Chapter 19 Electrical Activity of the Heart
1. Contraction of cardiac muscle cells is triggered by an electrical action potential.
2. The contractile machinery in cardiac muscle is similar to that in skeletal muscle.
3. Cardiac muscle forms a functional syncytium.
4. Cardiac contractions are initiated by action potentials that arise spontaneously in specialized pacemaker cells.
5. A system of specialized cardiac muscle cells initiates and organizes each heartbeat.
6. Cardiac action potentials are extremely long.
7. Membrane calcium channels play a special role in cardiac muscle.
8. The long duration of the cardiac action potential guarantees a period of relaxation (and refilling) between heartbeats.
9. Atrial cells have shorter action potentials than do ventricular cells.
10. Specialized ion channels cause cardiac pacemaker cells to depolarize to threshold and form action potentials.
11. Sympathetic and parasympathetic nerves act on cardiac pacemaker cells to increase or decrease the heart rate.
12. Cells of the atrioventricular node act as auxiliary pacemakers and also protect the ventricles from beating too fast.
13. Sympathetic nerves act on all cardiac cells to cause quicker, more forceful contractions.
14. Parasympathetic effects are opposite to those of sympathetic activation but are generally restricted to the sinoatrial node, atrioventricular node, and atria.
15. Dysfunction in the specialized conducting system leads to abnormalities in cardiac rhythm (arrhythmias).
16. Atrioventricular node block is a common cause of cardiac arrhythmias.
17. Cardiac tachyarrhythmias result either from abnormal action potential formation (by the sinoatrial node or ectopic pacemakers) or from abnormal action potential conduction (“reentry”).
18. Common antiarrhythmic drugs affect the ion channels responsible for the cardiac action potential.
Contraction of Cardiac Muscle Cells Is Triggered by an Electrical Action Potential
The heart is a muscular pump that propels blood through the blood vessels by alternately relaxing and contracting. As the heart muscle relaxes, the ventricles fill with venous blood. During cardiac contraction, some of this blood is ejected into the arteries. Cardiac contraction takes place in two stages: (1) the right and left atria begin to contract, and (2) after a delay of 50 to 150 milliseconds (msec), the right and left ventricles begin to contract. Atrial contraction helps to finish filling the ventricles with blood. The delay allows time for this “topping up” of ventricular volume. Ventricular contraction ejects blood out of the left ventricle into the aorta and out of the right ventricle into the pulmonary artery. After ventricular ejection, the heart relaxes, and the ventricles begin to refill. The entire contractile sequence is initiated and organized by an electrical signal, an action potential, which propagates from muscle cell to muscle cell, through the heart.
This chapter begins with a brief description of how cardiac muscle contracts, followed by a detailed description of the action potentials that initiate and organize the heart’s contractions. Several common electrical dysfunctions of the heart are then discussed.
Throughout this chapter, comparisons are made between cardiac and skeletal muscle (Table 19-1). In both cardiac and skeletal muscle, an electrical action potential in each muscle cell is necessary to trigger a contraction. The molecular mechanisms that carry out the contraction are also similar in both types of muscle. However, important differences exist between cardiac and skeletal muscle in the characteristics of the action potentials that initiate contractions.
Table 19-1 Sequence of Events in Contraction of Skeletal Muscle and Cardiac Muscle
| Skeletal muscle | Cardiac muscle |
|---|---|
| Action potential is generated in somatic motor neuron | Note: Action potentials in autonomic motor neurons are not needed to initiate heartbeats |
| Acetylcholine is released | Note: Neurotransmitters are not needed to make the heart beat |
| Nicotinic cholinergic receptors on muscle cell membrane are activated | Note: Activation of receptors is not needed. A completely isolated or denervated heart still beats |
| Ligand-gated Na+ channels in muscle membrane open | Pacemaker Na+ channels spontaneously open (and K+ channels close) in membranes of pacemaker cells |
| Muscle membrane depolarizes to threshold level for formation of action potential | Pacemaker cell membranes depolarize to threshold for formation of action potential |
| Action potential forms in muscle cell but does not enter other cells | Action potential forms in a pacemaker cell and then propagates from cell to cell throughout the whole heart |
| Note: Skeletal muscle cells do not have slow Ca2+ channels | During action potential, extracellular Ca2+ (“trigger” Ca2+) enters cell through “slow” Ca2+ channels |
| Action potential causes Ca2+ release from sarcoplasmic reticulum; Ca2+ binds to troponin | Entry of extracellular trigger Ca2+ causes release of more Ca2+ from sarcoplasmic reticulum; Ca2+ binds to troponin |
| Actin’s binding sites are made available for actin-myosin cross-bridge formation | Actin’s binding sites are made available for actin-myosin cross-bridge formation |
| Cross-bridge cycling generates contractile force between actin and myosin filaments | Cross-bridge cycling generates contractile force between actin and myosin filaments |
| Muscle contracts (brief “twitch”); Ca2+ is taken up by sarcoplasmic reticulum | Heart contracts (complete “beat” or “systole”); Ca2+ is taken up by sarcoplasmic reticulum or pumped back out of cell into extracellular fluid |
| Muscle relaxes | Heart relaxes |
The Contractile Machinery in Cardiac Muscle Is Similar to That in Skeletal Muscle
Cardiac muscle, as with skeletal muscle, has a striated appearance under the light microscope (Figure 19-1). These cross-striations have the same structural basis in cardiac and skeletal muscle. Each striated cardiac muscle fiber (muscle cell) is made up of a few hundred myofibrils. Each myofibril has a repetitive pattern of light and dark bands. The various bands and lines within a myofibril are given letter designations (A band, I band, Z disk). The alignment of these bands in adjacent myofibrils accounts for the striated appearance of the whole muscle fiber. Each repeating unit of myofibrillar bands is called a sarcomere. This name, which means “little muscle,” is apt because a single sarcomere constitutes the basic, contractile subunit of the cardiac muscle. By definition, a sarcomere extends from one Z disk to the next, a distance of approximately 0.1 mm, or 100 μm.
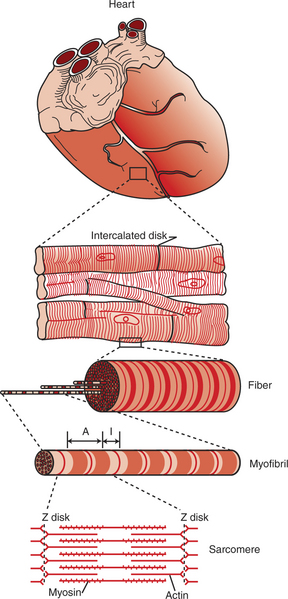
FIGURE 19-1 Under the light microscope, cardiac muscle fibers (muscle cells) are seen to be striated, similar to skeletal muscle. Electron microscopy reveals that the striations result from an orderly arrangement of actin (thin) filaments and myosin (thick) filaments into muscular subunits called sarcomeres (as shown in bottom drawing). Sarcomeres are the structural and functional subunits of cardiac muscle, as they are in skeletal muscle. Unlike skeletal muscle fibers, however, cardiac muscle fibers often branch, and they link end to end with neighboring fibers at structures called intercalated disks. Unseen within the intercalated disks are nexi, or gap junctions, which are minute cytoplasmic channels that allow action potentials to propagate from cell to cell.
As in skeletal muscle, each cardiac muscle sarcomere is composed of an array of thick and thin filaments. The thin filaments are attached to the Z disks; they interdigitate with the thick filaments. The thin filaments are composed of actin molecules. The thick filaments are composed of myosin molecules. In the presence of adenosine triphosphate (ATP) and calcium ions (Ca2+), myosin and actin interact in a series of steps called the cross-bridge cycle, which results in contraction and force generation in each sarcomere and therefore in the whole muscle cell (for details, see Figures 1-3, 1-4, 1-5, and 6-5).
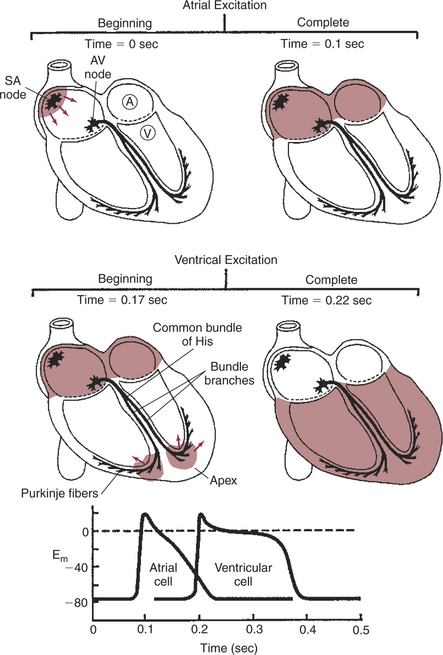
FIGURE 19-3 Heart is pictured at four instants during initiation of a normal contraction. Top left (time t = 0 sec), Pacemaker cell in the sinoatrial (SA) node has just reached threshold, and an action potential has begun to propagate outward across the atria. Top right (t = 0.1 sec), Action potential has reached all parts of both atria (all atrial cells are at the plateau of their action potentials). Middle left (t = 0.17 sec), Action potential has passed through the atrioventricular (AV) node and down the bundle branches and has just reached the ventricular apex. Middle right (t = 0.22 sec), Action potential has just finished propagating outward through the walls of both ventricles (all ventricular cells are at the plateau of their action potentials). Bottom, Graph shows the timing of action potentials in a left atrial cell (labeled A, top left) and a left ventricular cell (V, top left). Their locations make these among the last atrial and ventricular cells to be depolarized as action potentials propagate across the atria and ventricles, respectively. Em, Membrane potential in millivolts.
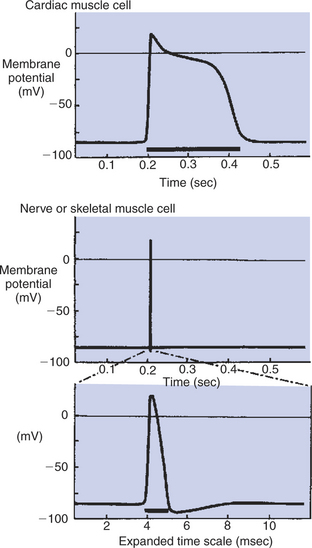
FIGURE 19-4 Action potentials in cardiac muscle cells (top) last 100 times longer than action potentials in nerve or skeletal muscle cells (middle). Bottom, The nerve or skeletal muscle action potential is shown on a greatly expanded time scale. The prolonged phase of depolarization in cardiac muscle cells is called the plateau of the action potential. The dark bars under each action potential indicate the length of the absolute refractory period.
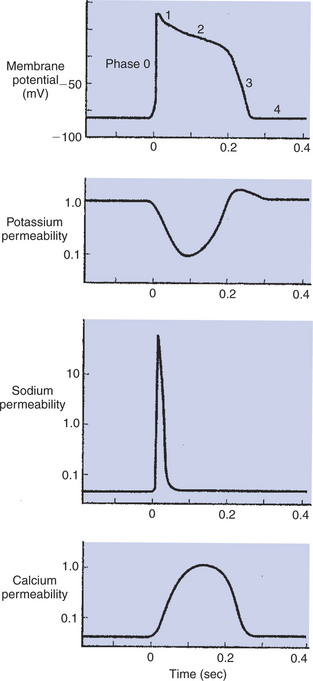
FIGURE 19-5 Membrane potential of a cardiac muscle cell (top) is determined by the relative permeabilities of the cell membrane to K+ (second from top), Na+ (second from bottom), and Ca2+ (bottom). At rest (left side of graphs), the cell is much more permeable to K+ than to Na+ or Ca2+. A cardiac action potential (middle of graphs) is produced by a characteristic sequence of changes in the permeabilities to K+, Na+, and Ca2+. The action potential ends when the permeabilities return to their resting state (right side of graphs). Phases 0 to 4 are discussed in the text.
Cardiac Muscle Forms a Functional Syncytium
Although the molecular basis of contraction is the same for cardiac and skeletal muscle, the two muscle types differ in regard to electrical linkages between neighboring cells, and this difference has important consequences. Individual skeletal muscle cells are electrically isolated (insulated) from one another, so action potentials cannot “jump” from one skeletal muscle cell to another. As described in Chapter 5, an action potential in a skeletal muscle cell is initiated only in response to an action potential in the somatic motor neuron that innervates the skeletal muscle cell. Each neural action potential causes release of the neurotransmitter acetylcholine, which activates nicotinic cholinergic receptors on the skeletal muscle cell, which in turn depolarizes the muscle cell to threshold for the formation of an action potential. Once formed, the action potential propagates along the length of that particular muscle cell and then stops. The muscle action potential causes the cell to contract. Neighboring cells may or may not contract at the same time, depending on whether or not action potentials are initiated in the neighboring cells by their motor neurons.
In contrast, cardiac muscle cells are electrically linked to one another. When an action potential is started in a single cardiac muscle cell, it propagates along the length of that cell. At specialized points of contact with neighboring cells, ionic currents then “jump the gap” and initiate action potentials in the neighboring cells. Because cardiac action potentials propagate from cell to cell through cardiac tissue, neighboring cardiac muscle cells all contract in synchrony, as a unit, and then they all relax. In this regard, cardiac muscle tissue behaves as if it were a single cell. Cardiac muscle is therefore said to form a functional syncytium (literally, “acts like same cell”).
The specialized cellular structures that allow cardiac action potentials to propagate from cell to cell are evident under the light microscope (see Figure 19-1). Cardiac muscle appears as an array of fibers (individual cardiac muscle cells) that are arranged approximately in parallel but with some branching. Adjacent cells are joined together by dark-appearing structures called intercalated disks. Electron microscopy has revealed that within these disks are tiny open channels between neighboring cells. These nexi, or gap junctions, provide points of contact between the intracellular fluid of adjacent cells. When an action potential depolarizes the cell on one side of an intercalated disk, positive ions flow through the gap junctions and into the neighboring cell. This local, ionic current depolarizes the neighboring cell to threshold for the formation of an action potential. In effect, an action potential propagates from cell to cell through the gap junctions that are located within the intercalated disks. Skeletal muscle does not have intercalated disks or nexi (gap junctions).
Cardiac Contractions Are Initiated by Action Potentials That Arise Spontaneously in Specialized Pacemaker Cells
Because cardiac muscle tissue forms a functional syncytium, and because a cardiac action potential leads to contraction, any one cardiac muscle cell can initiate a heartbeat. In other words, if a single cardiac muscle cell depolarizes to threshold and forms an action potential, that action potential will propagate from cell to cell, throughout the heart, and cause the whole heart to contract. Most cardiac muscle cells have the property of remaining stable at a resting membrane potential; they never form action potentials by themselves. However, a few specialized cardiac muscle cells have the property of depolarizing spontaneously toward the threshold for the formation of action potentials. When any one of these specialized cells reaches threshold and forms an action potential, a heartbeat results. Cardiac cells that depolarize spontaneously toward threshold are called pacemaker cells because they initiate heartbeats and therefore determine the rate, or pace, of the heart.
Although all spontaneously depolarizing cells in the heart are called pacemaker cells, only one pacemaker cell, the one that reaches threshold first, actually triggers a particular heartbeat. In the normal heart, the pacemaker cells that depolarize most quickly to threshold are located in the sinoatrial (SA) node. The SA node is in the right atrial wall, at the point where the venae cavae enter the right atrium.
Because it has spontaneously depolarizing pacemaker cells, the heart initiates its own muscle action potentials and contractions. Motor neurons are not necessary for initiating cardiac contractions, whereas they are needed for initiating skeletal muscle contractions. Motor neurons (sympathetic and parasympathetic) do affect the heart rate, by influencing the rapidity of the pacemaker cells’ depolarization to threshold level, but the heart continues to beat even without any sympathetic or parasympathetic influences. Thus a denervated heart still beats, whereas a denervated skeletal muscle remains relaxed (in fact, paralyzed). The ability of the heart to beat without neural input enables surgically transplanted hearts to function. When a donor’s heart is connected to a recipient’s circulation during cardiac transplantation, no nerves are attached to the heart. The pacemaker cells in the transplanted heart initiate its action potentials and contractions, just as they do in a normal heart. The only factor missing is control of the heart rate through cardiac sympathetic and parasympathetic nerves.
A System of Specialized Cardiac Muscle Cells Initiates and Organizes Each Heartbeat
Each normal heartbeat is initiated by an action potential that arises spontaneously in one of the pacemaker cells in the SA node (Figure 19-2). Once formed, the action potential propagates rapidly, from cell to cell, across the right and left atria, causing both atria to contract. Next, the action potential works its way, from cell to cell, through a special pathway of cardiac muscle cells that lies between the atria and the ventricles. This pathway consists of the atrioventricular (AV) node and the first part of the AV bundle, also called the bundle of His. The AV node and AV bundle provide the only route for the conduction of action potentials from the atria to the ventricles. Elsewhere, the atria and ventricles are separated by a layer of connective tissue, which can neither form nor propagate action potentials. In addition to providing the only conductive pathway between the atria and the ventricles, the AV node and the first part of the AV bundle have the special property of very slow conduction of action potentials. It takes 50 to 150 msec for an atrial action potential to travel through the AV node and the first part of the AV bundle; that is, it takes 50 to 150 msec for an atrial action potential to propagate into the ventricles. Slow conduction through the AV junction creates the delay between atrial and ventricular contractions.
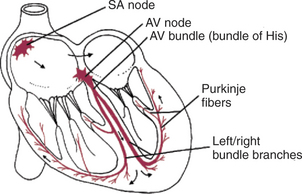
FIGURE 19-2 Specialized conduction system of the heart is responsible for the initiation and organization of cardiac contractions. The system is composed of specialized cardiac muscle fibers, not nerves. AV, Atrioventricular; SA, sinoatrial.
Once past the slowly conducting cells of the AV junction, the cardiac action potential enters a branching network of specialized cardiac cells that have the property of extremely rapid conduction of action potentials from cell to cell. This network begins with the AV bundle, which has slowly conducting cells in its first portion (connected to the AV node) and rapidly conducting cells beyond that. The AV bundle divides to form the rapidly conducting left and right bundle branches. At the ventricular apex, the bundle branches break up into a dispersed network of Purkinje fibers, which carry the action potential rapidly along the inner walls of both ventricles. The Purkinje fibers propagate action potentials into the normal ventricular muscle fibers within the inner walls (subendocardial layers) of both ventricles. From there, the action potentials propagate quite rapidly outward, from cell to cell, through the ventricular walls. As the action potential reaches each ventricular muscle fiber, that fiber contracts. The extremely rapid conduction of the cardiac action potential, from cell to cell, through the latter portion of the AV bundle, the bundle branches, and the Purkinje system results in a nearly synchronous contraction of all the fibers in both ventricles.
The SA and AV nodes, AV bundle, bundle branches, and Purkinje fibers are collectively called the specialized conduction system of the heart. This system is composed of specialized cardiac muscle cells, not nerves. The particular characteristics of the components in the specialized conduction system cause each heartbeat to follow a specific, patterned sequence. In a normal beat, both atria contract, almost simultaneously. Next, there is a brief pause (caused by slow conduction of the action potential through the AV node). The two ventricles then contract, almost simultaneously. Finally, the entire heart relaxes and refills.
Figure 19-3 reemphasizes the role of the specialized conduction system in initiating and organizing a normal cardiac contraction. In this “time lapse” illustration, atrial excitation begins at time t = 0, when one SA node cell has reached threshold and an action potential is just beginning to propagate out of the SA node and into regular atrial tissue. Within 0.1 second, the action potential has propagated completely across the right and left atria, and a coordinated contraction of both atria is just beginning. As the action potential propagates across the atria, it also depolarizes the first cells in the AV node, beginning at time t = 0.04 second. While the atria are in a depolarized (excited) state, the action potential is propagating slowly from cell to cell through the AV node and first part of the AV bundle. After traversing this slowly conducting region, the action potential is propagated rapidly through the remainder of the bundle of His and its branches. The action potential arrives at the ventricular apex at time t = 0.17 second. Note that it takes about 0.13 second (0.17 − 0.04 second) for the action potential to travel through the AV node and bundles; that is, 0.13 second represents a typical delay between atrial depolarization and ventricular depolarization. From the ventricular apex, the action potential is propagated rapidly throughout both ventricles by Purkinje fibers. Ventricular excitation (depolarization) is complete by time t = 0.22 second, and both ventricles contract. By this time the atria have repolarized to a resting state and are relaxing. After ventricular excitation and contraction, the ventricles relax, and the whole heart remains in a resting state until the next beat is originated by an SA node pacemaker cell.
Cardiac Action Potentials Are Extremely Long
Two major differences between action potentials in skeletal muscle and cardiac muscle have already been mentioned: First, cardiac muscle cells are electrically connected, whereas skeletal muscle cells are electrically isolated. Second, the heart has pacemaker cells, which form spontaneous action potentials, whereas skeletal muscle cells only depolarize and form action potentials when “commanded” to do so by motor neurons.
A third important difference between skeletal and cardiac action potentials is their duration (Figure 19-4). The entire action potential in a skeletal muscle lasts only 1 to 2 msec. A cardiac action potential lasts about 100 times longer (100-250 msec). Prolongation of the cardiac action potential is brought about by prolonged changes in the permeability of the cardiac muscle membrane to sodium (Na+), potassium (K+), and Ca2+ ions. Cardiac muscle cell membranes have Na+ and K+ channels similar to those found in skeletal muscle, but the timing of their opening and closing is different in cardiac muscle. In addition, cardiac cell membranes also have special Ca2+ channels that are not present in skeletal muscle. The movement of extracellular Ca2+ through cardiac Ca2+ channels has an especially important role in prolonging the cardiac action potential. The presence of Ca2+ channels and the important role of extracellular Ca2+ in the action potential is the fourth major difference between cardiac and skeletal muscle.
To understand the special significance of the membrane Ca2+ channels in cardiac muscle, it is useful to review the roles of K+ and Na+ channels in skeletal muscle and to emphasize some ways in which cardiac K+ and Na+ channels are similar to those in skeletal muscle. As explained in Chapter 4, many of the K+ channels in a neuron or skeletal muscle cell membrane are open when the cell is at rest, and most of the Na+ channels are closed. As a result, the resting cell is much more permeable to K+ than to Na+. The tendency of the positively charged K+ to leave the cell creates a resting membrane potential (polarization) in which the inside of the cell membrane is negative in comparison with the outside. The resting membrane potential is typically between −70 and −85 mV (see Figure 19-4, bottom). An action potential is created when something depolarizes the cell (makes it less negative inside). Depolarization to the threshold voltage for opening the voltage-gated Na+ channels allows an influx of extracellular Na+ into the cell. This rapid entry of positive ions causes the cell membrane to become positively charged on its inside surface. This positive membrane potential persists for only a moment, however, because the Na+ channels become inactivated very quickly, and the cell rapidly repolarizes toward its resting membrane potential. Repolarization is also promoted by the opening of additional K+ channels. In fact, this opening of extra K+ channels may cause neurons and skeletal muscles to become hyperpolarized (even more negative than normal) for a few moments at the end of each action potential (see Figure 19-4, bottom).
In a resting skeletal muscle cell, calcium ions are stored within the intracellular organelle called the sarcoplasmic reticulum. The occurrence of an action potential in the skeletal muscle cell causes Ca2+ to be released from the sarcoplasmic reticulum into the free intracellular fluid, which is called the cytosol. The increase in cytosolic Ca2+ concentration initiates muscle contraction (see Figure 1-5). The contraction initiated by a single action potential is very brief in skeletal muscle, because the cytosolic Ca2+ is rapidly pumped back into the sarcoplasmic reticulum by active transport, and the muscle relaxes. Note that the Ca2+ responsible for initiating skeletal muscle contraction comes entirely from the intracellular storage site, the sarcoplasmic reticulum. No extracellular Ca2+ enters the cell during the action potential, because skeletal muscle cells do not have membrane Ca2+ channels. In cardiac muscle, in contrast, membrane Ca2+ channels and the entry of extracellular Ca2+ into the cells play key roles in both action potentials and contractions.
Membrane Calcium Channels Play a Special Role in Cardiac Muscle
Figure 19-5 depicts a cardiac muscle cell action potential and the sequence of changes in K+, Na+, and Ca2+ permeability that are responsible for the action potential. As the time line begins (on the left side of each graph), the cardiac cell is at a normal, negative resting membrane potential of about −80 mV. The cardiac membrane potential is negative at rest for the same reason that skeletal muscle cells have negative resting membrane potentials: many K+ channels are open at rest, and most of the Na+ channels are closed. As a result, membrane permeability to K+ is much higher than membrane permeability to Na+ (see Figure 19-5, middle two graphs). In resting cardiac cells, the membrane Ca2+ channels are closed, so Ca2+ permeability is very low (see Figure 19-5, bottom); extracellular Ca2+ ions are prevented from entering the cardiac cells.
As in skeletal muscle, a cardiac action potential is created when the cell is depolarized to the threshold voltage for opening the voltage-gated Na+ channels. The rapid influx of extracellular Na+ into the cell causes the cell membrane to become positively charged on its inside surface (phase 0 in Figure 19-5, top). The Na+ channels inactivate very quickly, and the membrane begins to repolarize (phase 1). However, in cardiac muscle, repolarization is interrupted, and there is a prolonged plateau of depolarization, which lasts about 200 msec (phase 2). The plateau of the cardiac action potential is brought about by two conditions that do not occur in nerves or skeletal muscle fibers: (1) some K+ channels close, so K+ permeability decreases, and (2) many of the Ca2+ channels open, so Ca2+ permeability increases. Because the Ca2+ concentration is higher in the extracellular fluid than in the intracellular fluid, Ca2+ flows through the open Ca2+ channels and into the cytosol. The combination of reducing the exit of K+ from the cell and allowing the entrance of Ca2+ into the cell keeps the cell membrane in a depolarized state. After about 200 msec, the K+ channels reopen, and the Ca2+ channels close; K+ permeability increases, and Ca2+ permeability decreases. The combination of increasing the exit of K+ from the cell and shutting off the entrance of Ca2+ into the cell causes the cell to repolarize (phase 3) and eventually to reach its stable, negative resting membrane potential (phase 4).
The specialized Ca2+ channels in cardiac muscle cell membranes are called slow Ca2+ channels because they take much longer to open than the Na+ channels, and they stay open much longer. As shown in Figure 19-5, Na+ permeability increases and then decreases (Na+ channels open and then inactivate) within a few milliseconds. Ca2+ permeability, in comparison, is slow to increase (Ca2+ channels are slow to open) and Ca2+ permeability remains elevated for about 200 msec (the time Ca2+ channels stay open). In recognition of their much quicker responses, the Na+ channels in cardiac muscle are sometimes called fast Na+ channels.
The Ca2+ that enters a cardiac cell during an action potential triggers the release of additional Ca2+ from the sarcoplasmic reticulum. This process is called calcium-triggered calcium release. In less than 0.1 second, the concentration of free Ca2+ in the cytosol increases about 100-fold. As in skeletal muscle, this increase in cytosolic Ca2+ initiates concentration. When the Ca2+ channels close, at the conclusion of the action potential, most of the free, cytosolic Ca2+ is pumped back into the sarcoplasmic reticulum or pumped back across the cell membrane into the extracellular fluid. Both these processes involve active transport, because the Ca2+ is being pumped against its electrochemical gradient. Once the cytosolic Ca2+ concentration is returned to its low, resting level, the cardiac muscle relaxes. Figure 19-6 shows the relationship between action potentials and the resulting contractions in a cardiac muscle cell.
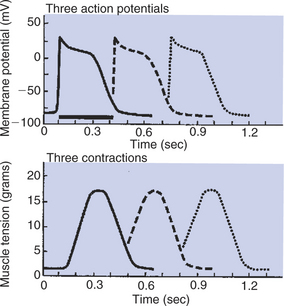
FIGURE 19-6 The first of three cardiac action potentials (solid line, top) causes a cardiac contraction (solid line, bottom). Note that the action potential and contraction have similar durations. The heavy horizontal bar under the first action potential shows the duration of the absolute refractory period. The dashed line and dotted line in the top graph show the earliest possible occurrence of a second and a third action potential, each occurring right after the absolute refractory period for the preceding action potential. The dashed line and dotted line in the bottom graph depict the corresponding cardiac contractions. Because of the long refractory period, each contraction is almost over before the earliest possible next contraction can begin. This guarantees a period of cardiac relaxation between contractions.
The Long Duration of the Cardiac Action Potential Guarantees a Period of Relaxation (and Refilling) Between Heartbeats
Na+ channels become inactivated at the peak of the cardiac action potential. Na+ cannot pass through an inactivated channel; therefore, as long as the Na+ channels remain inactivated, another action potential cannot occur. The inactivated state ends, and Na+ channels become susceptible to reopening, only when the cell membrane potential returns to its resting level. Thus, Na+ inactivation guarantees that the upstroke of a second action potential cannot occur until the first action potential is completed.
While the Na+ channels are inactivated, the cell is refractory (resistant) with regard to the formation of an action potential. The time after the beginning of one action potential during which another action potential cannot be initiated is called the absolute refractory period (or refractory period). Because Na+ inactivation lasts until the membrane potential returns to its resting level, the refractory period lasts about as long as an action potential. Thus the refractory period in a nerve or skeletal muscle cell lasts about 1 or 2 msec, whereas the refractory period in a cardiac muscle cell lasts 100 to 250 msec (see Figure 19-4).
The long refractory period in cardiac muscle guarantees a period of relaxation (and cardiac refilling) between cardiac contractions. Figure 19-6 (top) depicts the quickest possible succession of three action potentials in a cardiac muscle cell: the second action potential begins immediately after the conclusion of the refractory period for the first action potential. Likewise, the third action potential begins immediately after the conclusion of the refractory period for the second. The bottom graph shows the pattern of muscle contraction that results from the three action potentials. Note that contractile strength reaches a peak late in the plateau phase of each action potential, and that the contractile strength decreases (the muscle begins to relax) during the repolarization phase of each action potential. Because the next action potential cannot begin until the first one has ended, the cardiac muscle cell becomes partially relaxed before the earliest possible subsequent contraction can begin; that is, each action potential produces a cardiac contraction that is distinctly separated from the preceding contraction. Because of its long refractory period, cardiac muscle cannot sustain a continuous contraction. Thus the heart has a guaranteed period of relaxation (and refilling) between heartbeats.
The pattern of changes in muscle tension depicted in the bottom of Figure 19-6 corresponds closely to the changes in the cytosolic Ca2+ concentration. This makes sense, considering that the increase in cytosolic Ca2+ concentration during an action potential initiates muscle contraction, and the subsequent removal of Ca2+ from the cytosol permits the muscle to relax. Contractile tension decreases almost to its resting level during the repolarization phase of the action potential, because cytosolic Ca2+ concentration is reduced almost to its resting level by the time the action potential is over. In other words, during the repolarization phase of the action potential, active transport pumps move most of the free, cytosolic Ca2+ back into the sarcoplasmic reticulum or out into the extracellular fluid.
In skeletal muscle cells, an action potential lasts only 1 to 2 msec. The membrane is repolarized (and the refractory period is over) even before the release of Ca2+ from the sarcoplasmic reticulum is finished, and many milliseconds before the released Ca2+ is pumped back into the sarcoplasmic reticulum. As a result, the cytosolic Ca2+ concentration reaches its peak level after the action potential is over, and the contractile tension resulting from the action potential also reaches its peak after the action potential is over. Because a contractile twitch lasts much longer than the refractory period in skeletal muscle, several action potentials can occur during the time of a single contractile twitch. Multiple action potentials in quick succession cause cytosolic Ca2+ concentration to build to a high level and stay there. The resulting contractile tension is stronger than the tension that results from a single action potential, and it is sustained for a longer time. In effect, the muscle twitches caused by successive action potentials “fuse” together. This phenomenon is called temporal summation. Fusion and temporal summation are the mechanisms that permit graded and prolonged tension development in skeletal muscle. In contrast, the long refractory period in cardiac muscle cells prevents the fusion and summation of cardiac contractions. Each contraction of the heart (each heart beat) is followed immediately by a relaxation.
Atrial Cells Have Shorter Action Potentials Than Do Ventricular Cells
The previous description of cardiac ion channels, action potentials, and contractions is based on properties of normal ventricular cells. Atrial cells are basically similar, except that their action potentials are shorter than action potentials in ventricular cells. As with ventricular cells, atrial cells have fast Na+ channels that open briefly at the beginning of an action potential and then become inactivated. Likewise, atrial slow Ca2+ channels open during the action potential, and K+ channels close. The differences between atrial and ventricular cells are that atrial slow Ca2+ channels typically stay open a shorter time than those in ventricular cells, and atrial K+ channels stay closed for a shorter time. As a result, the plateau of an atrial cell’s action potential is shorter and not as “flat” as the plateau of a ventricular cell’s action potential (see Figure 19-3, bottom). As a consequence of having a shorter action potential, atrial cells have a shorter refractory period than do ventricular cells. Therefore the atrial cells are capable of forming more action potentials per minute than are ventricular cells; that is, the atria can “beat” faster than the ventricles. The implications of this difference are discussed later in this chapter.
Specialized Ion Channels Cause Cardiac Pacemaker Cells to Depolarize to Threshold and Form Action Potentials
As mentioned, the cardiac pacemaker cells of the SA node spontaneously depolarize to threshold and then form action potentials. The spontaneous depolarization is called a pacemaker potential, and it is the distinguishing feature of a pacemaker cell (Figure 19-7, top). The action potentials of cardiac pacemaker cells typically have a rounded appearance; they lack the very rapid (phase 0) depolarization seen in ventricular and atrial cells.
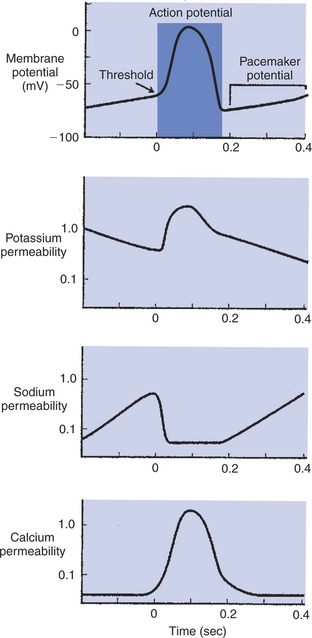
FIGURE 19-7 A cardiac pacemaker cell depolarizes spontaneously to threshold and initiates its own action potential (top). The spontaneous depolarization (called the pacemaker potential) is the result of a spontaneous, progressive decrease in K+ permeability (second from top) and an increase in Na+ permeability (second from bottom). An increase in Ca2+ permeability makes a late contribution to the depolarization toward threshold (bottom). Once threshold level is reached, an action potential is produced. The action potential is driven primarily by a large, prolonged increase in Ca2+ permeability. The absence of fast Na+ channels in pacemaker cells causes the upstroke of the pacemaker action potential to be much slower than that seen in nonpacemaker cells. (Compare with Figure 19-5.)
The spontaneous depolarizations and rounded action potentials are consequences of the particular ion channels found in pacemaker cells. Pacemaker cells lack the usual voltage-gated fast Na+ channels. Instead, these cells have pacemaker Na+ channels (also called “funny Na+ channels”), which close during an action potential and then begin to open again, spontaneously, once an action potential has finished. The spontaneous opening of the pacemaker Na+ channels causes a progressive increase in the cell’s Na+ permeability (see Figure 19-7, second from bottom). The increase in Na+ permeability allows Na+ to enter the cell from the extracellular fluid, which depolarizes the cell toward threshold. Pacemaker cells also have an unusual set of K+ channels, which participate in their spontaneous depolarization. At the end of one action potential, K+ permeability in pacemaker cells is quite high, because most K+ channels are open. Then some K+ channels begin to close. As K+ permeability decreases, less K+ leaves the cells, which makes the cells progressively less negatively charged inside. Ca2+ channels also make a small contribution to the pacemaker potential. Late in the pacemaker potential, just before a pacemaker cell reaches threshold, slow Ca2+channels begin to open, and Ca2+ permeability begins to increase. The resulting entry of Ca2+ into the cell speeds its final approach to threshold. Thus the pacemaker potential is caused by the opening of pacemaker Na+ channels, the closing of K+ channels, and (late in the process) the opening of Ca2+ channels. These spontaneous changes in Na+, K+, and Ca2+ channels in pacemaker cells are in contrast to the stable status of the ion channels in normal, resting ventricular or atrial cells.
Once threshold is reached in a pacemaker cell, an action potential occurs. The upstroke of the action potential is quite slow compared with the rapid, phase 0 depolarization in a normal ventricular or atrial cell, because there are no fast Na+ channels in pacemaker cells and therefore no sudden influx of Na+. The ion primarily responsible for the action potential in a pacemaker cell is Ca2+. Once threshold is reached, many of the cell’s slow Ca2+ channels open. The permeability to Ca2+ increases, and extracellular Ca2+ flows into the cell. The action potentials in pacemaker cells are often called slow action potentials, because they lack a rapid, phase 0 depolarization and because they are caused primarily by the opening of slow Ca2+ channels. In contrast, normal ventricular or atrial action potentials are called fast action potentials. Note, however, that all cardiac action potentials (even the “fast” ones) have a very long duration compared with action potentials in nerve or skeletal muscle cells.
Sympathetic and Parasympathetic Nerves Act on Cardiac Pacemaker Cells to Increase or Decrease the Heart Rate
Figure 19-8 shows how the neurotransmitters norepinephrine and acetylcholine affect the pacemaker cells of the heart. Norepinephrine exerts its effect by activating β-adrenergic receptors on the cell membranes of pacemaker cells. Activation of such receptors speeds up the ion channel changes that are responsible for the spontaneous depolarization of pacemaker cells. Because the pacemaker cells reach threshold more quickly in the presence of norepinephrine, there is a shorter interval between heartbeats. Therefore, heart rate is elevated above its intrinsic or spontaneous level.
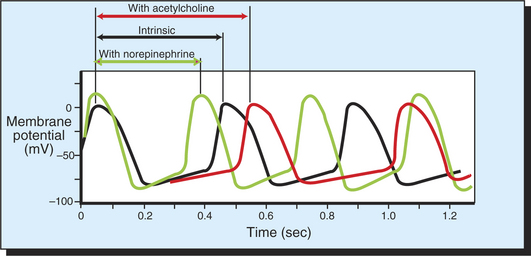
FIGURE 19-8 In the absence of neurohumoral influences, a pacemaker cell of the SA node spontaneously depolarizes to threshold and initiates a series of action potentials, three of which are shown by the black line. The interval between action potentials under these conditions determines the intrinsic, or spontaneous, heart rate. Acetylcholine decreases the rate of depolarization and therefore lengthens the interval between action potentials. Norepinephrine increases the rate of depolarization and therefore shortens the interval between action potentials.
Acetylcholine has the opposite effect. Acetylcholine activates muscarinic cholinergic receptors on the cell membranes of pacemaker cells, which slows the ion channel changes that are responsible for the pacemaker cell’s spontaneous depolarization. Acetylcholine makes it take longer for pacemaker cells to reach threshold. There is consequently a longer time between heartbeats, so the heart rate is slowed below its intrinsic or spontaneous rate.
Sympathetic neurons release norepinephrine at the SA node cells, and thus sympathetic nerve activity increases the heart rate. Epinephrine or norepinephrine, released from the adrenal glands and circulating in the bloodstream, has the same effect. Parasympathetic neurons release acetylcholine at the SA node cells, and thus parasympathetic activity decreases the heart rate. Figure 19-9 illustrates how sympathetic and parasympathetic neurons interact in the control of the heart rate. In the absence of norepinephrine and acetylcholine, the heart beats at its intrinsic rate. For a large dog, this rate is typically about 140 beats per minute (beats/min). Heart rates below the intrinsic rate are achieved by activation of parasympathetic neurons and release of acetylcholine. Accordingly, the graph indicates that parasympathetic activity is high during awake rest (heart rate of 90 beats/min) and very high during sleep (heart rate of 55 beats/min). Heart rates above the intrinsic rate occur during exercise or emotional arousal and are achieved by activation of the sympathetic nerves to the heart and release of norepinephrine (or by circulating epinephrine or norepinephrine). The highest possible level of sympathetic activity, and therefore the highest possible heart rate, occurs during maximal exercise or a defense alarm reaction (fear, “fight or flight” response).
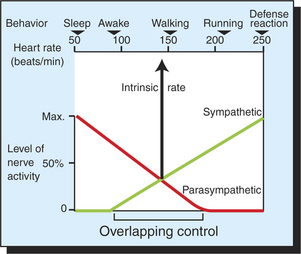
FIGURE 19-9 Upper scale shows that heart rate in a normal, large dog ranges from 50 to 250 beats/minute, depending on behavioral state. The graph shows that this wide range of heart rates is brought about by the interactions between sympathetic nerve activity, which speeds the heart above its intrinsic rate, and parasympathetic nerve activity, which slows the heart below its intrinsic rate. Sympathetic and parasympathetic nerves are simultaneously active over a considerable portion of the heart rate range (Overlapping control). Note that the heart beats at its intrinsic rate (about 140 beats/minute) either in the absence of any neural influence or when sympathetic and parasympathetic effects are equal and opposite.
Through variation in the levels of sympathetic and parasympathetic tone, the dog’s heart rate is adjusted, over a wide range, as appropriate for each behavioral situation. When both systems are partially active, the resulting heart rate represents the outcome of a “tug-of-war” between sympathetic action to increase the heart rate and parasympathetic action to decrease the heart rate. Typically, the sympathetic and parasympathetic systems are both partially active during awake states, ranging from quiet rest (heart rate about 90 beats/min) to moderate exercise (heart rate about 175 beats/min). Parasympathetic activity predominates in the lower part of this range, and sympathetic activity predominates in the higher part. When sympathetic activity and parasympathetic activity are equal, their effects cancel, and the heart rate is at its intrinsic (spontaneous) level. Simultaneous activation of sympathetic and parasympathetic neurons appears to give the nervous system tight control over the heart rate under a wide variety of behavioral conditions.
Cells of the Atrioventricular Node Act as Auxiliary Pacemakers and Also Protect the Ventricles from Beating Too Fast
As with SA node cells, the cells of the AV node normally exhibit pacemaker activity and slow action potentials. As shown in Figure 19-10, the AV node cells spontaneously depolarize toward threshold, but much more slowly than do the SA node cells. Therefore, under normal circumstances, the SA node cells reach threshold first and initiate an action potential, which then propagates from cell to cell across the atria and into the AV node. As this action potential enters the AV node, it encounters cells that are slowly, spontaneously depolarizing toward threshold. The arriving action potential quickly depolarizes these AV node pacemaker cells to threshold, and they form an action potential, which then propagates into the AV bundle and the ventricles. Thus, under normal conditions, each cardiac action potential is triggered by an SA node pacemaker cell, and the pacemaker activity of the AV node cells is irrelevant.
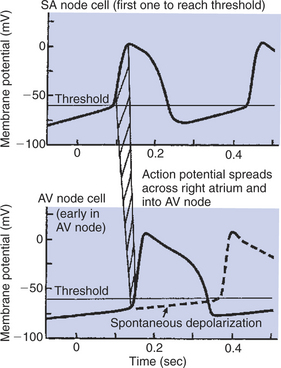
FIGURE 19-10 Both sinoatrial (SA) node cells and atrioventricular (AV) node cells exhibit pacemaker activity (spontaneous depolarization toward threshold). Normally the SA node cells depolarize more quickly and reach the threshold first (top). The resulting atrial action potential propagates into the AV node (as represented by hatched band) and depolarizes the AV node cells quickly to their threshold, causing them to form an action potential (solid line, bottom graph). However, if the SA node pacemaker cells become nonfunctional or if atrial action potentials are not conducted into the AV node, the AV node cells eventually depolarize to threshold and initiate action potentials on their own (dashed line, bottom graph). In this way the AV node cells serve as an auxiliary (emergency) ventricular pacemaker.
Under certain abnormal conditions, AV node pacemaker function becomes critical for survival. For example, if the SA node is damaged and does not depolarize to threshold, the spontaneous depolarization of the AV node pacemaker cells initiates action potentials and therefore cardiac contractions. If not for this auxiliary pacemaker function of AV node cells, the heart with a damaged SA node would not beat at all. The heart rate is characteristically very low when the AV node cells are initiating the heartbeats. Because the AV node pacemaker cells depolarize more slowly than normal SA node cells, the heart rate resulting from AV node pacemakers is very low, about 30 to 40 beats/min in a resting dog, compared with 80 to 90 beats/min when the SA node cells are the pacemakers. Nevertheless, if the SA node fails as a pacemaker, enough heartbeats are initiated by the AV node to sustain life temporarily. Thus, AV node cells are sometimes called the heart’s emergency pacemakers.
Another important feature of the AV node cells is that they have much longer refractory periods than do normal atrial cells. The long refractory period of AV node cells helps protect the ventricles from being stimulated to contract at rates that are too rapid for efficient pumping. This protective function of the AV node is critical to an animal’s survival when atrial action potentials are extremely frequent (see later discussion of atrial flutter/fibrillation). The long refractory period of the AV node cells plays an important role, even in a normal heart. Once a normal action potential reaches the ventricles, it is prevented from “circling back” (and reactivating the atria) by the prolonged refractory state of the AV node cells.
Table 19-2 summarizes the four important electrical characteristics of the AV node previously discussed. Note that three of these characteristics are influenced by the nervous system. As indicated in the table, sympathetic activity increases the conduction velocity of the AV node cells, shortens their refractory period, and speeds their auxiliary pacemaker activity. Parasympathetic activation has the opposite effects. These sympathetic and parasympathetic effects are appropriate for different heart rates. For example, when sympathetic activity is high and the SA node pacemakers are initiating heartbeats frequently, the whole process of cardiac contraction and relaxation must be sped up. Thus it is appropriate that sympathetic action also increases the velocity of action potential conduction through the AV node, which shortens AV delay. In addition, sympathetic activation shortens the AV node refractory period, which allows each of the frequent atrial action potentials to be conducted to the ventricles. Finally, sympathetic activation enhances AV node auxiliary pacemaker activity, which provides the animal with a high enough ventricular rate to cope with some stress, even if the SA node pacemaker has failed. Conversely, when parasympathetic activation causes the SA node pacemakers to decrease the heart rate, all aspects of cardiac contraction and relaxation can proceed at a more leisurely pace. Under these conditions it is appropriate for AV node conduction velocity to be slowed and the AV node refractory period to be lengthened.
Table 19-2 Electrical Characteristics of the Atrioventricular (AV) Node
| Characteristic (significance) | Sympathetic effect* | Parasympathetic effect† |
|---|---|---|
| Is the only conducting pathway between atria and ventricles (directs atrial action potentials into the rapidly conducting AV bundle and bundle branches) | — | — |
| Has a slow conduction velocity (creates AV delay) | Increases velocity (shortens AV delay) | Decreases velocity (lengthens AV delay) |
| Has a very long refractory period (protective effects: limits maximal rate to which atria can drive ventricles and prevents ventricular action potentials from reexciting atria) | Shortens refractory period (appropriate for high heart rates) | Lengthens refractory period (appropriate for low heart rates) |
| Spontaneously depolarizes to threshold (acts as auxiliary pacemaker) | Faster depolarization (speeds auxiliary pacemaker) | Slower depolarization (slows auxiliary pacemaker) |
Sympathetic Nerves Act on All Cardiac Cells to Cause Quicker, More Forceful Contractions
Sympathetic neurons release norepinephrine in all regions of the heart, not only at the SA and AV nodes, and all cardiac muscle cells have β-adrenergic receptors that are activated by norepinephrine. Circulating epinephrine or norepinephrine can also activate these same receptors. The effects of β-receptor activation on the SA and AV node cells have already been described (see Figure 19-8 and Table 19-2). In all other atrial and ventricular cells, β-receptor activation leads to higher, shorter action potentials and to stronger, quicker contractions. One reason for these effects is that activation of β receptors increases the number of Ca2+ channels that open during the plateau (phase 2) of an action potential, which increases the amount of extracellular Ca2+ that enters the cell. Because Ca2+ entry is the primary depolarizing influence during the plateau, increased Ca2+ entry raises the plateau (makes the membrane potential more positive). A secondary consequence is to shorten the action potential. The action potential becomes shorter because of a complicated effect of the elevated plateau on the K+ channels. Recall that K+ channels close at the beginning of a cardiac action potential and then, after a time, reopen (see Figure 19-5). Reopening of the K+ channels helps repolarize the cell to a resting state at the end of the action potential. The length of time before K+ channels reopen depends on the membrane voltage during the plateau of the action potential. Specifically, when the membrane potential is more positive than normal during the plateau, the K+ channels reopen sooner. This shortens the action potential and speeds repolarization. Overall, β-receptor activation makes each action potential higher and shorter. An action potential of higher amplitude propagates more quickly along each cell and from cell to cell, leading to faster conduction velocity. The shorter action potential means a shorter refractory period, which facilitates formation of more heartbeats per minute.
Because β-receptor activation opens more Ca2+ channels and increases the entry of extracellular Ca2+ into cardiac muscle cells during an action potential, it also increases the strength of the resulting contraction. The entry of more extracellular “trigger” Ca2+ creates a greater stimulus for the release of Ca2+ stores from the sarcoplasmic reticulum. Therefore the cytosolic Ca2+ concentration reaches an exceptionally high level during the action potential, which leads to a stronger contraction. The contraction also begins more quickly. In addition, the contraction is shorter, because β-receptor activation speeds up the pumps that move cytosolic Ca2+ back into the sarcoplasmic reticulum and out of the cell into the extracellular fluid. Thus, even though more Ca2+ than normal enters the cytosol during an action potential, its removal at the end of the action potential is quicker than normal. Overall, β-receptor activation makes each cardiac contraction stronger, quicker, and shorter.
In summary, sympathetic nerves act (1) on the SA node pacemaker cells to increase the heart rate, (2) on the AV node cells to increase the conduction velocity and shorten the AV delay, and (3) on all cardiac cells to shorten the refractory period and make each cardiac contraction stronger, quicker, and shorter. All these changes cause the heart to pump more blood at a higher pressure, which is an animal’s normal response during exercise or emotional arousal.
Because sympathetic effects on the heart are all brought about through activation of the β-adrenergic receptors on the cardiac muscle cells, the administration of a drug that activates β receptors (β-adrenergic agonist) has the same effects as sympathetic activation. Epinephrine and isoproterenol are two common β-adrenergic agonists. Conversely, the administration of a drug that binds to and blocks β receptors reduces all the effects of sympathetic activation. Propranolol and atenolol are common examples of such β-adrenergic antagonists. Examples of their use are provided later.
Parasympathetic Effects Are Opposite to Those of Sympathetic Activation but Are Generally Restricted to the Sinoatrial Node, Atrioventricular Node, and Atria
Parasympathetic nerves affect the heart by the release of acetylcholine, which activates muscarinic cholinergic receptors on cardiac muscle cells. Qualitatively, all the effects of parasympathetic activation are opposite to those of sympathetic activation, because the effects of activating muscarinic cholinergic receptors are opposite to the effects of activating β-adrenergic receptors. Parasympathetic nerves have very powerful effects on the SA node pacemaker cells (see Figure 19-8) and on the AV node cells (see Table 19-2). In addition, parasympathetic nerves exert strong, antisympathetic influences on all the atrial cells. However, parasympathetic nerves have relatively weak effects on the ventricular muscle cells, because very few ventricular cells receive direct parasympathetic innervation. By contrast, all ventricular muscle cells receive direct sympathetic innervation. In summary, the predominant parasympathetic influences are exerted at the SA node (to decrease the rate), at the AV node (to slow conduction and lengthen the refractory period), and on all supraventricular cells (to lengthen the refractory period and make their contractions weaker and slower).
Parasympathetic neurons do exert a curious, indirect effect on ventricular muscle cells. In the ventricles, parasympathetic neurons release their acetylcholine onto sympathetic neuron terminals. This acetylcholine activates muscarinic cholinergic receptors that are located on the sympathetic neuron terminals. The effect of this activation is to inhibit the release of norepinephrine from the terminals, which weakens the effects of sympathetic activation on ventricular cells.
Parasympathetic effects on the heart can be mimicked by the administration of a muscarinic cholinergic agonist (e.g., acetylcholine or muscarine) and blocked by the administration of a muscarinic cholinergic antagonist (e.g., atropine). Some therapeutic applications are mentioned later.
Dysfunction in the Specialized Conducting System Leads to Abnormalities in Cardiac Rhythm (Arrhythmias)
Cardiac arrhythmias result either from problems with the formation of action potentials or from problems with the propagation (conduction) of action potentials. One example of a problem with action potential formation has already been mentioned: sinus arrest, in which the SA node completely fails to form action potentials. In a patient with sinus arrest, the auxiliary pacemaker function of the AV node keeps the heart beating, although at an extremely low rate. Complete cessation of the SA node is the extreme case of the condition called sick sinus syndrome. In its more common and less extreme form, sick sinus syndrome is characterized by sluggish depolarization of the SA node pacemaker cells. Patients exhibit an abnormally slow heart rate at rest (bradycardia) and an insufficient increase in heart rate during exercise. Specifically, in sick sinus syndrome, the intrinsic sinus rate is abnormally low.
Even though the problem in sick sinus syndrome is intrinsic to the sinus itself, one treatment strategy is to administer a drug that blocks parasympathetic action on the heart (a cholinergic muscarinic antagonist, such as atropine). Table 19-3 illustrates the logic behind this treatment. In a normal, healthy large dog, the intrinsic rate of the heart is 140 beats/min. However, the heart rate at rest is about 90 beats/min because high parasympathetic tone normally slows the SA node pacemaker to a rate below its intrinsic rate. A drug that blocks parasympathetic effects on the heart would return the heart rate to 140 beats/min. A dog with a sick sinus has a low intrinsic heart rate, perhaps 80 beats/min. Parasympathetic tone makes the resting heart rate even lower, approximately 30 beats/min. A drug that blocks parasympathetic effects restores the heart rate to its intrinsic level, 80 beats/min. Therefore a dog with sick sinus syndrome treated with atropine has a heart rate that closely matches the rate of a normal resting dog.
Table 19-3 Treatment of Sick Sinus Syndrome by Blocking Parasympathetic Effects on Heart Rate with a Cholinergic Muscarinic Antagonist
| Heart rate | Normal dog (beats/min) | Dog with sick sinus syndrome (beats/min) |
|---|---|---|
| Intrinsic rate | 140 | 80 |
| Resting rate (with parasympathetic tone) | 90 | 30 |
| Rate after atropine | 140 | 80 |
Another possible therapeutic approach is to increase the heart rate by administering a drug that mimics the action of sympathetic nerves, specifically, a β-adrenergic agonist (e.g., isoproterenol). Enough isoproterenol would be given to increase the resting rate from 30 to 80 beats/min.
If drug treatment of sick sinus syndrome is ineffective, an alternative way to increase the heart rate is through the use of an artificial cardiac pacemaker. Such a device periodically applies electric shocks to the heart, which depolarize cardiac muscle to threshold. Shocks applied to the atria initiate atrial action potentials. If the AV node is functioning normally, these atrial action potentials are conducted to the ventricles, and the ventricles also contract. For temporary or emergency treatment, the pacemaker electrodes can be inserted intravenously (e.g., via the jugular vein) and advanced into the right atrial chamber. For long-term treatment, a battery-powered electrical stimulator can be surgically implanted under the patient’s skin and attached to electrodes that are either inserted into one of the heart’s chambers or attached to the outside surface of the heart.
Atrioventricular Node Block Is a Common Cause of Cardiac Arrhythmias
Whereas sick sinus syndrome exemplifies a dysfunction of action potential formation, AV node block is a common dysfunction of action potential conduction. If damage to the AV node prevents (blocks) conduction of atrial action potentials into the ventricles, the atria continue to beat at a rate determined by the SA node pacemaker cells. The ventricles also continue to beat, but at a much lower rate. In such a case the ventricular action potentials and contractions are being initiated by auxiliary pacemaker cells low in the AV node (i.e., below the level of the block). Because the AV node pacemaker cells depolarize more slowly than the SA node pacemaker cells, the ventricles in a resting dog with AV node block typically beat at only 30 to 40 beats/min. Furthermore, these ventricular beats are not synchronized with the atrial contractions.
Three degrees of severity of AV node block are recognized. Complete block of the AV node, in which no atrial action potentials are conducted to the ventricles, is called thirddegree AV node block. If action potentials are conducted sporadically from the atria to the ventricles, so that the AV node transmits some atrial action potentials but not all of them, the condition is called second-degree AV node block. In a patient with second-degree block, some atrial contractions are followed by ventricular contractions, and others are not. Strong parasympathetic activity can create or exaggerate second-degree AV node block because parasympathetic activity increases the refractory period of the AV node cells. For example, in quietly resting horses, parasympathetic activity is often so strong, and the AV node refractory period so long, that some atrial beats are not conducted to the ventricles. Therefore, if the pulse of a relaxed, resting horse is palpated, some “missing” ventricular contractions are likely to be noticed. During exercise the same horse does not show AV node block because parasympathetic activity has been decreased and sympathetic activity increased. Both these changes shorten the refractory period of the AV node and make it much more certain that every atrial action potential will be conducted to the ventricles.
Second-degree or third-degree AV node block often involves the electrical phenomenon known as decremental conduction. As mentioned, AV node cells have “slow” action potentials, characterized by a less rapid upstroke, a lower voltage amplitude, and a slower velocity of conduction than the action potentials in regular atrial or ventricular cells. All these differences make conduction of the action potential from cell to cell less reliable in the AV node than in regular atrial or ventricular tissue. When the AV node cells are in an electrically depressed state, an atrial action potential may simply die out within the AV node and may not be conducted to the ventricles. This fading and eventual stoppage of a cardiac action potential in a slowly conducting region is called decremental conduction.
The mildest degree of AV node block is first-degree block, in which every atrial action potential is transmitted to the ventricles, but the action potential propagates even more slowly than normal through the AV node. Therefore, in first-degree block, the delay between atrial contraction and ventricular contraction is abnormally long. Because the AV node conduction velocity can be slowed by parasympathetic activity and sped by sympathetic activity, the behavioral state of the patient characteristically influences the severity of first-degree block.
AV node block can be caused by cardiac trauma, toxins, viral or bacterial infections, ischemia, congenital heart defects, or cardiac fibrosis. AV node block is sometimes caused by inadvertent damage of AV node tissue during a surgical repair of a ventricular septal defect.
AV node block must be treated if the resulting ventricular rate is too low to maintain adequate blood flow to the body. Drugs that block parasympathetic actions on the heart (muscarinic cholinergic antagonists such as atropine) might reduce the AV node refractory period and decremental conduction sufficiently to overcome a blocked state. The same effect might be achieved with a drug that mimics the effect of sympathetic nerves by activating β-adrenergic receptors (e.g., isoproterenol) (see Table 19-2). If drug treatment fails to correct AV node block, an artificial pacemaker is needed. In the case of AV node block, the pacemaker needs to be applied to the ventricles; pacing the atria would not be beneficial because atrial action potentials would not be conducted to the ventricles.
Cardiac Tachyarrhythmias Result Either from Abnormal Action Potential Formation (by the Sinoatrial Node or Ectopic Pacemakers) or from Abnormal Action Potential Conduction (“Reentry”)
Tachyarrhythmias are arrhythmias in which the atrial rate or the ventricular rate (or both) is abnormally high. An occasional extra atrial or ventricular beat is called a precontraction or a premature beat. Occasional precontractions are common both in animals and in humans and usually have no clinical significance. If the precontractions become frequent or continuous, the condition is called tachycardia, which means “rapid heart.” Tachycardia is a clinically significant sign. The tachyarrhythmias result from abnormal pacemaker activity. The pacemaker initiating the rapid or “extra” beats can be the SA node itself. Alternatively, a region of abnormal cardiac muscle outside the SA node can act as a pacemaker by spontaneously depolarizing to threshold before the regular SA node pacemaker does. Any such region is called an ectopic pacemaker. Common causes of ectopic pacemaker activity include cardiac infection or trauma, reaction to a drug or toxin, electrolyte imbalances, myocardial ischemia, and myocardial infarction.
Tachycardia is a heart rate that is more rapid than is appropriate for the behavioral circumstances (e.g., 160 beats/ min in a resting dog). The tachycardias are named for the site of the pacemaker at which they originate. If the tachycardia appears to originate from the SA node pacemaker cells, the condition is called sinus tachycardia. If the tachycardia originates from an ectopic pacemaker within the atria, it is called atrial tachycardia. Atrial tachycardia is common in some canine breeds, including boxers and wolfhounds. Junctional tachycardias originate from ectopic pacemakers within the AV node or first part of the AV bundle. Supraventricular tachycardia is a collective term that encompasses sinus tachycardia, atrial tachycardia, and junctional tachycardia. If the ectopic pacemaker causing tachycardia is within the ventricles, the condition is called ventricular tachycardia. In this situation the ventricles beat at a rapid rate, as dictated by the ectopic ventricular pacemaker. In occasional patients, some of the action potentials initiated by an ectopic ventricular pacemaker may be conducted backward through the AV node and may cause atrial precontractions. Usually, however, the AV node does not conduct action potentials backward; the atria continue to beat at the rate dictated by the normal SA node pacemaker. In either case, ventricular contractions are not preceded in the normal way by atrial contractions. The major dysfunction associated with ventricular tachycardia is that the ventricles do not relax long enough between contractions for adequate filling, and this problem is exacerbated by the absence of appropriately timed atrial contractions.
An extremely rapid atrial tachycardia is called atrial flutter. Atrial flutter does not lead to ventricular flutter because of the long refractory period of the AV node cells; the AV node conducts some, but not all, of the frequent atrial depolarizations to the ventricles. This is an example of the AV node protecting the ventricles from beating at too rapid a rate. If atrial contractions become so rapid that they lose synchrony, the condition is called atrial fibrillation. Atrial fibrillation is characterized by the continuous, random passage of action potentials through the atria. Fibrillating atria appear to quiver; there is no effective, coordinated contraction, and no blood is pumped. Atrial fibrillation is common in horses and in certain breeds of dogs, including Dobermans. Atrial fibrillation usually does not lead to ventricular fibrillation because of the protective effect of the AV node. The ventricles continue to contract with a synchronized, effective pumping stroke, at a rate that is limited by the refractory period of the AV node.
Synchronous ventricular contractions are essential for life. If the synchrony of ventricular contractions is disrupted and the ventricles begin to fibrillate, ventricular pumping stops. In ventricular fibrillation, each tiny region of the ventricular wall contracts and relaxes at random, in response to action potentials that propagate randomly and continuously throughout the ventricles. The condition of ventricular fibrillation (“V-fib”) is synonymous with sudden cardiac death.
In most cases, ventricular fibrillation can be reversed only by electrical defibrillation. In this process a strong electrical current is passed briefly through the heart muscle. This current depolarizes all the cardiac cells simultaneously and holds them in a depolarized state for several milliseconds. It is hoped that when the current is turned off, all the cardiac muscle cells will simultaneously repolarize to a resting membrane potential, so that the normal pacemaker of the heart will have a chance to initiate beats in an organized and synchronized manner once again. Sometimes it works; however, if the cardiac problems that caused ventricular fibrillation to develop in the first place are still present, fibrillation is likely to recur. Usually, defibrillation is performed by placing stimulating electrodes (“paddles”) on either side of the thorax. Therefore the stimulating current passes through, and depolarizes, the skeletal muscles of the thorax as well as the cardiac muscle of the heart. The resulting, involuntary contraction of the skeletal muscles causes the patient to “jump” at the moment of defibrillation.
Ectopic pacemaker activity typically arises when a region of cardiac muscle develops the abnormal, twin properties of slow conduction of action potentials and an ability to conduct action potentials in only one direction. Figure 19-11 illustrates how a region of slow, one-way conduction in the wall of one cardiac chamber can initiate tachycardia. The process begins with a normally originating action potential being conducted in only one direction through the abnormal region of cardiac muscle. If the conduction through the abnormal muscle is so slow that all the normal muscle is past its refractory period by the time the action potential emerges from the abnormal region, the emerging action potential can trigger another action potential in the normal muscle. If this second action potential then propagates around the cardiac chamber and back into the abnormal region, a self-perpetuating cycle can develop. The action potential once again propagates slowly through the abnormal region, and once again it emerges from the abnormal region after the normal muscle is past its refractory period. The result is a sequence of reentrant action potentials that propagate through the normal cardiac muscle, each one initiating a contraction—an “extra” beat. The reentrant pathway does not necessarily have to be all the way around the circumference of a cardiac chamber. An ischemic or infarcted area of cardiac muscle can form the nonconducting center around which reentrant action potentials can travel. This passage of an action potential around and around a nonconducting center is called a circus movement. In order for the circus movement of the action potential to be self-regenerating, however, a portion of the circular, conducting pathway must have the twin properties of slow and one-way conduction. In effect, an area of slow, one-way conduction within a circular conducting pathway (and around a nonconducting center) functions as an ectopic pacemaker. Reentry of cardiac action potentials can lead to occasional precontractions, continuous tachycardia, or even fibrillation. In any of these cases, the resulting tachyarrhythmia is called a reentrant arrhythmia.
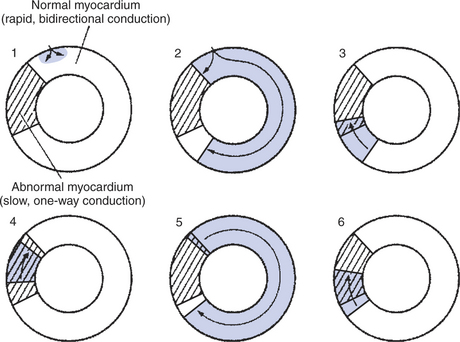
FIGURE 19-11 Cross section of a cardiac chamber (atrium or ventricle) is pictured at six different instants to illustrate how reentrant arrhythmias occur. The region of abnormal myocardium (cross-hatched area) conducts action potentials slowly and only in one direction (clockwise in this example). 1, Normal action potential has just entered this ring of tissue, and only the gray-shaded area is depolarized. 2, Action potential propagates rapidly in both directions through the normal cardiac tissue but is blocked from entering the abnormal myocardium in a counterclockwise direction. 3, The clockwise-going action potential can enter the abnormal region. 4, While the action potential propagates slowly, in a clockwise direction through the abnormal region, the normal cardiac tissue repolarizes to a resting state. 5, Action potential emerges from the abnormal region into normal cardiac tissue and propagates through the normal tissue for a second time. Meanwhile, the abnormal tissue repolarizes to a resting state. 6, Action potential begins to move slowly through the abnormal region for a second time. States 4, 5, and 6 repeat themselves. Thus the abnormal region functions as an ectopic pacemaker.
Common Antiarrhythmic Drugs Affect the Ion Channels Responsible for the Cardiac Action Potential
Whereas ventricular fibrillation is generally lethal without electrical defibrillation, other tachycardias can often be treated successfully with antiarrhythmic drugs. Because tachyarrhythmias result from extra cardiac action potentials, effective antiarrhythmic drugs must work by counteracting either the formation or the propagation of the extra action potentials.
Local anesthetics (e.g., quinidine, lidocaine) constitute one category of antiarrhythmic drugs. They act by binding to some of the fast Na+ channels in cardiac muscle cells and preventing them from opening. This counteracts membrane depolarization and action potential formation. In essence, blocking some of the Na+ channels raises the threshold for action potential formation. This tends to “quiet” ectopic pacemakers and also to stifle reentrant arrhythmias. Na+ channel blockers such as lidocaine or procaine (Novocain) are called “local anesthetics” because, when applied to sensory neurons, they prevent the propagation of neural action potentials that would signal pain to the brain. The cardiac, antiarrhythmic effect of local anesthetics is not the result of their blockage of pain pathways.
A second category of antiarrhythmic drugs is the calcium channel blockers. Examples include verapamil, diltiazem, and nifedipine. These drugs bind to slow Ca2+ channels and prevent them from opening, which decreases the entry of Ca2+ into cardiac muscle cells during an action potential. Because Ca2+ entry is the primary depolarizing influence during the plateau (phase 2) of the cardiac action potential, one major effect of a Ca2+ channel blocker is to lower the plateau (make the membrane potential less positive). A secondary consequence is to lengthen the action potential. The action potential is longer because of a complicated effect of the height of the plateau on K+ channels, as discussed earlier in connection with sympathetic effects on cardiac action potentials. Drugs that lengthen the cardiac action potential also lengthen the refractory period, which makes it less likely that early extra action potentials will be formed in ectopic pacemakers or that they will propagate even if they are formed.
The calcium channel blockers have especially strong effects on the cells of the SA and AV nodes. As mentioned, Ca2+ entry through slow Ca2+ channels is the main event in the slow action potentials of these cells. Not surprisingly, therefore, the amplitude of slow action potentials is greatly reduced by Ca2+ channel blockers, and these action potentials are also lengthened. Low-amplitude, long action potentials propagate very slowly from cell to cell, which decreases the likelihood that early extra action potentials will form or propagate in SA or AV node cells.
Calcium channel blockers are especially effective in protecting the ventricles from rapid rates in cases of persistent atrial flutter or fibrillation. By increasing the refractoriness and decreasing the conduction velocity of AV node cells, Ca2+ channel blockers cause many of the extra atrial action potentials to “die out” (through decremental conduction) in the AV node.
By reducing the entry of extracellular Ca2+ into cardiac muscle cells during an action potential, Ca2+ channel blockers not only suppress tachyarrhythmias, but also decrease the strength of cardiac contractions. Less entry of extracellular “trigger” Ca2+ means a less powerful stimulus for the release of stored Ca2+ from the sarcoplasmic reticulum. Therefore the cytosolic Ca2+ concentration does not increase as much as normal during the action potential, so there is a less force-ful contraction. Some clinical situations in which it is desirable to decrease cardiac contractility are discussed in Chapter 21.
The cardiac glycosides (e.g., digitalis) constitute a third category of antiarrhythmic drugs. Cardiac glycosides act by inhibiting the Na+,K+ pump in cell membranes. As mentioned in Chapters 1 and 4, the Na+,K+ pump is an antiport carrier that uses energy from ATP to transport Na+ out of cells and K+ into cells. The pump also indirectly supplies energy to a Na+,Ca2+ antiporter that helps to transport Ca2+ back out of cardiac cells after it enters during an action potential. Inhibition of the Na+,K+ pump with a cardiac glycoside has several important effects on cardiac function. The effects are listed here without much explanation because the mechanisms are quite complex. First, cardiac muscle cells do not repolarize fully at the end of an action potential; the resting membrane potential is not as negative as normal. As a consequence, some Na+ channels remain inactivated, which makes the cells somewhat refractory with regard to the formation of subsequent action potentials. This tends to quiet ectopic pacemakers. Second, effects on the central nervous system lead to an increase in parasympathetic tone. This slows the heart rate, quiets atrial ectopic pacemakers, slows conduction through the AV node, and increases the refractory period of AV node cells. The overall effect is to suppress ectopic atrial action potentials or cause extra atrial action potentials to “die out” in the AV node and not to be conducted to the ventricles. A third effect of cardiac glycosides is to allow more Ca2+ than normal to accumulate inside cardiac cells, resulting in the strength of cardiac contractions increasing. In summary, the cardiac glycosides are antiarrhythmic and also increase cardiac contractility.
Beta-adrenergic antagonists (e.g., propranolol) constitute a fourth class of antiarrhythmic drug. Beta blockers, as they are called, bind to some of the β-adrenergic receptors on cardiac cells and prevent their activation by norepinephrine from sympathetic nerves or by epinephrine and norepinephrine from the adrenal medulla. Sympathetic activation tends to promote tachyarrhythmias by increasing heart rate, shortening refractory period, and speeding conduction of action potentials, especially through the AV node. Beta blockers reduce these effects and therefore reduce the likelihood that extra action potentials will form or propagate. An additional effect of β blockers is to reverse sympathetic activation– induced increases in cardiac contractility.
In summary, of the four categories of drugs used to treat tachyarrhythmias, three also have pronounced effects on cardiac contractility. The calcium channel blockers and β blockers decrease cardiac contractility, whereas cardiac glycosides increase contractility. Local anesthetics have little effect on contractility. This variety of effects allows a clinician to select the type of antiarrhythmic drug that is best matched to each patient’s cardiac contractile state.
Electrical dysfunction of the heart has been discussed in considerable detail to illustrate how specific abnormalities in the specialized cardiac conduction system can result in specific and serious arrhythmias. Electrical dysfunction of the heart is encountered often in clinical practice, and its consequences are often serious or even lethal. Because electrical dysfunction is so important, Chapter 20 is devoted to an explanation of the electrocardiogram, which is the most commonly used tool for evaluating electrical dysfunction of the heart.
CLINICAL CORRELATIONS
Third-Degree Atrioventricular Block
History.
A 5-year-old male English bulldog has fainted several times during the past 3 weeks. On each occasion he collapses, is apparently unconscious for a few seconds, and then slowly recovers. These episodes occur most often during exertion. In general, he tends to be less active than normal, but he has no other obvious signs of illness.
Clinical Examination.
The dog is moderately obese. There are no obvious neurological deficits. His mucous membranes appear normal; they are pink, and the capillary refill time is normal (1.5 seconds). Auscultation of the chest reveals a slow, regular heart rate of 45 beats/min. The femoral pulse rate is also 45 beats/min and strong. Thoracic radiography reveals a mildly enlarged heart, but the radiographs are otherwise within normal limits.
The electrocardiogram (ECG) reveals a disparity between the atrial rate (atrial depolarizations occurring regularly, 140 times/min) and the ventricular rate (ventricular depolarizations occurring regularly, 45 times/min). There is no consistent time interval between the atrial and ventricular depolarizations.
Comment.
Voltage fluctuations are produced at the body surface by atrial and ventricular depolarizations (see Chapter 20). Electrocardiography detects these voltage fluctuations and produces a graph of voltage as a function of time. The ECG of this dog shows a complete dissociation between atrial and ventricular depolarizations, which provides definitive diagnostic evidence of complete (third-degree) AV node block. The dog’s atria are depolarizing 140 times/min in response to action potentials being initiated in the normal manner by pacemaker cells of the SA node. However, the atrial action potentials are not being conducted through the AV node. Ventricular action potentials are being initiated, at the slow rate of 45/min, by auxiliary pacemaker cells located below the blocked region of the AV node.
The low ventricular rate in this dog allows a longer-than-normal time for ventricular filling between beats. Therefore the volume of blood ejected with each beat (the stroke volume) is greater than normal. The increased stroke volume causes the femoral pulse to be very strong.
In a normal dog, sympathetic and parasympathetic nerves acting on the SA node pacemaker cells adjust the heart rate so that cardiac output is matched to the metabolic requirements of the body. In a dog with complete AV block, the ventricles do not respond to these autonomically mediated changes in SA node pacemaker rate. Typically, the rate of ventricular contractions is low at rest and cannot increase much during exercise. Therefore, cardiac output does not increase enough during exertion to meet the increased metabolic needs of exercising skeletal muscle. As a consequence, arterial blood pressure decreases. The decreased arterial pressure during exercise causes brain blood flow to fall below the level needed to sustain consciousness. The dog faints.
Treatment.
Drug therapy for AV node block involves either blocking the effects of parasympathetic nerves on the AV node (with a muscarinic cholinergic antagonist drug such as atropine) or mimicking the effects of sympathetic activation (with cautious use of a β-adrenergic agonist such as isoproterenol or dopamine). The rationale for these treatments is based on the following physiology: AV node block occurs because atrial action potentials “die out” in the AV node (decremental conduction). Parasympathetic activation increases the tendency for decremental conduction because parasympathetic nerves act on AV node cells to increase their refractory period and to decrease the velocity with which action potentials spread from cell to cell. Therefore, blocking parasympathetic effects sometimes (but rather rarely) reverses AV node block. Sympathetic nerves decrease the refractory period of AV node cells and increase their conduction velocity. Therefore, sympathetic activation decreases the tendency for decremental conduction; a sympathomimetic drug (one that mimics the effects of sympathetic activation) has the same effect. In addition, sympathetic nerves act directly on the ventricular auxiliary pacemaker cells to increase their rate. Therefore, even if a sympathomimetic drug does not unblock the AV node, it increases somewhat the ventricular rate.
Many cases of third-degree AV block cannot be managed effectively with drugs, so an artificial ventricular pacemaker must be installed. The procedure is straightforward; pacemaker electrodes can be inserted into the right ventricle through a systemic vein (e.g., external jugular) with only sedation and local anesthesia. The electrode wires are attached to a battery-powered pacemaker unit that is then implanted under the skin.
Berne RM, Levy MN. Cardiovascular physiology, ed 8. St Louis: Mosby, 2001.
Cote E, Ettinger SJ. Electrocardiography and cardiac arrhythmias. Ettinger SJ, Feldman EC. Textbook of veterinary internal medicine: diseases of the dog and cat, ed 6, St Louis: Saunders, 2005.
Katz AM. Physiology of the heart, ed 4. Baltimore: Lippincott Williams & Wilkins, 2005.
Lederer WJ. Cardiac electrophysiology and the electrocardiogram. In: Boron WF, Boulpaep EL. Medical physiology: a cellular and molecular approach. Philadelphia: Saunders, 2005.
Lilly LS. Pathophysiology of heart disease: a collaborative project of medical students and faculty, ed 4. Baltimore: Williams & Wilkins, 2006.
Milnor WR. Cardiovascular physiology. New York: Oxford University Press, 1990.
Opie LH. The heart: physiology, from cell to circulation, ed 4. Baltimore: Lippincott Williams & Wilkins, 2003.
Reece WO. Dukes’ physiology of domestic animals, ed 12. Ithaca, NY: Comstock Cornell University Press, 2004.
PRACTICE QUESTIONS
1. An increase in heart rate could result from:
2. In which of the following arrhythmias will there be more atrial beats per minute than ventricular beats?
3. The normal pathway followed by a cardiac action potential is to begin in the SA node and then propagate:
5. Which of the following types of drugs would be the best choice to treat a patient with both supraventricular tachycardia and inadequate cardiac contractility?
6. During which phase of a normal ventricular action potential is it most likely that fast Na+ channels are in an inactivated state, slow Ca2+ channels are open, and most K+ channels are closed?
7. Which of the following is true for both cardiac muscle and skeletal muscle?