Henry J. Binder and Adrian Reuben
In general, the digestive-absorptive processes for most of the constituents of our diet are highly efficient. For example, normal adult intestine absorbs ~95% of dietary lipid. However, we ingest most of the constituents of dietary food in a form that the intestine cannot readily absorb. Multiple digestive processes convert dietary food to a form that can be absorbed, primarily in the small intestine, but also, to a much smaller extent, in the colon.
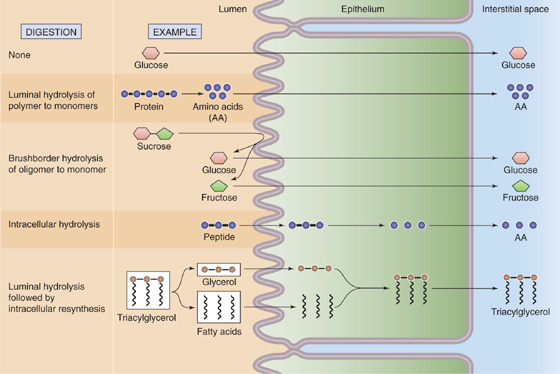
The digestive process—the enzymatic conversion of complex dietary substances to a form that can be absorbed—is initiated by the sight, smell, and taste of food. Although some digestion (that of carbohydrates) begins in the mouth and additional digestion may occur within the lumen of the stomach, most digestive processes occur in the small intestine. Digestion within the small intestine occurs either in the lumen, mediated by pancreatic enzymes, or at the small intestine brush border membrane (membrane digestion), mediated by brush border enzymes. Several different patterns of luminal, brush border, and cytosolic digestion exist (Fig. 45-1). Some of the dietary carbohydrate and protein that escape digestion and absorption in the small intestine are altered in the large intestine by bacterial enzymes to short-chain fatty acids that are absorbed by the colon.
Figure 45-1 General mechanisms of digestion and absorption. Digestion-absorption can follow any of five patterns. First, the substance (e.g., glucose) may not require digestion; the intestinal cells may absorb the nutrient as ingested. Second, a polymer (e.g., protein) may be digested in the lumen to its constituent monomers (e.g., amino acids) by pancreatic enzymes before absorption. Third, an oligomer (e.g., sucrose) is digested into its constituent monomers (e.g., monosaccharides) by brush border enzymes before absorption. Fourth, an oligomer (e.g., oligopeptide) may be directly absorbed by the cell and then broken down into monomers (e.g., amino acids) inside the cell. Finally, a substance (e.g., TAG) may be broken down into its constituent components before absorption; the cell may then resynthesize the original molecule.
The digestive processes for carbohydrates, proteins, and lipids result in the conversion of dietary nutrients to a chemical form for which intestinal absorptive processes exist. As a consequence, the digestive-absorptive processes for the several dietary constituents are closely integrated and regulated biological events that ensure survival. Multiple diseases can alter these digestive-absorptive processes and can thereby impair nutrient assimilation (i.e., the overall process of digestion and absorption). Because of the substantial segmental distribution of nutrient absorption along the gastrointestinal tract (Fig. 45-2), the clinical manifestations of disease (Table 45-1) often reflect these segmental differences.
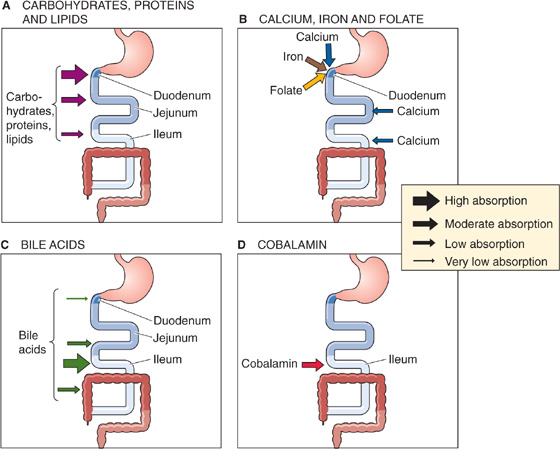
Figure 45-2 Sites of nutrient absorption. A, The entire small intestine absorbs carbohydrates, proteins, and lipids. However, the absorption is greatest in the duodenum, somewhat less in the jejunum, and much less in the ileum. The thickness of the arrows in the inset indicates the relative magnitude of total absorption at the indicated site in vivo. The maximal absorptive capacity of a specific segment under optimized experimental conditions (e.g., substrate concentrations) may be greater. B, Some substances are actively absorbed only in the duodenum. C, Bile acids are absorbed along the entire small intestine, but active absorption occurs only in the ileum. D, The vitamin cobalamin is absorbed only in the ileum.
Table 45-1 Major Gastrointestinal Diseases and Nutritional Deficiencies
Disease |
Organ Site of Predominant Disease |
Defects in Nutrient Digestion/Absorption |
Celiac sprue |
Duodenum and jejunum |
Fat absorption, lactose hydrolysis |
Chronic pancreatitis |
Exocrine pancreas |
Fat digestion |
Surgical resection of ileum; Crohn disease of ileum |
Ileum |
Cobalamin and bile acid absorption |
Primary lactase deficiency |
Small intestine |
Lactose hydrolysis |
We classify dietary carbohydrates into two major groups: (1) the monosaccharides (monomers) and (2) the oligosaccharides (short polymers) and polysaccharides (long polymers). The small intestine can directly absorb the monomers but not the polymers. Some polymers are digestible, that is, the body can digest them to form the monomers that the small intestine can absorb. Other polymers are nondigestible, or “fiber.” The composition of dietary carbohydrate is quite varied and is a function of culture. The diet of so-called developed countries contains considerable amounts of “refined” sugar and, compared with most developing countries, less fiber. Such differences in the fiber content of the Western diet may account for several diseases that are more prevalent in these societies (e.g., colon carcinoma and atherosclerosis). As a consequence, the consumption of fiber by the health-conscious public in the United States has increased during the past 2 decades. In general, increased amounts of fiber in the diet are associated with increased stool weight and frequency.
Approximately 45% to 60% of dietary carbohydrate is in the form of starch, which is a polysaccharide. Starch is a storage form for carbohydrates that is primarily found in plants, and it consists of both amylose and amylopectin. In contrast, the storage form of carbohydrates in animal tissues is glycogen, which is consumed in much smaller amounts. Amylose is a straight-chain glucose polymer that typically contains multiple glucose residues, connected by α-1, 4 linkages. In contrast, amylopectin is a massive branched glucose polymer that may contain 1 million glucose residues. In addition to the α-1, 4 linkages, amylopectin has frequent α-1, 6 linkages at the branch points. Amylopectins are usually present in much greater quantities (perhaps 4-fold) than amylose. Glycogen—the “animal starch”—also has α-1, 4 and α-1, 6 linkages like amylopectin. However, glycogen is more highly branched (i.e., α-1, 6 linkages).
Most dietary oligosaccharides are the disaccharides sucrose and lactose, which represent 30% to 40% of dietary carbohydrates. Sucrose is table sugar, derived from sugar cane and sugar beets, whereas lactose is the sugar found in milk. The remaining carbohydrates are the monosaccharides fructose and glucose, which make up 5% to 10% of total carbohydrate intake. There is no evidence of any intestinal absorption of either starches or disaccharides. Because the small intestine can absorb only monosaccharides, all dietary carbohydrate must be digested to monosaccharides before absorption. The colon cannot absorb monosaccharides.
Dietary fiber consists of both soluble and insoluble forms and includes lignins, pectins, and cellulose. These fibers are primarily present in fruits, vegetables, and cereals. Cellulose is a glucose polymer connected by β-1, 4 linkages, which cannot be digested by mammalian enzymes. However, enzymes from colonic bacteria may degrade fiber. This process is carried out with varying efficiency; pectins, gum, and mucilages are metabolized to a much greater degree than either cellulose or hemicellulose. In contrast, lignins, which are aromatic polymers and not carbohydrates, are not altered by microbial enzymes in the colonic lumen and are excreted unaltered in stool.
As we discuss later, the digestive process for dietary carbohydrates has two steps: (1) intraluminal hydrolysis of starch to oligosaccharides by salivary and pancreatic amylases (Fig. 45-3) and (2) so-called membrane digestion of oligosaccharides to monosaccharides by brush border disaccharidases. The resulting carbohydrates are absorbed by transport processes that are specific for certain monosaccharides. These transport pathways are located in the apical membrane of the small intestine villous epithelial cells.
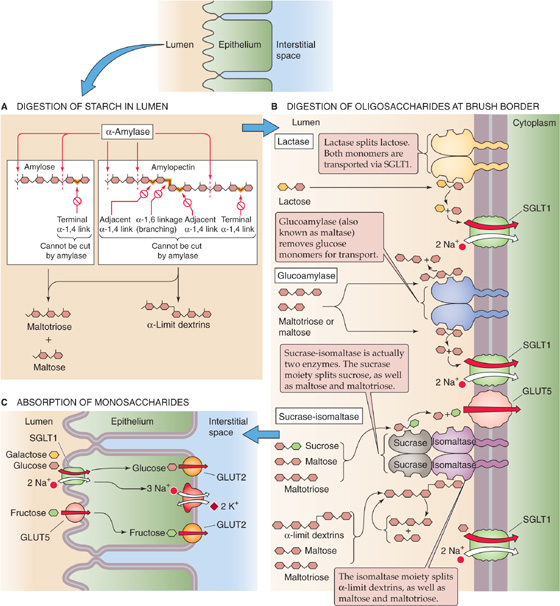
Figure 45-3 Digestion of carbohydrates to monosaccharides. A, Salivary and pancreatic α-amylase are endoenzymes. They can digest the linear internal α-1, 4 linkages between glucose residues, but they cannot break terminal α-1, 4 linkages (i.e., between the last two sugars in the chain). They also cannot split the α-1, 6 linkages at the branch points of amylopectin or the adjacent α-1, 4 linkages. As a result, the products of α-amylase action are linear glucose oligomers, maltotriose (a linear glucose trimer), maltose (a linear glucose dimer), and α-limit dextrins (which contain an α-1, 6 branching linkage). B, The brush border oligosaccharidases are intrinsic membrane proteins with their catalytic domains facing the lumen. The sucrase-isomaltase is actually two enzymes, and, therefore, four oligosaccharidases split the oligosaccharides produced by α-amylase into monosaccharides. C, SGLT1 is the Na+-coupled transporter that mediates the uptake of glucose or galactose from the lumen of the small intestine into the enterocyte. GLUT5 mediates the facilitated diffusion of fructose into the enterocyte. Once the monosaccharides are inside the enterocyte, GLUT2 mediates their efflux across the basolateral membrane into the interstitial space.
Both salivary and pancreatic acinar cells (see Chapter 43) synthesize and secrete α-amylases. Salivary and pancreatic amylases, unlike most of the pancreatic proteases that we discuss later, are secreted not in an inactive proenzyme form, but rather in an active form. Salivary and pancreatic α-amylases have similar enzymatic function, and their amino acid sequences are 94% identical. Salivary amylase in the mouth initiates starch digestion; in healthy adults, this step is of relatively limited importance. Salivary amylase is inactivated by gastric acid, but it can be partially protected by complexing with oligosaccharides.
Pancreatic α-amylase completes starch digestion in the lumen of the small intestine. Although amylase binds to the apical membrane of enterocytes, this localization does not provide any kinetic advantage for starch hydrolysis. Cholecystokinin (CCK) stimulates the secretion of pancreatic α-amylase by pancreatic acinar cells (see Chapter 43).
α-Amylase is an endoenzyme that hydrolyzes internal α-1, 4 linkages (Fig. 45-3A). α-Amylase does not cleave terminal α-1, 4 linkages, α-1, 6 linkages (i.e., branch points), or α-1, 4 linkages that are immediately adjacent to α-1, 6 linkages. As a result, starch hydrolysis products are maltose, maltotriose, and α-limit dextrins. Because α-amylase has no activity against terminal α-1, 4 linkages, glucose is not a product of starch digestion. The intestine cannot absorb these products of amylase digestion of starch, and thus further digestion is required to produce substrates (i.e., monosaccharides) that the small intestine can absorb by specific transport mechanisms.
The human small intestine has three brush border oligosaccharidases: lactase, glucoamylase (most often called maltase), and sucrase-isomaltase. These enzymes are all integral membrane proteins whose catalytic domains face the intestinal lumen (Fig. 45-3B). Sucrase-isomaltase is actually two enzymes—sucrase and isomaltase (also known as α-dextrinase or debranching enzyme)—bound together. Thus, four oligosaccharidases are present at the brush border. Lactase has only one substrate; it breaks lactose into glucose and galactose. The other three enzymes have more complicated substrate spectra. All cleave the terminal α-1, 4 linkages of maltose, maltotriose, and α-limit dextrins. In addition, each of these three enzymes has at least one other activity. Maltase can also degrade the α-1, 4 linkages in straight-chain oligosaccharides up to nine monomers in length. However, maltase cannot split either sucrose or lactose. The sucrase moiety of sucrase-isomaltase is required to split sucrose into glucose and fructose. The isomaltase moiety of sucrase-isomaltase is critical; it is the only enzyme that can split the branching α-1, 6 linkages of α-limit dextrins. (See Note: Oligosaccharidases)
The action of the four oligosaccharidases generates several monosaccharides. Maltose is hydrolyzed to two glucose residues, whereas the hydrolysis products of sucrose are glucose and fructose. The hydrolysis of lactose by lactase yields glucose and galactose. The activities of the hydrolysis reactions of sucrase-isomaltase and maltase are considerably greater than the rates at which the various transporters can absorb the resulting monosaccharides. Thus, uptake, not hydrolysis, is the rate-limiting step. In contrast, lactase activity is considerably less than that of the other oligosaccharidases and is rate limiting for overall lactose digestion-absorption.
The oligosaccharidases have a varying spatial distribution throughout the small intestine. In general, peak oligosaccharidase distribution and activity occur in the proximal jejunum (i.e., at the ligament of Treitz). Considerably less activity is noted in the duodenum and distal ileum, and none is reported in the large intestine. The distribution of oligosaccharidase activity parallels that of active glucose transport.
These oligosaccharidases are affected by developmental and dietary factors in different ways. In many nonwhite ethnic groups, as well as in almost all other mammals, lactase activity markedly decreases after weaning in the postnatal period. The regulation of this decreased lactase activity is genetically determined. The other oligosaccharidases do not decrease in the postnatal period. In addition, long-term feeding of sucrose upregulates sucrase activity. In contrast, sucrase activity is greatly reduced much more by fasting than is lactase activity. In general, lactase activity is both more susceptible to enterocyte injury (e.g., following viral enteritis) and is slower to recover from damage than is other oligosaccharidase activity. Thus, reduced lactase activity (as a consequence of both genetic regulation and environmental effects) has substantial clinical significance in that lactose ingestion may result in a range of symptoms in affected individuals (Fig. 45-4). (See Note: Lactose Intolerance)
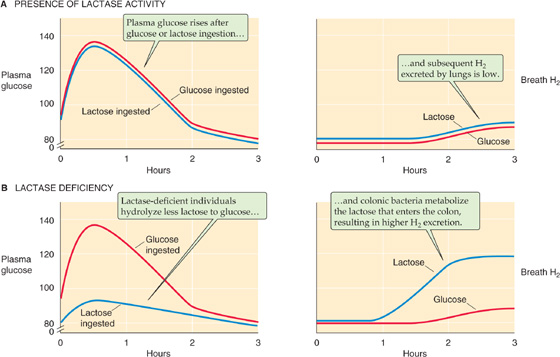
Figure 45-4 Effects of lactase deficiency on levels of glucose in the plasma and H2 in the breath. A, In an individual with normal lactase activity, blood glucose levels rise after the ingestion of either glucose or lactose. Thus, the small intestine can split the lactose into glucose and galactose and can absorb the two monosaccharides. At the same time, H2 in the breath is low. B, In an adult with low lactase activity, the rise in blood levels is less pronounced after ingesting lactose. Because the rise is normal after ingesting glucose, we can conclude that the difference is the result of lactase activity. Conversely, the individual with lactase deficiency excretes large amounts of H2 into the breath. This H2 is the product of lactose catabolism by colonic bacteria.
Lactase Deficiency
Primary lactase deficiency is extremely common in nonwhites, and it also occurs in some whites. Lactase activity decreases after weaning; the time course of its reduction is determined by hereditary factors. Ingestion of lactose in the form of milk and milk products by individuals with decreased amounts of small intestinal lactase activity may be associated with a range of gastrointestinal symptoms, including diarrhea, cramps, and flatus, or with no discernible symptoms. Several factors determine whether individuals with lactase deficiency experience symptoms after ingestion of lactose, including rate of gastric emptying, transit time through the small intestine, and, most importantly, the ability of colonic bacteria to metabolize lactose to short-chain fatty acids, CO2, and H2. Figure 45-4A shows the rise of plasma [glucose] following the ingestion of either lactose or glucose in adults with normal lactase levels. This figure also shows that the [H2] in the breath rises only slightly following the ingestion of either lactose or glucose in these individuals with normal lactase levels. Figure 45-4B shows that in individuals with primary lactase deficiency, the ingestion of lactose leads to a much smaller rise in plasma [glucose], although the ingestion of glucose itself leads to a normal rise in plasma [glucose]. Thus, no defect in glucose absorption per se is present, but simply a markedly reduced capacity to hydrolyze lactose to glucose and galactose. In lactase-deficient individuals, breath H2 is increased after lactose ingestion, because nonabsorbed lactose is metabolized by colonic bacteria to H2, which is absorbed into the blood and is subsequently excreted by the lungs. In contrast, the rise in breath H2 is normal after the ingestion of glucose in these individuals.
Treatment for symptomatic individuals with primary lactase deficiency is reduction or elimination of milk and milk products or the use of milk products treated with a commercial lactase preparation. No other defects in intestinal function or structure are associated with primary lactase deficiency.
The three monosaccharide products of carbohydrate digestion—glucose, galactose, and fructose—are absorbed by the small intestine in a two-step process involving their uptake across the apical membrane into the epithelial cell and their coordinated exit across the basolateral membrane (Fig. 45-3C). The Na/glucose transporter 1 (SGLT1) is the membrane protein responsible for glucose and galactose uptake at the apical membrane. The exit of all three monosaccharides across the basolateral membrane uses a facilitated sugar transporter (GLUT2). Because SGLT1 cannot carry fructose, the apical step of fructose absorption occurs by the facilitated diffusion of fructose through GLUT5. Thus, although two different apical membrane transport mechanisms exist for glucose and fructose uptake, a single transporter (GLUT2) is responsible for the movement of both monosaccharides across the basolateral membrane.
The uptake of glucose across the apical membrane through SGLT1 (Fig. 45-5A) represents active transport, because the glucose influx occurs against the glucose concentration gradient (see Chapter 5). Glucose uptake across the apical membrane is energized by the electrochemical Na+ gradient, which, in turn, is maintained by the extrusion of Na+ across the basolateral membrane by the Na-K pump. This type of Na+-driven glucose transport is an example of secondary active transport (see Chapter 5). Inhibition of the Na-K pump reduces active glucose absorption by decreasing the apical membrane Na+ gradient and thus decreasing the driving force for glucose entry.
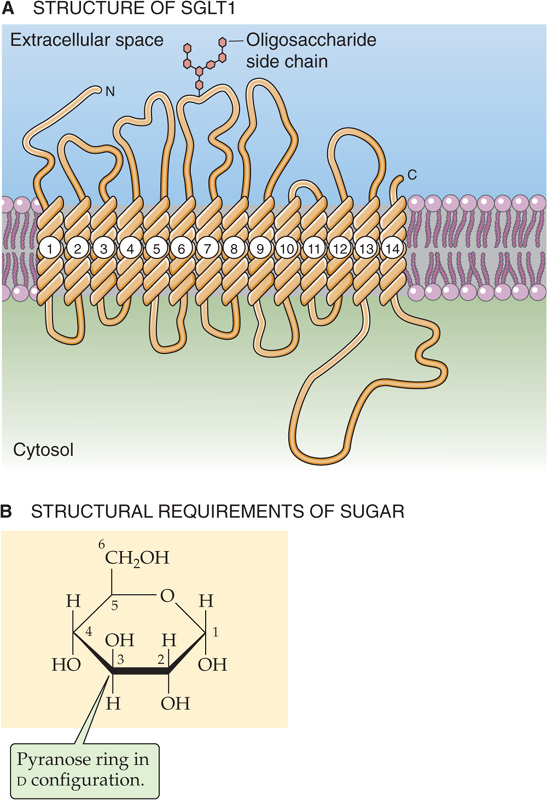
Figure 45-5 SGLT1. A, The SGLT family of proteins is believed to have 12 membrane-spanning segments. The deduced amino acid sequence has an open reading frame of 662 amino acids, predicting a molecular mass of 73 kDa. SGLT1 has a Na+-sugar stoichiometry of 2 : 1. B, SGLT1 transports only hexoses in a d-configuration and with a pyranose ring. This figure shows d-glucose; d-galactose is identical, except the H and OH on C-4 are inverted.
The affinity of SGLT1 for glucose is markedly reduced in the absence of Na+. The varied affinity of SGLT1 for different monosaccharides reflects its preference for specific molecular configurations. SGLT1 has two structural requirements for monosaccharides: (1) a hexose in a d-configuration and (2) a hexose that can form a six-membered pyranose ring (Fig. 45-5B). SGLT1 does not absorb l-glucose, which has the wrong stereochemistry, and it does not absorb d-fructose, which forms a five-membered ring. (See Note: Sodium/Glucose Cotransporters)
Glucose-Galactose Malabsorption
Molecular studies have been performed with jejunal mucosa from patients with so-called glucose-galactose malabsorption (or monosaccharide malabsorption). These individuals have diarrhea when they ingest dietary sugars that are normally absorbed by SGLT1. This diarrhea results from both reduced small intestine Na+ and fluid absorption (as a consequence of the defect in Na+-coupled monosaccharide absorption) and fluid secretion secondary to the osmotic effects of nonabsorbed monosaccharide. Eliminating the monosaccharides glucose and galactose, as well as the disaccharide lactose (i.e., glucose + galactose), from the diet eliminates the diarrhea. The monosaccharide fructose, which crosses the apical membrane through GLUT5, does not induce diarrhea. Early studies identified the abnormality in this hereditary disorder as a defect at the apical membrane that is presumably related to defective or absent SGLT1. Molecular studies of SGLT1 have revealed multiple mutations that result in single amino acid substitutions in SGLT1, each of which prevents the transport of glucose by SGLT1 in affected individuals. Patients with glucose-galactose malabsorption do not have glycosuria (i.e., glucose in the urine), because glucose reabsorption by the proximal tubule normally occurs through both SGLT1 and SGLT2 (see Chapter 36).
Early work showed that fructose absorption is independent of Na+ but has characteristics of both a carrier-mediated and a passive process. These observations show that the small intestine has separate transport systems for glucose and fructose. Subsequent studies established that facilitated diffusion is responsible for fructose absorption. Fructose uptake across the apical membrane is mediated by GLUT5 (see Chapter 5), a member of the GLUT family of transport proteins. GLUT5 is present mainly in the jejunum. (See Note: Facilitated Diffusion of Monosaccharides by the GLUT Transporters)
The efflux of glucose, fructose, and galactose across the basolateral membrane also occurs by facilitated diffusion. The characteristics of the basolateral sugar transporter, identified as GLUT2, are similar to those of other sugar transport systems in erythrocytes, fibroblasts, and adipocytes. GLUT2 has no homology to SGLT1 but is 41% identical to GLUT5, which is responsible for the uptake of fructose from the lumen.
With the exception of antigenic amounts of dietary protein that are absorbed intact, proteins must first be digested into their constituent oligopeptides and amino acids before being taken up by the enterocytes. Digestion-absorption occurs through four major pathways. First, several luminal enzymes (i.e., proteases) from the stomach and pancreas may hydrolyze proteins to peptides and then to amino acids, which are then absorbed (Fig. 45-6). Second, luminal enzymes may digest proteins to peptides, but enzymes present at the brush border digest the peptides to amino acids, which are then absorbed. Third, luminal enzymes may digest proteins to peptides, which are themselves taken up as oligopeptides by the enterocytes. Further digestion of the oligopeptides by cytosolic enzymes yields intracellular amino acids, which are moved by transporters across the basolateral membrane into the blood. Fourth, luminal enzymes digest dietary proteins to oligopeptides, which are taken up by enterocytes and moved directly into the blood. Overall protein digestion-absorption is very efficient; less than 4% of ingested nitrogen is excreted in the stool.
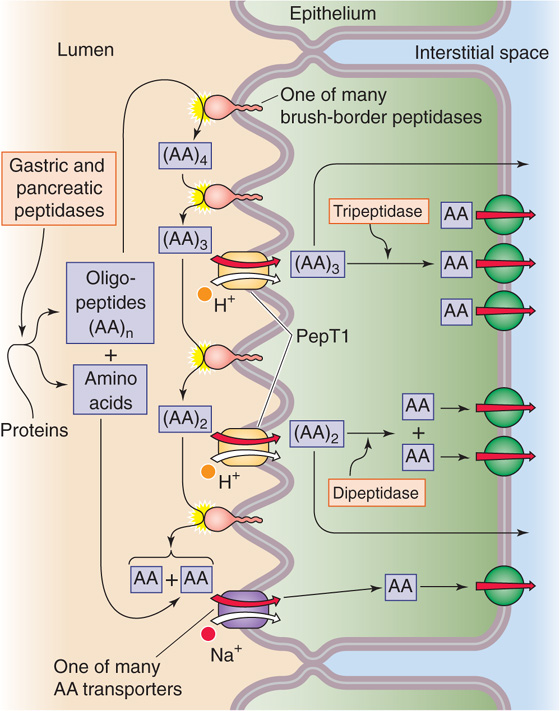
Figure 45-6 Action of luminal, brush border, and cytosolic peptidases. Pepsin from the stomach and the five pancreatic proteases hydrolyze proteins—both dietary and endogenous—to single amino acids, AA, or to oligopeptides, (AA)n. These reactions occur in the lumen of the stomach or small intestine. Various peptidases at the brush borders of enterocytes then progressively hydrolyze oligopeptides to amino acids. The amino acids are directly taken up by any of several transporters. The enterocyte directly absorbs some of the small oligopeptides through the action of the H+/oligopeptide cotransporter (PepT1). These small peptides are digested to amino acids by peptidases in the cytoplasm of the enterocyte. Several Na+-independent amino acid transporters move amino acids out of the cell across the basolateral membrane.
The protein that is digested and absorbed in the small intestine comes from both dietary and endogenous sources. Dietary protein in developed countries amounts to 70 to 100 g/day. This amount is far in excess of minimum daily requirements and represents 10% to 15% of energy intake. In contrast, dietary protein content in developing countries in Africa is often 50 g/day. Deficiency states are rare unless intake is markedly reduced.
Proteins are encoded by mRNA and consist of 20 amino acids. Nine of these amino acids are essential (see Chapter 58); that is, they are not synthesized in adequate amounts by the body and thus must be derived from either animal or plant protein sources. In addition, cells synthesize additional amino acids by post-translational modifications: γ-carboxyglutamic acid, hydroxylysine, 4-hydroxyproline, and 3-hydroxyproline. Protein digestion is influenced by the amino acid composition of the protein, by the source of protein, and by food processing. Thus, proteins rich in proline and hydroxyproline are digested relatively less completely. Cooking, storage, and dehydration also reduce the completeness of digestion. In general, protein derived from animal sources is digested more completely than plant protein.
In addition to dietary sources of protein, significant amounts of endogenous protein are secreted into the gastrointestinal tract, then conserved by protein digestion and absorption. Such endogenous sources represent ~50% of the total protein entering the small intestine and include enzymes, hormones, and immunoglobulins present in salivary, gastric, pancreatic, biliary, and jejunal secretions. A second large source of endogenous protein is desquamated intestinal epithelial cells as well as plasma proteins that the small intestine secretes.
Neonates can absorb substantial amounts of intact protein from colostrum (see Chapter 57) through the process of endocytosis. This mechanism is developmentally regulated and in humans remains active only until ~6 months of age. In adults, proteins are almost exclusively digested to their constituent amino acids and dipeptides and tripeptides or tetrapeptides before absorption. However, even adults absorb small amounts of intact proteins. These absorbed proteins can be important in inducing immune responses to dietary proteins.
Both gastric and pancreatic proteases, unlike the digestive enzymes for carbohydrates and lipids, are secreted as proenzymes that require conversion to their active form for protein hydrolysis to occur. The gastric chief cells secrete pepsinogen. We discuss the pH-dependent activation of pepsinogen in Chapter 42. The hydrolytic activity of pepsin is maximal at a pH of 1.8 to 3.5, and pepsin is irreversibly inactivated at a pH of less than 7. Pepsin is an endopeptidase with primary specificity for peptide linkages of aromatic and larger neutral amino acids. Although pepsin in the stomach partially digests 10% to 15% of dietary protein, pepsin hydrolysis is not absolutely necessary; patients with either total gastrectomies or pernicious anemia (who do not secrete acid and thus whose intragastric pH is always >7) do not have increased fecal nitrogen excretion.
Five pancreatic enzymes (Table 45-2) participate in protein digestion and are secreted as inactive proenzymes. Trypsinogen is initially activated by a jejunal brush border enzyme, enterokinase (enteropeptidase), by the cleavage of a hexapeptide, thereby yielding trypsin. Trypsinogen is also autoactivated by trypsin. Trypsin also activates the other pancreatic proteolytic proenzymes. The secretion of proteolytic enzymes as proenzymes, with subsequent luminal activation, prevents pancreatic autodigestion before enzyme secretion into the intestine.
Table 45-2 Pancreatic Peptidases
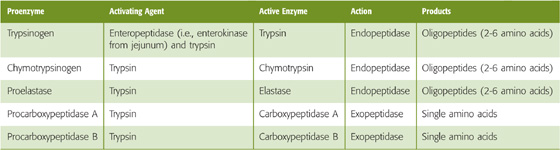
Pancreatic proteolytic enzymes are either exopeptidases or endopeptidases and function in an integrated manner. Trypsin, chymotrypsin, and elastase are endopeptidases with affinity for peptide bonds adjacent to specific amino acids, thus resulting in the production of oligopeptides with two to six amino acids. In contrast, the exopeptidases—carboxypeptidase A and carboxypeptidase B—hydrolyze peptide bonds adjacent to the carboxy terminus, thereby resulting in the release of individual amino acids. The coordinated action of these pancreatic proteases converts ~70% of luminal amino nitrogen to oligopeptides and ~30% to free amino acids.
Small peptides present in the small intestinal lumen after digestion by gastric and pancreatic proteases undergo further hydrolysis by peptidases at the brush border (Fig. 45-6). Multiple peptidases are present on both the brush border and in the cytoplasm of villous epithelial cells. This distribution of cell-associated peptidases stands in contrast to that of the oligosaccharidases, which are found only at the brush border. Because each peptidase recognizes only a limited repertoire of peptide bonds, and because the oligopeptides to be digested contain 24 different amino acids, large numbers of peptidases are required to ensure the hydrolysis of peptides.
As we discuss later, a transporter on the apical membrane of enterocytes can take up small oligopeptides, primarily dipeptides and tripeptides. Once inside the cell, these oligopeptides may be further digested by cytoplasmic peptidases. The brush border and cytoplasmic peptidases have substantially different characteristics. For example, the brush border peptidases have affinity for relatively larger oligopeptides (three to eight amino acids), whereas the cytoplasmic peptidases primarily hydrolyze dipeptides and tripeptides. Because the brush border and cytoplasmic enzymes often have different biochemical properties (e.g., heat lability and electrophoretic mobility), it is evident that the peptidases in the brush border and cytoplasm are distinct, independently regulated molecules.
Like the pancreatic proteases, each of the several brush border peptidases is an endopeptidase, an exopeptidase, or a dipeptidase and has affinity for specific peptide bonds. The exopeptidases are either carboxypeptidases, which release carboxy-terminal amino acids, or aminopeptidases, which hydrolyze the amino acids at the amino-terminal end. Cytoplasmic peptidases are relatively less numerous.
During the postnatal period, intestinal epithelial cells absorb protein by endocytosis, a process that provides a mechanism for transfer of passive immunity from mother to child. The uptake of intact protein by the epithelial cell ceases by the sixth month; the cessation of this protein uptake, called closure, is hormonally mediated. For example, administration of corticosteroids during the postnatal period induces closure and reduces the time that the intestine can absorb significant amounts of whole protein.
The adult intestine can absorb finite amounts of intact protein and polypeptides. Uncertainty exists regarding the cellular route by which these substances are absorbed, as well as the relationship of the mechanism of protein uptake in adults to that in neonates. Enterocytes can take up by endocytosis a small amount of intact protein, most of which is degraded in lysosomes (Fig. 45-7). A small amount of intact protein appears in the interstitial space. The uptake of intact proteins also occurs through a second, more specialized route. In the small intestine, immediately overlying Peyer’s patches (follicles of lymphoid tissue in the lamina propria), M cells replace the usual enterocytes on the surface of the gut. M cells have few microvilli and are specialized for protein uptake. They have limited ability for lysosomal protein degradation; rather, they package ingested proteins (i.e., antigens) in clathrin-coated vesicles, which they secrete at their basolateral membranes into the lamina propria. There, immunocompetent cells process the target antigens and transfer them to lymphocytes to initiate an immune response. Although protein uptake in adults may not have nutritional value, such uptake is clearly important in mucosal immunity and probably is involved in one or more disease processes.
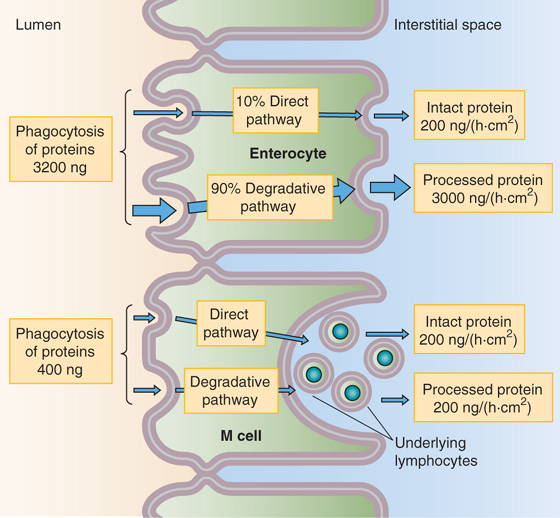
Figure 45-7 Absorption of whole proteins. Both enterocytes and specialized M cells can take up intact proteins. The more abundant enterocytes can endocytose far more total protein than can the M cells. However, the lysosomal proteases in the enterocytes degrade ~90% of this endocytosed protein. The less abundant M cells take up relatively little intact protein, but approximately half of this emerges intact at the basolateral membrane. There, immunocompetent cells process the target antigens and then transfer them to lymphocytes, thus initiating an immune response.
Virtually all absorbed protein products exit the villous epithelial cell and enter the blood as individual amino acids. Substantial portions of these amino acids are released in the lumen of the small intestine by luminal proteases and brush border peptidases and, as we discuss later, move across the apical membranes of enterocytes through several amino acid transport systems (Fig. 45-6). However, substantial amounts of protein are absorbed from the intestinal lumen as dipeptides, tripeptides, or tetrapeptides and are then hydrolyzed to amino acids by intracellular peptidases.
The transporter responsible for the uptake of luminal oligopeptides (Fig. 45-8A) is distinct from the various amino acid transporters. Furthermore, administering an amino acid as a peptide (e.g., the dipeptide glycylglycine) results in a higher blood level of the amino acid than administering an equivalent amount of the same amino acid as a monomer (e.g., glycine; Fig. 45-8B). One possible explanation for this effect is that the oligopeptide cotransporter, which carries multiple amino acids rather than a single amino acid into the cell, may simply be more effective than amino acid transporters in transferring amino acid monomers into the cell. This accelerated peptide absorption has been referred to as a kinetic advantage and raises the question of the usefulness of the enteral administration of crystalline amino acids to patients with impaired intestinal function or catabolic deficiencies. The evidence for a specific transport process for dipeptides, tripeptides, and tetrapeptides comes from direct measurements of oligopeptide transport, molecular identification of the transporter, and studies of the hereditary disorders of amino acid transport, cystinuria, and Hartnup disease.
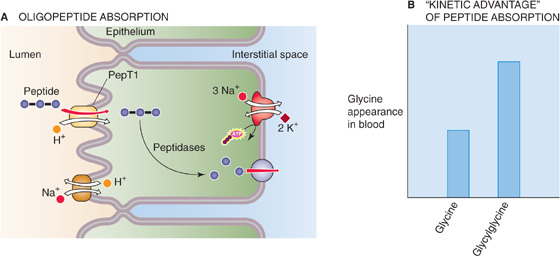
Figure 45-8 Absorption of oligopeptides. A, The H+/oligopeptide cotransporter PepT1 moves dipeptides, tripeptides, and tetrapeptides into the enterocyte, across the apical membrane. Peptidases in the cytoplasm hydrolyze the oligopeptides into their constituent amino acids, which then exit across the basolateral membrane through one of three Na+-independent amino acid transporters. B, If glycine is present in the lumen only as a free amino acid, then the enterocyte absorbs it only through apical amino acid transporters. However, if the same amount of glycine is present in the lumen in the form of the dipeptide glycylglycine, the rate of appearance of glycine in the blood is about twice as high. Thus, PepT1, which moves several amino acid monomers for each turnover of the transporter, is an effective mechanism for absorbing “amino acids.”
Oligopeptide uptake is an active process driven not by a Na+ gradient, but by a proton gradient. Oligopeptide uptake occurs through an H+/oligopeptide cotransporter known as PepT1 (SLC15A1; see Chapter 5), which is also present in the renal proximal tubule. PepT1 also appears to be responsible for the intestinal uptake of certain dipeptide-like antibiotics (e.g., oral amino-substituted cephalosporins). As noted earlier, after their uptake, dipeptides, tripeptides, and tetrapeptides are usually hydrolyzed by cytoplasmic peptidases to their constituent amino acids, the forms in which they are transported out of the cell across the basolateral membrane. Because peptides are almost completely hydrolyzed to amino acids intracellularly, few peptides appear in the portal vein. Proline-containing dipeptides, which are relatively resistant to hydrolysis, are the primary peptides present in the circulation.
Multiple amino acid transport systems have been identified and characterized in various nonepithelial cells. The absorption of amino acids across the small intestine requires sequential movement across both the apical and basolateral membranes of the villous epithelial cell. Although the amino acid transport systems have overlapping affinities for various amino acids, the general consensus is that at least seven distinct transport systems are present at the apical membrane (see Table 36-1); we discuss the basolateral amino acid transporters in the next section. Whereas many apical amino acid transporters are probably unique to epithelial cells, some of those at the basolateral membrane are probably the same as in nonepithelial cells.
The predominant apical amino acid transport system is system B0 (SLC6A19; see Table 36-1), and it results in Na+- dependent uptake of neutral amino acids. As is the case for glucose uptake, uphill movement of neutral amino acids is driven by an inwardly directed Na+ gradient that is maintained by the basolateral Na-K pump. The uptake of amino acids by system B0 is an electrogenic process and represents another example of secondary active transport. It transports amino acids with an l-stereo configuration and an amino group in the α position. System B0+ (SLC6A14) is similar to system B0 but has broader substrate specificity. System b0+ (SLC7A9/SLC3A1 dimer) differs from B0+ mainly in being independent of Na+.
Other carrier-mediated transport mechanisms exist for anionic (i.e., acidic), cationic (i.e., basic), β amino acids, and imino acids (see Table 36-1). Because the apical amino acid transporters have overlapping affinities for amino acids, and because of species differences as well as segmental and developmental differences among the transporters, it has been difficult to establish a comprehensive model of apical membrane amino acid transport in the mammalian small intestine.
Amino acids appear in the cytosol of intestinal villous cells as the result either of their uptake across the apical membrane or of the hydrolysis of oligopeptides that had entered the apical membrane (Fig. 45-6). The enterocyte subsequently uses ~10% of the absorbed amino acids for intracellular protein synthesis.
Movement of amino acids across the basolateral membrane is bidirectional; the movement of any one amino acid can occur through one or more amino acid transporters. At least five amino acid transporters are present in the basolateral membrane (see Table 36-1). Three amino acid transport processes on the basolateral membrane mediate amino acid exit from the cell into the blood and thus complete the process of protein assimilation. Two other amino acid transporters mediate uptake from the blood for the purposes of cell nutrition. The three Na+-independent amino acid transport systems appear to mediate amino acid movement out of the epithelial cell into blood. One of these, system y+ (SLC7A1), is also present on the apical membrane. The two Na+-dependent processes facilitate their movement into the epithelial cell. Indeed, these two Na+-dependent transporters resemble those that are also present in nonpolar cells.
In general, the amino acids incorporated into protein within villous cells are derived more from those that enter across the apical membrane than from those that enter across the basolateral membrane. In contrast, epithelial cells in the intestinal crypt derive almost all their amino acids for protein synthesis from the circulation; crypt cells do not take up amino acids across their apical membrane.
Lipids in the diet are derived from animals or plants and are composed of carbon, hydrogen, and a smaller amount of oxygen. Some lipids also contain small but functionally important amounts of nitrogen and phosphorus (Fig. 45-11). Lipids are typified by their preferential solubility in organic solvents, compared with water. A widely used indicator of the lipidic nature of a compound is its octanol-water partition coefficient, which for most lipids is between 104 and 107. The biological fate of lipids depends critically on their chemical structure as well as on their interactions with water and other lipids in aqueous body fluids (e.g., intestinal contents and bile). Thus, lipids have been classified according to their physicochemical interactions with water. Lipids may be either nonpolar and completely insoluble in water (e.g., cholesteryl esters and carotene) or polar and amphiphilic, that is, having both polar (hydrophilic) and nonpolar (hydrophobic) groups. Added in small amounts, polar lipids form stable or unstable monolayers on the surface of water (see Fig. 2-1C), whereas in bulk their physicochemical behavior varies from insolubility (as is the case with triacylglycerols [TAGs] and cholesterol) to the formation of various macroaggregates, such as liquid crystals and micelles. Less-soluble lipids are incorporated into the macroaggregates of the more-polar lipids and are thus stably maintained in aqueous solutions. The term fat is generally used to refer to TAG—formerly called triglyceride—but it is also used loosely to refer to lipids in general.
Defects in Apical Amino Acid Transport: Hartnup Disease and Cystinuria
Hartnup disease and cystinuria are hereditary disorders of amino acid transport across the apical membrane. These autosomal recessive disorders are associated with both small intestine and renal tubule abnormalities (see Chapter 36 for the box on hyperaminoacidurias) in the absorption of neutral amino acids in the case of Hartnup disease and of cationic (i.e., basic) amino acids and cystine in the case of cystinuria.
The clinical signs of Hartnup disease are most evident in children and include the skin changes of pellagra, cerebellar ataxia, and psychiatric abnormalities. In Hartnup disease, the absorption of neutral amino acids by system B0 (SLC6A19) in the small intestine is markedly reduced, whereas that of cationic amino acids is intact (Fig. 45-9).
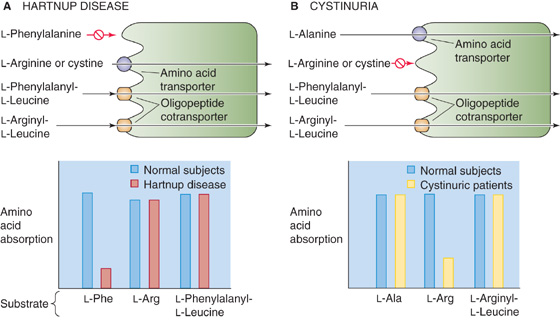
Figure 45-9 Genetic disorders of apical amino acid transport. A, In Hartnup disease, an autosomal recessive disorder, the apical system B0 (SLC6A19) is defective. As a result, the absorption of neutral amino acids such as l-phenylalanine, is reduced. (However, the absorption of l-cystine (i.e., Cys-S-S-Cys) and cationic (i.e., basic) amino acids (e.g., l-arginine) remains intact.) The enterocyte can absorb l-phenylalanine normally if the amino acid is present in the form of the dipeptide l-phenylalanyl-l-leucine, inasmuch as the oligopeptide cotransporter PepT1 is normal. B, In cystinuria, an autosomal recessive disorder, the apical system b0+ (SLC7A9/SLC3A1 dimer) is defective. As a result, the absorption of l-cystine (i.e., Cys-S-S-Cys) and cationic (i.e., basic) amino acids (e.g., l-arginine) is reduced. However, the absorption of amino acids that use system B0 (e.g., l-Ala) is normal. The enterocyte can absorb l-arginine normally if the amino acid is present in the form of the dipeptide l-arginyl-l-leucine.
The principal manifestation of cystinuria is the formation of kidney stones. In cystinuria, the absorption of cationic amino acids by system b0+ (SLC7A9/SLC3A1 dimer) is abnormal—as a result of mutations in SLC7A9 or SLC3A1—but absorption of neutral amino acids is normal.
Because neither of these diseases involves the oligopeptide cotransporter, the absorption of oligopeptides containing either neutral or cationic amino acids is normal in both diseases. Only 10% of patients with Hartnup disease have clinical evidence of protein deficiency (i.e., pellagra) commonly associated with defects in protein or amino acid absorption. The lack of evidence of protein deficiency is a consequence of the presence of more than one transport system for different amino acids, as well as a separate transporter for oligopeptides. Thus, oligopeptides containing neutral amino acids are absorbed normally in Hartnup disease, and oligopeptides with cationic amino acids are absorbed normally in cystinuria.
These two genetic diseases also emphasize the existence of amino acid transport mechanisms on the basolateral membrane that are distinct and separate from the apical amino acid transporters. Thus, in both Hartnup disease and cystinuria, oligopeptides are transported normally across the apical membrane and are hydrolyzed to amino acids in the cytosol, and the resulting neutral and cationic amino acids are readily transported out of the cell across the basolateral membrane.
Basolateral Amino Acid Transport Defects: Lysinuric Protein Intolerance
Lysinuric protein intolerance is a rare autosomal recessive disorder of amino acid transport across the basolateral membrane (Fig. 45-10). Evidence indicates impaired cationic amino acid transport and symptoms of malnutrition. It appears that the defect is in system y+L, which is located solely on the basolateral membrane. System y+L has two subtypes, y+LAT1 (SLC7A7/SLC3A2 dimer) and y+LAT2 (SLC7A6/SLC3A2 dimer). Mutations in the SLC7A7 gene (subtype y+LAT1) cause the disease lysinuric protein intolerance. Cationic amino acids are absorbed normally across the apical membrane in these patients. Unlike in Hartnup disease or cystinuria, in which the enterocytes can absorb the amino acid normally if it is presented as an oligopeptide, in lysinuric protein intolerance the enterocytes cannot absorb the amino acid regardless of whether the amino acid is “free” or is part of an oligopeptide. These observations are best explained by hypothesizing that the patients hydrolyze intracellular oligopeptides properly but have a defect in the transport of cationic amino acids across the basolateral membrane. This defect is present not only in the small intestine, but also in hepatocytes and kidney cells, and perhaps in nonepithelial cells as well.
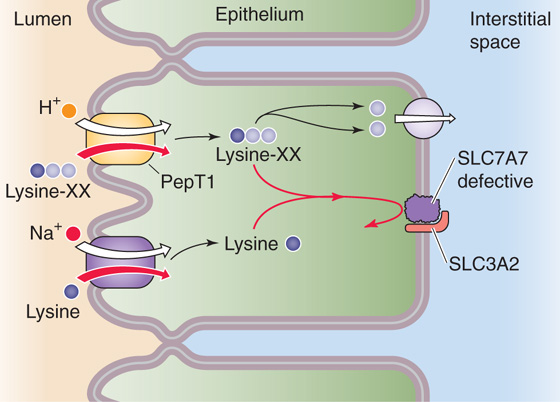
Figure 45-10 A genetic disorder of basolateral amino acid transport, lysinuric protein intolerance is an autosomal recessive defect in which the Na+-independent y+L amino acid transporter on the apical and basolateral membranes is defective. However, the absence of apical y+L (SLC7A6/SLC3A2 or SLC7A7/SLC3A2 dimers) does not present a problem because Na+-dependent amino acid transporters can take up lysine, and PepT1 can take up lysine-containing oligopeptides (lysine-XX). However, no other mechanism exists for moving lysine out of the enterocyte across the basolateral membrane.
Typical adult Western diets contain ~140 g of fat (providing ~55% of the energy), which is more than the recommended intake of less than 30% of total dietary calories (<70 g of fat). Of this fat, more than 90% is TAGs, which are commonly long-chain fatty acyl esters of glycerol, a trihydroxyl alcohol. The three esterification (i.e., acylation) positions on the glycerol backbone that are occupied by hydroxyl groups, are designated sn1-, sn2-and sn3-, according to a s tereochemical n umbering system adopted by an international committee on biochemical nomenclature (Fig. 45-11A–C). At body temperature, fats are usually liquid droplets. Newborn infants consume three to five times more lipid than adults, relative to body weight. Dietary fat is the body’s only source of essential fatty acids, and it acts as a vehicle for the absorption of fat-soluble vitamins (the handling of which is discussed in Chapter 46). Fat is also the major nutrient responsible for postprandial satiety. The ratio of saturated to unsaturated fatty acids in TAGs is high in animal fats and low in plant fats. Evidence indicates that in so-called developed countries, average fat intake is falling, as is the proportion of fat contributed by saturated fatty acids. Conversely, the proportion of polyunsaturated fats has risen. Milk and milk products contain 7% short-chain, 15% to 20% sn3-medium-chain, and 73% to 77% long-chain fatty acids. Fish contains unusual but metabolically important fatty acids (e.g., omega fatty acids), as well as wax esters.
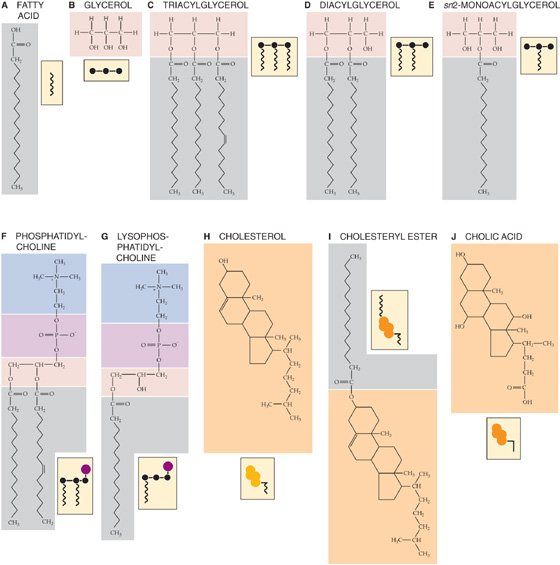
Figure 45-11 A to J, Chemical formulas of some common lipids. The example in A is stearic acid, a fully saturated fatty acid with 18 carbon atoms. B shows glycerol, a trihydroxy alcohol, with hydroxyl groups in positions sn1-, sn2-, and sn3-. In C, the left sn1-and center sn2-fatty acids are palmitic acid, a fully saturated fatty acid with 16 carbon atoms. The rightmost sn 3-fatty acid is palmitoleic acid, which is also a 16-carbon structure, but with a double bond between carbons 9 and 10. In F, the left sn1- fatty acid is palmitic acid (16 carbons, fully saturated), and the right sn 2-fatty acid is palmitoleic acid (16 carbons, double bond between carbons 9 and 10). In I, the example is the result of esterifying cholesterol and palmitic acid (16 carbons, fully saturated).
Approximately 5% (4 to 6 g/day) of dietary lipids come from cell membranes and are phospholipids. Most phospholipids are glycerophospholipids. They consist of a glycerol backbone that is esterified at the first two positions to fatty acids and at the third position to a phosphate that, in turn, is esterified to a head group (see Fig. 2-2). One of the glycerophospholipids, phosphatidylcholine (lecithin), is the predominant phospholipid (Fig. 45-11F). The other major class of membrane phospholipid is the sphingolipid, which has a serine rather than a glycerol backbone.
The diet contains ~0.5 g of unesterified cholesterol (also derived from animal cell membranes; Fig. 45-11H), whereas esterified cholesterol (Fig. 45-11I) is usually found only in liver or food made from blood products. Traces of lipovitamins and provitamins (e.g., carotene) are present in dietary fat, which may also contain lipid-soluble toxins and carcinogens from the environment. These undesirable lipid-soluble chemicals—which include nitrosamines, aflatoxins, and polycyclic hydrocarbons such as benzo(a)pyrene—are all found in margarine, vegetable oils, and other dietary fats. The diet also may contain skin lipids, which are chemically diverse and complex and are difficult to digest.
The bile secreted into the intestine (see Chapter 46) plays a key role in the assimilation of dietary lipids, as we explain later. This bile contains phospholipid (10 to 15 g/day)—also predominantly lecithin—and unesterified cholesterol (1 to 2 g/day). Quantitatively, these biliary lipids exceed those present in the diet by 2- to 4-fold. Membrane lipids from desquamated intestinal cells account for a further 2 to 6 g of lipid for digestion. Investigators have estimated that ~10 g/day of lipids are derived from dead bacteria. Most bacterial lipids are added in the colon.
The central process in the digestion of lipids is their hydrolysis in the aqueous milieu of the intestinal lumen. Lipid hydrolysis is catalyzed by lipases secreted by the glands and cells of the upper gastrointestinal tract. The products of lipolysis diffuse through the aqueous content of the intestinal lumen, traverse the so-called unstirred water layer and mucus barrier that line the intestinal epithelial surface, and enter the enterocyte for further processing. Because dietary lipids are insoluble in water, digestive lipases have evolved to act more efficiently at oil-water interfaces than on water-soluble substrates.
A key step preliminary to lipid digestion is the transformation of ingested solid fat and oil masses into an emulsion of fine oil droplets in water. The emulsification of dietary fats begins with food preparation (grinding, marinating, blending, and cooking), followed by chewing and gastric churning (see Chapter 42) caused by antral peristalsis against a closed pylorus. Emulsification of ingested lipids is enhanced when muscular movements of the stomach intermittently squirt the gastric contents into the duodenum and, conversely, when peristalsis of the duodenum propels the duodenal contents in retrograde fashion into the stomach through the narrow orifice of a contracted pylorus. The grinding action of the antrum also mixes food with the various digestive enzymes derived from the mouth and stomach. Intestinal peristalsis mixes luminal contents with pancreatic and biliary secretions. Together, these mechanical processes that reduce the size of the lipid droplets also dramatically increase their ratio of surface area to volume, thereby increasing the area of the oil-water interface.
The emulsion, produced by the mechanical processes just outlined, is stabilized by preventing the dispersed lipid particles from coalescing. This is achieved by coating the emulsion droplets with membrane lipids, denatured protein, dietary polysaccharides, certain products of digestion (e.g., fatty acids released by gastric lipase, and fatty acids and monoacylglycerols [MAGs] from intestinal and pancreatic digestion), and biliary phospholipids and cholesterol. Phospholipids and cholesterol are well suited as emulsion stabilizers because they dissolve neither in oil nor in water, but they have excellent interfacial solubilities at oil-water interfaces. Thus, they form a surface monomolecular layer on emulsion particles. The polar groups of the phospholipids project into the water; the charges of the polar groups and their high degree of hydration prevent coalescence of the emulsion particles. The core of the emulsion particle is composed of TAG, which also contains cholesteryl esters and other nonpolar or weakly polar lipids. A very small fraction of the TAG in the lipid particle localizes to the particle surface. The fat in breast milk is already emulsified by proteins and phospholipids incorporated into the surface of fat droplets during lactation. Foods such as sauces, ice cream, and puddings are stably emulsified during their preparation.
In some species, but not in humans, a small amount of lipid digestion begins in the mouth, mediated by lingual lipase. In the stomach, both lingual lipase that is swallowed and a gastric lipase secreted by gastric chief cells digest substantial amounts of lipid. Human gastric lipase is a 42-kDa glycoprotein whose secretion is stimulated by gastrin. Gastric lipase secretion is already well established in the neonatal period (unlike pancreatic lipase secretion), and thus gastric lipolysis is important for fat digestion in newborn infants. Gastric and lingual—as well as pharyngeal—lipases belong to a family of serine hydrolases that have acidic pH optima (pH 4), are stable in the acidic environment of the stomach, are resistant to digestion by pepsin, and are not inhibited by the surface layer of membrane lipids (emulsifiers) that envelopes TAG droplets. However, acid lipases are inactive at neutral pH and also are readily inactivated by pancreatic proteases (especially in the presence of bile salts) once they reach the small intestine.
In vivo, gastric lipase releases a single sn3-fatty acid from TAGs, thus leaving behind intact diacyclglycerols (Fig. 45-11D). The carboxyl groups of long-chain fatty acids, released from TAGs in the stomach, are protonated and insoluble at the acidic pH prevailing in the stomach. These fatty acids are not absorbed in the stomach, but rather they remain in the core of the TAG droplets, in whose emulsification they participate in the small intestine. Medium- and short-chain fatty acids are mainly protonated in gastric juice during feeding and passively move across the gastric mucosa into portal blood. (See Note: Gastric Absorption of Medium- and Short-Chain Fatty Acids)
In healthy adult humans, ~15% of fat digestion occurs in the stomach. In patients with pancreatic insufficiency, however, the lack of pancreatic proteases and HCO−3 in the duodenal lumen may permit the continued action of gastric lipase after the gastric contents leave the stomach and enter the duodenum. Extended gastric lipase activity partly alleviates fat malabsorption resulting from pancreatic disease and pancreatic lipase deficiency.
The process of fat digestion that begins in the stomach is completed in the proximal small intestine, predominantly by enzymes synthesized and secreted by pancreatic acinar cells (see Chapter 43), and is carried into the duodenum in the pancreatic juice. In humans, a lipase found in human milk—the so-called bile salt–stimulated milk lipase—also digests fat. A similar lipase is found in the milk of relatively few other animal species. Milk lipase is stable during passage through the acid environment of the stomach, yet it is active at the alkaline pH of the duodenum and jejunum, where it hydrolyzes diacylglycerols, MAGs, cholesteryl esters, and fat-soluble vitamin esters, as well as TAGs. Bile salts not only stimulate milk lipase activity but also protect the enzyme from proteolysis in the small intestine. Like gastric lipase, milk lipase is important for fat digestion in breast-fed infants.
Once the fatty acids generated in the stomach reach the duodenum, they trigger the release of CCK and gastric inhibitory polypeptide (GIP) from the duodenal mucosa. CCK stimulates the flow of bile into the duodenum by causing the gallbladder to contract and the sphincter of Oddi to relax (see Chapter 46). CCK also stimulates the secretion of pancreatic enzymes, including lipases and esterases (see Chapter 43). As we discuss later, long-chain fatty acids also facilitate the lipolytic action of pancreatic lipase.
The major lipolytic enzyme of pancreatic juice is a 48-kDa carboxylic esterase known as pancreatic lipase, sometimes referred to as TAG lipase or as colipase-dependent pancreatic lipase. In adults but not in infants, this enzyme, which is secreted into the duodenum in its active form in 1000-fold excess, is thought to effectively digest all dietary TAGs not hydrolyzed in the stomach. Full lipolytic activity of pancreatic lipase requires the presence of a small (10-kDa) protein cofactor called colipase, as well as an alkaline pH, Ca2+, bile salts, and fatty acids. The pancreas secretes colipase in the proform (i.e., procolipase), which has no intrinsic lipolytic activity. Trypsin cleaves procolipase into colipase and an N-terminal pentapeptide, enterostatin. This cleavage is important because the newly formed colipase is a cofactor of pancreatic lipase, as we discuss later. In addition, the N-terminal pentapeptide may partially control satiety. (See Note: Pancreatic Lipase)
Pancreatic lipase is active only at the oil-water interface of a TAG droplet. However, surface emulsifier components (e.g., phospholipids, protein) present at that interface inhibit lipase action. Bile salt micelles also inhibit lipolysis by displacing the lipase from the oil-droplet surface. Binding of colipase reverses this inhibition either by attaching first to the interface and serving as an anchor for the binding of the lipase or by first forming a colipase-pancreatic-lipase complex that then binds to the lipid interface. Colipase also can penetrate the phospholipid coating of the TAG emulsion. Bile salt micelles bring the colipase closer to the interface. Because bile salts are nearby, they can participate in the solubilization and removal of the products of lipolysis released from the emulsion droplet. Fatty acids have a biphasic effect. They enhance emulsification and augment lipolysis (probably by enhancing the binding of the colipase-lipase complex to the lipid interface). In contrast, their buildup causes product inhibition of lipase.
Studies of the crystal structure of pancreatic lipase have shown that when the enzyme is free in solution, the catalytic site of the lipase—located in a cleft in the molecule—is partly covered by a lid formed of loops of its peptide chain. The interaction of the colipase and lipase with the interface causes a conformational change in the lipase molecule that opens the lid, thereby allowing lipid substrate to diffuse to the now-exposed catalytic site of the enzyme.
Pancreatic lipase mainly hydrolyzes the ester bonds of TAGs at the first and third positions of the glycerol backbone. The end products of such reactions are two fatty acids and a single sn2-MAG (2-MAG) (Fig. 45-11E).
The pancreas secretes other enzymes that hydrolyze lipid esters. Carboxyl ester hydrolase is a pancreatic enzyme that is the same protein as bile salt–stimulated milk lipase. Like milk lipase, carboxyl ester hydrolase lacks substrate specificity and is active against a wide range of esters. Carboxyl ester hydrolase is probably the same enzyme as “pancreatic esterase,” “cholesterol esterase,” “lysophospholipase,” and others. Among the many products of reactions catalyzed by this enzyme are free cholesterol and free glycerol.
The pancreas also secretes phospholipase A2 (PLA2), which is active against glycerophospholipids (but not sphingolipids), from which it releases a single sn2-fatty acid to yield lysophospholipids (Fig. 45-11G). Pancreatic PLA2, secreted as a proenzyme, is effective at alkaline pH and requires bile salts for activity. PLA2 in the small intestine may also be derived from Paneth’s cells, whereas phospholipase found in the colon probably comes from anaerobic flora there. In contrast to the human intestinal lipases, bacterial lipases are nonspecific with respect to substrates, have neutral or slightly acidic pH optima, are not inhibited by bile acids, and do not require cofactors. In the human colon, both TAGs and phospholipids are totally hydrolyzed by bacteria. Fecal fat is thus generally present as fatty acid soaps and sterols. Even in severe fat malabsorption, intact acylglycerols are rarely found in the stools. The chemical test for stool fat, Sudan III staining, must be done in an acid environment because it depends on the property of Sudan III dye to partition into “oils” that contain protonated fatty acids.
After their secretion in pancreatic juice and bile, respectively, the various activated pancreatic lipases and biliary bile salts, lecithin, and cholesterol adsorb to the surface of the emulsion droplets arriving from the stomach (Fig. 45-12A). The lipolytic products—MAGs, fatty acids (including long-chain species from gastric lipolysis) that are now ionized at duodenal pH 5.5 to 6.5, lysolecithin, and cholesterol—act as additional emulsifiers. As surface TAGs are hydrolyzed, they are replaced by TAGs from the core of the emulsion particle. As the emulsion droplets become progressively smaller, their surface area increases, thereby increasing the rate of hydrolysis. Initially, a crystalline Ca2+-fatty acid soap phase forms near the surface of the TAG droplet, until the local free Ca2+ is depleted. At the same time, a multilamellar liquid crystalline layer of fatty acids, MAGs, lysolecithins, cholesterol, and possibly bile salts builds up on the surface of the emulsion particle. This liquid crystalline layer buds off as a multilamellar liquid crystal vesicle (Fig. 45-12B), which consists of several lipid bilayers. Bile salt micelles transform these multilamellar vesicles into unilamellar vesicles (Fig. 45-12C), which are single-lipid bilayers, and then into mixed micelles (Fig. 45-12D) composed of bile salts and mixed lipids (i.e., fatty acids, MAGs, lysophospholipids, and cholesterol).
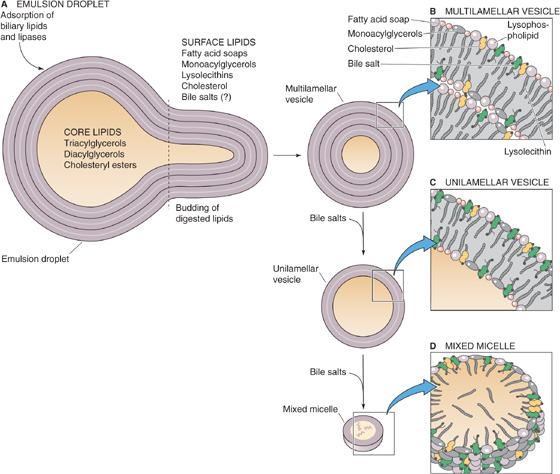
Figure 45-12 The breakdown of emulsion droplets to mixed micelles. A, The core of the emulsion droplet contains TAGs, diacylglycerols, and cholesteryl esters. On the surface are fatty acids, MAGs, lysolecithins, and cholesterol. Adsorbed to the surface are pancreatic lipase and possibly bile salts. As the lipases hydrolyze the TAGs at the surface, the TAGs from the core replace them, thus causing the droplet to shrink. B, A multilamellar liquid-crystalline layer of fatty acids, MAGs, lysolecithins, cholesterol, and bile salts builds up on the surface of the emulsion droplet and causes a small piece to bud off as a multilamellar vesicle. C, The addition of more bile salts to the multilamellar vesicle thins out the lipid coating and converts the multilamellar vesicle to a unilamellar vesicle. D, Further addition of bile salts leads to formation of a mixed micelle, in which hydrophobic lipid tails face inward and polar head groups face outward.
Continued digestion by PLA2 and other esterases can still occur on the mixture of aggregates now present. If intestinal contents taken from humans during fat digestion are centrifuged, three phases separate. An oily layer floats on top and contains fat droplets and lipolytic products that have not been solubilized by bile salts. A middle layer contains lipid vesicles, mixed lipid-bile salt micelles, simple bile salt micelles, and lipid monomers. Finally, a pellet contains debris and precipitated Ca2+ soaps of fatty acids. Whereas most fat absorption in health is from the micellar phase of digested lipid, in situations in which intraluminal bile salt concentrations are low (e.g., in newborns and patients with obstructive jaundice), lipid absorption can occur from vesicles.
To reach the interior of the enterocyte, lipolytic products must cross several barriers. These include (1) the mucous gel layer that lines the intestinal epithelial surface, (2) the unstirred water layer (disequilibrium zone) contiguous with the enterocyte’s apical membrane, and (3) the apical membrane itself. Although the mucous gel that lines the intestine is 95% water, its interstices provide a barrier to the free diffusion—from the bulk phase to the unstirred water layer—of lipid macroaggregates, particularly the various vesicles that exist in equilibrium with the mixed micelles and monomers. Because of this diffusion barrier, the unstirred water lying adjacent to the enterocyte’s apical membrane is not in equilibrium with the bulk phase of water in the lumen. According to calculations based on the diffusion of various probes under different experimental conditions and luminal fluid flow rates, it was originally estimated that the unstirred water layer was several hundred microns thick and posed a significant barrier to the diffusion of lipid nutrients to the enterocyte brush border. It is now thought that the unstirred water layer is likely only ~40 μm thick and does not constitute a major absorptive barrier. For short- and medium-chain fatty acids, which are readily soluble in water, diffusion of these monomers through the unstirred water layer to the enterocyte is efficient. As fatty acid chain length increases, the monomer’s solubility in water decreases, whereas its partitioning into micelles increases. It is true that the diffusion of a single monomer through the aqueous barriers is speedier than that of a single micelle or vesicle. However, mixed-lipid micelles act as a reservoir to give the aqueous solution such a high effective concentration of fatty acids and other lipid products that the diffusion of these micelles is the most efficient mechanism for bringing lipolytic products to the enterocytes. Calculations suggest that, compared with monomers dissolved in water, vesicular solubilization increases the “concentration” of long-chain fatty acids near the enterocyte’s brush border membrane by a factor of 100,000 and micellar solubilization by a factor of 1,000,000.
When the fatty acid/bile salt mixed micelles reach the enterocyte surface, they encounter an acidic microclimate generated by Na-H exchange at the brush border membrane. It is postulated that fatty acids now become protonated and leave the mixed micelle to enter the enterocyte, either by nonionic diffusion (see Chapter 36) of the uncharged fatty acid, or by collision and incorporation of the fatty acid into the cell membrane, or by carrier-mediated transport through fatty acid translocase (FAT/CD36) (Fig. 45-13). A plasma membrane fatty acid–binding protein (FABPpm) appears to enhance the translocation. Similarly, unesterified cholesterol and lysophospholipids must leave the micelle carrier to enter the enterocyte as monomers. Investigators have suggested that cholesterol derived from bile is better absorbed than is dietary cholesterol. After the entry of lipids into enterocytes, the remaining bile salts return to the lumen and are then absorbed passively throughout the small intestine and through active transport in the distal ileum (see Chapter 46). As with fatty acids, 2-MAGs, lysophospholipids, and cholesterol traditionally were assumed to enter the enterocyte by simple diffusion across the apical plasma membrane of the brush border villi. More recently, however, in addition to carriers for fatty acids, membrane proteins have been identified in both enterocytes and hepatocytes that may be responsible for the transfer of fatty acids, phospholipids, and cholesterol across their respective cell membranes. As well as providing a mechanism for facilitated or active absorption of the various products of lipid digestion, such carriers may yet be therapeutic targets for inhibiting lipid absorption. The drug ezetimibe, which lowers plasma cholesterol by interfering with its absorption and not its synthesis, appears to impede cholesterol uptake by enterocytes by inhibiting the brush border Niemann-Pick C-1–like-1 (NPC1L1) protein. NPC1L1 may yet prove to be the putative intestinal cholesterol transporter.
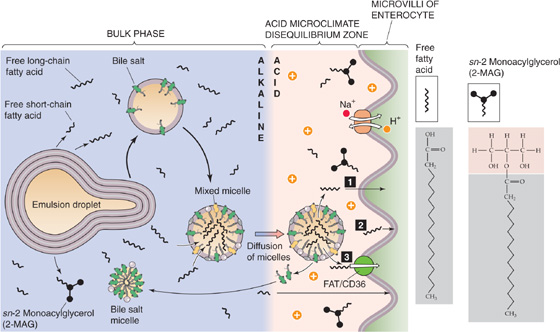
Figure 45-13 Micellar transport of lipid breakdown products to the surface of the enterocyte. Mixed micelles carry lipids through the acidic unstirred layer to the surface of the enterocyte. 2-MAG, fatty acids, lysophospholipids, and cholesterol leave the mixed micelle and enter an acidic microenvironment created by an apical Na-H exchanger. The acidity favors the protonation of the fatty acids. The lipids enter the enterocyte by (1) nonionic diffusion, (2) incorporation into the enterocyte membrane (collision), or (3) carrier-mediated transport.
The assimilation of fats, thus far, has been a process of disassembly of energy-dense, water-insoluble lipid macromolecular aggregates into monomers for intestinal absorption. The enterocyte elegantly reverses this process (Fig. 45-14). After absorbing long-chain fatty acids, MAGs, lysophospholipids, and cholesterol, the enterocyte re-esterifies them and assembles the products with specific apolipoproteins (or simply apoproteins) into emulsion-like particles called chylomicrons. The enterocyte then exports the chylomicrons to the lymph (chyle) for ultimate delivery to other organs through the bloodstream.
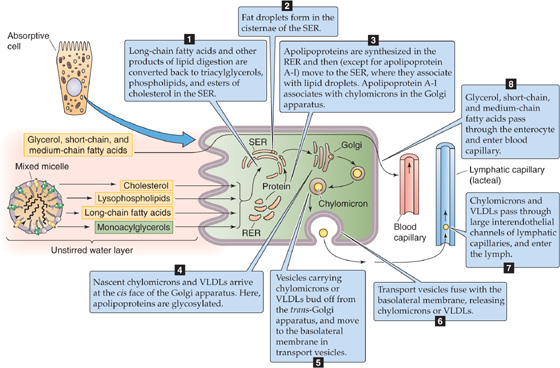
Figure 45-14 Re-esterification of digested lipids by the enterocyte and the formation and secretion of chylomicrons. The enterocyte takes up short- and medium-chain fatty acids and glycerol and passes them unchanged into the blood capillaries. The enterocyte also takes up long-chain fatty acids and 2-MAG and resynthesizes them into TAG in the SER. The enterocyte also processes cholesterol into cholesteryl esters and lysolecithin into lecithin. The fate of these substances, and the formation of chylomicrons, is illustrated by steps 1 to 8.
Chylomicrons are the largest of the five lipoprotein particles in the bloodstream. (The other lipoprotein particles—very-low-density lipoproteins (VLDLs), intermediate-density lipoproteins (IDLs), low-density lipoproteins (LDLs), and high-density lipoproteins (HDLs)—are discussed in Chapter 46 and Table 46-5). With an average diameter of ~250 nm (range, 75 to 1200 nm), chylomicrons consist primarily of TAGs, with smaller amounts of phospholipids, cholesteryl esters, cholesterol, and various apolipoproteins.
The first step in the enterocyte’s reformation of TAGs is for long-chain fatty acids to bind to a 12-kDa cytosolic protein called fatty acid–binding protein. The concentration of fatty acid–binding protein in the intestine is highest in regions that absorb fats, namely, the villi of the proximal jejunal enterocytes. Fatty acid–binding protein preferentially binds long-chain (rather than medium- or short-chain) fatty acids, thus minimizing both reflux back into the intestinal lumen and toxic damage to the enterocyte. Fatty acid–binding protein also ensures transfer of fatty acids to the smooth endoplasmic reticulum (SER) of the enterocyte. The re-esterification to form TAGs occurs within the SER. After a meal, enterocytes mainly use the MAG pathway to re-esterify absorbed fatty acids to absorbed 2-MAG. During fasting, enterocytes mainly use the phosphatidic acid pathway to esterify fatty acids that enter from the bloodstream. The necessary phosphatidic acid may arise either from glycerol-3-phosphate—itself derived from the metabolism of glucose or amino acids—or from the breakdown of bile lecithin that enters from the intestinal lumen. Both the MAG and the phosphatidic acid pathways depend on the activation of the fatty acid to acyl coenzyme A (acyl CoA), catalyzed by acyl CoA synthase. Long-chain fatty acids are the preferred substrate for this enzyme. The net effect of this series of reactions is the very rapid formation of TAGs in the SER, which maintains low fatty acid concentrations in the enterocyte. TAGs and fat droplets may be seen in the cisternae of the SER on electron microscopy. The enterocyte also esterifies both cholesterol and lysolecithin. (See Note: Regeneration of Triacylglycerols (TAGs) inside of Enterocytes; Regeneration of Triacylglycerols (TAGs) inside of Enterocytes; Acyl CoA Synthase; Intestinal Esterification of Cholesterol and Synthesis of Lecithin)
Besides the lipids, the other components of the chylomicron are the various apolipoproteins (see Table 46-5), which the enterocyte synthesizes in the rough endoplasmic reticulum (RER). These apolipoproteins, with the exception of apolipoprotein A-I, move to the lumen of the SER, where they associate with newly synthesized TAGs (Fig. 45-14). Apolipoprotein A-I associates with chylomicrons in the Golgi apparatus. Besides incorporating apolipoproteins, the packaging of nascent chylomicrons involves adding esterified cholesterol and a surface coating of lecithin and other phospholipids. It is thought that vesicles derived from the SER carry nascent chylomicrons to the cis face of the Golgi apparatus, where they fuse and deliver their contents internally. Enzymes in the Golgi apparatus glycosylate the apolipoproteins. Vesicles carrying processed chylomicrons bud off from the trans face of the Golgi and move toward the basolateral plasma membrane of the enterocyte. There they fuse with the basolateral membrane and leave the enterocyte.
The foregoing discussion focused on the digestion and absorption of long-chain TAGs. The handling of TAGs with medium-length fatty acid chains is very different. In the first place, the uptake into the enterocyte of fatty acids and MAGs derived from medium-chain TAGs does not depend on the presence of either mixed micelles or bile salts. Moreover, the enterocyte does not re-esterify the medium-chain fatty acids but instead transfers them directly into the portal blood. As a result, medium-chain TAGs are suitable fat substitutes for feeding patients with fat malabsorption.
As we have described, vesicles carrying mature chylomicrons discharge their contents outside the enterocyte through exocytosis at the basolateral membrane. Chylomicrons are too large to pass through the fenestrae of blood capillaries, and thus they enter lymph through the larger interendothelial channels of the lymphatic capillaries. In both the fed and fasted states, the intestine secretes VLDLs, which are smaller (30 to 80 nm) than chylomicrons. These lipoprotein particles are of similar protein and lipid composition to chylomicrons (see Table 46-5), but they are synthesized independently and carry mainly endogenous (as opposed to dietary) lipids. The lymph lacteals originate in the tips of the villi and discharge their contents into the cisternae chyli. Lymph flows from the cisternae chyli to the thoracic duct, to enter the blood circulation through the left subclavian vein. The protein and lipid composition of both chylomicrons and VLDLs are modified during their passage through lymph and on entry into the blood.
The process of lipid digestion and absorption has great reserve capacity and many redundancies. For example, the mechanical disruption of food is accomplished in several ways by the mouth, stomach, and proximal intestine. Many of the digestive lipases have overlapping functions, and pancreatic lipase, in particular, is secreted in great excess. Much of the small intestine is not used for fat absorption in healthy individuals. Nonetheless, fat malabsorption does occur in many disease states. A logical classification for these disorders can be devised based on knowledge of the normal physiology. Thus, fat malabsorption may occur because of impairments in intraluminal digestion, intraluminal dispersion, mucosal penetration, or transport from enterocyte to the blood circulation. Within each major category are subdivisions. Frequently, the pathophysiology of these disorders is mixed (i.e., more than one step in the digestive or absorptive process is deranged), but nonetheless the defective components can be identified, and appropriate therapy can be given.
Table 45-3 summarizes the fat-soluble vitamins A (see Chapter 15), D (see Chapter 52), E, and K (see Chapter 17). As a class, these vitamins rely on the lipid absorption process discussed in the preceding subchapter. Although individual fat-soluble vitamins, once digested and absorbed, have somewhat specific fates according to their chemical nature, they have numerous overlapping physical properties that determine their similar handling in the intestinal lumen, and uptake and processing by enterocytes. In contrast to their water-soluble counterparts (discussed later), fat-soluble vitamins do not form classical coenzyme structures or prosthetic groups with soluble apoproteins. Fat-soluble vitamins can also be stored in fat depots in the body. Each of the fat-soluble vitamins is really a family of related compounds, some of which are esters.
Table 45-3 Vitamins
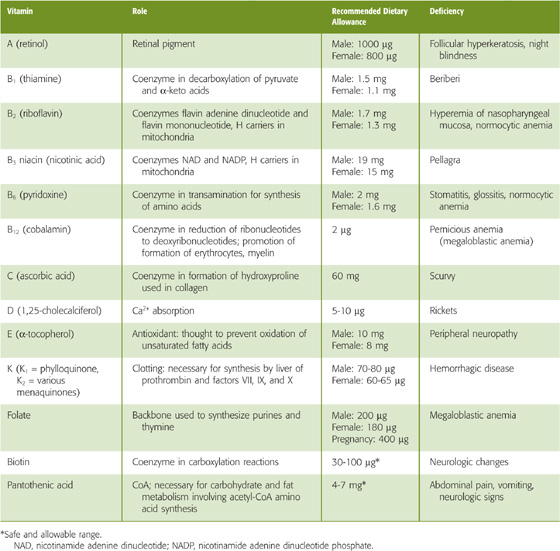
After ingestion, fat-soluble vitamins are released from their association with proteins by the acidity of gastric juice or by proteolysis. In addition, carboxyl ester hydrolases (found in pancreatic juice and in the mucosal brush border) liberate free vitamins from their esters. In the proximal small intestine, fat-soluble vitamins incorporate with other lipid products into emulsion droplets, vesicles, and mixed micelles, which ferry them to the enterocyte surface for uptake. The absorption efficiency of fat-soluble vitamins varies from 50% to 80% for A, D, and K to only 20% to 30% for vitamin E. Other ingestants—including dietary components and drugs and their carrier vehicles—can modify the absorption of fat-soluble vitamins. For example, high doses of vitamin A impair the absorption of vitamins E and K, whereas high doses of vitamin E enhance the absorption of vitamin A.
Enterocytes take up fat-soluble vitamins by simple diffusion or through transporters. After entry into the enterocyte, fat-soluble vitamins diffuse to the SER either as free molecules or attached to carrier proteins, such as a cellular retinol-binding protein in the case of vitamin A or retinol. In the SER, the vitamins associate with lipid droplets that form nascent chylomicrons and VLDLs, which then translocate through the Golgi and secretory vesicles for exocytosis into lymph. During passage through the enterocyte, vitamin A and tiny amounts of vitamin D are esterified with long-chain fatty acids, but vitamin E and K are not. Once in the systemic blood circulation, the fat-soluble vitamins A, D, E, and K enter the liver by receptor-mediated endocytosis of chylomicrons or remnant chylomicrons, as discussed in Chapter 46.
Fat-soluble vitamin deficiency occurs in various fat malabsorption states, including those induced by malabsorptive bariatric surgery, drugs (e.g., orlistat) that impair TAG hydrolysis, drugs (e.g., cholestyramine) that bind bile acids, and reduction of bile acids by impaired hepatobiliary function or by unabsorbable dietary fat substitutes. Fat-soluble vitamin deficiency can also result from impaired hepatic function. The consequences can include blindness and other irreversible eye disorders (vitamin A), bone demineralization and resorption (vitamin D), neurologic, neuromuscular and erythrocyte aberrations (vitamin E), and both hemorrhagic and hypercoagulable states (vitamin K). (See Note: Bariatric Surgery)
Treatment of fat-soluble vitamin deficiency includes water-miscible emulsions of vitamins A and E, which can enter enterocytes without special handling in the intestinal lumen. These compounds then move into portal blood together with small amounts of the more polar forms of some of the vitamins, such as retinoic acid in the case of vitamin A and menadione in the case of vitamin K.
Folate is also referred to as folic acid, or pteroylmonoglutamate (PteGlu1). As we discuss later, the reduced form of folate—tetrahydrofolate (THF)—is a cofactor in biochemical reactions involving the transfer of 1-carbon fragments. The recommended dietary allowance (RDA) for folate is 200 μg for men and 180 μg for women (Table 45-3), but it is more than doubled in pregnant women (see Chapter 56). THF is essential for the synthesis of thymine and purines, which are critical components of DNA. Thus, folate deficiency compromises DNA synthesis and cell division, an effect that is most clinically noticeable in the bone marrow, where the turnover of cells is rapid. Because RNA and protein synthesis are not impaired, large red blood cells called megaloblasts are produced. The resultant megaloblastic anemia can become quite severe if untreated. Megaloblastic cells also may be seen in other organs with rapid cell turnover, such as the small intestine. Folic acid supplementation during pregnancy also reduces the risk of neural tube defects.
The medicinal form of folate is PteGlu1, a monoglutamate. Figure 45-15A shows the structure of PteGlu1 and also illustrates how folate can act as a methyl acceptor or donor in the interconversion of serine to glycine. Dietary folate exists in several forms, much of it as folate polyglutamate, or PteGlu7 (Fig. 45-15B), which is widely available in the diet, particularly in spinach, beans, and liver. The intestinal absorption of PteGlu7 requires deconjugation by a brush border peptidase to PteGlu1, which then enters the enterocyte through a transporter (Fig. 45-15C). This deconjugation is catalyzed by folate conjugase, a zinc-activated exopeptidase present in the brush border. This enzyme removes glutamate residues from PteGlu7 in a stepwise fashion before absorption of PteGlu1. This stepwise hydrolysis of the polyglutamate chain of PteGlu7 is the rate-limiting step in folate digestion-absorption. Both folate deconjugation and absorption occur only in the proximal small intestine and are maximally active at a pH of 5.
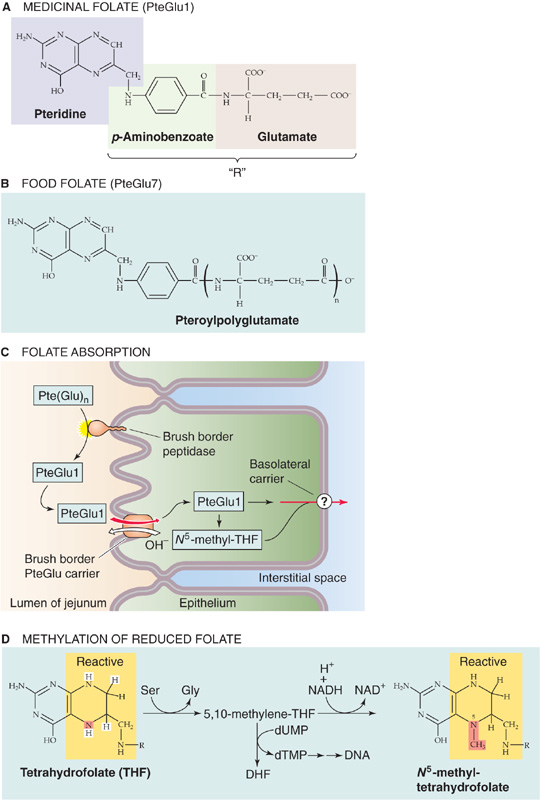
Figure 45-15 Folate deconjugation and absorption. A, Tetrahydrofolate has three parts: the biologically active pteridine moiety, a p-aminobenzoate, and a glutamate. PteGlu1 is the oxidized form of folate and is biologically inactive. B, Dietary folate is similar to medicinal folate but has several glutamate residues. PteGlu7 is also oxidized and inactive. C, In the proximal small intestine, a brush border peptidase sequentially removes all but the last of the glutamate residues from dietary folate. The enterocyte then absorbs the resulting PteGlu1 using a folate-OH exchanger. Once inside the enterocyte, the PteGlu1 exists across the basolateral membrane through an unknown transporter. The enterocyte may reduce some of the PteGlu1 to DHF and then to THF, the biologically active form of folate. The enterocyte may then methylate some of the THF to form N5-methyl-THF. D, After the cell has reduced PteGlu1 to THF by adding the four highlighted hydrogens, it first converts THF to 5, 10-methylene-THF, thus breaking down serine to glycine in the process. This 5, 10-methylene-THF is the methyl donor in the conversion of the nucleotide deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) in the synthesis of DNA. A second reaction converts this 5, 10-methylene-THF to N5-methyl-THF, which can then act as a methyl donor in the synthesis of methionine (see Fig. 45-16B). NAD, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide (reduced form).
PteGlu1 absorption is saturable, shows substrate specificity, and is markedly enhanced at an acid pH. Folate absorption represents an apical membrane anion exchange process, in which folate uptake is linked to the efflux of OH− across the apical membrane (i.e., folate-OH exchange). The mechanism of folate movement out of the epithelial cell across the basolateral membrane is not understood.
The PteGlu1 taken up by the enterocyte is not biologically active. The enzyme difolate reductase acts on PteGlu1 to first form dihydrofolate (DHF) and then the biologically active derivative THF. The cell then converts THF to 5, 10-methylene THF (Fig. 45-15D), the form of folate needed for DNA synthesis. The cell also can transform this 5, 10-methylene THF to N5-methyl THF, which—as we discuss in the next section—can act as a methyl donor in the synthesis of methionine. The circulating, storage, and active forms of folate constitute various reduced (DHF and THF) and methylated derivatives of THF. The liver is the primary site at which dietary pteroylglutamates are reduced and methylated, although the intestinal epithelium may make a small contribution to these reactions.
Cobalamin, or vitamin B12 (Fig. 45-16A), is synthesized only by microorganisms, not by mammalian cells. The primary source of cobalamin in humans is the ingestion of animal products—meat, fish, shellfish, eggs, and (to a limited extent) milk. Cobalamin is not present in vegetables or fruit. Therefore, strict vegetarians are at risk of developing dietary cobalamin deficiency.
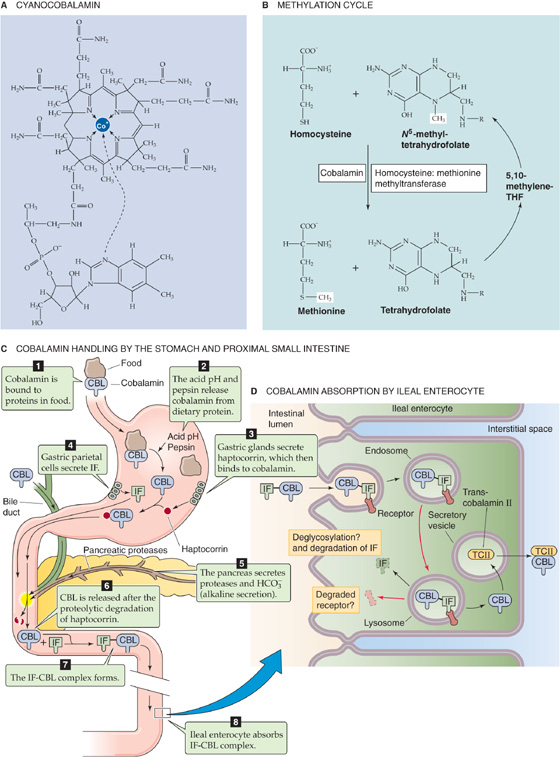
Figure 45-16 Cobalamin and the role of IF in the absorption of cobalamin. A, Cyanocobalamin. B, Methylation cycle. Cobalamin is the coenzyme for the enzyme homocysteine : methionine methyltransferase, which transfers a methyl group from N5-methyltetrafolate to homocysteine, thereby forming methionine and tetrahydrofolate. C, Steps 1 to 8 show the fate of dietary cobalamin (CBL). Steps 4 to 8 show the role of IF. In addition, bile carries cobalamin into the duodenum. D, The intrinsic-factor/cobalamin complex is thought to be endocytosed. The cobalamin is liberated within the enterocyte by mechanisms that have not been established. Within the enterocyte, cobalamin binds to transcobalamin II (TCII), which is required for cobalamin’s exit from the enterocyte.
Cobalamin’s primary function is to serve as a coenzyme for homocysteine : methionine methyltransferase (Fig. 45-16B), which transfers a methyl group from methyltetrafolate to homocysteine, thereby converting homocysteine to methionine. Methionine is an essential amino acid and in an altered form serves as an important donor of methyl groups in several important enzymatic reactions. If cobalamin is deficient and methionine levels fall, then the body converts its stores of intracellular folate (e.g., PteGlu1, THF, 5, 10-methylene THF) into N5-methyl THF (Fig. 45-15D) in an effort to produce more methionine. As a result, 5, 10-methylene THF (the form of folate needed for DNA synthesis) falls, an effect that explains why folate and cobalamin deficiencies cause identical hematologic abnormalities (i.e., megaloblastic anemia). In addition, cobalamin deficiency causes various neurologic and psychological abnormalities that are not part of the syndrome of folate deficiency. Some of these abnormalities may be linked to deficient activity of methylmalonyl CoA mutase, another cobalamin-dependent coenzyme.
Cobalamin reaches the stomach bound to proteins in ingested food. In the stomach, pepsin and the low gastric pH release the cobalamin from the ingested proteins (Fig. 45-16C). The now-free cobalamin binds to haptocorrin (formerly known as “R” type binder), a glycoprotein secreted by the salivary and gastric glands. The parietal cells of the stomach secrete a second protein, intrinsic factor (IF), crucial for the absorption of cobalamin. However cobalamin and IF do not interact in the acidic milieu of the stomach. Rather, gastric acidity enhances the binding of cobalamin to haptocorrin. When this cobalamin-haptocorrin complex reaches the duodenum, the haptocorrin is degraded by pancreatic proteases (Fig. 45-16C).
After the release of cobalamin from the cobalamin-haptocorrin complex in the proximal small intestine—made alkaline by the secretion of HCO−3 from the pancreas and duodenum—both dietary cobalamin and cobalamin derived from bile bind to IF. The cobalamin-IF complex is highly resistant to enzyme degradation. As noted earlier, the gastric parietal cells secrete IF, a 45-kDa glycoprotein. Histamine, acetylcholine, and gastrin stimulate gastric acid secretion (see Chapter 42), and they also stimulate IF secretion. Although IF secretion parallels proton secretion, three important differences exist. First, similar to pepsinogen secretion, histamine triggers an IF release that peaks within minutes and then continues at a reduced rate. This secretory pattern is related to the secretion of preformed IF; histamine has no effect on IF synthesis. Second, although cAMP is important in IF secretion, a role for intracellular Ca2+ has not yet been established. Third, H2 histamine-receptor antagonists block IF secretion, but omeprazole, an inhibitor of the parietal cell H-K pump, does not affect IF secretion.
The next step in the absorption of cobalamin is the binding of the cobalamin-IF complex to specific receptors on the apical membranes of enterocytes in the ileum. Cobalamin without IF neither binds to ileal receptors nor is absorbed. The binding of the cobalamin-IF complex is selective and rapid and requires Ca2+, but it is not energy dependent. The enterocyte next internalizes the cobalamin-IF complex in a process that is energy dependent but has not been well characterized. Inside the cell, cobalamin and IF dissociate; lysosomal degradation may play a role here. Within the enterocyte, cobalamin binds to another transport protein—transcobalamin II—which is required for cobalamin’s exit from the enterocyte. The cobalamin exits the ileal enterocyte across the basolateral membrane bound to transcobalamin II, possibly by an exocytotic mechanism. The transcobalamin II-cobalamin complex enters the portal circulation, where it is delivered to the liver for storage and for secretion into the bile.
Total body cobalamin stores are large (~5 mg), particularly when compared with the daily rate of cobalamin absorption and loss. (The daily cobalamin requirement for normal adults is only 2 micrograms.) The load of cobalamin presented to the small intestine is derived about equally from two sources: the diet and biliary secretions. The latter is the result of the enterohepatic circulation (see Chapter 46) of cobalamin; after its absorption, cobalamin is delivered throughout the body, and the excess is secreted by the liver into the bile, where it once again can be reabsorbed by the small intestine and recirculated.
Cobalamin deficiency has many possible causes. As already mentioned, a strict vegetarian diet is deficient in cobalamin. In pernicious anemia, a disorder seen primarily in elderly persons, the absence of gastric parietal cells results in the absence of gastric acid and IF secretion. Consequently, cobalamin absorption is markedly reduced, and cobalamin deficiency develops. Other causes of cobalamin deficiency include related problems in the intestine. Bacterial overgrowth in the small intestine as a result of stasis (e.g., multiple jejunal diverticulosis) can be associated with cobalamin deficiency as a consequence of bacterial binding and metabolism of cobalamin. Crohn disease affecting the ileum and ileal resection are other possible causes of cobalamin malabsorption and deficiency as a result of an absence of ileal receptors for the cobalamin-IF complex.
Pernicious Anemia
The close relationship between acid and gastrin release is clearly manifested in individuals with impaired acid secretion. In pernicious anemia, atrophy of the gastric mucosa in the corpus and an absence of parietal cells result in the lack in the secretion of both gastric acid and IF. Many patients with pernicious anemia exhibit antibody-mediated immunity against their parietal cells, and many of these patients also produce anti-IF autoantibodies.
Because IF is required for cobalamin absorption in the ileum, the result is impaired cobalamin absorption. In contrast, the antrum is normal. Moreover, plasma gastrin levels are markedly elevated as a result of the absence of intraluminal acid, which normally triggers gastric D cells to release somatostatin (see Chapter 42), which, in turn, inhibits antral gastrin release (see Chapter 42 for the box titled Gastrinoma or Zollinger-Ellison Syndrome). Because parietal cells are absent, the elevated plasma gastrin levels are not associated with enhanced gastric acid secretion.
The clinical complications of cobalamin deficiency evolve over a period of years. Patients develop megaloblastic anemia (in which the circulating red blood cells are enlarged), a distinctive form of glossitis, and neuropathy. The earliest neurologic findings are those of peripheral neuropathy, as manifested by paresthesias and slow reflexes, as well as impaired senses of touch, vibration, and temperature. If untreated, the disease will ultimately involve the spinal cord, particularly the dorsal columns, thus producing weakness and ataxia. Memory impairment, depression, and dementia can also result. Parenteral administration of cobalamin reverses and prevents the manifestations of pernicious anemia, but it does not influence parietal cells or restore gastric secretion of either IF or intraluminal acid.
The physiological importance and complex regulation of Ca2+ and vitamin D are discussed in Chapter 52. The Ca2+ load presented to the small intestine comprises dietary sources and digestive secretions. Most of the dietary Ca2+ (~1000 mg/day) comes from milk and milk products (see Fig. 52-1), but not all of it is bioavailable. For example, only very little of the Ca2+ present in leafy vegetables is absorbed because of the concomitant presence of oxalate, a salt that binds Ca2+ and reduces its availability for absorption. The small intestine absorbs ~500 mg/day of Ca2+, but it also secretes ~325 mg/day of Ca2+. Thus the net uptake is ~175 mg/day.
Active, transcellular uptake of Ca2+ occurs only in the epithelial cells of the duodenum, but Ca2+ is absorbed by passive, paracellular diffusion throughout the small intestine. More Ca2+ is absorbed in the jejunum and ileum by diffusion than in the duodenum by active transport; this difference arises largely because the duodenum has a smaller total surface area and because the flow of Ca2+-containing fluid through the duodenum is faster (Fig. 45-17).
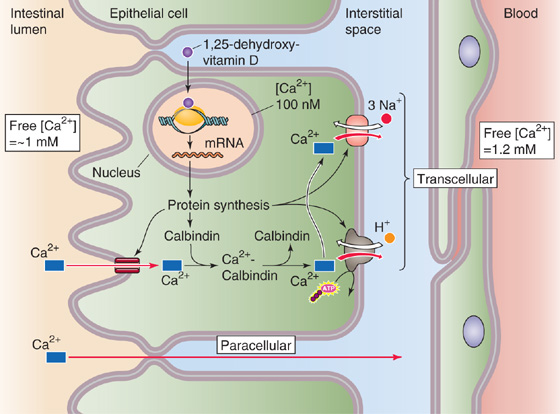
Figure 45-17 Active Ca2+ uptake in the duodenum. The small intestine absorbs Ca2+ by two mechanisms. The passive, paracellular absorption of Ca2+ occurs throughout the small intestine. This pathway predominates, but it is not under the control of vitamin D. The second mechanism—the active, transcellular absorption of Ca2+—occurs only in the duodenum. Ca2+ enters the cell across the apical membrane through a channel. Inside the cell, the Ca2+ is buffered by binding proteins, such as calbindin, and is also taken up into intracellular organelles, such as the endoplasmic reticulum. The enterocyte then extrudes Ca2+ across the basolateral membrane through a Ca2+ pump and an Na-Ca exchanger. Thus, the net effect is Ca2+ absorption. The active form of vitamin D (1,25 dihydroxyvitamin D) stimulates all three steps of transcellular Ca2+ absorption.
The active transport of Ca2+ across the villous epithelial cells of the duodenum is transcellular and is under the control of vitamin D—primarily through genomic effects (see Chapter 4). Transcellular Ca2+ absorption involves three steps. The uptake of Ca2+ across the apical membrane occurs through Ca2+ channels, driven by the electrochemical gradient between the lumen and the cell. Cytosolic Ca2+ then binds to a protein called calbindin, which buffers intracellular Ca2+. This step is important because it allows unbound (i.e., free) intracellular Ca2+ to remain rather low despite large transcellular fluxes of Ca2+. A Ca2+ pump and an Na-Ca exchanger on the basolateral membrane then extrude the Ca2+ from the cell into the interstitial fluid. The active form of vitamin D—1,25-dihydroxyvitamin D—stimulates all three steps of the transcellular pathway, but its most important effect is to enhance the second step by increasing the synthesis of calbindin.
The passive absorption of Ca2+ throughout the small intestine occurs through the paracellular pathway, which is not under the control of vitamin D. Ca2+ absorption is also enhanced by low plasma [Ca2+] and during pregnancy and lactation. Absorption tends to diminish with aging.
Vitamin D itself is a fat-soluble vitamin that is absorbed mainly in the jejunum. Of course, the skin synthesizes vitamin D3 from cholesterol in a process that requires ultraviolet light (see Chapter 52). Thus, dietary vitamin D (both vitamin D2 and D3) is most important in regions of the world that do not receive much sunlight and during long, dark winters.
Mg2+ is an important intracellular ion that is required as an enzyme cofactor—many enzymes using ATP actually require that the ATP be complexed with Mg2+—and is critical for neurotransmission and muscular contractions. Mg2+ deficiency can affect neuromuscular, cardiovascular, and gastrointestinal function. Mg2+ is also important for the proper secretion of, and end-organ response to, parathyroid hormone. Thus, Mg2+ depletion is typically associated with hypocalcemia.
Hemochromatosis
Hereditary hemochromatosis (HH) is a relatively common inherited disorder (3 to 5 persons per 1000 of northwestern European ancestry are homozygous) in which the body absorbs excessive iron from the diet. This autosomal recessive disease becomes clinically significant only in homozygotes. If left untreated, HH can be fatal. The excess iron is stored in the liver, where it reaches toxic concentrations. Cirrhosis eventually results, and the risk of hepatoma is greatly increased. Iron also ultimately accumulates in other organ systems, where it causes pancreatic damage (diabetes mellitus), bronze pigmentation of the skin, pituitary and gonadal failure, arthritis, and cardiomyopathy. The disease hardly ever becomes apparent before the individual enters the third decade of life. Women are relatively protected as long they are premenopausal, because their monthly menstrual flow keeps total body iron stores relatively normal.
The diagnosis can be made by detection of elevated iron and transferrin-saturation levels and elevated ferritin. A liver biopsy is confirmatory.
Treatment, reminiscent of the medieval approach to disease, is to remove (phlebotomize) one or even several units of blood (1 unit = 500 mL) from the patient on a regular basis until the iron overload is corrected, as evidenced by normal plasma ferritin levels. Afterward, most patients require phlebotomy only once every few months to maintain their low iron stores.
The HFE gene that is associated with HH is related to the major histocompatibility complex (MHC) class I family. More than 80% of patients with HH are homozygous for a missense mutation (C282Y) in HFE. However, the role of this mutated gene in hemochromatosis is not yet clear because the penetrance of hemochromatosis in individuals with the C282Y allele is very low (<5%). Attention has also focused on hepcidin, a recently identified hepatic protein that appears to play a critical role in the regulation, by body iron stores, of duodenal iron absorption.
Because increased body iron stores reduce iron absorption, hemochromatosis represents an inappropriately high rate of iron absorption relative to body iron stores. Because hepcidin downregulates both DMT1 expression and iron absorption, hemochromatosis may represent a defect in the regulation of hepcidin, but the relationship between the HFE gene and hepcidin is not known. It is possible that a mutated HFE gene causes an inappropriately low hepatic hepcidin expression, thus resulting in hemochromatosis.
The relationship between hepcidin and iron absorption has also been identified in the anemia of inflammation, caused by a reduction in duodenal iron absorption. In this setting, the cytokine interleukin-6 stimulates hepcidin expression, which, in turn, results in decreased DMT1 expression, ferroportin activity, and iron absorption.
Mg2+ is widely available in different foods but is present in particularly large amounts in green vegetables, cereals, and meats. The RDA for Mg2+ (Table 45-3) in young adults is ~350 mg/day for men and ~280 mg/day for women. The Mg2+ load to the small intestine is derived from both dietary sources and digestive secretions.
Mg2+ absorption by the gastrointestinal tract is not yet well understood, but it appears to differ substantially from the absorption of the other key divalent cation, Ca2+, in three important respects. First, an active transport process for Mg2+ absorption appears to exist in the ileum, rather than in the duodenum, as is the case for Ca2+. Second, 1,25-dihydroxyvitamin D does not consistently increase Mg2+ absorption. Third, patients with increased intestinal Ca2+ absorption (e.g., absorptive hypercalciuria) have normal Mg2+ absorption. Along with active Mg2+ absorption in the ileum, the rest of the small intestine absorbs Mg2+ passively.
Iron plays several critical roles in human physiology, both in the heme groups of the cytochromes and as a key component of the oxygen-carrying heme moieties of hemoglobin and myoglobin. The most important complication of iron depletion is anemia. Iron overload produces hemochromatosis, a not uncommon genetic disease (see the box titled Hemochromatosis).
Dietary iron takes two major forms: iron that is part of a heme moiety and iron that is not. These two types of dietary iron are absorbed by distinctly different mechanisms (Fig. 45-18). Overall iron absorption is low; 10% to 20% of ingested iron is absorbed. Heme iron is absorbed more efficiently than nonheme iron. Body stores of iron depend almost exclusively on iron absorption because no regulated pathway for iron excretion exists. Except in menstruating women, who require ~50% more iron in their diets, very little iron is lost from the body. Dietary iron comes primarily from meat—especially liver and fish—as well as vegetables. The RDA for iron (Table 45-3) in young adults is ~10 mg/day for men and ~15 mg/day for women.
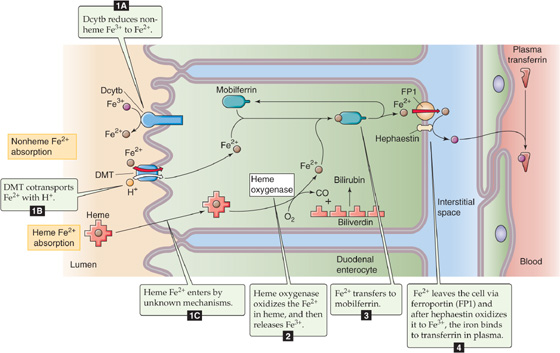
Figure 45-18 Absorption of nonheme and heme iron in the duodenum. The absorption of nonheme iron occurs almost exclusively as Fe2+, which crosses the duodenal apical membrane through DMT1, driven by a H+ gradient, which is maintained by Na-H exchange. Heme enters the enterocyte by an unknown mechanism. Inside the cell, heme oxygenase releases Fe3+, which is then reduced to Fe2+. Cytoplasmic Fe2+ then binds to mobilferrin for transit across the cell to the basolateral membrane. Fe2+ probably exits the enterocyte through basolateral ferroportin. The ferroxidase activity of hephaestin converts Fe2+ to Fe3+ for carriage in the blood plasma bound to transferrin.
Nonheme Iron Nonheme iron may be either ferric (Fe3+) or ferrous (Fe2+). Ferric iron tends to form salt complexes with anions quite easily and thus is not readily absorbed; it is not soluble at pH values higher than 3. Ferrous iron does not complex easily and is soluble at pH values as high as 8. Ascorbic acid (vitamin C) forms soluble complexes with iron and reduces iron from the ferric to the ferrous state, thereby enhancing iron absorption. Tannins, present in tea, form insoluble complexes with iron and lower its absorption.
Iron movement does not occur passively but requires one or more proteins to facilitate its movement into and out of cells (especially enterocytes, hepatocytes, and macrophages), as well as for intracellular binding. The absorption of nonheme iron is restricted to the duodenum. The enterocyte takes up nonheme iron across the apical membrane through the divalent metal transporter DMT1 (SLC11A2), which cotransports Fe2+ and H+ into the cell (see Chapter 5). DMT1, as well as the oligopeptide cotransporter that we discussed earlier, is unusual in being energized by the inwardly directed H+ gradient. DMT1 also efficiently absorbs a host of other divalent metals, including several that are highly toxic (e.g., Cd2+, Pb2+). In the case of dietary ferric iron, the ferric reductase Dcytb—which is related to cytochrome b—presumably reduces Fe3+ to Fe2+ at the extracellular surface of the apical membrane before uptake through DMT1.
Fe2+ moves into the cytoplasm of the enterocyte, where it binds to mobilferrin, an intracellular protein that ferries the Fe2+ to the basolateral membrane. The enterocyte then translocates the Fe2+ across the basolateral membrane, possibly through ferroportin transporter (FP1, also known as IREG1). The mRNA encoding FP1 has an iron-responsive element (see Chapter 4) in its 5′ untranslated region; thus, an increase in intracellular iron levels would be expected to decrease FP1 synthesis. Following the exit of Fe2+ from the enterocyte through FP1, the ferroxidase hephaestin—a homologue of the plasma protein ceruloplasmin, which carries copper (see Chapter 46)—apparently oxidizes the Fe2+ to Fe3+, which then binds to plasma transferrin (see Chapter 2) for carriage in the blood.
Once in the circulation, nonheme iron bound to transferrin is ultimately deposited in all the tissues of the body, but it has a particular predilection for the liver and reticuloendothelial system. Inside these cells, it binds to the protein apoferritin to form ferritin, the major storage form of iron. Smaller amounts of storage iron exist in an insoluble form called hemosiderin.
Heme Iron Derived from myoglobin and hemoglobin, heme iron is also absorbed by duodenal epithelial cells. Heme iron enters the cells either by binding to a brush border protein or through an endocytotic mechanism. Inside the cell, heme oxygenase enzymatically splits the heme iron, thus releasing free Fe3+, CO, and biliverdin (see Fig. 46-6). The cell reduces the biliverdin to bilirubin, which the liver eventually excretes in bile (see Chapter 46 for the box on jaundice). The enterocyte reduces the Fe3+ to Fe2+, which it then handles in the same manner as nonheme iron.
Iron absorption is tightly regulated by the size of existing body iron stores. In physiologically normal subjects, iron absorption is limited but is markedly increased in states of iron deficiency, caused most often by gastrointestinal bleeding or excessive menstrual flow. For example, the expressions of DMT1 and FP1 increase in iron deficiency. Conversely, an increase in iron stores modestly reduces iron absorption.
The molecular mechanisms by which iron stores regulate iron absorption are incompletely understood but play a role in the pathophysiology of hemochromatosis. Recent attention has focused on a 25–amino acid peptide secreted by hepatic Kupffer’s cells, hepcidin, which appears to down-regulate duodenal iron absorption by regulating DMT1 activity as well as other proteins in the iron-responsive pathways (e.g., FP1). Hepcidin likely is a negative regulator of iron absorption because mice that fail to express hepcidin have elevated body iron stores, whereas mice with enhanced hepcidin expression have profound iron deficiency.
Nutritionists recommend that the daily intake of carbohydrate versus fat should not differ with age, gender, or activity level. Of the total caloric intake in a Western diet, 55% to 60% is typically carbohydrate, 25% to 30% is fat, and the remaining 10% to 15% is protein.
The requirements for total caloric intake vary among individuals and depend on certain factors, including a person’s ability to use and store energy (efficiency) and the daily activity level. Differences in the efficiency of energy use among individuals are the result, in part, of variations in muscle mass but also of genetic factors. Because adipose tissue has a low metabolic rate, people with a large fat mass require less caloric intake per kilogram of body weight. Stated differently, the requirement for energy intake is greater per kilogram of lean body mass than per kilogram of total body mass (which includes fat). Thus, men generally require a greater daily caloric intake per kilogram of body weight than do women, who have relatively less muscle and more fat.
Activity level is the primary factor determining the daily energy requirement, assuming a stable body weight. For example, in a steady state, athletes must consume more than nonathletes not only because of a higher muscle-to-fat ratio but also because of a higher energy expenditure. Manual labor necessitates greater energy intake than does sedentary activity, again to maintain energy balance and body weight. Excess intake over output causes weight gain over time. Using the conversion of 9.4 kcal/g, 1 kg of fat stores 9400 kcal, sufficient energy to carry the BMR for 4 days.
Eating a low-carbohydrate diet (e.g., the Atkins diet) leads to an accelerated breakdown of tissue protein and thus to the wasting of muscle and other vital tissues, as well as the breakdown of fat. The breakdown of both protein and fat leads to an accumulation of ketone bodies in the blood (from the conversion of amino acids and fatty acids to acetyl CoA).
In addition to serving as a source of energy, fatty acids are also important for membrane structure (see Chapter 2), as well as signal transduction by pathways such as those involving diacylglycerols and arachidonic acid (see Chapter 3). In mammalian cells, fatty acid synthase produces two major fatty acids: palmitic and oleic acids. Palmitic acid, which has 16 carbons and is fully saturated, is referred to as 16 : 0. Oleic acid has 18 carbons and a single cis double bond between carbons 9 and 10. This unsaturated fatty acid is referred to as 18 : 1 cis-Δ9. Because mammals cannot insert double bonds beyond carbon 9, they need two fatty acids in the diet: linoleate (18 : 2 cis-Δ9 Δ12) and linolenate (18 : 3 cis-Δ9 Δ12 Δ15). These essential fatty acids serve as precursors for other unsaturated fatty acids, including arachidonic acid, which is a precursor to prostaglandins, leukotrienes, and thromboxanes.
The current recommendations favoring low fat intake are based on the view that a high fat intake is associated with chronic diseases such as atherosclerosis and non–insulin-dependent diabetes (see Chapter 51 for the box on sulfonylureas). However, fats have a positive side. In addition to the roles we have discussed in this chapter, fats enhance satiety and aid in the absorption of certain vitamins. Finally, the so-called ω-3 polyunsaturated fatty acids—in which the first double bond is three carbons from the terminal methyl group (or “ω carbon”)—appear to protect against cardiovascular disease and some forms of cancer.
The diet must contain the nine essential amino acids (see Table 58-2) because the body cannot synthesize them (see Chapter 46). Eleven other amino acids are necessary for protein synthesis, but the body can synthesize their carbon skeletons from intermediates of carbohydrate metabolism. Vegetarian diets can meet the protein needs of the body, provided the protein contains adequate amounts of essential amino acids. Food protein is “scored” based on its content of essential amino acids, compared with that of a reference protein, usually egg protein, which is given a score of 100. For example, a food containing protein with a score of 40 for threonine, 80 for phenylalanine, and 100 for lysine—all three of which are essential amino acids—receives a protein score of 40 because, relative to the standard, threonine is present in the lowest amount.
Protein intake is most important to meet the needs for tissue maintenance and repair, for muscle and neural function, and to maintain host defense mechanisms. The daily requirement for protein intake depends on one’s nutritional status. The average human needs ~0.6 g of protein per kilogram of body weight per day to maintain nitrogen balance. The RDA of protein is ~0.8 g/kg of body weight for adults, ~1 g/kg for adolescents, and ~2 g/kg in the first 6 months of life. Pregnant and lactating women require extra protein intake to ensure adequate fetal development and milk production. Athletes require more than 1 g/kg to maintain a greater lean body mass and to fuel a highly active metabolism. Well-balanced but larger meals usually provide adequate protein for those with a greater need. Burn victims, patients recovering from surgery, and patients with disorders of protein absorption all require increased daily protein intake.
The distribution of amino acids required for protein accretion by growing infants and children is different than that for tissue maintenance. Moreover, the requirements change throughout development. The child uses 25% of amino acid intake for protein accretion at 6 months of age but only 10% by 18 months of age. More than 40% of a child’s protein intake must consist of essential amino acids versus only 20% for an adult.
Proteins play a key role in host defenses. For example, proteins provide the structural backbone for skin and mucus. Protein synthesis is essential for phagocytes and lymphocytes that are responsible for antibody and cell-mediated immunity. The skin, lungs, and intestinal tract are the main structural defenses against invading organisms. In both the lungs and the gastrointestinal tract, mucus (containing glycoproteins) coats the surface of these passageways and aids in defending against disease by catching most foreign particles. Protein-depleted individuals, regardless of age, have impaired immune competence. Protein depletion limits the availability of amino acids for synthesis of the cellular proteins of the immune system, including glutathione, mucus glycoproteins, and metallothreonine. The acute-phase response to invading organisms is suboptimal in a protein-deficient state.
The impaired immune competence of patients with AIDS is a function of poor nutrition in addition to the effects of the virus itself. During infection, the body mobilizes amino acids to synthesize proteins for defense against invading organisms. Thus, improving protein and energy intake may be beneficial for some patients with AIDS.
Aside from being the backbone of proteins, amino acids play a variety of physiological roles. For example, arginine is a precursor to nitric oxide (see Chapter 3). Glutamate is a major excitatory neurotransmitter in the brain, whereas glycine is a major inhibitory neurotransmitter (see Chapter 13). Glutamine is a major source of NH3 production in the kidney (see Chapter 39), and it also regulates protein turnover in muscle. The decrease in muscle glutamine concentration that occurs during trauma and infection is associated with a general decline of muscle function. Research on anorectic patients shows that the most important factor affecting muscle function is insufficient nutrient intake. Increasing total nutrient intake in these subjects by total parenteral nutrition increases muscle function before having a measurable effect on muscle mass.
Vitamins and minerals do not provide energy but are essential for such functions as metabolism, immune competence, muscle force production, and blood clotting.
Minerals Table 45-4 lists the essential minerals. The current recommendations for daily mineral intake are based on a mix of balance studies and usual dietary intakes in the United States. The recommendation for copper, for example, is based on balance studies, whereas those for manganese, chromium, and molybdenum are based on dietary intakes.
Table 45-4 Essential Minerals
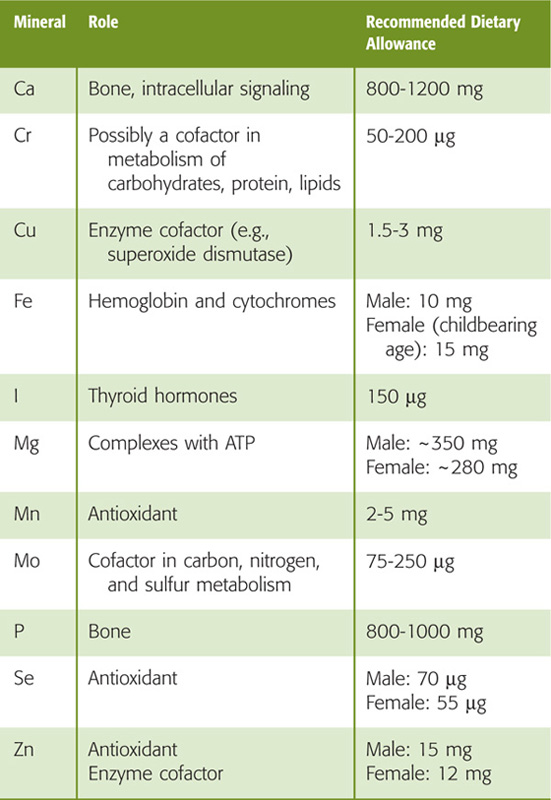
Assigning daily mineral intakes is problematic because some methods of determining mineral status do not always expose functional deficiencies. For example, a frank deficiency in Ca2+ intake leads to bone loss even though blood Ca2+ levels remain normal. Iron deficiency is difficult to detect because no clinical signs appear until iron stores are depleted. It is difficult to base recommendations simply on absorption because, for some minerals, absorption varies with intake. For example, copper deficiency or toxicity is unlikely in humans because absorption is inversely related to intake. Furthermore, interactions among minerals must be taken into account when establishing recommendations.
The goal is to base recommendations on scientific evidence. Radioisotopes can be used to monitor storage, absorption, and excretion. Another approach is to establish the physiological role of the mineral and then determine the mineral intake required for maintaining that physiological role. However, because of redundancy in function, it is often difficult to assess which mineral is deficient when function is compromised. For example, both iron and copper are involved in energy metabolism at the level of the electron transport chain. The blood clotting cascade involves Ca2+, copper, and vitamin K. Zinc, selenium, and manganese all have antioxidant activities.
Vitamins Even though vitamins are not energy sources themselves, they play an integral role as cofactors in many metabolic processes. Some vitamins are involved in group transfer reactions, such as decarboxylations and carboxylations in fatty acid and glucose metabolism and transaminations in amino acid metabolism. Vitamins act as oxidizing and reducing agents in the generation of ATP, as well as antioxidants to quench free radicals produced as a byproduct of oxidation.
Of the 13 identified vitamins, RDAs have been established for 11 (Table 45-3). Safe and allowable ranges are estimated for the remaining two—biotin and pantothenic acid. Recommendations differ widely among countries. Lower recommendations are generally based on scientific evidence. For each vitamin, a person’s nutritional status falls into one of five categories: deficient, marginal, satisfactory, excessive, and toxic. Although “marginal” and “excessive” are not usually associated with overt clinical signs, people whose vitamin status falls into one of these categories are at increased risk of various diseases.
In the past, recommendations for the intake of vitamins and minerals were based largely on levels necessary to promote normal growth and development. However, the role of vitamins and minerals in optimizing body function and in promoting longevity is becoming an area of intense interest both to researchers and to the general public. Older people generally have a less vigorous immune response than do young people, in large part because of deficiencies in iron, zinc, and vitamin C. Correcting these deficiencies improves immune competence significantly. In addition, older people with poor dietary habits may have inadequate intake of Ca2+, vitamin D, and other nutrients involved in bone deposition and strength (see Chapter 52), thus putting them at greater risk for hip fracture.
Mineral deficiencies usually do not occur without extreme abnormalities in diet, and even then, a mineral deficiency may not impair function (Table 45-4). However, deficiencies of almost every vitamin can cause functional impairment (Table 45-3).
A current controversy surrounds the use of so-called mega-doses of certain vitamins and minerals. Such excessive intake has mixed effects. Slight excesses of vitamins A and E, zinc, and selenium are associated with an enhanced immune response, especially for patients with burns, trauma, and sepsis. In these conditions, “excess” intake may not be an excess at all but rather the intake that meets the greater need. Increased intake of fruits and vegetables—which contain a variety of vitamins as well as “fiber”—clearly decreases the risk of various cancers. However, efforts to link these effects specifically to dietary carotenoid levels have failed to show a correlation; indeed, the excessive intake of β-carotene supplements may even increase the risk of some cancers. Antioxidants (e.g., β-carotene, vitamins C and E) quench peroxyl radicals, suppress tumor growth, and decrease atherosclerotic lesions in rabbits. Enhanced vitamin E intake lowers the risk of coronary heart disease, by nearly one half.
The excessive intake of certain minerals and vitamins may compromise the immune response. Excess vitamin E intake in infants may increase the risk of infection, possibly by quenching superoxide radicals that are important for killing bacteria. Excess lipids can impair the immune response, too. High intake of saturated and polyunsaturated fatty acids leads to decreases in cell-mediated immunity. Excessive Ca2+ intake interferes with the ability to use iron, zinc, and Mg2+, whereas high dietary copper affects zinc absorption and excretion.
Because the kidneys readily excrete water-soluble vitamins in the urine, toxicity from excessive intake is not common. Because fat-soluble vitamins are not easily excreted in the urine, it is easier to develop toxicity for these vitamins. In particular, polar bear liver, a component of the Inuit diet, contains extraordinarily high levels of vitamin A (35,000 IU/g versus the RDA of 5000 IU), which can lead to acute hypervitaminosis A and death.
Books and Reviews
Daniel H: Molecular and integrative physiology of intestinal peptide transport. Annu Rev Physiol 2004; 66:361-384.
Farrell JJ: Digestion and absorption of nutrients and vitamins. In Feldman M, Friedman LS, Sleisenger MH (ed): Gastrointestinal and Liver Disease, vol 2, 7th ed. Philadelphia: WB Saunders, 2002, pp 1715-1750.
Hentze MW, Muckenthaler MU, Andrews NC: Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004; 117:285-297.
Palacin M, Estevez R, Bertran J, Zorzano A: Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev 1998; 78:969-1054.
Verrey F, Ristic Z, Romeo E, et al: Novel renal amino acid transporters. Annu Review Physiol 2005; 67:557-572.
Wright EM: The intestinal Na+/glucose cotransporter. Annu Rev Physiol 1993; 55:575-589.
Journal Articles
Clarke DC, Miskovic D, Han XX, et al: Overexpression of membrane-associated fatty acid binding protein (FABPpm) in vivo increases fatty acid sarcolemmal transport and metabolism. Physiol Genomics 2004; 17:31-37.
Feder JN, Gnirke A, Thomas W, et al: A novel MHC class I–like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13:399-408.
Fei YJ, Kanai Y, Nussberger S, et al: Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature 1994; 368:563-566.
Gunshin H, Mackenzie B, Berger UV, et al: Cloning and characterization of a proton-coupled mammalian metal ion transporter. Nature 1997; 388:482-488.
Shiau YF: Mechanisms of intestinal fatty acid uptake in the rat: The role of an acidic microclimate. J Physiol 1990; 421:463-474.