Chapter 18 Development of Eyes and Ears
Development of Eyes and Related Structures
The eyes are derived from four sources:
The neuroectoderm of the forebrain differentiates into the retina, the posterior layers of the iris, and the optic nerve. The surface ectoderm forms the lens of the eye and the corneal epithelium. The mesoderm between the neuroectoderm and the surface ectoderm gives rise to the fibrous and vascular coats of the eye. Neural crest cells migrate into the mesenchyme and differentiate into the choroid, sclera, and corneal endothelium. Early eye development results from a series of inductive signals.
Homeobox-containing genes, including the transcription regulator Pax6, fibroblast growth factors, and other inducing factors play an important role in the molecular development of the eye (see Chapter 21).
The first evidence of eye development is at 22 days when optic grooves (sulci) appear in the neural folds at the cranial end of the embryo (Fig. 18-1A and B). As the neural folds fuse to form the forebrain, the optic grooves evaginate (protrude) from the future diencephalon to form hollow diverticula—optic vesicles—that project from the wall of the forebrain into the adjacent mesenchyme (Fig. 18-1C). The cavities of the optic vesicles are continuous with the cavity of the forebrain. Formation of the optic vesicles is induced by the mesenchyme adjacent to the developing brain.
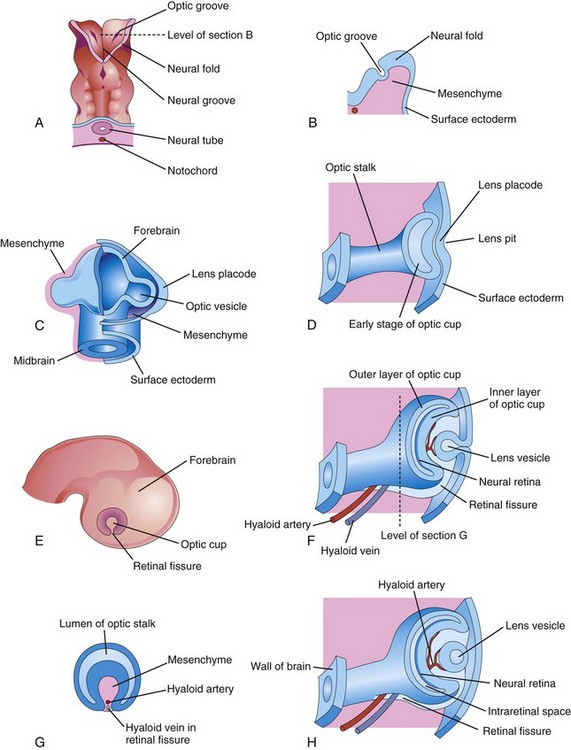
FIGURE 18–1 Illustrations of the early stages of eye development. A, Dorsal view of the cranial end of an embryo at approximately 22 days, showing the optic grooves, the first indication of eye development. B, Transverse section of a neural fold showing the optic groove in it. C, Schematic drawing of the forebrain of an embryo at approximately 28 days, showing its covering layers of mesenchyme and surface ectoderm. D, F, and H, Schematic sections of the developing eye, illustrating successive stages in the development of the optic cup and lens vesicle. E, Lateral view of the brain of an embryo at approximately 32 days, showing the external appearance of the optic cup. G, Transverse section of the optic stalk, showing the retinal fissure and its contents. Note that the edges of the retinal fissure are growing together, thereby completing the optic cup and enclosing the central artery and vein of the retina in the optic stalk and cup.
As the optic vesicles grow, their distal ends expand and their connections with the forebrain constrict to form hollow optic stalks (Fig. 18-1D). The vesicles soon come in contact with the surface ectoderm. Concurrently, the surface ectoderm adjacent to the vesicles thickens to form lens placodes, the primordia of the lenses (Fig. 18-1C). Formation of placodes is induced by the optic vesicles after the surface ectoderm has been conditioned by the underlying mesenchyme. An inductive message passes from the vesicles, stimulating the surface ectodermal cells to form the lens primordia. The lens placodes invaginate as they sink deep to the surface ectoderm, forming lens pits (Figs. 18-1D and 18-2). The edges of the pits approach each other and fuse to form spherical lens vesicles (Figs. 18-1F and H), which gradually lose their connection with the surface ectoderm.
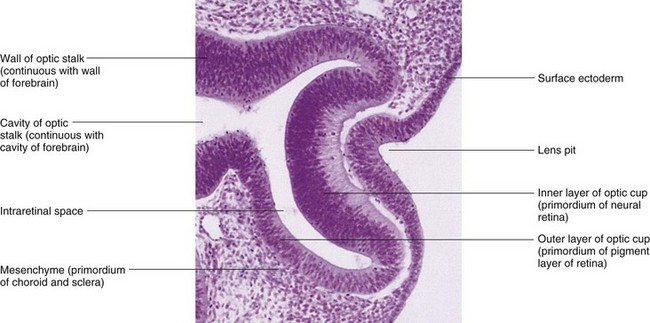
FIGURE 18–2 Photomicrograph of a sagittal section of the eye of an embryo (x200) at approximately 32 days. Observe the primordium of the lens (invaginated lens placode), the walls of the optic cup (primordium of retina), and the optic stalk (primordium of optic nerve).
(From Moore KL, Persaud TVN, Shiota K: Color Atlas of Clinical Embryology, 2nd ed. Philadelphia, WB Saunders, 2000.)
As the lens vesicles are developing, the optic vesicles invaginate to form double-walled optic cups, which consist of two layers that are connected to the developing brain by the optic stalks (Figs. 18-1E and F and 18-2). The optic cup becomes the retina and the optic stalk becomes the optic nerve. The lens and part of the cornea develop from the ectoderm and mesoderm. The opening of each optic cup is large at first, but the rim of the cup infolds around the lens (Fig. 18-3A). By this time, the lens vesicles have lost their connection with the surface ectoderm and have entered the cavities of the optic cups (Fig. 18-4). Linear grooves—retinal fissures (optic fissures)—develop on the ventral surface of the optic cups and along the optic stalks (Figs. 18-1E to H and 18-3A to D). The center of the optic cup, where the retinal fissure is deepest, forms the optic disc, where the neural retina is continuous with the optic stalk (Figs. 18-2 and18-3C and D). The developing axons of the ganglion cells pass directly into the optic stalk and convert it into the optic nerve. Myelination of nerve fibers (formation of a sheath around the fibers) begins during the later part of fetal development and during the first postnatal year.
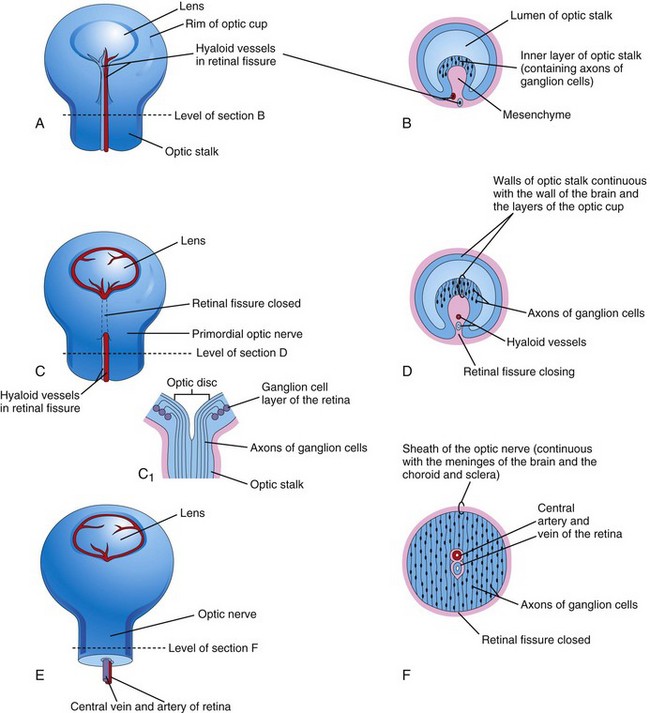
FIGURE 18–3 Illustrations of the closure of the retinal fissure and formation of the optic nerve. A, C, and E, Views of the inferior surface of the optic cup and stalk, showing progressive stages in the closure of the retinal fissure. C1, Schematic sketch of a longitudinal section of a part of the optic cup and stalk, showing the optic disc and axons of ganglion cells of the retina growing through the optic stalk to the brain. B, D, and F, Transverse sections of the optic stalk showing successive stages in closure of the retinal fissure and formation of the optic nerve. Note that the lumen of the optic stalk is gradually obliterated as axons of ganglion cells accumulate in the inner layer of the optic stalk as the optic nerve forms.
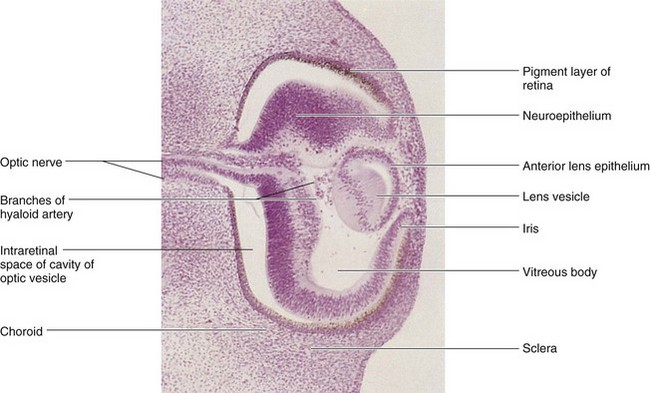
FIGURE 18–4 Photomicrograph of a sagittal section of the eye of an embryo (x100) at approximately 44 days. Observe that it is the posterior wall of the lens vesicle that forms the lens fibers. The anterior wall does not change appreciably as it becomes the anterior lens epithelium.
(From Nishimura H [ed]: Atlas of Human Prenatal Histology. Tokyo, Igaku-Shoin, 1983.)
The retinal fissures contain vascular mesenchyme from which hyaloid blood vessels develop (Fig. 18-3B and C). The hyaloid artery, a branch of the ophthalmic artery, supplies the inner layer of the optic cup, the lens vesicle, and the mesenchyme in the cavity of the optic cup (Figs. 18-1H and 18-3C). The hyaloid vein returns blood from these structures. As the edges of the retinal fissure fuse, the hyaloid vessels are enclosed within the primordial optic nerve (Fig. 18-3C to F). Distal parts of the hyaloid vessels eventually degenerate, but proximal parts persist as the central artery and vein of retina (see Fig. 18-9D).
Development of Retina
The retina develops from the walls of the optic cup, an outgrowth of the forebrain (Figs. 18-1C to F and 18-2).The walls of the cup develop into the two layers of the retina: the outer, thin layer of the cup becomes the pigment layer of retina, and the inner, thick layer differentiates into the neural retina. The proliferation and differentiation of retinal precursor cells are regulated by forkhead transcription factors. By the sixth week, melanin appears in the retinal pigment epithelium (Fig. 18-4).
During the embryonic and early fetal periods, the two layers of the retina are separated by an intraretinal space (Fig. 18-4 and see Fig. 18-9A and B); which is derived from the cavity of the optic cup. This space gradually disappears as the two layers of the retina fuse (see Fig. 18-8D), but the fusion is not firm. Because the optic cup is an outgrowth of the forebrain, the layers of the optic cup are continuous with the wall of the brain (Fig. 18-1H).
Under the influence of the developing lens, the inner layer of the optic cup proliferates to form a thick neuroepithelium (Figs. 18-2 and 18-4). Subsequently, the cells of this layer differentiate into the neural retina, the light-sensitive region of the retina. This region contains photoreceptors (rods and cones) and the cell bodies of neurons (e.g., bipolar and ganglion cells). Fibroblast growth factor signaling regulates retinal ganglion cell differentiation.
Because the optic vesicle invaginates as it forms the optic cup, the neural retina is “inverted,” that is, light-sensitive parts of the photoreceptor cells are adjacent to the outer retinal pigment epithelium. As a result, light traverses the thickest part of the retina before reaching the photoreceptors; however, because the neural retina is thin and transparent, it does not form a barrier to light. The axons of ganglion cells in the superficial layer of the neural retina grow proximally in the wall of the optic stalk (Figs. 18-3B to D and 18-4). As a result, the cavity of the optic stalk is gradually obliterated as the axons of the many ganglion cells form the optic nerve (Fig. 18-3F).
The optic nerve is surrounded by three sheaths that evaginated with the optic vesicle and stalk; consequently, they are continuous with the meninges of the brain.
Cerebrospinal fluid (CSF) is present in the subarachnoid space between the intermediate and inner sheaths of the optic nerve.
Myelination of the axons within the optic nerves begins in the late fetal period. After the eyes have been exposed to light for approximately 10 weeks, myelination is complete, but the process normally stops short of the optic disc, where the optic nerves leave the eyeballs. Normal neonates can see, but not too well because they are farsighted; they respond to changes in illumination and are able to fixate points of contrast. Visual acuity improves rapidly over the first year of infancy to almost normal adult levels.
Birth Defects of the Eyes
Coloboma
Coloboma results when the optic fissure fails to close completely, leaving a gap in eye structures. These defects can occur in any ocular structure from the cornea to the optic nerve. The eyelid may be involved but it is caused by other mechanisms. Retinochoroidal coloboma is characterized by a localized gap in the retina, usually inferior to the optic disc. The defect is bilateral in most cases.
Coloboma of the iris is a defect in the inferior sector of the iris or a notch in the pupillary margin, giving the pupil a keyhole appearance (Fig. 18-5). The defect may be limited to the iris or it may extend deeper and involve the ciliary body and retina. The defect may be caused by environmental factors, but a simple coloboma is frequently hereditary and is transmitted as an autosomal dominant characteristic.

Detachment of the Retina
This defect occurs when the inner and outer layers of the optic cup fail to fuse during the fetal period to form the retina and obliterate the intraretinal space (Figs. 18-3 and 18-9). It occurs in conjunction with other syndromes such as Down syndrome and Marfan syndrome (a connective tissue multisystemic disorder). The separation of the neural and pigment layers of the retina may be partial or complete. Retinal detachment may result from unequal rates of growth of the two retinal layers; as a result, the layers of the optic cup are not in perfect apposition. Sometimes the layers of the optic cup appear to have fused and separated later; such secondary detachments usually occur in association with other defects of the eye and head or trauma.
Knowledge about eye development makes it clear that when there is a detached retina, it is not a detachment of the entire retina because the retinal pigment layer remains firmly attached to the choroid—vascular layer of the eyeball (see Fig. 18-9D). The detachment is at the site of adherence of the outer and inner layers of the optic cup. Although separated from the pigment layer of the retina, the neural retina retains its blood supply (central artery of retina), derived from the embryonic hyaloid artery (see Fig. 18-9A and D).
Postnatally, the pigment layer normally becomes fixed to the choroid, but its attachment to the neural retina is not firm; hence, a detached retina may follow a blow to the eye or occur spontaneously. As a result, fluid accumulates between the pigment and neural layers, and vision is impaired.
Cyclopia
In this rare anomaly, the eyes are partially or completely fused, forming a single median eye enclosed in a single orbit (Fig. 18-6). There is usually a tubular nose (proboscis) superior to the eye. Cyclopia (single midline eye) and synophthalmia (fusion of eyes) represent a spectrum of ocular defects. These severe eye defects are associated with other craniocerebral defects that are incompatible with life. Cyclopia appears to result from severe suppression of midline cerebral structures—holoprosencephaly (see Chapter 17)—that develop from the cranial part of the neural plate. Cyclopia is transmitted by recessive inheritance.
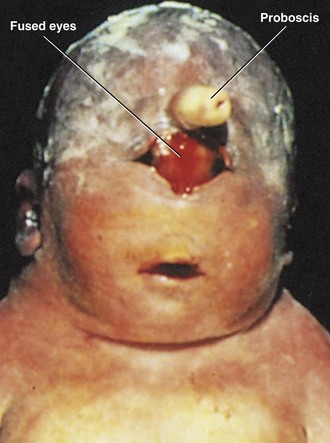
FIGURE 18–6 Male neonate with cyclopia (synophthalmia). Cyclopia (fusion of eyes) is a severe, uncommon birth defect of the face and eye, associated with a proboscis that represents the nose. The white substance covering his head is vernix caseosa—a normal fatty protective covering.
(Courtesy of Dr. Susan Phillips, Department of Pathology, Health Sciences Centre, Winnipeg, Manitoba, Canada.)
Microphthalmia
Congenital microphthalmia is a heterogeneous group of eye defects. The eye may be very small and associated with other ocular defects (e.g., such as a facial cleft; see Chapter 9, and trisomy13; see Chapter 20), or it may be a normal-appearing rudimentary eye. The affected side of the face is underdeveloped and the orbit is small.
Severe microphthalmia results from arrested development of the eye before or shortly after the optic vesicle has formed in the fourth week. The eye is essentially underdeveloped and the lens does not form. If the interference with development occurs before the retinal fissure closes in the sixth week, the eye is larger, but the microphthalmos is associated with gross ocular defects. When eye development is arrested in the eighth week or during the early fetal period, simple microphthalmos results (small eye with minor ocular abnormalities). Some cases of microphthalmos are inherited. The hereditary pattern may be autosomal dominant, autosomal recessive, or X-linked. Most cases of simple microphthalmia are caused by infectious agents (e.g., rubella virus, Toxoplasma gondii, and herpes simplex virus) that cross the placental membrane during the late embryonic and early fetal periods (see Chapter 20).
Anophthalmia
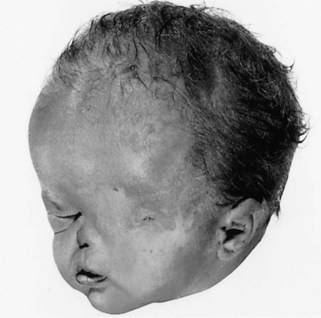
Anophthalmia (unilateral or bilateral) denotes absence of the eye, which is rare. The eyelids form, but no eyeball develops (Fig. 18-7). The formation of the orbit relies on stimulation from the developing eye, so orbit defects are always present. This severe defect is usually accompanied by other severe craniocerebral defects. In primary anophthalmos, eye development is arrested early in the fourth week and results from failure of the optic vesicle to form. In secondary anophthalmos, development of the forebrain is suppressed and absence of the eye or eyes is one of several associated defects.
Development of Ciliary Body
The ciliary body is a wedge-shaped extension of the choroid. Its medial surface projects toward the lens, forming ciliary processes (see Fig. 18-9C and D). The pigmented portion of the ciliary epithelium is derived from the outer layer of the optic cup, which is continuous with the pigment layer of the retina (Figs. 18-8 and 18-9D). The nonvisual retina is the non-pigmented, ciliary epithelium, which represents the anterior prolongation of the neural retina in which no neural elements develop (see Fig. 18-11). The ciliary muscle—the smooth muscle of the ciliary body that is responsible for focusing the lens—and the connective tissue in the ciliary body develop from mesenchyme located at the edge of the optic cup in the region between the anterior scleral condensation and the ciliary pigment epithelium.
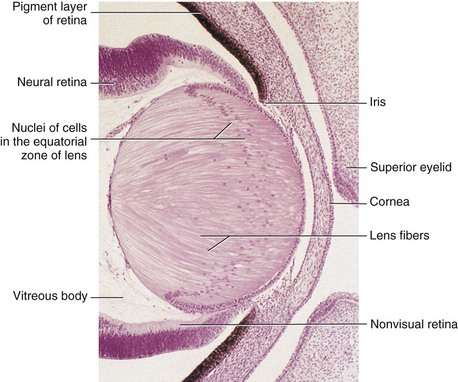
FIGURE 18–8 Sagittal section of part of the developing eye of an embryo (x280) at approximately 56 days. The lens fibers have elongated and obliterated the cavity of the lens vesicle. Note that the inner layer of the optic cup has thickened to form the primordial neural retina, and that the outer layer is heavily pigmented, which is the primordium of the pigment layer of the retina.
(From Moore KL, Persaud TVN, Shiota K: Color Atlas of Clinical Embryology, 2nd ed. Philadelphia, WB Saunders, 2000.)
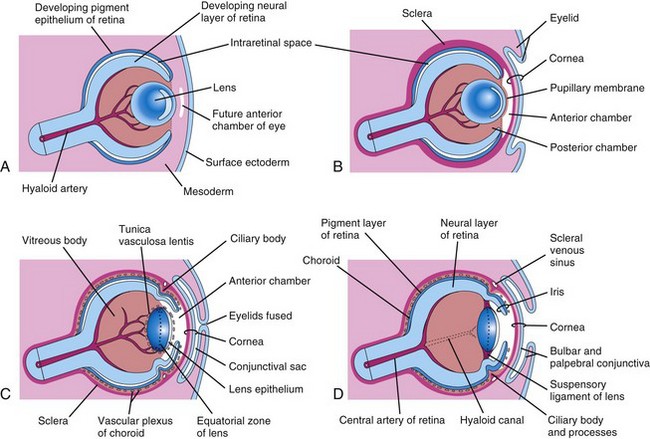
FIGURE 18–9 Diagrammatic drawings of sagittal sections of the eye, showing successive developmental stages of development of the lens, retina, iris, and cornea. A, At 5 weeks. B, At 6 weeks. C, At 20 weeks. D, Neonate. Note that the retina and optic nerve are formed from the optic cup and optic stalk (Fig. 18-1C).
Development of Iris
The iris develops from the rim of the optic cup (Fig. 18-3A), which grows inward and partially covers the lens (see Figs. 18-7 and 18-9). The two layers of the optic cup remain thin in this area. The epithelium of the iris represents both layers of the optic cup; it is continuous with the double-layered epithelium of the ciliary body and with the retinal pigment epithelium and neural retina. The connective tissue framework (stroma) of the iris is derived from neural crest cells that migrate into the iris. The dilator pupillae and sphincter pupillae muscles of the iris are derived from neuroectoderm of the optic cup. They appear to arise from the anterior epithelial cells of the iris. These smooth muscles result from a transformation of epithelial cells into smooth muscle cells.
Color of the Iris
The iris is typically light blue or gray in most neonates. The iris acquires its definitive color after birth as pigmentation occurs during the first 6 to 10 months. The concentration and distribution of pigment-containing cells—chromatophores—in the loose vascular connective tissue of the iris determine eye color. If the melanin pigment is confined to the pigmented epithelium on the posterior surface of the iris, the iris appears blue. If melanin is also distributed throughout the stroma (supporting tissue) of the iris, the eye appears brown. Iris heterochromia can also result from changes to the sympathetic innervations to the eye.
Congenital Aniridia
In this rare anomaly, there is a lack of iris tissue or almost complete absence of the iris. This defect results from an arrest of development at the rim of the optic cup during the eighth week (Fig. 18-3A). The defect may be associated with glaucoma, cataracts, and other eye abnormalities. Aniridia may be familial, the transmission being dominant or sporadic. Mutation of the Pax6 gene results in aniridia.
Development of Lens
The lens develops from the lens vesicle, a derivative of the surface ectoderm (Fig. 18-1F and H). The anterior wall of the vesicle, composed of cuboidal epithelium, becomes the subcapsular lens epithelium (Fig. 18-9C). The nuclei of the tall columnar cells forming the posterior wall of the lens vesicle undergo dissolution. These cells lengthen considerably to form highly transparent epithelial cells, the primary lens fibers. As these fibers grow, they gradually obliterate the cavity of the lens vesicle (Figs. 18-9A to C, 18-10, and 18-11). Pax 6 and Sox 2 are required for the induction of the lens. The transcription factors Pitx3, GATA-3 and FoxE3, regulate the formation and differentiation of the lens fibers.
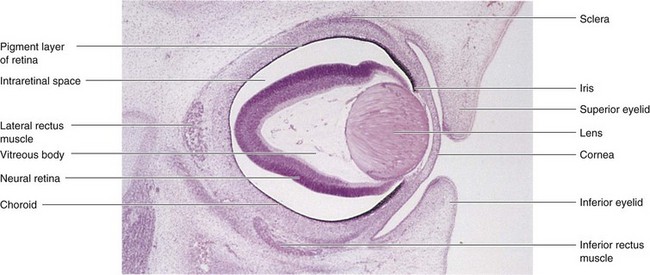
FIGURE 18–10 Photomicrograph of a sagittal section of the eye of an embryo (x50) at approximately 56 days. Observe the developing neural retina and pigment layer of the retina. The intraretinal space disappears when these two layers of the retina fuse.
(From Moore KL, Persaud TVN, Shiota K: Color Atlas of Clinical Embryology, 2nd ed. Philadelphia, WB Saunders, 2000.)
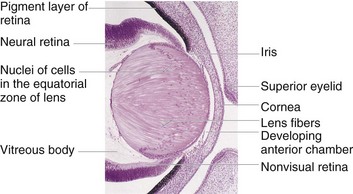
FIGURE 18–11 Photomicrograph of a portion of the developing eye of an embryo at the end of the embryonic period. Observe that the lens fibers have elongated and obliterated the cavity of the lens vesicle.
(From Moore KL, Persaud TVN, Shiota K: Color Atlas of Clinical Embryology, 2nd ed. Philadelphia, WB Saunders, 2000.)
The rim of the lens is called the equatorial zone because it is located midway between the anterior and posterior poles of the lens (Figs. 18-9C and 18-11). The cells in the equatorial zone are cuboidal; as they elongate, they lose their nuclei and become secondary lens fibers. These new lens fibers are added to the external sides of the primary lens fibers. Although secondary lens fibers continue to form during adulthood and the lens increases in diameter, the primary lens fibers must last a lifetime.
The developing lens is supplied with blood by the distal part of the hyaloid artery (Figs. 18-4 and 18-9); however, it becomes avascular in the fetal period when this part of the hyaloid artery degenerates. Thereafter, the lens depends on diffusion from the aqueous humor (watery fluid) in the anterior chamber of the eye (Fig. 18-9C), which bathes its anterior surface, and from the vitreous humor in other parts. The developing lens is invested by a vascular mesenchymal layer, the tunica vasculosa lentis. The anterior part of this capsule is the pupillary membrane (Fig. 18-9B). The pupillary membrane develops from the mesenchyme posterior to the cornea in continuity with the mesenchyme developing in the sclera. The part of the hyaloid artery that supplies the tunica vasculosa lentis disappears during the late fetal period. As a result, the tunica vasculosa lentis and pupillary membrane degenerate (Fig. 18-9C and D); however, the lens capsule produced by the anterior lens epithelium and the lens fibers persists. This capsule represents a greatly thickened basement membrane and has a lamellar structure because of its development. The former site of the hyaloid artery is indicated by the hyaloid canal in the vitreous body (Fig. 18-9D), which is usually inconspicuous in the living eye.
The vitreous body forms within the cavity of the optic cup (Figs. 18-4 and 18-9C). It is composed of vitreous humor, the fluid component of the vitreous body. The primary vitreous humor is derived from mesenchymal cells of neural crest origin, which secrete a gelatinous matrix, a surrounding substance, called the primary vitreous body. The primary vitreous humor is surrounded later by a gelatinous secondary vitreous humor, which is generally believed to arise from the inner layer of the optic cup. The secondary vitreous humor consists of primitive hyalocytes (vitreous cells), collagenous material, and traces of hyaluronic acid.
Persistent Pupillary Membrane
Remnants of the pupillary membrane, which cover the anterior surface of the lens during the embryonic period and most of the fetal period (Fig. 18-9B), may persist as web-like strands of connective tissue or vascular arcades over the pupil in neonates, especially in premature infants. This tissue seldom interferes with vision and tends to atrophy. Very rarely the entire pupillary membrane persists, giving rise to congenital atresia of the pupil (absence of opening of the pupil); surgery or laser treatment is needed in some cases to provide an adequate pupil.
Persistence of Hyaloid Artery
The distal part of the hyaloid artery normally degenerates as its proximal part becomes the central artery of the retina (Fig. 18-9C and D). If the distal part of the hyaloid artery persists, it may appear as a freely moving, nonfunctional vessel or as a worm-like structure projecting from the optic disc (Fig. 18-3C). Sometimes the hyaloid artery remnant may appear as a fine strand traversing the vitreous body. A remnant of the artery may also form a cyst. In unusual cases, the entire distal part of the artery persists and extends from the optic disc through the vitreous body to the lens. In most of these unusual cases, the eye is microphthalmic (very small).
Development of Aqueous Chambers
The anterior chamber of the eye develops from a cleft-like space that forms in the mesenchyme located between the developing lens and cornea (Figs. 18-4, 18-9A to C, and 18-11). The mesenchyme superficial to this space forms the substantia propria (transparent connective tissue) of the cornea and the mesothelium of the anterior chamber. After the lens is established, it induces the surface ectoderm to develop into the epithelium of the cornea and conjunctiva.
The posterior chamber of the eye develops from a space that forms in the mesenchyme posterior to the developing iris and anterior to the developing lens. When the pupillary membrane disappears and the pupil forms (Fig. 18-9C and D), the anterior and posterior chambers of the eye are able to communicate with each other through the scleral venous sinus (Fig. 18-9D). This vascular structure encircling the anterior chamber of the eye is the outflow site of aqueous humor from the anterior chamber to the venous system.
Congenital Glaucoma
Abnormal elevation of intraocular pressure in neonates usually results from abnormal development of the drainage mechanism of the aqueous humor during the fetal period (Fig. 18-12). Intraocular tension rises because of an imbalance between the production of aqueous humor and its outflow. This imbalance may result from abnormal development of the scleral venous sinus (Fig. 18-9D). Congenital glaucoma is genetically heterogeneous (includes a number of phenotypes that are similar but are actually determined by different genotypes), but the condition may also result from a rubella infection during early pregnancy (see Chapter 20). Mutations in the CYP1B1 gene are associated with approximately 85% of cases of congenital glaucoma.
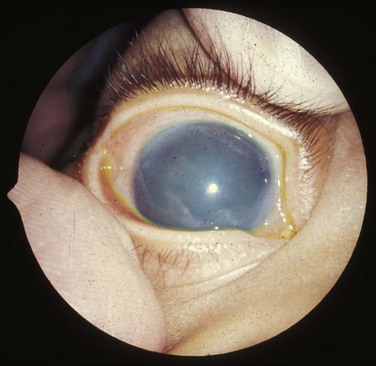
FIGURE 18–12 Clouding of the cornea caused by congenital glaucoma. The clouding may also result from infection, trauma, or metabolic disorders.
(Reprinted from Otolaryngologic Clinics of North America 40(1), Guercio J, Martyn L, Congenital malformations of the eye and orbit, 113-140, Copyright 2007, with permission from Elsevier.)
Congenital Cataracts
In this condition, the lens is opaque and frequently appears grayish white. Without treatment, blindness results. Many lens opacities are inherited, dominant transmission being more common than recessive or sex-linked transmission. Some congenital cataracts are caused by teratogenic agents, particularly the rubella virus (Fig. 18-13), that affect early development of the lenses. The lenses are vulnerable to rubella virus between the fourth and seventh weeks, when primary lens fibers are forming (see Chapter 20). Cataract and other ocular defects caused by the rubella virus could be completely prevented if immunity to rubella were conferred on all women of reproductive age.
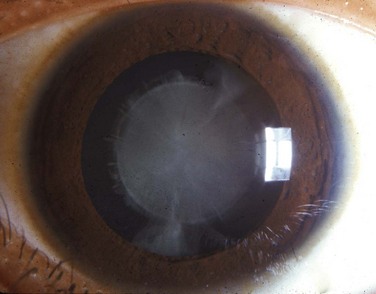
FIGURE 18–13 Typical appearance of a child with a congenital cataract that may be caused by the rubella virus. Cardiac defects and deafness are other birth defects common to this infection.
(Reprinted from Otolaryngologic Clinics of North America 40(1), Guercio J, Martyn L, Congenital malformations of the eye and orbit, 113-140, Copyright 2007, with permission from Elsevier.)
Physical agents, such as radiation, can also damage the lens and produce cataracts. Another cause of cataract is an enzymatic deficiency—congenital galactosemia. These cataracts are not present at birth, but may appear as early as the second week after birth. Because of the enzyme deficiency, large amounts of galactose from milk accumulate in the infant’s blood and tissues, causing injury to the lens and resulting in cataract formation.
Development of Cornea
The cornea is induced by the lens vesicle. The inductive influence results in transformation of the surface ectoderm into the transparent, multilayered avascular cornea. The cornea is formed from three sources, the:
Edema of Optic Disc
The relationship of the sheaths of the optic nerve to the meninges of the brain and the subarachnoid space is important clinically. An increase in CSF pressure (often resulting from increased intracranial pressure) slows venous return from the retina, causing papilledema (fluid accumulation) of the optic disc. This occurs because the retinal vessels are covered by pia mater and lie in the extension of the subarachnoid space that surrounds the optic nerve.
Development of Choroid and Sclera
The mesenchyme surrounding the optic cup (largely of neural crest origin) reacts to the inductive influence of the retinal pigment epithelium by differentiating into an inner vascular layer, the choroid, and an outer fibrous layer, the sclera (Fig. 18-9C and D). The sclera develops from a condensation of mesenchyme external to the choroid and is continuous with the stroma (supporting tissue) of the cornea. Toward the rim of the optic cup, the choroid becomes modified to form the cores (central masses) of the ciliary processes (Fig. 18-9D), consisting chiefly of capillaries supported by delicate connective tissue. The first choroidal blood vessels appear during the 15th week; by the 23rd week, arteries and veins can be easily distinguished.
Development of Eyelids
The eyelids develop during the sixth week from mesenchyme derived from neural crest cells, and from two cutaneous folds of the surface ectoderm that grow over the cornea (Fig. 18-9B and C). The eyelids begin to adhere to one another during the eighth week and remain adherent until the 26th to the 28th week (Fig. 18-9C). While the eyelids are adherent, there is a closed conjunctival sac anterior to the cornea. As the eyelids open, the bulbar conjunctiva is reflected over the anterior part of the sclera and the surface epithelium of the cornea (Fig. 18-9D). The palpebral conjunctiva lines the inner surface of the eyelids.
The eyelashes and glands in the eyelids are derived from the surface ectoderm in a manner similar to that described for other parts of the integument (see Chapter 19). The connective tissue and tarsal plates (fibrous plates in the eyelids) develop from mesenchyme in the developing eyelids. The orbicularis oculi muscle is derived from mesenchyme in the second pharyngeal arch (see Chapter 9) and is supplied by its nerve (CN VII).
Congenital Ptosis of Eyelid
Drooping of the superior (upper) eyelids at birth is relatively common (Fig. 18-14). Ptosis (blepharoptosis) may result from failure of normal development of the levator palpebrae superioris muscle. Congenital ptosis may result from prenatal injury or dystrophy (defective nutrition) of the superior division of the oculomotor nerve (CN III), which supplies this muscle. If ptosis is associated with inability to move the eyeball superiorly, there is also failure of the superior rectus muscle of the eyeball to develop normally. Congenital ptosis may be transmitted as an autosomal dominant trait. Vision can be affected if the margin of the eyelid partially or completely covers the pupil; early surgical correction would be indicated.
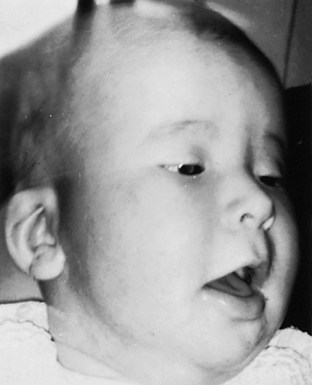
FIGURE 18–14 A child with congenital bilateral ptosis. Drooping of the superior eyelids usually results from abnormal development or failure of development of the levator palpebrae superioris, the muscle that elevates the eyelids. The infant is contracting the frontalis muscle of the forehead in an attempt to raise the eyelids.
(From Avery ME, Taeusch HW Jr: Schaffer’s Diseases of the Newborn, 5th ed. Philadelphia, WB Saunders, 1984.)
Coloboma of Eyelid
Large defects of the eyelid (palpebral colobomas) are uncommon. A coloboma is usually characterized by a small notch in the superior eyelid, but the defect may involve almost the entire lid. Palpebral colobomas appear to result from local developmental disturbances in the formation and growth of the eyelids. Drying and ulceration of the cornea can result from lower eyelid coloboma.
Cryptophthalmos
Cryptophthalmos, a rare disorder, results from congenital absence of the eyelids; as a result, skin covers the eyes. The eyeball is small and defective and the cornea and conjunctiva usually do not develop. Fundamentally, the defect means absence of the palpebral fissure (slit) between eyelids; usually there is varying absence of eyelashes and eyebrows and other eye defects. Cryptophthalmos is an autosomal recessive condition that is usually part of the cryptophthalmos syndrome that includes urogenital anomalies.
Development of Lacrimal Glands
At the superolateral angles of the orbits, the lacrimal glands develop from a number of solid buds from the surface ectoderm. The lacrimal ducts drain into the lacrimal sac and eventually into nasolacrimal duct. The glands are small at birth and do not function fully until approximately 6 weeks; hence, neonates do not produce tears when they cry. Tears are often not present with crying until 1 to 3 months.
Development of Ears
The ears are composed of three anatomic parts:
The external and middle parts of the ears are concerned with the transference of sound waves to the internal ears, which convert the waves into nerve impulses and registers changes in equilibrium.
Development of Internal Ear
The internal ears are the first of the three parts of the ears to develop. Early in the fourth week, a thickening of surface ectoderm, the otic placode, appears on each side of the myelencephalon, the caudal part of the hindbrain (Fig. 18-15A and B). Inductive signals from the paraxial mesoderm and notochord stimulate the surface ectoderm to form the placodes (see Chapter 4; Fig. 4-9). Each otic placode soon invaginates and sinks deep to the surface ectoderm into the underlying mesenchyme. In so doing, it forms an otic pit (Fig. 18-15C and D). The edges of the pit soon come together and fuse to form an otic vesicle—the primordium of the membranous labyrinth (Figs. 18-15E to G and 18-16). The otic vesicle soon loses its connection with the surface ectoderm, and a diverticulum grows from the vesicle and elongates to form the endolymphatic duct and sac (Fig. 18-17A to E).
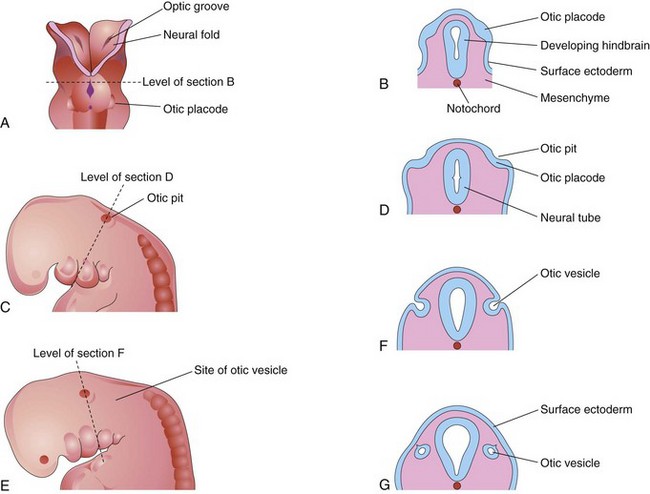
FIGURE 18–15 Drawings illustrating early development of the internal ear. A, Dorsal view of an embryo at approximately 22 days, showing the otic placodes. B, D, F, and G, Schematic coronal sections illustrating successive stages in the development of otic vesicles. C and E, Lateral views of the cranial region of embryos, at approximately 24 and 28 days, respectively.
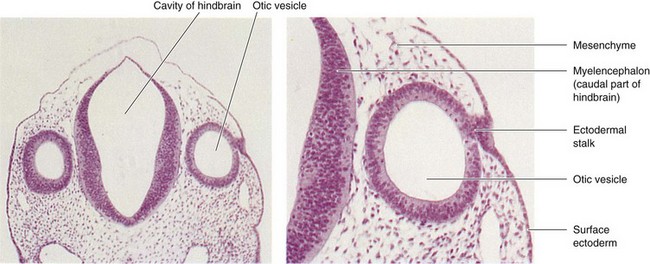
FIGURE 18–16 Left, Photomicrograph of a transverse section of an embryo (x55) at approximately 26 days. Observe the otic vesicles, the primordia of the membranous labyrinths, which give rise to the internal ears. Right, Higher magnification of the right otic vesicle (x120). Note the ectodermal stalk, which is still attached to the remnant of the otic placode. The otic vesicle will soon lose its connection with the surface ectoderm.
(From Nishimura H [ed]: Atlas of Human Prenatal Histology. Tokyo, Igaku-Shoin, 1983.)
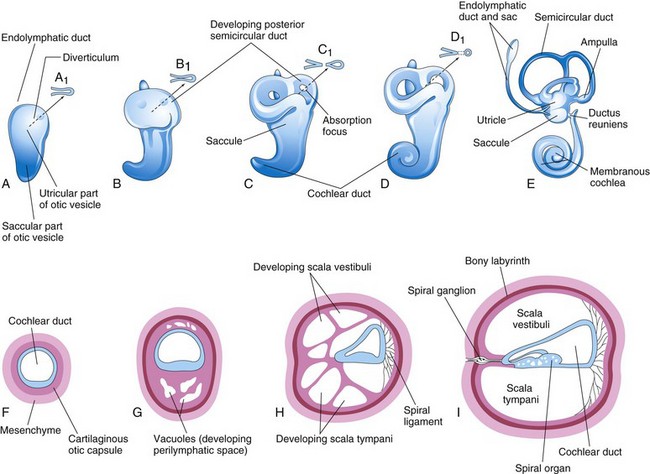
FIGURE 18–17 Drawings of the otic vesicles showing the development of the membranous and bony labyrinths of the internal ear. A to E, Lateral views showing successive stages in the development of the otic vesicle into the membranous labyrinth from the fifth to eighth weeks. A to D, Diagrammatic sketches illustrating the development of a semicircular duct. F to I, Sections through the cochlear duct showing successive stages in the development of the spiral organ and the perilymphatic space from the 8th to the 20th weeks.
Two regions of the otic vesicles are recognizable:
Three disc-like diverticula grow out from the utricular parts of the primordial membranous labyrinths. Soon the central parts of these diverticula fuse and disappear (Fig. 18-17B to E). The peripheral unfused parts of the diverticula become the semicircular ducts, which are attached to the utricle and are later enclosed in the semicircular canals of the bony labyrinth. Localized dilatations, the ampullae, develop at one end of each semicircular duct. Specialized receptor areas—cristae ampullares—differentiate in the ampullae and the utricle and saccule (maculae utriculi and sacculi).
From the ventral saccular part of the otic vesicle, a tubular diverticulum—the cochlear duct—grows and coils to form the membranous cochlea (Fig. 18-17C to E). Tbx1 expression in the mesenchyme surrounding the otic vesicle regulates the formation of the cochlear duct by controlling retinoic acid activity. A connection of the cochlea with the saccule, the ductus reuniens, soon forms. The spiral organ (of Corti) differentiates from cells in the wall of the cochlear duct (Fig. 18-17F to I). Ganglion cells of the vestibulocochlear nerve (cranial nerve [CN] VIII) migrate along the coils of the membranous cochlea and form the spiral ganglion (of cochlea). Nerve processes extend from this ganglion to the spiral organ, where they terminate on the hair cells. The cells in the spiral ganglion retain their embryonic bipolar condition.
Inductive influences from the otic vesicle stimulate the mesenchyme around the otic vesicle to condense and differentiate into a cartilaginous otic capsule (Fig. 18-17F). Recent studies indicate that the Pax2 gene is required for formation of the spiral organ of Corti and the spiral ganglion. Transforming growth factor β1 may play a role in modulating epithelial-mesenchymal interaction in the internal ear and in directing the formation of the otic capsule or bony labyrinth.
As the membranous labyrinth enlarges, vacuoles appear in the cartilaginous otic capsule and soon coalesce to form the perilymphatic space. The membranous labyrinth is now suspended in perilymph (fluid in the perilymphatic space). The perilymphatic space, related to the cochlear duct, develops two divisions, the scala tympani and scala vestibuli (Fig. 18-17H and I). The cartilaginous otic capsule later ossifies to form the bony labyrinth of the internal ear. The internal ear reaches its adult size and shape by the middle of the fetal period (20–22 weeks).
Development of Middle Ear
Development of the tubotympanic recess (Fig. 18-18B) from the first pharyngeal pouch is described in Chapter 9. The proximal part of the tubotympanic recess forms the pharyngotympanic tube (auditory tube). The distal part of the recess expands and becomes the tympanic cavity (Fig. 18-18C), which gradually envelops the small bones of the middle ear—auditory ossicles (malleus, incus, and stapes), their tendons and ligaments, and the chorda tympani nerve. Development of the ossicles is described in Chapter 9. These structures receive a more or less complete epithelial investment. It has been suggested that, in addition to apoptosis (programmed cell death) in the middle ear, an epithelium-type organizer located at the tip of the tubotympanic recess probably plays a role in the early development of the middle ear and tympanic membrane.
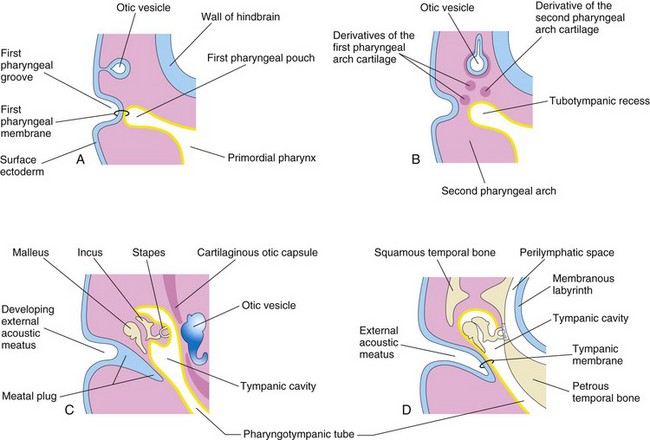
FIGURE 18–18 Schematic drawings illustrating development of the external and middle parts of the ear. Observe the relationship of these parts of the ear to the otic vesicle, the primordium of the internal ear. A, At 4 weeks, illustrating the relation of the otic vesicle to the pharyngeal apparatus. B, At 5 weeks, showing the tubotympanic recess and pharyngeal arch cartilages. C, Later stage, showing the tubotympanic recess (future tympanic cavity and mastoid antrum) beginning to envelop the ossicles. D, Final stage of ear development showing the relationship of the middle ear to the perilymphatic space and the external acoustic meatus. Note that the tympanic membrane develops from three germ layers: surface ectoderm, mesenchyme, and endoderm of the tubotympanic recess.
During the late fetal period, expansion of the tympanic cavity gives rise to the mastoid antrum, located in the petromastoid part of the temporal bone. The mastoid antrum is almost adult size at birth; however, no mastoid cells are present in neonates. By 2 years of age, the mastoid cells are well developed and produce conical projections of the temporal bones, the mastoid processes. The middle ear continues to grow through puberty. The tensor tympani muscle, attached to the malleus, is derived from mesenchyme in the first pharyngeal arch and is innervated by trigeminal nerve (CN V), the nerve of this arch. The stapedius muscle is derived from the second pharyngeal arch and is supplied by facial nerve (CN VII), the nerve of this arch. The signaling molecules FGF-8, endothelin-1, and Tbx1 are involved in middle ear development.
Development of External Ear
The external acoustic meatus, the passage of the external ear leading to the tympanic membrane, develops from the dorsal part of the first pharyngeal groove. The ectodermal cells at the bottom of this funnel-shaped tube proliferate to form a solid epithelial plate, the meatal plug (Fig. 18-18C). Late in the fetal period, the central cells of this plug degenerate, forming a cavity that becomes the internal part of the external acoustic meatus (Fig. 18-18D). The meatus, relatively short at birth, attains its adult length in approximately the ninth year.
The primordium of the tympanic membrane is the first pharyngeal membrane, which forms the external surface of the tympanic membrane. In the embryo, the pharyngeal membrane separates the first pharyngeal groove from the first pharyngeal pouch (Fig. 18-18A). As development proceeds, mesenchyme grows between the two parts of the pharyngeal membrane and differentiates into the collagenic fibers in the tympanic membrane.
To summarize, the tympanic membrane develops from three sources:
The auricle (pinna), which projects from the side of the head, develops from mesenchymal proliferations in the first and second pharyngeal arches—auricular hillocks—surrounding the first pharyngeal groove (Fig. 18-19A). As the auricle grows, the contribution by the first arch is reduced. The lobule (earlobe) of the auricle is the last part of the auricle to develop. The auricles begin to develop at the base of the neck (Fig. 18-19A and B). As the mandible develops, the auricles assume their normal position at the side of the head (Fig. 18-23).
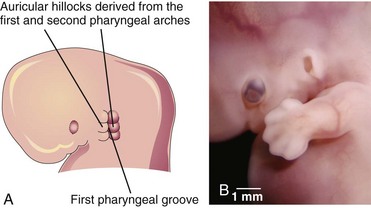
FIGURE 18–19 Illustration of the development of the auricle, the part of the external ear that is not within the head. A, At 6 weeks. Note that three auricular hillocks are located on the first pharyngeal arch and three on the second arch. B, Photograph of a 7-week embryo. Note the developing external ear.
(B, Courtesy of Dr. Brad Smith, University of Michigan, Ann Arbor, Michigan.)
The parts of the auricle derived from the first pharyngeal arch are supplied by its nerve, the mandibular branch of the trigeminal nerve; the parts derived from the second arch are supplied by cutaneous branches of the cervical plexus, especially the lesser occipital and greater auricular nerves. The nerve of the second pharyngeal arch, the facial nerve, has few cutaneous branches; some of its fibers contribute to the sensory innervation of the skin in the mastoid region and probably in small areas on both aspects of the auricle.
Congenital Deafness
Because formation of the internal ear is independent of development of the middle and external ears, congenital impairment of hearing may be the result of maldevelopment of the sound-conducting apparatus of the middle and external ears, or of the neurosensory structures of the internal ear. Approximately 3 in every 1000 newborns have significant hearing loss, of which there are many subtypes.
Most types of congenital deafness are caused by genetic factors and many of the genes responsible have been identified. Mutations in the GJB2 gene are responsible for approximately 50% of nonsyndromic recessive hearing loss. Congenital deafness may be associated with several other head and neck defects as a part of the first arch syndrome (see Chapter 9). Abnormalities of the malleus and incus are often associated with this syndrome. A rubella infection during the critical period of development of the internal ear, particularly the seventh and eighth weeks, can cause defects of the spiral organ and deafness. Congenital fixation of the stapes results in conductive deafness in an otherwise normal ear. Failure of differentiation of the annular ligament, which attaches the base of the stapes to the oval window (fenestra vestibuli), results in fixation of the stapes to the bony labyrinth.
Auricular Abnormalities
Severe defects of the external ear are rare, but minor deformities are common. There is a wide variation in the shape of the auricle. Almost any minor auricular defect may occasionally be found as a usual feature in a particular family. Minor defects of the auricles may serve as indicators of a specific pattern of congenital anomalies. For example, the auricles are often abnormal in shape and low-set in infants with chromosomal syndromes (Fig. 18-20) such as trisomy 18 and in infants affected by maternal ingestion of certain drugs (e.g., trimethadione).
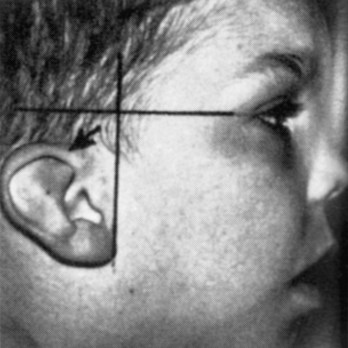
FIGURE 18–20 Low-set slanted ear. This designation is made when the margin of the auricle or helix (arrow) meets the cranium at a level inferior to the horizontal plane through the corner of the eye.
(From Jones KL: Smith’s Recognizable Patterns of Human Malformation, 5th ed. Philadelphia, WB Saunders, 1996.)
Auricular Appendages
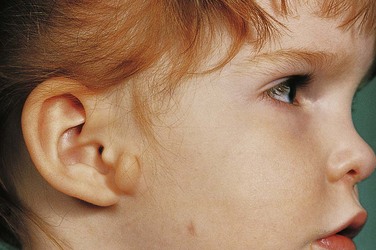
Auricular appendages (skin tags) are common and may result from the development of accessory auricular hillocks (Fig. 18-21). The appendages usually appear anterior to the auricle, more often unilaterally than bilaterally. The appendages, often with narrow pedicles, consist of skin but may contain some cartilage.
Absence of the Auricle
Anotia (absence of the auricle) is rare but is commonly associated with the first pharyngeal arch syndrome (see Chapter 9). Anotia results from failure of mesenchymal proliferation.
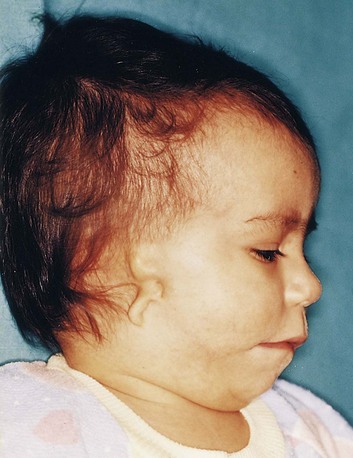
Microtia
Microtia (a small or rudimentary auricle) results from suppressed mesenchymal proliferation (Fig. 18-22). This defect often serves as an indicator of associated birth defects, such as an atresia of the external acoustic meatus (80% of cases) and middle ear anomalies. The cause can be both genetic and environmental.
Preauricular Sinuses and Fistulas
Pit-like cutaneous depressions or shallow sinuses are occasionally located in a triangular area anterior to the auricle (Fig. 18-23). The sinuses are usually narrow tubes or shallow pits that have pinpoint external openings. Some sinuses contain a vestigial cartilaginous mass.
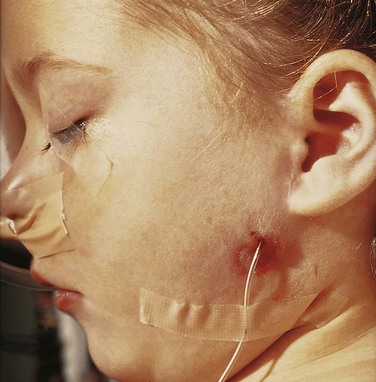
FIGURE 18–23 A child with an auricular fistula relating to the first pharyngeal arch. Note the external orifice of the fistula below the auricle and the upward direction of the catheter (in sinus tract) toward the external acoustic meatus.
(Courtesy of Dr. Pierre Soucy, Division of Paediatric General Surgery, Children’s Hospital of Eastern Ontario, Ottawa, Ontario, Canada.)
Preauricular sinuses may be associated with internal anomalies, such as deafness and kidney malformations. The embryologic basis of auricular sinuses is uncertain but it may relate to incomplete fusion of the auricular hillocks or to abnormal mesenchymal proliferation and defective closure of the dorsal part of the first pharyngeal groove. Most of this pharyngeal groove normally disappears as the external acoustic meatus forms. Other auricular sinuses appear to represent ectodermal folds that are sequestered during formation of the auricle. Bilateral preauricular sinuses are typically familial. The majority of sinuses are asymptomatic and have only minor cosmetic importance; however, they can become infected. Auricular fistulas (narrow canals) connecting the preauricular skin with the tympanic cavity or the tonsillar fossa (see Fig. 9-10F ) are extremely rare.
Atresia of External Acoustic Meatus
Atresia (blockage) of this canal results from failure of the meatal plug to canalize (Fig. 18-18C and Fig. 18-25). Usually the deep part of the meatus is open, but the superficial part is blocked by bone or fibrous tissue. Most cases are associated with the first arch syndrome (see Chapter 9). Often abnormal development of both the first and second pharyngeal arches is involved. The auricle is also severely affected and defects of the middle and/or internal ear are sometimes present. Atresia of the external acoustic meatus can occur bilaterally or unilaterally and usually results from autosomal dominant inheritance.
Absence of External Acoustic Meatus
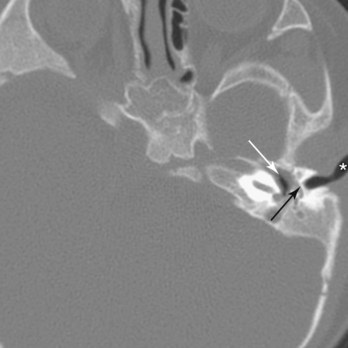
Absence of the external acoustic meatus is rare; usually the auricle is normal (Fig. 18-24). This defect results from failure of inward expansion of the first pharyngeal groove and failure of the meatal plug to disappear (Fig. 18-18C ).
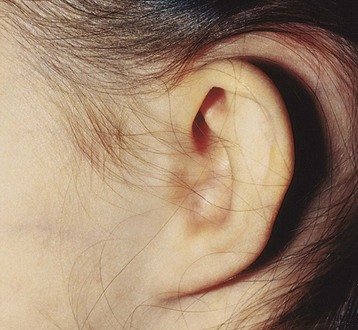
FIGURE 18–24 A child with no external acoustic meatus; however, the auricle is normal. Observe the absence of the opening of the external acoustic meatus. A computed tomography scan revealed normal middle and internal ear structures.
(Courtesy of Dr. A.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, University of Manitoba, Winnipeg, Manitoba, Canada.)
Congenital Cholesteatoma
This is a fragment of keratinized epithelial cells that is retained after birth. The “rest” appears as a white cyst-like structure medial to and behind the tympanic membrane. It may be that the rest consists of cells from the meatal plug that was displaced during its canalization (Fig. 18-18C ). It has been suggested that congenital cholesteatoma may originate from an epidermoid formation that normally involutes by 33 weeks gestation. Choleseatomas can exhibit growth and invasion of neighboring bone.
Summary of Development of Eyes
The first indication of the eyes is the optic grooves in the neural folds at the cranial end of the embryo; the grooves form at the beginning of the fourth week. The grooves deepen to form hollow optic vesicles that project from the forebrain. The optic vesicles contact the surface ectoderm and induce development of the lens placodes.
Summary of Development of Ears
Clinically Oriented Problems
Case 18–1
An infant was born blind and deaf with congenital heart disease. The mother had a severe viral infection early in her pregnancy.
Case 18–3
An infant was born with small, multiple calcifications in the brain, microcephaly, and microphthalmia. The mother was known to consume very rare meat.
Case 18–4
A mentally deficient female infant had low-set, malformed ears; a prominent occiput; and rocker-bottom feet. A chromosomal abnormality was suspected.
References and Suggested Reading
Artunduaga MA. A classic twin study of external ear malformations, including microtia. N Engl J Med. 2009;361:1216.
Barishak YR. Embryology of the Eye and Its Adnexa, ed 2. Basel, Switzerland: Karger; 2001.
Bauer PW, MacDonald CB, Melhem ER. Congenital inner ear malformation. Am J Otol. 1998;19:669.
Box J, Chang W, Wu DK. Patterning and morphogenesis of the vertebrate ear. Int J Dev Biol. 2007;51:521.
Braunstein EM, Monks DC, Aggarwal VS, et al. Tbx1 and Brn4 regulate retinoic acid metabolic genes during cochlear morphogenesis. BMC Dev Biol. 2009;9:31.
Carlson BM. Human Embryology and Developmental Biology, ed 4. St. Louis: Mosby-Year Book; 2009.
Davis N, Mor E, Ashley-Padan R. Roles for Dicer1 in the patterning and differentiation of the optic cup neuroepithelium. Development. 2011;138:127.
FitzPatrick DR, van Heyningen V. Developmental eye disorders. Curr Opin Genet Dev. 2005;15:348.
Graw J. Eye development. Curr Top Dev Biol. 2010;90:343.
Haddad JJr. The ear. In Behrman RE, Kliegman RM, Jenson HB, editors: Nelson Textbook of Pediatrics, ed 17, Philadelphia: Elsevier/Saunders, 2004.
Jason R, Guercio BS, Martyn LJ. Congenital malformations of the eye and orbit. Otolaryngol Clin North Am. 2007;40:113.
Jones KL. Smith’s Recognizable Patterns of Human Malformation, ed 6. Philadelphia: WB Saunders; 2005.
Kozmik Z. Pax genes in eye development and evolution. Curr Opin Genet Dev. 2005;15:430.
Litsiou A, Hanson S, Streit A. A balance of FGF, BMP and WNT signalling positions the future placode territory in the head. Development. 2005;132:4051.
Moore KL, Dalley AF, Agur AMR. Clinically Oriented Anatomy, ed 6. Baltimore: Williams & Wilkins; 2010.
Olitsky SE, Nelson LB. Disorders of the eye. In Behrman RE, Kliegman RM, Jenson HB, editors: Nelson Textbook of Pediatrics, ed 17, Philadelphia: Elsevier/Saunders, 2004.
O’Rahilly R. The early development of the otic vesicle in staged human embryos. J Embryol Exp Morphol. 1963;11:741.
O’Rahilly R. The prenatal development of the human eye. Exp Eye Res. 1975;21:93.
Paquette LB, Jackson HA, Yavare CJ, et al. In utero eye development documented by fetal MR imaging. Am J Neuroradiol. 2009;30:1787.
Porter CJW, Tan SW. Congenital auricular anomalies: Topographic anatomy, embryology, classification, and treatment strategies. Plast Reconstr Surg. 2005;115:1701.
Rodriguez-Vázquez JF. Development of the stapes and associated structures in human embryos. J Anat. 2005;207:165.
Sellheyer K. Development of the choroid and related structures. Eye. 1990;4:255.
Smith AN, Radice G, Lang RA. Which FGF ligands are involved in lens induction? Dev Biol. 2010;337:195.
Toriello HV, Reardon W, Gorlin RJ. Hereditary Hearing Loss and Its Syndromes, ed 2. Oxford: Oxford University Press; 2004.
Wilson E, Saunders R, Trivedi R. Pediatric Ophthalmology: Current Thought and a Practical Guide. New York: Springer; 2008.