Chapter 14 Drug development
Introduction
Introduction
Drug development comprises all the activities involved in transforming a compound from drug candidate (the end-product of the discovery phase) to a product approved for marketing by the appropriate regulatory authorities. Efficiency in drug development is critical for commercial success, for two main reasons:
• Development accounts for about two-thirds of the total R&D costs. The cost per project is very much greater in the development phase, and increases sharply as the project moves into the later phases of clinical development. Keeping these costs under control is a major concern for management. Failure of a compound late in development represents a lot of money wasted.
• Speed in development is an important factor in determining sales revenue, as time spent in development detracts from the period of patent protection once the drug goes to market. As soon as the patent expires, generic competition sharply reduces sales revenue.
Despite a high level of awareness in the pharmaceutical industry of the need to reduce the money and time spent on development, both have actually increased significantly over the last two decades (see Chapter 22). This is mainly due to external factors, particularly the increased stringency applied by regulatory authorities in assessing the safety and efficacy of new compounds (see Chapter 20). The development burden is, therefore, tending to increase, thereby increasing the need for companies to improve their performance in this area in order to remain profitable and competitive.
The nature of drug development
Drug discovery, as described in Section 2, is invariably an exploration of the unknown, and successful projects may end up with compounds quite different from what had originally been sought: there is a large component of ‘unplannability’. In contrast, drug development has a very clear-cut goal: to produce the drug in a marketable form, and to gain regulatory permission to market it for use in the target indication(s) as quickly as possible. The work required to do this falls into three main parts, respectively technical, investigative and managerial:
• Technical development – solving technical problems relating to the synthesis and formulation of the drug substance, aimed mainly at ensuring the quality of the end-product:
• Investigative studies – establishing the safety and efficacy of the product, including assessment of whether it is pharmacokinetically suitable for clinical use in man:
 Coordination – managing quality control, logistics, communication and decision making in a large multidisciplinary project to ensure high-quality data and to avoid unnecessary delays:
Coordination – managing quality control, logistics, communication and decision making in a large multidisciplinary project to ensure high-quality data and to avoid unnecessary delays:An important distinction between the technical and investigative aspects of development is that, in tackling technical problems, it is assumed that a solution does exist, and so the team’s task is to find and optimize it as quickly as possible, whereas in assessing safety and efficacy it cannot be assumed that the compound reaches the required standards – rather, the object is to discover this as quickly and cheaply as possible. In other words, technical development is essentially an exercise in problem solving, whereas clinical and toxicological development is a continuing investigation of the properties of the compound. Although technical problems, such as an unacceptably complex and poor-yielding synthesis route, or difficulty in developing a satisfactory formulation, can result in abandonment of the project, this is relatively uncommon. Failure on account of the drug’s biological properties, such as toxicity, poor efficacy or unsatisfactory pharmacokinetics, is, however, very common, and largely accounts for the fact that only some 10% of compounds entering Phase I clinical trials are eventually marketed. An important aspect of the management of drug-development projects, therefore, is to establish firm ‘no-go’ criteria, and to test the compound against them as early as possible.
Development proceeds along much more clearly defined lines than discovery, and is consequently more ‘plannable’, particularly the non-clinical studies, where standard experimental protocols exist for most of the work that needs to be carried out. This applies also in Phase I clinical studies. Delays can nevertheless occur if unexpected findings emerge, for example poor oral absorption in humans, or species-specific toxic effects, which require additional work to be carried out before clinical trials can proceed. If the drug has a completely novel mechanism of action this often prolongs the technical phase whilst off-target effects are explored (sometimes at the insistence of the regulatory authority).
Beyond Phase I, the route to be followed is generally much less well charted, and success depends to a much greater extent on strategic decisions by the project team as to which clinical indications should be investigated (see Chapter 17). They will need to assess, for example, whether recruiting patients to the trial will be easy or difficult, what exclusion criteria should apply, what clinical outcome measures should be used, and how long the treatment and assessment periods will need to be. To achieve registration as quickly as possible, it may, for example, be expedient to select a relatively low-market, but quick-to-test, clinical indication for the initial trials, and to run these trials in parallel with more prolonged trials in the major indication. Careful attention needs to be given to the patient group selected for the trial, so as to maximize the chance of success in obtaining a clear-cut result. Experience shows that inconclusive clinical trials resulting from poor decisions of this sort are a common cause of failure or delay in drug development. Where the indication allows this an adaptive trial design may allow a more efficient evaluation of the drug (see Chapter 17).
Components of drug development
Figure 14.1 summarizes the main activities involved in developing a typical synthetic compound. It shows the main tasks that have to be completed before the compound can be submitted for regulatory approval, but needs to be translated into an operational plan (Figure 14.2) that will allow the project to proceed as quickly and efficiently as possible. It is obvious that certain tasks have to be completed in a particular order. For example, a supply of pure compound, prepared in an acceptable formulation, has to be available before Phase I clinical studies can begin. Animal toxicity data must also be available before the compound can be given to humans. Deciding on the dosage schedule to be used in efficacy trials requires knowledge of the pharmacokinetics and metabolism of the compound in humans. Because the data generated will be included in the final registration proposal, it is essential that each part of the work should be formally reported and ‘signed off’ by the group responsible and archived for future reference. A typical development project is likely to involve several hundred individuals, expert in different disciplines and working on different aspects of the project, and coordinating their work is a complex and demanding task. For this reason, most companies assign specialist project managers to this task. Their role is to design a project plan, based on input from the experts involved, to monitor progress and to adapt the plan accordingly. As well as being good organizers, project managers need to be excellent communicators, diplomatic, and with a good understanding of the scientific and technological aspects of the project. Figure 14.2 is a much-simplified outline of a project plan of the development of a typical orally active drug. Each ‘task’, represented by an arrow, starts and ends at a circular symbol (representing an ‘event’), and decision points are marked by diamond symbols. This type of graphical format, which is widely used as a project management tool and implemented in many commercially available software packages, is known as a PERT (project evaluation and review technique) chart. By assigning times – shortest possible, maximum, and expected – to each task, the timing of the whole project can be assessed and the critical path – i.e. the sequence of tasks that need to be completed on time in order to avoid an overall delay – defined. In Figure 14.2 the process has been reduced to a bare minimum to allow representation on a single page; in practice, each of the ‘tasks’ shown (e.g. develop formulations, perform Phase I studies, etc.) needs to be further subdivided into a series of subtasks and timings to enable the project to be planned and monitored at the operational level. The complete diagram for a typical drug development project will be of such size and complexity as to frighten all but the most hardened project management professionals. Software tools, fortunately, are available which allow the project to be viewed in different ways, such as Gantt charts, which are barcharts set against a calendar timescale, showing the expected start and completion dates for each task, many of which will be running simultaneously on any given date1.
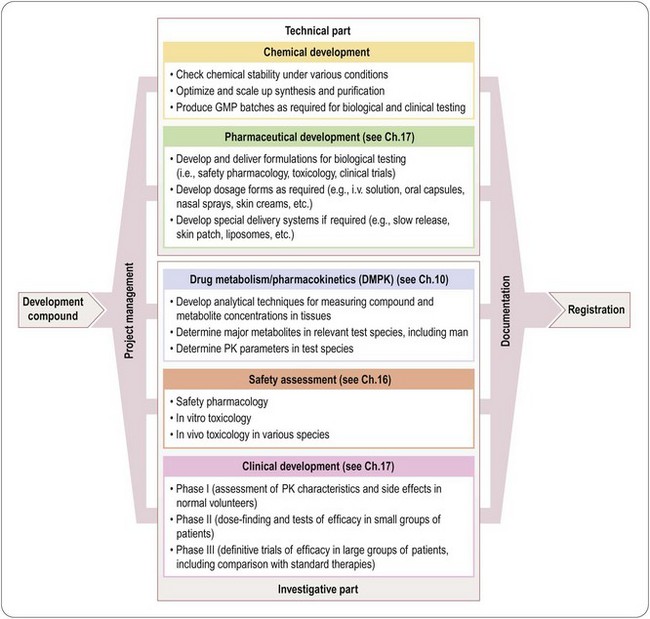
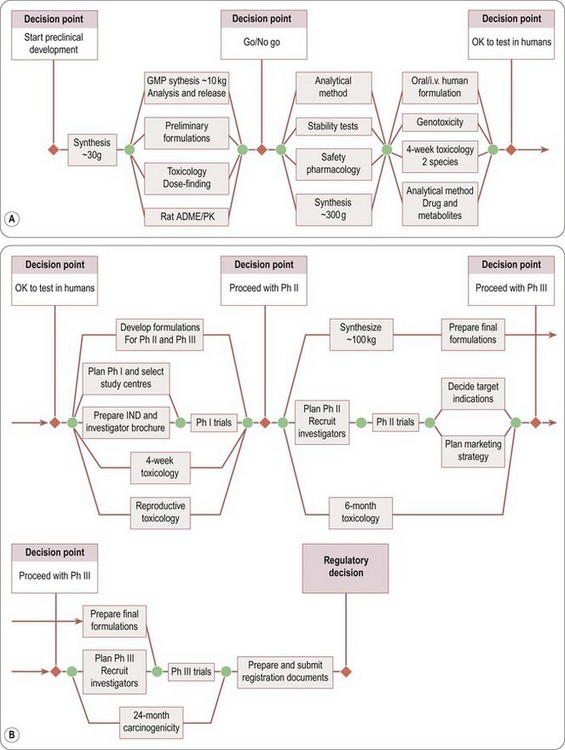
Fig. 14.2 Simplified flowchart showing the main activities involved in drug development. The nodes indicated by circles represent the start and finish points of each activity, and the diagram indicates which activities need to be completed before the next can begin. By assigning timescales to each activity, the planned overall development time can be determined and critical path activities identified. (A) Preclinical development. (B) Clinical development.
In this section of the book we outline the main technical and experimental parts of the work that goes into drug development, namely toxicology, pharmaceutical development and clinical studies. Chapter 19 discusses the principles underlying the patenting of drugs. Chapter 20 describes how regulatory bodies go about evaluating new compounds for registration, and Chapter 21 presents an introduction to the principles of pharmaceutical marketing. Chemical development, covering the specialized technical aspects of producing the drug substance economically and safely on a large scale, as well as the control measures needed to ensure consistent high quality of the final product, is beyond the scope of this book (see Gadamasetti, 2007; Repic, 1998 for a full account of this subject).
The interface between discovery and development
For the purposes of this book, drug development is presented as an operation separate from discovery and following on from it, but the distinction is actually not clear-cut. Increasingly, as has been stressed in Chapters 9 and 10, activities previously undertaken during development are taking place earlier, as an integral part of the discovery process. The emphasis on the ‘druggability’ of leads (Chapter 9) reflects a concern for focusing on structures that are least likely to have unsatisfactory pharmacokinetic, toxicological or stability problems. Whereas in vitro tests of absorption and metabolism were traditionally performed on individual compounds during the development phase, they are now being incorporated into medium-throughput screens in the discovery phase (Chapter 10), as are in vitro toxicity tests. Formulation work also often begins during the discovery phase, particularly if the characteristics of the lead compounds suggest that specialized formulations are likely to be required for use in pharmacological profiling of compounds in vivo. Furthermore, the selection of a single compound for full development will sometimes be deferred until data from Phase I trials on several candidates have been obtained. Thus, the preliminary work needed before Phase I, and the Phase I trials themselves, will need to be performed on a group of candidate compounds. This is clearly more expensive than choosing the development compound before Phase I, but may be justified as a strategy for reducing the risk of failure at a later (and more expensive) stage.
Decision points
The decision to advance a drug candidate into early development is the first of several key strategic decision points in the history of the drug development project. The timing, nomenclature and decision-making process vary from company to company, and Figure 14.3 shows a typical scheme, developed by Novartis:
• The early selection point (ESP) is the decision to take the drug candidate molecule into early (preclinical) development. The proposal will normally be framed by the drug discovery team and evaluated by a research committee, which determines whether the criteria to justify further development have been met. After this checkpoint, responsibility normally passes to a multidisciplinary team with representatives from research, various development functions, patents, regulatory affairs and marketing, under the direction of a professional project manager. In a large multinational company the team will have international representation, and the development plan will be organized to meet global development standards as far as possible.
• The decision to develop in man (DDM) controls entry of the compound into Phase I, based on the additional information obtained during the preclinical development phase (i.e. preliminary toxicology, safety pharmacology, pharmacokinetics, etc.). An important task once this decision point is passed is normally the production of a sufficient quantity (usually 2–5 kg) of clinical-grade material. Passing this decision point takes the project into Phase I and Phase IIa clinical studies, described in more detail in Chapter 17, which are designed to reveal whether the drug has an acceptable pharmacokinetic and side-effect profile in normal volunteers (Phase I), and whether it shows evidence of clinical efficacy in patients (Phase IIa). For drugs acting by novel mechanisms, Phase IIa provides the all-important first ‘proof of concept’, on which the decision whether or not to proceed with the serious business of full development largely rests2.
• The full development decision point (FDP) is reached after the Phase I and Phase IIa (‘proof-of-concept’) studies have been completed, this being the first point at which evidence of clinical efficacy in man is obtained. It is at this point that the project becomes seriously expensive in terms of money and manpower, and has to be evaluated strictly in competition with other projects. Evaluation of the likely commercial returns as well as the chances of successful registration and the time and cost of the ‘pivotal’ Phase III studies, are, therefore, important considerations at this point.
• The submission decision point (SDP) is the final decision to apply for registration, based on a check that the amount and quality of the data submitted are sufficient to ensure a smooth passage through the regulatory process. Hold-ups in registration can carry serious penalties in terms of the cost and time required to perform additional clinical studies, as well as loss of confidence among financial analysts, who will have been primed to expect a new product to bring in revenues according to the plan as originally envisaged.
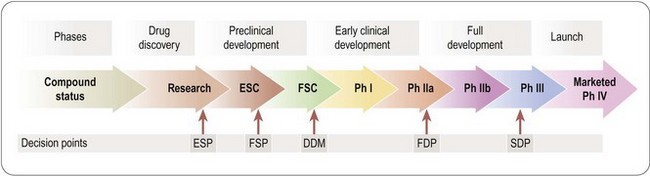
Fig. 14.3 Strategic decision points in drug development. ESP, early selection point; FSP, final selection point; DDM, decision to develop in man; FDP, full development decision point; SDP, submission decision point.
A couple of aphorisms are often applied to drug development, namely:
• In research, surprise = discovery; in development, surprise = disaster
• It is as valuable to stop a project as to carry one forward.
Like most aphorisms they contain a grain of truth, but only a small one. With regard to the first, equating surprise with disaster applies, if at all, only to the technical parts of development, not to the investigational parts, which are, as discussed above, a continuation of research into the properties of the drug. Surprises in this arena can be good or bad for the outcome of the project. Finding, to the company’s surprise, that sildenafil (Viagra) improved the sex life of trial subjects, set the development project off in a completely new, and very successful, direction.
The value attached to stopping projects reflects the frustration – commonly felt in large research organizations – that projects that have little chance of ending in success tend to carry on, swallowing resources, through sheer inertia, sustained mainly by the reluctance of individuals to abandon work to which they may have devoted many years of effort. In practice, of course, the value of stopping a project depends only on the possibility of redeploying the resources to something more useful, i.e. on the ‘opportunity cost’. If the resources used cannot be redeployed, or if no better project can be identified, there is no value in stopping the project. What is certain is that it is only by carrying projects forward that success can be achieved. Despite the aphorism, it is no surprise that managers who regularly lead projects into oblivion achieve much less favourable recognition than those who bring them to fruition!
The need for improvement
As emphasized elsewhere in this book, innovative new drugs are not being registered as fast as the spectacular developments in biomedical science in the last decades of the 20th century had led the world to expect. The rate of new drug approvals in recent years has shown a disheartening decline and there is little sign of the anticipated surge (see Chapter 22), despite a steadily rising R&D spend. This worrying problem was analysed in a 2004 report by the FDA, which lays the blame firmly on the failure of the development process to keep up with advances in biomedicine. The report comments: ‘the applied sciences needed for medical product development have not kept pace with the tremendous advances in the basic sciences’. The report outlines a historical success rate (i.e. chance of reaching the market) for new compounds entering Phase I clinical trials of 14% and comments that this figure did not improve between 1985 and 2000. Furthermore, a recent analysis cited in this report shows that the cost of development, per compound registered, almost doubled in 2000–2002 compared with 1995–2000, whereas the cost of discovery changed very little. In their view, too little effort is being made to develop an improved ‘product development toolkit’ that places more reliance on early laboratory data, and relies less on animal models and clinical testing in assessing safety and efficacy. The FDA and other regulatory bodies hold a large amount of data which could be used to analyse in a much more systematic way than has so far been done by the predictive value of particular laboratory tests in relation to clinical outcome. New screening technologies and computer modelling approaches need to be brought into the same frame. Currently, extrapolation from laboratory and animal data to the clinical situation relies largely on biological intuition – it is assumed, for example, that a compound that does not cause hepatotoxicity in animals is unlikely to do so in man – but there may well be cheaper and quicker tests that would be at least as predictive. There are many other examples, in the FDA’s view, where new technologies offer the possibility of replacing or improving existing procedures, with substantial savings of money and time. The task is beyond the capabilities of any one pharmaceutical company, but needs collaboration and funding at the national or international level. The FDA has continued to issue reports aimed at improving the regulatory process and reducing delays in approval of important new drugs (see CDER 2011; FDA, 2010).
The remaining chapters in this section give a simple overview of the main activities involved in drug development. Griffin and O’Grady (2002) describe the drug development process in more detail.
CDER. Identifying CDER’s science and research needs – report July 2011, 2011. p. 24
FDA Report. Innovation stagnation: challenge and opportunity on the critical path to new medical products. www.fda.gov/oc/initiatives/criticalpath/whitepaper.html, 2004.
FDA Report. Advancing regulatory science for public health. www.fda.gov/scienceresearch/specialtopics/regulatoryscience/ucm228131.htm, 2010.
Gadamasetti K, ed. Process chemistry in the pharmaceutical industry. Chichester: CRC Press/Taylor and Francis; 2007;vol. 2:520.
Griffin JP, O’Grady JO. The textbook of pharmaceutical medicine, 4th ed. London: BMJ Books; 2002.
Repic O. Principles of process research and chemical development in the pharmaceutical industry. New York: John Wiley; 1998.