or tethering.
Myelomeningocele develops during the first 28 days of pregnancy when the neural tube fails to close and fuse at some point along its length. It may be detected prenatally or at birth, accounts for 90% of spinal cord lesions, and may be located at any point along the spinal column. Usually the sac is encased in a fine membrane that is prone to tears through which cerebrospinal fluid (CSF) leaks. In other instances the sac may be covered by dura, meninges, or skin, in which case there is rapid and spontaneous epithelialization. The largest number (75%) of myelomeningoceles occur in the lumbar or lumbosacral area (Fig. 11-3). The location and magnitude of the defect determine the nature and extent of neurologic impairment. When the defect is below the second lumbar vertebra, the nerves of the cauda equina are involved, giving rise to symptoms such as flaccid, areflexic partial paralysis of the lower extremities and varying degrees of sensory deficit. Unlike a spinal cord injury, the degree of deficit is not necessarily uniform on both sides but may vary between extremities, depending on the compromise to specific nerves from malformation or tethering.
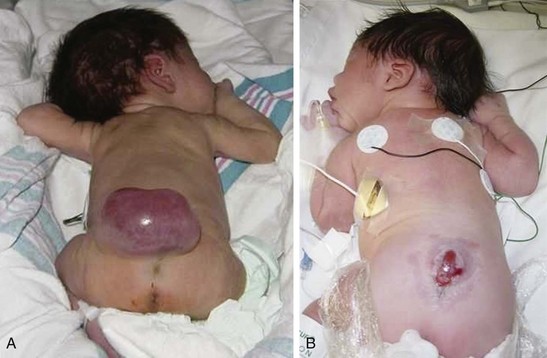
Fig. 11-3 A, Myelomeningocele with intact sac before surgery. B, Myelomeningocele with ruptured sac. (Courtesy Dr. Robert C. Dauser, Neurosurgery, Baylor College of Medicine, Houston.)
 Critical Thinking Exercise—Myelomeningocele
Critical Thinking Exercise—Myelomeningocele
The anomaly most frequently associated with myelomeningocele is hydrocephalus; approximately 80% of children with SB develop hydrocephalus (Kinsman and Johnston, 2007). Although present at birth, hydrocephalus may not be apparent until shortly thereafter, or after the primary closure of the opening on the back. Careful monitoring of head circumference, fontanel tension, and ventricular size by head ultrasonography can indicate its presence. Hydrocephalus can occur because the NTD itself disrupts the flow of CSF. In many cases Chiari malformation (type II) is responsible (see p. 411). Type II Chiari malformation (a downward herniation of the brain into the brainstem) is present, though asymptomatic, in many children with SB. It can, however, adversely affect respiratory function, causing episodic apnea. Other clinical symptoms of problematic Chiari malformation include stridor, hoarse cry from vocal cord paralysis, feeding difficulties, aspiration pneumonia, and, in older children, upper extremity spasticity. The appearance of such symptoms should not be taken for granted; immediate referral is required to prevent further neurologic deterioration.
Pathophysiology
The pathophysiology of SB is best understood when related to the normal formative stages of the nervous system. At approximately 20 days of gestation a decided depression, the neural groove, appears in the dorsal ectoderm of the embryo. During the fourth week of gestation the groove deepens rapidly, and its elevated margins develop laterally and fuse dorsally to form the neural tube. Neural tube formation begins in the cervical region near the center of the embryo and advances in both directions—caudally and cephalically—until by the end of the fourth week of gestation the ends of the neural tube, the anterior and posterior neuropores, close.
Most authorities believe the primary defect in neural tube malformations is a failure of neural tube closure. However, some evidence indicates that the defects are a result of splitting of the already closed neural tube as a result of an abnormal increase in CSF pressure during the first trimester.
Clinical Manifestations
The manifestations of SB vary widely according to the degree of the spinal defect. The defect is readily apparent on inspection. The degree of neurologic dysfunction is directly related to the anatomic level of the defect and thus the nerves involved. Sensory disturbances usually parallel motor dysfunction. The upper level of sensory and motor impairment can be determined by observation of the infant’s response to a pinprick over the legs and trunk. The infant responds to the sensory stimulus with limb movement, arousal, and crying. When withdrawal activity is used to determine the lowest level of spinal cord function, the response to pinprick should begin above the lesion.
Defective nerve supply to the bladder affects both sphincter and detrusor tone, which often causes constant dribbling of urine or produces overflow incontinence. This can often be mistaken for normal voiding patterns in the newborn. Some infants with SB, however, are able to void in a stream and achieve complete bladder emptying with each void.
Frequently the infant has poor anal sphincter tone and poor anal skin reflex, which result in lack of bowel control and sometimes rectal prolapse. Avoid taking rectal temperatures in affected infants. Because bowel sphincter function is frequently affected, the thermometer can cause irritation and rectal prolapse.
If the defect is below the third sacral vertebra, the infant has no motor impairment, but may have saddle anesthesia with bladder and anal sphincter paralysis.
Sometimes the denervation to the muscles of the lower extremities produces joint deformities in utero. These are primarily flexion or extension contractures, talipes valgus or varus contractures, kyphosis, lumbosacral scoliosis, and hip dislocations. The extent and severity of these associated orthopedic deformities again depend on the degree of nerve involvement. Most flexion deformities result from the pull of stronger, fully innervated muscles acting without the counterpull of their nonfunctioning paralyzed antagonists. See Box 11-2 for summary of clinical manifestations of SB cystica and occulta.
Diagnostic Evaluation
The diagnosis is made on the basis of clinical manifestations and examination of the meningeal sac. Diagnostic measures used to evaluate the brain and spinal cord include MRI, ultrasonography, CT, and myelography.
Laboratory examinations are used primarily to determine causative organisms in the major complications of myelomeningocele: meningitis and urinary tract infections. Infants with urinary incontinence require urinalysis, culture, and evaluation of blood urea nitrogen and creatinine clearance.
Prenatal Detection: It is possible to determine the presence of some major open NTDs prenatally. Ultrasonographic scanning of the uterus and elevated maternal concentrations of α-fetoprotein (AFP, or MS-AFP), a fetal-specific γ-1-globulin, in amniotic fluid may indicate the presence of anencephaly or myelomeningocele. (See Chapter 5.) The optimum time for performing these diagnostic tests is between 16 and 18 weeks of gestation, before AFP concentrations normally diminish and in sufficient time to permit a therapeutic abortion. It is recommended that such diagnostic procedures, as well as genetic counseling, be considered for all mothers who have borne an affected child, and testing is offered to all pregnant women (Kirkham, Harris, and Grzybowski, 2005). In addition, elective prelabor cesarean birth may result in less motor dysfunction. Chorionic villus sampling is also a method for prenatal diagnosis of NTDs; however, it carries certain risks (skeletal limb depletion) and is not recommended before 10 weeks of gestation.
Early surgical closure of the myelomeningocele sac through fetal surgery has been evaluated in relation to prevention of injury to the exposed spinal cord tissue and the improvement of neurologic and urologic outcomes in the affected child. Currently the Management of Myelomeningocele Study, a clinical trial supported by the National Institute of Health, is evaluating outcomes of fetal surgical correction of myelomeningocele at three sites in the United States; the results are expected to be published in 2011. The overall mortality rate from fetal surgery has been reported to be 4% to 6%, and complications include oligohydramnios, preterm delivery, and a smaller birth weight (Kaufman, 2004; Sutton, 2008).
Therapeutic Management
Management of the child who has a myelomeningocele requires a multidisciplinary team approach involving the specialties of neurology, neurosurgery, pediatrics, urology, orthopedics, rehabilitation, physical therapy, occupational therapy, and social services, as well as intensive nursing care in a variety of specialty areas. The collaborative efforts of these specialists focus on (1) the myelomeningocele and the problems associated with the defect—hydrocephalus, paralysis, orthopedic deformities, and genitourinary (GU) abnormalities; (2) possible acquired problems that may or may not be associated, such as Chiari II malformation, meningitis, seizures, hypoxia, and hemorrhage; and (3) other abnormalities, such as cardiac or gastrointestinal (GI) malformations. Many hospitals have routine outpatient care by multidisciplinary teams to provide the complex follow-up care needed for children with myelodysplasia.
Initial Care: Care of the newborn involves preventing infection; performing a neurologic assessment, including observation for associated anomalies; and dealing with the impact of the anomaly on the family. Although meningoceles are repaired early, especially if the sac is in danger of rupturing, the philosophy regarding skin closure of myelomeningocele varies. Most authorities believe that early closure, within the first 24 to 72 hours, offers the most favorable outcome. Surgical closure within the first 24 hours is recommended if the sac is leaking CSF (Kinsman and Johnston, 2007). Early closure, preferably in the first 12 to 18 hours, not only prevents local infection and trauma to the exposed tissues but also avoids stretching other nerve roots (which may occur as the meningeal sac expands during the first hours after birth), thus preventing further motor impairment. Broad-spectrum antibiotics are initiated, and neurotoxic substances such as povidone-iodine are avoided at the malformation.
A variety of neurosurgical and plastic surgical procedures are employed for skin closure without disturbing the neural elements or removing any portion of the sac. The objective is satisfactory skin coverage of the lesion and meticulous closure. Wide excision of the large membranous covering may damage functioning neural tissue.
Associated problems are assessed and managed by appropriate surgical and supportive measures. Shunt procedures provide relief from imminent or progressive hydrocephalus (see p. 409). When diagnosed, ventriculitis, meningitis, and urinary tract infection are treated with vigorous antibiotic therapy and supportive measures. Surgical intervention for Chiari II malformation is indicated only when the child is symptomatic (i.e., high-pitched crowing cry, stridor, respiratory difficulties, oral-motor difficulties, upper extremity spasticity).
Improved surgical techniques do not alter the major physical disability and deformity or chronic urinary tract infections that affect the quality of life for these children. Superimposed on these physical problems are the disorder’s effects on family life and finances and on school and hospital services.
Musculoskeletal Considerations: According to most orthopedists, musculoskeletal problems that will affect later locomotion should be evaluated early and treatment, where indicated, instituted without delay. Neurologic assessment determines the neurosegmental level of the lesion, spasticity and progressive paralysis, potential for deformity, and functional expectations. Orthopedic and musculoskeletal management includes preventing joint contractures, correcting the existing deformity, preventing or minimizing effects of motor and sensory deficits, preventing skin breakdown, and obtaining the best possible function of affected lower extremities. Common musculoskeletal problems requiring attention in SB include deformities of the knees, hips, feet, and spine; fractures and insensate skin further complicate orthopedic care. Other problems that may occur later include kyphosis and scoliosis (Lazzaretti and Pearson, 2010). Because children with this condition often have decreased sensitivity in lower extremities, preventive skin care is important. A high percentage (60%) of children seen in a wound clinic for skin breakdown had myelomeningocele at birth (Samaniego, 2003).
The status of the neurologic deficit remains the most important factor in determining the child’s ultimate functional abilities; however, many children with lumbar and sacral myelomeningocele are able to achieve functional ambulation (Kinsman and Johnston, 2007). With technologic advances, a variety of lightweight orthoses, including braces, special “walking” devices, and custom-built wheelchairs, are available to provide mobility to children with spinal cord lesions (see also Chapter 39). Early in infancy, intervention with passive range-of-motion exercises, positioning, and stretching exercises may help decrease the incidence of muscle contractures (Brown, 2001). Corrective surgical procedures, when indicated, are best initiated at an early age so that the child will not lag significantly behind age-mates in developmental progress. Where little hope exists for lower extremity function, surgery is seldom recommended unless it will improve sitting position in a wheelchair and function for activities of daily living and mobility.
Physical therapy and musculoskeletal management of children with myelomeningocele is a continual process to achieve optimum function and ambulation when possible. Problems such as type II Chiari malformation, hydrocephalus, and a tethered spinal cord can complicate expectations.
Management of Genitourinary Function: Myelomeningocele is one of the most common causes of neuropathic (neurogenic) bladder dysfunction among children. Myelomeningocele affects approximately 1 in 1000 infants born in the United States, and as many as 90% experience subsequent voiding dysfunction. In infants the goal of treatment is to preserve renal function. In older children the goal is to preserve renal function and achieve optimum urinary continence. Urinary incontinence is a chronic, often debilitating problem for the child. In addition, the neuropathic bladder may produce urinary system distress, characterized by symptomatic urinary tract infections, ureterohydronephrosis, vesicoureteral reflux, or renal insufficiency. The characteristics of bladder dysfunction in children vary according to the level of the neurologic lesion and the influence of bony growth and development of the spine. In addition, the presence of type II Chiari malformation and subsequent hydrocephalus has the potential to affect bladder function, although spinal influences predominate.
During infancy, urinary incontinence is normally physiologic, but urinary system distress may occur. Ongoing urologic monitoring is essential. Evidence is growing that early intervention, based on evaluation during the neonatal period and before complications occur, has the following benefits: (1) improves bladder function, (2) reduces the subsequent risk of urinary system distress, and (3) reduces the need for reconstructive surgery of the lower urinary tract. Ultrasonography of the bladder and ureters and routine urinalysis (and urine cultures when indicated) are used to detect urinary system distress before renal function is compromised. In addition, urodynamic testing is used to identify bladder dysfunction that predisposes the child to urinary system distress (Gray and Moore, 2009). These conditions include high pressure detrusor hyperreflexia (reflex contractions of the detrusor muscle) with vesicosphincter dyssynergia (incoordination of detrusor and sphincter muscles), low bladder wall compliance (poor distensibility of the bladder wall causing increased intravesical pressures during urine filling and storage), or detrusor areflexia (absence of detrusor contractions caused by the spinal defect).
Infants may have one of several predominant neuropathic bladder disorders. Detrusor contractions associated with vesicosphincter dyssynergia are particularly common. Some infants are able to empty the bladder efficiently despite incoordination between the sphincter mechanism and detrusor, but the majority experience chronic residual urine, urinary tract infections, or more serious types of urinary system distress. A minority of infants have poor detrusor contraction strength or detrusor areflexia. This condition is particularly damaging to the urinary system when it coexists with low bladder wall compliance and an elevated detrusor leak point pressure. Low bladder wall compliance occurs when collagen or fibrosis causes stiffening of the bladder wall. This stiffened bladder wall raises intravesical pressures, obstructing the bladder, ureters, and, ultimately, the nephron. The impact of low bladder wall compliance is directly related to the influence of the bladder outlet. Among children with myelodysplasia, the urethral muscles are typically weakened, and collagen replaces much of the muscle tissue. As a result, the sphincter is fixed, so that it neither closes efficiently to prevent urinary leakage nor opens well to allow urinary flow with a detrusor contraction. When the magnitude of the pressure required to drive urine across the abnormal sphincter is greater than 40 cm H2O (the detrusor leak point pressure) and the compliance of the bladder wall is low (<10 cm H2O), the risk of urinary system distress is high.
In contrast, a small number of infants experience effective detrusor contractions without vesicosphincter dyssynergia. Effective bladder evacuation is likely among this group, and the incidence of urinary system distress during the first year of life is low.
As the child grows, detrusor hyperreflexia is often replaced by deficient detrusor contraction strength and stress urinary incontinence (SUI) (leakage produced by physical exertion). The bladder wall is often poorly compliant (producing chronically elevated intravesical pressures), and the bladder outlet, while incompetent, obstructs the outflow of urine. When the detrusor leak point pressure exceeds 40 cm H2O, the child is predisposed to chronic urinary leakage and urinary distress symptoms, including recurrent urinary tract infections and reflux. When the detrusor leak point pressure is lower than 40 cm H2O, urinary leakage is more severe, although the risk of urinary system distress is lessened. Thus the child with more severe urinary incontinence is less predisposed than the “drier” child to serious urinary tract infections.
Infants with myelomeningocele and a neurogenic bladder who are not at risk for urinary system distress are managed by diaper containment and watchful waiting. The infant empties the bladder into a diaper, the urine is routinely monitored for infection, and the upper urinary tracts are monitored for evidence of urinary system distress (dilation of the ureters, renal pelves, or collecting systems) via serial ultrasonography.
In contrast, children with evidence of urinary system distress, or those considered at risk based on early urodynamic testing, are placed on clean intermittent catheterization (CIC), typically in combination with an antispasmodic medication such as oxybutynin or propantheline (Gray and Moore, 2009; de Jong, Chrzan, Klijn, et al, 2008). Anticholinergic medications are prescribed because they reduce detrusor muscle tone and reduce bladder pressures during both urine filling and storage and during micturition. CIC is not intended to prevent spontaneous voiding. Instead, it ensures routine, regular bladder evacuation, further preventing deleterious elevation of intravesical pressures. Usually, the parents learn to catheterize the infant every 4 hours during the day and once each night. Follow-up evaluation, consisting of serial ultrasonography and urinalysis, is completed every 3 to 6 months as indicated.
Infants with significant urinary system distress and hostile neuropathic bladder dysfunction at birth sometimes require temporary urinary diversion to ensure adequate urine outflow and prevent further damage to the upper urinary tracts. A vesicostomy is a relatively simple procedure wherein the anterior bladder wall is brought to the abdominal wall, creating a small stoma for urinary drainage. Urine is contained via a diaper, but double diapering or use of a larger diaper that can be placed higher on the abdomen is necessary for adequate urine containment. Meticulous skin care is necessary because the perineal skin is exposed to continuous urinary leakage.
Among older children the quest for continence typically begins with a CIC program. The parents learn the procedure, and teach the child to self-catheterize as soon as possible, usually by 6 years of age (Gray and Moore, 2009). The child with detrusor hyperreflexia and dyssynergia often responds well to antispasmodic medications and CIC. In contrast, the child with poor bladder wall compliance and SUI often requires a combination of antispasmodic medications to reduce intravesical filling pressures and an asympathetic agonist (such as imipramine, pseudoephedrine, or phenylpropanolamine) to enhance sphincter competence. Unfortunately, the combination of medications and CIC is typically only partially effective, and more aggressive interventions are often required to render the neuropathic bladder both continent and free from its predisposition toward producing urinary system distress.
When the child cannot attain continence by conservative measures, surgery is considered. Augmentation enterocystoplasty (or gastrocystoplasty) is a surgical procedure that increases bladder capacity, reverses or halts the negative effects of the poorly compliant bladder wall, and reduces harmfully high bladder pressures caused by detrusor hyperreflexia with vesicosphincter dyssynergia. A detubularized segment of large or small bowel or a wedge of the fundus of the stomach has been used to successfully augment bladder capacity. The choice of segment varies according to the surgeon’s preference and the status of the patient’s urinary and GI systems. Large and small bowel segments produce significant volumes of mucus that may clog catheters used for CIC. Augmentation with the stomach produces less mucus, and its acidic secretions may reduce the urinary system’s predisposition to infection. The bladder must be irrigated to decrease mucus within the bladder; this also decreases the possible complications of infection, stones, and bladder perforation.
Even though augmentation of the bladder may improve or resolve urinary leakage related to detrusor hyperreflexia or urinary system distress caused by low bladder wall compliance, the SUI produced by the abnormal sphincter mechanism typically persists. Several surgical procedures help correct this intrinsic sphincter deficiency. The Mitrofanoff procedure uses the appendix to provide an alternative route for intermittent catheterization. The appendix is removed from the colon and used to create a continent conduit between the abdominal wall and the bladder. The resulting stoma is relatively small and produces minimum mucus. The ureter may be used as an alternative to the appendix for some children. If the appendix is insufficient, a segment of tapered intestine, ileum, or colon may be used to create a conduit (Monti tube) (Gray and Moore, 2009; Mitrofanoff and Liard, 2001). CIC through the easily accessible abdominal route fosters greater independence in children, especially in those unable to transfer from wheelchair to toilet to perform CIC.
When intrinsic sphincter deficiency produces only mild stress urinary leakage, the construction of a Mitrofanoff route alone may be sufficient to achieve continence between catheterization episodes. However, when SUI is more severe, a suburethral sling or suburethral collagen injection is used to alleviate intrinsic sphincter deficiency.
The suburethral sling is a slip of fascia or synthetic material that is placed below the proximal third of the urethra. The sling may be placed in a fashion that uses only slight tension to obstruct the urethra and prevent SUI. The sling may be used for both boys and girls, and the procedure can be completed at the same time the augmentation enterocystoplasty is constructed. After augmentation enterocystoplasty and placement of a suburethral sling, the patient can expect to evacuate the bladder by CIC of the appendiceal Mitrofanoff route or the urethra if a Mitrofanoff route has not been constructed.
Suburethral injection of glutaraldehyde cross-linked (GAX) collagen also may be used to alleviate or prevent SUI caused by intrinsic sphincter deficiency. Collagen is used to bulk or expand the urethral tissue, promoting coaptation (approximation) of the mucosa. The collagen implant complements the urethra’s ability to form a watertight seal, rather than obstructing the urethral lumen. Collagen may be injected using different approaches. Transurethral collagen is injected through the working channel of a cystoscope. Transperineal collagen is directed underneath the urethra using a needle inserted through the perineal skin. In this case the location of the urethra is confirmed by simultaneous cystoscopic visualization of the urethra. The antegrade approach requires creation of a suprapubic cystostomy tract. A flexible cystoscope is then inserted through the cystostomy tract, and collagen is injected into the proximal urethra. Multiple injections may be required to achieve optimum continence. Subsequent injections may be required when the collagen is dissipated or resorbed by the body over a period of years.
The artificial urinary sphincter provides another alternative for the management of intrinsic sphincter deficiency in the child with myelomeningocele. The device consists of a urethral cuff, abdominal reservoir, and control pump. In the activated position, the cuff is filled, and the pressure of this cuff closes the urethral lumen. During micturition, the control pump is used to baffle fluid from the urethral cuff to the abdominal reservoir, opening the urethra for micturition or catheterization. However, because of the significant risk for infection, need for revision with growth, and mechanical failure, the popularity of the artificial urinary sphincter has declined.
Because of advances in neurogenic bladder management, adolescents and young adults with myelomeningocele and neurogenic bladders have been followed for up to 30 years without evidence of deterioration in renal function. Nevertheless, urinary and fecal incontinence are common, and these conditions lead to significant, and sometimes devastating, problems with growth and developmental tasks, including establishing independence and social and intimate relationships. This observation underscores the need to aggressively manage both continence and the threat of urinary system distress from an early age and to establish an expectation of social continence critical to providing these patients with the skills they need to thrive as adolescents and adults. Newborns with SB and normal urodynamics require close follow-up care during the first several years of life to prevent deterioration in urodynamic status as a result of neurologic deterioration.
Bowel Control: Some degree of fecal continence can be achieved in most children with myelomeningocele with diet modification, regular toilet habits, and prevention of constipation and impaction. It is frequently a lengthy process. Dietary fiber supplements (recommended 10 g/day), laxatives, suppositories, or enemas aid in producing regular evacuation. Older children and adolescents seeking more independence may attain bowel continence and higher quality of life after undergoing an antegrade continence enema procedure (Doolin, 2006). In a procedure similar to the Mitrofanoff, the appendix or ileum is used to create a catheterizable channel with attachment of the proximal end to the colon. The distal end of the channel exits through a small abdominal stoma. Every 1 or 2 days, a catheter is passed through the stoma, allowing enema solution to be instilled directly into the colon; this is called an antegrade colonic irrigation. After administration of the enema solution, the child sits on the toilet for 30 to 60 minutes as stool is flushed out through the rectum. Frequency of enemas and volume of solution used to completely evacuate the bowel vary among individuals.
Prognosis: The early prognosis for the child with myelomeningocele depends on the neurologic deficit present at birth, including motor ability, bladder innervation, and associated neurologic anomalies. Early surgical repair of the spinal defect, antibiotic therapy to reduce the incidence of meningitis and ventriculitis, prevention of urinary system dysfunction, and early detection and correction of hydrocephalus have significantly increased the survival rate and quality of life in such children. Many children with SB achieve partial independent living and gainful employment. Reports of survival rates vary, and many include adults who were born before medical advances and surgical techniques seen in the past 25 years. Coordinated care for adults with SB is essential; however, multidisciplinary adult care is often inadequate (Lazzaretti and Pearson, 2010). In children and adolescents with SB the achievement of urinary continence is associated with improved self-concept and esteem, especially among girls (Moore, Kogan, and Parekh, 2004). This chronic condition has an array of associated complications, including hydrocephalus and shunt malfunctions, scoliosis, bowel and bladder management issues, latex allergy, and epilepsy. However, based on current medical knowledge and ethical considerations, aggressive, early management is favored for the child with myelomeningocele.
Prevention: The Centers for Disease Control and Prevention (2009) continues to affirm that 50% to 70% of NTDs can be prevented by daily consumption of 0.4 mg of folic acid among women of childbearing age. The data indicate that serum folate concentrations among women of childbearing age decreased 16% from 2003 to 2004 in all ethnic groups studied. Lowest serum folate levels were seen in non-Hispanic Caucasians in 2003 to 2004; however, overall serum folate levels remained below recommended levels in non-Hispanic African-Americans during all three periods studied (Centers for Disease Control and Prevention, 2007). These results indicate that nurses and other health care workers have an important task in disseminating information that may decrease the incidence of birth defects in children by promoting maternal consumption of folic acid.*
To ensure adequate daily intake of the recommended amount of folic acid, women must take a folic acid supplement, eat a fortified breakfast cereal containing 100% of the recommended dietary allowance of folic acid (e.g., Kellogg’s Product 19, General Mills Total, Multigrain Cheerios Plus), or increase their consumption of fortified foods (cereal, bread, rice, grits, pasta) and foods naturally rich in folate (green, leafy vegetables and citrus fruits). For women who have had a previous pregnancy affected by NTDs, folic acid intake is increased to 4 mg under supervision of a practitioner beginning 1 month before a planned pregnancy and continuing through the first trimester. Supplementation of 4 mg of folate should not be given solely in multivitamin preparations because of the risk of overdose of other vitamins. The only population in which folic acid has not been effective in decreasing the incidence of NTDs is in women taking antiepileptic medications during pregnancy (Finnell, Gould, and Spiegelstein, 2003).
Nursing Care Management
The basic needs of the infant with a myelomeningocele are essentially the same as for any newborn infant. (See Chapter 8.) Special needs related to the defect and potential complications are discussed in the following section. As the child matures, the problems increase and involve all aspects of daily living; therefore care is directly related to the child’s habilitation at each stage of development.
Assessment
At the time of delivery an examination is performed to assess the intactness of the membranous cyst. During transport to the nursery, make every effort to prevent trauma to this protective covering. In addition to the routine assessment of the newborn (see Chapter 8), assess the infant for the level of neurologic involvement. Note movement of extremities or skin response, especially an anal reflex that might provide clues to the degree of motor or sensory impairment.
Care of the Myelomeningocele Sac: The infant is usually placed in an incubator or radiant warmer so that temperature can be maintained without clothing or covers that might irritate the CNS lesion. When an overhead warmer is used, the dressings over the defect require more frequent moistening because of the dehydrating effect of the radiant heat. Before surgical closure the myelomeningocele is kept from drying by the application of a sterile, moist, nonadherent dressing. The moistening solution is usually sterile normal saline. Dressings are changed frequently (every 2 to 4 hours), and the sac is closely inspected for leaks, abrasions, irritation, and signs of infection. The sac must be carefully cleansed if it becomes soiled or contaminated. Sometimes the sac ruptures during delivery or transport, and any opening in the sac greatly increases the risk of infection to the CNS.
Positioning: One of the most important and challenging aspects of early care of the infant with myelomeningocele is positioning. Before surgery the infant remains in the prone position to minimize tension on the sac and the risk of trauma. The prone position allows for optimum positioning of the legs, especially in cases of associated hip dysplasia. A variety of aids, including diaper rolls, pads, or specially designed frames and appliances, are available to maintain the desired position.
The prone position affects other aspects of the infant’s care. For example, in this position the infant is more difficult to keep clean, pressure areas are a constant threat, and feeding becomes a problem. The infant’s head is turned to one side for feeding. Fortunately, most defects are repaired early, and the infant can be held for feeding soon after surgery. Physical therapy consultation may be necessary for difficult positioning problems. Speech-language pathologist consultation may be needed for difficulty with oral-motor skills that may indicate complications caused by a Chiari malformation.
General Care: Diapering the infant may be contraindicated until the defect has been repaired and healing is well advanced or epithelialization has taken place. The padding beneath the diaper area is changed as needed to keep the skin dry and free of irritation. When the nurse detects urinary retention (the bladder is still an abdominal organ in early infancy), CIC is employed. Because the bowel sphincter is frequently affected, there may be continual passage of stool, often misinterpreted as diarrhea, which is a constant irritant to the skin and a source of infection to the spinal lesion.
Areas of sensory and motor impairment are subject to skin breakdown and therefore require meticulous care. The infant may be placed on a pressure-reducing mattress or mattress to prevent pressure on the knees and ankles. (See Skin Care, Chapter 10, and Maintaining Healthy Skin, Chapter 27.)
Gentle range-of-motion exercises are carried out to prevent contractures, and stretching of contractures is performed when indicated. However, these exercises may be restricted to the foot, ankle, and knee joint. When the hip joints are unstable, stretching against tight hip flexors or adductor muscles, which act much like bowstrings, may aggravate a tendency toward subluxation. A physical therapy consultation is often necessary to develop a multidisciplinary plan to prevent long-term complications.
Some infants with unrepaired myelomeningocele are unable to be held in the arms and cuddled as unaffected infants are, so their need for tactile stimulation is met by caressing, stroking, and other comfort measures. To facilitate handling and reduce parental anxiety, the infant can recline on a pillow placed in the parent’s lap. Black-and-white drawings or geometric shapes can be placed within the infant’s view, and other stimulation usually provided for infants is appropriate. All infants respond to pleasant sounds. (See Developmental Outcome, Chapter 10.)
Ophthalmic complications may occur in children with SB and hydrocephalus. The appearance of a squint, other ocular motility, or papilledema usually denotes hydrocephalus and is reported. Ophthalmologic follow-up care, particularly in children with shunts, is generally included in the multidisciplinary care plan.
Postoperative Care: Postoperative care for the infant with myelomeningocele involves the same basic care as for any postsurgical infant: monitoring vital signs, weight, and intake and output; maintaining body temperature; assessing and relieving pain; providing nourishment; and observing for signs of infection. The wound is managed according to the surgeon’s directions, and general care is continued as preoperatively.
The prone position is maintained after operative closure, although many neurosurgeons allow a side-lying or partial side-lying position unless it aggravates a coexisting hip dysplasia or permits undesirable hip flexion. This offers an opportunity for position changes, which reduces the risk of pressure sores and facilitates feeding. Once the effects of anesthesia have subsided and the infant is alert, feedings may resume unless there are other anomalies or associated complications.
Nursing assessments are carried out for implementation of comfort measures in the postoperative period. The infant can be held upright against the body, taking care to avoid pressure on the operative site. In the case of an unusually large defect, skin grafting may be required for wound closure; the infant must then be kept prone postoperatively with as little movement as possible to prevent tension on the skin graft.
The nurse can assist in determining the extent of neuromuscular involvement. Note movement of the extremities or skin response, especially an anal reflex, that might provide clues to the degree of motor or sensory status. Measure head circumference daily (see Chapter 6), and examine the fontanels for signs of tension or bulging. The nurse is also alert to early signs of infection, such as elevated or decreased temperature (axillary), irritability, and lethargy, and to signs of increased intracranial pressure (ICP). Urinary catheterization may be needed for urine retention. Although it may not have been a problem preoperatively, swelling around the operative site may cause transient urine retention, which resolves in 2 to 5 days.
Family Support and Home Care: As soon as the parents are able to cope with the infant’s condition, encourage them to become involved in care. They need to learn how to continue at home the care that has been initiated in the hospital: positioning, feeding, skin care, and range-of-motion exercises when appropriate. Parents also need to learn CIC technique when prescribed. The family needs to know the signs of complications and how to reach assistance when needed.
As the child grows and develops, parents need guidance to encourage and stimulate the infant to accomplish age-appropriate developmental tasks within the limits imposed by the disabilities. Upper limb movement can be stimulated early by placing the infant on the floor in a prone position with toys within reach. Activities that encourage body consciousness, such as rolling over and pulling to a sitting position, are encouraged at the appropriate times. Creeping and crawling help the child explore the environment. The parents may need help to modify appliances and activities normally expected of a growing child. A standing table, frame, or parapodium is helpful for a variety of activities, and it is best for the child to begin supported weight bearing and standing as close as possible to the expected time for standing to occur.
It is important for the family to understand the nature of sensory deficit in a child with a spinal defect. The child will be insensitive to pressure or other sources of tissue injury. Therefore the family must be alert to hot or cold items that could cause thermal injury to tissues and remember to inspect the skin regularly for signs of pressure, especially over bony prominences. Because of sensory impairment, the child is unaware of bladder discomfort. Therefore signs of urinary tract infections may go unnoticed. Urinary tract infection is often considered when the child becomes ill.
The long-range planning with and support of the parents and newborn begin in the hospital and extend throughout childhood and even into young adulthood. The life expectancy of children with SB extends well into adulthood; therefore planning should involve long-term goals and plans for optimum function as an adult. Long-range planning goals should include a discussion of achievement of functional mobility, urinary continence, and as much bowel continence as much physically possible. Discussion about aspects of adulthood such as having a mate, sexual relationships, and bearing and rearing children is important and should not be overlooked (Barker, Saulino, and Caristo, 2002; Rowe and Jadhav, 2008). The unique service needs of adolescents with SB as they attempt to gain independence from family and establish a life of their own has not been adequately addressed in the literature yet is slowly emerging (Buran, McDaniel, and Brei, 2002; Rowe and Jadhav, 2008). Advances in neurology, orthopedics, and urology have enabled adolescents to progress into adulthood with fewer deficits than observed in previous decades; one key factor is the recognition of subtle signs of neurologic deterioration and rapid intervention (Rowe and Jadhav, 2008).
Nurses assume an important role as a central member of the health team. As a coordinator, the nurse reviews information with the family, takes responsibility for family teaching, and acts as a liaison between inpatient and outpatient services. The child may require numerous hospitalizations over the years, and each one will be a source of stress to which the younger child is especially vulnerable.
Changes in functional ability, particularly in the lower extremities, bowel, or bladder, may indicate the presence of a tethered cord, one that is bound down or restricted in an abnormal position by scar tissue. These symptoms usually occur after a growth spurt and can best be detected with MRI. Tethering can be repaired surgically but, unfortunately, may recur. Hydromyelia, a dilation of the central canal of the spinal cord and elevated fluid accumulation, may occur with SB; common symptoms include rapidly developing scoliosis, upper extremity weakness, spasticity, and lower extremity ascending motor strength changes (Barker, Saulino, and Caristo, 2002).
Habilitation involves solving not only problems of self-help and locomotion but also the most distressing problem of incontinence, which threatens the child’s social acceptability. Assistance in preparing the child and the school regarding the special needs of children with disabilities helps the parents provide a better initial adjustment to broader social experiences. Numerous organizations and agencies offer assistance and support to children and families (see Family-Centered Care box). The Spina Bifida Association* provides services and support for families of children with spinal lesions.
 FAMILY-CENTERED CARE
FAMILY-CENTERED CARE
Additional References on Spina Bifida for Families and Professionals
Kriegsman KH, Zaslow EL, D’Zmura-Rechsteiner J: Taking charge: teenagers talk about life and physical disabilities, Bethesda, Md, 1992, Woodbine House.
Lutkenhoff M, editor: Children with spina bifida: a parent’s guide, Bethesda, Md, 1999, Woodbine House.
Lutkenhoff M, Oppenheimer S, editors: Spinabilities: a young person’s guide to spina bifida, Bethesda, Md, 1997, Woodbine House.
Rowley-Kelly F, Reigel D: Teaching the student with spina bifida, Baltimore, 1993, Paul H Brooks.
Sandler A: Living with spina bifida: a guide for families and professionals, Chapel Hill, NC, 1997, University of North Carolina Press.
The multiple aspects of care of the child with a disability are discussed in Chapter 22. Complex problems associated with partial or complete lower extremity paralysis are discussed in Chapters 39 and 40 and include bowel and bladder control; orthopedic appliances; and the observation and management of complications, especially urinary tract infections (see Chapter 30) and pressure necrosis (see Wounds, Chapter 18).
Latex Allergy
Latex allergy, or latex hypersensitivity, was identified as being a serious health hazard when a report linked intraoperative anaphylaxis with latex in children with SB. Latex, a natural product derived from the rubber tree, is used in combination with other chemicals to give elasticity, strength, and durability to many products. Children with SB are at high risk for developing latex allergy because of repeated exposure to latex products during surgery and procedures. Children with chronic renal failure are also at an increased risk for latex allergy, and efforts should be made to decrease their exposure to latex products (Dehlink, Prandstetter, Eiwegger, et al, 2004). Allergic reactions range from urticaria, wheezing, watery eyes, and rashes to anaphylactic shock. More severe reactions tend to occur when latex comes in contact with mucous membranes, wet skin, the bloodstream, or an airway. There also can be cross-reactions to a number of foods (e.g., banana, avocado, kiwi, chestnut).
Latex hypersensitivity reactions have been diagnosed in infants. Symptoms included wheezing, facial swelling, facial rash, or anaphylaxis (Kimata, 2004). Allergic reactions to latex protein can also occur when the substance is transferred to food by food handlers wearing latex gloves, prompting several states to pass legislation that prohibits the use of latex gloves in food service. In addition to patients with SB, high-risk populations include patients with urogenital anomalies or multiple surgeries and health care workers. See Box 11-3 for medical conditions associated with risk of latex allergy.
The most important goals are prevention of latex sensitivity and identification of children with a known hypersensitivity (see Nursing Care Guidelines box). High-risk and latex-allergic individuals must be managed in a latex-free environment. Take care that they do not come in direct or secondary contact with products or equipment containing latex at any time during medical treatment. Allergy testing can identify latex sensitivity with varying success. Skin prick testing and provocation testing carry the risk of allergic reaction or anaphylaxis. Several commercially available assays can be useful in confirming latex sensitivity. To date, none of these tests demonstrates complete diagnostic reliability, and they should not be the sole determinant of the presence or absence of an allergic response to latex.
 NURSING CARE GUIDELINES
NURSING CARE GUIDELINES
Identifying Latex Allergy
• Does your child have any symptoms, such as sneezing, coughing, rashes, or wheezing, when handling rubber products (balloons, tennis or Koosh balls, adhesive bandage strips) or when in contact with rubber hospital products, such as gloves or catheters?
• Has your child ever had an allergic reaction during surgery?
• Does your child have a history of rashes, asthma, or allergic reactions to medication or foods, especially milk, kiwi, bananas, or chestnuts?
• How would you identify or recognize an allergic reaction in your child?
• What would you do if an allergic reaction occurred?
• Has anyone ever discussed latex or rubber allergy or sensitivity with you?
• Has the child had any allergy testing?
• When did the child last come in contact with any type of rubber product? Were you present?
Modified from Romanczuk A: Latex use with infants and children: it can cause problems, MCN 18(4):208-212, 1993.
The radioallergosorbent test (RAST) has been used to measure the serum level of latex-specific immunoglobulin E. The RAST has been shown to be 90% to 95% sensitive. Administration of antihistamines and steroids (dexamethasone) before and after surgery to reduce the possibility of a serious reaction remains controversial because it may interfere with healing.
Because children who have SB are prone to develop sensitivity to latex, reducing exposure, from birth on, may decrease the chance of allergy development. Nonlatex products lists are available to parents and health care workers; these products may be substituted for those containing latex. In the health care arena it is important to use products with the lowest potential risk of sensitizing patients and staff members. The FDA has proposed user labeling for latex-containing devices that come into contact directly or indirectly with live human tissue.*
The identification of those sensitive to latex is best accomplished through careful screening of all patients. During the health interview with the parent or child, ask all patients, not only those at risk, about sensitivity to latex. Be certain this is a routine part of all preoperative and preprocedural histories. Stress the importance of the allergy history to all personnel (e.g., phlebotomists). (See Nursing Care Guidelines box for questions related to latex allergy.) Children with latex hypersensitivity should carry some form of allergy identification, such as a Medic-Alert bracelet. Education programs regarding latex hypersensitivity are aimed at those who care for high-risk groups, such as children with SB, and may include relatives, school nurses, teachers, child care workers, and baby-sitters. In addition to educating caregivers about the child’s exposure to medical products that contain latex, nurses need to inform them of common nonmedical latex objects such as water toys, pacifiers, and plastic storage bags.† Items brought to the hospital, such as floral bouquets, are also screened for latex toys or balloons. Parents should also receive literature explaining signs and symptoms of latex hypersensitivity and appropriate emergency treatment. (See Anaphylaxis, Chapter 29.)
Hydrocephalus
Hydrocephalus is not a single disease entity but rather a group of conditions, resulting from disturbances in the dynamics of cerebral circulation and CSF, which may be caused by various conditions. The advent of MRI and CT scanning has provided valuable information about the pathophysiology of hydrocephalus. The causes of hydrocephalus are diverse and include either congenital (myelomeningocele, intrauterine viral infection [cytomegalovirus, toxoplasmosis], aqueduct stenosis) or acquired conditions such as intraventricular hemorrhage, tumor, CSF infection, or head injury (Kestle, 2003; Kinsman and Johnston, 2007).
Case Study—Hydrocephalus with Myelomeningocele
Pathophysiology
To appreciate the condition, an understanding of the dynamics of CSF and the relationship between the various structures that make up the ventricular and subarachnoid spaces is necessary (Fig. 11-4). The two mechanisms by which CSF is formed are secretion by the choroid plexuses and lymphatic-like drainage by the extracellular fluid of the brain. CSF circulates throughout the ventricular system and is then absorbed within the subarachnoid spaces by a mechanism that is not entirely clear.
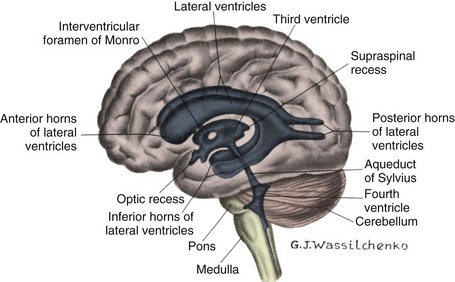
Fig. 11-4 Cerebral ventricular system. (From Thompson JM, McFarland G, Hirsch JE, et al: Mosby’s clinical nursing, ed 5, St Louis, 2002, Mosby.)
Ventricular Circulation: The fluid flows from the lateral ventricles through the foramen of Monro to the third ventricle, where it combines with fluid secreted into the third ventricle. From there CSF flows through the aqueduct of Sylvius into the fourth ventricle, where more fluid is formed; it then leaves the fourth ventricle by way of the lateral foramen of Luschka and the midline foramen of Magendie and flows into the cisterna magna. From there CSF flows to the cerebral and cerebellar subarachnoid spaces, where it is absorbed. A large portion is absorbed through the arachnoid villi, but the sinuses, veins, brain substance, and dura also participate in absorption.
Mechanisms of CSF Fluid Imbalance: The causes of hydrocephalus vary, but the result is either (1) impaired absorption of CSF fluid within the subarachnoid space, obliteration of the subarachnoid cisterns, or malfunction of the arachnoid villi (nonobstructive or communicating hydrocephalus); or (2) obstruction to the flow of CSF through the ventricular system (obstructive or noncommunicating hydrocephalus) (Kinsman and Johnston, 2007). The terms communicating and noncommunicating hydrocephalus traditionally referred to obstructive and nonobstructive types of hydrocephalus when pneumoencephalography was used to establish the diagnosis. Because other diagnostic methods are now used, the terms may be used only as a reference point in the diagnosis. Other authorities suggest that hydrocephalus be classified according to the cause as either congenital or acquired hydrocephalus (Rudy, 2005). Rarely, a tumor of the choroid plexus causes increased CSF secretion. Any imbalance of secretion and absorption causes an increased accumulation of CSF in the ventricles, which become dilated (ventriculomegaly) and compress the brain substance against the surrounding rigid bony cranium. When this occurs before fusion of the cranial sutures, it causes enlargement of the skull, as well as dilation of the ventricles. In children under 10 to 12 years of age, previously closed suture lines, especially the sagittal suture, may become diastatic, or opened. The cranial sutures do not permanently fuse until approximately the age of 12 years or later.
Most cases of hydrocephalus are a result of developmental malformations. Although the defect usually is apparent in early infancy, it may become evident at any time from the prenatal period to late childhood or early adulthood. Other causes include neoplasms, CNS infections (e.g., meningitis, encephalitis), and trauma (e.g., shaken baby syndrome). An obstruction to the normal flow can occur at any point in the CSF pathway to produce increased pressure and dilation of the pathways proximal to the site of obstruction. Table 11-3 describes the most frequent sites of obstruction and the consequences.
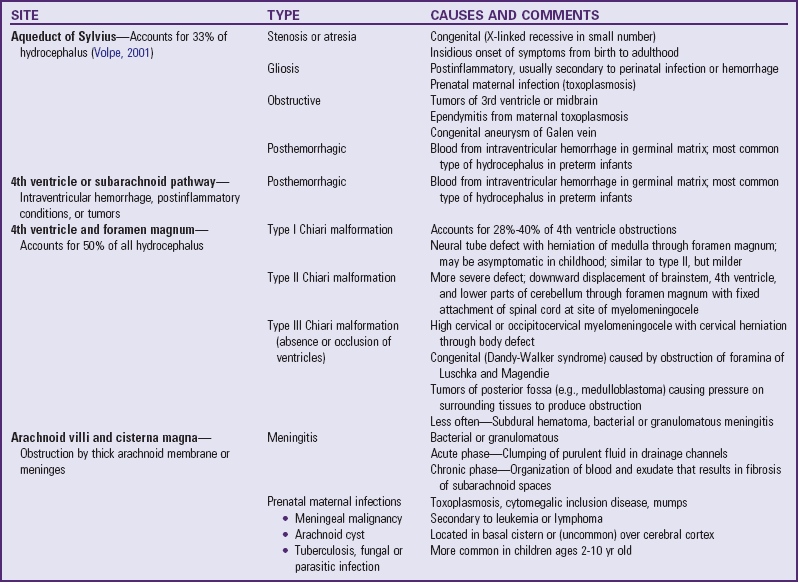
Developmental defects (e.g., Chiari malformation [see following discussion], aqueduct stenosis, aqueduct gliosis, and atresia of the foramina of Luschka and Magendie [Dandy-Walker malformation]) account for most cases of hydrocephalus from birth to 2 years of age. Dandy-Walker malformation involves a cystic expansion of the fourth ventricle and subsequent obstruction of CSF flow resulting in hydrocephalus (Kinsman and Johnston, 2007). Hydrocephalus is so often associated with myelomeningocele that all such infants should be observed for its development. In the remainder of cases there is a history of intrauterine infection (toxoplasmosis, cytomegalovirus), hemorrhage (posthemorrhagic hydrocephalus in preterm infant), and neonatal meningoencephalitis (bacterial or viral). In older children hydrocephalus is most often a result of intracranial masses (vascular anomalies, cysts, tumors), preexisting developmental defects, intracranial infections, trauma, or hemorrhage.
Chiari Malformations: A Chiari malformation is a brain defect involving posterior fossa contents. Table 11-3 describes the major types. The type II Chiari malformation, seen almost exclusively with myelomeningocele, is characterized by herniation of a small cerebellum, medulla, pons, and fourth ventricle into the cervical spinal canal through an enlarged foramen magnum. The resulting obstruction of CSF flow causes the hydrocephalus.
Clinical Manifestations
The three factors that influence the clinical picture in hydrocephalus are the acuity of onset, timing of onset, and associated structural malformations. In infancy, before closure of the cranial sutures, head enlargement (increasing occipitofrontal circumference [OFC]) is the predominant sign, whereas in older infants and children the lesions responsible for hydrocephalus produce other neurologic signs through pressure on adjacent structures.
Infancy: In infants with hydrocephalus, the head grows at an abnormal rate, although the first signs may be bulging fontanels with or without head enlargement (Fig. 11-5). The anterior fontanel is tense, often bulging, and nonpulsatile. Scalp veins are dilated and markedly so when the infant cries. With the increase in intracranial volume, the bones of the skull become thin and the sutures become palpably separated to produce the cracked-pot sound (Macewen sign) on percussion of the skull. In severe cases there may be frontal protrusion, or frontal bossing, with depressed eyes, and the eyes may be rotated downward, producing a setting-sun sign, in which the sclera may be visible above the iris. Pupils are sluggish, with unequal response to light.
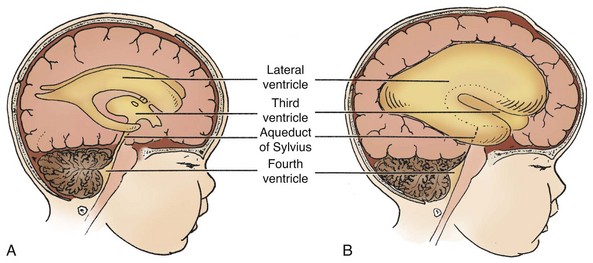
Fig. 11-5 Hydrocephalus: a block in flow of cerebrospinal fluid. A, Patent cerebrospinal fluid circulation. B, Enlarged lateral and third ventricles caused by obstruction of circulation—stenosis of aqueduct of Sylvius.
The infant is irritable and lethargic, feeds poorly, and may display changes in level of consciousness, opisthotonos (often extreme), and lower extremity spasticity. The infant cries when picked up or rocked and quiets when allowed to lie still. Early infantile reflexes may persist, and normally expected responses may not appear, indicating failure in the development of normal cortical inhibition.
Infants with Chiari malformation may exhibit behaviors that reflect cranial nerve dysfunction as a result of brainstem compression, including swallowing difficulties, stridor, apnea, aspiration, respiratory difficulties, and arm weakness.
The preterm infant with posthemorrhagic hydrocephalus may not exhibit any clinical signs and symptoms other than a gradual increase in head circumference. Alternatively, the nurse may note subtle seizure activity and alternating levels of consciousness. Assess ventricular size by ultrasonography or CT scanning in preterm infants at high risk for intraventricular hemorrhage.
If hydrocephalus is allowed to progress, development of lower brainstem functions is disrupted, as manifested by difficulty in sucking and feeding and a shrill, brief, high-pitched cry. Eventually the skull becomes enlarged, and the cortex is destroyed. If the hydrocephalus is rapidly progressive, the infant may display emesis, somnolence, seizures, and cardiopulmonary distress.
Childhood: The signs and symptoms in early to late childhood are caused by increased ICP, and specific manifestations are related to the focal lesion. Most commonly resulting from posterior fossa neoplasms and aqueduct stenosis, the clinical manifestations are primarily those associated with space-occupying lesions (i.e., headache on awakening with improvement following emesis or upright posture, papilledema, strabismus, and extrapyramidal tract signs such as ataxia [see Chapter 36]). As with infants, the child is irritable, lethargic, apathetic, confused, and often incoherent. In one of the congenital defects with later onset (by age 3 months), the Dandy-Walker syndrome, characteristic manifestations are a bulging occiput, nystagmus, ataxia, and cranial nerve palsies.
Manifestations of Chiari malformation in children over 3 years of age are related to spinal cord dysfunction rather than brainstem compression as observed in infants. Scoliosis proximal to the level of the myelomeningocele (usually associated with Chiari malformation) and development of upper extremity spasticity, which may progress to weakness and atrophy, are common. Cranial nerve deficits are rare.
Diagnostic Evaluation
Antenatal diagnosis of hydrocephalus is possible with fetal ultrasonography as early as 14 to 15 weeks of gestation. Delivery is not currently recommended until fetal lung maturity has been achieved.
In infancy the diagnosis of hydrocephalus is based on head circumference that crosses one or more percentile lines on the head measurement chart within a period of 2 to 4 weeks and on associated neurologic signs that are progressive. However, other diagnostic studies are needed to localize the site of CSF obstruction. Routine daily head circumference measurements are carried out in infants with myelomeningocele, hemorrhage, or intrauterine viral or CNS infections. In evaluation of a preterm infant, specially adapted head circumference charts are consulted to distinguish abnormal head growth from rapid but normal head growth.
The primary diagnostic tools for detecting hydrocephalus in older infants and children are CT and MRI (Fig. 11-6). Mild sedation is usually required because the child must remain absolutely still for an accurate picture. Diagnostic evaluation of children who have symptoms of hydrocephalus after infancy is similar to that employed in those with a suspected intracranial tumor. In the neonate, echoencephalography is useful in comparing the ratio of lateral ventricle to cortex. Sometimes isotope ventriculograms are used to assess the flow and patency of existing shunts and to evaluate the size of the ventricles.
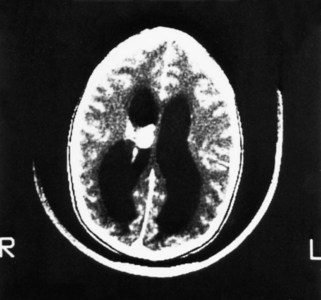
Problems in differential diagnosis are related to the child whose head circumference is greater than the 95th percentile but whose head growth parallels the normal growth curve. It is sometimes valuable to measure the parental OFC to detect a possible normal familial characteristic (benign familial megalencephaly). (See Table 37-2 for diagnostic tests for neurologic evaluation.)
Therapeutic Management
The treatment of hydrocephalus is directed toward (1) relief of ventricular pressure, (2) treatment of the cause of the ventriculomegaly, (3) treatment of associated complications, and (4) management of problems related to the effect of the disorder on psychomotor development. The treatment is, with few exceptions, surgical.
Medical therapy has been largely disappointing and inadequate. Many newborn infants with progressive cranial enlargement secondary to intracranial hemorrhage demonstrate spontaneous stabilization and resolution. Serial lumbar punctures and medications have been used with varying success but are recommended only until the preterm infant is stable enough to tolerate major surgery. The administration of acetazolamide and isosorbide or furosemide is somewhat beneficial in decreasing the production of CSF in selected cases of slowly progressive disease. The medication reduces the ICP until spontaneous arrest of hydrocephalus takes place or as a temporary measure when surgery is contraindicated.
Surgical Treatment: Improved neurosurgical techniques have established surgical treatment as the therapy of choice in almost all cases of hydrocephalus. This is accomplished by direct removal of an obstruction, such as resection of a neoplasm, cyst, or hematoma, or, in rare instances of fluid overproduction, by choroid plexus extirpation (plexectomy or electric coagulation). However, most children require a shunt procedure that provides primary drainage of the CSF from the ventricles to an extracranial compartment, usually the peritoneum.
Most shunt systems consist of a ventricular catheter, a flush pump, a unidirectional flow valve, and a distal catheter. All are radiopaque for easy visualization after placement, and all are tested for accuracy before insertion. A reservoir is frequently added to allow direct access to the ventricular system for administration of medications and removal of fluid. In all models the valves are designed to open at a predetermined intraventricular pressure and close when the pressure falls below that level, thus preventing backflow of fluid. Most shunts now in use have differential pressure and adjustable programmable valves with capability for changing the pressures with an external magnet, thus avoiding additional surgery (Chiafery, 2006; Kestle, 2003).
The standard procedure for many years has been the ventriculoperitoneal (VP) shunt, especially in neonates and young infants (Fig. 11-7). There is greater allowance for excess tubing, which minimizes the number of revisions needed as the child grows. Since it requires repeated lengthening, the ventriculoatrial (VA) shunt (ventricle to right atrium) is reserved for older children who have attained most of their somatic growth and children with abdominal pathologic conditions. The VA shunt is contraindicated in children with cardiopulmonary disease or elevated CSF protein.
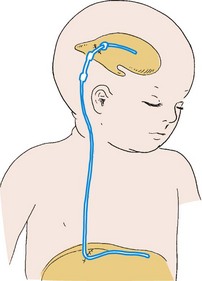
Animation—Ventriculoperitoneal Shunt
Although placement of ventricular shunts (into the amniotic sac) in utero for ventricular enlargement is possible, results have not been as promising as shunting soon after birth (Golden and Bönnemann, 2007).
The initial shunt is placed when indicated on the basis of individual assessment. The timing of revisions varies widely. In most instances revisions are performed when physical signs indicate shunt malfunction (i.e., signs of elevated ICP). Sometimes revisions are planned for specific times during development. The initial success rate is relatively high. However, shunts are associated with complications that interfere with continued shunt function or that threaten the child’s life.
Endoscopic third ventriculostomy (ETV) is a procedure that has potential for allowing greater independence from VA or VP shunting in children with hydrocephalus. In this procedure a small opening is made in the floor of the third ventricle, allowing CSF to flow freely through the previously blocked ventricle, thus bypassing the aqueduct of Sylvius. Children with SB, anatomic ventricular malformations, posthemorrhagic hydrocephalus (preterm infants), and postinfectious hydrocephalus are reportedly poor candidates for this procedure (Drake, 2008), as are children with bleeding disorders and those who have had previous radiotherapy. Reports of the success of ETV in children vary (see Research Focus box). Complications include CSF leak, hemorrhage from a perforated basilar artery, meningitis, cranial nerve injury, obstruction, and hypothalamic injury (Drake, 2008).
 RESEARCH FOCUS
RESEARCH FOCUS
Endoscopic Ventriculostomy Success Based on Age
Several studies have reported that the younger the child, the lower the success rate of the endoscopic ventriculostomy; infants less than 6 months to 1 year of age reportedly have the lowest success rates (Drake, 2008; Kadrian, van Gelder, Florida, et al, 2008; Wagner and Koch, 2005), whereas others report success in infants as well as older children (Faggin, Bernardo, Stieg, et al, 2009; Fritsch, Kienke, Ankerman, et al, 2005).
Complications: The major complications of VP shunts are infection and malfunction. All shunts are subject to mechanical difficulties, such as kinking, plugging, or separation and migration of tubing. Malfunction is most often caused by mechanical obstruction either within the ventricles from particulate matter (tissue or exudate) or at the distal end from thrombosis or displacement as a result of growth. Functional obstruction of a shunt’s antisiphon device remains a common complication. Shunt malfunctions are reported to be 40% at 1 year and 50% at 2 years (Drake, 2008; Kestle, 2003). In a large study occurring over 13 years, at least one shunt revision was required in 58.5% of children; the median time to shunt failure or malfunction was 88 days (Mittler, 2005). The child with a shunt obstruction often is seen in an emergency visit with clinical manifestations of increased ICP, frequently accompanied by worsening neurologic status.
The most serious complication, shunt infection, can occur at any time, but the period of greatest risk is 1 to 2 months after placement. Shunt infection rates are reported to be approximately 10% (Drake, 2008). The infection may be a result of intercurrent infections at the time of shunt placement. Infections include sepsis, bacterial endocarditis, wound infection, shunt nephritis, meningitis, and ventriculitis. Brain abscess associated with colonic perforation and infection with a gram-negative enteric organism suggests an ascending shunt infection in a child who has a VP shunt. Meningitis and ventriculitis are of greatest concern because any complicating CNS infection is a significant predictor of subnormal intellectual outcome. Infection is treated with antibiotics administered intravenously or intrathecally for a minimum of 7 to 10 days. Antibiotic-impregnated shunts may decrease infection rates (Drake, 2008). A persistent infection may require removal of the shunt until the infection is controlled; however, this practice has been recently challenged (Drake, 2008). External ventricular drainage (EVD), or external ventriculostomy, is used until CSF is sterile. EVD allows removal of CSF from a tube placed in the child’s ventricle that flows by gravity into a collection device.
The primary reasons for inserting an EVD include unstable status, increased ICP that is difficult to stabilize, or infection from an existing VP shunt. The EVD may drain CSF intermittently or continuously according to need. The EVD is a closed system made up of transparent pliable tubing, a collection bag, and, at times, a drip chamber between the tubing and the collection bag. The EVD is placed at the level of the child’s external auditory meatus with the head at a 20- to 30-degree elevation, depending on physician preference. Elevating the EVD above this level decreases the flow of CSF, and placing the device below the level of the external meatus increases the flow. Ambulation or sitting up in bed or chair usually requires that the tubing be clamped to prevent imbalance in CSF drainage. In addition, the EVD is a closed sterile system and should be handled as such in relation to emptying the device or changing the scalp dressing. Accurate and frequent documentation of the incision site; amount, color, and consistency of drainage into the device; and the child’s vital and neurologic signs are an important part of the nursing care.
Complications related to an EVD include infection, meningitis, hemorrhage, obstruction, malfunction (Ngo, Ranger, Singh, et al, 2009), and, in some cases, tentorial herniation as a result of imbalance in CSF drainage (Pope, 1998). In preterm infants with intraventricular hemorrhage requiring CSF drainage, a ventricular access drain may be temporarily inserted. This system is similar to the EVD but has an access port for frequent taps and can be used to administer antibiotics and thrombolytic agents such as urokinase.
Another serious shunt-related complication is subdural hematoma caused by too rapid reduction of ICP and size. This usually can be averted by careful assessment of ICP before insertion of the shunt and use of correct valvular pressure. Other complications that may occur include peritonitis, abdominal abscesses, perforation of abdominal organs by catheter or trocar (at the time of insertion), fistulas, hernias, and ileus. Children often need shunt lengthening as body growth occurs. This procedure usually involves replacing the distal catheter below the valve during the toddler period and again before the growth acceleration of puberty.
Prognosis: The prognosis of children with treated hydrocephalus depends largely on the cause of the dilated ventricles before shunt placement and the amount of irreversible brain damage before shunting (Kinsman and Johnston, 2007). For example, malignant tumors have a high mortality rate regardless of other complicating factors.
Survivors have a high incidence of subnormal intellectual capacity, and a large majority have major physical and disabling neurologic handicaps such as ataxia, spastic diplegia, poor fine motor coordination, and perceptual deficits. Some children demonstrate aggressive or delinquent behavior, and those with myelomeningocele are likely to have accelerated pubertal development as a result of increased gonadotropin secretion with increased ICP (Kinsman and Johnston, 2007). Depression requiring treatment occurred in 45% of children in one survey; in the same survey 54% of the children with hydrocephalus required four or more shunt revisions (Gupta, Park, Solomon, et al, 2007).
Surgically treated hydrocephalus in patients with little or no evidence of irreversible brain damage has a survival rate of about 90%, with most deaths occurring within the first year of treatment. Those with poor outcomes included children shunted for posthemorrhagic hydrocephalus or meningitis, with 40% and 30%, respectively, showing cognitive impairment (Kestle, 2003). Of the surviving children, approximately two thirds are intellectually normal. The presence of additional medical problems in infancy, including ocular defects, is the most significant variable associated with a high likelihood of neurocognitive impairment. Most children who require shunting must depend on the shunt for the remainder of their life.
Nursing Care Management
The infant with suspected or confirmed hydrocephalus is observed carefully for signs of increasing ventricular size and increasing ICP. In infants the head is measured daily at the point of largest measurement—the OFC. (See Chapter 6 for technique.) To avoid the likelihood of wide discrepancies, the point at which the measurements are taken is indicated on the head with a marking pen. Fontanels and suture lines are palpated for size, signs of bulging, tenseness, and separation. Irritability, lethargy, seizure activity, and altered vital signs and feeding behavior may indicate an advancing pathologic condition.
In older children, who are usually admitted to the hospital for elective or emergency shunt revision, the most valuable indicators of increasing ICP are an alteration in the child’s level of consciousness, complaint of headache, and changes in interaction with the environment. Changes are identified by observing and comparing present behavior with customary behavior, sleep patterns, developmental capabilities, and habits obtained through a detailed history and a baseline assessment. This baseline information serves as a guide for postoperative assessment and evaluation of shunt function.
The nurse is responsible for preparing the child for diagnostic tests such as MRI or a CT scan and for assisting with procedures such as a ventricular tap, which is often performed to relieve excessive pressure and to obtain CSF during the preoperative period. Sedation is required because the child must remain absolutely still during diagnostic testing. A variety of drugs are available for sedation. (See Chapter 27 for preparing children for procedures.) If surgery is anticipated, IV infusions should not be placed in a scalp vein.
Postoperative Care: In addition to routine postoperative care and observation, the infant or child is positioned carefully on the unoperated side to prevent pressure on the shunt valve. The child remains flat to help avert complications resulting from too rapid reduction of intracranial fluid. The surgeon indicates the position to be maintained and the extent of activity allowed.
The nurse continues observation for signs of increased ICP, which indicate obstruction of the shunt. Neurologic assessment includes pupil dilation (pressure causes compression or stretching of the oculomotor nerve, producing dilation on the same side as the pressure) and blood pressure (hypoxia to the brainstem causes variability in these vital signs). The nurse also observes for abdominal distention because CSF may cause peritonitis or a postoperative ileus as a complication of distal catheter placement.
Because infection is the greatest hazard of the postoperative period, nurses are continually on the alert for the usual manifestations of CSF infection, including elevated temperature, poor feeding, vomiting, decreased responsiveness, and seizure activity. There may be signs of local inflammation at the operative sites and along the shunt tract. Antibiotics are administered by the IV route as ordered, and the nurse may also need to assist with intraventricular instillation. Inspect the incision site for leakage, and test any suspected drainage for glucose, an indication of CSF.
Family Support: Specific needs and concerns of parents during periods of hospitalization are related to the reason for the child’s hospitalization (shunt revision, infection, diagnosis) and the diagnostic and surgical procedures to which the child must be subjected. Parents may have little understanding of anatomy; therefore they need further explanation and reinforcement of information that was given to them by the physician and neurosurgeon, including information about what to expect. They are especially frightened of any procedure that involves the brain, and the fear of disability or brain damage is real and pervasive. Nurses can calm their anxiety with explanations of the rationale underlying the various nursing and medical activities such as positioning or testing and by simply being available and willing to listen to their concerns.
To prepare for the child’s discharge and home care, instruct the parents on how to recognize signs that indicate shunt malfunction or infection. Active children may have injuries, such as a fall, that can damage the shunt, and the tubing may pull out of the distal insertion site or become disconnected during normal growth. Contact sports should be avoided, but swimming or tennis is appropriate. A helmet may be worn to protect the head in case of a fall or injury when outside play is vigorous (Chiafery, 2006). It is also important for the nurse to encourage families to enroll infants and toddlers with hydrocephalus into an early childhood development program. Depending on the degree of initial damage and the underlying cause, many children have normal intellectual development.
The management of hydrocephalus in a child is a demanding task for both the family and health professionals, and helping the family cope with the child’s difficulties is an important nursing responsibility. Children with hydrocephalus have lifelong special health care needs. The nurse can provide optimum primary health care, including advice on immunizations, treatments for common infectious conditions, or child care and school precautions. The overall aim is to establish realistic goals and an appropriate educational program that will assist the child in achieving the maximum potential. Families can be referred to community agencies for support and guidance. The National Hydrocephalus Foundation (NHF)* and the Hydrocephalus Association† provide information on the condition for families, and the NHF assists interested groups in establishing local organizations.
Anticipatory guidance will prepare parents for possible problems and help them avoid being overprotective of the child. Few restrictions need be placed on the child’s activities (mainly contact sports), and the child is encouraged to live as would any other youngster of the same age and abilities. Parents need support and encouragement in coping with the child and problems the child may encounter in relationships with peers and others. Reactions of other children when the child has a noticeably enlarged head or requires shaving at times of revision are stressful for both the child and the parents. (See Chapter 22 for problems and coping with a child with a disability.)
Cranial Deformities
In the normal newborn the cranial sutures are separated by membranous seams several millimeters wide. For the first few hours to 1 to 2 days after birth, the cranial bones are highly mobile, which allows the bones to mold and overlap one another, adjusting the circumference of the head to accommodate to the changing shape and character of the birth canal. The principal sutures in the infant’s skull are the sagittal, coronal, and lambdoid sutures, and the major soft areas at the juncture of these sutures are the anterior and posterior fontanels. (See Fig. 8-7.)
After birth, growth of the skull bones occurs in a direction perpendicular (at right angles) to the line of the suture, and normal closure occurs in a regular and predictable order. Although the age at which closure takes place varies widely in individual children, solid union of all sutures is not completed until late childhood. Normally, sutures and fontanels are ossified by the following ages:
• Eight weeks—Posterior fontanel closed
• Six months—Fibrous union of suture lines and interlocking of serrated edges
Closure of a suture before the expected time inhibits the perpendicular growth. Since normal increase in brain volume requires expansion, the skull is forced to grow in a direction parallel to the fused suture. This alteration in skull growth always distorts the head shape when the underlying brain growth is normal. The small head with closed sutures and a normal shape is a result of deficient brain growth; the suture closure is secondary to this brain growth failure. Failure of brain growth is not secondary to suture closure.
Various types of cranial deformities are encountered in early infancy. These include the enlarged head with frontal protrusion, or bossing (characteristic of hydrocephalus); the parietal bossing that is seen in chronic subdural hematoma; the small head; and a variety of skull deformities (Fig. 11-8). Some occur during prenatal development. In others, head circumference is usually within normal limits at birth, and the deviation from normal development becomes apparent with advancing age.
Fig. 11-8 Craniostenosis. Abnormal head configuration resulting from premature closing of cranial sutures.
Microcephaly
Primary (genetic) microcephaly reflects a small brain and may be caused by an autosomal recessive disorder or a chromosomal abnormality (Kinsman and Johnston, 2007). Secondary (nongenetic) microcephaly can result from a variety of insults that occur during the third trimester of pregnancy, the perinatal period, or early infancy (Kinsman and Johnston, 2007). These stimuli may be irradiation (especially between 4 and 20 weeks of gestation), maternal infection (notably toxoplasmosis, rubella, or cytomegalovirus [see Maternal Infections, Chapter 10]), or chemical agents. Infection, trauma, metabolic disorders, and anoxia are all capable of causing decreased brain growth. Secondary microcephaly may also occur as a result of maternal diabetes and maternal hyperphenylalanemia. Fetal exposure to alcohol and tobacco use was shown to result in a 2.6-fold increase in the risk of isolated secondary microcephaly compared with other causes, including syndromes (Krauss, Morrissey, Winn, et al, 2003). Microcephaly is defined as an OFC greater than 3 standard deviations below the mean for age and sex (Kinsman and Johnston, 2007).
In both types the neurologic manifestations range from decerebration, complete unresponsiveness, and autistic behavior to mild motor impairment, educable neurocognitive impairment, and mild hyperkinesis. There appears to be a decided relationship between microcephaly and cognitive delays of varying degrees; however, not all children with microcephaly have cognitive delays.
Nursing Care Management
There is no specific treatment. Nursing care is supportive and may be directed toward helping parents adjust to rearing a child with cognitive impairment when this condition is present. (See Chapter 24.)
CraniosTENosis
Craniostenosis is defined as the premature closure at birth of one or more cranial sutures (Kinsman and Johnston, 2007). The clinical picture depends on which sutures close, the duration of the closure process, and the success or failure of the other sutures to compensate by expansion (see Fig. 11-8). The condition may be divided according to the number of sutures involved; primary craniostenosis involves only one suture, whereas secondary craniostenosis involves two or more sutures (Kinsman and Johnston, 2007). Focal hydrodynamic mechanisms are involved in the compensatory skull changes seen in craniostenosis. Brain atrophy and an underlying motor delay account for the position-induced skull changes. Craniostenosis is also a common feature of children with Crouzon, Apert (Box 11-4), Pfeiffer, Carpenter, and Jackson-Weiss syndromes (Kinsman and Johnston, 2007). Potential risk factors for craniostenosis include maternal Caucasian race, advanced maternal age, male infant, use of nitrosatable drugs, fertility treatments, and certain paternal occupations (agriculture, repairmen, mechanics, forestry). Recently genetic mutations have been linked to the occurrence of craniostenosis, and in such cases genetic counseling is advised (Merritt, 2009). Diagnosis is established with CT scan, and MRI is useful in identifying accompanying brain abnormalities. Increased ICP is more frequent in children with more than one prematurely fused suture.
The most common form of craniostenosis is premature closure of the sagittal suture with resulting elongation of the skull in the anteroposterior direction. A similar head shape occurs as a result of postnatal position maintenance in some preterm infants; however, in this case there is no premature closure of sutures. Craniostenosis may cause an increase in ICP, which may or may not cause cognitive delays but can result in progressive papilledema, optic atrophy, and eventual blindness. Other complications include facial asymmetry and malocclusion.
Trigonocephaly, or metopic craniostenosis, represents a premature closure of the metopic suture in utero, a congenital problem that is familial and may not require surgical treatment. The metopic suture occurs where the right and left frontal bones meet on the forehead. Craniostenosis of the metopic suture may be an autosomal dominantly inherited disorder not associated with functional brain or other abnormalities.
Therapeutic Management
Treatment involves surgical excision of long bars of bone (strip craniectomy) along or parallel to the fused suture. Various surgical procedures are employed in an effort to release the fused suture and direct growth. Surgery is performed to achieve the best possible cosmetic effect and, in severe cases, to relieve cerebral pressure symptoms and complications. The advised timing of suture release is before 6 months of age for best cosmetic and neurodevelopmental results. An endoscopic strip craniectomy results in less blood loss than other types of craniectomy, and length of stay is decreased (Merritt, 2009).
Nursing Care Management
Nursing care primarily involves the early identification of persistent cranial molding weeks after regular birth molding would have resolved and referral for follow-up evaluation. In the postoperative period, nursing care includes observation for changes in neurologic status, hemorrhage, or infection.
Because of the type of bone surgery involved with craniostenosis, blood loss can be large. Therefore the nurse carefully monitors hematocrit and hemoglobin. Parents may also wish to provide a compatible blood donor for their infant. Nurses need to inform and guide parents through this blood bank procedure. With endoscopic surgery, blood loss is minimized, but the child must wear a cranial molding helmet for several months (Merritt, 2009). Instructions regarding compliance with the helmet and skin care are essential.
Most children have substantial swelling of the eyelids postoperatively; careful handling and talking to the child may help calm fears while the eyelids are swollen shut. Eye care should be limited to gentle cleansing with a moist cloth. Pain management measures should be instituted in infants experiencing postoperative pain as they would be for older children or adults. Fluids and adequate hydration are essential. Oral feedings resume as soon as possible for hydration and for the infant’s nutritive sucking needs.
Early surgical management of craniostenosis allows proper expansion of the brain and the creation of an acceptable appearance. Parents require special support and education during this time, especially from other parents whose infants have undergone similar operations. The nurse can serve as a liaison for this type of parental support.
Craniofacial Abnormalities
Craniofacial abnormalities are those deformities involving the skull and facial bones. They have a low incidence rate in the population, but their effects can be psychologically devastating to affected children and their families. Box 11-4 lists deformities caused by abnormal growth of cranial bone(s).
Most craniofacial anomalies are compatible with life, and all efforts are made to help the child and family live as normal a life as anyone else. Advances in microscopic, orthopedic, neurologic, and plastic reconstructive surgery techniques have made it possible for children with craniofacial anomalies not only to survive beyond childhood, but also to live a fulfilling life without the social stigmas of past decades. These children, however, may continue to face erroneous assumptions of cognitive impairment because of their appearance. Craniofacial multidisciplinary teams are dedicated to helping the child and family achieve optimum potential for intellectual growth, physical competence, and social acceptance.
Therapeutic Management
Craniofacial surgical correction involves peeling the patient’s face away from the skull and remolding the understructures. Parts can be brought together, the skull reshaped and remodeled, and bone fragments removed or reshaped. Bone segments from the child’s hip or ribs may be used to reshape the skull or facial features. The procedures are performed at various ages, depending on the anomaly, in craniofacial centers specializing in this pediatric problem. The timing of surgery is before school entry and is determined on an individual basis to ensure normal growth and development. Depending on the abnormality, other surgeries are performed, such as mandibular and digit correction. After surgery, continued growth conforming to the inborn abnormality is unlikely.
Nursing Care Management
Direct nursing efforts toward preparation for surgery (often several surgical procedures over time), postoperative care similar to care of any child with cranial surgery, and support of the child and family. Frequently this child and family must adjust to the unfamiliar body image, which may be as traumatic as the previous deformity. A helmet may be worn to protect the operative site and bone grafts for 6 months to 2 years. Follow-up care is important.
Pierre Robin Sequence
Pierre Robin sequence (PRS) is a defect characterized by retroposition of the tongue and mandible, which often results in neonatal respiratory and feeding problems. The condition has an incidence of 1 in 8500 to 14,000 live births, and about one half of those with PRS have other congenital anomalies (Buchenau, Urschitz, Sautermeister, et al, 2007). The tongue may be large (glossoptosis) and frequently falls over the neonate’s airway, causing occlusion and respiratory distress. In severe upper airway obstruction a tracheostomy may be required. From a lateral view the infant’s lower jaw can be seen to be positioned posterior (micrognathia) to the upper jaw. PRS may be diagnosed in the nursery when the infant has apnea and cyanosis, due primarily to the upper airway obstruction. The neonate is positioned to facilitate an open airway, and the practitioner is notified immediately. A tongue-lip adhesion is a common surgical procedure that repositions the tongue anteriorly (Bijnen, Don Griot, Mulder, et al, 2009). Surgical mandibular distraction is another technique used to correct the defect (Dauria and Marsh, 2008). There are usually no associated neurocognitive defects in isolated PRS.
Positional Plagiocephaly
Since the Back to Sleep campaign began in 1992 advocating nonprone sleeping for infants to prevent sudden infant death syndrome (SIDS), an increase in the incidence of positional plagiocephaly has been observed (American Academy of Pediatrics, 2005; Littlefield, Saba, and Kelly, 2004). Prevalence of positional plagiocephaly at 4 months is reported to range from slightly less than 20% to as much as 48% (Robinson and Proctor, 2009). The term plagiocephaly connotes an oblique or asymmetric head; positional plagiocephaly, deformational plagiocephaly, or nonsynostotic plagiocephaly implies an acquired condition that occurs as a result of cranial molding during infancy (Hummel and Fortado, 2005). Because the infant’s sutures are not closed, the skull is pliable and, when the infant is placed on the back to sleep, the posterior occiput flattens over time (Fig. 11-9, A). A typical bald spot develops, which is usually transient. As a result of prolonged pressure on one side of the skull, that side becomes misshapen; mild facial asymmetry may develop. The sternocleidomastoid muscle may tighten on the preferential side, and torticollis may also develop. Congenital or acquired torticollis may cause plagiocephaly; other causes of deformational plagiocephaly include certain craniofacial syndromes. This discussion centers only on plagiocephaly caused by supine sleeping position.
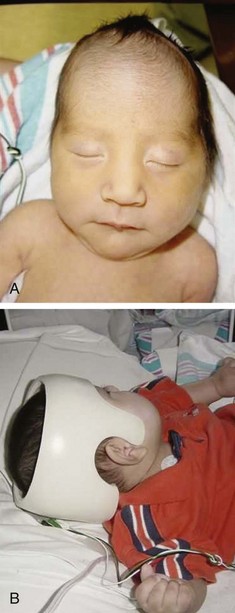
Fig 11-9 A, Plagiocephaly. B, Helmet used to correct plagiocephaly. (Courtesy Dr. Gerardo Cabrera-Meza, Department of Neonatology, Baylor College of Medicine, Houston.)
Therapeutic Management
Treatment of torticollis and plagiocephaly initially involves exercises to loosen the tight muscle and switching head position sides during feeding, carrying, and sleep. If the plagiocephaly is not resolved within 4 to 8 weeks of physical therapy, a customized helmet may be worn to decrease the pressure on the affected side of the skull (Fig. 11-9, B). If no improvement occurs with physical therapy or a molded helmet over a given period, the infant may be referred to a pediatric neurosurgeon or craniofacial surgeon. In one study repositioning was not as helpful in reducing plagiocephaly as was the use of an orthotic helmet. Those treated with a helmet were older and had a longer treatment period, leading the authors to conclude that early detection and orthotic intervention were likely to be more successful (Graham, Gomez, Halberg, et al, 2005).
The helmet is worn 23 hours a day for a prescribed period (usually 3 months). Repositioning and physical therapy are said to be more effective when used before the infant can roll over or move his or her head alone (i.e., before approximately 3 to 4 months of age) (Robinson and Proctor, 2009). Reports of developmental delay in infants with positional plagiocephaly (nonsynostotic) vary in regard to outcomes, but current studies do not conclusively prove that such infants are at higher risk for developmental delays (Robinson and Proctor, 2009).
Nursing Care Management
Minor skull flattening is not considered significant, but parents should learn to prevent plagiocephaly by altering the infant’s head position during sleep. Infants should be placed prone on a firm surface during awake time (tummy time), which prevents plagiocephaly and facilitates development of upper shoulder girdle strength; the latter helps in the progressive development of movements such as rolling over and starting to rise up on all fours, which are precursors to crawling and eventually walking. Thirty minutes of supervised tummy time per day in infants younger than 6 months of age is recommended (Robinson and Proctor, 2009).
Despite the perceived increase in the incidence of positional plagiocephaly, the supine sleeping position is still recommended because it has led to a significant decrease in loss of infant lives from SIDS (American Academy of Pediatrics, 2005). Additional measures to prevent plagiocephaly include avoiding excessive time spent in car restraint seats or infant seats and bouncers. Alternating the infant’s head position for sleep times can also prevent unilateral molding. When a nurse or parent notices plagiocephaly, a consultation with the primary practitioner is recommended to evaluate the head shape and ascertain the need for early intervention.
Nurses are in a unique position in well-child care settings to encourage parents to follow guidelines for preventing plagiocephaly, to demonstrate alternating head placement for sleeping, to demonstrate sternocleidomastoid muscle exercises (as appropriate to the condition), and to encourage tummy time for infants during awake periods. Most important, nurses should continue to encourage parents to place the infant in a supine sleep position despite the development of plagiocephaly. Parents should not become so alarmed by plagiocephaly that they abandon supine sleeping position for the infant but should consult with the practitioner for further advice.
Skeletal Defects
This discussion is limited to those defects in development that are most common, that are amenable to therapy, and that involve nurses to a considerable extent. Less common defects and disorders are described in Table 11-4.
Developmental Dysplasia of the Hip
The broad term developmental dysplasia of the hip (DDH) describes a spectrum of disorders related to abnormal development of the hip that may develop at any time during fetal life, infancy, or childhood. A change in terminology from congenital hip dysplasia and congenital dislocation of the hip to DDH more properly reflects the varying onset and types of hip abnormalities in which there is a shallow acetabulum, subluxation, or dislocation (Box 11-5).
Case Study—Developmental Dysplasia of the Hip
The incidence of hip instability of some kind is approximately 1 to 1.5 per 1000 live births (Hosalkar, Horn, Friedman, et al, 2007). Approximately 30% to 50% of infants with DDH were born breech. The left hip is involved in 60% of cases, the right hip in 20%, and both hips in 20%. Eighty percent of those affected are female. Caucasian children have a higher incidence of DDH than other ethnic groups (Maher, Salmond, and Pellino, 2002).
Etiology and Pathophysiology
The cause of DDH is unknown, but certain factors such as gender, birth order, family history, intrauterine position, delivery method, joint laxity, and postnatal positioning affect the risk of DDH. Predisposing factors associated with DDH are divided into three broad categories: (1) physiologic factors, including maternal hormone secretion and intrauterine positioning; (2) mechanical factors; and (3) genetic factors. Mechanical factors involve breech presentation, multiple fetuses, oligohydramnios, and large infant size, as well as continued maintenance of the hips in adduction and extension, causing a dislocation (see Cultural Competence box). Genetic factors account for a higher incidence (6%) of DDH in siblings of affected infants, and an even greater incidence (36%) of recurrence if a sibling and one parent were affected.
 CULTURAL COMPETENCE
CULTURAL COMPETENCE
Developmental Dysplasia of the Hip and Infant Care Practices
A striking relationship exists between the development of dislocation and methods of handling infants. Among cultures with the highest incidence of dislocation (Navajo Indians and Canadian Natives [formerly referred to as Eskimos]), newly born infants are tightly wrapped in blankets or other swaddling material or are strapped to cradle boards. In cultures where mothers carry infants on their backs or hips in the widely abducted straddle position, such as in the Far East and Africa, the disorder is virtually unknown.
DDH may be further categorized into two major groups: (1) typical, in which the infant is neurologically intact; and (2) teratologic, which involves a neuromuscular defect such as arthrogryposis or myelodysplasia. The teratologic forms usually occur in utero and are much less common.
Fig. 11-10 illustrates three degrees of DDH. They are also outlined in Box 11-5.
Clinical Manifestations
The diagnosis of DDH should be made in the newborn period if possible, since treatment initiated before 2 months of age achieves the highest rate of success. In the newborn period dysplasia usually appears as hip joint laxity rather than as outright dislocation. Subluxation and the tendency to dislocate can be demonstrated by the Ortolani or Barlow test. With the infant quiet and relaxed in the supine position on a firm surface and the legs facing the examiner, the hips are flexed (not forced) at right angles and the knees are flexed. The examiner places the middle finger of each hand over the greater trochanter and the thumbs on the inner side of the thigh at a point opposite the lesser trochanter. The knees are carried to midabduction, and each hip joint is submitted, one at a time, first to forward pressure exerted behind the trochanter and then to backward pressure exerted from the thumbs in front as the opposite joint is held steady. If the femoral head can be felt to slip forward into the acetabulum on pressure from behind, it is dislocated (Ortolani test) (Fig. 11-11, D).
Fig. 11-11 Signs of developmental dysplasia of hip. A, Asymmetry of gluteal and thigh folds. B, Limited hip abduction, as seen in flexion. C, Apparent shortening of femur, as indicated by level of knees in flexion. D, Ortolani clunk (if infant is <4 weeks of age). E, Positive Trendelenburg sign (if child is weight bearing).
Sometimes an audible “clunk” can be heard on exit or entry of the femur out of or into the acetabulum. If, on pressure from the front, the femoral head is felt to slip out over the posterior lip of the acetabulum and immediately slips back in place when pressure is released, the hip is said to be dislocatable or “unstable” (Barlow test). The audible clunk is not pathologic and occurs as a result of breaking surface tension across the hip joint, knee rotation, snapping of gluteal tendons, or patellofemoral motion.
 NURSING ALERT
NURSING ALERT
The Barlow and Ortolani tests should be performed only by an experienced clinician to prevent damage to the hip. If these tests are performed too vigorously in the first 2 days of life, when the hip subluxates freely, persistent dislocation may occur.
The Ortolani and Barlow tests are most reliable from birth to 2 or 3 months of age. Adduction contractures develop at about 6 to 10 weeks, and the Ortolani sign disappears. After this time the most sensitive test is limited hip abduction (Fig. 11-11, B). Other signs are shortening of the thigh on the affected side (Galleazzi sign) (Fig. 11-11, C), asymmetric thigh and gluteal folds (Fig. 11-11, A), and broadening of the perineum (in bilateral dislocation). Weight bearing may precipitate a transition from subluxation to dislocation in unrecognized cases.
A common cause of decreased hip abduction in the newborn is pelvic obliquity in which one side of the pelvis is higher than the other. This is caused by intrauterine positioning and will resolve on its own.
In the older infant and child the affected leg is shorter than the other, with telescoping or piston mobility; that is, the head of the femur can be felt to move up and down in the buttock when the extended thigh is first pushed toward the child’s head and then pulled distally. Instability of the hip on weight bearing delays walking and produces a characteristic limp, waddling gait and toe walking. When the child stands first on one foot and then on the other (holding onto a chair, rail, or someone’s hands), bearing weight on the affected hip, the pelvis tilts downward on the normal side instead of upward as it would with normal stability (positive Trendelenburg sign) (Fig. 11-11, E). The practitioner should test the child for at least 30 seconds. In both unilateral and bilateral dislocations the greater trochanter is prominent and appears above a line from the anterosuperior iliac spine to the tuberosity of the ischium. The child with bilateral dislocations has marked lordosis and a peculiar waddling gait.
Diagnostic Evaluation
DDH is often not detected at the initial examination after birth; thus all infants should be carefully monitored for hip dysplasia at follow-up visits throughout the first year of life. The primary diagnostic tools in the newborn period are the assessment techniques just described.
Radiographic examination in early infancy is not reliable because ossification of the femoral head does not normally take place until the third to sixth month of life. However, the cartilaginous head can be visualized directly with real-time high-resolution ultrasonography. There has been debate regarding the role of ultrasound technology in diagnosing DDH in early infancy, and widespread newborn screening has been suggested. However, numerous studies reveal this approach has a high rate of false positives and subsequent overtreatment. Once the proximal femoral epiphysis ossifies (usually 4 to 6 months), and in older children, radiographic examination is useful in confirming the diagnosis (Hosalkar, Horn, Friedman, et al, 2007). An upward slope in the roof of the acetabulum (the acetabular angle) greater than 40 degrees with upward and outward displacement of the femoral head is a frequent finding in older children. CT scan may be useful to assess the position of the femoral head relative to the acetabulum after closed reduction and casting.
Therapeutic Management
Treatment begins as soon as the condition is recognized because early intervention is more favorable to the restoration of normal bony architecture and function. The longer treatment is delayed, the more severe the deformity, the more difficult the treatment, and the less favorable the prognosis. The treatment varies with the child’s age and the extent of the dysplasia. The goal of treatment is to obtain and maintain a safe, congruent position of the hip joint to promote normal hip joint development.
Newborn to 6 Months: The hip joint is maintained by dynamic splinting in a safe position with the proximal femur centered in the acetabulum in an attitude of flexion. A variety of abduction devices are available for maintaining the femur in the acetabulum. Of these, the Pavlik harness is the most widely used device, and with time, motion, and gravity the hip works into a more abducted, reduced position (Fig. 11-12). The rate of reduction is about 95% effective when a Pavlik harness is used on a full-time basis for 6 weeks (Hosalkar, Horn, Friedman, et al, 2007). The harness is a dynamic splint that is worn continuously until the hip is clinically and radiographically stable, usually about 3 to 5 months. It is highly effective when the device is well constructed, follow-up care is adequate, and the parents follow instructions in its use. The Pavlik harness does not rigidly immobilize the hip but acts to prevent hip extension or adduction. Because of the infant’s rapid growth rate, the straps should be checked every 1 to 2 weeks for adjustments. Improper positioning may cause vascular or nerve damage.
Fig. 11-12 Child in Pavlik harness. (From Ball JW: Mosby’s pediatric patient teaching guides, St Louis, 1998, Mosby.)
When adduction contracture is present, other devices (such as skin traction) are employed to slowly and gently stretch the hip to full abduction, after which wide abduction is maintained until stability is attained. When maintaining stable reduction is difficult, a hip spica cast is applied and changed periodically to accommodate the child’s growth. After 3 to 6 months, sufficient stability is acquired to allow transfer to a removable protective abduction brace. The duration of treatment depends on development of the acetabulum but usually lasts less than a year.
Six to 18 Months: In this age-group the dislocation may not be recognized until the child begins to stand and walk, when attendant shortening of the limb and contractures of hip adductor and flexor muscles become apparent. Gradual reduction by traction may be used for approximately 3 weeks. An alternative to traction is the closed surgical reduction with casting. An individualized home traction program may be developed for the child preoperatively to decrease the length of hospitalization and keep the child in the home environment. Written directions should be provided to increase compliance with preoperative care.
The child who has initial traction treatment then undergoes an attempted closed reduction of the hip under general anesthesia; an arthrogram is used to confirm reduction. If the hip is not reducible, an open operative reduction is performed. After reduction, the child is placed in a hip spica cast for 2 to 4 months until the hip is stable, at which time a flexion-abduction brace is applied.
Older Child: Correction of the hip deformity in the older child is inherently more difficult than in the preceding age-groups because secondary adaptive changes and other etiologic factors (such as rheumatoid arthritis or nonambulatory cerebral palsy) complicate the condition. Operative reduction, which may involve preoperative traction, tenotomy of contracted muscles, and any one of several innominate osteotomy procedures designed to construct an acetabular roof, is usually required. After cast removal and before weight bearing is permitted, range-of-motion exercises help restore movement. Other rehabilitative measures may include muscle strengthening, a period of crutch walking, and gait training. Successful reduction and reconstruction become increasingly difficult after the age of 4 years and are usually impossible or inadvisable after age 6 because of severe shortening and contracture of muscles and deformity of the femoral and acetabular structures.
Hip dislocation is often observed in older children with SB and reportedly does not significantly interfere with ambulation. Reduction of the affected hip is not worthwhile in most of these children unless the dislocation is bilateral or there is significant flexion contracture (Rowe and Jadhav, 2008).
Nursing Care Management
Nurses are in a unique position to detect DDH in the newborn. During the infant assessment process and routine nurturing activities, the nurse inspects the hips and extremities for any deviations from normal. Observation for unequal gluteal and thigh folds is routine, and nurses who have been educated to perform the Ortolani and Barlow tests should refer the infant with a positive test result to the practitioner. The ambulatory child who displays a limp or waddling gait is referred for evaluation. This may indicate an orthopedic or neurologic problem. The nurse also assesses nonambulatory children with cerebral palsy for evidence of dislocation.
The former practice of double- or triple-diapering for DDH is controversial, but is generally not recommended because it promotes hip extension, thus exacerbating improper hip development.
Observations during routine care, such as diapering, provide an opportunity to observe the infant for limited movement and a wide perineum, which is an indication to assess for leg shortening, unequal gluteal and thigh folds, and limited abduction.
Care of the Child: The primary nursing goal is teaching parents to apply and maintain the reduction device. The Pavlik harness allows for easy handling of the infant and usually produces less apprehension in the parent than heavy braces and casts. It is important that parents understand the correct use of the appliance, which may or may not allow for its removal during bathing. If the infant has a harness that is not removed, a sponge bath is recommended.
The following instructions for preventing skin breakdown with the harness are stressed:
• Always put an undershirt (or a shirt with extensions that close at the crotch) under the chest straps and put knee socks under the foot and leg pieces to prevent the straps from rubbing the skin.
• Check frequently (at least two or three times a day) for red areas under the straps and the clothing.
• Gently massage healthy skin under the straps once a day to stimulate circulation. In general, avoid lotions and powders because they can cake and irritate the skin.
The parents are permitted to pad shoulder straps at pressure points if desired, but unbuckling or removal is determined on the basis of the family’s level of understanding and the degree of hip deformity. In general, parents are not encouraged to adjust the harness without supervision. The practitioner should examine the child before any adjustment is attempted to ascertain that the hips are in correct position before the harness is resecured. Problems reported by parents included difficulty applying the harness after the bath, carrying the child, skin-crease dermatitis, feet slipping, and difficulty in clothing the child (Hassan, 2009). These findings highlight the importance of close follow up within the first few weeks of treatment and written take-home instructions for the parents to follow in managing the Pavlik harness.
The major challenges in the care of an infant or child in a cast or other device are related to maintaining the device and adapting nurturing activities to meet the patient’s needs. Generally, treatment and follow-up care of these children are carried out in a clinic, practitioner’s office, or outpatient unit. Hospitalization may be necessary for cast application or brace fitting but seldom exceeds 24 to 48 hours. Longer hospitalization is required for open reduction. (See also The Child in a Cast, Chapter 39.)
Casts offer more challenging nursing problems, since they cannot be removed for routine care. Care of an infant or small child with a cast requires nursing innovation to reduce irritation and to maintain cleanliness of both the child and the cast, particularly in the perineum and sacrum. The importance of spica cast care should be emphasized when providing instructions. The life of the cast should be prolonged to hold the legs and hips in proper position postoperatively and prevent an unnecessary cast change.
Chapter 39 discusses cast care and observation, so these are not elaborated on here. However, since DDH is almost the only reason for application of casts in early infancy, some of the problems specific to that age-group are mentioned.
Parents need to learn the proper care of the cast (or brace) and need help to devise means for maintaining cleanliness. A disposable newborn diaper is tucked beneath the entire perineal opening of the cast. A larger (toddler-size) diaper can be applied and fastened over the small diaper and cast. The goal is to absorb the urine while avoiding urine saturation or soiling of the cast.
For tightly fitting casts, transparent film dressing can be cut into strips as for petaling (see Chapter 39), and one edge can be applied to the cast edge and the other directly to the perineum; this forms a continuous waterproof bridge between the perineum and the cast to prevent leakage. An additional advantage to using this dressing material is that it keeps both the skin and the cast dry while allowing observation of the skin beneath the dressing.
Older infants and small children may stuff bits of food, small toys, or other items under the cast. Alert parents to this possibility so they can initiate suitable preventive measures, such as placing clothing over the cast.
Feeding the infant in a hip spica cast or brace offers problems of positioning. Very young infants can be fed in the supine position with the head elevated, and, with the infant’s hips and legs supported on a pillow at the side, the parent can cuddle the infant during feeding. A somewhat similar position can be used for breast-feeding (i.e., with the infant supported on pillows or held in a “football” hold facing the mother with the legs behind her). An alternate position is to hold the infant upright on the mother’s lap with the infant’s legs astride the mother’s legs.
Infants who are able to sit up can be fed in a feeding table or modified high chair. A padded tilt board with an adjustable chair may be adapted for the child in a hip spica cast. The table or chair provides an excellent place for the child to play in an upright position. The child’s transportation in a safe car seat is also a vital consideration. Many hospitals have a child passenger safety program. A loan program for the appropriate automotive safety restraint or referral to an agency that provides this service may be offered by the discharge coordinator nurse or social worker.*
Family Support and Home Care: It is important for nurses, parents, and other caregivers to understand that children with DDH need to be involved in all the activities of any child in the same age-group. Toys are chosen that can be used in a prone position on the floor or in the seats devised for feeding and other activities. Confinement in a cast or appliance should not exclude children from family (or unit) activities. They can be held astride a lap for comfort and transported to areas of activity. The child may be allowed to walk in a cast or brace. An adapted wheelchair or stroller can offer mobility to the older infant or child. (See Chapter 39 for further discussion of care of a child in a spica cast.)
Congenital Clubfoot
Congenital clubfoot is a complex deformity of the ankle and foot that includes forefoot adduction, midfoot supination, hindfoot varus, and ankle equinus. Also referred to as talipes equinovarus (TEV), congenital clubfoot involves bone deformity and malposition with soft tissue contracture (Fig. 11-13). This condition requires early evaluation and treatment for optimum correction. TEV is the most frequently occurring type of clubfoot (approximately 95%), although other variations may be seen and are generally described according to the position of the ankle and foot (Box 11-6). Unilateral clubfoot may occur as an isolated defect or in association with other disorders or syndromes, such as a chromosomal disorder, arthrogryposis (a generalized immobility of the joints), cerebral palsy, or SB.
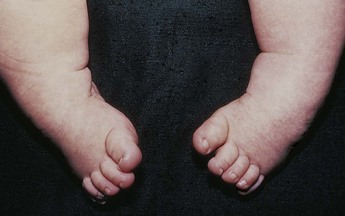
Fig. 11-13 Bilateral congenital talipes equinovarus (congenital clubfoot) in 2-month-old infant. (From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 5, St Louis, 2007, Mosby.)
The incidence of clubfoot in the general population is 1 to 2 per 1000 live births, with boys affected twice as often as girls. Bilateral clubfeet occur in 50% of the cases (Hosalkar, Spiegel, and Davidson, 2007). A positive family history is associated with increased incidence. Incidence varies with geographic location, with the lowest incidence in China and the highest in Polynesia.
Clubfoot may be further divided into three categories: (1) positional clubfoot (also called transitional, mild, or postural clubfoot), which is believed to occur primarily from intrauterine crowding and responds to simple stretching and casting; (2) syndromic (or teratologic) clubfoot, which is associated with other congenital abnormalities such as myelomeningocele (myelodysplasia) or arthrogryposis and is a more severe form of clubfoot that is often resistant to treatment; and (3) congenital clubfoot, also referred to as idiopathic or true clubfoot, which may occur in an otherwise normal child and has a wide range of rigidity and prognosis. The third category may be detected in utero by ultrasonography and is the most common type of TEV seen.
The mild, or positional, clubfoot may correct spontaneously or may require passive exercise or serial casting. The infant has no bony abnormality, but may have tightness and shortening of the soft tissues medially and posteriorly. The teratologic clubfoot usually requires surgical correction and has a high incidence of recurrence. The congenital clubfoot almost always requires surgical intervention because of the bony abnormality.
Pathophysiology
The exact cause of clubfoot remains unknown. However, there is a strong familial tendency, with a 1 in 10 chance that a parent with clubfoot will have an affected offspring. Other possible theories as to the cause of clubfoot include arrested fetal developmental of skeletal and soft tissue during gestational weeks 9 to 10, when foot development occurs; abnormal neuromuscular dysfunction or muscle abnormalities; and possibly a defect in the primary germ plasma, resulting in ankle dysplasia. Cases of clubfoot have been observed as a possible result of distal limb amniotic banding, a condition in which the amnion forms constrictive bands around a limb in utero, cutting off the circulation to the limb and resulting in further abnormal or arrested development.
Diagnostic Evaluation
Clubfoot is readily apparent at birth if it has not been detected antenatally. Once it is detected, a careful yet comprehensive physical assessment of the affected foot (or feet) should be carried for appropriate decisions to be made regarding treatment and discussion of possible treatment plans and prognosis with the parents. The affected foot (or feet) is usually smaller and shorter, with an empty heel pad and a transverse plantar crease. When the defect is unilateral, the affected limb is usually shorter and some calf atrophy may be present. Anteroposterior and lateral (maximum dorsiflexion) radiographs are useful in determining the type and severity of clubfoot. Ultrasonography may also be used. The nurse should perform a thorough hip examination on all infants with a clubfoot. An increased risk of hip dysplasia is associated with clubfoot deformities.
Therapeutic Management
The goal of treatment for clubfoot is to achieve a painless, plantigrade, and stable foot. Once the diagnosis is established, treatment is ideally initiated in the newborn period and involves three stages: (1) correction of the deformity, (2) maintenance of the correction until normal muscle balance is regained, and (3) follow-up observation to avert possible recurrence of the deformity.
Serial casting begins immediately or shortly after birth. Successive casts allow for gradual stretching of skin and tight structures on the medial side of the foot (Fig. 11-14). Manipulation and casting are repeated frequently (every few days for 1 to 2 weeks, then at 1- to 2-week intervals) to accommodate the rapid growth of early infancy. The extremity or extremities are casted until maximum correction is achieved, usually within 8 to 12 weeks. A Denis Browne splint may be used to manage feet that correct with casting and manipulation. A radiograph or ultrasound is then evaluated to see the relationship of the bones to each other. Failure to achieve normal alignment by 3 months indicates the need for surgical intervention, which may take place between 6 and 12 months of age. The foot (or feet) is immobilized postoperatively for approximately 6 to 12 weeks, and the child is allowed to walk after the cast is removed.
Fig. 11-14 Feet casted for correction of bilateral congenital talipes equinovarus. (From Brashear HR, Raney RB: Handbook of orthopaedic surgery, ed 10, St Louis, 1986, Mosby.)
Surgical intervention for clubfoot involves pin fixation and the release of tight joints and tendons. Casting of the affected foot and leg is performed, and after 2 or 3 months a varus-prevention brace is used to maintain correction. With severe deformities, repeated surgical tendon or joint releases may be necessary.
In recent years several nonsurgical approaches to clubfoot have been reintroduced. These methods involve daily or weekly manipulation and stretching of tissues with either casting (Ponseti manipulation) or taping and splinting of the affected extremity (French physical therapy). A percutaneous Achilles tendon lengthening may be performed before the final casting at 6 to 7 weeks to correct the equinus deformity. With the French physical therapy treatment, a continuous passive motion machine may be used several hours daily to stretch and strengthen involved muscle groups (Faulks and Luther, 2005).
Prognosis: Some feet respond to treatment readily; some respond only to prolonged, vigorous, and sustained efforts; and the improvement in others remains disappointing even with maximum effort. Parents should realize that outcomes are not always predictable and depend on the severity of the deformity; age of the child at initial intervention; compliance with treatment protocols; and development of bones, muscles, and nerves. Surgical intervention may not restore the ankle to an entirely normal state, with the affected foot and leg remaining smaller and thinner than the unaffected side. Ankle range of motion after surgery may be even less than it was preoperatively. Many children with surgically corrected clubfoot, however, are able to walk without a limp and run and play.
Nursing Care Management
Nursing care of the child with nonsurgical correction of clubfoot is the same as for any child who has a cast or corrective splint. (See Chapter 39.) The child spends considerable time in a corrective device, so nursing care plans include both long- and short-term goals. Careful observation of skin and circulation is particularly important in young infants because of their normally rapid growth rate.
Since treatment and follow-up care are handled in the orthopedist’s office, clinic, or outpatient department, parent education and support are important in nursing care of these children. Parents need to understand the diagnosis, the overall treatment program, the importance of regular cast changes, and the role they play in the long-term effectiveness of the therapy. Nursing responsibilities include reinforcing and clarifying the orthopedic surgeon’s explanations and instructions, providing emotional support, teaching parents about care of the cast or appliance (including vigilant observation for potential problems; see discussion of cast care on p. 423 and in Chapter 39), and encouraging parents to facilitate normal development within the limitations imposed by the deformity or therapy.
Metatarsus Adductus and Metatarsus Varus
Metatarsus adductus, sometimes also referred to as metatarsus varus when the forefoot is adducted and supinated (Hosalkar, Spiegel, and Davidson, 2007), is probably the most common congenital foot deformity. In most instances it is a result of abnormal intrauterine positioning and is usually detected at birth. It can occur bilaterally in 50% of newborns, and 10% of affected children will also have DDH. The deformity is characterized by medial adduction of the toes and forefoot, frequently associated with inversion, and convexity of the lateral border of the (kidney-shaped) foot. Metatarsus adductus can be divided into three categories: type I, in which the forefoot is flexible and corrects easily with manipulation; type II, in which the forefoot is only partially flexible and corrects passively past neutral position but only to neutral position with active manipulation; and type III, in which the forefoot is rigid and will not stretch to neutral position with manipulation. Unlike TEV, with which it is often confused, the angulation occurs at the tarsometatarsal joint, while the heel and ankle remain in a neutral position. Ankle range of motion is normal. This deformity often causes a pigeon-toed gait.
Management depends on the rigidity and type of the deformity. Correction can usually be accomplished by gentle manipulation and passive stretching of the foot with types I and II, which the parent is taught to perform. Repeated and consistent stretching is continued for the first 6 weeks, after which the treatment is based on the flexibility of the foot. With type III, the child usually requires serial manipulation and casting to correct the defect. Casting is performed every 1 to 2 weeks for 6 to 8 weeks after which a corrective shoe or orthosis may be used. Surgical correction is rarely required for the condition unless there is residual deformity at 4 years of age, at which time soft tissue release and serial casting are performed. Older children with deformity may require more extensive surgery (Hosalkar, Spiegel, and Davidson, 2007).
Nursing Care Management
The nursing role primarily involves identifying the defect so that early therapy and instruction of the parents can begin. The nurse teaches the parents how to hold the heel firmly and to stretch the forefoot. If casting is needed, the nurse instructs the parents in cast care and observation. (See Chapter 39.)
Skeletal Limb Deficiency
Congenital limb deficiencies, or disruption defects, are manifested by destruction of a previously normal body part. Others may involve the loss of functional capacity of varying degrees. They are characterized by underdevelopment of skeletal elements of the extremities. The range of malformation can extend from minor defects of the digits to serious abnormalities such as amelia (absence of an entire extremity) or meromelia (partial absence of an extremity), which includes phocomelia (seal limbs), an interposed deficiency of long bones with relatively good development of hands and feet attached at or near the shoulder or the hips. Most disruption defects are primary defects of development of the limb, but prenatal destruction of the limb can occur, such as full or partial amputation of a limb in utero from constriction of an amniotic band (amniotic band syndrome).
Pathophysiology
Limb deficiencies are caused by both heredity and environment, and they can originate at any stage of limb development. Formation of limbs may be suppressed at the time of limb bud formation, or there may be interference in later stages of differentiation and growth. Heredity appears to play a prominent role, and prenatal environmental insults have been implicated in a number of cases, such as the well-publicized thalidomide tragedy in the late 1950s and early 1960s, which demonstrated a clear relationship between the time of exposure of the pregnant woman to the antiemetic drug and the presence and type of limb deformity in the newborn.
Therapeutic Management
An expert in dysmorphology, usually a genetic specialist, should evaluate children with congenital limb deficiencies. An extensive prenatal history and a pedigree are obtained. Extensive laboratory evaluation may be required.
The child with a limb deficiency is fitted with prosthetic devices whenever possible, and a functional replacement should be applied at the earliest possible stage of development in an attempt to match the infant’s motor readiness. This favors natural progression of prosthetic use. For example, an infant with an upper extremity deficiency is fitted with a simple passive device such as a mitten prosthesis between 3 and 6 months of age, when limb exploration is active, sitting is beginning (with the extremities needed for support), and bilateral hand activities are encouraged. Lower limb prostheses are applied when the infant is ready to pull to a standing position.
In preparation for prosthetic devices, surgical modification is often necessary to ensure the most favorable use of the device, since severe deformity can interfere with its effective use. Phocomelic digits are preserved for controlling switches of externally powered appliances in the upper extremities. Digits (in both the upper and lower extremities) provide the child with surfaces for tactile exploration and stimulation. Prostheses are replaced to accommodate the child’s growth and increasing capabilities.
Nursing Care Management
Prosthetic application, training and habilitation are most successfully carried out in a center that specializes in meeting the special needs of these children, especially very young children and those with multiple amputations. It involves a prosthetist, who specializes in the development, fitting, and maintenance of prosthetic limbs, and other health care workers such as physical and occupational therapists. Specialized limb deficiency clinics are most helpful to parents and provide an introduction to support groups for both parents and affected children. Parents must encourage the child in making age-commensurate adjustments to the environment. Although these children need assistance, overprotection may produce overdependency with later maladjustment to school and other situations.
Parents of children with limb deficiencies should always be referred for genetic counseling.