CHAPTER 37
Gerhard Giebisch and Erich Windhager
The distribution of K+ in the body differs strikingly from that of Na+. Whereas Na+ is largely extracellular, K+ is the most abundant intracellular cation. Some 98% of the total body K+ content (~50 mmol/kg body weight) is inside cells; only 2% is in the extracellular fluid (ECF). The body tightly maintains the plasma [K+] at 3.5 to 5.0 mM.
Table 37-1 summarizes the most important physiological functions of K+ ions. A high [K+] inside cells and mitochondria is essential for maintaining cell volume, for regulating intracellular pH and controlling cell-enzyme function, for DNA and protein synthesis, and for cell growth. The relatively low extracellular [K+] is necessary for maintaining the steep K+ gradient across cell membranes that is largely responsible for the membrane potential of excitable and nonexcitable cells. Therefore, changes in extracellular [K+] can cause severe disturbances in excitation and contraction. As a general rule, either doubling the normal plasma [K+] or reducing it by half results in severe disturbances in skeletal and cardiac muscle function. The potentially life-threatening disturbances of cardiac rhythmicity that result from a rise of plasma [K+] are particularly important (see the box titled Hyperkalemia and the Heart).
Table 37-1 Physiological Role of K+ Ions
A. Roles of Intracellular K+ |
|
Cell-volume maintenance |
Net loss of K+ → cell shrinkage |
|
Net gain of K+ → cell swelling |
Intracellular pH regulation |
Net loss of K+ → cell acidosis |
|
Net gain of K+ → cell alkalosis |
Cell enzyme functions |
K+ dependence of enzymes (e.g., some ATPases, succinic dehydrogenase) |
DNA/protein synthesis, growth |
Lack of K+ → reduction of protein synthesis, stunted growth |
B. Roles of Transmembrane [K+] Ratio |
|
Resting cell membrane potential |
Reduced [K+]i/[K+]o → membrane depolarization |
|
Increased [K+]i/[K+]o → membrane hyperpolarization |
Neuromuscular activity |
Low plasma K+: muscle weakness, muscle paralysis, intestinal distention, respiratory failure |
|
High plasma K+: increased muscle excitability; later, muscle weakness (paralysis) |
Cardiac activity |
Low plasma K+: slowed conduction of pacemaker activity, arrhythmias |
|
High plasma K+: conduction disturbances, ventricular arrhythmias, and ventricular fibrillation |
Vascular resistance |
Low plasma K+: vasoconstriction |
|
High plasma K+: vasodilation |
Chronic K+ depletion leads to several metabolic disturbances. These include the following: (1) inability of the kidney to form concentrated urine; (2) a tendency to develop metabolic alkalosis; and, closely related to this acid-base disturbance, (3) a striking enhancement of renal ammonium excretion (see Chapters 38 and 39).
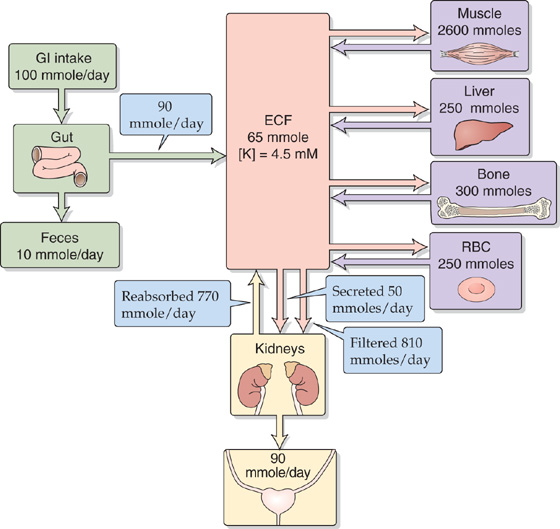
Figure 37-1 illustrates processes that govern K+ balance and the distribution of K+ in the body: (1) gastrointestinal (GI) intake, (2) renal and extrarenal excretion, and (3) the internal distribution of K+ between the intracellular and extracellular fluid compartments. The first two processes accomplish external K+ balance (i.e., body versus environment), whereas the last achieves internal K+ balance (i.e., intracellular versus extracellular fluids).
Figure 37-1 Distribution and balance of K+ throughout the body. Intracellular K+ concentrations are similar in all tissues in the four purple boxes. The values in the boxes are approximations. RBC, red blood cell.
External K+ Balance The relationship between dietary K+ intake and K+ excretion determines external K+ balance. The dietary intake of K+ is approximately equal to that of Na+, 80 to 120 mmol/day. This K+ intake is more than the entire K+ content of the ECF, which is only ~70 mmol. For the plasma K+ content to remain constant, the body must excrete K+ through renal and extrarenal mechanisms at the same rate as K+ ingestion. Moreover, because dietary K+ intake can vary over a wide range, it is important that these K+ excretory mechanisms be able to adjust appropriately to variable K+ intake. The kidney is largely responsible for K+ excretion, although the GI tract plays a minor role. The kidneys excrete 90% to 95% of the daily K+ intake; the colon excretes 5% to 10%. Although the colon can adjust its K+ excretion in response to some stimuli (e.g., adrenal hormones, changes in dietary K+, decreased capacity of the kidneys to excrete K+), the colon—by itself—is incapable of increasing K+ secretion sufficiently to maintain external K+ balance.
Internal K+ Balance Maintaining normal intracellular and extracellular [K+] requires not only the external K+ balance just described, but also the appropriate distribution of K+ within the body. Most of the K+ is inside cells—particularly muscle cells, which represent a high faction of body mass—with smaller quantities in liver, bone, and red blood cells. The markedly unequal distribution between the intracellular and extracellular K+ content has important quantitative implications. Of the total intracellular K+ content of ~3 mol, shuttling as little as 1% to or from the ECF would cause a 50% change in extracellular [K+], with severe consequences for neuromuscular function (Table 37-1).
What happens when the body is presented with a K+ load? By far, the most common source of a K+ load is dietary K+. When one ingests K+ salts, both the small intestine and the colon (see Chapter 44) absorb the K+. In addition to external sources of K+, substantial amounts of K+ may enter the ECF from damaged tissues (see the box titled Clinical Implications: Evaluating Hypokalemia and Hyperkalemia in the Patient). Such K+ release from intracellular to extracellular fluid can lead to a severe, even lethal, increase in plasma [K+] (i.e., hyperkalemia). However, even a large meal presents the body with a K+ load that could produce hyperkalemia if it were not for mechanisms that buffer and ultimately excrete this K+.
Some four fifths of an ingested K+ load temporarily moves into cells, so that plasma [K+] rises only modestly, as shown in the upper panel of Figure 37-2. Were it not for this translocation, plasma [K+] could reach dangerous levels. The transfer of excess K+ into cells is rapid and almost complete after an hour (lower panel of Fig. 37-2, gold curve). With a delay, the kidneys begin to excrete the surfeit of K+ (lower panel of Fig. 37-2, brown curve), leaching from the cells the excess K+ that they had temporarily stored.
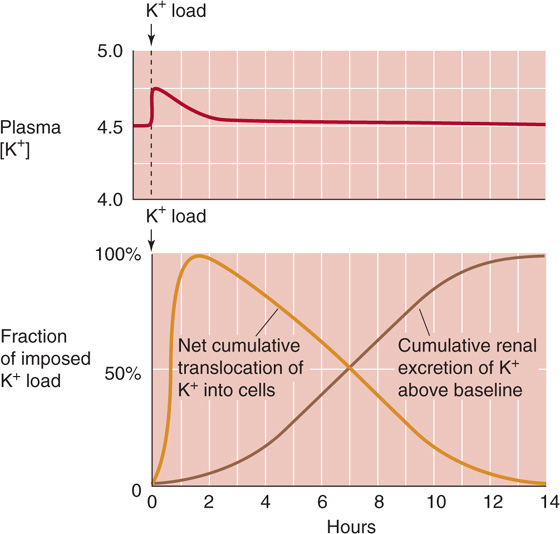
Figure 37-2 K+ handling following an acute K+ load. (Data from Cogan MG: Fluid and Electrolytes: Physiology and Pathophysiology. Norwalk, CT: Appleton & Lange, 1991.)
What processes mediate the temporary uptake of K+ into cells during K+ loading? As shown in Figure 37-3, the hormones insulin, epinephrine (a β-adrenergic agonist), and aldosterone all promote the transfer of K+ from extracellular to intracellular fluid through the ubiquitous Na-K pump. Indeed, the lack of insulin or a deficient renin-angiotensin-aldosterone system can significantly compromise tolerance to K+ loading and can predispose to hyperkalemia. Similarly, administering β-adrenergic blockers (in treatment of hypertension) impairs sequestration of an acute K+ load. (See Note: Hormonal Response to Acute K+ Loading)
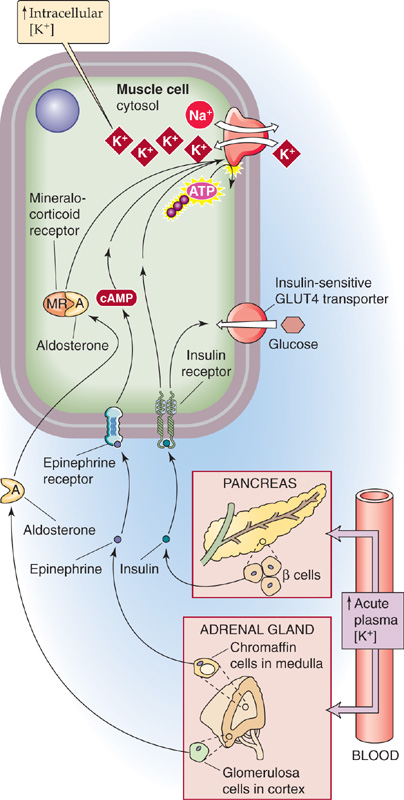
Figure 37-3 K+ uptake into cells in response to high plasma [K+].
Acid-base disturbances also affect internal K+ distribution. As a rule, acidemia leads to hyperkalemia as tissues release K+. One can think of this K+ release as an “exchange” of intracellular K+ for extracellular H+, although a single transport protein generally does not mediate this “exchange” (see Chapter 28). Extracellular acidosis probably leads to loss of cellular K+ because the resulting decrease in intracellular pH lessens the binding of K+ to nondiffusible intracellular anions (Fig. 37-4). Moreover, intracellular acidosis compromises both the Na-K pump and the Na/K/Cl cotransporter (NKCC2), both of which move K+ into cells. Interestingly, for the same degree of acidemia, mineral acids produce a greater degree of hyperkalemia than do organic acids.
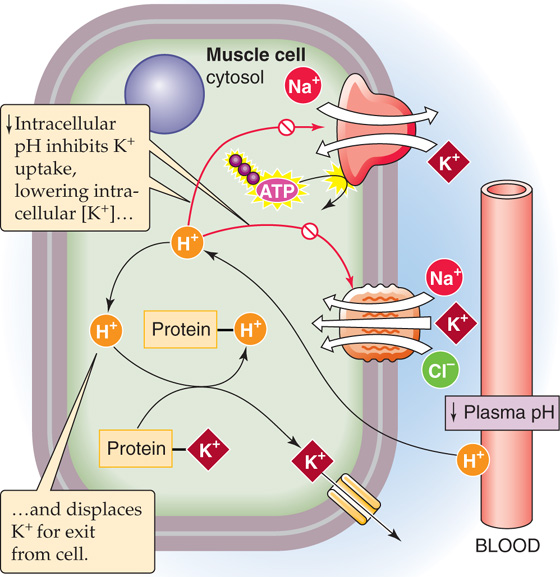
Figure 37-4 Effect of acidosis on K+ uptake into cells.
Alkalemia causes cells to take up K+, thus leading to hypokalemia. Although the mechanism is not known, high HCO−3 levels—even in the absence of extracellular pH changes—induce hypokalemia by stimulating K+ transfer into cells. Another example of this apparent exchange of K+ for H+ is the effect of K+ on acid-base homeostasis. As discussed in Chapters 28 and 39, K+ depletion causes intracellular acidosis and extracellular alkalosis.
In some clinical conditions, an increase of the extracellular osmolality induces the transfer, not only of water, but also of K+ into the extracellular space. The rise in plasma K+ following infusion of hypertonic mannitol solutions is an example of this phenomenon.
At a normal glomerular filtration rate (GFR) and at physiological levels of plasma [K+], the kidney filters ~800 mmol/day of K+, far more than the usual dietary intake of 80 to 120 mmol/day. Therefore, to achieve K+ balance, the kidneys normally need to excrete 10% to 15% of the filtered K+. Under conditions of low dietary K+ intake, the kidneys excrete 1% to 3% of filtered K+, so that—with a normal-or low-K+ diet—the kidneys could achieve K+ balance by filtration and reabsorption alone. Considering only the filtered K+ load and external K+ balance, we would have no reason to suspect that the kidneys would be capable of K+ secretion. However, with a chronically high dietary K+, when the kidneys must rid the body of excess K+, urinary K+ excretion may exceed 150% of the total amount of filtered K+. Therefore, even if the tubules reabsorbed none of the filtered K+, they must be capable of secreting an amount equivalent to at least 50% of the filtered K+ load.
As discussed later, even in the absence of a large dietary K+ load, K+ secretion by the tubules is an important component of urinary K+ excretion. Therefore, K+ handling is a complex combination of K+ filtration at the glomerulus as well as both K+ reabsorption and secretion by the renal tubules.
Figure 37-5 summarizes the pattern of K+ transport along the nephron under conditions of low or normal/high K+ intake. In either case, the kidney filters K+ in the glomerulus and then extensively reabsorbs it along the proximal tubule (~80%) and the loop of Henle (~10%), so that only ~10% of the filtered K+ enters the distal convoluted tubule (DCT). Moreover, in either case, the medullary collecting duct (MCD) reabsorbs K+. The K+ handling depends critically on dietary K+ in five nephron segments: the DCT, the connecting tubule (CNT), the initial collecting tubule (ICT), the cortical collecting tubule (CCT), and the MCD.
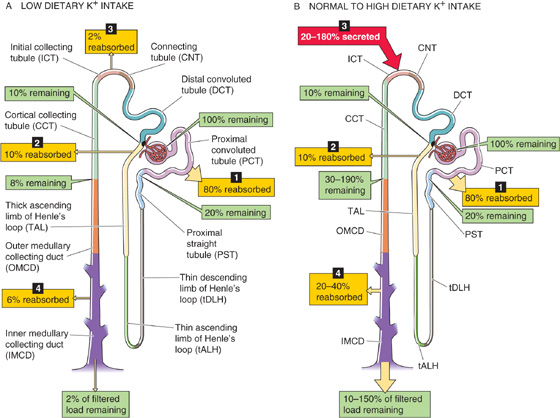
Figure 37-5 K+ handling along the nephron. In A and B, the numbered yellow boxes indicate the fraction of the filtered load that various nephron segments reabsorb, whereas the red box in B indicates the fraction of the filtered load secreted by the ICT and CCT. The green boxes indicate the fraction of the filtered load that remains in the lumen after these segments.
Low Dietary K+ When the body is trying to conserve K+, the classic distal tubule (i.e., DCT, CNT, and ICT) and CCT all reabsorb K+, so that only a small fraction of the filtered load (1% to 3%) appears in the urine (Fig. 37-5A). In states of K+ depletion, this additional K+ reabsorption can be lifesaving, by retrieving from the tubule lumen precious K+ that escaped reabsorption along the proximal tubule and loop of Henle. Despite the degree to which the kidneys can enhance K+ reabsorption, they cannot restrict K+ loss in the urine as effectively as they can restrict Na+ loss. Therefore, a negative K+ balance and hypokalemia may develop when K+ intake has been abnormally low for prolonged periods of time.
Normal or High Dietary K+ When external K+ balance demands that the kidneys excrete K+, the ICT, CCT, and the more proximal portion of the MCD secrete K+ into the tubule lumen (Fig. 37-5B). Together, these segments, known as the distal K+ secretory system, account for most of the urinary excretion of K+. It is also this distal K+ secretory system that responds to many stimuli that modulate K+ excretion. Even at normal rates of K+ excretion (10% to 15% of the filtered load), the proximal tubules and loop of Henle first absorb very large amounts of K+ (~90% of the filtered load), so that the K+ appearing in the urine may largely represent K+ secreted by more distal segments of the nephron.
The kidney traps K+ in the medullary interstitium, with the interstitial [K+] being highest at the tip of the papilla and falling toward the cortex. This medullary K+ trapping is the result of three steps along the nephron. First, because interstitial [K+] rises toward the tip of the papilla, juxtamedullary nephrons, whose long loops of Henle dip into the inner medulla (see Chapter 33), secrete K+ passively into the thin descending limb of the loop of Henle (tDLH). Indeed, analysis of fluid collected from the hairpin bend of the long loops of Henle of juxtamedullary nephrons shows that the amount of K+ delivered to the collection site at the hairpin bend can exceed not only the amount of K+ present at the end of the proximal tubule, but also the amount of K+ filtered. This K+ secretion by the tDLH is the first step of a process known as medullary K+ recycling (Fig. 37-6).
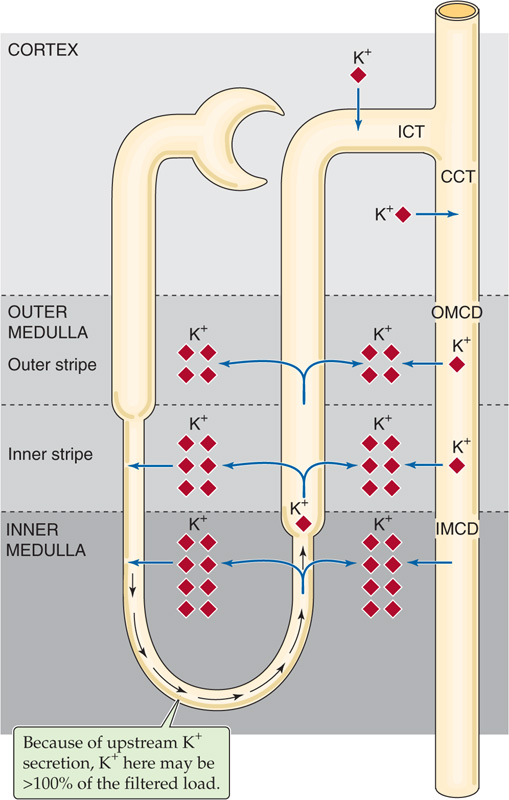
Figure 37-6 Medullary recycling of K+ by juxtamedullary nephrons.
The second step of medullary K+ recycling is K+ reabsorption by the thin (tALH) and thick (TAL) ascending limbs, which deposit K+ in the medullary interstitium. This newly deposited interstitial K+ contributes to the high interstitial [K+]. Together, the tALH and TAL of a juxtamedullary loop reabsorb more K+ than the descending limb secretes, so that net K+ reabsorption occurs along the loop, thereby contributing to medullary K+ trapping.
The third step of medullary K+ recycling is the reabsorption of K+ by the MCDs. Regardless of whether the distal K+ secretory system (ICT, CCT, early MCD) reabsorbs K+ (Fig. 37-5A) or secretes K+ (Fig. 37-5B), the MCDs reabsorb some K+, thereby contributing to medullary K+ trapping.
One would think that—with respect to K+ excretion—medullary K+ recycling is inefficient because K+ exits the ascending limb and MCD only to re-enter the nephron upstream, in the tDLH. However, medullary recycling and concomitant K+ trapping may be important in maximizing the excretion of K+ when K+ intake is high. Under these conditions, K+ secretion by the distal K+ secretory system is intense, so that luminal [K+] in the MCD may rise to 200 mM or higher. Thus, enhanced K+ trapping in the medullary interstitium minimizes the [K+] difference between the MCD lumen and its peritubular environment, thus reducing the passive loss of K+ from the MCD.
The proximal tubule reabsorbs most of the filtered K+ through two paracellular mechanisms (Fig. 37-7A): solvent drag and electrodiffusion. Solvent drag (see Chapter 20) occurs along the entire length of the proximal tubule. Active Na+ transport (see Chapter 35) drives net fluid reabsorption across the proximal tubule epithelium through the paracellular shunt pathway, thereby entraining K+ along with the fluid. Indeed, one of the distinguishing features of proximal tubule K+ reabsorption is its strong dependence on net fluid reabsorption. Thus, interventions that depress fluid reabsorption almost always inhibit K+ reabsorption.
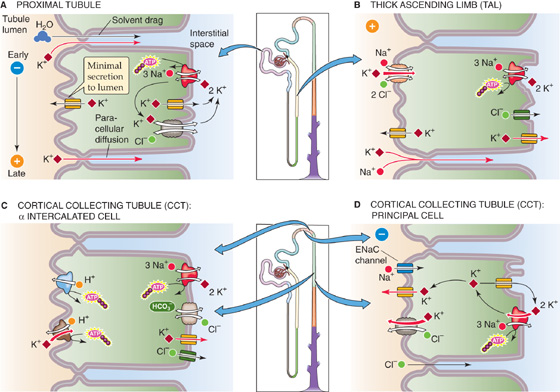
Figure 37-7 Cellular models of K+ transport along the nephron.
As fluid flows down the proximal tubule, the luminal voltage shifts from negative to positive (see Chapter 35). By the late proximal tubule, the transepithelial voltage is sufficiently positive to provide a favorable gradient for K+ electrodiffusion (see Chapter 6) through the low-resistance paracellular pathway, from lumen to blood.
Although the proximal tubule reabsorbs K+ by paracellular pathways, the proximal tubule has several cellular pathways for K+ movement that do not directly participate in K+ reabsorption (Fig. 37-7A): (1) a basolateral Na-K pump, a feature common to all tubule cells; (2) apical and basolateral K+ channels; and (3) a basolateral K/Cl cotransporter (KCC).
The K+ conductance of the basolateral membrane greatly exceeds that of the apical membrane, and it appears that different channels are responsible for the K+ conductances in these two membranes. The open probability of the basolateral K+ channel increases sharply with the turnover rate of the Na-K pump. Accordingly, most of the K+ taken up by the Na-K pump recycles across the basolateral membrane by K+ channels and a KCC and does not appear in the lumen. This basolateral K+ channel belongs to the class of inwardly rectifying K+ channels (ROMK) characterized by ATP sensitivity: a decrease in [ATP]i enhances channel activity (see Chapter 7). This regulation by [ATP]i is most likely involved in the coupling between basolateral Na-K pumps and K+ channels. An increase in pump rate—which could occur when increased apical Na+ entry increases [Na+]i—would lower [ATP]i, relieving inhibition of the K+ channels. Were it not for this crosstalk between the basolateral Na-K pump and apical K+ channels, the K+ content of proximal tubule cells would fluctuate dramatically during alterations in net Na+ transport.
In contrast to the high state of activity of the basolateral K+ channels, apical K+ channels are largely quiescent under normal conditions. They appear to become active, however, when tubule cells swell, possibly when entry of Na+ from the lumen increases rapidly. These channels, which may be activated by membrane stretch, then allow K+ to leave the cell, a process that causes cells to shrink toward their original volume (a volume-regulatory decrease; see Chapter 5).
Even if the apical K+ channels were open more often, uptake of K+ across the apical membrane would not occur, because the electrochemical K+ gradient favors K+ movement from cell to lumen. Because K+ cannot enter across the apical membrane, no transcellular reabsorption of K+ can take place. Thus, proximal tubule K+ reabsorption must occur exclusively by the paracellular pathway.
As discussed earlier, the tDLHs of the loop of Henle, particularly those originating from juxtamedullary nephrons, secrete K+ (Fig. 37-6). This secretion of K+ from the medullary interstitium into the tDLH lumen is passive, driven by the high [K+] of the medullary interstitium and made possible by a substantial K+ permeability. Both the tDLH lumen and the interstitium become increasingly K+ rich toward the tip of the papilla.
In the tALH, K+ moves from lumen to interstitium passively, by the paracellular pathway. The major driving force for this passive reabsorption of K+ is the lumen-to-interstitium K+ gradient, which becomes progressively larger as the fluid moves toward the cortex because interstitial [K+] becomes progressively lower toward the cortex.
The TAL of the loop of Henle reabsorbs K+ by both paracellular and transcellular mechanisms (Fig. 37-7B). The lumen-positive voltage, together with the relatively high paracellular K+ permeability, allows passive K+ reabsorption to occur by the paracellular pathway. The lumen-positive voltage develops because of a substantial difference in the ion permeabilities of the apical and basolateral membranes. The apical membrane is K+ selective, so that the apical membrane potential depends mainly on the cell-to-lumen [K+] gradient. In contrast, the TAL basolateral membrane is permeable to both K+ and Cl−. Hence, the basolateral membrane potential lies between the equilibrium potentials of Cl− (~–50 mV) and K+ (~–90 mV), so that it is less negative than if the basolateral membrane were permeable only to K+. Because the apical membrane potential is more negative than the basolateral membrane potential, the transepithelial voltage is lumen positive. This potential provides a substantial driving force for the passive, paracellular reabsorption of cations such as K+ and Na+. Half of the K+ reabsorbed by the TAL exits by this paracellular pathway, with the lumen-positive potential being the main driving force. (See Note: Electrical Profile across an Epithelial Cell in the Thick Ascending Limb)
The other half of K+ reabsorption by the TAL occurs by a transcellular pathway, using the apical NKCC2 (see Chapter 5). The major evidence for such a mechanism is the mutual interdependence of Na+, Cl−, and K+ transport. Removing any of the three cotransported ions from the lumen abolishes transcellular K+ reabsorption. However, because inhibiting the NKCC2 also abolishes the lumen-positive potential (see Chapter 35), this inhibition also blocks passive, paracellular K+ reabsorption.
The apical NKCC2 is a typical example of a “secondary” active transporter (see Chapter 5). Expressed in electrical terms, the apical membrane potential is some 10 to 12 mV more positive than the K+ equilibrium potential, so that K+ would tend to diffuse passively from cell to lumen. Because the combined inward gradients of Na+ and Cl− across the apical membrane greatly exceed that of K+, the energy is adequate for the coupled uptake of all three ion species. Ultimately, a primary active transporter (i.e., Na-K pump) at the basolateral membrane drives the secondary active transport of K+ in the apical membrane. Inhibiting the basolateral Na-K pump leads to an increase in [Na+]i and [Cl−]i, so that apical Na/K/Cl transport ceases. A characteristic feature of the NKCC2 is its sensitivity to a class of diuretics that have their main site of action in the TAL. Therefore, administering so-called loop diuretics such as furosemide or bumetanide blocks net reabsorption of Na+, Cl−, and K+.
The apical K+ conductance mentioned earlier—which mainly represents a K+ channel called ROMK—is also important for the function of the NKCC2 (Fig. 37-7B). The major function of the apical K+ channel is to provide a mechanism for recycling much of the K+ from cell to lumen, so that luminal [K+] does not fall so low as to jeopardize Na/K/Cl cotransport (see Chapter 35). Nevertheless, some of the K+ entering the cell by the NKCC2 exits across the basolateral membrane, thereby accounting for transcellular K+ reabsorption. The presence of apical K+ channels also explains why inhibiting Na/K/Cl cotransport—either with loop diuretics (e.g., furosemide) or by deleting from the lumen of any of the cotransported ions—causes the TAL to engage in net K+ secretion. Active K+ uptake no longer opposes the K+ leak from the cell to lumen.
Ultrastructurally, the ICT and CCT are nearly identical (see Chapter 33); they are made up of ~70% principal cells (which secrete K+) and ~30% intercalated cells (some of which reabsorb K+). As discussed earlier, the ICT, CCT, and MCD reabsorb K+ in response to K+ depletion (Fig. 37-5A). This K+ reabsorption is a transcellular process (Fig. 37-7C), mediated by α intercalated cells, a subpopulation of intercalated cells that are interspersed among the principal cells. K+ reabsorption occurs in two steps: (1) an active step mediated by an apical ATP-driven H-K pump (see Chapter 5), similar to that present at the apical membrane of parietal cells in gastric glands (see Chapter 42); and (2) a passive step mediated by a basolateral K+ channel, which allows K+ to leak out. The α intercalated cells are also responsible for H+ secretion (see Chapter 39). In K+ depletion, a condition in which the abundance of H-K pumps increases dramatically, is often associated with accelerated secretion of H+ and the development of hypokalemic alkalosis.
The early portion of the classic distal tubule (i.e., DCT) secretes K+ but at a relatively low rate. However, a high rate of K+ secretion into the tubule fluid is one of the distinguishing features of late portions of the classic distal tubule (i.e., CNT and ICT) and of the CCT. Of the two cell types in the ICT and CCT, the principal cell secretes K+, by a transcellular process (Fig. 37-7D). The three key elements of the principal cell are (1) an Na-K pump for active K+ uptake at the basolateral membrane, (2) a relatively high (and variable) apical K+ permeability, and (3) a favorable electrochemical driving force for K+ exit across the apical membrane. In addition, K+ may move from cell to lumen through the apical KCC (see Chapter 5). The net effects are the active movement of K+ from blood to cell and the passive movement of K+ from cell to lumen. Changes in any of the three elements can affect the secretion of K+.
The capacity for K+ secretion diminishes from the cortical to the MCD. Indeed, the MCD is responsible for K+ reabsorption, which contributes to medullary K+ recycling (Fig. 37-6). This K+ loss from the MCD lumen can occur by passive movement by the paracellular pathway—which has a significant K+ permeability—driven by a favorable K+ concentration gradient. The luminal [K+] of the MCD is high for two reasons. First, the segments just upstream (i.e., distal K+ secretory system) may have secreted K+. Second, the continuing reabsorption of fluid in the medullary collecting tubules, particularly in the presence of high arginine vasopressin (AVP) levels, further increases luminal [K+]. In addition to passive K+ reabsorption, apical H-K pumps (similar to those in α intercalated cells in Fig. 37-7C) may mediate active K+ reabsorption, especially during low K+ intake.
Table 37-2 summarizes factors that modulate K+ secretion. These may be grouped into luminal and peritubular (i.e., bathing basolateral membrane) factors.
Table 37-2 Luminal and Peritubular Factors That Modulate K+ Secretion by the Distal K+-Secretory System*
LUMINAL FACTORS |
|
Stimulators |
Inhibitors |
↑ Flow rate |
↑ [K+] |
↑ [Na+] |
↑ [Cl−] |
↓ [Cl−] |
↑ [Ca2+] |
↑ [HCO−3] |
Ba2+ |
Negative luminal voltage |
Amiloride |
Diuretics acting upstream of ICT/CCT |
|
PERITUBULAR FACTORS |
|
Stimulators |
Inhibitors |
↑ K+ intake |
↓ pH |
↑ [K+] |
Epinephrine |
↑ pH |
|
Aldosterone |
|
AVP |
|
* ICT, CCT, and proximal portion of MCD.
One of the most potent stimuli of K+ secretion is the rate of fluid flow along the distal K+ secretory system (i.e., ICT and CCT). Under almost all circumstances, an increase in luminal flow increases K+ secretion (Fig. 37-8), and a similar relationship holds between final urine flow and K+ excretion. Accordingly, the increased urine flow that occurs with extracellular volume expansion, osmotic diuresis, or administration of several diuretic agents (e.g., acetazolamide, furosemide, thiazides) leads to enhanced K+ excretion (kaliuresis). Almost uniformly, increased urinary flow is also associated with increased Na+ excretion (natriuresis), so that both solutes appear in increased amounts in the urine.
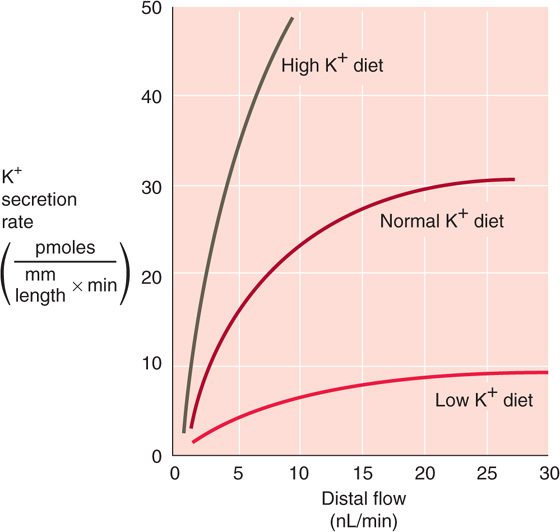
Figure 37-8 Effect of flow (i.e., Na+ delivery) on K+ excretion. (Data from Stanton BA, Giebisch G: Renal potassium transport. In Windhager E [ed]: Handbook of Physiology: Renal Physiology, pp 813-874. New York: Oxford University Press, 1992.)
The strong flow dependence of distal K+ secretion is a consequence of the high apical K+ permeability of principal cells (Fig. 37-7D). When luminal flow is low, then as K+ moves from the principal cell to the lumen, luminal [K+] rises rapidly to a steady-state level, thereby inhibiting further K+ diffusion from the cell. Thus, total K+ secretion is relatively low. When luminal flow is high, it sweeps newly secreted K+ downstream. The resulting fall in luminal [K+] steepens the K+ gradient across the apical membrane and consequently increases passive K+ flux from cell to lumen.
Increased luminal flow also increases the Na+ delivery to tubule cells, thus raising luminal [Na+] and enhancing Na+ uptake. This incremental supply of Na+ to the principal cell stimulates its Na-K pump, increases basolateral K+ uptake, and further increases K+ secretion. The Na+ sensitivity of K+ secretion is most pronounced when luminal [Na+] is lower than 35 mM, as is often the case in the CCT.
The apical step of K+ secretion in the ICT and CCT occurs by diffusion of K+ from the principal cell to the lumen, a process that depends on the apical electrochemical K+ gradient (Fig. 37-7D). Increases in luminal [Na+]—enhancing apical Na+ entry through epithelial Na+ channels (ENaCs) (see Chapter 35)—depolarize the apical membrane, favoring the exit of K+ from cell to lumen (i.e., K+ secretion). Conversely, a fall in luminal [Na+] hyperpolarizes the apical membrane, thereby inhibiting K+ secretion.
The diuretic amiloride (see Chapter 35) has the same effect on K+ secretion as decreasing luminal [Na+]. By blocking apical ENaCs, amiloride hyperpolarizes the apical membrane and reduces the electrochemical gradient for K+ secretion. Thus, amiloride is a K+-sparing diuretic.
Lowering luminal Cl−—replacing it with an anion (e.g., SO2–4 or HCO−3) that the tubule reabsorbs poorly—promotes K+ loss into the urine, independent of changes in the lumen-negative voltage. Underlying this effect may be the KCC in the apical membrane of the principal cell (Fig. 37-7D). Lowering luminal [Cl−] increases the cell-to-lumen Cl− gradient, which presumably stimulates K/Cl cotransport and thus K+ secretion.
When luminal [Cl−] in the DCT is low, thiazide diuretics may indirectly inhibit K+ secretion. These drugs inhibit Cl− uptake by a luminal Na/Cl cotransporter in the DCT (see Chapter 35), thus presenting a higher luminal [Cl−] to the downstream K+ secretory sites in the ICT and CCT. This elevation in luminal [Cl−] secondarily inhibits K+ efflux from the principal cell through the KCC (Fig. 37-7D) and thus reduces K+ secretion. (See Note: Paradoxical Inhibition of K+ Secretion by Thiazide Diuretics in Low-Cl-States)
Both mineralocorticoids and glucocorticoids cause kaliuresis (see Chapter 50). Primary hyperaldosteronism leads to K+ wasting and hypokalemia, whereas adrenocortical insufficiency (i.e., deficiency of both mineralocorticoids and glucocorticoids) leads to K+ retention and hyperkalemia.
Mineralocorticoids Aldosterone, the main native mineralocorticoid, induces K+ secretion in the ICT and CCT, particularly when its effects are prolonged. Aldosterone and desoxycorticosterone acetate (DOCA), a powerful synthetic mineralocorticoid, increase the transcription of genes that enhance Na+ reabsorption and, secondarily, K+ secretion in the principal cells of the ICT and CCT (see Chapter 35). Three factors act in concert to promote K+ secretion. First, over a period of a few hours, aldosterone increases the basolateral K+ uptake by stimulating the Na-K pump. Over a few days, elevated aldosterone also leads to a marked amplification of the area of the basolateral membrane of principal cells, as well as to a corresponding increase in the number of Na-K pump molecules. Conversely, adrenalectomy causes a significant reduction in basolateral surface area and Na-K pumps in principal cells. Second, mineralocorticoids stimulate apical ENaCs, thus depolarizing the apical membrane and increasing the driving force for K+ diffusion from cell to lumen. Third, aldosterone increases the K+ conductance of the apical membrane. (See Note: Acute Effects of IV Aldosterone)
Considering its mechanisms of action, we could expect aldosterone to stimulate both K+ secretion and Na+ reabsorption. Therefore, one would think that the renal excretion of K+ and Na+ should always be inversely related. However, the extent to which aldosterone increases K+ excretion depends strongly on Na+ excretion. For example, when Na+ intake and excretion are low, the Na+ retention induced by aldosterone reduces luminal flow, and Na+ delivery to such low levels that K+ excretion fails to increase. In contrast, under high-flow conditions (e.g., with a high Na+ load or following administration of diuretics that act on the TAL or the DCT), aldosterone increases K+ excretion.
Long-term administration of mineralocorticoids leads to a sequence of events known as aldosterone escape. Aldosterone leads to Na+ retention and hence volume expansion. Eventually, the volume expansion causes proximal Na+ reabsorption to fall (see Fig. 34-11), thereby increasing Na+ delivery to the ICT and CCT and ultimately raising Na+ excretion toward pre-aldosterone levels. Thus, the kidney can escape the Na+-retaining effect of aldosterone, albeit at the price of expanding the extracellular volume. The situation is different with respect to K+, because the increased Na+ delivery to the distal K+ secretory system continues to stimulate K+ secretion. Once the body has become depleted of K+, the aldosterone-stimulated K+ secretion eventually wanes, and the distal nephron may return to net K+ reabsorption.
Glucocorticoids Under physiological conditions, glucocorticoids enhance K+ excretion (Fig. 37-9). Corticosterone and dexamethasone, a synthetic glucocorticoid, produce this effect largely by increasing flow along the distal K+ secretory system (i.e., ICT and CCT). Because glucocorticoids increase the GFR and probably lower the water permeability of the distal K+ secretory system, they increase the amounts of fluid and Na+ remaining in these segments. These two flow effects, by themselves, increase K+ secretion. Indeed, when one perfuses the distal K+ secretory system at a constant rate, glucocorticoids have no effect on K+ transport. In addition, administering unphysiologically high doses of glucocorticoids may stimulate K+ transport directly as glucocorticoids bind to mineralocorticoid receptors; despite the presence of 11β-hydroxysteroid dehydrogenase in aldosterone target cells (see Chapter 35).
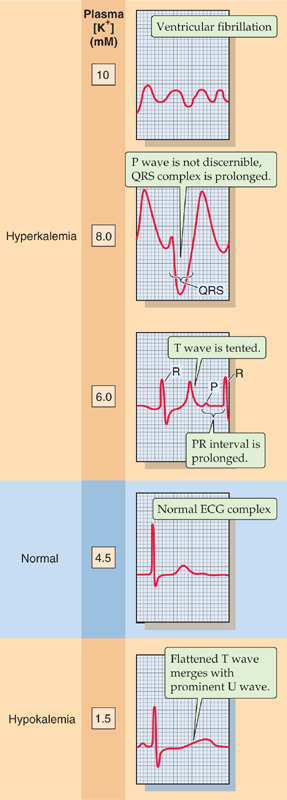
Figure 37-9 Effect of changes in plasma [K+] on the ECG. These five traces show recordings from the precordial V4 lead.
Hyperkalemia and the Heart
An electrocardiogram (ECG)—a measure of the electrical activity of the myocardial cells (see Chapter 21)—can be clinically useful for detecting rising plasma [K+].
As plasma [K+] begins to rise (Fig. 37-9, 6 mM), the T wave becomes tall and peaked, assuming a symmetric, “tented” shape. The PR interval becomes prolonged, and the P wave gradually flattens. Eventually, the P wave disappears (Fig. 37-9, 8 mM). Moreover, the QRS complex, which represents depolarization of the ventricles, widens and merges with the T wave, forming a sine-wave pattern. At higher concentrations, ventricular fibrillation, a lethal arrhythmia, may develop (Fig. 37-9, 10 mM). These changes on the ECG do not always occur in this precise order, and some patients with hyperkalemia may progress very rapidly to ventricular fibrillation. Any change in the ECG resulting from hyperkalemia requires immediate clinical attention.
Decreases in plasma [K+] also affect the ECG. For example, moderate decreases in [K+] to ~3 mM cause the QT interval to lengthen and the T wave to flatten. Lower values of [K+] lead to the appearance of a U wave (Fig. 37-9, 1.5 mM), which represents the delayed repolarization of the ventricles.
Dietary K+ Loading An increase in dietary K+ intake causes, after some delay, an increase in urinary K+ excretion (Fig. 37-5B). If the period of high-K+ intake is prolonged, a condition of tolerance (K+ adaptation) develops in which the kidneys become able to excrete large doses of K+—even previously lethal doses—efficiently, with only a small rise in plasma [K+].
The kaliuresis following an acute or chronic K+ load is the result of increased K+ secretion in the ICT and CCT (compare the three curves in Fig. 37-8 at a single flow), a process that occurs by three mechanisms. First, a transient rise in plasma [K+], even if maintained for only short periods, is an effective stimulus for K+ excretion. The mechanism is increased K+ uptake across the basolateral membrane of principal cells by the Na-K pump, a response shared by most of the cells of the body. As is the case with mineralocorticoid stimulation, the ultrastructure of the ICT and CCT correlates with changes in the rate of K+ transport induced by high dietary K+. Therefore, a high-K+ diet—independent of the effects of aldosterone—causes surface amplification of the basolateral membrane of principal cells. Second, the increased plasma [K+] stimulates glomerulosa cells in the adrenal cortex to synthesize and release aldosterone (see Chapter 55). This mineralocorticoid is a potent stimulus for K+ secretion both in the kidney by principal cells (see earlier) and in the colon (see Chapter 44). Third, acute K+ loading inhibits proximal Na+ and fluid reabsorption and increases the flow and Na+ delivery to the distal K+ secretory system, processes that stimulate K+ secretion.
Dietary K+ Deprivation In response to K+ restriction, the kidneys retain K+ (Fig. 37-5A). The rate of urinary K+ excretion may fall to 1% to 3% of the filtered load, a finding reflecting the action of two mechanisms at the level of the ICT and CCT. First, the low plasma [K+] suppresses K+ secretion by the principal cells (Fig. 37-8, lower curve) both by reducing basolateral K+ uptake by principal cells and by reducing aldosterone secretion. Second, the low plasma [K+] stimulates K+ reabsorption by intercalated cells through H-K pumps (Fig. 37-7C), thus causing the [K+] of the final urine to fall sharply, sometimes to levels lower than that of the plasma. In addition, the MCD responds to K+ depletion by increasing its reabsorption of K+, by enhancing both the activity of its apical K-H pump and its paracellular K+ permeability.
Whereas states of high K+ secretion (i.e., aldosterone or high-K+ diet) amplify the basolateral membrane area of principal cells, K+ deprivation amplifies the apical membrane of α intercalated cells. Moreover, K+ depletion causes an increased incorporation of rod-shaped particles in the apical membrane of α intercalated cells, and the density of these particles correlates well with enhanced K+ reabsorption. These morphological changes probably underlie the increased active K+ reabsorption and H+ secretion mediated by H-K pumps.
Acid-base disturbances have marked effects on renal K+ transport. In general, either metabolic alkalosis or respiratory alkalosis leads to increased K+ excretion (see Chapter 27). Conversely, acidosis reduces K+ excretion, although this response is more variable than that to alkalosis. Changes in systemic acid-base parameters affect K+ transport mainly by acting on the distal K+ secretory system.
As discussed, alkalosis leads to hypokalemia, owing to K+ uptake by cells throughout the body. Despite this fall in plasma [K+], alkalosis stimulates K+ secretion in the distal K+ secretory system, thereby worsening the hypokalemia. Conversely, K+ secretion falls in acidosis despite the shift of K+ from cells to ECF and the concomitant rise in plasma [K+].
The cellular events underlying the renal response to acid-base disturbances most likely involve effects of pHi on both the basolateral Na-K pump and the apical K+ channels of the principal cells in the ICT and CCT. Tubule perfusion studies with 42K indicate that decreasing the extracellular pH—which also decreases pHi—inhibits the Na-K pump and thus K+ secretion. Even more important is that the decrease in pHi also reduces the permeability of the apical K+ channels, which are exquisitely sensitive to acidification (Fig. 37-10). The reverse changes occur in alkalosis.
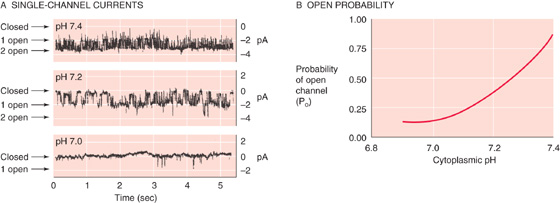
Figure 37-10 Effect of intracellular acidosis on K+-channel activity in the apical membrane of a principal cell. In A, the recordings show single-channel K+ currents from inside-out patches, with different pH values on the “cytoplasmic” side. As shown in B, channels are almost never open at pH values at 7.0 or lower. (Data from Wang W, Geibel J, Giebisch G: Am J Physiol 1990; 259:F494-F502.)
The changes in tubule flow (i.e., fluid delivery to the distal K+ secretory system) that occur in acid-base disturbances may modulate the effects of pH changes per se. For example, metabolic alkalosis increases the flow by delivering an HCO−3-rich solution to the ICT and CCT. By itself, this increased flow enhances K+ secretion, and thus potentiates the effect of alkalosis per se. In contrast, acidosis also increases distal flow, in this case by inhibiting proximal fluid reabsorption, with the consequence that the tendency of increased flow to increase K+ secretion opposes the effect of acidosis per se to decrease it. Therefore, the net effect of acidosis on K+ excretion is variable.
By both extrarenal and renal mechanisms, epinephrine lowers K+ excretion. First, epinephrine enhances K+ uptake by extrarenal tissues (see Chapter 27), thereby lowering plasma [K+] and reducing the filtered K+ load. Second, catecholamines directly inhibit K+ secretion in nephron segments downstream of the ICT.
Although it is not a major regulator of K+ excretion, AVP, also known as antidiuretic hormone [ADH]), can stimulate the distal K+ secretory system by two mechanisms (Fig. 37-7D): (1) AVP increases the apical Na+ conductance, thus depolarizing the apical membrane and providing a larger driving force for K+ efflux from cell to lumen; and (2) AVP increases apical K+ permeability. However, AVP also reduces urine flow and thus inhibits K+ secretion. Therefore, the two opposing effects—a direct stimulation of K+ secretion and a flow-related reduction of K+ secretion—may cancel each other.
Clinical Implications: Evaluating Hypokalemia and Hyperkalemia in the Patient
We can directly apply the physiological principles discussed in this chapter to understanding the common causes and treatment of hypokalemia and hyperkalemia.
Hypokalemia
The body can lose K+ from three sites: the kidneys, the GI tract, and the skin. Renal losses occur most commonly in patients taking diuretics (e.g., furosemide and thiazides) that act at sites upstream to the distal K+ secretory system (i.e., ICT, CCT, and proximal portion of MCD). K+ losses can also occur in individuals with renal tubule disorders or alterations in the renin-angiotensin-aldosterone system (e.g., hyperaldosteronism). Because gastric and intestinal secretions have a high [K+], vomiting and severe diarrhea are common causes of hypokalemia. Significant K+ depletion through the skin can occur in two situations: (1) strenuous exercise on a hot, humid day can cause dehydration and hypokalemia from the loss of many liters of perspiration; and (2) severe and extensive burns can result in the transudation of vast amounts of K+-containing fluid through the skin. In both cases, the lost fluid is K+ rich compared with plasma.
Low plasma [K+] can also develop as the result of altered K+ distribution within the body, without any net loss of whole-body K+. Common causes include alkalosis, a catecholamine surge (e.g., as during any acute stress to the body, such as an acute myocardial infarction), and excessive insulin administration during the treatment of diabetic ketoacidosis.
Patients may develop hypokalemia because of inadequate dietary K+ intake. Patients receiving large amounts of intravenous saline (which lacks K+) may also waste K+. Some persons in rural areas ingest clay (a behavior called pica), which binds K+ in the GI tract and prevents K+ absorption.
Hyperkalemia
Probably the most common cause of a high laboratory value for plasma [K+] is pseudohyperkalemia, a falsely elevated value that results from traumatic hemolysis of red blood cells during blood drawing. Red blood cells release their intracellular stores of K+ in the blood sample, and the laboratory reports a falsely elevated plasma [K+]. Patients with myeloproliferative disorders associated with greatly increased numbers of platelets or white blood cells can also show a falsely elevated [K+] because of K+ released during clot formation within the blood sample tube.
Excessive intake of K+ rarely causes hyperkalemia in persons with healthy kidneys. However, even in these individuals, a large K+ bolus can cause transient hyperkalemia (Fig. 37-2, top), often from an ill-advised intravenous line. Altered distribution of K+ can lead to elevated plasma levels in patients with acidosis and, rarely, as a result of β-adrenergic blockade. Digitalis, a cardiac glycoside that inhibits the Na-K pump (see Chapter 5), is another rare cause of hyperkalemia resulting from K+ redistribution. Massive breakdown of cells can release large amounts of K+ into the extracellular space. This release can occur in patients with intravascular hemolysis (e.g., from a mismatched blood transfusion), burns, crush injuries, rhabdomyolysis (massive muscle destruction such as can be seen with trauma or sepsis), GI bleeding with subsequent intestinal absorption of K+-rich fluid, or destruction of tumor tissue or leukemic blood cells by chemotherapy.
Impaired renal excretion of K+ is the primary cause of a sustained increase in plasma [K+]. Hypoaldosteronism, high doses of amiloride, and distal renal tubular acidosis can be responsible, but advanced renal failure itself from any of a myriad number of disorders is the most common cause.
The treatment of hyperkalemia depends on the severity of the problem. For severe hyperkalemia (i.e., [K+] > 8 mM) with accompanying ECG changes, the physician immediately infuses calcium gluconate intravenously to counter the electrophysiological effects of the hyperkalemia. (Ca2+ raises the threshold for action potentials and lessens membrane excitability). One may also simultaneously administer NaHCO3, glucose, and often insulin to move some of the K+ from the extracellular to the intracellular space. These are all temporizing measures. To remove the excess K+ from the body, the physician administers a nonabsorbable Na-K cation exchange resin (e.g., sodium polystyrene sulfonamide) either orally or as an enema. The resin binds the K+ and carries it out of the body through the GI tract. However, the resin takes several hours to work. Thus, for patients with hyperkalemia and ECG changes, the temporizing measures buy time for the resin to act. For patients with end-stage renal failure, dialysis is often necessary to return the plasma [K+] to normal.
It should be apparent from the foregoing discussion that the net effect of a specific disturbance on K+ excretion in the final urine often represents a result of two or more interacting factors. Changes in flow often indirectly amplify or attenuate the direct response of the distal K+ secretory system to a given stimulus. Figure 37-11 shows four examples of attenuating effects. The first three (acidosis, volume expansion, and high water intake) are characterized by the competition between a direct inhibitory effect on K+ secretion and an indirect stimulatory effect of increased flow (Fig. 37-11A through C).
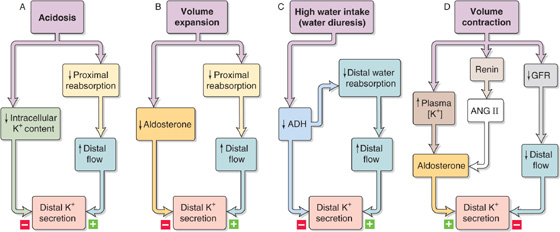
Figure 37-11 A to D, Interaction of opposing factors on K+ secretion.
Extracellular volume contraction (Fig. 37-11D) has three effects. First, it decreases distal flow and thus inhibits the distal K+ secretory system. Second, volume contraction increases angiotensin II (ANG II) levels, a process that, in turn, increases aldosterone release and hence K+ secretion. Third, volume contraction also produces a small rise in plasma [K+], the magnitude of which may normally not evoke much of an increase in aldosterone release. However, in the presence of elevated ANG II, even such a small increase in plasma [K+] is a potent stimulus for aldosterone release (see Chapter 50). Thus, the potentiation between K+ and ANG II ensures continued K+ excretion, even when low fluid delivery to the distal K+ secretory system would have the opposite effect.
In some instances, the direct actions of an agent and flow-related effects may have additive effects. Three examples follow.
First, metabolic alkalosis—as discussed—directly stimulates the distal K+ secretory system. In addition, the delivery of the poorly reabsorbable HCO−3 in metabolic alkalosis increases distal flow, thereby potentiating K+ excretion.
Second, hyperkalemia directly stimulates the distal K+ secretory system. In addition, because increased plasma [K+] inhibits Na+ and fluid reabsorption in the proximal tubule, distal flow increases, again potentiating K+ excretion.
Third, administration of diuretics that act on a site upstream of the ICT and CCT increases Na+ delivery to the distal K+ secretory system and thereby stimulates K+ secretion. In addition, the volume contraction induced by the diuretic raises aldosterone levels, again potentiating K+ excretion. Hypokalemia is thus a possible harmful side effect of diuretic treatment.
Books and Reviews
Giebisch G: Challenges to potassium metabolism: Internal distribution and external balance. Wien Klin Wochenschr 2004; 116:353-366.
Hebert SC, Desir G, Giebisch G, Wang W: Molecular diversity and regulation of renal potassium channels. Physiol Rev 2005; 85:319-371.
Jamison RL: Potassium recycling. Kidney Int 1987; 31:695-703.
Wingo CS, Cain BD: The renal H-K-ATPase: Physiological significance and role in potassium homeostasis. Annu Rev Physiol 1993; 55:323-347.
Journal Articles
Greger R, Schlatter E: Presence of luminal K+, a prerequisite for active NaCl transport in the cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflugers Arch 1981; 392:92-94.
Gu RM, Wei Y, Falck JR, et al: Effects of protein tyrosine kinase and protein tyrosine phosphatase on apical K+ channels in the TAL. Am J Physiol 2001; 281:C1188-C1195.
Ho K, Nichols CG, Lederer WJ, et al: Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 1993; 362:31-38.
Liu W, Xu S, Woda C, et al: Effect of flow and stretch on the [Ca2+]i, response of principal and intercalated cells in cortical collecting duct. Am J Physiol 2003; 285:F998-F1012.
Malnic G, Klose R, Giebisch G: Micropuncture study of renal potassium excretion in the rat. Am J Physiol 1964; 206:674-686.
Stokes J: Consequences of potassium recycling in the renal medulla: Effects on ion transport by the medullary thick ascending limb of Henle’s loop. J Clin Invest 1982; 70:219-229.
Wade JB, O’Neil RG, Pryor JL, Boulpaep EL: Modulation of cell membrane area in renal cortical collecting tubules by corticosteroid hormones. J Cell Biol 1979; 81:439-445.
Wang W, Geibel J, Giebisch G: Regulation of small conductance K channel in the apical membrane of rat cortical collecting tubule. Am J Physiol 1990; 259:F494-F502.