Chapter 21 Common Signaling Pathways Used During Development
During the process of embryonic development, undifferentiated precursor cells differentiate and organize into the complex structures found in functional adult tissues. This intricate process requires cells to integrate many different cues, both intrinsic and extrinsic, for development to occur properly. These cues control the proliferation, differentiation, and migration of cells to determine the final size and shape of the developing organs. Disruption of these signaling pathways can result in human developmental disorders and birth defects. These key developmental signaling pathways are also frequently co-opted in the adult by diseases such as cancer.
Given the diverse changes that occur during embryogenesis, it may appear that there should also be a correspondingly diverse set of signaling pathways that regulate these processes. In contrast, the differentiation of many different cell types is regulated through a relatively restricted set of molecular signaling pathways:

Epigenetics: These are heritable changes in gene function that do no result from a change in DNA sequence. Examples of epigenetic modifications are histone acetylation and DNA methylation.
Stem cells: Stem cells in the embryo can give rise to all cells and tissues in the developing organism. Adult stem cells maintain tissues in the mature organism. These types of stem cells and induced pluripotent stem cells (iPS) are potential sources of cells for regeneration and/or repair of injured or degenerating cells and organs.
Intercellular Communication
During embryonic development, cells receive signals from their external environment and communicate with neighboring cells. This communication directs the cell to undergo different processes such as proliferation, differentiation, and migration. Two classes of proteins required for intercellular communication are discussed: gap junctions and cell adhesion molecules.
Gap Junctions
Gap junctions are a means for cells to directly communicate with one another; this is known as gap junction intercellular communication (GJIC). Although the pore size of the channels vary, only small molecules (e.g., second messengers, ions such as calcium, and ATP) less than 1 kiloDalton (kDa) can pass through, so most proteins and nucleic acids are excluded. In the nervous and cardiac systems, gap junctions help to establish electrical cell coupling (“electrical” synapse).
Although the function of gap junctions is quite straightforward, the structure of these intercellular channels is complex and highly regulated throughout development (Fig. 21-1). Each gap junction is composed of two hemichannels known as connexons. Each connexon is hexameric and, as such, is made up of six individual connexin subunits. An individual connexin (Cx) molecule consists of four transmembrane domains. There are more than 20 different connexin molecules in vertebrates. Cellular and tissue functional diversity of gap junctions results from whether individual connexons are the same (homotypic) or different (heterotypic) and if each connexon is made from the same or different connexin molecules (homomeric or heteromeric), respectively.
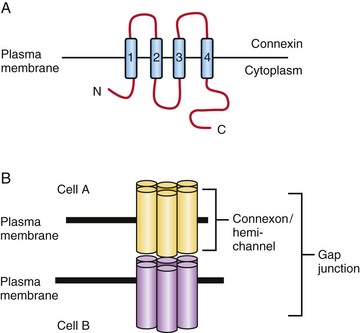
FIGURE 21–1 Gap junction intercellular communication. A, The connexin molecule consists of four transmembrane domains, two extracellular domains and its N- and C-termini are cytoplasmic. B, Connexons, or hemi-channels, are hexameric structures consisting of 6 connexin subunits. A gap junction can be formed from two homophilic or heterophilic connexons. Small molecules (including ions and ATP) less than 1 kDa can pass through an open gap junction.
Early in development, GJIC is important in the distribution of ions and other molecules essential for regionalization prior to the establishment of distinct boundaries and compartments. Their importance has been demonstrated in the developing chick hindbrain (rhombencephalon) by dye transfer and electrical coupling studies.
Some of the better characterized connexins include Cx43 (heart, brain), Cx45 (heart, pancreas), Cx32 (myelin), and Cx36 (pancreas, brain). In this nomenclature system, the number following Cx refers to the molecular weight in kDa. Mutations in Cx genes have been described and result in diseases such as the hereditary peripheral neuropathy X-linked Charcot-Marie-Tooth disease (Cx32).
Cell Adhesion Molecules
Cell adhesion molecules have large extracellular domains which interact with extracellular matrix (ECM) components or adhesion molecules on neighboring cells. These molecules often contain a transmembrane segment and a short cytoplasmic domain, which regulates intracellular signaling cascades. Two classes of molecules that have important roles during embryonic development are discussed: cadherins and members of the Immunoglobulin superfamily (IgSF) of cell adhesion molecules.
Cadherins
Cadherins are critical for embryonic morphogenesis as they regulate the separation of cell layers (endothelial and epidermal), cell migration, cell sorting, establishment of well-defined boundaries, synaptic connections, and in the growth cones of neurons. These properties result from cadherins mediating the interaction between the cell and its extracellular milieu (neighboring cells and extracellular matrix [ECM]). Cadherins were originally classified by their site of expression. For example, E-cadherin (Epithelial cadherin) is highly expressed in epithelial cells whereas N-cadherin (Neural cadherin) is highly expressed in neural cells. Cadherins mediate homophilic, calcium-dependent binding. A typical cadherin molecule has a large extracellular domain, a transmembrane domain and an intracellular tail (Fig. 21-2). The extracellular domain contains five extracellular repeats (EC repeats) and has four Ca2+-binding sites. Cadherins form dimers that interact with cadherin dimers in adjacent cells. These complexes are found clustered in adherens junctions, which result in the formation of a tight barrier between epithelial or endothelial cells. Via its intracellular domain, cadherins bind to p120-catenin, β-catenin, and α-catenin. These proteins connect cadherin to the cytoskeleton. E-cadherin expression is lost as epithelial cells transition to mesenchymal cells (this is known as the epithelial to mesenchymal transition [EMT]). EMT is required for the formation of neural crest cells during development and the same process also occurs during tumor development.
Immunoglobulin Superfamily
There are over 700 members of the immunoglobulin superfamily (IgSF) of cell adhesion molecules in the human genome. This large family of proteins is involved in a wide variety of cellular processes. One member of this class, Neural Cell Adhesion Molecule (NCAM), is an abundant protein in the brain and has three isoforms that result from alternative splicing. It has a large extracellular domain that contains five immunoglobulin (Ig) repeats and two fibronectin domains as well (Fig. 21-2). This region mediates the calcium independent homophilic binding of NCAM to itself and heterophilic binding to other cell adhesion molecules (L1 and TAG-1), receptor tyrosine kinases (Fibroblast Growth Factor Receptor) or the ECM. Ligand binding induces intracellular signaling via the Fyn and FAK intracellular kinases. NCAM undergoes a unique post-translational modification, polysialylation (PSA). PSA-NCAM is abundant early in neural development and becomes restricted to areas of neural plasticity and migration in the adult. It is believed that polysialylation decreases the adhesiveness of NCAM and thus facilitates migration. NCAM regulates neurite outgrowth, axonal path finding, survival, and plasticity.
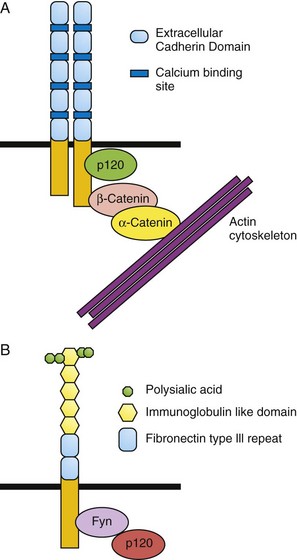
FIGURE 21–2 Structure of cadherin and neural cell adhesion molecule (NCAM). A, The cadherin extracellular domain contains four calcium-binding sites and five repeated domains called extracellular cadherin domains (ECD). Each cadherin molecule forms a homodimer. On the intracellular domain, cadherin bind directly to p120 catenin and to β-catenin, which binds to α-catenin. This complex links the cadherin molecules to the actin cytoskeleton. B, Extracellularly, NCAM contains five immunoglobulin (Ig) repeats and two fibronectin-III domains. The fifth Ig repeat is modified by polysialyation, which decreases the adhesiveness of the NCAM molecule. Intracellular signaling is transmitted by the Fyn and Fak kinases.
Morphogens
Extrinsic signals guide the differentiation and migration of cells during development and thereby dictate the morphology and function of developing tissues (see Chapter 5). Many of these morphogens are found in concentration gradients in the embryo, and different morphogens can be expressed in opposing gradients in the dorsal/ventral, anterior/posterior, and medial/lateral axes. The fate of a specific cell can be determined by its location along these gradients. Cells can be attracted or repelled by morphogens depending on the set of receptors expressed on their surface.
Retinoic Acid
The anterior (rostral, head)/posterior (caudal, tail), or anteroposterior (AP) axis of the embryo is crucial for determining the correct location for structures such as limbs and for the patterning of the nervous system. For decades, it has been clinically evident that alterations in the level of vitamin A (retinol) in the diet (excessive or insufficient amounts) can lead to the development of congenital malformations (see Chapters 17 and 20). The bioactive form of vitamin A is retinoic acid that is formed by the oxidation of retinol to retinal by retinol dehydrogenases and the subsequent oxidation of retinal by retinal aldehyde dehydrogenase. Free levels of retinoic acid can be further modulated by cellular retinoic acid–binding proteins that sequester retinoic acid. As well, retinoic acid can be actively degraded into inactive metabolites by enzymes such as CYP26 (Fig. 21-3). Normally, retinoic acid acts to “posteriorize” the body plan. Therefore, either excessive retinoic acid or inhibition of its degradation leads to a truncated body axis where structures have a more posterior nature. In contrast, insufficient retinoic acid or defects in the enzymes such as retinal aldehyde dehydrogenase will lead to a more anteriorized structure. At a molecular level, retinoic acid binds to its receptors inside the cell and activates them. Retinoic acid receptors are transcription factors, and therefore their activation will regulate the expression of downstream genes. Crucial targets of retinoic acid receptors in development are the Hox genes. Due to their profound influence on early development, retinoids are powerful teratogens, especially during the first trimester.
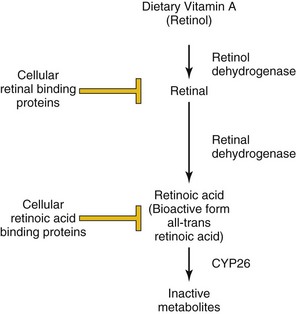
FIGURE 21–3 Regulation of retinoic acid metabolism and signaling. Dietary retinol (vitamin A) is converted to retinal via the action of retinol dehydrogenases. The concentration of free retinal is controlled by the action of cellular retinal–binding proteins. Similarly, retinal is converted to retinoic acid by retinal dehydrogenases, and its free level is modulated by sequestration by cellular retinoic acid–binding proteins and degradation by CYP26. The bioactive form of retinoic acid is all-trans retinoic acid.
Transforming Growth Factor β/Bone Morphogenetic Protein
Members of the TGF-β superfamily include TGF-β, BMPs, activin, and nodal. These molecules contribute to the establishment of dorsoventral patterning, cell fate decisions, and formation of specific organs, including the nervous system, kidneys, skeleton, and blood (see Chapters 5, 16, and 17). In humans, there are three TGF-β isoforms (TGF-β1, TGF-β2, and TGF-β3). Binding of these ligands to heterotetrameric (four subunit) complexes, consisting of specific type I (inactive kinase domain) and type II TGFβ receptor subunit (TβR-II) (constitutively active) transmembrane serine-threonine kinase receptors, results in intracellular signaling events (Fig. 21-4). When TGF-β ligands bind to their respective membrane-bound type II receptor, a type I receptor is recruited and transphorylated, and its kinase domain is activated, subsequently phosphorylating intracellular receptor-associated Smad proteins (R-Smads). The Smad proteins are a large family of intercellular proteins that are divided into three classes: receptor-activated (R-Smads, Smads 1–3, 5, 8), common-partner (co-Smads, Smad4), and inhibitory Smads (I-Smads, Smad6, Smad7). R-Smad/Smad4 complexes translocate to the nucleus and regulate target gene transcription by interacting with other proteins or as transcription factors by directly binding to DNA. TβR-I activation is a highly regulated process that involves membrane-anchored co-receptors and other receptor-like molecules that can sequester ligands and prevent their binding to respective TβR-II receptors. Dominant negative forms of TβR-II have inactive kinase domains and cannot transphosphorylate TβR-I, thereby blocking downstream signaling events. The diversity of TGF-β ligand, TβR-I and TβR-II, coreceptor, ligand trap, and R-Smad combinations contributes to particular developmental and cell-specific processes, often in combination with other signaling pathways.
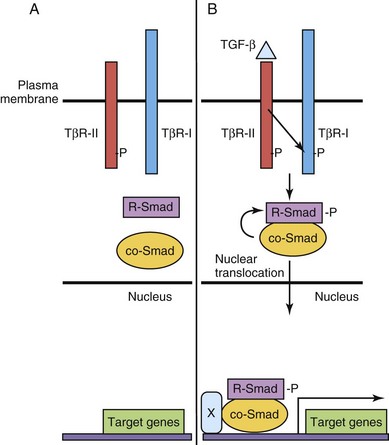
FIGURE 21–4 Transforming growth factor β (TGF-β)/Smad signaling pathway. A, The type II TGF-β receptor subunit (TβR-II) is constitutively active. B, Upon binding of ligand to TβR-II, a type I receptor subunit is recruited to form a heterodimeric receptor complex and the TβR-I kinase domain is transphosphorylated (-P). Signaling from the activated receptor complex phosphorylates R-Smads, which then bind to a co-Smad, translocate from the cytoplasm to the nucleus, and activate gene transcription with cofactor(s) (X).
Hedgehog
Sonic hedgehog (Shh) was the identified first mammalian ortholog of the Drosophila gene hedgehog. Shh and other related proteins, desert hedgehog and Indian hedgehog, are secreted morphogens critical to early patterning, cell migration, and differentiation of many cell types and organ systems (see Chapter 5). Cells have variable thresholds for response to the secreted Shh signal. The primary receptor for Shh is Patched (PTCH in human, PTC family in mouse), a 12-transmembrane domain protein that, in the absence of Shh, inhibits Smoothened (Smo), a seven-transmembrane domain G protein–linked protein, and downstream signaling to the nucleus. However, in the presence of Shh, Ptc inhibition is blocked and downstream events follow, including nuclear translocation of Gli (Gli1, Gli2, Gli3), with transcriptional activation of target genes, such as Ptc-1, Engrailed, and others (Fig. 21-5).
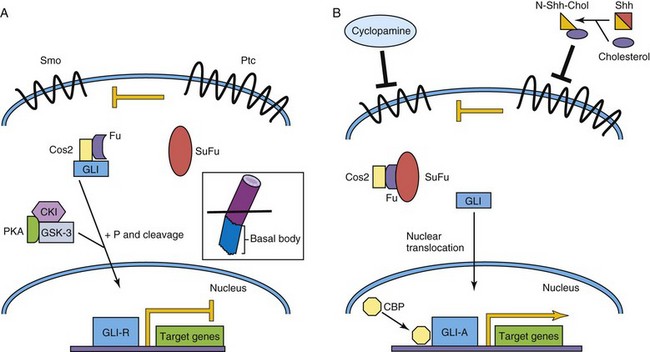
FIGURE 21–5 Sonic hedgehog/Patched signaling pathway. A, The Patched (Ptc) receptor inhibits signaling from the Smoothened (Smo) receptor. In a complex with Costal-2 (Cos2) and Fused (Fu), Gli is modified to become a transcriptional repressor, Gli-R. B, Sonic hedgehog (Shh) is cleaved and cholesterol is added to its N-terminus. This modified Shh ligand inhibits the Ptc receptor, permitting Smo signaling, and ultimately activated Gli (Gli-A) translocates to the nucleus to activate target genes with CBP. In vertebrates, Shh signaling takes place in primary cilia (inset). CBP, cyclic AMP–binding protein; CKI, casein kinase I; GSK-3, glycogen synthase kinase-3; P, phosphate group; PKA, protein kinase A; SuFu, suppressor of Fused.
The Shh protein is modified post-translationally by the addition of cholesterol and palmitate moieties to the N- and C-termini, respectively. These lipid modifications affect Shh’s association with the cell membrane, formation of Shh multimers, and, of significance, affect the movement of Shh, altering its tissue distribution and concentration gradients. One of the best explained activities of Shh activity in vertebrate development is the role of Shh in patterning the ventral neural tube (see Chapters 4 and 17). Shh is secreted at high levels by the notochord. The concentration of Shh is highest in the floor plate of the neural tube and lowest in the roof plate, where members of the TGF-β family are highly expressed. The cell fates of four ventral interneuron classes and motor neurons are determined by relative Shh concentrations and by a combinatorial code of homeobox and basic HLH (bHLH) genes.
The requirement of Shh pathway signaling for many developmental processes is underscored by the discovery of human mutations of members of the Shh pathway and the corresponding phenotypes of genetically modified mice, in which members of the Shh pathway are either inactivated (loss of function/knockout) or over-expressed (gain of function). Mutations of SHH and PTCH have been associated with holoprosencephaly, a congenital brain defect resulting in the fusion of the two cerebral hemispheres, anophthalmia or cyclopia (see Chapter 18) and dorsalization of forebrain structures; in sheep, this defect can also result from exposure to the teratogen cyclopamine, which disrupts Shh signaling (see Fig. 21-5). Of interest, some patients with severe forms of the inborn error of cholesterol synthesis, the autosomal recessive Smith-Lemli-Opitz syndrome, have holoprosencephaly (see Chapter 20). GLI3 mutations are associated with autosomal dominant polydactyly syndromes (see Chapter 16), such as Greig’s and Pallister-Hall syndromes. Gorlin’s syndrome, often due to germline PTCH mutations, is a constellation of congenital malformations mostly affecting the epidermis, craniofacial structures (see Chapter 9), and the nervous system. These patients are significantly predisposed to basal cell carcinomas, especially after radiation therapy, and a smaller proportion will develop malignant brain tumors (medulloblastomas) during childhood. Somatic mutations of PTCH, SUFU, and SMO have also been identified in patients with sporadic medulloblastomas not associated with Gorlin’s syndrome.
In vertebrates, the Shh pathway is linked to primary cilia (inset, Fig. 21-5) and their constituent intraflagellar transport (IFT) and basal body proteins. IFT proteins act upstream of the GLI activator (GLI-A) and repressor (GLI-R) proteins and are necessary for their production. Mutations involving genes encoding basal body proteins, such as Talpid3 and oral-facial-digital syndrome 1 (Ofd1), affect Shh signaling in knockout mice. A group of human cilia-related diseases called the ciliopathies, includes rare genetic diseases as well as more common disorders such as autosomal recessive polycystic kidney disease.
Wnt/β-Catenin Pathway
The Wnt-secreted glycoproteins are vertebrate orthologs of the Drosophila gene Wingless. Similar to the other morphogens previously discussed, the 19 Wnt family members control several processes during development, including establishment of cell polarity, proliferation, apoptosis, cell fate specification, and migration. Wnt signaling is very complex and three signaling pathways have been elucidated to date—the classic or “canonical” β-catenin-dependent pathway is discussed here (Fig. 21-6). Specific Wnts bind to one of 10 Frizzled (Fzd) seven transmembrane domain cell surface receptors, and with low-density lipoprotein receptor–related-protein (LRP5/LRP6) co-receptors, thereby activating downstream intracellular signaling events. β-catenin plays an integral role in canonical Wnt signaling. In the absence of Wnt binding, in a protein complex with adenomatous polyposis coli (APC) and axin, cytoplasmic β-catenin is phosphorylated by glycogen synthase kinase (GSK-3) and targeted for degradation. In the presence of Wnts, GSK-3 is itself phosphorylated by Dishevelled (Dvl), and thus inactivated; it cannot phosphorylate β-catenin. β-catenin is stabilized, accumulates in the cytoplasm, and translocates to the nucleus where it activates target gene transcription, in a complex with T-cell factor (TCF) transcription factors. The many β-catenin/TCF target genes include vascular endothelial growth factor (VEGF), c-myc, and matrix metalloproteinases.
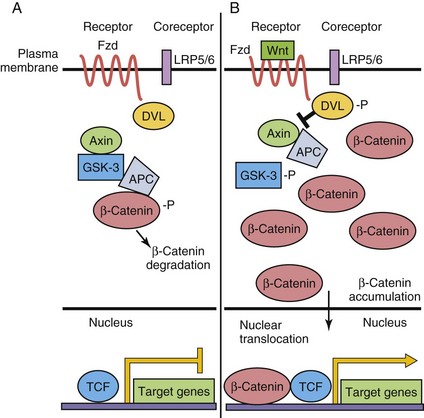
FIGURE 21–6 Wnt/β-catenin canonical signaling pathway. A, In the absence of Wnt ligand binding to Frizzled (Fzd) receptor, β-catenin is phosphorylated (-P) by a multiprotein complex and targeted for degradation. Target gene expression is repressed by T-cell factor (TCF). B, When Wnt binds to the Fzd receptor, LRP coreceptors are recruited, Dishevelled (DVL) is phosphorylated, and β-catenin then accumulates in the cytoplasm. Some β-catenin enters the nucleus to activate target gene transcription. APC, adenomatous polyposis coli; GSK-3, glycogen synthase kinase-3; LRP, lipoprotein receptor–related-protein.
There are several noncanonical Wnt signaling pathways; some share Frizzled receptors. However, all of these pathways are distinguished from the canonical Wnt pathway by not involving β-catenin stabilization, degradation, and/or nuclear translocation. One of the best-studied noncanonical Wnt signaling pathways is known as the Wnt-cGMP/Ca2+ pathway and acts through phospholipase C (PLC) to increase intracellular calcium concentrations, thereby activating protein kinase C (PKC) and/or calmodulin-dependent kinase II (CamKII), resulting in a myriad of downstream effects.
Dysregulated Wnt signaling is a prominent feature in many developmental disorders and cancer. A Frizzled (FZD9) gene is present in the Williams-Beuren syndrome deletion region. LRP5 mutations are found in the osteoporosis-pseudoglioma syndrome. Dvl2 knockout mice have malformations of the cardiac outflow tract, abnormal somite segmentation, and neural tube defects. Similar to the Shh pathway, canonical Wnt pathway mutations (in β-catenin, APC, and axin1 genes) have been described in children with medulloblastoma. Moreover, somatic APC mutations are common (~50%) in adults with sporadic colorectal carcinomas and germline APC mutations are a feature of familial adenomatous polyposis and Turcot’s syndrome (multiple colorectal adenomas and increased frequency of primary brain tumors).
Receptor Tyrosine Kinases
Common Features
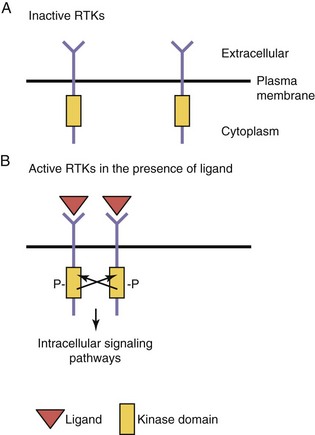
Growth factors, such as insulin, epidermal growth factor, nerve growth factor and other neurotrophins, and members of the platelet-derived growth factor family, bind to cell surface transmembrane receptors found on target cells. These receptors, members of the RTK superfamily, have three domains: (1) an extracellular ligand binding domain, (2) a transmembrane domain, and (3) an intracellular kinase domain (Fig. 21-7). These receptors are found as monomers in the quiescent or unbound state, but upon ligand binding, these receptor units dimerize. This process of dimerization brings the two intracellular kinase domains into close proximity such that one kinase domain can phosphorylate and activate the other receptor (transphosphorylation). Transphosphorylation is required to fully activate the receptors, which then initiate a series of intracellular signaling cascades. The mechanism of transphosphorylation requires both receptor subunits within a dimer to have functional kinase domains for signal transduction to occur. If there is an inactivating mutation of one receptor subunit’s kinase domain, the functional consequence will be to abolish signaling through a heterodimer resulting from the combination of wild-type and mutant receptor subunits (a dominant negative mode of action). Such a mutation in the kinase domain of the VEGF receptor 3 (VEGFR-3) results in the autosomal dominantly inherited lymphatic disorder called Milroy disease.
Regulation of Angiogenesis by Receptor Tyrosine Kinases
Growth factors generally promote cellular proliferation, migration, and survival (i.e., are antiapoptotic). Dysregulation of RTKs or their downstream signaling components are frequently found in human cancers. During embryogenesis, signaling through RTKs is crucial for normal development and affects many different processes such as the growth of new blood vessels (see Chapter 4), cellular migration, and neuronal axonal guidance.
Endothelial cells are derived from a progenitor cell (the hemangioblast) that can give rise to both the hematopoietic cell lineage and endothelial cells. The early endothelial cells proliferate and eventually coalesce to form the first primitive blood vessels. This process is termed vasculogenesis. After the first blood vessels are formed, they undergo intensive remodeling and maturation into the mature blood vessels in a process called angiogenesis. This maturation process involves the recruitment of vascular smooth muscle cells to the vessels that stabilize them. Vasculogenesis and angiogenesis are both dependent on the function of two distinct RTK classes, members of the VEGF and Tie receptor families. VEGF-A is essential for endothelial and blood cell development. VEGF-A knockout mice fail to develop blood or endothelial cells and die at early embryonic stages. The heterozygous VEGF-A mice have severe defects in their vasculature demonstrating that VEGF-A gene dose is important (haploinsufficiency). A related molecule, VEGF-C, was shown to be crucial for the development of lymphatic endothelial cells. VEGF-A signals through two receptors, VEGFR-1 and VEGFR-2, which are expressed by endothelial cells. VEGF-A signals predominantly through VEGFR-2 in order for vasculogenesis to properly occur in the embryo.
The process of angiogenic refinement depends on the function of the angiopoietin/Tie2 signaling pathway. Tie2 is a RTK that is specifically expressed by endothelial cells and angiopoietin 1 and angiopoietin 2 are its ligands that are expressed by the surrounding vascular smooth muscle cells. This represents a paracrine signaling system where receptor and ligand are expressed in adjacent cells. Both the VEGF/VEGFR-2 and angiopoietin/Tie2 signaling pathways are co-opted by tumors to stimulate growth of new blood vessels, which stimulates their growth and metastasis. This demonstrates how normal signaling pathways in the developing human can be reused for disease processes, such as cancer, in the adult.
Notch-Delta Pathway
The Notch signaling pathway is integral for cell fate determination, including maintenance of stem-cell niches, proliferation, apoptosis, and differentiation. These processes are essential for all aspects of organ development through regulation of lateral and inductive cell–cell signaling. Notch proteins are single transmembrane receptors (Notch 1–4) that interact with membrane-bound Notch (see Chapter 5) ligands (Delta-like ligands, Dll-1, Dll-3, Dll-4) and Serrate-like ligands, Jagged-1, Jagged-2) on adjacent cells (Fig. 21-8). Ligand-receptor binding triggers proteolytic events, some mediated by secretases, leading to the release of the Notch intracellular domain (NICD). When the NICD translocates to the nucleus, a series of intranuclear events culminates in the induction of expression of hairy enhancer of split (Hes), an HLH transcription factor that maintains the progenitor state by repressing proneural basic HLH genes.
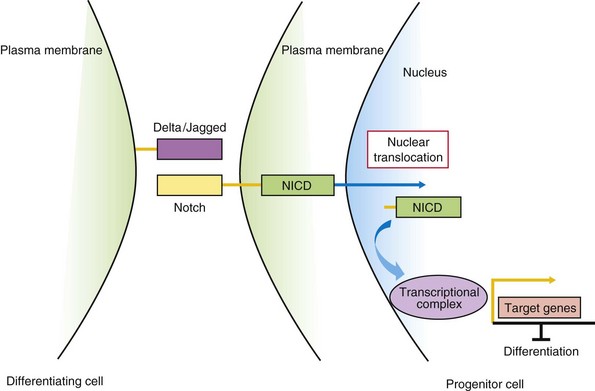
FIGURE 21–8 Notch/Delta signaling pathway. In progenitor cells (right), activation of Notch signaling leads to cleavage of the Notch intracellular domain (NICD). Proteases such as γ-secretase mediate this cleavage event. NICD translocates to the nucleus, binds to a transcriptional complex, and activates target genes, such as Hes1, that inhibit differentiation. In differentiating cells (left), Notch signaling is not active.
The process of lateral inhibition ensures that in a population of cells with equivalent developmental potential, there are the correct numbers of two distinct cell types. In the initial cell-cell interaction, the progenitor cell responding to the Notch-ligand Delta, through a negative feedback mechanism, reduces its own expression of Delta, with Notch receptor signaling maintaining the cell as an uncommitted progenitor. However, the adjacent cell maintains Delta expression levels with reduced Notch signaling and differentiation, mediated by, for example, proneural HLH genes. Inductive signaling with other surrounding cells expressing morphogens may overcome the cell’s commitment to a neural cell fate (default state) to an alternative glial cell fate. Understanding the function of the Notch-Delta signaling pathway in mammalian development has been assisted by loss-of-function studies in the mouse. Evidence of JAGGED1 or NOTCH-2 mutations in Alagille syndrome (arteriohepatic dysplasia), with liver, kidney, cardiovascular, ocular, and skeletal malformations, and NOTCH-3 gene mutations in the CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) adult vascular degenerative disease, with a tendency to early-age onset of stroke-like events, support the importance of the Notch signaling pathway in embryonic and postnatal development, respectively.
Pharmacological manipulation of the Notch signaling pathway may be a means to treat human diseases. For example, gamma secretase inhibitors (GSI) are in the midst of clinical trials for such diverse disorders as Alzheimer disease, pulmonary hypertension and cancer. For the former, gamma secretase is also a protease required for the production of amyloid-β protein in the brain. Some of the GSI under development are non-selective, whereas others spare the Notch signaling pathway.
Transcription Factors
Transcription factors belong to a large class of proteins that regulate the expression of many target genes, either through activation or repression mechanisms. Typically, a transcription factor will bind to specific nucleotide sequences in the promoter/enhancer regions of target genes and regulate the rate of transcription of its target genes via interacting with accessory proteins. Recently, it was demonstrated that transcription factors can both activate or repress target gene transcription depending on the cell in which they are expressed, the specific promoter, the chromatin context, and the developmental stage. In addition, some transcription factors do not need to bind to DNA to regulate transcription, but may bind to other transcription factors already bound to the promoter DNA thereby regulating transcription. Also, they may bind and sequester other transcription factors from their target genes, thus repressing their transcription. The transcription factor superfamily is composed of many different classes of proteins. Three examples of this diverse family of proteins are discussed: Hox/Homeobox, Pax, and bHLH transcription factors.
Hox/Homeobox Proteins
The Hox genes were first discovered in the fruit fly, Drosophila melanogaster. Mutations in these genes of the HOM-C complex lead to dramatic phenotypes (homeotic transformation) such as the Antennapedia gene in which legs instead of antennae sprout from the head. The order of the Hox genes along the AP axis is faithfully reproduced in their organization at the level of the chromosome. In humans, the order of the Hox genes along the AP axis and chromosomal location is conserved as well. Defects in HOXA1 have been shown to impair human neural development, and mutations in HOXA13 and HOXD13 result in limb malformations (see Chapter 16).
All the HOX genes contain a 180-base pair sequence, the homeobox, which encodes a 60–amino-acid homeodomain composed of three α helices. The third (recognition) helix binds to DNA sites that contain one or more TAAT/ATTA tetranucleotide-binding motifs in the promoters of their target genes. The homeodomain is the most conserved region of the protein and is highly conserved across evolution, whereas other regions of the protein are not as well conserved. Mutations in the DNA binding region of the homeobox gene NKX2.5 are associated with cardiac atrial-septal defects and mutations in ARX are associated with the central nervous system malformation syndrome lissencephaly (see Chapter 17).
Pax Genes
All the Pax genes contain conserved bipartite DNA-binding motifs called the Pax (or paired) domain, and most Pax family members also contain a homeodomain. PAX proteins have been shown to both activate and repress transcription of target genes. The D. melanogaster ortholog of Pax6, eyeless, was shown to be essential for eye development because the homozygous mutant flies had no eyes. In gain-of-function experiments, ectopic expression of eyeless led to the formation of additional eyes. In fruit flies, eyeless is clearly a master regulator of eye development. Eyeless shares a high degree of sequence conservation with its human ortholog PAX6. PAX6 was shown to be associated with ocular malformations such as aniridia (absence of the iris) and Peter’s anomaly. In human eye diseases, the level of PAX6 expression seems to be crucial as patients with only one functional copy (haploinsufficiency) have ocular defects and patients without PAX6 function are anophthalmic (see Chapter 18). This concept of haploinsufficiency is a recurring theme for many different transcription factors and corresponding human malformations.
PAX3 and PAX7 encode both homeodomain and Pax DNA-binding domains. The human childhood cancer alveolar rhabdomyosarcoma results from a translocation that results in the formation of a chimeric protein wherein PAX3 or PAX7 (including both DNA domains) is fused to the strong activating domains of the Forkhead family transcription factor FOXO1A. The autosomal dominant human disease Waardenburg’s syndrome type I has been demonstrated to result from mutations in the PAX3 gene. Patients with this syndrome have hearing deficits, ocular defects (dystopia canthorum), and pigmentation abnormalities best typified by a white forelock.
Basic Helix-Loop-Helix Transcription Factors
The bHLH genes are a class of transcription factors that regulate cell fate determination and differentiation in many different tissues during development. At a molecular level, bHLH proteins contain a basic (positively charged) DNA-binding region that is followed by two α helices that are separated by a loop. The α helices have a hydrophilic and a hydrophobic side (amphipathic). The hydrophobic side of the helix is a motif for protein-protein interactions between different members of the bHLH family. This domain is the most conserved region of the bHLH proteins across different species. bHLH proteins often bind other bHLHs (heterodimerize) to regulate transcription. These heterodimers are composed of tissue-specific bHLH proteins bound to ubiquitously expressed bHLH proteins. The powerful prodifferentiation effect of bHLH genes can be repressed by several different mechanisms. For example, inhibitors of differentiation (Id) proteins are HLH proteins that lack the basic DNA-binding motif. When Id proteins heterodimerize with specific bHLH proteins, they prevent binding of these bHLH proteins to their target gene promoter sequences (called E-boxes). Growth factors, which tend to inhibit differentiation, increase the level of Id proteins that sequester bHLH proteins from their target promoters. As well, growth factors can stimulate the phosphorylation of the DNA-binding domain of bHLH proteins, which inhibits their ability to bind to DNA. bHLH genes are crucial for the development of tissues such as muscle (MyoD/Myogenin) and neurons (NeuroD/Neurogenin) in humans (see Chapter 15). MyoD expression was shown to be sufficient to transdifferentiate several different cell lines into muscle cells, demonstrating that it is a master regulator of muscle differentiation. Studies of knockout mice confirmed that MyoD and another bHLH Myf5 are crucial for the differentiation of precursor cells into primitive muscle cells (myoblasts). The differentiation of these myoblasts into fully differentiated muscle cells is controlled by myogenin. Similarly, Mash1 (ASCL1) and Neurogenin1 (NEUROD3) are proneural genes that regulate the formation of neuroblasts from the neuroepithelium (see Chapter 17). Mouse models have shown that these genes are crucial for the specification of different subpopulations of precursors in the developing central nervous system. For example, Mash1 knockout mice had defects in forebrain development, whereas Neurogenin1 knockout mice had defects in cranial sensory ganglia and ventral spinal cord neurons. Specification of these neuroblasts is regulated by other proneural genes known as NeuroD and Math5 (ATOH7). Muscle and neuronal differentiation (see Chapters 15 and 17) are controlled by a cascade of bHLH genes that function at early and at late stages of cellular differentiation. As well, both differentiation pathways are inhibited via signaling through the Notch pathway.
Epigenetics
Our understanding of the role of epigenetic modifications in regulating embryonic development has greatly expanded in recent years. Epigenetics differs from genetics in that it represents the study of heritable changes in the function of genes that cannot be explained by any underlying changes in DNA sequence. This classic definition of epigenetics has now expanded to also include the study of modifications such as histone acetylation and phosphorylation, in which although gene expression is impacted, these modifications are not necessarily inherited. In this chapter, two powerful mechanisms of epigenetic regulation, histone acetylation and DNA methylation are discussed. Disorders of chromatin remodeling include Rett, Rubinstein-Taybi, alpha-thalassemia/X-linked mental retardation syndromes, and various cancers.
Histone Acetylation
Histones are the positively charged nuclear proteins around which genomic DNA is coiled to tightly pack it within the nucleus. Modification of these proteins is a common pathway by which transcription factors regulate the activity of their target promoters. One such modification is acetylation. DNA is less tightly bound to acetylated histones, thus allowing for more open access of transcription factors and other proteins to the promoters of their target genes. Histone acetylation status is controlled by genes such as histone acetyl transferases, which add acetyl groups, and histone deacetylases, which remove acetyl groups. Transcription factors can modify histone acetylation by either recruiting histone acetyl transferases or by recruiting histone deacetylases (Fig. 21-9). Phosphorylation of histones also leads to an opening of the chromatin structure and activation of gene transcription.
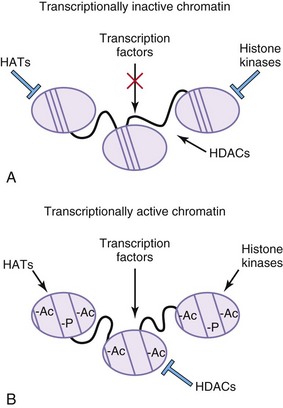
FIGURE 21–9 Epigenetic modifications alter transcriptional properties of chromatin. A, In areas of transcriptionally inactive chromatin, the DNA is tightly bound to the histone cores. The histones are not acetylated or phosphorylated. Histone deacetylases (HDACs) are active, whereas histone acetyl transferases (HATs) and histone kinases are inactive. DNA is highly methylated (Me). B, In areas of transcriptionally active chromatin, the DNA is not as tightly bound to the histone cores and the DNA in unmethylated. The histone proteins are acetylated (Ac) and phosphorylated (-P). HDACs are inactive, whereas HATs and histone kinases are active.
DNA Methylation
In contrast to the dynamic mechanism of histone modifications, DNA methylation is used for the long-term repression of genes. Cytosine residues are rapidly methylated at GC dinucleotides following embryo implantation by enzymes called DNA methyltransferases. During embryonic development, pluripotency genes, which are expressed in embryonic stem cells, are repressed as the cells differentiate. This repression is maintained via methylation of these loci in differentiated cells. This methylation state is only erased in the primordial germ cells to enable re-expression in stem cells. DNA methylation is also used by the body for effective repression of viral genomes that are integrated into our own. These repressive marks are not reset in the primordial germ cells and inherited by the progeny. In cancer, tumor suppressor genes are frequently inactivated by DNA methylation that allows for uncontrolled cellular growth.
Stem Cells: Differentiation Versus Pluripotency
Stem cells (Fig. 21-10) have the property of self-renewal through symmetric (vertical) or asymmetric (horizontal) cell divisions, and under specific conditions in the embryo and adult, can give rise to all of the differentiated cell types in the body (totipotent or pluripotent). Several types of stem cell populations have been characterized: embryonic stem cells (ESCs), adult stem cells, and cancer stem cells. ESCs, derived from the inner cell mass of the blastula, are pluripotent and can give rise to all differentiated cell types from the ectoderm, endoderm, and mesoderm, the primary germ layers, but do not contribute to extra-embryonic tissues. ESCs express several transcription factors, such as Sox2 and Oct-4, that repress differentiation.
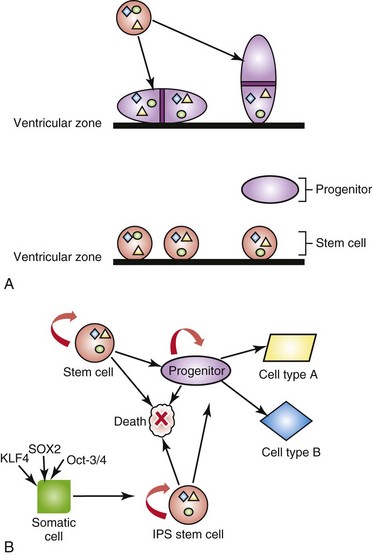
FIGURE 21–10 Neural stem cells and induced pluripotent stem cells. A, Adult or embryonic stem cells can either divide symmetrically, giving rise to two equivalent daughter stem cells, (“vertical” cell division; the plane of mitosis is perpendicular to the ventricular surface) or asymmetrically, giving rise to a daughter stem cell and a nervous system progenitor cell (“horizontal” cell division: the plane of mitosis is parallel to the ventricular surface). In this example, the progenitor cell does not retain the nuclear or cytoplasmic factors (colored shapes) that remain in the stem cell; however, the progenitor cell expresses new proteins (e.g. receptor tyrosine kinases) in its plasma membrane. B, Stem cells and induced pluripotent stem cells (iPS) have the capacity for self-renewal, cell death and/or to become progenitors. Progenitor cells have a more limited capacity for self-renewal, but also can differentiate into various cell types or undergo cell death. Adult, differentiated somatic cells, such as skin fibroblasts, can be reprogrammed into iPS with the introduction of the master transcription factors SOX2, Oct-3/4, or KLF4.
Adult stem cells are found in relative abundance in differentiated tissues and organs that undergo rapid regeneration, such as the bone marrow, hair follicles, and the intestinal mucosal epithelium. However, there are “nests” of adult stem cells in many other tissues, including those that have been previously considered nonregenerative, such as the central nervous system and retina; these stem cell populations are small and located in the subventricular zone and ciliary margins, respectively. Hematopoietic stem cells derived from bone marrow, peripheral blood, and umbilical cord sources are now routinely used to treat primary immunodeficiencies, various inherited metabolic disorders and as a “rescue” strategy following myeloablative cancer treatments.
Cancer stem cells (CSC) are under intense study since it has become evident through the study of leukemias and solid tumors (e.g. colorectal cancer, malignant gliomas) that a small population of these cells, identified by various cell surface markers (such as CD133 in solid tumors), are often resistant to cancer treatments such as radiation or chemotherapy. Investigators are focusing their efforts on eradicating the CSC population in addition to standard therapies to result in higher cure rates.
One can harness the power of stem cells to repair degenerative disorders such as Parkinson’s disease and tissues severely damaged by ischemia (stroke) and trauma (spinal cord injury). However, researchers have been limited by the available sources of stem cells from the embryo or adult. Hence, there has been tremendous interest in de-differentiating somatic cells such as epithelial cells and fibroblasts from adults into induced pluripotent stem cells (iPS). Recent studies have identified several key master transcription factors (Fig. 21-10, panel B), such as Oct4, Sox2, and KLF4, or Nanog, that can reprogram differentiated cells into pluripotent cells. These iPS cells can be manipulated using nonviral means of gene delivery and have the potential to treat the majority of human diseases where cell regeneration may restore structure and/or function.
Summary of Common Signaling Pathways Used During Development
References and Suggested Reading
Abate-Shen C. Deregulated homeobox gene expression in cancer: Cause or consequence? Nat Rev Cancer. 2002;2:777.
Appel B, Eisen JS. Retinoids run rampant: Multiple roles during spinal cord and motor neuron development. Neuron. 2003;40:461.
Ausio J, Levin DB, De Amorim GV, et al. Syndromes of disordered chromatin remodeling. Clin Genet. 2003;64:83.
Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118-132.
Charron F, Tessier-Lavigne M. Novel brain wiring functions for classical morphogens: A role as graded positional cues in axon guidance. Development. 2005;132:2251.
Chizhikov VV, Millen KJ. Roof plate–dependent patterning of the vertebrate dorsal central nervous system. Dev Biol. 2005;277:287.
Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2006;438:937.
Dellovade T, Romer JT, Curran T, et al. The hedgehog pathway and neurological disorders. Annu Rev Neurosci. 2006;29:539.
De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer Disease. Nat Rev Neurol. 2010;6:99.
Eisenberg LM, Eisenberg CA. Wnt signal transduction and the formation of the myocardium. Dev Biol. 2006;293:305.
Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433.
Goetz SC, Anderson KV. The primary cilium: a signaling centre during vertebrate development. Nat Rev Genet. 2010;11:331.
Gumbiner BM. Regulation of Cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622.
Hooper JE, Scott MP. Communicating with hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306.
Kageyama R, Ohtsuka T, Hatakeyama J, et al. Roles of bHLH genes in neural stem cell differentiation. Exp Cell Res. 2005;306:343.
Kaufman DS. Toward clinical therapies using hematopoietic cells derived from human pluripotent stem cells. Blood. 2009;114:3513.
Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216.
Laird DW. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2009;20:92.
Larsson J, Karlsson S. The role of Smad signaling in hematopoiesis. Oncogene. 2005;24:5676.
Lee JE. Basic helix-loop-helix genes in neural development. Curr Opin Neurobiol. 1997;7:13.
Li F, Chong ZZ, Maiese K. Winding through the WNT pathway during cellular development and demise. Histol Histopathol. 2006;21:103.
Li Z, Wang H, Eyler CE, et al. Turning cancer stem cells inside out: An exploration of glioma stem cell signaling pathways. J Biol Chem. 2009;284:16705.
Lohela M, Bry M, Tammela T, et al. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;2:154.
Louvi A, Artavanis-Tsakonas S. Notch signaling in vertebrate neural development. Nat Rev Neurosci. 2006;7:93.
Lupo G, Harris WA, Lewis KE. Mechanisms of ventral patterning in the vertebrate nervous system. Nat Rev Neurosci. 2006;7:103.
Marino S. Medulloblastoma: Developmental mechanisms out of control. Trends Mol Med. 2005;11:17.
Marlétaz F, Holland L, Laudet V, et al. Retinoic acid signaling and the evolution of chordates. Int J Biol Sci. 2006;2:38.
Park CB, Dufort D, Elsevier Trophoblast Research Award Lecture. The multifaceted role of nodal signaling during mammalian reproduction. Placenta 32 (supplement B). 2011;25:S125.
Parker MH, Seale P, Rudnicki MA. Looking back to the embryo: Defining transcriptional networks in adult myogenesis. Nat Rev Genet. 2003;4:497.
Pearson JC, Lemons D, McGinnis W. Modulating Hox gene functions during animal body patterning. Nat Rev Genet. 2005;6:893.
Robson EJ, He SJ, Eccles MR. A PANorama of PAX genes in cancer and development. Nat Rev Cancer. 2006;6:52.
Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425.
Sahni V, Kessler JA. Stem cell therapies for spinal cord injury. Nat Rev Neurol. 2010;6:363.
Staal FJT, Clevers HC. Wnt signaling and haematopoiesis: A Wnt–Wnt situation. Nat Rev Immunol. 2005;5:21.
Stecca B, Ruiz i Altaba A. Brain as a paradigm of organ growth: Hedgehog-Gli signaling in neural stem cells and brain tumors. J Neurobiol. 2005;64:476.
Sylvie J, Ellen C, Kris V. The role of Wnt in cell signaling and cell adhesion during early vertebrate development. Front Biosci. 2011;17:2352.
Tian T, Meng AM. Nodal signals pattern vertebrate embryos. Cell Mol Life Sci. 2006;63:672.
Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog-Patched-Gli pathway in human development and disease. Am J Hum Genet. 2000;67:1047.
Wan M, Cao X. BMP signaling in skeletal development. Biochem Biophys Res Commun. 2005;328:651.
Wigle JT, Eisenstat DD. Homeobox genes in vertebrate forebrain development and disease. Clin Genet. 2008;73:212.
Yoon K, Gaiano N. Notch signaling in the mammalian central nervous system: Insights from mouse mutants. Nat Neurosci. 2005;8:709.
Zhu AJ, Scott MP. Incredible journey: How do developmental signals travel through tissue? Genes Dev. 2004;18:2985.