Eugene J. Barrett
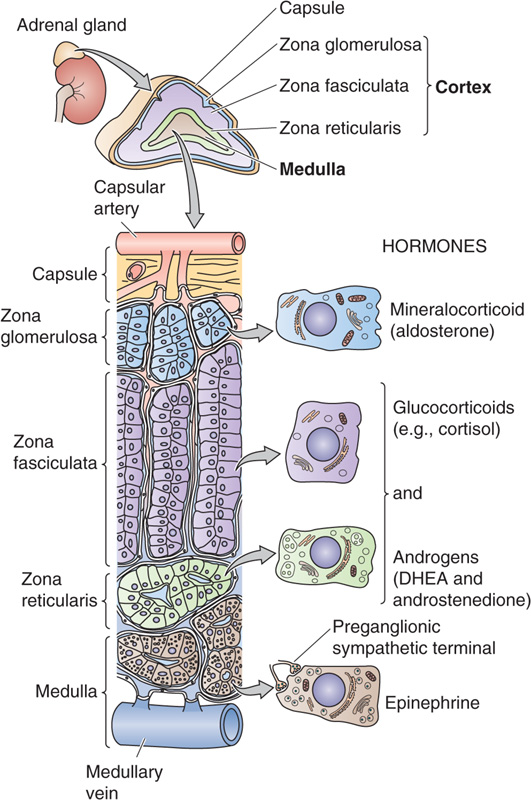
The human adrenal glands, each weighing only ~4 g, are located above the upper pole of each kidney in the retroperitoneal space. They produce four principal hormones: cortisol, aldosterone, epinephrine (adrenaline), and norepinephrine. Each adrenal gland is composed of an inner medulla and an outer cortex (Fig. 50-1). Embryologically, the cortex is derived from mesoderm, whereas the medulla is derived from neural crest cells (see Chapter 10) that migrate into the developing cortex. The cortex produces two principal steroid hormones, cortisol and aldosterone, as well as several androgenic steroids. The medulla produces epinephrine and norepinephrine.
Figure 50-1 Anatomy of the adrenal gland. An adrenal gland sits on each kidney. The adrenal gland is actually two glands—the cortex and the medulla. The adrenal cortex comprises three layers that surround the medulla. The outermost layer contains the glomerulosa cells that secrete aldosterone, and the two inner layers of cortex (fasciculata and reticularis) synthesize cortisol and sex steroids. The blood supply enters the cortex in the subcapsular region and flows through anastomotic capillary beds while coursing through both the cortex and the medulla. The adrenal medulla contains chromaffin cells that secrete epinephrine and a small amount of norepinephrine.
The adrenal cortex can be further divided into three cellular layers: the glomerulosa layer near the surface, the fasciculata layer in the midcortex, and the reticularis layer near the cortical-medullary junction. Aldosterone, the main mineralocorticoid in humans, is made in the glomerulosa cell layer. Cortisol, the principal glucocorticoid, is made in the fasciculata and reticularis layers. Although both cortisol and aldosterone are made by enzymatic modification of cholesterol and are structurally quite similar, their actions on the body differ dramatically. Cortisol is considered a glucocorticoid because it was recognized early on to increase plasma glucose levels; deficiency of cortisol can result in hypoglycemia. Aldosterone is considered a mineralocorticoid because it promotes salt and water retention by the kidney. The activities of these two hormones overlap, particularly at high hormone levels, but this distinction is still very useful in identifying their most obvious functions. The adrenal cortex also synthesizes and secretes androgenic steroids, which to a large extent appear to be byproducts of cortisol production.
In the adrenal medulla, chromaffin cells produce epinephrine or adrenaline, a catecholamine that is synthesized from the amino acid tyrosine. Although the primary product of the medulla is epinephrine, it also produces variable amounts of the epinephrine precursor norepinephrine. These catecholamines are distinct from the steroid hormones both structurally and functionally.
Steroid hormones are divided into three major classes based on their actions: glucocorticoids, mineralocorticoids, and sex steroids. Cortisol is the prototypical, naturally occurring glucocorticoid. The ability of cortisol to increase plasma [glucose] largely results from its ability to enhance mobilization of amino acids from proteins in many tissues and to enhance the ability of the liver to convert these amino acids into glucose and glycogen by activating gluconeogenesis.
Only a small difference can be seen in the structures of cortisol and aldosterone (Fig. 50-2): aldosterone lacks the –OH group at position 17 but has an aldehyde (aldo) group at position 18. Despite the seemingly minor chemical difference, aldosterone at physiological concentrations has virtually no glucocorticoid activity.
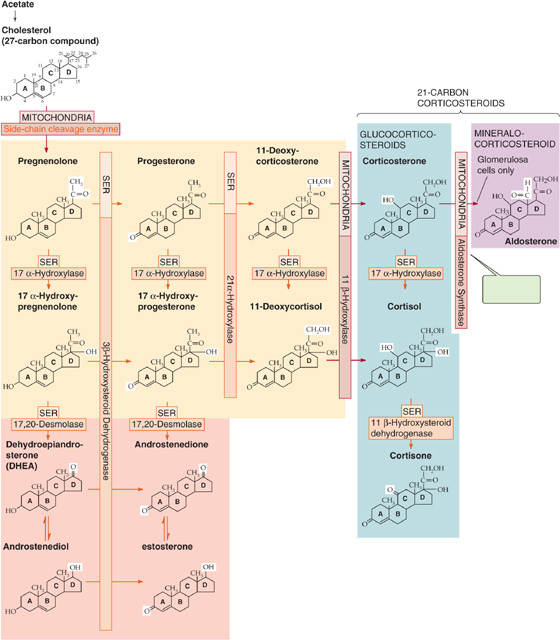
Figure 50-2 Biosynthesis of adrenal steroids. This schematic summarizes the synthesis of the adrenal steroids—the mineralocorticoid aldosterone and the glucocorticoid cortisol—from cholesterol. The individual enzymes are shown in the horizontal and vertical boxes; they are located in either the SER or the mitochondria. The SCC enzyme that produces pregnenolone is also known as 20, 22-desmolase. The chemical groups modified by each enzyme are highlighted in the reaction product. If the synthesis of cortisol is prevented by any one of several dysfunctional enzymes, other steroid products may be produced in excess. For example, a block in 21 α-hydroxylase diminishes production of both cortisol and aldosterone and increases production of the sex steroids. Certain of these pathways are shared in the biosynthesis of the androgens (see Fig. 54-5) and estrogens (see Fig. 55-9).
Target Tissues Although classified as a glucocorticoid, cortisol affects more than the principal glucose regulatory tissues, namely, the liver, fat, and muscle. Most body tissues, including bone, skin, other viscera, hematopoietic and lymphoid tissue, and the central nervous system (CNS), are target sites for glucocorticoid action. Although cortisol is the primary glucocorticoid in humans, in other species (e.g., the rat), corticosterone is the major glucocorticoid.
Actions Glucocorticoids have numerous actions other than their ability to raise plasma glucose levels. These actions are described later but include potent immunosuppressive and anti-inflammatory activity, effects on protein and fat metabolism, behavioral effects on the CNS, and important effects on calcium and bone metabolism. Some of the diverse physiological effects of the glucocorticoids can be appreciated from studies of clinical states of excess glucocorticoid secretion (Cushing syndrome) and glucocorticoid deficiency (Addison disease). The multiple actions of glucocorticoids, in particular, their “anti-inflammatory” action on leukocytes, led to the development of numerous synthetic analogues that are more potent, have a longer half-life, and are more selective in their specific glucocorticoid actions than are cortisol or corticosterone. Table 50-1 lists some of these compounds and indicates their relative potency as mineralocorticoids and glucocorticoids.
Table 50-1 Relative Potency* of Glucocorticoid and Mineralocorticoid Analogues
Compound |
Glucocorticoid Effect |
Mineralocorticoid Effect |
Cortisol |
1 |
1.5 |
Prednisone |
3-4 |
0.5 |
Methylprednisone |
10 |
0.5 |
Dexamethasone |
20 |
1 |
Fludrocortisone |
12 |
125 |
*Relative potency is determined by a combined consideration of the compound’s biological half-life and its affinity for the glucocorticoid or mineralocorticoid receptor.
As discussed in Chapter 47, most of the well-characterized actions of glucocorticoids result from their genomic actions to influence (either positively or negatively) the transcription of a variety of genes through GREs. However, glucocorticoids also exert nongenomic actions (see Chapter 47) that occur promptly (0 to 3 hours) and are not inhibited by blockade of gene transcription.
Cushing Syndrome and Addison Disease
Glucocorticoid excess is most commonly seen clinically in individuals receiving glucocorticoids for treatment of a chronic inflammatory or neoplastic disorder. Less commonly, individuals overproduce cortisol either because of a primary cortisol-producing adrenal tumor or secondary to a pituitary tumor that produces ACTH, which, in turn, stimulates excess cortisol production from normal adrenal glands. In either case, the cortisol excess causes a constellation of symptoms and signs including truncal adiposity (abdomen, neck, facies), hypertension, loss of subcutaneous adipose and connective tissue in the extremities with associated easy bruising, loss of bone mineral, muscle weakness and wasting, and hyperglycemia. This constellation is referred to as Cushing syndrome after the famous American neurosurgeon who characterized this disorder. Specific therapy is based on identifying whether the clinical picture arises from a tumor in the adrenal or the pituitary gland and then removing the culprit. When the pituitary gland is responsible, the disorder is referred to as Cushing disease. In the case of patients receiving glucocorticoid therapy, the signs and symptoms of Cushing syndrome are carefully monitored, and efforts made to minimize these side effects. Unfortunately, all glucocorticoid drugs with anti-inflammatory actions also produce these other side effects.
Like glucocorticoid excess, glucocorticoid deficiency—or more accurately adrenal insufficiency (which includes both glucocorticoid and mineralocorticoid)—can produce an array of symptoms and signs. Although tuberculosis was once a common cause of primary adrenal insufficiency, today autoimmune adrenal disease is the most common cause. Failure of adrenal cortical hormone secretion leads to increases in circulating concentrations of ACTH as well as other products of POMC. Two of these products (α-MSH and γ-MSH) cause skin hyperpigmentation. The lack of glucocorticoid predisposes to hypoglycemia. The combined absence of glucocorticoid and mineralocorticoid leads to hypotension, whereas aldosterone deficiency leads to hyperkalemia. Before the development of glucocorticoid and mineralocorticoid therapy, this disorder was uniformly fatal.
Synthesis of cortisol, as with all steroid hormones, starts with cholesterol (Fig. 50-2). Like other cells producing steroid hormones, the adrenal gland has two sources of cholesterol (see Chapter 47): (1) it can import cholesterol from circulating cholesterol-containing low-density lipoprotein (LDL cholesterol) by means of LDL receptor–mediated endocytosis (see Chapter 2), or (2) it can synthesize cholesterol de novo from acetate (see Fig. 46-16). Although both pathways provide the steroid nucleus needed for cortisol and aldosterone synthesis, circulating LDL is quantitatively more important.
In the adrenal gland, cholesterol is metabolized through a series of five reactions to make either cortisol or aldosterone. Except for 3β-hydroxysteroid dehydrogenase (3β-HSD), the enzymes responsible belong to the family of cytochrome P-450 oxidases (Table 50-2) and are located within the cells in either the mitochondria or the smooth endoplasmic reticulum (SER).
Table 50-2 Cytochrome P-450 Enzymes Involved in Steroidogenesis*
Enzyme |
Synonym |
Gene |
Cholesterol side chain cleavage |
P-450SSC |
CYP11A1 |
11β-Hydroxylase |
P-450c11 |
CYP11B1 |
17α-Hydroxylase |
P-450c17 |
CYP17 |
17, 20-Desmolase |
P-450c17 |
CYP17 |
21α-Hydroxylase |
P-450c21 |
CYP21A2 |
Aldosterone synthase |
P-450aldo |
CYP11B2 |
Aromatase |
P-450arom |
CYP19 |
*P-450arom catalyzes a reaction essential for the production of estrogens (see Chapter 55).
1. The pathway for cortisol and aldosterone synthesis begins in the mitochondria, where the cytochrome P-450 side chain–cleavage (SCC) enzyme (also called 20, 22-desmolase or P-450SSC) removes the long side chain (carbons 22 to 27) from the carbon at position 20 of the cholesterol molecule (27 carbon atoms). This enzyme, or the supply of substrate to it, appears to be the rate-limiting step for the overall process of steroid hormone synthesis.
2. The product of the SCC-catalyzed reaction is pregnenolone (21 carbon atoms), which exits the mitochondrion. The SER enzyme 3β-HSD (not a P-450 enzyme) oxidizes the hydroxyl group at position 3 of the A-ring to a ketone to form progesterone.
3. A P-450 enzyme in the SER, 17α-hydroxylase (P-450c17), then adds a hydroxyl group at position 17 to form 17α-hydroxyprogesterone. However, as shown in Figure 50-2, an alternative path to 17α-hydroxyprogesterone may be used: the 17α-hydroxylase may first add a hydroxyl group at position 17 of pregnenolone and form 17α-hydroxypregnenolone, which can then be converted to 17α-hydroxyprogesterone by the aforementioned 3β-HSD.
4. In the SER, 21α-hydroxylase (P-450c21) adds a hydroxyl at carbon 21 to produce 11-deoxycortisol.
5. In the mitochondria, 11β-hydroxylase (P-450c11) adds yet another hydroxyl, this time at position 11, to produce cortisol.
The enzymes represented by the vertical bars in Figure 50-2, as well as SCC, are present in all three cellular layers of the adrenal cortex. However, 17α-hydroxylase is not substantially present in the glomerulosa layer. Thus, only the fasciculata and reticularis layers can synthesize cortisol.
The cells of the fasciculata and reticularis layers of the adrenal cortex can synthesize androgens. These cells convert 17α-hydroxypregnenolone and 17α-hydroxyprogesterone into the adrenal androgens dehydroepiandrosterone and androstenedione. The enzyme that catalyzes this reaction is called 17, 20-desmolase; however, it turns out to be the same SER enzyme as the 17α-hydroxylase that produced the 17α-hydroxypregnenolone and 17α-hydroxyprogesterone in the first place. The androgens formed by the adrenal are far less potent than either testosterone or dihydrotestosterone. However, peripheral tissue can use 17-ketosteroid reductase to convert androstenedione to testosterone (see Chapter 54). In this manner, the adrenal can contribute significant amounts of circulating androgen.
The cortisol synthesized by the adrenal cortex diffuses out of the cells and into the blood plasma. There, ~90% of the cortisol is transported bound to corticosteroid-binding globulin (CBG), also known as transcortin, which is made in the liver. Transcortin is a 383–amino acid glycoprotein whose affinity for cortisol is ~30-fold higher than for aldosterone. An additional ~7% of the circulating cortisol is bound to albumin. Thus, only 3% to 4% of the circulating cortisol is free.
The clearance of cortisol from the body depends principally on the liver and kidney. An early step is the formation of an inactive metabolite, cortisone, by the action of 11β-hydroxysteroid dehydrogenase (11β-HSD). This 11β-HSD pathway (Fig. 50-2) is not part of the adrenal’s normal formation of cortisone. Rather, one of the two 11β-HSD isozymes (11β-HSD1) is highly expressed in certain glucocorticoid target tissues, including liver and both subcutaneous and visceral adipose tissue. The enzyme is quite reversible. Indeed, when glucocorticoids were first developed as pharmaceutical agents, it was cortisone that was used to treat patients suffering from a variety of inflammatory disorders (e.g., rheumatoid arthritis). For some time, investigators thought that cortisone was the active principle. Only later did it become apparent that the body must convert the administered cortisone to cortisol, which is the biologically active agent. Because excess cortisol produces insulin resistance and many features of the metabolic syndrome (e.g., glucose intolerance, hypertension, dyslipidemia; see Chapter 51)—and 11β-HSD1 is expressed abundantly in adipose tissue—an interesting hypothesis is that increased 11β-HSD1 activity in adipose tissue locally produces cortisol and thus promotes the development of insulin resistance.
21α-Hydroxylase Deficiency
Mutations can affect one or more of the enzyme steps in steroid hormone synthesis and can produce unique clinical syndromes that are a direct result of failure to manufacture a particular hormone, accumulation of excessive amounts of precursor steroids, or both. The most common of these enzymatic disorders is 21α-hydroxylase deficiency. From Figure 50-2, we would predict that deficiency of this enzyme would lead to inadequate production of both glucocorticoid and mineralocorticoid hormones, which is indeed what occurs. Affected infants are ill with symptoms of “salt losing” (hypotension, dehydration) and glucocorticoid deficiency (hypoglycemia). The natural reaction of the body is to attempt to overcome this deficiency by increasing the secretion of pituitary ACTH, which stimulates the synthesis of cortisol and aldosterone. ACTH also causes growth of the adrenal gland. However, if the mutant enzyme is totally inactive, no cortisol or aldosterone synthesis will occur, although all other enzymes of the pathway involved in glucocorticoid and mineralocorticoid synthesis will be expressed in increased amounts. The result is greater than normal activity of the SCC enzyme, 3β-HSD, 17α-hydroxylase, and 11β-hydroxylase, and the net effect is increased synthesis of both precursor molecules and adrenal androgens. The combination of inadequate production of glucocorticoids and mineralocorticoids, excessive production of androgens, and enhanced growth of the adrenal gland is the classical clinical syndrome of salt-losing, virilizing congenital adrenal hyperplasia. In female patients, the presence of excessive adrenal androgen in utero results in ambiguous genitalia at birth, a condition that should alert the pediatrician to the potential diagnosis.
The second 11β-HSD isozyme (11β-HSD2) is expressed highly in the renal distal tubule and collecting duct (see Fig. 35-13B). The 11-βHSD2 in these cells catalyzes an essentially irreversible conversion of cortisol to cortisone. This breakdown of cortisol allows aldosterone to regulate the relatively nonspecific mineralocorticoid receptor (MR) without interference from cortisol.
The multiple hydroxylation reactions that convert cholesterol to cortisol result in a hydrophilic compound that, unlike cholesterol, is soluble in plasma, yet lipophilic enough to cross the plasma membrane of target tissue without requiring a membrane transporter. Cortisol, like all steroid hormones, binds to intracellular receptors within target cells (see Chapter 3). Virtually all nucleated tissues in the body contain receptors for glucocorticoids. The glucocorticoid receptor (GR) is primarily located in the cytoplasm, where in its unbound form it is complexed to a chaperone protein (i.e., the heat shock protein hsp90, among others; see Fig. 4-16A). Binding of cortisol causes the chaperone to dissociate from the GR and thus allows the cortisol-GR complex to translocate to the nucleus. There, the cortisol-receptor complex associates with glucocorticoid response elements (GREs) on the 5′ untranslated region of multiple genes to either enhance or diminish gene expression (see Chapter 4).
GRs are structurally similar to the receptors for mineralocorticoids, sex steroids, vitamin D, vitamin A, and thyroid hormone. These receptors, either homodimers or heterodimers, belong to the superfamily of nuclear receptors that contains domains A through F (see Chapter 3). The middle, or C, region is responsible for DNA binding through two “zinc fingers” (see Chapter 4), whereas the E region, located toward the C terminus, binds cortisol. Activity of the glucocorticoid-receptor complex requires dimerization of two identical receptor complexes (i.e., the GR functions as a homodimer) at the near-palindromic nucleotide site of the GRE on the chromatin. Most actions of glucocorticoids are expressed by modulation of gene transcription. One exception is the acute feedback effect of cortisol to block the release of preformed adrenocorticotropic hormone (ACTH) in the secretory granules of pituitary corticotrophs. This glucocorticoid effect is demonstrable within seconds to minutes and may relate to an as yet undefined effect of glucocorticoid on membrane trafficking.
Although glucocorticoids are named for their ability to increase hepatic glucose and glycogen synthesis, they affect many somatic tissues. In liver, cortisol induces the synthesis of enzymes that are involved in the metabolism of amino acids, thus facilitating their conversion to carbohydrates through gluconeogenesis. In muscle, cortisol stimulates the breakdown of muscle protein, thus providing amino acid substrate to the circulation and subsequently to the liver. Similarly, cortisol induces mobilization of fat from subcutaneous adipose tissue. Fatty acids supplied to the circulation afford an alternative fuel to glucose and increase the availability of glucose. For unknown reasons, although fat is mobilized from the extremities, some is also deposited centrally (see the description of moon facies in the box titled Therapy with Glucocorticoids).
Cortisol has other actions that are not clearly related to its glucocorticoid action, including effects on the immune system, among which is an acute action to promote neutrophil release from the bone marrow into the systemic circulation. In humans, cortisol also diminishes the circulating lymphocyte count, partly as a result of sequestration of lymphocytes in the reticuloendothelial system (spleen, thymus, and bone marrow). At high concentrations, glucocorticoids cause the lysis of lymphocytes in some species. Glucocorticoids also act on the cellular elements of trabecular bone (see Chapter 52) in that they decrease the ability of osteoblasts to synthesize new bone. They also interfere with absorption of Ca2+ from the gastrointestinal tract. In addition, glucocorticoids act on the CNS and can cause a variety of effects, including alterations in mood and cognition.
As summarized in Figure 50-3, regulation of the synthesis and secretion of cortisol begins with the release of corticotropin-releasing hormone (CRH) from hypothalamic neurons, as part of either a normal, daily circadian rhythm or a centrally driven stress response. CRH stimulates the release of ACTH, also called corticotropin, from the anterior pituitary. ACTH directly stimulates the adrenal fasciculata and reticularis layers to produce and secrete cortisol. Circulating cortisol exerts negative feedback control on the release of both ACTH and CRH.
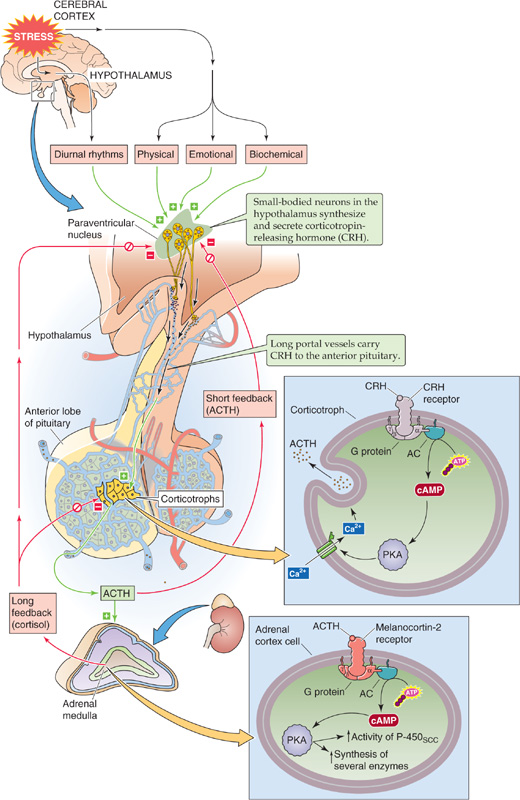
Figure 50-3 The hypothalamic-pituitary-adrenocortical axis. Small-bodied neurons in the paraventricular nucleus of the hypothalamus secrete CRH, a 41–amino acid peptide that reaches the corticotrophs in the anterior pituitary through the long portal veins. CRH binds to a GPCR on the corticotroph membrane, triggering the adenylyl cyclase/cAMP/PKA (AC/cAMP/PKA) pathway. The activation of L-type Ca2+ channels leads to an increase in [Ca2+]i that rapidly leads to the release of preformed ACTH. CRH also increases gene transcription and synthesis of the ACTH precursor, POMC. After its release by corticotrophs, ACTH binds to MC2Rs on the cell membranes in all three layers of the adrenal cortex. This receptor triggers the AC/cAMP/PKA pathway, thus rapidly enhancing the conversion of cholesterol to pregnenolone and more slowly increasing the synthesis of several proteins that are needed for cortisol synthesis. The cerebral cortex can stimulate the hypothalamic neurons to increase their secretion of CRH. Cortisol exerts negative feedback at the level of both the pituitary and hypothalamus. In addition, ACTH produced by the corticotrophs negatively feeds back on the hypothalamic neurons in a short loop.
Therapy with Glucocorticoids
The variety of glucocorticoid actions on body tissue is well illustrated by considering some of the clinically observed effects of hypercortisolism in patients receiving glucocorticoid drugs. Most strikingly, the entire body habitus changes. Body fat is centripetally distributed from the extremities to the face and trunk. This change is apparent on the trunk by an increase in supraclavicular and dorsal interscapular fat. It also causes a rounding of the face called the moon facies, which is a result of increasing subcutaneous fat in the cheeks and submandibular region. Conversely, the wasting of fat (and some supporting tissues) in the extremities produces thinning of the skin and fragility of cutaneous blood vessels. Direct effects of glucocorticoids on bone, as well as their inhibition of intestinal Ca2+ absorption, produce osteopenia, which can be manifested as osteoporosis, frequently with pathologic fractures. The interference with normal immune function increases both the frequency and severity of infections. Rare malignancies can develop. Wasting of muscle tissue results in generalized weakness that is usually most prominent in the proximal muscles of the lower extremities. Finally, as would be expected from a glucocorticoid, patients are glucose intolerant. After an oral glucose challenge (see Chapter 51), blood glucose levels become higher than normal because glucocorticoid antagonizes the action of insulin. Not infrequently, frank diabetes develops (see Chapter 51 for the box on diabetes mellitus). When cortisol is overproduced endogenously (from tumors producing either ACTH or cortisol), hypertension is common. The hypertension most likely results from the weak mineralocorticoid action of cortisol. Exogenous synthetic glucocorticoid therapy rarely produces hypertension because most of these drugs lack the mineralocorticoid activity of the endogenous hormone.
CRH CRH is a 41–amino acid neuropeptide made by small-bodied neurons in the paraventricular nucleus of the hypothalamus (see Chapter 47). The structure of CRH is highly conserved among species. In humans, in addition to the hypothalamus, CRH is present in several tissues, including the pancreas and the testes, as well as throughout the CNS, where it serves as a neurotransmitter. The hypothalamic neurons synthesize and release CRH through the classic secretory pathway (see Chapter 2). Neurons store CRH in secretory vesicles located in synaptic terminals in the median eminence of the hypothalamus and can release CRH acutely in the absence of new synthesis. After release into the interstitial fluid of the median eminence, CRH enters the hypophyseal portal venous plexus and travels to the anterior pituitary.
CRH Receptor CRH arriving in the anterior pituitary binds to a G protein–coupled receptor (GPCR) on the cell membrane of corticotroph cells. Hormone binding activates Gαs, which, in turn, stimulates adenylyl cyclase and raises [cAMP]i (see Chapter 3). Subsequent stimulation of protein kinase A (PKA) activates L-type Ca2+ channels and thus leads to an increase in [Ca2+]i. This rise in [Ca2+]i rapidly leads to the exocytosis of preformed ACTH. Over a much longer time, CRH receptor activation also leads to increased gene transcription and synthesis of the ACTH precursor (discussed later).
Arginine Vasopressin Although CRH is the major regulator of ACTH secretion, the paraventricular nuclei also make another hormone, arginine vasopressin (AVP; see Chapter 47). AVP is also a potent ACTH secretagogue. AVP probably plays a physiological role in the regulation of ACTH secretion in various stress states.
ACTH A 39–amino acid peptide hormone secreted by the corticotroph cells of the anterior pituitary, ACTH can also be produced by ectopic sources, particularly by small cell carcinomas of the lung. Pituitary corticotrophs synthesize ACTH by complex post-translational processing of a large precursor protein (i.e., a preprohormone) called proopiomelanocortin (POMC). POMC is the precursor not only for ACTH but also for a variety of peptide hormones (Fig. 50-4). In the anterior pituitary, POMC yields a long N-terminal peptide, a joining (J) peptide, ACTH, and β-lipotropin (β-LPH). During fetal life and pregnancy, the intermediate pituitary lobe—a small wedge of tissue between the more familiar anterior and posterior lobes—processes the same POMC in a very different manner to yield a different array of peptides: a short N-terminal peptide, γ-melanocyte–stimulating hormone (γ-MSH), J peptide, α-MSH, corticotropin-like intermediate lobe peptide (CLIP), γ-LPH, and β-endorphin. Other cells—such as the appetite-controlling POMC neurons in the hypothalamus (see Chapter 48)—can also synthesize POMC. The melanocortins include ACTH as well as α-, β-, and γ-MSH. The melanocortins bind to a family of five GPCRs, the melanocortin receptors (MC1R to MC5R).
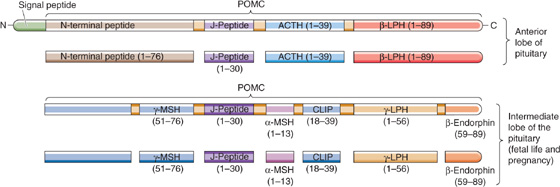
Figure 50-4 The primary gene transcript is the preprohormone POMC. The processing of POMC yields a variety of peptide hormones. This processing is different in the anterior and intermediate lobes of the pituitary. In the anterior pituitary, POMC yields a long N-terminal peptide, a joining (J) peptide, ACTH, and β-LPH. In the intermediate pituitary, the same POMC yields a short N-terminal peptide, γ-MSH, J peptide, α-MSH, CLIP, γ-LPH, and β-endorphin. Metabolism by the intermediate lobe is only important during fetal life and pregnancy. (Data from Young JB, Landsberg L: Catecholamines and the adrenal medulla. In Wilson JD, Foster DW, Kronenberg HM, Larsen PR (eds): Williams Textbook of Endocrinology, 9th ed, pp 665-728. Philadelphia: WB Saunders, 1998.)
α-MSH and γ-MSH act on MC1R receptors in melanocytes to increase the dispersion of pigment granules. In some patients who greatly overproduce ACTH, hyperpigmentation is a prominent clinical finding. Whether this hyperpigmentation is the result of increased production of MSH, increased production of β-LPH (which also has MSH activity), or the melanotropic action of ACTH per se remains uncertain. β-LPH and γ-LPH mobilize lipids from adipocytes in animals, although their physiological role in humans is unclear. β-Endorphin has potent opioid actions in the CNS (see Chapter 13), but its physiological actions (if any) in the systemic circulation are not known.
ACTH Receptor In the adrenal cortex, ACTH binds to MC2R on the plasma membranes of all three steroid-secreting cell types. However, because only the cells in the fasciculata and reticularis layers have the 17α-hydroxylase needed for synthesizing cortisol (Fig. 50-2), these cells are the only ones that secrete cortisol in response to ACTH. ACTH appears to have few other actions at physiological concentrations. MC2R is coupled to a heterotrimeric G protein and stimulates adenylyl cyclase (see Chapter 3). The resulting increase in [cAMP]i activates PKA, which phosphorylates a variety of proteins. A rapid effect of ACTH is to stimulate the rate-limiting step in cortisol formation, that is, the conversion of cholesterol to pregnenolone through the SCC enzyme. In addition, ACTH—over a longer time frame—increases the synthesis of several proteins needed for cortisol synthesis: (1) each of the P-450 enzymes involved in cortisol synthesis (Fig. 50-2), (2) the LDL receptor required for the uptake of cholesterol from blood (see Chapter 2), and (3) the 3-hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase that is the rate-limiting enzyme for cholesterol synthesis by the adrenal (see Chapter 45).
Thus, ACTH promotes the acute synthesis of cortisol—and as discussed later, aldosterone to a lesser extent—by the adrenal and increases the content of adrenal enzymes involved in steroidogenesis. In the absence of pituitary ACTH, the fasciculata and reticularis layers of the adrenal cortex atrophy. The glomerulosa layer does not atrophy under these conditions because in addition to ACTH, angiotensin II (ANG II) and high levels of K+ are trophic factors that act on the glomerulosa layer. The atrophy of the fasciculata and reticularis layers occurs routinely in people treated with glucocorticoid drugs and leaves the person with an iatrogenic form of adrenal insufficiency when use of the drug is abruptly discontinued. Conversely, chronic stimulation of the adrenal by ACTH, such as can occur with pituitary tumors (Cushing disease) or with the simple physiological ACTH excess that can occur with chronic stress, can increase the weight of the adrenals several-fold.
Cortisol exerts negative feedback control on the very axis that stimulates its secretion (Fig. 50-3), and it does so at the level of both the anterior pituitary and hypothalamus.
Feedback to the Anterior Pituitary In the corticotrophs of the anterior pituitary, cortisol acts by binding to a cytosolic receptor, which then moves to the nucleus, where it binds to GREs and modulates gene expression and thus inhibits the synthesis of both the CRH receptor and ACTH. Even though, as seen earlier, the POMC gene yields multiple secretory products, cortisol is the main regulator of the transcription of POMC. In addition, elevated levels of cortisol in plasma inhibit the release of presynthesized ACTH stored in vesicles.
Feedback to the Hypothalamus The negative feedback of cortisol on the CRH-secreting neurons of the hypothalamus is less important than that on the corticotrophs discussed earlier. Plasma cortisol decreases the mRNA and peptide levels of CRH in paraventricular hypothalamic neurons. Cortisol also inhibits the release of presynthesized CRH. Synthetic glucocorticoids have a similar action.
Control by a Higher CNS Center CRH-secreting neurons in the hypothalamus are under higher CNS control, as illustrated by two important features of the hypothalamic-pituitary-adrenocortical axis: (1) the circadian and pulsatile nature of ACTH and cortisol secretion and (2) integration of signals from higher cortical centers that modulate the body’s responses to a variety of stressors.
The pituitary secretes ACTH with a circadian rhythm. The suprachiasmatic nucleus of the hypothalamus, which lies above the optic chiasm and receives input from the retina, controls the circadian rhythms of the body. Indeed, blind people lose their circadian rhythms. Input from hypothalamic nuclei to the corticotrophs—through both CRH and ADH—appears to modulate the circadian secretion of ACTH and thus the circadian secretion of cortisol as well. As is the case for other hypothalamic releasing hormones, CRH is released in pulses. As a result, superimposed on the circadian rhythm of ACTH is the pulsatile secretion of ACTH, as shown in Figure 50-5. The greatest ACTH secretory activity occurs in the early morning and diminishes late in the afternoon and early evening. The mechanism by which hypothalamic neurons generate pulses of secretory activity is not understood.
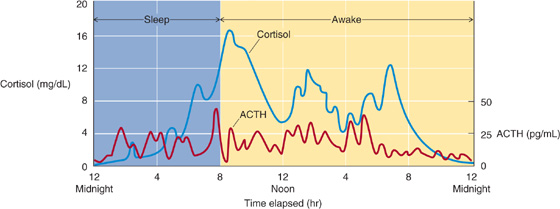
Figure 50-5 Rhythm of ACTH and cortisol. The corticotrophs release ACTH in a circadian rhythm, greater in the early morning hours and less late in the afternoon and early evening. Superimposed on the circadian rhythm is the effect on the corticotrophs of the pulsatile secretion of CRH by the hypothalamus. Thus, ACTH levels exhibit both circadian and pulsatile behavior. Although both ACTH and cortisol are secreted episodically, the duration of the ACTH bursts is briefer, reflecting the shorter half-life of ACTH in plasma. (Data from Young JB, Landsberg L: Catecholamines and the adrenal medulla. In Wilson JD, Foster DW, Kronenberg HM, Larsen PR (eds): Williams Textbook of Endocrinology, 9th ed, pp 665-728. Philadelphia: WB Saunders, 1998.)
Other evidence of higher CNS control is the enhanced CRH secretion—and thus the enhanced ACTH secretion—that occurs in response to physical, psychological, and biochemical stress. An example of biochemical stress is hypoglycemia, which stimulates the secretion of both CRH and ACTH and thus leads to an increased release of cortisol that tends to raise blood glucose levels.
The increase in ACTH secretion that occurs nocturnally and with stress appears to be the result of an increased amplitude of the secretory CRH burst, rather than an increased frequency of secretion episodes. Because the half-life of cortisol is much longer than that of ACTH, the period of the pulsatile changes in cortisol is longer and the magnitude of the excursions is damped in comparison with those of ACTH.
Aldosterone determines extracellular volume by controlling the extent to which the kidney excretes or reabsorbs the Na+ filtered at the renal glomerulus. Na+ in the extracellular space retains water—it is the primary osmotically active particle in the extracellular space—and thus the amount of Na+ that is present determines the volume of extracellular fluid (see Chapter 5). The extracellular volume is itself a prime determinant of arterial blood pressure (see Chapter 23), and therefore aldosterone plays an important role in the maintenance of blood pressure.
The effects of aldosterone on salt balance determine the extracellular volume and should not be confused with the effects of AVP (also known as antidiuretic hormone or ADH). AVP regulates the free water balance of the body (see Chapter 40). Water freely passes across cell membranes and thus affects the concentration of Na+ and other solutes throughout the body (see Chapter 5). Unlike aldosterone, AVP makes only a small contribution to the maintenance of extracellular volume; instead, AVP regulates serum osmolality and hence the Na+ concentration. Thus, to a first approximation, one can think of aldosterone as the primary regulator of extracellular volume because of its effect on renal Na+ reabsorption and AVP as the primary regulator of plasma osmolality because of its effect on free water balance.
As is the case for cortisol, the adrenal cortex synthesizes aldosterone from cholesterol by using P-450 enzymes in a series of five steps. The initial steps in the synthesis of aldosterone from cholesterol follow the same synthetic pathway that cortisol-secreting cells use to generate progesterone (Fig. 50-2). Because glomerulosa cells are the only ones that contain aldosterone synthase, these cells are the exclusive site of aldosterone synthesis.
1. The cytochrome P-450 SCC enzyme (P-450SSC) produces pregnenolone from cholesterol. This enzyme—or the supply of substrate to it—appears to be the rate-limiting step for the overall process of steroid hormone synthesis.
2. The SER enzyme 3β-HSD, which is not a P-450 enzyme, oxidizes pregnenolone to form progesterone.
3. Because glomerulosa cells have minimal 17α-hydroxylase (P-450c17), they do not convert progesterone to 17α-hydroxyprogesterone. Instead, glomerulosa cells use a 21α-hydroxylase (P-450c21) in the SER to hydroxylate the progesterone further at position 21 and to produce 11-deoxycorticosterone (DOC).
4. In the mitochondria, 11β-hydroxylase (P-450c11) adds an —OH at position 11 to produce corticosterone. This pair of hydroxylations in steps 3 and 4 are catalyzed by the same two enzymes that produce cortisol from 17α-hydroxyprogesterone.
5. The glomerulosa cells—but not the fasciculata and reticularis cells—also have aldosterone synthase (P-450aldo), which first adds an —OH group to the methyl at position 18 and then oxidizes this hydroxyl to an aldehyde group, hence the name aldosterone. This mitochondrial P-450 enzyme, also called 18-methyloxidase, is an isoform of the same 11β-hydroxylase (P-450c11) that catalyzes the DOC-to-corticosterone step. In fact, aldosterone synthase can catalyze all three steps between DOC and aldosterone: 11β-hydroxylation, 18-methyl hydroxylation, and 18-methyl oxidation.
As with cortisol, no storage pool of presynthesized aldosterone is available in the glomerulosa cell for rapid secretion. Thus, secretion of aldosterone by the adrenal is limited by the rate at which the glomerulosa cells can synthesize the hormone. Although ACTH also stimulates the production of aldosterone in the glomerulosa cell, increases in extracellular [K+] and the peptide hormone ANG II are physiologically more important secretagogues. These secretagogues enhance secretion by increasing the activity of enzymes acting at rate-limiting steps in aldosterone synthesis. These enzymes include the SCC enzyme, which is common to all steroid-producing cells, and aldosterone synthase, which is unique to glomerulosa cells and is responsible for formation of the C-18 aldehyde.
Once secreted, ~37% of circulating aldosterone remains free in plasma. The rest weakly binds to CBG (~21%) and albumin (~42%).
The major action of aldosterone is to stimulate the kidney to reabsorb Na+ and water and to enhance K+ secretion. Aldosterone has similar actions on salt and water transport in the colon, salivary glands, and sweat glands. MRs are also present in the myocardium, liver, brain, and other tissues, but the physiological role of mineralocorticoids in these latter tissues is unclear.
Aldosterone, like cortisol and all the other steroid hormones (see Chapter 4), acts by modulating gene transcription. In the kidney, aldosterone binds to both low- and high-affinity receptors. The low-affinity receptor appears to be identical to the GR. The high-affinity receptor is a distinct MR; it has homology to the GR, particularly in the zinc finger region involved in DNA binding. Surprisingly, MR in the kidney has a similar affinity for aldosterone and cortisol. Because cortisol normally circulates at much higher concentrations than does aldosterone (5 to 20 μg/dL versus 2 to 8 ng/dL), the biological effect of aldosterone on any potential target would be expected to be greatly overshadowed by that of cortisol. (Conversely, aldosterone has essentially no significant glucocorticoid action because aldosterone binds only weakly to its low-affinity receptor—that is, the GR.)
How then do the renal tubule cells avoid sensing cortisol as a mineralocorticoid? As noted earlier, the cells that are targets for aldosterone, particularly the distal convoluted tubule and the initial cortical collecting duct of the kidney, contain an enzyme (11β-hydroxysteroid dehydrogenase 2) that converts cortisol to cortisone, which has a very low affinity for the MR (see Fig. 35-13B). Unlike 11β-HSD1, which reversibly interconverts cortisone with cortisol, 11β-HSD2 cannot convert cortisone back to cortisol. As a result, locally within the target cell, the ratio of cortisol to aldosterone is much smaller than the cortisol dominance seen in plasma. In other words, 11β-HSD2 is so effective at removing cortisol from the cytosol of aldosterone target tissues that cortisol behaves as only a weak mineralocorticoid despite the high affinity of cortisol for the so-called MR. Thus, the presence of 11β-HSD2 effectively confers aldosterone specificity on the MR.
In the target cells of the renal tubule, aldosterone increases the activity of several key proteins involved in Na+ transport (see Chapter 35). It increases transcription of the Na-K pump, thus augmenting distal Na+ reabsorption. Aldosterone also raises the expression of apical Na+ channels and an Na/K/Cl cotransporter. The net effect of these actions is to increase Na+ reabsorption and K+ secretion. The enhanced K+ secretion (see Chapter 37) appears to occur as a secondary effect to the enhanced Na+ reabsorption. However, the stoichiometry between Na+ reabsorption and K+ secretion in the distal tubule is not fixed.
Aldosterone regulates only that small fraction of renal Na+ reabsorption that occurs in the distal tubule and collecting duct. Although most Na+ reabsorption occurs in the proximal tubule by aldosterone-independent mechanisms, loss of aldosterone-mediated Na+ reabsorption can result in significant electrolyte abnormalities, including life-threatening hyperkalemia and, in the absence of other compensatory mechanisms, hypotension. Conversely, excess aldosterone secretion produces hypokalemia and hypertension.
Hyperaldosteronism is an uncommon cause of hypertension. Primary aldosteronism can result from either an isolated adrenal adenoma or bilateral adrenal hyperplasia; even more rarely, adrenal carcinoma can produce excess aldosterone. (See Note: Licorice as a Cause of Apparent Mineralocorticoid Excess)
Three secretagogues are known for the aldosterone synthesized in the glomerulosa cells of the adrenal cortex. The most important is ANG II, which is a product of the renin-angiotensin cascade. An increase in plasma [K+] is also a powerful stimulus for aldosterone secretion. Third, just as ACTH promotes cortisol secretion, it also promotes the secretion of aldosterone, although this effect is weak.
Renin-Angiotensin The renin-angiotensin-aldosterone axis is introduced in the discussion of the control of extracellular fluid volume in Chapter 40. The liver synthesizes and secretes a very large protein called angiotensinogen, which is an α2-globulin (Fig. 50-6). Renin, which is synthesized by the granular cells (or juxtaglomerular cells) of the juxtaglomerular apparatus (JGA) in the kidney (see Chapter 33), is the enzyme that cleaves this angiotensinogen to form ANG I, a decapeptide. Finally, angiotensin-converting enzyme (ACE) cleaves the ANG I to form the octapeptide ANG II. ACE is present in both the vascular endothelium of the lung (~40%) and elsewhere (~60%). In addition to its role as a potent secretagogue for aldosterone, ANG II exerts powerful vasoconstrictor actions on vascular smooth muscle (see Chapter 23). ANG II has a short half-life (<1 minute) because plasma aminopeptidases further cleave it to the heptapeptide ANG III. (See Note: Metabolism of the Angiotensins)
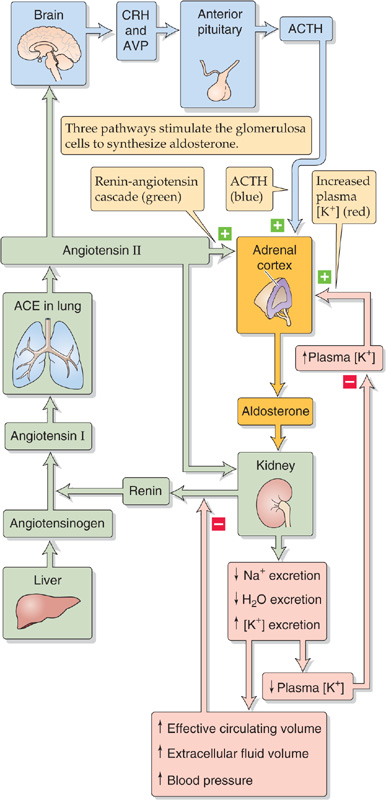
Figure 50-6 Control of aldosterone secretion. Three pathways, shown in three different colors, stimulate the glomerulosa cells of the adrenal cortex to secrete aldosterone.
On the plasma membrane of the glomerulosa cell, ANG II binds to the AT1 receptor (type 1 ANG II receptor), which couples through the Gαq-mediated pathway to phospholipase C (PLC). Stimulation of PLC leads to the formation of diacylglycerol (DAG) and inositol 1, 4, 5-triphosphate (IP3; see Chapter 3). DAG activates protein kinase C (PKC). IP3 triggers the release of Ca2+ from intracellular stores, thus causing a rise in [Ca2+]i, which activates Ca2+-dependent enzymes such as PKC and Ca2+–calmodulin-dependent protein kinases. These changes lead to depolarization of the glomerulosa cell’s plasma membrane, opening of voltage-activated Ca2+ channels, and a sustained increase in Ca2+ influx from the extracellular space. This rise in [Ca2+]i is primarily responsible for triggering the synthesis (i.e., secretion) of aldosterone. Aldosterone secretion increases because the rise in [Ca2+]i facilitates the production of pregnenolone either by directly increasing the activity of SCC or by enhancing the delivery of cholesterol to the SCC enzyme in the mitochondria (Fig. 50-2). In addition, increased [Ca2+]i also stimulates aldosterone synthase and in this manner enhances the conversion of corticosterone to aldosterone.
Treating Hypertension by Attacking the Renin-Angiotensin-Aldosterone Axis
Because the renin-angiotensin-aldosterone axis plays an important role in maintaining extracellular volume and arterial pressure, pharmacologists have sought ways to disrupt this system to treat hypertension. Several agents acting on different parts of the axis are available. Spironolactone is a diuretic that directly antagonizes the effects of aldosterone on the renal tubule. It is a weak diuretic, but it is particularly useful in treating patients with ascites. For patients with hypertension or congestive heart failure, it is usually added to one of the more common thiazide diuretics (see Chapter 35) to prevent K+ wasting.
ACE inhibitors have been available for many years and are among the safest and best-tolerated of all antihypertensive medications. The first to be marketed in the United States was captopril, but many others, with longer half-lives that allow once-daily administration, are now available. These drugs also improve the quality of life and survival of patients with heart failure. More recently, drugs have been developed that specifically block the AT1 receptor. These drugs offer a good alternative for those patients who cannot tolerate ACE inhibitors, usually because of a chronic cough, the most common side effect of these drugs.
Potassium An increase in extracellular K+ ([K+]o) has a more direct action on the glomerulosa cell (Fig. 50-6). High [K+]o depolarizes the plasma membrane and opens voltage-gated Ca2+ channels. The result is an influx of Ca2+ and a rise in [Ca2+]i that stimulates the same two steps as ANG II—production of pregnenolone from cholesterol and conversion of corticosterone to aldosterone. Unlike the situation for ANG II, the [Ca2+]i increase induced by high [K+]o does not require activation of PLC or release of Ca2+ from the intracellular stores. Because increased [K+]o and ANG II both act by raising [Ca2+]i, they act synergistically on the glomerulosa cell.
ACTH ACTH has only a minor effect on aldosterone secretion by glomerulosa cells (Fig. 50-6). ACTH regulates aldosterone secretion by a pathway that is distinct from that of ANG II or high [K+]o. As noted earlier for the fasciculata and reticularis cells, MC2R in the glomerulosa cell is coupled through a heterotrimeric G protein to adenylyl cyclase. Increases in ACTH raise [cAMP]i and activate PKA, which phosphorylates large numbers of cytosolic proteins. At some as yet undefined level, these changes stimulate Ca2+ influx across the plasma membrane and enhance the synthesis and secretion of aldosterone. ACTH also enhances mineralocorticoid activity by a second mechanism: stimulation of the fasciculata cells to secrete cortisol, corticosterone, and DOC, all of which have weak mineralocorticoid activity. The hypertension seen in individuals who oversecrete ACTH appears to be mediated by the excess synthesis of these weak mineralocorticoids. Neither ANG II nor hyperkalemia affects the cAMP pathway triggered by ACTH.
The feedback regulation exerted by aldosterone is indirect and occurs through its effects of both increasing salt retention (i.e., extracellular volume) and decreasing [K+]o.
Renin-Angiotensin Axis As discussed in Chapter 40, a decrease in effective circulating volume stimulates the granular cells of the JGA of the kidney to increase their synthesis and release of renin, which increases the generation of ANG II and, therefore, aldosterone (Fig. 50-6). The JGA is located at the glomerular pole of the nephron, between the afferent and efferent arterioles, where the early distal tubule comes in close proximity with its own glomerulus (Fig. 50-7). Histologically, the JGA comprises specialized epithelial cells of the distal tubule called macula densa cells, as well as specialized smooth muscle cells of the afferent arteriole, which are called granular cells or juxtaglomerular cells. Macula densa cells and granular cells communicate by means of an extracellular matrix.
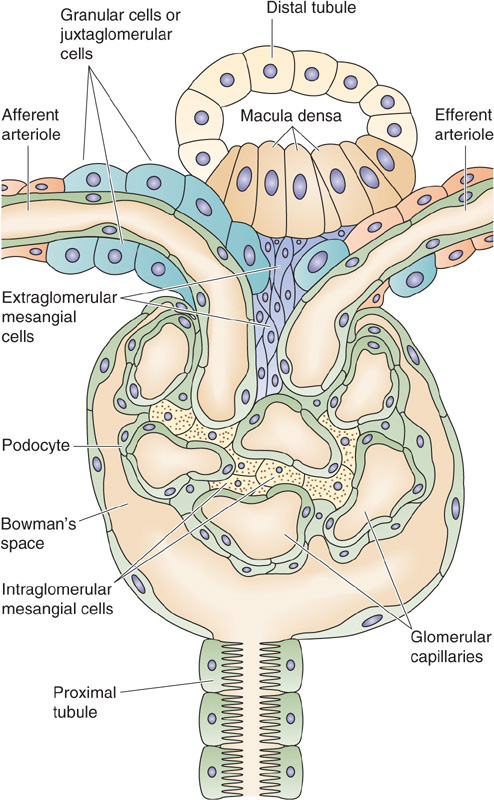
Figure 50-7 Structure of the JGA.
Decreases in effective circulating volume—or the associated decreases in systemic arterial pressure—stimulate renin release from the JGA in two ways (see Chapter 40): first, decreased systemic arterial blood pressure stimulates the baroreceptor reflex, which triggers medullary control centers to increase sympathetic outflow to the JGA. The result of both α-and β-adrenergic stimulation is enhanced renin secretion. Second, “renal” baroreceptors in the afferent arteriole—possibly the granular cells themselves—respond to a fall in pressure of the afferent arteriole (i.e., decreased stretch in the arteriolar wall) by increasing the secretion of renin. Thus, both mechanisms enhance renin release and lead to increased levels of ANG II and aldosterone. ANG II negatively feeds back on renin release by directly inhibiting renin release by the kidney (short-loop feedback). ANG II also negatively feeds back on renin release by acutely increasing blood pressure (see Chapter 23), a process that reduces the stimuli to the aforementioned two baroreceptor pathways. Finally, aldosterone negatively feeds back on renin release more slowly by enhancing renal Na+ reabsorption (see Chapter 35) and thus increasing effective circulating blood volume and blood pressure. Therefore, ANG II and aldosterone complete the regulatory feedback circuit that governs the secretion of aldosterone.
Potassium High plasma [K+] stimulates the glomerulosa cell in the adrenal cortex to synthesize and release aldosterone, which, in turn, stimulates the principal cells of the renal collecting duct to reabsorb more Na+ and excrete more K+ (Fig. 50-6). This excretion of K+ causes plasma [K+] to fall toward normal. As a result, stimulation of glomerulosa cells declines, aldosterone secretion falls, and the negative feedback loop is completed. This sequence of events (i.e., hyperkalemia → aldosterone secretion → K+ excretion) probably plays a vital role in preventing wide swings in plasma [K+] in response to episodic dietary intake of large K+ loads.
Role of Aldosterone in Normal Physiology What, then, is the role of aldosterone in normal physiology? Presumably, the salt- and water-retaining properties of aldosterone are of greatest value in meeting the environmental stresses associated with limited availability of salt, water, or both. Such conditions are not prevalent in most Western societies, but they still exist in many developing countries and were probably universal in previous periods of human evolutionary history. In healthy, normotensive humans, blockade of aldosterone generation with ACE-inhibiting drugs reduces ANG II production and markedly decreases plasma aldosterone, but it causes only slight decreases in total body Na+ and blood pressure. Redundant mechanisms for maintaining blood pressure probably prevent a larger blood pressure decrease. The reason for the mild effect on Na+ balance is probably an adaptive increase in salt intake; indeed, patients with adrenal cortical insufficiency frequently crave dietary salt. In contrast to the minor effects of low aldosterone on blood pressure and Na+ balance in physiologically normal people, the effects of blocking aldosterone production can be catastrophic for K+ balance; low aldosterone can result in life-threatening hyperkalemia.
Role of Aldosterone in Disease Aldosterone does play important roles in several pathologic conditions. For example, in many patients with hypertension, ACE inhibitors are effective in reducing blood pressure, a finding implying that their renin-angiotensin-aldosterone axis was overactive. In hypotension, as occurs with hemorrhage or dehydration, aldosterone secretion increases, thus increasing effective circulating volume and blood pressure.
Aldosterone secretion also increases in congestive heart failure. However, the increased salt retention in this setting is inappropriate because it results in worsening edema formation. In this case, the increase in circulating aldosterone occurs despite preexisting volume overload. The problem in congestive heart failure is that the JGA does not perceive the very real volume overload as an increase in effective circulating volume. Indeed, the reduced cardiac output in heart failure diminishes renal glomerular filtration, partly because of decreased arterial blood pressure and partly because of enhanced sympathetic nervous system activity, which constricts the afferent arterioles of the kidney. As a result, the kidney inappropriately assumes that the extracellular volume is decreased and stimulates the renin-angiotensin-aldosterone system.
Spontaneous increases in aldosterone synthesis can occur in patients with tumors of the glomerulosa cell, a disorder called hyperaldosteronism or Conn syndrome. Hypertension and hypokalemia frequently develop in these patients. As would be expected from feedback regulation of the reninangiotensin-aldosterone system, the plasma renin concentration is characteristically suppressed in this form of hypertension.
In addition to its renal effects, aldosterone increases fibrosis within certain tissues, including myocardium and blood vessel wall. Agents that block the aldosterone receptor (e.g., spironolactone or eplerenone) have been used successfully to antagonize these actions of aldosterone and to improve clinical outcomes in patients with heart failure. This effect appears to be independent of any diuretic effect of these agents.
In Chapter 47, I describe cells of the hypothalamus as neuroendocrine because they are part of the CNS and appear anatomically as neural tissue, yet they release peptide hormones (e.g., CRH) into the blood that act downstream on the pituitary—a classic endocrine function. The adrenal medulla is similar in many ways. The cells of the medulla, termed chromaffin cells because the catecholamines that they contain stain avidly with chromium, derive from neural crest cells (see Chapter 10) and migrate into the center of the adrenal cortex, which is derived from the mesoderm. The adrenomedullary cells synthesize and secrete epinephrine and—to a lesser extent—norepinephrine. Norepinephrine is the neurotransmitter of the sympathetic division of the autonomic nervous system (see Chapter 14). Both the norepinephrine and epinephrine made in the adrenal medulla enter the circulation and act on distal tissues just like other hormones.
Chromaffin cells are the structural and functional equivalents of the postganglionic neurons in the sympathetic nervous system (see Chapter 14). The preganglionic sympathetic fibers of the splanchnic nerves, which release acetylcholine (ACh), are the principal regulators of adrenomedullary hormone secretion.
The vascular supply to the adrenal medulla is also unusual. The medulla receives vascular input from vessels that begin in a subcapsular plexus of the adrenal cortex. The vessels then branch into a capillary network in the cortex only to merge into small venous vessels that branch into a second capillary network within the medulla. This portal blood supply (originating at the entrance to the adrenal) exposes the adrenal medulla to the highest concentrations of glucocorticoids and mineralocorticoids of all somatic tissues.
Investigators first appreciated nearly a century ago that extracts of the adrenal medulla have a powerful pressor effect. Subsequent work showed that the catecholamines—dopa, dopamine, norepinephrine, and epinephrine—are all made in the adrenal medulla. Norepinephrine is found in many other somatic tissues in amounts that roughly parallel the extent of sympathetic innervation of the tissue. In other words, the norepinephrine in these other tissues is not made there but is derived from the sympathetic nerve endings in them. Epinephrine, the principal product of the adrenal medulla, is made only in the adrenal medulla.
Dopamine, norepinephrine, and epinephrine are all synthesized from the amino acid tyrosine. Figure 50-8A summarizes the reactions involved in the synthesis of epinephrine. Figure 50-9 illustrates the cellular localization of the four enzymatic reactions, as well as the three critical transport steps that shuttle the reactants and products to their proper location:
1. The activity of the first enzyme in the pathway, tyrosine hydroxylase, which converts tyrosine to L-dopa, is rate limiting for overall synthesis. This enzyme is located within the cytosol of adrenal medullary cells, as well as in sympathetic nerve terminals and in specific cells within the CNS.
2. The cytosolic enzyme amino acid decarboxylase converts L-dopa to dopamine in numerous tissues, including the adrenal medulla.
3. A catecholamine-H+ exchanger (VMAT1) moves the dopamine into membrane-enclosed dense-core vesicles called chromaffin granules.
4. Dopamine β-hydroxylase converts dopamine to norepinephrine by hydroxylating the β carbon. This β-hydroxylase is localized to the inner surface of the membrane of granules within the adrenal medulla and sympathetic nerves. In the nerve terminals of postganglionic sympathetic neurons, the synthetic pathway terminates at this step, and the granules store the norepinephrine for later secretion. However, the cells of the adrenal medulla convert the norepinephrine to epinephrine in three final steps.
5. Norepinephrine formed in the secretory granules moves out into the cytosol.
6. The cytosolic enzyme phenylethanolamine-N-methyltransferase (PNMT) transfers a methyl group from S-adenosylmethionine to norepinephrine, thus creating epinephrine. This enzyme is present only in the cytosol and the adrenal medulla.
7. The secretory granules in the adrenal medulla take up the newly synthesized epinephrine. The same VMAT1 catecholamine-H+ exchanger noted in step 3 appears to mediate this uptake of epinephrine. The proton gradient is maintained by an H+ pump (i.e., a vacuolar-type HATPase) within the secretory vesicle membrane (see Chapter 5). Thus, in the adrenal medulla, the secretory granules store both epinephrine and norepinephrine before secretion.
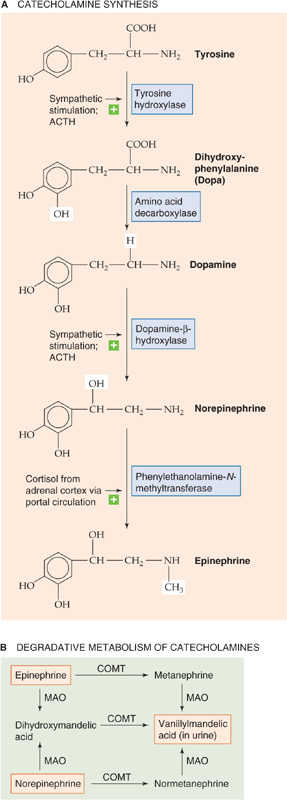
Figure 50-8 A and B, Synthesis and degradation of catecholamines. In A, the horizontal arrows indicate enhancement of the reaction. MAO, monoamine oxidase.
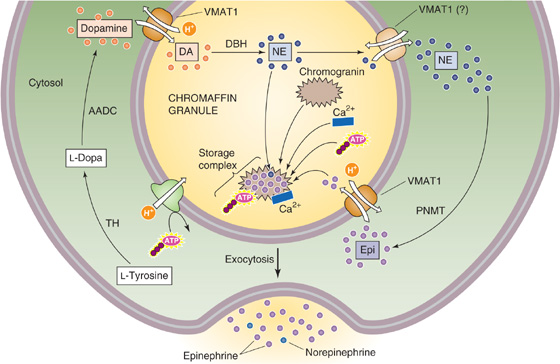
Figure 50-9 Cellular view of catecholamine synthesis. The chromaffin cell synthesizes and stores epinephrine in a sequence of four enzymatic and three transport steps. AADC, amino acid decarboxylase; DA, dopamine; DBH, dopamine β-hydroxylase; Epi, epinephrine; NE, norepinephrine; TH, tyrosine hydroxylase.
Epinephrine synthesis is under control of the CRH-ACTH-cortisol axis at two levels. First, ACTH stimulates the synthesis of dihydroxyphenylalanine (Dopa) and norepinephrine. Second, cortisol transported from the adrenal cortex by the portal circulation to the medulla upregulates PNMT in chromaffin cells. The result is synergy between the CRH-ACTH-cortisol axis and the sympathetic-epinephrine axis. Thus, the stress that is sensed and propagated by the CRH-ACTH-cortisol axis sustains the epinephrine response.
Similar to the secretory granules that are present in the postganglionic sympathetic neurons, the secretory granules of the adrenal medulla contain very high concentrations of catecholamines (≤0.5 M). These catecholamines—along with ATP and Ca2+—bind to granular proteins called chromogranins and thus are not osmotically active in these storage vesicles. Chromogranins are what make dense-core vesicles dense. In humans, the dominant chromogranin is chromogranin B. The release of catecholamines is initiated by CNS control. ACh released from preganglionic neurons in the splanchnic nerves acts on nicotinic ACh receptors to depolarize the postganglionic chromaffin cells. This depolarization triggers the opening of voltage-gated Ca2+ channels, a process that raises [Ca2+]i and triggers the exocytotic release of epinephrine. The secretion of adrenal catecholamines is accompanied by the release of ATP and the granule proteins. The release of chromogranin A has been used as a marker of adrenal medullary activity. In the circulation, the catecholamines dissociate from the binding complex and are free to act on target tissues.
The early description of the fight-or-flight response to stress (see Chapter 14) exemplifies the central control of adrenomedullary function. An organism faced with a severe external threat responds with centrally driven release of adrenal hormones, as well as activation of other aspects of the sympathetic division. This response includes increases in heart rate and contractility, mobilization of fuel stores from muscle and fat, piloerection, pupillary dilatation, and increased sphincter tone of the bowel and bladder. Each response is in some way adapted to deal with the perceived threat successfully. This combined neuroendocrine response is activated within seconds. The secreted catecholamines act very quickly after reaching their target tissues.
The biological actions of catecholamines are very brief, lasting only ~10 seconds in the case of epinephrine. Circulating catecholamines are degraded first by the enzyme catecholamine-O-methyltransferase (COMT), which is present in high concentrations in endothelial cells and the heart, liver, and kidneys (Fig. 50-8B). COMT converts epinephrine to metanephrine, as well as norepinephrine to normetanephrine. A second enzyme, monoamine oxidase, converts these metabolites to vanillylmandelic acid (VMA). The liver and also the gut then conjugate these compounds to sulfate or glucuronide (see Chapter 46) to form derivatives that the kidney excretes in the urine. Measurement of the concentration of catecholamines, metanephrines, and VMA in the urine provides a measure of the total adrenal catecholamine production by both the adrenal medulla and the sympathetic system.
Many of the hormones already discussed have a unique receptor on the cell surface (e.g., insulin, parathyroid hormone, growth hormone, thyroid-stimulating hormone) or within the cell (e.g., thyroid hormones or cortisol). Other hormones (e.g., AVP) and neurotransmitters (e.g., ACh and glutamate) may bind to more than one type of receptor, each acting through a different signal transduction process. The situation for the catecholamines is even more complex. Epinephrine and norepinephrine can each bind to more than one type of adrenergic receptor or adrenoceptor, all of which are GPCRs. Conversely, individual adrenoceptors can generally bind both epinephrine and norepinephrine—albeit with different affinities.
About a half century ago, investigators found that epinephrine could stimulate both vasodilatory and vasoconstrictor responses in the same vascular bed, depending on the epinephrine concentration. This property, together with the observation that certain drugs could selectively block one or the other of these effects, led to the designation of α-and β-adrenergic receptors. This dichotomy proved too simple when it was observed that the actions of some drugs that have pure α or pure β activity could be blocked in some tissues but not in others; this variability in response suggested that the tissue response is determined by a subtype of α or β receptor.
It is now clear that at least three types of β and two types of α receptors exist (see Table 14-2), as well as subtypes within these major classes. These receptors differ in primary structure and in the types of G proteins that associate with the receptor. The several β receptors are coupled to stimulatory heterotrimeric G proteins (Gαs) that stimulate adenylyl cyclase and thus increase levels of cAMP, the principal intracellular mediator of β activation. The α2 adrenoceptors are coupled to other G proteins (Gαi) that inhibit adenylyl cyclase and thus lower [cAMP]i in target tissues. The α1 receptors are coupled to yet another heterotrimeric G protein (Gαq) that either activates PLC or, in some cases, appears to alter the activity of a plasma membrane ion channel directly (see Chapter 3). The net effect of α1 stimulation is to increase the concentration of IP3 and [Ca2+]i in target tissues.
Recognition of the diversity of adrenoceptor subtypes has led to a panoply of pharmacological agents that block or stimulate one or the other of these receptor subtypes. Some drugs are nonspecific and affect several receptor subtypes; others specifically block only a single subtype. The clinical value of a particular drug depends on its spectrum of activity. Thus, for example, an agent that selectively blocks the vasoconstrictor response to norepinephrine could be a very useful antihypertensive agent, but its utility would be compromised if it also blocked the ability of bladder smooth muscle to contract.
The actions of the autonomic nervous system (see Chapter 14) in the control of blood pressure and heart rate (see Chapter 22), sweating (see Chapter 59), micturition (see Chapter 32), and airway resistance (see Chapter 27) are discussed in more detail in other chapters, as indicated. Here, I mention only some of the unique actions attributed to adrenal catecholamine release that integrate several bodily functions as part of the stress response. These adrenally mediated activities do not occur in isolation but are usually accompanied by generalized noradrenergic sympathetic discharge.
In response to the stress of simple exercise (see Chapters 25 and 60), blood flow to muscle is increased; circulating epinephrine appears to be important in this response. Circulating epinephrine also relaxes bronchial smooth muscle to meet the demand for increased ventilation and, when combined with the increased blood flow, increases oxygen delivery to the exercising muscle. Similarly, to sustain muscular activity, particularly early in exercise, epinephrine acting through the β adrenoceptor activates the degradation of muscle glycogen to provide a ready fuel source for the contracting muscle (see Chapter 3). Epinephrine also activates lipolysis in adipose tissue to furnish free fatty acids for more sustained muscular activity if needed. In liver, as in muscle, epinephrine activates glycogenolysis, thus maintaining the supply of glucose in the blood.
In addition to enhancing blood flow and ventilation, the integrated response to exercise increases fuel availability by decreasing insulin levels. Circulating epinephrine, acting through a β adrenoceptor, stimulates the secretion of insulin (see Chapter 51). However, during exercise, local autonomic innervation, acting by means of an α adrenoceptor of the pancreas, inhibits this effect, so insulin levels fall. The net effects are to promote glycogenolysis and to allow muscle to increase its work while maintaining glycemia so that brain function is not impaired. The fleeing human must not only run, but know where to run.
Unlike other glandular tissue, no endocrine feedback loop governs the secretion of adrenal medullary hormones. Control of catecholamine secretion resides within the CNS. This principle can be illustrated by the changes in epinephrine secretion that occur with even mild hypoglycemia. Decreases in blood glucose to less than ~3.5 mM (normally, ~5.5 mM) are sensed by the CNS, which triggers a central sympathetic response that increases the firing of preganglionic fibers in the celiac plexus. This sympathetic outflow suppresses endogenous insulin secretion by the α-adrenergic mechanism noted earlier, thus promoting an increase in plasma [glucose]. This sympathetic outflow to the adrenal medulla also triggers a major release of epinephrine that, through β adrenoreceptors in the liver, stimulates increased hepatic glycogenolysis. This response helps to restore plasma [glucose] to normal. Restoration of normoglycemia diminishes central sympathetic outflow.
Pheochromocytoma
The dramatic biological effects of catecholamines are well illustrated by patients with pheochromocytoma. A pheochromocytoma is a relatively uncommon tumor caused by hyperplasia of either adrenal medullary tissue or extra-adrenal chromaffin tissue that failed to involute after birth. These tumors can be benign or malignant. They make catecholamines, just like the normal medulla, except in an unregulated fashion. Patients with pheochromocytomas typically have a plethora of symptoms, as would be expected from such a wide-ranging hormonal system. Paroxysmal (sudden outburst) hypertension, tachycardia, headache, episodes of sweating, anxiousness, tremor, and glucose intolerance usually dominate the clinical findings. The key to the diagnosis of this disorder is a careful history, evidence on physical examination of excessive adrenergic tone, and laboratory detection of increased amounts of urinary catecholamines and their metabolites. When chemical evaluation of the urinary metabolites confirms the presence of a pheochromocytoma, it is often possible to localize the tumor to one or the other adrenal gland and to resect the tumor. Rarely, both glands are affected, and bilateral adrenalectomy is necessary. Such patients must subsequently receive glucocorticoid and mineralocorticoid replacement. No therapy is routinely given to replace the adrenal medullary function. It is not clear whether these individuals react less well to external stimuli that would trigger the fight-or-flight response.
Books and Reviews
Autelitano DJ, Lundblad JR, Blum M, Roberts JL: Hormonal regulation of POMC gene expression. Annu Rev Physiol 1989, 51:715-726.
Burnstein KL, Cidlowski JA: Regulation of gene expression by glucocorticoids. Annu Rev Physiol 1989; 51:683-699.
Fitzsimons JT: Angiotensin, thirst, and sodium appetite. Physiol Rev 1998; 78:583-686.
Funder JW: Glucocorticoid and mineralocorticoid receptors: Biology and clinical relevance. Annu Rev Med 1997; 48:231-240.
Young JB, Landsberg L: Catecholamines and the adrenal medulla. In Wilson JD, Foster DW, Kronenberg HM, Larsen PR (eds): Williams Textbook of Endocrinology, 9th ed, pp 665-728. Philadelphia: WB Saunders, 1998.
Journal Articles
Fevold HR: Regulation of the adrenal cortex secretory pattern by adrenocorticotropin. Science 1967; 156:1753-1755.
Gordon RD: Primary aldosteronism. J Endocrinol Invest 1995; 18:495-511.
Smith GW, Aubry JM, Dellu F, et al: Corticotropin releasing factor receptor 1–deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron 1998; 20:1093-1102.
Turnbull AV, Rivier C: Corticotropin-releasing factor (CRF) and endocrine responses to stress: CRF receptors, binding protein, and related peptides. Proc Soc Exp Biol Med 1997; 215:1-10.
Weitzman ED, Fukushima D, Nogeire C, et al: Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. J Clin Endocrinol Metab 1971; 33:14-22.