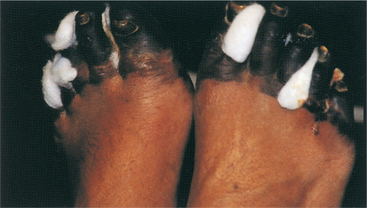
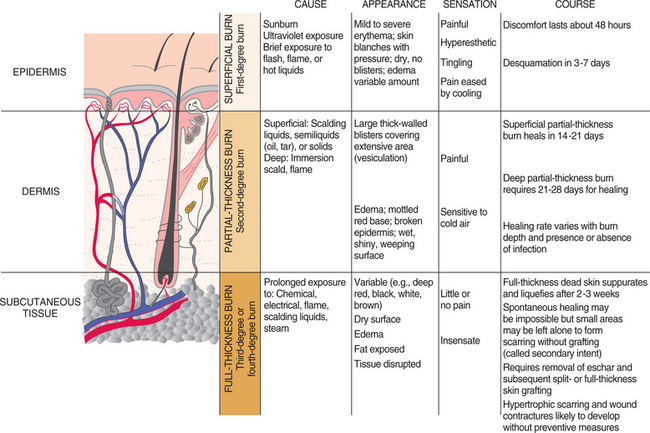
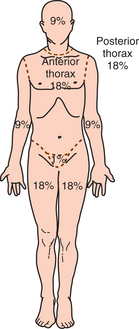
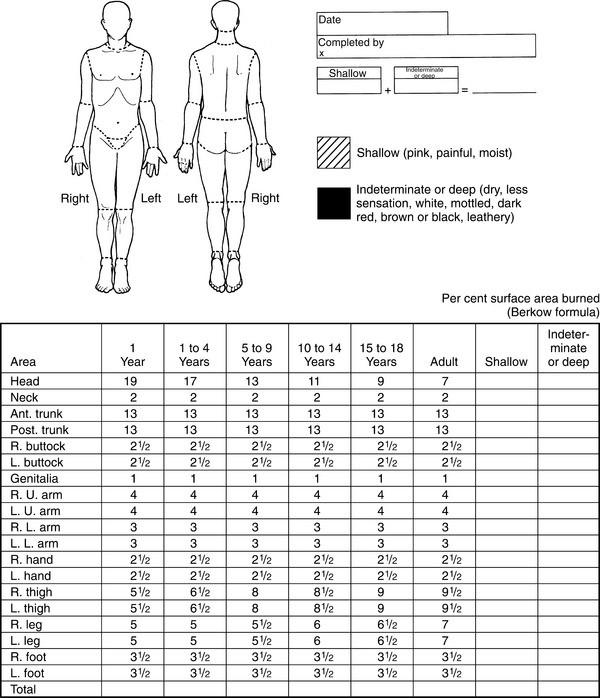
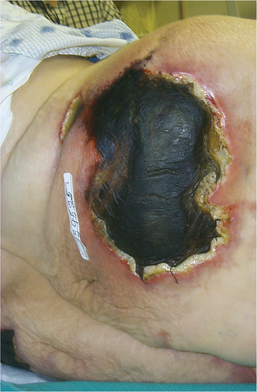
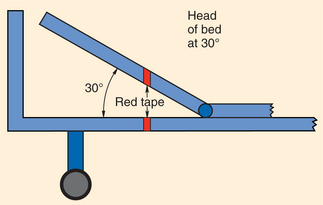
1. Aboulafia, DM. The epidemiologic, pathologic, and clinical features of AIDS-associated pulmonary Kaposi’s sarcoma. Chest. 2000;117(4):1128–1145.
2. Akula, SM, Ford, PW, Whitman, AG, et al. B-Raf-dependent expression of vascular endothelial growth factor-A in Kaposi Sarcoma associated herpesvirus-infected human B cells. Blood. 2005;105(11):4516–4522.
3. Arasteh, K, Hannah, A. The role of vascular endothelial growth factor (VEGF) in AIDS-related Kaposi’s sarcoma. Oncologist. 2000;5(suppl 1):28–31.
4. Atkinson, W, Wolfe, C, Humiston, S, et al. Epidemiology and prevention of vaccine-preventable diseases. Bethesda, MD: Centers for Disease Control and Prevention, Department of Health and Human Services, 2000.
5. Baddour, LM. Breast cellulitis complicating breast conservation therapy. J Intern Med. 1999;245(1):5–9.
6. Bagel, J. Establishing a practical and effective psoriasis treatment center. Dermatol Clin. 2000;18(2):349–357.
7. Ballard, T, Lagorio, S, De Angelis, G, et al. Cancer incidence and mortality among flight personnel: a meta-analysis. Aviat Space Environ Med. 2000;71(3):216–224.
8. Belsito, DV. The diagnostic evaluation, treatment, and prevention of allergic contact dermatitis in the new millennium. J Allergy Clin Immunol. 2000;105(3):409–420.
9. Bergstrom, N. Treatment of pressure ulcers. Bethesda, MD: Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research, 1994. [Clinical practice guideline no. 15].
10. Bergstrom, N, Braden, B, Kemp, M, et al. Multi-site study of incidence of pressure ulcers and the relationship between risk level, demographic characteristics, diagnoses, and prescription of preventive interventions. J Am Geriatr Soc. 1996;44(1):22–30.
11. Berlowitz, DR, Anderson, JJ, Ash, AS, et al. Reducing random variation in reported rates of pressure ulcer development. Med Care. 1998;36(6):818–825.
12. Bianchi, DW. Fetomaternal cell trafficking: a new cause of disease? Am J Med Genet. 2000;91(1):22–28.
13. Blettner, M, Zeeb, H, Auvinen, A, et al. Mortality from cancer and other causes among male airline cockpit crew in Europe. Int J Cancer. 2003;106(6):946–952.
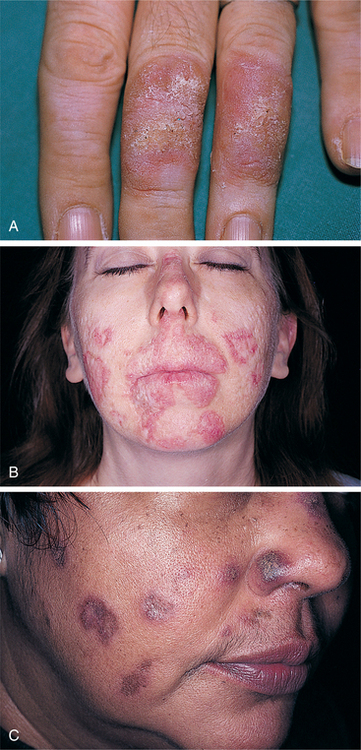
14. Bodemer, C, Belon, M, Hamel-Teillac, D, et al. Scleroderma in children: a retrospective study of 70 cases. Ann Dermatol Venereol. 1999;126(10):691–694.
15. Bouffard, D, Wong, TY, Hernandez, M, Mihm, MO. Suspicion of early desmoplastic melanoma. Melanoma Lett. 1993;11(3):1.
16. Brinckerhoff, LH, Thompson, LW, Slingluff, CL. Melanoma vaccines. Curr Opin Oncol. 2000;12(2):163–173.
17. Brown, CD, Zitelli, JA. The prognosis and treatment of true local cutaneous recurrent malignant melanoma. Dermatol Surg. 1995;21(4):285–290.
18. Brown, CW, Marschall, SF. Connective tissue update: focus on dermatomyositis. Consultant. 1999;39(10):2867–2875.
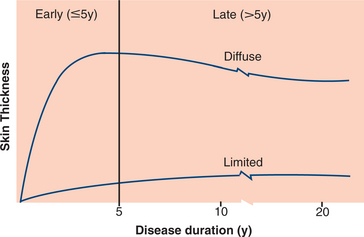
19. Bryan, C, Knight, C, Black, CM. Prediction of five-year survival following presentation with scleroderma: development of a simple model using three disease factors at first visit. Arthritis Rheum. 1999;42(12):2660–2665.
20. Cafiero, F, Peressini, A, Percivale, PL, et al. Selective lymph node dissection in patients with intermediate thickness melanoma: our experience. Anticancer Res. 2000;20(1B):497–500.
21. Callaghan, S. Skin considerations with silicone-type interfaces. Adv Phys Ther. 1996;7:22–23.
22. Cantwell, A, Infection with bacteria as the cause of scleroderma. Physician’s Page. 2008;5(1):5–8. Available on-line at www.roadback.org Accessed April 26, 2008.
23. Carrougher, GJ. Burn care and therapy. St Louis: Mosby, 1998.
24. Cathomas, G. Human herpesvirus 8: a new virus discloses its face. Virchows Arch. 2000;436(3):195–206.
25. Cella, G Road Back Foundation. Antibiotic Therapy for Rheumatic Diseases. Patient survey by Harris Interactive, Inc., 2006. Available on-line at www.roadback.org Accessed August 14, 2006.
26. Centers for Medicare and Medicaid Services. Principles of documentation, November 12, 2005. Available on-line at www.cms.hhs.gov/manuals/downloads/som107_exhibit_007a.pdf Accessed August 14, 2006.
27. Cerovac, S, Roberts, AH. Burns sustained by hot bath and shower water. Burns. 2000;26(3):251–259.
28. Chang D, Hackshaw D, Dattarck, et al: Frostbite, March 2006. Available on-line at www.emedicine.com Accessed April 26, 2008.
29. Chosidow, O. Scabies and pediculosis. Lancet. 2000;355(9206):819–826.
30. Clements, PJ, Furst, DE, Wong, WK, et al. High-dose versus low-dose D-penicillamine in early diffuse systemic sclerosis: analysis of a two-year, double-blind, randomized, controlled clinical trial. Arthritis Rheum. 1999;42(6):1194–1203.
31. Collins, N. The difference between albumin and prealbumin. Adv Skin Wound Care. 14(5), 2001. [235-235].
32. Davis M: Top ten contact dermatitis allergens identified in Mayo Clinic Study. Available on-line at www.mayoclinic.org Accessed March 3, 2006.
33. D’Cruz, D. Autoimmune diseases associated with drugs, chemicals and environmental factors. Toxicol Lett. 2000;112-113:421–432.
34. Deep tissue injury. Washington, D.C., 2002, National Pressure Ulcer Advisory Panel. Available on-line at www.npuap.org/DOCS/DTI.doc Accessed April 26, 2008.
35. Descamps, V. HIV-1 infected patients with toxic epidermal necrolysis: an occupational risk for healthcare workers. Lancet. 1999;353(9167):1855–1856.
36. deVries, TJ, Fourkour, A, Punt, CJ, et al. Melanoma-inhibiting activity (MIA) mRNA is not exclusively transcribed in melanoma cells: low levels of MIA mRNA are present in various cell types and in peripheral blood. Br J Cancer. 1999;81(6):1066–1170.
37. Diaz, LA, Giudice, GJ. End of the century overview of skin blisters. Arch Dermatol. 2000;136(1):106–112.
38. Driscoll, J. Integumentary management of the patient with multiple traumatic injuries. Acute Care Perspect. 1999;7(3):1. [3-4, 16-18].
39. Elgart, ML. Current treatments for scabies and pediculosis. Skin Therapy Lett. 2000;5(1):1–3.
40. Englert, H, Small-McMahon, J, Davis, K, et al. Systemic sclerosis prevalence and mortality in Sydney 1974-1988. Aust N Z J Med. 1999;29(1):42–50.
41. Feldman, SR, Koo, JY, Menter, A, et al. Decision points for the initiation of systemic treatment for psoriasis. J Am Acad Dermatol. 2005;53(1):101–107.
42. Fink, CA, Bates, MN. Melanoma and ionizing radiation: is there a causal relationship? Radiat Res. 2005;164(5):701–710.
43. Frances, C. Smoker’s wrinkles: epidemiological and pathologic considerations. Clin Dermatol. 1998;16(5):565–570.
44. Frantz, RA. Measuring prevalence and incidence of pressure ulcers. Adv Wound Care. 1997;10(1):21–24.
45. Fuller, J. Cover up and clean up to prevent deadly infections. Nursing2005. 2005;35(1):31.
46. Gallego, H, Crutchfield, CE, Lewis, EJ, et al. Report of an association between discoid lupus erythematosus and smoking. Cutis. 1999;63(4):231–234.
47. Gang, RK, Sanyal, SC, Bang, RL, et al. Staphylococcal septicaemia in burns. Burns. 2000;26(4):359–366.
48. Gascon, P, Schwartz, RA. Kaposi’s sarcoma: new treatment modalities. Dermatol Clin. 2000;18(1):169–175.
49. Geisse, J. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol. 2004;50(5):722–733.
50. Generini, S, Fiori, G, Moggi, PA, et al. Systemic sclerosis: a clinical overview. Adv Exp Med Biol. 1999;455:73–83.
51. Generini, S, Matucci, CM. Raynaud’s phenomenon and vascular disease in systemic sclerosis. Adv Exp Med Biol. 1999;455:93–100.
52. Ghossein, RA, Carusone, L, Bhattacharya, S. Review: polymerase chain reaction detection of micrometastases and circulating tumor cells: application to melanoma, prostate, and thyroid carcinomas. Diagn Mol Pathol. 1999;8(4):165–175.
53. Gnann, JW, Pellett, PE, Jaffe, HW. Human herpesvirus 8 and Kaposi’s sarcoma in persons infected with human immunodeficiency virus. Clin Infect Dis. 2000;30(suppl 1):S72–S76.
54. Green, A. Daily sunscreen application and beta-carotene supplementation in prevention of basal-cell and squamous-cell carcinomas of the skin: a randomized controlled trial. Lancet. 1999;354(9180):723–729.
55. Griffiths, CEM. Aging of the skin. In: Tallis RC, Fillit HM, Brocklehurst JC, eds. Brocklehurst’s textbook of geriatric medicine and gerontology. ed 5. New York: Churchill Livingstone; 1998:1293–1298.
56. Gulin, J, Korn, JH. Systemic sclerosis: challenges in diagnosis and management. J Musculoskel Med. 1999;16:288–300.
57. Habif, TP. Skin disease: diagnosis and treatment. St Louis: Mosby, 2000.
58. Haldorsen, R, Reitan, JB, Tveten, U. Cancer incidence among Norwegian airline pilots. Scand J Work Environ Health. 2000;26(2):106–111.
59. Halpern A: P.T.s can help patients by recognizing signs of skin disorders. Presented at the Hospital of the University of Pennsylvania (HUP), 1993.
60. Harrison, RA, Haque, AU, Roseman, JM, et al. Socioeconomic characteristics and melanoma incidence. Ann Epidemiol. 1998;8(5):327–333.
61. Healy, E, Flannagan, N, Ray, A, et al. Melanocortin-1-receptor gene and sun sensitivity in individuals without red hair. Lancet. 2000;355(9209):1072–1073.
62. Henry, KD. Effect of physical therapy on a patient with dermatomyositis. Phys Ther Case Reports. 1999;2(4):157–161.
63. Hess, CT, Salcido, R. Wound care: clinical guide. Springhouse, PA: Springhouse Publishing, 1999.
64. HHS (U.S. Department of Health and Human Services). Pressure ulcer treatment. Rockville, MD: The Department, 1995. [AHCPR publication no. 95-0652].
65. HHS (U.S. Department of Health and Human Services). Pressure ulcers in adults: predilection and prevention. Rockville, MD: The Department, 1992. [AHCPR publication no. 92-0047].
66. Higgins, E. Alcohol, smoking, and psoriasis. Clin Exp Dermatol. 2000;25(2):107–110.
67. Hill, LL, Ouhtit, A, Loughlin, SM, et al. Fas ligand: a sensor for DNA damage critical in skin cancer etiology. Science. 1999;285(5429):898–900.
68. Hofer, MF. Atopic dermatitis: the first allergic step in children. Rev Med Suisse Romande. 2000;120(3):263–267.
69. Iscovich, J, Boffetta, P, Franceschi, S, et al. Classic Kaposi sarcoma: epidemiology and risk factors. Cancer. 2000;88(3):500–517.
70. Jemal, A, Siegel, MPH, Ward, E, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56(2):106–130.
71. Jewell, ML, McCauliffe, DP. Patients with cutaneous lupus erythematosus who smoke are less responsive to antimalarial treatment. J Am Acad Dermatol. 2000;42(6):983–987.
72. Josty, IC, Narayanan, V, Dickson, WA. Burns in patients with epilepsy: changes in epidemiology and implications for burn treatment and prevention. Epilepsia. 2000;41(4):453–456.
73. Kabat-Zinn, J, Wheeler, E, Light, T, et al. Influence of a mindfulness mediation based stress reduction intervention on rates of skin clearing in patients with moderate to severe psoriasis. Psychosom Med. 1998;60(5):625–632.
74. Kahaleh, MB, LeRoy, EC. Autoimmunity and vascular involvement in systemic sclerosis (SSc). Autoimmunity. 1999;31(3):195–214.
75. Kalka, K, Merk, H, Mukhtar, H. Photodynamic therapy in dermatology. J Am Acad Dermatol. 2000;42(3):389–413.
76. Kendall, K: Personal communication, Gulf Breeze, FL, 2000, Medical Center for Continuing Education.
77. Kohen, R. Skin antioxidants: their role in aging and in oxidative stress new approaches for their evaluation. Biomed Pharmacother. 1999;53(4):181–192.
78. Koo, J. Systemic sequential therapy of psoriasis: a new paradigm for improved therapeutic results. J Am Acad Dermatol. 1999;41(3, pt 2):S25–S28.
79. Koo, J, Liao, W. Update on psoriasis therapy: a perspective from the USA. Keio J Med. 2000;49(1):20–25.
80. Koo, JY, Bagel, J, Sweetser, MT, et al. Alefacept in combination with ultraviolet B phototherapy for the treatment of chronic plaque psoriasis: results from an open-label, multicenter study. J Drugs Dermatol. 2006;5(7):623–628.
81. Koo, JYM. New developments in topical sequential therapy for psoriasis. Skin Therapy Lett. 2005;10(9):1–4.
82. Krogstad, AL, Lonnroth, P, Larson, G, et al. Capsaicin treatment induces histamine release and perfusion changes in psoriatic skin. Br J Dermatol. 1999;141(1):87–93.
83. Langner, I, Blettner, M, Gundestrup, M, et al. Cosmic radiation and cancer mortality among airline pilots: results from a European cohort study (ESCAPE). Radiat Environ Biophys. 2004;42(4):247–256.
84. Le, C, Morales, A, Trentham, DE. Minocycline in early diffuse scleroderma. Lancet. 1998;352(9142):1755–1756.
85. Leung, DY. Atopic dermatitis: new insights and opportunities for therapeutic intervention. J Allergy Clin Immunol. 2000;105(5):860–876.
86. Livingston Wuerthele-Caspe, V, Brodkin, E, Mermod, C. Etiology of scleroderma: preliminary clinical report. J Med Soc N J. 1947;44:256–259.
87. Loehne, H. Pulsatile lavage with suction. In Sussman C, Bates-Jensen BM, eds.: Wound care notebook: a collaborative practice manual for physical therapists and nurses, ed 3, Ambler, PA: Lippincott Williams & Wilkins, 2006.
88. Loehne H, Streed SA, Gaither B, et al: Aerosolization of microorganisms during pulsatile lavage with suction, 1999.
89. Luedtke-Hoffmann, KA, Schafer, DS. Pulsed lavage in wound cleansing. Phys Ther. 2000;80(3):292–300.
90. Lund, CC, Browder, NC. The estimation of areas of burn. Surg Gynecol Obstet. 1994;79:352–358.
91. Maklebust, J, Sieggreen, M. Pressure ulcers: guidelines for prevention and nursing management, ed 2. Springhouse, PA: Springhouse Publishing, 1996.
92. Maragakis, LL, Cosgrove, SE, Song, X, et al. An outbreak of multidrug-resistant Acinetobacter baumannii associated with pulsatile lavage wound treatment. JAMA. 2004;292(24):3006–3011.
93. McCulloch JM, Kloth L, eds. Wound healing: alternatives in management (contemporary perspectives in rehabilitation). Philadelphia: FA Davis, 1994.
94. McGill, V, Kowal-Vern, A, Gamelli, RL. Outcome for older burn patients. Arch Surg. 2000;135(3):320–325.
95. McGwin, G, Chapman, V, Rousculp, M. The epidemiology of fire-related deaths in Alabama, 1992-1997. J Burn Care Rehabil. 2000;21(1, pt 1):75–83.
96. Medsger, TA. Systemic sclerosis (scleroderma). In: Stein JH, ed. Internal medicine. ed 4. St Louis: Mosby; 1994:2443–2449.
97. Mitsuyasu, RT. Update on the pathogenesis and treatment of Kaposi’s sarcoma. Curr Opin Oncol. 2000;12(2):174–180.
98. Morgan J: Kaposi’s sarcoma. DermNet NZ: The dermatology resource, New Zealand, 2005. Available on-line at www.dermnetnz.org Accessed August 10, 2006.
99. Morton, D. Therapeutic vaccine for disseminated melanoma prolongs survival. J Clin Oncol. 2002;20:4549–4554.
100. Morton, DL, Ollila, MD, Eddy, C, et al. Cytoreductive surgery and adjuvant immunotherapy: a new management paradigm for metastatic melanoma. CA Cancer J Clin. 1999;49:101–116.
101. Muller, MG, Borgstein, PJ, Pijpers, R, et al. Reliability of the sentinel node procedure in melanoma patients: analysis of failures after long-term follow-up. Ann Surg Oncol. 2000;7(6):461–468.
102. Murphy, JC, Banwell, PE, Roberts, AH, et al. Frostbite: pathogenesis and treatment. J Trauma. 2000;48(1):171–178.
103. Navid, F, Furman, WL, Fleming, M, et al. The feasibility of adjuvant interferon alpha-2b in children with high-risk melanoma. Cancer. 2005;103(4):780–787.
104. Nicol, NH. Managing atopic dermatitis in children and adults. Nurse Pract. 2000;25(4):58–70.
105. Nietert, PJ, Sutherland, SE, Silver, RM, et al. Is occupational organic solvent exposure a risk factor for scleroderma? Arthritis Rheum. 1998;41(6):1111–1118.
106. Nietert, PJ, Sutherland, SE, Silver, RM, et al. Solvent oriented hobbies and the risk of systemic sclerosis. J Rheumatol. 1999;26(11):2369–2372.
107. Ortonne, JP. Recent developments in the understanding of the pathogenesis of psoriasis. Br J Dermatol. 1999;140(suppl 54):1–7.
108. O’Sullivan, SB, Schmitz, TJ. Physical rehabilitation: assessment and treatment, ed 5. Philadelphia: FA Davis, 2006.
109. Otley, CC, Zitelli, JA. Review of sentinel lymph node biopsy and systemic interferon for melanoma: promising but investigational modalities. Dermatol Surg. 2000;26(3):177–180.
110. Owen-Schaub, L, Chan, H, Cusack, JC, et al. Fas and fas ligand interactions in malignant disease. Int J Oncol. 2000;17(1):5–12.
111. Oxman, MN. A vaccine to prevent herpes zoster and postherpetic neuralgia. N Engl J Med. 2005;352(22):2271–2284.
112. Pacifico, MD, Grover, R, Richman, PI, et al. Development of a tissue array for primary melanoma with long-term follow-up: discovering melanoma cell adhesion molecule as an important prognostic marker. Plast Reconstr Surg. 2005;115(2):367–375.
113. Peters, BP, Weissman, FG, Gill, MA. Pathophysiology and treatment of psoriasis. Am J Health Syst Pharm. 2000;57(7):645–659.
114. Ponten, J. How do skin cancers get their start? Skin Cancer Found J. 1999;17:34–35. [94].
115. Pruitt, BA. The evolutionary development of biologic dressings and skin substitutes. J Burn Care Rehabil. 1997;18:S2–S5.
116. Pruitt, BA, Mason, AD, Goodwin, CW. Epidemiology of burn injury and demography of burn care facilities. Probl Gen Surg. 1990;7(2):235–251.
117. Pruitt, BA, McManus, AT, Kim, SH, et al. Burn wound infections: current status. World J Surg. 1998;22(2):135–145.
118. Pukkala, E, Aspholm, R, Auvinen, A, et al. Cancer incidence among 10,211 airline pilots: a Nordic study. Aviat Space Environ Med. 2003;74(7):699–706.
119. Rafnsson, V, Hrafnkelsson, J, Tulinius, H. Incidence of cancer among commercial airline pilots. Occup Environ Med. 2000;57(3):175–179.
120. Ranieri, JM, Wagner, JD, Wenck, S, et al. The prognostic importance of sentinel lymph node biopsy in thin melanoma. Ann Surg Oncol. 2006;13(7):927–932.
121. Ranieri, JM, Wagner, JD, Wiebke, EA, et al. Lack of prognostic importance of reverse-transcriptase polymerase chain reaction detection of circulating messenger RNA in patients with melanoma. Plast Reconstr Surg. 2005;115(4):1058–1063.
122. Reamy, BV. Frostbite: review and current concepts. J Am Board Fam Pract. 1998;11(1):34–40.
123. Rees, JL, Birch-Machin, M, Flanagan, N, et al. Genetic studies of the human melanocortin-1 receptor. Ann N Y Acad Sci. 1999;885:134–142.
124. Reimann, S, Luger, R, Metze, D. Topical administration of capsaicin in dermatology for treatment of itching and pain. Hautarzt. 2000;51(3):164–172.
125. Richard R, Staley M, eds. Burn care and rehabilitation: principles and practice. Philadelphia: FA Davis, 1994.
126. Richard, R, Ward, RS. Splinting strategies and controversies. J Burn Care Rehabil. 2005;26(5):392–396.
127. Rigel, DS. Recent advances in diagnostic techniques. Skin Cancer Found J. 1998;16:36–37.
128. Rigel, DS, Rigel, EG, Rigel, AC. Effects of altitude and latitude on ambient UVB radiation. J Am Acad Dermatol. 1999;40(1):114–116.
129. Surgery. Worldwide Melanoma Update. 1997;1:11. [J Rivers, ed.].
130. Rockwell, WB, Ehrlich, HP. Fibrinolysis inhibition in human burn blister fluid. J Burn Care Rehabil. 1990;11:1–6.
131. Rolewski, SL. Clinical review: topical retinoids. Dermatol Nurs. 2003;15(5):447–450. [459-465].
132. Romagnolo, SC, Benedetto, AV. Rosacea in a new light. SKINmed. 2005;4(1):47–48.
133. Rupp, JF, Kaplan, DL. Pruritus: causes cures, part 1. Consultant. 1999;39(11):3157–3160.
134. Sabir, AM, Werth, VP. Cutaneous manifestations of lupus erythematosus. J Musculoskel Med. 1999;16:482–491.
135. Sallis, R, Chassay, CM. Recognizing and treating common cold-induced injury in outdoor sports. Med Sci Sports Exerc. 1999;31(10):1367–1373.
136. Schindl, A, Schindl, M, Pernerstorfer-Schon, H, et al. Low intensity laser irradiation in the treatment of recalcitrant radiation ulcers in patients with breast cancer. Photodermatol Photoimmunol Photomed. 2000;16(1):34–37.
137. Seibold, JR, Korn, JH, Simms, R, et al. Recombinant human relaxin in the treatment of scleroderma: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2000;132(11):871–879.
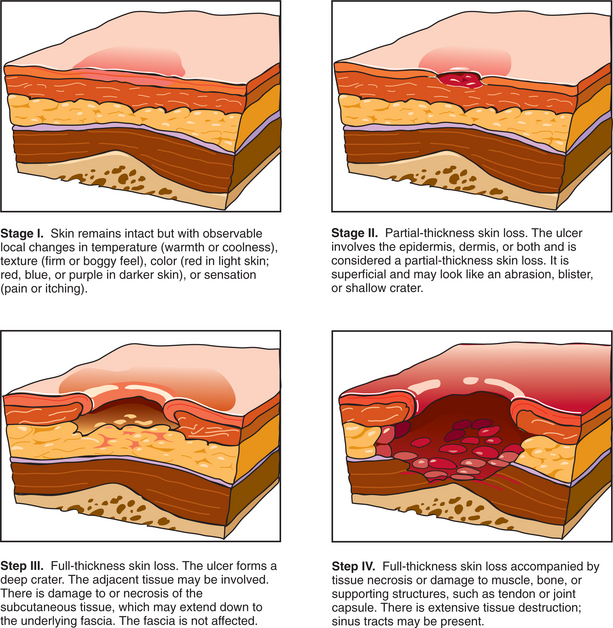
137a. Shea, JD, Pressure sores: classification and management. Clin Orthop Relat Res. 1975(112):89–100.
138. Sheridan, RL, Remensnyder, JP, Schnitzer, JJ, et al. Current expectations for survival in pediatric burns. Arch Pediatr Adolesc Med. 2000;154(3):245–249.
139. Sicherer, SH, Leung, DY. Advances in allergic skin disease, anaphylaxis, and hypersensitivity reactions to foods, drugs, and insects. J Allergy Clin Immunol. 2005;116(1):153–163.
140. Sidbury, R, Hanifin, JM. Old, new, and emerging therapies for atopic dermatitis. Dermatol Clin. 2000;18(1):1–11.
141. Skin Cancer Physicians, Skin cancer net. Am Acad Dermatol 2006. Available on-line at www.skincarephysicians.com Accessed August 10, 2006.
142. Soltani, MH, Pichardo, R, Song, Z, et al. Microtubule-associated protein 2, a marker of neuronal differentiation, induces mitotic defects, inhibits growth of melanoma cells, and predicts metastatic potential of cutaneous melanoma. Am J Pathol. 2005;166(6):1841–1850.
143. Stallone, G, Schena, A, Infante, B, et al. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N Engl J Med. 2005;352(13):1317–1323.
144. Stern, RS, Liebman, EJ, Vakeva, L. Oral psoralen and ultraviolet-A light (PUVA) treatment of psoriasis and persistent risk of nonmelanoma skin cancer: PUVA follow-up study. J Natl Cancer Inst. 1998;90(17):1278–1284.
145. Stratton, SP, Dorr, RT, Alberts, DS. The state-of-the-art in chemoprevention of skin cancer. Eur J Cancer. 2000;36(10):1292–1297.
146. Su, CW, Lohman, R, Gottlieb, LJ. Frostbite of the upper extremity. Hand Clin. 2000;16(2):235–247.
147. Sussman C, Bates-Jensen BM, eds. Wound care: a collaborative practice manual for physical therapists and nurses, ed 2, Gaithersburg, MD: Aspen Publishers, 2001.
148. Tanaka, A, Lindor, K, Ansari, A, et al. Fetal microchimerisms in the mother: immunologic implications. Liver Transpl. 2000;6(2):138–143.
149. Thiboutot, DM. Acne and rosacea. Dermatol Clin. 2000;18(1):63–71.
150. Thissen, MR, Schroeter, CA, Neumann, HA. Photodynamic therapy with delta-aminolaevulinic acid for nodular basal cell carcinomas using a prior bulking technique. Br J Dermatol. 2000;142(2):338–339.
151. Thomas, JM, Patocskai, EJ. The argument against sentinel node biopsy for malignant melanoma: its use should be confined to patients in clinical trials. BMJ. 2000;321(7252):3–4.
152. Trefzer, U, Weingart, G, Chen, Y, et al. Hybrid cell vaccination for cancer immune therapy: first clinical trial with metastatic melanoma. Int J Cancer. 2000;85(5):618–626.
153. Trentham, DE, Minocycline in early diffuse scleroderma SSc the next step. Physician’s Page. 2008;3(2):3. Available on-line at www.roadback.org Accessed April 26, 2008.
154. Unglaub, F, Woodruff, S, Demir, E, et al. Patients with epilepsy: a high-risk population prone to severe burns as a consequence of seizures while showering. J Burn Care Rehabil. 2005;26(6):526–528.
155. Valenta, R, Seiberler, S, Natter, S, et al. Autoallergy: a pathogenetic factor in atopic dermatitis? J Allergy Clin Immunol. 2000;105(3):432–437.
156. Wagner, JD, Corbett, L, Park, HM, et al. Sentinel lymph node biopsy for melanoma: experience with 234 consecutive procedures. Plast Reconstr Surg. 2000;105(6):1956–1966.
157. Weisinger, GF, Quittan, M, Nuhr, M, et al. Aerobic capacity in adult dermatomyositis/polymyositis patients and healthy controls. Arch Phys Med Rehabil. 2000;81(1):1–5.
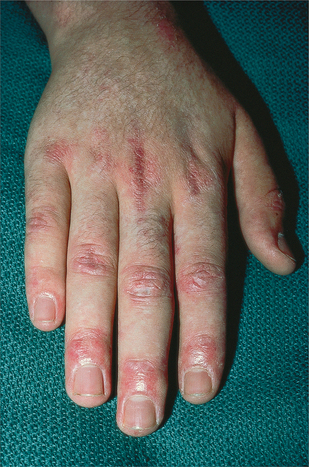
158. Wigley, FM. Systemic sclerosis and related syndromes. B. Clinical features. In: Klippel JH, ed. Primer on the rheumatic diseases. ed 11. Atlanta: Arthritis Foundation; 1997:267–272.
159. Williams, H, Flohr, C. How epidemiology has challenged three prevailing concepts about atopic dermatitis. J Allergy Clin Immunol. 2006;118(1):209–213.
160. Winchester, R. Psoriatic arthritis and the spectrum of syndromes related to the SAPHO (synovitis, acne, pustulosis, hyperostosis, and osteitis) syndrome. Curr Opin Rheumatol. 1999;11(4):251–256.
161. Wolf, SE, Herndon, DN. Burn care. Austin, TX: Landes Bioscience Publishing, 1999.
162. Wong, SL. The role of sentinel lymph node biopsy in the management of thin melanoma. Am J Surg. 2005;190(2):196–199.
163. Wooldridge, WE. Psoriasis, joint swelling, and draining plaques. J Musculoskel Med. 1996;13(7):61–62.
164. Yarchoan R: Bevacizumab in treating patients with Kaposi’s sarcoma. National Cancer Institute, Clinical Trial NCT00058136, June 2006.
165. Young, AJ, O’Brien, C, Sawka, MN, et al. Physiological problems associated with wearing NBC protective clothing during cold weather. Aviat Space Environ Med. 2000;71(2):184–189.
166. Yowler, CJ, Mozingo, DW, Ryan, JB, et al. Factors contributing to delayed extremity amputation in burn patients. J Trauma. 1998;45(3):522–526.
167. Zandi, S, Shamsadini, S, Zahedi, MJ, et al. Helicobacter pylori and rosacea. East Mediterr Health J. 2003;9(1-2):167–171.
168. Zitelli, JA, Brown, CD, Hanusa, BH. Surgical margins for excision of primary cutaneous melanoma. J Am Acad Dermatol. 1997;37(3, pt 1):422–429.