Health Problems of the Newborn

http://evolve.elsevier.com/wong/ncic

Birth of a Child with a Physical Defect, Ch. 11
Blood Specimens, Ch. 27
The Child with Cognitive, Sensory, or Communication Impairment, Ch. 24
Congenital Adrenogenital Hyperplasia, Ch. 38
Diaper Dermatitis, Ch. 13
Family-Centered Care of the Child with Chronic Illness or Disability, Ch. 22
Genetic Evaluation and Counseling, Ch. 5
Genetic Screening, Ch. 5
Health Promotion of the Newborn and Family, Ch. 8
The High-Risk Newborn and Family, Ch. 10
Infants of Diabetic Mothers, Ch. 10
Birth Injuries
 Several factors predispose an infant to birth injuries. Maternal factors include uterine dysfunction that leads to prolonged or precipitous labor, preterm or postterm labor, and cephalopelvic disproportion. Injury may result from dystocia caused by fetal macrosomia, multifetal gestation, abnormal or difficult presentation (not caused by maternal uterine or pelvic conditions), and congenital anomalies. Intrapartum events that can result in scalp injury include the use of intrapartum monitoring of fetal heart rate and collection of fetal scalp blood for acid-base assessment. Obstetric birth techniques can cause injury. Forceps birth, vacuum extraction, version and extraction, and cesarean birth are potential contributory factors. Often more than one factor is present, and multiple predisposing factors may be related to a single maternal condition.
Several factors predispose an infant to birth injuries. Maternal factors include uterine dysfunction that leads to prolonged or precipitous labor, preterm or postterm labor, and cephalopelvic disproportion. Injury may result from dystocia caused by fetal macrosomia, multifetal gestation, abnormal or difficult presentation (not caused by maternal uterine or pelvic conditions), and congenital anomalies. Intrapartum events that can result in scalp injury include the use of intrapartum monitoring of fetal heart rate and collection of fetal scalp blood for acid-base assessment. Obstetric birth techniques can cause injury. Forceps birth, vacuum extraction, version and extraction, and cesarean birth are potential contributory factors. Often more than one factor is present, and multiple predisposing factors may be related to a single maternal condition.
Many injuries are minor and resolve spontaneously in a few days; others, although minor, require some degree of intervention. Still others can be serious or even fatal. Part of the nurse’s responsibility is to identify such injuries so that appropriate interventions can be initiated as soon as possible. Birth injuries are classified according to the type of body structure involved (Box 9-1).
Soft Tissue Injury
Infants may sustain various types of soft tissue injury during birth, primarily in the form of bruises and abrasions secondary to dystocia. Soft tissue injury usually occurs when there is some degree of disproportion between the presenting part and the maternal pelvis (cephalopelvic disproportion). Box 9-2 lists common types of soft tissue injury. The use of forceps to facilitate a difficult vertex delivery may produce discoloration or abrasions with the same configuration as the forceps on the sides of the neonate’s face. Petechiae or ecchymoses may be observed on the presenting part after a breech or brow delivery. After a difficult or precipitous delivery, the sudden release of pressure on the head can produce scleral hemorrhages or generalized petechiae over the face and head. Petechiae and ecchymoses may also appear on the head, neck, and face of an infant born with a nuchal cord, giving the infant’s face a cyanotic appearance. A well-defined circle of petechiae and ecchymoses may also appear on the occipital region of the newborn’s head when a vacuum suction cup is applied during delivery. Rarely, lacerations occur during cesarean section.
These traumatic lesions generally fade spontaneously and without treatment within a few days. However, petechiae may be a manifestation of an underlying bleeding disorder and are evaluated.
Nursing Care Management
Nursing care is directed primarily toward assessing the injury, maintaining asepsis of the area to prevent breakdown and infection, and providing an explanation and reassurance to the parents. The nurse records an accurate description of the injury (e.g., extent of petechiae) to facilitate subsequent comparative nursing evaluations.
Regardless of how benign the injury, parents may be concerned and mourn the loss of the expected “perfect” infant. Explanations of the cause and treatment, if any, need to be thorough and repeated frequently. If the injury is temporarily disfiguring, such as extensive facial bruising, nurses can demonstrate acceptance of the child through their example of sensitive, personal care.
Head Trauma
Head trauma that occurs during the birth process is usually benign but occasionally results in more serious injury. The injuries that produce serious trauma, such as intracranial hemorrhage and subdural hematoma, are discussed in relation to neurologic disorders in the newborn. (See Chapters 10 and 37.) Skull fractures are discussed with other fractures sustained during the birth process. The three most common types of extracranial hemorrhagic injury are caput succedaneum, subgaleal hemorrhage, and cephalhematoma.
Caput Succedaneum
The most commonly observed scalp lesion is caput succedaneum, a vaguely outlined area of edematous tissue situated over the portion of the scalp that presents in a vertex delivery (Fig. 9-1, A). The swelling consists of serum and/or blood that has accumulated in the tissues above the bone. Typically the swelling extends beyond the bone margins (or sutures) and may be associated with overlying petechiae or ecchymosis. It is present at or shortly after birth. No specific treatment is necessary, and the swelling subsides within a few days.

Subgaleal Hemorrhage
Subgaleal hemorrhage is bleeding into the subgaleal compartment (Fig. 9-1, B). The subgaleal compartment is a potential space that contains loosely arranged connective tissue. It is located beneath the galea aponeurosis, the tendinous sheath that connects the frontal and occipital muscles and forms the inner surface of the scalp. The injury occurs as a result of forces that compress and then drag the head through the pelvic outlet. The bleeding extends beyond bone, often posterior into the neck, and continues after birth, with the potential for serious complications and morbidity.
Early detection of the hemorrhage is vital; serial head circumference measurements and inspection of the back of the neck for increasing edema and a firm mass are essential. A boggy fluctuant mass over the scalp that crosses the suture line and moves as the baby is repositioned is an early sign of subgaleal hemorrhage (Doumouchtsis and Arulkumaran, 2006). Other signs include pallor, tachycardia, a forward and lateral positioning of the newborn’s ears as the hematoma extends posteriorly, and increasing head circumference (Mangurten, 2006). Computed tomography (CT) or magnetic resonance imaging is useful in confirming the diagnosis. Replacement of lost blood and clotting factors is required in acute cases of hemorrhage. Monitoring the infant for changes in level of consciousness and a decrease in the hematocrit is also key to early recognition and management. An increase in serum bilirubin levels may occur as a result of the degrading blood cells within the hematoma.
Cephalhematoma
A cephalhematoma forms when blood vessels rupture during labor or delivery to produce bleeding into the area between the bone and its periosteum. The injury occurs most often with primiparous women and is often associated with forceps delivery and vacuum extraction. Unlike caput succedaneum, the boundaries of the cephalhematoma are distinguishable and do not extend beyond the limits of the bone (Fig. 9-1, C). The cephalhematoma may involve one or both parietal bones but rarely affects the occipital and frontal bones. The swelling is usually minimum or absent at birth and increases in size on the second or third day. Blood loss is usually not significant.
No treatment is indicated for uncomplicated cephalhematoma. Most lesions are absorbed within 2 weeks to 3 months. Lesions that result in severe blood loss to the area or that involve an underlying fracture require further evaluation. Hyperbilirubinemia may result during resolution of the hematoma. A local infection can develop and is suspected when swelling suddenly increases.
Nursing Care Management
Nursing care involves assessment and observation of the common scalp injuries and vigilance in observing for possible associated complications such as skin breakdown, infection, or, rarely, acute blood loss and hypovolemia. Because caput and cephalhematoma injuries resolve spontaneously, parents need reassurance of their usual benign nature.
Fractures
Fracture of the clavicle, or collarbone, is the most common birth injury. It is often associated with difficult vertex or breech deliveries of infants of greater-than-average size. Further examination usually reveals crepitus (the coarse, crackling sensation produced by the rubbing together of fractured bone fragments), and radiographs usually reveal a complete fracture with overriding of the fragments. A palpable spongy mass, representing localized edema and hematoma, is also a sign of a fractured clavicle.
The newborn with a fractured clavicle may have no symptoms, but the nurse should suspect a fracture if an infant has limited use of the affected arm, malpositioning of the arm, an asymmetric Moro reflex, or focal swelling or tenderness or cries when the arm is moved. Eliciting the scarf sign (extending arm across chest toward opposite shoulder) for assessment of gestational age is contraindicated if a fractured clavicle is suspected.
In neonates, fractures of long bones, such as the femur or the humerus, are difficult to detect by radiographic examination. Although osteogenesis imperfecta is a rare finding, assess a newborn infant with a fracture for other evidence of this congenital disorder.
Fractures of the neonatal skull are uncommon. The bones, which are less mineralized and more compressible than bones in older infants and children, are separated by membranous seams that allow the head contour to adjust to the birth canal during delivery. Skull fractures usually follow a prolonged, difficult delivery or forceps extraction. Most fractures are linear, but some may be visible as depressed indentations that compress or decompress like a Ping-Pong ball. Management of depressed skull fractures is controversial; many resolve without intervention. Nonsurgical elevation of the indentation using a hand breast pump or vacuum extractor has been reported (Mangurten, 2006). Surgery may be required in the presence of bone fragments or signs of increased intracranial pressure. A similar finding in neonates is craniotabes, which is usually benign or may be associated with prematurity, rickets, or hydrocephalus. In this condition the cranial bone(s) moves freely on palpation and may be easily compressed.
Nursing Care Management
Often no intervention is prescribed other than proper body alignment, careful dressing and undressing of the infant, and handling and carrying techniques that support the affected bone. If the infant has a fractured clavicle, it is important to support the upper and lower back rather than pull the infant up from under the arms. Occasionally, for immobilization and relief of pain, the arm on the side of the fractured clavicle may be abducted at more than 60 degrees with the elbow flexed at more than 90 degrees for 7 to 10 days (Mangurten, 2006).
Linear skull fractures usually require no treatment. A Ping-Pong–type fracture may require decompression by surgical intervention. The infant is carefully observed for signs of neurologic complications. The parents of an infant with a fracture of any bone should be involved in caring for the infant during hospitalization as part of discharge planning for care at home. Evaluate any newborn who is large for gestational age and delivered vaginally for a fractured clavicle. The newborn with a fractured clavicle may have no symptoms, but suspect a fracture if the infant has limited use of the affected arm, malpositioning of the arm, an asymmetric Moro reflex, or focal swelling or tenderness or cries in pain when the arm is moved.
Paralyses
Pressure on the facial nerve (the seventh cranial nerve) during delivery may result in injury to the nerve. The primary clinical manifestations are loss of movement on the affected side, such as an inability to completely close the eye, drooping of the corner of the mouth, and absence of wrinkling of the forehead and nasolabial fold (Fig. 9-2). The paralysis is most noticeable when the infant cries. The mouth is drawn to the unaffected side, the wrinkles are deeper on the normal side, and the eye on the involved side remains open.
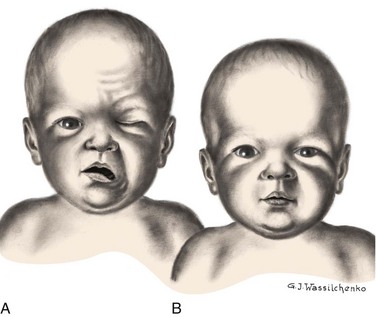
Fig. 9-2 A, Paralysis of right side of face 15 minutes after forceps delivery. Absence of movement on affected side is especially noticeable when infant cries. B, Same infant 24 hours later.
No medical intervention is necessary. The paralysis usually disappears spontaneously in a few days but may take as long as several months.
Brachial Palsy
Brachial plexus injury results from forces that alter the normal position and relationship of the arm, shoulder, and neck. Erb palsy (Erb-Duchenne paralysis) is caused by damage to the upper plexus and usually results from stretching or pulling away of the shoulder from the head, as might occur with shoulder dystocia or with a difficult vertex or breech delivery. Other identified risk factors include an infant with birth weight of over 4000 g (8.8 lb), a second stage of labor of less than 15 minutes, maternal body mass index greater than 29, and a vacuum-assisted extraction (Hudic, Fatusic, Sinanovic, et al, 2006). The less common lower plexus palsy, or Klumpke palsy, results from severe stretching of the upper extremity while the trunk is relatively less mobile.
The clinical manifestations of Erb palsy are related to the paralysis of the affected upper extremity and muscles. The arm hangs limp alongside the body. The shoulder and arm are adducted and internally rotated. The elbow is extended, and the forearm is pronated, with the wrist and fingers flexed; a grasp reflex may be present because finger and wrist movement remain normal, but the Moro reflex is absent (Tappero, 2009) (Fig. 9-3). In lower plexus palsy the muscles of the hand are paralyzed, with consequent wrist drop and relaxed fingers. In a third and more severe form of brachial palsy, total plexus injury, the entire arm and hand are paralyzed and hang limp and motionless at the side. The Moro reflex is absent on the affected side for all forms of brachial palsy.
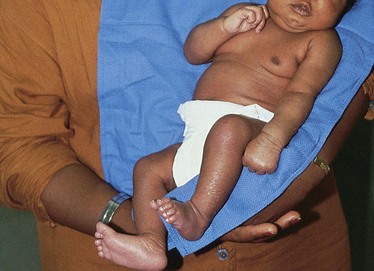
Fig. 9-3 Left-sided brachial plexus (Erb-Duchenne) palsy. Note extended, internally rotated arm and pronated wrist on affected side.
Treatment of the affected arm is aimed at preventing contractures of the paralyzed muscles and maintaining correct placement of the humeral head within the glenoid fossa of the scapula. Complete recovery from stretched nerves usually takes 3 to 6 months. Full recovery is expected in 88% to 92% of infants (Paige and Moe, 2006). However, avulsion of the nerves (complete disconnection of the ganglia from the spinal cord that involves both anterior and posterior roots) results in permanent damage. For those injuries that do not improve spontaneously by 3 months, surgical intervention may be needed to relieve pressure on the nerves or to repair the nerves with grafting (Joyner, Soto, and Adam, 2006). In some cases injection of botulinum toxin A into the pectoralis major muscle may be effective in reducing muscle contractures after birth-related brachial plexus injuries (Price, Ditaranto, Yaylali, et al, 2007).
Phrenic Nerve Paralysis
Phrenic nerve paralysis results in diaphragmatic paralysis as demonstrated by ultrasonography, which shows paradoxic chest movement and an elevated diaphragm. Initially, chest radiography may not demonstrate an elevated diaphragm if the neonate is receiving positive pressure ventilation. The injury sometimes occurs in conjunction with brachial palsy. Respiratory distress is the most common and important sign of injury. Because injury to the phrenic nerve is usually unilateral, the lung on the affected side does not expand and respiratory efforts are ineffectual. Breathing is primarily thoracic, and cyanosis, tachypnea, or complete respiratory failure may be seen. Pneumonia and atelectasis on the affected side may also occur.
Nursing Care Management
Nursing care of the infant with facial nerve paralysis involves aiding the infant in sucking and helping the mother with feeding techniques. A comprehensive evaluation of the infant’s oral motor skills by an infant feeding specialist is recommended to develop an effective multidisciplinary feeding regimen. Because part of the mouth cannot close tightly around the nipple, the use of a soft rubber nipple with a large hole may be helpful but should be used carefully to prevent choking. The infant may require partial gavage feeding and supplemental oral stimulation with a minimum amount of formula to prevent aspiration. Breast-feeding is not contraindicated, but the mother will need assistance in helping the infant grasp and compress the areolar area.
If the lid of the eye on the affected side does not close completely, instill artificial tears as needed to prevent drying of the conjunctiva, sclera, and cornea. The lid is often taped shut to prevent injury. If the infant requires eye care at home, teach the parents the procedure for administering eye drops before the infant is discharged from the nursery. (See Chapter 27.)
Nursing care of the newborn with brachial palsy is concerned primarily with proper positioning of the affected arm. The affected arm should be gently immobilized on the upper abdomen; passive range-of-motion exercises of the shoulder, wrist, elbow, and fingers are initiated at 7 to 10 days of age (Joyner, Soto, and Adam, 2006). Wrist flexion contractures may be prevented with the use of supportive splints. In dressing the infant, give preference to the affected arm. Undressing begins with the unaffected arm, and redressing begins with the affected arm to prevent unnecessary manipulation and stress on the paralyzed muscles. Teach parents to use the “football” position when holding the infant and to avoid picking the child up from under the axillae or by pulling on the arms.
The infant with phrenic nerve paralysis requires the same nursing care as any infant with respiratory distress. The family’s emotional needs are also an important part of nursing care; the family needs reassurance regarding the neonate’s progress toward an optimal outcome. Follow-up care is also essential because of the extended length of recovery. Parents may wish to contact the Brachial Plexus Palsy Foundation and visit the website for further information.*
Dermatologic Problems in the Newborn
Erythema toxicum neonatorum, also known as flea bite dermatitis or newborn rash, is a benign, self-limiting eruption that usually appears within the first 2 days of life. The 1- to 3-mm lesions are firm, pale yellow or white papules or pustules on an erythematous base, which resemble flea bites. Erythema toxicum may appear as one or two isolated “flea bites” or as multiple lesions; the rash commonly disappears from one location and reappears elsewhere hours later. The rash appears most commonly on the face, proximal extremities, trunk, and buttocks, but it may be located anywhere on the body except the palms and soles. The rash may be more obvious during crying episodes. There are no systemic manifestations, and successive crops of lesions heal without pigmentation changes. The rash usually lasts approximately 5 to 7 days.
The cause is unknown. However, a smear of the pustule shows numerous eosinophils and a relative absence of neutrophils. Obtain bacterial, fungal, or viral cultures when the diagnosis is questionable. Although no treatment is necessary, parents are usually concerned about the rash and need to be reassured of its benign and transient nature.
Candidiasis
Candidiasis, also known as moniliasis, is not uncommon in the newborn. Candida albicans, the organism usually responsible, may cause disease in any organ system. It is a yeastlike fungus (produces yeast cells and spores) that can be acquired from a maternal vaginal infection during delivery; by person-to-person transmission (especially from poor hand-washing technique); or from contaminated hands, bottles, nipples, or other articles. Mucocutaneous, cutaneous, and disseminated candidiasis are observed in this age-group. It is usually a benign disorder in the neonate and is often confined to the oral and diaper regions. (See Diaper Dermatitis, Chapter 13.)
Oral candidiasis (thrush) is characterized by white adherent patches on the tongue, palate, and inner aspects of the cheeks. Oral candidiasis can be distinguished from coagulated milk when attempts to remove the patches with a tongue blade are unsuccessful. The infant may refuse to suck or may feed poorly because of pain in the mouth. This condition tends to be acute in the newborn (rarely appears in first week of life) and chronic in older infants and young children. Thrush appears when the oral flora is altered as a result of antibiotic therapy or poor hand washing by the infant’s caregiver. Although the disorder is usually self-limiting, spontaneous resolution may take as long as 2 months, during which time lesions may spread to the larynx, trachea, bronchi, and lungs and along the gastrointestinal tract.
The disease is treated with good hygiene, application of a fungicide, and correction of any underlying disturbance. The source of infection, usually the mother, should be treated to prevent reinfection. Topical application of 1 ml of nystatin (Mycostatin) over the surfaces of the oral cavity four times a day or every 6 hours is usually sufficient to prevent spread of the disease or prolongation of its course. Several other drugs may be used, including amphotericin B (Fungizone), clotrimazole (Lotrimin, Mycelex), fluconazole (Diflucan), or miconazole (Monistat, Micatin) given intravenously, orally, or topically. To prevent relapse, therapy should be continued for at least 2 days after the lesions disappear (Lawrence and Lawrence, 2005). Gentian violet solution may be used in addition to one of the antifungal drugs in chronic cases of oral thrush; however, the former does not treat gastrointestinal Candida organisms and may irritate the oral mucosa.
Nursing Care Management
Direct nursing care toward preventing spread of the infection and correct application of the prescribed topical medication. For candidiasis in the diaper area, teach the caregiver to keep the diaper area clean and to apply the medication to affected areas as prescribed. (See Diaper Dermatitis, Chapter 13.) Older infants can introduce Candida organisms into their mouths with hands contaminated by contact with diaper dermatitis.
In cases of oral thrush, administer nystatin after feedings. Distribute the medication over the surface of the oral mucosa and tongue with an applicator or syringe; the remainder of the dose is deposited in the mouth to be swallowed by the infant to treat any gastrointestinal lesions. In addition to good hygienic care, other measures to control thrush include rinsing the infant’s mouth with plain water after each feeding before applying the medication and boiling reusable nipples and bottles for at least 20 minutes after a thorough washing (spores are heat resistant). Boil pacifiers for at least 20 minutes once daily, and treat the nipples of breast-feeding mothers to prevent reinfection. If the mother is breast-feeding, simultaneous treatment of the infant and mother is recommended if either is infected (Lawrence and Lawrence, 2005).
Herpes
Neonatal herpes is one of the most serious viral infections in the newborn, with a mortality rate of up to 60% in infants with disseminated disease. The disease may be classified according to the following types: (1) skin, eye, and mouth; (2) localized central nervous system (CNS) disease; or (3) disseminated infection involving multiple sites such as the lungs, liver, adrenal glands, CNS, skin, eyes, and mouth. Approximately 86% to 90% of herpes simplex virus (HSV) transmission occurs during delivery. The rash appears as vesicles or pustules on an erythematous base. Clusters of lesions are common. The lesions ulcerate and crust over rapidly. Fetal scalp monitoring sites are commonly the primary site of infection. The risk of infection during vaginal birth in the presence of genital herpes is estimated to be as high as 57% with active primary infection at term (Brown, Wald, Morrow, et al, 2003). However, in up to 80% of cases of neonatal HSV infection, the mother has no history or symptoms of infection at the time of birth, but serologic testing reveals evidence of the herpes virus (Kimberlin, 2005).
Most infants with neonatal herpes eventually develop this characteristic rash, but up to 20% of neonates with disseminated disease do not develop a skin rash (Kimberlin, 2007). Ophthalmologic clinical findings include chorioretinitis and microphthalmia; neurologic involvement such as microcephaly and encephalomalacia may also develop (Kimberlin, 2007). Disseminated infections may involve virtually every organ system, but the liver, adrenal glands, and lungs are most commonly affected. In HSV meningitis infants develop multiple lesions of cortical hemorrhagic necrosis. It can occur alone or with oral, eye, or skin lesions. The presenting symptoms, which may occur in the second to fourth week of life, include lethargy, poor feeding, irritability, and local or generalized seizures.
Infants with CNS and disseminated disease have a much higher mortality rate than those initially seen with skin, eye, or mouth disease. Neonatal HSV may be difficult to detect in the early newborn period, and nonspecific signs such as irritability, fever, poor feeding, or lethargy may be seen. When the diagnosis is delayed, mortality may be high even with antiviral therapy, and long-term irreversible complications such as seizures, blindness, and psychomotor and learning delays are not uncommon.
Nursing Care Management
Neonates with herpes virus or suspected infection (as a result of exposure) should be carefully evaluated for clinical manifestations. The absence of skin lesions in the neonate exposed to maternal herpes virus does not indicate absence of disease. Institute Contact Precautions (in addition to Standard Precautions) according to American Academy of Pediatrics and American College of Obstetricians and Gynecologists (2007) guidelines or hospital protocol. It is recommended that swabs of the mouth, nasopharynx, conjunctivae, rectum, and any skin vesicles be obtained from the exposed neonate. In addition, obtain urine, stool, blood, and cerebrospinal fluid specimens for culture. Antiviral therapy with acyclovir is initiated if the cultures are positive or if there is strong suspicion of herpes infection (American Academy of Pediatrics, 2009b).
Early recognition and treatment with antiviral therapy are key to the prevention of serious and often fatal complications. Closely evaluate for the disease infants who are seen in the first 5 or 6 weeks of life with the nonspecific signs of poor feeding, lethargy, fever, and irritability, with or without the characteristic rash.
Bullous Impetigo
Bullous impetigo is an infectious superficial skin condition most often caused by various strains of Staphylococcus aureus. Bullous vesicular lesions erupt on previously untraumatized or intact skin. The lesions may appear on any body surface and sometimes become widespread, but the usual distribution involves the buttocks, perineum, trunk, face, and extremities. The neonatal form may appear first in the diaper region (Morelli, 2007). They vary in size from a few millimeters to several centimeters, contain turbid fluid, and are easily ruptured (Morelli, 2007). The bullae rupture in 1 or 2 days, leaving a superficial red, moist, denuded area with little crusting. In some cases the condition may be mistaken for thermal injury or staphylococcal scalded skin syndrome (SSSS). Bullous impetigo lesions develop on intact skin, whereas lesions of SSSS spread systemically from an original infection site. There is no cutaneous sensitivity with bullous impetigo, and the Gram stain and blister cultures are positive for staphylococci.
Treatment usually involves the administration of oral antibiotics and topical application of mupirocin (Bactroban). Systemic treatment with erythromycin may be required if the lesions are near the mouth or in the event of abscess formation. Recovery is usually rapid and uneventful.
Nursing Care Management
Once the diagnosis is suspected, the infant is isolated until therapy is instituted to prevent spread of the infection to other infants. Persons who have come in contact with the infant are investigated to determine a possible source of the infecting organism. Scrutinize other infants who have mutual contacts for early detection of any infection. Instruct parents and other visitors regarding precautions for the prevention of infection, especially through hand washing and Standard Precautions. (See Infection Control, Chapter 27.)
To prevent older infants from scratching the lesions, the arms may need to be confined by using elbow restraints, by pulling the undershirt sleeves over the hands and securing the openings with tape, or by applying mittens. If restraints of any kind are used, the infant is allowed freedom of movement at supervised times. Rocking, cuddling, and holding during feeding are essential components of care.
Birthmarks
Discolorations of the skin are common findings in the newborn infant. (See discussion on skin assessment under Physical Assessment, Chapter 8.) Most, such as mongolian spots or telangiectatic nevi, involve no therapy other than reassuring parents of the benign nature of these discolorations. However, some can be the manifestation of a disease that suggests further examination of the child and other family members (e.g., multiple flat, light brown café-au-lait spots often characterize the autosomal dominant hereditary disorder neurofibromatosis and are common findings in Albright syndrome).
Darker or more extensive lesions demand further inspection. Excision of the lesion is recommended when feasible or for biopsy. Such lesions include the reddish brown solitary nodule that appears on the face or upper arm and usually represents a spindle and epithelioid cell nevus (juvenile melanoma); a giant pigmented nevus (bathing trunk nevus), a dark brown to black irregular plaque that is at risk of transformation to malignant melanoma; and the dark brown or black macules that become more numerous with age (junctional or compound nevi).
Vascular birthmarks may be divided into vascular malformations and vascular tumors (hemangiomas). Experts now recommend labeling vascular tumors as hemangiomas of infancy or infantile hemangiomas to differentiate them from other vascular tumors and malformations. Hemangiomas may be further classified as localized, segmental, or multifocal (Miller and Frieden, 2005). Localized superficial hemangiomas tend to appear early in infancy and spontaneously resolve without therapy within several years, whereas the segmental variety is more likely to cause complications such as ulceration and vital organ compromise and to involve developmental defects. Multifocal hemangiomas are less likely to be associated with the complications seen with the segmental variety (Miller and Frieden, 2005). This discussion focuses only on the more common hemangiomas of infancy.
Vascular stains (malformations) are permanent lesions that are present at birth and are initially flat and erythematous. Any vascular structure—capillary, vein, artery, or lymphatic—may be involved. The two most common vascular stains are port-wine stains (nevus flammeus) and transient macular stains such as the stork bite or salmon patch, usually located on the glabella or nape of the neck. Port-wine lesions are pink, red, or, rarely, purple stains of the skin that thicken, darken, and proportionately enlarge as the child grows (Fig. 9-4, A).
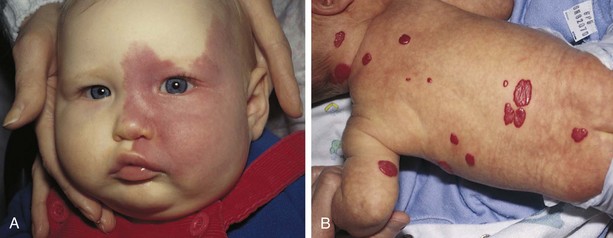
Fig. 9-4 A, Port-wine stain. B, Strawberry hemangioma. (From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 4, St Louis, 2002, Mosby.)
Port-wine stains may also be associated with structural malformations, such as glaucoma or leptomeningeal angiomatosis (tumor of blood or lymph vessels in the pia arachnoid, or Sturge-Weber syndrome) or bony or muscular overgrowth (Klippel-Trénaunay-Weber syndrome). Monitor children with port-wine stains on the eyelids, forehead, cheeks, or extremities for these syndromes with periodic ophthalmologic examination, neurologic imaging, and measurement of extremities.
The treatment of choice for port-wine stains is the flashlamp pulsed dye laser. The child’s skin is reported to respond to therapy and have fewer side effects than in adults (Stier, Glick, and Hirsch, 2008). A series of treatments is usually needed (see Atraumatic Care box). The treatments can significantly lighten or completely clear the lesions with almost no scarring or pigment change.
Infantile hemangiomas, also sometimes referred to as strawberry or capillary hemangiomas, are benign cutaneous tumors that involve only capillaries. These are often not apparent at birth but may appear within a few weeks as an erythematous patch, enlarge considerably during the first year of life and then begin to involute spontaneously. It may take 5 to 12 years for complete resolution. As many as 50% of patients may be left with residual findings such as telangiectasia, redundant fatty tissue, or skin atrophy (Alster and Railan, 2006). These hemangiomas are bright red, rubbery nodules with a rough surface and a well-defined margin (Fig. 9-4, B). A relationship has been established between infantile hemangiomas and placental tissue (Metry, 2004). One study demonstrated that low birth weight was the most significant risk factor for infantile hemangioma (Drolet, Swanson, Frieden, et al, 2008).
Cavernous venous hemangiomas involve deeper vessels in the dermis and have a bluish red color and poorly defined margins. These latter forms may be associated with the trapping of platelets (Kasabach-Merritt syndrome) and subsequent thrombocytopenia.
Hemangiomas may also occur as part of the PHACE syndrome:
P—Posterior fossa brain malformation
H—Hemangiomas (segmental cervicofacial)
Most cavernous venous hemangiomas are large defects and are located on the face (Miller and Frieden, 2005). This neurocutaneous syndrome is diagnosed by the presence of a facial hemangioma in addition to either one or several of the other associated conditions; clinical outcomes vary according to the organs involved.
Although many localized superficial hemangiomas require no treatment because of their high rate of spontaneous involution, some vision and airway obstruction may necessitate therapy. Ulceration is a common complication, especially when the hemangioma is perineal or perioral. This may result in pain, bleeding, infection, and scarring. The pulsed dye laser can effectively reduce some hemangiomas; systemic prednisone administered for 2 to 3 weeks or longer may also deter further growth. Optional treatments may include interferon alfa, imiquimod, vincristine, bleomycin, cyclophosphamide, becaplermin, debulking surgery, and no treatment (Pandey, Gangopadhyay, and Upadhyay, 2008; Stier, Glick, and Hirsch, 2008).
Nursing Care Management
Birthmarks, especially those on the face, are upsetting to parents. Families need an explanation of the type of lesion, its significance, and possible treatment.* They can benefit from seeing photographs of other infants before and after treatment for port-wine stains or after the passage of time for hemangiomas. Pictures taken to follow the involution process may further help parents gain confidence that progress is taking place.
If laser therapy is performed, the lesion will have a purplish black appearance for 7 to 10 days, after which the blackness will fade and give way to redness with an eventual lightening of the treated area. During the treatment phase caution parents to avoid any trauma to the lesion or picking at the scab. Trim the infant’s fingernails as an added precaution. Washing the area gently with water and dabbing it dry is adequate, although in some cases a topical antibiotic ointment may be used. Do not give any salicylates during the treatment phase because they decrease the effects of the therapy. Keep the infant out of the sun for several weeks and then protected with a sunscreen of at least SPF 15. Complications associated with laser treatment include possible secondary infection, keloid or pyogenic granuloma formation, localized dermatitis, and hyperpigmentation or hypopigmentation.
Problems Related to Physiologic Factors
The term hyperbilirubinemia refers to an excessive level of accumulated bilirubin in the blood and is characterized by jaundice, or icterus, a yellowish discoloration of the skin and other organs. Hyperbilirubinemia is a common finding in the newborn and in most instances is relatively benign. However, in extreme cases, it can indicate a pathologic state.
 Critical Thinking Case Study—Hyperbilirubinemia
Critical Thinking Case Study—Hyperbilirubinemia
Hyperbilirubinemia may result from increased unconjugated or conjugated bilirubin. The unconjugated form (Table 9-1) is the type most commonly seen in newborns. The following discussion of hyperbilirubinemia is limited to unconjugated hyperbilirubinemia.
TABLE 9-1
COMPARISON OF MAJOR TYPES OF UNCONJUGATED HYPERBILIRUBINEMIA*

*Table depicts patterns of jaundice in term infants; patterns in preterm infants will vary according to factors such as gestational age, birth weight, and illness.
Pathophysiology
Bilirubin is one of the breakdown products of hemoglobin that results from red blood cell (RBC) destruction. When RBCs are destroyed, the breakdown products are released into the circulation, where the hemoglobin splits into two fractions: heme and globin. The globin (protein) portion is used by the body, and the heme portion is converted to unconjugated bilirubin, an insoluble substance bound to albumin.
In the liver the bilirubin is detached from the albumin molecule and, in the presence of the enzyme glucuronyl transferase, is conjugated with glucuronic acid to produce a highly soluble substance, conjugated bilirubin glucuronide, which is then excreted into the bile. In the intestine, bacterial action reduces the conjugated bilirubin to urobilinogen, the pigment that gives stool its characteristic color. Most of the reduced bilirubin is excreted through the feces; a small amount is eliminated in the urine (Fig. 9-5).
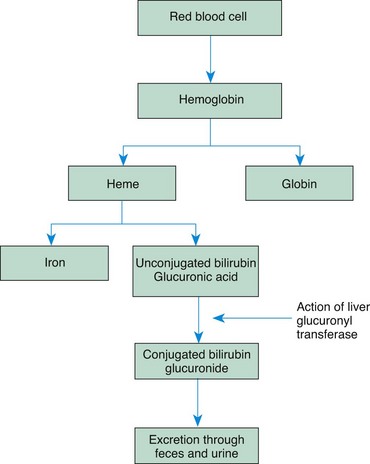
Normally the body is able to maintain a balance between the destruction of RBCs and the use or excretion of by-products. However, when developmental limitations or a pathologic process interferes with this balance, bilirubin accumulates in the tissues to produce jaundice. Possible causes of hyperbilirubinemia in the newborn are:
• Physiologic (developmental) factors (prematurity)
• An association with breast-feeding or breast milk
• Excess production of bilirubin (e.g., hemolytic disease, biochemical defects, bruises)
• Disturbed capacity of the liver to secrete conjugated bilirubin (e.g., enzyme deficiency, bile duct obstruction)
• Combined overproduction and underexcretion (increased hemolytic process)
• Some conditions or disease states (e.g., glucose-6-phosphate dehydrogenase [G6PD] deficiency, hypothyroidism, galactosemia, infant of a diabetic mother)
• Genetic predisposition to increased production (Native Americans, Asians)
The first two causes, physiologic factors and an association with breast-feeding, are discussed in the following sections; the third major cause, hemolytic disease, is presented on p. 295.
Complications: Unconjugated bilirubin is highly toxic to neurons; therefore an infant with severe hyperbilirubinemia is at risk of developing bilirubin encephalopathy, a term that describes varying degrees of CNS damage resulting from the deposition of unconjugated bilirubin in brain cells. Kernicterus describes the yellow staining of the brain cells that may result in bilirubin encephalopathy. The damage occurs when the serum concentration reaches toxic levels, regardless of cause. There is evidence that a fraction of unconjugated bilirubin crosses the blood-brain barrier in neonates with physiologic hyperbilirubinemia. When certain pathologic conditions exist in addition to elevated bilirubin levels, the infant has an increased permeability of the blood-brain barrier to unconjugated bilirubin and, thus, potential irreversible damage. The exact level of serum bilirubin required to cause damage is not yet known.
Multiple factors contribute to bilirubin neurotoxicity; therefore serum bilirubin levels alone do not predict the risk of CNS injury. Factors that enhance the development of bilirubin encephalopathy include acidosis, lowered serum albumin levels, intracranial infections such as meningitis, and abrupt fluctuations in blood pressure. In addition, any condition that increases the metabolic demands for oxygen or glucose (e.g., fetal distress, hypoxia, hypothermia, or hypoglycemia) also increases the risk of CNS damage despite lower serum levels of bilirubin. The administration of hypertonic solutions such as glucose and sodium bicarbonate in acutely ill infants, which causes a sudden rise in serum osmolality, has also been a contributing factor in the development of bilirubin encephalopathy.
The signs of bilirubin encephalopathy are those of CNS depression or excitation. Prodromal symptoms consist of decreased activity, lethargy, irritability, hypotonia, and seizures. Later these subtle findings are followed by development of athetoid cerebral palsy, cognitive delay, and deafness (Sgro, Shah, and Campbell, 2005). Long-term effects include evidence of neurologic damage, such as cognitive impairment, attention deficit hyperactivity disorder, delayed or abnormal motor movement (especially ataxia or athetosis), behavior disorders, perceptual problems, or sensorineural hearing loss.
Physiologic Jaundice
The most common cause of hyperbilirubinemia is the relatively mild and self-limited physiologic jaundice, or icterus neonatorum. Unlike hemolytic disease of the newborn (HDN) (see p. 295), physiologic jaundice is not associated with any pathologic process. Although almost all newborns experience elevated bilirubin levels, only about half demonstrate observable signs of jaundice.
Critical Thinking Exercise—Jaundice
Two phases of physiologic jaundice have been identified in full-term infants. In the first phase, bilirubin levels of formula-fed Caucasian and African-American infants gradually increase to approximately 5 to 6 mg/dl by 3 to 4 days of life, then decrease to a plateau of 2 to 3 mg/dl by the fifth day (Blackburn, 2007). Bilirubin levels maintain a steady plateau state in the second phase without increasing or decreasing until approximately 12 to 14 days, at which time levels decrease to the normal value of 1 mg/dl (Blackburn, 2007). This pattern varies according to racial group, method of feeding (breast versus bottle), and gestational age. In preterm formula-fed infants, serum bilirubin levels may peak as high as 10 to 12 mg/dl at 5 or 6 days of life and decrease slowly over a period of 2 to 4 weeks (Blackburn, 2007).
As noted above, infants of Asian descent (as well as Native Americans) have mean bilirubin levels almost twice those seen in Caucasians or African-Americans. An increased incidence of hyperbilirubinemia occurs in newborns from certain geographic areas, particularly areas around Greece (see Cultural Competence box). These populations may have G6PD deficiency, which can cause acute hemolytic anemia. Hyperbilirubinemia also develops in a small number of newborns with Crigler-Najjar syndrome, an inherited disorder in which there is an absence of glucuronyl transferase. Infants with metabolic disorders such as galactosemia or hypothyroidism may also develop hyperbilirubinemia.
 CULTURAL COMPETENCE
CULTURAL COMPETENCE
Risk Factors for Hyperbilirubinemia
Neonates of East Asian ethnicity (China, Taiwan, Macao, Hong Kong, Japan, and Korea) are at higher risk for high mean serum bilirubin levels than neonates of any different ethnic origin. The apparent reason for this is the increased presence of certain genes in the East Asian population that modulate bilirubin metabolism in the liver (Watchko, 2009). Exclusive breast-feeding is another risk factor for neonatal hyperbilirubinemia. Watchko (2009) stresses that a combination of risk factors increases the newborn’s likelihood of developing hyperbilirubinemia. Therefore infants of East Asian mothers who are breast-feeding should be carefully evaluated during the early neonatal period (first week of life) for elevated serum bilirubin levels.
Mechanisms Involved in Physiologic Jaundice: On average, newborns produce twice as much bilirubin as do adults because of higher concentrations of circulating erythrocytes and a shorter life span of RBCs (only 70 to 90 days, in contrast to 120 days in older children and adults). In addition, the liver’s ability to conjugate bilirubin is reduced because of limited production of glucuronyl transferase. Newborns also have a lower plasma-binding capacity for bilirubin because of lower albumin concentrations than older children. Normal changes in hepatic circulation following birth may contribute to excessive demands on liver function.
Normally, conjugated bilirubin is reduced to urobilinogen by the intestinal flora and excreted in feces. However, the relatively sterile and less motile newborn bowel is initially less effective in excreting urobilinogen. In the newborn intestine the enzyme β-glucuronidase is able to convert conjugated bilirubin into the unconjugated form, which is subsequently reabsorbed by the intestinal mucosa and transported to the liver. This process, known as enterohepatic circulation or enterohepatic shunting, is accentuated in the newborn and is thought to be a primary mechanism in physiologic jaundice (Blackburn, 2007). Feeding (1) stimulates peristalsis and produces more rapid passage of meconium, thus diminishing the amount of reabsorption of unconjugated bilirubin; and (2) introduces bacteria to aid in the reduction of bilirubin to urobilinogen. Colostrum, a natural cathartic, facilitates meconium evacuation.
Jaundice in Breast-Feeding Infants
Breast-feeding is associated with an increased incidence of jaundice. Two types have been identified. Breast-feeding–associated jaundice (early-onset jaundice) begins at 2 to 4 days of age and occurs in approximately 12% to 35% of breast-fed newborns (Blackburn, 2007). The jaundice is related to the process of breast-feeding and probably results from decreased caloric and fluid intake by breast-fed infants before the milk supply is well established, since fasting is associated with decreased hepatic clearance of bilirubin. A decrease in milk (fluid) intake may result in dehydration, which also concentrates the circulating bilirubin in the blood; however, supplemental fluids such as glucose water or water do not enhance bilirubin excretion and may delay the excretion process.
Breast milk jaundice (late-onset jaundice) begins around the fourth day and occurs in 2% to 4% of breast-fed infants (Blackburn, 2007). Rising levels of bilirubin peak during the second week and gradually diminish. Despite high levels of bilirubin that may persist for 3 to 12 weeks, these infants are well. The jaundice may be caused by factors in the breast milk (pregnanediol, fatty acids, and β-glucuronidase) that either inhibit the conjugation or decrease the excretion of bilirubin. Less frequent stooling by breast-fed infants may allow extended time for reabsorption of bilirubin from the intestine via the enterohepatic route described above (see Table 9-1).
Clinical Manifestations
The most obvious sign of hyperbilirubinemia is jaundice, the yellowish discoloration primarily of the sclera, nails, or skin. As a rule, jaundice that appears within the first 24 hours is caused by HDN, sepsis, or one of the maternally derived diseases such as diabetes mellitus or infections. Jaundice that appears on the second or third day, peaks on the third to fifth day, and declines on the fifth to seventh day is usually the result of physiologic jaundice; as noted above, this pattern may vary according to ethnic origin. The intensity of the jaundice is not always related to the degree of hyperbilirubinemia; therefore serum bilirubin levels are necessary.
Diagnostic Evaluation
Total serum bilirubin is measured to determine the degree of hyperbilirubinemia. Normal values of unconjugated bilirubin are 0.2 to 1.4 mg/dl. In the newborn, levels must exceed 5 mg/dl before jaundice (icterus) is observable. However, evaluation of jaundice is not based solely on serum bilirubin levels, but also on the timing of the appearance of clinical jaundice; gestational age at birth; age in days since birth; family history, including maternal Rh factor; evidence of hemolysis; feeding method; infant’s physiologic status; and progression of serial serum bilirubin levels. The following criteria are indicators of pathologic jaundice that warrant further investigation as to the cause. It is not an all-inclusive list; other factors are also evaluated:
• Appearance of clinical jaundice within 24 hours of birth
• Persistent clinical jaundice over 2 weeks in full-term, formula-fed infant
• Total serum bilirubin levels over 12.9 mg/dl (term infant) or over 15 mg/dl (preterm infant); upper limit for breast-fed infant: 15 mg/dl
• Increase in serum bilirubin by 5 mg/dl/day
• Direct bilirubin exceeding 1.5 to 2 mg/dl
• Total serum bilirubin level over 95th percentile for age (in hours) on hour-specific risk nomogram (Fig. 9-6, A)
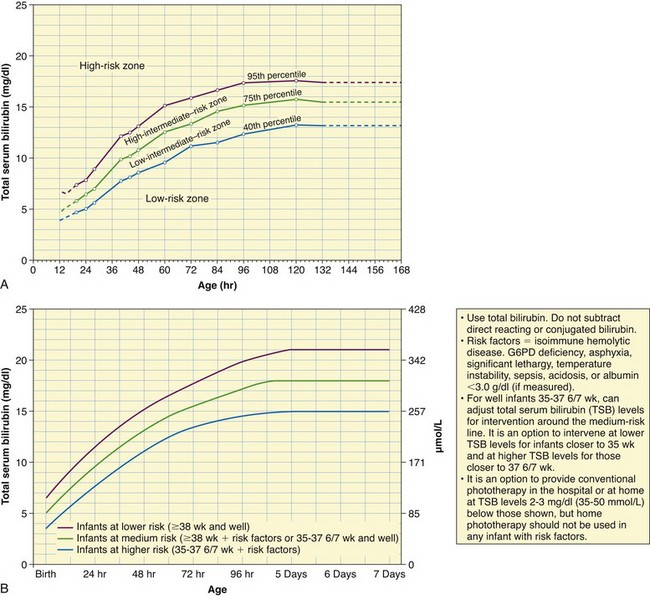
Fig. 9-6 A, Nomogram for designation of risk in 2840 well newborns at 36 or more weeks of gestational age with birth weight of 2000 g (4.4 lb) or more, or 35 or more weeks of gestational age and birth weight of 2500 g (5.5 lb) or more, based on the hour-specific serum bilirubin values. (This nomogram should not be used to represent the natural history of neonatal hyperbilirubinemia.) B, Guidelines for phototherapy in hospitalized infants of 35 or more weeks of gestation. (A, From Bhutani VK, Johnson L, Sivieri EM: Predictive ability of a predischarge hour-specific serum bilirubin for subsequent significant hyperbilirubinemia in healthy term and near-term newborns, Pediatrics 103(1):6-14, 1999. B, From American Academy of Pediatrics, Subcommittee on Hyperbilirubinemia: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114(1):297-316, 2004.)
Following are risk factors that may place the term infant at high risk for hyperbilirubinemia: maternal race (e.g., Asian or Asian American), gestational age 35 to 36 weeks, significant bruising, cephalhematoma or significant bruising, exclusive breastfeeding, blood group incompatibility or hemolytic disease such as G6PD, and history of sibling with hyperbilirubinemia (Watchko, 2009).
Noninvasive monitoring of bilirubin via cutaneous reflectance measurements (transcutaneous bilirubinometry, or TcB) allows for repetitive estimations of total serum bilirubin and, when used correctly, may decrease the need for invasive monitoring. With shorter maternity stays, the value of transcutaneous bilirubin measurements as a screening tool for evaluating the need for obtaining serum bilirubin levels or closely monitoring the infant has received considerable attention. Some TcB monitors provide accurate measurements within 2 to 3 mg/dl in most neonatal populations at serum levels below 15 mg/dl (American Academy of Pediatrics, 2004). Regardless of the screening instrument chosen, it is important to note that, to date, no transcutaneous bilirubin meter measures the total serum bilirubin level, and they must be used according to published guidelines as screening tools, not as predictors of need for therapy. Multiple readings over time at a consistent site (e.g., sternum, forehead) are more valuable than a single reading. Once phototherapy has been initiated, TcB is no longer useful as a screening tool.
The use of hour-specific serum bilirubin levels to predict newborns at risk for rapidly rising levels has now become an official recommendation by the Academy of Pediatrics (2004) for monitoring healthy neonates at more than 35 weeks of gestation before discharge from the hospital. Using a nomogram (see Fig. 9-6, A) with three designated risk levels (high, intermediate, or low risk) of hour-specific total serum bilirubin values assists in determining which newborns might need further evaluation before and after discharge. Universal bilirubin screening based on hour-specific total serum bilirubin, performed possibly at the same time as other routine newborn metabolic screening (e.g., phenylketonuria [PKU], galactosemia), has been recommended (American Academy of Pediatrics, 2004; Bhutani, Johnson, and Keren, 2004). The hour-specific bilirubin risk nomogram is used to determine the infant’s risk for developing hyperbilirubinemia requiring medical treatment or closer screening. Some experts have recently recommended universal hour-specific screening in combination with the clinical risk factors listed above, as well as targeted follow-up to prevent further cases of kernicterus (Maisels, Bhutani, Bogen, et al, 2009).
It is now recommended that healthy late-preterm and term infants (>35 weeks of gestation) receive follow-up care and assessment of bilirubin within 3 days of discharge, if discharged at less than 24 hours, and a risk assessment with the hour-specific nomogram; likewise, newborns discharged at 24 to 47.9 hours should receive follow-up evaluation within 4 days, and those discharged between 48 and 72 hours should receive follow up within 5 days (American Academy of Pediatrics, 2004). The guidelines for monitoring and treating neonatal hyperbilirubinemia are published extensively elsewhere, and the reader is referred to the American Academy of Pediatrics 2004 reference for an in-depth overview of management guidelines.
End-tidal carbon monoxide levels (measured in exhaled breath) may be of value in determining the presence of hemolysis and the rate of heme degradation and bilirubin production in some infants. These determinations are often useful in determining the need for surveillance during the first week of life (American Academy of Pediatrics, 2004).
Therapeutic Management
The primary goals in the treatment of hyperbilirubinemia are to prevent bilirubin encephalopathy and, as in any blood group incompatibility, to reverse the hemolytic process (see p. 288). The main form of treatment involves the use of phototherapy. Exchange transfusion is generally used for reducing dangerously high bilirubin levels that occur with hemolytic disease.
The pharmacologic management of hyperbilirubinemia with phenobarbital has centered primarily on the infant with hemolytic disease and is most effective when given to the mother several days before delivery. Phenobarbital promotes (1) hepatic glucuronyl transferase synthesis, which increases bilirubin conjugation and hepatic clearance of the pigment in bile; and (2) protein synthesis, which may increase albumin for more bilirubin binding sites. However, the use of phenobarbital in either the antenatal or the postnatal period has not proved to be as effective as other treatments in reducing bilirubin. Bilirubin production in the newborn can be decreased by inhibiting heme oxygenase—an enzyme needed for heme breakdown (to biliverdin)—with metalloporphyrins, especially tin protoporphyrin and tin mesoporphyrin. The use of heme-oxygenase inhibitors provides a preventive approach to hyperbilirubinemia (Dennery, 2005).
Healthy late-preterm and full-term infants with jaundice may also benefit from early initiation of feedings and frequent breast-feeding. These preventive measures are aimed at promoting increased intestinal motility, decreased enterohepatic shunting, and normal bacterial flora in the bowel to effectively enhance the excretion of unconjugated bilirubin.
Phototherapy: Phototherapy consists of exposing the infant’s skin to an appropriate light source. Light promotes bilirubin excretion by photoisomerization, which alters the structure of bilirubin to a soluble form (lumirubin).
For phototherapy to be effective, the infant’s skin must be fully exposed to an adequate amount of light or irradiance. When serum bilirubin levels are rapidly increasing or approximating critical levels, double or intensive phototherapy is recommended. This technique often involves the application of phototherapy with lights above the infant and another source of light (e.g., fiberoptic mattress) under the infant (Stokowski, 2006). The goal is to increase irradiance to the 430 to 490 nm band, which provides best results (American Academy of Pediatrics, 2004). Available commercial phototherapy delivery systems are numerous and include halogen spotlights, light-emitting diodes, fluorescent tubes or bank lights, and fiberoptic blankets (Stokowski, 2006). The color of the infant’s skin does not influence the efficacy of phototherapy. Best results occur within the first 24 to 48 hours of treatment. Phototherapy alone is not effective in the management of hyperbilirubinemia when levels are at a critical level or are rising rapidly; it is designed primarily for the treatment of mild to moderate hyperbilirubinemia.
The American Academy of Pediatrics (2004) practice parameter guidelines provide suggestions for initiating phototherapy (Fig. 9-6, B) and for implementing exchange transfusion in infants 35 weeks of gestation or more. The initiation of phototherapy should always be based on individual clinical judgment rather than serum bilirubin levels alone.
Preterm infants presumably have a higher risk of developing pathologic jaundice at lower serum bilirubin levels than healthy full-term infants because of associated illness factors that may alter the blood-brain barrier’s susceptibility to bilirubin. However, evidence-based treatment protocols, especially for preterm infants weighing less than 1500 g (3.3 lb), have not been established (Maisels and McDonough, 2008). Carefully evaluate each infant with other illness and risk factors in mind, rather than depending on absolute values for all infants in a specific group.
Phototherapy has not been found to cause long-term adverse effects. The effectiveness of treatment is determined by a decrease in total serum bilirubin levels. Concurrently, the infant’s total physical status is assessed continually because the suppression of jaundice by phototherapy may mask signs of sepsis, hemolytic disease, or hepatitis.
Management of Breast-Feeding Jaundice: Recommendations for prevention and management of early-onset jaundice in breast-fed infants are to monitor for early stooling; initiate early and frequent breast-feeding; and discourage the use of dextrose water, formula, or water. The infant’s weight, voiding, and stooling should be evaluated along with the breast-feeding pattern (Lawrence and Lawrence, 2005).
Bilirubin levels are monitored in late-onset jaundice, and treatment options vary. If the serum bilirubin levels remain above 16 mg/dl for more than 24 hours, obtain a bilirubin reading 2 hours after breast-feeding, which may then be interrupted for 10 to 12 hours (provide fluid and calories during this time) and repeat levels drawn; with a serum bilirubin level decrease of 2 mg/dl or more and levels below 15 mg/dl, the infant may resume breast-feeding. If levels do not drop significantly, further evaluation is necessary (Lawrence and Lawrence, 2005). It is not within the scope of this text to discuss the full spectrum of treatment possibilities; therefore consult other sources. Whenever possible, offer parents the option of continuing breast-feeding, provided that the jaundiced infant is closely monitored for additional contributing factors. Home phototherapy and continued breast-feeding are options for the family with a jaundiced newborn.
Prognosis: Early recognition and treatment of neonatal hyperbilirubinemia prevent unnecessary medical therapies, parent-infant separation, breast-feeding disruption and possibly failure, and bilirubin encephalopathy. The characteristic features of bilirubin encephalopathy include sensorineural hearing loss, dental enamel hypoplasia, gaze paralysis, athetosis (involuntary writhing movements), and delayed motor skills; intellectual impairment is reported to be mild.
Nursing Care Management
Part of the routine physical assessment includes observing for evidence of jaundice at regular intervals. Jaundice is most reliably assessed by observing the infant’s skin color from head to toe and the color of the sclerae and mucous membranes. Applying direct pressure to the skin, especially over bony prominences such as the tip of the nose or the sternum, causes blanching and allows the yellow stain to be more pronounced. Also, bilirubin (especially at high levels) is not uniformly distributed in skin. The nurse observes the infant in natural daylight for a true assessment of color.
The transcutaneous bilirubin meter is a useful screening device to detect neonatal jaundice in full-term infants. Because phototherapy reduces the accuracy of the instrument, its value is limited to assessments made before the initiation of phototherapy. Blood samples are also taken for the measurement of bilirubin in the laboratory.
In many cases, jaundice may appear after discharge in the term and late-preterm infant. A careful history from the parents may reveal significant familial patterns of hyperbilirubinemia (older siblings of the infant). Other considerations in assessment include the family’s ethnic origin (e.g., higher incidence in Asian infants); type of delivery (e.g., induction of labor); and infant characteristics such as weight loss after birth, gestational age, sex, and bruising. Assess the method and frequency of feeding.
Basic nursing care of the infant with hyperbilirubinemia differs from that of any newborn infant only in management of specific therapy (see Nursing Care Plan). (See Nursing Care of the Newborn and Family, Chapter 8, and Nursing Care of High-Risk Newborns, Chapter 10.)
 NURSING CARE PLAN
NURSING CARE PLAN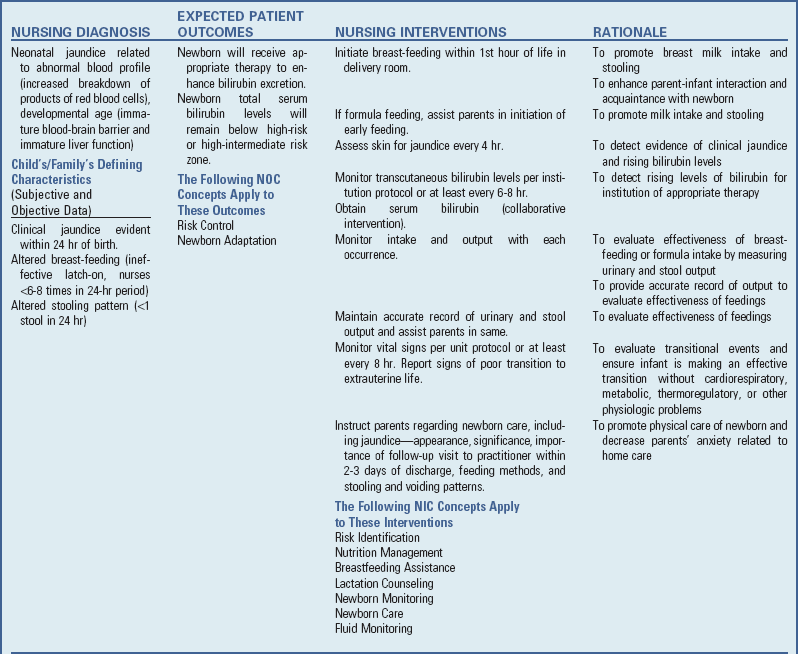

Prevention of physiologic and breast-feeding jaundice may be possible with early introduction of feedings and frequent nursing without water supplementation. Make every effort to provide an optimum thermal environment to reduce metabolic needs.
Nursing Care Plan—The Newborn with Jaundice
Phototherapy: The infant who receives phototherapy is placed under the light source, exposing as much skin surface as possible, and repositioned frequently to expose all body surface areas to the light. Once phototherapy has been initiated, frequent (every 6 to 12 hours) serum bilirubin levels are necessary because visual and transcutaneous assessments of jaundice are no longer considered valid.
The nurse institutes several precautions to protect the infant during phototherapy. An opaque mask shields the infant’s eyes to prevent exposure to the light (Fig. 9-7). The eye shield should be properly sized and positioned to cover the eyes completely but prevent any occlusion of the nares. The infant’s eyelids are closed before the mask is applied because the corneas may become excoriated if they come in contact with the dressing. The nurse checks the newborn’s eyes at least every 4 to 6 hours for evidence of discharge, excessive pressure on the lids, or corneal irritation. Remove eye shields during feedings, which provide the opportunity for visual and sensory stimulation.
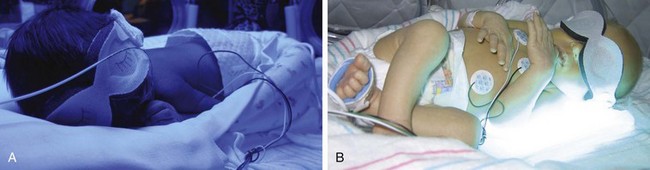
Fig. 9-7 A, Infant receiving phototherapy; note nested boundaries for comfort and eye protection. B, Newborn laying on phototherapy light source, which may be used with overhead lights to provide intensive phototherapy. (Courtesy E. Jacobs, Texas Children’s Hospital, Houston.)
Monitor infants who are in an open crib receiving phototherapy for temperature instability, since phototherapy may cause an increase in the body temperature. The distance between the phototherapy light source and the infant must be maintained as outlined by the manufacturer’s guidelines; halogen lights placed too close to the infant’s skin may cause burns (Stokowski, 2006). Maintaining the infant in a flexed position with rolled blankets along the sides of the body helps maintain heat and provides comfort.
Accurate charting is another important nursing responsibility and includes (1) times that phototherapy is started and stopped, (2) proper shielding of the eyes, (3) type of phototherapy unit (by manufacturer), (4) number of lamps, (5) distance between surface of lamps and infant, (6) use of phototherapy in combination with an incubator or open bassinet, (7) photometer measurement of light intensity (microwatts), and (8) side effects.
Side Effects of Phototherapy: Minor side effects for which the nurse should be alert include loose, greenish stools; transient skin rashes; mild hyperthermia; increased metabolic rate; and priapism. Dehydration and electrolyte disturbances, such as hypocalcemia, are uncommon yet may occur. To prevent or minimize these effects, the nurse monitors the temperature to detect early signs of hypothermia or hyperthermia, and observes the skin for evidence of dehydration and drying, which can lead to excoriation and breakdown. Oily lubricants or lotions are not used on the skin to prevent increased tanning. Full-term and late-preterm infants receiving phototherapy may require additional fluid volume or feedings to compensate for insensible and intestinal fluid loss. Because phototherapy enhances the excretion of unconjugated bilirubin through the bowel, loose stools may indicate accelerated bilirubin excretion. Frequent stooling can cause perianal irritation; therefore meticulous skin care, especially keeping the skin clean and dry, is essential.
Once phototherapy is permanently discontinued, there is often a subsequent increase in the serum bilirubin level, often called the “rebound effect.” This is usually transient and resolves without resuming therapy.
Another reaction to phototherapy is the bronze-baby syndrome, in which the serum, urine, and skin turn grayish brown several hours after the infant is placed under the light. This reaction is probably caused by retention of a bilirubin breakdown product of phototherapy, possibly copper porphyrin. The syndrome almost always occurs in infants who have elevated conjugated hyperbilirubinemia and some degree of cholestasis. The browning generally resolves after discontinuation of phototherapy.
Family Support: Parents need reassurance concerning their infant’s progress. The nurse explains all the procedures to familiarize them with the benefits and risks. Reassure parents that the naked infant under the bilirubin light is warm and comfortable. Remove eye shields and turn off phototherapy when the parents are visiting to facilitate the attachment process. Also reassure parents that the neonate is accustomed to darkness after months of intrauterine existence and benefits a great deal from auditory and tactile stimulation (see Family-Centered Care box).
 FAMILY-CENTERED CARE
FAMILY-CENTERED CARE
Phototherapy and Parent-Infant Interaction
The traditional use of phototherapy has evoked concerns regarding a number of psychobehavioral issues, including parent-infant separation, potential social isolation, decreased sensorineural stimulation, altered biologic rhythms, altered feeding patterns, and activity changes. Parental anxiety is greatly increased, particularly at the sight of the newborn blindfolded and under special lights. The interruption of breast-feeding for phototherapy is a potential deterrent to successful maternal-infant attachment and interaction.
Because research has demonstrated that bilirubin catabolism occurs primarily within the first few hours of the initiation of phototherapy, there is increased support for the removal of the infant from treatment for feeding and holding. The benefits of stopping phototherapy for parental feeding and holding outweigh concerns related to the clearance of bilirubin in the healthy full-term newborn with mild to moderate hyperbilirubinemia. Home phototherapy offers an additional opportunity to foster parent-infant attachment.
The initiation of any treatment requires informed consent by the parents; however, in the case of phototherapy, parents may feel considerable anxiety when nurses use such words as “kernicterus” and “possible harm to the brain” to describe possible effects of hyperbilirubinemia. It is imperative that nurses remain sensitive to parents’ feelings and information needs during this process. An important nursing intervention is the assessment of the parents’ understanding of the treatment involved and clarification of the nature of the therapy.
One of the most important nursing interventions is recognition of breast-feeding jaundice. Lack of familiarity among health professionals has caused many newborns prolonged hospitalization, termination of breast-feeding, and unnecessary phototherapy. Care of the new mother may include supporting successful and frequent breast-feeding. Parents also need reassurance of the benign nature of the jaundice and encouragement to resume breast-feeding if temporary cessation is prescribed. Unfortunately, jaundice increases the risk of breast-feeding being discontinued and development of the vulnerable child syndrome—the parents’ belief that their child has suffered a “close call” and is vulnerable to serious injury.
Discharge Planning and Home Care: With short hospital stays, mothers and infants may be discharged before evidence of jaundice is present. It is imperative that the nurse discuss signs of jaundice with the mother because any clinical symptoms will probably appear at home. Teach parents to evaluate the number of voids and evidence of adequate breast-feeding once the infant is home and encourage them to bring the newborn to the hospital, clinic, or primary care practitioner if there are indications of hyperbilirubinemia. Breast-feeding mother-infant dyads must receive appropriate guidance and assistance with breast-feeding to ensure the infant is receiving an adequate amount of breast milk and that stooling is occurring. A follow-up visit to the health care practitioner within 2 or 3 days after discharge to evaluate feeding and elimination patterns and jaundice is important in the posthospital care of the full-term newborn (see Diagnostic Evaluation, p. 290, for follow-up recommendations).
If home phototherapy is instituted, the hospital, durable medical equipment company representative, or home health care nurse is usually responsible for teaching family members and assessing their abilities to implement the treatment safely and in a timely manner. General guidelines for home care preparation and education are discussed in Chapters 8, 25, and 26. Written instructions and supervision of care—especially the application of eye shields, if needed—are essential. The minor side effects of phototherapy are reviewed, and parents may need instruction in taking axillary temperatures and recording times and amounts of feedings and the number of wet diapers and stools.
Regardless of how benign the disorder or the therapy, parents need support and understanding. In jaundice associated with breast-feeding, follow-up blood studies are usually required to assess the progress of the jaundice. If temporary cessation of breast-feeding is prescribed, teach mothers to pump the breasts every 2 to 3 hours to maintain lactation; the expressed milk is properly stored for use after breast-feeding is resumed. Nurses should take measures to help the mother achieve successful breast-feeding, including consultation with a lactation specialist on an outpatient basis.
Hemolytic Disease of the Newborn
Hyperbilirubinemia in the first 24 hours of life is most often the result of hemolytic disease of the newborn (HDN), an abnormally rapid rate of RBC destruction. Anemia caused by this destruction stimulates the production of RBCs, which in turn provides increasing numbers of cells for hemolysis. Major causes of increased erythrocyte destruction are isoimmunization (primarily RhD) and ABO incompatibility.
Blood Incompatibility
The membranes of human blood cells contain a variety of antigens, also known as agglutinogens, substances capable of producing an immune response if recognized by the body as foreign. The reciprocal relationship between antigens on RBCs and antibodies in the plasma causes agglutination (clumping). In other words, antibodies in the plasma of one blood group (except the AB group, which contains no antibodies) produce agglutination when mixed with antigens of a different blood group. In the ABO blood group system the antibodies occur naturally. In the Rh system the person must be exposed to the Rh antigen before significant antibody formation takes place and causes a sensitivity response known as isoimmunization.
Rh Incompatibility (Isoimmunization): The Rh blood group consists of several antigens (with D being the most prevalent). For simplicity, only the terms Rh positive (presence of antigen) and Rh negative (absence of antigen) are used in this discussion. (See Autosomal Inheritance Patterns, Chapter 5.) The presence or absence of the naturally occurring Rh factor determines the blood type.
Ordinarily, no problems are anticipated when the Rh blood types are the same in both mother and fetus or when the mother is Rh positive and the infant is Rh negative. Difficulty may arise when the mother is Rh negative and the infant is Rh positive. Although the maternal and fetal circulations are separate, there is evidence of a bidirectional trafficking of fetal RBCs and cell-free DNA to the maternal circulation (Moise, 2007). More commonly, however, fetal RBCs enter into the maternal circulation at the time of delivery. The mother’s natural defense mechanism responds to these alien cells by producing anti-Rh antibodies.
Under normal circumstances, this process of isoimmunization has no effect on the fetus during the first pregnancy with an Rh-positive fetus, since the initial sensitization to Rh antigens rarely occurs before the onset of labor. However, with the increased risk of fetal blood being transferred to the maternal circulation during placental separation, maternal antibody production is stimulated. During a subsequent pregnancy with an Rh-positive fetus, these previously formed maternal antibodies to Rh-positive blood cells enter the fetal circulation, where they attach to and destroy fetal erythrocytes (Fig. 9-8). Multiple gestations, abruptio placentae, placenta previa, manual removal of the placenta, and cesarean delivery increase the incidence of transplacental hemorrhage and subsequent isoimmunization (Moise, 2008).
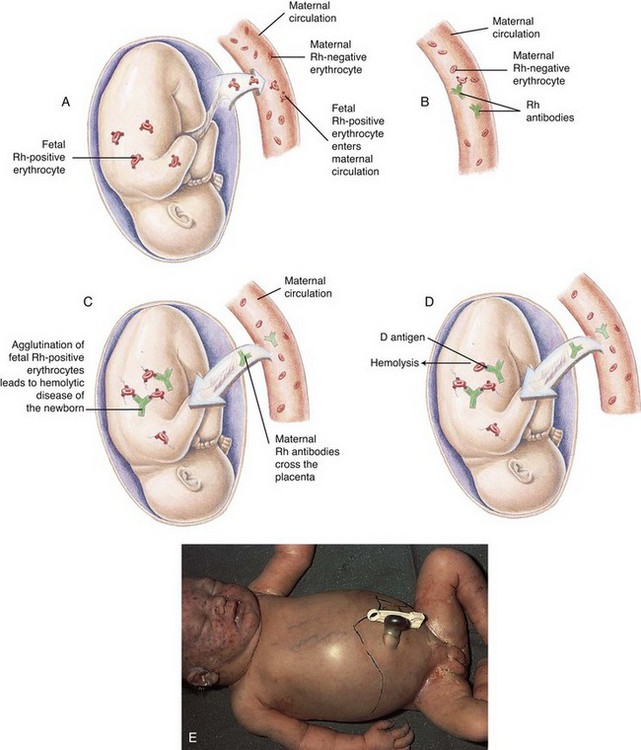
Fig. 9-8 Hemolytic disease of the newborn (HDN). A, Before or during delivery, Rh-positive erythrocytes from the fetus enter the blood of an Rh-negative woman through a tear in the placenta. B, The mother is sensitized to the Rh antigen and produces Rh antibodies. Because this usually happens after delivery, there is no effect on the fetus in the first pregnancy. C, During a subsequent pregnancy with an Rh-positive fetus, Rh-positive erythrocytes cross the placenta, enter the maternal circulation, and (D) stimulate the mother to produce antibodies against the Rh antigen. The Rh antibodies from the mother cross the placenta, causing agglutination and hemolysis of fetal erythrocytes, and HDN develops (E). (From McCance K, Huether S: Pathophysiology: the biological basis for disease in adults and children, ed 6, St Louis, 2010, Mosby.)
Because the condition begins in utero, the fetus attempts to compensate for the progressive hemolysis by accelerating the rate of erythropoiesis. As a result, immature RBCs (erythroblasts) appear in the fetal circulation; hence the term erythroblastosis fetalis.
The development of maternal sensitization to Rh-positive antigens exhibits wide variability. Sensitization may occur during the first pregnancy if the woman previously received an Rh-positive blood transfusion. No sensitization may occur in situations in which a strong placental barrier prevents transfer of fetal blood into the maternal circulation. Approximately 10% to 15% of sensitized mothers have no hemolytic reaction in the newborn. In addition, some Rh-negative women, even though exposed to Rh-positive fetal blood, are immunologically unable to produce antibodies to the foreign antigen.
In the most severe form of erythroblastosis fetalis (hydrops fetalis), the progressive hemolysis causes fetal hypoxia; cardiac failure; generalized edema (anasarca); and fluid effusions into the pericardial, pleural, and peritoneal spaces (hydrops). The fetus may be delivered stillborn or in severe respiratory distress. Maternal Rh immunoglobulin (RhIg) administration, early intrauterine detection of fetal anemia by ultrasonography (serial Doppler assessment of the peak velocity in the fetal middle cerebral artery), and subsequent treatment by fetal blood transfusions or high-dose intravenous immunoglobulin (IVIG) have dramatically improved the outcome of affected fetuses (Moise, 2008).
ABO Incompatibility: Hemolytic disease can also occur when the major blood group antigens of the fetus are different from those of the mother. The major blood groups are A, B, AB, and O. The incidence of these blood groups varies according to race and geographic location. In the North American Caucasian population, 46% have type O blood, 42% have type A blood, 9% have type B blood, and 3% have type AB blood.
The presence or absence of antibodies and antigens determines whether agglutination will occur. Antibodies in the plasma of one blood group (except the AB group, which contains no antibodies) will produce agglutination (clumping) when mixed with antigens of a different blood group. Naturally occurring antibodies in the recipient’s blood cause agglutination of a donor’s RBCs. The agglutinated donor cells become trapped in peripheral blood vessels, where they hemolyze, releasing large amounts of bilirubin into the circulation.
The most common blood group incompatibility in the neonate is between a mother with O blood group and an infant with A or B blood group. Hemolysis due to anti-A is more common than for anti-B (see Table 9-2 for possible ABO incompatibilities). Naturally occurring anti-A or anti-B antibodies already present in the maternal circulation cross the placenta and attach to fetal RBCs, causing hemolysis. Usually the hemolytic reaction is less severe than in Rh incompatibility; however, rare cases of hydrops have been reported (Black and Maheshwari, 2009). Unlike the Rh reaction, ABO incompatibility may occur in the first pregnancy. The risk of significant hemolysis in subsequent pregnancies is thought to be unchanged from the first.
Clinical Manifestations
Jaundice appears shortly after birth (during the first 24 hours), and serum levels of unconjugated bilirubin rise rapidly. Anemia results from the hemolysis of large numbers of erythrocytes, and hyperbilirubinemia and jaundice result from the liver’s inability to conjugate and excrete the excess bilirubin. Most newborns with HDN are not jaundiced at birth. However, hepatosplenomegaly and varying degrees of hydrops may be evident. If the infant is severely affected, hydrops, anemia, and hypovolemic shock are apparent. Hypoglycemia may occur as a result of pancreatic cell hyperplasia.
Diagnostic Evaluation
Early identification and diagnosis of RhD sensitization is important in the management and prevention of fetal complications. A maternal antibody titer (indirect Coombs test) should be drawn at the first prenatal visit. Genetic testing allows early identification of paternal zygosity at the RhD gene locus, thus allowing earlier detection of the potential for isoimmunization and avoiding further maternal or fetal testing (Moise, 2008). Amniocentesis can be used to test the fetal blood type of a woman whose antibody screen is positive; the use of polymerase chain reaction may determine the fetal blood type, hemoglobin, hematocrit, and presence of maternal antibodies. Chorionic villus sampling has drawbacks that preclude its use, including possible spontaneous abortion of the fetus and fetomaternal hemorrhage, which would make the situation worse. With either method the determination of an Rh-negative fetus requires no further treatment. The detection of cell-free fetal DNA in the maternal plasma of RhD–negative women to detect an RhD-positive fetus has been used successfully in the United Kingdom; however, this technology is not yet available in the United States (Finning, Martin, and Daniels, 2009; Moise, 2008). Such testing would negate the necessity of amniocentesis for fetal blood type.
Ultrasonography is an important adjunct in the detection of isoimmunization. Alterations in the placenta, umbilical cord, and amniotic fluid volume, as well as the presence of fetal hydrops, can be detected with high-resolution ultrasonography, allowing early, noninvasive treatment before the development of erythroblastosis. Serial Doppler ultrasonography of fetal middle cerebral artery peak velocity is often used to detect and measure fetal hemoglobin and, subsequently, fetal anemia (Moise, 2008). Erythroblastosis fetalis caused by Rh incompatibility can also be assessed by evaluating rising anti-Rh antibody titers in the maternal circulation (indirect Coombs test) or by testing the optical density of amniotic fluid (delta OD 450 test), because bilirubin discolors the fluid. The disease in the newborn is suspected on the basis of the timing and appearance of jaundice (see Table 9-1) and can be confirmed postnatally by detecting antibodies attached to the circulating erythrocytes of affected infants (direct Coombs test, or direct antiglobulin test). The Coombs test is routinely performed on cord blood samples from infants born to Rh-negative mothers.
Therapeutic Management
The primary aim of therapeutic management of isoimmunization is prevention. Postnatal therapy usually entails phototherapy for mild cases and exchange transfusion for more severe forms. In severe cases of hydrops, aggressive interventions such as pericardial and pleural fluid aspiration, mechanical ventilatory support, and inotrope therapy may be required for stabilization. Although phototherapy may control bilirubin levels in mild cases, the hemolytic process may continue, causing severe anemia (if untreated) between 7 and 21 days of life.
Prevention of Rh Isoimmunization: The administration of RhIg, a human gamma-globulin concentrate of anti-D, to all unsensitized Rh-negative mothers after delivery or abortion of an Rh-positive infant or fetus prevents the development of maternal sensitization to the Rh factor. The injected anti-Rh antibodies destroy (by subsequent phagocytosis and agglutination) fetal RBCs passing into the maternal circulation before the mother’s immune system can recognize them. Because the immune response is blocked, anti-D antibodies and memory cells (which produce the primary and secondary immune responses, respectively) are not formed. The inhibition of memory cell formation is especially important because memory cells provide long-term immunity by initiating a rapid immune response once the antigen is reintroduced (McCance and Huether, 2010).
To be effective, RhIg (such as RhoGAM) must be administered to unsensitized mothers within 72 hours (but possibly as long as 3 to 4 weeks) after the first delivery, miscarriage, or abortion, and repeated after subsequent pregnancies. The administration of RhIg at 26 to 28 weeks of gestation further reduces the risk of Rh isoimmunization. RhIg is not effective against existing Rh-positive antibodies in the maternal circulation. RhIg is administered intramuscularly, not intravenously, and only to Rh-negative women with a negative Coombs test—never to the infant or father.
The use of a heme-oxygenase inhibitor, such as tin mesoporphyrin (given intramuscularly to the newborn), has proved effective in preventing neonatal hyperbilirubinemia.
Maternal administration of high-dose IVIG, alone or in combination with plasmapheresis, decreases the fetal effects of RhD isoimmunization (Moise, 2002; Urbaniak, 2008).
Studies have also demonstrated the effectiveness of neonatal IVIG at decreasing the severity of RBC destruction (hemolysis) in HDN and subsequent development of jaundice; IVIG administered to the neonate attacks the maternal cells that destroy neonatal RBCs, slowing the progression of bilirubin production (Mundy, 2005). This therapy, often used in conjunction with phototherapy, may decrease the necessity for exchange transfusion (Gottstein and Cooke, 2003).
Intrauterine Transfusion: Infants of mothers already sensitized may be treated by intrauterine transfusion, which consists of infusing blood into the fetus’s umbilical vein. The need for therapy is based on the antenatal diagnosis of isoimmunization by determining the optical density of amniotic fluid (by amniocentesis) as an index of fetal hemolysis, or by serial ultrasonography, which may detect the presence of fetal hydrops and anemia as early as 16 weeks of gestation. With ultrasound technology fetal transfusion may be accomplished directly via the umbilical vein, infusing Rh O-negative packed RBCs to raise the fetal hematocrit to 40% to 50%; fetal movement and transfusion risks are minimized by administering a muscle-paralyzing drug for temporary fetal paralysis. The frequency of intrauterine transfusions may vary according to institution yet may be as often as every 2 weeks until the fetus reaches pulmonary maturity at approximately 37 to 38 weeks of gestation (Moise, 2002, 2008).
Exchange Transfusion: Exchange transfusion, in which the infant’s blood is removed in small amounts (usually 5 to 10 ml at a time) and replaced with compatible blood (Rh-negative blood), is a standard mode of therapy for treatment of severe hyperbilirubinemia that is unresponsive to phototherapy, and it is the treatment of choice for severe hyperbilirubinemia and hydrops caused by Rh incompatibility. Exchange transfusion removes the sensitized erythrocytes, lowers the serum bilirubin level to prevent bilirubin encephalopathy, corrects the anemia, and prevents cardiac failure. Indications for exchange transfusion include rapidly increasing serum bilirubin levels and hemolysis despite aggressive phototherapy. The criteria for exchange transfusion in preterm infants vary according to associated illness factors. The American Academy of Pediatrics (2004) practice parameter guidelines provide suggestions for initiating phototherapy and exchange transfusion in infants 35 weeks of gestation or more. An infant born with hydrops fetalis or signs of cardiac failure is a candidate for immediate exchange transfusion with fresh whole blood.
For exchange transfusion, fresh whole blood is typed and cross-matched to the mother’s serum. The amount of donor blood used is usually double the infant’s blood volume, which is approximately 85 ml/kg body weight. The double-volume exchange transfusion replaces approximately 85% of the neonate’s blood.
An exchange transfusion is a sterile surgical procedure. A catheter is inserted into the umbilical vein and threaded into the inferior vena cava. Depending on the infant’s weight, 5 to 10 ml of blood is withdrawn within 15 to 20 seconds, and the same volume of donor blood is infused until the targeted volume (double the estimated blood volume) is reached. If the donor blood has been citrated (addition of citrate phosphate dextrose adenine to prevent coagulation), calcium gluconate may be given after the infusion of each 100 ml of donor’s blood to prevent hypocalcemia.
ABO Incompatibility: The treatment for ABO hemolytic disease is early detection and implementation of phototherapy for the reduction of hyperbilirubinemia. The initial diagnosis is often more difficult because the direct Coombs test may be negative or weakly reactive. The presence of jaundice within the first 24 hours, elevated serum bilirubin levels, RBC spherocytosis, and increased erythrocyte production is diagnostic of ABO incompatibility. In some centers IVIG transfusions are used in combination with phototherapy to treat ABO incompatibility. Exchange transfusion is not commonly required for ABO incompatibility except when phototherapy fails to decrease bilirubin concentrations.
Prognosis: The severe anemia of isoimmunization may result in stillbirth, shock, congestive heart failure, poor feeding, or poor weight gain. Perinatal survival rates for HDN are reported to be above 90% as a result of early prenatal detection and intrauterine management (Moise, 2008). Complications from exchange transfusion are uncommon; however, close monitoring is imperative.
Nursing Care Management
The initial nursing responsibility is recognizing jaundice. The possibility of hemolytic disease can be anticipated from the prenatal and perinatal history. Prenatal evidence of incompatibility, maternal blood type O, and a positive Coombs test are cause for increased vigilance for early signs of jaundice in an infant.
If an exchange transfusion is required, the nurse prepares the infant and the family and assists the practitioner with the procedure. The infant must remain NPO (“nothing by mouth”) during the procedure; therefore a peripheral infusion of dextrose and electrolytes is established. The nurse documents blood volumes exchanged, including the amount of blood withdrawn and infused, the time of each procedure, and the cumulative record of the total volume exchanged. The nurse also evaluates vital signs frequently (monitored electronically during the procedure) and correlates them with the removal and infusion of blood. If signs of cardiac or respiratory problems occur, the procedure is stopped temporarily and resumed once the infant’s cardiorespiratory function stabilizes. The nurse also observes for signs of transfusion reaction (temperature instability, hypotension, tachycardia, bradycardia, rash) and maintains adequate neonatal thermoregulation, blood glucose levels, and fluid balance.
Family Support: Parents often feel guilty because they think they have caused the blood incompatibility. Parents should never be made to feel responsible or negligent. The nurse encourages them to express their thoughts. The nurse should praise parents for actions they took to prevent any problems, such as frequent antepartum examinations and blood tests.
Hypoglycemia
Neonatal hypoglycemia has many recognized causes; the following discussion primarily focuses on transient neonatal hypoglycemia.
Hypoglycemia is present when the newborn’s blood glucose concentration is lower than the body’s requirement for cellular energy and metabolism. However, the precise definition of hypoglycemia for every newborn in regard to gestational age, birth weight, metabolic needs, and illness or wellness state remains unknown. Cornblath, Hawdon, Williams, and colleagues (2000) have suggested an operational threshold at which interventions to increase serum glucose levels should be instituted to prevent serious effects. For the healthy full-term infant, born after an uneventful pregnancy and delivery, recommendations are to monitor glucose levels only in the presence of risk factors (see following) or clinical manifestations of hypoglycemia; in these infants a plasma glucose of less than 45 mg/dl (2.5 mmol/L) requires intervention. Healthy full-term, breast-fed newborns may not fit into this category because human milk appears to provide adequate substrate (Cornblath, Hawdon, Williams, et al, 2000). Hoseth, Joergensen, Ebbesen, and colleagues (2000) evaluated blood glucose levels in healthy full-term, breast-fed infants and found significant hypoglycemia in only 2 of the 223 infants during the first 4 days of life (see Research Focus box). Maternal tobacco use, method of delivery, and anesthetics did not affect the infants’ glucose levels.
 RESEARCH FOCUS
RESEARCH FOCUS
Early Breast-Feeding in Infants of Women with Gestational Diabetes
Chertok, Raz, Shoham, and colleagues (2009) reported on a study of 84 infants of women with gestational diabetes who were breast-fed in the delivery room; these infants had higher mean blood glucose levels than those who were not breast-fed in the delivery room and also in comparison to infants fed formula for the first feeding. The researchers postulated that early breast-feeding may facilitate glycemic stability in infants born to women with gestational diabetes.
In infants who are at risk for altered metabolism as a result of maternal illness factors (diabetes [gestational or otherwise], pregnancy-induced hypertension, terbutaline administration) or newborn factors (perinatal hypoxia, infection, hypothermia, polycythemia, congenital malformations, hyperinsulinism, smallness for gestational age, fetal hydrops, late-preterm birth), close observation and monitoring of blood glucose levels within 2 to 3 hours of birth are recommended. If the newborn has a blood glucose below 36 mg/dl (2.0 mmol/L), intervention such as breast- or bottle-feeding should be instituted; if levels remain low despite feeding, intravenous dextrose is warranted. In such infants the treatment should be aimed at maintaining the blood glucose levels above 45 mg/dl (2.5 mmol/L) (Cornblath, Hawdon, Williams, et al, 2000). Blood glucose levels for infants with severe hyperinsulinism may need to be higher (60 mg/dl [3.3 mmol/L]) to prevent serious effects. Hypoglycemia in preterm infants requires further study, but it has been suggested that values be maintained above 47 mg/dl (2.6 mmol/L) (Cornblath, Hawdon, Williams, et al, 2000). Cowett and Loughead (2002) concluded that plasma glucose levels below 45 mg/dl (2.5 mmol/L) are suboptimal for either the term or preterm infant and appropriate therapy should be implemented in such infants.
The decision of when to treat the hypoglycemic newborn is not based on a single plasma glucose value but on a number of clinical factors (Rozance and Hay, 2006; Sperling and Menon, 2004).
Pathophysiology
After birth the infant must supply nutrients to meet energy requirements for maintaining body temperature, respiration, muscular activity, and regulation of blood glucose. Glucose comes primarily from glycogen stores deposited in the liver, heart, and skeletal muscles during the last trimester of pregnancy.
The brain is especially dependent on adequate glucose supply for appropriate function. There is evidence of a major shift in energy metabolism from glucose to carbohydrate in newborns during the first several hours of life; hence the importance of providing adequate energy substrate. Although newborns demonstrate the ability to use ketones and amino acids as energy substrate, there are certain limitations. Infants with severe hyperinsulinism are unable to compensate metabolically and require more glucose than normal. Conditions that decrease the availability of substrate or prevent appropriate metabolism of available substrate place the infant at risk for hypoglycemia. These include intrauterine growth failure, prematurity, maternal diabetes, maternal use of hypoglycemic drugs, maternal administration of tocolytics such as terbutaline and ritodrine, intrapartum administration of glucose, perinatal hypoxia, infection, hypothermia, polycythemia, fetal hydrops, inborn errors of metabolism (IEMs) such as galactosemia, certain congenital malformations, endocrine disorders, abnormal extrauterine transition, and failure to receive adequate perinatal nutrition.
Both transient neonatal hypoglycemia and recurrent hypoglycemia are based on conditions with decreased hepatic glucose production. Transient neonatal hypoglycemia is associated with intrapartum glucose administration, terbutaline administration, gestational diabetes, intrauterine growth restriction, perinatal stress or asphyxia, prematurity, cold stress, polycythemia, and size that is large for gestational age. Recurrent hypoglycemia is observed in neonates with excessive insulin production or hyperinsulinism and includes infants with IEMs, Beckwith-Wiedmann syndrome, nesidioblastosis, Rh isoimmunization, and certain rare endocrine disorders (Cowett and Loughead, 2002; Miclic, 2008; Sperling, 2007).
Clinical Manifestations
The signs of hypoglycemia are usually vague and often indistinguishable from those observed in other newborn conditions, such as hypocalcemia, septicemia, CNS disorders, or cardiorespiratory problems. Because the brain depends on glucose for energy, cerebral signs such as jitteriness, tremors, twitching, weak or high-pitched cry, lethargy, hypotonia, limpness, seizures, and coma are prominent. Other clinical manifestations are cyanosis, apnea, rapid and irregular respirations, sweating, eye rolling, and refusal to feed. The symptoms often are transient but recurrent.
Diagnostic Evaluation
Diagnosis is confirmed by direct analysis of blood glucose concentration. Two consecutive specimens of blood should be analyzed because of the many factors that can affect readings.
Point-of-care blood glucose monitors in neonatal care must be accurate, rapid, and inexpensive; must demonstrate reliability with neonatal hematocrit ranges; must accept small blood volumes; and must provide reliable data for diagnosing neonatal hypoglycemia and hyperglycemia (Desphande and Platt, 2005; Sirkin, Jalloh, and Lee, 2002). Blood glucose measurements with a reagent strip such as Dextrostix or Chemstrip bG, read manually or with a glucose reflectance meter, are considered inaccurate and unreliable (Desphande and Platt, 2005).
Strict quality monitoring, regular calibration, and adherence to strict protocols are necessary to ensure accuracy. The most accurate method is the laboratory analysis of serum glucose. Blood specimens may be obtained from heel, arterial, or venous punctures. (See Atraumatic Care box, p. 254 [Chapter 8].)
Proper handling of the specimen is essential because storage at room temperature increases glycolysis. Accurate readings can be facilitated by storing the blood sample on ice to slow cellular metabolism or by removing the RBCs through centrifugation.
Therapeutic Management
Intravenous infusion of glucose is one method of treating hypoglycemia. In full-term infants who are borderline hypoglycemic and clinically asymptomatic, the early institution of milk feeding (breast or formula) may reestablish normoglycemia, thus avoiding the need for intravenous glucose. Milk feeding likely stimulates ketogenesis and facilitates gluconeogenesis (Sperling and Menon, 2004). Infants who are at increased risk for developing hypoglycemia should have their blood glucose measured within 1 hour after birth. The procedure should be repeated every 1 to 2 hours for the first 6 to 8 hours, then every 4 to 6 hours for 2 days.
Oral glucose feedings are often used as a treatment for hypoglycemia in healthy newborns. However, formula and breast milk are just as effective. Hypoglycemia is preventable in most instances by the initiation of early feeding in healthy, asymptomatic term newborns (Cowett and Loughead, 2002). Breast-fed infants should be put to breast as soon as possible after delivery. (See Infants of Diabetic Mothers, Chapter 10, for management of hypoglycemia related to transient hyperinsulinemia.)
A systematic review by the Joanna Briggs Institute (2007) concluded that early and exclusive breast-feeding is safe and adequate to meet the metabolic needs of healthy term infants; such infants do not require glucose monitoring, nor do they require supplemental feeding with dextrose water or other milk substitutes. The study further stressed the importance of maintaining newborn thermoregulation to prevent hypoglycemia.
Nursing Care Management
Much of the nursing responsibility for the infant with hypoglycemia involves identification of the problem through careful observation of physical status. Another concern is to reduce environmental factors, such as cold stress and respiratory difficulty, which predispose the infant to the development of a decreased blood glucose level.
An intravenous glucose infusion is required for infants with symptomatic hypoglycemia who are unable to tolerate oral feedings, infants who are unable to maintain adequate glucose levels with oral feedings, and infants with profound hypoglycemia. An initial bolus infusion of 2 ml/kg of 10% dextrose over 1 minute followed by a continuous dextrose infusion of 6 to 8 mg/kg is appropriate therapy for the hypoglycemic infant (Miclic, 2008). Major nursing objectives include preventing, anticipating, and recognizing potential dangers of concentrated dextrose infusion. Too-rapid infusion of the hypertonic solution can cause circulatory overload, hyperglycemia, and intracellular dehydration. Maintaining the ordered flow rate with an intravenous pump and checking and charting hourly intake decrease the chance of such problems.
The infusion is administered through a peripheral vein to increase hemodilution of the concentrated solution and prevent irritation of the vessel walls. Extravasation of the fluid into the surrounding area can cause tissue sloughing. Termination of the glucose solution must be gradual to prevent hypoglycemia caused by hyperinsulinism.
Because hypoglycemia may be a symptom of some other underlying pathophysiologic process, parents are usually concerned about their infant’s progress, particularly because these infants do not feed well or demonstrate behaviors that are typical of healthy infants. Nurses need to be aware of parents’ thoughts, allow them to express their feelings, and update them on the infant’s progress.
Hyperglycemia
Hyperglycemia in the newborn is usually defined as a blood glucose concentration greater than 125 mg/dl in the full-term infant or greater than 150 mg/dl in the preterm infant. Those affected are usually low-birth-weight infants who are unable to tolerate intravenous glucose infusions at the usual rate. The glucose intolerance is probably related to general immaturity of the usual regulatory mechanisms. Increased blood glucose levels may also occur in infants with sepsis or decreased insulin sensitivity (such as infants with transient diabetes mellitus), infants receiving methylxanthines, and infants who are stressed (e.g., infants with respiratory distress syndrome, infants undergoing surgical procedures).
Hyperglycemia is usually asymptomatic but detected on routine screening. Most often, hyperglycemia is treated by reducing the infant’s glucose intake. Untreated hyperglycemia may result in an osmotic diuresis with subsequent fluid volume loss and dehydration; if severe, it may result in an intraventricular hemorrhage as a result of fluid shifts in the CNS (Blackburn, 2007). Insulin infusion is sometimes administered to very low–birth-weight infants who require but are unable to tolerate intravenous dextrose solutions with concentrations greater than 5 g/dl.
Nursing Care Management
Monitor blood glucose frequently, especially in the infant receiving insulin. This requires numerous heel sticks, and sites should be rotated to minimize tissue damage. (See Blood Specimens, Chapter 27, and the Atraumatic Care box titled “Heel Punctures” in Chapter 8.) Carefully measure urinary output to detect any evidence of glycosuria and possible osmotic diuresis.
As in the care of all infants, give parents a careful explanation of the therapy, provide frequent progress reports, and support them to reduce anxiety. (See Nursing Care of High-Risk Newborns, Chapter 10.)
Hypocalcemia
As with many conditions in the neonate, hypocalcemia is difficult to differentiate from other disorders (sepsis, meningitis, narcotic withdrawal, hypoglycemia), and the etiology may be ill defined. The incidence is highest at two times during the neonatal period. Early-onset hypocalcemia, which appears within the first 24 to 48 hours, is the more common form and typically affects the preterm or small-for-gestational-age infant and any infant who has experienced perinatal asphyxia. Preterm infants may have hypocalcemia as a result of inadequate calcium intake, increased calcitonin levels, possibly resistance to parathyroid hormone activation preventing calcium removal from bones, acidosis, and decreased vitamin D intake and absorption (Blackburn, 2007). An infant born to a diabetic mother may also experience early hypocalcemia, possibly as a result of relative maternal hyperparathyroidism and transient neonatal hypoparathyroidism. Symptoms include jitteriness, prolonged QT interval, apnea, cyanotic episodes, a high-pitched cry, and abdominal distention.
Late-onset hypocalcemia, which is not apparent until after the first 3 or 4 days of life, is referred to as cow’s milk–induced hypocalcemia or neonatal tetany. Although uncommon in developed countries, it may be observed in well-nourished infants who are fed modified cow’s milk, such as evaporated milk formula. Cow’s milk, which has a high phosphorus content, produces hyperphosphatemia and a resultant hypocalcemia by increasing calcium deposition in the bone and soft tissues. Late hypocalcemia may also be seen in infants with intestinal malabsorption, hyperinsulinemia, hypoparathyroidism, or hypomagnesemia.
The manifestations of neonatal tetany reflect neuromuscular irritation: twitching, tremors, irritability, high-pitched cry, tachycardia, and rarely seizures (Blackburn, 2007). The preterm neonate with hypocalcemia may be asymptomatic. Neonatal tetany is rarely seen in industrialized countries because of the prevalent use of commercial formula or human milk as the newborn’s primary source of nutrition.
In some preterm neonates, rickets and osteopenia may occur as a result of calcium or phosphorus deficiency in association with vitamin D deficiency, which prevents adequate intestinal absorption of minerals (Blackburn, 2007). Very low–birth-weight infants, infants with chronic conditions such as chronic lung disease (bronchopulmonary dysplasia), and infants on prolonged diuretic management are at particular risk for developing rickets and osteopenia (Blackburn, 2007). There have been reported cases of vitamin D deficiency in healthy breast-fed infants who received minimal ultraviolet light exposure or whose breast-feeding mother had a diet deficient in vitamin D; hence the American Academy of Pediatrics’ (2008a) recommendation for vitamin D supplementation (400 international units/day, oral) in all newborns exclusively breast-fed.
Diagnostic Evaluation
Diagnosis of hypocalcemia is confirmed with serum electrolyte determinations. Normal infant serum calcium values are usually in the range of 7.0 to 8.0 mg/dl (1.75 to 2.0 mmol/L) (Blackburn, 2007).
In full-term infants hypocalcemia is indicated at total serum calcium levels below 7.8 to 8 mg/dl (1.95 to 2.0 mmol/L) or ionized calcium levels (the biologically important fraction of calcium) below 3.0 to 4.4 mg/dl (1.1 mmol/L). In preterm infants a lower limit of 7.0 mg/dl (1.8 mmol/L) is considered hypocalcemic (Blackburn, 2007). Most clinicians consider the serum ionized calcium to be the best standard for monitoring blood calcium activity.
Therapeutic Management
In most instances early-onset hypocalcemia is temporary and resolves in 1 to 3 days. Restoration of a normal calcium level is facilitated by early feedings, physiologic correction of hypoparathyroidism, and, sometimes, administration of calcium supplements.
Treatment of hypocalcemia involves intravenous administration of 10% calcium gluconate. The drug is administered slowly over 10 to 30 minutes or as a continuous infusion; intravenous administration should not exceed 100 mg/min (Custer and Rau, 2009). Rapid intravenous calcium administration may cause cardiac dysrhythmias and circulatory collapse. The heart rate and blood pressure should be electronically monitored. Take care to ascertain that the infusion device is positioned within the vein because extravasation into surrounding tissue causes local necrosis, calcification, and sloughing. Intramuscular administration of calcium gluconate is contraindicated because it precipitates in the tissue, causing necrosis. If the infant can tolerate oral fluids, oral doses of calcium are given with formula. Adequate intake of vitamin D and phosphorus is imperative, especially in extremely low– and very low–birth-weight infants. Exercise caution in the use of oral calcium salts because of their hypertonicity and subsequent effects on the bowel of at-risk infants.
Nursing Care Management
Nursing care of the infant with hypocalcemia is directed toward identifying infants at risk for early hypocalcemia, observing for clinical manifestations in such infants, and administering supplemental calcium, vitamin D, and phosphorus. Monitor the infant continuously during intravenous infusions. Calcium gluconate can cause tissue necrosis and scar formation; therefore it is recommended that superficial veins such as those on the scalp be avoided. To prevent tissue necrosis, carefully observe the infusion site and changes it as needed. Calcium gluconate is also incompatible with a number of drugs, most notably sodium bicarbonate.
The nurse also observes for signs of acute hypercalcemia (vomiting, bradycardia). If such symptoms occur, discontinue the injection or infusion and notify the practitioner. Institute seizure precautions because seizures are common.
To prevent late-onset hypocalcemia, the nurse provides preventive care and anticipatory guidance regarding the correct use of an infant formula containing the appropriate balance of calcium and phosphorus and assists the parent in planning for meeting the growing infant’s nutritional needs through an affordable commercial infant formula.
If the infant is discharged on formula feedings supplemented with calcium, teach the parents the correct procedure for diluting the mineral in the formula and advise them to use only the prescribed formula. Also teach parents to observe for any signs of hypocalcemia or hypercalcemia in the infant receiving supplemental calcium.
Hemorrhagic Disease of the Newborn
Hemorrhagic disease of the newborn, or vitamin K deficiency bleeding, is a bleeding disorder that occurs as a result of a vitamin K deficiency. Hemorrhagic disease may be classified according to appearance as early, classic, or late onset. Newborns’ vitamin K stores are virtually absent and prothrombin activity is moderately deficient, decreases until approximately 72 hours after birth, and then begins to increase. Consequently, vitamin K–dependent coagulation factors (II, VII, IX, X) are significantly reduced. In addition, the newborn’s relatively sterile intestinal tract is unable to synthesize the vitamin until feedings have begun.
Signs and symptoms of hemorrhagic disease typically appear within hours of birth and can include oozing from the umbilicus or circumcision site, bloody or black stools, hematuria, ecchymoses on the skin and scalp, epistaxis, or bleeding from punctures. Classic hemorrhagic disease usually occurs 1 to 7 days after birth. Signs and symptoms are the same as those seen with early-onset disease. Diagnosis is confirmed by findings of prolonged prothrombin time and partial thromboplastin time accompanied by normal platelet count and fibrinogen level.
Infants born to mothers who are taking antiepileptic drugs phenytoin and phenobarbital may have a severe form of hemorrhagic disease of the newborn and should be carefully evaluated and treated accordingly. The bleeding is most severe within the first 24 hours of life and may require treatment with intravenous vitamin K and fresh frozen plasma (Stoll, 2007).
A late form of hemorrhagic disease (late onset) appears at approximately 2 to 12 weeks of age. This form occurs in totally or predominantly breast-fed infants who did not receive adequate vitamin K prophylaxis at birth. Although vitamin K levels in breast milk appear to be lower than in cow’s milk–based formulas, previous studies indicate that hemorrhagic disease occurred in infants who were exclusively breast-fed and who did not receive the standard prophylaxis at birth or were given a single dose of oral vitamin K (Lawrence and Lawrence, 2005). Manifestations of late-onset disease include evidence of intracranial hemorrhage; deep ecchymoses; and bleeding from the gastrointestinal tract, mucous membranes, skin punctures, or surgical incisions.
Therapeutic Management
The goal of management is prevention of hemorrhagic disease of the newborn with prophylactic administration of vitamin K. In the United States, intramuscular administration of vitamin K (phytonadione [AquaMEPHYTON, Mephyton]) in a dose of 0.5 to 1 mg once during the first few hours after birth is standard practice. The current recommendation to prevent hemorrhagic disease in infants who are breast-fed is to provide intramuscular vitamin K at birth and for the mother to have a well-balanced diet (American Academy of Pediatrics, 2009a). The administration of oral vitamin K is currently not recommended to prevent neonatal hemorrhagic disease (Stoll, 2007).
In newborns with hemorrhagic disease, treatment is the same as the preventive measures except that the vitamin may be given intravenously to prevent a hematoma at an intramuscular site. Bleeding usually ceases within 2 to 4 hours of vitamin K administration.
Nursing Care Management
Nursing care primarily involves prevention through careful administration of the vitamin into the vastus lateralis or ventrogluteal (not dorsogluteal) muscle. In instances in which this procedure is not routinely carried out (e.g., home births or emergency deliveries), the nurse observes for signs of the bleeding disorder and notifies the practitioner for appropriate diagnosis and treatment. Encourage breast-feeding mothers to increase their intake of foods containing vitamin K; the best sources are green vegetables, especially broccoli.
Inborn Errors of Metabolism
IEMs include a large number of inherited diseases caused by interruptions in the various pathways involved in the metabolism of protein, carbohydrates, or lipids. Fig. 9-9 is a conceptual model representing the multiple interactions in a metabolic pathway in health and disease. Metabolic pathways are series of biochemical reactions by which substrates are sequentially converted into other by-products, aiming at a final end-product that can be successfully used or eliminated by the body. Each of these transformations is mediated by an enzyme, which is under the control of a specific structural gene. Fig. 9-9, A, represents the normal metabolic conversion of substance A into end-product K, via by-products B, C, D, etc. Enzymes b and c, whose synthesis is controlled by genes β and δ, catalyze steps B → C and C → D, respectively. A feedback loop ensures physiologic levels of K. A gene mutation may result in qualitative or quantitative changes in the enzyme, resulting in a different (and therefore ineffective) enzyme, or in the decreased synthesis of the enzyme, including its total absence. This ineffective (or missing) enzyme will interrupt the pathway, resulting in accumulation of the by-product that immediately precedes the blockage, as well as lack of the by-products beyond the blockage. IEMs can occur as a result of such accumulations or absences of an essential by-product.
Fig. 9-9 Metabolic pathway. A, Normal metabolic pathway. B, Mutation of gene β. C, Mutation of gene δ. D, Genetic alteration of enzyme d. A, Substance; B-F, by-products; K, end-product; b, c, and, d, enzymes; β and δ, genes. (Adapted from da Cunha MF: Genetic basis of human disease. In Bullock BA, Henze RL, editors: Focus on physiology, Philadelphia, 2000, Lippincott.)
In Fig. 9-9, B, a mutation in gene β prevents normal synthesis of enzyme b, creating an interruption of the pathway between B and C. Assuming a continuous uptake of A, two outcomes ensue: accumulation of B, and depletion of all products beyond the block (C, D, … K). An example of this situation is Tay-Sachs disease (or GM2 gangliosidosis). In this disease, a mutation in the HexA gene (here exemplified by β) causes the lack of the enzyme hexosaminidase A (HexA), represented by b. Absence of b creates an accumulation of ganglioside GM2 (B)—lipids, in nerve cells, resulting in the clinical manifestations of this progressive neurologic disorder because the lipids are not broken down by the cellular liposomes.
Fig. 9-9, C, depicts a mutation in gene δ, which causes depletion of enzyme c and interrupts the metabolic conversion of C into D. In this instance, an alternative pathway C → E → F is opened. This event may have two outcomes: product F may eventually be converted into K, with no significant clinical consequences; or F may represent the end-point to the alternative pathway. If accumulation of F reaches toxic levels, a disease process may occur. Such is the case with PKU, an IEM that creates an intolerance to the amino acid phenylalanine. The missing enzyme (c, in this case), which results from mutation in the phenylalanine hydroxylase (PAH) gene (δ), is phenylalanine hydroxylase. The alternative pathway leads to the formation and accumulation of phenylketones (F). In combination with phenylalanine deficiency, an excessive amount of phenylketones contributes to the postnatal completion of myelination of nerves, resulting in profound cognitive impairment.
Fig. 9-9, D, represents a genetic alteration of the enzyme d, which is involved in the feedback control of synthesis of K. As a result, K may accumulate to toxic levels. The prototype genetic disease here is Lesch-Nyhan syndrome, in which d is the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT) and K is uric acid. A deficiency in HGPRT and accumulation of uric acid to extremely high levels will result in the development of cognitive impairment and self-mutilation tendency.
The mode of inheritance in IEMs is almost always autosomal recessive. The heterozygote has one gene with a normal effect and is still able to produce the enzyme in sufficient amounts to carry out the metabolic function under normal circumstances. Therefore the heterozygote does not exhibit symptoms of the disorder. The homozygote, who inherits a defective gene from both parents, has no functioning enzyme and thus is clinically affected.
Individually, different IEMs are rare; collectively they account for a significant proportion of health problems in children. It is becoming possible to detect and screen for an increasing number of IEMs—to detect the disease in the heterozygote, the newborn, and the fetus and to identify heterozygotes at risk for having a child with an IEM. With most IEMs, early diagnosis and prompt treatment are essential to prevent a relentless course of physical and mental deterioration. Prenatal diagnosis provides for special care of the infant immediately after birth. Neonatal screening is useful in detecting many disorders after a few days of life, but it is less helpful in detecting symptoms early in the neonatal period. Nurses caring for neonates must be certain that screening is performed, especially in infants who are discharged early, are born at home, or are in neonatal intensive care units. (See Genetic Screening, Chapter 5.)
A list of conditions routinely screened in each state can be found at http://genes-r-us.uthscsa.edu/nbsdisorders.pdf. Nurses also need to make certain that the neonate has a primary care provider and current home address and telephone number documented on the newborn screening blood spot card. Nurses should instruct parents to ask about newborn screening test results at their newborn’s first well-child visit. Most screening tests require a heel puncture to obtain sufficient blood to completely cover circles on special blotting paper. A new screening test, tandem mass spectrometry, has the potential to identify up to 40 IEMs. With tandem mass spectrometry, earlier identification of IEMs may prevent further developmental delays and morbidities in affected children. (See Atraumatic Care box, p. 254, for measures to reduce the pain of lancing and squeezing the heel, and see Newborn Screening for Disease, Chapter 8.)
Some nonspecific manifestations—including lethargy, poor feeding, vomiting, diarrhea, hypoglycemia, metabolic acidosis, respiratory distress, apnea, hypothermia, coma, and seizures—occur in a wide variety of genetic and acquired disorders. The time of onset may be important. Most IEMs produce no symptoms during the first 24 hours of life. Other manifestations that may indicate an IEM include jaundice, hepatomegaly, unusual odor (sweat, urine, feces), abnormal eating patterns (food aversions, vomiting after eating certain foods), coarse facial features, macroglossia (enlarged tongue), abnormal hair, dysmorphic features, and abnormal eye findings (e.g., cataracts, retinal changes). A family history of neonatal deaths (within the same sibling group, or among family members) alerts the observer to the possibility of an IEM. The initial recognition of signs that might indicate an IEM is the responsibility of health professionals, including nurses.
Although there are many categories of IEMs, only three are discussed here because they can be identified in the neonatal period and because treatment has been reasonably successful. They are examples of (1) disorders of protein metabolism (PKU), (2) disorders of carbohydrate metabolism (galactosemia), and (3) disorders of hormone synthesis (congenital hypothyroidism). (Table 5-4 outlines other IEMs.)
Phenylketonuria
PKU, a genetic disease inherited as an autosomal recessive trait, is caused by an absence of the enzyme phenylalanine hydroxylase needed to metabolize the essential amino acid phenylalanine. The prevalence of PKU varies widely in the United States because different states have different definition criteria for what constitutes hyperphenylalaninemia and PKU. The reported figures for PKU range from 1 per 19,000 to 1 per 13,500 live births. The disease has a wide variation of incidence by ethnic groups. The disease is most prevalent among individuals of Northern European ancestry, American Indians, and Alaskan Natives, whereas African-American, Hispanic, Jewish, and Asian individuals account for the lowest frequencies (Hellekson, 2001). Among African-Americans, for instance, the incidence of PKU is 1 per 50,000 live births (McPhee and Ganong, 2006).
Classic PKU is at one end of a spectrum of conditions that involve defects of amino acids phenylalanine and tyrosine metabolism known as hyperphenylalaninemia. Within the spectrum of hyperphenylalaninemia are conditions with varying degrees of severity depending on the degree of enzyme deficiency (Rezvani, 2007). Because other forms are the result of a deficiency of other enzymes and are diagnosed and treated differently, the following discussion of PKU is limited to the severe, classic form.
Pathophysiology
In PKU the hepatic enzyme phenylalanine hydroxylase, which controls the conversion of phenylalanine to tyrosine, is absent. This results in the accumulation of phenylalanine in the bloodstream and urinary excretion of abnormal amounts of its metabolites, the phenyl acids (Fig. 9-10). One of these phenyl ketones, phenylpyruvic acid, gives urine the characteristic musty odor associated with this disease and is responsible for the term phenylketonuria.
Amino acids produced by the metabolism of phenylalanine are absent in PKU. One of these, tyrosine, is needed to form the pigment melanin and the hormones epinephrine and thyroxine. Decreased melanin production results in similar phenotypes of most children with PKU: blond hair, blue eyes, and fair skin that is particularly susceptible to eczema and other dermatologic problems. Children with a genetically darker skin color may be red haired or brunette.
Severe hyperphenylalaninemia (>360 to 600 mmol/L) causes progressive damage to the developing brain with severe consequences: defective myelination, cystic degeneration of the gray and white matter, and disturbances in cortical lamination. Cognitive impairment occurs before the metabolites are detected in the urine and will progress if ingested phenylalanine levels are not lowered.
Clinical Manifestations
Clinical manifestations of PKU include growth failure (failure to thrive); frequent vomiting; irritability; hyperactivity; and unpredictable, erratic behavior. Older children commonly display bizarre or schizoid behavior patterns such as fright reactions, screaming episodes, head banging, arm biting, disorientation, failure to respond to strong stimuli, and spasticity or catatonia-like positions. Many of the severely cognitively impaired children have seizures, and approximately 80% of untreated persons with PKU demonstrate abnormal electroencephalographs, regardless of whether overt seizures occur.
Diagnostic Evaluation*
The objective in diagnosing or treating the disorder is to prevent cognitive impairment. The most commonly used test for screening newborns is the Guthrie bacterial inhibition assay for phenylalanine in the blood. Bacillus subtilis, present in the culture medium, grows if the blood contains an excessive amount of phenylalanine. The normal range of blood phenylalanine concentration in newborns is 0.5 to 1 mg/dl. The Guthrie test detects serum phenylalanine levels greater than 4 mg/dl (normal value is 1.6 mg/dl). Only fresh heel blood, not cord blood, can be used for the test.
Newborn screening tests are mandatory in all 50 U.S. states. The screening test is most reliable if the blood sample is taken after the infant has ingested a source of protein. Because of early discharge of newborns, recommendations for screening include (1) collecting the initial specimen as close as possible to discharge and no later than 7 days after birth, (2) obtaining a subsequent sample by 2 weeks of age if the initial specimen is collected before the newborn is 24 hours old, and (3) designating a primary care provider to all newborns before discharge for adequate newborn follow-up screening (Kaye, Committee on Genetics, Accurso, et al, 2006). In several states a second newborn screening is performed when the infant is 1 to 2 weeks old, on the basis that a maximum number of children with genetic disorders will be identified (American Academy of Pediatrics, 2008b). After a positive screen, diagnostic testing must be performed promptly (Kaye, Committee on Genetics, Accurso, et al, 2006).
When collecting the specimen, avoid “layering” the blood specimen on the special Guthrie paper. Layering is placing one drop of blood on top of the other or overlapping the specimen. Best results are obtained by collecting the specimen with a pipette from the heel stick and spreading the blood uniformly over the blot paper.
A major concern is that a significant number of infants are not rescreened for PKU after early discharge and are at risk for a missed or delayed diagnosis. Give special consideration to screening infants born at home who have no hospital contact, as well as infants adopted internationally.
Because of the possibility of variant forms of hyperphenylalaninemia, a natural dietary protein challenge test is recommended after approximately 3 months of dietary treatment to confirm the diagnosis of classic PKU.
Therapeutic Management*
Treatment of PKU involves the restriction of dietary protein. As with most genetic disorders, because the genetic enzyme is intracellular, systemic administration of phenylalanine hydroxylase is of no value. Phenylalanine cannot be totally eliminated because it is an essential amino acid in tissue growth. Therefore dietary management must meet two criteria: (1) meet the child’s nutritional need for optimum growth, and (2) maintain phenylalanine levels within a safe range.
Professionals agree that infants with PKU who have blood phenylalanine levels higher than 10 mg/dl should begin treatment to establish metabolic control as soon as possible, ideally by 7 to 10 days of age (Kaye, Committee on Genetics, Accurso, et al, 2006). Most clinicians now agree that, to achieve optimum metabolic control and outcome, a restricted phenylalanine diet, including medical foods and low-protein products, most likely will be medically required for virtually all individuals with classic PKU for their entire life (Kaye, Committee on Genetics, Accurso, et al, 2006). Such life-time reduction of phenylalanine intake is necessary to prevent neuropsychologic and cognitive deficits, since even a mild hyperphenylalaninemia (>1.2 mmol/L) would produce such effects. To evaluate the effectiveness of dietary treatment, frequent monitoring of blood phenylalanine and tyrosine levels is necessary.
The diet is calculated to allow 20 to 30 mg of phenylalanine per kilogram of body weight per day, which should maintain serum phenylalanine levels between 2 and 8 mg/dl. Significant brain damage is likely to occur when levels are greater than 11 to 15 mg/dl. In the United States a level of 2 to 6 mg/dl is recommended for children 12 years and younger and 2 to 10 mg/dl for older persons (Kaye, Committee on Genetics, Accurso, et al, 2006). For optimum growth to occur, the diet begins no later than 3 weeks of age.
Because all natural food proteins contain approximately 15% phenylalanine, specially prepared milk substitutes are prescribed for the infant (Table 9-3). Some of these products are made from specially treated enzymatic casein hydrolysate, which provides only 0.4% phenylalanine (28.5 mg/8 oz). They also contain minerals and vitamins to provide a balanced nutritional formula. Tyrosine and several other amino acids are supplied in the formula. Because of the low phenylalanine content of breast milk, total or partial breast-feeding may be possible with close monitoring of phenylalanine levels (Lawrence and Lawrence, 2005). Diet substitutes for older children, such as Phenyl-Free 2 (Mead Johnson) and Phenex-2 (Ross), contain no phenylalanine and allow for greater exchanges with natural low-phenylalanine foods in the diet, leading to a more normal diet.
TABLE 9-3
FORMULAS FOR INFANTS AND TODDLERS WITH METABOLIC CONDITIONS*†
| FORMULA (MANUFACTURER) | COMMENTS AND NUTRITIONAL CONSIDERATIONS |
| Phenyl-Free 1 (Mead Johnson) | Phenylalanine free; use for infants and toddlers with PKU; iron fortified; powder; phenylalanine from breast milk, infant formula, or other foods required to support growth; lactose free |
| Phenyl-Free 2 (Mead Johnson) | Phenylalanine free; use for children >1 yr of age and adults with PKU; iron fortified; powder; permits increased supplementation with normal foods; higher protein content than Phenyl-Free 1 |
| Phenex-1 (Ross) | Phenylalanine free; powder; use for infants and toddlers with PKU |
| Phenex-2 (Ross) | Phenylalanine free; powder; use for children and adults with PKU |
| Phenyl-Free 2 HP (Mead Johnson) | Phenylalanine free; powder; use for children and adults with PKU; permits increased supplementation with normal foods |
| Similac Isomil Advance | Lactose-free, soy-based formula for infants with galactosemia; Similac Go & Grow for children 9-24 mo of age |
| RCF (Ross) | Carbohydrate-free soy formula with iron; may be used in children with carbohydrate intolerance or galactosemia |
| Enfamil ProSobee Lipil (Mead Johnson) | Soy-based, lactose-free formula for infants with galactosemia; contains AA and DHA (LCPUFAs) (see also Chapter 8, Infant Formulas) |
| Enfamil Next Step ProSobee Lipil (Mead Johnson) | Soy-based, lactose-free formula for infants with galactosemia; contains AA and DHA (for infants ≥9 mo old) |
AA, Arachidonic acid; DHA, docosahexaenoic acid; LCPUFAs, long-chain polyunsaturated fatty acids; PKU, phenylketonuria.
*Ross Laboratories and Mead Johnson manufacture several specialty formulas for metabolic disorders for infants. For a comprehensive list of metabolic disease formulas, contact the manufacturers.
†This is not an exhaustive list of metabolic formulas or companies that offer such products. Reader is advised to consult primary care practitioner for additional information and specific guidelines for feeding children with special dietary needs.
A low-phenylalanine diet begins as soon as possible after birth and continues throughout life. Adherence to this diet can be especially challenging in adolescence and adulthood. To evaluate the effectiveness of dietary treatment, frequent monitoring of blood phenylalanine and tyrosine levels is necessary. Achieving optimum outcomes also relies on periodic monitoring of intellectual, neurologic, behavioral, and neuropsychologic parameters (National Institutes of Health Consensus Development Conference Statement, 2001). Because phenylalanine levels greater than or equal to 20 mg/dl in mothers with PKU affect the normal embryologic development of the fetus, women with PKU who are not on life-long diet must resume a low-phenylalanine diet before pregnancy. The Centers for Disease Control and Prevention (2002) reported that women who do not adhere to a strict diet before and during pregnancy deliver infants with a 93% risk for cognitive impairment and 72% risk for microcephaly.†
Prognosis: Although many individuals with treated PKU manifest no cognitive and behavioral deficits, many comparisons of individuals with PKU to controls show lower performance on IQ tests, with larger differences in other cognitive domains. However, their performance is still in the average range. Evidence for differences in behavioral adjustment is inconsistent despite anecdotal reports suggesting greater risk for internalizing psychopathology and attention disorders. In addition, there are insufficient data on the effects of phenylalanine restriction over many decades of life (Kaye, Committee on Genetics, Accurso, et al, 2006). Total bone mineral density is considerably lower in children who are on a low-phenylalanine diet, even though calcium, phosphorus, and magnesium intakes are higher than normal.
Currently, treatment for many genetic diseases aims at modifying the phenotypical expression of those conditions. In the case of PKU, early childhood treatment with a reduced phenylalanine diet is highly successful at removing the possibility of cognitive impairment. However, PKU-rescued adults (i.e., those treated with reduction of phenylalanine in early childhood) remain unable to metabolize the amino acid and will always exhibit phenylalaninemia. For this reason, life-long low-phenylalanine diet is recommended (Moyle, Fox, Bynevelt, et al, 2007). PKU-rescued women who do not maintain low phenylalanine levels tend to produce children with cognitive and developmental impairments. Although indirectly related to the maternal PKU genotype, this is due to the fact that embryos are developing in a uterine environment with excessive concentrations of blood phenylalanine. To avoid this possibility, PKU-rescued women contemplating motherhood need to be placed again on low-phenylalanine diet for the duration of the pregnancy.
Recently sapropterin dihydrochloride, or BH4, has been used successfully in persons with hyperphenylalaninemia to decrease circulating phenylalanine levels. BH4 was given in weight-based daily oral doses of 5 to 20 mg/kg/day and was shown to effectively reduce phenylalanine levels in persons 8 years old and older (Lee, Treacy, Crombez, et al, 2008). Additional data indicate that BH4 may be safely tolerated by children as young as 4 years of age; in this study children were able to increase phenylalanine daily intake slightly because of the blood level reductions caused by the drug (Trefz, Burton, Longo, et al, 2009). It is important to note that those taking BH4 in these studies remained on a phenylalanine diet restriction.
Nursing Care Management
The principal nursing considerations involve teaching the family regarding the dietary restrictions. Although the treatment may sound simple, the task of maintaining such a strict dietary regimen is demanding, especially for older children and adolescents. Foods with low phenylalanine levels (e.g., some vegetables [except legumes]; fruits; juices; and some cereals, breads, and starches) must be measured to provide the prescribed amount of phenylalanine. Most high-protein foods, such as meat and dairy products, are either eliminated or restricted to small amounts.
Maintaining the diet during infancy presents few problems. Parents can introduce solid foods such as cereal, fruits, and vegetables as usual to the infant. Difficulties arise as the child gets older. A decreased appetite and refusal to eat may reduce intake of the calculated phenylalanine requirement. The child’s increasing independence may inhibit absolute control of what he or she eats. Either factor can result in decreased or increased phenylalanine levels. During the school years, peer pressure becomes a major force in deterring the child from eating the prescribed foods or abstaining from high-protein foods such as milkshakes or ice cream. Limitations of this diet are best illustrated by an example: a  -lb hamburger may provide a 2-day phenylalanine allowance for a school-age child. Illness and growth spurts increase the body’s need for this essential amino acid. Adolescence is a particularly difficult period, and limiting foods containing phenylalanine in adolescents with PKU is challenging. Special camps to educate adolescent girls with PKU regarding appropriate food intake demonstrated short-term effects in decreasing blood phenylalanine levels (Singh, Kable, Guerrero, et al, 2000). However, studies show a gradual decline in diet compliance with consequent increases in blood phenylalanine levels during early adolescence and young adulthood (Walter and White, 2004).
-lb hamburger may provide a 2-day phenylalanine allowance for a school-age child. Illness and growth spurts increase the body’s need for this essential amino acid. Adolescence is a particularly difficult period, and limiting foods containing phenylalanine in adolescents with PKU is challenging. Special camps to educate adolescent girls with PKU regarding appropriate food intake demonstrated short-term effects in decreasing blood phenylalanine levels (Singh, Kable, Guerrero, et al, 2000). However, studies show a gradual decline in diet compliance with consequent increases in blood phenylalanine levels during early adolescence and young adulthood (Walter and White, 2004).
The assistance of a registered dietitian is essential. Parents need a basic understanding of the disorder and practical suggestions regarding food selection and preparation.* A number of support groups for parents of children with PKU are available nationwide. Many Internet resources also contain valuable information regarding dietary counseling and food options. Meal planning is based on an exchange list. As soon as children are old enough, usually by early preschool, they should be involved in the daily calculation, menu planning, and formula preparation. A computer voice-activated calculator, cards, or colored beads can help children keep track of the daily allowance of phenylalanine foods. A system of goal setting, self-monitoring, contracts, and rewards can promote compliance in adolescents.
Family Support†: In addition to the problems related to a child with a chronic disorder (see Chapter 22), the parents have the burden of knowing they are carriers of the defect and must make serious decisions regarding future children. Prenatal testing is now available to detect the PAH mutation in heterozygotes. Genetic counseling is especially important to ensure that the heterozygote couple understands their recurrence risk. Genetic counseling is needed for the person with PKU when he or she is of reproductive age to learn about the chances of having a child with PKU or a child affected by a high phenylalanine in-utero environment. (See Role of Nurses in Genetic Counseling and Referral, Chapter 5.)
Galactosemia
Galactosemia is a rare autosomal recessive disorder that results from various gene mutations leading to three distinct enzymatic deficiencies. The most common type of galactosemia (classic galactosemia) results from a deficiency of a hepatic enzyme, galactose 1-phosphate uridyltransferase (GALT), and affects approximately 1 of 47,000 births. The other two varieties of galactosemia involve deficiencies in the enzymes galactokinase (GALK) and galactose 4′-epimerase (GALE); these are extremely rare disorders (Kaye, Committee on Genetics, Accurso, et al, 2006). All three enzymes (GALT, GALK, and GALE) are involved in the conversion of galactose into glucose (Fig. 9-11).

Accumulation of activated 1-phosphate metabolites of galactose (and fructose) is extremely toxic to various tissues, especially in the kidneys, liver, and nervous system. As galactose 1-phosphate accumulates in the blood, a series of abnormalities develop. Hepatic dysfunction leads to cirrhosis, resulting in jaundice in the infant by the second week of life. The spleen subsequently becomes enlarged as a result of portal hypertension. Cataracts are usually recognizable by 1 or 2 months of age; cerebral damage, manifested by the symptoms of lethargy and hypotonia, is evident soon afterward. Infants with galactosemia appear normal at birth, but on ingestion of milk (which has a high lactose content) they begin to show progressive symptoms, including vomiting, diarrhea, and weight loss (Askin and Diehl-Jones, 2003). Escherichia coli sepsis is another common initial clinical sign (Kaye, Committee on Genetics, Accurso, et al, 2006; Kishnani and Chen, 2007). Death during the first month of life is frequent in untreated infants.
Diagnostic Evaluation*
Diagnosis is made on the basis of the infant’s history, physical examination, galactosuria, increased levels of galactose in the blood, or decreased levels of uridine diphosphate–galactose transferase activity in erythrocytes. The infant may display characteristics of malnutrition and dehydration; decreased muscle mass and body fat may be evident. Newborn screening for this disease is required in all states (http://genes-r-us.uthscsa.edu/nbsdisorders.pdf). Heterozygotes can also be identified, since they have significantly lower levels of the essential enzyme. Although asymptomatic, such individuals have been noted to spontaneously dislike and therefore limit the ingestion of galactose-containing foods.
Therapeutic Management
During infancy, treatment consists of eliminating all milk and lactose-containing formula, including breast milk. Traditionally, lactose-free formulas are used, with soy-protein formula being the feeding of choice (see Table 9-3). However, recent research suggests that elemental formula (galactose-free) may be more beneficial than soy formulas (Zlatunich and Packman, 2005). The American Academy of Pediatrics (2009a) recommends the use of soy formula for infants diagnosed with galactosemia. As the infant progresses to solids, only foods low in galactose should be consumed. Certain fruits are high in galactose, and some dietitians recommend that they be avoided. Nurses should give food lists to the family to ensure appropriate foods are chosen.
If galactosemia is suspected, implement supportive treatment and care, including monitoring for hypoglycemia, liver failure, bleeding disorders, and E. coli sepsis (Kaye, Committee on Genetics, Accurso, et al, 2006).
Prognosis: Follow-up studies of children treated from birth or within the first 2 months of life after symptoms appear have found long-term complications, such as ovarian dysfunction, cataracts, abnormal speech, cognitive impairment, growth restriction, and motor delay (Kaye, Committee on Genetics, Accurso, et al, 2006; Lashley, 2002). These findings have revealed that eliminating sources of galactose does not significantly improve the outcome. New therapeutic strategies, such as enhancing residual transferase activity, replacing depleted metabolites, or using gene replacement therapy, are needed to improve the prognosis for these children.
Nursing Care Management†
Nursing interventions are similar to those for PKU and other IEMs, except that dietary restrictions are easier to maintain because many more foods are allowed. However, reading food labels carefully for the presence of any form of lactose, especially dairy products, is mandatory. Many drugs, such as some of the penicillin preparations, contain lactose as filler and also must be avoided. Unfortunately, lactose is an unlabeled ingredient in many pharmaceuticals. Therefore instruct parents to ask their local pharmacist about galactose content of any over-the-counter or prescription medication.
Congenital Hypothyroidism
Congenital hypothyroidism (CH) (formerly called by the undesirable term cretinism), an inborn error of thyroid metabolism, occurs in neonates who are born without the ability to synthesize adequate amounts of thyroid hormone. Results of screening tests in the United States indicate that CH occurs in approximately 1 of every 4000 to 1 of every 3000 newborns (Kaye, Committee on Genetics, Accurso, et al, 2006). Infants with Down syndrome have a much higher rate (1 in 140 newborns) of either permanent or transient forms of the disorder. Also, a higher incidence of other congenital abnormalities has been observed in infants with CH.
A number of etiologic factors are implicated in CH, and the condition may be permanent or transient. Permanent CH can result from defective thyroid gland development, an enzymatic defect in thyroxine synthesis (primary disease), or (rarely) pituitary dysfunction (secondary disease). Transient hypothyroidism results from intrauterine transfer of goiter-inducing substances (such as the antithyroid drugs), which inhibit thyroid hormone secretion. Although self-limiting, this type is potentially fatal because the infant’s thyroid is unable to produce its own hormones once the maternal supply is terminated. In addition, regardless of etiology, a large goiter in a neonate may cause total obstruction of the airway. Many preterm infants have hypothyroidism (hypothyroxinemia) at birth as a result of hypothalamic and pituitary immaturity. However, this type is considered transient and often requires no treatment. Infants born prior to 28 weeks gestation may require temporary thyroid hormone replacement.
Clinical Manifestations
The severity of the disorder depends on the amount of thyroid tissue present and able to produce the necessary levels of thyroid hormones. Usually the newborn does not exhibit obvious signs of hypothyroidism immediately after birth because of the exogenous source of prenatal thyroid hormone supplied by the maternal circulation. However, subtle signs such as poor feeding, lethargy, prolonged neonatal jaundice, respiratory difficulty, cyanosis, constipation, hoarse cry, large fontanels, and bradycardia may be seen within the first few weeks of life. In addition, infants with CH may be born postterm (42 weeks). Clinical manifestations may be delayed in infants with a functional remnant of thyroid gland; infants with some types of familial hypothyroidism; and breast-fed infants, who may not display symptoms until weaned.
Classic features of untreated CH usually appear after approximately 6 weeks of life and include typical facial features (depressed nasal bridge, short forehead, puffy eyelids, and large tongue); thick, dry, mottled skin that feels cold to the touch; coarse, dry, lusterless hair; abdominal distention; umbilical hernia; hyporeflexia; bradycardia; hypothermia; hypotension with narrow pulse pressure; anemia; and widely patent cranial sutures. Bone age is greatly delayed from birth. The most serious consequence is delayed development of the nervous system, which leads to severe cognitive impairment. The severity of the intellectual deficit is related to the degree of hypothyroidism and the duration of the condition before treatment. Other nervous system manifestations include slow, awkward movements and abnormal deep tendon reflexes (often referred to as being “hung-up” because the relaxation phase after the contraction is slow), hypotonia, spasticity, speech disorders, fine motor incoordination, and strabismus.
Diagnostic Evaluation
Diagnosis is aimed at early identification of the disorder to prevent the serious impact on mental development and stunted physical growth as a result of delayed treatment. Mean IQ is reported to be proportional to the age when treatment is initiated (Kaye, Committee on Genetics, Accurso, et al, 2006). Neonatal screening consists of an initial filter-paper blood-spot thyroxine (T4) measurement followed by measurement of thyroid-stimulating hormone (TSH) in infants with low T4 values. Newborn screening is mandatory in all 50 U.S. states. Although it is best to obtain a heel stick blood sample for the test between 2 and 4 days of age, specimens are usually taken within the first 24 to 48 hours or before discharge as part of concurrent screening for other metabolic defects. Early screening can result in overdiagnosis (false positives), but this is preferable to missing the diagnosis.
For screening results that show a low level of T4 (<10%), obtain TSH levels, and if these are elevated (>40 mU/L), further tests to determine the cause of the disease should be carried out (American Academy of Pediatrics and American Thyroid Association, 2006). Additional tests include serum measurement of T4, triiodothyronine (T3) resin uptake, free T4, and thyroid-bound globulin. Tests of thyroid gland function (thyroid scan and uptake) usually involve oral administration of a radioactive isotope of iodine (131I) and measurement of iodine uptake by the thyroid, usually within 24 hours. Patients with CH have low T4, T3, and free T4 levels and decreased thyroid uptake of 131I. Skeletal radiography is employed to assess bone age.
In the newborn, thyroid function studies are elevated in comparison with values in older children. Thus it is important to document the timing of the tests. In preterm and sick full-term infants, thyroid function tests are usually lower than in healthy full-term infants; a repeat T4 and TSH may be evaluated after 30 weeks (corrected age) in newborns born before that time and after resolution of the acute illness in the sick full-term infant.
Therapeutic Management
Treatment involves life-long thyroid hormone replacement therapy that begins as soon as possible after diagnosis to abolish all signs of hypothyroidism and to reestablish normal physical and mental development. The drug of choice is synthetic levothyroxine sodium (Synthroid or Levothroid). Regular measurement of T3, T4, and TSH levels is important in ensuring optimum treatment. Optimum dosage of l-thyroxine should be able to maintain blood TSH concentration between 0.5 and 2.0 mU/L during the first 3 years of life (American Academy of Pediatrics and American Thyroid Association, 2006). Bone age surveys are also performed to ensure optimum growth.
Prognosis: If treatment begins shortly after birth (by 2 weeks of age) and is consistently maintained, normal physical growth and intelligence are possible. The most significant factor adversely affecting eventual intelligence appears to be inadequate treatment, which may be related to nonadherence.
Nursing Care Management
The most important nursing objectives include collecting an adequate specimen and identifying the disorder early. The integrity of the blood specimen must be maintained for the test to be accurate; overlaying of blood on the designated spot may produce inaccurate results. Blood is applied to only one side of the paper so that complete saturation is obtained. Keep the specimen paper dry and avoid excessive heat exposure.
Nurses caring for neonates must be certain that screening is performed, especially in infants who are preterm, discharged early, or born at home. Although the screening test is specific, some children may not be identified, and nurses in community health need to be aware of the earliest signs of the disorder. Parental remarks about an unusually “quiet and good” baby together with any of the early physical manifestations should lead to a suspicion of hypothyroidism, which requires a referral for specific tests.
Once the diagnosis is confirmed, parents need an explanation of the disorder and the necessity of life-long treatment. The nurse must stress the importance of compliance with the drug regimen for the child to achieve normal growth and development. Because the drug is tasteless, it can be crushed and added to formula, breast milk, water, or food. However, do not administer soy, fiber, or iron with the medication. If a dose is missed, twice the dose should be given the next day. Unless there are maternal contraindicative factors, breast-feeding is acceptable in infants with hypothyroidism (Lawrence and Lawrence, 2005). Parents also need to be aware of signs indicating overdose, such as rapid pulse, dyspnea, irritability, insomnia, fever, sweating, and weight loss. Ideally they should know how to count the pulse and be instructed to withhold a dose and consult their practitioner if the pulse rate is above a certain value. Signs of inadequate treatment are fatigue, sleepiness, decreased appetite, and constipation.
If the diagnosis was delayed past early infancy, the chance of permanent cognitive impairment is great. Parents need the same guidance in caring for their child as do others who have an offspring with cognitive impairment. (See Chapter 24.) They need an opportunity to discuss their feelings regarding late recognition of the disorder. Although treatment will not reverse the intellectual deficit, it may prevent further damage. Genetic counseling is important, especially if the disorder is caused by an inborn error of thyroid hormone synthesis, which is autosomal recessive. (See Chapter 5 for a discussion of genetic counseling.)
Problems Caused by Perinatal Environmental Factors
Prenatal environmental effects from chemicals such as alcohol, medications, or drugs; infectious disease; or radiation or other environmental factors may be regarded as nongenetic causes of congenital anomalies because these substances can produce congenital structural, functional, or growth defects. An agent that produces congenital malformations or increases their incidence is called a teratogen.
The relationship of the fetal and maternal circulations allows for the interchange of chemical substances across the placental membrane. Many drugs have been suspected of producing congenital malformations, and some have been definitely implicated. Some of the most recognized teratogenic drugs include alcohol, tobacco, antiepileptic medications (valproic acid, phenytoin), isotretinoin (Accutane), lithium, methotrexate, cocaine, and diethylstilbestrol. (See Chapter 10, Drug-Exposed Infants and Fetal Alcohol Syndrome or Alcohol-Related Birth Defects.) The limited metabolic capabilities of the fetal liver and its immature enzyme and transport systems render the unborn child ill equipped for maintaining homeostasis when chemical disturbances are imposed by the mother or the environment. This includes both substances produced by the mother in response to a disease state (such as diabetes) and exogenous substances ingested or inhaled by the mother.
The teratogenic effect of drugs is not believed to affect developing tissue until day 15 of gestation, when tissue differentiation begins to take place. Before that time, drugs usually have little effect because they are believed to have an insignificant affinity for undifferentiated tissue. Also, until implantation takes place, at approximately 7 days after conception, the embryo is not exposed to maternal blood that contains the drug. However, some drugs may affect the uterine lining, making it unsuitable for implantation. Drugs administered between days 15 and 90 may produce an effect if the tissue for which the drug has an affinity is in the process of differentiation at that time. After 90 days, when differentiation is complete, most fetal tissues are relatively resistant to teratogenic effects of drugs. However, the impact on ongoing neurologic development is not known.
Nursing Care Management
Caution expectant mothers against ingesting any medication without first consulting a practitioner. To help ensure that fewer women will inadvertently take some chemical that might harm the fetus, medication labels are now required to include information regarding the possible teratogenic effects. Excessive use of some commonplace drugs, such as alcohol, valproic acid, and isotretinoin, produces characteristic malformations in the fetus.
Nurses should be aware of Birth Defect Research for Children, Inc.,* which offers help and information to families with children with defects caused by maternal exposure to drugs, chemicals, radiation, or other environmental agents.
Radiation
Ionizing radiation in large doses has been shown to be both mutagenic and teratogenic in humans. Pelvic irradiation of pregnant women—from natural background radiation that is present everywhere in varying degrees, from occupational exposure, or from diagnostic or therapeutic procedures—is believed to be hazardous to the embryo, although the extent of teratogenicity and the exact dosage required to induce somatic change is not yet known. The risk of untoward effects to an irradiated fetus whose mother is undergoing diagnostic radiation by CT is reported to be the same as that of the general population, or 1% to 3% (Ratnapalan, Bona, and Koren, 2003). Patel, Reede, Katz, and colleagues (2007) affirm that, although there is a theoretical risk of carcinogenesis when the fetus is exposed to diagnostic radiation, with the radiation levels currently used there are no known risks for congenital malformations or cognitive impairment. These authors present an extensive review of diagnostic radiation methods used for pregnant and lactating mothers. Radiation may damage the conceptus at any time during its prenatal existence, and it is known that rapidly dividing and differentiating cells, such as those of the embryo, have increased radiosensitivity. As with other teratogens, the type of effect produced is closely correlated with the stage of development at which the radiation exposure occurs.
To help prevent the possibility of radiation damage, it is advisable (1) to avoid unnecessary radiation exposure, such as elective radiographs to the pelvis and abdomen, in women of childbearing age except during the 2 weeks immediately after menstruation; (2) to ascertain whether pregnancy is a possibility; and (3) to advise both men and women who have lower abdominal or pelvic radiographs to avoid conception for several months. Pregnant women should avoid radioactive iodine exposure, since iodine has an affinity for fetal thyroid tissue and can lead to developmental problems such as fetal goiter, microcephaly, intrauterine growth restriction, malignancy, and death. Women should also avoid the use of nonradioactive iodides in vaginal douche solutions, vaginal suppositories containing povidone-iodine (Betadine), and iodinized drugs for asthmatics during pregnancy (Blackburn, 2007).
The occurrence of childhood cancer as a result of prenatal or preconceptual radiation exposure has been reported to be unfounded (Mettler and Stazzone, 2007).
Key Points
• Problems of the newborn may be attributed to birth injuries, transient metabolic illnesses, and IEMs.
• The forces of labor and delivery may cause soft tissue injury, head trauma, fractures, and paralysis.
• The most common forms of paralysis in the newborn are facial nerve, brachial plexus, and phrenic nerve palsies.
• Common skin problems of the newborn include erythema toxicum; candidiasis; bullous impetigo; and birthmarks, especially port-wine stains and hemangiomas.
• Because of their immature physiologic status, infants may be predisposed to hyperbilirubinemia, hypoglycemia, hyperglycemia, and hypocalcemia.
• In the newborn hyperbilirubinemia may result from excess production of bilirubin, decreased capacity of the liver to conjugate bilirubin, and/or deconjugation of bilirubin in the neonatal intestine (enterohepatic shunting).
• The primary treatment of hyperbilirubinemia is phototherapy.
• HDN is characterized by abnormally rapid destruction of RBCs as a result of blood incompatibility between mother and fetus.
• Hypoglycemia can often be prevented by initiating early feedings in the healthy asymptomatic newborn.
• Hemorrhagic disease of the newborn is characterized by oozing from the umbilicus or circumcision site, bloody or black stools, hematuria, ecchymoses, and epistaxis.
• The most significant IEMs are CH, PKU, and galactosemia.
• Thyroid replacement medication is required to treat CH.
• Life-long dietary control is the treatment of choice for PKU and galactosemia to prevent serious neurobehavioral deficits.
• Perinatal environmental factors such as chemicals, drugs ingested in pregnancy, and radiation may have fetal teratogenic effects.
References
Alster, TS, Railan, D. Laser treatment of vascular birthmarks. J Craniofac Surg. 2006;17(4):720–723.
American Academy of Pediatrics. Pediatric nutrition handbook, ed 6. Elk Grove Village, Ill: The Academy; 2009.
American Academy of Pediatrics. Red book: 2009 report of the Committee on Infectious Diseases, ed 28. Elk Grove, Ill: The Academy; 2009.
American Academy of Pediatrics. Prevention of rickets and vitamin D deficiency in infants, children, and adolescents. Pediatrics. 2008;122(5):1142–1148.
American Academy of Pediatrics, Newborn Screening Authoring Committee. Newborn screening expands: recommendations for pediatricians and medical homes—implications for the system. Pediatrics. 2008;121(2):192–217.
American Academy of Pediatrics, Subcommittee on Hyperbilirubinemia. Clinical practice guideline: management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114(1):297–316.
American Academy of Pediatrics, American College of Obstetricians and Gynecologists. Guidelines for perinatal care, ed 6. Elk Grove Village, Ill: The Academy; 2007.
American Academy of Pediatrics, American Thyroid Association. Update of newborn screening and therapy for congenital hypothyroidism. Pediatrics. 2006;117(6):2290–2303.
Askin, DF, Diehl-Jones, B. Liver, part 3, Pathophysiology of liver dysfunction. Neonat Netw. 2003;22(3):5–15.
Bhutani, VK, Johnson, LH, Keren, R. Diagnosis and management of hyperbilirubinemia in the term neonate: for a safer first week. Pediatr Clin North Am. 2004;51(4):843–861.
Black, LV, Maheshwari, A. Disorders of the fetomaternal unit: hematologic manifestations in the fetus and neonate. Semin Perinatol. 2009;33(1):12–19.
Blackburn, S. Maternal, fetal, and neonatal physiology: a clinical perspective, ed 3. St Louis: Saunders; 2007.
Brown, ZA, Wald, A, Morrow, RA, et al. Effect of serologic status and cesarean delivery on transmission rates of herpes simplex virus from mother to infant. JAMA. 2003;289:203–209.
Centers for Disease Control and Prevention. Barriers to dietary control among pregnant women with phenylketonuria—United States, 1998-2000. MMWR. 2002;51(5):117–120.
Chertok, IR, Raz, I, Shoham, I, et al. Effects of early breastfeeding on neonatal glucose levels of term infants born to women with gestational diabetes. J Hum Nutr Diet. 2009;22(2):166–169.
Cornblath, M, Hawdon, JM, Williams, AF, et al. Controversies regarding definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics. 2000;105(5):1141–1145.
Cowett, RM, Loughead, JL. Neonatal glucose metabolism: differential diagnoses, evaluation, and treatment of hypoglycemia. Neonat Netw. 2002;21(4):9–18.
Custer JW, Rau RE, eds. Harriet Lane handbook, 18 edition, St. Louis: Mosby, 2009.
Dennery, PA. Metalloporphyrins for the treatment of neonatal jaundice. Curr Opin Pediatr. 2005;17(2):167–169.
Desphande, S, Platt, MW. The investigation and management of neonatal hypoglycaemia. Semin Fetal Neonat Med. 2005;10(4):351–361.
Doumouchtsis, SK, Arulkumaran, S. Head injuries after instrumental vaginal deliveries. Curr Opin Obstet Gynecol. 2006;18(2):129–134.
Drolet, BA, Swanson, EA, Frieden, IJ, et al. Infantile hemangiomas: an emerging health issue linked to an increased rate of low birth weight infants. J Pediatr. 2008;153(5):712–715.
Finning, K, Martin, P, Daniels, G. The use of maternal plasma for prenatal RhD blood group genotyping. Methods Mol Biol. 2009;496:143–157.
Gottstein, R, Cooke, RW. Systematic review of intravenous immunoglobulin in haemolytic disease of the newborn. Arch Dis Child Fetal Neonatal Ed. 2003;88(1):F6–F10.
Hellekson, KL. Practice guidelines: NIH consensus statement on phenylketonuria. Am Fam Physician. 2001;63(7):1430–1432.
Hoseth, E, Joergensen, A, Ebbesen, F, et al. Blood glucose levels in a population of healthy, breast fed, term infants of appropriate size for gestational age. Arch Dis Child Fetal Neonatal Educ. 2000;83(2):F117–F119.
Hudic, I, Fatusic, Z, Sinanovic, O, et al. Etiological risk factors for brachial plexus palsy. J Matern Fetal Neonatal Med. 2006;19(10):655–661.
Joanna Briggs Institute. Management of asymptomatic hypoglycaemia in healthy term neonates for nurses and midwives. Nurs Standard. 2007;22(8):35–38.
Joyner, B, Soto, MA, Adam, HM. Brachial plexus injury. Pediatr Rev. 2006;27(6):238–239.
Kaye, CI, Committee on GeneticsAccurso, F, et al. Newborn screening fact sheets. Pediatrics. 2006;118(3):e934–e963.
Kimberlin, DW. Herpes simplex virus infections of the newborn. Semin Perinatol. 2007;31(1):19–25.
Kimberlin, DW. Herpes simplex virus infections in neonates and early childhood. Semin Pediatr Infect Dis. 2005;16:271–281.
Kishnani, PS, Chen, YT. Defects in galactose metabolism. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Lashley, FR. Newborn screening: new opportunities and new challenges. Newborn Infant Nurs Rev. 2002;2(4):228–242.
Lawrence, RA, Lawrence, RM. Breastfeeding: a guide for the medical profession, ed 6. St Louis: Mosby; 2005.
Lee, P, Treacy, EP, Crombez, E, et al. Safety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuria. Am J Med Genet A. 2008;146A(22):2851–2859.
Maisels, MJ, Bhutani, VK, Bogen, D, et al. Hyperbilirubinemia in the newborn infant ≥35 weeks’ gestation: an update with clarifications. Pediatrics. 2009;124(4):1193–1198.
Maisels, MJ, McDonough, AF. Phototherapy for neonatal jaundice. N Engl J Med. 2008;358(9):920–928.
Mangurten, HH. Birth injuries. In Fanaroff AA, Martin RJ, eds.: Neonatal-perinatal medicine: diseases of the fetus and infant, ed 8, St Louis: Mosby, 2006.
McCance, K, Huether, S. Pathophysiology: the biological basis for disease in adults and children, ed 6. St Louis: Mosby; 2010.
McPhee, SJ, Ganong, WF. Pathophysiology of disease: an introduction to clinical medicine, ed 5. New York: Lang Medical Books; 2006.
Metry, D. Update on hemangiomas of infancy. Curr Opin Pediatr. 2004;16(4):373–377.
Mettler, FA, Stazzone, MM. Environmental health hazards: pediatric radiation injuries. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Miclic, TL. Neonatal glucose homeostasis. Neonat Netw. 2008;27(3):203–207.
Miller, T, Frieden, IJ. Hemangioma: new insights and classification. Pediatr Ann. 2005;34(3):179–187.
Moise, KJ. Management of rhesus alloimmunization in pregnancy. Obstet Gynecol. 2008;112(1):164–176.
Moise, KJ. Red cell alloimmunization. In Gabbe SG, Niebyl JR, Simpson KL, eds.: Obstetrics: normal and problem pregnancies, ed 5, London: Churchill Livingstone, 2007.
Moise, KJ. Management of rhesus alloimmunization in pregnancy. Obstet Gynecol. 2002;100(3):600–611.
Morelli, JG. Cutaneous bacterial infections. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Moyle, JJ, Fox, AM, Bynevelt, M, et al. A neuropsychological profile of off-diet adults with phenylketonuria. J Clin Exp Neuropsychol. 2007;29(4):436–441.
Mundy, CA. Intravenous immunoglobulin in the management of hemolytic disease of the newborn. Neonat Netw. 2005;24(6):17–24.
National Institutes of Health Consensus Development Conference Statement. Phenylketonuria: screening and management, Oct 16-18, 2000. Pediatrics. 2001;108(4):972–982.
Paige, PL, Moe, PC. Neurologic disorders. In Merenstein GB, Gardner SL, eds.: Handbook of neonatal intensive care, ed 6, St Louis: Mosby, 2006.
Pandey, A, Gangopadhyay, An, Upadhyay, VD. Evaluation and management of infantile hemangioma: an overview. Ostomy Wound Manage. 2008;54(5):16–18. [20, 22-26, 28-29].
Patel, SJ, Reede, DL, Katz, DS, et al. Imaging the pregnant patient for nonobstetric conditions: algorithms and radiation dose considerations. Radiographics. 2007;27(6):1705–1722.
Price, AE, Ditaranto, P, Yaylali, I, et al. Botulinum toxin type A as an adjunct to the surgical treatment of the medial rotation deformity of the shoulder in birth injuries of the brachial plexus. J Bone Joint Surg Br. 2007;89(3):327–329.
Ratnapalan, S, Bona, N, Koren, G. Ionizing radiation during pregnancy. Can Fam Physician. 2003;49:873–874.
Rezvani, I. Defects in metabolism of amino acids. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Rozance, PJ, Hay, WH. Hypoglycemia in newborn infants: features associated with adverse outcomes. Biol Neon. 2006;90(2):74–86.
Sgro, M, Shah, V, Campbell, D. Canadian Paediatric Surveillance Program: challenge of early discharge: newborn assessment for jaundice. available at www.cps.ca/English/Surveillance/CPSP/Resources/NHS.htm, 2005. [(accessed June 28, 2007)].
Singh, RH, Kable, JA, Guerrero, NV, et al. Impact of a camp experience on phenylalanine levels, knowledge, attitudes, and health beliefs relevant to nutrition management of phenylketonuria in adolescent girls. J Am Diet Assoc. 2000;100(7):797–803.
Sirkin, A, Jalloh, T, Lee, L. Selecting an accurate point-of-care testing system: clinical and technical issues and implications in neonatal blood glucose monitoring. J Spect Pediatr Nurs. 2002;7(3):104–112.
Sperling, MA. Hypoglycemia. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Sperling, MA, Menon, RK. Differential diagnosis and management of neonatal hypoglycemia. Pediatr Clin North Am. 2004;51(3):703–723.
Stier, MF, Glick, SA, Hirsch, RJ. Laser treatment of pediatric vascular lesions: port wine stains and hemangiomas. J Am Acad Dermatol. 2008;58(2):261–285.
Stokowski, LA. Fundamentals of phototherapy for neonatal jaundice. Adv Neonat Care. 2006;6(6):303–312.
Stoll, BJ. Blood disorders. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Tappero, E. Musculoskeletal system assessment. In Tappero E, Honeyfield MA, eds.: Physical assessment of the newborn: a comprehensive approach to the art of physical examination, ed 4, Santa Rosa, Calif: NICU Ink, 2009.
Trefz, FK, Burton, BK, Longo, N, et al. Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double-blind, placebo-controlled study. J Pediatr. 2009;154(5):700–707.
Urbaniak, SJ. Noninvasive approaches to the management of RhD hemolytic disease of the fetus and newborn. Transfusion. 2008;48(1):12–19.
Walter, JH, White, FJ. Blood phenylalanine control in adolescents with phenylketonuria. Int J Adolesc Med Health. 2004;16(1):41–45.
Watchko, JF. Identification of neonates at risk for hazardous hyperbilirubinemia: emerging clinical insights. Pediatr Clin North Am. 2009;56(3):671–687.
Zlatunich, CO, Packman, S. Galactosaemia: early treatment with an elemental formula. J Inherit Metab Dis. 2005;28:163–168.
*210 Spring Haven Circle, Royersford, PA 19468; www.brachialplexuspalsyfoundation.org.
*Information is available from Birthmarks and Hemangiomas InterNETwork Support Group, http://members.tripod.com/~Michelle_G/SPTGP.html; and Vascular Birthmarks Foundation, 877-VBF-4646; www.birthmark.org.
*Always refer patient to a genetic metabolic specialist. For a reference list, visit the American Society of Human Genetics website, www.ashg.org.
*A resource for dietary management is Acosta PB, Yannicelli S: The Ross metabolic formula system nutrition support protocols, ed 4, Columbus, Ohio, 2001, Abbott Nutrition; 800-227-5767; http://abbottnutrition.com.
†For more information, contact American Society of Human Genetics, 9650 Rockville Pike, Bethesda, MD 20814; 301-634-7300, 866-HUM-GENE; www.ashg.org.
*A helpful resource is Schuett V, editor: Low protein cookery for phenylketonuria, ed 3, Madison, Wis, 1997, University of Wisconsin Press.
†National support groups include the Children’s PKU Network, which offers a variety of support services: 3790 Via de la Valle, Suite 120, Del Mar, CA 92014; 800-377-6677; e-mail: PKUnetwork@aol.com; www.pkunetwork.org; and the National PKU Alliance, Christine Brown, Executive Director, PO Box 501, Tomahawk, WI 54487; 715-437-0477; www.npkua/org.
*Always refer patients to a genetic metabolic specialist. For a reference list visit the American Society of Human Genetics website, www.ashg.org.
†Information and support for parents can be found at the American Liver Foundation, www.liverfoundation.org; and at Parents of Galactosemic Children, Inc., PO Box 2401, Mandeville, LA 74070-2401; 866-900-PGC1; www.galactosemia.org.
*800 Celebration Ave., Suite 225, Celebration, FL 34747; 407-466-8304; www.birthdefects.org.