Chapter 20 Human Birth Defects
We ought not to set them aside with idle thoughts or idle words about “curiosities” or “chances.” Not one of them is without meaning; not one that might not become the beginning of excellent knowledge, if only we could answer the question—why is it rare, or being rare, why did it in this instance happen?
Birth defects (congenital anomalies) are developmental disorders present at birth. Birth defects are the leading cause of infant mortality and may be structural, functional, metabolic, behavioral, or hereditary.
Classification of Birth Defects
The most widely used reference guide for classifying birth defects is the International Classification of Diseases; however, no single classification has universal appeal. Each is limited, having been designed for a particular purpose. Attempts to classify human birth defects, especially those that result from errors of morphogenesis (development of form), reveal the frustration and obvious difficulties in the formulation of concrete proposals that could be used in medical practice. A practical classification system for birth defects that takes into consideration the time at onset of the injury, possible etiology, and pathogenesis is now widely accepted among clinicians.
Teratology: Study of Abnormal Development
Teratology is the branch of science that studies the causes, mechanisms, and patterns of abnormal development. A fundamental concept in teratology is that certain stages of embryonic development are more vulnerable to disruption than others. Until the 1940s, it was generally believed that human embryos were protected from environmental agents, such as drugs, viruses and chemicals by their extraembryonic/fetal membranes (amnion and chorion) and their mothers’ uterine and abdominal walls.
In 1941, the first well-documented cases were reported that an environmental agent (rubella virus) could produce severe birth defects, such as cataracts, cardiac defects, and deafness, if the rubella infection was present during the critical period of development of the eyes, heart, and ears. In the 1950s, severe limb defects and other developmental disruptions were found in infants of mothers who had consumed a sedative called thalidomide during early pregnancy. These discoveries, more than six decades ago, focused worldwide attention on the role of drugs and viruses in the etiology (causes) of human birth defects. It is estimated that 7% to 10% of human birth defects result from the disruptive actions of drugs, viruses, and environmental toxins.
More than 20% of infant deaths in North America are attributed to birth defects. Major structural defects; for example, spina bifida cystica, a severe type of vertebral defect in which part of the neural tube fails to fuse (see Chapter 17, Fig. 17-15), are observed in approximately 3% of neonates. Additional birth defects can be detected after birth; thus, the incidence reaches approximately 6% in 2-year-olds and 8% in 5-year-olds.
The causes of birth defects are often divided into:
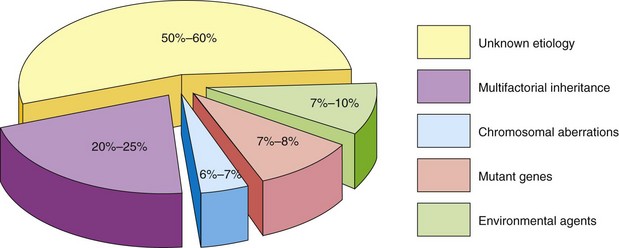
For 50% to 60% of birth defects, the etiology is unknown (Fig. 20-1). The defects may be single or multiple and of major or minor clinical significance. Single minor defects are present in approximately 14% of neonates. Defects of the external ears, for example, are of no serious medical significance, but they may indicate the possible presence of associated major defects. For example, the presence of a single umbilical artery alerts the clinician to the possible presence of cardiovascular and renal anomalies. Ninety percent of infants with three or more minor defects also have one or more major defects. Of the 3% born with clinically significant defects, 0.7% have multiple major defects; most of these infants die during infancy. Major developmental defects are much more common in early embryos (10%–15%); however, most of them abort spontaneously during the first 6 weeks. Chromosome abnormalities are present in 50% to 60% of early, spontaneously aborted embryos.
Birth Defects Caused by Genetic Factors*
Numerically, genetic factors are the most important causes of birth defects. It has been estimated that they cause approximately one third of all defects (Fig. 20-1). Nearly 85% of all defects have no known causes. Any mechanism as complex as mitosis or meiosis may occasionally malfunction. Chromosomal abnormalities or aberrations are present in 6% to 7% of zygotes (single-cell embryos).
Many of these early abnormal embryos never undergo normal cleavage and become blastocysts. In vitro studies of cleaving zygotes less than 5 days old have revealed a high incidence of abnormalities. More than 60% of day 2 cleaving zygotes were found to be abnormal. Many defective zygotes, blastocysts, and 3-week-old embryos abort spontaneously, and the overall frequency of chromosome abnormalities in these embryos is at least 50%.
Two kinds of changes occur in chromosome complements: numerical and structural. The changes may affect the sex chromosomes and/or the autosomes (chromosomes other than sex chromosomes). In some instances, both kinds of chromosomes are affected. Persons with chromosome abnormalities usually have characteristic phenotypes (morphologic characteristics), such as the physical characteristics of infants with Down syndrome (see Fig. 20-6). They often look more like other persons with the same chromosome abnormality than their own siblings (brothers or sisters). This characteristic appearance results from genetic imbalance. Genetic factors initiate defects by biochemical or other means at the subcellular, cellular, or tissue level. The abnormal mechanisms initiated by the genetic factors may be identical or similar to the causal mechanisms initiated by a teratogen (e.g., a drug).
Numerical Chromosomal Abnormalities
In the United States, approximately one in 120 live-born infants has a chromosomal abnormality. Numerical aberrations of chromosomes usually result from nondisjunction, an error in cell division in which there is failure of a chromosomal pair or two chromatids of a chromosome to disjoin during mitosis or meiosis. As a result, the chromosomal pair or chromatids pass to one daughter cell and the other daughter cell receives neither (Fig. 20-2). Nondisjunction may occur during maternal or paternal gametogenesis (see Chapter 2). The chromosomes in somatic cells are normally paired; they are called homologous chromosomes (homologs). Normal human females have 22 pairs of autosomes plus two X chromosomes, whereas normal males have 22 pairs of autosomes plus one X chromosome and one Y chromosome.
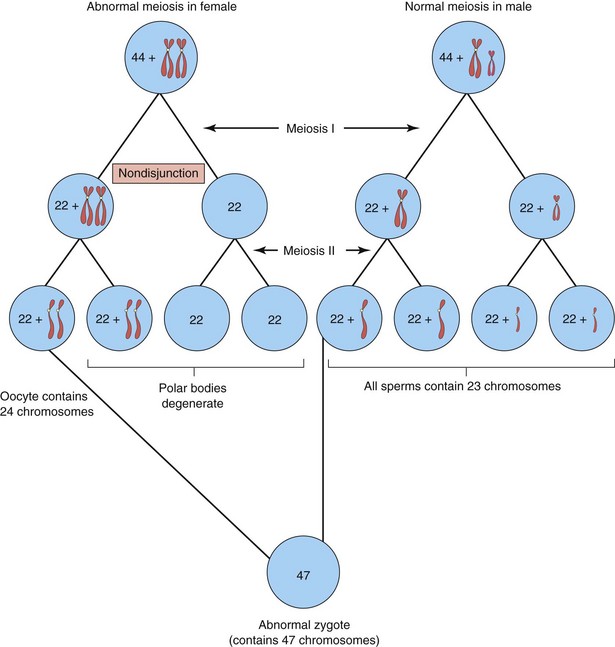
FIGURE 20–2 Diagram showing nondisjunction of chromosomes during the first meiotic division of a primary oocyte resulting in an abnormal oocyte with 24 chromosomes. Subsequent fertilization by a normal sperm produces a zygote with 47 chromosomes—aneuploidy—deviation from the human diploid number of 46.
Glossary of Teratologic Terms
A birth defect is a structural abnormality of any type; however, not all variations of development are anomalies. Anatomic variations are common; for example, bones vary among themselves, not only in their basic shape, but in lesser details of surface structure. There are four clinically significant types of birth defect: malformation, disruption, deformation, and dysplasia.
Other descriptive terms are used to describe infants with multiple defects and terms have evolved to express causation and pathogenesis.
Whereas a sequence is a pathogenetic and not a causal concept, a syndrome often implies a single cause, such as trisomy 21 (Down syndrome). In both cases, however, the pattern of defects is known or considered to be pathogenetically related. In the case of a sequence, the primary initiating factor and cascade of secondary developmental complications are known. For example, the Potter sequence, attributed to oligohydramnios, results from either renal agenesis or leakage of amniotic fluid. An association, in contrast, refers to statistically, not pathogenetically or causally related defects. One or more sequences, syndromes, or field defects may very well constitute an association.
Dysmorphology is an area of clinical genetics that is concerned with the diagnosis and interpretation of patterns of structural defects. Recurrent patterns of birth defects are the hallmarks of syndrome recognition. Identifying these patterns in individuals has resulted in improved understanding of the etiology and pathogenesis of these conditions.
Inactivation of Genes
During embryogenesis, one of the two X chromosomes in female somatic cells is randomly inactivated and appears as a mass of sex chromatin (see Chapter 6). Inactivation of genes on one X chromosome in somatic cells of female embryos occurs during implantation. X inactivation is important clinically because it means that each cell from a carrier of an X-linked disease has the mutant gene causing the disease, either on the active X chromosome or on the inactivated X chromosome that is represented by sex chromatin. Uneven X inactivation in monozygotic (identical twins) twins is one reason given for discordance for a variety of birth defects. The genetic basis for discordance is that one twin preferentially expresses the paternal X and the other the maternal X.
Aneuploidy and Polyploidy
Changes in chromosome number represent either aneuploidy or polyploidy. Aneuploidy is any deviation from the human diploid number of 46 chromosomes. An aneuploid is an individual who has a chromosome number that is not an exact multiple of the haploid number of 23 (e.g., 45 or 47). A polyploid is an individual who has a chromosome number that is a multiple of the haploid number of 23 other than the diploid number (e.g., 69; see Fig. 20-10).
The principal cause of aneuploidy is nondisjunction during cell division (Fig. 20-2), resulting in an unequal distribution of one pair of homologous chromosomes to the daughter cells. One cell has two chromosomes and the other has neither chromosome of the pair. As a result, the embryo’s cells may be hypodiploid (45, X0 as in Turner syndrome [Figs. 20-3 to 20-5]), or hyperdiploid (usually 47, as in trisomy 21 or Down syndrome [Fig. 20-6]). Embryos with monosomy—missing a chromosome—usually die. Approximately 99% of embryos lacking a sex chromosome (45, X0 as in Turner syndrome) abort spontaneously (Fig. 20-5).
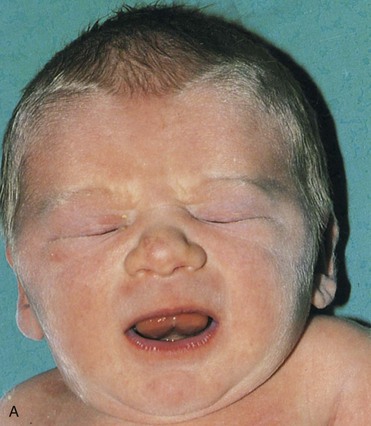
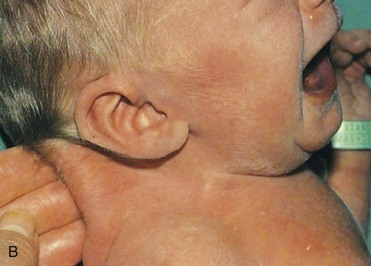
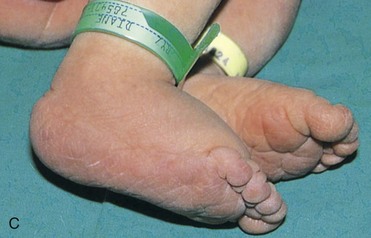
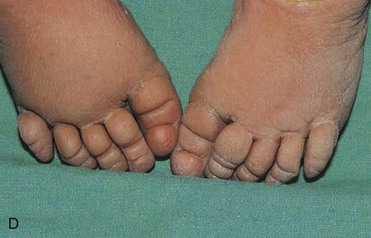
FIGURE 20–3 Female infant with Turner syndrome (45, X0). A, Face of the infant with Turner syndrome. B, Lateral view of infant’s head and neck showing a short webbed neck, prominent ears. These infants have defective gonadal development (and gonadal dysgenesis). C, Infant’s feet showing the characteristic lymphedema (puffiness and swelling), a useful diagnostic sign. D, Lymphedema of toes, a condition that usually leads to nail underdevelopment (hypoplasia).
(Courtesy of Dr. A.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada.)
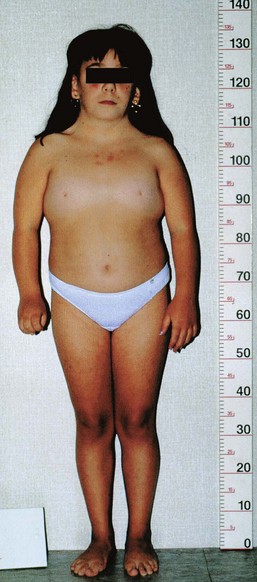
FIGURE 20–4 Turner syndrome in a 14-year-old girl. Note the features of the syndrome: short stature, webbed neck, absence of sexual maturation, broad shield-like chest with widely spaced nipples, and lymphedema of the hands and feet.
(Courtesy of Dr. F. Antoniazzi and Dr. V. Fanos, Department of Pediatrics, University of Verona, Verona, Italy.)
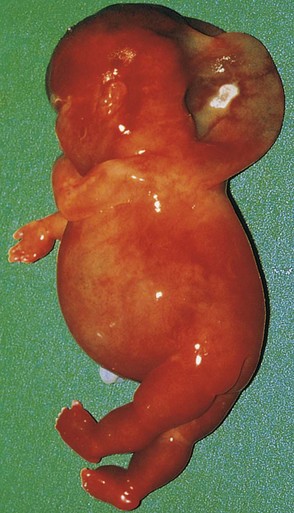
FIGURE 20–5 Female fetus (16 weeks) with Turner syndrome. Note the excessive accumulation of watery fluid (hydrops) and the large cystic hygroma (lymphangioma) in the posterior head and cervical region. The hygroma causes the loose neck skin and webbing seen postnatally (see Fig. 20-3B).
(Courtesy of Dr. A.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada.)

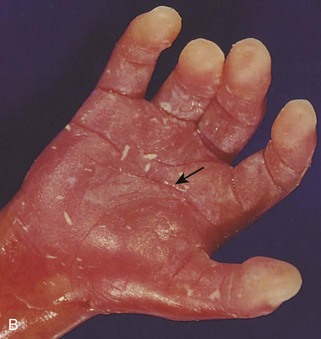

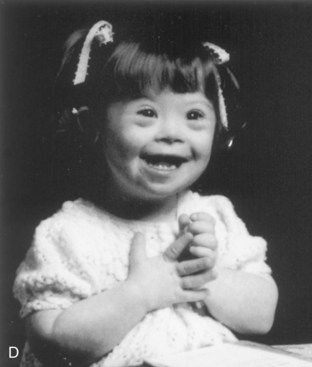
FIGURE 20–6 A, Anterior view of a female fetus (16.5 weeks) with Down syndrome. B, Hand of fetus. Note the single, transverse palmar flexion crease (simian crease, arrow) and the clinodactyly (incurving) of the fifth digit. C, Anterior view of the faces of dizygotic male twins that are discordant for Down syndrome (trisomy 21). The one on the right is smaller than the unaffected twin. The twin on the right developed from a zygote that contained an extra 21 chromosome. Note the characteristic facial features of Down syndrome in this infant: upslanting palpebral fissures, epicanthal folds, and flat nasal bridge. D, A 2- -year-old girl with Down syndrome.
-year-old girl with Down syndrome.
(A and B, Courtesy of Dr. D.K. Kalousek, Department of Pathology, University of British Columbia, Vancouver, British Columbia, Canada; C and D, Courtesy of Dr. A.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada.)
Turner Syndrome
Approximately 1% of monosomy X female embryos survive; the incidence of 45, X0 (Turner syndrome) in female neonates is approximately 1 in 8000 live births. Half of the affected individuals have 45, X0; the other half have a variety of abnormalities of a sex chromosome. The phenotype of Turner syndrome is female (Figs. 20-3 and 20-4). Secondary sexual characteristics do not develop in 90% of affected females, and hormone replacement is required.
Phenotype refers to the morphologic characteristics of an individual as determined by the genotype and the environment in which it is expressed. The monosomy X chromosome abnormality is the most common cytogenetic abnormality observed in fetuses that abort spontaneously (Fig. 20-5); it accounts for approximately 18% of all abortions caused by chromosome abnormalities. The error in gametogenesis (nondisjunction) that causes monosomy X (Turner syndrome), when it can be traced, is in the paternal gamete (sperm) in approximately 75% of cases; that is, it is the paternal X chromosome that is usually missing. The most frequent chromosome constitution in Turner syndrome is 45, X0; however, nearly 50% of these people have other karyotypes (the chromosome characteristics of an individual cell or a cell line).
Trisomy of Autosomes
The presence of three chromosome copies in a given chromosome pair is called trisomy. Trisomies are the most common abnormalities of chromosome number. The usual cause of this numerical error is meiotic nondisjunction of chromosomes (Fig. 20-2), resulting in a gamete with 24 instead of 23 chromosomes and subsequently in a zygote with 47 chromosomes.
Trisomy of the autosomes is associated with three main syndromes (Table 20-1):
Table 20–1 Trisomy of Autosomes
| CHROMOSOMAL ABERRATION/SYNDROME | INCIDENCE | USUAL CLINICAL MANIFESTATIONS |
|---|---|---|
| Trisomy 21 (Down syndrome)* (see Fig. 20-6) |
1 : 800 | Mental deficiency; brachycephaly, flat nasal bridge; upward slant to palpebral fissures; protruding tongue; transverse palmar flexion crease; clinodactyly of fifth digit; congenital heart defects; gastrointestinal tract abnormalities |
| Trisomy 18 syndrome (Edwards syndrome)† (see Fig.20-7) |
1 : 8000 | Mental deficiency; growth retardation; prominent occiput; short sternum; ventricular septal defect; micrognathia; low-set malformed ears, flexed digits, hypoplastic nails; rocker-bottom feet |
| Trisomy 13 syndrome (Patau syndrome)† (see Fig. 20-8) |
1 : 12,000 | Mental deficiency; severe central nervous system malformations; sloping forehead; malformed ears, scalp defects; microphthalmia; bilateral cleft lip and/or palate; polydactyly; posterior prominence of the heels |
* The importance of this disorder in the overall problem of mental deficiency is indicated by the fact that persons with Down syndrome represent 10% to 15% of institutionalized mental defectives. The incidence of trisomy 21 at fertilization is greater than at birth; however, 75% of embryos are spontaneously aborted and at least 20% are stillborn.
† Infants with this syndrome rarely survive beyond 6 months.
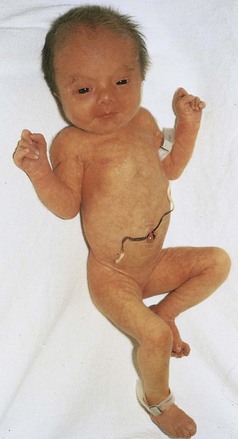
FIGURE 20–7 Female neonate with trisomy 18. Note the growth retardation, clenched fists with characteristic positioning of the fingers (second and fifth ones overlapping the third and fourth ones), short sternum, and narrow pelvis.
(Courtesy of Dr. A.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada.)
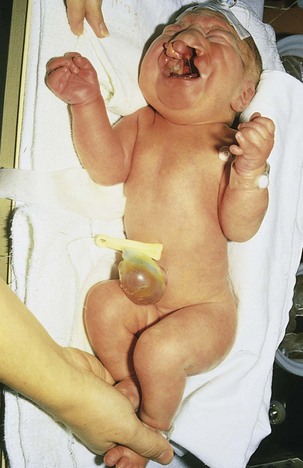
FIGURE 20–8 Female neonate with trisomy 13. Note the bilateral cleft lip, low-set malformed left ear, and polydactyly (extra digits). A small omphalocele (herniation of viscera into umbilical cord) is also present.
(Courtesy of Dr. A.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada.)
Infants with trisomy 13 and trisomy 18 are severely malformed and mentally deficient; they usually die early in infancy. More than half of trisomic embryos spontaneously abort early. Trisomy of the autosomes occurs with increasing frequency as maternal age increases. For example, trisomy 21 occurs once in approximately 1400 births in mothers ages 20 to 24 years, but once in approximately 25 births in mothers 45 years and older (Table 20-2). Trisomy 13 is the most common chromosomal abnormality in neonates.
Table 20–2 Incidence of Down Syndrome in Neonates
| MATERNAL AGE (YEARS) | INCIDENCE |
|---|---|
| 20–24 | 1 : 1400 |
| 25–29 | 1 : 1100 |
| 30–34 | 1 : 700 |
| 35 | 1 : 350 |
| 37 | 1 : 225 |
| 39 | 1 : 140 |
| 41 | 1 : 85 |
| 43 | 1 : 50 |
| 45+ | 1 : 30 |
Errors in meiosis occur with increasing maternal age and the most common aneuploidy seen in older mothers is trisomy 21. Because of the current trend of increasing maternal age, it has been estimated that by the end of this decade, children born to women older than 34 years will account for 39% of infants with trisomy 21. Translocation or mosaicism occurs in approximately 5% of the affected children. Mosaicism, two or more cell types containing different numbers of chromosomes (normal and abnormal), leads to a less severe phenotype and the IQ of the child may be nearly normal.
Trisomy of Sex Chromosomes
This type of trisomy is a common disorder (Table 20-3); however, because there are no characteristic physical findings in infants or children, this disorder is not usually detected until puberty (Fig. 20-9). Sex chromatin studies were useful in the past for detecting some types of trisomy of the sex chromosomes because two masses of sex chromatin are present in nuclei of XXX females (trisomy X), and nuclei of XXY males (Klinefelter syndrome) contain a mass of sex chromatin (see Chapter 6). Today, diagnosis is best achieved by chromosome analysis or other molecular cytogenetic techniques.
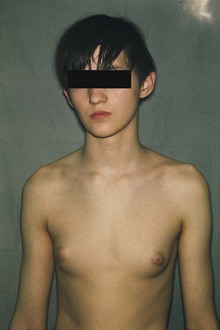
FIGURE 20–9 Adolescent male with Klinefelter syndrome (XXY trisomy). Note the presence of breasts; approximately 40% of males with this syndrome have gynecomastia (development of breasts) and small testes.
(Courtesy of Children’s Hospital, Winnipeg, Manitoba, Canada.)
Tetrasomy and Pentasomy
Tetrasomy and pentasomy of the sex chromosomes also occur. Persons with these abnormalities have four or five sex chromosomes, respectively; the following chromosome complexes have been reported in females: 48, XXXX and 49, XXXXX; and in males: 48, XXXY, 48, XXYY, 49, XXXYY, and 49, XXXXY. The extra sex chromosomes do not accentuate sexual characteristics; however, usually the greater the number of sex chromosomes present, the greater the severity of mental deficiency and physical impairment.
Mosaicism
A person who has at least two cell lines with two or more different genotypes (genetic constitutions) is a mosaic. Either the autosomes or sex chromosomes may be involved. Usually the defects are less serious than in persons with monosomy or trisomy; for instance, the features of Turner syndrome are not as evident in 45, X/46, XX mosaic females as in the usual 45, X females. Mosaicism usually results from nondisjunction during early cleavage of the zygote (see Chapter 2). Mosaicism resulting from loss of a chromosome by anaphase lagging also occurs; the chromosomes separate normally but one of them is delayed in its migration and is eventually lost.
Triploidy
The most common type of polyploidy is triploidy (69 chromosomes). Triploid fetuses have severe intrauterine growth retardation with severe head-body disproportion (Fig. 20-10). Although triploid fetuses can be live-born, they do not survive very long. Triploidy most frequently results from fertilization of an oocyte by two sperms (dispermy). Failure of one of the meiotic divisions (see Chapter 2), resulting in a diploid oocyte or sperm, could also account for some cases. Triploid fetuses account for approximately 20% of chromosomally abnormal spontaneous abortions.
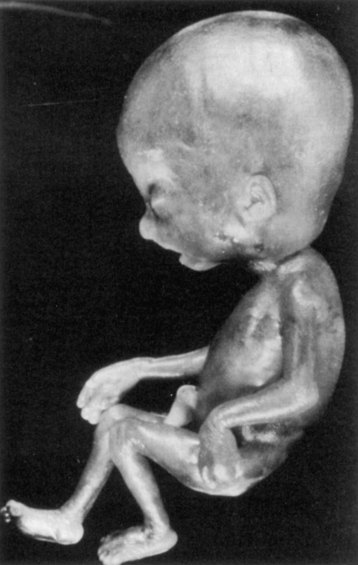
FIGURE 20–10 Triploid fetus (69 chromosomes) illustrating severe head-to-body disproportion. Triploid fetuses account for nearly 20% of chromosomally abnormal miscarriages.
(From Crane JP: Ultrasound evaluation of fetal chromosome disorders. In Callen PW [ed]: Ultrasonography in Obstetrics and Gynecology, 3rd ed. Philadelphia, WB Saunders, 1994.)
Tetraploidy
Doubling of the diploid chromosome number from 46 to 92 (tetraploidy) probably occurs during the first cleavage division of the zygote (see Chapter 2). Division of this abnormal zygote subsequently results in an embryo with cells containing 92 chromosomes. Tetraploid embryos abort very early and often all that is recovered is an empty chorionic sac (once called a “blighted embryo”).
Structural Chromosomal Abnormalities
Most structural chromosomal abnormalities result from chromosome breakage, followed by reconstitution in an abnormal combination (Fig. 20-11). Chromosome breakage may be induced by various environmental factors, such as ionizing radiation, viral infections, drugs, and chemicals. The type of structural chromosome abnormality depends on what happens to the broken pieces. The only two aberrations of chromosome structure that are likely to be transmitted from a parent to an embryo are structural rearrangements, such as inversion and translocation. Overall, structural abnormalities of chromosomes are present in about 1 in 375 neonates.
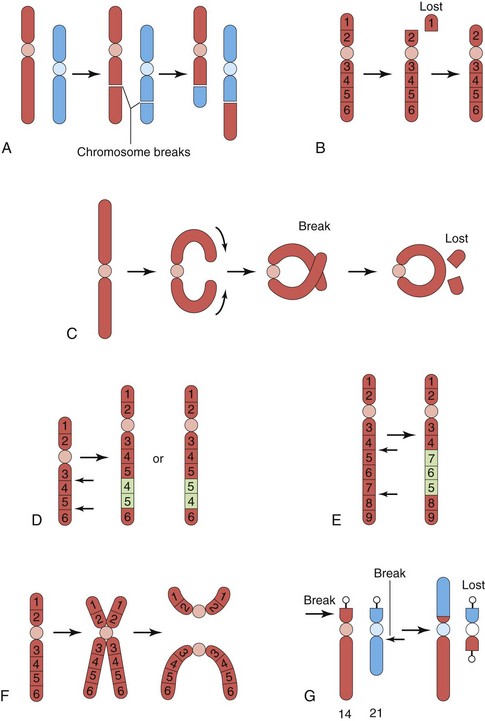
FIGURE 20–11 Diagrams illustrating various structural chromosomal abnormalities. A, Reciprocal translocation. B, Terminal deletion. C, Ring chromosome. D, Duplication. E, Paracentric inversion. F, Isochromosome. G, Robertsonian translocation. Arrows indicate how the structural abnormalities are produced.
(Modified from Nussbaum RL, McInnes RR, Willard HE: Thompson and Thompson Genetics in Medicine, 6th ed. Philadelphia, WB Saunders, 2004)
Translocation
This abnormality is the transfer of a piece of one chromosome to a nonhomologous chromosome. If two nonhomologous chromosomes exchange pieces, it is called a reciprocal translocation (Fig. 20-11A and G). Translocation does not necessarily cause abnormal development. Persons with a translocation between a number 21 chromosome and a number 14 chromosome, for example (Fig. 20-11G), are phenotypically normal. Such persons are called balanced translocation carriers; they have a tendency, independent of age, to produce germ cells with an abnormal translocation chromosome. Three to 4% of infants with Down syndrome have translocation trisomies, that is, the extra 21 chromosome is attached to another chromosome.
Deletion
When a chromosome breaks, part of it may be lost (Fig. 20-11B). A partial terminal deletion from the short arm of chromosome 5 causes the cri du chat syndrome (Fig. 20-12). Affected infants have a weak cat-like cry, microcephaly (small neurocranium), severe mental deficiency, and congenital heart disease. A ring chromosome is a type of deletion chromosome from which both ends have been lost, and the broken ends have rejoined to form a ring-shaped chromosome (Fig. 20-11C). Ring chromosomes are rare but they have been found for all chromosomes. These abnormal chromosomes have been described in persons with 45, X (Turner syndrome), trisomy 18 (Edwards syndrome), and other structural chromosomal abnormalities.
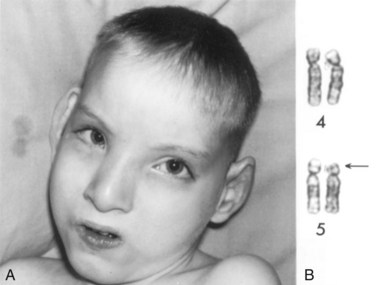
FIGURE 20–12 A, Male child with cri du chat syndrome (cat-like cry). Note microcephaly (small neurocranium) and hypertelorism (increased distance between orbits). B, Partial karyotype of this child showing a terminal deletion of the short arm (end) of chromosome number 5. The arrow indicates the site of the deletion.
(A, From Gardner EJ: Principles of Genetics, 5th ed. New York, John Wiley & Sons, 1975. B, Courtesy of the late Dr. M. Ray, Department of Human Genetics, University of Manitoba, Winnipeg, Manitoba, Canada.)
Microdeletions and Microduplication
High-resolution banding techniques have allowed detection of very small interstitial and terminal deletions in a number of chromosomal disorders. An acceptable resolution of chromosome banding on a routine analysis reveals 550 bands per haploid set, whereas high-resolution chromosome banding reveals up to 1300 bands per haploid set. Because the deletions span several contiguous genes, these disorders, as well as those with microduplications, are referred to as contiguous gene syndromes (Table 20-4). Two examples are:
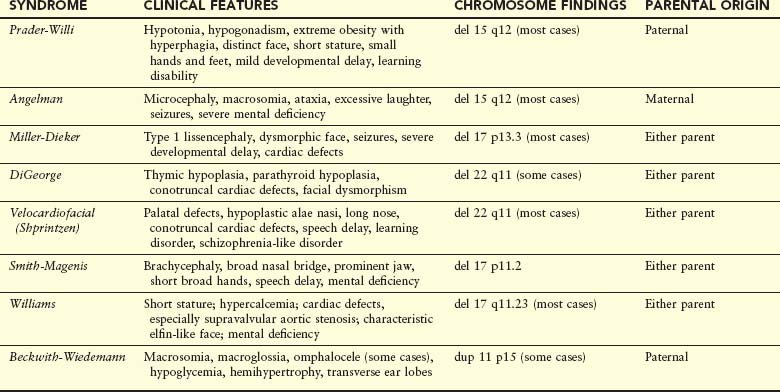
PWS and AS are often associated with a visible deletion of band q12 on chromosome 15. The clinical phenotype is determined by the parental origin of the deleted chromosome 15. If the deletion arises in the mother, AS occurs; if passed on by the father, the child exhibits the PWS phenotype. This suggests that the phenomenon of genetic imprinting, whereby differential expression of genetic material is dependent on the sex of the transmitting parent.
Molecular Cytogenetics
Several new methods for merging classic cytogenetics with DNA technology have facilitated a more precise definition of chromosome abnormalities, location, or origins, including unbalanced translocations, accessory or marker chromosomes, and gene mapping. One new approach to chromosome identification is based on fluorescent in situ hybridization (FISH), whereby chromosome-specific DNA probes can adhere to complementary regions located on specific chromosomes. This allows improved identification of chromosome location and number in metaphase spreads or even in interphase cells. FISH techniques using interphase cells may soon obviate the need to culture cells for specific chromosome analysis, such as in the case of prenatal diagnosis of fetal trisomies.
Studies using subtelomeric FISH probes in individuals with mental deficiency of unknown etiology, with or without birth defects, have identified submicroscopic chromosome deletions or duplications in 5% to 10% of these individuals. Alterations in DNA sequence copy number are present in solid tumors, and are found in association with developmental abnormalities and/or mental deficiency.
Comparative genomic hybridization (CGH) can detect and map these changes in specific regions of the genome. Microarray-based CGH (array comparative genomic hybridization) is now being used to identify genomic rearrangements in individuals who had been previously considered to have mental deficiency or multiple birth defects of unknown etiology, despite normal test results from traditional chromosome or gene analysis. Thus, these investigations have become important in the routine evaluation of patients with previously unexplained mental deficiency, autism, and multiple congenital anomalies.
Duplications
These abnormalities may be represented as a duplicated part of a chromosome, within a chromosome (Fig. 20-11D), attached to a chromosome or as a separate fragment. Duplications are more common than deletions and are less harmful because there is no loss of genetic material. However, there is often a resulting clinical effect on the phenotype leading to either mental impairment or birth defects in individuals with chromosome duplication. Duplication may involve part of a gene, a whole gene, or a series of genes.
Inversion
This is a chromosomal aberration in which a segment of a chromosome is reversed. Paracentric inversion is confined to a single arm of the chromosome (Fig. 20-11E), whereas pericentric inversion involves both arms and includes the centromere. Carriers of pericentric inversions are at risk of having offspring with abnormalities because of unequal crossing over and malsegregation at meiosis.
Isochromosomes
The abnormality resulting in isochromosomes occurs when the centromere divides transversely instead of longitudinally (Fig. 20-11E). An isochromosome is a chromosome in which one arm is missing and the other is duplicated. An isochromosome appears to be the most common structural abnormality of the X chromosome. Persons with this abnormality are often short in stature and have other stigmata of Turner syndrome. These characteristics are related to the loss of an arm of an X chromosome.
Birth Defects Caused by Mutant Genes
Seven to 8% of birth defects are caused by gene defects (Fig. 20-1). A mutation, usually involving a loss or change in the function of a gene, is any permanent, heritable change in the sequence of genomic DNA. Because a random change is unlikely to lead to an improvement in development, most mutations are deleterious and some are lethal.
The mutation rate can be increased by a number of environmental agents, such as large doses of ionizing radiation. Defects resulting from gene mutations are inherited according to mendelian laws; consequently, predictions can be made about the probability of their occurrence in the affected person’s children and other relatives. An example of a dominantly inherited birth defect—achondroplasia (Fig. 20-13)—results from a G-to-A transition mutation at nucleotide 1138 of the cDNA in the fibroblast growth factor receptor 3 gene on chromosome 4p. Other defects, such as congenital suprarenal hyperplasia (see Fig. 20-18) and microcephaly, are attributed to autosomal recessive inheritance. Autosomal recessive genes manifest themselves only when homozygous; as a consequence, many carriers of these genes (heterozygous persons) remain undetected.
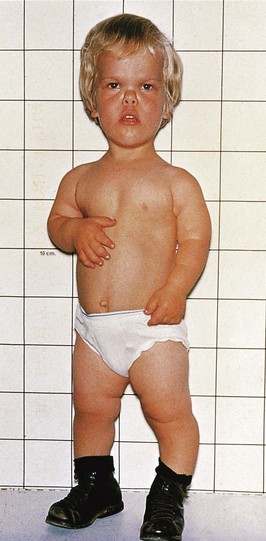
FIGURE 20–13 A young boy with achondroplasia showing short stature, short limbs and fingers, normal length of trunk, bowed legs, a relatively large head, prominent forehead, and depressed nasal bridge.
(Courtesy of Dr. A.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada.)
Fragile X syndrome is the most commonly inherited cause of moderate mental deficiency (Fig. 20-14). It is one of more than 200 X-linked disorders associated with mental impairment. The fragile X syndrome has a frequency of 1 in 1500 male births and may account for much of the excess of males in the mentally deficient population. Diagnosis of this syndrome can be confirmed by chromosome analysis demonstrating the fragile X chromosome at Xq27.3, or by DNA studies showing an expansion of CGG nucleotides in a specific region of the FMR-1 gene.

FIGURE 20–14 Siblings with fragile X syndrome. A, An 8-year-old mentally deficient boy exhibiting a relatively normal appearance with a long face and prominent ears. B, His 6-year-old sister who also has this syndrome. She has a mild learning disability and similar features of long face and prominent ears. Note the strabismus (crossed right eye). Although an X-linked disorder, sometimes female carriers have expression of the disease.
(Courtesy of Dr. A.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada.)
Several genetic disorders have been confirmed to be caused by expansion of trinucleotides in specific genes. Other examples include myotonic dystrophy, Huntington chorea, spinobulbar atrophy (Kennedy syndrome), Friedreich ataxia, and others. X-linked recessive genes are usually manifest in affected (hemizygous) males, and occasionally in carrier (heterozygous) females, for example, fragile X syndrome (Fig. 20-14).
The human genome comprises an estimated 20,000 to 25,000 genes per haploid set or 3 billion base pairs. Because of the Human Genome Project and international research collaboration, many disease- and birth defect–causing mutations in genes have been and will continue to be identified. Most genes will be sequenced and their specific function determined.
Understanding the cause of birth defects will require a better understanding of gene expression during early development. The majority of genes that are expressed in a cell are expressed in a wide variety of cells and are involved in basic cellular metabolic functions, such as nucleic acid and protein synthesis, cytoskeleton and organelle biogenesis, and nutrient transport and other cellular mechanisms. These genes are referred to as housekeeping genes. The specialty genes are expressed at specific times in specific cells and define the hundreds of different cell types that make up the human organism. An essential aspect of developmental biology is regulation of gene expression. Regulation is often achieved by transcription factors that bind to regulatory or promoter elements of specific genes.
Epigenetic regulation refers to changes in phenotype (appearance) or gene expression caused by mechanisms other than changes in the underlying DNA sequence. The mechanisms of epigenetic change are not entirely clear, but it is believed that modifying transcriptional factors, DNA methylation, or histone modification may also be key in altering developmental events. Several birth defects may be the result of altered gene expression due to environmental factors, such as stress or altered nutrition rather than due to changes in DNA sequences.
Genomic imprinting is an epigenetic process whereby the female and male germ lines confer a sex-specific mark on a chromosome subregion, so that only the paternal or maternal allele of a gene is active in the offspring. In other words, the sex of the transmitting parent will influence expression or nonexpression of certain genes in the offspring (Table 20-4). This is the reason for PWS and AS, in which case the phenotype is determined by whether the microdeletion is transmitted by the father (PWS) or the mother (AS). In a substantial number of cases of PWS and AS, as well as several other genetic disorders, the condition arises from a phenomenon referred to as uniparental disomy. In the situation with PWS and AS, both chromosomes 15s originate from only one parent. PWS occurs when both chromosomes 15s are derived from the mother, and AS occurs when both are paternally derived. The mechanism for this is believed to begin with a trisomic conceptus, followed by a loss of the extra chromosome in an early postzygotic cell division. This results in a “rescued” cell, in which both chromosomes have been derived from one parent.
Uniparental disomy has involved several chromosome pairs. Some are associated with adverse clinical outcomes involving chromosome pairs 6 (transient neonatal diabetes mellitus), 7 (Silver-Russel syndrome), and 15 (PWS and AS), whereas others (1 and 22) are not associated with any abnormal phenotypic effect.
Homeobox genes are a group of genes found in all vertebrates. They have highly conserved sequences and order. They are involved in early embryonic development and specify identity and spatial arrangements of body segments. Protein products of these genes bind to DNA and form transcriptional factors that regulate gene expression. Disorders associated with some homeobox mutations are described in Table 20-5.
Table 20–5 Examples of Disorders in Humans Associated with Homeobox Mutations
| NAME | CLINICAL FEATURES | GENE |
|---|---|---|
| Waardenburg syndrome (type I) | White forelock, lateral displacement of medial canthi of the eyes, cochlear deafness, heterochromia, tendency to facial clefting, autosomal dominant inheritance | HuP2 gene in humans, homologous to mouse Pax3 gene |
| Synpolydactyly (type II syndactyly) | Webbing and duplication of fingers, supernumerary metacarpal, autosomal dominant inheritance | HOX D 13 mutation |
| Holoprosencephaly (one form) | Incomplete separation of lateral cerebral ventricles, anophthalmia or cyclopia, midline facial hypoplasia or clefts, single maxillary central incisors, hypotelorism, autosomal dominant inheritance with widely variable expression | HPE 3 (sonic hedgehog) mutation gene that is homologous to the Drosophila segment polarity gene hedgehog |
| Schizencephaly (type II) | Full-thickness cleft within the cerebral ventricles often leading to seizures, spasticity, and mental deficiency | Germline mutation in the EMX2 homeobox gene, homologous to the mouse EMX2 |
Developmental Signaling Pathways
Normal embryogenesis is regulated by several complex signaling cascades (see Chapter 21). Mutations or alterations in any of these signaling pathways can lead to birth defects. Many pathways are cell autonomous and only alter the differentiation of that particular cell, as seen in proteins produced by HOX A and HOX D gene clusters (in which mutations lead to a variety of limb defects). Other transcriptional factors act by influencing the pattern of gene expression of adjacent cells. These short-range signal controls can act as simple on-off switches (paracrine signals); others, termed morphogens, elicit many responses depending on their level of expression with other cells.
One such developmental signaling pathway is initiated by the secreted protein called sonic hedgehog (Shh) that sets off a chain of events in target cells, resulting in activation and repression of target cells by transcription factors in the Gli family. Perturbations (disturbances) in the regulation of the Shh-Patched-Gli (Shh-Ptch-Gli) pathway lead to several human diseases, including some cancers and birth defects.
Shh is expressed in the notochord, the floor plate of the neural tube, the brain, and other regions, such as the zone of polarizing activity of the developing limbs, and the gut. Sporadic and inherited mutations in the human Shh gene leads to holoprosencephaly (see Chapter 17, Fig. 17-40), a midline defect of variable severity involving abnormal central nervous system (CNS) septation, facial clefting, single central incisor, hypotelorism, or a single cyclopic eye (see Chapter 18, Fig. 18-6). Shh protein needs to be processed to an active form and is modified by the addition of a cholesterol moiety. Defects in cholesterol biosynthesis, such as in the autosomal recessively inherited disorder Smith-Lemli-Opitz syndrome, share many features, particularly brain and limb defects reminiscent of Shh-pathway diseases. This suggests that Shh signaling may play a key role in several genetic disorders.
The three Gli genes identified as transcriptional factors are in the Shh-Ptch-Gli pathway. Mutations in the Gli3 gene have been implicated in several autosomal dominantly inherited disorders, including Greig cephalopolysyndactyly syndrome (deletions or point mutations); Pallister-Hall syndrome with hypothalamic hamartomas, central or postaxial polydactyly, among other defects of the face, brain, and limbs (frameshift or nonsense mutations); simple familial postaxial polydactyly type A and B, as well as preaxial polydactyly type IV (nonsense, missense, and frameshift mutations).
A comprehensive, authoritative, and daily updated listing of all known human genetic disorders and gene loci can be found on the Internet (Online Mendelian Inheritance in Man [OMIM]): McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University [Baltimore, MD], and National Center for Biotechnology information, National Library of Medicine [Bethesda, MD], 2006; www.ncbi.nlm.nih.gov/omim/).
Birth Defects Caused By Environmental Factors
Although the human embryo is well protected in the uterus, many environmental agents known as teratogens cause developmental disruptions after maternal exposure to them (Table 20-6). A teratogen is any agent that can produce a birth defect (congenital anomaly) or increase the incidence of a defect in the population. Environmental factors, such as infections and drugs, may simulate genetic conditions (e.g., when two or more children of normal parents are affected).
Table 20–6 Some Teratogens Known to Cause Human Congenital Anomalies or Birth Defects
| AGENTS | MOST COMMON BIRTH DEFECTS |
|---|---|
| Drugs | |
| Alcohol | Fetal alcohol syndrome (FAS): intrauterine growth restriction (IUGR), mental deficiency, microcephaly, ocular anomalies, joint abnormalities, short palpebral fissures |
| Androgens and high doses of progestogens | Varying degrees of masculinization of female fetuses: ambiguous external genitalia resulting in labial fusion and clitoral hypertrophy |
| Aminopterin | IUGR; skeletal defects; malformations of the central nervous system (CNS), notably meroencephaly (most of the brain is absent) |
| Cocaine | IUGR, prematurity, microcephaly, cerebral infarction, urogenital defects, neurobehavioral disturbances |
| Diethystilbestrol | Abnormalities of uterus and vagina, cervical erosion and ridges |
| Isotretinoin (13-cis-retinoic acid) | Craniofacial abnormalities; neural tube defects (NTDs), such as spina bifida cystica; cardiovascular defects; cleft palate; thymic aplasia |
| Lithium carbonate | Various defects, usually involving the heart and great vessels |
| Methotrexate | Multiple defects, especially skeletal, involving the face, cranium, limbs, and vertebral column |
| Misoprostol | Limb abnormalities, ocular and cranial nerve defects, autism spectrum disorder |
| Phenytoin | Fetal hydantoin syndrome: IUGR, microcephaly, mental deficiency, ridged frontal suture, inner epicanthal folds, eyelid ptosis, broad depressed nasal bridge, phalangeal hypoplasia |
| Tetracycline | Stained teeth, hypoplasia of enamel |
| Thalidomide | Abnormal development of limbs, e.g., meromelia (partial absence) and amelia (complete absence); facial defects; systemic anomalies, e.g., cardiac, kidney, and ocular defects |
| Trimethadione | Development delay, V-shaped eyebrows, low-set ears, cleft lip and/or palate |
| Valproic acid | Craniofacial anomalies, NTDs, cognitive abnormalities, often hydrocephalus, heart and skeletal defects |
| Warfarin | Nasal hypoplasia, stippled epiphyses, hypoplastic phalanges, eye anomalies, mental deficiency |
| Chemicals | |
| Methylmercury | Cerebral atrophy, spasticity, seizures, mental deficiency |
| Polychlorinated biphenyls | IUGR, skin discolorization |
| Infections | |
| Cytomegalovirus | Microcephaly, chorioretinitis, sensorineural hearing loss, delayed psychomotor/mental development, hepatosplenomegaly, hydrocephaly, cerebral palsy, brain (periventricular) calcification |
| Herpes simplex virus | Skin vesicles and scarring, chorioretinitis, hepatomegaly, thrombocytopenia, petechiae, hemolytic anemia, hydranencephaly |
| Human parvovirus B19 | Fetal anemia, nonimmune hydrops fetalis, fetal death |
| Rubella virus | IUGR, postnatal growth retardation, cardiac and great vessel abnormalities, microcephaly, sensorineural deafness, cataract, microphthalmos, glaucoma, pigmented retinopathy, mental deficiency, neonate bleeding, hepatosplenomegaly, osteopathy, tooth defects |
| Toxoplasma gondii | Microcephaly, mental deficiency, microphthalmia, hydrocephaly, chorioretinitis, cerebral calcifications, hearing loss, neurologic disturbance |
| Treponema pallidum | Hydrocephalus, congenital deafness, mental deficiency, abnormal teeth and bones |
| Venezuelan equine encephalitis virus | Microcephaly, microphthalmia, cerebral agenesis, CNS necrosis, hydrocephalus |
| Varicella virus | Cutaneous scars (dermatome distribution), neurologic defects (limb paresis [incomplete paralysis]), hydrocephaly, seizures, etc.), cataracts, microphthalmia, Horner syndrome, optic atrophy, nystagmus, chorioretinitis, microcephaly, mental deficiency, skeletal anomalies (hypoplasia of limbs, fingers, and toes, etc.), urogenital anomalies |
| Radiation | |
| High levels of ionizing radiation | Microcephaly, mental deficiency, skeletal anomalies, growth retardation, cataracts |
The important principle is that “not everything that is familial is genetic.” The organs and parts of an embryo are most sensitive to teratogenic agents during periods of rapid differentiation (Fig. 20-15). Environmental factors cause 7% to 10% of birth defects (Fig. 20-1). Because biochemical differentiation precedes morphologic differentiation, the period during which structures are sensitive to interference by teratogens often precedes the stage of their visible development by a few days.
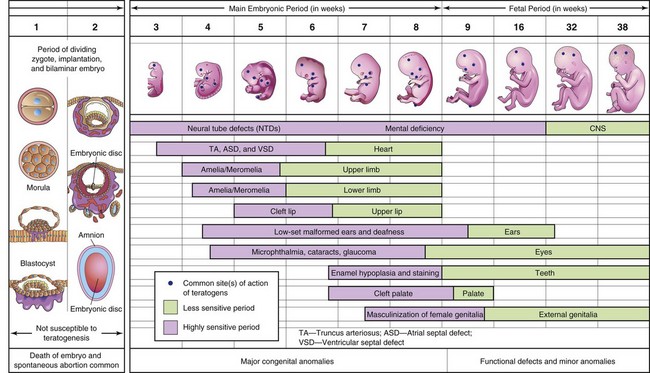
FIGURE 20–15 Schematic illustration of critical periods in human prenatal development. During the first 2 weeks of development, the embryo is usually not susceptible to teratogens; a teratogen either damages all or most of the cells, resulting in death of the embryo, or damages only a few cells, allowing the conceptus to recover and the embryo to develop without birth defects. Mauve denotes highly sensitive periods when major birth defects may be produced (e.g., amelia, absence of limbs, neural tube defects, spina bifida cystica). Green indicates stages that are less sensitive to teratogens when minor defects may be induced (e.g., hypoplastic thumbs).
Teratogens do not appear to be effective in causing defects until cellular differentiation has begun; however, their early actions (e.g., during the first 2 weeks) may cause death of the embryo. The exact mechanisms by which drugs, chemicals, and other environmental factors disrupt embryonic development and induce abnormalities still remain obscure. Even thalidomide’s mechanisms of action on the embryo are a “mystery,” and more than 20 hypotheses have been postulated to explain how this hypnotic agent disrupts embryonic development.
Many studies have shown that certain hereditary and environmental influences may adversely affect embryonic development by altering fundamental processes, such as the intracellular compartment, surface of the cell, extracellular matrix, and fetal environment. It has been suggested that the initial cellular response may take more than one form (genetic, molecular, biochemical, or biophysical), resulting in different sequences of cellular changes (cell death, faulty cellular interaction-induction, reduced biosynthesis of substrates, impaired morphogenetic movements, and mechanical disruption). Eventually, these different types of pathologic lesion could possibly lead to the final defect (intrauterine death, developmental defects, fetal growth retardation, or functional disturbances) through a common pathway.
Rapid progress in molecular biology is providing more information on the genetic control of differentiation, as well as the cascade of events involved in the expression of homeobox genes and pattern formation. It is reasonable to speculate that disruption of gene activity at any critical stage could lead to a developmental defect. This view is supported by recent experimental studies that showed that exposure of mouse and amphibian embryos to the teratogen retinoic acid (Vitamin A) altered the domain of gene expression and disrupted normal morphogenesis. Retinoic acid is now known to be a highly teratogenic. Researchers are now directing increasing attention to the molecular mechanisms of abnormal development in an attempt to understand better the pathogenesis of birth defects.
Principles of Teratogenesis
When considering the possible teratogenicity of an agent, such as a drug or chemical, three important principles must be considered:
Critical Periods of Human Development
The stage of development of an embryo when an agent, such as a drug or virus, is present determines its susceptibility to a teratogen (Fig. 20-15). The most critical period in development is when cell division, cell differentiation, and morphogenesis are at their peak. Table 20-7 indicates the relative frequencies of birth defects for certain organs.
Table 20–7 Incidence of Major Defects in Human Organs at Birth*
| ORGAN | INCIDENCE |
|---|---|
| Brain | 10 : 1000 |
| Heart | 8 : 1000 |
| Kidneys | 4 : 1000 |
| Limbs | 2 : 1000 |
| All other | 6 : 1000 |
| Total | 30 : 1000 |
* Data from Connor JM, Ferguson-Smith MA: Essential Medical Genetics, 2nd ed. Oxford, Blackwell Scientific Publications, 1987.
The critical period for brain development is from 3 to 16 weeks, but its development may be disrupted after this because the brain is differentiating and growing rapidly at birth. Teratogens may produce mental deficiency during the embryonic and fetal periods (Fig. 20-15).
Tooth development continues long after birth (see Chapter 19); hence, development of permanent teeth may be disrupted by tetracyclines from 18 weeks (prenatal) to 16 years (see Fig. 19-20E).
The skeletal system also has a prolonged critical period of development extending into childhood; hence, the growth of skeletal tissues provides a good gauge of general growth.
Environmental disturbances during the first 2 weeks after fertilization may interfere with cleavage of the zygote and implantation of the blastocyst, and/or cause early death and spontaneous abortion of the embryo; however, disturbances during the first 2 weeks are not known to cause birth defects (Fig. 20-15). Teratogens acting during the first 2 weeks either kill the embryo, or their disruptive effects are compensated for by powerful regulatory properties of the early embryo. Most development during the first 2 weeks is concerned with the formation of extraembryonic structures such as the amnion, umbilical vesicle, and chorionic sac (see Chapters 2 and 3).
Development of the embryo is most easily disrupted when the tissues and organs are forming (Figs. 20-15 and 20-16). During this organogenetic period (fourth to eighth weeks; see Chapter 5), teratogens may induce major birth defects. Physiologic defects, for example, minor morphologic defects of the external ears, and functional disturbances such as mental deficiency are likely to result from disruption of development during the fetal period. Some microorganisms, such as Toxoplasma gondii, are known to cause serious birth defects, particularly of the brain and eyes, when they infect the fetus (see Figs. 20-22 and 20-23 and Table 20-6).
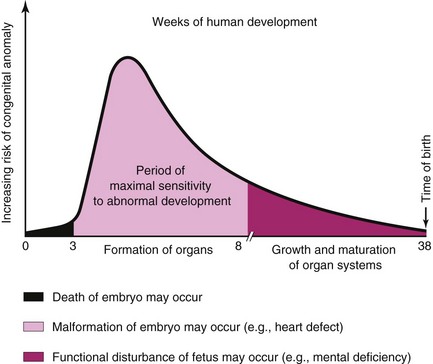
FIGURE 20–16 Schematic illustration showing the increasing risk of birth defects developing during organogenesis.
Each tissue, organ, and system of an embryo has a critical period during which its development may be disrupted (Fig. 20-15). The type of birth defect produced depends on which parts, tissues, and organs are most susceptible at the time the teratogen is active. The following examples illustrate that teratogens may affect different organ systems that are developing at the same time:
Early in the critical period of limb development, thalidomide causes severe defects such as meromelia, absence of part of the upper and/or lower limbs (see Fig. 20-20). Later in the sensitive period, thalidomide causes mild to moderate limb defects, such as hypoplasia of radius and ulna.
Embryologic timetables, such as shown in Figure 20-15, are helpful when considering the cause of a human birth defect; however, it is wrong to assume that defects always result from a single event occurring during the critical period, or that one can determine from these tables the day on which the defect was produced. All one can state is that the teratogen would have had to disrupt development before the end of the most critical period of the tissue, part, or organ concerned. For example, the critical period for limb development is 24 to 36 days after fertilization.
Dose of the Drug or Chemical
Animal research has shown that there is a dose-response relationship for teratogens; however, the dose used in animals to produce defects is often at levels much higher than human exposures. Consequently, animal studies are not readily applicable to human pregnancies. For a drug to be considered a human teratogen, a dose-response relationship has to be observed, that is, the greater the exposure during pregnancy, the more severe the phenotypic effect.
Genotype (Genetic Constitution) of the Embryo
There are numerous examples in experimental animals and several suspected cases in humans that show that there are genetic differences in response to a teratogen. Phenytoin, for example, is a well-known human teratogen (Table 20-6). Five to 10% of embryos exposed to this anticonvulsant medication develop the fetal hydantoin syndrome (see Fig. 20-19). Approximately one third of exposed embryos, however, have only some of the birth defects, and more than half of the embryos are unaffected. It appears, therefore, that the genotype of the embryo determines whether a teratogenic agent will disrupt its development.
Human Teratogens
Awareness that certain agents can disrupt prenatal development offers the opportunity to prevent some birth defects; for example, if women are aware of the harmful effects of drugs such as alcohol, environmental chemicals (e.g., polychlorinated biphenyls), and some viruses, most of them will not expose their embryos to these teratogenic agents.
The general objective of teratogenicity testing of drugs, chemicals, food additives, and pesticides is to identify agents that may be teratogenic during human development, and to alert physicians and pregnant women of their possible danger to their embryos/fetuses.
Proof of Teratogenicity
To prove that an agent is a teratogen, it must be shown that the frequency of defects is increased above the spontaneous rate in pregnancies in which the mother is exposed to the agent (prospective approach), or that malformed infants have a history of maternal exposure to the agent more often than normal children (retrospective approach). Both types of data are difficult to obtain in an unbiased form. Case reports are not convincing unless both the agent and type of defect are so uncommon that their association in several cases can be judged not coincidental.
Drug Testing in Animals
Although testing of drugs in pregnant animals is important, the results are of limited value for predicting drug effects in human embryos. Animal experiments can suggest only that similar effects may occur in humans. If a drug or chemical produces teratogenic effects in two or more species, the probability of potential human hazard must be considered to be high; however, the dose of the drug has also to be considered.
Drugs as Teratogens
Drugs vary considerably in their teratogenicity. Some teratogens (e.g., thalidomide) cause severe disruption of development if administered during the organogenetic period, the fourth to eighth weeks. Other teratogens cause mental and growth restriction and other defects if used excessively throughout development (e.g., alcohol).
The use of prescription and nonprescription drugs during pregnancy is surprisingly high. Forty to 90% of women consume at least one drug during pregnancy. Several studies have indicated that some pregnant women take an average of four drugs, excluding nutritional supplements, and approximately half of these women take them during the highly sensitive period (Fig. 20-15). Drug consumption also tends to be higher during the critical period of development among heavy smokers and drinkers. Despite this, less than 2% of birth defects are caused by drugs and chemicals. Only a few drugs have been positively implicated as human teratogenic agents (Table 20-6).
Although only 7% to 10% of defects are caused by recognizable teratogens (Fig. 20-1), new agents continue to be identified. It is best for women to avoid using all medication during the first trimester, unless there is a strong medical reason for its use, and then only if it is recognized as reasonably safe for the human embryo. The reason for this caution is that, even though well-controlled studies of certain drugs (e.g., marijuana) have failed to demonstrate a teratogenic risk to human embryos, they affect the embryo (e.g., fetal decreased length and birth weight).
Cigarette Smoking
Maternal smoking during pregnancy is a well-established cause of intrauterine growth restriction (IUGR). Despite warnings that cigarette smoking is harmful to the embryo/fetus, some women continue to smoke during their pregnancies. In heavy cigarette smokers, premature delivery is twice as frequent as in mothers who do not smoke, and their infants weigh less than normal (see Chapter 6, Fig. 6-11). Low birth weight (<2000 g) is the chief predictor of infant death.
In a case–control study, there was a modest increase in the incidence of infants with conotruncal heart defects and limb deficiencies associated with both maternal and paternal smoking. Moreover, there is some evidence that maternal smoking may cause urinary tract anomalies, behavioral problems, and decreased physical growth. Nicotine constricts uterine blood vessels, causing a decrease in uterine blood flow, lowering the supply of oxygen and nutrients available to the embryo/fetus from the maternal blood in the intervillous space of the placenta. The resulting deficiency impairs cell growth and may have an adverse effect on mental development. High levels of carboxyhemoglobin, resulting from cigarette smoking, appear in the maternal and fetal blood and may alter the capacity of the blood to transport oxygen. As a result, chronic fetal hypoxia (low oxygen levels) may occur and affect fetal growth and development. Maternal smoking is also associated with smaller brain volumes in preterm infants.
Alcohol
Alcoholism affects 1% to 2% of women of childbearing age. Both moderate and high levels of alcohol intake during early pregnancy may result in alterations in growth and morphogenesis of the embryo/fetus. Infants born to chronic alcoholic mothers exhibit a specific pattern of defects, including prenatal and postnatal growth deficiency, mental deficiency, and other defects (Fig. 20-17 and Table 20-6).
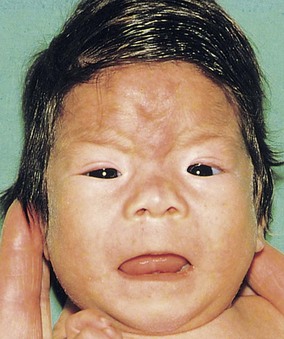
FIGURE 20–17 Fetal alcohol syndrome in an infant. Note the thin upper lip, elongated and poorly formed philtrum (vertical groove in medial part of upper lip), short palpebral fissures, flat nasal bridge, and short nose.
(Courtesy of Dr. A.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada.)
Microcephaly (small neurocranium), short palpebral fissures, epicanthal folds, maxillary hypoplasia, short nose, thin upper lip, abnormal palmar creases, joint defects, and congenital heart disease are also present in most infants. This pattern of defects—fetal alcohol syndrome (FAS)—is detected in 1 to 2 infants per 1000 live births.
The incidence of FAS is related to the population studied. Clinical experience is often necessary to make an accurate diagnosis of FAS because the physical defects in affected children are nonspecific. Nonetheless, the overall pattern of clinical features present is unique, but may vary from subtle to severe.
Maternal alcohol abuse is now thought to be the most common cause of mental deficiency. Moderate maternal alcohol consumption (1–2 oz of alcohol per day) can result in cognitive impairment and behavioral problems. The term fetal alcohol effects (FAE) was introduced after recognition that many children exposed to alcohol in utero had no external dysmorphic features, but had neurodevelopmental impairments.
The preferred term for the whole range of prenatal alcohol effects is fetal alcohol spectrum disorder (FASD). It is estimated that the general population prevalence of fetal alcohol spectrum disorder may be as high as 1%. The susceptible period of brain development spans the major part of gestation (Fig. 20-15); therefore, the safest advice is total abstinence from alcohol during pregnancy.
Androgens and Progestogens
The terms progestogens and progestins are used for substances, natural or synthetic, that induce some or all the biologic changes produced by progesterone, a hormone secreted by the corpus luteum of the ovaries that promotes and maintains a gravid endometrium (see Chapter 7). Some of these substances have androgenic (masculinizing) properties that may affect the female fetus, producing masculinization of the external genitalia (Fig. 20-18). The incidence of birth defects varies with the hormone and the dose. Preparations that should be avoided are the progestins, ethisterone, and norethisterone. From a practical standpoint, the teratogenic risk of these hormones is low. Progestin exposure during the critical period of development is also associated with an increased prevalence of cardiovascular defects, and exposure of male fetuses during this period may double the incidence of hypospadias (see Chapter 12, Fig. 12-42).
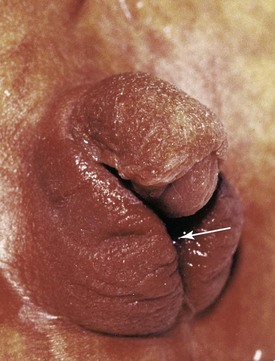
FIGURE 20–18 Masculinized external genitalia of a 46, XX female infant. Observe the enlarged clitoris and fused labia majora. The virilization was caused by excessive androgens produced by the suprarenal glands during the fetal period (congenital adrenal hyperplasia). The arrow indicates the opening of the urogenital sinus.
(Courtesy of Dr. Heather Dean, Department of Pediatrics and Child Health, University of Manitoba, Winnipeg, Manitoba, Canada.)
Many women use contraceptive hormones—birth control pills. Oral contraceptives containing progestogens and estrogens, taken during the early stages of an unrecognized pregnancy, are suspected of being teratogenic agents, but the results of several epidemiologic studies are conflicting. The infants of 13 of 19 mothers who had taken progestogen-estrogen birth control pills during the critical period of development exhibited the VACTERL syndrome. The acronym VACTERL stands for Vertebral, Anal, Cardiac, Tracheal, Esophageal, Renal, and Limb anomalies. As a precaution, use of oral contraceptives should be stopped as soon as pregnancy is suspected or detected because of these possible teratogenic effects.
Diethylstilbestrol (DES) is a human teratogen. Both gross and microscopic congenital abnormalities of the uterus and vagina have been detected in women who were exposed to DES in utero. Three types of lesions were observed: vaginal adenosis, cervical erosions, and transverse vaginal ridges. A number of young women ages 16 to 22 years have developed adenocarcinoma of the vagina after a common history of exposure to this synthetic estrogen in utero. However, the probability of cancers developing at this early age in females exposed to DES in utero now appears to be low. The risk of cancer from DES exposure in utero is estimated to be less than 1 in 1000.
Males who were exposed to DES in utero, after maternal treatment before the 11th week of gestation, had a higher incidence of genital tract anomalies, including epididymal cysts and hypoplastic (underdeveloped) testes. However, fertility in the men exposed to DES in utero seems to be unaffected. Expression of the homeobox gene HOXA10 is altered following in utero exposure to DES.
Antibiotics
Tetracyclines cross the placental membrane and are deposited in the embryo’s bones and teeth at sites of active calcification. As little as 1 g/day of tetracycline during the third trimester of pregnancy can produce yellow staining of the deciduous and/or permanent teeth (see Chapter 19, Fig. 19-20E). Tetracycline therapy during the fourth to ninth months of pregnancy may also cause tooth defects (e.g., enamel hypoplasia), yellow to brown discoloration of the teeth, and diminished growth of long bones. Calcification of the permanent teeth begins at birth and, except for the third molars, is complete by 7 to 8 years of age; hence, long-term tetracycline therapy during childhood can affect the permanent teeth.
Deafness has been reported in infants of mothers who have been treated with high doses of streptomycin and dihydrostreptomycin as antituberculosis agents. More than 30 cases of hearing deficit and eighth cranial nerve damage have been reported in infants exposed to streptomycin derivatives in utero. Penicillin has been used extensively during pregnancy and appears to be harmless to the human embryo and fetus.
Anticoagulants
All anticoagulants except heparin cross the placental membrane and may cause hemorrhage in the embryo or fetus. Warfarin and other coumarin derivatives are antagonists of vitamin K. Warfarin is used for the treatment of thromboembolitic disease and in patients with artificial heart valves. Warfarin is definitely a teratogen; there are reports of infants with hypoplasia of the nasal cartilage, stippled epiphyses, and various CNS defects whose mothers took this anticoagulant during the critical period of their embryo’s development. The period of greatest sensitivity is between 6 and 12 weeks after fertilization. Second- and third-trimester exposure may result in mental deficiency, optic atrophy, and microcephaly. Heparin is not a teratogen; furthermore, it does not cross the placental membrane (see Chapter 7, Fig. 7-7).
Anticonvulsants
Approximately 1 in 200 pregnant women is epileptic and requires treatment with an anticonvulsant. Of the anticonvulsant drugs available, there is strong evidence that trimethadione is a teratogen. The main features of the fetal trimethadione syndrome are prenatal and postnatal growth retardation, developmental delay, V-shaped eyebrows, low-set ears, cleft lip and/or palate, and cardiac, genitourinary, and limb defects. Use of this drug is contraindicated during pregnancy.
Phenytoin is definitely a teratogen (Fig. 20-19). Fetal hydantoin syndrome occurs in 5% to 10% of children born to mothers treated with phenytoins or hydantoin anticonvulsants. The usual pattern of defects consists of IUGR, microcephaly, mental deficiency, ridged frontal suture, inner epicanthal folds, eyelid ptosis, broad depressed nasal bridge, nail and/or distal phalangeal hypoplasia, and hernias.
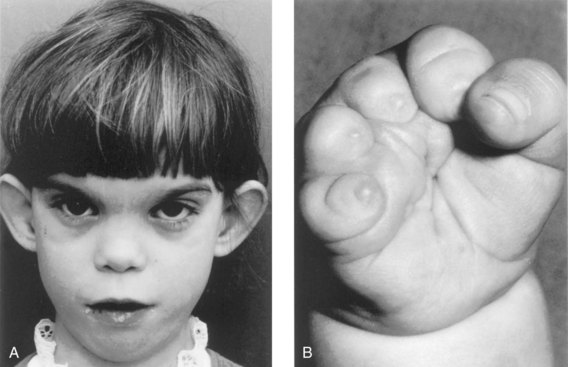
FIGURE 20–19 Fetal hydantoin syndrome in a young girl. A, She has a learning disability due to microcephaly and mental deficiency. Note the large ears, wide space between the eyes (hypertelorism), epicanthal folds, and short nose. Her mother has epilepsy and ingested Dilantin throughout her pregnancy. B, Right hand of the girl with severe digital hypoplasia (short fingers) born to a mother who took Dilantin throughout her pregnancy.
(A, Courtesy of Dr. A.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada. B, From Chodirker BN, Chudley AE, Reed MH, Persaud TVN; Am J Med Genet 27:373,
Valproic acid has been the drug of choice for the management of different types of epilepsy; however, use of it in pregnant women has led to a pattern birth defects consisting of craniofacial, heart, limb defects, and postnatal cognitive developmental delay. There is also an increased risk of neural tube defects (e.g., spina bifida cystica; Chapter 17, Fig. 17-15). Phenobarbital is considered to be a safe antiepileptic drug for use during pregnancy. Magnesium sulfate and diazepam are also widely used for seizure prophylaxis.
Antineoplastic Agents
With the exception of the folic acid antagonist aminopterin, few well-documented reports of teratogenic effects are available for assessment. Because the data available on the possible teratogenicity of antineoplastic drugs are inadequate, it is recommended that they should be avoided, especially during the first trimester of pregnancy.
Tumor-inhibiting chemicals are highly teratogenic because these agents inhibit mitosis in rapidly dividing cells. The use of aminopterin during the embryonic period often results in intrauterine death of the embryos, but the 20% to 30% of those that survive are severely malformed. Busulfan and 6-mercaptopurine administered in alternating courses throughout pregnancy have produced multiple severe abnormalities, but neither drug alone appears to cause major defects (Table 20-6).
Methotrexate, a folic acid antagonist and a derivative of aminopterin, is a potent teratogen that produces major birth defects. It is most often used as a single agent or in combination therapy for neoplastic diseases; however, it may also be indicated in patients with severe rheumatic diseases including rheumatoid arthritis. Multiple skeletal and other birth defects were found in an infant born to a mother who attempted to terminate her pregnancy by taking methotrexate.
Corticosteroids
Low doses of corticosteroids, including cortisone and hydrocortisone, do not induce cleft palate or any other defect in human embryos. Because of the risks of fetal bleeding and premature closure of the ductus arteriosus, nonsteroidal anti-inflammatory drugs should not be taken during the last few weeks of pregnancy.
Angiotensin-Converting Enzyme Inhibitors
Exposure of the fetus to angiotensin-converting enzyme inhibitors as antihypertensive agents causes oligohydramnios, fetal death, hypoplasia of the bones of the calvaria, IUGR, and renal dysfunction. During early pregnancy, the risk to the embryo is apparently less and there is no indication to terminate a pregnancy. Because of the high incidence of serious perinatal complications, it is recommended that angiotensin-converting enzyme inhibitors not be prescribed during pregnancy.
Insulin and Hypoglycemic Drugs
Insulin is not teratogenic in human embryos except possibly in maternal insulin coma therapy. Hypoglycemic drugs (e.g., tolbutamide) have been implicated, but evidence of their teratogenicity is weak; there is no convincing evidence that oral hypoglycemic agents (particularly sulfonylureas) are teratogenic in human embryos.
The incidence of birth defects (e.g., sacral agenesis) is increased two to three times in the offspring of diabetic mothers; approximately 40% of all perinatal deaths of diabetic infants are the result of birth defects. Women with insulin-dependent diabetes mellitus may significantly decrease their risk of having infants with birth defects by achieving good control of their disease before conception.
Retinoic Acid (Vitamin A)
Isotretinoin (13-cis-retinoic acid), used for treating severe cystic acne, is a known human teratogen. The critical period for exposure appears to be from the third week to the fifth week. The risk of spontaneous abortion and birth defects after exposure is high. The most common major defects observed are craniofacial dysmorphism (microtia, micrognathia), cleft palate and/or thymic aplasia, cardiovascular defects, and neural tube defects. Postnatal longitudinal follow-up of children exposed in utero to isotretinoin revealed significant neuropsychological impairment.
Vitamin A is a valuable and necessary nutrient during pregnancy, but long-term exposure to large doses is unwise. Pregnant women should avoid high levels of vitamin A because an increased risk of birth defects among the offspring of women who took more than 10,000 IU of vitamin A daily has been reported.
Analgesics
Aspirin (acetylsalicylic acid or ASA) and acetaminophen are commonly used during pregnancy for the relief of fever or pain. Clinical trials suggest that large doses are potentially harmful to the embryo or fetus. The use of single-ingredient acetaminophen during the first trimester does not appear to increase the risk of major birth defects. Although epidemiologic studies indicate that aspirin is not a teratogenic agent, large doses should be avoided, especially during the first trimester.
Thyroid Drugs
Potassium iodide in cough mixtures and large doses of radioactive iodine may cause congenital goiter. Iodides readily cross the placental membrane and interfere with thyroxin production. They may also cause thyroid enlargement and cretinism (arrested physical and mental development and dystrophy of bones and soft parts).
Maternal iodine deficiency may also cause congenital cretinism. Pregnant women have been advised to avoid douches or creams containing povidone-iodine because it is absorbed by the vagina, enters the maternal blood, and may be teratogenic. Propylthiouracil interferes with thyroxin formation in the fetus and may cause goiter. The administration of antithyroid substances for the treatment of maternal thyroid disorders may cause congenital goiter if the mother is given the substances in excess of the amount required to control the disease.
Tranquilizers
Thalidomide is a potent teratogen and it has been estimated that nearly 12,000 infants were born with defects caused by this drug. The characteristic presenting feature is meromelia, but the defects ranged from amelia (absence of limbs) through intermediate stages of development (rudimentary limbs) to micromelia (abnormally small and/or short limbs). Phocomelia (“seal limbs”), a type of meromelia, is present in some of these individuals (e.g., see Fig. 20-20).
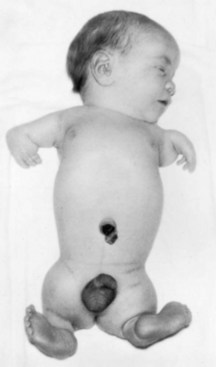
FIGURE 20–20 Neonate male infant showing typically malformed limbs (meromelia—limb reduction) caused by thalidomide ingested by his mother during the critical period of limb development.
(From Moore KL: Manit Med Rev 43:306, 1963.)
Thalidomide also caused anomalies of other organs, for example, absence of the external and internal ears, hemangioma on the forehead, heart defects, and anomalies of the urinary and alimentary systems. It is well established clinically that the period when thalidomide caused congenital anomalies was 20 to 36 days after fertilization. This sensitive period coincides with the critical periods for the development of the affected parts and organs (Figs. 20-15 and 20-16). Thalidomide is currently used for the treatment of leprosy and several autoimmune diseases. It is absolutely contraindicated in women of childbearing age.
Psychotropic Drugs
Lithium is the drug of choice for the long-term maintenance of patients with bipolar disorders; however, it has caused birth defects, mainly of the heart and great vessels, in infants born to mothers given the drug early in pregnancy. Although lithium carbonate is a known human teratogen, the U.S. Food and Drug Administration has stated that the agent may be used during pregnancy if “in the opinion of the physician the potential benefits outweigh the possible hazards.”
Benzodiazepines, such as diazepam and oxazepam, are frequently prescribed for pregnant women. These drugs readily cross the placental membrane and their use during the first trimester of pregnancy is associated with craniofacial anomalies in neonates. Selective serotonin reuptake inhibitors (SSRIs) are used to treat depression during pregnancy. Recent reports warn of an increased risk of atrial and ventricular septal defects, persistent pulmonary hypertension, and neurobehavioral disturbances in infants exposed to SSRIs in utero. The mechanism is believed to be SSRIs blocking catecholamine transport, which affects placental blood flow.
Illicit Drugs
Several currently popular “street drugs” are used for their hallucinogenic properties. There is no evidence that marijuana is a human teratogen; however, there is an indication that marijuana use during the first 2 months of pregnancy affects fetal length and birth weight. In addition, sleep and electroencephalographic patterns in neonates exposed prenatally to marijuana were altered.
Second only to marijuana, cocaine is the most widely used illicit drug among women of childbearing age. Reports dealing with the prenatal effects of cocaine include spontaneous abortion, prematurity, IUGR, microcephaly, cerebral infarction, urogenital anomalies, neurobehavioral disturbances, and neurologic abnormalities.
Methadone, used for the treatment of heroin addiction, is considered to be a “behavioral teratogen,” as is heroin. Infants born to narcotic-dependent women maintained on methadone therapy were found to have CNS dysfunction, lower birth weights, and smaller head circumferences than nonexposed infants. There is also concern about the long-term postnatal developmental effects of methadone. The problem, however, is difficult to resolve because other drugs are often used in combination with methadone, and heavy use of alcohol and cigarettes is prevalent among narcotic-dependent women.
Maternal use of methamphetamine, a sympathetic nervous system stimulant, results in small for gestational age fetuses with neurobehavioral changes.
Environmental Chemicals as Teratogens
There is increasing concern about the possible teratogenicity of environmental chemicals, including industrial and agricultural chemicals, pollutants, and food additives. Most of these chemicals have not been positively implicated as teratogens in humans.
Organic Mercury
Infants of mothers whose main diet during pregnancy consists of fish containing abnormally high levels of organic mercury acquire fetal Minamata disease—neurologic and behavioral disturbances resembling cerebral palsy. Severe brain damage, mental deficiency, and blindness have been detected in infants of mothers who received methylmercury in their food.
Similar observations have been made in infants whose mothers ate pork that became contaminated when the pigs ate corn grown from seeds sprayed with a mercury-containing fungicide. Methylmercury is a teratogen that causes cerebral atrophy, spasticity, seizures, and mental deficiency.
Lead
Abundantly present in the workplace and environment, lead passes through the placental membrane and accumulates in embryonic and fetal tissues. Prenatal exposure to lead is associated with increased abortions, fetal defects, IUGR, and functional deficits. Several reports have indicated that children born to mothers who were exposed to subclinical levels of lead revealed neurobehavioral and psychomotor disturbances.
Polychlorinated Biphenyls
These teratogenic chemicals produce IUGR and skin discoloration. The main dietary source of polychlorinated biphenyls in North America is probably sport fish caught in contaminated waters. In Japan and Taiwan, the teratogenic chemical was detected in contaminated cooking oil.
Infectious Agents as Teratogens
Throughout prenatal life, the embryo and fetus are endangered by a variety of microorganisms. In most cases, the assault is resisted; however, in some cases, abortion or stillbirth occurs.
In others, the infants are born with IUGR, birth defects, or neonatal diseases (Table 20-6). The microorganisms cross the placental membrane and enter the embryonic and fetal bloodstream. Because there is a propensity for the CNS to be affected, the fetal blood–brain barrier also apparently offers little resistance to microorganisms.
Rubella (German Measles)
The virus that causes rubella, a communicable disease, is the prime example of an infective teratogen. In cases of primary maternal infection during the first trimester of pregnancy, the overall risk of embryonic/fetal infection is approximately 20%. The rubella virus crosses the placental membrane and infects the embryo/fetus. The clinical features of congenital rubella syndrome are cataracts (see Chapter 18, Fig. 18-13), cardiac defects, and deafness; however, the following abnormalities are occasionally observed: mental deficiency, chorioretinitis, glaucoma (see Chapter 18, Fig. 18-12), microphthalmia, and tooth defects. The earlier in pregnancy the maternal rubella infection occurs, the greater is the danger that the embryo will be malformed.
Most infants have birth defects if the disease occurs during the first 4 to 5 weeks after fertilization. This period includes the most susceptible organogenetic periods of the eyes, internal ears, heart, and brain (Fig. 20-15). The risk of defects from a rubella infection during the second and third trimesters is low (approximately 10%), but functional defects of the CNS (mental deficiency) and internal ears (hearing loss) may result if infection occurs during the late fetal period. There is no evidence of fetal defects after the fifth gestational month; however, infections may produce chronic disease and dysfunction of the eyes, ears, and CNS. Because of widespread immunization against rubella virus, fewer infants are affected.
Cytomegalovirus
Infection with cytomegalovirus (CMV) is the most common viral infection of the fetus, occurring in approximately 1% of neonates. Most pregnancies end in spontaneous abortion when the infection occurs during the first trimester. It is the leading cause of congenital infection with morbidity at birth. Neonates infected during the early fetal period usually show no clinical signs and are identified through screening programs. CMV infection later in pregnancy may result in severe birth defects: IUGR, microphthalmia, chorioretinitis, blindness, microcephaly, cerebral calcification, mental deficiency, deafness, cerebral palsy, and hepatosplenomegaly. Of particular concern are cases of asymptomatic CMV infection, which are often associated with audiologic, neurologic, and neurobehavioral disturbances in infancy.
Herpes Simplex Virus
Maternal infection with herpes simplex virus in early pregnancy increases the abortion rate by threefold, and infection after the 20th week is associated with a higher rate of prematurity. Infection of the fetus with this virus usually occurs very late in pregnancy, probably most often during delivery. The congenital defects that have been observed in newborns include cutaneous lesions, microcephaly, microphthalmia, spasticity, retinal dysplasia, and deficiency.
Varicella (Chickenpox)
Varicella and herpes zoster (shingles) are caused by the same virus, varicella-zoster virus, which is highly infectious. Maternal varicella infection during the first two trimesters of pregnancy causes the following birth defects: skin scarring, muscle atrophy, hypoplasia of limbs, rudimentary digits, eye and brain damage, and mental deficiency. There is a 20% chance of these or other defects when the infection occurs during the critical period of development (Fig. 20-15). After 20 weeks of gestation, there is no proven teratogenic risk.
Human Immunodeficiency Virus
This retrovirus causes acquired immunodeficiency syndrome (AIDS). There is conflicting information on the fetal effects of in utero infection with human immunodeficiency virus. Some of the birth defects reported are growth failure, microcephaly, and specific craniofacial features. Most cases of transmission of the virus from mother to fetus probably occur at the time of delivery. Breast-feeding increases the risk of transmitting the virus to the neonate. Preventing the transmission of the virus to women and their infants is of obvious importance because of the potential effects.
Toxoplasmosis
Toxoplasma gondii, an intracellular parasite, was named after the gondi, a North African rodent in which the organism was first detected. This parasite may be found in the bloodstream, tissues, or reticuloendothelial cells, leukocytes, and epithelial cells.
Maternal infection is usually acquired by:
It is thought that the soil and garden vegetables may become contaminated with infected animal feces carrying oocysts (encapsulated zygote in the life cycle of sporozoan protozoa). Oocysts can also be transported to food by flies and cockroaches.
The T. gondii organism crosses the placental membrane and infects the fetus (Figs. 20-21 and 20-22), causing destructive changes in the brain (intracranial calcifications) and eyes (chorioretinitis) that result in mental deficiency, microcephaly, microphthalmia, and hydrocephaly. Fetal death may follow infection, especially during the early stages of pregnancy.
FIGURE 20–21 Chorioretinitis of congenital ocular toxoplasmosis induced by Toxoplasma infection. A, Necrotizing cicatricial lesion of macula (arrow). B, Satellite lesion around and adjacent to necrotizing cicatricial main lesion (arrows). C, Recrudescent lesion adjacent to large necrotizing cicatricial main lesion (arrows).
(From Yokota K: Congenital anomalies and toxoplasmosis. Congenit Anom (Kyoto) 35:151, 1995.)

FIGURE 20–22 Congenital cerebral defectsinduced by Toxoplasma infection. The diagnostic images were obtained at 2 years and 9 months of age. A, Plain CT (computed tomography) scan. The lateral ventricles are moderately dilated. Multiple calcified foci are apparent in the brain parenchyma (arrows 1), and along the ventricular wall (arrow 2). B, MRI (magnetic resonance imaging), T1 WI (400/22, 0.5 T). The cortical gyri are widened on the left side and the cortex is thickened in the left frontal lobe (arrow) compared with corresponding structure on the right. C, MRI, T2 WI (2,500/120, 0.5 T). The left frontal lobe shows abnormal hypointensity (arrow).
(From Yokota K: Congenital anomalies and toxoplasmosis, Congenit Anom (Kyoto) 35:151, 1995.)
Mothers of congenitally defective infants are often unaware of having had toxoplasmosis, the disease caused by the parasitic organism. Because animals (cats, dogs, rabbits, and other domestic and wild animals) may be infected with this parasite, pregnant women should avoid them and the eating of raw or poorly cooked meat from them (e.g., rabbits). In addition, unpasteurized milk should be avoided.
Congenital Syphilis
The incidence of congenital syphilis is steadily increasing with more cases now than in any of the past 2 decades. One in 10,000 live-born infants in the United States is infected. Treponema pallidum, the small, spiral microorganism that causes syphilis, rapidly crosses the placental membrane as early as 6 to 8 weeks of development. The fetus can become infected at any stage of the disease or at any stage of pregnancy.
Primary maternal infections (acquired during pregnancy) nearly always cause serious fetal infection and birth defects; however, adequate treatment of the mother kills the microorganisms, thereby preventing them from crossing the placental membrane and infecting the fetus.
Secondary maternal infections (acquired before pregnancy) seldom result in fetal disease and birth defects. If the mother is untreated, stillbirths occur in approximately one fourth of cases. Only 20% of all untreated pregnant women will deliver a normal infant at term.
Early fetal manifestations of untreated maternal syphilis are congenital deafness, abnormal teeth and bones, hydrocephalus, and mental deficiency. Late fetal manifestations of untreated congenital syphilis are destructive lesions of the palate and nasal septum, dental abnormalities (centrally notched, widely spaced peg-shaped upper central incisors, known as Hutchinson teeth), and facial defects (frontal bossing, including protuberance or swelling, saddle nose, and poorly developed maxilla).
Radiation as a Teratogen
Exposure to high levels of ionizing radiation may injure embryonic cells, resulting in cell death, chromosome injury, mental deficiency, and deficient physical growth. The severity of the embryonic damage is related to the absorbed dose of radiation, the dose rate, and the stage of embryonic or fetal development when the exposure to radiation occurs.
In the past, large amounts of ionizing radiation (hundreds to several thousand rads) were given inadvertently to embryos and fetuses of pregnant women who had cancer of the cervix. In all cases, their embryos were severely malformed or killed. Growth retardation, microcephaly, spina bifida cystica (see Chapter 17, Fig. 17-15), pigment changes in the retina, cataracts, cleft palate, skeletal and visceral abnormalities, and mental deficiency have been observed in infants who survived after receiving high levels of ionizing radiation. Development of the CNS was nearly always affected.
Observations of Japanese atomic bomb survivors and their children suggest that 8 to 16 weeks after fertilization is the period of greatest sensitivity for radiation damage to the brain, resulting in severe mental deficiency. By the end of the 16th week, most neuronal proliferation is completed, after which the risk of mental deficiency decreases. It is generally accepted that large doses of radiation (>25,000 mrad) are harmful to the developing CNS. There is no conclusive proof that human birth defects have been caused by diagnostic levels of radiation. Scattered radiation from a radiographic examination of a region of the body that is not near the uterus (e.g., thorax, sinuses, teeth) produces a dose of only a few millirads, which is not teratogenic to the embryo. For example, a pelvic CT in the third trimester of pregnancy results in a whole-body dose to her fetus of approximately 1 to 5 rads. If the embryonic radiation exposure is 5 rads or less, the radiation risks to the embryo are minuscule; however, it is prudent to be cautious during diagnostic examinations of the pelvic region in pregnant women (radiographic examinations and medical diagnostic tests using radioisotopes) because they result in exposure of the embryo to 0.3 to 2 rads. The recommended limit of maternal exposure of the whole body to radiation from all sources is 500 mrad (0.005 Gy) for the entire gestational period.
Maternal Factors as Teratogens
Approximately 4% of pregnant women have diabetes. Poorly controlled diabetes mellitus in the mother, particularly during embryogenesis, is associated with an increased rate of spontaneous miscarriages and a two- to threefold higher incidence of birth defects. Neonates of diabetic mothers are usually large (macrosomia), with prominent fat pads over the upper back and lower jaw. These infants are at an increased risk for brain anomalies, skeletal defects, sacral agenesis, and congenital heart defects, in addition to neonatal metabolic complications, respiratory distress syndrome, and neurodevelopmental abnormalities.
Phenylketonuria (autosomal recessive inherited inborn error of metabolism) occurs in one per 10,000 infants born in the United States. If untreated, women who are homozygous for phenylalanine hydroxylase deficiency—phenylketonuria—and those with hyperphenylalaninemia are at a higher risk of having an offspring with microcephaly, cardiac defects, mental deficiency, and IUGR. The brain damage and mental deficiency can be prevented if the phenylketonuric mother is placed on a phenylalanine-restricted diet before and during pregnancy.
The risk of neural tube defects (see Chapter 17, Fig. 17-17) is greater in the offspring of mothers with low levels of folic acid and vitamin B12.
Mechanical Factors as Teratogens
Amniotic fluid absorbs mechanical pressures, thereby protecting the embryo from most external trauma. A significantly reduced quantity of amniotic fluid (oligohydramnios) may result in mechanically induced deformation of the limbs (see Chapter 7); for example, hyperextension of the knee. Congenital dislocation of the hip and clubfoot may be caused by mechanical forces, particularly in a malformed uterus. Such deformations may be caused by any factor that restricts the mobility of the fetus, thereby causing prolonged compression in an abnormal posture. Intrauterine amputations or other anomalies caused by local constriction during fetal growth may result from amniotic bands—rings formed as a result of rupture of the amnion during early pregnancy (see Chapter 7, Fig. 7-21).
Birth Defects Caused By Multifactorial Inheritance
Common birth defects (e.g., cleft lip with or without cleft palate) have familial distributions consistent with multifactorial inheritance (see Fig. 20-1). Multifactorial inheritance may be represented by a model in which “liability” to a disorder is a continuous variable determined by a combination of genetic and environmental factors, with a developmental threshold dividing individuals with the defect from those without it (Fig. 20-23).
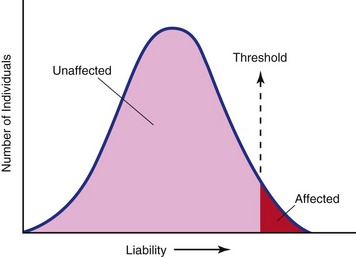
FIGURE 20–23 Multifactorial threshold model. Liability to a trait is distributed normally with a threshold dividing the population into unaffected and affected classes.
(From Thompson MW, McInnes RR, Willard HF: Thompson & Thompson Genetics in Medicine, 5th ed. Philadelphia, WB Saunders, 1991.)
Multifactorial traits are often single major defects, such as cleft lip, isolated cleft palate, neural tube defects (e.g., meroencephaly, spina bifida cystica), pyloric stenosis, and congenital dislocation of the hips. Some of these defects may also occur as part of the phenotype in syndromes determined by single-gene inheritance, chromosome abnormality, or an environmental teratogen.
The recurrence risks used for genetic counseling of families having birth defects, determined by multifactorial inheritance, are empirical risks based on the frequency of the defect in the general population and in different categories of relatives. In individual families, such estimates may be inaccurate because they are usually averages for the population rather than precise probabilities for the individual family.
Summary of Human Birth Defects
Clinically Oriented Problems
Case 20–1
A physician was concerned about the drugs a woman said she was taking when she first sought medical advice during her pregnancy.
Case 20–2
During a pelvic examination, a 38-year-old woman learned that she was pregnant. The physician was concerned about her age inasmuch as it was her first pregnancy.
Case 20–3
A pregnant woman asked her physician whether there are any drugs considered safe during early pregnancy.
Case 20–4
A 10-year-old girl contracted a rubella infection (German measles) and her mother was worried that the child might develop cataracts and heart defects.
Case 20–5
A pregnant woman who has two cats that often “spent the night out” was told by a friend that she should avoid close contact with her cats during her pregnancy. She was also told to avoid flies and cockroaches.
Discussion of these problems appears at the back of the book.
References and Suggested Reading
Bale JFJr. Fetal infections and brain development. Clin Perinatol. 2009;36:639.
Baley JE, Toltzis P. Viral infections. In Martin RJ, Fanaroff AA, Walsh MC, editors: Fanaroff and Martin’s Neonatal–Perinatal Medicine: Diseases of the Fetus and Infant, 8th ed, Philadelphia: Mosby, 2006.
Berry RJ, Bailey L, Mulinarae J, et al. Fortification of flour with folic acid. Food Nutr Bull. 2010;31(Suppl 1):S22.
Biencowe H, Cousens S, Modell B, et al. Folic acid to reduce neonatal mortality from neural tube disorders. Int J Epidemiol. 2010;39(Suppl 1):i110.
Briggs GG, Freeman RK, Yaffe SJ. Drugs in Pregnancy and Lactation, 8th ed. Baltimore: Williams & Wilkins; 2008.
Callen PW, editor. Ultrasonography in Obstetrics and Gynecology, 5th ed, Philadelphia: WB Saunders, 2008.
Centers for Disease Control and Prevention. Improved national prevalence estimates for selected major birth defects—United States, 1999–2001 (MMWR 54:1301, 2006). JAMA. 2006;295:618.
Chudley AE, Hagerman RJ. The fragile X syndrome. J Pediatr. 1987;110:821.
Drugan A, Isada NB, Evans MI. Prenatal diagnosis in the molecular age—indications, procedures, and laboratory techniques. In MacDonald MG, Seshia MMK, Mullett MD, editors: Avery’s Neonatology, Pathophysiology & Management of the Newborn, 6th ed, Philadelphia: Lippincott Williams & Wilkins, 2005.
Einfeld SL, Brown R. Down syndrome—new prospects for an ancient disorder. JAMA. 2010;303:2525.
Frey KA. Male reproductive health and infertility. Prim Care. 2010;37:643.
Gianicolo EA, Cresci M, Ait-Ait L, et al. Smoking and congenital heart disease: The epidemiological and biological link. Curr Pharm Des. 2010;16:2572.
Hales B. DNA repair disorders causing malformations. Curr Opin Genet Dev. 2005;15:234.
Hall JG. Chromosomal clinical abnormalities. In Behrman RE, Kliegman RM, Jenson HB, editors: Nelson Textbook of Pediatrics, 17th ed, Philadelphia: WB Saunders, 2004.
Hampton T. Researchers discover a range of factors undermine sperm quality, male fertility. JAMA. 2005;294:2829.
Hudgins L, Cassidy SB. Congenital anomalies. In Martin RJ, Fanaroff AA, Walsh MC, editors: Fanaroff and Martin’s Neonatal–Perinatal Medicine: Diseases of the Fetus and Infant, 8th ed, Philadelphia: Mosby, 2006.
Jones KL. Smith’s Recognizable Patterns of Human Malformation, 6th ed. Philadelphia: Elsevier/Saunders; 2005.
Kriebs JM. Changing the paradigm. HIV in pregnancy. J Perinat Neonat Nurs. 2006;20:71.
Malik S, Cleves MA, Honein MA, et al. Maternal smoking and congenital heart defects. Pediatr. 2008;121:e81.
Medicode, Inc. Medicode’s Hospital and Payer: International Classification of Diseases, Clinical Modification, ICD-9-CM 2006, vols 1–3. Salt Lake City: Medicode; 2006.
Nussbaum RL, McInnes RR, Willard HF. Thompson & Thompson Genetics in Medicine [rev rep], 6th ed. WB Saunders, Philadelphia, 2004.
Persaud TVN. Environmental Causes of Human Birth Defects. Springfield, IL: Charles C Thomas; 1990.
Rajaraman P, Simpson J, Neta G, et al. Early life exposure to diagnostic radiation and ultrasound scans and risk of childhood cancer: case control study. BMJ. 2011:342. d472 doi:10.1136/bmj.d472 (online)
Rasmussen SA, Erickson JD, Reef SE, et al. Principles and practice of teratology for the obstetrician. Clin Obstet Gynecol. 2009;51:106.
Reef SE, Strebel P, Dabbagh A, et al. Progress toward control of rubella and prevention of congenital rubella syndrome—worldwide, 2009. J Infect Dis. 2011;204(Suppl 1):S24-S27.
Richardson GA, Goldschmidt L, Willford J. Continued effects of prenatal cocaine use: preschool development. Neurotoxicol Teratol. 2009;31:325.
Sackett C, Weller RA, Weller EB. Selective serotonin reuptake inhibitor use during pregnancy and possible neonatal complications. Curr Psychiatry Rep. 2009;11:253.
Schwarz EB, Maselli J, Norton M, et al. Prescription and teratogenic medications in United States ambulatory practices. Am J Med. 2005;118:1240.
Shiota K, Uwabe C, Nishimura H. High prevalence of defective human embryos at the early postimplantation period. Teratology. 1987;35:309.
Society of Obstetricians and Gynaecologists of Canada. Alcohol use in pregnancy consensus clinical guidelines. J Obstet Gynaecol Can. 2010;32(Suppl 3):S1.
Spranger J, Benirschke K, Hall JG, et al. Errors of morphogenesis, concepts and terms. J Pediatr. 1982;100:160.
Weindling AM. Offspring of diabetic pregnancy: Short-term outcomes. Semin Fetal Neonatal Med. 2009;14:101.
Weiner CP. Drugs for Pregnant and Lactating Women, 2nd ed. Philadelphia: WB Saunders; 2009.
Yolton K, Khoury J, Xu Y, et al. Low-level prenatal exposure to nicotine and infant neurobehavior. Neurotoxicol Teratol. 2009;31:356.