Hereditary Influences on Health Promotion of the Child and Family
Heredity and Environment in Human Disease
Variable Patterns of Gene Expression and Inheritance
Disorders of the Intrauterine Environment

http://evolve.elsevier.com/wong/ncic

Abnormal Sexual Development, Ch. 11
Birth of a Child with a Physical Defect, Ch. 11
Cleft Lip and Cleft Palate, Ch. 11
Communicating with Families, Ch. 6
Cranial Deformities, Ch. 11
Cystic Fibrosis, Ch. 32
Defects in Physical Development, Ch. 11
Down Syndrome, Ch. 24
Family-Centered Care of the Child with Chronic Illness or Disability, Ch. 22
Fetal Alcohol Syndrome, Ch. 11
Fragile X Syndrome, Ch. 24
Guidelines for Communication and Interviewing, Ch. 6
Hypertrophic Pyloric Stenosis, Ch. 33
Malformations of the Central Nervous System, Ch. 11
Multiple Births, Ch. 3
Muscular Dystrophies, Ch. 40
Phenylketonuria, Ch. 9
Retinoblastoma, Ch. 36
Skeletal Defects, Ch. 11
Genetic Influences on Health
The twentieth century was a time of intense work and discovery in the field of medical genetics (McKusick, 2002), or the study of human hereditary disease. The modern era of medical genetics began with the discovery of inborn errors of metabolism, launched by the work of Archibald Garrod when, in 1902, he discovered alkaptonuria. This would be the precursor of modern biochemical genetics. In the second half of the twentieth century, pioneers Beadle and Tatum laid the foundation of molecular genetics when they discovered the pathway of genetic information from deoxyribonucleic acid (DNA) to ribonucleic acid (RNA) to protein. In 1956, Ingram discovered the molecular defect responsible for sickle cell disease. From that point forward, research in medical genetics focused on identifying the protein (enzyme) products of specific genes and relating them to disease processes. Cytogenetics, the study of chromosome disorders, started in 1952 with Lejeune’s discovery of the genetic basis of Down syndrome. In recent times, molecular-based knowledge and technologies have been greatly accelerated by the Human Genome Project (HGP), which is rapidly identifying genes and DNA variations associated with disease.
The HGP, an international collaborative research program that spanned 13 years, accomplished its end objective in 2003 when researchers uncovered the sequence of the gene-rich areas of the genome. The genetic information obtained through the HGP represented a major step toward understanding human genes and opened new horizons in every aspect of medicine and biology with a major impact on health promotion, disease prevention, and treatment (Collins, Green, Guttmacher, et al, 2003).
Much effort has focused on identifying gene variations. As a human species, we are 99.9% alike; however, it may be the 0.1% variation that will help us understand the genetic risk for illnesses. Researchers hope to devise treatment strategies that are specific to an individual’s genetic predisposition or molecular mechanism of disease. Another outcome of the HGP is the study of how inheritance affects the body’s response to medications, a field called pharmacogenetics or pharmacogenomics. The clinical application of this allows for individualized medication selection and dosing to improve efficacy and safety. In addition, drugs can be developed that target specific molecular and cellular disease mechanisms.
The information garnered from the HGP has affected areas beyond the realm of science, as it began to affect human life and ethical decision making. It is not surprising that part of the total budget for the project was allotted to the Ethical, Legal, and Social Implications (ELSI) program to deal with the new genetic information. Research by the ELSI program has focused on issues such as genetic discrimination, privacy, education, informed consent, and DNA banking. It will continue to look at new issues generated by increasing knowledge of the human genome.
Nurses and other health care providers are increasingly faced with incorporating genetic-genomic information into their practice. In response to this need, the Consensus Panel on Genetic/Genomic Nursing Competencies was established in 2006. This independent panel of nurse leaders from clinical, research, and academic settings established essential minimal competencies necessary for nurses to deliver competent genetic- and genomic-focused nursing care. The American Association of Colleges of Nursing overwhelmingly endorsed the revised baccalaureate essentials document, which identified genetics and genomics as strong forces influencing the role of nurses in patient care. This chapter provides foundational information to help nurses begin using genetics and genomics information and technology when caring for children and families (Box 5-1).
Heredity and Environment in Human Disease
Before the discoveries of the HGP, the total number of human genes had been estimated at approximately 50,000 to 100,000. It is now known that the human genome consists of 20,000 to 25,000 genes, a surprisingly low number for a sophisticated species (International Human Genome Sequencing Consortium, 2004).
Genes are segments of DNA that contain genetic information necessary to control a certain physiologic function or characteristic. These segments are often referred to as sites, or loci, indicating a physical or “geographic” location on a chromosome. Genetic disorders may result from gene mutations (single-gene, polygenic, or mitochondrial disorders) or chromosome abnormalities. Genes that encode proteins are termed structural genes. Mutations in structural genes may have significant qualitative and quantitative effects on the synthesis of the corresponding protein, with potential clinical consequences. Proteins can be classified as structural (or constitutive) proteins, and those that affect the metabolism of other molecules or substrates (enzymes). Table 5-1 summarizes the effects of protein disorders on selected genetic diseases. Such alterations in an individual genome may have been inherited from a parent or may represent an event that is new to that person and may be the first case in that family (new mutation). It is therefore erroneous to consider all genetic disorders as having a positive family history.

In earlier times, human diseases were thought to be either clearly genetic or typically environmental. However, the observation that some genetic disorders are congenital (present at birth) whereas others are expressed later in life has led scientists to conclude that many, if not most, diseases are caused by a genetic predisposition that can be activated by an environmental trigger. The concept of complex (or multifactorial) diseases emerged from this thought. Examples of such interactions are found in single-gene disorders, such as phenylketonuria (PKU) and sickle cell disease, and multifactorial conditions, such as cancer and neural tube defects (NTDs). PKU is a disorder resulting from the (genetically determined) absence of an enzyme that metabolizes the amino acid phenylalanine. However, the deleterious effects in the infant are expressed only after sufficient ingestion of phenylalanine-containing substances, such as milk (environmental trigger). Even in the case of a “classic” genetic condition, such as sickle cell disease, its acute symptoms are precipitated by certain conditions such as lowered oxygen tension, infection, or dehydration.
Cancer is another example of genetic-environment interplay and explains the difference between inherited conditions and somatic cell genetic disorders. A normal somatic cell (any body cell other than the ova and sperm) may become a cancer cell after acquiring a series of gene changes. This process is the typical “genetic” cause of cancer. In a small subset of families, a mutation in a gene normally involved in regulation of cell growth, DNA repair, or cell death (apoptosis) is transmitted through the germ cells (ova and sperm). Children who inherit the genetic mutation will have it in all of their somatic cells, making them more susceptible to subsequent genetic changes in one or more cells that may transform into cancer cells. An increasing number of tests are available to identify at-risk family members who have inherited a cancer-susceptibility genetic mutation. However, this type of testing in children is controversial. Beyond the genetic component of cancer, there is little dispute that environmental insult, such as tobacco smoking, sun exposure, and radiation, can be carcinogenic. Such environmental triggers are capable of spontaneously creating noninherited mutations in genes that regulate cell growth and cell response to cell abnormalities that can eventually lead to malignant transformation.
Evidence is growing that genes play an important role in human susceptibility and resistance to infection even in cases with a clear environmental cause of the infectious disease. Evidence for this genetic element in resistance gained heightened recognition during the first decade of the acquired immunodeficiency (AIDS) epidemic. Researchers discovered that adults with a specific deletion in both copies of their CCR5 genes did not become infected with human immunodeficiency virus (HIV) despite repeated exposure. Later it was found that children exposed in utero to HIV typically had a significantly delayed onset of disease if at least one of their CCR5 genes had the specific mutation (Romiti, Colognesi, Cancrini, et al, 2000). Understanding the mechanism of resistance associated with CCR5 mutation led to a novel molecular therapy (Wilkin, Su, Kuritzkes, et al, 2007).
Congenital Anomalies
Embryogenesis and fetal development are an intricate and precisely timed series of events in which all parts must be properly integrated to ensure a coordinated whole. Insults during development or abnormalities in differentiation or in the proper timing of organogenesis may result in a variety of congenital anomalies. Congenital anomalies, or birth defects, occur in 2% to 4% of all live-born children and are often classified as deformations, disruptions, dysplasias, or malformations. Deformations are often caused by extrinsic mechanical forces on normally developing tissue. Club foot is an example of a deformation often caused by uterine constraint. Disruptions result from the breakdown of previously normal tissue. Congenital amputations caused by amniotic bands (fibrous strands of amnion that wrap around different body parts during development) are examples of disruption anomalies (Zieve, Juhn, and Eltz, 2009). Dysplasias result from abnormal organization of cells into a particular tissue type. Congenital abnormalities of the teeth, hair, nails, or sweat glands may be manifestations of one of the more than 100 different ectodermal dysplasia syndromes (National Foundation for Ectodermal Dysplasia, 2009). Malformations are abnormal formations of organs or body parts resulting from an abnormal developmental process. Most malformations occur before 12 weeks of gestation. Cleft lip, an example of a malformation, occurs at approximately 5 weeks of gestation when the developing embryo naturally has two clefts in the area. Normally between 5 and 7 weeks, cells rapidly divide and migrate to fill in those clefts. If there is an abnormality in this developmental process, the embryo is left with either a unilateral or bilateral cleft lip that may also involve the palate.
The types of anomalies that can result from genetic or prenatal environmental causes can be major structural abnormalities with serious medical, surgical, or quality-of-life consequences, or they can be minor anomalies or normal variants with no serious consequences, such as a sacral dimple, an extra nipple, or a café-au-lait spot. Congenital anomalies can occur in isolation, such as congenital heart defect, or multiple anomalies may be present. A recognized pattern of anomalies resulting from a single specific cause is called a syndrome (e.g., Down syndrome or fetal alcohol syndrome). A nonrandom pattern of malformations for which a cause has not been determined is called an association (e.g., VACTERL [vertebral defects, anal atresia, cardiac defect, tracheoesophageal fistula, and renal and limb defects] association). When a single anomaly leads to a cascade of additional anomalies, the pattern of defects is referred to as a sequence. Pierre Robin sequence begins with the abnormal development of the mandible, resulting in abnormal placement of the tongue during development. The normal developmental process for the palate is prevented because the tongue obstructs the migration of the palatal shelves toward the midline and a cleft palate remains. Consequently, infants born with Pierre Robin sequence have a recessed mandible and an abnormally placed tongue and are at risk for obstructive apnea. NTDs, cleft lip and palate, deafness, congenital heart defects, and cognitive impairment are examples of congenital malformations that can occur in isolation or as part of a syndrome, association, or sequence and can have different causes, such as single-gene or chromosome abnormalities, prenatal exposures, or multifactorial causes.
Genetic Disorders
Genetic disorders can be caused by chromosome abnormalities as seen in Turner syndrome, Down syndrome, or velo-cardio-facial syndrome (VCFS); single-gene mutations as seen in sickle cell anemia, neurofibromatosis, or Duchenne muscular dystrophy; a combination of genetic and environmental factors as seen in NTDs or maturity-onset diabetes in the young; and mitochondrial DNA (mtDNA) mutations as seen in nonsyndromic deafness susceptibility due to aminoglycoside sensitivity. Whereas numeric or structural chromosome aberrations automatically involve large groups of genes, a small gene mutation does not alter chromosome structure and number. Alterations in single genes (single-gene disorders) or in many genes (polygenic disorders) may represent too small a lesion to cause an identifiable alteration in chromosomal structure. Human nucleated somatic cells contain approximately 25,000 genes distributed, in the form of tightly coiled DNA molecules, along 46 chromosomes. Since human chromosomes vary in size, the larger the chromosome, the greater the number of genes carried.
Both numeric and structural abnormalities of autosomes (all chromosomes except the X and Y chromosomes) account for a variety of syndromes usually characterized by cognitive deficiencies. A few are associated with a group of characteristics that clearly indicate the precise chromosome anomaly. Nurses often note dysmorphic facial features, behavioral characteristics such as an unusual cry and poor feeding behavior, and other neurologic manifestations such as hypotonia or abnormal reflex responses, which may alert them to these and other chromosome abnormalities.
Numeric Chromosome Abnormalities
With the exception of brief periods of gametogenesis, human beings are diploid individuals, and human somatic cells are diploid (a cell that contains two copies of each chromosome). A diploid chromosome number in humans is represented by the notation 2n = 46. A haploid chromosome constitution (n = 23) is found in germ cells, the male and female gametes (sperm and ova). Somatic cells contain 44 autosomes (the 22 pairs of chromosomes that do not greatly influence sex determination at conception) and two sex chromosomes, XX in females and XY in males. For the purpose of cytogenetic studies, chromosomes are usually displayed in a karyotype, the laboratory-made arrangement of specially prepared chromosomes according to their size and centromere position. The location of the centromere allows the classification of human chromosomes as acrocentric, submetacentric, and metacentric chromosomes (Fig. 5-1).
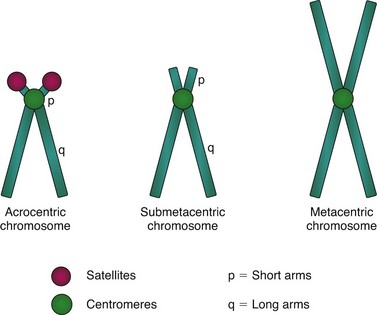
Fig. 5-1 Chromosome classification by centromere position. Positioning of centromeres results in chromosomes with extremely short arms (acrocentric chromosomes), relatively short arms (submetacentric chromosomes), or arms of equal length (metacentric chromosomes).
Numeric chromosome abnormalities occur whenever entire chromosomes are added or deleted. The addition of one or more chromosomes to each pair (increments of the haploid number, 23) will result in triploid cells with 69 chromosomes (46 + 23), or tetraploid cells with 92 chromosomes (46 + 23 + 23), and so on. The product of this uniform addition of chromosomes to all the original pairs is termed euploidy, a euploid cell being one whose chromosome number is a multiple of 23.
Individuals who are triploid (3n = 69) have a genetic imbalance of such magnitude that the few who are carried to term have severe multiple abnormalities that limit their life span to a few hours or days. On the other hand, one chromosome may be added to or lost from one of the pairs, creating a condition of aneuploidy. When one chromosome is added to a pair, the embryo, fetus, or child is described as having a trisomy, and the total chromosome number is 47. Most fetuses that have an autosomal trisomy are not live born. Fetuses with trisomy 21, trisomy 18, and trisomy 13 may be live born. When one chromosome is lost from the pair, the fetus is described as having a monosomy, and the total chromosome number in somatic cells is 45. The loss of a chromosome and its related complement of genes is overall more detrimental than the addition of a chromosome. The only monosomy compatible with life is monosomy X (Turner syndrome); yet most 45,X pregnancies spontaneously miscarry (Sybert and McCauley, 2004).
The most common cause of alteration in the number of chromosomes is a misdistribution of chromosomes during mitosis or meiosis. As somatic cells multiply by mitosis, each daughter cell receives the same chromosome number as the mother cell. This equitable chromosome sharing in anaphase is due to a phenomenon termed disjunction, by which chromatids of each chromosome separate and migrate to opposite poles of the cell. Disruption of this orderly chromosome distribution occurs in nondisjunction, which can occur during both mitosis and meiosis. In mitosis, failure of chromatids to separate properly during anaphase will result in daughter cells with different chromosome numbers (e.g., 45 and 47, instead of 46 and 46). Of those, the 45-chromosome monosomic cells will tend to degenerate and die, but those with the extra chromosome (trisomic) will continue to divide and generate a complete line of trisomic cells.
When mitotic nondisjunction occurs during embryonic development (Fig. 5-2), the trisomic cell line proliferates concomitantly with the normal cell line, and an individual with mosaicism for that particular chromosome result. Mosaicism, therefore, results in an individual (mosaic) with two or more genetically different cell populations. The chromosomal notation for a male with mosaic type of Down syndrome, for example, is 46,XY/47,XY,+21). The slash (/) indicates a dual cell population in which one has the normal chromosomal constitution (46,XY), while the other carries an extra chromosome 21 and has a total chromosome number of 47. The percentage, or level, of mosaicism depends on the stage of embryonic development in which the cell division error occurs. If it occurs at the first cell division after fertilization, the level of mosaicism may be as high as 50%. If the cell division error occurs in later development, the abnormal cells may be localized to one cell type, such as the brain tissue or germ cell line (ovaries or testes). The extent of clinical manifestations is determined by the type of tissues that contain cells with abnormal chromosome numbers and the percentage of affected cells, and may vary from near normal to a fully manifested syndrome.
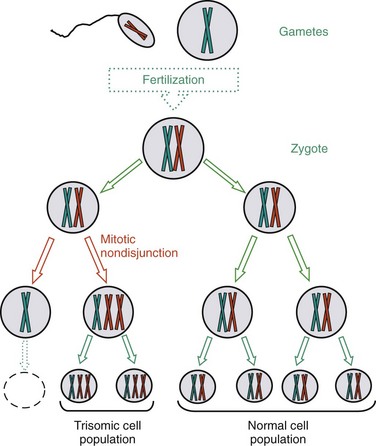
Fig. 5-2 Mitotic nondisjunction resulting in individual with different cell populations (mosaicism). This event occurs during embryonic development, after normal zygote was formed by fertilization of two normal gametes. Only one chromosome pair is represented. As represented here, mitotic nondisjunction and uneven chromosome distribution result in some cell populations with the extra chromosome, whereas other cell lines have the normal chromosome complement.
Meiotic nondisjunction (Fig. 5-3) is a major cause of aneuploidy, an abnormal chromosome pattern in which the total number of chromosomes is not a multiple of the haploid number, 23. Nondisjunction can occur during meiosis I and II, during both oogenesis and spermatogenesis, resulting in gametes with aneuploid chromosome number (e.g., 22 or 24, instead of 23). As in the case of somatic cells, gametes lacking a chromosome are not likely to survive, but gametes with an extra chromosome are more often viable. Fertilization of an aneuploid gamete with a normal gamete will produce an aneuploid zygote. The most common aneuploidies in humans are trisomies.
Pathophysiology Review
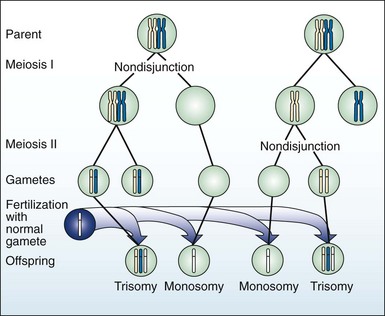
Autosome Aneuploidies
Examples of numeric alterations affecting the autosomes include some of the most common trisomies found in humans: trisomy 21 (Down syndrome), trisomy 18 (Edwards syndrome), and trisomy 13 (Patau syndrome) (Table 5-2).
TABLE 5-2
PARTIAL LIST OF CHROMOSOMAL GENETIC DISORDERS

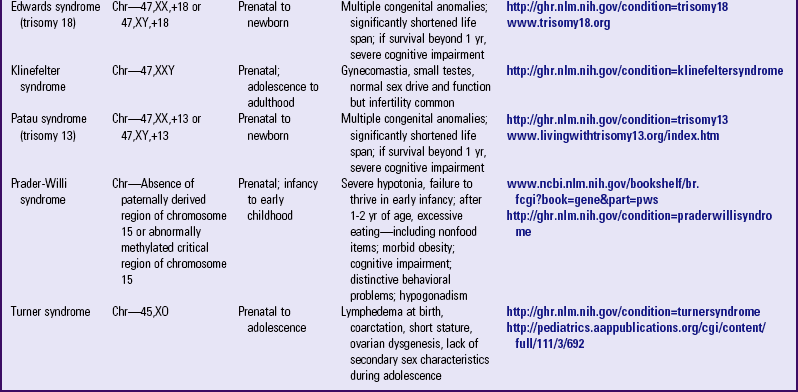
AD, Autosomal dominant; Chr, chromosomal.
*Support groups are listed on most websites. However, before referring families to support groups, particularly for families that discovered the diagnosis prenatally, carefully review the support group and describe its focus to the couple so they can make an informed decision about whether to visit it.
Trisomy 21: Down syndrome affects 1 in 800 to 1 in 1000 live births and is the most common aneuploidy compatible with life expectancy into adulthood. Physical and cognitive abnormalities vary. Intelligence quotient (IQ) range is typically mild to moderate impairment. In spite of modern medical developments, life expectancy is still shortened, with 20% dying in the first decade, and 50% by age 60 years. Adults with Down syndrome are also more likely to develop Alzheimer disease; more than 75% of those over age 60 are affected with the disease (Rimoin, Connor, Pyeritz, et al, 2002).
The chromosomal constitution of Down syndrome is variable, with three possible configurations (Lashley, 2005):
1. Trisomy—The nomenclature for a female with trisomy is 47,XX+21 and for a male with trisomy is 47,XY,+21. Trisomy encompasses 92% of all cases of Down syndrome. The extra chromosome 21 is unattached and segregates freely during meiosis. The risk for this type of Down syndrome increases linearly with increasing maternal age (from 1 : 1500 live births for mothers age 20 years, to 1 : 50 live births for mothers over age 45) (Jones, 2006); however, because young women have more babies, about 75% of babies with trisomy 21 are born to younger mothers.
2. Translocation Down syndrome—The accepted nomenclature for a male with Down syndrome due to robertsonian translocation between acrocentric chromosomes 14 and 21 is 46,XY,t(14;21). Translocation (discussed later) accounts for approximately 4% of all male and female Down syndrome cases. The majority of cases are sporadic (without family history), but about 25% have one balanced translocation carrier parent. When one such carrier and a partner with normal chromosomes reproduce, their theoretical chances of producing a live-born child with Down syndrome are 33%, but the actual observed risk is approximately 15% if the mother is the carrier and less than 10% if the father is the carrier. The observed chance of producing a live-born child who is a balanced translocation carrier approaches 50%. Because chromosome 21 is an acrocentric chromosome, it is possible for the translocation to be with both chromosome 21s. A carrier mother [45,XX,t(21;21)] or father [45,XY,t(21;21)] of the translocation would have a 100% chance of producing a child with Down syndrome, since the other parent would normally contribute one chromosome 21. This latter situation is one of the rare examples in genetics where an abnormality is passed on to all living progeny.
3. Mosaic Down syndrome—The nomenclature for a female with mosaic Down syndrome is 46,XX/47,XX+21. This rarer type of Down syndrome can occur in males and females. It results from mitotic nondisjunction during early embryonic development of a normal zygote. Children with this type have mixed cell populations, some with the normal karyotype, others with the extra chromosome. Contrary to what one might expect, children with mosaic Down syndrome do not necessarily have a better developmental outcome than those with free trisomy type. The proportion of trisomic cells in various tissues and organs play a role in the child’s developmental potential and syndrome-associated potential health problems.
Sex Chromosome Aneuploidies
Alterations in number may also involve the sex chromosomes (see Table 5-2). The possible mechanisms by which sex chromosome abnormalities may occur are the same as those previously described (i.e., prefertilization nondisjunction during one of the meiotic divisions of gametogenesis in either parent or in the early postfertilization divisions of the zygote). An alteration in the number of sex chromosomes usually does not produce the profound effects that are associated with the autosomal trisomies. Intelligence may be normal or low normal, or the child may have some learning disabilities, but moderate or severe cognitive impairment is less common. Some of the most common genetic disorders caused by sex chromosome aneuploidies are Klinefelter, XYY, triple-X female, and Turner syndromes.
47,XXY: Klinefelter syndrome is the most common of all sex chromosome aneuploidies. Physical abnormalities include elements of decreased masculinization, such as gynecomastia; hypogonadism (with sterility resulting from degeneration of seminiferous tubules); and increased pubis-to-sole length, reflecting elongated lower limbs (Fig. 5-4). Mental development is normal in most cases, with a mean full-scale IQ between 85 and 90. Cognitive difficulties tend to be in expressive language, auditory processing, and auditory memory. Chromosome mosaicism (46,XY/47,XXY) rarely occurs and results in individuals with milder manifestations than their trisomic counterparts. Overall, the phenotype of Klinefelter syndrome is highly variable, making it difficult, in the absence of chromosome studies, to make a prepubertal clinical diagnosis.
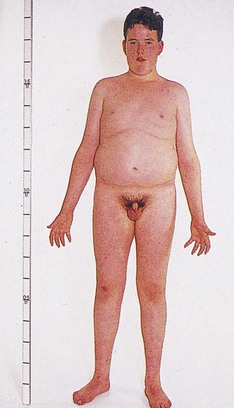
Fig. 5-4 Klinefelter syndrome. This young man exhibits many characteristics of Klinefelter syndrome: small testes, some development of the breasts, sparse body hair, and long limbs. This syndrome results from the presence of two or more X chromosomes with one Y chromosome (genotypes XXY or XXXY, for example). (From Patton KT, Thibodeau GA: Anatomy and physiology, ed 7, St Louis, 2010, Mosby.)
47,XYY: This genotype was reported in the early 1960s by Patricia Jacobs, a Scottish cytogeneticist who detected an increased frequency of double-Y men among inmates of penal institutions in Great Britain. In early reports an extra Y chromosome was reported as being responsible for an individual’s increased tendency toward aggression against property (as opposed to aggression against humans). A detailed statistical analysis by Digamber Borgaonkar (Johns Hopkins University) of more than 200 cases later revealed that the only correlates with an extra Y chromosome were tall stature (>6 feet) and skin disorders, such as persistent adulthood acne. That large study found no significant correlations between XYY and cognitive impairment and aggressive tendencies, and the syndrome remains today a scientific curiosity. A majority of children produced by XYY fathers have normal chromosomal constitution, probably reflecting a selective advantage of normal haploid gametes over aneuploid ones.
45,XO:  Turner syndrome, originally described clinically as ovarian dysgenesis (with gonads consisting of streaks of connective tissue and devoid of germ cells), is an example of a monosomy that is compatible with life. Clinical manifestations are variable in expression. Intellectually, verbal IQ exceeds performance IQ. There is no prepubertal growth spurt, and girls with Turner syndrome are generally infertile. It is common practice to administer female hormones around the time that puberty would occur to provide the girl with Turner syndrome some secondary sex characteristics; however, the female hormones may further stunt growth and must be used judiciously. The child’s growth is usually normal until 3 years of age and then slows, gradually drifting away from the normal growth curve. Treatment for the decreased growth velocity includes growth hormone and anabolic steroids. Mosaicism also occurs in Turner syndrome (e.g., 46,XX/45,XO), resulting in milder expression of the phenotype. Girls with Turner syndrome may have difficulty with peer relationships and with understanding social cues. They may exhibit behavioral problems, especially immature, socially isolated behavior. Most, however, lead productive lives and function as independent adults.
Turner syndrome, originally described clinically as ovarian dysgenesis (with gonads consisting of streaks of connective tissue and devoid of germ cells), is an example of a monosomy that is compatible with life. Clinical manifestations are variable in expression. Intellectually, verbal IQ exceeds performance IQ. There is no prepubertal growth spurt, and girls with Turner syndrome are generally infertile. It is common practice to administer female hormones around the time that puberty would occur to provide the girl with Turner syndrome some secondary sex characteristics; however, the female hormones may further stunt growth and must be used judiciously. The child’s growth is usually normal until 3 years of age and then slows, gradually drifting away from the normal growth curve. Treatment for the decreased growth velocity includes growth hormone and anabolic steroids. Mosaicism also occurs in Turner syndrome (e.g., 46,XX/45,XO), resulting in milder expression of the phenotype. Girls with Turner syndrome may have difficulty with peer relationships and with understanding social cues. They may exhibit behavioral problems, especially immature, socially isolated behavior. Most, however, lead productive lives and function as independent adults.
 Critical Thinking Exercise—Turner Syndrome
Critical Thinking Exercise—Turner Syndrome47,XXX: A relatively common condition (1 : 1000 live female births), females with triple-X display a normal phenotype, with an increased risk of learning disabilities when compared with their euploid sisters. Gynecologic complications include delayed menarche and premature menopause. As with XXY men, the offspring of XXX women is largely normal, indicative of a selective advantage of euploid gametes.
Structural Chromosome Abnormalities
Chromosomes are subject to structural alterations resulting from breakage and rearrangement. Chromosome breakage has long been recognized as a significant source of genetic abnormalities. Many clastogens (chromosome-breaking agents) have been identified, including physical (e.g., ionizing radiation), chemical (e.g., chlorpromazine), and biologic (e.g., viral infections) agents. Chromosome breakage can also result from many nonspecific causes, such as influenza. These breaks are usually restricted to somatic cells and are temporary. Chromosome breakage becomes significant when it is permanent (or long lasting) and when these permanent changes, in addition to appearing in somatic cells, are also present in germ cells and thus have the potential of being transmitted to the offspring.
A chromosome deletion occurs when chromosome breakage results in loss of the broken fragment at a chromosome’s terminal end or within the chromosome. Chromosome deletions often have significant clinical impact, as in a chromosome 5 terminal deletion that results in cri du chat syndrome. Chromosome breakage can create unstable end points (“sticky ends”), which predispose the chromosomes to a variety of rearrangements of the fragments. A relatively rare structural abnormality that can occur as a result of chromosomal “sticky ends” is a ring chromosome. If a break occurs in the terminal end of both arms of a chromosome, the ends may fuse together, forming a circle. Like any structural alteration of a chromosome, the clinical manifestations depend on which genes are lost.
A more common rearrangement resulting from chromosome breakage is a translocation, which occurs when a chromosomal fragment reunites with another, nonhomologous chromosome. Two types of translocations have clinical significance: reciprocal translocations and robertsonian translocations. In a reciprocal translocation, breaks occur in two different chromosomes and the fragments are mutually exchanged, resulting in derivative chromosomes. Robertsonian translocations occur when the short arms of two acrocentric chromosomes (pairs 13 to 15 and pairs 21 and 22) break off and the remaining long arms fuse at the centromere, forming a “single chromosome” (Fig. 5-5). Both types result in individuals who have the correct amount of genetic information (although “rearranged”), and therefore no clinical manifestations are expected. These persons are termed balanced translocation carriers. These asymptomatic, balanced translocation carriers (either male or female) may pass the translocation to their offspring in a balanced or unbalanced form, depending on how the chromosomes segregate to the gametes. If it is passed in the unbalanced form, the combination is often lethal, and an early spontaneous abortion occurs. The chance of having a live-born child with birth defects associated with the unbalanced translocation depends on the quantity and role of the missing or additional genetic material. Approximately 5% of cases of repeated spontaneous abortion (two or more) can be attributed to a balanced translocation carrier parent.
Pathophysiology Review
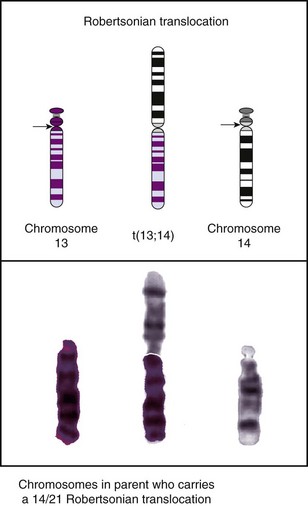
Fig. 5-5 Translocation. In a robertsonian translocation the long arms of two acrocentric chromosomes (13 and 14) fuse, forming a single chromosome. (From Jorde LB, Carey JC, Bamshad MJ, et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
Some structural chromosome abnormalities are too small to reliably visualize under a light microscope but are still clinically relevant. Fragile, or weak, sites associated with expanded triplet repeats (described later in the chapter) have been identified on both the autosomes and the X chromosome. A classic example is fragile X syndrome. Contiguous gene syndromes are disorders characterized by a microdeletion or microduplication of smaller chromosome segments, which may require special analysis techniques or molecular testing to detect (Brown, 2003). Microdeletion syndromes, such as VCFS, are more common than microduplication syndromes.
46,XX,del(5p) or 46,XY,del(5p): Cri du chat, or cat’s cry, syndrome is a rare (1 : 50,000 live births) (Jorde, Carey, Bamshad, et al, 2003) chromosome deletion syndrome resulting from loss of the small arm of chromosome 5. In early infancy this syndrome manifests with a typical but nondistinctive facial appearance, often a “moon-shaped” face with wide-spaced eyes (hypertelorism) (Fig. 5-6). As the child grows, this feature is progressively diluted, and by age 2 years the child is indistinguishable from age-matched controls. Profound cognitive impairment persists throughout their short life; many die in infancy. Typical of this disease is a crying pattern that is abnormal and catlike. At times it sounds like an angry cat, at others like a soft mewing sound. This is a result of a laryngeal atrophy that improves with age. By age 3 years the crying pattern is still abnormal, but it acquires a normal pitch and loses its catlike quality.

Fragile X Syndrome: Fragile X syndrome acquired its name from the fact that, in special cell culture conditions, the affected X chromosome may display a gap in its terminal portion. However, it is important to recognize that this is an X-linked condition caused by an expanded triplet repeat (described later in the chapter) with increased prevalence among males (approximately 1 : 4000 males and 1 : 6000 females). Clinical features include cognitive impairment and a typical facial appearance, with an elongated face and large ears (Gardner and Sutherland, 2004).
Velo-Cardio-Facial Syndrome: VCFS, sometimes called DiGeorge syndrome, is the most common (1 : 2000) microdeletion syndrome. It is caused by a specific microdeletion within the long arm of chromosome 22 (22q11.2 deletion) (Shprintzen, 2008). Manifestations of this condition are variable, with approximately 180 different possible clinical features described. Although no one feature is found in every patient, cognitive impairment is common and can range from full-scale measured IQ in the borderline low-normal range with characteristic learning disabilities to mild cognitive impairment (Antshel, Fremont, Kates, et al, 2008). Although most patients’ deletion is caused by a sporadic event, those with the condition can transmit the microdeletion in an autosomal dominant manner. Therefore their chances of producing a child with VCFS are 50% with each pregnancy.
Chromosome Instability Syndromes: Chromosome instability syndromes are a heterogeneous group of genetic disorders characterized by a high frequency of chromosome breakage observed in vitro. They include ataxia telangiectasia, Fanconi anemia, and xeroderma pigmentosum. These syndromes are associated with decreased immune function and an increased incidence of cancer.
Single-Gene Disorders
Chromosome anomalies typically affect large numbers of genes; however, a single-gene disorder is caused by an abnormality within a gene or in a gene’s regulatory region. Single-gene disorders display a mendelian pattern of dominant or recessive inheritance that was first delineated in the mid-nineteenth century by Gregor Mendel’s experiments with plants. Single-gene disorders can affect all body systems and may have mild to severe expressions. Since single-gene disease involves very short DNA segments within a chromosome, they cannot be detected by chromosome analysis and demand specific and sophisticated molecular detection methods, such as DNA-based techniques (Table 5-3).
TABLE 5-3
SELECT TECHNIQUES OF DNA ANALYSIS AND DISEASE DETECTION
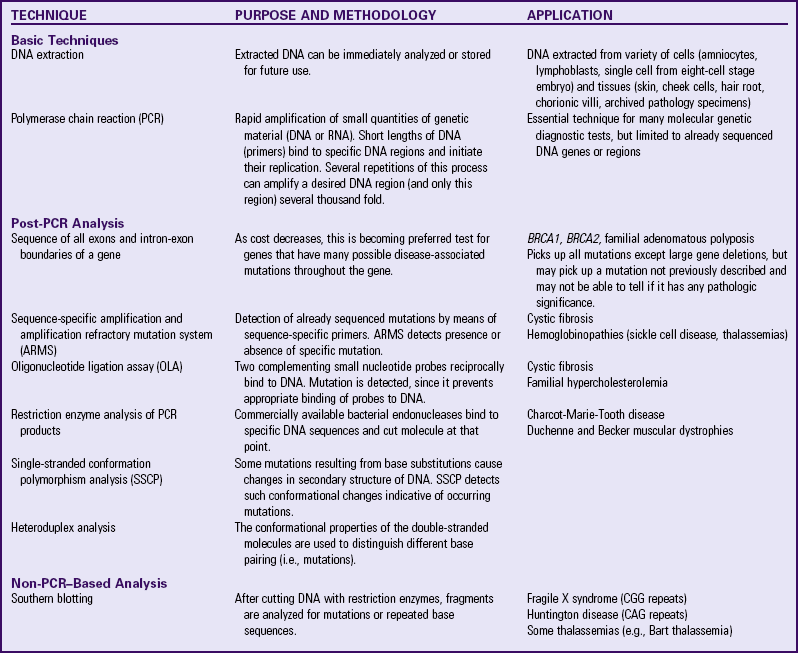
CAG, Cytosine-adenine-guanine; CGG, cytosine-guanine-guanine; DNA, deoxyribonucleic acid; RNA, ribonucleic acid.
Mendelian inheritance laws allow for risk prediction in single-gene disorders; however, phenotypic expression may be altered by incomplete penetrance or variable expressivity of the responsible allele. An allele is said to have reduced or incomplete penetrance in a population when a proportion of persons who possess that allele do not express the phenotype. An allele is said to have variable expressivity when individuals possessing that allele display the features of the syndrome in various degrees, from mild to severe. If a person expresses even the mildest possible phenotype, the allele is penetrant in that individual.
Autosomal Inheritance Patterns
Autosomal Dominant Inheritance: General characteristics of autosomal dominant inheritance are presented in Box 5-2. A clear understanding of transmission of autosomal inheritance patterns requires the understanding of a few basic facts. First, most genetic diseases are rare. The probability that two affected persons will mate is very low for most genetic disorders (with the exception of societal selection, as in the case of achondroplasia). Second, depending on the disease, if one parent is affected, he or she is much more likely to be heterozygous (have one mutant allele) than homozygous (have two mutant alleles). Usually, an individual with two dominant mutant alleles will experience physical or mental abnormalities at a much more severe level.
Those assumptions being accepted, two questions remain: (1) what are the chances of transmitting the mutant allele to the offspring? and (2) what are the risks of the offspring being affected? If a gene has only two alleles, one normal and one mutant, then three possible allele combinations exist: normal/normal, normal/mutant, and mutant/mutant. In autosomal dominant conditions, only persons with normal/normal combination will be disease free, assuming that the mutant allele is 100% penetrant. Fig. 5-7 shows the relationships between genotypes and phenotypes for an autosomal dominant trait. Considering that genetic diseases are rare and that persons who are homozygous for the mutated allele are more severely affected, it is most likely that an affected individual who is heterozygous for the mutant allele will mate with a genotypically normal partner (Fig. 5-8, A). The outcomes of these matings are best expressed by the use of Punnett squares. Also depicted (Fig. 5-8, B) is the mating of a homozygote for the mutated gene with a genotypically normal partner. The result of these matings is one of the few instances in medical genetics in which all progeny have a 100% chance of being affected, assuming that the mutant allele they receive is 100% penetrant. These matings, however, are extremely rare.
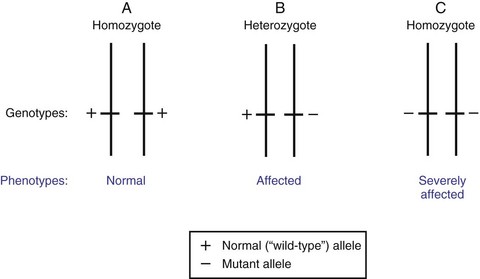
Fig. 5-7 Dominant inheritance pattern. Schematic representation of the three possible allelic arrangements of a gene with two alleles. Depicted here are genotypes and possible phenotypes for a trait transmitted by a dominant gene. The presence of a single copy of the mutant allele in heterozygous person (B) is sufficient to express the phenotype in question. Double dose of the mutated allele, in homozygous person (C), results in more severe expression of the phenotype.
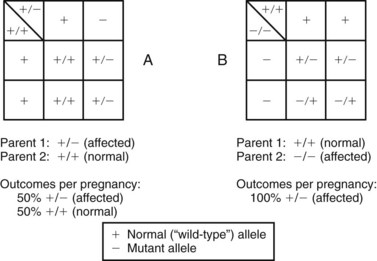
Fig. 5-8 Determination of mating outcomes in autosomal dominant inheritance obtained with Punnett squares. A, Possible outcomes of the mating of an affected heterozygous individual (+/−) with a normal partner (+/+). B, Mating of an affected homozygous individual (−/−) and a normal partner (+/+).
Many children diagnosed with an autosomal dominant disorder have a positive family history of the disease. In other instances, that child may represent the first occurrence of that condition in the family. In the latter case, the event may be due to a new mutation (fresh mutation) in that child or to the presence of the mutation only in the germ cells of a healthy parent. The birth of other affected children indicates the second possibility. The range of expression of autosomal dominant genes is highly variable, from minor manifestations (e.g., polydactyly), to severe, debilitating, and life-threatening disease (e.g., neurofibromatosis). Depending on the degree of disability the condition imposes on the individual and the ability to procreate, the mutated gene either will be eliminated or will continue to be passed on through several generations. In addition, in diseases that have a late age of onset (e.g., Huntington disease), a person with a disease-associated mutation may be healthy and asymptomatic during childbearing years and be unaware of the risk of passing on the mutant allele to offspring. Consequently, the mutant allele continues to be passed on through several generations.
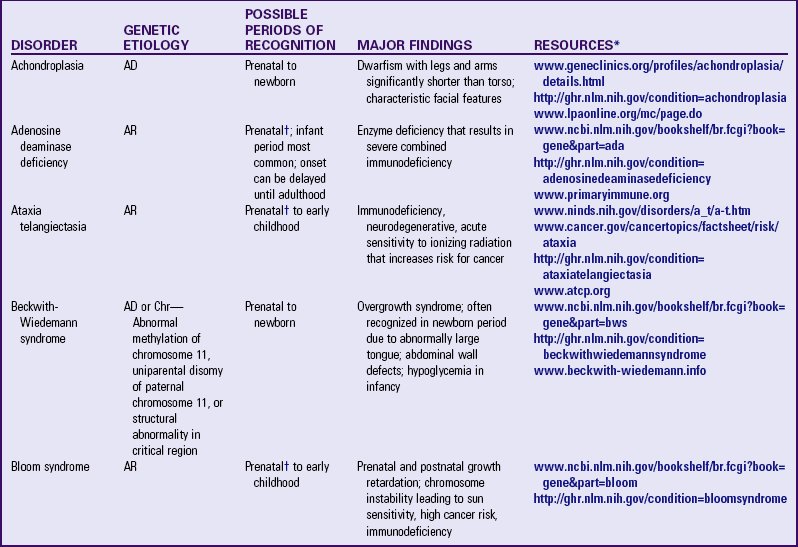
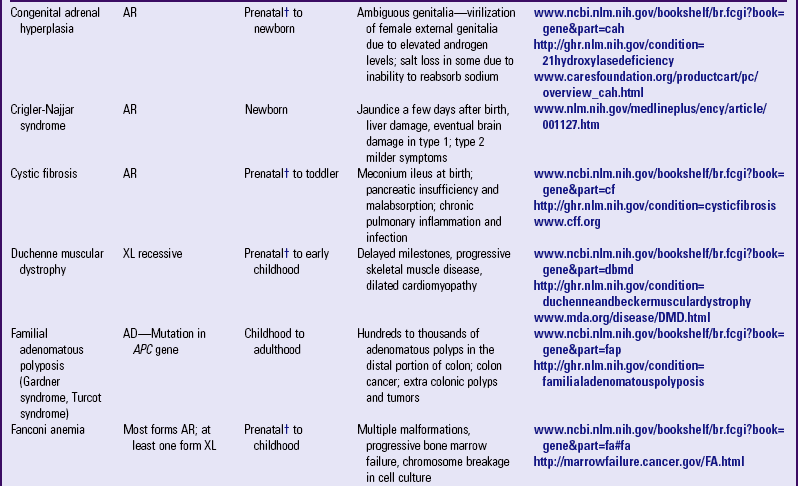
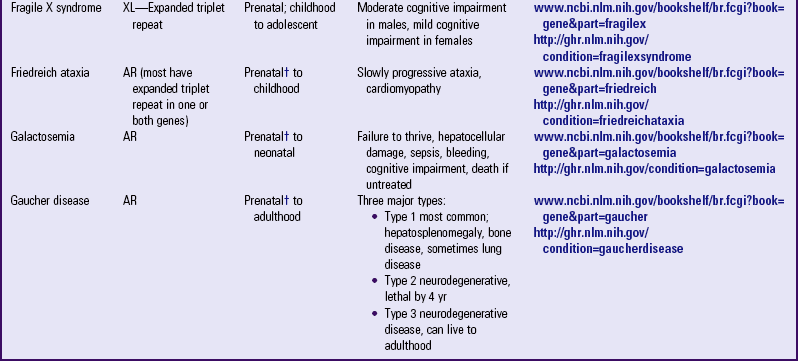
Other examples of autosomal dominant disorders include achondroplasia, neurofibromatosis, and Marfan syndrome. An idealized pedigree for autosomal dominant inheritance is found in Fig. 5-9 and discussed in the Nursing Care Guidelines box (p. 111). The gene mutation associated with achondroplasia is considered 100% penetrant. The pedigree in the figure demonstrates that although an affected parent has a 50% chance with each pregnancy of transmitting the gene mutation associated with achondroplasia, 50% of offspring do not necessarily inherit the gene mutation. Selected examples of autosomal dominant disorders are found in Table 5-4.
TABLE 5-4
PARTIAL LIST OF MENDELIAN INHERITED GENETIC DISORDERS
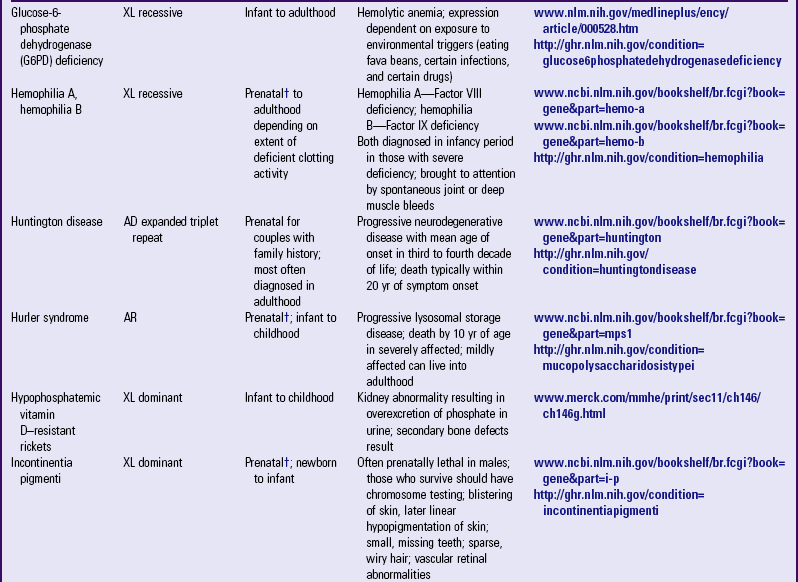
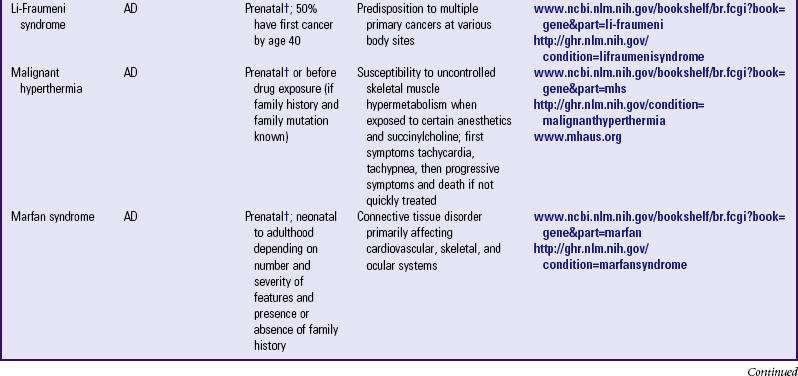
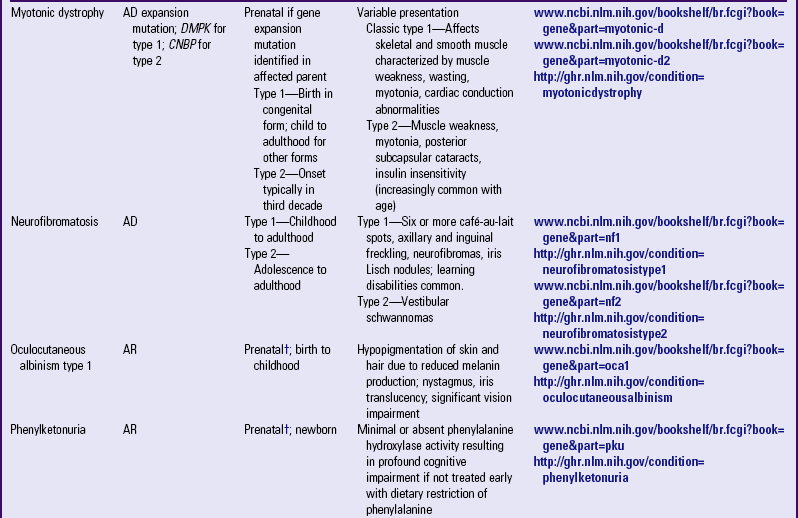
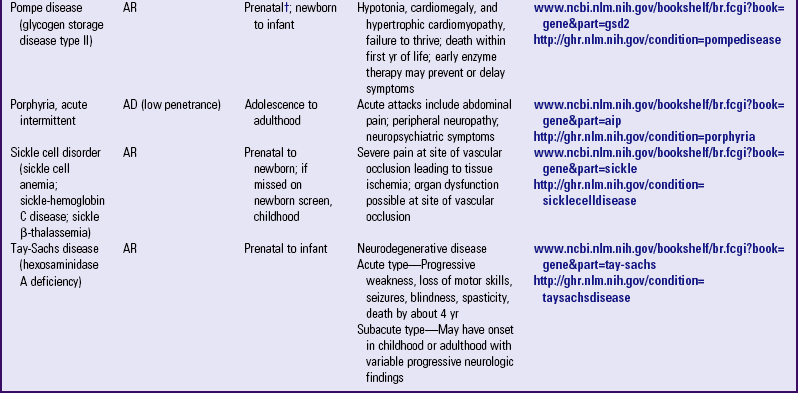
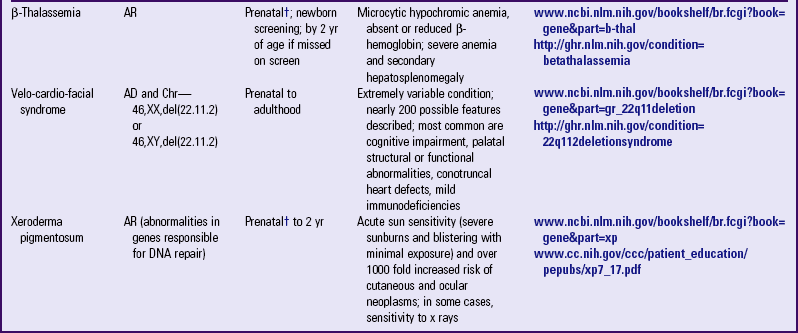
AD, Autosomal dominant; AR, autosomal recessive; Chr, chromosomal; DNA, deoxyribonucleic acid; XL, X-linked.
*Support groups are listed on most websites. However, before referring families to support groups, particularly for families that discovered the diagnosis prenatally, carefully review the support group and describe its focus to the couple so they can make an informed decision about whether to visit it.
†Mutation(s) in affected family member or in carrier parents need to be known before prenatal testing can be informative.
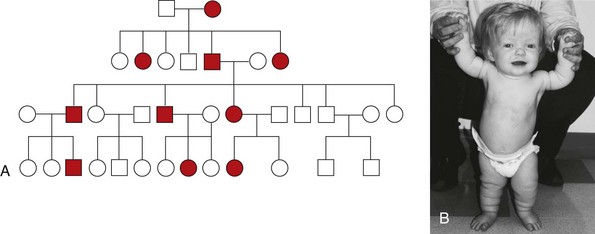
Fig. 5-9 Pedigree for achondroplasia. A, Pedigree showing the transmission of an autosomal dominant disease. B, Achondroplasia. This girl has short limbs relative to trunk length. She also has a prominent forehead, low nasal root, and redundant skin folds in the arms and legs. (B, From Jorde LB, Carey JC, Bamshad MJ, et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
Autosomal Recessive Inheritance: General characteristics of autosomal recessive inheritance are presented in Box 5-3. Children who display an autosomal recessive disorder are always homozygous for that trait (both the maternally and paternally inherited alleles contain disease-associated mutations). This is due to the fact that a recessive allele is one whose phenotypic expression occurs only when both genes have disease-associated mutations. Although this makes the alleles homozygous because both alleles are recessive, the disease-associated mutation may be different in each allele. When this is the case, the pair of alleles is more accurately referred to as compound heterozygous for the recessive trait. In the heterozygote, a recessive allele is “masked” by the wild type (normal) allele, which is dominant (Fig. 5-10). Whereas the possible pregnancy outcomes in autosomal dominant pattern (see Fig. 5-7) are children who are either affected (if the gene mutation is fully penetrant) or unaffected when completely free of the gene mutation, in autosomal recessive inheritance a third possibility arises, that of a heterozygous carrier (see Fig. 5-10, B). These are individuals who are clinically normal (or nearly normal), but who are at risk of having offspring who are affected.
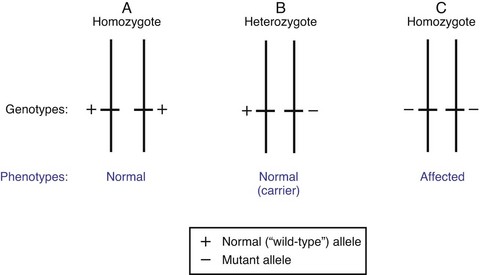
Fig. 5-10 Recessive inheritance pattern. Schematic representation of the three possible allelic arrangements of a gene with two alleles. Depicted here are genotypes and possible phenotypes for a trait transmitted by a recessive gene. The presence of a single copy of the mutant allele in the heterozygous person (B) results in a phenotypically normal individual who carries the mutant allele. The affected phenotype is only expressed in presence of a double dose of the mutated allele (C).
Identification of such carriers is of paramount importance for genetic counseling. In the case of an unaffected couple who produces a child with a recessive disease, identification is easy; since they each must contribute a mutant allele, they must be considered obligate carriers, even in the absence of specific carrier testing for that gene. Specific tests to detect heterozygous carriers of a variety of genetic diseases are available, but it would be impractical and certainly not cost-effective to use all available tests to screen all prospective parents without specific risk factors. Genetic screening for carriers is usually limited to populations at risk, either because they belong to a high-risk group for a certain disorder (e.g., Ashkenazi Jews and Tay-Sachs disease) or because of positive family history. Carriers for some specific disorders can be identified by specific tests before conception, by means of routine screening, or after conception by means of prenatal diagnosis (e.g., Tay-Sachs disease, cystic fibrosis [CF], sickle cell disorder). It is estimated that each person carries from three to eight mutated genes for a severe recessive disease. For example, 1 in 25 persons in the United States and Northern European populations carries a recessive gene mutation for CF. In the African-American population, 1 in 10 persons is a carrier for the sickle cell gene mutation. For PKU, the carrier rate in the general population is 1 in 50.
However, since genetic diseases are rare, the probability of the mating of two persons who carry the same gene in the same allelic configuration is very small. The chances are increased if mating occurs among persons who select a mate because of geographic, ethnic, or religious restrictions or blood relationship (consanguinity). For example, 1 in 30 Ashkenazi Jews is a carrier of a gene mutation associated with Tay-Sachs disease, and 1 in 3600 live births would be affected without preconception screening. For comparison, the frequency of Tay-Sachs disease outside this population is 100 times smaller (i.e., 1 in 360,000 live births).
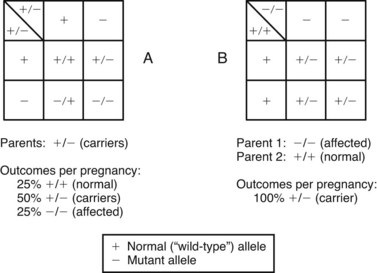
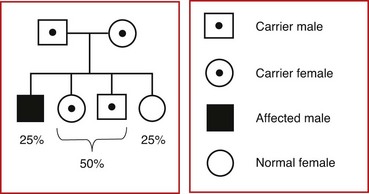
Other examples of autosomal recessive disorders include the thalassemias, congenital adrenal hyperplasia, and galactosemia. Fig. 5-11 illustrates two situations involving autosomal recessive traits. Fig. 5-11, A, depicts the most common occurrence in autosomal recessive disorders: the mating of two carrier parents with each pregnancy carrying a 25% chance of producing an affected child. Fig. 5-11, B, reflects the mating of an affected parent with a genotypically normal partner, in which 100% of the offspring will be carriers. An idealized pedigree representing the mating of two heterozygous (asymptomatic) carriers and its possible outcomes in each and every pregnancy is shown in Fig. 5-12. Selected examples of autosomal recessive disorders are found in Table 5-4.
Sex-Linked Inheritance Patterns
The transmission of genes located on one of the sex chromosomes (either X or Y) is termed sex-linked inheritance. However, few genes have been found on the Y chromosome, so frequently the terms sex-linked and X-linked are interchangeably (and incorrectly) used. The testicular organizing region of the Y chromosome determines the formation of testes and the development of male sexual structures during embryonic growth. A gene for hairy ears, with high prevalence in southern Asia, has also been located on the Y chromosome. Inheritance of Y-linked genes follows a father-to-son, or male-to-male, pattern. It is important to remember that men give their X chromosomes to their daughters (and their Y to their sons), so in analyzing a family pedigree, male-to-male transmission of a gene rules out X-linked inheritance.
Women have two homologous X chromosomes; therefore inheritance of genes located on the X chromosome (X-linked inheritance) follows the same pattern as that for autosomal genes. However, in the case of males, the X and Y chromosomes have small areas of homology and therefore do not pair side-by-side during meiosis. Because of this, men are hemizygous for all genes on the X chromosome that do not have a homologous site on the Y chromosome, and their alleles are represented as single copies. Therefore, in the inheritance of an X-linked gene, the single-copy presence of its normal, or its mutant, allele will result in the expression of the normal or mutant phenotype, respectively. This is true for both dominant and recessive X-linked diseases in males (Fig. 5-13).
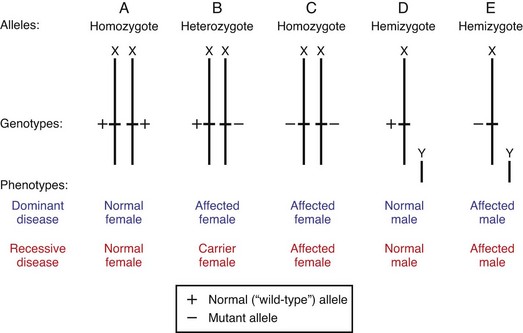
Fig. 5-13 X-linked inheritance pattern. A to C, Possible allelic arrangements of an X-linked gene in females. Note that phenotypic expression of an X-linked gene in women is typically similar to that of an autosomal gene. However, unequal X inactivation could result in more active X chromosomes with the mutation and could result in symptoms. D and E, Uneven pairing of X and Y chromosomes in males, and its phenotypic expressions. Note that there are no carrier males, since the phenotype is determined solely by the characteristic of X-linked allele. Hemizygous males are either normal or affected.
X-Linked Recessive Inheritance: In females the alleles of an X-linked recessive gene behave as the alleles of any autosomal recessive gene: the effect of the abnormal allele is “hidden” by the normal (dominant) allele. Therefore females who have a disease-associated mutation in both members of the gene pair will express the phenotype. Although it is rare for females to express the phenotype if only one member of the gene pair carries a disease-associated mutation, it is possible due to X inactivation.
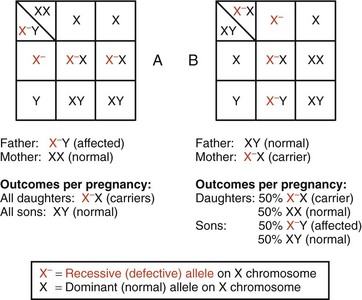
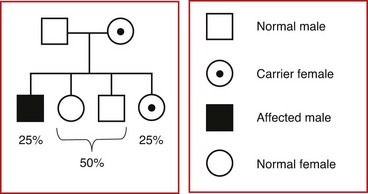
Soon after fertilization, it is normal for one X chromosome in females to be inactivated through a natural process called methylation. This occurs in each cell in the early blasotcyst stage, and whether the paternal or maternal X is inactivated is random in each cell. At the time this occurs, the X that has been inactivated will remain so through all subsequent cell divisions. Females who express a phenotype may do so due to the inactivation of a greater proportion of X chromosomes carrying the normal allele in the tissues or organs associated with the disorder. Examples of X-linked recessive disorders include hemophilia types A and B and Duchenne muscular dystrophy. Fig. 5-14 illustrates the outcomes of each pregnancy between an affected man and a normal woman (see Fig. 5-14, A), and between a normal man and a carrier woman (see Fig. 5-14, B). An idealized pedigree depicting the mating between a genetically normal man and a carrier woman, and its possible outcomes per pregnancy, is shown in Fig. 5-15. The primary characteristics of X-linked recessive inheritance are listed in Box 5-4.
X-Linked Dominant Inheritance: X-linked dominant inheritance (see Fig. 5-13) is rare. The main characteristics of X-linked dominant inheritance are (1) both males and females can be affected, but females, because of the random nature of X inactivation, are usually less severely affected than males; (2) affected men do not transmit the defective allele to their sons; and (3) all daughters of an affected man are affected and have a 50% chance of passing on the defective allele to their sons and daughters. Examples of X-linked dominant disorders are hypophosphatemic vitamin D–resistant rickets and incontinentia pigmenti. Fig. 5-16 illustrates the outcomes of each pregnancy between an affected man and an unaffected woman (see Fig. 5-16, A) and between an unaffected man and an affected woman (see Fig. 5-16, B). An idealized pedigree representing the mating of an affected man with a genetically normal woman is shown in Fig. 5-17.
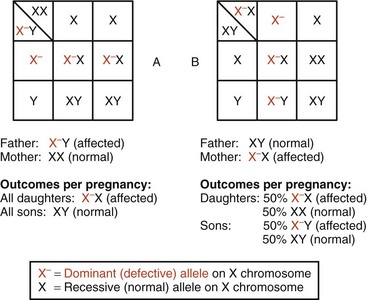
Fig. 5-16 X-linked dominant inheritance: Punnett squares. A, Possible outcomes of mating of affected male with normal female. B, Mating of normal male and affected female. Percentages shown refer to outcome possibilities in each pregnancy.

Fig. 5-17 X-linked dominant inheritance: typical pedigree. The idealized pedigree on the left represents possible outcomes of mating of affected male and normal female. Percentages depicted express risks for each pregnancy resulting from this mating. Since the mutant allele is carried in the X chromosome, an affected male will transfer it to 100% of his daughters.
Variable Patterns of Gene Expression and Inheritance
A number of variables have been observed that explain or modify basic inheritance patterns and the effects of chromosome abnormalities. Some of these variations have been recognized for some time; others are newly discovered phenomena that explain some apparent contradictions in the established patterns of inheritance. Also, some disorders have been reported to follow more than one inheritance pattern in different families (e.g., a classically recessive disorder may occasionally be reported to be following a dominant or X-linked pattern in other families). This phenomenon is known as locus heterogeneity (King, Rotter, and Motulsky, 2002).
The most notable of these gene variations is mutation. As discussed above, mutations are heritable changes in the DNA sequence of a gene. Mutations can result from a substitution of bases (point mutations) or the insertion or deletion of bases (Korf, 2000). Some genes have a high mutation rate, and various forms of the resulting disease may have varying expression. One example is the CFTR gene, in which more than 1000 different CF-associated mutations have been identified (the vast majority being point mutations or small deletions). Mutations can occur in somatic cells or in germ cells. Somatic cell mutations are passed on to the daughter cells of the mutated cell, but are not transmitted to the offspring. Germ cells mutations affect the gametes and are hereditary. Mutation rates are not constant for all genes, so some diseases may occur with much greater frequency than others.
Variable expression is an important concept that describes differences in the extent and severity of phenotype. There is a continuum of expression for any affected person from very mild to severe clinical manifestations. For those with very mild manifestations, it may take an expert clinician to identify the condition. For example, a parent of a child with classic neurofibromatosis may exhibit only a few “birth marks” that a medical geneticist, genetics advanced practice nurse, or genetic counselor would recognize as café-au-lait spots, one of the manifestations of neurofibromatosis.
The discovery of expansion mutations in some genetic disorders has helped explain their variation in inheritance patterns and clinical expression (Nelson, 1996). Within genes are sequences of DNA nucleotide repeats. A normal gene has a certain number of these repeats; for example, the FMR1 gene on the X chromosome usually has about 5 to 40 repeats. When FMR1 has 59 to 200 repeats, that area of the gene can become unstable and gain further repeats during meiosis. Females who have this number of repeats are considered carriers of a premutation for fragile X syndrome. A person with fragile X has a full mutation allele with hundreds to thousands of repeats. Because fragile X syndrome is an X-linked disorder, females are less likely to be affected because of the presence of a normal allele on the homologous X chromosome. These expanding repeats have also been found to occur in autosomal dominant disorders such as myotonic dystrophy and in autosomal recessive disorders such as Friedreich ataxia (Chamberlain, 1996). Expansion mutations that have a tendency to further expand when transmitted from one generation to the next can display phenotypic anticipation. Pedigrees display anticipation when individuals in successive generations develop the disorder at an earlier age and/or with more severe manifestations.
Genomic imprinting and uniparental disomy are two genetic phenomena that consider the parental origin of genetic information (i.e., maternally or paternally derived). The concept of genomic imprinting refers to modification, in some instances, of genetic material, resulting in phenotypic differences based on whether the genes and chromosomes were derived from the mother or the father. Genomic imprinting is exhibited during pregnancy, when paternally derived chromosomes seem to positively influence placental development and maternally derived chromosomes seem to positively influence fetal development. This phenomenon also occurs in some genetic disorders, such as Prader-Willi and Angelman syndromes. In both these disorders about two thirds of affected individuals have a deletion of the same segment of chromosome 15. However, the clinical manifestations of Prader-Willi and Angelman syndromes are markedly different. If the deletion occurs on the paternally derived chromosome 15, the child exhibits Prader-Willi syndrome; if the deletion occurs on the maternally derived chromosome 15, the child manifests Angelman syndrome (Jorde, Carey, Bamshad, et al, 2003). Prader-Willi syndrome is characterized by failure to thrive and central hypotonia in the newborn and infancy period with later insatiable hunger that can lead to morbid obesity during childhood. Children with Prader-Willi syndrome also have cognitive dysfunction, typical dysmorphic features, behavioral disturbances, hypothalamic hypogonadism, and short stature. In contrast, Angelman syndrome includes severe cognitive impairment, characteristic facies, abnormal (puppetlike) gait, and paroxysms of inappropriate laughter. Children with Angelman syndrome are usually nonverbal, although they may vocalize (Fridman, Varela, Kok, et al, 2000).
In some cases both copies of a chromosome pair are determined to have come from one parent, either the mother or the father, instead of one from each; this phenomenon is called uniparental disomy (Fridman, Varela, Kok, et al, 2000). An example of uniparental disomy was reported with CF, in which both chromosomes, each with a mutant recessive gene, came from the carrier mother; the father was not a carrier. In cases that appear to be nonpaternal, uniparental disomy may be a factor. One of several theories about uniparental disomy is that the chromosome pair was originally a trisomy and the father’s chromosome was randomly eliminated, leaving two copies of the mother’s chromosome. Because the chromosomes appear as a normal “pair,” diagnosis of this situation is only possible with molecular (DNA) techniques. Uniparental disomy has also been reported with Beckwith-Wiedemann syndrome, which is characterized by overgrowth and hypoglycemia at birth (see Table 5-2).
Mitochondrial Disorders
The nucleus is not the only site of genetic information. Extranuclear DNA is found in a cytoplasmic cellular organelle, the mitochondrion, whose primary function in cellular metabolism is the production of energy. Mutations in mtDNA also account for nonmendelian inheritance patterns. Inheritance of traits contained in mtDNA is exclusively maternal, since only the mitochondria from the ovum are transmitted to the zygote. Mitochondria are not contained within the sperm head.
An additional complexity in mitochondrial inheritance results from the fact that, during mitosis, mitochondria are randomly distributed among the daughter cells, so that both normal and mutated mitochondria may be found in the same cell, a phenomenon known as heteroplasmy (Korf, 2000). This leads to variable dosages of mutated mtDNA between tissues and organs. This variation in mutation load leads to a highly variable spectrum of clinical manifestations, and individuals with the same mtDNA mutation may range from symptom free, to mildly affected, to severely impaired. Symptoms may include seizures, pancreatitis, and metabolic disease. Different manifestations may be seen in different members of the same family. Heteroplasmy complicates the use of prenatal diagnosis. Once the mtDNA mutation is identified in the mother, her pregnancies can be tested for the same mutation. However, the mutation load identified in sampled fetal tissue (chorionic villi or amniocytes) may not correspond to other fetal tissues, the mutational load of which will continue to change during development due to random mitotic segregation of cytoplasmic organelles. Consequently, it is not possible to predict the unborn child’s phenotype based on prenatal test results.
mtDNA mutations are responsible for various childhood diseases (Table 5-5), such as Leigh syndrome (movement disorder, respiratory dyskinesia, regression, hypotonia, seizures, and failure to thrive). Because mitochondrial disorders have such variability of expression, determining the diagnosis can be confusing. However, when a child has an unexplained constellation of symptoms, a mitochondrial disorder should be considered. Examples of mitochondrial syndromes include Kearns-Sayre syndrome (external ophthalmoplegia, pigmentary retinopathy, heart block, ataxia, increased cerebrospinal fluid protein); myoclonic epilepsy with ragged red fibers, or MERRF (myoclonic epilepsy, myopathy, dementia); and MELAS (mitochondrial encephalomyopathy, lactic acidosis, strokelike episodes) (Korf, 2000). Nuclear DNA (nDNA) mutations can also cause disorders of the mitochondria, so not all “mitochondrial disorders” are caused by mutations in mtDNA.
TABLE 5-5
PARTIAL LIST OF MITOCHONDRIAL GENETIC DISORDERS
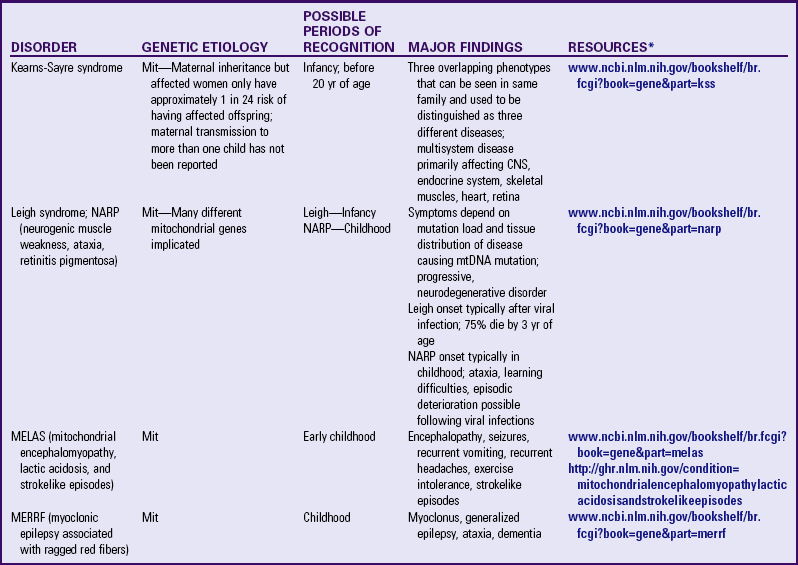
CNS, Central nervous system; Mit, mitochondrial; mtDNA, mitochondrial deoxyribonucleic acid.
*Support groups are listed on most websites. However, before referring families to support groups, particularly for families that discovered the diagnosis prenatally, carefully review the support group and describe its focus to the couple so they can make an informed decision about whether to visit it.
Hereditary Cancer Predisposition Genes
The process of carcinogenesis implies permanent changes in the DNA of the targeted cell. Early observations recognized genetic influences in cancer: (1) certain types of cancer occur more frequently within certain families (breast, colon, ovarian, some leukemias); (2) some well-defined genetic disorders show a predisposition to various malignancies (familial adenomatous polyposis and colon cancer, Bloom syndrome and lymphomas); (3) many chromosome abnormalities occur frequently with malignancies (Philadelphia chromosome in chronic myelogenous leukemia); and (4) certain chromosome aneuploidies predispose the person to cancer (Down syndrome and acute leukemias).
The discovery of oncogenes in the early 1970s marked the beginning of a new era in the field of cancer genetics (Box 5-5). Initially thought to be carried exclusively by retroviruses, oncogenes were later identified as natural genes that existed in all mammals. Early investigations suggested that oncogenes were found in inactive form (i.e., no correlation with cancer could be identified in this state). In reality, oncogenes are normally involved in cell growth and division. A mutation in an oncogene can disrupt this normal process and transform a normal cell into one that has uncontrolled cell growth or division, predisposing the cell to further mutations that eventually transform the cell into one that is malignant.
Since the discovery of oncogenes, two other classes of genes associated with cancer development have been identified: tumor suppressor genes and mismatch repair genes. Tumor suppressor genes normally inhibit cell growth and division. A mutation in these genes can interfere with this normal function and lead to uninhibited cell growth and division. Among this group is p53 (sometimes called TP53), whose germ cell (or hereditary) mutations are associated with Li-Fraumeni syndrome and whose somatic cell mutations (sporadic) result in various malignancies, such as bladder cancer. Li-Fraumeni syndrome is inherited as an autosomal dominant trait that predisposes children and young adults to the development of various tumors, including osteosarcoma; soft tissue sarcomas; breast, brain, and adrenocortical carcinomas; and leukemia.
Mismatch repair genes normally function by recognizing and repairing DNA errors that occur during replication or mutations that are caused by external agents such as ultraviolet light or chemical exposure. Xeroderma pigmentosum is a classic example of inherited predisposition to cancers caused by a germline mutation in one of several possible mismatch repair genes. Infants and children with xeroderma pigmentosum are at considerable risk for skin cancers triggered by sun and ultraviolet light exposure.
Multifactorial (Complex) Disorders
A number of frequently encountered diseases and defects show an increased incidence in some families but have no clear-cut affected-unaffected classification. Although the incidence is higher than would be expected by chance, no specific mode of inheritance can be identified. In some, environmental factors, including the prenatal environment, appear to play an important role. These conditions are classified as multifactorial disorders, in which a genetic susceptibility and specific environmental agents interact to produce a disease state. Multifactorial disorders include NTDs, cleft lip and cleft palate, many congenital heart defects, congenital hip dislocation, and pyloric stenosis.
Recurrence risks for multifactorial conditions are empirically based on observed recurrence within a population. In general, recurrence risk is usually low (<10%). For example, NTDs occur in 1 out of 1000 births in the general population. The recurrence risk after the first affected child is 2 or 3 out of 100 births (American Academy of Pediatrics, 1999). Advances in genetics are enhancing knowledge of multifactorial inheritance and familial risks (King, Rotter, and Motulsky, 2002).
Disorders of the Intrauterine Environment
The intrauterine environment can have a profound and permanent effect on the developing fetus, with or without chromosome or single-gene abnormalities. Intrauterine growth retardation, for example, can occur with many genetic syndromes, such as Down, Russell-Silver, Prader-Willi, and Turner syndromes (Rimoin, Connor, Pyeritz, et al, 2002), or it can be caused by nongenetic factors such as maternal alcohol ingestion. Placental abnormalities are increasingly being found to be the etiologic factor in neurodevelopmental disorders (such as cerebral palsy and cognitive impairment) that were previously attributed to asphyxia during delivery (Bos, Einspieler, and Prechtl, 2001).
Teratogens, agents that cause birth defects when present in the prenatal environment, account for the majority of adverse intrauterine effects not attributable to genetic factors. Types of teratogens include drugs (phenytoin [Dilantin], warfarin [Coumadin], isotretinoin [Accutane]), chemicals (ethyl alcohol, cocaine, lead), infectious agents (rubella, cytomegalovirus), physical agents (maternal ionizing radiation, hyperthermia), and metabolic agents (maternal PKU). Many of these teratogenic exposures and the resulting effects are completely preventable, such as ingestion of alcohol resulting in fetal alcohol syndrome or fetal alcohol effects, which causes severe birth defects, including cognitive impairment. The incidence of fetal alcohol syndrome is estimated at 5.2 per 10,000 live births (American Academy of Pediatrics, 2000). (See Chapter 11.)
Cytogenetic Diagnostic Techniques
Chromosomes are usually studied under light microscopy. After a significant number of cells are prepared so the chromosomes can be visualized during mitotic metaphase under a light microscope and imaged through a computer, their chromosomes are displayed in a karyotype, arranged according to their size, position of their centromeres, and banding patterns. A relatively recent advance called chromosome painting uses DNA probes attached to different colored fluorescent dyes to identify chromosomes by their color (Fig. 5-18). Robotic cell harvesters and computerized imaging systems have drastically cut down on manual labor; however, expert technicians and cytogeneticists are still necessary to analyze and interpret karyotypes whether they are traditionally stained or fluorescently colored. The most common human cells used for chromosomes studies are peripheral blood leukocytes obtained by venipuncture and cells obtained by biopsy from a variety of tissues, such as skin (including fetal skin cells), chorionic villi, and bone marrow.
Pathophysiology Review
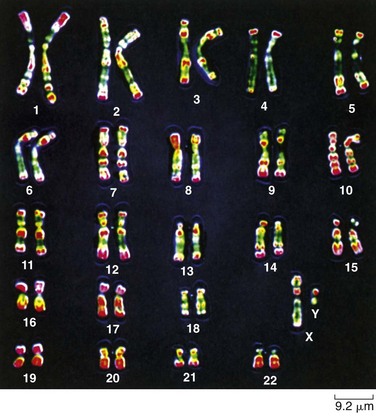
Fig. 5-18 Human karyotype. (From Raven PH, Johnson G, Singer S, et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
Numeric abnormalities such as trisomies and gross structural anomalies such as deletions, duplications, and translocations of larger segments of genetic material may be detectable by karyotype analysis. Large chromosome structural anomalies may be visualized with the help of basic staining techniques, and various types of banding procedures (e.g., Giemsa, or G, banding; centromeric, or C, banding; replication banding). Very small changes, such as microdeletions, microduplications, and fragile sites, usually require special molecular techniques such as fluorescent in situ hybridization (FISH) which uses fluorescent-labeled single-stranded DNA probes designed to attach to specific areas of chromosomes. For example, the FISH test used to diagnose VCFS contains probes that adhere to 22q11.2. Although persons with VCFS have two chromosome 22s, the FISH probe attaches to the specific region on only one of the chromosomes, since on the other the region of interest is deleted. A more recent test done in some cytogenetics laboratories, comparative genomic hybridization, uses molecular probes to detect up to thousands of submicroscopic imbalances throughout the genome (Edelmann and Hirschhorn, 2009).
Molecular Diagnostic Techniques
Information derived from the HGP has led to significant advances in the diagnosis of genetic disease. Identification of single-gene mutations responsible for some genetic disorders is now possible as increasing numbers of genes are being mapped (located on a specific chromosome or segment of a chromosome). In some conditions, such as CF, sickle cell disease, Huntington disease, and fragile X syndrome, the specific gene is known and tests are available to detect many of the specific disease-associated mutations. For some disorders, such as familial adenomatous polyposis, sequencing of the related gene may be necessary because mutations can be found anywhere in the gene and there are not any particular common mutations to target. Gene sequencing is more labor intensive and expensive than targeted DNA mutation testing. A regularly updated Internet source for available genetic tests can be found at GeneTests at the University of Washington, Seattle (www.genetests.org). At the time of this writing, GeneTests listed nearly 600 laboratories that provided clinical genetic testing for more than 1400 different genetic disorders.
Predisposition Genetic Testing
Molecular diagnostic techniques have enabled predisposition testing, which is the identification of gene mutations associated with genetic disorders in asymptomatic individuals. Depending on the penetrance of the allele(s), these may be considered presymptomatic or susceptibility tests.
Familial adenomatous polyposis is one of several overlapping conditions associated with mutation in the APC gene. These mutations are considered virtually 100% penetrant, resulting in hundreds to thousands of adenomatous polyps and eventual colon cancer. Once the APC mutation has been identified in an affected family member, at-risk family members can be tested for the same mutation. Because of the high penetrance, such testing in asymptomatic individuals would be considered presymptomatic testing. APC mutation testing in at-risk children is done to identify those who need colon screening and to identify polyp formation early so cancer-preventive surgical decisions can be made (Lynch, Lynch, Lynch, et al, 2008).
Hereditary breast and ovarian cancer has primarily been associated with mutations in either BRCA1 or BRCA2. Penetrance of mutations in these genes has been reported to be as high as 85% for breast cancer and lower for other associated cancers (ovarian, prostate, pancreatic, gastrointestinal, and melanoma). Once the BRCA1 or BRCA2 mutation has been identified in an affected family member, asymptomatic at-risk family members can be tested for the same mutation. Onset and type of cancer varies between members of a family identified with a mutation. In addition, although some members may develop one or more of the associated cancers, other mutation-carrying members may die in their eighties without ever developing cancer (Nusbaum and Isaacs, 2007; Fossland, Stroop, Schwartz, et al, 2009). Therefore testing for mutations in BRCA1 and BRCA2 in an asymptomatic family member is considered susceptibility testing. This type of testing is discouraged in at-risk children until they are cognitively and emotionally able to make an informed decision about testing for themselves (American Academy of Pediatrics, 2001).
Therapeutic Management of Genetic Disease
Therapy for genetic disease is currently aimed at correcting the phenotypic expression of gene abnormalities, and therefore the major goal of therapy is modification of the internal or external environment to correct or minimize the effects of the genetic defect. However, the HGP has opened doors to genotype intervention, and clinical applications of gene manipulation techniques are currently being tested.
Phenotype Modification
Examples of currently used intervention aimed at modifying the phenotypic expression of genetic disorders follow.
Surgical Management: Surgical repair of structural defects has made it possible to prolong life in a number of multifactorial disorders, such as congenital heart disease and NTDs. Numerous facial and limb deformities can be corrected by plastic and reconstructive techniques. In the case of familial adenomatous polyposis, the colon is surgically removed to prevent colon cancer that would eventually develop in one or more of the countless polyps that line the colon. The use of splenectomy in several hereditary disorders of blood cells prevents the trapping and destruction of abnormal blood cells in that organ. Early diagnosis and enucleation in retinoblastoma have reduced the mortality from this malignant eye tumor. Fetal surgery may also be performed for some life-threatening anomalies such a diaphragmatic hernia. Corrective surgery for children born with ambiguous genitalia (e.g., girls with congenital adrenal hyperplasia), although widely used, has been recently the focus of controversy that involves the issue of parental versus affected individual (future) preferences.
Diet Modification: For disorders in which an enzyme deficiency causes a toxic accumulation of a substance or its by-products, restricting the intake of foods containing that substance may prevent irreversible damage from the improper metabolism of these compounds. This dietary control is lifelong and requires a high level of adherence. Examples in infants and children include the low-phenylalanine diet prescribed for PKU, elimination of dairy products containing lactose for hereditary lactase deficiency, avoidance of foods containing or producing galactose for galactosemia, and a diet low in branched-chain amino acids for maple syrup urine disease. Women with PKU who have not maintained dietary control must reinstitute a strict low-phenylalanine diet before conception and maintain it throughout pregnancy to prevent a high risk of adverse fetal effects (see Chapter 9).
Preconception and prenatal folic acid supplementation has been shown to reduce the incidence or recurrence of NTDs (see Chapter 11). Therefore in 1992 the U.S. Public Health Service recommended that all women capable of becoming pregnant should consume 0.4 mg of folic acid per day. This recommendation was added to the existing mandate by the U.S. Food and Drug Administration that, as of 1998, folic acid would be added to enriched grain products. This would add 0.1 mg folic acid to the daily diet of the average person. Since that time, the incidence of NTDs has declined by 19% (Honein, Paulozzi, Mathews, et al, 2001). Women who have previously had a pregnancy affected by an NTD are advised to take 4.0 mg of folic acid per day beginning 1 month before conception and continuing throughout the first trimester of pregnancy. The risk of recurrence of an NTD in another pregnancy is reduced by 72% in women following this regimen.
Metabolic Manipulation: In some deficiency diseases, supplying the missing product that cannot be synthesized prevents undesirable effects. For example, thyroid hormone is prescribed to prevent the damaging effects of hypothyroidism, and providing the missing blood factors prevents life-threatening and debilitating hemorrhages in persons with different types of hemophilia. Other examples are insulin for diabetes mellitus, growth hormone for growth hormone deficiency, and corticosteroids for adrenogenital syndrome.
Removal of toxic substances that accumulate in vital tissues as a result of a hereditary disease can prevent disabling complications. Some of the deleterious effects of hemochromatosis, a hereditary disorder characterized by an excess accumulation of iron in the liver, heart, and pancreas, can be reduced with the removal of iron by administration of chelating agents or periodic therapeutic phlebotomies.
Diet supplements can be given when the individual is unable to synthesize or effectively utilize substances needed as cofactors in metabolism. This includes the use of vitamin B12 in persons with pernicious anemia, whose absorption of this vitamin is impaired. Vitamin and cofactor supplementation may be used to treat mitochondrial disease. Phenobarbitone can be used to enhance hepatic metabolism of unconjugated bilirubin in children with Crigler-Najjar syndrome type 2, thus helping prevent brain damage (kernicterus) and jaundice (Scriver, Beaudet, Sly, et al, 2001).
Avoidance of Drugs or Other Harmful Substances: In drug-induced disease, such as glucose-6-phosphate dehydrogenase (G6PD) deficiency and the porphyrias, avoidance of the drugs that precipitate a reaction is a simple preventive measure. These include aspirin and quinine-based antimalarial medications in the case of G6PD deficiency, and ethanol, barbiturates, anticonvulsants, sulfonamides, and oral contraceptives in the case of acute intermittent porphyria (Scriver, Beaudet, Sly, et al, 2001). Some anesthetic agents may precipitate symptoms of malignant hyperthermia and myotonic dystrophy.
Immunologic Prevention: The administration of immunoglobulin to Rh-negative mothers after the birth of an Rh-positive infant is effective in preventing Rh-antibody formation, which causes hemolytic disease of the newborn in subsequent births.
Transplantation: Better control of tissue incompatibility problems is improving the survival of children undergoing replacement of nonfunctioning organs with normal organs. Transplanted organs include the kidneys in hereditary polycystic kidneys, the heart in severe cardiac myopathy, the liver in hepatic atresia, and bone marrow.
Gene Product Replacement: Recombinant DNA technology makes it possible to produce large quantities of certain gene products. Such technology avoids the infectious contamination risk that accompanied extraction of gene products from mass quantities of human blood or tissue.
The administration of gene products is especially applicable when the missing product is a circulatory peptide or protein, as in coagulation factor VIII or IX in hemophilia type A and type B, respectively (Saenko and Pipe, 2006). If the gene product is membrane bound, as is the case for enzymes deficient in various lysosomal storage diseases, the natural enzyme product may need to be purposefully altered so that it can enter the cell through a naturally occurring cell receptor. Gaucher disease was the first lysosomal disorder to be successfully treated in this manner, and lessons learned in the process have led to more recent enzyme replacement therapies for disorders such as Pompe disease and Hurler syndrome (Bailey, 2008). If the missing gene product is a substance that acts within a metabolic pathway of a specific organ protected by a barrier, such as the enzyme hexosaminidase A in Tay-Sachs disease (blood-brain barrier), injectable products become ineffective and replacement is more problematic. The introduction of a foreign gene product may eventually trigger an immune reaction, as has been observed in hemophilia when the recipient develops antibody against the clotting factor, greatly reducing the efficiency of an otherwise successful therapy.
Gene Transfer: Gene transfer can be achieved through introduction of a normal gene by a harmless virus vector. For example, introduction of a normally functioning gene may correct an abnormal phenotype resulting from the absence of that gene. Introduction of a toxic gene may cause apoptosis of a cancer cell. Introduction of antigen or cytokine gene can stimulate the immune system to eliminate malignant cells. This approach has been attempted in humans with the transfer of normal gene copies of β-hemoglobin into bone marrow cells in an effort to treat a form of β-thalassemia; it has also been used in sickle cell disease and in adenosine deaminase deficiency. Gene therapy research has suffered a number of setbacks in clinical research, and the search for safer, more effective ways to transfer genes into human cells continues.
Environmental Modification
Inherited diseases or defects with no therapeutic modality can be modified to enhance the quality of life for the affected individual. Some examples of environmental manipulation include hearing aids for children with congenital hearing loss, glasses or vision enhancers such as books in enlarged print or Braille for the visually impaired, mobilizing devices such as braces and wheelchairs for persons with muscle and bone impairment, prosthetic devices for those with limb deficiencies, and infant stimulation programs to maximize the potential of children who are developmentally delayed. Environmental manipulation also includes protection from the damaging effects of the environment, such as exposure to sunlight, which can cause skin fragility and blistering in persons with porphyrias and increases the risk of skin cancers in patients with xeroderma pigmentosum and oculocutaneous albinism (Kingston, 2002).
Impact of Hereditary Disorders on the Family
Genetic testing can be broadly divided into two categories: diagnostic testing and screening. Tests to detect a disease-associated genetic mutation in symptomatic individuals are rapidly assuming greater importance in management of genetic disorders as more genes and mutations are identified and techniques are developed for easy application of the tests. Dramatic improvements in testing capabilities also provide opportunities to identify disease-associated mutations in people before they become symptomatic. With improved technology, mass screening for numerous genetic disorders will probably become routine. However, to be truly effective, screening programs depend on thorough education of both health professionals and the public regarding these programs and the limitations and implications of testing. The religious, moral, ethical, and legal issues revolving around screening and prenatal diagnosis are extensive and change over time (Farrell, Certain, and Farrell, 2001).
Purposes of Screening
Genetic screening is presumptive identification of an unrecognized genetic predisposition for future disease in individuals or their progeny for which preventive or disease course–altering interventions exist. In general, genetic screening targets populations, whereas genetic diagnostic testing targets individuals. The first corollaries of any genetic screening intervention must be voluntary participation, equal access to all, and confidentiality (both in conducting the tests and in handling records and results). In addition, education and counseling about tests and procedures must be an integral part of any screening program. Attention must be paid to quality control of all aspects of testing and laboratory procedures.
Genetic screening has three purposes: (1) to provide for early recognition of a disease, before symptoms occur, for which effective intervention and therapy exist (e.g., PKU); (2) to identify carriers of a genetic disease for the purpose of maximizing parenthood planning options (e.g., Tay-Sachs disease); and (3) to obtain population data on frequency, spectrum, and natural history of genetic variations not currently known to be associated with disease.
Screening for genetic disorders can occur during various times in a person’s life: (1) preconception screening of selected populations for heterozygous carriers (e.g., Ashkenazi Jewish carrier screening panel) (Gross, Pletcher, Monaghan, et al, 2008); (2) screening of relatives of known carrier or affected individuals within a family, for the purpose of reproductive decision making; (3) postconceptional (prenatal) screening (e.g., maternal serum screen for identifying risks for NTDs and chromosome abnormalities); and (4) newborn screening (e.g., panel for PKU in all newborns, as mandated by law in all U.S. states).
Newborn Screening: Newborn screening began in the 1960s when Guthrie devised the blood spot test to screen for PKU. Newborn screening for PKU was begun in all states, and currently each state offers screening for a growing number of disorders such as hemoglobinopathies, galactosemia, hypothyroidism, and congenital adrenal hypoplasia. In the past, decisions about which disorder to include were based on the following criteria:
• The disease occurs with a significant frequency.
• An inexpensive and reliable method of testing exists.
• There is effective treatment and intervention.
• If untreated, the baby will die or be severely developmentally impaired.
Because the actual disorders screened by individual states varied considerably, the American College of Medical Genetics (ACMG) was commissioned by the Maternal Child Health Bureau of the Health Resources and Services Administration to develop a uniform screening panel. The ACMG task force used the following minimum criteria when considering which disorders to include (American College of Medical Genetics Newborn Screening Expert Group, 2006):
• It can be identified at a phase (24 to 48 hours after birth) at which it would not ordinarily be clinically detected.
• A test with appropriate sensitivity and specificity is available for it.
• There are demonstrated benefits of early detection, timely intervention, and efficacious treatment of the condition being tested.
A panel of 29 core conditions and 25 secondary conditions was recommended by ACMG in its 2006 report (Sweetman, Millington, Therrell, et al, 2006). With advancing technology the number of conditions screened by individual states can be expected to change. A regularly updated list of conditions offered in each state’s newborn screening panel can be found at the National Newborn Screening and Genetics Resource Center website (http://genes-r-us.uthscsa.edu).
Because many of the conditions tested for are metabolic disorders, it is important that the first newborn screening blood sample be obtained at least 24 hours after the first protein feeding or within the first 72 hours of life. Infants found to be screen positive (thus at high risk) for a metabolic condition such as PKU or galactosemia are immediately referred to a medical center for testing to confirm the diagnosis and initiate treatment. Of course, this relies on a coordinated follow-up program that includes well-informed primary care providers and nurses who can locate newborns with abnormal screening results and facilitate the necessary follow-up visits.
Screening for Reproductive Information: Screening for heterozygotes (carriers) can detect unaffected persons with certain variant genes who, when they mate with an individual who carries a similar variant gene, are at high risk of producing an affected offspring. These individuals are thus provided with the information they need for making decisions about family planning. Carriers of a number of diseases can be detected by laboratory tests, but, because of the rarity of these diseases, mass screening is not feasible except in persons or populations known to be at risk. Persons at risk include close relatives of those with an inborn error of metabolism or other detectable disorder, and certain ethnic populations known to have a high incidence of a specific disease, such as sickle cell anemia in African-Americans, Tay-Sachs disease in Ashkenazi Jews, and thalassemia in people of Mediterranean ancestry.
This type of screening is sometimes controversial. Screening for carrier status for CF is one such situation that has the potential to create ethical dilemmas. Children with CF have a disease-associated mutation in each of their CFTR genes. More than 1000 different CFTR mutations have been identified, yet carrier testing for CF typically analyzes less than 100 of the possible CFTR mutations. Therefore, after screening, a couple may feel safe to proceed with pregnancy yet still have a child with CF. Carrier status screening has influenced marriage partner selection. Some carriers have made life choices based on inaccurate understanding of the implications of carrier status (e.g., thinking they would get the disease or that they were at high risk for having an affected child regardless of whether the partner is also a carrier). These misunderstandings and their consequences underscore the importance of careful education before decision making regarding carrier screening.
Carrier testing for children is generally not recommended, since the information is for reproductive purposes. Instead, parents are counseled that the child should make a decision about carrier testing when she or he is old enough to make an informed choice. Yet newborn screening tests reveal carrier status for certain conditions such as CF and sickle cell. Careful counseling is necessary with carrier screening to ensure that individuals understand the limitations of testing and the implications of results. Even with careful counseling, there may be significant misunderstanding or misuse of information (Ciske, Haavisto, Laxova, et al, 2001; Farrell, Kosorok, Rock, et al, 2001).
Screening for Epidemiologic Information: Public health officials may use screening as a method for monitoring the incidence of diseases or malformations in a population to detect environmental or other causes that might significantly influence the incidence of the disorders. For example, geographic and socioeconomic variations in the incidence of NTDs eventually led to research that determined that folic acid supplementation of 0.4 mg/day in women of childbearing age reduces NTD occurrence by as much as 50% (Mills and Signore, 2004).
Significance of Screening to Families
Mass screening programs have received mixed acceptance from health professionals and the general public. Various groups have raised objections concerning the accuracy of the screening tests. An ideal genetic screening test must have high sensitivity (ability to detect true positives) and specificity (ability to detect true negatives); it must yield rapid results, be safe, and be cost-effective; it should cause minimum physical and emotional discomfort to all involved. Other reasons for controversy include health professionals’ lack of knowledge about the testing purposes and implications of results, the public cost of testing (if government funded), and the psychologic implications of learning about carrier status. However, with several well-organized or legislated programs proving their value in preventing disease or of the damaging effects of disease, an increasing number of programs are gaining acceptance and support.
Potential benefits of carrier screening include facilitating genetic counseling and reproductive planning and providing useful information to other at-risk family members. Newborn screening provides for early detection and treatment initiation, maximizing quality of life. Yet realizing these benefits requires that couples and parents be given adequate information to make informed decisions about whether to pursue screening. For newborn screening, parent choice about testing may be limited by state regulations. Some states allow exemptions from newborn screening in certain situations, such as objections on religious grounds if there is a conflict with religious practices and beliefs of an established church. Regardless, parents need to be educated about the purpose; benefits, risks, and limitations of newborn screening; and the implications of abnormal results (Spahis and Bowers, 2006). Focus groups with parents have indicated that this type of education is preferred during the prenatal period (Campbell and Ross, 2003).
The social stigma of being the carrier of a “defective” gene may be a side effect of screening. In some families such knowledge is embarrassing and damaging to self-esteem. Teenagers may be especially vulnerable to the effects of knowing they carry a specific disease-associated gene mutation at a time when identity formation and peer approval are extremely important. Cultural views regarding this knowledge can have profound effects on the members of some ethnic groups. In some cases, social status within the cultural group can be impaired.
Probably the most important area for nursing practice is teaching, or the ability to convey clear, precise, and unambiguous information to clients. Families need to understand why the screening is proposed, what the results mean, and how the family can interpret false-positive and false-negative results (Baroni, Anderson, and Mischler, 1997). Anxiety is greater when families have not received sufficient information about the screening or testing process and its significance for their health. Families also have a right to know who assumes the cost of additional testing or retesting—the family or the state. The nurse is a valuable resource in ensuring that families are aware of alternatives and in helping them make the best decision (Spahis and Bowers, 2006).
Prenatal Testing
Pediatric nurses may encounter couples who had prenatal testing and are preparing for the anticipated care of their unborn child. Such couples may be making primary care arrangements; meeting professionals in the newborn intensive care unit; meeting with surgeons to learn about the surgeries their newborn will need in the first few days, weeks, or months of life; and talking with other parents who have children with the same condition. Thus it is important that pediatric nurses have a general understanding of the capabilities, limitations, and risks of prenatal testing.
Advances in technology have greatly increased the spectrum of available prenatal screening and diagnostic options. Although screening options are available for all pregnant women, prenatal diagnostic testing is reserved for pregnancies considered at increased risk for less than healthy outcomes. The purposes of prenatal testing include identifying congenital anomalies and genetic disorders that may need intervention during pregnancy or soon after delivery; enabling parents to prepare for the birth of a child who will require long-term medical or developmental interventions; and enabling parents to make decisions about placing their child for adoption or terminating the pregnancy. However, even normal prenatal test results do not guarantee the birth of a healthy baby. When referring a family for prenatal testing, the nurse can benefit from general guidelines, some of which are delineated in Box 5-6.
Prenatal Screening Tests
Biochemical maternal serum screening is used to detect pregnancies at increased risk for certain congenital anomalies and chromosome disorders. In the first trimester, maternal serum can be analyzed for free β-human chorionic gonadotropin (hCG) and pregnancy-associated plasma protein A. These values, in combination with nuchal fold translucency measurement, can be used to identify pregnancies at high risk for chromosome anomalies, particularly aneuploidy. In such situations, the couple is offered diagnostic testing, which may include chorionic villi sampling (CVS) or amniocentesis, depending on the fetus’s gestational age. Risk for neural tube and abdominal wall defects is screened during the second trimester when alpha-fetoprotein (AFP) is detectable in maternal serum. Also during the second trimester, hCG, unconjugated estriol, and inhibin A can be measured to screen for aneuploidies. High-risk pregnancies can be further evaluated with diagnostic ultrasound, amniocentesis, or fetal cord blood sampling (American College of Obstetrics and Gynecology, 2007).
Prenatal Diagnostic Procedures
Recommendations for diagnostic testing may be triggered by preexisting risks (e.g., previous child with a genetic disorder), personal characteristics (e.g., member of subpopulation at risk for certain genetic diseases), or a newly identified risk following prenatal screening for the current pregnancy (see Box 5-6). Specific risk factors are usually identified in the family history, previous pregnancy outcomes, or the mother’s medical history. Ethnic risk factors are based on a higher carrier frequency for certain genetic diseases in selected populations. Indications for prenatal diagnosis when any of these risk factors is present are based on a greater than general population risk that a congenital anomaly or genetic disorder will occur in the pregnancy. The specific risk is, of course, different for each situation.
Invasive prenatal diagnostic tests include CVS, amniocentesis, and fetal blood sampling. CVS can be performed between 10 and 12 weeks of gestation. Using either a transcervical or transabdominal approach guided by ultrasound, the clinician obtains a small biopsy of the chorionic villi. Cells from the sample can be grown for chromosome and single-gene disorder studies. Contamination of the sample with maternal cells can compromise the accuracy of test results. Amniocentesis is usually performed at 14 to 18 weeks of gestation under ultrasound guidance. Fetal skin cells that have sloughed off and are present in the amniotic fluid can be isolated and cultured for chromosome or single-gene analyses. AFP can be measured in the amniotic fluid and is a much more reliable indicator of an NTD or abdominal wall defect than maternal serum AFP. An additional test for an enzyme specific to neural tissue, acetylcholinesterase, may be done; if this test is positive in the presence of an elevated AFP level, it is diagnostic of an open NTD. A diagnostic ultrasound clarifies the extent and location of the NTD or abdominal wall defect. A less frequently used test is fetal blood sampling, in which blood is drawn from the umbilical vein. This procedure can be performed after 18 weeks of gestation and is usually done for prenatal evaluation of fetal hematologic abnormalities, inborn errors of metabolism, fetal infection, and rapid chromosome analysis (Rodeck, 2004).
A few additional diagnostic tests are infrequently used. Fetal biopsy is sometimes used to diagnose certain genetic skin disorders and metabolic disorders when DNA studies are unavailable or are uninformative. Fetal echocardiography may be performed for further diagnosis when a cardiac defect is noted on ultrasound. Research is under way to improve prenatal diagnostic testing.
A relatively new option for parents at risk for having a child with a genetic disorder is preimplantation genetic diagnosis. Through in vitro (test tube) fertilization, the embryo can be tested at the six- to eight-cell stage for specific genetic disorder. Normal or an unaffected carrier embryo or embryos are implanted. This technique has been used for Tay-Sachs disease, CF, and some X-linked disorders (Kent-First, 2000). It has been used by a couple who wished not only to avoid the birth of another child affected with Fanconi anemia, but also to have a healthy sibling who could provide a human lymphocyte antigen match for an affected sibling who needed stem cell transplantation (Verlinsky, Rechitsky, Schoolcraft, et al, 2001).
Genetic Evaluation and Counseling
In recent years the role of genetics as an etiologic agent in disease and disability has assumed a more prominent place in the nursing care of infants and children. The expanded recognition of genetic diseases and disorders and an increasingly well-informed public are creating a justified demand for genetic evaluation, diagnosis, information regarding risks to present and future generations, and access to available therapies. Unfortunately, however, persons who need expert genetic counseling often make uninformed decisions on their own or are the victims of well-meaning but equally uninformed relatives, acquaintances, or paraprofessionals. Nurses involved in infant and child care continually encounter families that have a risk of transmitting a disorder to an offspring, as well as children who may have an undiagnosed genetic disorder that needs expert evaluation.
It is a nurse’s responsibility to learn basic genetic principles, to be alert to situations in which families could benefit from genetic evaluation and counseling, to know about special services that can help manage and support affected children, and to be familiar with facilities in their areas where these services are available. In this way, nurses will be able to direct individuals and families to needed services and be active participants in the genetic evaluation and counseling process. Early identification of a genetic disorder allows anticipation of associated conditions and implementation of available preventive measures and therapy to avoid potential complications and to enhance the child’s health. It may also prevent the unexpected birth of another affected child in the immediate or extended family.
Pediatric Indications for Genetic Consultation
The ACMG has produced guidelines for genetics consultation (Pletcher, Toriello, Noblin, et al, 2007). The indications can be broadly divided into preconception-prenatal, pediatric, and adult. The indications that are relevant for newborns through adolescents are:
• Family history of hereditary diseases, birth defects, or developmental problems
• Family history of sudden cardiac death or early-onset cancer
• Family history of mental illness
• Abnormal genetic test result ordered by a nongenetics professional who lacks the knowledge and experience to discuss implications of results
• Progressive neurologic condition
• Major congenital anomaly (e.g., cleft lip, congenital heart defect, limb abnormality, spina bifida)
• Pattern of major or minor anomalies suggestive of genetic disorder
• Congenital or early-onset hearing loss or vision loss
• Cognitive impairment or autism
• Abnormal sexual maturation or delayed puberty
• Abnormally tall or short stature when compared with other family members
• Excessive bleeding or excessive clotting
• Parental requests that child be evaluated by a genetics professional
Genetic evaluation for diagnostic purposes may occur at any point in the life span. In the newborn period, birth defects and abnormal newborn screen results are obvious reasons for referral. Beyond the newborn period, indicators for referral include metabolic disorders, developmental delays, growth delays, behavioral problems, cognitive delays, abnormal or delayed sexual development, and medical problems known to be associated with genetic diseases. For example, a preschooler with hyperactivity and autistic-like behaviors may need evaluation for fragile X syndrome, and a 17-year-old girl with primary amenorrhea and short stature should be evaluated for Turner syndrome (Gardner and Sutherland, 2004).
With so many recent advances in genetic testing, it is not unusual for a child or adult with longstanding medical problems, including cognitive impairment, to be referred for reevaluation of his or her condition as a possible genetic disorder that might not have been diagnosable a few years earlier, such as microdeletion disorders or single-gene mutations. If a genetic diagnosis is made, the patient is usually referred back to the primary care physician with recommendations for routine management.
Persons may not be affected themselves but may request genetic counseling about the heritability of a trait. A young couple contemplating childbearing may be concerned about a disorder in one of their families or may seek advice because they are related. A couple who are both members of a population at risk for certain genetic diseases may wish to determine whether they carry an associated gene mutation (e.g., African-Americans and sickle cell anemia, Ashkenazi Jews and Tay-Sachs disease, or persons of Mediterranean ancestry and thalassemia). A couple planning adoption might seek genetic evaluation and counseling regarding a prospective child.
More often, persons who inquire about the possibility of recurrence of a disease or disorder have a child, or had a child who died, with a genetic disease or disorder. They are concerned about the likelihood of having another, similarly affected child and may want to know what reproductive choices are available. They might seek this advice before initiating another pregnancy or after the mother is already pregnant. If prenatal diagnostic testing is not done, the history of a condition in an older sibling, such as galactosemia, alerts health personnel to initiate specific and thorough testing for the condition in a newborn. In this way, early therapy can be initiated, thus minimizing or eliminating the effects of the disease or defect.
Parents need to know the risk in their particular situation and how it relates to the random risk for any prospective parents. When families understand the risks involved, they can make informed decisions regarding family planning and available prenatal testing. Additional counseling is necessary if abnormal results are obtained so the couple can decide to continue or terminate the pregnancy. They need considerable support for either decision (Veach, LeRoy, and Bartels, 2003). Parents may also have concerns about risks to unaffected siblings and about reproductive implications for their affected and unaffected offspring.
Genetic Services
Comprehensive genetic services consist of a group of specialists, which may include clinical geneticists, advanced practice nurses in genetics, genetic counselors, psychologists, biochemical geneticists, cytogeneticists, molecular geneticists, nurses, social workers, and other auxiliary personnel. The services are most often under the leadership of a physician trained in medical genetics, who assumes responsibility for the medical aspects of the problem. Although genetic centers were typically considered a resource for genetic evaluation, diagnosis, and counseling, a growing number of centers are managing the health care needs of patients with complex, rare genetic disorders. Some centers specialize in treatment of genetic disorders (e.g., enzyme replacement therapy for lysosomal storage disorders). Most centers are associated with a large medical center, which may have extensive outreach programs with satellite clinics throughout adjacent urban and rural areas. Numerous specialty clinics also deal with specific genetic disorders (such as CF, muscular dystrophy, hemophilia, or craniofacial anomalies) and provide their own genetic counseling services. Unfortunately, these units are concentrated in and around large metropolitan areas. As a result, counseling is not always accessible to a large number of people who could benefit from the service.
Unlike a medical prognosis, which predicts the outcome of a disease for an individual, a genetic prognosis may have implications for others as well, including members of the immediate family, other relatives, and future offspring. During a genetic evaluation or follow-up visit, a genetics professional obtains or updates the patient’s family history and pedigree, developmental history, and medical history (including pregnancy, labor, and delivery information); performs a physical examination, including dysmorphic features; and orders appropriate testing such as biochemical, cytogenetic, or molecular procedures. A genetic diagnosis may or may not be made initially. Genetic evaluation and counseling are time consuming and labor intensive. The pediatric nurse can facilitate the referral and help the family understand the sometimes lengthy process in arriving at a diagnosis. Nurses can also collect preliminary histories, including family history, and help parents prepare questions for the counseling session (Consensus Panel on Genetic/Genomic Nursing Competencies, 2006).
Genetic counseling is a communication process that deals with the human problems associated with the occurrence, or risk of occurrence, of a genetic disorder in a family. This process involves an attempt by one or more appropriately trained persons to help the individual or family:
• Comprehend the medical facts, including the diagnosis, the probable course of the disorder, and the available management
• Appreciate the way heredity contributes to the disorder and the risk of recurrence in specified relatives
• Understand the options for dealing with the risk of recurrence
• Choose the course of action that seems appropriate to them in view of their risk and family goals and act in accordance with that decision
• Make the best possible adjustment to the disorder in an affected family member and to the risk of recurrence of that disorder
Estimation of Risks
Effective genetic counseling based on a diagnosis or known risk factors requires a thorough evaluation of each situation. The genetics nurse or counselor derives risks of occurrence or recurrence from information acquired through a thorough family history, including known genetic information. A careful, detailed family history not only provides a picture of the proband (the affected person, or index case) in relation to other family members, but also may identify others who are similarly affected or who might be presymptomatic or at risk of producing affected children. Analyzing the pattern of affected family members can assist in confirming a tentative diagnosis or in determining the level of risk in multifactorial inheritance.
An accurate diagnosis is essential to provide specific risk figures. There are more than 10,000 known inherited disorders, many of which have similar clinical manifestations but different modes of inheritance. For example, symptoms in the early stages of severe X-linked muscular dystrophy appear much like those of the milder autosomal recessive and autosomal dominant varieties, autosomal recessive neurogenic muscular atrophies, and nongenetic poliomyelitis. The significance of the risks related to each type of disorder is readily apparent. For disorders with an unknown or multifactorial cause, recurrence risks are termed empiric, meaning they are based on observations of recurrence in similar situations, rather than theoretic, meaning they are based on mendelian inheritance patterns.
Communicating Risks
When explaining reproductive risk estimates, the genetics nurse or counselor does not attempt to make recommendations or decisions for patients. The nurse or counselor communicates comprehensive and current information about the nature of the disorder, the extent of risk involved, the probable consequences, and alternative solutions. The nurse then supports the informed patients regarding their decisions about whether to pursue preconception or prenatal testing and what to do in response to test results. However, genetics professionals recognize that recommendations do have a place when genetic testing is used to identify risk for diseases or disorders that have available treatments that can prevent, delay, or improve symptoms. Children who test positive for a mutation in the APC gene associated with familial adenomatous polyposis will eventually develop colon polyps, and eventually one or more will become cancerous. Therefore genetics professionals recommend specific screening measures for these children; when polyps are discovered, the professionals recommend a surgical consult to discuss colectomy options. As with any health care encounter, shared decision making with parents is the goal.
It is helpful to explain risks in different ways and to use examples to aid in understanding the meaning of probabilities. Most people do not have an adequate knowledge of genetics and human biology to fully comprehend these complex concepts. Words and concepts that can be used include percentages, chance, odds, and likelihood. For example, if a 40-year-old woman has a 1 : 112 risk of having a child with Down syndrome, other ways to explain it include, “You have about a 1% chance of having a baby with Down syndrome,” or “Out of 112 women your age having a baby, odds are that one of them will have a child with Down syndrome.” Games of probability can also be used, such as flipping coins, baseball pools, and lotteries.
Nurses can contact a family before genetic consultation to assess the family’s needs and attempt to reduce their anxiety. The telephone contact can also be used to obtain a family history and explain the clinic procedures. Many families are concerned about such things as whether they will be required to undress, whether blood is to be drawn, whether they can accompany the child during the visit, or whether they will be told what to do about reproduction. Families who know what to expect are able to gain more from a genetics consultation.
 NURSING ALERT
NURSING ALERT
Families may misunderstand probability, even when it is fully explained. The nurse should impress on them that each pregnancy is an independent event. Often parents who are told that a recessive disorder carries a 1 : 4 risk of recurrence incorrectly reason that because they already have one affected child, the next three will be unaffected. Chance has no memory; the risk is 1 : 4 for each and every pregnancy.
Role of Nurses in Genetics
All nurses need to be prepared to use genetic and genomic information and technology when providing care. Nearly 50 nursing organizations endorsed essential minimum competencies necessary for nurses to deliver competent genetic and genomic focused nursing care (Consensus Panel on Genetic/Genomic Nursing Competencies, 2006). The professional practice domains include applying/integrating genetic knowledge into nursing assessment; identifying and referring clients who may benefit from genetic information or services; identifying genetics resources and services to meet clients’ needs; and providing care and support before, during, and after providing genetic information and/or services. Often a nurse is the first one to recognize the need for genetic evaluation by identifying an inherited disorder in a family history or by noting physical, cognitive, or behavioral abnormalities when performing a nursing assessment (Box 5-7).
Nurses who specialize in genetics are guided by the Genetics/Genomics Nursing Scope and Standards of Practice (International Society of Nurses in Genetics, 2006). Genetics nurses at the basic level tend to work in clinics that focus on services for patients with specific single-gene disorders such as CF, sickle cell disease, muscular dystrophy, and hemophilia. They may also work in genetics clinics. Genetics nurses in advanced practice work in a variety of specialty clinics or genetics centers. These nurses evaluate a patient’s medical, developmental, prenatal, birth, and family histories; perform physical examinations, including dysmorphology examination; order tests and diagnostic procedures and evaluate their results; prescribe therapies that are within their scope of practice; and evaluate patient outcomes. Genetics nurses collaborate with teams that include medical geneticists and genetic counselors.
This chapter highlights the role of all nurses who care for children and their families. The Essential Nursing Competencies and Curricula Guidelines for Genetics and Genomics (Consensus Panel on Genetic/Genomic Nursing Competencies, 2006) is used as a framework for discussion (Box 5-8).
Nursing Assessment: Applying and Integrating Genetic and Genomic Knowledge
Family health history is an important tool to identify individuals and families at increased risk for disease, risk factors for disease (such as obesity), and inheritance patterns of diseases. Because of its importance, all nurses need to be able to elicit family history information and document the collected information in pedigree format.
When eliciting a family health history, nurses should collect information about all family members within a minimum of three generations. This process usually takes 20 to 30 minutes. When possible, it is best to include both parents in the interview to elicit information about relatives on both sides of the family. Medical records, birth and death records, family Bibles, and photograph albums are helpful resources, and persons being interviewed should be instructed to bring such items if they are available. It may be necessary to consult other members of the family. The level of education and the degree of understanding vary widely among informants and influence their reliability. The informants may be reticent, particularly if they view the disorder as something to be ashamed of or in some way threatening. Sometimes true relationships may be concealed, such as adoption or misattributed paternity.
Skillful interviewing is necessary to obtain essential, but often embarrassing or private information. Since the parents may not be married, they should be addressed as couples or partners and asked about other unions that may have produced a pregnancy. In eliciting a birth history from the mother and father, the nurse should specifically ask about abortions, miscarriages, and stillbirths in addition to live births. To identify all members of the family tree, it is best to ask about pregnancies even of young teenagers. When inquiring about family diseases, the nurse might ask the question in different ways. For example, if a person denies any cognitive impairment in the family, asking other questions, such as about learning problems, being in special education classes, and failing or not completing school, may uncover a family history.
The family history is recorded in the form of a pedigree chart or family tree (in some disciplines termed a genogram), using standard symbols to indicate persons, relationships, and significant details related to them (Bennett, Steinhaus, Uhrich, et al, 1995) (Fig. 5-19). Construction of a pedigree begins with the affected child (proband, index case, or original patient) and all of the mother’s pregnancies (Fig. 5-20, A). Next, the maternal family history is explored in a similar manner (Fig. 5-20, B); then information is gathered about relatives on the father’s side, as well as any children or pregnancies that may have occurred through the father’s previous unions (Fig. 5-20, C) (see Nursing Care Guidelines box). It is important at this point to determine whether the couple might be related in any way, although contrary to popular belief, this is usually a concern only for first cousins or closer relatives. The first-cousin risk for birth defects is about 5% compared with the general population risk of 2% to 3%. The primary risk increase is for autosomal recessive disorders, in which the chance that two individuals carry the same rare, deleterious gene is increased if they are related.
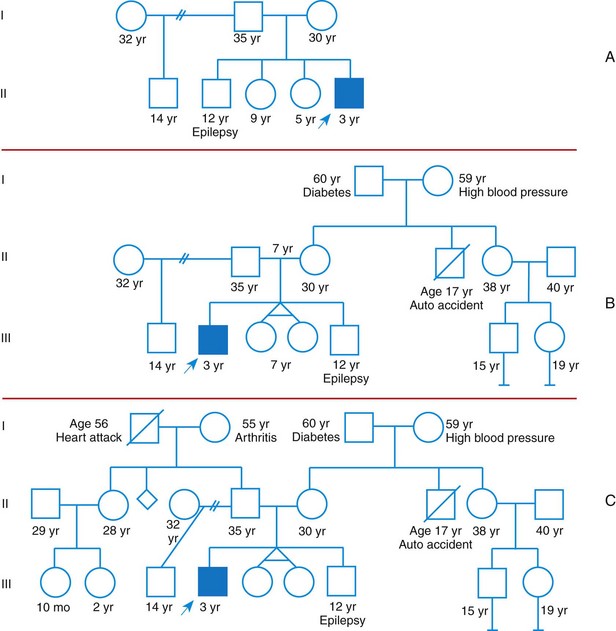
Fig. 5-20 Construction of a pedigree. A, Proband, siblings, and parents. B, Maternal relatives. C, Paternal relatives added.
 NURSING CARE GUIDELINES
NURSING CARE GUIDELINES
Pedigree Construction
1. Begin diagram in the middle of a large sheet of paper.
2. Represent males by a square placed to the left and females by a circle placed to the right.
3. Represent the proband (index case, original patient) with an arrow (if the counselee or patient is different, place a C under that person’s symbol).
4. Use a horizontal line between a square and a circle for a mating or marriage.
5. Suspend offspring vertically from the mating line and place in order of birth with oldest to the left (regardless of sex).
6. Symbolize generations by Roman numerals, with the earliest generation at the top.
7. Include three generations: grandparents, parents, offspring, siblings, aunts, uncles, and first cousins of proband.
8. Include name of each person, their age, health problems, and date and cause of death. Because of privacy concerns, include only first names of the family you are interviewing.
For a detailed description of pedigree construction, see Nelson-Anderson DL, Waters CV: Genetic connections: a guide to documenting your individual and family health history, Washington, Mo, 1995, Sonters.
The nurse solicits information not only about other affected family members but also about (1) births (live birth, stillbirths, and abortions [especially spontaneous abortions], including gestational age of pregnancy); (2) infertility problems; (3) matings (legally sanctioned, consanguineous, multiple, unwed, and other complex relationships); (4) health of family members, including any other genetic diseases or disorders or birth defects; and (5) death and causes of death, including early infant deaths. Sometimes the place of birth and ethnic background are significant. For example, the carrier frequency for Tay-Sachs disease is higher in Ashkenazi Jews from Eastern Europe than in Jews from other geographic regions. Also, when a pedigree chart is being evaluated, a sister’s death in infancy from a congenital heart defect might be genetically significant, whereas a healthy sibling’s death from drowning at age 1 year would not. Information concerning first-degree relatives is most important and should be complete.
Time is recognized as one of the biggest barriers to collecting a detailed family health history (Rich, Burke, Heaton, et al, 2004). In response to this known barrier, departments within the U.S. Department of Health and Human Services collaborated to develop and launch the surgeon general’s My Family Health Portrait (https://familyhistory.hhs.gov/fhh-web/home.action). Nurses can play an important role in teaching families how to access and use this web-based tool and encourage them to share the collected family history with their health care providers. The updated version of My Family Health Portrait allows the code to be downloaded so that patients’ family histories can be imported into electronic medical records. The Genetic Alliance has an online booklet about how to gather family history that is available in English, Spanish, and Chinese (www.geneticalliance.org/ksc_assets/tools/book1ga_ll022309.pdf). The National Human Genome Research Institute’s Education and Community Involvement Branch (www.genome.gov/27026369) funded several family history demonstration projects that produced materials to help people, particularly the medically underserved, use My Family Health Portrait and talk with health care professionals about the health implications of their family history. Nurses need to be aware of these resources and help families use them.
In addition to family history, nurses caring for children and families need to collect pregnancy, labor and delivery, perinatal, medical, and developmental histories. Although it is common for genetics nurses to obtain all of these histories before or during an initial genetics consultation, not all nurses are expected to obtain all of these assessment data from each patient during a pediatric visit. Electronic medical records are making it more practical to construct a comprehensive set of histories even when many health care professionals contribute only a portion of the total history.
All nurses are taught to perform physical assessments, but they are seldom taught to recognize minor anomalies and dysmorphology that may suggest a genetic disorder. Yet nurses are keen in recognizing delays in development, behavior differences, and global appearances that raise concern that a newborn, infant, child, or adolescent needs further evaluation. Although dysmorphology is beyond the scope of this chapter, readers are encouraged to review the January 2009 issue of American Journal of Medical Genetics (Carey, 2009). Drawings and photographs of normal and abnormal morphologic characteristics are provided for the head, face, and extremities, together with accepted dysmorphology terminology. Nurses knowledgeable in dysmorphology are able to articulate specific concerns about a child’s appearance rather than relying on the outdated and offensive phrase, “funny looking kid.” When a major anomaly is identified, nurses should raise suspicion that the child could have additional congenital anomalies. When three or more minor anomalies are identified, nurses should suspect the possibility of an underlying syndrome. However, it is important to consider the biologic parents’ physical appearance, development, and behavior when considering the relevance of the child’s combination of minor anomalies.
Identification and Referral
Nurses have an important role in identifying patients and families who have, or are at risk for developing or transmitting, a genetic condition (see Box 5-7 for assessment clues to genetic disorders). Once family and patients concerns are identified, they may need help identifying and accessing necessary services. Nurses need to become familiar with the genetic services in their areas. As mentioned before, many services tend to be in large metropolitan areas. A regularly updated resource for locating genetics clinics can be found at www.genetests.org (click on Clinic Directory tab). Contact information for specific genetics professionals can be found at the following websites: geneticists, www.acmg.net (click on Find a Geneticist); genetics nurses, www.isong.org (call or e-mail office); and genetic counselors, www.nsgc.org (click on Find a Counselor). In addition, state health departments either offer services or can help identify health professionals with specialty training in genetics.
When facilitating genetics consultations, nurses should share with the genetics professional the findings in the histories they collected that triggered the consultation. Nurses can also help the referral process by determining and communicating the family’s initial concerns, their state of knowledge about the reason for referral, and their attitudes and beliefs concerning genetics.
Providing Education, Care, and Support
Maintaining contact with the family or making a referral to a health care practice or an agency that can provide a sustained relationship is critical. As mentioned, it is becoming more common for genetics health care professionals to provide regular follow up and management, particularly for children with rare genetic disorders. However, some families choose not to have follow-up visits with genetic experts.
Regardless of whether families choose to receive continued care with a genetics center, clinic, or professional, nurses can help patients and families process and clarify the information they receive during a genetics visit. Misunderstanding of this information can have many causes, including cultural differences, the disparity of knowledge between the counselor and the family, and the heightened emotion surrounding genetic counseling. Family members have difficulty absorbing all the information presented during a genetics evaluation and counseling session. Knowing this, genetics professionals write and send clinic summary letters to families. The nurse may need to help the family understand terminology in the letter, help them identify and articulate remaining questions or areas of clarification, and coach them through the process of accessing genetics health professionals to get remaining questions and concerns answered. Information often needs to be repeated several times before the family understands the content and its implications.
Nurses must assess for and address parents’ feelings of guilt about carrying “bad genes” or having “made my child sick.” Depending on the type of cytogenetic disorder, the nurse may be able to absolve the parents of guilt by explaining the random nature of segregation during both gamete formation and fertilization and that errors in cell division unique to the pregnancy in question are not likely to happen again and are not inherited. If the condition is a mendelian-inherited or mitochondrial disorder, it is important to assess parents’ understanding of recurrence risk, help them understand the chances that a subsequent pregnancy will not be affected, and ensure they have been given information about their options for future children (preimplantation diagnosis, use of donor egg or sperm, prenatal diagnosis, or adoption). Families often try to reason that some unrelated event caused the abnormality (e.g., a fall, a urinary tract infection, or “one glass of wine”) before the mother was aware that she was pregnant. These misconceptions need to be assessed and dispelled.
After a genetics visit, and sometimes before the visit, parents often use the Internet to find answers to their questions. During the initial genetics evaluation, a diagnosis may not be possible. Instead, findings in medical, developmental, and family histories lead the professional to order genetic tests and other diagnostic procedures. Diagnoses under consideration are discussed briefly with the parents. Some parents are satisfied with the brief information and do not care to find out more until the actual diagnosis is established. Other parents go home and seek as much information as they can about the diagnoses under consideration. The information they find can be terrifying and overwhelming and inaccurate or misleading. Nurses can play an important role in helping parents identify reliable, accurate resources for information at whatever time they desire it. It is also important to stress that everything that is described for a genetic condition may not be relevant to their child. Before the follow-up genetics visit when test and procedure results are discussed, nurses can help parents identify and write down the questions and concerns they need addressed before leaving the clinic.
Once a genetics diagnosis is made or a genetic predisposition to a delayed-onset disorder is identified, nurses need to have frequent contact with patients and families as they attempt to incorporate recommended therapies or disease-prevention strategies into their daily lives. For example, a disorder such as PKU requires conscientious diet management; therefore it is important to make certain that the family understands and follows instructions and is able to navigate the health care system to access the essential formula and low-phenylalanine food products. An infant evaluated for cleft palate and cardiac defect and subsequently found to have VCFS requires surgical intervention for the congenital malformations. The infant also benefits from early intervention services and eventually an individualized education plan in school, since developmental delay and eventual learning problems are common.
Initial and ongoing assessment of the family’s coping abilities, resources, and support systems is vital to determine their need for additional assistance and support. As with any family who has a child with chronic health care needs, nurses must teach the family to become the child’s advocate. Nurses can help families locate agencies and clinics specializing in a specific disorder or its consequences that can provide services (e.g., equipment, medication, and rehabilitation), educational programs, and parent support groups. Referral to local and national support groups or contact with a local family that has a child with the same condition can be helpful for new parents. Privacy and confidentiality are imperative, and both families must give permission before their contact information is given (International Society of Nurses in Genetics, 2006). Nurses can also be instrumental in helping parents start a support group when none is available.
Parental attachment and adjustment to the baby can be supported and facilitated by nursing interventions. Assessing the parents’ understanding of the child’s disorder and providing simple and truthful explanations can help them begin to understand their child’s health issues. Guiding the parents in recognizing their child’s cues, responses, and strengths can be helpful even for experienced parents. A caring attitude conveys the value of their child and, by extension, their value as parents. The nurse can help the parents identify their strengths as a family and identify support that is available to them.
Giving birth to and raising a child with a genetic disorder is not necessarily a lifetime burden. It is important for nurses to ask parents to describe their experience raising their child with a particular genetic condition. What has been the impact on their family? While parents may initially experience negative outcomes, such as shock, emotional distress, and grief, families can adapt and thrive. Resources for managing stress and restoring balance in the lives of families affected by a genetic condition can help. Van Riper’s (2007) research has identified nursing interventions that can promote resilience and adaptation in families of children with Down syndrome. Van Riper’s recommendations are useful for families of children with any type of genetic disorder:
• Recognize multiple stressors, strains, and transitions in their lives (e.g., unmet family needs).
• Discuss and implement strategies for reducing family demands (e.g., setting priorities and reducing the number of outside activities family members are involved in).
• Identify and use individual, family, and community resources (e.g., humor, family flexibility, supportive extended family, respite care, local support groups, and Internet resources).
• Expand the range and efficacy of their coping strategies (e.g., increase the use of active strategies such as reframing, mobilize their ability to acquire and accept help, and decrease the use of passive appraisal).
• Encourage the use of an affirming style of family problem-solving communication (e.g., one that conveys support and caring and exerts a calming influence).
Some families do struggle after learning their child has a genetic disorder. Families may feel ashamed of a hereditary disorder and seek to blame their partner for transmitting a faulty gene or chromosome. Intrafamilial strife, hostility, and marital or couple disharmony, sometimes to the point of family disintegration, can occur. Nurses should be alert for evidence of risk factors that indicate poor adjustment (e.g., child abuse, divorce, or other maladaptive behaviors). Referral to psychosocial professionals for crisis intervention may be necessary.
Key Points
• Most if not all disease processes have a genetic component.
• Genetic diseases may be caused by chromosome abnormalities, gene mutations, or mtDNA mutations. In addition, expression of a genetic disease are often influenced by environmental factors.
• Congenital anomalies—errors of morphogenic development—may arise at any stage of development and demonstrate wide variability in causative factors. Environmental teratogens and maternal disease may also disrupt fetal development, leading to birth defects.
• Chromosome disorders are caused by abnormalities in either chromosome structure or number.
• Alterations in chromosome number occur as a result of unequal distribution of genetic material during gamete formation (meiotic nondisjunction) or early cell division of the zygote (mitotic nondisjunction).
• Genes can be located on an autosome or a sex chromosome. Their mode of expression can be dominant or recessive.
• Disorders caused by single-gene mutations are distributed in families according to predictable mendelian principles of inheritance, but recognition of the pattern may be influenced by variable expressivity and penetrance (frequency of expression). In addition, a genetic disorder can be phenotypically expressed by seemingly unrelated manifestations.
• Most current interventions for genetic disease aim at correcting or modifying the phenotypic expression.
• The objectives of genetic testing are to detect the presence of disease in individuals, detect unaffected carriers of a disease, and monitor the incidence of disease or malformations in a population.
• Prenatal testing is aimed at both screening (ultrasound and maternal biochemical marker testing) and diagnosis (amniocentesis, ultrasound, CVS, and fetal cord blood sampling).
• A genetic counseling goal is to provide individuals and families with information needed to make informed decisions about a course of action that is most appropriate to them.
• Competent nursing practice includes applying and integrating genetic and genomic knowledge during nursing assessment; when identifying and referring patients and families who may benefit from genetic services; when identifying resources for patients and families; and when providing education, care, and support.
References
American Academy of Pediatrics. Ethical issues with genetic testing in pediatrics. Pediatrics. 2001;107(6):1451–1455.
American Academy of Pediatrics, Committee on Substance Abuse and Committee on Children with Disabilities. Fetal alcohol syndrome and alcohol-related neurodevelopmental disorders. Pediatrics. 2000;106(2 Pt 1):358–361.
American Academy of Pediatrics. Folic acid for the prevention of neural tube defects. Pediatrics. 1999;104(2):325–327.
American College of Medical Genetics Newborn Screening Expert Group. Newborn screening: toward a uniform screening panel and system—executive summary. Genet Med. 2006;8(5 Suppl):1S–11S.
American College of Obstetrics and Gynecology. Screening for fetal chromosomal abnormalities: clinical management guidelines for obstetrician-gynecologists. Obstet Gynecol. 2007;109(1):217–227.
Antshel, KM, Fremont, W, Kates, WR, et al. The neurocognitive phenotype in velo-cardio-facial syndrome: a developmental perspective. Dev Disabil Res Rev. 2008;14(1):43–51.
Bailey, L. An overview of enzyme replacement therapy for lysosomal storage diseases. Online J Issues Nursing. 13(1), 2008. [Manuscript 3].
Baroni, MA, Anderson, YE, Mischler, E. Cystic fibrosis newborn screening: impact of early screening results on parenting stress. Pediatr Nurs. 1997;23(2):143–151.
Bennett, RL, Steinhaus, KA, Uhrich, SB, et al. Recommendations for standardized human pedigree nomenclature. Am J Med Genet. 1995;56:745–752.
Bos, AF, Einspieler, C, Prechtl, HF. Intrauterine growth retardation, general movements, and neurodevelopmental outcome: a review. Dev Med Child Neurol. 2001;43(1):61–68.
Brown, SM. Essentials of medical genomic. Hoboken, NJ: Wiley-Liss; 2003.
Campbell, E, Ross, LF. Parental attitudes regarding newborn screening of PKU and DMD. Am J Med Genet. 2003;120A(2):209–214.
Carey, JC. Elements of morphology: standard terminology. editor. Am J Med Genet A. 2009;149A(1):1–127.
Chamberlain, S. Friedreich’s in relief. Nat Genet. 1996;12(4):344–345.
Ciske, D, Haavisto, A, Laxova, A, et al. Genetic counseling and neonatal screening for cystic fibrosis: an assessment of the communication process. Pediatrics. 2001;107(4):699–705.
Collins, FS, Green, ED, Guttmacher, AE, et al. A vision for the future of genomics research: a blueprint for the genomic era. Nature. 2003;422:1–13.
Consensus Panel on Genetic/Genomic Nursing Competencies. Essential nursing competencies and curricula guidelines for genetics and genomics. Silver Spring, Md: American Nurses Association; 2006.
Edelmann, L, Hirschhorn, K. Clinical utility of array CGH for the detection of chromosomal imbalances associated with mental retardation and multiple congenital anomalies. Ann N Y Acad Sci. 2009;1151:157–166.
Farrell, M, Certain, I, Farrell, P. Genetic counseling and risk communication services of newborn screening programs. Arch Pediatr Adolesc Med. 2001;155(2):120–126.
Farrell, P, Kosorok, MR, Rock, MJ, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth: Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics. 2001;107(1):1–13.
Fossland, VS, Stroop, JB, Schwartz, RC, et al. Genetic issues in patients with breast cancer. Surg Oncol Clin North Am. 2009;18(1):53–71. [viii].
Fridman, C, Varela, MC, Kok, F, et al. Paternal UPD15: further genetic and clinical studies in four Angelman syndrome patients. Am J Med Genet. 2000;92(5):322–327.
Gardner, RJ, Sutherland, GR. Chromosome abnormalities and genetic counseling, ed 3. New York: Oxford University Press; 2004.
Gross, SJ, Pletcher, BA, Monaghan, KG, et al. Carrier screening in individuals of Ashkenazi Jewish descent. Genet Med. 2008;10(1):54–56.
Honein, M, Paulozzi, LJ, Mathews, TJ, et al. Impact of folic acid fortification of the US food supply on the occurrence of neural tube defects. JAMA. 2001;285(23):2981–2986.
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945.
International Society of Nurses in Genetics. Genetics/genomics nursing: scope and standards of practice. Silver Springs, Md: American Nurses Association; 2006.
Jones, KL. Smith’s recognizable patterns of human malformation. Philadelphia: Saunders; 2006.
Jorde, LB, Carey, JC, Bamshad, MJ, et al. Medical genetics, ed 3. St. Louis: Mosby; 2003.
Kent-First, M. The critical and expanding role of genetics in assisted reproduction. Prenatal Diagnosis. 2000;20(7):536–551.
King, RA, Rotter, JI, Motulsky, AG. Approach to genetic basis of common diseases. In King RA, Rotter JI, Motulsky AG, eds.: The genetic basis of common diseases, ed 2, New York: Oxford, 2002.
Kingston, HM. ABC of clinical genetics, ed 3. London: BMJ Books; 2002.
Korf, BR. Human genetics: a problem-based approach, ed 2. Cambridge, Mass: Blackwell Science; 2000.
Lashley, FR. Clinical genetics in nursing practice. New York: Springer; 2005.
Lynch, HT, Lynch, JF, Lynch, PM, et al. Hereditary colorectal cancer syndromes: molecular genetics, genetic counseling, diagnosis and management. Fam Cancer. 2008;7(1):27–39.
McKusick, VA. History of medical genetics. In: Rimoin DL, Connor MJ, Pyeritz RE, et al, eds. Principles and practice of medical genetics. New York: Churchill Livingstone, 2002.
Mills, JL, Signore, C. Neural tube defect rates before and after food fortification with folic acid. Birth Defects Res A Clin Mol Teratol. 2004;70:844–845.
National Foundation for Ectodermal Dysplasia, What is ED?. Mascoutah, Ill: The Foundation; 2009. available at http://nfed.org/about_ed_faq.asp [(accessed December 15, 2009)].
Nelson, DL. Allelic expansion underlies many genetic diseases. Growth Genet Horm. 1996;12(1):1–4.
Nusbaum, R, Isaacs, C. Management updates for women with a BRCA1 or BRCA2 mutation. Molec Diagn Ther. 2007;11(3):133–144.
Pletcher, BA, Toriello, HV, Noblin, SJ, et al. Indications for genetic referral: a guide for healthcare providers. Genet Med. 2007;9(6):385–389.
Rich, EC, Burke, W, Heaton, CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med. 2004;19(3):273–280.
Rimoin DL, Connor MJ, Pyeritz RE, et al, eds. Emery and Rimoin’s principles and practice of medical genetics. London: Churchill Livingstone, 2002.
Rodeck, CH. Fetal blood sampling: historical perspectives. NeoReviews. 2004;5(6):e229–e231.
Romiti, ML, Colognesi, C, Cancrini, C, et al. Prognostic value of a CCR5 defective allele in pediatric HIV-1 infection. Mol Med. 2000;6(1):28–36.
Saenko, EL, Pipe, SW. Strategies towards a longer acting factor VIII. Haemophilia. 2006;12(Suppl 3):42–51.
Scriver CR, Beaudet AL, Sly WS, et al, eds. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001.
Shprintzen, RJ. Velo-cardio-facial syndrome: 30 years of study. Dev Disabil Res Rev. 2008;14(1):3–10.
Spahis, JK, Bowers, NR. Navigating the maze of newborn screening. MCN Am J Matern Child Nurs. 2006;31(3):190–196.
Sweetman, L, Millington, DS, Therrell, BL, et al. Naming and counting disorders (conditions) included in newborn screening panels. Pediatrics. 2006;117(5 Pt 2):S308–S314.
Sybert, VP, McCauley, E. Turner’s syndrome. N Engl J Med. 2004;351:1227–1238.
Van Riper, M. Families of children with Down syndrome: responding to “a change in plans” with resilience. J Pediatr Nurs. 2007;22(2):116–128.
Veach, PM, LeRoy, BS, Bartels, DM. Facilitating the genetic counseling process: a practice manual. New York: Springer; 2003.
Verlinsky, Y, Rechitsky, S, Schoolcraft, W, et al. Preimplantation diagnosis for Fanconi anemia combined with HLA matching. JAMA. 2001;285(24):3130–3133.
Wilkin, JZ, Su, Z, Kuritzkes, DR, et al. HIV type 1 chemokine coreceptor use among antiretroviral-experienced patients screened for a clinical trial of a CCR5 inhibitor: AIDS Clinical Trial Group A5211. Clin Infect Dis. 2007;44(4):591–595.
Zieve, D, Juhn, G, Eltz, DR. Amniotic constriction bands. available at www.nlm.nih.gov/medlineplus/ency/article/001579.htm, 2009. [(accessed June 9, 2009)].
Ethical, Legal, and Social Issues (ELSI). www.genome.gov/PolicyEthics (NIH site) and www.ornl.gov/hgmis/elsi/elsi.html (Department of Energy site) (program developed to study the issues surrounding the availability of genetic information)
Family Village. www.familyvillage.wisc.edu (information and resources for families with disabilities and persons who provide services for them)
GeneTests. www.genetests.org (genetic testing resource; requires password to enter the site; easy to obtain)
Genetic Alliance. www.geneticalliance.org (international coalition of individuals, professionals, and genetic support organizations)
Genetic Home Reference. http://ghr.nlm.nih.gov (handbook on basic human and clinical genetics concepts, summaries of a variety of genetic conditions, excellent glossary)
Health on the Net Code of Conduct. www.hon.ch/HONcode/Conduct.html (describes the Health on the Net Foundation code of conduct for medical and health websites, which addresses the reliability and credibility of information)
International Society of Nurses in Genetics. www.isong.org (provides information about various genetics education programs, instructional resources, conferences targeting nurses)
National Cancer Institute. www.cancer.gov (provides accurate and up-to-date cancer information)
National Coalition for Health Professional Education in Genetics. www.nchpeg.org (promotes access to human genetics education resources for health professionals in a variety of specialties and disciplines)
National Human Genome Research Institute. http://genome.gov (information on genome research and its ethical, legal, and social implications)
National Organization for Rare Disorders. www.rarediseases.org (a federation of voluntary health organizations dedicated to helping people with rare “orphan” diseases)
OMIM, Online Mendelian Inheritance in Man. www.ncbi.nlm.nih.gov/Omim (up-to-date summaries of human genes and genetic disorders)
United Mitochondrial Disease Foundation. www.umdf.org (provides information about mitochondrial defects, including professional and lay materials)