Edward J. Masoro
Biomedical science paid surprisingly little attention to a remarkable change in human biology during the 20th century—the marked increase in human life expectancy in developed nations. For example, in the United States, life expectancy for men progressively increased from 47.9 years in 1900 to 74.5 years in 2002, and for women, it increased from 50.7 years in 1900 to 79.9 years in 2002. Not until 1974 did the United States establish the National Institute on Aging (NIA) in the National Institutes of Health. The NIA has had a major impact in the United States and throughout the world in the promotion of research on aging and on the development of geriatric medicine. (See Note: Life Expectancy)
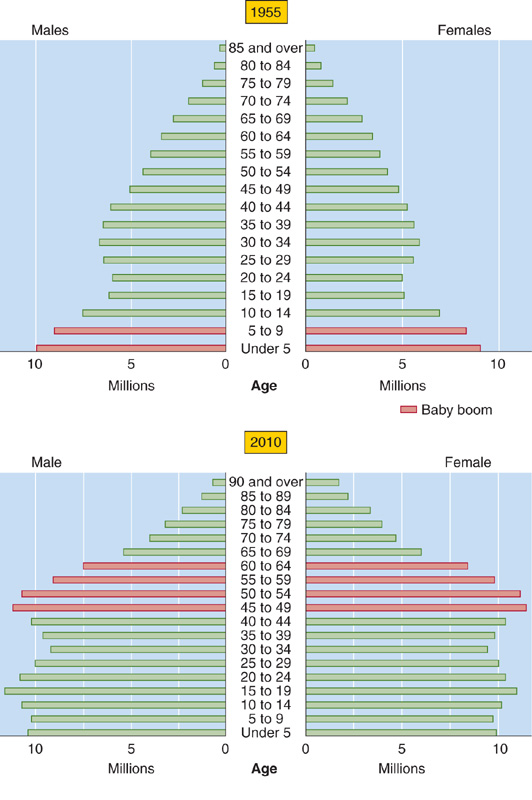
The fraction of the U.S. population 65 years of age or older was only 4% in 1900 but 13% in 1990. This trend in age structure is projected to continue (Fig. 62-1). Moreover, because women have a greater life expectancy, they comprised 70.5% of the population that was more than 80 years old in 1990 in developed nations.
Figure 62-1 The age structure of the 1955 U.S. population and the projected age structure of the 2010 U.S. population. (From Tauber C: Sixty-Five Plus in America. Washington, DC: U. S. Bureau of the Census, 1992, rev. 1993.)
The shift in age structure of the U.S. population during the 20th century depended only modestly on an increase in life expectancy from birth. More important was the progressive decrease in birth rates, which led the elderly to become an ever-increasing fraction of the population, particularly in developed nations. Indeed, the effect of the post-World War II “Baby Boom Generation” on population age structure is clearly apparent in Figure 62-1. However, because birth rates are unlikely to fall much further, future changes in the age structure of the U.S. population will depend mainly on further projected increases in life expectancy. (See Note: Caspaces)
The age of an organism usually refers to the length of time the individual has existed. Biogerontologists and members of the general public alike usually use aging to mean the process of senescence. For example, we may say that a person is young for her age, an expression meaning that the processes of senescence appear to be occurring slowly in that person. Aging—the synonym for senescence that we use throughout this chapter—is the progressive deteriorative changes, during the adult period of life, which underlie an increasing vulnerability to challenges and thereby decrease the ability of the organism to survive.
Biogerontologists distinguish biological age from chronologic age. Although we easily recognize the biological aging of family members, friends, and pets, it would be helpful to have a quantitative measure of the rate of aging of an individual. Biomarkers of aging—morphologic and functional changes that occur with time in the adult organism—could in principle serve as a measure of senescent deterioration. Alas, a generally agreed on panel of biomarkers of aging has yet to emerge, so it is currently impossible to quantitate the aging of individuals.
In contrast to the aging of individuals, it has long been possible to measure the rate of aging of populations. In 1825, Benjamin Gompertz, a British actuary, published a report on human age-specific death rate—the fraction of the population entering an age interval (e.g., 60 to 61 years of age) that dies during the age interval. Gompertz found that, after early adulthood, the age-specific death rate increases exponentially with increasing adult age. The same is true for other human populations (Fig. 62-2) and for many animal populations. Based on the assumption that the death rate reflects the vulnerability caused by senescence, it has generally been accepted that the slope in Figure 62-2 reflects the rate of population aging. Although gompertzian and related analyses had long been viewed as the “gold standard” for measuring population aging, some biogerontologists have challenged this approach.
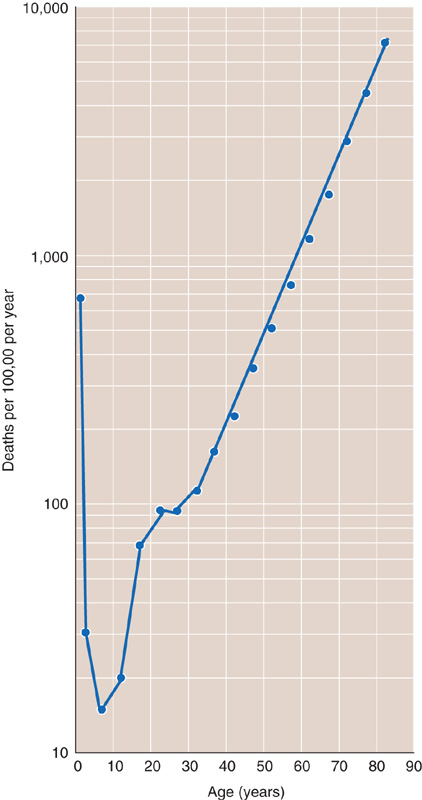
Figure 62-2 Age-specific mortality for the U.S. population (men and women) for 2002. Data are projections from the 2000 U.S. census.
Most evolutionary biologists no longer accept the once popular belief that aging is an evolutionary adaptation with a genetic program similar to that for development. The current view is that aging evolved by default and reflected the absence of forces of natural selection that would otherwise eliminate mutations that promote senescence. For example, consider a cohort of a species that reaches reproductive maturity at age X. At that age, all members of the cohort will be involved in generating progeny. Furthermore, assume that this species is evolving in a hostile environment—the case for most species. As the age of this cohort increases past X, fewer and fewer members survive, so that all members of the cohort die before exhibiting senescence. In this cohort, genes with detrimental actions—expressed only at advanced ages—would not be subjected to natural selection. If we now move the progeny of our cohort to a highly protective environment, many may well live to ages at which the deleterious genes can express their effects, thereby giving rise to the aging phenotype. This general concept led to three genetic mechanisms, discussed in the following paragraphs. These mechanisms are not mutually exclusive, and each has experimental support. (See Note: Peter Medawar)
In 1952, Peter Medawar proposed a variant of the foregoing model, now referred to as the mutation-accumulation mechanism. He proposed that most deleterious mutations in gametes will result in progeny that are defective during most of life, and natural selection removes such genes from the population. However, a very few mutated genes will not have deleterious effects until advanced ages, and natural selection would fail to eliminate such genes.
George Williams proposed another variant in 1957. He postulated that the genes with deleterious actions in late life actually increase evolutionary fitness in early adulthood. Natural selection will strongly favor such alleles because they promote the ability of the young adult to generate progeny and because they have a negative impact only after reproduction—antagonistic pleiotropy. In this situation, aging is a byproduct of natural selection.
In 1977, Tom Kirkwood proposed the Disposable Soma Theory, according to which the fundamental life role of organisms is to generate progeny. Natural selection would apportion the use of available energy between reproduction and body (i.e., somatic) maintenance, to maximize the individual’s lifetime yield of progeny. As a consequence, less energy is available for somatic maintenance than is needed for indefinite survival. This theory further proposes that a hostile environment increases the fraction of energy expended in reproduction and leaves a smaller fraction for somatic maintenance.
Measuring the effects of aging on the human physiology presents investigators with a difficulty—the subjects’ life span is greater than the investigator’s scientific life span.
Cross-Sectional Design The usual approach to the foregoing difficulty is a cross-sectional design in which investigators study cohorts with several different age ranges (e.g., 20 to 29 year olds, 30 to 39 year olds) over a brief period (e.g., a calendar year). However, this design suffers from two serious potential confounders. One is the cohort effect; that is, different cohorts have had different environmental experiences. For example, in studies of the effects of aging on cognition, a confounding factor could be that younger cohorts have had the benefit of a relatively higher level of education. If aware of a potential confounder, the investigator may be able to modify the study’s design to avoid the confounder.
The second potential confounder is selective mortality—individuals with risk factors for diseases that cause death at a relatively young age are underrepresented in older age groups. For example, in a study on the effect of age on plasma lipoproteins, mortality at a young age from cardiovascular disease would preferentially eliminate individuals with the highest low-density lipoprotein levels.
Longitudinal Design To circumvent the confounders encountered in cross-sectional designs, investigators can repeatedly study a subject over a significant portion of his or her lifetime. However, this longitudinal design has other problems. Long-term longitudinal studies require a special organizational structure that can outlive an individual investigator and complete the study. Even shorter longitudinal studies are very costly. Some problems are inherent in the course of longitudinal studies, including the effect of repeated measurements on the function assessed, changes in subjects’ lifestyle (e.g., diet), dropout of subjects from the study, and changes in professional personnel and technology.
Age-associated diseases are those that do not cause morbidity or mortality until advanced ages. Examples are coronary artery disease, stroke, many cancers, type 2 diabetes, osteoarthritis, osteoporosis, cataracts, Alzheimer disease, and Parkinson disease. These are either chronic diseases or acute diseases that result from long-term processes (e.g., atherogenesis).
Most gerontologists have held the view that age-associated diseases are not an integral part of aging. These gerontologists developed the concept of primary and secondary aging to explain why age-associated diseases occur in almost all elderly people. Primary aging refers to intrinsic changes occurring with age, unrelated to disease or environmental influences. Secondary aging refers to changes caused by the interaction of primary aging with environmental influences or disease processes.
In contrast, some gerontologists adhere to the following view, expressed by Robin Holliday: “The distinction between age-related changes that are not pathological and those that are pathological is not at all fundamental.” Moreover, the genetic mechanisms proposed for the evolution of aging may apply equally to the processes underlying both primary and secondary aging.
In this major section, I consider three major classes of cellular and molecular processes that may be proximate causes of organismic aging: (1) damage caused by oxidative stress and other factors, (2) inadequate repair of damage, and (3) dysregulation of cell number. No single one of these processes is the underlying mechanism of aging. The basic mechanism of aging is likely to be the long-term imbalance between damage and repair. During growth and development, the genetic program not only creates a complex structure, but also repairs damaged molecules that arise in the process. Following development is a brief adult period when damage and repair are in balance, and then begins long-term imbalance in favor of damage.
The factors underlying the imbalance vary among species and among individuals within species, as a result of both genetic and environmental variability. For example, oxidative stress is one of many damaging processes that underlie aging, and the genome of the animal as well as the environment will determine the extent to which it is an important causal factor.
One gram of tissue from a small mammal has a higher resting metabolic rate (RMR; see Chapter 58) than the same mass of tissue from a larger mammal (e.g., a human). Because smaller mammals have a shorter life span than humans, Max Rubner reported in 1908 that a gram of tissue from diverse domestic animals and humans has similar lifetime energy expenditure. Based on these findings, Raymond Pearl in 1928 proposed that organisms have a finite amount of a “vital principle” that they deplete at a rate proportional to the rate of energy expenditure. However, later experimental evidence did not support this Rate of Living Theory of Aging.
Reactive O2 Species As illustrated in Figure 62-3A, reactive oxygen species (ROS) include molecules such as hydrogen peroxide (H2O2), neutral free radicals such as the hydroxyl radical (•OH), and anionic radicals such as the superoxide anion radical (O2.−). Free radicals have an unpaired electron in the outer orbital, shown in red in Figure 62-3A. These free radicals are extremely unstable because they react with a target molecule to capture an electron and thus become a stable molecule with only paired electrons in the outer shell. However, the target molecule left behind becomes a free radical, initiating a chain reaction that continues until two free radicals meet to create a product with a covalent bond. ROS—particularly •OH, which is the most reactive of them all—have the potential to damage important biological molecules, such as proteins, lipids and DNA. However, ROS also play important physiological roles in the oxidation of iodide anions by thyroid peroxidase in the formation of thyroid hormone (see Chapter 49), as well as in the destruction of certain bacteria by NADPH oxidase and myeloperoxidase in phagocytic cells. Finally, the highly reactive signaling molecule nitric oxide (see Chapter 3) is a free radical (Fig. 62-3A). (See Note: Physiological Roles of Reactive Oxygen Species (ROS); Nitric Oxide (NO))
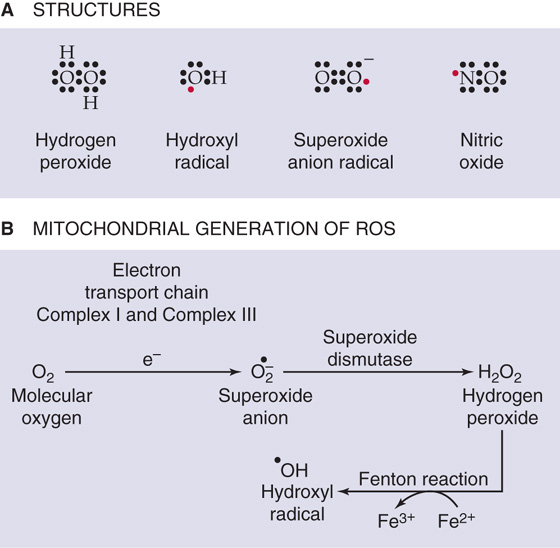
Figure 62-3 ROS. A, Structures. B, Mitochondrial generation.
ROS can also form as the result of ionizing radiation. Quantitatively, the most important source of ROS is the mitochondrial electron transport chain (see Chapter 5).
Complex I and complex III of the electron transport chain generate O2.− as byproducts (Fig. 62-3B). The enzyme superoxide dismutase (SOD) converts O2.− to H2O2, which, in turn, can yield the highly reactive •OH.
Only a small fraction of the oxygen (<1%) used in aerobic metabolism generates ROS. However, even that amount would be lethal in the absence of protective mechanisms. Fortunately, organisms have two potent antioxidant defenses. The major defense is enzymatic, specifically SODs, catalase, and glutathione peroxidase (Fig. 62-4). In addition, low-molecular-weight antioxidants, such as vitamins C and E, play a minor role in the defense against the metabolically produced radicals.
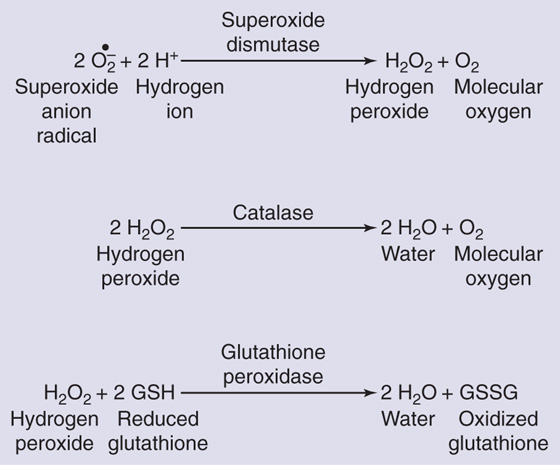
Figure 62-4 Enzymatic defenses against ROS. SODs eliminate the O2.− but generate H2O2, which, as shown in Figure 62-3B, can yield the highly reactive •OH through the Fenton reaction. The H2O2 is eliminated by catalase or glutathione peroxidase, which yield relatively nonreactive products: water, molecular O2, and oxidized glutathione.
Because these defense mechanisms are not fully protective, some investigators have suggested that ROS may cause the molecular damage observed in aging. According to the oxidative stress theory, an imbalance between the production and removal of ROS is the major cause of aging. (See Note: Free Radical Theory of Aging)
Glycation and Glycoxidation Glycation refers to nonenzymatic reactions between the carbonyl groups of reducing sugars (e.g., glucose) and the amino groups of macromolecules (e.g., proteins, DNA) to form advanced glycation end products (AGEs). Figure 62-5 shows an interaction of open-chain D-glucose with a lysine residue on a protein, yielding a Schiff base, and water. The Schiff base undergoes an intramolecular rearrangement to form an open-chain Amadori compound that undergoes the Amadori rearrangement to form a ring structure called an Amadori product. In cooking, Amadori products undergo a series of further reactions to produce polymers and copolymers called melanoidins, which give a brown color to cooked food. In humans, the Amadori product can undergo a series of intramolecular and intermolecular rearrangements that include oxidation (glycoxidation) to form AGE molecules. For example, the Amadori product in Figure 62-5 can either form carboxymethyllysine or react with an arginine residue on the same or a different protein to form a cross-link called pentosidine. (See Note: Maillard Reaction)
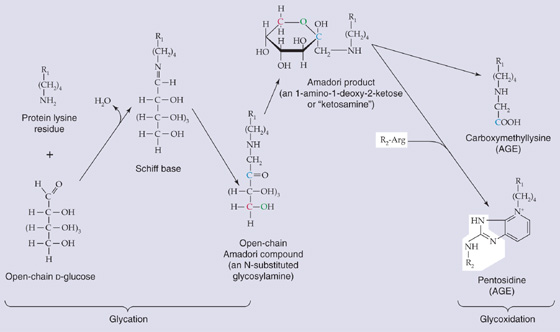
Figure 62-5 Examples of glycation, glycoxidation, and the formation of AGEs. R1 and R2, two different proteins or two different domains of the same protein.
The formation of AGEs is especially important for long-lived proteins, and it appears to play a role in the long-term complications of diabetes. The similarity between the aging phenotype and that of the diabetic patient led Anthony Cerami to propose the Glycation Hypothesis of Aging. Although glucose is not the only reducing sugar involved in glycation, it is an important one. Thus, the level of glycemia is a major factor in glycation, and periods of hyperglycemia are probably the reason glycation—including the glycation of hemoglobin—is enhanced in patients with diabetes. Proteins containing AGEs exhibit altered structural and functional properties. For example, AGE formation in lens proteins of the eye probably contribute to age-associated opacification. Moreover, with advancing age, the increased stiffness of collagen in connective tissues (e.g., blood vessels; see Chapter 19) may also, in part, contribute to AGE-mediated collagen cross-links. AGE-induced DNA damage may lead to alterations in genomic function.
Mitochondrial Damage Because mitochondria are the major source of ROS, they are also likely to be a major target of oxidative damage. Damage to mitochondrial DNA (mtDNA) increases greatly with age because, unlike genomic DNA, mtDNA is not protected by histones (see Chapter 4). According to the Mitochondrial Theory of Aging, the damage to mtDNA reduces ability of the mitochondria to generate ATP, and this decreased production of ATP results in the loss of cell function and hence aging.
Somatic Mutations Damage to genomic DNA and mtDNA can occur as the result of radiation and other environmental agents, such as toxic chemicals. In recent years, oxidative stress has been recognized as a major source of DNA damage. Cells can repair much of the damage to DNA, and the level of damage is in a steady state between damaging and repair processes. According to the DNA Damage Theory of Aging, accumulated DNA damage interferes with DNA replication and transcription, thereby impairing the ability of cells to function and causing aging. Moreover, this loss of function increases as the steady-state level of DNA damage increases. Although oxidative stress results in DNA damage, it is not clear that DNA damage and mutations in somatic cells are sufficient to cause the organismic functional deterioration that characterizes the aging phenotype.
Many biogerontologists believe that agents that cause damage throughout life are not really the cause of aging. Rather, they believe that aging occurs because of the progressive age-associated loss in the ability to repair such damage. (See Note: Age-Associated Inadequacy of Repair Processes)
DNA Repair As noted earlier, the steady-state level of damaged DNA depends on the balance between damaging and repair processes. The DNA Repair Theory of Aging proposes that DNA repair declines with advancing age and eventually falls, the steady-state level of DNA damage consequently rises, and the integrity of the genome is thereby compromised. Investigators have tested the effects of aging on only some of the cell’s multiple repair pathways in some species. For example, evidence suggests that the repair of nucleotide excision decreases with advancing age in laboratory rodents.
Protein Turnover In addition to oxidative stress and non-enzymatic glycation, many other processes occur, including deamidation, racemization, and isomerization. These many different alterations result in changes in the secondary and tertiary structures as well as aggregation and fragmentation of protein molecules, all of which can interfere with protein function. Protecting the organism from an excessive accumulation of altered proteins are proteolytic degradation and subsequent biosynthetic replacement—protein turnover.
The rate of total body protein turnover in humans decreases with age. Thus, the average lifetime of most, but not all protein, species increases with age. Especially susceptible to damage are the long-lived proteins in the extracellular matrix, particularly collagen and elastin, which with increasing age undergo changes such as oxidation, glycation, and cross-linking. For the cells embedded in the matrix, these changes probably alter properties such as proliferation, migration, and the response to cytokines. It is possible that an increased level of altered proteins results in loss of functional proteins and may contribute to aging.
Membrane Deterioration Because of the high level of polyunsaturated fatty acids (i.e., those with many double bonds) in the phospholipid bilayer of many membranes, oxidative processes generate lipid peroxides that accumulate with age in the membranes. Moreover, lipid peroxides can undergo cleavage to yield reactive lipid aldehydes that further damage the membrane, including its proteins. Although the cell rapidly replaces membrane lipids with new molecules, these replacements alter the membranes. Thus, the number of double bonds in the fatty acids gradually falls, and the ratio of cholesterol to phospholipid in the membrane lipids gradually rises. Therefore, with increasing age, membrane fluidity (see Chapter 2) gradually decreases. It is possible that these age-associated changes may interfere with important membrane functions, such as the barrier function, transport, and signaling processes.
For most cell types, the number of cells remains nearly constant over much of adult life. An imbalance in favor of cell division results in hyperplasia (see Chapter 48), such as occurs in the prostate of elderly men, or in neoplasia (i.e., formation of new, abnormal cells), a disease process that increases in frequency with age. An imbalance in favor of cell removal results in a reduction of cell number, such as occurs with age in some skeletal muscles. Of course, in cell types that are truly postmitotic in adult life, any age-associated loss of cells results in a decrease in number.
Limitations in Cell Division In 1961, Leonard Hayflick and Paul Moorhead reported that human fibroblasts in culture could divide only a limited number of times, a phenomenon known as the Hayflick limit. This concept also applies to many other somatic cell types in culture. Although Hayflick hypothesized that this limited cell proliferation in culture was a “test tube” model of aging, more recent findings indicated that the in vitro cell culture system falls short as a valid model of organismic aging. Nevertheless, intensive study of the Hayflick limit led to consideration of the role of telomeres in aging.
Telomeres are elements at the ends of linear chromosomes and are composed of repeated specific DNA sequences and associated proteins. In the late 1980s, Calvin Harley found that the telomeres of human cells in culture shortened with each mitotic division. When the telomeres shorten to a critical length, the cell can no longer divide—a probable basis of the Hayflick limit. Such cells also exhibit other functional changes.
Are the telomere findings in culture systems relevant to organismic senescence? A reduction in telomere length could play a role for cell types that exhibit an age-associated decrease in cell number. Clearly, a reduction in telomere length cannot be a factor in the aging of cells that are truly postmitotic during adult life. Telomeres do not shorten in the germline, which, unlike somatic cells, contains significant levels of telomerase, an enzyme that catalyzes the extension of telomere length. Cancer cells are also rich in telomerase.
Although not associated with a major dysregulation of cell number, aging may impair the burst in proliferation that is needed to meet certain challenges. For example, with increasing age, the immune system is less effective in protecting the organism from infection. An important factor may be that an antigenic challenge may trigger a proliferation of T lymphocytes, perhaps because of an age-associated decrease in the length of T-lymphocyte telomeres.
Cell Removal Necrosis and apoptosis are two major processes by which the body loses cells. Necrosis, a cellular response to severe trauma, is manifested by uncontrolled breakdown of cellular structure, cell lysis, and an inflammatory response. The morphologic characteristics of necrosis are cell swelling and loss of membrane integrity.
Apoptosis is programmed cell death (see Chapter 2). It plays a key role in organogenesis and tissue renewal, and it also occurs in response to relatively mild damage. Apoptosis requires ATP, is gene driven, and is characterized by preservation of organelles, maintenance of membrane integrity, absence of inflammation, cell shrinkage, and fragmentation of the cell into multiple membrane-enclosed apoptotic bodies. Macrophages or neighboring cells remove the apoptotic bodies by phagocytosis. (See Note: Apoptosis)
Three interacting pathways (see Fig. 25-1C) lead to apoptosis (Fig. 62-6). First, in the extrinsic pathway, extracellular signals bind to cell surface receptors—death receptors (DRs)—of the tumor necrosis factor receptor (TNFR) family, which are examples of receptors that act at least in part through regulated proteolysis (see Table 3-1). The DRs include Fas, TNFR1, DR3, DR4, and DR5, all of which have a cytosolic death domain. In the case of the receptor Fas, the homotrimeric ligand called FasL binds to three Fas molecules and results in the formation of a trimer of ligand-receptor complexes. This clustering allows the death domains on Fas to bind to death domains of an intracellular adapter protein called FADD (Fas-associated death domain). Death-effector domains of FADD recruit several copies of procaspase 8, the aggregation of which leads to the autoproteolytic cleavage of procaspase 8, thereby releasing active caspase 8. This initiator caspase is a member of the caspase family of proteolytic enzymes. Relatively small numbers of caspase 8 molecules, by proteolytic cleavage, activate much larger numbers of effector caspases, including caspases 3 and 7, as well as another initiator caspase called caspase 9. This amplified cascade results in the proteolysis of numerous cytosolic and nuclear proteins and leads to apoptosis. One of the cytosolic caspase targets is a gelsolin, which, when cleaved, severs actin filaments and leads to a loss of normal cell shape. One of the nuclear caspase targets is inhibitor of caspase-activated DNase (ICAD), which normally binds to and thereby inactivates caspase-activated DNase (CAD). Cleavage of ICAD releases active CAD, which then severs chromosomal DNA. (See Note: Caspaces)
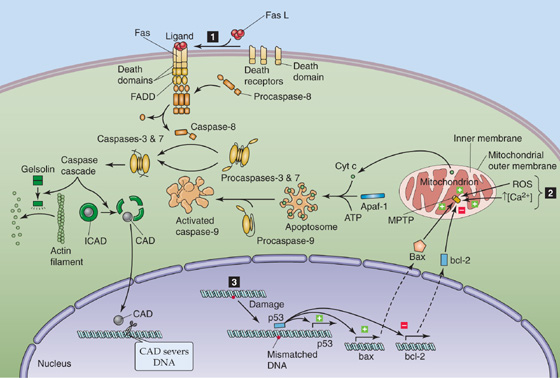
Figure 62-6 Apoptotic signaling pathways.
The second pathway involves damage to the mitochondria (Fig. 62-6), triggered by such agents as ROS, increases in [Ca2+] in the mitochondrial matrix, and caspases. The result is the opening of a large pore in the mitochondrial inner membrane—the mitochondrial permeability transition pore (MPTP)—followed by mitochondrial swelling, rupture of the outer mitochondrial membrane, and release of cytochrome c into the cytosol. There, the apoptotic protease-activating factor (Apaf-1) complexes with the cytochrome c and ATP, to form a wheel-like structure that contains seven of each molecule—an apoptosome. This structure recruits seven procaspase 9 molecules and results in the formation of active caspase 9, thus committing the cell to apoptosis.
The third pathway is triggered by damage to nuclear DNA (Fig. 62-6). The tumor suppressor p53, a nuclear protein, recognizes certain base-pair mismatches. In cases of modest DNA damage, p53 increases the transcription of p21, which, in turn, halts the cell cycle. In cases of more severe DNA damage, p53 upregulates its own transcription. In addition, p53 increases the transcription of the proapoptotic protein bax (Bcl–2-associated X protein) and decreases the transcription of the antiapoptotic protein bcl-2 (B-cell chronic lymphocytic lymphoma/lymphoma 2). The increase in the ratio of bax to bcl-2 appears to activate MPTP, thereby precipitating the events leading to apoptosis described in the second pathway. Finally, p53 also upregulates Fas and thus reinforces the first pathway.
Dysregulation of apoptosis promotes aging. Failure of apoptosis to remove damaged cells could result in abnormal function or increase the risk of cancer. Excess apoptosis would unnecessarily decrease cell number.
The age-related changes just described at cellular and molecular levels can also manifest as deterioration of entire physiological systems. Although I discuss typical age-related changes in physiological systems, the extent of change among individuals may range from barely perceptible to very marked. Indeed, a subset of individuals shows minimal physiological deterioration—these people have undergone “successful” aging. Many individuals show marked deterioration with age in all physiological systems, whereas other individuals exhibit little or no deterioration in one or more systems. Although the nature of the aging process is similar in the two sexes—except, of course, for the reproductive system—important quantitative differences exist. For example, women lose bone mass much faster with increasing age than do men. Because of the great reserve capacity or redundancy of some physiological systems, the effect of aging on a physiological process is often not apparent until either the individual faces an unusual challenge or function has fallen to less than some critical level.
Women reach peak height by age 16 to 17 years and men by 18 to 19 years. After these peaks, height starts to decline, primarily because of compression of the cartilaginous disks between the vertebrae and loss of vertebral bone. This decline begins at ~20 years of age in women and at 25 years of age in men. By the age of 70 years, height has fallen 2.5% to 5% lower than peak level.
In most Americans, body mass increases until middle age in both sexes and begins to decrease after age 70 years. Fat-free mass is defined as body mass minus adipose tissue fat mass, and lean body mass is defined as fat-free mass minus both bone mass and non–adipose tissue fat mass. Both fat-free mass and lean body mass progressively decrease over most of adult life in both sexes. Although a sedentary lifestyle may contribute to this loss, lifelong athletes also show a progressive age-associated loss in fat-free mass and lean body mass.
Adipose tissue fat mass increases with adult age, but the extent differs markedly among individuals. Although a sedentary lifestyle may be a factor, even physically fit individuals who do not exhibit an age-associated increase in body mass show a small but progressive increase in adipose tissue fat mass (in parallel with the aforementioned decrease in fat-free body mass). In addition, the distribution of body fat changes with increasing age, with an accumulation of fat around abdominal viscera and in abdominal subcutaneous tissue. At the same time, a decrease occurs in the extremities and the face; facial fat loss can give rise to the gaunt look that characterizes many elderly persons.
Skin As is clearly evident from cosmetic advertisements, most of us use the skin as an indicator of aging. Intrinsic aging is manifest in skin areas protected from the sun, such as the buttocks. The additional damage caused by long-term exposure to the sun’s ultraviolet radiation is called photoaging.
In intrinsic aging, the thickness of the epidermis (see Chapter 15) decreases slightly, with no change in the outermost epidermal layer, the stratum corneum. The rate of generation of keratinocytes, which end their lives as the stratum corneum, slows with age, thereby increasing the dwell time of stratum corneum components. The decreasing number of melanocytes reduces photoprotection, and the decreasing number of Langerhans’ cells reduces immune surveillance.
Intrinsic aging of the dermis affects mainly the extracellular matrix. The amount of elastin and collagen decreases, and their structure changes. Glycosaminoglycan composition also changes. As a result, the dermis thins by ~20% and becomes stiffer, less malleable, and thus more vulnerable to injury. (See Note: Effect of Aging on Glycosaminoglacan Composition)
Photoaging increases the extent of most intrinsic age changes in both the epidermis and dermis, and it has additional effects. For example, photoaging causes coarse wrinkles, which occur minimally or not at all because of intrinsic aging.
Aging also reduces the number and function of sweat glands as well as the production of sebum by sebaceous glands. The number of active melanocytes in hair follicles decreases, resulting in graying of hair. Nail growth also slows with increasing age.
Skeletal Muscle A steady loss in skeletal muscle mass—sarcopenia—occurs with aging, particularly beyond 50 years, and it primarily reflects a loss of number and, to a lesser extent, size of muscle fibers. The sarcopenia partly results from inactivity, but it is also caused by a progressive loss of the motor neurons innervating type II motor units, which are recruited less frequently. With loss of their motor nerve, affected muscle fibers either atrophy and die or become innervated by a sprout that emerges from a healthy axon nearby. This process of reinnervation ultimately results in larger motor units and thus a decrement in fine motor control (see Chapter 9). The reduction in muscle strength and power is often a major cause of disability in elderly persons. However, strength training in elderly persons can increase the size of the fibers and can thereby increase muscle mass.
Bone Remodeling of bone occurs throughout adult life; it involves the coordinated activity of osteoclasts, which resorb bone, and osteoblasts, which form bone. Until middle age, bone resorption and formation are in balance. However, starting in middle age, resorption exceeds formation, thus leading to a progressive loss in bone mass. In women, bone loss accelerates during the first few years following menopause. Bone loss can progress to osteoporosis, defined by the World Health Organization as a bone mineral density 2.5 standard deviations or more lower than mean values for young adults (see Chapter 52). Osteoporosis, a major problem in geriatric medicine, carries a heightened risk of bone fractures.
Synovial Joints Synovial joints permit free movement of the bones linked to them. With increasing adult age, joint flexibility declines, mainly because of the aging of articular cartilage. This cartilage thins and exhibits altered mechanical features, including decreases in tensile stiffness, fatigue resistance, and strength. These changes are partly the result of decreased water content. Aging impairs the function of chondrocytes, increases the cross-linking of collagen, and causes a loss of proteoglycans. The age-related changes in joint cartilage undoubtedly play a major role in the development of osteoarthritis.
It is a common misconception that advancing age causes marked deterioration in the nervous system. However, in the absence of neurodegenerative disorders such as Alzheimer disease and Parkinson disease, impairment of the nervous system with age is much less severe.
Sensory Functions Most sensory systems exhibit some deterioration with age. Sensitivity to touch decreases, as do the abilities to sense vibration and to distinguish two spatially distinct points of contact. Proprioception, including the vestibular system of the inner ear, also deteriorates somewhat. As discussed in Chapter 59, the loss of thermoregulatory ability, a serious problem for many elderly people, occurs in part because of an impaired ability to sense heat and cold.
Hearing loss, particularly of high-frequency sound, is almost an invariable consequence of advancing age. This impairment is usually caused by loss of hair cells of the organ of Corti (see Chapter 15), but it can also stem from loss of nerve cells of the auditory nerve or from reduced blood supply to the cochlea. A deficit in central processing can make it difficult for some elderly people to distinguish spoken words from background noise.
Vision also deteriorates with increasing age. A progressive loss in the power of accommodation (presbyopia) occurs during adult life. Almost all elderly persons have a reduced number of retinal cones, lessened ability to alter pupil size in response to light intensity, and decreased ability of retinal rods to adapt to low-intensity light (see Chapter 15). In addition, age-associated diseases—cataracts, glaucoma, and macular degeneration—can markedly decrease vision in many elderly persons.
The ability to detect and discriminate among sweet, sour, salty, and bitter taste qualities deteriorates somewhat at advanced ages, along with a marked reduction in olfaction (see Chapter 15). Because taste involves both gustation and olfaction, many elderly persons live in a world of “pastel” food flavors.
Motor Functions A major effect of aging is the slowing of reaction time: the time elapsed between the stimulus and the motor response. This delay is observable in simple responses and becomes more pronounced as the complexity of the response increases (e.g., the need to make a choice among responses). Thus, a hallmark of nervous system aging is the slowing of central processing. One result is that elderly people tend to execute movements more slowly than do young people.
The ability to maintain posture and balance deteriorates with increasing age. Slowing of central processing is a factor, but decreased muscle strength and deterioration of vision and proprioception also play important roles. Not surprisingly, elderly persons have a high incidence of falls. Even when capable of walking at normal speeds, healthy elderly people tend to walk more slowly than the young and take shorter and more frequent steps. This walking pattern is less taxing for a person with knee and ankle joints that are less flexible, aids in maintaining balance, and enables a deteriorating sensory system to monitor hazards more effectively.
Cognitive Functions Although most people generally believe that cognitive functions (e.g., intelligence, memory, learning) decline with advancing age, the cognitive decline is not marked in the absence of dementia. The decline that does occur in healthy elderly persons may reflect the slowing of central processing. The capacity to use knowledge is not decreased in the healthy aged, but the ability to solve novel problems does decline. Certain types of memory deteriorate with advancing age, such as remembering where the car keys were left, but other types are not lost, such as retrieving conceptual information. Older people are capable of learning, but they do so less quickly than younger people.
Atherosclerosis can cause marked deterioration of cardiovascular function in elderly persons, and chronic obstructive pulmonary disease (see Chapter 27) can do the same for pulmonary function. However, in the absence of such diseases, age-associated changes in these physiological systems are modest.
Cardiovascular Function As discussed in Chapter 19, aging decreases the distensibility of arteries. The decreased compliance elevates systolic pressure, slightly decreases diastolic pressure, and thus widens pulse pressure. Afterload (see Chapter 22), the resistance to ejection of blood from the left ventricle, increases with advancing age, primarily because of reduced arterial compliance. The increased afterload causes thickening of the left ventricular wall, which involves an increase in size but not number of myocytes.
Preload, the end-diastolic volume of blood in the left ventricle (see Chapter 22), does not change with age in subjects at rest. Although early diastolic filling falls, a compensatory increase in left atrial contraction enhances late-diastolic filling. Many elderly persons suffer from postural hypotension (see Chapter 25) because of age-associated blunting of the arterial baroreceptor reflex.
Pulmonary Function The strength and endurance of the respiratory muscles decrease with age, primarily because of atrophy of type IIa muscle fibers. Lung volumes—both static volume and forced expiratory volume (see Chapter 27) gradually decrease with age. With age, small airways have an increased tendency to collapse (atelectasis) because of degeneration of the collagen and elastin support structure. This situation results in impaired ventilation of dependent lung regions, ventilation-perfusion mismatch (see Chapter 31), and reduced resting arterial PO2.
Exercise Maximal O2 uptake ( O2max) declines progressively with aging in physically trained individuals and even more so in untrained individuals of the same chronologic age. Decreasing muscle mass and reduced cardiovascular and pulmonary function probably contribute to the decline in O2max, and the relative importance of each factor varies among individuals.
O2max) declines progressively with aging in physically trained individuals and even more so in untrained individuals of the same chronologic age. Decreasing muscle mass and reduced cardiovascular and pulmonary function probably contribute to the decline in O2max, and the relative importance of each factor varies among individuals.
The cardiovascular system of elderly persons responds to exercise differently than in the young. For a given increase in cardiac output, heart rate rises less and stroke volume rises more in elderly people. Because the heart is less responsive to adrenergic stimulation, the increase in stroke volume is primarily the result of the Frank-Starling mechanism. Thus, during exercise, the left ventricular end-diastolic and end-systolic volumes increase, and maximal left ventricular ejection fraction falls.
Elderly persons exhibit a decrease in the pulmonary diffusing capacity and alveolar capillary volume, and ventilation-perfusion mismatch increases. These alterations in pulmonary function have been implicated in the decrease in O2max.
The ability of the body to respond to physical conditioning decreases with aging. Nevertheless, skeletal muscle and the cardiovascular system remain responsive to exercise into the 10th decade of life.
Cross-sectional studies show that glomerular filtration rate (GFR) starts to decline at 30 years of age and thereafter falls linearly with age. However, longitudinal analysis revealed that one third of the participants of the Baltimore Longitudinal Study of Aging exhibited the GFR decline predicted from cross-sectional analysis, one third had a steeper decline, and one third had no decline at all. Thus, an age-associated decline in GFR is not inevitable.
Cross-sectional studies indicate that renal tubule transport functions decrease with age. The kidneys do not respond as effectively to changes in sodium load, do not dilute or concentrate urine as effectively, and also have a somewhat impaired ability to excrete potassium, phosphate, and acid.
The capacity and compliance of the urinary bladder decrease with advancing age, and the number of uninhibited contractions increases, thus making it more difficult to postpone voiding. The rate of bladder emptying decreases, and the residual bladder volume after voiding increases. (See Note: Effect of Aging on Bladder Function)
Although gastrointestinal problems are the second most common reason for hospital admission of elderly patients, the gastrointestinal system functions in healthy elderly persons about as well as in the young. Although a loss of ability to secrete gastric acid was previously thought to be part of aging, it is now clear that this loss is limited to persons infected with Helicobacter pylori (see the box in Chapter 42 for more on that topic). The loss of skeletal muscle at both ends of the gastrointestinal tract can lead to minor age-related decreases in function (i.e., chewing, swallowing, fecal continence). Minor decreases occur in secretion by exocrine glands. Liver mass and hepatic blood flow, as well as the clearance of certain drugs, decrease significantly. Moreover, elderly people experience a delay in hepatic regeneration following damage. (See Note: Effect of Aging on the Gastrointestinal Tract)
Total energy expenditure decreases with age, primarily because of decreases in physical activity. An age-associated decrease in the RMR (see Chapter 58) reflects a decrease in fat-free mass; that is, the RMR per kilogram of fat-free mass does not decrease.
Endocrine Pancreas The impaired glucose tolerance (see Chapter 51) that usually occurs with aging is caused by increased insulin resistance, which, in turn, results mainly from increased adiposity. However, aging per se does play a small role. An increase in serum low-density lipoproteins (see Chapter 46) occurs in both genders with advancing age.
Pituitary Aging diminishes peak concentrations of hormone generated by the pulsatile action of somatotrophs. Greatly reduced plasma insulin-like growth factor 1 (IGF-1) concentrations result (see Chapter 48).
Adrenal Cortex The basal, circadian, and stimulated secretion of cortisol exhibits little age-related change. Aldosterone secretion is also well preserved. In contrast, the plasma concentration of the adrenal cortical hormone dehydroepiandrosterone (see Chapter 54) decreases markedly with increasing age.
Thyroid Gland Thyroid function appears to be unaffected by age into the ninth decade of life. However, in centenarians, plasma thyroid-stimulating hormone (TSH) levels may decline because of decreased secretion, and free triiodothyronine levels may fall because of impaired 5′/3′-deiodinase (see Chapter 49). Plasma parathyroid hormone levels increase with advancing age.
Gonads Reproductive ability in women abruptly ceases at ~50 years of age with the occurrence of the menopause (see Chapter 55). Men do not undergo an abrupt change in reproductive function during middle age. However, a progressive decrease in male reproductive and related functions does occur, often referred to as the andropause (see the box on testosterone and aging in Chapter 54).
Slowing the aging process and thereby extending life have been human goals throughout recorded history and probably in preliterate times as well. The marked increase in human life expectancy in the 20th century could be viewed as achieving this goal. However, much of that increase results from prevention of premature deaths related to infections and other environmental hazards. It remains to be established how much, if any, of the increase relates to slowing the aging process. Indeed, over the centuries, the life span of the oldest of the old in human populations has changed little, although the fraction of the population reaching these advanced ages has increased significantly. In contrast, both environmental and genetic manipulations can markedly extend the maximal life span of a population in several other species.
Restricting the food intake of rats, starting soon after weaning, increases both mean and maximum life span of several strains of rats and of both genders, as first reported by Clive McCay in 1935. The marked increase in rat longevity is graphically illustrated by the survival curves in Figure 62-7. Manipulation of the components (protein, fat, carbohydrate, minerals, and vitamins) of a purified rat diet revealed that the life-extending action of food restriction resulted from caloric restriction, rather than from a specific dietary component. (See Note: Clive McCay (1920-1967))
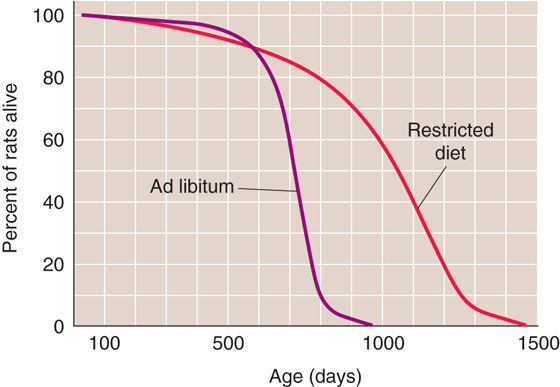
Figure 62-7 Survival curves for a population of 115 male F344 rats fed ad libitum and 115 male F344 rats with restricted food intake (i.e., 60% of the ad libitum intake) starting at 6 weeks of age. Both the median length of life and the maximum length of life are markedly greater in the restricted population. (Data from Yu BP, Masoro EJ, Murata I, et al: J Gerontol 1982; 37:130-141.)
Reducing food intake also extends the life of mice, hamsters, dogs, fish, several invertebrate animal species, and yeast. Studies of rhesus monkeys, still in progress, indicate that food restriction may also extend the life of nonhuman primates.
Does food restriction extend life by slowing aging processes? This is a difficult question to answer because of the lack of consensus on how to measure the aging of individuals or populations. However, for two reasons, most biogerontologists believe that food restriction does retard the aging processes. First, compared with rodents of the same age who were fed ad libitum, rodents on food-restriction regimens maintained physiological processes more like those of young animals. Second, food restriction delayed the onset or the progression of most age-associated diseases, including neoplastic, degenerative, and immune diseases.
During the past 70 years, many hypotheses have been proposed for the biological mechanisms underlying the life-prolonging action of food restriction, but none is firmly established. McCay proposed that caloric restriction extended life by retarding growth. However, later observations showed that food restriction, even when initiated in young adult rats or middle-aged mice, significantly extended life. Another early hypothesis was that food restriction extends life by markedly reducing adipose fat mass. However, it is possible to dissociate the effects of food restriction on longevity from the effects on fat mass in rats and mice.
Currently, the most popular view is that food restriction extends life by decreasing oxidative stress. Indeed, food restriction does decrease the accumulation of age-associated oxidative damage. Food restriction also causes a sustained reduction in plasma glucose levels, which could reduce glycation and glycoxidation. Food restriction also causes marked and sustained reductions in plasma levels of insulin and IGF-1. Genetic studies strongly indicate that decreasing insulin-like signaling (see Chapters 48 and 51) extends life. Thus, decreasing the levels of insulin and IGF-1 may well play an important role in life extension.
Another theory of the antiaging action of caloric restriction is hormesis, defined as the beneficial effects resulting from cellular responses to mild repeated stress, which would stimulate maintenance and repair processes and thereby retard aging. Food restriction, at the level that extends life, is a mild stress repeated daily. Moreover, food-restricted rodents have an increased ability to cope with acute, intense damaging agents, such as surgery, toxic chemicals, and high environmental temperatures.
Caloric restriction is an example of environmental factors that determine longevity. It is also clear that genetics has a major role. For example, the large difference in life span among species (from <100 days in Drosophila melanogaster to <5 years in mice to >100 years in humans) is primarily, if not exclusively, the result of genetic differences. Moreover, selective breeding within a species can produce populations that differ significantly in longevity.
Longevity probably depends on multiple genes. Thus, it was a surprise when Friedman and Johnson reported in 1988 that mutation of the age-1 gene of the nematode worm, Caenorhabditis elegans, resulted in a marked increase in longevity. The age-1 gene encodes a phosphatidylinositol-3-kinase, which is a component of the insulin-like signaling pathway. C. elegans, like other invertebrates, does not have separate signaling pathways for insulin and IGF-1, as do mammals (see Chapters 48 and 51). The mutation of age-1 in the Friedman and Johnson study caused some loss of function—that is, it was a weak mutation. Among other single-gene manipulations found to extend the life of C. elegans, D. melanogaster, Saccharomyces cerevisiae, and mice, many but not all involve a partial loss of function of the insulin-like signaling pathway.
Ames dwarf mice have a recessive point mutation in the Prop-1 gene, which results in the inability of the pituitary to produce growth hormone (GH), TSH, and prolactin. These mice have low levels of thyroid hormone, IGF-1, and insulin. Significantly, these dwarf mice have an increased life span compared with littermates not homozygous for this mutation.
Further support for a role of reduced insulin-like signaling in life extension comes from studies of mice with a knockout of the GHR/BP gene, which encodes the GH receptor and its proteolytic cleavage product, GH-binding protein. This knockout mouse exhibits growth retardation, high plasma GH levels, low plasma IGF-1 levels, and significant life extension. Reducing the expression of the insulin receptor by 85% to 90% in the adipose tissue of mice resulted in significant life extension. Finally, overexpression of the Klotho gene in mice increased the plasma level of Klotho protein, which suppressed insulin and IGF-1 signaling and extended the life of the mice.
In yeast, overexpression of the sir2 gene increases the level of a sirtuin protein called Sir2 and extends the replicative life of S. cerevisiae. Sir2 is a deacetylase that stabilizes ribosomal DNA. Sir2 may play a role in other species. Indeed, stimulating Sir2 orthologues extends the life span of C. elegans, D. melanogaster, and human cell lines. However, it is not yet clear whether sirtuin proteins play an important role in the life-prolonging action of caloric restriction.
The practice of antiaging medicine is becoming popular and plays an important role in preventing the occurrence and progression of certain age-associated diseases. For example, exercise and diet can reduce the incidence of coronary heart disease, stroke, and type 2 diabetes. However, some practitioners of antiaging medicine, as well as suppliers of pharmaceuticals and nutriceuticals, claim to have “magic bullets” that slow or even reverse aging. The magic bullets include antioxidants (e.g., vitamins E and C), amino acids (e.g., methionine), drugs (e.g., deprenyl), and hormones (e.g., melatonin, dehydroepiandrosterone, GH, estrogen and testosterone). No credible evidence indicates that any of these agents will reverse or even slow human aging. Aside from the question of efficacy is the possibility of long-term adverse effects of these magic bullets. Combined estrogen and progestin therapy is a case in point. Although hailed for relieving the symptoms of menopause, this hormone replacement therapy was long in use before a well-designed study uncovered its harmful effects on the cardiovascular system. In light of the animal studies that strongly indicate that insulin-like signaling promotes aging, the current use of recombinant GH is of concern. Although GH increases lean body mass in elderly persons, well-designed studies will be needed before GH is used as an antiaging agent.
Books and Reviews
Finkel T, Holbrook NJ: Oxidants, oxidative stress, and the biology of aging. Nature 2000; 408:239-247.
Kim S-H, Komiker P, Campisi J: Telomeres, aging, and cancer: In search of a happy ending. Oncogene 2002; 21:503-511.
Liang H, Masoro EJ, Nelson JF, et al: Genetic mouse models of extended lifespan. Exp Gerontol 2003; 38:1353-1364.
Masoro EJ: Caloric Restriction: A Key to Understanding and Modulating Aging. Amsterdam: Elsevier, 2002.
Tauber C: Sixty-Five Plus in America. Washington, DC: U. S. Bureau of the Census, 1992, rev. 1993
Zhang Y, Herman B: Ageing and apoptosis. Mech Ageing Dev 2002; 123:245-260.
Journal Articles
Cristofalo VJ, Allen RG, Pignolo RJ, et al: Relationship between donor age and the replicative lifespan of human cells in culture: A reevaluation. Proc Natl Acad SciUS A 1998; 95:10620-10625.
Hughes KA, Alipaz JA, Drnevich JM, Reynolds RM: A test of evolutionary theories of aging. Proc Natl Acad Sci U S A 2002; 99:14286-14291.
Kurosu H, Yamamoto M, Clark JD, et al: Suppression of aging in mice by the hormone Klotho. Science 2005; 309:1829-1833.
Lindeman RD, Tobin J, Shock NW: Longitudinal studies on the rate of decline in renal function with age. J Am Geriatr Soc 1985; 33:278-285.
Pongor S, Ulrich PC, Bencsath FA, Cerami A: Aging of proteins: Isolation and identification of a fluorescent chromophore from the reaction of polypeptides with glucose. Proc Natl Acad Sci U S A 1984; 81:2684-2688.
Yu BP, Masoro EJ, Murata I, et al: Life span of SPF Fischer 344 male rats fed ad libitum or restricted diets: Longevity, growth, lean body mass, and disease. J Gerontol 1982; 37:130-141.