Hematologic Disorders
LYMPHOID HYPERPLASIA
The lymphoid tissue of the body plays an important role in the recognition and processing of foreign antigens, such as viruses, fungi, and bacteria. In addition, the lymphoid tissue has a protective function through a variety of direct and indirect mechanisms. In responding to antigenic challenges, lymphoid cells proliferate, thus increasing their numbers, to combat the offending agent more effectively. This proliferation results in enlargement of the lymphoid tissue, which is seen clinically as lymphoid hyperplasia.
CLINICAL FEATURES: Lymphoid hyperplasia may affect the lymph nodes, the lymphoid tissue of Waldeyer’s ring, or the aggregates of lymphoid tissue that are normally scattered throughout the oral cavity, particularly in the oropharynx, the soft palate, the lateral tongue, and the floor of the mouth. When lymphoid hyperplasia affects the lymph nodes, usually the site that the lymph node drains can be identified as a source of active or recent infection. In the head and neck region, the anterior cervical chain of lymph nodes is most commonly involved, although any lymph node in the area may be affected.
With acute infections, the lymphadenopathy appears as enlarged, tender, relatively soft, freely movable nodules. Chronic inflammatory conditions produce enlarged, rubbery firm, nontender, freely movable nodes. Sometimes these chronic hyperplastic lymph nodes may be difficult to distinguish clinically from lymphoma, and a history of a preceding inflammatory process and lack of progressive enlargement are helpful clues that are consistent with a reactive process. Another condition, however, that should be considered in the differential diagnosis of multiple, persistently enlarged, nontender lymph nodes is human immunodeficiency virus (HIV) infection (see page 271).
Tonsillar size is variable from one person to the next, but lymphoid tissue is normally more prominent in younger individuals, usually reaching its peak early during the second decade of life and gradually diminishing thereafter. Some patients have such large tonsils that it seems as if they would occlude the airway (so-called kissing tonsils). Often, however, these patients have no symptoms and are unaware of a problem. As long as the large tonsils are symmetrical and asymptomatic (Fig. 13-1), it is likely that they are normal for that particular patient. Tonsillar asymmetry is a potentially serious sign that should be evaluated further to rule out the presence of a metastatic tumor or lymphoma.
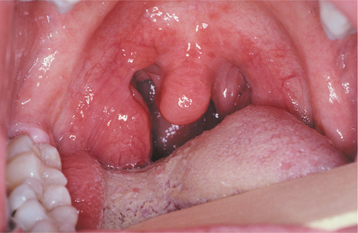
Fig. 13-1 Lymphoid hyperplasia. The large tonsil observed in this patient represents a benign hyperplasia of the lymphoid cells. If significant asymmetry is observed, further investigation may be warranted to rule out the possibility of lymphoma.
Hyperplastic intraoral lymphoid aggregates appear as discrete, nontender, submucosal swellings, usually less than 1 cm in diameter, which may appear normal or dark pink in color if the aggregate is deeper; they may have a creamy yellow-orange hue if the collection of lymphocytes is closer to the surface (Figs. 13-2 and 13-3). Lymphoid hyperplasia commonly involves the posterior lateral tongue, where it may appear somewhat ominous. The enlargement is usually bilaterally symmetrical, however, which helps to distinguish the condition from a malignancy. The buccal lymph node may also become hyperplastic and appear as a nontender, solitary, freely movable nodule, usually less than 1 cm in diameter, within the substance of the cheek. Infrequently, a more diffuse lymphoid hyperplasia involves the posterior hard palate, producing a slowly growing, nontender, boggy swelling with an intact mucosal surface and little color change. These palatal lesions may be clinically impossible to distinguish from extranodal lymphoma and would, therefore, necessitate biopsy.
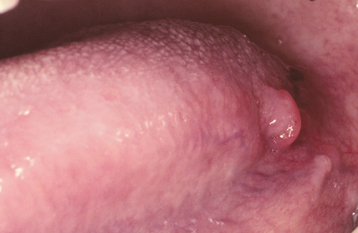
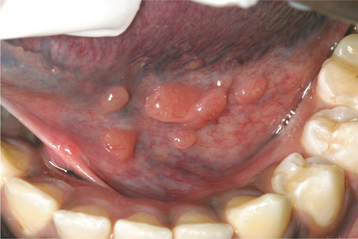
HISTOPATHOLOGIC FEATURES: The microscopic features of lymphoid hyperplasia include sheets of small, well-differentiated lymphocytes with numerous interspersed, sharply demarcated collections of reactive lymphoblasts called germinal centers. The cells that comprise the germinal centers are primarily transformed B lymphocytes that may demonstrate numerous mitoses. Macrophages can also be identified by the presence of phagocytized material (tingible bodies) in their cytoplasm as they engulf nuclear debris from the proliferating lymphocytes. In some instances, immunohistochemical studies and clonality assays must be performed to rule out the possibility of follicular lymphoma.
TREATMENT AND PROGNOSIS: Once the diagnosis of lymphoid hyperplasia is confirmed, no treatment is usually required because it is a completely benign process. For those patients with palatal lymphoid hyperplasia that may interfere with a dental prosthesis, complete excision of the lesion is recommended.
HEMOPHILIA
Hemophilia (hemo = blood; philia = loving) represents a variety of bleeding disorders associated with a genetic deficiency of any one of the clotting factors of the blood (Table 13-1). This condition was common in certain European royal families, many of whom carried an X-linked hereditary deficiency of either factor VIII or factor IX. Consequently, as a result of inbreeding, a significant proportion of the male members of these families had hemophilia. In the days before blood transfusions and clotting factor replacement therapy, many of these patients died as a direct result of, or from the complications of, uncontrolled hemorrhage. It is not known whether these people had factor VIII or factor IX deficiency, because all of the affected individuals died before the definitive diagnostic studies were developed to determine precisely which deficiency was present. Because hemophilia A (factor VIII deficiency) is the most significant and widely recognized form of hemophilia and accounts for 80% to 85% of the bleeding diatheses associated with a specific clotting factor deficiency, most of this discussion centers on that entity. Its estimated prevalence in the United States is 1 in 10,000 persons (or 1 in 5000 males).

As previously mentioned, a deficiency of factor IX or hemophilia B (Christmas disease) also may be encountered. Hemophilia B is similar to hemophilia A in its presentation, being transmitted in an X-linked fashion. Hemophilia B is much less common than hemophilia A, occurring with a prevalence of 1 in 60,000 (or 1 in 30,000 males). The term Christmas disease was obtained from the surname of the first person, a Canadian boy, who was identified as having hemophilia B in 1952.
Another clotting disorder that is sometimes seen, von Willebrand’s disease, is the result of a genetic deficiency of a plasma glycoprotein called von Willebrand’s factor. This glycoprotein aids in the adhesion of platelets at a site of bleeding, and it also binds to factor VIII, acting as a transport molecule. Von Willebrand’s disease is a genetically heterogeneous condition, with several subtypes currently identified, and it may be transmitted in an autosomal dominant or recessive pattern. It is the most common of the inherited bleeding disorders, affecting an estimated 1 in 800 to 1000 persons. However, many cases of von Willebrand’s disease are mild and may be clinically insignificant.
CLINICAL FEATURES: Hemophilia A is an X-linked disorder. Females typically carry the trait, but it is expressed primarily in males. Approximately 1 in 5000 males is born with this genetic disease, with about 30% of the cases representing new mutations. Failure of normal hemostasis after circumcision is typically one of the first signs that a bleeding disorder is present.
The severity of the bleeding disorder depends on the extent of the clotting factor deficiency. Hemophilia A is a heterogeneous disorder that is caused by any one of a variety of mutations associated with the gene for factor VIII. Because the mutations occur at different sites in the factor VIII gene (more than 900 different mutations have been identified), a clinical spectrum of deficiency of factor VIII is seen. This results in varying degrees of disease expression, with those mutations affecting more significant or larger portions of the factor VIII gene causing more severe clinical disease. Not all patients have an absolute lack of the particular clotting factor; rather, the deficiency may be a percentage of the normal value in a given patient. For example, a patient with only 25% of normal factor VIII levels may be able to function normally under most circumstances; one with less than 5% commonly manifests a marked tendency to bruise with only minor trauma.
In infants, oral lacerations and ecchymoses that involve the lips and tongue are a frequent occurrence as a result of the common falls and bumps experienced by this age group. If not treated appropriately, then such lacerations may result in significant blood loss in more severely affected patients. Sometimes deep hemorrhage occurs during normal activity and may involve the muscles, soft tissues, and weight-bearing joints (hemarthrosis), especially the knees (Fig. 13-4). The result of such uncontrolled bleeding is the formation of scar tissue as the body removes the extravasated blood. This often causes a crippling deformity of the knee joints secondary to arthritis and ankylosis. Sometimes the tissue hemorrhage results in the formation of a tumorlike mass, which has been called pseudotumor of hemophilia. Such lesions have been reported in the oral regions.
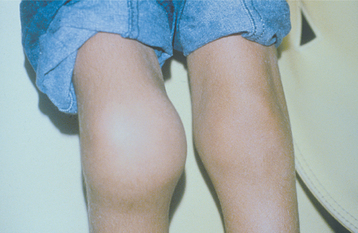
Fig. 13-4 Hemophilia. The enlargement of the knees of this patient with factor VIII deficiency is due to repeated episodes of bleeding into the joints (hemarthrosis). Inflammation and scarring have resulted.
An increased coagulation time (delay in blood clotting), of course, is the hallmark feature of this group of conditions. Uncontrollable or delayed hemorrhage may result from any laceration; this includes surgical incisions, dental extractions, and periodontal curettage (Fig. 13-5). Measurements of the platelet count, bleeding time, prothrombin time (PT), and partial thromboplastin time (PTT) should be ordered as screening tests for any patient with a possible bleeding disorder.
TREATMENT AND PROGNOSIS: The treatment of clotting factor deficiencies essentially consists of replacement therapy with the appropriate clotting factor. Whether treatment is instituted depends on the severity of the clotting factor deficiency.
Patients who have greater than 25% of normal values of factor VIII may function normally. For patients with mild hemophilia (5% to 40% of normal levels of factor VIII), no special treatment is typically required for normal activities. If surgery is to be performed, then clotting factor replacement therapy may be indicated.
For patients with severe deficiencies (<1% of normal levels of factor VIII), injections with the clotting factor must be performed as soon as a hemorrhagic episode occurs to prevent such complications as the crippling joint deformities of the knees.
The use of aspirin is strictly contraindicated because of its adverse effect on blood platelet function. Severe hemorrhage may result if these patients use aspirin-containing medications.
Genetic counseling should be provided to these patients and their families to help them understand the mechanism of inheritance. Using molecular techniques, women who are carriers can be confirmed. In addition, affected male fetuses can now be identified, and the severity of the factor VIII mutation can be assessed.
Optimal dental care is strongly encouraged for these patients to prevent oral problems that might require surgery. If oral or periodontal surgery is necessary, then consultation with the patient’s physician is mandatory. The patient is usually prepared for the procedure by the administration of clotting factor just before the surgery. With an extensive surgical procedure, additional doses of clotting factor may be needed subsequently. In addition, epsilon-aminocaproic acid (EACA), an antifibrinolytic agent that inhibits clot degradation, should be given 1 day before the surgery and continued for 7 to 10 days afterward. Alternative therapy for patients who have levels of factor VIII greater than 5% of normal is desmopressin, which can be given just before surgery. This drug causes the release of bound factor VIII, producing a temporary increase in the plasma levels of the clotting factor. Desmopressin may also be used to manage most patients affected by type 1 von Willebrand’s disease, which represents approximately 70% to 80% of the cases of that disorder.
Although it saved many lives, clotting factor replacement therapy has also resulted in a tragic complication for many of these patients. Cryoprecipitation, the traditional method of concentrating clotting factors from the serum, also resulted in the concentration of several viruses, including the hepatitis viruses and human immunodeficiency virus (HIV). Currently more than 40% of hemophilia A and B patients in the United States are estimated to be infected with hepatitis C virus. In addition, as many as 80% to 90% of hemophiliac patients treated with multiple doses of factor VIII cryoprecipitate were infected with HIV. The methods of preparing the clotting factors have been modified to eliminate the risk of acquiring HIV from the preparation; however, many hemophiliac patients who were infected developed acquired immunodeficiency syndrome (AIDS). Recombinant DNA technology now provides a source of factor VIII that is manufactured by inserting the human factor VIII gene into bacteria that then synthesize the protein. Therefore, this product can now be manufactured without contamination by any viral organisms, and young people affected by hemophilia have minimal risk of contracting these infections.
Other problems must occasionally be confronted, however. Approximately 6% of patients with hemophilia A may develop antibodies directed against factor VIII, and this is a very serious complication. Because the antibodies react with the factor VIII molecule, the result is an inhibition of the activity of the clotting factor, and these patients are once more faced with the prospect of uncontrolled bleeding. Patients with factor IX deficiency can develop similar inhibitory antibodies to factor IX, but this appears to occur much less frequently. Attempts to induce immune tolerance may help some individuals, although more immediate care has generally centered on bypassing the factor VIII–related portion of the clotting cascade by administration of recombinant factor VIIa. Research has shown this approach to be effective, although costly.
PLASMINOGEN DEFICIENCY (LIGNEOUS CONJUNCTIVITIS; HYPOPLASMINOGENEMIA)
Plasminogen deficiency is a rare autosomal recessive condition that is caused by any one of several mutations of the gene responsible for the production of plasminogen, the precursor to plasmin. In the clotting cascade, factors are activated that lead to the development of a clot; however, simultaneously serum proteins such as plasminogen are converted to plasmin, which is responsible for degrading the clot. Without the formation of plasmin, the clot tends to grow and persist despite having performed its original hemostatic function. The result of plasminogen deficiency is a buildup of fibrin, deposited as irregular plaques and nodules that primarily affect mucosal surfaces. Involvement of the conjunctival mucosa is characterized by the formation of thick, firm plaques, for which the term ligneous conjunctivitis has been used (ligneous means “woodlike”). Even though this condition was initially described in the nineteenth century, it was during the late 1990s that an explanation for the majority of these cases was provided. Similar lesions have been produced in mice that have been genetically manipulated to create knock-out mutations of the plasminogen gene.
CLINICAL FEATURES: The most striking aspect of plasminogen deficiency is the development of thick, creamy yellow to erythematous, firm plaques and nodules involving primarily the conjunctival mucosa of the upper eyelid. Typically the condition is detected during the first decade of life, but lesions can develop later as well. Even though this is an autosomal recessive condition, there is a tendency for the disease to present more often in women, although the reason for this is unknown.
In addition to the conjunctival lesions, other mucosal surfaces can be affected, including the oral mucosa, laryngeal mucosa, and vaginal mucosa. In a recent series of 50 patients with this condition, ocular lesions were documented in 80%, gingival lesions in 34%, respiratory tract lesions in 16%, and vaginal lesions in 8%. Laryngeal mucosal involvement often includes the vocal cords, which will typically cause a raspy, hoarse voice.
Oral lesions of plasminogen deficiency primarily involve the gingivae, presenting as patchy ulcerated papules and nodules with a very irregular surface (Fig. 13-6). These lesions may be few in number or distributed diffusely in all quadrants, and they tend to wax and wane in severity.
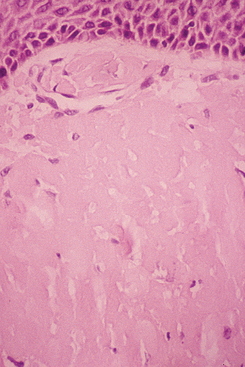
HISTOPATHOLOGIC FEATURES: The microscopic features of the lesions associated with this condition can be very confusing for the pathologist who is not familiar with the disease. The accumulation of fibrin appears as diffuse sheets of acellular eosinophilic material that bears a close resemblance to amyloid (Fig. 13-7). Special stains for amyloid (such as Congo red) are negative, however, because this material represents fibrin. Confirmation that the eosinophilic material is fibrin can be done using the Fraser-Lendrum histochemical staining method. Variable numbers of inflammatory cells are seen, and granulation tissue is usually seen adjacent to the fibrin deposits.
TREATMENT AND PROGNOSIS: Treatment of plasminogen deficiency remains a problem. Damage to the mucosal tissues, including surgical trauma, should be minimized to reduce the likelihood of fibrin accumulation. Careful, thorough oral hygiene practices should be encouraged to diminish the effect of local inflammation. Sporadic reports describe resolution of the conjunctival lesions with either topical or systemic plasminogen; however, this agent is not available commercially. Some patients have experienced spontaneous regression of their lesions over time. Currently topical heparin combined with prednisone may be the most reasonable approach until replacement plasminogen is marketed or gene therapy is feasible. Interestingly, these patients do not have any unusual problems with intravascular thrombus formation, and their lifespan does not appear to be shortened.
ANEMIA
Anemia is a general term for either a decrease in the volume of red blood cells (hematocrit) or in the concentration of hemoglobin. This problem can result from a number of factors, including a decreased production of erythrocytes or an increased destruction or loss of erythrocytes. Laboratory studies, such as the red blood cell (RBC) count, hematocrit, hemoglobin concentration, mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), and mean corpuscular hemoglobin concentration (MCHC), can help indicate the probable cause of the anemia.
Rather than being a disease itself, anemia is often a sign of an underlying disease, such as renal failure, liver disease, chronic inflammatory conditions, malignancies, or vitamin or mineral deficiencies. The diverse causes and complexity of the problem of anemia are presented in Box 13-1.
CLINICAL FEATURES: The symptoms of anemia are typically related to the reduced oxygen-carrying capacity of the blood, which is a result of the reduced numbers of erythrocytes. Symptoms such as tiredness, headache, or lightheadedness are often present.
Pallor of the mucous membranes may be observed in severe cases of anemia. The palpebral conjunctiva is often the site where this paleness is most easily appreciated, but the oral mucosa may show similar signs.
SICKLE CELL ANEMIA
Sickle cell anemia is one of the more severe genetic disorders of hemoglobin synthesis (hemoglobinopathies). Because of the mutational substitution of a thymine molecule for an adenine in DNA, the codon is altered to code for the amino acid valine rather than glutamic acid in the β-globin chain of hemoglobin. This results in a hemoglobin molecule that, in the deoxygenated state, is prone to molecular aggregation and polymerization. Consequently, the red blood cells of patients with sickle cell anemia have a marked tendency to undergo deformation from the normal biconcave disk shape to a rigid-and-curved (sickle) shape. Because the genes for hemoglobin synthesis are codominant, if only one allele is affected, then only 40% to 50% of that patient’s hemoglobin will be abnormal. Such a patient is simply a carrier and is said to have sickle cell trait, a condition that has no significant clinical manifestations in most everyday circumstances. Some sickling may be precipitated under certain conditions, however, particularly with low-oxygen tensions associated with exercise or high altitudes.
This abnormal gene has persisted in the human race perhaps because it confers a degree of resistance to the malarial organism. As a result, the gene is seen most frequently in populations, such as African, Mediterranean, and Asian, who reside in areas where malaria is endemic. In the United States, nearly 2.5 million people (approximately 8% of the black population) carry this trait.
Unfortunately, in patients who inherit two alleles that code for sickle hemoglobin, the red blood cells contain primarily sickle hemoglobin, which results in the condition called sickle cell disease. In the United States, about 1 of every 350 to 400 blacks is born with this disease. Such patients are often susceptible to the problems associated with abnormal red blood cell morphology. The sickled erythrocytes are more fragile than normal and they tend to block the capillaries because of their shape and adherence properties. As a result, these patients have a chronic hemolytic anemia and many difficulties related to reduced blood flow to organs and tissues, which produces ischemia, infarction, and tissue death.
CLINICAL AND RADIOGRAPHIC FEATURES: Virtually any tissue or organ may be affected in sickle cell disease. The clinical spectrum of involvement can vary tremendously, with approximately one third of patients exhibiting severe manifestations. Perhaps the most dramatic sign of this disease is the sickle cell crisis, a situation in which the sickling of the erythrocytes becomes severe. Hypoxia, infection, hypothermia, or dehydration may precipitate a crisis; however, for most crises there is no identifiable predisposing factor. Patients who experience a crisis suffer extreme pain from ischemia and infarction of the affected tissue. The long bones, lungs, and abdomen are among the most commonly affected sites, and each episode lasts 3 to 10 days. Pulmonary involvement, known as acute chest syndrome, is particularly serious, and one large study indicated that this is frequently precipitated by fat embolism or community-acquired pneumonia. Some patients may experience such crises monthly; others may go for 1 year or longer without problems. Often fever accompanies the crisis; therefore, infection must be considered in the differential diagnosis.
Patients with sickle cell disease are susceptible to infections, especially those caused by Streptococcus pneumoniae, probably because of the destruction of the spleen at an early age by repeated infarctions. Such infections are the most common cause of death among children affected by sickle cell disease in the United States.
Other problems include delayed growth and development in most patients. Impaired kidney function and ocular abnormalities develop secondary to the damage caused by vaso-occlusive episodes in the capillary networks of those organs. If the patient lives long enough, then renal failure may eventually develop. In addition, approximately 5% to 8% of these patients will experience central nervous system (CNS) damage in the form of a stroke, which occurs at an average age of about 8 years.
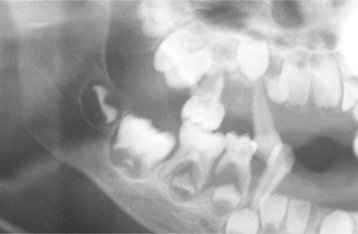
The oral radiographic features of sickle cell disease are relatively nonspecific. They consist of a reduced trabecular pattern of the mandible because of increased hematopoiesis occurring in the marrow spaces. Occasionally, a “hair-on-end” appearance is seen on the skull radiograph, although this is less prominent than that seen in thalassemia (Fig. 13-8). Other oral problems that have been reported include an increased prevalence of osteomyelitis of the mandible, prolonged paresthesia of the mandibular nerve, and asymptomatic pulpal necrosis.
HISTOPATHOLOGIC FEATURES: In homozygous sickle cell disease, a peripheral blood smear shows a peculiar curved distortion of the erythrocytes, resembling a sickle or boomerang shape.
TREATMENT AND PROGNOSIS: The patient experiencing a sickle cell crisis should be managed with supportive care, including fluids, rest, and appropriate analgesic therapy (usually narcotic preparations). It is important, but often difficult, to rule out the possibility of infection.
All 50 states now screen for this hemoglobin disorder as part of their newborn infant health care system to identify affected individuals as soon as possible so that appropriate therapy can be instituted. For children with a diagnosis of sickle cell disease, continuous prophylactic penicillin therapy is indicated until at least 5 years of age. In addition, the child should be given polyvalent pneumococcal vaccinations. Situations that might precipitate a crisis, such as strenuous exercise, dehydration, or exposure to cold, should be avoided. For adults with relatively severe disease, hydroxyurea has been approved for treatment. This drug increases the fetal form of hemoglobin (hemoglobin F), which may inhibit polymerization of hemoglobin S and may also reduce the adherence of erythrocytes to the vessel walls. Unfortunately, hydroxyurea has a number of potential side effects and should be used judiciously. Bone marrow transplantation is curative, but this is a procedure with multiple potential complications and is used primarily for severely affected patients having a histocompatibility antigen (HLA)-matched donor sibling. Only about 1% of sickle cell anemia patients currently meet these criteria.
When surgery is necessary, local anesthesia, if possible, is usually preferred. If general anesthesia is indicated, then precautions should be taken to avoid conditions that might induce a crisis, such as hypoxia, vascular stasis, acidosis, infection, reduced body temperature, or dehydration.
For patients who have either the sickle cell trait or the disease, genetic counseling is appropriate. DNA diagnostic techniques have been used for several years to assess whether a fetus is affected by sickle cell disease, permitting consideration of termination of the pregnancy. Molecular evaluation of the DNA from a single cell obtained from an embryo that was fertilized in vitro has allowed selection of a nonaffected embryo for uterine implantation. For parents who are carriers of the sickle cell trait, this is one method to ensure that their offspring do not have sickle cell disease.
Although the mortality rate for sickle cell disease in developed countries has improved dramatically over the past few years, the prognosis is variable because of the wide spectrum of disease activity. Those who are severely affected, however, often are quite disabled because of the many complications of the disease and have a decreased life span.
THALASSEMIA
Thalassemia represents a group of disorders of hemoglobin synthesis that are characterized by reduced synthesis of either the α-globin or β-globin chains of the hemoglobin molecule. As in those with sickle cell trait, people who carry the trait for one of the forms of thalassemia seem to be more resistant to infection by the malarial organism; an increased frequency of these genes is seen in Mediterranean, African, Indian, and Southeast Asian populations. Because the original cases were reported from the region of the Mediterranean Sea, the name thalassemia was given, derived from the Greek word thalassa, meaning “sea.” The thalassemias are considered to be among the most common inherited conditions that affect humans.
An understanding of the structure and synthesis of hemoglobin is helpful in explaining the pathophysiology of these conditions. The hemoglobin molecule is a tetramer that is composed of two α chains and two β chains; if one of the chains is not being made in adequate quantities, then the normal amount of hemoglobin cannot be made. Furthermore, the excess globin chains accumulate within the erythrocyte, further compromising the structure and function of the cell. These abnormal erythrocytes are recognized by the spleen and selected for destruction (hemolysis). In addition, there is evidence of ineffective erythropoiesis caused by premature cell death of erythrocyte precursors in the bone marrow because of activation of apoptotic mechanisms. The net result is that the patient has hypochromic, microcytic anemia.
Because two genes code for the β chain and four genes code for the a chain, the degree of clinical severity in these conditions can vary considerably. The severity depends on which specific genetic alteration is present and whether it is heterozygous or homozygous. In the heterozygous state, an adequate amount of normal hemoglobin can be made and the affected patient experiences few signs or symptoms. In the homozygous state, however, the problems are often severe or even fatal. In addition, variations in the severity of the clinical presentation may be a reflection of the specific alteration in the genetic code, because more than 200 different mutations have been documented for β-thalassemia alone.
CLINICAL AND RADIOGRAPHIC FEATURES:
β-THALASSEMIA: If only one defective gene for the β-globin molecule is inherited (thalassemia minor), no significant clinical manifestations are usually present.
When two defective genes for the β-globin molecule are inherited, the patient is affected with thalassemia major, also called Cooley’s anemia or Mediterranean anemia. The disease is usually detected during the first year of life because a severe microcytic, hypochromic anemia develops when fetal hemoglobin synthesis ceases after 3 to 4 months of age. The red blood cells that are produced are extremely fragile and survive for only a few days in the peripheral circulation.
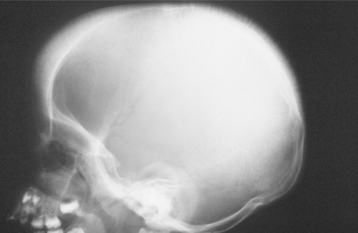
In an attempt to maintain adequate oxygenation, the rate of hematopoiesis (despite being ineffective) is greatly increased (up to 30 times normal), resulting in massive bone marrow hyperplasia, as well as hepatosplenomegaly and lymphadenopathy because of extramedullary hematopoiesis. The bone marrow hyperplasia may affect the jaws especially, producing an altered trabecular pattern and marked, painless enlargement of the mandible and maxilla (Fig. 13-9). This results in a characteristic “chipmunk” facies and causes reduced size or obliteration of the paranasal sinuses. Frontal bossing is also present, and a skull radiograph may show a prominent “hair-on-end” appearance of the calvaria (Fig. 13-10). Generalized maturational delay of the patient is typically seen. Delayed development of the dentition also has been described, with the teeth showing a mean delay of approximately 1 year compared with a matched population.
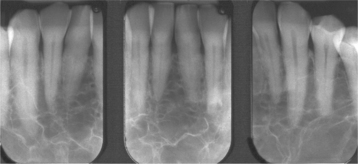
Fig. 13-9 Thalassemia. Periapical radiographs of the anterior mandible showing reduced trabeculation because of increased hematopoiesis. (Courtesy of Dr. José Luis Tapia.)
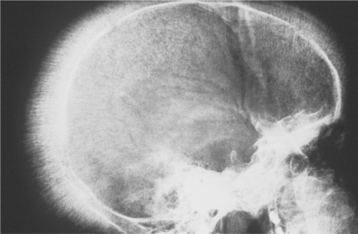
Fig. 13-10 Thalassemia. Lateral skull radiograph depicting the characteristic “hair-on-end” appearance in a patient with thalassemia.
Without therapy, tissue hypoxia worsens and serious bacterial infections with pneumococcal organisms often develop. Eventually, high-output cardiac failure occurs; many patients die by 1 year of age as a result of infection or heart problems.
α-THALASSEMIA: Because four α-globin genes may be affected, α-thalassemia has a broader spectrum of involvement than does β-thalassemia.
With the alteration of only one gene, no disease can be detected. With the inheritance of two altered genes, the condition is known as a-thalassemia trait. These patients have a mild degree of anemia and microcytosis that is usually not clinically significant. With three altered genes, the term hemoglobin H (HbH) disease is applied. Patients have problems with hemolytic anemia and splenomegaly. For patients with severe hemolysis, splenectomy may be indicated.
The homozygous state, in which all four genes are abnormal, causes severe generalized fetal edema, a condition that has been termed hydrops fetalis. Hydrops fetalis is not specific for α-thalassemia and can be seen as a manifestation of other diseases, such as severe Rh incompatibility. Infants with α-thalassemia who are affected by this problem typically die within a few hours of birth.
TREATMENT AND PROGNOSIS: Thalassemia major is treated today primarily by means of blood transfusions. These should be administered every 2 to 3 weeks to simulate the normal hematologic state. Unfortunately, with repeated blood transfusions, iron overload inevitably develops because of the constant infusion of exogenous red blood cells. This is a serious problem, and often death is due to hemochromatosis, an abnormal deposition of iron throughout the tissues of the body. The heart, liver, and endocrine glands are particularly affected by the toxic accumulation of iron. To combat this problem, an iron-chelating agent, deferoxamine (also known as desferrioxamine), must be given. If such therapy is used steadfastly, patients with β-thalassemia may have a relatively normal life span; however, problems may arise with patient compliance because this medication must be infused parenterally over several hours for at least 250 nights each year. Hematopoietic stem cell transplantation has also been used with considerable success for individuals who are relatively young, have little organ damage, and have an HLA-matched donor.
Clinicians can now identify α-thalassemia, with its attendant hydrops fetalis (historically considered a fatal condition), in utero by molecular testing, and the fetus can be given intrauterine umbilical vein transfusions. An 80% survival rate has been reported for these infants, although they will require either lifelong transfusion therapy or hematopoietic stem cell transplantation.
For patients who have developed an abnormal facial appearance caused by thalassemia, surgical correction can be performed in many cases. Prevention of thalassemia also is desirable, either by screening for carriers of the genetic trait or by prenatal diagnosis.
APLASTIC ANEMIA
Aplastic anemia is a rare, life-threatening hematologic disorder that is characterized by failure of the hematopoietic precursor cells in the bone marrow to produce adequate numbers of all types of blood cells. A significant amount of evidence supports the concept that most cases of aplastic anemia represent an immune-mediated disease caused by cytotoxic T lymphocytes that target differentiating hematopoietic cells in the marrow. As a result, the hematopoietic stem cells do not seem to undergo normal maturation despite normal or increased levels of cytokines, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), which normally induce the production and maturation of several types of white blood cells.
Although the underlying trigger for the immune-mediated destruction of the hematopoietic cells is unknown, some cases of aplastic anemia are associated with exposure to certain environmental toxins (e.g., benzene), treatment with certain drugs (especially the antibiotic chloramphenicol), or infection with certain viruses (particularly non-A, non-B, non-C, non-G hepatitis). It is possible that the abnormal immune response is perhaps initiated by such exogenous stimuli in certain instances. A few genetic disorders, such as Fanconi’s anemia and dyskeratosis congenita (see page 746), also are associated with an increased frequency of aplastic anemia.
CLINICAL FEATURES: Because all of the formed elements of the blood are decreased in patients with aplastic anemia, the initial symptoms may be related to any one or several of the deficiencies. The erythrocyte deficiency produces signs and symptoms related to a decreased oxygen-carrying capacity of the blood; therefore, patients may experience fatigue, lightheadedness, tachycardia, or weakness. The platelet deficiency (thrombocytopenia) is seen as a marked tendency for bruising and bleeding, which affects a variety of sites. Retinal and cerebral hemorrhages are some of the more devastating manifestations of this bleeding tendency. Deficiency of white blood cells (neutropenia, leukopenia, or granulocytopenia) is the most significant complication of this disease, predisposing the patient to bacterial and fungal infections that often are the cause of death.
The oral findings related to thrombocytopenia include gingival hemorrhage (Fig. 13-11), oral mucosal petechiae, purpura, and ecchymoses. The oral mucosa may appear pale because of the decreased numbers of red blood cells. Oral ulcerations associated with infection, particularly those that involve the gingival tissues, may be present. Minimal erythema is usually associated with the periphery of the ulcers. Gingival hyperplasia has also been reported in association with aplastic anemia.
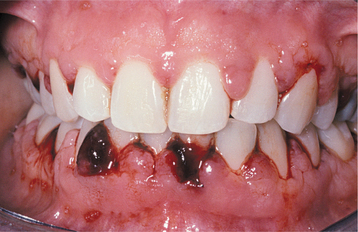
HISTOPATHOLOGIC FEATURES: A bone marrow biopsy specimen usually demonstrates a relatively acellular marrow with extensive fatty infiltration. The histopathologic features of an oral ulceration in a patient with aplastic anemia show numerous microorganisms in addition to a remarkable lack of inflammatory cells in the ulcer bed.
DIAGNOSIS: The diagnosis of aplastic anemia is usually established by laboratory studies. A pancytopenia is characterized by at least two of the following findings:
TREATMENT AND PROGNOSIS: The course for patients with aplastic anemia is unpredictable. For the milder forms of the disease, spontaneous recovery of the marrow may occur in some instances; progression to severe aplastic anemia may be seen in others. Generally, in severe cases, the chances of spontaneous recovery are slim. If a particular environmental toxin or drug is associated with the process, then withdrawal of the offending agent may sometimes result in recovery.
The treatment is initially supportive. Appropriate antibiotics are given for the infections that develop, and transfusions of packed red blood cells or platelets are administered for symptomatic treatment of anemia and bleeding problems, respectively.
Definitive therapy for aplastic anemia is to replace the defective marrow with normal marrow, either by bone marrow transplantation or peripheral blood stem cell transplantation from a matched donor. Patients must be carefully selected; patients younger than 40 years of age and those with an HLA-matched donor (usually a sibling) have the best prognosis, but unfortunately only about 30% of patients meet these criteria.
For those patients who would not be a good prospect for bone marrow transplantation because of their advanced age or no matched donor, immunosuppressive therapy is recommended. Antithymocyte globulin (in the United States) or antilymphocyte globulin (in Europe) combined with cyclosporine produces a response in the majority of these patients. Compared with treatment results from only 25 years ago, the prognosis for this condition has markedly improved. In the past, for patients with severe aplastic anemia treated with only antibiotics and transfusions, the mortality rate was greater than 80% in the first year after the diagnosis. Currently, an overall long-term survival of 75% of these patients can be achieved with either bone marrow transplant or immunosuppressive therapy. However, even if the disease is controlled, then these patients remain at risk for recurrent marrow aplasia and are at increased risk for acute leukemia.
NEUTROPENIA
Neutropenia refers to a decrease in the number of the circulating neutrophils below 1500/mm3 in an adult. It is often associated with an increased susceptibility of the patient to bacterial infections. Clinicians must be aware of this disorder because infection of the oral mucosa may be the initial sign of the disease. Interestingly, several ethnic groups, including patients of African and Middle Eastern background, will consistently have neutrophil counts that would qualify as neutropenia (as low as 1200/mm3), yet these individuals are otherwise healthy. This finding has been termed benign ethnic neutropenia, and it appears to have no effect on the health of the patient because neutrophil counts respond to bacterial challenge.
A decrease in neutrophils may be precipitated by several mechanisms, most of which involve decreased production or increased destruction of these important inflammatory cells. When infections are noted in infancy and neutropenia is detected, the problem is usually the result of a congenital or genetic abnormality, such as Schwachman-Diamond syndrome, dyskeratosis congenita (see page 746), cartilage-hair syndrome, or severe congenital neutropenia. If the neutropenia is detected later in life, it usually represents one of the acquired forms. Many acquired neutropenias have an unknown cause; however, others are clearly associated with various causes. A decreased production of neutrophils and the other formed elements of the blood may result from the destruction of the bone marrow by malignancies, such as leukemia (see page 587), or by metabolic diseases, such as Gaucher disease (see page 818), and osteopetrosis (see page 615).
Many drugs may affect neutrophil production, either through direct toxic effects on the bone marrow progenitor cells or by unknown idiosyncratic mechanisms. These drugs include the following:
• Anticancer chemotherapeutic agents (e.g., nitrogen mustard, busulfan, chlorambucil, cyclophosphamide)
Nutritional deficiencies of vitamin B12 or folate, which may be a consequence of malabsorption syndromes, can inhibit neutrophil production.
A variety of viral and bacterial infections not only may reduce production of neutrophils but also seem to increase their destruction, typically at the sites of infection. Viral infections that have been implicated include the following:
Numerous bacterial infections, such as typhoid, tuberculosis, brucellosis, and tularemia, may also cause neutropenia. The increased destruction of neutrophils by an autoimmune mechanism also occurs in such disorders as systemic lupus erythematosus (SLE), in which autoantibodies directed against the neutrophil are produced.
CLINICAL FEATURES: Most patients with neutropenia have some form of bacterial infection rather than a viral or fungal infection, particularly if the other elements of the immune system (lymphocytes, plasma cells, and monocytes) are still intact. Staphylococcus aureus and gram-negative organisms seem to cause the most problems for patients with neutropenia. The suppuration and abscess formation normally associated with such infections may be markedly reduced because of the lack of neutrophils. The most common sites of infection include the middle ear, the oral cavity, and the perirectal area. When neutrophil counts drop below 500/mm3, however, pulmonary infections often develop.
The oral lesions of neutropenia consist of ulcerations that usually involve the gingival mucosa, probably because of the heavy bacterial colonization of this area and the chronic trauma that it receives. These ulcers characteristically lack an erythematous periphery, although this finding has been variable. Premature periodontal bone loss with exfoliation of the deciduous dentition has been described.
HISTOPATHOLOGIC FEATURES: A biopsy specimen of a neutropenic ulceration usually shows a reduced number or the absence of neutrophils. Bacterial invasion of the host tissue may be apparent in some instances.
TREATMENT AND PROGNOSIS: Infections related to neutropenia are managed with appropriate antibiotic therapy. The patient should be encouraged to maintain optimal oral hygiene to decrease the bacterial load in the oral cavity. Studies using recombinant human granulocyte colony-stimulating factor (G-CSF), a cytokine that promotes the growth and differentiation of neutrophils, have shown remarkable results. Patients with severe neutropenia have a significant increase in neutrophil counts and resolution of infections after treatment with this agent. Patients who do not respond to G-CSF may have to be considered for hematopoietic stem cell transplantation, depending on the severity of the neutropenia and subsequent infections.
AGRANULOCYTOSIS
Agranulocytosis is a condition in which the cells of the granulocytic series, particularly neutrophils, are absent. As in other disorders of the formed elements of the blood, agranulocytosis may occur as a result of decreased production or increased destruction or use of these cells. Although some cases are idiopathic, most are induced by exposure to one of several drugs. Some drugs, such as the anticancer chemotherapeutic agents, induce agranulocytosis by inhibiting the normal mitotic division and maturation of the hematopoietic stem cells. In other instances, the drugs trigger an immunologic reaction that results in the destruction of granulocytes. Rarely, agranulocytosis may be a congenital syndrome (congenital agranulocytosis, Kostmann syndrome) that results from a decreased level of the cytokine granulocyte colony-stimulating factor (G-CSF).
CLINICAL FEATURES: Agranulocytosis typically develops within a few days after a person ingests the offending drug. Because of the lack of granulocytes (especially neutrophils), bacterial infections often develop and patients may show signs and symptoms of malaise, sore throat, swelling, fever, chills, bone pain, pneumonia, and shock. The erythrocyte and platelet counts are usually normal or only slightly depressed.
Oral lesions are common and include necrotizing, deep, punched-out ulcerations of the buccal mucosa, tongue, and palate. The gingivae are especially susceptible to infection, often resembling the pattern of necrotizing ulcerative gingivitis (NUG) (see page 157).
HISTOPATHOLOGIC FEATURES: Microscopic examination of a biopsy specimen from one of the oral ulcerations in agranulocytosis characteristically shows abundant bacterial organisms, both on the surface and within the tissue. The host inflammatory response is relatively sparse, with few granulocytes, particularly neutrophils, seen in the ulcer bed.
TREATMENT AND PROGNOSIS: If the clinician believes that a particular drug has caused the agranulocytosis, the medication should be discontinued as soon as is reasonably possible. In many instances, the granulocyte count returns to normal within 10 to 14 days after cessation of the offending agent. For patients who have agranulocytosis secondary to cancer chemotherapy, oral hygiene should be meticulous to foster an immaculate oral environment. In addition, the use of chlorhexidine-containing mouth rinses seems to reduce the severity of the oral lesions. Active infections are treated with appropriate antibiotic medications.
If the agranulocytosis is related to cancer treatment, the white blood cell count usually returns to normal after a period of weeks. For patients whose granulocyte counts do not recover, administration of G-CSF or granulocyte-macrophage colony-stimulating factor (GM-CSF) may be beneficial. The overall mortality rate for this condition in the past was 20% to 30%, although cytokine therapy and the newer broad-spectrum antibiotics have improved the outlook for these patients.
CYCLIC NEUTROPENIA (CYCLIC HEMATOPOIESIS)
Cyclic neutropenia is a rare idiopathic hematologic disorder that is characterized by regular periodic reductions in the neutrophil population of the affected patient. The underlying cause seems to be a mutation of the neutrophil elastase (ELA2) gene, resulting in arrested development of neutrophils at the promyelocyte stage within the marrow. This mutation is also associated with premature apoptosis of these myeloid precursor cells. The best estimated frequency of this disease in the population is about 1 in 1 million. Although an autosomal dominant pattern of inheritance has been described in a few cases, most examples of cyclic neutropenia are isolated.
Symptoms usually begin in childhood and tend to correlate with the neutrophil counts. When the neutrophil count is at its nadir (i.e., lowest point), the patient experiences problems with infection. As the neutrophil count rises toward normal, the signs and symptoms abate. Very low neutrophil counts usually are present for 3 to 6 days, and blood monocyte and eosinophil levels are typically increased when the neutrophil count is depressed. Even when the neutrophil count is at its peak, the levels are often less than normal.
CLINICAL AND RADIOGRAPHIC FEATURES: The signs and symptoms of cyclic neutropenia occur in rather uniformly spaced episodes, which usually have a 21-day cycle. Patients typically complain of recurrent episodes of fever, anorexia, cervical lymphadenopathy, malaise, pharyngitis, and oral mucosal ulcerations. Other gastrointestinal mucosal areas, including the colon, rectum, and anus, may be affected by recurrent ulcerations.
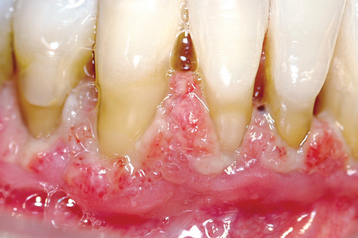
The oral ulcerations develop on any oral mucosal surface that is exposed to even minor trauma, particularly the lips, tongue, buccal mucosa, and oropharynx (Fig. 13-12). An erythematous halo is variably present at the periphery of the ulcers. The gingiva is the most severely affected region of the oral cavity. Severe periodontal bone loss with marked gingival recession and tooth mobility are also characteristic (Fig. 13-13).
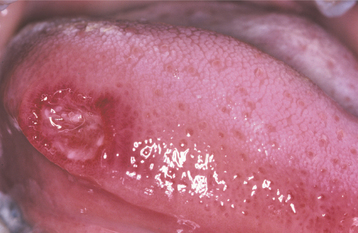
Fig. 13-12 Cyclic neutropenia. Ulceration of the lateral tongue is typical of the lesions associated with cyclic neutropenia. (From Allen CM, Camisa C: Diseases of the mouth and lips. In Sams WM, Lynch P, editors: Principles and practice of dermatology, ed 2, New York, 1996, Churchill Livingstone.) Churchill Livingstone
DIAGNOSIS: The diagnosis of cyclic neutropenia should be established by sequential complete blood counts (typically two to three times per week for 8 weeks) to determine whether cycling of the neutrophil levels occurs. The neutrophil count should be less than 500/mm3 for 3 to 5 days during each of at least three successive cycles to make this diagnosis.
HISTOPATHOLOGIC FEATURES: The histopathologic features of cyclic neutropenia are similar to those of the other neutropenic and granulocytopenic ulcerations if the biopsy is performed during the nadir of the neutrophil count.
TREATMENT AND PROGNOSIS: Supportive care for the patient with cyclic neutropenia includes antibiotic therapy for significant infections that might occur while the neutrophil count is at its lowest. Unfortunately, this approach cannot be considered a permanent treatment. Other methods that have been used with marginal success include splenectomy, corticosteroid therapy, and nutritional supplementation. Studies have shown that administration of the cytokine granulocyte colony-stimulating factor (G-CSF) several times weekly seems to correct the lack of production of neutrophils. This treatment results in a decrease in the time of neutropenia from 5 days to 1 day, which improves the clinical course of the disease. The cycles are reduced from 18 to 21 days to 11 to 13 days, and the severity of mucositis and infection are reduced.
Supportive care in the form of optimal oral hygiene should be maintained to reduce the number and severity of oral infections and improve the prognosis of the periodontal structures. Fortunately, for many of these patients, the severity of symptoms related to cyclic neutropenia seems to diminish after the second decade of life, despite the fact that the cycling of the neutrophils continues.
THROMBOCYTOPENIA
Thrombocytopenia is a hematologic disorder that is characterized by a markedly decreased number of circulating blood platelets (formed elements derived from megakaryocyte precursors in the bone marrow). Platelets are necessary for hemostasis and clot formation. A platelet count of 200,000 to 400,000/mm3 is considered normal. The decrease in platelets may be the result of the following:
REDUCED PLATELET PRODUCTION: Reduced production of platelets may be the result of various causes, such as infiltration of the bone marrow by malignant cells or the toxic effects of cancer chemotherapeutic drugs. In such instances, decreases in the other formed elements of the blood are also seen.
INCREASED PLATELET DESTRUCTION: Increased destruction of platelets may be caused by an immunologic reaction, which is often precipitated by any one of more than 100 different drugs; heparin is one of the most common offending agents. This type of reaction is typically idiosyncratic and, therefore, not related to the dose of the drug. Similarly, autoantibodies directed against platelets, specifically certain surface glycoproteins, may on rare occasions be induced by viral infection or vaccination. In addition, certain systemic diseases may have thrombocytopenia as a component, such as systemic lupus erythematosus and HIV infection. Increased destruction may also occur by nonimmunologic means because of increased consumption of platelets associated with abnormal blood clot formation. This occurs in patients with conditions such as thrombotic thrombocytopenic purpura (TTP).
SEQUESTRATION IN THE SPLEEN: Under normal conditions, one third of the platelet population is sequestered in the spleen. Consequently, conditions that cause splenomegaly (e.g., portal hypertension secondary to liver disease, splenic enlargement secondary to tumor infiltration, splenomegaly associated with Gaucher disease) also cause larger numbers of platelets to be taken out of circulation. Regardless of the cause, the result for the patient is a bleeding problem because normal numbers of platelets are not available for proper hemostasis.
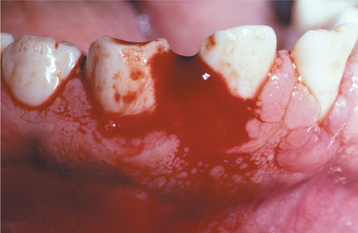
CLINICAL FEATURES: Clinical evidence of thrombocytopenia is not usually seen until the platelet levels drop below 100,000/mm3. The severity of involvement is directly related to the extent of platelet reduction. The condition often is initially detected because of the presence of oral lesions. Minor traumatic events are continuously inflicted on the oral mucosa during chewing and swallowing of food. The small capillaries that are damaged during this process are normally sealed off with microscopic thrombi. In a patient with thrombocytopenia, however, the thrombi do not form properly. This results in a leakage of blood from the small vessels. Clinically, this usually produces pinpoint hemorrhagic lesions known as petechiae. If a larger quantity of blood is extravasated, then an ecchymosis or bruise results (Fig. 13-14). With even larger amounts of extravasated blood, a hematoma (hemat = blood; oma = tumor) will develop (Fig. 13-15). Spontaneous gingival hemorrhage often occurs in these patients, as does bleeding from sites of minor trauma.
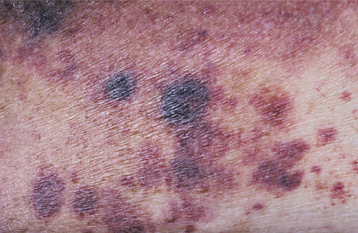
Fig. 13-14 Thrombocytopenia. The bruising (purpura) seen on this patient’s forearm is a result of reduced platelet count secondary to myelodysplasia, a preleukemic bone marrow disorder.
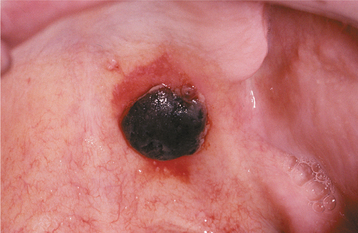
Fig. 13-15 Thrombocytopenia. This dark palatal lesion represents a hematoma caused by a lack of normal coagulation, characteristic of thrombocytopenia.
Similar hemorrhagic events occur throughout the body. With severe thrombocytopenia (<10,000 platelets/mm3), massive bleeding from the gastrointestinal or urinary tract may be fatal. Epistaxis is often present in these patients, and hemoptosis indicates signifi-cant pulmonary hemorrhage. Intracranial hemorrhage is also a potentially fatal complication of severe thrombocytopenia.
Special types of thrombocytopenia include idiopathic (immune) thrombocytopenic purpura (ITP) and TTP. ITP usually occurs during childhood, classically after a nonspecific viral infection. The symptoms of thrombocytopenia appear quickly and may be severe. Most cases, however, resolve spontaneously within 4 to 6 weeks, and 90% of patients recover by 3 to 6 months.
TTP is a serious disorder of coagulation and is thought to be caused by some form of endothelial damage that appears to trigger the formation of numerous thrombi within the small blood vessels of the body.
HISTOPATHOLOGIC FEATURES: Gingival biopsy may be performed for diagnostic purposes in patients with suspected TTP. Approximately 30% to 40% of such biopsy specimens show the presence of fibrin deposits in the small vessels. These deposits are more readily appreciated after staining the tissue section using the periodic acid-Schiff (PAS) method.
TREATMENT AND PROGNOSIS: If the clinician believes the thrombocytopenia to be drug-related, the drug should be discontinued immediately. In most instances, the platelet count returns to normal after several days. Platelet transfusions and corticosteroid therapy may be necessary if life-threatening hemorrhage occurs. As mentioned earlier, ITP often resolves spontaneously, but those cases that are more severe may require corticosteroid therapy or intravenous immunoglobulin (IVIG) therapy. For some forms of thrombocytopenia, such as TTP, the patient’s prognosis is relatively guarded. In the past, the condition was almost uniformly fatal, although the outlook has improved since therapy with plasmapheresis or plasma exchange transfusions became available. More than 70% of these patients now survive with proper treatment.
POLYCYTHEMIA VERA (PRIMARY POLYCYTHEMIA; POLYCYTHEMIA RUBRA VERA; PRIMARY ACQUIRED ERYTHROCYTOSIS)
Polycythemia vera is a rare idiopathic hematologic disorder that is best thought of as an increase in the mass of the red blood cells. Uncontrolled production of platelets and granulocytes, however, is often seen concurrently, and most authorities feel that this condition represents a relatively nonaggressive myeloprolifera-tive disorder. Researchers believe the overproduction is related to the abnormal behavior of a single progenitor marrow stem cell, which begins multiplying without regard to the normal regulatory hormones, such as erythropoietin. This gives rise to a group or clone of unregulated cells that then produce the excess numbers of these formed elements of the blood at two to three times the normal rate. These cells generally function in a normal fashion.
CLINICAL FEATURES: Polycythemia vera typically affects older adults. The median age at diagnosis is 60 years. Only 5% of cases are diagnosed before the age of 40 years. No sex predilection is seen, and the annual incidence estimates of the condition have ranged widely, from 0.2 to 28.0 cases per million population. Recent evidence suggests that an acquired mutation of one of the tyrosine kinase genes, Janus kinase 2 (JAK2), may play a significant role in the development of this disorder, because more than 95% of patients with polycythemia vera have been shown to have this mutation.
The initial symptoms of the disease are nonspecific and include the following:
A ruddy complexion may be evident on physical examination. One relatively characteristic complaint, described in about 40% of affected patients, is that of generalized pruritus (itching), particularly after bathing, without evidence of a rash.
The problems caused by thrombus formation, which would be expected with the increased viscosity of the blood and the increased platelet numbers, include transient ischemic attacks, cerebrovascular accidents, and myocardial infarctions. Hypertension and splenomegaly are also common.
A peculiar peripheral vascular event called erythromelalgia affects the hands and feet. Patients experience a painful burning sensation accompanied by erythema and warmth. This may eventually lead to thrombotic occlusion of the vessels that supply the digits. Digital gangrene and necrosis may result. Erythromelalgia is probably caused by excessive platelets, and its onset seems to be precipitated by exercise, standing, or warm temperatures.
Strangely enough, these patients may also have problems with excess hemorrhage. Epistaxis and ecchymoses are often a problem, and gingival hemorrhage has been described.
TREATMENT AND PROGNOSIS: With the initial diagnosis of polycythemia vera, an immediate attempt is made to reduce the red blood cell mass. The first treatment is usually phlebotomy, with as much as 500 mL of blood removed daily. If thrombotic events are an immediate problem, then treatment with low-dose aspirin should be started. To control the platelet levels, anagrelide hydrochloride, a selective inhibitor of megakaryocyte maturation and platelet production, may be prescribed. Antihistamines are used to help control the symptoms of pruritus.
Long-term management may include intermittent phlebotomy, although myelosuppressive therapy has also been advocated. Each has disadvantages. An increased risk of thrombosis is associated with phlebotomy, and an increased risk of leukemia is associated with some chemotherapeutic drugs. Hydroxyurea is one chemotherapeutic agent that may not pose an increased risk of leukemia, however, because it acts as an antimetabolite and does not appear to have any mutagenic properties. Nevertheless, in 2% to 10% of patients with polycythemia vera, acute leukemia ultimately develops.
Overall, the prognosis is fair; patients with polycythemia vera survive an average of 10 to 12 years after the diagnosis, if treated. Given the fact that the median age at diagnosis is 60 years, the majority of affected patients do not seem to have a markedly higher death rate compared with their unaffected peers.
LEUKEMIA
Leukemia represents several types of malignancies of hematopoietic stem cell derivation. The disease begins with the malignant transformation of one of the stem cells, which initially proliferates in the bone marrow and eventually overflows into the peripheral blood of the affected patient. Problems arise when the leukemic cells crowd out the normal defense cell and erythrocyte precursors. In the United States, approximately 2.5% of all cancers are leukemia, and 3.9% of deaths from cancer can be attributed to this disease.
Leukemias are usually classified according to their histogenesis and clinical behavior. Therefore, the broad categories would be acute or chronic (referring to the clinical course) and myeloid or lymphocytic/lymphoblastic (referring to the histogenetic origin). Myeloid leukemias can differentiate along several different pathways; thus they produce malignant cells that usually show features of granulocytes or monocytes, and less frequently, erythrocytes or megakaryocytes.
Acute leukemias, if untreated, run an aggressive course and often result in the death of the patient within a few months. Chronic leukemias tend to follow a more indolent course, although the end result is the same. One of the greatest successes in cancer treatment has been achieved in acute lymphoblastic leukemia of childhood, a condition that used to be uniformly fatal but now is often capable of being controlled.
Leukemias are probably the result of a combination of environmental and genetic factors. Certain syndromes are associated with an increased risk. These genetic disorders include the following:
In addition, certain types of leukemia show specific chromosomal abnormalities. The first chromosomal abnormality to be detected was found in patients with chronic myeloid leukemia, and this malignancy was characterized by a genetic alteration called the Philadelphia chromosome. This abnormality represents a translocation of the chromosomal material between the long arms of chromosomes 22 and 9. This rearrangement of the genetic material occurs in such a fashion as to fuse the breakpoint cluster region (bcr) gene with the Abelson (c-abl) oncogene, producing an entirely new gene: bcr-abl. This gene is continuously transcribed, and the resulting protein product, a tyrosine kinase, causes the uncontrolled proliferation of the leukemic cells. Identifying such pathogenetic mechanisms has opened up an entirely new field of chemotherapy that targets specific molecular mechanisms of carcinogenesis. A variety of other genetic alterations in the bone marrow stem cells has been associated with the myelodysplasia syndromes, a group of disorders that appear to represent early stages in the evolution of acute myeloid leukemia. As the genetic alterations accumulate in the stem cells, the chances of the patient developing leukemia increase.
Some environmental agents are associated with an increased risk of leukemia, but their overall contribution to the leukemia problem is thought to be less than 5%. Exposure to pesticides, benzene, and benzene-like chemicals has been associated with an increased risk of developing leukemia. Ionizing radiation has also been implicated; this was documented by the increased frequency of chronic myeloid leukemia in the survivors of the atomic bomb blasts at Hiroshima and Nagasaki during World War II. Viruses have also been shown to produce leukemia, although this is not a common finding. The most thoroughly studied is the retrovirus known as human T-cell leukemia/lymphoma virus type 1 (HTLV-1), which is transmitted by contaminated blood from infected to uninfected individuals. This virus can cause a relatively rare form of malignancy of T lymphocytes, which may present as a leukemia or non-Hodgkin’s lymphoma (see page 595). Most cases have been identified in parts of the Caribbean, central Africa, and southwestern Japan.
As knowledge about this group of diseases increases, the fact that the leukemias are diverse and complex cannot be overlooked. For example, eight distinct subtypes of acute myeloid leukemia have now been identified, and each subtype has a different treatment approach and prognosis. Because of the complexity of this area, the discussion is limited to those aspects of leukemia that are more directly related to the oral or head and neck region.
CLINICAL FEATURES: If all types of leukemia are considered, this condition occurs at a rate of 13 cases per 100,000 population annually. Slightly more males than females are affected. The myeloid leukemias generally affect an adult population; acute myeloid leukemia affects a broader age range, which includes children. Chronic myeloid leukemia shows a peak incidence during the third and fourth decades of life. Acute lymphoblastic leukemia, in contrast, occurs predominantly in children and represents one of the more common childhood malignancies. Chronic lymphocytic leukemia, the most common type of leukemia, primarily affects older adults.
Many of the clinical signs and symptoms of leukemia are related to the marked reduction in the numbers of normal white and red blood cells, a phenomenon that results from the crowding out of the normal hematopoietic stem cells by the malignant proliferation (myelophthisic anemia). Because of the reduced red blood cell count and subsequent reduction in oxygen-carrying capacity of the blood, patients complain of fatigue, easy tiring, and dyspnea on mild exertion. The malignant cells may also infiltrate other organs and often cause splenomegaly, hepatomegaly, and lymphadenopathy.
Leukemic patients may also complain of easy bruising and bleeding, problems that are caused by a lack of blood platelets (thrombocytopenia), the result of megakaryocytes being crowded out of the marrow. Petechial hemorrhages of the posterior hard palate and the soft palate may be observed, and these may be accompanied by spontaneous gingival hemorrhage, especially with platelet counts less than 10,000 to 20,000/mm3. Because disturbances in stem cell differentiation accompany the myelodysplasia syndromes, thrombocytopenia is often present in these patients, and gingival hemorrhage has been reported in this setting. Serious hemorrhagic complications may result from bleeding into the central nervous system or the lungs.
A fever associated with infection may be the initial sign of the leukemic process. Perirectal infections, pneumonia, urinary tract infections, and septicemia are common infectious complications. The microorganisms that are typically involved include gram-negative bacteria, gram-positive cocci, and certain Candida species.
Ulceration of the oral mucosa is often present as a result of the impaired ability of the host to combat the normal microbial flora. Usually, the gingival mucosa is the most severely affected because of the abundant bacteria normally present around the teeth. The neutropenic ulcers that are produced are typically deep, punched-out lesions with a gray-white necrotic base. Oral candidiasis is often a complication of leukemia, involving the oral mucosa diffusely. Herpetic infections are the most common viral lesions, and these may involve any area of the oral mucosa rather than being confined to the keratinized mucosa, as in immunocompetent patients.
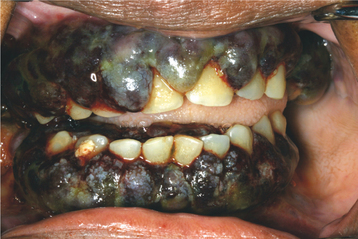
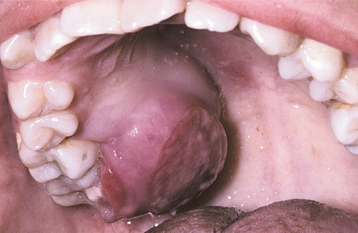
Occasionally, the leukemic cells infiltrate the oral soft tissues and produce a diffuse, boggy, nontender swelling that may or may not be ulcerated. This occurs most frequently with the myelomonocytic types of leukemia, and it may result in diffuse gingival enlargement (Figs. 13-16 and 13-17) or a prominent tumorlike growth (Fig. 13-18). The tumorlike collection of leukemic cells is known as granulocytic sarcoma or extramedullary myeloid tumor, and historically the term chloroma has been used because it is often greenish (chlor = green; oma = tumor) on fresh-cut sections. Other oral manifestations include infiltration of the periapical tissues, simulating periapical inflammatory disease both clinically and radiographically.
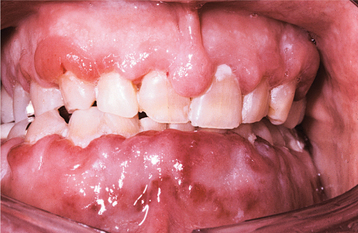
Fig. 13-16 Leukemia. Diffuse gingival enlargement, as depicted in this photograph, may occur in leukemic patients, particularly in those with monocytic leukemia. This older man had a history of myelodysplasia for several years before the development of leukemia.
HISTOPATHOLOGIC FEATURES: Microscopic examination of leukemia-affected tissue shows diffuse infiltration and destruction of the normal host tissue by sheets of poorly differentiated cells with either myelomonocytic characteristics or lymphoid features.
DIAGNOSIS: The diagnosis is usually established by confirming the presence of poorly differentiated leukemic cells in the peripheral blood and bone marrow. Bone marrow biopsy is normally performed in conjunction with the peripheral blood studies because some patients may go through an aleukemic phase in which the atypical cells are absent from the circulation. Classifying the type of leukemia requires establishing the immunophenotype by using immunohistochemical markers to identify cell surface antigens expressed by the tumor cells. Immunohistochemical confirmation of certain characteristic enzymes (e.g., myeloperoxidase, lysozyme) is necessary to identify and classify the myeloid leukemias. In addition, cytogenetic and molecular characterization of the lesional cells is typically necessary. In many cases, the results of these various studies will be significant because the patient’s prognosis is directly affected.
TREATMENT AND PROGNOSIS: The treatment of a patient with leukemia consists of various forms of chemotherapy; the type of leukemia dictates the chemotherapeutic regimen. In most cases the purpose of chemotherapy is to destroy as many of the atypical cells as possible in a short time, thus inducing a remission. For this reason, this technique has been termed induction chemotherapy. Usually, this phase of chemotherapy requires high doses of toxic chemotherapeutic agents; often, the patient experiences a number of unpleasant side effects during treatment. Once remission has been induced, this state must be maintained. This is the purpose of maintenance chemotherapy, which typically requires lower doses of chemotherapeutic drugs given over a longer period.
If the bcr-abl fusion is identified in the leukemic cells of a patient with chronic myeloid leukemia, then treatment with a tyrosine kinase inhibitor is appropriate. The first tyrosine kinase inhibitor to be developed and marketed was imatinib mesylate, and a significant proportion of patients will respond dramatically to this therapy. Imatinib must be taken continuously, because relapses develop quickly if the drug is stopped. Other tyrosine kinase inhibitors are currently being investigated as well.
Newer treatments for chronic lymphocytic leukemia include monoclonal antibodies directed against cell surface antigens, such as CD20, a B-lymphocyte antigen. Rituximab is one such agent that is now being investigated in the treatment of hematologic malignancies of B-cell differentiation.
Drug therapy may be combined with radiation therapy to the CNS because the chemotherapeutic drugs often do not cross the blood-brain barrier effectively. Therefore, the leukemic cells may survive in this site and cause a relapse of the leukemia. Direct intrathecal infusion of the chemotherapeutic agent may be performed to circumvent the problem of the blood-brain barrier. If this strategy succeeds in inducing a remission, then a bone marrow transplant may be considered as a therapeutic option, particularly for the types of leukemia that tend to relapse. This option often is reserved for patients younger than 45 years of age because the success rate is less favorable in older patients.
Supportive care is often necessary if these patients are to survive their leukemia. For patients with bleeding problems, transfusions with platelets may be necessary. If severe anemia is present, packed red blood cells may be required. Infections, of course, should be evaluated with respect to the causative organism, and appropriate antibiotics must be prescribed. Support must be maintained from an oral perspective because many of these patients experience infections of the oral mucosa during the course of their disease. Optimal oral hygiene should be encouraged, and aggressive investigation of any oral complaint should be performed as soon as possible to prevent potentially serious oral infectious complications.
The prognosis of a particular patient depends on a number of variables, including the type of leukemia, the age of the patient, and the cytogenetic alterations associated with the disease. In children with acute lymphoblastic leukemia, more than 80% of these patients are now considered to be cured after appropriate treatment. In an adult with the same diagnosis, even though the rate of initial remission induction is 80%, the 5-year survival rate is generally much lower in most reported series.
Patients younger than 60 years of age with acute myeloid leukemia have a 5-year survival rate of approximately 40% today. This form of leukemia in a patient older than 60 years, however, has a much poorer prognosis, with less than a 10% chance of survival seen in that population. Similarly, patients with a previous history of myelodysplasia have an unfavorable prognosis.
Even though an indolent period is experienced with chronic myeloid leukemia, eventually the neoplastic cells undergo a process known as blast transformation, in which they become less differentiated, proliferate wildly, and cause the patient’s death within 3 to 6 months. In the past, the 5-year survival rate for chronic myeloid leukemia was in the 20% range. Today, most centers are reporting 5-year survival rates of approximately 80%, a dramatic improvement presumably because of the effect of tyrosine kinase inhibitor therapy. Additional factors that may play a role in improved survival include diagnosis of the disease at an earlier stage and the availability of better supportive care. Attempts to control chronic myeloid leukemia by bone marrow transplantation from an HLA-matched donor have resulted in 5-year survival rates of 60% to 70% in younger patients with this disease. This may be an option for those patients who do not respond to tyrosine kinase inhibitor therapy.
Chronic lymphocytic leukemia is considered to be incurable, but its course is highly variable and depends on the stage of the disease. Patients with limited disease have an average survival time of more than 10 years. Those with more advanced disease survive an average of only 2 years.
LANGERHANS CELL HISTIOCYTOSIS (HISTIOCYTOSIS X; LANGERHANS CELL DISEASE; IDIOPATHIC HISTIOCYTOSIS; EOSINOPHILIC GRANULOMA; LANGERHANS CELL GRANULOMA; LANGERHANS CELL GRANULOMATOSIS)
The term histiocytosis X was introduced as a collective designation for a spectrum of clinicopathologic disorders characterized by proliferation of histiocyte-like cells that are accompanied by varying numbers of eosinophils, lymphocytes, plasma cells, and multinucleated giant cells. The distinctive histiocytic cells present in this lesion have been identified as Langerhans cells, and the condition is now designated as Langerhans cell histiocytosis. Langerhans cells are dendritic mononuclear cells normally found in the epidermis, mucosa, lymph nodes, and bone marrow. These cells process and present antigens to T lymphocytes. For many years, researchers have debated whether Langerhans cell histiocytosis represents a nonneoplastic condition or a true neoplasm. Studies examining the clonality of the lesional cells of this condition have shown this to be a monoclonal proliferation, a finding that is more consistent with a neoplastic process.
CLINICAL AND RADIOGRAPHIC FEATURES: The clinicopathologic spectrum traditionally considered under the designation of Langerhans cell histiocytosis includes the following:
• Monostotic or polyostotic eosinophilic granuloma of bone—solitary or multiple bone lesions without visceral involvement
• Chronic disseminated histiocytosis—a disease involving bone, skin, and viscera (Hand-Schüller-Christian disease)
• Acute disseminated histiocytosis—a disease with prominent cutaneous, visceral, and bone marrow involvement occurring mainly in infants (Letterer-Siwe disease)
It is difficult to categorize many patients into one of these classic designations because of overlapping clinical features. The often-cited Hand-Schüller-Christian triad—bone lesions, exophthalmos, and diabetes insipidus—is present in only a few patients with chronic disseminated disease. It is widely believed that the traditional designations of Hand-Schüller-Christian and Letterer-Siwe disease serve no useful purpose and should be discontinued. Many cases reported as Letterer-Siwe disease in the older literature probably included obscure infections, immunodeficiency syndromes, and malignant histiocytic lesions. Pulmonary Langerhans cell histiocytosis has also been described, but this probably is unrelated to the condition that affects the jaws. Patients who develop pulmonary Langerhans cell histiocytosis are usually adults with a history of smoking, and clonality studies suggest that this is probably a reactive process.
Although Langerhans cell histiocytosis may be encountered in patients over a wide age range, more than 50% of all cases are seen in patients younger than age 15. Although some series have reported a male predilection, overall the sexes appear to be equally affected. Bone lesions, either solitary or multiple, are the most common clinical presentation. Lesions may be found in almost any bone, but the skull, ribs, vertebrae, and mandible are among the most frequent sites. Children younger than age 10 most often have skull and femoral lesions; patients older than age 20 more often have lesions in the ribs, shoulder girdle, and mandible. Adult patients with solitary or multiple bone lesions may have lymphadenopathy but usually do not have significant visceral involvement.
The jaws are affected in 10% to 20% of all cases. Dull pain and tenderness often accompany bone lesions. Radiographically, the lesions often appear as sharply punched-out radiolucencies without a corticated rim, but occasionally an ill-defined radiolucency is seen. Bone involvement in the mandible usually occurs in the posterior areas, and a characteristic “scooped out” appearance may be evident when the superficial alveolar bone is destroyed. The resulting bone destruction and loosening of the teeth clinically may resemble severe periodontitis (Fig. 13-19). Extensive alveolar involvement causes the teeth to appear as if they are “floating in air” (Fig. 13-20).
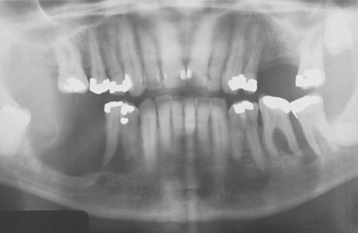
Fig. 13-19 Langerhans cell histiocytosis. Severe bone loss in the mandibular molar regions that resembles advanced periodontitis. (Courtesy of Dr. James White.)
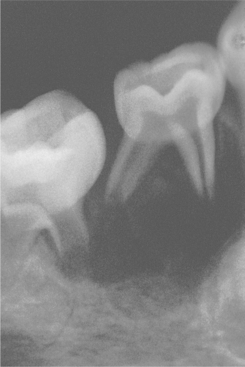
Fig. 13-20 Langerhans cell histiocytosis. Periapical radiograph showing marked bone loss involving the mandibular teeth in a young girl, resulting in a “floating-in-air” appearance of the teeth.
Ulcerative or proliferative mucosal lesions or a proliferative gingival mass may develop if the disease breaks out of bone (Fig. 13-21). Occasionally, this process may involve only the oral soft tissues. Lesions also can occur within the body of the mandible or maxilla, where they may simulate a periapical inflammatory condition.
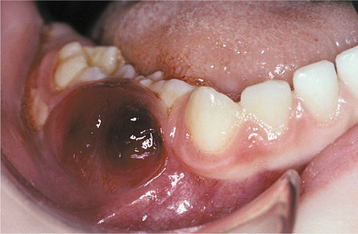
Fig. 13-21 Langerhans cell histiocytosis. Clinical photograph of the same patient shown in Fig. 13-20. The lesion has broken out of bone and produced this soft tissue mass.
HISTOPATHOLOGIC FEATURES: The bone lesions of patients with Langerhans cell histiocytosis show a diffuse infiltration of large, pale-staining mononuclear cells that resemble histiocytes. These cells have indistinct cytoplasmic borders and rounded or indented vesicular nuclei. Varying numbers of eosinophils are typically interspersed among the histiocyte-like cells (Fig. 13-22). Plasma cells, lymphocytes, and multinucleated giant cells are often seen, and areas of necrosis and hemorrhage may be present.
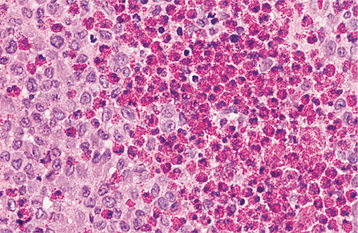
Fig. 13-22 Langerhans cell histiocytosis. There is a diffuse infiltrate of pale-staining Langerhans cells intermixed with numerous red granular eosinophils.
The identification of lesional Langerhans cells is necessary to confirm the diagnosis. Because Langerhans cells cannot be differentiated from other histiocytes by routine histologic staining, additional diagnostic methods are required. Electron microscopic evaluation of lesional tissue has been the gold standard for many years because, ultrastructurally, Langerhans cells contain rod-shaped cytoplasmic structures known as Birbeck granules, which differentiate them from other mononuclear phagocytes (Fig. 13-23). Most laboratories now rely on immunohistochemical procedures to identify the lesional Langerhans cells because of their immunoreactivity with antibodies directed against either CD-1a or CD-207, the latter marker being even more specific for Langerhans cells. To a lesser extent, the lesional cells have S-100 protein immunoreactivity, and they also may show affinity for peanut agglutinin (PNA).
TREATMENT AND PROGNOSIS: Accessible bone lesions, such as those in the maxilla and mandible, are usually treated by curettage. Low doses of radiation may be used for less accessible bone lesions, although the potential for induction of malignancy secondary to this treatment is a concern in younger patients. Intralesional injection with corticosteroid agents has also been reported to be effective in some patients with localized bone lesions. Infrequently, the apparent spontaneous regression of localized Langerhans cell histiocytosis has been reported. The prognosis for bone lesions in the absence of significant visceral involvement is generally good; however, progression or dissemination of the disease may occur, particularly for patients who have three or more bones affected.
Chronic disseminated disease is often associated with considerable morbidity, but few patients die as a result of the disease. Because of the relative rarity of disseminated cases, the ideal treatment has yet to be identified. Single-agent chemotherapy using prednisolone, etoposide, vincristine, or cyclosporine has produced a good response in a significant percentage of such patients, although recurrence is typically seen in over half of the cases. A combination of vincristine and prednisone seems to reduce this risk of recurrence. The acute disseminated form of the disease seen in infants and young children may not respond to these more conservative approaches, and multiple chemotherapeutic agents are given in that situation. Diffuse involvement with compromise of multiple organs is associated with a poor prognosis and is often fatal. In general, the prognosis is poorer for patients in whom the first sign of the disease develops at a very young age and somewhat better for patients who are older at the time of onset.
HODGKIN’S LYMPHOMA (HODGKIN’S DISEASE)
Hodgkin’s lymphoma represents a malignant lymphoproliferative disorder, although for many years the exact nature of the process was poorly understood. The difficulty in comprehending the character of the condition is reflected in the relatively noncommittal term Hodgkin’s disease, which was used for decades and still may be heard today. Perhaps one reason why Hodgkin’s lymphoma was not easily understood is that, unlike most malignancies, the neoplastic cells (Reed-Sternberg cells) make up only about 1% to 3% of the cells in the enlarged lymph nodes that characterize this condition. Current evidence regarding the histogenesis of the Reed-Sternberg cell points to a B-lymphocyte origin. Certainly, the disease can cause death if appropriate therapy is not instituted, although the treatment of this malignancy is one of the few major success stories in cancer therapy during the past 20 years. In the United States, Hodgkin’s lymphoma is about one sixth as common as non-Hodgkin’s lymphoma; approximately 8000 cases are diagnosed annually. Although the cause of this disease is unknown, epidemiologic and molecular studies have linked Epstein-Barr virus (EBV) infection to a significant percentage of these lesions.
CLINICAL FEATURES: Hodgkin’s lymphoma almost always begins in the lymph nodes, and any lymph node group is susceptible. The most common sites of initial presentation are the cervical and supraclavicular nodes (70% to 75%) or the axillary and mediastinal nodes (5% to 10% each). The disease initially appears less than 5% of the time in the abdominal and inguinal lymph nodes.
Overall, a male predilection is observed, and a bimodal pattern is noted with respect to the patient’s age at diagnosis. One peak is observed between 15 and 35 years of age; another peak is seen after the age of 50.
The usual presenting sign is the identification by the patient of a persistently enlarging, nontender, discrete mass or masses in one lymph node region (Fig. 13-24). In the early stages, the involved lymph nodes are often rather movable; as the condition progresses, the nodes become more matted and fixed to the surrounding tissues. If it is untreated, then the condition spreads to other lymph node groups and eventually involves the spleen and other extralymphatic tissues, such as bone, liver, and lung. Oral involvement has been reported, but it is rare. In about 30% of patients with Hodgkin’s disease, other systemic signs and symptoms may be present, such as weight loss, fever, night sweats, and generalized pruritus (itching). The absence of these systemic signs and symptoms is considered to be better in terms of the patient’s prognosis, and this information is used in staging the disease. Patients who have no systemic signs are assigned to category A and those with systemic signs to category B.
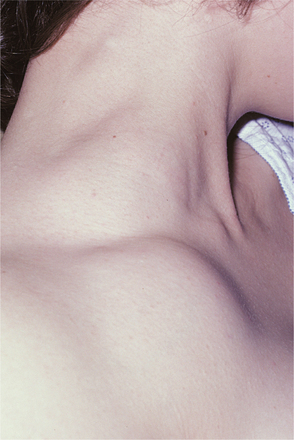
Fig. 13-24 Hodgkin’s lymphoma. The prominent supraclavicular and cervical masses represent Hodgkin’s lymphoma.
The staging of Hodgkin’s lymphoma is important for planning treatment and estimating the prognosis for a given patient. The staging procedure typically includes confirmation of the pathologic diagnosis, careful history and physical examination, abdominal and thoracic computed tomography (CT) scans or magnetic resonance imaging (MRI) studies, chest radiographs, and routine hematologic studies (e.g., complete blood count, serum chemistries, erythrocyte sedimentation rate). Evaluation of the extent of disease involvement using positron emission tomography (PET) scans is becoming part of the standard protocol, particularly at large institutions. Lymphangiography, gallium scan, bone marrow biopsy, exploratory laparotomy, and splenectomy may be necessary if the information that they would provide might have an effect on staging or treatment. A summary of the staging system for Hodgkin’s lymphoma is presented in Table 13-2.
Table 13-2
Ann Arbor System for Classi. cation of Hodgkin’s Lymphoma
| Stage | Defining Features |
| I | Involvement of a single lymph node region (I) or a single extralymphatic organ or site (IE) |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm (II) or one or more lymph node regions with an extralymphatic site (IIE) |
| III | Involvement of lymph node regions on both sides of the diaphragm (III), possibly with an extralymphatic organ or site (IIIE), the spleen (IIIS), or both (IIISE) |
| IV | Diffuse or disseminated involvement of one or more extralymphatic organs (identified by symbols), with or without associated lymph node involvement |
| A: Absence of systemic signs | |
| B: Presence of fever, night sweats, and/or unexplained loss of 10% or more of body weight during the 6- month period before diagnosis |
Adapted from DeVita VT, Hubbard SM: Hodgkin’s disease, N Engl J Med 328:560-565, 1993.
HISTOPATHOLOGIC FEATURES: Hodgkin’s lymphoma is recognized to comprise two main forms, (1) nodular lymphocyte–predominant Hodgkin’s lymphoma and (2) classical Hodgkin’s lymphoma, the latter of which is divided into five subtypes. Although this group of diseases has certain features in common, current immunohistochemical and molecular biologic techniques have allowed distinctions to be made among the various types. The common features include effacement of the normal nodal architecture by a diffuse, often mixed, infiltrate of inflammatory cells that is interspersed with large, atypical neoplastic lymphoid cells. In the case of classical Hodgkin’s lymphoma, this atypical cell is known as a Reed-Sternberg cell (Fig. 13-25). The Reed-Sternberg cell is typically binucleated (“owl-eye” nuclei), although it may be multinucleated (“pennies on a plate”), with prominent nucleoli. The malignant cell in nodular lymphocyte–predominant Hodgkin’s lymphoma is the “popcorn cell,” which is so-named because of the resemblance of the nucleus to a kernel of popped corn. The pathologist must see one of these types of distinctive atypical cells to make a diagnosis of Hodgkin’s lymphoma, although their presence does not automatically imply that diagnosis, because similar cells may be seen in certain viral infections, especially infectious mononucleosis. To summarize, Hodgkin’s lymphoma is currently classified in the following manner:
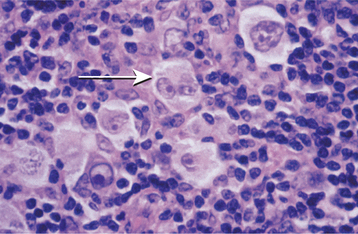
Fig. 13-25 Hodgkin’s lymphoma. This high-power photomicrograph shows the characteristic Reed-Sternberg cell (arrow) of Hodgkin’s lymphoma, identified by its “owl-eye” nucleus.
• Nodular lymphocyte–predominant Hodgkin’s lymphoma, or
• Classical Hodgkin’s lymphoma (comprising five histopathologic subtypes):
These names describe the most prominent histopathologic feature of each type, and specific epidemiologic and prognostic characteristics are associated with each type.
Nodular lymphocyte–predominant Hodgkin’s lymphoma constitutes 4% to 5% of all cases of Hodgkin’s lymphoma in the United States. In the past, this form was probably combined with the lymphocyte-rich subtype, but the presence of the characteristic popcorn cells is a significant clue to the diagnosis.
Lymphocyte-rich classical Hodgkin’s lymphoma represents about 6% of all cases. Sheets of small lymphocytes with few Reed-Sternberg cells characterize this form.
The nodular sclerosis subtype makes up 60% to 80% of cases and occurs more frequently in females during the second decade of life. This type gets its name from the broad fibrotic bands that extend from the lymph node capsule into the lesional tissue. Reed-Sternberg cells in the nodular sclerosis form appear to reside in clear spaces and, therefore, are referred to as lacunar cells.
The mixed cellularity form accounts for about 15% to 30% of the cases and is characterized by a mixture of small lymphocytes, plasma cells, eosinophils, and histiocytes with abundant Reed-Sternberg cells.
The lymphocyte depletion subtype, the most aggressive type, makes up less than 1% of the cases in recent reports. Before modern immunohistochemical techniques, many examples of large cell lymphoma or anaplastic T-cell lymphoma were undoubtedly included in this category. In this form of Hodgkin’s lymphoma, numerous bizarre giant Reed-Sternberg cells are present, with few lymphocytes.
Occasionally, examples of Hodgkin’s lymphoma are encountered that really do not fit the criteria for any of the known subtypes, and these are designated as unclassifiable.
TREATMENT AND PROGNOSIS: The treatment of Hodgkin’s lymphoma depends on the stage of involvement. Patients who had limited disease (stages I and II) often were managed by local radiation therapy alone. Recent treatment trends, however, combine less extensive radiotherapy fields with milder multiagent chemotherapy regimens to maximize disease control and minimize long-term complications of therapy. Patients with stage III or IV disease require chemotherapy; radiation therapy is used conjointly if significant mediastinal involvement or residual disease is detected. For many years a regimen known as MOPP (mechlorethamine, Oncovin, procarbazine, prednisone) was widely used to treat Hodgkin’s lymphoma. Because significant long-term side effects can be associated with this chemotherapy, another regimen known as ABVD (Adriamycin, bleomycin, vinblastine, dacarbazine [DTIC]) is now used most often, particularly for early and intermediate stage disease, because it has fewer complications.
Before modern cancer therapy was developed for Hodgkin’s lymphoma, the 5-year survival rate was only 5%. The prognosis for this disease is fairly good today; the best treatment results occur in those who present in the early stages. Patients with stage I and II disease often have an 80% to 90% relapse-free 10-year survival rate; those with stage III and IV disease have a 55% to 75% 10-year survival rate.
The histopathologic subtype also influences the response to therapy. Patients with the lymphocyte-predominant and nodular sclerosis forms have the best prognosis, whereas those with the mixed cellularity form have a less favorable prognosis. In the past, researchers believed that the lymphocyte depletion form had a poor prognosis. However, with newer immunohistochemical studies, clinicians now realize that many of these cases were misdiagnosed; therefore, the available data are probably not reliable. In most instances, however, the stage of disease now plays a more important role in determining the patient’s prognosis than does the histopathologic subtype.
After 15 years posttreatment, patient mortality is due more often to the complications of therapy: either secondary malignancy or cardiovascular disease. Currently, research is focused on the development of treatment regimens that continue to have a superior cure rate, while simultaneously decreasing the risk of treatment-related complications.
NON-HODGKIN’S LYMPHOMA
The non-Hodgkin’s lymphomas include a diverse and complex group of malignancies of lymphoreticular histogenesis and differentiation. In most instances, they initially arise within lymph nodes and tend to grow as solid masses. This is in contrast to lymphocytic leukemias (see page 587), which begin in the bone marrow and are characterized by a large proportion of malignant cells that circulate in the peripheral blood. The non-Hodgkin’s lymphomas most commonly originate from cells of the B-lymphocyte series, with an estimated 85% of European and American lymphoid neoplasms having this derivation. Tumors with a T-lymphocyte derivation are less common, whereas true histiocyte-derived lymphomas are even rarer.
The microscopic appearance of the lesional cells was used in the past to classify the tumors as either lymphocytic or histiocytic. With the development of modern immunologic techniques, however, it is now known that many of the lesions that had been classified as histiocytic were in fact neoplasms composed of transformed B lymphocytes. In the early 1980s, a group of American pathologists devised a classification scheme, known as the Working Formulation for Clinical Use, which may still be referred to in the United States. Based on this classification, lymphomas were broadly grouped into three categories:
Unfortunately, the Working Formulation has been shown to be somewhat limited in its utility and accuracy. Many lesions that have been recently defined are not included in this classification. For these reasons, an international study group in the early 1990s devised a new method of categorizing the lymphomas, known as the REAL (revised European-American lymphoma) classification. With this system, a combination of histopathologic features, immunologic cell surface markers, and gene rearrangement studies are used to organize this group of neoplasms. Recently the World Health Organization (WHO) revised its lymphoma classification system to conform to a slightly modified version of the REAL classification (Box 13-2). The latter two classifications appear to be more precise than the Working Formulation, and currently most pathologists in the United States categorize lymphomas according to the modified REAL system, although some of the more sophisticated molecular studies may not be available at smaller laboratories.
More than 58,000 cases of non-Hodgkin’s lymphoma are diagnosed in the United States annually; approximately one third of this number will die of the disease each year. For reasons that are currently unclear, the incidence of this malignancy seems to be rising in the United States. The prevalence of lymphoma is increased in patients who have immunologic problems, such as congenital immunodeficiencies (e.g., Bloom syndrome, Wiskott-Aldrich syndrome, common variable immunodeficiency), AIDS, organ transplantation, and autoimmune disease (e.g., Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis).
Viruses may play a role in the pathogenesis of at least some of these lesions. For example, Epstein-Barr virus (EBV) has been implicated, but not proven, to be an etiopathogenic agent in Burkitt’s lymphoma (see page 600), a type of high-grade, small, noncleaved B-cell lymphoma. In addition, EBV may be related to lymphomas developing in the setting of immunosuppression after solid organ or bone marrow transplant (resulting in the condition known as posttransplant lympho-proliferative disorder) or in association with AIDS (see page 272). Human herpesvirus 8 (HHV-8) has not only been associated with Kaposi’s sarcoma but also with primary body cavity lymphoma and some cases of plasmablastic lymphoma. A blood-borne human retrovirus called human T-cell leukemia/lymphoma virus type I (HTLV-1) has been shown to cause an aggressive form of peripheral T-cell lymphoma among certain populations in the Caribbean, central Africa, and southwest Japan.
Even bacteria have been shown to induce the formation of so-called mucosa-associated lymphoid tissue (MALT) lymphoma of the stomach. Antibiotic treatment of Helicobacter pylori infection of the stomach lining often results in complete regression of this low-grade lymphoma.
CLINICAL AND RADIOGRAPHIC FEATURES: Non-Hodgkin’s lymphoma occurs primarily in adults, although children may be affected, particularly by the more aggressive intermediate- and high-grade lymphomas. The condition most commonly develops in the lymph nodes, but so-called extranodal lymphomas are also found. In the United States, approximately 20% to 40% of lymphomas develop in an extranodal site, but in Asian countries such as Korea and Japan, nearly half of all lymphomas are extranodal.
With a nodal presentation, the patient usually is aware of a nontender mass that has been slowly enlarging for months. The lesion typically involves a local lymph node collection, such as the cervical, axillary, or inguinal nodes; one or two freely movable nodules are noticed initially. As the malignancy progresses, the nodes become more numerous and are fixed to adjacent structures or matted together (Fig. 13-26). Gradually, the process involves other lymph node groups, and invasion of adjoining normal tissues occurs.
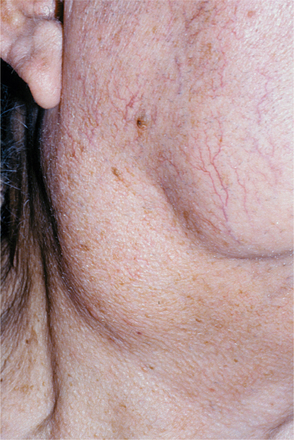
Fig. 13-26 Non-Hodgkin’s lymphoma. The matted, nontender lymph node enlargement in the lateral cervical region represents a common presentation of lymphoma.
In the oral cavity, lymphoma usually appears as extranodal disease. Although the oral lesions of lymphoma are often a component of more widely disseminated disease, at times the lymphoma begins in the oral tissues and has not spread to other sites. The malignancy may develop in the oral soft tissues or centrally within the jaws. Soft tissue lesions appear as nontender, diffuse swellings; they most commonly affect the buccal vestibule, posterior hard palate, or gingiva (Figs. 13-27 and 13-28). Such swellings characteristically have a boggy consistency. The lesion may appear erythematous or purplish, and it may or may not be ulcerated. Patients who wear a denture that contacts the lesional site often complain that their denture does not fit because it feels too tight.
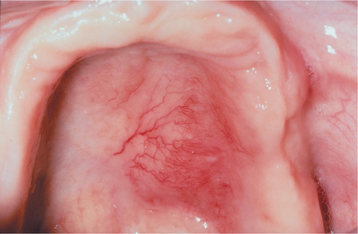
Fig. 13-27 Non-Hodgkin’s lymphoma. One of the frequent locations of extranodal lymphoma in the head and neck area is the palate, where the tumor appears as a nontender, boggy swelling. Note the overlying telangiectatic blood vessels, a feature often seen with malignancy.
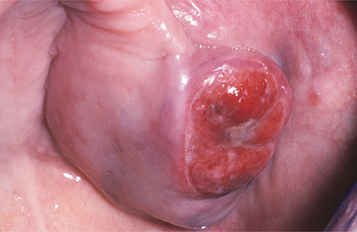
Lymphoma of bone may cause vague pain or discomfort, which might be mistaken for a toothache. The patient may complain of paresthesia, particularly with a mandibular lesion (so-called numb chin syndrome). Radiographs usually show an ill-defined or ragged radiolucency, although in the early stages, the radiographic changes may be subtle or nonexistent. If untreated, then the process typically causes expansion of the bone, eventually perforating the cortical plate and producing a soft tissue swelling. Such lesions have been mistaken for a dental abscess, although a significant amount of pain is not present in most cases.
Clinical staging to determine the extent to which the disease has spread is an important factor in assessing the prognosis for a particular patient. The staging evaluation should include a history, physical examination, complete blood count, liver function studies, routine chest radiographs, CT scans of the pelvic and abdominal regions, lymphangiography, and bone marrow biopsy. The staging system for Hodgkin’s lymphoma (see Table 13-2) has been widely adopted for use with the non-Hodgkin’s lymphomas.
HISTOPATHOLOGIC FEATURES: Non-Hodgkin’s lymphomas are histopathologically characterized by a proliferation of lymphocytic-appearing cells that may show varying degrees of differentiation, depending on the type of lymphoma. Low-grade lesions consist of well-differentiated small lymphocytes. High-grade lesions tend to be composed of less differentiated cells. All lymphomas grow as infiltrative, broad sheets of relatively uniform neoplastic cells that usually show little or no evidence of lesional tissue necrosis (Figs. 13-29 and 13-30). In some lesions, particularly those of B-lymphocyte origin, a vague semblance of germinal center formation may be seen (i.e., a nodular or follicular pattern). Other lymphomas show no evidence of such differentiation, and this pattern is termed diffuse. If the lymphoma arises in a lymph node, then the tumor destroys the normal architecture of the node. An extranodal lymphoma destroys the normal adjacent host tissue by infiltrating throughout the area. In the oral cavity, diffuse large B-cell lymphoma, which is considered to be a high-grade lymphoma, is the most common diagnosis, comprising approximately 60% of the cases.
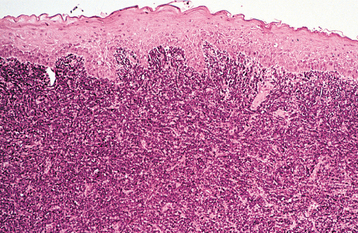
Fig. 13-29 Non-Hodgkin’s lymphoma. This low-power photomicrograph shows a diffuse infiltration of the subepithelial connective tissue by lymphoma.
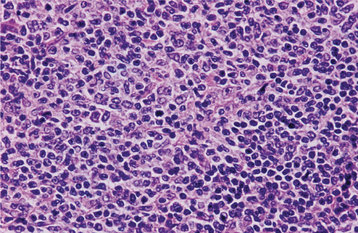
Fig. 13-30 Non-Hodgkin’s lymphoma. This high-power photomicrograph shows lesional cells of lymphoma, consisting of a population of poorly differentiated cells of the lymphocytic series with minimal cytoplasm.
Standard of care demands that appropriate immunohistochemical and cytogenetic studies be performed for a tumor diagnosed as lymphoma. In general, these studies can become quite involved and, therefore, are beyond the scope of this text.
TREATMENT AND PROGNOSIS: The treatment of a patient with non-Hodgkin’s lymphoma is based on several factors, including the stage and grade of the lymphoma, the overall health of the patient, and the patient’s pertinent past medical history. The patient’s health must be considered because many of the chemotherapeutic regimens are quite debilitating. Surgical management is not usually indicated.
Low-grade lymphomas are perhaps the most controversial in terms of treatment. Some authorities recommend no particular treatment because these tumors are slow growing and tend to recur despite chemotherapy. Given the fact that low-grade lymphomas arise in older adults and the median survival without treatment is 8 to 10 years, many clinicians opt for a “watch and wait” strategy, treating the patient only if symptoms develop. Unfortunately, approximately 40% of low-grade lymphomas eventually transform to a high-grade lymphoma, leading to the patient’s demise. Because these low-grade lymphomas have been considered “incurable,” new treatments are being investigated. Most of these lesions are of B-cell differentiation, so treatment strategies using monoclonal antibodies directed against CD20, a B-cell surface antigen, are now being evaluated. Rituximab is one of the agents being examined in clinical trials.
For the intermediate-grade and high-grade lymphomas, the treatment of localized disease consists of radiation plus chemotherapy. With more advanced and disseminated disease, chemotherapy alone usually is implemented. Multiagent chemotherapy is used routinely, and new combinations are being evaluated continuously. Unfortunately, although the response rate of many lesions is good and much progress has been made in this area, the cure rate is not high. For intermediate-grade lesions, a failure rate of 30% to 50% can be expected. High-grade lymphomas are associated with a 60% mortality rate at 5 years after diagnosis and treatment.
MYCOSIS FUNGOIDES (CUTANEOUS T-CELL LYMPHOMA)
From its name, one might think that mycosis fungoides is a fungal infection. The early dermatologists who first recognized mycosis fungoides knew that this was not the case; however, they still thought the disease resembled a fungal condition. Thus this term has persisted. This condition, in fact, represents a lymphoma that is derived from T lymphocytes, specifically the T-helper (CD4+) lymphocyte. With modern diagnostic techniques, clinicians now know that there are several types of cutaneous lymphomas, each having specific T-lymphocyte or B-lymphocyte differentiation patterns. Even though mycosis fungoides is the most common of these cutaneous lymphomas, it is still a relatively rare malignancy; only about 400 new cases are diagnosed in the United States annually. This condition exhibits a peculiar property called epidermotropism (i.e., a propensity to invade the epidermis of the skin). Oral involvement, although infrequent, may also be present.
CLINICAL FEATURES: Mycosis fungoides is a condition that usually affects middle-aged adult men; there is a 2:1 male-to-female ratio and a mean age at diagnosis of 55 to 60 years. The disease progresses through three stages, usually over the course of several years.
The first stage, known as the eczematous (erythematous) stage, is often mistaken for psoriasis of the skin because of the well-demarcated, scaly, erythematous patches that characterize these lesions. Patients may complain of pruritus. With time, the erythematous patches evolve into slightly elevated, red lesions (plaque stage). These plaques tend to grow and become distinct papules and nodules. At this time, the disease has entered the tumor stage (Fig. 13-31). Visceral involvement is also seen at this point.
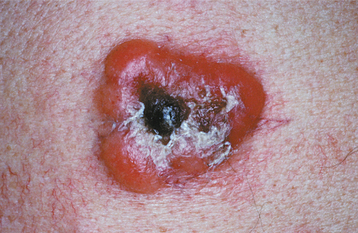
Fig. 13-31 Mycosis fungoides. In the tumor stage of the disease, patients with mycosis fungoides have ulcerated nodules of the skin. (From Damm DD, White DK, Cibull ML et al: Mycosis fungoides: initial diagnosis via palatal biopsy with discussion of diagnostic advantages of plastic embedding, Oral Surg Oral Med Oral Pathol 58:413-419, 1984.)
Approximately 35 cases of mycosis fungoides with oral involvement have been reported. The most commonly affected sites are the tongue, hard and soft palates, and gingiva (Fig. 13-32). The buccal mucosa, tonsils, lips, sinuses, and nasopharynx may also be affected. The oral lesions present as erythematous, indurated plaques or nodules that are typically ulcerated. Generally, these lesions appear late in the course of the disease and develop after the cutaneous lesions.
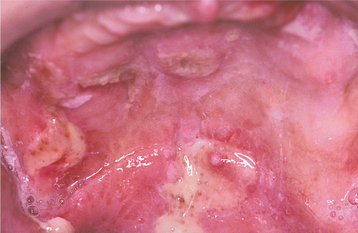
Fig. 13-32 Mycosis fungoides. The ulcerated palatal lesions represent a rare example of oral mucosal involvement by mycosis fungoides.
Sézary syndrome is an aggressive expression of mycosis fungoides that essentially represents a dermatopathic T-cell leukemia. The patient has a generalized exfoliative erythroderma, as well as lymphadenopathy, hepatomegaly, and splenomegaly. The lung, kidneys, and CNS can also be involved. This condition follows a fulminant course and typically results in the patient’s death within a short period of time; the median survival for this form of the disease is 2 to 3 years.
ECZEMATOUS STAGE: The early stages of mycosis fungoides may be difficult to diagnose histopathologically because of the subtle changes that characterize the initial lesions. A psoriasiform pattern of epithelial alteration is seen, with parakeratin production and elongation of the epithelial rete ridges. Scattered, slightly atypical lymphocytic cells may be seen in the connective tissue papillae, but such features are often mistaken for an inflammatory process.
PLAQUE STAGE: With the development of the plaque stage, a more readily identifiable microscopic pattern emerges. Examination of the surface epithelium reveals infiltration by atypical lymphocytic cells, which are sometimes referred to as mycosis cells or Sézary cells. These atypical lymphocytes classically form small intraepithelial aggregates termed Pautrier’s microabscesses (Fig. 13-33). The lesional cells have an extremely unusual nucleus because of the marked infolding of the nuclear membrane, which results in what is termed a cerebriform nucleus. This feature can best be appreciated when viewed in special semithin, plastic-embedded microscopic sections (Fig. 13-34). The diagnosis of mycosis fungoides can be confirmed by demonstrating positivity for CD4 (a cell surface marker for T-helper cells) in the lesional cell population. In addition, T-cell receptor gene rearrangement analysis should identify a monoclonal population of T lymphocytes. A mixed infiltrate of eosinophils, histiocytes, and plasma cells may be observed in the subepithelial connective tissue.
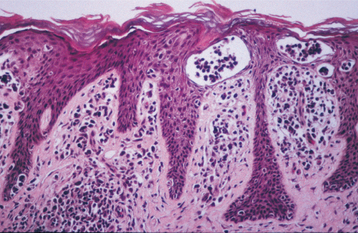
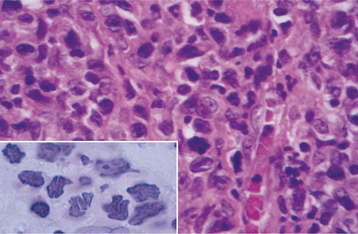
TUMOR STAGE: As the condition progresses to the tumor stage, the diffuse infiltration of the dermis and epidermis by atypical lymphocytic cells makes it easier to identify as a malignant process. Other types of lymphoma would enter into the histopathologic differential diagnosis.
Immunohistochemical studies demonstrating a T-helper phenotype, combined with the T-cell receptor gene rearrangement studies, would help to distinguish the malignant infiltrate from other lymphomas and establish the diagnosis of mycosis fungoides. Examination of the peripheral blood of a patient with Sézary syndrome shows circulating atypical lymphoid cells.
TREATMENT AND PROGNOSIS: Topical nitrogen mustard, topical carmustine, superpotent topical corticosteroids, electron beam therapy, or photochemotherapy (PUVA [8-methoxy-psoralen + ultraviolet A]) are effective in controlling mycosis fungoides during the early stages. Ultimately, the topical forms of therapy fail, and aggressive chemotherapy is necessary, particularly if there is visceral involvement. Newer agents that may be added to the chemotherapy regimen include monoclonal antibodies directed against the cell surface marker CD52, certain retinoid compounds, and specific interferon compounds. Another new agent that may be used for advanced disease is known as denileukin diftitox, which is derived from diphtheria toxin and targets the interleukin-2 receptor on the neoplastic lymphocytes. If Sézary syndrome develops, then extracorporeal photopheresis or chemotherapy is used as a treatment modality. Extracorporeal photopheresis involves the ingestion of the photoactive drug 8-methoxypsoralen, followed by the removal of a portion of the patient’s blood and a separation of red and white blood cells. The red blood cells are returned to the patient immediately. The white blood cells are irradiated outside the body (extracorporeal) with ultraviolet A. These altered white cells are then infused back into the patient. Their altered state may help generate an immunologic response to the patient’s own abnormal lymphocytes.
Although mycosis fungoides is not considered to be curable, the disease is usually slowly progressive. As a result, there is a median survival time of 8 to 10 years, and patients may die of causes unrelated to their lymphoma. Once the disease progresses beyond the cutaneous involvement, the course becomes much worse. The patient usually dies of organ failure or sepsis within 1 year.
BURKITT’S LYMPHOMA
Burkitt’s lymphoma is a malignancy of B-lymphocyte origin that represents an undifferentiated lymphoma. It was named after the missionary doctor, Denis Burkitt, who first documented the process. In the original report, this type of lymphoma was described in young African children, and it seemed to have a predilection for the jaws. Because it was seen frequently in sub-Saharan Africa, the term African Burkitt’s lymphoma has been applied to the disease. In addition, there is increased prevalence in other areas of the world, such as northeastern Brazil, and some investigators now refer to such tumors arising in these areas of increased prevalence as endemic Burkitt’s lymphoma. Re-searchers believe this malignancy is related pathogenetically to Epstein-Barr virus (EBV), because more than 90% of the tumor cells, particularly in the African type, show expression of EBV nuclear antigen, and affected patients have elevated antibody titers to EBV. Characteristic cytogenetic chromosomal translocations, which may also be responsible for neoplastic transformation, have also been described. Tumors with a similar histomorphology, commonly referred to as sporadic or American Burkitt’s lymphoma, have been observed in other countries where the neoplasm is usually first detected as an abdominal mass. Some HIV-related lymphomas may also have the microscopic features of Burkitt’s lymphoma, and these lesions have been designated immunodeficiency-associated Burkitt’s lymphoma. Similar tumors have been reported in other immunodeficiency settings, such as in patients who have received allografts or have a congenital immunodeficiency syndrome.
CLINICAL AND RADIOGRAPHIC FEATURES: As many as 50% to 70% of the cases of endemic Burkitt’s lymphoma present in the jaws. The malignancy usually affects children (peak prevalence, about 7 years of age) who live in Central Africa, and a male predilection is usually reported. The posterior segments of the jaws are more commonly affected, and the maxilla is involved more commonly than the mandible (a 2:1 ratio). Sometimes all four quadrants of the jaws show tumor involvement.
The tendency for jaw involvement seems to be age related; nearly 90% of 3-year-old patients have jaw lesions, in contrast to only 25% of patients older than age 15. Sporadic Burkitt’s lymphoma tends to affect patients over a greater age range than is noted for the African tumor. Although the abdominal region is typically affected, jaw lesions have been reported in sporadic Burkitt’s lymphoma (Fig. 13-35).
Fig. 13-35 Burkitt’s lymphoma. This patient had documented American Burkitt’s lymphoma involving the abdominal region. The retromolar swelling represents oral involvement with the malignancy.
The growth of the tumor mass may produce facial swelling and proptosis. Pain, tenderness, and paresthesia are usually minimal, although marked tooth mobility may be present because of the aggressive destruction of the alveolar bone. Premature exfoliation of deciduous teeth and enlargement of the gingiva or alveolar process may also be seen.
The radiographic features are consistent with a malignant process and include a radiolucent destruction of the bone with ragged, ill-defined margins (Fig. 13-36). This process may begin as several smaller sites, which eventually enlarge and coalesce. Patchy loss of the lamina dura has been mentioned as an early sign of Burkitt’s lymphoma.
HISTOPATHOLOGIC FEATURES: Burkitt’s lymphoma histopathologically represents an undifferentiated, small, noncleaved B-cell lymphoma. The lesion invades as broad sheets of tumor cells that exhibit round nuclei with minimal cytoplasm. Each tumor nucleus often has several prominent nucleoli, and numerous mitoses are seen. Immunohistochemical studies using markers that identify proliferating cells (e.g., Ki-67) typically indicate that almost 100% of the tumor cells are in the process of replicating. On viewing the lesion on low-power magnification, a classic “starry-sky” pattern is often appreciated—a phenomenon that is caused by the presence of macrophages within the tumor tissue (Fig. 13-37). These macrophages have abundant cytoplasm, which microscopically appears less intensely stained in comparison with the surrounding process. Thus these cells tend to stand out as “stars” set against the “night sky” of deeply hyperchromatic neoplastic lymphoid cells (Fig. 13-38).
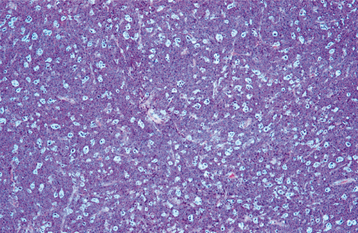
Fig. 13-37 Burkitt’s lymphoma. This low-power photomicrograph shows the classic “starry-sky” appearance, a pattern caused by interspersed histiocytic cells with abundant cytoplasm (“stars”) set against a background of malignant, darkly staining lymphoma cells (“night sky”).
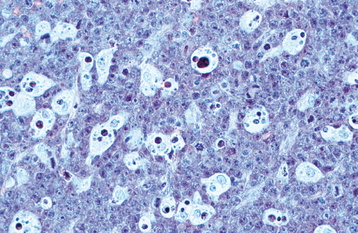
Fig. 13-38 Burkitt’s lymphoma. This high-power photomicrograph demonstrates the undifferentiated, small, dark lesional cells with numerous histiocytes.
Because the histopathologic features of Burkitt’s lymphoma can appear similar to some cases of diffuse large B-cell lymphoma, standard of care now dictates that, in addition to immunohistochemical studies, molecular genetic analysis of the tumor tissue should be performed. This distinction is important because these two malignancies are treated differently. Burkitt’s lymphoma is characterized by one of several specific translocations, the most common being t(8;14), that results in overexpression of the oncogene c-myc, an event that presumably drives the neoplastic proliferation.
TREATMENT AND PROGNOSIS: Burkitt’s lymphoma is an aggressive malignancy that usually results in the death of the patient within 4 to 6 months after diagnosis if it is not treated. Treatment generally consists of an intensive chemotherapeutic regimen, which emphasizes the use of high doses of cyclophosphamide. More than 90% of the patients respond to this treatment.
The prognosis for Burkitt’s lymphoma in the past was poor, with a median survival time of only 10½ months. More recent trials with more intensive, multi-agent chemotherapeutic protocols have shown an 85% to 95% event-free (no evidence of recurrence) survival rate 3 to 5 years after treatment for patients with stage I or II disease. Even for advanced stage (III and IV) Burkitt’s lymphoma, the event-free survival has improved to 75% to 85%.
EXTRANODAL NK/T-CELL LYMPHOMA, NASAL-TYPE (ANGIOCENTRIC T-CELL LYMPHOMA; MIDLINE LETHAL GRANULOMA; IDIOPATHIC MIDLINE DESTRUCTIVE DISEASE; POLYMORPHIC RETICULOSIS; MIDLINE MALIGNANT RETICULOSIS; ANGIOCENTRIC IMMUNOPROLIFERATIVE LESION)
Also known as angiocentric T-cell lymphoma, extranodal NK/T-cell lymphoma, nasal-type is a rare process that is characterized clinically by aggressive, nonrelenting destruction of the midline structures of the palate and nasal fossa. For many decades, the nature of this process has been controversial, a fact that can readily be appreciated by the wide variety of terms by which it has been called. In actuality, many of the cases reported as “midline lethal granuloma” in the past represented a wide variety of immunologic (e.g., Wegener’s granulomatosis) and infectious (e.g., tertiary syphilis) diseases. The term midline lethal granuloma should be used only as a descriptive designation of a destructive midline condition, and thorough diagnostic evaluation, including biopsy and culture, is necessary to make a definitive diagnosis. Once the other causes of midline destruction have been eliminated, the consensus among most investigators is that this disorder should be classified as a natural killer (NK) T-cell lymphoma, based on modern cytogenetic, immunologic, and molecular studies. The difficulty in distinguishing among these destructive disorders can be appreciated by the fact that lymphomatoid granulomatosis, which until recently was considered part of this T-cell lymphoma spectrum, has now been determined to be an Epstein-Barr virus (EBV)-driven proliferation of B lymphocytes.
Even though extranodal NK/T-cell lymphoma often does not have the classic histopathologic features of lymphoma microscopically, it behaves in a malignant fashion and responds to the same treatments to which lymphomas respond. For reasons that are unclear, this condition is seen with greater frequency in Asian, Guatemalan, and Peruvian populations.
CLINICAL FEATURES: Extranodal NK/T-cell lymphoma is typically observed in adults. The initial signs and symptoms are often localized to the nasal region and include nasal stuffiness or epistaxis. Pain may accompany the nasal symptoms. Swelling of the soft palate or posterior hard palate may precede the formation of a deep, necrotic ulceration, which usually occupies a midline position. This ulceration enlarges and destroys the palatal tissues, which typically creates an oronasal fistula (Fig. 13-39). Secondary infection may complicate the course of the disease, and life-threatening hemorrhage is a potential problem in some instances.
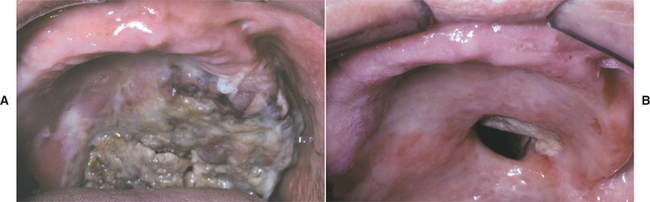
HISTOPATHOLOGIC FEATURES: Histopathologic examination of one of these lesions shows a mixed infiltrate of a variety of inflammatory cells, often arranged around blood vessels (angiocentric) (Fig. 13-40). The lesional process appears to invade and destroy the normal tissue in the area. Necrosis is often present in some areas of the lesion, presumably secondary to infiltration of the blood vessels by the tumor cells. Large, angular, lymphocytic cells with an atypical appearance are usually identified as a component of the cellular infiltrate. Immunohistochemical evaluation of this infiltrate often shows that the large atypical cells mark with antibodies directed against T-lymphocyte antigens. Molecular genetic studies typically show monoclonal gene rearrangements of the T-lymphocyte receptor, consistent with a lymphoreticular malignancy.
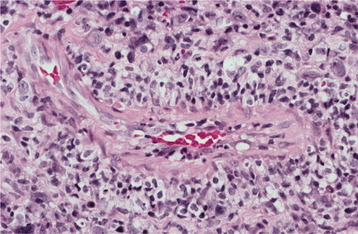
TREATMENT AND PROGNOSIS: Without treatment, extranodal NK/T-cell lymphoma is a relentlessly progressive, highly destructive process that ultimately leads to the patient’s death by secondary infection, massive hemorrhage, or infiltration of vital structures in the area. Lesions that are localized usually respond to radiation therapy, a feature that is similar to that of T-cell lymphomas of other sites. Approximately 4500 cGy is required to control the disease, and many of these patients show no evidence of recurrence or dissemination of the lesion. Five-year sur-vival rates in some series have been reported to be as high as 85%. For patients with more disseminated disease, combination chemotherapy is indicated, and a less favorable prognosis can be expected, with 30% to 50% 5-year survival generally reported.
MULTIPLE MYELOMA
Multiple myeloma is a relatively uncommon malignancy of plasma cell origin that often appears to have a multicentric origin within bone. The cause of the condition is unknown, although sometimes a plasmacytoma (see page 606) may evolve into multiple myeloma. This disease makes up about 1% of all malignancies and 10% to 15% of hematologic malignancies. If metastatic disease is excluded, then multiple myeloma accounts for nearly 50% of all malignancies that involve the bone. Nearly 20,000 cases are diagnosed annually in the United States.
The abnormal plasma cells that compose this tumor are typically monoclonal. The abnormal cells probably arise from a single malignant precursor that has undergone uncontrolled mitotic division and has spread throughout the body. Because the neoplasm develops from a single cell, all the daughter cells that comprise the lesional tissue have the same genetic makeup and produce the same proteins. These proteins are the immunoglobulin components that the plasma cell would normally produce, although in the case of this malignant tumor the immunoglobulins are not normal or functional. The signs and symptoms of this disease result from the uncontrolled proliferation of the tumor cells and the uncontrolled manufacture of their protein products.
CLINICAL AND RADIOGRAPHIC FEATURES: Multiple myeloma is typically a disease of adults, with men being affected slightly more often than women. The median age at diagnosis is between 60 and 70 years, and it is rarely diagnosed before age 40. For reasons that are not understood, the disease occurs twice as frequently in blacks as whites, making this the most common hematologic malignancy among black persons in the United States.
Bone pain, particularly in the lumbar spine, is the most characteristic presenting symptom. Some patients experience pathologic fractures caused by tumor destruction of bone. They may also complain of fatigue as a consequence of myelophthisic anemia. Petechial hemorrhages of the skin and oral mucosa may be seen if platelet production has been affected. Fever may be present as a result of neutropenia with increased susceptibility to infection. Metastatic calcifications may involve the soft tissues and are thought to be caused by hypercalcemia secondary to tumor-related osteolysis.
Radiographically, multiple well-defined, punched-out radiolucencies or ragged radiolucent lesions may be seen in multiple myeloma (Fig. 13-41). These may be especially evident on a skull film. Although any bone may be affected, the jaws have been reported to be involved in as many as 30% of cases. The radiolucent areas of the bone contain the abnormal plasma cell proliferations that characterize multiple myeloma.
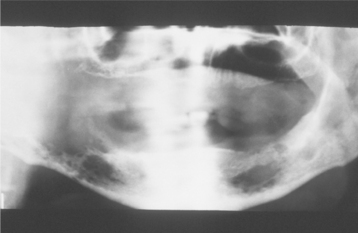
Fig. 13-41 Multiple myeloma. Multiple myeloma affecting the mandible. The disease produced several radiolucencies with ragged, ill-defined margins. (Courtesy of Dr. Joseph Finelli.)
Renal failure may be a presenting sign in these patients because the kidneys become overburdened with the excess circulating light chain proteins of the tumor cells. These light chain products, which are found in the urine of 30% to 50% of patients with multiple myeloma, are called Bence Jones proteins, after the British physician who first described them in detail.
Approximately 15% of patients with multiple myeloma show deposition of amyloid (see page 822) in various soft tissues of the body, and this may be the initial manifestation of the disease. Amyloid deposits are due to the accumulation of the abnormal light chain proteins. Sites that are classically affected include the oral mucosa, particularly the tongue. The tongue may show diffuse enlargement and firmness or may have more of a nodular appearance. Sometimes the nodules are ulcerated. Another area that is commonly affected is the periorbital skin, with the amyloid deposits appearing as waxy, firm, plaquelike lesions (see Fig. 17-7 on page 823).
HISTOPATHOLOGIC FEATURES: Histopathologic examination of the lesional tissue in multiple myeloma shows diffuse, monotonous sheets of neoplastic, variably differentiated, plasmacytoid cells that invade and replace the normal host tissue (Fig. 13-42). Mitotic activity may be seen with some frequency. The monoclonality of the plasma cell population can be demonstrated using antibodies directed against the lambda and kappa light chain components of the immunoglobulin molecule. In a neoplastic proliferation of plasma cells, virtually all of the lesional cells will mark with only one of these antibodies. In contrast, a reactive plasma cell infiltrate will show a mixture of lambda- and kappa-producing plasma cells. Occasionally, deposition of amyloid may be observed in association with the neoplastic cells. Like other types of amyloid, this material appears homogeneous, eosinophilic, and relatively acellular. It stains metachromatically with crystal violet and shows an affinity for Congo red, demonstrating apple-green birefringence on viewing with polarized light. A biopsy specimen of bone marrow from a patient with multiple myeloma should show at least 10% atypical plasma cells making up the marrow cell population.
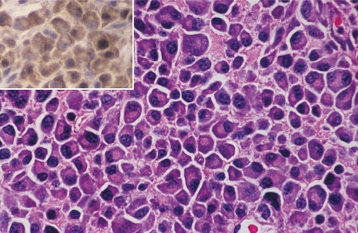
Fig. 13-42 Multiple myeloma. This high-power photomicrograph reveals sheets of malignant plasma cells with eccentric nuclei and stippled nuclear chromatin. Immunohistochemical studies (inset) show a uniform reaction of the lesional cells for antibodies directed against kappa light chains, indicating a monoclonal neoplastic proliferation.
DIAGNOSIS: Although the histopathologic and radiographic findings strongly suggest a diagnosis of multiple myeloma, screening of the serum or urine by protein electro-phoresis should be performed. If an abnormality is detected, then this should be confirmed by protein immunoelectrophoresis, which is a more sensitive test, as an additional parameter to establish the diagnosis. The serum and urine protein immunoelectrophore-sis should show the presence of myeloma protein (M-protein). This represents the massive overproduction of one abnormal immunoglobulin by the neoplastic clone of plasma cells, thus this feature is termed monoclonal gammopathy. This monoclonal protein consists of two heavy chain polypeptides of the same immunoglobulin (Ig) class (IgA, IgG, IgM, IgD, or IgE) and one of two light chain polypeptides of the same class (kappa or lambda). Occasionally, the neoplastic cells produce only the light chain component.
TREATMENT AND PROGNOSIS: The goals of treatment related to multiple myeloma include not only controlling the malignancy but also making the patient comfortable and prolonging the patient’s survival. Initial attempts to control multiple myeloma generally consist of chemotherapy. An alkylating agent, such as melphalan or cyclophosphamide, is often used in conjunction with prednisone, and approximately 60% of patients will respond initially to this regimen. However, virtually all patients eventually experience relapse of their disease. More aggressive chemotherapeutic regimens and bone marrow transplantation, either autologous or allogeneic, may be considered in otherwise healthy patients under the age of 55 to 65 years, but these individuals comprise a minority of multiple myeloma patients. Addition of thalidomide to the treatment protocol may improve survival; however, ongoing studies are evaluating the effect of this drug. Radiation therapy is useful only as palliative treatment for painful bone lesions. Any one of several bisphosphonate medications (clodronate, pamidronate, or zoledronic acid) can be prescribed to reduce the possibility of myeloma-related fracture with its attendant pain, but these medications do not appear to increase survival. A small percentage of these patients may experience the complication of bisphosphonate-related osteonecrosis of the jaws (see page 299).
The prognosis is considered poor, but younger patients tend to fare better than older ones. Pretreatment serologic studies examining the levels of b2-microglobulin and albumin should be performed. With lower levels of b2-microglobulin (<3.5 mg/L) and higher levels of albumin (>35 g/L), the median survival is approximately 5 years. In contrast, when serum b2-microglobulin levels are >5.5 mg/L, the 5-year survival rate falls to about 2½ years. A median survival time of about 3 to 4 years can be expected after the onset of symptoms. In the past, a 10% 5-year survival rate was typical; the prognosis today has improved only slightly. Most hematology and oncology centers report a 5-year survival rate of 25%. With aggressive chemotherapy and bone marrow transplantation, the 5-year survival rate may be improved to as high as 50%; however, only a small percentage of multiple myeloma patients can reasonably tolerate this treatment. This approach seems to hold the most promise for control of this aggressive disease, however, and some centers have indicated that as many as 20% of their patients may survive for longer than 10 years.
PLASMACYTOMA
The plasmacytoma is a unifocal, monoclonal, neoplastic proliferation of plasma cells that usually arises within bone. Infrequently, it is seen in soft tissue, in which case, the term extramedullary plasmacytoma is used. Some investigators believe that this lesion represents the least aggressive part of a spectrum of plasma cell neoplasms that extends to multiple myeloma. Therefore, the plasmacytoma is important because it may ultimately give rise to the more serious problem of multiple myeloma.
CLINICAL AND RADIOGRAPHIC FEATURES: The plasmacytoma usually is detected in an adult male, with an average age at diagnosis of 55 years. The male-to-female ratio is 3:1. Most of the lesions present centrally within a single bone, and the spine is the most commonly involved site. About one third of the cases are reported in that location. The initial symptoms often relate to swelling or bone pain; occasionally, however, this lesion is detected on routine radiogra-phic examination. The extramedullary plasmacytoma appears as a relatively nondescript, well-circumscribed, nontender soft tissue mass. An even stronger male predilection is seen with this lesion, approaching a 6:1 male-to-female ratio. Approximately 80% to 90% of extramedullary plasmacytomas develop in the head and neck region, and such lesions have been reported in the tonsils, the nasopharynx, the paranasal sinuses, the nose, and the parotid gland.
Radiographically, the lesion may be seen as a well-defined, unilocular radiolucency with no evidence of sclerotic borders or as a ragged radiolucency similar to the appearance of multiple myeloma (Fig. 13-43). No other lesions should be identifiable by a skeletal survey or careful physical examination, however.
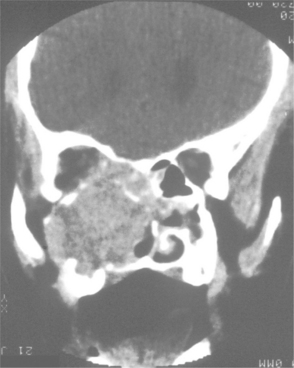
HISTOPATHOLOGIC FEATURES: The histopathologic features of the plasmacytoma are identical to those of multiple myeloma. Sheets of plasma cells show varying degrees of differentiation. Immunohistochemical studies demonstrate that these plasma cells are monoclonal. As many as 25% to 50% of these patients also show a monoclonal gammopathy on evaluation by serum protein immunoelectrophoresis, although the amount of abnormal protein is much less than that seen with multiple myeloma. Solitary plasmacytoma also differs from multiple myeloma in that no evidence of plasma cell infiltration should be seen by a random bone marrow biopsy, and the patient should not show signs of anemia, hypercalcemia, or renal failure. Immunohistochemically, extramedullary plasmacytoma appears to differ from its intrabony counterparts in that it shows a marked decrease or lack of immunoreactivity for antibodies directed against cyclin D1 and CD56.
TREATMENT AND PROGNOSIS: Plasmacytomas are usually treated with radiation therapy, and typically a dose of at least 4000 cGy is delivered to the tumor site. A few lesions have been surgically excised with good results, although this is not the preferred treatment in most instances. Unfortunately, when patients with plasmacytoma of bone are observed on a long-term basis, most will eventually develop multiple myeloma. About 50% show evidence of disseminated disease within 2 to 3 years. However, one third of these patients will not have symptoms of multiple myeloma for as long as 10 years. Extramedullary plasmacytoma seems to have a much better prognosis, with only 30% of these patients showing progression to multiple myeloma and 70% having a 10-year disease-free period after treatment.
BIBLIOGRAPHY
Bradley, G, Main, JHP, Birt, BD, et al. Benign lymphoid hyperplasia of the palate. J Oral Pathol. 1987;16:18–26.
Harsany, DL, Ross, J, Fee, WE. Follicular lymphoid hyperplasia of the hard palate simulating lymphoma. Otolaryngol Head Neck Surg. 1980;88:349–356.
Kolokotronis, A, Dimitrakopoulos, I, Asimaki, A. Follicular lymphoid hyperplasia of the palate: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96:172–175.
Menasce, LP, Shanks, JH, Banerjee, SS, et al. Follicular lymphoid hyperplasia of the hard palate and oral mucosa: report of three cases and a review of the literature. Histopathology. 2001;39:353–358.
Napier, SS, Newlands, C. Benign lymphoid hyperplasia of the palate: report of two cases and immunohistochemical profile. J Oral Pathol Med. 1990;19:221–225.
Wright, J, Dunsworth, A. Follicular lymphoid hyperplasia of the hard palate: a benign lymphoproliferative process. Oral Surg Oral Med Oral Pathol. 1983;55:162–168.
Blanco-Carrion, J, Liñares-Gonzalez, A, Batalla-Vazquez, P, et al. Morbidity and economic complications following mucogingival surgery in a hemophiliac HIV-infected patient: a case report. J Periodontol. 2004;75:1413–1416.
Cahill, MR, Colvin, BT. Haemophilia. Postgrad Med J. 1997;73:201–206.
Dunn, AL, Abshire, TC. Recent advances in the management of the child who has hemophilia. Hematol Oncol Clin North Am. 2004;18:1249–1276.
Federici, AB. Clinical diagnosis of von Willebrand disease. Haemophilia. 2004;10(suppl 4):169–176.
Federici, AB, Berntorp, E, Lee, CA. The 80th anniversary of von Willebrand’s disease: history, management and research. Haemophilia. 2006;12:563–572.
Heiland, M, Weber, M, Schmelzle, R. Life-threatening bleeding after dental extraction in a hemophilia A patient with inhibitors to factor VIII: a case report. J Oral Maxillofac Surg. 2003;61:1350–1353.
Izumi, Y, Taniguchi, T, Maruyama, Y, et al. Effective periodontal treatment in a patient with type IIA von Willebrand’s disease: report of a case. J Periodontol. 1999;70:548–553.
Kitchens, CS. Approach to the bleeding patient. Hematol Oncol Clin North Am. 1992;6:983–989.
Laguna, P, Klukowska, A. Management of oral bleedings with recombinant factor VIIa in children with haemophilia A and inhibitor. Haemophilia. 2005;11:2–4.
Lee, CA, Lillicrap, D, Astermark, J. Inhibitor development in hemophiliacs: the roles of genetic versus environmental factors. Semin Thromb Hemost. 2006;32(suppl 2):10–14.
Lofqvist, T, Nilsson, IM, Berntorp, E, et al. Haemophilia prophylaxis in young patients—a long-term follow-up. J Intern Med. 1997;241:395–400.
Mannucci, PM. Treatment of von Willebrand’s disease. N Engl J Med. 2004;351:683–694.
Morimoto, Y, Yoshioka, A, Shima, M, et al. Intraoral hemostasis using a recombinant activated factor VII preparation in a hemophilia A patient with inhibitor. J Oral Maxillofac Surg. 2003;61:1095–1097.
Pruthi, RK. Hemophilia: a practical approach to genetic testing. Mayo Clin Proc. 2005;80:1485–1499.
Sutor, AH. Desmopressin (DDAVP) in bleeding disorders of childhood. Semin Thromb Hemost. 1998;24:555–566.
Drew, AF, Kaufman, AH, Kombrinck, KW, et al. Ligneous conjunctivitis in plasminogen-deficient mice. Blood. 1998;91:1616–1624.
Gokbuget, AY, Mutlu, S, Scully, C, et al. Amyloidaceous ulcer-ated gingival hyperplasia: a newly described entity related to ligneous conjunctivitis. J Oral Pathol Med. 1997;26:100–104.
Schott, D, Dempfle, C-E, Beck, P, et al. Therapy with a purified plasminogen concentrate in an infant with ligneous conjunctivitis and homozygous plasminogen deficiency. N Engl J Med. 1998;339:1679–1686.
Schuster, V, Seregard, S. Ligneous conjunctivitis. Surv Ophthalmol. 2003;48:369–388.
Scully, C, Gokbuget, AY, Allen, C, et al. Oral lesions indicative of plasminogen deficiency (hypoplasminogenemia). Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001;91:334–337.
Tefs, K, Gueorguieva, M, Klammt, J, et al. Molecular and clinical spectrum of type I plasminogen deficiency: a series of 50 patients. Blood. 2006;108:3021–3026.
Watts, P, Suresh, P, Mezer, E, et al. Effective treatment of ligneous conjunctivitis with topical plasminogen. Am J Ophthalmol. 2002;133:451–455.
Beddall, A. Anaemias. Practitioner. 1990;234:713–715.
Brown, RG. Determining the cause of anemia: general approach, with emphasis on microcytic hypochromic anemias. Postgrad Med. 1991;89:161–170.
Groopman, JE, Itri, LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91:1616–1634.
Hoggarth, K. Macrocytic anaemias. Practitioner. 1993;237:331–335.
Hoggarth, K. Microcytic anaemias. Practitioner. 1993;237:338–341.
Means, RT. Advances in the anemia of chronic disease. Int J Hematol. 1999;70:7–12.
Provan, D. Mechanisms and management of iron deficiency anaemia. Br J Haematol. 1999;105(suppl 1):19–26.
Remuzzi, G, Ingelfinger, JR. Correction of anemia—payoffs and problems. N Engl J Med. 2006;355:2144–2146.
Tefferi, A. Anemia in adults: a contemporary approach to diagnosis. Mayo Clin Proc. 2003;78:1274–1280.
Ashley-Koch, A, Yang, Q, Olney, RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a human genome epidemiology review. Am J Epidemiol. 2000;151:839–845.
Brosco, JP, Seider, MI, Dunn, AC. Universal newborn screening and adverse medical outcomes: a historical note. Ment Retard Dev Disabil Res Rev. 2006;12:262–269.
Crowley, JJ, Sarnaik, S. Imaging of sickle cell disease. Pediatr Radiol. 1999;29:646–661.
Kelleher, M, Bishop, K, Briggs, P. Oral complications associated with sickle cell anemia. A review and case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;82:225–228.
Mehta, SR, Afenyi-Annan, A, Byrns, PJ, et al. Opportunities to improve outcomes in sickle cell disease. Am Fam Physician. 2006;74:303–310. 313-314
Okpala, IE. New therapies for sickle cell disease. Hematol Oncol Clin North Am. 2005;19:975–987.
Steinberg, MH. Management of sickle cell disease. N Engl J Med. 1999;340:1021–1030.
Stuart, MJ, Nagel, RL. Sickle-cell disease. Lancet. 2004;364:1343–1360.
Vichinsky, EP, Neumayr, LD, Earles, AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med. 2000;342:1855–1865.
Xu, K, Shi, ZM, Veeck, LL, et al. First unaffected pregnancy using preimplantation genetic diagnosis for sickle cell anemia. JAMA. 1999;281:1701–1706.
Cannell, H. The development of oral and facial signs in beta-thalassaemia major. Br Dent J. 1988;164:50–51.
Clarke, GM, Higgins, TN. Laboratory investigation of hemoglobinopathies and thalassemias: review and update. Clin Chem. 2000;46:1284–1290.
Drew, SJ, Sachs, SA. Management of the thalassemia-induced skeletal facial deformity. J Oral Maxillofac Surg. 1997;55:1331–1339.
Hazza’a, AM, Al-Jamal, G. Dental development in subjects with thalassemia major. J Contemp Dent Pract. 2006;7:63–70.
Hazza’a, AM, Al-Jamal, G. Radiographic features of the jaws and teeth in thalassemia major. Dentomaxillofac Radiol. 2006;35:283–288.
Joshi, D-D, Nickerson, HJ, McManus, MJ. Hydrops fetalis caused by homozygous α-thalassemia and Rh antigen alloimmunization: report of a survivor and literature review. Clin Med Res. 2004;2:228–232.
Olivieri, NF. The beta-thalassemias. N Engl J Med. 1999;341:99–109.
Richer, J, Chudley, AE. The hemoglobinopathies and malaria. Clin Genet. 2005;68:332–336.
Riggs, DR. The thalassemia syndromes. Q Rev Med. 1993;86:559–564.
Rund, D, Rachmilewitz, E. β-thalassemia. N Engl J Med. 2005;353:1135–1146.
Schrier, SL, Angelucci, E. New strategies in the treatment of the thalassemias. Annu Rev Med. 2005;56:157–171.
Tyler, PA, Madani, G, Chaudhuri, R, et al. The radiological appearances of thalassaemia. Clin Radiol. 2006;61:40–52.
Bacigalupo, A, Brand, R, Oneto, R, et al. Treatment of acquired severe aplastic anemia: bone marrow transplantation compared with immunosuppressive therapy—the European Group for Blood and Marrow Transplantation experience. Semin Hematol. 2000;37:69–80.
Ball, SE. The modern management of severe aplastic anaemia. Br J Haematol. 2000;110:41–53.
Brodsky, RA, Jones, RJ. Aplastic anemia. Lancet. 2005;365:1647–1656.
Fonseca, R, Tefferi, A. Practical aspects in the diagnosis and management of aplastic anemia. Am J Med Sci. 1997;313:159–169.
Horowitz, MM. Current status of allogeneic bone marrow transplantation in acquired aplastic anemia. Semin Hematol. 2000;37:30–42.
Keohane, EM. Acquired aplastic anemia. Clin Lab Sci. 2004;17:165–171.
Luker, J, Scully, C, Oakhill, A. Gingival swelling as a manifestation of aplastic anemia. Oral Surg Oral Med Oral Pathol. 1991;71:55–56.
Marsh, JCW. Hematopoietic growth factors in the pathogenesis and for the treatment of aplastic anemia. Semin Hematol. 2000;37:81–90.
Schrezenmeier, H, Raghavachar, A, Heimpel, H. Granulocyte-macrophage colony-stimulating factor in the sera of patients with aplastic anemia. Clin Investig. 1993;71:102–108.
Sepúlveda, E, Brethauer, U, Rojas, J, et al. Oral manifestations of aplastic anemia in children. J Am Dent Assoc. 2006;137:474–478.
Socie, G, Henry-Amar, M, Bacigalupo, A, et al. Malignant tumors occurring after treatment of aplastic anemia. N Engl J Med. 1993;329:1152–1157.
Young, NS. Acquired aplastic anemia. Ann Intern Med. 2002;136:534–546.
Young, NS, Calado, RT, Scheinberg, P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–2519.
Alexander, SW, Pizzo, PA. Current considerations in the management of fever and neutropenia. Curr Clin Top Infect Dis. 1999;19:160–180.
Ancliff, PJ. Congenital neutropenia. Blood Rev. 2003;17:209–216.
Boxer, LA, Hutchinson, R, Emerson, S. Recombinant human granulocyte-colony-stimulating factor in the treatment of patients with neutropenia. Clin Immunol Immunopathol. 1992;62:S39–S46.
Dale, DC. Immune and idiopathic neutropenia. Curr Opin Hematol. 1998;5:33–36.
Haddy, TB, Rana, SR, Castro, O. Benign ethnic neutropenia: what is a normal absolute neutrophil count? J Lab Clin Med. 1999;133:15–22.
Hakki, SS, Aprikyan, AAG, Yildirim, S, et al. Periodontal status in two siblings with severe congenital neutropenia: diagnosis and mutational analysis of the cases. J Periodontol. 2005;76:837–844.
James, RM, Kinsey, SE. The investigation and management of chronic neutropenia in children. Arch Dis Child. 2006;91:852–858.
Vandendries, ER, Drews, RE. Drug-associated disease: hematologic dysfunction. Crit Care Clin. 2006;22:347–355.
Welte, K, Zeidler, C, Dale, DC. Severe congenital neutropenia. Semin Hematol. 2006;43:189–195.
Zaromb, A, Chamberlain, D, Schoor, R, et al. Periodontitis as a manifestation of chronic benign neutropenia. J Periodontol. 2006;77:1921–1926.
Bergman, OJ. Oral infections in haematological patients. Dan Med Bull. 1992;39:15–29.
Carey, PJ. Drug-induced myelosuppression: diagnosis and management. Drug Safety. 2003;26:691–706.
Deas, DE, Mackey, SA, McDonnell, HT. Systemic disease and periodontitis: manifestations of neutrophil dysfunction. Periodontol 2000. 2003;32:82–104.
Glasser, L, Duncan, BR, Corrigan, JJ. Measurement of serum granulocyte-colony-stimulating factor in a patient with congenital agranulocytosis (Kostmann’s syndrome). Am J Dis Child. 1991;145:925–928.
Kuipers, EJ, Vellenga, E, de Wolf, JTM, et al. Sulfasalazine-induced agranulocytosis treated with granulocyte-macrophage colony stimulating factor. J Rheumatol. 1992;19:621–622.
Pisciotta, AV. Drug-induced agranulocytosis: peripheral destruction of polymorphonuclear leukocytes and their marrow precursors. Blood Rev. 1990;4:226–237.
Salama, A, Mueller-Eckhardt, C. Immune-mediated blood cell dyscrasias related to drugs. Semin Hematol. 1992;29:54–63.
Aprikyan, AAG, Liles, WC, Boxer, LA, et al. Mutant elastase in pathogenesis of cyclic and severe congenital neutropenia. J Pediatr Hematol Oncol. 2002;24:784–786.
Baer, PN, Iacono, VJ. Cyclic neutropenia: report of a case with a 15-year follow-up. Periodontal Clin Investig. 1994;16:14–19.
Boxer, LA, Hutchinson, R, Emerson, S. Recombinant human granulocyte-colony-stimulating factor in the treatment of patients with neutropenia. Clin Immunol Immunopathol. 1992;62:S39–S46.
Dale, DC, Bolyard, AA, Aprikyan, A. Cyclic neutropenia. Semin Hematol. 2002;39:89–94.
Haurie, C, Dale, DC, Mackey, MC. Cyclical neutropenia and other periodic hematological disorders: a review of mechanisms and mathematical models. Blood. 1998;92:2629–2640.
Kinane, D. Blood and lymphoreticular disorders. Periodontol 2000. 1999;21:84–93.
Nakai, Y, Ishihara, C, Ogata, S, et al. Oral manifestations of cyclic neutropenia in a Japanese child: case report with a 5-year follow-up. Pediatr Dent. 2003;25:383–388.
Pernu, HE, Pajari, UH, Lanning, M. The importance of regular dental treatment in patients with cyclic neutropenia. Follow-up of 2 cases. J Periodontol. 1996;67:454–459.
Bolton-Maggs, PHB. Idiopathic thrombocytopenic purpura. Arch Dis Child. 2000;83:220–222.
Chong, BH. Clinical aspects of thrombocytopenias. Aust Fam Physician. 1994;23:1463–1464. [1468-1470, 1473].
George, JN. Thrombotic thrombocytopenic purpura. N Engl J Med. 2006;354:1927–1935.
Lusher, JM. Screening and diagnosis of coagulation disorders. Am J Obstet Gynecol. 1996;175:778–783.
Medeiros, D, Buchanan, GR. Idiopathic thrombocytopenic purpura: beyond consensus. Curr Opin Pediatr. 2000;12:4–9.
Murrin, RJA, Murray, JA. Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev. 2006;20:51–60.
Rogers, GM. Overview of platelet physiology and laboratory evaluation of platelet function. Clin Obstet Gynecol. 1999;42:349–359.
Thompson, CC, Tacke, RB, Woolley, LH, et al. Purpuric oral and cutaneous lesions in a case of drug-induced thrombocytopenia. J Am Dent Assoc. 1982;105:465–467.
Wazny, LD, Ariano, RE. Evaluation and management of drug-induced thrombocytopenia in the acutely ill patient. Pharmacotherapy. 2000;20:292–307.
Campbell, PJ, Green, AR. The myeloproliferative disorders. N Engl J Med. 2006;355:2452–2466.
Cao, M, Olsen, RJ, Zu, Y. Polycythemia vera: new clinicopatho-logic perspectives. Arch Pathol Lab Med. 2006;130:1126–1132.
Johansson, P. Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. 2006;32:171–173.
Lengfelder, E, Merx, K, Hehlmann, R. Diagnosis and therapy of polycythemia vera. Semin Thromb Hemost. 2006;32:267–275.
Silver, RT. Treatment of polycythemia vera. Semin Thromb Hemost. 2006;32:437–442.
Tefferi, A, Spivak, JL. Polycythemia vera: scientific advances and current practice. Semin Hematol. 2005;42:206–220.
Abbott, BL. Recent advances in chronic lymphocytic leukemia. Cancer Invest. 2006;24:302–309.
Amin, KS, Ehsan, A, McGuff, HS, et al. Minimally differentiated acute myelogenous leukemia (AML-M0) granulocytic sarcoma presenting in the oral cavity. Oral Oncol. 2002;38:516–519.
Barrett, AP. Gingival lesions in leukemia: a classification. J Periodontol. 1984;55:585–588.
Bhatia, S, Robison, LL. Epidemiology of leukemia and lymphoma. Curr Opin Hematol. 1999;6:201–204.
Chapple, ILC, Saxby, MS, Murray, JA. Gingival hemorrhage, myelodysplastic syndromes, and acute myeloid leukemia. A case report. J Periodontol. 1999;70:1247–1253.
Estey, E, Döhner, H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907.
Faderl, S, Talpaz, M, Estrov, Z, et al. Chronic myelogenous leukemia: biology and therapy. Ann Intern Med. 1999;131:207–219.
Farhi, DC, Rosenthal, NS. Acute lymphoblastic leukemia. Clin Lab Med. 2000;20:17–28.
Heaney, ML, Golde, DW. Myelodysplasia. N Engl J Med. 1999;340:1649–1660.
Hollsberg, P, Hailer, DA. Pathogenesis of diseases induced by human lymphotropic virus type I infection. N Engl J Med. 1993;328:1173–1182.
Keating, MJ. Chronic lymphocytic leukemia. Semin Oncol. 1999;26(suppl 14):107–114.
Krause, JR. Morphology and classification of acute myeloid leukemias. Clin Lab Med. 2000;20:1–16.
Larson, RA. Management of acute lymphoblastic leukemia in older patients. Semin Hematol. 2006;43:126–133.
Lowenberg, B, Downing, JR, Burnett, A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062.
Menasce, LP, Banerjee, SS, Beckett, E, et al. Extra-medullary myeloid tumour (granulocytic sarcoma) is often misdiagnosed: a study of 26 cases. Histopathology. 1999;34:391–398.
Peterson, DE, Gerad, H, Williams, LT. An unusual instance of leukemic infiltrate: diagnosis and management of periapical tooth involvement. Cancer. 1983;51:1716–1719.
Pui, C-H, Evans, WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–178.
Quintás-Cardama, A, Cortes, JE. Chronic myeloid leukemia: diagnosis and treatment. Mayo Clin Proc. 2006;81:973–988.
Rhee, D, Myssiorek, D, Zahtz, G, et al. Recurrent attacks of facial nerve palsy as the presenting sign of leukemic relapse. Laryngoscope. 2002;112:235–237.
Shanafelt, TD, Byrd, JC, Call, TG, et al. Narrative review: initial management of newly diagnosed, early-stage chronic lymphocytic leukemia. Ann Intern Med. 2006;145:435–447.
Sollecito, TP, Draznin, J, Parisi, E, et al. Leukemic gingival infiltrate as an indicator of chemotherapeutic failure following monoclonal antibody therapy: a case report. Spec Care Dentist. 2003;23:108–110.
Stoopler, ET, Pinto, A, Alawi, F, et al. Granulocytic sarcoma: an atypical presentation in the oral cavity. Spec Care Dentist. 2004;24:65–69.
Vibhute, P, Carneiro, E, Genden, E, et al. Palatal enlargement in chronic lymphocytic leukemia. AJNR Am J Neuroradiol. 2006;27:1649–1650.
Vural, F, Ozcan, MA, Ozsan, GH, et al. Gingival involvement in a patient with CD56+ chronic myelomonocytic leukemia. Leuk Lymphoma. 2004;45:415–418.
Weirda, WG, Kipps, TJ. Chronic lymphocytic leukemia. Curr Opin Hematol. 1999;6:253–261.
Yee, KWL, O’Brien, SM. Chronic lymphocytic leukemia: diagnosis and treatment. Mayo Clin Proc. 2006;81:1105–1129.
Azouz, EM, Saigal, G, Rodriguez, MM, et al. Langerhans’ cell histiocytosis: pathology, imaging and treatment of skeletal involvement. Pediatr Radiol. 2005;35:103–115.
Bartnick, A, Friedrich, RE, Roeser, K, et al. Oral Langerhans cell histiocytosis. J Craniomaxillofac Surg. 2002;30:91–96.
Cleveland, DB, Goldberg, KM, Greenspan, JS, et al. Langerhans’ cell histiocytosis. Report of three cases with unusual oral soft tissue involvement. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;82:541–548.
Dagenais, M, Pharoah, MJ, Sikorski, PA. The radiographic characteristics of histiocytosis X: a study of 29 cases that involve the jaws. Oral Surg Oral Med Oral Pathol. 1992;74:230–236.
Eckardt, A, Schultze, A. Maxillofacial manifestations of Langerhans cell histiocytosis: a clinical and therapeutic analysis of 10 patients. Oral Oncol. 2003;39:687–694.
Hartman, KH. A review of 114 cases of histiocytosis X. Oral Surg Oral Med Oral Pathol. 1980;49:38–54.
Hicks, J, Flaitz, CM. Langerhans cell histiocytosis: current insights in a molecular age with emphasis on clinical oral and maxillofacial pathology practice. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:S42–S66.
Howarth, DM, Gilchrist, GS, Mullan, BP, et al. Langerhans cell histiocytosis: diagnosis, natural history, management, and outcome. Cancer. 1999;85:2278–2290.
Key, SJ, O’Brien, CJ, Silvester, KC, et al. Eosinophilic granuloma: resolution of maxillofacial bony lesions following minimal intervention. Report of three cases and a review of the literature. J Craniomaxillofac Surg. 2004;32:170–175.
Manfredi, M, Corradi, D, Vescovi, P. Langerhans-cell histiocytosis: a clinical case without bone involvement. J Periodontol. 2005;76:143–147.
Mitomi, T, Tomizawa, Noda, T. Tooth development included in the multifocal jaw lesions of Langerhans cell histiocytosis. Int J Paediatr Dent. 2005;15:123–126.
Muzzi, L, Prato, GPP, Ficarra, G. Langerhans’ cell histiocytosis diagnosed through periodontal lesions: a case report. J Periodontol. 2002;73:1528–1533.
Putters, TF, de Visscher, JGAM, van Veen, A, et al. Intralesional infiltration of corticosteroids in the treatment of localized Langerhans’ cell histiocytosis of the mandible. Report of known cases and three new cases. Int J Oral Maxillofac Surg. 2005;34:571–575.
Scolozzi, P, Lombardi, T, Monnier, P, et al. Multisystem Langerhans’ cell histiocytosis (Hand-Schüller-Christian disease) in an adult: a case report and review of the literature. Eur Arch Otorhinolaryngol. 2004;261:326–330.
Yousem, SA, Colby, TV, Chen, Y-Y, et al. Pulmonary Langerhans’ cell histiocytosis. Molecular analysis of clonality. Am J Surg Pathol. 2001;25:630–636.
Advani, RH, Horning, SJ. Treatment of early-stage Hodgkin’s disease. Semin Hematol. 1999;36:270–281.
Aisenberg, AC. Problems in Hodgkin’s disease management. Blood. 1999;93:761–779.
Ansell, SM, Armitage, JO. Management of Hodgkin lymphoma. Mayo Clin Proc. 2006;81:419–426.
Connors, JM. State-of-the-art therapeutics: Hodgkin’s lymphoma. J Clin Oncol. 2005;23:6400–6408.
Diehl, V, Josting, A. Hodgkin’s disease. Cancer J Sci Am. 2000;6(suppl 2):S150–S158.
Harris, NL. Hodgkin’s disease: classification and differential diagnosis. Mod Pathol. 1999;12:159–176.
Herrin, HK. The oral implications of Hodgkin’s disease. Gen Dent. 1999;47:572–575.
Jaffe, ES. Introduction: Hodgkin’s lymphoma—pathology, pathogenesis, and treatment. Semin Hematol. 1999;36:217–219.
Küppers, R, Hansmann, M-L. The Hodgkin and Reed/Sternberg cell. Int J Biochem Cell Biol. 2005;37:511–517.
Küppers, R, Yahalom, J, Josting, A. Advances in biology, diagnostics, and treatment of Hodgkin’s disease. Biol Blood Marrow Transplant. 2006;12:66–76.
Re, D, Küppers, R, Diehl, V. Molecular pathogenesis of Hodgkin’s lymphoma. J Clin Oncol. 2005;23:6379–6386.
Yencha, MW. Primary parotid gland Hodgkin’s lymphoma. Ann Otol Rhinol Laryngol. 2002;111:338–342.
Ando, M, Matsuzaki, M, Murofushi, T. Mucosa-associated lymphoid tissue lymphoma presented as diffuse swelling of the parotid gland. Am J Otolaryngol. 2005;26:285–288.
Armitage, JO. Staging non-Hodgkin lymphoma. CA Cancer J Clin. 2005;55:368–376.
Chan, JKC, Banks, PM, Cleary, ML, et al. A revised European-American classification of lymphoid neoplasms proposed by the International Lymphoma Study Group. Am J Clin Pathol. 1995;103:543–560.
Chang, C-C, Rowe, JJ, Hawkins, P, et al. Mantle cell lymphoma of the hard palate: a case report and review of the differential diagnosis based on the histomorphology and immunophenotyping pattern. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96:316–320.
Cheson, BD. What is new in lymphoma? CA Cancer J Clin. 2004;54:260–272.
Evans, LS, Hancock, BW. Non-Hodgkin lymphoma. Lancet. 2003;362:139–146.
Folk, GS, Abbondanzo, SL, Childers, EL, et al. Plasmablastic lymphoma: a clinicopathologic correlation. Ann Diagn Pathol. 2006;10:8–12.
Hennessy, BT, Hanrahan, EO, Daly, PA. Non-Hodgkin lymphoma: an update. Lancet Oncol. 2004;5:341–353.
Isaacson, PG, Du, M-Q. MALT lymphoma: from morphology to molecules. Nature Rev Cancer. 2004;4:644–653.
Kolokotronis, A, Konstantinou, N, Christakis, I, et al. Localized B-cell non-Hodgkin’s lymphoma of the oral cavity and maxillofacial region: a clinical study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:303–310.
Krause, JR, Shahidi-Asl, M. Molecular pathology in the diagnosis and treatment of non-Hodgkin’s lymphomas. J Cell Mol Med. 2003;7:494–512.
Knowles, DM. Immunodeficiency-associated lymphoproliferative disorders. Mod Pathol. 1999;12:200–217.
Lu, P. Staging and classification of lymphoma. Semin Nucl Med. 2005;35:160–165.
Mealey, BL, Tunder, GS, Pemble, CW. Primary extranodal malignant lymphoma affecting the periodontium. J Periodontol. 2002;73:937–941.
Mishima, K, Matsuoka, H, Yamada, E, et al. Application of the polymerase chain reaction for the diagnosis of malignant lymphoma of the nasal and oral cavities. J Oral Pathol Med. 1998;27:43–47.
Mounter, PJ, Lennard, AL. Management of non-Hodgkin’s lymphomas. Postgrad Med J. 1999;75:2–6.
Ostrowski, ML, Unni, KK, Banks, PM, et al. Malignant lymphoma of bone. Cancer. 1986;58:2646–2655.
Rizvi, MA, Evens, AM, Tallman, MS, et al. T-cell non-Hodgkin lymphoma. Blood. 2006;107:1255–1264.
Sandlund, JT, Downing, JR, Crist, WM. Non-Hodgkin’s lymphoma in childhood. N Engl J Med. 1996;334:1238–1248.
Shine, NP, Blake, SP, O’Leary, G. Extranodal lymphoma: clinical presentation and diagnostic pitfalls. Hosp Med. 2005;66:341–345.
Solomides, CC, Miller, AS, Christman, RA, et al. Lymphomas of the oral cavity: histology, immunologic type, and incidence of Epstein-Barr virus infection. Hum Pathol. 2002;33:153–157.
Tomich, CE, Shafer, WG. Lymphoproliferative disease of the hard palate: a clinicopathologic entity. Oral Surg Oral Med Oral Pathol. 1975;39:754–768.
Van der Waal, RIF, Huijgens, PC, van der Valk, P, et al. Characteristics of 40 primary extranodal non-Hodgkin lymphomas of the oral cavity in perspective of the new WHO classification and the International Prognostic Index. Int J Oral Maxillofac Surg. 2005;34:391–395.
Vega, F, Lin, P, Medeiros, J. Extranodal lymphomas of the head and neck. Ann Diagn Pathol. 2005;9:340–350.
Abel, EA, Wood, GS, Hoppe, RT. Mycosis fungoides: clinical and histologic features, staging evaluation, and approach to treatment. CA Cancer J Clin. 1993;43:93–115.
Berti, E, Tomasini, D, Vermeer, MH, et al. Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas: a distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155:483–492.
Chua, MS-T, Veness, MJ. Mycosis fungoides involving the oral cavity. Australas Radiol. 2002;46:336–339.
Damm, DD, White, DK, Cibull, ML, et al. Mycosis fungoides: initial diagnosis via palatal biopsy with discussion of diagnostic advantages of plastic embedding. Oral Surg Oral Med Oral Pathol. 1984;58:413–419.
Hata, T, Aikoh, T, Hirokawa, M, et al. Mycosis fungoides with involvement of the oral mucosa. Int J Oral Maxillofac Surg. 1998;27:127–128.
Kerl, H, Cerroni, L. Primary cutaneous B-cell lymphomas: then and now. J Cutan Pathol. 2006;33(suppl 1):1–5.
Kim, EJ, Hess, S, Richardson, SK, et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005;115:798–812.
Kim, YH, Hoppe, RT. Mycosis fungoides and the Sézary syndrome. Semin Oncol. 1999;26:276–289.
Scarisbrick, JJ. Staging and management of cutaneous T-cell lymphoma. Clin Exp Dermatol. 2006;31:181–186.
Sirois, DA, Miller, AS, Harwick, RD, et al. Oral manifestations of cutaneous T-cell lymphoma: a report of eight cases. Oral Surg Oral Med Oral Pathol. 1993;75:700–705.
Wain, EM, Setterfield, J, Judge, MR, et al. Mycosis fungoides involving the oral mucosa in a child. Clin Exp Dermatol. 2003;28:499–501.
Wright, JM, Balciunas, BA, Muus, JH. Mycosis fungoides with oral manifestations: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol. 1981;51:24–31.
Zackheim, HS. Cutaneous T cell lymphoma: update of treatment. Dermatology. 1999;199:102–105.
Zic, J, Arzubiaga, C, Salhaney, KE, et al. Extracorporeal photopheresis for the treatment of cutaneous T-cell lymphoma. J Am Acad Dermatol. 1992;27:729–736.
Cairo, MS, Sposto, R, Perkins, SL, et al. Burkitt’s and Burkitt-like lymphoma in children and adolescents: review of the Children’s Cancer Group experience. Br J Haematol. 2003;120:660–670.
Dave, SS, Fu, K, Wright, GW, et al. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med. 2006;354:2431–2442.
Ferry, JA. Burkitt’s lymphoma: clinicopathologic features and differential diagnosis. Oncologist. 2006;11:375–383.
Hummel, M, Bentink, S, Berger, H, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354:2419–2430.
Sandlund, JT, Downing, JR, Crist, WM. Non-Hodgkin’s lymphoma in childhood. N Engl J Med. 1996;334:1238–1248.
Tsui, SHC, Wong, MH, Lam, WY. Burkitt’s lymphoma presenting as mandibular swelling—report of a case and review of publications. Br J Oral Maxillofac Surg. 2000;38:8–11.
U ar, DA, Bozkaya, S, Karaca, I, et al. Childhood craniofacial Burkitt’s lymphoma presenting as maxillary swelling: report of a case and review of literature. J Dent Child. 2006;73:45–50.
ar, DA, Bozkaya, S, Karaca, I, et al. Childhood craniofacial Burkitt’s lymphoma presenting as maxillary swelling: report of a case and review of literature. J Dent Child. 2006;73:45–50.
Extranodal NK/T-Cell Lymphoma, Nasal Type
Al-Hakeem, DA, Fedele, S, Carlos, R, et al. Extranodal NK/T-cell lymphoma, nasal type. Oral Oncol. 2007;43:4–14.
Batsakis, JG, Luna, MA. Midfacial necrotizing lesions. Semin Diagn Pathol. 1987;4:90–116.
Koom, WS, Chung, EJ, Yang, W-I, et al. Angiocentric T-cell and NK/T-cell lymphomas: radiotherapeutic viewpoints. Int J Radiat Oncol Biol Phys. 2004;59:1127–1137.
Lee, PY, Freeman, NJ, Khorsand, J, et al. Angiocentric T-cell lymphoma presenting as lethal midline granuloma. Int J Dermatol. 1997;36:419–427.
Mosqueda-Taylor, A, Meneses-Garcia, A, Zarate-Osorno, A, et al. Angiocentric lymphomas of the palate: clinico-pathological considerations in 12 cases. J Oral Pathol Med. 1997;26:93–97.
Pagano, L, Gallamini, A, Fianchi, L, et al. NK/T-cell lymphomas “nasal type”: an Italian multicentric retrospective survey. Ann Oncol. 2006;17:794–800.
Patel, V, Mahajan, S, Kharkar, V, et al. Nasal extranodal NK/T-cell lymphoma presenting as a perforating palatal ulcer: a diagnostic challenge. Indian J Dermatol Venereol Leprol. 2006;72:218–221.
Vidal, RW, Devaney, K, Ferlito, A, et al. Sinonasal malignant lymphomas: a distinct clinicopathological category. Ann Otol Rhinol Laryngol. 1999;108:411–419.
Wu, SM, Min, Y, Ostrzega, N, et al. Lymphomatoid granulomatosis: a rare mimicker of vasculitis. J Rheumatol. 2005;32:2242–2245.
Yong, W, Zheng, W, Zhu, J, et al. Midline NK/T-cell lymphoma nasal-type: treatment outcome, the effect of l-asparaginase based regimen, and prognostic factors. Hematol Oncol. 2006;24:28–32.
Baldini, L, Radaelli, F, Chiorboli, O, et al. No correlation between response and survival in patients with multiple myeloma treated with vincristine, melphalan, cyclophosphamide, and prednisone. Cancer. 1991;68:62–67.
Denz, U, Haas, PS, Wäsch, R, et al. State of the art therapy in multiple myeloma and future perspectives. Eur J Cancer. 2006;42:1591–1600.
Dimopoulos, MA, Moulopoulos, A, Smith, T, et al. Risk of disease progression in asymptomatic multiple myeloma. Am J Med. 1993;94:57–61.
George, ED, Sadovsky, R. Multiple myeloma: recognition and management. Am Fam Physician. 1999;59:1885–1894.
Hogan, MC, Lee, A, Solberg, LA, et al. Unusual presentation of multiple myeloma with unilateral visual loss and numb chin syndrome in a young adult. Am J Hematol. 2002;70:55–59.
Kyle, RA, Rajkumar, SV. Treatment of multiple myeloma: an emphasis on new developments. Ann Med. 2006;38:111–115.
Lebowitz, RA, Morris, L. Plasma cell dyscrasias and amyloidosis. Otolaryngol Clin North Am. 2003;36:747–764.
Ludwig, H, Advances in biology and treatment of multiple myeloma. Ann Oncol 2005;(suppl 2):ii106–ii112.
Palumbo, A, Bertola, A, Musto, P, et al. Oral melphalan, prednisone, and thalidomide for newly diagnosed patients with myeloma. Cancer. 2005;104:1428–1433.
Riedel, DA, Pottern, LM. The epidemiology of multiple myeloma. Hematol Oncol Clin North Am. 1992;6:225–247.
Smith, A, Wisloff, F, Samson, D. Guidelines on the diagnosis and management of multiple myeloma 2005. Br J Haematol. 2005;132:410–451.
Dimopoulos, MA, Kiamouris, C, Moulopoulos, LA. Solitary plasmacytoma of bone and extramedullary plasmacytoma. Hematol Oncol Clin North Am. 1999;13:1249–1257.
González, J, Elizondo, J, Trull, JM, et al. Plasma-cell tumours of the condyle. Br J Oral Maxillofac Surg. 1991;29:274–276.
Kremer, M, Ott, G, Nathrath, M, et al. Primary extramedullary plasmacytoma and multiple myeloma: phenotypic differences revealed by immunohistochemical analysis. J Pathol. 2005;205:92–101.
Majumdar, S, Raghavan, U, Jones, NS. Solitary plasmacytoma and extramedullary plasmacytoma of the paranasal sinuses and soft palate. J Laryngol Otol. 2002;116:962–965.
Meis, JM, Butler, JJ, Osborne, BM, et al. Solitary plasmacytomas of bone and extramedullary plasmacytomas: a clinicopathologic and immunohistochemical study. Cancer. 1987;59:1475–1485.
Nofsinger, YC, Mirza, N, Rowan, PT, et al. Head and neck manifestations of plasma cell neoplasms. Laryngoscope. 1997;107:741–746.
Rothfield, RE, Johnson, JT, Slavrides, A. Extramedullary plasmacytoma of the parotid. Head Neck. 1990;12:352–354.