The Peripheral Nervous System
The peripheral nervous system (PNS) includes somatic motor and sensory components of cranial and spinal nerves arising from neurons whose cell bodies are located within the brainstem and spinal cord or lie in dorsal root ganglia. In addition, peripheral aspects of the autonomic nervous system (ANS) also contribute to axons found in peripheral nerves. Axons from the three components extend from the cell bodies to form peripheral nerves. Disorders of the PNS can be broadly divided into neuropathies, in which the pathology is confined to the nerve, and myopathies, in which the pathology occurs in muscle. Disorders of the PNS can be subdivided further according to site of anatomic involvement.46
Signs and symptoms of PNS involvement relate to the motor and sensory systems, as well as the ANS. Motor involvement, termed lower motor neuron (LMN) involvement, occurs when any of the following sites is affected:
• Cell body of the alpha motor neuron (anterior horn cell) located within the spinal cord or brainstem
• Axons that arise from the anterior horn cell that form spinal and peripheral nerves and cranial nerves
Sensory fibers of the PNS will show involvement if a lesion occurs in the dorsal root ganglion where the cell body is located or in the nerve root proximal to the ganglia, or distally in fibers of the peripheral nerve (Fig. 39-1). Similarly, when the ANS preganglionic or postganglionic motor fibers are involved, involuntary motor function of organs will be affected, and when sensory ANS fibers are affected, unconscious sensory functions (such as baroreceptors signaling arterial pressure, receptors within organs signaling irritants, distention, hypoxia, and so forth) will have transmission into the central nervous system (CNS) altered.
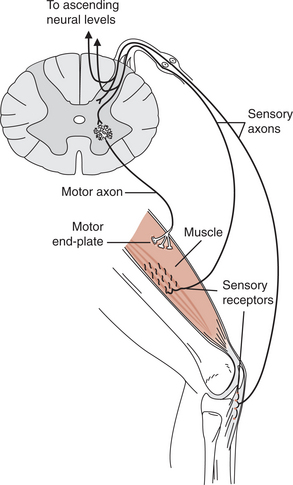
Figure 39-1 Potential sites of involvement in the peripheral nervous system. Motor: motor neuron cell body, axon, motor endplate, muscle fiber. Sensory: cell body in ganglion, axon, sensory receptor.
STRUCTURE
Nerves in the PNS are supported and covered by three connective tissue coverings that act like a tube surrounding the nerve. The inner-most covering is the endoneurium, which surrounds each individual axon. The middle layer, or perineurium, envelopes groups, or fascicles, of axons and is responsible for maintaining the blood-nerve barrier. The outer-most layer, or epineurium, surrounds the entire nerve and provides cushioning for the entire nerve.131 The surface of an axon is formed by a phospholipid membrane called the axolemma. Lying between the axolemma and the endoneurium are Schwann cells (Fig. 39-2). Throughout life, axonal-Schwann cell molecular signaling occurs. In large diameter axons (greater than 1 μm), the Schwann cell receives a signal to wrap its membrane around the axon, thus creating myelin. In small diameter axons, the Schwann cell merely envelopes and supports nonmyelinated fibers. Myelin not only provides electrical insulation essential for rapid saltatory conduction of the axon potential but also affects axonal properties. The presence of myelin causes sodium channels to cluster at the nodes of Ranvier, thus reinforcing efficient saltatory conduction.97 In the smallest axons, Schwann cells do not make myelin but do provide support for these unmyelinated fibers, whose action potentials are conducted by local circuit conduction (Table 39-1). Within a peripheral nerve, only about 25% of the fibers are myelinated.52
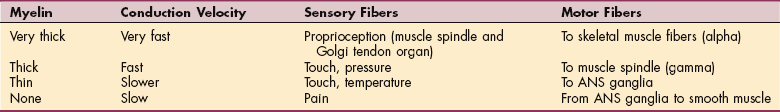
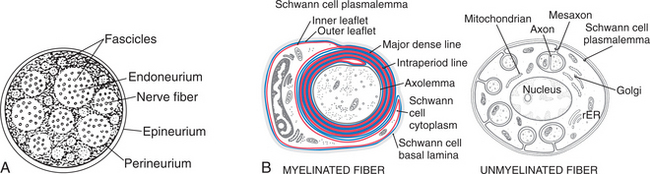
Figure 39-2 A, Cross–section of a peripheral nerve showing connective tissue coverings. Externally, the nerve is enveloped by the epineurium; internally, individual axons are surrounded by endoneurium. The perineurium surrounds groups of axons, termed fascicles. B, Although the majority of the fibers are unmyelinated, they are still associated with supporting Schwann cells. In myelinated fibers (left), each Schwann cell forms a myelin internode whose borders are formed by the nodes of Ranvier. Schwann cell sheath supporting unmyelinated fibers (right). (A, From Wildsmith JAW: Peripheral nerve and local anesthetic drugs, Br J Anaesth 58:692, 1986. Reproduced by permission. B, From Haines DE: Fundamental neuroscience for basic and clinical applications, ed 3, Edinburgh, 2006, Churchill Livingstone.
Normal propagation of the action potential also requires sufficient energy, supplied by a vascular plexus interlaced between connective tissue layers. Each peripheral nerve receives an artery that penetrates the epineurium; this artery’s branches extend into the perineurium as arterioles, and branches from the arterioles enter the endoneurium as capillaries. Vessels supplying peripheral nerves appear coiled when a limb is in a shortened position, but uncoiled after movement so that neural vascular supply is not impaired with a limb’s normal excursion. This rich vascular supply makes peripheral nerves relatively resistant to ischemia.156
PERIPHERAL NERVOUS SYSTEM CHANGES WITH AGING
Changes that occur in the PNS may be considered as one component of a continuum that relates to normal growth and development, or the changes may represent a combination of pathologic processes superimposed on the normal aging process. Because of the difficulty of studying human peripheral nerves in vivo, experimental animals have been used to assess the effects of aging.
Age does not affect the size or number of fascicles, but the perineurium and epineurium do thicken with age and the endoneurium often becomes fibrosed with increased collagen. Even with these changes, the cross-sectional area decreases slightly with age because there is a reduced number of unmyelinated and myelinated fibers. Ventral root fibers controlling motion are more affected than dorsal root fibers controlling sensation. Blood vessels to nerves may become atherosclerotic with aging, and occlusion may contribute to loss of nerve fibers. The prevalence of peripheral neuropathies seen in older people has been attributed to this vascular pathology.
Decreases in protein production are hypothesized to cause myelin deterioration.164 When individual myelinated fibers are examined, shorter internodes are seen, suggesting that a demyelinating–remyelinating process occurs with aging. This structural alteration in peripheral nerve myelination may be reflected in diminished appreciation of vibratory sense.
ANS dysfunction is more common in the elderly. This dysfunction may be related to the changes seen in the nervous system in the elderly. Cell bodies show chromatolysis, as well as an accumulation of lipofuscins, representing a diminished ability of the cell to rid itself of toxins. Loss of cell bodies has been observed in the sympathetic ganglia, along with a loss of unmyelinated fibers in peripheral nerves. Sympathetic control of dermal vasculature shows an age-related decline that leads to a diminished wound repair efficiency. In an aging animal model, transcutaneous nerve stimulation (TENS) improved the vascular response. Peripheral activity of sympathetic nerves were affected by the low frequency electrical stimulation.77
When the motor endplate is examined, age-related changes have occurred, but these changes are seen as early as the third decade of life and are not reported in all muscles. When sensory receptors have been evaluated, density and morphology have been found to be altered in the elderly. Altered axonal myelination creates slowing of nerve conduction velocities (NCVs) in the elderly. In addition, the loss of fibers decreases the amplitude of the potential. Simultaneous with the decreased protein production is a decrease in intraaxonal transport by cytoskeletal elements in the peripheral nerve. Electromyographic (EMG) studies of elderly people without evidence of neurologic disorders of the PNS show loss of motor units, as well as signs of reinnervation. Morphologic changes observed in people over 60 years of age are manifested by decreased strength and sensory changes.108
Healthy elderly, with no evidence of neurologic disease, may provide a clinical history suggestive of peripheral neuropathy. This includes numbness and tingling in the hands and feet along with mild, diffuse weakness—especially in the distal muscles of the hand. Sensory alterations may lead to poor balance and gait instability. On examination, sensory thresholds are increased.
The cause of an aging neuropathy can be attributed to a combination of factors. First, loss of both motor and sensory cell bodies; second, a dying-back condition, suggesting neurons can metabolically support a limited number of fibers or receptors, similar to that seen in other systemic neuropathies; and last, over the course of a lifetime, chronic compression of the peripheral nerves or repetitive trauma may have damaged the nerves. All these factors, combined with coexisting medical conditions, atherosclerosis, and nutritional deficiencies, may create this neuropathy of aging.
When the aging PNS is damaged, wallerian degeneration is delayed and regeneration takes longer because secretion of trophic factors is slower than in younger individuals. Density of regenerating axons is less. In a partial nerve injury, collateral sprouting is reduced, further limiting recovery of function.164
RESPONSE TO INJURY
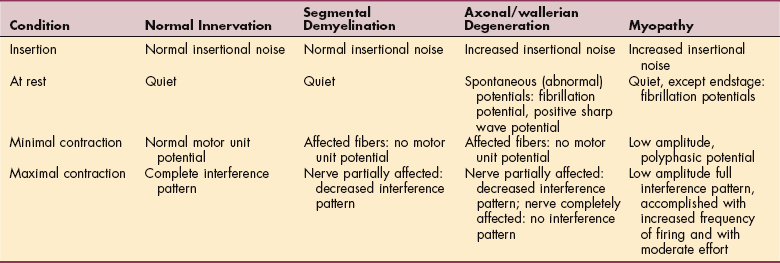
Peripheral nerve damage occurs by any one of several causal conditions: heredity, trauma, infections, toxins, and metabolism.26 When either motor or sensory nerves are affected, there is a limited response to injury, regardless of the cause. Either fibers demyelinate or fibers degenerate. Segmental demyelination occurs when nerves are subject to external compression or disease. Degeneration occurs in any peripheral nerve disorder that directly affects the axon, including physical injury (crush, stretch, or laceration), as well as disease.
Loss of myelin, typically in segments, leaves the axon intact but bare where the myelin is lost. This is called segmental demyelination. More severe involvement causes axonal degeneration, distal to the lesion (termed anterograde, or wallerian, degeneration14,73), that begins immediately after involvement and is completed over a period of a few weeks. Neuropathic diseases that affect the axon or its cell body causing axonal degeneration typically affect the longest nerve fibers first (a length-dependent process), with signs and symptoms beginning distally and spreading proximally as the disease progresses. Because nerves in the legs are longer, the feet and lower legs are involved long before the fingers and hands. Those conditions that affect only myelin cause segmental demyelination in both sensory and motor fibers. Thus disruption of the conduction of the action potential from proprioceptors and mechanoreceptors causes sensory changes. Those neuropathies that affect myelin cause demyelination of motor nerves to muscle and preganglionic fibers of the ANS create weakness, proprioceptive and tactile changes, and autonomic involvement by disrupting conduction of the action potential.
CLASSIFICATION OF NERVE INJURY
Traumatic injury to peripheral nerves from mechanical involvement secondary to compression, ischemia, and stretching can be classified using one of two systems based on the structural and functional changes that occur. Seddon140 initially divided nerve injury into three categories: neurapraxia, axonotmesis, and neurotmesis (Fig. 39-3). Sunderland153 divided this classification into five categories, based on axonal and connective tissue covering involvement.
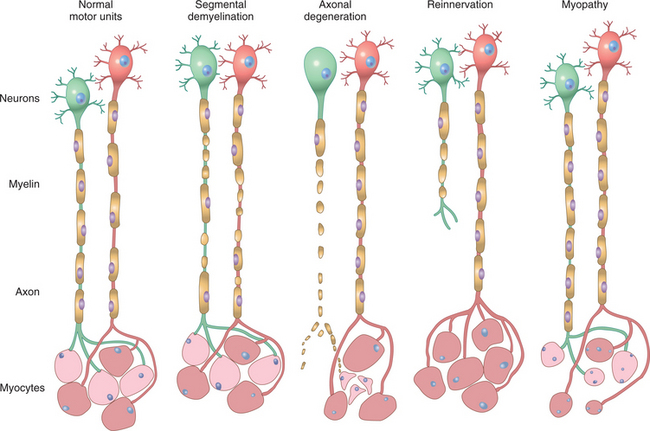
Figure 39-3 Types of nerve involvement and recovery that occur in peripheral nerves. A, Illustration of two adjacent normal myelinated nerves. B, Segmental demyelination. Several internodes of myelin have demyelinated, but the axon remain intact. The repair process for segmental demyelination occurs rapidly because Schwann cells divide and remyelinate the bare portion of the axon. Shorter internodal distance occurs with remyelination, thus nerve conduction velocity may not return to normal, even though muscle contracts normally. C, Illustration of axonal degeneration. The axon and myelin have degenerated, but the connective tissue covering remains intact in an axonotmesis. In neurotmesis, the connective tissue covering is disrupted at the lesion site. Signs of chromatolysis occur in the cell body after axonotmesis and neurotmesis. Note that muscle atrophies rapidly because it has lost the trophic influence from the nerve cell body. D, The repair process for axonotmesis and neurotmesis is more complex. Growth cones from the proximal axon must cross the lesion site and regrow down the connective tissue channels and reestablish a motor endplate or sensory connection before remyelination occurs. In partial nerve injuries, while the injury axon is regrowing, adjacent motor units sprout collateral fibers, leading to expansion of the size of this (red) motor unit. E, In myopathic conditions, scattered muscle fibers in adjacent motor units are small (degenerating or regenerating), while the neurons and axons are normal. (From Kumar V, Fausto N, Abbas A: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, WB Saunders.)
Neurapraxia involves segmental demyelination, which slows or blocks conduction of the action potential at the point of demyelination in a myelinated nerve. Neurapraxias often occur after nerve compression that induces mild ischemia in nerve fibers. When segmental demyelination occurs because of disease, the response may be termed a myelinopathy. Conduction of the action potential is normal above and below the point of compression, and because the axon remains intact, muscle does not atrophy. Axonotmesis occurs when the axon has been damaged, but the connective tissue coverings that support and protect the nerve remain intact. Prolonged compression that produces an area of infarction and necrosis causes an axonotmesis. In the presence of disease, wallerian degeneration creates an axonopathy, which is analogous to an axonotmesis. Neurotmesis, the most severe axonal loss, is the complete severance of the axon, as well as the disruption of its supporting connective tissue coverings (endoneurium, perineurium, and/or epineurium) at the site of injury. Neurotmesis is caused by gunshot or stab wounds or avulsion injuries that disrupt a section of the nerve or entire nerve. When axonal continuity is lost (either axonotmesis or neurotmesis), axons distal to the lesion degenerate (wallerian degeneration). Because muscle fibers innervated by the axon depend on the nerve cell body as a source of nourishment or trophic control, when axons degenerate, muscle fibers rapidly atrophy (Table 39-2).

If segmental demyelination has occurred, molecular signaling to remaining Schwann cells causes them to begin dividing mitotically. Newborn Schwann cells move to envelope the denuded segment of nerve and once these cells are in place they will begin to form myelin (Fig. 39-3, B). The potential for regeneration after axonal/Wallerian degeneration is possible as long as the nerve cell body remains viable; new axons can sprout from the proximal end of damaged axons (Fig. 39-3, C). However, successful functional regeneration requires that the proximal and distal ends of the connective tissue tube are aligned. This occurs in an axonotmesis because the connective tissue coverings remain intact. In a neurotmesis, without surgical intervention, recovery is less likely because the proximal end of the endoneurium is not approximated to the distal endoneurium. Without surgery, axonal sprouts often enter nearby soft tissue and form a neuroma, or axonal regrowth occurs down the incorrect endoneurial tube, rendering reinnervation nonfunctional.164 Once the axon has established a distal contact either with muscle or sensory receptor, remyelination will begin. When partial axonal degeneration occurs, adjacent noninvolved axons will produce collateral sprouts that will innervate muscle fibers before the damaged axons have time to grow and reinnervate those muscle fibers. This results in an enlarged motor unit for the neuron that has collateral sprouts (Fig. 39-3, D) Numerous reports in the literature link various molecular factors to nerve regeneration and healing following repair.116,155
CLASSIFICATION OF NEUROPATHY
Neuropathies include a wide variety of causes and can be classified in many ways, including the rate of onset, type and size of nerve fibers involved, distribution pattern, or pathology (Table 39-3). For example, when a single peripheral nerve is affected the result is a mononeuropathy, which is commonly a result of trauma. The term polyneuropathy indicates involvement of several peripheral nerves. A radiculoneuropathy indicates involvement of the nerve root as it emerges from the spinal cord, and polyradiculitis indicates involvement of several nerve roots and occurs when infections create an inflammatory response.
Table 39-3
Causes of Peripheral Neuropathies and Myopathies and Their Effects
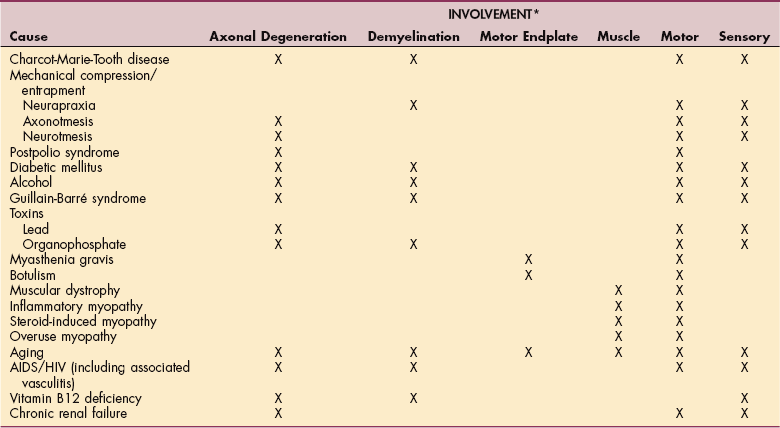
AIDS, Acquired immunodeficiency syndrome; HIV, human immunodeficiency virus.
*X indicates the most common types of involvement for each cause.
In addition to involvement of the peripheral nerve, the motor endplate or muscle itself may be involved in a peripheral disorder. Involvement of muscle, termed myopathy, follows a different clinical pattern than nerve. When muscle is involved, the disorder typically is reflected by proximal weakness, wasting, and hypotonia without sensory impairments (see Chapter 23).173
SIGNS AND SYMPTOMS OF PERIPHERAL DYSFUNCTION
The presence of signs and symptoms aid in the localization of the level or levels of involvement. Loss of sensory function will follow a peripheral nerve distribution if that is the anatomic region involved, or it will follow a dermatomal pattern when the spinal nerve or dorsal root ganglia (cell body) has been affected (Fig. 39-4).
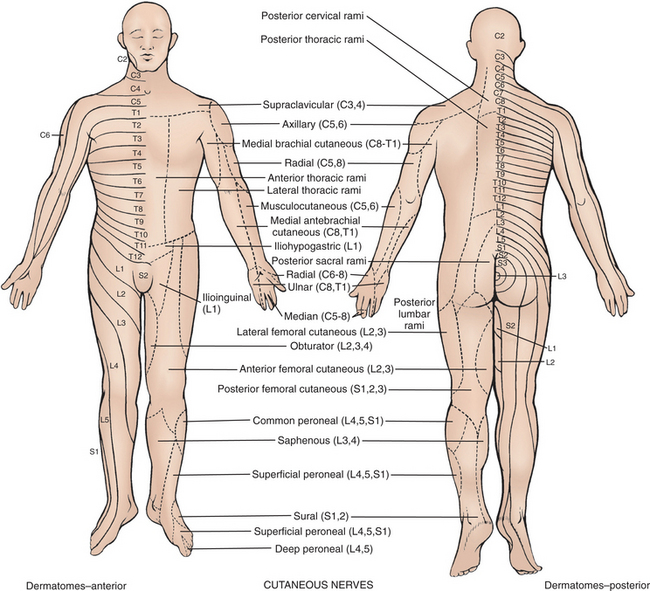
Figure 39-4 Dermatomal (right side of body, anterior and posterior) and peripheral sensory nerve (left side of body, anterior and posterior) patterns. (From Auerbach PS: Wilderness medicine, ed 5, St. Louis, Mosby, 2007.)
Similarly, when a peripheral nerve has motor involvement, paresis or paralysis will occur in muscles innervated by that nerve distal to the lesion. When spinal motor nerves are involved, weakness occurs in all the muscles receiving axons from that spinal level (a myoto- mal pattern). Individuals with only peripheral nerve involvement will have no signs or symptoms of CNS dysfunction.
Although differences occur in symptom evolution and in progression and severity of a neuropathy, a classic pattern of involvement would occur as follows. Involvement of sensory fibers is reflected by distal sensory deficits with the longest nerves in the body involved first. The first noticeable features of neuropathies are often sensory and consist of tingling, prickling, burning, or bandlike dysesthesias and paresthesias in the feet. When more than one nerve is involved, the sensory loss follows a glove-and-stocking distribution that is attributed to the dying-back of the longest fibers in all nerves from distal to proximal (Fig. 39-5).
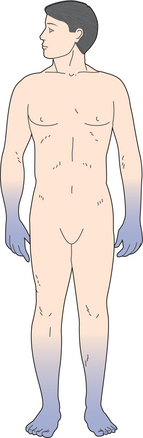
Figure 39-5 A stocking and glove pattern of sensory loss occurs in polyneuropathy. A gradient of greater distal loss tapering to less proximal involvement is seen.
The most common symptoms of motor nerve involvement include distal weakness and abnormalities of tone (hypotonicity or flaccidity). When clients are asked to walk on their heels or toes, weakness of dorsiflexors or plantarflexors, respectively, becomes apparent. Deep tendon reflexes (DTRs) are diminished or absent, and distal-most DTRs will be affected first. In the presence of axonal degeneration, rapid atrophy occurs, along with electrophysiologic changes (Tables 39-4 and 39-5). Prolonged paralysis gives rise to secondary complications like contracture formation and edema.
Table 39-4
Normal Nerve Conduction Velocities And Distal Latencies*


*In general, in the upper extremities, nerve conduction velocity for motor fibers averages about 60 m/sec. Investigators have reported values ranging from 45 to 75 m/sec. In the lower extremity, the normal range for motor nerve conduction is in the 40-to 50-m/sec range. Distal latency is a time value, reported in milliseconds (m/sec), that it takes for an evoked potential to be propagated along the nerve and recorded from either the muscle (motor) or the skin (sensory).
Adapted from Dyck PJ, Thomas PK (eds): Peripheral neuropathy, ed 3, Philadelphia, 1993, WB Saunders.
In addition to weakness and hypotonia, a diagnosis of any one of the muscle diseases may be associated with muscle tenderness or cramping. Classically, the motor involvement in a myopathy is opposite to that of a neuropathy. In a myopathy, the weakness tends to be proximal; in a neuropathy, motor symptoms tend to first occur distally.
Finally, because the nerve fibers from the ANS are also located in peripheral nerves, they, too, are subject to the effects of trauma or disease. Preganglionic fibers are myelinated and can be affected by segmental demyelination. In the presence of axonal degeneration, changes will occur in vascular control and sweating. For example, when a person has sustained a laceration of the median nerve in the region of the hand that lacks innervation, autonomic involvement creates smooth skin that does not sweat or wrinkle, or when a neuropathy has a systemic metabolic cause, the person may develop hypotension with cardiac irregularities.26
PATHOGENESIS AND DIAGNOSIS OF PERIPHERAL DYSFUNCTION
Trauma, inherited disorders, environmental toxins, and nutritional disorders may affect the myelin (myelinopathy), axon (axonopathy), or cell body of a peripheral nerve. The anatomic region or regions affected determine the severity of the involvement and the amount of function lost (see Table 39-2). Although the phenotype of peripheral dysfunction (i.e., physical characteristics/traits) remains unchanged, much of the recent research in pathophysiology of these disorders has delved into genetic and molecular causes and consequences for what occurs. Findings in these areas may allow development of treatments aimed at altering cellular problems.
Because the nervous system is the means of signaling from the CNS to the muscle, conduction of the action potential is affected in neuropathies and myopathies. In most disorders, electrophysiologic studies are used to determine where and how the nerve or muscle may be affected.
HEREDITARY NEUROPATHIES
Hereditary neuropathies were once considered rare, genetically determined disorders; however, recent studies reflect, in some cases, that these represent 43% of undiagnosed neuropathies.154 Hereditary neuropathies can be divided into two broad categories: those in which neu- ropathy is the primary disorder and those in which neuropathy is part of a greater multisystem disorder.9 This section concentrates on the first group, which includes Charcot-Marie-Tooth disease and its related hereditary polyneuropathies.
Charcot-Marie-Tooth Disease
Charcot-Marie-Tooth (CMT) disease, also known as hereditary motor and sensory neuropathy (HMSN) or peroneal muscular atrophy, is the most common inherited disorder affecting motor and sensory nerves. It was originally described by three neurologists, Jean Martin Charcot, Pierre Marie, and Howard Henry Tooth, in the 1880s. Initially the disorder involves the fibular (peroneal) nerve and affects muscles in the foot and lower leg. It later progresses to the muscles of the forearms and hands, making activities like buttoning or writing difficult. CMT is a genetically heterogeneous group of disorders with the same clinical phenotype, characterized by distal limb muscle wasting and weakness, usually with skeletal deformities, distal sensory loss, and abnormalities of DTRs.118
Incidence
Of the neuropathies, CMT is relatively common; it is estimated that 1 in 2500 persons in the United States has some form of CMT. Onset may occur in childhood or adulthood.108
Etiology
CMT is a genetically heterogenous neuropathy that is inherited as autosomal dominant, autosomal recessive, or X-linked pattern.170 Over fifty loci defects on chromosomes have been identified though deoxyribonucleic acid (DNA) testing.9,112,146 These chromosomal defects create either duplication, deletion, or point mutations in the genetic code for proteins that are involved in the process of myelination. CMT1 is the most common autosomal dominant pattern and is subdivided into three forms: CMT1A, 1B, and 1C. CMT1A accounts for 70% of all CMT1 cases and is caused by a DNA duplication on chromosome 17 for peripheral myelin protein 22 (PMP22), creating segmental demyelination of the fibular (peroneal) nerve.128 A less common form, CMT2, has had chromosomal abnormalities mapped to chromosomes 1, 8, and X. On chromosome 1, CMT2 is associated with a mutation in human myelin protein zero (P0), which has been associated recently with axonal dysfunction. This second form of CMT is associated with axonal degeneration. CMT2 has an onset that varies between the second and seventh decades and has less involvement in the small muscles of the hands than CMT1.24
Pathology
Mutations in proteins (PMP, P0, and connexin) associated with Schwann cell myelination create extensive demyelination along with a hypertrophic onion bulb formation in which demyelinated axons are surrounded by Schwann cells and their processes as remyelination is attempted. The onion bulb formation creates palpable, enlarged peripheral nerves. CMT2 is associated with genetic mutations that disrupt neurofilament assembly and thus affect axonal transport, creating axonal involvement.9
Clinical Manifestations
Although the two major types of CMT have differing chromosomal etiologies, it is nearly impossible to tell CMT1 from CMT2 clinically. In all autosomal dominant disorders, there are degrees of genetic dominance. The presence of symptoms are not all-or-none but are graded, with differing degrees of signs and symptoms among family members who have inherited the defective gene. This is termed variable expressivity. In CMT1 some members of a family with the genetic mutation may have greater signs of the disorder than others who have only minor involvement.95 In the X-linked form of CMT, men are affected and have signs of both demyelination and axonal degeneration are evident.
CMT is a slowly progressive disorder and although CMT1 begins in childhood, the actual onset may be difficult to determine. Clinical signs of CMT include distally symmetric muscle weakness, atrophy, and diminished DTRs. Feet have pes cavus (high arch) deformities and hammer toes (Fig. 39-6). Because of the muscles affected, the client will have weakness of the dorsiflexors and evertors (peroneal musculature) and will ambulate with a footdrop (steppage) gait pattern. As CMT progresses, involvement will be seen distally in the upper extremities. Weakness and wasting of the intrinsic muscles of the hand occurs, followed by progressive wasting in the forearms. Because CMT1 demyelinates peripheral nerves, proprioception is lost in the feet and ankles, and cutaneous sensation is diminished in the foot and lower legs. Sensory loss is minimal in CMT2. Sensory symptoms can include tingling and burning in the feet and legs, as well as impaired proprioception.177
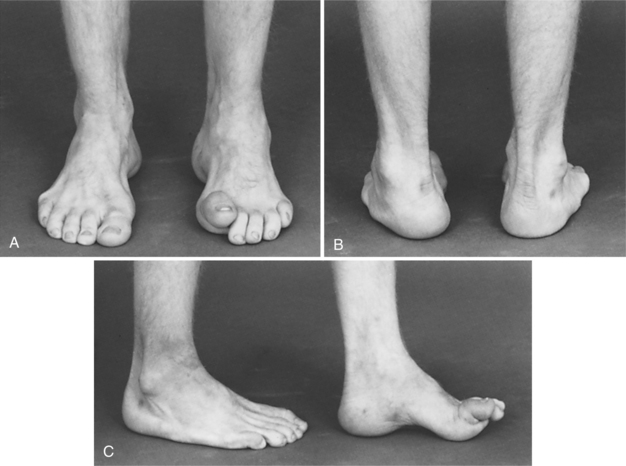
Figure 39-6 Pes cavus foot deformity in Charcot-Marie-Tooth disease. A, Clawing of left great toe. B, Left foot varus deformity. C, Cavus deformity with hammer toes. (From Canale ST: Campbell’s operative orthopaedics, ed 10, St Louis, 2003, Mosby.)
As muscle atrophy progresses below the knee, the appearance of the client’s legs takes on the shape of an inverted champagne bottle because normal muscle bulk is maintained above the knees.
MEDICAL MANAGEMENT
CMT is diagnosed by history and clinical examination, hereditary picture, electrophysiologic studies, and nerve biopsy. Most recently, because of the sensitivity and specificity of genetic studies, the diagnosis of CMT can be confirmed using gel electrophoresis to detect duplication, deletions, or sequence variations in genes.9 Although CMT1 produces demyelination, electrophysiologic testing reveals underlying axonal degeneration. Slowed motor nerve conduction does not have a linear correlation with the clinical severity of the disease.83
Both motor and sensory NCVs will be slowed in CMT194 but are normal or only slightly slowed in CMT2. Abnormalities of electrophysiologic studies in CMT2 will be a decreased amplitude of the potential, indicating axonal loss. The nerve biopsy is abnormal and will demonstrate either a demyelinating or axonal degenerative process.
TREATMENT.
Because CMT is an inherited disorder, there is no specific treatment to alter its course. Treatment is symptomatic to ensure that function is maintained in a safe manner. Footdrop and hand deformities can be helped by orthotic devices. Because the possibility of skin ulceration exists when tactile sensation and proprioception are affected, skin care precautions should be followed when total contact orthoses are used (see Chapter 10). To prevent contractures clients should be instructed in range of motion (ROM) exercises. Whether strengthening exercises can be used to counteract the effects of CMT has not been addressed; however, the long-term effects would be of little benefit in the presence of ongoing axonal degeneration. In a study examining the effects of weakness in CMT, results have found that individuals with CMT tend to be obese and have poor exercise tolerance. It is unknown whether exercise interventions can improve body composition and function.17
Studies using animal models have reported that antiprogesterone therapy combined with ascorbic acid have a positive effect on CMT1A. Although stem cell and gene therapy have been considered, the most promising treatment is pharmacologic therapies targeting the genetic mutation.112
PROGNOSIS.
CMT is a slowly progressive disorder; if unmanaged, contracture formation resulting from weakness will create further gait abnormalities, with clients reporting an increased number of falls. In the upper extremities, clients may develop problems with writing and handling objects. Individuals with CMT should be cautioned that some medications have been reported to cause an exacerbation of CMT. A database of the drugs that should be avoided is maintained by CMT North America. Among the identified medications are several anticancer drugs, including: vincristine, cisplatin, carboplatin, and taxoids.172
MECHANICAL INJURIES: COMPRESSION AND ENTRAPMENT SYNDROMES
The proximity of peripheral nerves to bony, muscular, and vascular structures can cause entrapment neuropathies characterized by changes in sensation and motor function, resulting from chronic neural compression. Another mechanical injury occurs as a result of traction on a nerve. As tension exceeds 10% to 20% of the axon’s resting length, the axon’s internal slack within fascicles is eliminated and structural damage occurs.144
Carpal Tunnel Syndrome
Carpal tunnel syndrome (CTS) is the most common entrapment neuropathy in the United States. It results from compression of the median nerve within the carpal tunnel at the wrist. CTS is characterized by general signs and symptoms of neuropathies: pain, tingling, numbness, paresthesia (Fig. 39-7), and later, muscular weakness in the distribution of the median nerve.
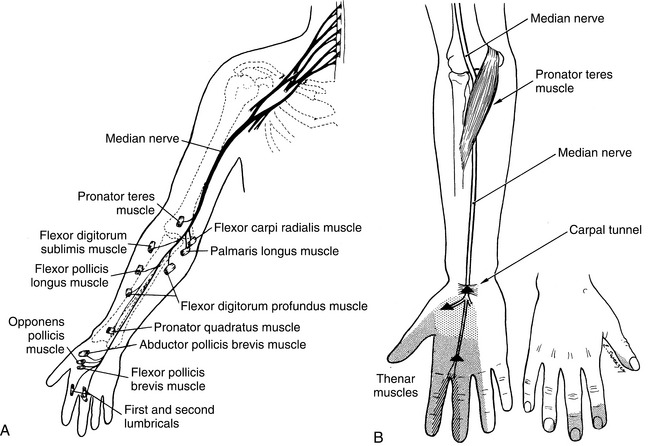
Figure 39-7 A, Median nerve course and motor innervation. B, The point of compression of the median nerve as it passes through the carpal tunnel. The lightly stippled area shows the sensory supply of the palmar cutaneous branch, which arises proximal to the carpal tunnel and thus is spared in the carpal tunnel syndrome. The densely stippled zone represents the cutaneous sensory area of the median nerve distal to the carpal tunnel. (A from Canale ST: Campbell’s operative orthopaedics, ed 10, St Louis, 2003, Mosby.; B from Noble J: Textbook of primary care medicine, ed 3, St Louis, 2001, Mosby.)
Incidence
Incidence in the United States is nearly 3.5 cases per 1000 individuals per year, and prevalence is estimated at 2.1%. As a condition, it produces one of the largest number of lost workdays among occupations. Nearly 70% of all CTS cases occur in women. Approximately 500,000 surgeries annually are performed for CTS. The incidence of surgery peaks in the 45-to 55-year-old group in women and in the over-65-year-old group in men.
Etiology
Although CTS is associated with occupational activities, any disorder that increases the volume of the contents of the carpal tunnel or that decreases the volume of the carpal tunnel will create a sustained rise in pressure within the tunnel that impinges on the median nerve. This includes synovial proliferation in rheumatoid arthritis, edema from local and systemic infections, congestive heart failure, pregnancy, and tumors. Callus formation after fracture, as well as malalignment of fractures, may reduce the volume of the canal. CTS is also more 2.5 times more likely in obese individuals (body mass index >29).39 Although some investigators161 have hypothesized that a compressive lesion located more proximally (thoracic outlet syndrome or cervical radiculopathy) on a nerve may predispose it to further injury (CTS), more recent examinations of clients with CTS have refuted this “double crush” hypothesis.84,161
Risk Factors
Also at risk for developing CTS are people with rheumatoid tenosynovitis, edema, pregnancy, hypothyroidism, and post-Colles’ fractures (Box 39-1).78,151 Although CTS has been reported in several occupations, because of the quality of the research, the convincing link between work and CTS is now questioned.39 In examining occupational studies, the literature identifies studies that report greatest incidence of CTS in frozen food workers and butchers. These support a positive association between a combination of factors: force and repetition and/or force and posture Although the job of a computer operator has been linked to CTS, when symptoms of paresthesia are rigorously assessed, CTS and other musculoskeletal pain disorders associated with long-term keyboarding can be as alleviated with 5 minute breaks every hour.151 Patients over the age of 63 years have a different pattern of risk factors for CTS than younger patients. This suggests that CTS in the elderly population may have different underlying pathogenetic mechanisms.10
Pathogenesis
In the carpal tunnel where there are 10 structures in a constrained compartment, normal tissue pressures are 7 to 8 mm Hg (Fig. 39-8). In CTS, these pressures rise above 30 mm Hg, when wrist flexion or extension occurs. Pressures go as high as 90 mm Hg when the wrist is fully flexed and up to 79.5 mm Hg when the wrist is extended.39 Pressure this great produces ischemia in the nerve. Ischemia accounts for the nocturnal symptoms or those that occur with wrist flexion. Unrelieved compression creates an initial neurapraxia with segmental demyelination of axons. Because the axons have lost their myelin padding they are more vulnerable, so that unrelieved compression can create an axonotmesis in which axon continuity is lost and wallerian degeneration occurs.
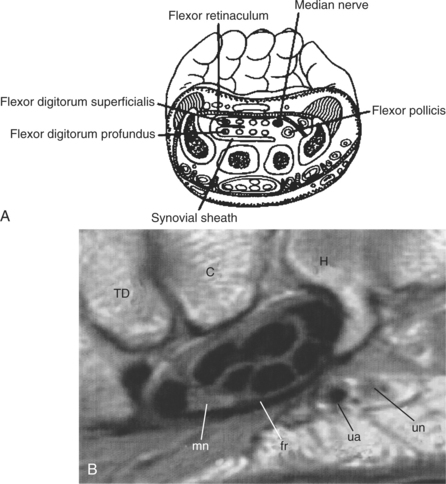
Figure 39-8 A, Cross-section of the carpal tunnel at the wrist. Contents of the tunnel include the tendon of the flexor pollicis longus (FPL), the four tendons of the flexor digitorum profundus (FDP), the four tendons of the flexor digitorum superficialis (FDS), and the median nerve. B, Carpal tunnel. TD, Trapezoid; C, capitate; H, hamate; mn, median nerve; fr, flexor retinaculum; ua, ulnar artery; un, ulnar nerve. (A from Noble J: Textbook of primary care medicine, ed 3, St Louis, 2001, Mosby.; B from Yu JS, Habib PA: Normal MR imaging anatomy of the wrist and hand, Radiol Clin North Am 44 (4):569-581, 2006.)
Clinical Manifestations
Persons with CTS experience sensory symptoms in the median nerve distribution (see Fig. 39-7). Pain may be located distally in the forearm or wrist and radiate into the thumb, index, and middle fingers. It may also radiate into the arm, shoulder, and neck. Comparing self-reported symptoms recorded on the Katz hand diagram allows symptoms to be assessed as classic, probable, possible, or unlikely to be CTS.25 Nocturnal pain is the hallmark of CTS. Even in the early stages of CTS most people will report being awakened by painful numbness in the middle of the night. Sensory symptoms usually precede motor symptoms. Diminished 2-point discrimination, diminished ability to perceive vibration, and elevation of threshold in Semmes-Weinstein monofilament testing routinely occur. Thenar weakness is seen in advanced cases. In nearly half of all cases, symptoms occur bilaterally. If CTS goes untreated, symptoms escalate into persistent pain with atrophy of the thenar musculature and the person will have a loss of grip strength. The combined loss of grip strength, inability to pinch, and sensory loss causes clumsiness in the hands.27 Because conditions that impinge nerve fibers in the neck (radiculopathy) or in the thoracic outlet also cause sensory symptoms that are referred to the hand, it is important to ascertain that the symptoms are related to CTS (see Box 39-1).
MEDICAL MANAGEMENT
The diagnosis of CTS is considered in any person with hand or wrist pain, numbness, and weakness and must be distinguished from a cervical radiculopathy or ulnar neuropathy.27 Diagnosis is determined by history, physical examination, and specialized tests. Provocation tests are used to replicate CTS symptoms. Phalen’s test, in which the wrist is flexed to 90 degrees for 1 minute (Fig. 39-9, A); Tinel’s test, or wrist percussion over the carpal tunnel (Fig. 39-9, B); and the carpal compression test (pressure is applied by the examiner by pressing his or her thumbs at the wrist over the flexor retinaculum) are all deemed positive when pain, numbness, and paresthesia are produced. The flick sign is a positive indicator of CTS when the client demonstrates what he/she does to relieve symptoms. Ask “What do you do with your hand(s) when your symptoms are the worst?” and the client demonstrates a flicking movement of the hand that looks similar to the motion seen in shaking a thermometer.25 When tests available to diagnose CTS have been compared to the gold standard of NCV, varying degrees of reliability have been reported. Most recently, Tinel’s test has sensitivity of 0.90 and specificity of 0.81, and Phalen’s test to reproduce symptoms only has a sensitivity of 0.85 and specificity of 0.79.85
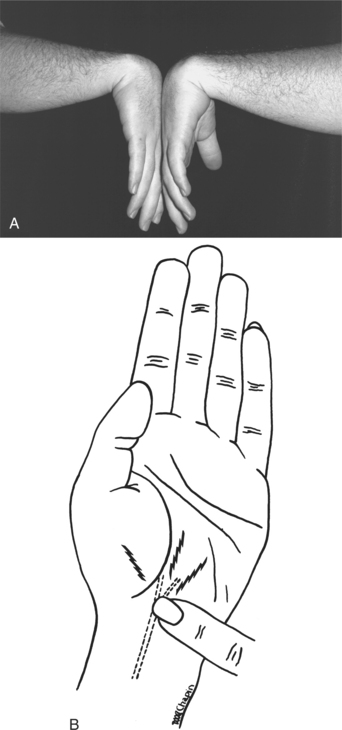
Figure 39-9 A, Phalen’s test. Patients maximally flex both wrists and hold the position for 1-2 minutes. If symptoms of numbness or paresthesia within the median nerve distribution are reproduced, the test is positive. B, Tinel’s sign in carpal tunnel syndrome. (A from Frontera WR, Silver JK: Essentials of physical medicine and rehabilitation, Philadelphia, 2002, Hanley and Belfus; B from Noble J: Textbook of primary care medicine, ed 3, St Louis, 2001, Mosby.)
The gold standard to confirm CTS is NCV testing. Distal motor and sensory latencies and sensory NCV across the carpal tunnel are most frequently administered. Changes in the sensory conduction across the wrist are reportedly the most sensitive indicator of CTS.174 A modified NCV technique, termed inching, has been shown to provide greater sensitivity and specificity for precise localization of anatomic entrapment for carpal tunnel.142 Although NCV testing generally provides the benchmark for CTS, a negative NCV study alone does not exclude the possibility of CTS. There are other imaging methods that are helpful in diagnosing CTS. Although magnetic resonance imaging (MRI) has also been found effective in identifying anomalies in the carpal tunnel, including altered tendon position, altered nerve position, swelling of the median nerve, and thickening of the tendon sheath to aid in establishing the diagnosis of CTS,5 but controversy exists over its use in diagnosis175 because of its variable sensitivity and specificity.180 Some believe that ultrasonography is more helpful in estimating the severity of symptoms and nerve conduction deficit.37,76,87
TREATMENT.
There is no universally accepted treatment for CTS. Although many studies have been conducted over the years, there are few well-controlled investigations that demonstrate the most effective treatment intervention. Thus many approaches are used for symptom management.44 For clients with mild symptoms, demonstrated by only subjective and objective sensory symptoms, conservative management is generally instituted. Medical management of mild symptoms includes steroid injection into the carpal canal to provide initial relief of symptoms. Early management also addresses ergonomic measures and modification of the client’s occupation. Alternative computer keyboards have been evaluated and three configurations are reported to promote a more neutral wrist position than use of a regular keyboard.101 Wearing of wrist splints to immobilize the wrist near neutral to minimize carpal tunnel pressures and client education are also instituted. These conservative approaches provide symptom relief up to 6 months.55 However, the long-term use of antiinflammatory medications and immobilization demonstrated a cure rate of only 18%. Relapse was noted within 1 year. Most recently, injection of methylprednisolone proximal to the tunnel has resulted in symptom relief for 77% of those treated when reassessed after 1 month. Fifty percent of those treated reported prolonged relief (at least 1 year) after injection.24
Surgical intervention is advocated for persons without resolution of symptoms following a traditional conservative approach for 2 to 3 months. Surgery is also indicated in untreated persons whose symptoms have lasted longer than 1 year and who demonstrate both motor and sensory NCV involvement or in persons with denervation as evidenced by fibrillation potentials on EMG. Release of the transverse carpal ligament is commonly performed and is usually successful. Complications fall into two categories: errors in diagnosis or surgical technique. Newer surgical techniques (flexor tenosynovectomy with transverse carpal ligament division, endoscopic release of the ligament, and neurolysis of the median nerve) are performed through limited incisions and require less exposure and less manipulation of the nerve than the classic open techniques. Seventy-six percent of the surgical cases experience return of normal 2-point discrimination and up to 70% have normal muscle strength return.73 However, a systematic review of surgical procedures has identified that newer procedures are no more effective than traditional approaches. In offering symptom relief, conflicting evidence exists about whether endoscopic release allows earlier return to work than open tunnel release.139 After surgery, nerve and tendon gliding techniques are advocated to reduce scarring, adhesions, and subsequent formation of fibrotic tissue.161
PROGNOSIS.
Prognosis relates directly to the severity of the nerve entrapment at diagnosis, clinical cause, and mode of treatment.
Sciatica
Sciatica is a radiculopathy occurring most often in individuals between the ages of 40 and 60 years in which the nerve root is affected, most typically by compression. Of those developing lumbosacral radiculopathy, 10% to 25% develop symptoms that last more than 6 weeks. Less commonly, sciatica may occur in the presence of abscess, blood clots, or tumors. Sciatica may be mistaken for intermittent claudication or low back pain without discogenic involvement and is one of the most common conditions managed in primary care settings.
Pathogenesis
The epidural space is innervated by a meningeal branch of the spinal nerve, the recurrent sinuvertebral nerve. Arising from the dorsal root ganglion, this nerve enters through the intervertebral foramen, divides into ascending and descending branches to blood vessels, and supplies the posterior longitudinal ligament, the superficial anulus fibrosis, anterior dura mater, and dural sleeve.102 In animal studies, the sinovertebral nerve responded to high threshold mechanical stimuli. Conduction velocity for fibers in the nerve corresponded to types III and IV, which lead researchers to correlate nerve function with nociception.141 Herniation of the intervertebral disk can impinge on the nerve root or structures innervated by the recurrent sinuvertebral nerve to cause pain.
Clinical Manifestations
In addition to low back pain, when sensory fibers are affected, pain will radiate into one or both legs. One of the reasons the motor and sensory nerves are affected so easily in a radiculopathy is the pressure occurs in an area where CNS connective tissue coverings meet the protective tissue coverings of the peripheral nerve, leaving that region of the nerve “at risk.”72 Coughing, sitting, and sneezing worsens the pain. For further information, see Chapter 27. Both clinical and experimental studies have shown that adjacent nerve roots may be affected when the lumbar disc herniates. Inflammatory chemical mediators released into the epidural space affect nearby nerve roots, without any direct compression of those roots.113
MEDICAL MANAGEMENT
Both radiologic tests and electrophysiologic studies are ordered. Various specific tests have been reported to provide reliable results. MRI is preferred to computed tomography (CT) scanning for lumbar spine imaging; however, because 60% of people without back symptoms have disk bulging on MRI, protrusion and bulges may not correlate with symptoms.7 A screening EMG examination of only four muscles in the leg identified over 89% of surgically confirmed.32 Others have noted that the H-reflex has provided better predictive value than standard motor and sensory nerve conductions radiculopathies.2 Just as radiologic studies are not sufficient alone to distinguish sciatica neither is electrophysiologic testing.
TREATMENT.
The effectiveness of medications has been reported as disappointing. Selective epidural injection of steroids at target nerve roots through the intervertebral foramina has offered short-term benefit for pain relief, as has the use of nonsteroidal antiinflammatory drugs (NSAIDs).21 Also unclear are the long-term effects of chemonucleolysis, which has been reported to be less effective than discectomy.50
PROGNOSIS.
Subjects who were evaluated 1 year after discectomy had recovery in unmyelinated and small myelinated fibers; the function of larger myelinated fibers did not improve. This provides a physiologic rationale for residual motor and sensory involvement.113
Idiopathic Facial Paralysis/Bell’s Palsy
Bell’s palsy is a common clinical condition in which the facial nerve is unilaterally affected. Bell’s palsy affects 20 of 100,000 people each year. Although any age group can be affected, it is most common in persons between the ages of 15 and 45 years.65
Etiology and Pathogenesis
The cause of Bell’s palsy is uncertain; however, evidence is increasing that indicates that the primary cause of Bell’s palsy is a latent herpes virus (herpes simplex type 1/herpes zoster) that has been reactivated.19,122 Days before Bell’s palsy onset, the client may recall experiencing severe pain in the area of the mastoid or a sensation of fullness in the ear. Pain suggests that this disorder is a product of an inflammatory response. Because the facial nerve lies in the auditory canal, any agent that causes inflammation and swelling creates a compression that initially causes demyelination. However, if the inflammatory response is more fulminating, ischemia will cause an axonal degeneration.
In addition, centrally located structures, such as acoustic neuromas (tumor), can produce unilateral paralysis in the face by impinging on the facial nerve as it emerges from the brainstem; however, these tend to produce a slowly progressive paralysis.
Risk Factors
People with diabetes mellitus and pregnant women19 have an increased incidence of Bell’s palsy.
Clinical Manifestations
A unilateral facial paralysis develops rapidly, often overnight. Paralysis of the muscles of facial expression on one side creates an asymmetrical facial appearance (Fig. 39-10). The corner of the mouth droops, the nasolabial fold is flattened, and the palpebral fissure is widened because the eyelid does not close. In addition to the motor fibers providing innervation for facial musculature, the facial nerve also innervates the stapedius muscle of the middle ear and the sensory and autonomic fibers, which innervate for taste and lacrimation and salivation, respectively. Therefore involvement of these fibers may produce additional signs and symptoms to those of facial paralysis. If the lesion is proximal to where the fibers of the chorda tympani enter the facial nerve, the client will experience loss of taste on the affected side. In a similar fashion, if the autonomic fibers are involved, the client will experience dry eye (lack of tearing) and will produce less but thicker saliva. Some clients report that sounds are louder than normal because the stapes bone of the middle ear is less able to accommodate sound when the stapedius muscle’s innervation is lost.
MEDICAL MANAGEMENT
Ask the client to wrinkle the forehead, close the eyes tightly, smile, and whistle while you observe for facial asymmetry. In addition to the clinical presentation and history, electrodiagnostic tests can be used to demonstrate whether the lesion is one of demyelination or axonal degeneration. However, EMG as a diagnostic tool is only helpful after the nerve has degenerated; therefore testing is most accurate after 1 week. Tests of facial nerve excitability will also indicate whether the paralysis is complete.
The LMN involvement of the facial nerve can be differentiated from an upper motor neuron (UMN) involvement of this nerve because with UMN involvement the client can close the eye and wrinkle the forehead but cannot smile voluntarily. With LMN involvement, the client is unable to close the eye, wrinkle the forehead, or smile voluntarily.
TREATMENT.
Since the outcome (demyelination or degeneration) is unknown initially, prophylactic administration of high-dose corticosteroids for 5 days, followed by a tapered dose for 5 days, has been advocated. For more severe involvement, this treatment is reported to help prevent permanent damage. Treatment should begin as soon as possible and no later than 10 days after onset of signs of paralysis. The association that has been discovered between herpes simplex virus and Bell’s palsy suggests that treatment with antiviral medications, such as acyclovir or acyclovir paired with corticosteroids, may aid in recovery.63 Patients who received a combined treatment of acyclovir (antiviral) with prednisolone (corticosteroid) had a recovery rate of 95.7% (better than corticosteroids alone, 89%). Treated within 3 days on onset of paralysis, a 100% recovery has been reported; the rate of recovery drops to 86% when treatment was delayed until day 4). Benefits of antiviral medications or nerve root decompression have not been established definitely.57,64 Yet controversies exist related to the medical management of Bell’s palsy. One systematic review reports that available evidence does not show significant effects of corticosteroids,133 whereas a metaanalysis found that corticosteroids provided both clinically and statistically significant recovery of motor function in facial nerve–innervated musculature.126 Studies of Bell’s palsy in children have indicated that there is no supporting evidence for the use of steroids or antiviral medications in children.134
To protect the cornea, the client should cover the eye with a patch or glasses and use artificial tears. Other palliative treatments, such as gentle massage and gentle heat, may also be used.1
PROGNOSIS.
Ninety-four percent of individuals with incomplete involvement make a full recovery, generally within 3 weeks. For complete involvement, 75% recover normal motor function, although the time course of recovery is longer.65 Factors associated with a poorer outcome include age greater than 60 years, presence of systemic comorbidities such as diabetes mellitus and hypertension, and symptoms indicating a lesion with autonomic involvement.16 Plastic surgery, using fascial slings to replace active muscle contraction, can help restore facial function when recovery does not occur. Another complication that can occur during recovery is a phenomenon called motor synkinesis (crocodile tears), which occurs when motor fibers of the facial nerve cross-innervate the autonomic branch of the greater superficial petrosal nerve. When muscles of the face contract, tears appear. This has been noted up to 1 year after the start of treatment.148
Tardy Ulnar Palsy/Retroepicondylar Palsy
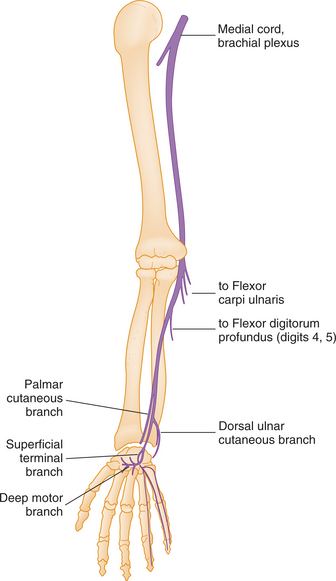
The ulnar nerve arises from the lower trunk of the brachial plexus and carries fibers from C8 and T1 nerve roots. At the elbow, it passes behind the medial epicondyle and then passes between the two heads of the flexor carpi ulnaris through the forearm to the wrist (Fig. 39-11). The distal portion of the nerve enters the palm by crossing the flexor retinaculum and divides into a superficial and deep branch in the hand.
Etiology
Because of its anatomic location, ulnar nerve palsy is a common complication of fractures in the region of the elbow. A late or tardy ulnar palsy may occur years after a fracture and is associated with callus formation or a valgus deformity of the elbow. These produce a gradual stretching of the nerve in the ulnar groove of the medial epicondyle.
Risk Factors
A similar type of tardy ulnar palsy occurs with repeated trauma for relatively long periods of time in clients with a shallow ulnar groove at the elbow. Ulnar neuropathy from entrapment at the elbow is the second most frequent upper extremity neuropathy (after carpal tunnel).
Pathogenesis
The mechanism of injury compressing the ulnar nerve has been attributed to recurrent microtrauma associated with fracture and fibrous bands or recurrent cubital subluxations, as well as entrapment at the entrance or exit of the cubital tunnel.92 Elbow flexion aggravates symptoms. Compression will initially cause a neurapraxia with demyelination of the nerve; if the pressure goes unrelieved, this will progress to an axonotmesis with denervation occurring below the level of the elbow.
Clinical Manifestations
Expect a clawhand deformity with metacarpophalangeal (MCP) extension and interphalangeal (IP) flexion of the ring and little fingers because of the unopposed action of the extensor muscle group and paralysis of the third and fourth lumbricals that normally flex the MCPs and extend the IPs (Fig. 39-12). Flattening of the hypothenar eminence along with abduction of the little finger coincides with weakness of the palmaris brevis and abductor digiti minimi. Marked atrophy of the interossei on the dorsal surface of the hand with guttering between the extensor tendons indicates the presence of denervation. Abduction and adduction movements of the fingers are impaired. Paralysis of the flexor carpi ulnaris (FCU) produces a radial deviation of the hand when wrist flexion is attempted. Sensory loss is variable, but impaired sensation may be expected involving the little finger and the ulnar aspect of the ring finger and along the ulnar aspect of the palm of the hand to the wrist. Occasionally, sensory symptoms extend proximally to the wrist (see Fig. 39-12).
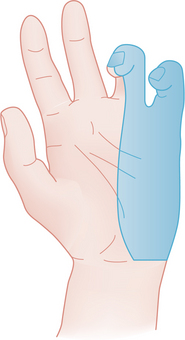
Figure 39-12 Clawing of the ring and little fingers (hyperextension of the metacarpophalangeal joint and flexion of the interphalangeal joints) from unopposed action of extensor musculature combined with paralysis of the intrinsic muscles of the hand occurs when there is involvement of the ulnar nerve. Shaded area represents ulnar nerve sensory distribution in the hand. (From Marx RS, Hockberger RS, Walls RM: Rosen’s emergency medicine: concepts and clinical practice, ed 6, St Louis, 2006, Mosby.)
MEDICAL MANAGEMENT
Percussion of or bending the elbow can replicate sysmptoms.92 NCV studies are helpful only when sufficient nerve damage has occurred to produce definite strength or sensory changes in the hand. NCVs are slowed through the involved region but are relatively normal above and below the epicondyle. Electromyography reports slowing of sensory or motor NCV across the elbow, prolonged conduction (termed a latency) to the FCU, along with changes in amplitude, duration, or shape of the sensory potential across the elbow. Sensory fibers were affected first. Detection of an abnormal latency requires accurate measurement of ulnar nerve segment length.106
TREATMENT.
Mild entrapments are managed conservatively; moderate and severe compression require surgery. To relieve the compression, either decompression, the preferred method (medial epicondylectomy), or transposition of the ulnar nerve to the anterior aspect of the elbow is performed.13,150 Symptomatically, the clawhand deformity should be treated with a splint that blocks MCP hyperextension (lumbrical bar) and allows the extensor digitorum to extend the IP joints.
PROGNOSIS.
Results of surgery are normally good when the individual has not had a chronic tardy ulnar involvement. Decompression surgery should have complete res- toration of function quickly, but recovery after transposition surgery may take up to 6 months. Surgery to treat chronic involvement (over 3 months) may have a less certain restoration of function.150 After nerve transposition, most NCVs at follow-up are improved. However, the magnitude of change in the motor conduction velocity does not correlate well with clinical improvement. One factor that has been identified to effect outcome is body mass index; increased body weight is related slightly to patient’s perception of poorer improvement.110
Thoracic Outlet Syndrome
Because clients diagnosed with thoracic outlet syndrome (TOS) have vague symptoms or symptoms that are difficult to interpret, TOS remains a controversial diagnosis. Because this disorder is complex and poorly defined, many clients have been labeled neurotic.
Definition
TOS is an entrapment syndrome caused by pressure from structures in the thoracic outlet on fibers of the brachial plexus at some point between the interscalene triangle and the inferior border of the axilla. In addition, vascular symptoms can occur because of pressure on the subclavian artery (Fig. 39-13).
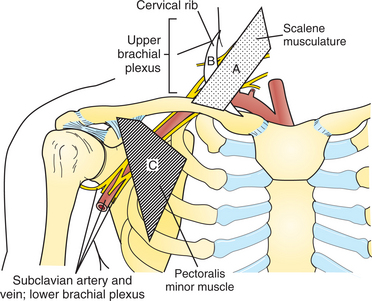
Figure 39-13 Schematic relationship of structures in development of thoracic outlet syndrome (TOS). Compression of the neurovascular bundle can occur with: (A) hypertrophy of scalene musculature impinging on structures lying between middle and anterior scalene; (B) the presence of cervical rib or fibrous bands between the cervical and first rib; or (C) compression by pectoralis minor during hyperabduction. (From Rakel RE: Textbook of medicine, ed 7, Philadelphia, 2007, Saunders.)
Etiology
The anatomy of the region of the thoracic outlet is extremely complex. Spinal nerve roots of the brachial plexus interact with surrounding bony ribs, muscles, and tendons (subclavius, anterior and middle scalene, and pectoralis minor) and the vascular supply (subclavian artery and vein) to the region. In addition to neurologic structures becoming entrapped, arterial and venous structures also may be affected individually or in combination. Thus multiple specialists may be involved in a person’s care. Practically, TOS can be divided into three groups: neurogenic (compression of brachial plexus), vascular (compression of subclavian artery and/or vein), and disputed (nonspecific TOS with chronic pain and symptoms of brachial plexus involvement).11,68
Risk Factors
Postural changes associated with growth and development, trauma to the shoulder girdle, and body composition have all been identified as contributing to the development of TOS. The human upright posture has contributed to the development of TOS because gravity pulls on the shoulder girdle creating traction on the structures. Additionally, congenital factors that affect the bony structures, such as a cervical rib or fascial bands, also compress the neurovascular bundle.
Pathogenesis
Chronic compression of nerve roots or proximal plexus and arteries between the clavicle and first rib or impinging musculature results in edema and ischemia in the nerves (see Fig. 39-13). This compression initially creates a neurapraxia in which the axons are preserved, but segmental demyelination occurs. After loss of myelin the axons are more vulnerable to unrelieved compression. The neurapraxia can progress to an axonotmesis in which axon continuity is lost and wallerian degeneration occurs.
Clinical Manifestations
Signs and symptoms reflect the structures that have been compressed. When the nerves are compressed, most people report paresthesias and pain in the arm; most often these are nocturnal. Other symptoms may include pain, tingling, and paresis. If the upper nerve plexus is involved (C5 to C7), pain is reported in the neck; this may radiate into the face (sometimes with ear pain) and anterior chest, as well as over the scapulae. Symptoms may also extend over the lateral aspect of the forearm into the hand. If the lower plexus is compromised (C7 to T1), pain and numbness occur in the posterior neck and shoulder, medial arm and forearm, and radiate into the ulnarly innervated digits of the hand. Weakness is usual in the muscles corresponding to nerve root innervation, and atrophy occurs in severe cases. Vascular symptoms may include coldness, edema in the hand or arm, Raynaud’s phenomenon (cyanosis), fatigue in hand and arm, and superficial vein distention in the hand.(Fig. 39-14).
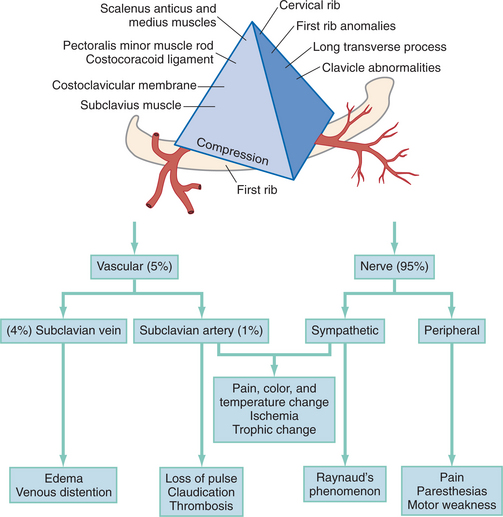
Figure 39-14 Relationship of thoracic outlet abnormalities and impairments. (From Marx RS, Hockberger RS, Walls RM: Rosen’s emergency medicine: concepts and clinical practice, ed 6, St Louis, 2006, Mosby.)
The clinical presentation usually relates to posture and activities that aggravate symptoms. Overhead and lifting activities, along with movements of the head, produce symptoms in the upper plexus.
MEDICAL MANAGEMENT
Provocative tests are used to elicit symptoms of TOS, but these tests have a high false-positive response. Maneuvers are performed bilaterally, and the pulse is monitored to note a change in its quality. Based on the belief that the anterior scalene compresses the neurovascular bundle, the individual is positioned to elicit the symptoms. However, mere obliteration of the peripheral pulse does not necessarily mean that TOS exists as an entrapment problem; sensory symptoms must be reproduced. For persons with a vascular component, blood pressure may differ from side to side. Although there is no universally accepted reliable diagnostic test for TOS, Adson’s maneuver (Fig. 39-15) appears among the most effective. Several other maneuvers with a positional component of the head, shoulder, or arm have been found to compress vascular or neural structures and thus evoke symptoms. These tests include the Allen’s, Wright’s, Halsted’s, costoclavicular, Roos/elevated arm stress test (EAST), and provocative elevation tests (Table 39-6).96 The sensitivity and specificity of Adson’s test improve when used in combination with the hyperabduction test (symptom replication), the Wright’s test (symptom replication), or the Roos test (Table 39-7).51
Table 39-6
Special Tests and Patterns of Positive Findings That Characterize Thoracic Outlet Syndrome*
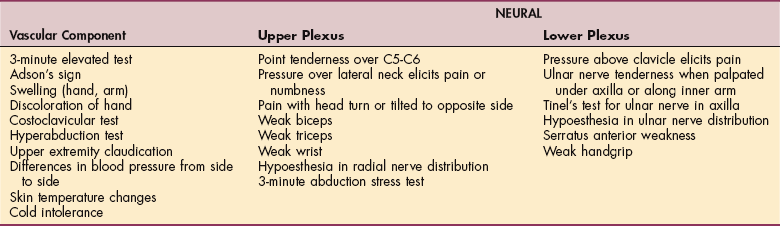
*With the use of special tests, patterns of position objective findings may help characterize thoracic outlet syndrome.
From Goodman CC, Synder TEK: Differential diagnosis for physical therapists: screening for referral, Philadelphia, 2007, WB Saunders.
Table 39-7
Diagnostic Utility of Tests for Thoracic Outlet Syndrome
| Provocation Test | Sensitivity | Specificity |
| Adson’s | 0.79 | 0.76 |
| Hyperabduction (HA)—pulse abolition (HAp) | 0.84 | 0.4 |
| Adson’s + HAs (symptom replication) | 0.72 | 0.88 |
| Adson’s ++ Wright’s | 0.54 | 0.94 |
| Adson’s ++ Roos | 0.72 | 0.82 |
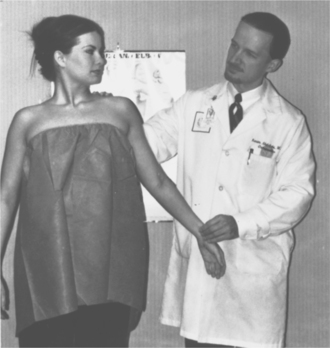
Figure 39-15 Adson’s test is one of many diagnostic tests used to examine the upper extremity to determine presence of thoracic outlet syndrome: arterial or neurologic. Hold patient’s arm in slight abduction while palpating the radial pulse. Ask the patient to inhale and hold the breath while extending the neck and rotating toward the affected side. Adson’s test is positive if the patient reports paresthesias or if the pulse fades away. (From DeLee JC, Drez D, Miller MD: DeLee and Drez’s orthopaedic sports medicine, ed 2, Philadelphia, 2003, WB Saunders.)
Radiographic Tests.
Radiographic procedures are used to identify bony abnormalities. Presence of a cervical rib may indicate that the nerve has been compressed; however, presence of the rib alone does not necessarily replicate symptoms. Plane films are used to distinguish between a C7 to T1 discogenic lesion and TOS.
Electrophysiologic Studies.
Because symptoms of TOS are related to neural compression, electrophysiologic studies are valuable in documenting the presence of neuropathy. NCV allows the examiner to pinpoint the lesion, either because of a change in amplitude or a slowing in conduction velocity (Box 39-2). Other, more refined electrophysiologic techniques, including somatosensory evoked potentials and F waves, are used to confirm a diagnosis of nerve root entrapment.
DIFFERENTIAL DIAGNOSIS.
TOS must be distinguished from other disorders with similar symptoms. These include cervical radiculopathy, reflex sympathetic dystrophy, tumors of the apex of the lung (Pancoast’s tumor, see Chapter 15), and ulnar nerve compression at either elbow or wrist. The sensory pattern of TOS distinguishes it from an ulnar neuropathy such as tardy ulnar palsy. Because the nerve roots are affected, the sensory changes extend above the hand and wrist into the forearm in TOS and follow a dermatomal pattern. Myofascial pain patterns may also mimic TOS symptoms.
TREATMENT.
Management is divided into conservative and surgical approaches. The initial treatment of the person with TOS is conservative when symptoms are mild to moderate in severity. Postural and breathing exercises and gentle stretching are the cornerstones of the initial conservative program. This is followed by strengthening exercises for shoulder girdle musculature, especially the trapezius, levator scapulae, and rhomboids. Initially, overhead exercises should be avoided because they tend to evoke symptoms. Therapists are cautioned against forceful stretching to mobilize the first rib.89
Surgical management of TOS is reserved for cases that are refractory to postural and exercise correction and those with vascular compromise.20 Once the decision for surgical intervention has been made, the physician must select a procedure and the anatomic approach. There are at least six different surgical procedures and six different anatomic approaches (Box 39-3). In scalenotomy the muscle is detached from the first rib; unfortunately, with this approach a high percentage of people experience recurring symptoms. Scalenectomy, removal of the scalene muscle, is advocated for people who have had recurrence of their symptoms. Clavicle resection is indi- cated primarily when the clavicle is damaged. When scalenectomy, with or without first rib resection, is the surgical approach used, its 5-year success rate is about 70%.135
PROGNOSIS.
After surgery, 70% of cases have a good or excellent response using a supraclavicular or transaxillary resection of the first rib. Improvement in pain symptoms ranges from 70% to 80%, some patients require occasional analgesics, and 10% note no improvement. In individuals with signs and symptoms and electrophysiologic changes consistent with classic TOS, no improvement in strength is noted when atrophy was present before surgery.20 Complications during surgery include pneumothorax, nerve compression, and transient winging of the scapula because the upper digitations of the serratus are detached.
A 4-year follow-up reported no significant difference in return to work or symptom severity when the first rib was resected compared to a conservative, nonoperative approach.86 Factors that are associated with long-term disability include preoperative depression, single status, and less than high school education.6
Saturday Night Palsy/Sleep Palsy
Saturday night palsy is associated with radial nerve compression in the arm. It results from direct pressure against a firm object and typically follows deep sleep on the arm with compression of the radial nerve at the spiral groove of the humerus in a person who is sleeping after becoming intoxicated. Sleep palsy has also been associated with lipoma compressing the radial nerve.43 If the radial nerve is compressed in the axilla, the damage is often referred to as a crutch palsy.
Clinical Manifestations
Symptoms of radial nerve paralysis depends on the level of the lesion. The more proximal the involvement, the more extensive the paralysis. When involvement occurs in the axilla, weakness occurs in elbow extension (triceps), elbow flexion (brachioradialis), and supination (supinator). If the nerve is damaged in the upper arm the triceps is spared. In addition, in both instances there will be paralysis of wrist extensors and the extensors of the fingers and thumb, diminishing grip strength. Sensory loss with radial nerve involvement is variable. If present, it is typically confined to the dorsum of the hand but may extend to the dorsum of the forearm.
MEDICAL MANAGEMENT
Diagnosis is by history, clinical examination, and electrophysiologic examination. This type of paralysis is usually classified as a neurapraxia or conduction block, signifying demyelination. There is slowing of nerve conduction in both motor and sensory fibers across the lesion site.
TREATMENT.
Medical management is aimed at asymptomatic management. A cock-up splint is used to maintain the wrist in an extended position until return of function.
PROGNOSIS.
If a neurapraxia is reported, normal conduction can be anticipated within a few months because the paralysis is related to a focal demyelination.59
Morton’s Neuroma
Morton’s neuroma is a common entrapment neuropathy in the forefoot, also called interdigital perineural fibroma (IPF), and most often involving the third toe interspace.
Definition and Etiology
No incidence or prevalence for Morton’s neuroma has been published in any study. However, the average age of individuals diagnosed with Morton’s neuroma is reported between 45 and 60 years, with women affected 5: 1 more than men. Bilateral involvement is uncommon.18
Pathogenesis
Three common digital nerves, two arising from the medial plantar nerve, and third from the lateral plantar nerve, pass between divisions of the plantar aponeurosis where each bifurcates into two interdigital nerves. The first common digital nerve supplies adjacent sides of the great and second toe, those of the second common digital nerve supply adjacent sides of the second and third toes, and the sides of the third and fourth toes are supplied by the third common digital nerve (Fig. 39-16). Mechanical irritation resulting from intrinsic factors, such as diminished intermetatarsal head distance90 and poor foot mechanics (excessive pronation during gait) that pulls the nerve more medially than normal and taut as the toes extend during terminal stance, and extrinsic factors, such as high heels in which the weight is transferred onto the forefoot, maintaining the nerve in a taut condition; narrow toe box on shoe that creates a greater compression in the area; and thin-soled shoes where ground forces interact with the deep transverse metatarsal ligament, causing compression in this confined space, have been implicated as contributing to this condition. Additional inflammatory conditions, such as arthritis, and activities that involve application of repetitive forces to the plantar nerves, such as jogging on a hard surface, produce shear forces that can irritate the nerve.
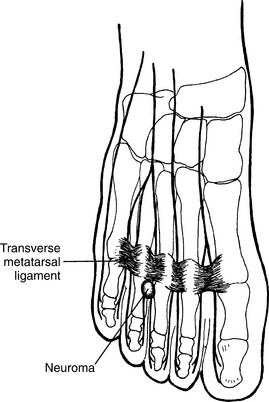
Figure 39-16 Morton’s neuroma involves the common digital nerve. The most frequent location is between third and fourth metatarsals. (From Frontera WR, Silver JK: Essentials of physical medicine and rehabilitation, Philadelphia, 2002, Hanley and Belfus.) Hanley and Belfus
Entrapment produces some or all of the following histopathology: thickening of the endoneurium, hyalization of endoneurial vessels, thickened perineurium, and demyelination of nerve fibers.18,137
Clinical Manifestations
Symptoms include burning, tingling, or sharp lancinating pain in one of the interspaces of the forefoot that occurs while walking. Pain may radiate into adjacent toes or proximally into the foot. Individuals may state that they must stop, remove their shoe, and massage their foot to relieve the symptoms. At its worst, the person may be apprehensive about stepping with the involved foot. Symptoms occur paroxysmally over many years.
MEDICAL MANAGEMENT
Typically, history and clinical examination have been used to diagnose this disorder. Two tests that provoke symptoms include plantar palpation of the involved space at the metatarsal heads as mediolateral compression is applied to the metatarsal heads (Mulder’s sign) and dorsiflexion of the involved toe producing symptoms and plantarflexion of the toe relieving them (Lasègue’s sign).45 The reported positive predictive values of these clinical tests vary widely. Recently, sonography and MRI have been used to assess the presence of Morton’s neuroma. Whereas the sensitivity for predicting the presence of Morton’s neuroma is reported at 0.79 and 0.86, respectively, the specificity of both sonography and MRI is 1.0145 and has been used to diagnose Morton’s neuromas.125,157
Differential diagnoses considered would include metatarsal stress fractures, metatarsalgia, and metatarsal phalangeal derangement.
TREATMENT.
Conservative, nonoperative management is directed at pressure relief and involves use of a soft orthosis (insoles) or metatarsal pad. These may provide symptom relief as long as the shoes the person is wearing have a wider toe box and a lower heel. If symptoms continue, injection of a local anesthetic or corticosteroid from the dorsal direction may be helpful. Finally, surgical treatment involves either neural decompression by releasing the intermetatarsal ligament or neurectomy, proximal to the location of the neuroma to allow retraction of the plantar nerve away from the weight-bearing surface.
PROGNOSIS.
A systematic review of these interventions reports that for studies in which orthoses have been used, 45% to 50% of the participants reported pain relief of more than 50% up to 1 year postintervention. For various surgical approaches, pain relief of more than 50% occurred in 65% to 100% of patients up to 3 years postsurgery.159
Neurotmesis
A neurotmesis occurs after total loss of axon and connective tissue continuity; the nerve is severed.
Pathogenesis
When the axon is lacerated, wallerian degeneration occurs distally and proximally the cell body also responds to the trauma. It swells and undergoes chromatolysis. The ribosomes that normally make protein for the cell disperse throughout the cytoplasm. Chromatolysis reflects a change in the metabolic priority of the cell as it switches from daily needs to a repair mode. Distally the axon begins to degenerate and myelin fragments within 12 hours of the lesion (see Fig. 39-3, C). This material is removed by macrophages responding to the inflammatory process.
As long as the cell body remains viable, a regenerative process begins with sprouting of a growth cone as soon as new cytoplasm is synthesized and transported down the axon from the cell body (see Fig. 39-3, D). As the growth cone grows, it releases proteases that dissolve material and permit the axon to enter the tissue more easily. Filopodia, which are fingerlike projections extending from the growth cone, sample the environment searching for chemical and tactile cues to guide the regenerating axon; however, because the tactile cues provided by the endoneurium are absent, many times these fibers become misguided and form a neuroma. The standard used to anticipate return of function is based on a growth rate of 1 mm a day or an inch a month. In reality, this is an average reflecting the delays that occur while the growth cone crosses the repair site and makes connection with sensory end organs or motor endplate. Growth occurs faster nearer the lesion site (3 mm/day) and slower as the length of the axon increases (1 mm/day).115
Clinical Manifestations
The degree of involvement relates to the nerve involved and its level of involvement. In any case, an immediate flaccid paralysis occurs in muscles distal to the lesion. Rapid atrophy ensues because of loss of the trophic influences of the nerve that innervated the muscle fibers. Sensory function is also lost below the level of the lesion.
MEDICAL MANAGEMENT
History and clinical examination are used to diagnose neurotmesis. In addition, electrophysiologic studies may be performed after a week. EMG will demonstrate the presence of fibrillation potentials and positive sharp waves, indicating denervation of muscle fiber. EMG can be used to determine whether the lesion is complete or partial.
TREATMENT.
Surgical management is needed to suture the connective tissue bundles together to guide the regenerating growth cone. Various microsurgical techniques (cable and interfascicular grafts) are used to try and direct the axon into the appropriate fascicle by restoring connective tissue continuity. After complete axonal transection, the neuron undergoes a number of degenerative processes, followed by attempts at regeneration. A distal growth cone seeks out connections with the degenerated distal fiber. The current surgical standard is epineurial repair with nylon suture. To span gaps that primary repair cannot bridge without excessive tension, nerve-cable interfascicular autografts are employed. Unfortunately, results of nerve repair to date have been no better than fair, with only 50% of patients regaining useful function. There is much ongoing research regarding pharmacologic agents, immune system modulators, enhancing factors, and entubulation chambers. Clinically applicable developments from these investigations will continue to improve the results of treatment of nerve injuries.88 Ideally, a primary repair will be carried out; operative delays lead to shrinkage and fibrosis of the distal connective tissue support structures.66
Other treatments are symptomatic. For the therapist this means splinting to support structures. Use of electrical stimulation to maintain muscle bulk is controversial; recent studies have shown that the chemical signal guiding the nerve (neural cell adhesion molecule [NCAM]) disappears when denervated muscle receives electrical stimulation.136 However, muscle bulk is maintained for up to 4 weeks. Because denervated skin does not wrinkle after it has been soaked in water, this has been used to evaluate denervation patterns.123
PROGNOSIS.
Recovery after neurotmesis is dependent on whether the nerve was repaired and the length of nerve that must be regenerated. Following transection, muscles atrophy rapidly, and after 2 years, they have undergone irreversible changes and have become fibrotic. If reinnervation occurs after 1 year, function is poor; with a delay of 18 to 24 months there is no hope for return of function.
METABOLIC NEUROPATHIES
Definition
A consensus conference has agreed that a detailed definition of diabetic neuropathy (DN) is “a descriptive term meaning a demonstrable disorder, either clinically evident or subclinical, that occurs in the setting of diabetes mellitus without other causes for peripheral neuropathy.”3,90 DN is a common complication associated with diabetes mellitus (see Chapter 11) comprised of a heterogenous group of progressive syndromes with diverse clinical manifestations. Neuropathies may be focal or diffuse and involve the autonomic or somatic PNS.11,12,167 Typically, the involvement occurs in a distally, symmetric pattern, termed diabetic polyneuropathy, although single, focal nerve involvement may be seen.
Incidence
In the United States, diabetes mellitus affects over 20 million people, and this number is expected to increase by 5% every year. The prevalence of DN is greater (54%) in type I diabetes (insulin-dependent diabetes mellitus [IDDM]) than the prevalence of DN in type 2 diabetes (noninsulin-dependent diabetes mellitus [NIDDM]), which is 30%. The most reliable estimates from clinical studies report that DN, although present in individuals with diabetes lasting longer than 25 years, is present in 7% of people within 1 year of diagnosis with diabetes.147
Etiology
DN is probably caused by the chronic metabolic disturbances that affect nerve cells and Schwann cells in diabetes. For years, hyperglycemia was considered the sole cause of these secondary complications of diabetes. Although consequences of hyperglycemia include elevated levels of sorbitol and fructose, which coincides with deficiencies of sodium-potassium and adenosine triphosphate (ATP) that alter the function of peripheral nerves, chronic hyperglycemia leads to abnormalities in microcirculation, creating endothelia capillary changes and local ischemia that affect the nerve. Excess sorbitol also damages Schwann cells. Most recently, researchers have suggested that alterations in insulin levels alter its regulatory roles in gene-regulation of neurotrophic factors, cell-cell adhesion molecules, and modification of proteins.147
Risk Factors
Although hyperglycemia is not directly attributed to damaging nerve fibers causing DN, it is a contributing factor. Conversely, some people develop neuropathies when glycemic control is good. A clear relationship does exist between duration of diabetes and development of DN. After the onset of the neuropathy, control of hyperglycemia is known to enhance the possibility of regeneration of fibers. Although studies have confirmed a genetic predisposition to diabetes, they have not confirmed such a predisposition to development of DN nor has a familial tendency been reported. Up to 50% of all people with diabetes never develop symptoms of neuropathy.
Pathogenesis
Many hypotheses exist for the pathogenesis of this disorder. The metabolic effect of hyperglycemia exposes nerves and their associated Schwann cells to glucose. The most prominent change in DN is loss of both myelinated and unmyelinated axons. Nerves are affected distally more than proximally. Subtle changes have been reported at the nodes of Ranvier in nerves of people with DN. This is associated with slowing of the NCV.166
A number of studies suggest that vascular changes affect peripheral nerves in diabetes. Evidence demonstrates endoneurial microvascular thickening. In the sural nerve, this has resulted in increased numbers of closed capillaries, which are believed to cause multifocal regions of ischemia and hypoxia in the nerve, resulting in an axonal degeneration.
Another explanation for the development of diabetic neuropathy proposes that the concentration of nerve growth factor (NGF), which has a structure that is molecularly and physiologically similar to insulin, is reduced. Since NGF acts as a trophic factor, its reduction also reduces nutrition to the nerve.
Clinical Manifestations
DN has been classified in a number of ways: presumed etiology, pathologic features, anatomic location, and a mixture of these. The most recent classification reflects the disturbances that occur in DN (Box 39-4)
Rapidly Reversible Neuropathy:
Hyperglycemic Neuropathy.: Hyperglycemic neuropathy occurs in individuals with poorly controlled diabetes, and in those who have been newly diagnosed, rapidly reversible nerve conduction abnormalities have been reported. These abnormalities are accompanied by distally symmetric sensory changes such as burning, paresthesias and tenderness in the feet and legs. Symptoms disappear when the individual’s blood sugar is controlled, although abnormalities in nerve conduction may persist.11
Generalized Symmetric Polyneuropathies:
Acute Sensory Neuropathy.: Hallmarks of acute sensory neuropathy, are the rapid onset of severe burning pain, deep aching pain, a sudden sharp “electric shock–like” sensation, and hypersensitivity of the feet that is often worse at night. Signs of this painful DPN are relatively normal: normal motor examinations in which the tendon reflexes are normal or reduced at the ankle. The patient may have no or only mild symmetric sensory loss, with allodynia. Testing procedures to confirm allodynia should apply the following concepts: apply a phasic stimulus (rub) to various parts of the body and ask the person whether burning occurs in a nearby region; Application of a cotton ball or Semmes-Weinstein monofilament is not applied long enough for the slow summation required for allodynia. Place a towel on the patient’s body and wait for a period of time before asking whether the cover creates pain; Do NOT use a sharp-dull to test for allodynia because it is pain from nonpainful stimuli. Noci- ceptive stimuli are perceived normally in acute sensory neuropathy. Electrophysiologic studies (NCV) may be normal or show minor changes. If the person can achieve and maintain stable blood glucose, recovery can occur within 1 year, even with severe symptoms.11
Chronic Sensorimotor Neuropathy.: Chronic sensorimotor neuropathy, or diabetic polyneuropathy (DPN), is the most common type of DN and up to 50% of patients may develop this condition. Typically, its onset is insidious, but occasionally signs and symptoms appear acutely. DPN’s clinical features include sensory loss, occasionally with selective fiber type involvement. Small fiber involvement leads to burning pain, and paresthesias, such as those described for acute sensory neuropathy, are more profound at night in the feet and lower legs (stocking pattern). Large fiber involvement results in painless paresthesia with impaired vibration, proprioception, touch, and pressure along with loss of ankle DTRs. Clients may report that they feel as if they were walking on cotton or clouds. In DPN, motor weakness is mild (presence of hammer toes and/or pes cavus) with wasting of small muscles in the feet and hands in more advanced cases. The presence of pronounced motor involvement implies that this is not DPN. DPN may be accompanied by clinical or subclinical autonomic involvement that can include cardiovascular and sympathetic disturbances resulting in sweating, orthostatic hypotension, and resting tachycardia (>100 bpm at rest).11
Autonomic Neuropathy.: Sympathetic and parasympathetic involvement may occur in both type I and type II diabetes; however, in type 2 diabetes, parasympathetic functions are more affected. After 10 to 15 years, 30% of patients have subclinical manifestations of autonomic involvement. Major manifestations associated with autonomic involvement are shown in Box 39-5.127
Mononeuropathies.: Mononeuropathies in the limbs or cranial nerves may occur in diabetes less often than the generalized, symmetric patterns. The median, ulnar, and peroneal nerves are most commonly affected in limb focal neuropathies. The somatic division of the oculomotor nerve is most commonly involved.
MEDICAL MANAGEMENT
The diagnosis is based on the history, clinical examination, electrodiagnostic studies, quantitative sensory evaluation, and autonomic function testing. Diagnosis of DN should not be based on a single symptom, sign, or test; a minimum of two abnormalities (signs and symptoms from NCV, sensory, or autonomic tests) has been recommended.3 Tools required for the sensory examination include a 128 Hz tuning fork to assess vibration and a 1 gm monofilament for touch. Autonomic functions can initially be assessed by blood pressure and hear rate response at rest, in standing and with exercise. Because diabetes is a common disorder and because neuropathies may be related to other causes, mere association of neuropathic signs and symptoms in a person with diabetes is not sufficient to diagnose DN. Other causes must be excluded.61
Sensory, motor, and F responses are important to assess nerve function at baseline and intermittently at follow-up visits. The most common electrical changes is a reduced amplitude in the sensory action potential (SNAP), which suggests axonal degeneration. A recent report found that a high percentage of newly diagnosed patients with type II diabetes have reduced SNAP in upper extremity nerves.130 Slowing of sensory and/or motor NCV suggests a demyelinating neuropathy, and pronounced slowing suggests that an alternative diagnosis should be explored.8 Sensory fibers are generally affected first before motor fibers.
TREATMENT.
Management is divided into general and specific measures. General measures include control of hyperglycemia165 and specific measures address the symptomatic management of the disorders (see section on Diabetes Mellitus in Chapter 11). Because there is evidence that further complications can be reduced by maintaining control of the diabetes, this is one specific area addressed by health care professionals. In addition, specific drug therapies are being evaluated. Currently, studies on medications and biochemical factors, such as gangliosides and NGFs, are being conducted and although some show promise. Tricyclic antidepressants are used alone or in combination with fluphenazine to treat painful neuropathies and gabapentin or carbamazepine is an efficacious approach to the management of pain in focal neuropathies.165 Although topical capsaicin has been recommended for allodynia, a systematic review concluded that it had moderate to poor efficacy in the management of chronic pain.103 Angiotensin-converting enzyme (ACE) inhibitors act on the vascular dysfunction and prevent the development and progression of diabetic neuropathy.100 In a systematic analysis of seven qualifying studies, researchers found vitamin B12 (either as B12 complex or methylcobalamin, one of two coenzyme forms of B12) had beneficial effects on pain and paresthesia more than electrophysiologic changes. Methylcobalamin improved autonomic symptoms in three studies.152
If the person has a painful DN, an algorithm has been developed that begins with physical modalities to manage pain. This is combined with simple analgesics. Further management may include a trial of topical or benign drugs.166 A common complication of DN is the development of neuropathic foot ulcers. When great toe and ankle joint mobility is limited greater forefoot pressures occur during gait, which may place patients with diabetes (type I or II) at risk for development of metatarsal ulceration.179 In type II diabetes, early detection of diabetic neuropathy along with prophylactic foot care regimens has led to fewer foot ulcerations and amputations.143 Institution of foot care procedures is essential (see Special Implications for the Therapist discussion of foot care in diabetes in Chapter 11). With the development of a footdrop gait, orthotic devices should be considered for the person’s safety. The type of orthosis and shoe construction prescribed should be carefully considered based on the sensory picture and the person’s ability to demonstrate appropriate foot care.
PROGNOSIS.
DN is a slowly progressive disorder. Because it is a metabolic disorder, other systems are often affected. Estimates are that over 50% of nontraumatic amputations in the United States are performed in diabetic clients. The presence of autonomic involvement is associated with an increased mortality risk.
Alcoholic Neuropathy
Peripheral neuropathies, typically with distally symmetrical involvement, appear in alcoholics after years of chronic alcohol abuse.
Etiology and Risk Factors
Although the exact pathogenesis of alcoholic neuropathy remains unclear,168 lesions affecting the peripheral nerves have been attributed to both the direct toxic effects of alcohol on nerve and nutritional deficiencies in thiamine and other B vitamins from poor dietary habits. However, there is more recent evidence that neither age nor nutritional status play a part in development of alcoholic neuropathies. Rather, alcohol-related neuropathies appear to be due to the total lifetime accumulation of ethanol.38 Patients exhibiting alcoholic neuropathy were divided into those with and without a coexisting thiamine deficiency. Researchers reported that patients without thiamine deficiency tended to have a more slowly progressive disorder in which sensory symptoms were dominant, primarily pain or a burning sensation. Along with these symptoms, the nerve biopsy demonstrated greater small fiber axonal loss. Those with alcoholic neuropathy with thiamine deficiency had large fiber involvement with segmental demyelination, and an acutely progressive motor dominant pattern along with loss of superficial and deep sensation was noted. These findings further support the view that alcohol directly effects nerve fibers.82
Pathogenesis
The exact pathogenesis of alcoholic neuropathy remains unclear. Segmental demyelination and axonal degeneration have been described in persons with alcoholic polyneuropathy; these differences may relate to the presence of vitamin deficiencies, as noted above. Changes occur distally at first and become more marked and proximal.54
Clinical Manifestations
Mild forms of alcoholic neuropathy exhibit minor loss of muscle bulk, diminished ankle reflexes, impaired sensation in the feet, and aching in the calves. Distal sensory changes include pain, paresthesia and numbness in a symmetric stocking-glove pattern. In addition, vibratory perception is impaired. It begins insidiously and progresses slowly; occasionally the onset may occur acutely. In the most advanced cases, symptoms involve all four extremities. Weakness and atrophy of distal musculature should be anticipated, with lower extremity involvement greater than upper extremity. Bilateral footdrop is observed during gait, and a wristdrop contributes to a diminished grip strength because of the client’s inability to extend the wrist. Both of these features are often combined with varying amounts of peripheral weakness in other muscles.
MEDICAL MANAGEMENT
The diagnosis is made by history, clinical examination, and electrodiagnostic testing showing loss of action potential amplitude (sensory and motor). Although diet is no longer implicated as a contributing factor in the development of alcoholic neuropathies, diet to improve nutritional status, along with vitamin supplements and abstinence from alcohol, is the treatment of choice. All other treatment is symptomatic. Orthotic devices, such as ankle-foot orthoses (AFOs) and cock-up splints, are used to manage weakness and improve function. Medications for sensory changes include carbamazepine, salicylates, and amitriptyline.
PROGNOSIS.
If the client totally abstains from alcohol, mild improvement can be expected, but recovery is slow (months to years) and incomplete when axonal degeneration has occurred.117 Therefore, to anticipate the outcome, review the client’s electrophysiologic studies to determine whether demyelination or degeneration is present.
Compression neuropathies, such as Saturday night palsy or peroneal nerve compression, may result from a bout of chronic alcohol intoxication where prolonged pressure compromises nerve function. Excessive alcohol intake may also produce rhabdomyolysis which produces proximal muscle weakness, swelling, and pigmented urine. Rhabdomyolysis occurs as product of renal failure after drinking.
Chronic Renal Failure
Neurologic.: Alteration of CNS and PNS function often occurs with chronic renal failure associated with uremia. CNS involvement (uremic encephalopathy) is manifested by recent memory loss, inability to concentrate, perceptual errors, and decreased alertness. Uremic toxins contribute to atrophy and demyelination of both sensory and motor nerves of the PNS. The lower extremities are much more commonly affected than the upper extremities; neurologic changes are typically symmetric and can also be manifested as peripheral neuropathy or restless leg syndrome, which is more pronounced at rest.
Anemia
CNS symptoms can develop in cases of severe pernicious anemia, whereas neuropathy is observed in early cases of B12 deficiency, allowing for early identification. The findings typically consist of a symmetric sensory neuropathy that begins in the feet and lower legs, although it rarely may involve the upper extremities, especially fine motor coordination of the hands. This upper extremity neuropathy may clinically manifest as problems with deteriorating handwriting. Affected individuals may also describe moderate pain or paresthesias of the extremities, especially the feet. The person may interpret the neuropathy as difficulty with locomotion when in fact they are experiencing the loss of proprioception. The affected individual may need to hold on to the wall, countertops, or furniture at home as a result of difficulties maintaining balance. There may be an associated positive Romberg sign. Loss of motor function is a late manifestation of B12 deficiency. Although a symmetric neuropathy is the usual pattern, B12 deficiency occasionally presents as a unilateral neuropathy and/or bilateral but asymmetric neuropathy. Rarely, subacute degeneration of the spinal cord caused by vitamin B12 deficiency can occur in pernicious anemia, characterized by pyramidal and posterior column deficits. CNS manifestations may include headache, drowsiness, dizziness, fainting, slow thought processes, decreased attention span, apathy, depression, and irritability.
INFECTIONS/INFLAMMATIONS
Overview and Definition
Guillain-Barré syndrome (GBS) was originally described by and named for the French neurologists who published case reports describing a syndrome of flaccid paralysis, areflexia, and albuminocytologic dissociation. More recently, the syndrome has been viewed as having distinct subtypes with varying distributions worldwide. Since the virtual elimination of poliomyelitis, GBS is the most common cause of rapidly evolving motor paresis and paralysis and sensory deficits. Individuals affected with GBS typically reach maximal weakness within 2 to 3 weeks but spend weeks to months recovering. The most common form of GBS is also known as acute inflammatory demyelinating polyradiculoneuropathy (AIDP).
Incidence
Annual incidence varies from 1 to 2 cases per 100,000 people. Although GBS occurs at all ages, peaks in frequency can be seen in young adults and in the fifth through the eighth decades. Occurrence is slightly greater for men than women and for whites more than blacks. Some researchers have noted a seasonal relationship associated with infections.