Chapter 17 Drugs Affecting Urine Formation
Renal Physiology and Drug Therapy
Extracellular Fluid
The physiology of body fluids and the role of the kidney are also addressed in Chapters 16 and 18 The volume of extracellular fluid (ECF) is determined primarily by total body sodium content for two reasons: First, sodium is the major constituent of ECF; secondly, active transport mechanisms control sodium in intracellular and extracellular compartments.1 Control of ECF involves cardiovascular, renal, and central nervous systems. Because these systems are closely integrated, one system can compensate for the failure of another system, thus ensuring that salt and water excretion remain appropriate even with changes in blood pressure. Among the compensatory mechanisms is autoregulation of renal blood flow through efferent arteriolar resistance. Because regulatory mechanisms adjust both short- and long-term sodium and water transport rates, small increases in mean arterial blood pressure can cause a marked increase in sodium excretion, ultimately changing total body sodium.1 However, a check-and-balance system exists whereby if sodium balance becomes negative, as sodium concentration in the ECF decreases, causing thirst and water intake to decrease accordingly until ECF sodium concentration normalizes. A positive balance does the opposite, resulting in an increase in water intake. These integrated systems maintain a sodium and water excretion that equals that of intake minus any lost through nonrenal mechanisms (e.g., feces, sweat).
Renal Transport of Fluids and Electrolytes
The proximal tubules are responsible for 66% of the sodium and glomerular filtrate reabsorbed by the kidney. The primary pathway of fluid and electrolyte reabsorption in the renal tubule begins in the lumen and progresses through the cell and the interstitial fluid and into the capillary. Two water-permeable cell membranes are traversed during reabsorption. In the proximal tubule, active sodium ion reabsorption from the lumen into the cell generates an osmotic gradient in the lumen that leads to an almost simultaneous movement of water into the cell. Sodium reabsorption that begins with entry across the luminal membrane continues with movement across the basolateral membrane into the interstitial space (Figure 17-1). Basolateral movement is the energy-dependent process, fueled by a Na/K-dependent ATPase located on the basolateral membrane. The exchange rate of two K+ for each three Na+ entering the interstitial fluid provides an electrochemical gradient that favors passive entry of Na+ from the lumen into the cell. The concentration gradient generated by the basolateral movement determines the rate of sodium movement from the lumen. Chloride (and to a lesser degree, other anions) follows sodium, maintaining electroneutrality of the reabsorbate and a slight electronegativity of the cell compared with the luminal contents. The concentration gradient also favors passive movement of potassium from the interstitium into the cell, where it is used to continue the Na-K exchange. Although movement of sodium from the lumen into the cell is passive (albeit at the cost of an active basolateral efflux), sodium reabsorption into the cell is facilitated by three additional entry mechanisms: diffusion with chloride (quantitatively the most important), co-transport with uncharged or acidic anions, and countertransport with hydrogen ions (important to acid–base regulation and the site of carbonic anhydrase action). Although transport of sodium, water, and other electrolytes raises interstitial pressure, movement into peritubular capillaries facilitates continued reabsorption. Bicarbonate processing also occurs in the proximal tubule.
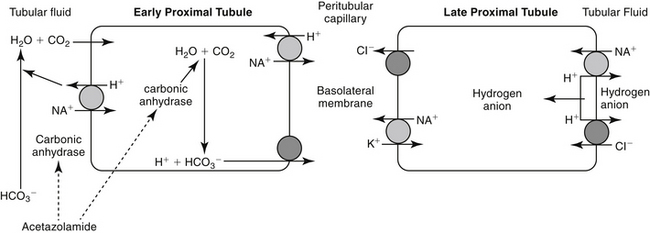
Figure 17-1 Movement of fluids and electrolytes into the proximal tubule and predominant sites of action of selected diuretics. The proximal tubule is the primary site of fluid resorption. Sodium enters the renal tubular cell by several mechanisms, including concentration-dependent passive diffusion, concentration-independent active transport, and countertransport with hydrogen. This latter mechanism yields bicarbonate and, although it occurs spontaneously, is also catalyzed by carbonic anhydrase. As sodium, chloride, and water are resorbed, the concentration of the remaining solute in the urine increases, causing an osmotic draw of fluid back into the tubule. Consequently, resorption of fluids is self-limiting to some degree. Dashed lines reflect inhibited actions, and solid lines reflect stimulation.
Unlike the proximal tubule, cells of the descending limb of the loop of Henle do not appear to be equipped with specialized transporting systems. The cells are relatively impermeable to sodium, chloride, and potassium but are permeable to water. Water moves from the lumen into the interstitium, causing electrolyte concentrations to progressively increase in the lumen until a maximum is reached at the bend. Diuretic drugs do not appear to be active in the descending portion of the tubule. In the ascending limb of the loop, chloride is transported “uphill,” achieving intracellular concentrations that are higher than predicted (based on the Nernst equation).2 Chloride movement occurs by a Na (downhill), K+-2Cl– (uphill) co-transport system, with Na+, K+-ATPase in the peritubular membrane providing the energy source. High intracellular chloride concentration facilitates chloride movement into the interstitium. The ion transports in the ascending loop of Henle are critical for proper function of the countercurrent mechanism in the renal medulla. The ascending loop of Henle tubule is not permeable to water, and fluid in the lumen becomes progressively diluted (Figure 17-2). About 25% of sodium-chloride reabsorption and 40% of potassium reabsorption occurs in the ascending loop of Henle, although only about 15% of the filtrate is reabsorbed in this region.2 In contrast to the proximal tubules, Na+-H+ exchange does not appear to occur in the loop of Henle, and little if any bicarbonate is processed.
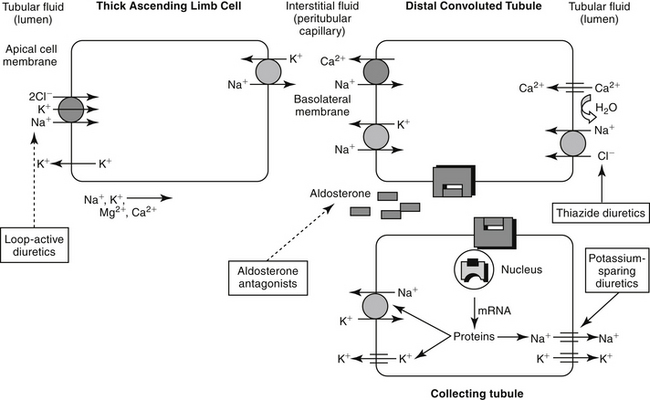
Figure 17-2 Movement of fluids and electrolytes in the thick ascending loop of Henle, the distal tubule, and the collecting ducts. Sites of action of selected diuretics also are indicated (dashed lines reflect inhibited actions, and solid lines reflect stimulation). In the ascending loop of Henle, electrolytes, but not water, are able to pass from the lumen into the cells. Up to 25% of the sodium and 40% of potassium is resorbed by way of a countertransport mechanism across a concentration gradient. In addition, chloride is actively transported into the cell. In the distal tubule, aldosterone plays a major role in fluid and electrolyte transport. Aldosterone mediates uptake of luminal sodium; sodium is exchanged for potassium, which is excreted into the lumen. Because of its location distal to most diuretics, efficacy of diurectics may be markedly decreased in high aldosterone states (e.g., congestive heart failure, liver disease). Aldosterone acts through steroid receptors. Like most steroidal compounds, the effects of aldosterone depend on nuclear transcription of effector proteins.
Reabsorption of water and electrolytes in the distal tubule and collecting ducts is variable. Sodium and chloride are reabsorbed against a concentration gradient. Because the early distal tubule is impermeable to water, an unfavorable concentration gradient is generated that limits the effectiveness of a sodium pump. Sodium and potassium contents in the urine are closely regulated by an aldosterone-sensitive mechanism in the distal late tubule (see Figure 17-2). Aldosterone signals the synthesis of a protein that increases the sodium and potassium permeability of the luminal membrane. As sodium moves in, potassium simultaneously moves from the interstitium into the cell. Electrogenic movement of sodium into the cell causes an electronegativity in the lumen that attracts potassium from the cell. Whether potassium is reabsorbed or excreted is determined primarily by plasma potassium, which in turn tends to depend on dietary intake.2
In the medullary collecting ducts, only small amounts of sodium chloride and potassium are reabsorbed. Antidiuretic hormone increases permeability of the cell membrane to water, which moves from the lumen, following the previously established medullary osmotic gradient. Less than 5% of filtered sodium is reabsorbed in the distal tubule and medullary collecting system under normal conditions.2
Atrial Natriuretic Hormone
Atrial natriuretic hormone (ANH), a “natriuretic factor” first described in 1984, is involved in the control of ECF. It is synthesized from a prohormone and stored as a peptide in granules in atrial myocardial cells. Concentrations increase above baseline when either the ECF expands, blood pressure increases, or dietary salt intake increases. Renal blood flow and glomerular filtration are subsequently increased. Sodium excretion also increases, presumably by a direct tubular action. Peripheral vasodilation can result in decreased blood pressure. Effects occur rapidly but are not sustained, suggesting that ANH is a mechanism that can restore equilibrium rapidly.2
Reduction of ECF is beneficial in conditions associated with inappropriate fluid retention such as in certain cardiac, renal, and liver diseases. Although the cause of ECF expansion differs in each disease, the commonality is salt and water retention. Because sodium is the primary cationic constituent of ECF, sufficient renal excretion of sodium ultimately reduces ECF.
Diuretic Therapy
Therapeutic Use of Diuretics
Three strategies exist for movement of inappropriate fluid accumulation (edema): correction of the underlying disease (often not possible), restriction of dietary or other sodium intake, and administration of diuretics. Of these, diuretics remain the cornerstone for treatment of edema or volume overload, particularly that which is life threatening.1 Although diuretics increase the rate of urine formation, their therapeutic indications include maintenance of urine flow; mobilization and reduction of inappropriate ECF stores, such as that manifested as edema or ascites; correction of specific ion imbalances; reduction in the rate of intraocular fluid formation; reduction of blood pressure; and reduction of pulmonary capillary wedge pressure.2
Targets of Diuretic Therapy
Diuretics are classified by their mechanism of action and include the loop diuretics, carbonic anhydrase inhibitors, thiazides, osmotic diuretics, and potassium-sparing diuretics (Figure 17-3, Table 17-1). Rational selection of diuretics relies on an appreciation of their mechanisms of action. With the exception of the osmotic diuretics and carbonic anhydrase inhibitors (the latter targets sodium bicarbonate), each class targets sodium or chloride reabsorption of tubular cells, effectively preventing the establishment of the normal ion gradient by renal tubular cells.1‑3 Diuretics that increase net urinary excretion of sodium chloride or sodium bicarbonate also are referred to as natriuretic. The efficacy and use of each class of diuretics varies with their site of action and the mechanism by which sodium reabsorption is inhibited. The mechanisms of sodium resorption that are targeted by diuretic therapy vary in location within the renal tubule and include electrogenic passive diffusion (proximal and late distal tubule and collecting system); exchange with hydrogen (generated by the actions of carbonic anhydrase and bicarbonate reabsorption); co-transport with glucose, organic acids, and phosphate (proximal tubule); reabsorption along with chloride reabsorption (late proximal tubule); and co-transport with chloride and potassium (by way of the thick ascending limb of the loop of Henle, which results in formation of medullary interstitial hyperosmolarity).
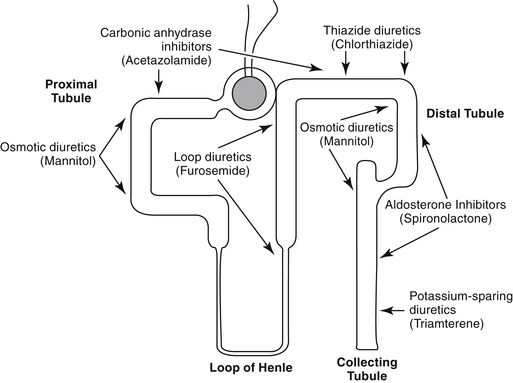
Principles of Diuretic Therapy Use
Several principles can guide diuretic therapy4: (1) The pattern of electrolyte excretion varies with the class of diuretic. (2) Maximal response to each drug class is limited by the site of action of the diuretic; as such, assuming drug delivery to the site of action has been appropriate, diuretic failure will occur to all members of the same drug class. class. (3) The combination of two (or more) diuretics with different mechanisms of action should cause additive and may cause synergistic effects.2
When used to reduce ECF volume, the selection of the most appropriate diuretic should be based on the cause of ECF volume retention. Diuretic selection for the patient with acute renal failure also should take into account the ability of the diuretic to reach the target tissue despite reduction in renal blood flow. The impact of the direct or indirect actions of the diuretic on systemic sequelae beyond decreased ECF volume (e.g., metabolic acidosis, hypokalemia) should be considered. Several diuretics also influence renal physiology by virtue of their effects on renal vasculature and may be preferred during states of reduced renal blood flow. Finally, several diuretics target physiologic processes that are not unique to the kidney (i.e., carbonic anhydrase inhibitors) and thus may be used therapeutically for reasons other than diuresis.
Factors Limiting Response to Diuretics
Two types of tolerance to diuretic therapy have been described in human patients. Short-term tolerance, or “braking,” occurs after the first dose and probably reflects a response to protect intravascular volume. Restoration of diuretic-induced loss of volume will resolve this type of tolerance. Long-term administration of loop diuretics can cause tolerance that reflects hypertrophy of the distal nephron in response to prolonged exposure to increased solute concentration. Sodium reabsorption increases accordingly, decreasing diuresis. Because thiazides target regions of the nephron that hypertrophy, a combination of thiazides with loop-acting diuretics results in a synergistic diuretic response in some human patients.
Several other factors may negatively affect response to diuretic therapy. Most diuretics are present at physiologic pH as uncharged molecules or organic ions and reach the renal tubular cell by active tubular secretion. The degree of ionization can affect the rapidity with which drugs are transported to renal receptors. Declining renal blood flow can preclude drug delivery to the site of diuretic action. For drugs that must reach distal sites (e.g., thiazides), it may be necessary to double doses to achieve a clinical response. For each diuretic a threshold (in drug concentration) must be reached before diuresis will occur. Lack of response may reflect simply an underdose for that patient, and dose titration is indicated. With renal disease characterized by proteinuria, many diuretics will remain bound to plasma proteins present in the tubular lumen and thus will remain inactive. Administration of the diuretic with albumin appears to facilitate response to diuretics in human patients with edema associated with the nephrotic syndrome. Resistance to diuretic therapy also may reflect the presence of another drug that decreases response. For example, nonsteroidal antiinflammatory drugs (NSAIDs) can alter intrarenal prostaglandin regulatory mechanisms, and a number of drugs compete for active tubular secretion of the diuretic into the tubular lumen.
In addition to their direct tubular effects, all diuretics indirectly influence renal tubular function. Accommodation to the effects of a diuretic in a normal animal may result in the loss of any pharmacologic effect several days after the start of diuretic therapy. Response to any diuretic will be modulated by internal homeostatic mechanisms that normally direct body fluid volumes and osmolar concentrations. For example, if a diuretic fails to cause a net sodium excretion (such as occurs with mannitol), ECF contraction will increase the concentration of electrolytes, stimulating water intake and replenishment of the lost volume.2 Refractoriness to thiazides can develop rapidly as a result of salt-retaining mechanisms being activated. Edema (ascites) associated with liver disease may not respond to diuretic therapy because signals from the cirrhotic liver indicate a depleted rather than an exaggerated ECF.2
Finally, refractoriness to diuretic therapy may reflect pharmacokinetic changes. The oral bioavailability of some diuretics (e.g., furosemide) is quite variable in human patients and unpredictable in the individual patient. It may be necessary to try several doses before the most effective one is found. Some diuretics (spironolactone, amiloride) are more effective after metabolism by the liver, although this also may be species dependent. Although the elimination half-life of several diuretics is sufficiently long to allow twice-daily elimination (in human patients), several of the loop diuretics are characterized by an elimination half-life of 2 to 3 hours. For such drugs the pharmacologic effect is decreased once the drug is no longer present. Rebound reabsorption of sodium by the nephrons has been described in such cases, suggesting that constant-rate intravenous infusion may provide better response. In addition, in patients that are not responding well, small increases in diuresis in the presence of the drug potentially will be magnified to clinically significant increases if the response is continuous.
Refractoriness to diuretic therapy can be approached by cage rest (which may improve renal circulation), an increase in the dose of the diuretic, intravenous administration, the use of a more effective diuretic (such as a loop-active diuretic), or a decrease in interval such that the drug is present at the site for a longer period of time (constant infusion). For constant-rate intravenous infusion, a loading dose (full to double dose) should be given, followed by a maintenance dose given each hour. Continued refractoriness should lead to the addition of a second diuretic that works through a different yet complementary mechanism of action (e.g., thiazides with a loop-acting drug).
Diuretics
Osmotic Diuretics
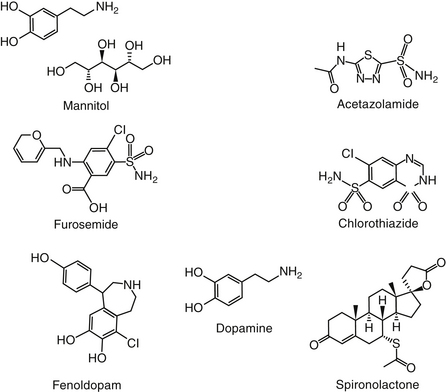
Osmotic diuretics increase the osmolality of extracellular fluid, enhancing flow of water from tissues to interstitial fluid and plasma. For a solute to act as an osmotic diuretic, it must be freely filtered at the glomerulus, not reabsorbed by the renal tubule, and be pharmacologically and metabolically inert. Mannitol (Figure 17-4) is the most commonly used osmotic diuretic; others include urea, glycerol, and isosorbide. As the concentration of an osmotic diuretic increases in the renal tubular lumen, osmotic forces overcome the movement of water with sodium into the renal tubular cell. Eventually, as water retention in the urine increases, sodium concentration decreases and passive sodium reabsorption is reduced. Sodium loss is, however, relatively small. Although mannitol appears to work throughout the renal tubule, the principal site of action appears to be the loop of Henle (see Figure 17-3).1 Mannitol is distributed to ECF and thus extracts water from intracellular compartments, increasing ECF, decreasing blood viscosity, and inhibiting renin release.1 The impact on intracellular compartments is therapeutically beneficial in patients with cerebral edema and glaucoma.1 Renal blood flow increases, removing NaCl and urea from the renal medulla and decreasing renal medullary tonicity. Medullary tonicity also may be reduced further by a prostaglandin-mediated increase in renal medullary blood flow. Mannitol is not absorbed after oral administration. In humans it is characterized by an elimination half-life of approximately 1 hour.
KEY POINT 17-2
Osmotic diuretics will be useful for reducing intracellular edema only if they surround but do not enter the target cell.
The major adverse effect of osmotic diuretics is increased ECF, which can be detrimental in patients with pulmonary edema or cardiac failure. Hyponatremia resulting from water extraction causes headaches, nausea, and vomiting in human patients.1 In contrast, loss of water in excess of electrolytes can cause hypernatremia and dehydration. Osmotic diuretics generally are contraindicated in anuria of severe renal disease or in patients that are not responsive to test doses. Mannitol and urea also are contraindicated for patients with active cranial bleeding. Glycerin (but not mannitol) can be metabolized, causing hyperglycemia.
Mannitol is most commonly used to treat acute renal failure resulting from an acute reduction in glomerular filtration or acute changes in renal tubular permeability. Mannitol provides protection to the tubules in that it attenuates reduction in glomerular filtration rate (GFR) associated with acute tubular nephrosis if the drug is administered before an ischemic insult.1 Efficacy (experimentally) for treatment of nephrotoxicity, however, is documented only when administered before the toxin; clinical efficacy is even less obvious. Mannitol is particularly indicated for treatment of toxic nephrosis because the concentration of the toxin in the urine will be reduced by the osmotic draw of water by the solute. In contrast to diuretics that act on tubular segments, osmotic diuretics usually maintain their effect in the oliguric state that accompanies acute renal failure because they will continue to be filtered by the glomerulus. If the tubular cell becomes permeable, however, as may occur with certain toxins or prolonged tubular ischemia, the osmotic diuretics may lose their efficacy. Yet in patients with acute tubular nephrosis, mannitol may convert an oliguric patient to a nonoliguric state.1
Mannitol is distributed to ECF and thus is not effective in movement of fluids from interstitial tissues. ECF volume will initially increase and may prove detrimental to the patient with decompensated cardiac function. Plasma osmolality increases after treatment with mannitol. Cerebrospinal fluid (CSF) and aqueous humor formation subsequently decrease. Whether mannitol will cross the blood–brain barrier is unclear. However, because mannitol is distributed into interstitial fluids but does not penetrate cell membranes, intracellular edema will also be reduced. As such, mannitol is used to treat selected causes of cerebral edema associated with increased intracellular fluid volume. The use of mannitol for treatment of acute brain injury is discussed in Chapter 27. A Cochrane review failed to find conclusive evidence regarding its efficacy, although the number of eligible clinical trials for review was limited.5 Mannitol can be used to decrease brain mass before neurosurgery. The use of mannitol to facilitate diagnosis of ureteral obstruction has been described.6
Carbonic Anhydrase Inhibitors
Two types of carbonic anhydrase are located in the proximal tubule, both targeted by carbonic anhydrase inhibitors: type II, located in the cytoplasm; and type IV, located in the luminal and basolateral membranes (see Figure 17-1). In the lumen, H+ (generated from the Na+-H+ transporter) reacts with HCO3− to form H2CO3, which, in the presence of brush border carbonic anhydrase, rapidly decomposes to water and CO2. The CO2 rapidly diffuses into the tubular cell, where it reacts with water to form H2CO3. This reaction normally proceeds slowly but is markedly accelerated by carbonic anhydrase in the cytoplasm. Because intracellular H+ is low (because of Na+-H+ co-transport), H2CO3 spontaneously ionizes to form H+ and HCO3−. An electrochemical gradient moves Na+HCO3– into the interstitial space, with water following. Chloride becomes concentrated in the lumen and diffuses down its gradient into the interstitium (see Figure 17-1).1 Carbonic anhydrase inhibitors target both the cytoplasmic and membrane-bound carbonic anhydrase, completely impairing NaHCO3 reabsorption in the proximal tubule (see Figure 17-3). The collecting tubule is a secondary target. As a result, urine concentrations of bicarbonate increase, and up to 35% of the filtered load is excreted. Because hydrogen ions are not generated by the conversion of bicarbonate to CO2 and H2O, they are not available for exchange with sodium. Thus the amount of acid and ammonia excreted in urine also decreases. The loss of titratable acid and ammonia secretion in the collecting duct results in an increase in urinary pH to approximately 8 and the potential development of metabolic acidosis.1 At least 65% of sodium bicarbonate is reabsorbed through carbonic anhydrase–independent mechanisms. Sodium and chloride not reabsorbed in the proximal tubule are delivered to the loop of Henle, where most of the chloride and sodium subsequently are absorbed. Up to 70% of potassium is excreted because of the increased sodium load. Bicarbonate remains in the urine, contributing to alkalinity. Ultimately, however, much of the bicarbonate that remains in the proximal tubular lumen is resorbed distally in the nephron by mechanisms that are not well understood.
Several sequelae result from the diuretic mechanism of carbonic anhydrase inhibitors. First, metabolic acidosis develops as bicarbonate is lost in the proximal tubules. Second, the filtered load of HCO3− decreases to the point that the uncatalyzed (spontaneous) reaction between CO2 and water leads to HCO3 reabsorption. This in turn decreases the response of the renal tubule to carbonic anhydrase inhibitors. Thus the diuretic effect of these drugs is self-limiting. However, in patients refractory to alternative diuretic therapy, the combination of acetazolamide with diuretics that block sodium resorption distally may result in marked naturiesis. Thus carbonic anhydrase inhibitors may be useful for combination therapy.1 In the distal tubule, as sodium is reabsorbed, potassium excretion markedly increases. Carbonic anhydrase is located in other tissues. Aqueous humor and CSF formation are both decreased by carbonic anhydrase inhibitors. Accordingly, the major indication for their use is glaucoma. The effect in the eye is direct and is not influenced by metabolic acidosis. The effect of carbonic anhydrase inhibition in the brain is not as well understood and may result from both the direct effects and the development of metabolic acidosis. Although carbonic anhydrase also is located in the gastric mucosa, only large doses of inhibitors reduce gastric acid secretion. Carbonic anhydrase activity in red blood cells will be impaired, causing an increase in CO2 levels in peripheral tissues and decreased levels in expired air. Carbonic anhydrase can increase delivery of solutes to the macula densa. Tubuloglomerular feedback may be triggered, increasing afferent arteriolar resistance and reducing renal blood flow and GFR.
Side effects of carbonic anhydrase inhibitors are not common. Large doses may cause drowsiness. Side effects can also result from urinary alkalinization or metabolic acidosis. Hepatic encephalopathy can be induced as renal ammonia is diverted from the urine. Precipitation of calcium phosphate may cause calculus formation. Respiratory or metabolic acidosis can be worsened. Carbonic anhydrase inhibitors are contraindicated in patients with cirrhosis or other causes of hepatic encephalopathy or conditions associated with acidosis. The impact of carbonic anhydrase inhibitors on urinary pH can reduce the rate of excretion of weak bases.1
Acetazolamide (Figure 17-4) is a potent, reversible inhibitor of carbonic anhydrase. Its primary indication in veterinary medicine is for the treatment of glaucoma with the intent to decrease aqueous humor formation. It is used less commonly to control CSF formation in patients with hydrocephalus or other causes of increased cerebral fluid pressure. Acetazolamide is characterized by some antiepileptic activity, although tolerance rapidly develops to this effect. Occasionally, acetazolamide might be used to alkalinize the urine. Indications might be for selected causes of crystalluria and to facilitate excretion of weakly acidic drugs, such as phenobarbital, and salicylate. Because its efficacy is self-limiting, combination with sodium bicarbonate may be indicated if persistent urinary alkalinization is desired.
Acetazolamide is orally bioavailable, with peak concentrations occurring within 2 hours after administration. The drug is eliminated by active tubular secretion with some passive reabsorption. In humans the drug is totally eliminated in 24 hours. The drug is relatively safe, although metabolic acidosis may occur (is is usually self-limiting). In patients with hepatic encephalopathy, alkaline urine may increase the amount of urinary ammonia reabsorbed because a greater proportion will be un-ionized. This may result in exacerbation of neurologic dysfunction, and this drug should not be used in patients with severe hepatic dysfunction. Acetazolamide decreases iodide uptake by the thyroid gland (perhaps similar to other sulfonamide antibiotics) and should be avoided in hypothyroid patients or patients undergoing thyroid testing. Whether the drugs can render a euthyroid patient hypothyroid is not known. Drugs that depend on urine acidity are less effective when used with acetazolamide. This includes urinary antiseptics such as methenamine, which is rarely if ever indicated in veterinary medicine; efficacy of weakly acidic antibiotics might also be impaired, whereas that of weak bases will be enhanced.
Methazolamide is among the orally administered carbonic anydrase inhibitors used to treat glaucoma in dogs. Its impact on intraocular pressure and aqueous humor flow rate was described in Beagles receiving a single dose of 25 or 50 mg (10 dogs per group; 5 control) followed by multiple twice-daily dosing for 9 days.7 A diurnal difference in intraocular pressure was measured in all animals, being highest in the morning. At 25 and 50 mg, intraocular pressure decreased in the morning but increased in the evening compared with baseline. Compared with baseline, aqueous humor flow rate increased in both groups. Although response to 50 mg (compared with 25) was greater for both intraocular pressure and aqueous humor flow rate, the difference was not significant, perhaps reflecting a small sample size.
Thiazide Diuretics
The thiazide diuretics, represented by chlorothiazide (see Figure 17-4), were developed to enhance the potency of carbonic anhydrase inhibitors.1 Although most do inhibit carbonic anhydrase to some degree, their efficacy as diuretics reflects their ability to directly inhibit sodium chloride co-transport in the distal tubule, perhaps by competing with chloride for binding (see Figure 17-2).1 Newer diuretics have been developed that act at the same site as the thiazides but are not thiazides; the term thiazide-like diuretics is applied to these latter drugs. The primary site of action of these drugs also is the distal tubule (see Figure 17-3), a site characterized by avid binding for thiazide diuretics. Although some action has also been described in the proximal tubule, this may reflect the weak carbonic anhydrase action of these drugs. Compared with other diuretics, the thiazide diuretics are less effective in causing sodium excretion because close to 90% of reabsorption of sodium from the urine has occurred by the time passage through the distal and collecting tubules is complete. Because the tubular site of potassium secretion is distal to the site of thiazide action, potassium excretion is increased and more sodium is reabsorbed. Thiazides have variable effects on calcium excretion, with excretion decreasing with chronic administration. Thiazides cause magnesium excretion, and the potential advent of hypomagnesia is being recognized in humans receiving thiazides long term.1
The thiazides vary in their oral bioavailability, with that of chlorothiazide being the poorest (10% bioavailable in humans). The degree of protein binding varies among the drugs, and thus delivery to the kidneys via glomerular filtration may be limited. Active tubular secretion of the drugs can be antagonized by probenecid. Likewise, the elimination half-life of the drugs is also variable, with that of chlorothiazide being very short (1.5 hours in humans). For hydrochlorothiazide bioavailability (in humans) is 65%, whereas the elimination half-life is 2.5 hours. Thus the duration of diuretic effect is quite variable. The potency also is variable, with chlorothiazide 10 times more potent than hydrochlorothiazide. Thus doses also are quite variable.
The thiazide diuretics are characterized by a wide safety margin, and clinical toxicity is rare. As with the loop diuretics, most serious toxicities reflect overzealous use. Volume depletion, hypotension, hyponatremia, hypochloremia, hypomagnesemia, and hypercalcemia have been reported. Potassium depletion can be clinically significant, particularly in patients with primary or secondary hyperaldosteronism. Oral potassium supplementation is indicated for patients that become hypokalemic; this is particularly important for patients receiving digoxin because the risk of digoxin toxicity is enhanced in the hypokalemic patient. Alternatively, the thiazides can be used in combination with a potassium-sparing diuretic. Like the carbonic anhydrase inhibitors, thiazides may exacerbate neurologic dysfunctions associated with cirrhotic liver disease. Thiazides appear to diminish the effects of insulin, particularly in the hypokalemic patient, and should not be given to patients with diabetes mellitus.
Thiazides are involved in a number of drug interactions. They can diminish the effectiveness of anticoagulants, uricosuric drugs (for treatment of gout), and insulin and may increase the effects of anesthetics, digitalis glycosides, lithium, and vitamin D. Thiazides do provide additive or synergistic effects when combined with loop diuretics. Efficacy of the thiazides is decreased by NSAIDs and methenamines (because of alkalinization of urine). Hypokalemia induced by the thiazides may be worsened by amphotericin B or corticosteroids. Quinidine has reportedly caused lethal drug interactions with thiazides by causing ventricular tachycardia; however, this may reflect thiazide-induced hypokalemia.1
Thiazides are used primarily in the treatment of early congestive heart failure. Although they may be less effective as diuretics, thiazides, in contrast to most other diuretics, minimally affect the composition of ECF. Thiazides may directly decrease glomerular filtration, particularly after intravenous administration. Thus they should not be given to patients with compromised renal function. Thiazides decrease renal excretion of calcium and are contraindicated in patients with hypercalcemia. In contrast, bromide excretion may be facilitated, and thiazides may be useful for treatment of bromide toxicity.
Drugs That Interfere with Renal Epithelial Sodium Transport
Potassium-Sparing Diuretics
Diuretics that are not associated with kaliuresis include the aldosterone antagonists (see later discussion), triamterene, and amiloride.1 The primary indication for the latter drugs is their ability to spare potassium wasting. Because they cause only a small amount of sodium wasting, however, they usually are combined with another diuretic. Both triamterene and amiloride impair electrogenic sodium reabsorption in the late distal tubule and the collecting systems, after much sodium reabsorption has already occurred. Because the normal electrical potential across the tubular epithelium is lost, the driving force for potassium secretion is reduced. The potassium-sparing effects of these drugs are most effective in the patient whose potassium excretion has markedly increased (i.e., by hyperaldosteronism or therapy with potassium-wasting diuretics). Like the thiazides, amiloride decreases calcium excretion into the urine. The impact of either triamterene or amiloride on renal hemodynamics is minimal.
The drugs are orally bioavailable (50% to 60% in human patients). Both triamterene and amiloride are actively secreted into the proximal tubule (the route by which they reach their site of action), although a portion of triamterene undergoes hepatic metabolism to a metabolite that is active in humans. Both liver disease and hepatic disease can increase the risk of adverse reactions to triamterene. The most serious toxicity is hyperkalemia that is more likely to occur in a patient receiving angiotensin-converting enzyme inhibitors or aldosterone inhibitors. As such, the drugs are contraindicated in patients at risk for or already have developed hyperkalemia. NSAIDs and dietary potassium intake increase the risk of hyperkalemia. As a pteridine derivative, triamterene is a weak folic acid antagonist. Folic acid deficiency leading to megaloblastic anemia has been reported in human patients with cirrhosis.1 Triamterene also can reduce glucose tolerance. Amiloride, like triamterene, can cause gastrointestinal upset (vomiting, diarrhea). In general, these drugs are not effective diuretics unless combined with another diuretic and, usually, thiazides.
Aldosterone Antagonists
Aldosterone and mineralocorticoid agonists cause sodium and water retention in exchange for potassium and hydrogen excretion.1 Spironolactone (see Figure 17-2) competitively antagonizes the actions of aldosterone and other mineralocorticoids by binding to the receptor such that it is not active. The site of action is limited to the late distal tubule and the collecting duct (see Figure 17-3). Aldosterone causes sodium and water retention by interacting with mineralocorticoid receptors. Interaction with specific DNA sequences results in the expression of multiple gene products called aldosterone-induced proteins. The proteins appear to activate or increase the expression of preexisting yet “silent” sodium channels and pumps in the cell membrane. Sodium moves into the cell from the luminal membrane, causing an electronegative lumen that is conducive to potassium excretion. As such, spironolactone (see Figure 17-4) is effective as a diuretic only in the presence of aldosterone, and efficacy will be impaired in the presence of high concentrations of aldosterone. In addition to several segments of the nephron, aldosterone receptors occur in the colon and salivary glands. Unlike the thiazides and carbonic anhydrase inhibitors, spironolactone causes calcium excretion in the urine. Spironolactone has no effect on renal hemodynamics. However, it has a variety of effects on the diseased myocardium. The recently discovered beneficial effects of spironolactone on both acute myocardial damage8 and chronic cardiac remodeling9 are briefly discussed in Chapter 14 Spironolactone conceivably should be the drug of choice in high aldosterone states such as congestive heart failure or portal hypertension, but its efficacy is less than that of the loop-acting diuretics. Consequently, sole therapy with spironolactone occurs early in the respective syndrome or in combination with other diuretics. Supporting its early use in patients with congestive heart failure is the recognition that, in these patients, spironolactone promotes magnesium and potassium retention, increases uptake of myocardial norepinephrine, attenuates formation of myocardial fibrosis, and decreases mortality associated with both progressive ventricular dysfunction and malignant ventricular arrhythmias.10,11 Indeed, its apparent positive effects of decreasing mobidity and mortality rates led to early termination of a clinical trial examining these effects in humans.10
KEY POINT 17-3
Because of its site of action, spironolactone might enhance the efficacy of any naturietic that acts proximal to its site of action in the distal tubule (see Figure 17-3).
Spironolactone is 60% to 70% bioavailable (in humans), is highly protein bound, and undergoes extensive first-pass metabolism and enterohepatic circulation.1 In humans spironolactone is characterized by a short half-life (1.4 hours). Its active metabolite (canrenone, which is available outside of the United States as canrenoate, a prodrug of canrenone); however, has an elimination half-life that is much longer (16.5 hours). It is not clear what proportion of the drug is metabolized to the active metabolite in dogs and cats. Spironolactone (or other mineralocorticoid receptors) are the only diuretics that do not require access to the tubular lumen in order to cause diuresis (although the active metabolite apparently does).1
The most serious toxicity of spironolactone, like other potassium-sparing diuretics, is hyperkalemia, which is more likely to occur when the drug is given in combination with potassium-wasting diuretics and oral potassium supplementation. The contraindications and side effects that characterize the potassium-sparing diuretics also pertain to spironolactone. In addition, spironolactone has induced metabolic acidosis in patients with cirrhosis. Diarrhea, gastritis, gastric bleeding, and peptic ulceration have been reported in human patients receiving the drug; consequently, it is contraindicated in patients with gastric ulceration. Central nervous system adverse effects such as drowsiness, lethargy, ataxia, and confusion have been reported in human patients. Androgen side effects (e.g., gynecomastia impotence) have been reported in human patients. Finally, the ability of spironolactone to induce malignancy has been raised.1 Aspirin (salicylates) reduce the efficacy of spironolactone by competing for active tubular secretion. Spironolactone alters the elimination of digitalis glycosides.
Spironolactone is most commonly used to control edema associated with hyperaldosteronism. It is generally, however, combined with either a thiazide or a loop-acting diuretic. Edema associated with hypertension and liver disease are among the more common indications. Spironolactone is the drug of choice for treatment of edema associated with chronic liver disease (secondary to hyperaldosteronism) in human patients.1 Rarer indications include syndromes associated with potassium depletion.
High-Ceiling Diuretics
The efficacy of diuretics acting on the proximal tubule is limited by the marked reabsorptive capacity of the thick ascending limb of the loop of Henle. Diuretics that act beyond the loop of Henle also are limited in efficacy because only a small percentage of the filtrate reaches that area. In contrast, drugs that act at the loop of Henle tend to be very effective.1 The term high ceiling refers to the peak diuretic effect of these drugs, which far surpasses that of other diuretics. The primary site of action is the thick ascending limb of the loop of Henle, where the drugs bind to and impair the luminally located Na+K+2Cl− co-transport mechanism (see Figure 17-2). Because of their site of action, these drugs also are referred to as loop diuretics.1 Two isoforms of the Na+K+Cl− co-transport enzyme exist with the second isoform located in the apical membrane of the thick ascending loop. The second is located in many body tissues.12 The efficacy of high-ceiling loop diuretics reflects the large amount of filtrate that reaches this region of the kidney and the lack of a very efficient reabsorptive region beyond the loop. Ethacrynic acid is a phenoxyacetate derivative, and furosemide is a sulfonamide derivative (see Figure 17-4). Furosemide is the member of this class used in small animal patients. Abbott and Kovacic12 reviewed the use of furosemide in veterinary medicine.
Furosemide causes a profound increase in Na+ and Cl– urinary excretion. Ultimately, as ECF decreases in response to furosemide, glomerular filtration decreases and proximal tubular reabsorption of sodium is enhanced. Species differences should be expected in response to furosemide. Dogs respond across a broad therapeutic range. Cats respond in a narrower range, but with a more rapid and intense saluresis and diuresis compared with dogs.12 Doses in cats greater than 10 mg/kg, administered intramuscularly, may be associated with lethargy and decreased appetite that may last 24 to 48 hours after dosing. In dogs the dose above which intolerance occurs is 50 mg/kg, administered intramuscularly. Blood pressure will decline in both species at these doses, with hypotension more profound in cats.12 Morbidity at higher doses may reflect volume contraction, hypotension, electrolyte abnormalities, and activation of neurohumoral endocrine responses. At massive doses furosemide inhibits carbonic anhydrase activity in the proximal tubule, but this minimally affects diuretic actions at normal doses. Like the thiazides, the mechanism of action of furosemide is proximal to the site of Na+ and K+ exchange; thus potassium excretion is increased. Furosemide also markedly enhances the excretion of calcium and magnesium but does not increase calcium reabsorption in the distal tubule, as do the thiazides. Thus furosemide is indicated for treatment of hypercalcemia, although care must be taken to replace sodium and chloride losses. Because furosemide increases the excretion of acid and ammonia in the distal nephron, diuretic-induced metabolic alkalosis may develop. This can be exacerbated if ECF is rapidly mobilized such that its volume contracts. The kidney’s ability to excrete a concentrated urine during hydropenia or a dilute urine during states of water diuresis is impaired by furosemide.
KEY POINT 17-4
In addition to its loop-diuretic effects, furosemide is useful for a variety of other physiologic effects, particularly in the vasculature
Furosemide will increase renal blood flow if volume depletion is avoided. The effect is, however, variable. NSAIDs attenuate the diuretic response to loop diuretics, perhaps by altering prostaglandin-mediated increases in blood flow. Loop diuretics block tubuloglomerular feedback, presumably by inhibiting transport of NaCl to the macula densa.1 Because of this effect, they also are powerful stimulants of renin release; prostaglandin (prostacyclin) release may be involved in this response. During states of volume depletion, renin activation also may reflect stimulation of the sympathetic nervous system and of intrarenal baroreceptor mechanisms.
Loop diuretics, particularly furosemide, increase venous capacity, possibly because of prostaglandin release. Although this effect is short lived, it enhances the initial diuretic response. This effect is likely to be blunted or inhibited by NSAIDs, including newer drugs that are more potent toward COX-2 compared with COX-1. Decreased peripheral vascular resistance also has been reported.12 The hemodynamic effects of furosemide may be of particular benefit in the patient with pulmonary edema: The capacity of veins increases, and left ventricular filling pressures subsequently decrease. Impaired electrolyte transport in other tissues generally is clinically irrelevant, with the exception of the endolymph, which may contribute to the ototoxicity characteristic of this group of diuretics. The pulmonary effects may contribute to potential efficacy in the treatment of the acute respiratory distress syndrome or acute lung injury (the latter when combined with albumin). In the dog furosemide dilates gastrointestinal vascular beds.12
Furosemide appears to be renoprotective in states of ischemic damage; this may reflect a reduction in oxygen consumption in response to inhibition of co-transport. However, it also has antioxidant effects and protects against free radicals at low doses (0.1 mg/kg/day in rats).12 Nonetheless, the use of furosemide in acute renal failure has not been consistently demonstrated to reduce patient mortality rates in humans.12
Furosemide has a variety of actions other than vascular in the lungs. It attenuates bronchospasm in response to a number of stimuli and stimulates bronchodilation. Furosemide inhibits laryngeal irritant receptors in anesthetized dogs. Clinical signs of dyspnea may be relieved with inhaled furosemide.12
Interestingly, furosemide appears to have anticonvulsant properties. Its mechanism may reflect an increase in threshold and decreased seizure propagation. Intraneuronal chloride may be maintained; this may reflect interaction with potassium chloride co-transport enzymes. Interaction may impart neuroprotection. Seizure-induced cellular swelling may be reduced.12 Furosemide may reduce production of CSF. The target enzymes are present in the choriod plexus. A 50% reduction in CSF has been reported in cats treated with 50 mg/kg intravenously. However, furosemide should not be used to reduce CSF or intracerebral pressure in the presence of or at a dose sufficient to induced hypotension.12 Furosemide has been associated with a decrease in intracranial pressure but is more effective if it follows a dose of mannitol.12 In contrast to thoracic lymph flow, furosemide appears to decrease ascitic transudate from selected neoplasms.
Furosemide is rapidly absorbed from the gastrointestinal tract. Bioavailability is only about 60%, however, ranging from 10% to 100% in the individual patient. Thus several different oral doses must be given before therapeutic failure can be assumed. Intravenous administration is indicated in patients for whom lack of response due to oral absorption must be avoided. Alternatively, a drug that is 100% bioavailable after oral administration (e.g., bumetanide or torsemide in human patients) should be given. Furosemide is highly protein bound, which limits delivery through the glomerulus, but it is able to reach its site of action by active tubular secretion.
The most common toxicity associated with furosemide therapy generally reflects overzealous administration, resulting in altered electrolyte and fluid balance. Depletion of body sodium can cause hypotension, reduced GFR, circulatory collapse, and thromboembolic episodes. In the patient with liver disease characterized by activation of the renin–angiotensin–aldosterone system, furosemide can induce hepatic encephalopathy. Continued Na+ delivery results in continued exchange for H+ and K+, causing hypochloremic alkalosis. Hypokalemia likewise can occur, particularly in patients with insufficient dietary intake or patients receiving digoxin therapy. Hypomagnesemia and hypocalcemia are less common, albeit possible, consequences of overzealous furosemide therapy.1 Furosemide depletes total body iodide, which may be of benefit in states of hyperiodism.12 Because of its impact in the developing fetus, furosemide is not recommended in pregnant or lactating animals. Furosemide increases lymph flow through the thoracic duct in dogs at 8 to 10 mg/kg. Accordingly, it might be avoided in animals with pleural effusion. Ototoxicity can lead to tinnitus (in human patients), deafness, vertigo, and (described in humans) “a sense of fullness in the ears.” Ototoxicity most commonly occurs with rapid intravenous administration, more with ethacrynic acid than with furosemide, and generally is reversible. Contraindications for furosemide therapy include severe Na+ or volume depletion, hypersensitivity to sulfonamides, and anuria that has not responded to a test dose of furosemide. In human patients furosemide should not be used during pregnancy. Chronic use of furosemide may result in renal hypertophy.12 The impact of furosemide on mucociliary transport rates is controversial, with some studies showing a detrimental effect.12 Avoidance of furosemide in patients with inflammatory lung disease, particularly that which is associated with airway secretions, is prudent. Although it contains a sulfur moiety, furosemide does not contain an aryl amine (see Chapters 4 and 7), and patients that are allergic to sulfonamides are not likely to show a cross-reactivity to furosemide. However, cross-allergy has been reported, albeit rarely, in humans. The sulfur moiety may play a role in pancreatitis, which has been reported rarely in humans.12 Furosemide may directly inhibit insulin secretion; however, this effect is species specific, occurring in dogs and humans but not mice.
Drug interactions involving furosemide may become clinically important. Furosemide is chemically incompatible with a number of drugs (demonstrated for diazepam, dopamine, dobutamine, metoclopramide, and morphine). As such, its use as a constant-rate infusion should be through its own line and not “piggybacked” onto lines containing other drugs. When administered as a constant-rate infusion, furosemide will likely have to be diluted. Stability has been demonstrated for at least 8 hours when diluted fivefold (to a concentration of 10 mg/mL) in either 5% dextrose, lactated Ringer’s solution, 0.9% saline, or sterile water.13 Dilution to 5 mg/mL with water or 5% dextrose is also apparently safe, but 0.9% saline or lactated Ringer’s solution may result in cloudiness. Presumed precipitation with the latter probably reflects decreased solution pH.
Because it is highly protein bound (in humans), furosemide may compete with other drugs for protein-binding sites; clinically relevant interactions have been attributed to competition. For example, anticoagulant activity may be enhanced in the presence of furosemide. Other drugs known to interact with furosemide, perhaps because of competition for protein binding, include propranolol and lithium (both characterized by higher plasma drug concentrations). Competitive binding may affect thyroid function tests; initial displacement of thyroxine, causing higher free concentrations, may negatively inhibit thyroid-stimulating hormone.12 The freed thyroxine will be cleared more rapidly and thus may not be interpreted as high. It is possible that total thyroxine will decrease; the low thyroid-stimulating hormone may be interpreted as hypothyroid.
Potential ototoxicity induced by furosemide is enhanced by aminoglycosides (synergistic) and cisplatin. The risk of cardiac arrhythmias induced by digoxin also are increased by furosemide. Both NSAIDs and probenecid block diuretic response to furosemide, the former perhaps resulting from attenuation of response to renal prostaglandins and the latter from competition for active tubular secretion. In contrast, the thiazides act synergistically with furosemide to induce diuresis. The nephrotoxic potential of other drugs (e.g., cephalosporins, aminoglycosides) is enhanced by furosemide.
Furosemide is used for a variety of indications (see Table 17-1). Treatment of hypertension is limited to animals that have not responded to other therapies. Among the most common indications is edema of a variety of causes that reflect increased hydrostatic pressure and potentially decreased oncotic draw. However, edema associated with vascular permeability generally is not an indication; fluid loss in such cases generally is at the cost of decreases in total body water.
Furosemide is the drug of choice for the management of acute pulmonary edema. Its use in the long-term management of sodium retention might be postponed until other, less effective diuretics become ineffective. Edema associated with the nephrotic syndrome appears to respond only to loop diuretics. For the patient with refractory edema, furosemide can be combined with other diuretics, preferably potassium sparing, such as the thiazides or spironolactone. Doses much higher than normal may be necessary to induce diuresis in patients with renal disease for two reasons. First, renal tubular function is abnormal, and response to furosemide may be impaired. Second, drug delivery to the site of action may be decreased. Administration as a constant-rate infusion (0.66 mg/kg load followed by 0.66 mg/kg/hr) will induce more diuresis, natriuresis, and calciuresis but less kaliuresis compared with intermittent bolus administration.14 An advantage of constant-rate infusion is avoidance of toxic concentrations that have been associated with ototoxicy in humans. Because of its effects on iodine depletion, it may be useful in the cat with hyperthyroidism before radioiodine treatment, facilitating uptake, as has been demonstrated in humans receiving 40 mg/kg/day for 5 days. Its use as an adjunct to surgery or to control thyroid storm in hyperthyroid cats warrants consideration.
Proteinuria caused by glomerular nephritis may also reduce the efficacy of furosemide because it binds to protein in the urine. The massive doses necessary for acute renal failure may cause hepatotoxicity. Once oliguria has been definitively established, furosemide therapy should be discontinued. Because loop diuretics interfere with the kidney’s ability to produce a concentrated urine, when combined with hypertonic saline, furosemide can be used to treat life-threatening hyponatremia. The use of furosemide to facilitate diagnosis of ureteral obstruction also has been described.6
Pressor Agents
Drugs that increase cardiac output should, by virtue of increased renal blood flow and glomerular filtration, increase urine output. Among the pressors used to support myocardial depression and hypotension is the positive inotrope dopamine (see Chapter 14). Dopamine receptors in the renal vasculature of several species induce renal vasodilation, with the effect accomplished at low doses of dopamine. Higher doses cause release of epinephrine and subsequent renal arterial vasoconstriction. Dobutamine does not have a similar effect in the renal vasculature. Consequently, dopamine may be the preferred inotropic drug in the face of hypotension that threatens renal function. However, Sigrist15 has retrospectively reviewed the role of dopamine in acute renal failure, including meta-analyses assessment in humans. In humans many of the studies cited for use have been implemented in animal models or healthy individuals. Because mortality does not change, the incidence of side effects no longer justifies its use. The lack of consistent response in the presence of disease may reflect receptor desensitization; further, profoundly ill patients are likely to realize changes in pharmacokinetics and pharmacodynamics that increase the risk of adversity. Finally, it is not clear if increase urine production stimulated through hemodynamic changes is in fact a positive response or if urine production is an indicator of improvement. Effects in animals are more likely to be realized in animal models of acute renal failure when administered in advance of the insult.
KEY POINT 17-5
Dopamine receptor activity of the feline kidney is different from that of the canine kidney and accordingly is less responsive to dopamine.
Species differences exist in the renal response to dopamine. Although dopamine receptors are present in the feline renal cortex, their numbers are lower compared with those of other species.16 The cat is among the species for which diuretic doses of dopamine do not cause renal arterial vasodilation, perhaps because of fewer dopamine receptors in the renal vasculature. Higher doses of dopamine do increase diuresis and natriuresis.17
Fenoldopam (see Figure 17-4) is a dopamine DA-1 agonist with no α or β adrenergic receptor effects used for treatment of emergency hypertension in humans. It also stimulates systemic vasodilation, natriuresis, and diuresis in human patients with renal disease.18 Renoprotection has been described, supporting its use in acute renal failure.19,20 In a dog exsanguination21 or aortic clamp (n = 8 Labrador Retrievers)19 models, fenoldopam (0.1 μg/kg/min) maintained renal blood flow before, during, and after induction of acute hypovolemia.21 Zimmerman-Pope and coworkers22 attempted to study the impact of fenoldopam on renal function after nephrotomy but found that the model insufficiently affected renal function. Unlike dopamine, feline kidneys respond to fenoldopam. It exhibited a 300-fold greater affinity for feline dopamine receptor, compared with dopamine, suggesting that feline vasculature contains the receptors but lacks dopamine.17 Wohl and colleagues23 found no effect of low-dose (3 μg/kg/min) dopamine on indices of renal diuresis (urine output, renal blood flow, sodium excretion, fractional excretion, or creatinine clearance) in anesthetized healthy cats (n = 12). Arterial blood pressure did transiently decrease. In contrast, Simmons and coworkers20 found that feline kidneys responded to fenaldopam. Ketamine and diazepam were used to induce anesthesia for instrumentation purposes only; healthy awake cats were studied (n = 6). A 12-hour pretreatment assessment of baseline (an infusion of 2.2 mL/kg/hr of saline) was followed by fenoldopam infusion over 2 hours (0.5 μg/kg/min). Parameters were then studied for 12 hours. Fenoldopam increased urine output, sodium excretion, and fractional excretion (with comparable changes in urine specific gravity); creatinine clearance also increased after a transient decrease. The effects were delayed: GFR was increased at 6 hours after fenoldopam administration, coinciding with increases in urine output. Central venous pressure simultaneously decreased. Studies in humans thus far have not clearly demonstrated a beneficial effect of fenoldopam in patients with acute renal failure; further studies are indicated.
The use of Diuretics in Selected Clinical Conditions
Congestive Heart Failure
The intial diuretic selection for human patients with mild congestive heart failure is a thiazide; however, most patients will require a loop diuretic. In human patients the rate of oral absorption of diuretics is slowed, requiring longer to maximal response. Delivery of loop diuretics to their site of action is normal, and doses do not necessarily need to be increased unless there is evidence of renal insufficiency. Renal response to diuretic therapy may be decreased, however, requiring more exposure to drugs. Although a dose increase may be indicated, a decrease in interval may be more likely to cause a response. A thiazide diuretic should be added to therapy if dietary salt retention coupled with a loop diuretic have not been effective. Attention should be paid to ensure that hypokalemia and volume depletion do not occur with this combination. The use of spironolactone may increase as its effects on cardiac remodeling are further described.
Cirrhosis
Ascites that accompanies cirrhotic liver disease generally reflects a state of hyperaldosteronism and subsequent sodium and water retention. Therefore spironolactone is the diuretic of choice for human patients. Although the amount of diuresis can be expected to be only moderate, this is desirable because greater diuresis may negatively affect intravascular volume. In human patients repeated large-volume paracentesis minimizes the need for more potent diuretics. The active metabolites of spironolactone allow once-daily dosing, although 3 to 4 days must elapse before full pharmacologic effects are realized. Insufficient response to spironolactone indicates the need for an additional diuretic; spironolactone should be continued. For human patients a thiazide is used initially, and a loop diuretic is used in place of the thiazide only if response has been inadequate. The decreased response to a loop diuretic in a patient with cirrhosis is not understood but does not reflect decreased drug delivery. Rather, the tubular cells do not respond maximally. Although higher doses may be of benefit, decreasing the interval may be more likely to increase response.
Nephrotic Syndrome
Response to diuretics in patients with the nephrotic syndrome may be less than ideal if hypoalbuminemia is sufficient to decrease binding of the diuretic (e.g., <2 g/dL). Unbound drug will diffuse into tissues (i.e., volume of distribution will increase), removing the drug from the site of action in the renal tubule. In such patients addition of albumin to the therapeutic regimen will increase response. Binding to albumin in tubular fluid also decreases response and is more likely to occur when urine albumin concentrations exceed 4 g/dL. Dose increases (twofold to threefold) may help compensate for increased tubular binding of the diuretic. Because tubules in patients with nephrotic syndrome may not respond to drugs in general as well as those in the normal patient, decreasing the interval (such that the duration of exposure is longer) also may increase response.
Drugs That Alter Renal Conservation of Water: Antidiuretic Hormone
Antidiuretic hormone (ADH) is released by the posterior pituitary in response to increased plasma osmolality (as little as 2%, or 280 mOsm/kg) and depleted ECF volume. The latter might occur, for example, resulting from acute causes such as hypovolemia, sodium depletion, and hemorrhage or chronic causes such as cardiac failure, hepatic cirrhosis with ascites, hypothyroidism, and excessive use of diuretics. Antidiuresis involves the hypothalamus, neurohypophysis, posterior pituitary, and kidney. Neurons of osmoreceptors, baroreceptors, and higher cerebral centers stimulate the hypothalamus. Calcium-mediated degranulation results in the release of ADH as well as oxytocin and other mediators. A number of chemical mediators are also associated with the release of ADH, including angiotensin II, prostaglandins, and acetylcholine. Inhibitors of release include opioids, atrial natriuretic peptide, and γ-aminobutyric acid.
The actions of ADH are receptor mediated. At least two receptors have been identified. Both receptors are located in the kidney, although the specific tissue site varies. Glomerular, vasa recta, and interstitial medullary receptors (V1) participate in the control of the GFR, medullary blood flow, and prostaglandin synthesis, respectively. The predominant effect of ADH occurs in the collecting duct and is mediated by V2 receptors. In the presence of ADH, the collecting ducts of the cortex and medulla become permeable to water, which follows the osmotic drag.
A number of therapeutic drugs can alter ADH secretion either directly or indirectly. Drugs that alter the osmolality of urine may indirectly alter the secretion of ADH. Drugs that stimulate ADH secretion include the vinca alkaloids, cyclophosphamide, tricyclic antidepressants, and isoproterenol. Inhibitors of ADH secretion include ethanol, mineralocorticoids, and glucocorticoids. The effect of mineralocorticoids results from volume depletion that accompanies sodium loss. Inhibition by glucocorticoids, on the other hand, probably results from both a central effect and cardiac effects. Drugs that inhibit prostaglandin synthesis also will facilitate the action of ADH. Lithium is an example of a drug that inhibits the effects of ADH.
Drugs that Alter Urinary pH
Urinary acidification or alkalinization depends on normal renal function. Changes in urine pH are at best modest, and the effect on systemic acid–base status is equally modest. In the face of renal deficiency, the use of acidifying salts may be harmful. Changes in urinary pH are implemented to enhance efficacy of a drug in the urine, to enhance solubility of a drug or other solute in the urine, to facilitate urinary excretion of a toxin, or to promote an unfavorable environment for microbial growth. Excretion of an acidic compound is likely to be enhanced if the pKa of the compound is within the range of 3 to 7.5; for a basic compound a pka 7.5 to 10.5 is necessary.
Urinary Acidifiers
Ammonium chloride is a urinary acidifier. Ammonium ion (NH4+) serves as a proton donor. Ammonia (NH3) formed by the kidney is excreted in an acid urine as the ammonium ion. In states of acidosis, renal production of ammonia is stimulated, increasing the concentration of a proton acceptor, thus buffering urinary acid by allowing secretion of protons in tubular fluid. The ammonium of orally administered ammonium chloride is converted by the liver to urea, freeing hydrogen ion, which subsequently decreases bicarbonate. Thus efficacy depends on hepatic conversion of ammonia to urea. Of the urinary acidifiers, ammonium chloride probably provides the most consistent changes in pH. The use of ammonium chloride is contraindicated in hepatic insufficiency.
Acetohydroxamic acid (5 mg/kg every 8 hr orally) is structurally similar to urea that acts as a potent and irreversible inhibitor of bacterial urease. It has been used in humans as adjuvant therapy to treat urinary tract infections associated with urease-producing bacteria (e.g, Staphylococcus, Klebsiella, Corynebacterium urealyticum,Pseudomonas, and Providencia). Approximately 15% of patients have had laboratory findings characteristic of a hemolytic anemia; accordingly, care might be particularly taken in cats.
Other urinary acidifiers include DL-methionine, ethylenediamine dihydrochloride, and sodium acid phosphate. Methemoglobinemia has been reported in cats receiving phosphate-containing urinary acidifiers.
Cysteine Urolithiasis
Cystinuria also is generally accompanied by increased urinary excretion of lysine, arginine, and ornithine in the urine. Cystine is a non-esssential amino acid composed of two cysteine amino acids joined at their sulfur atoms. It can be synthesized in the body from the essential amino acid, methionine. Cystine solubility in urine decreases as pH increases, predisposing to urolith formation. Cysteine urolithiasis represents a small proportion of uroliths in the dog (1% to 22%).24 Cystinuria appears to decline with age in dogs with uroliths.24 Therapies have been variable in choice and effect. Dietary management has included low methionine or sodium diets, the latter reducing the transport of cysteine into the urine in return for sodium.24 Pharmacologic management has included conversion of cysteine to a more soluble compound with the heavy metal chelator D-penicillamine or tioproinin. However, side effects limit the successful use of D-penicillamine. The use of tioproinin has been reported in an open study of 88 dogs with cystinuria and uroliths.24 Dogs in which uroliths had been removed were treated with 30 mg/kg, and dogs with uroliths present received 40 mg/kg orally every 12 hours. Treatment duration ranged from 1 month to 13 years (mean 2.8 years), with the median less than 1 year. Stones resolved in 63% of dogs receiving tioproinin, with the mean time to dissolution being 1.6 months. However, uroliths re-formed in 12 of the dogs (between 3 to 36 months) receiving tioproinin prophylactically at 30 mg/kg, with dissolution occurring in five of those when the dose was increased to 40 mg/kg. Side effects occurred in 12% of animals, with the most common being myopathy related to chewing or swallowing and aggression. Disconcertingly, proteinuria (n = 3) and thrombocytopenia (n = 4) also were reported. The authors emphasized the potential importance of lifelong therapy.
Because the potential for cysteine crystal formation is enhanced in an acidic urinary pH, one strategy for prevention of cysteine uroliths has been an increase in urinary pH. Urine pH should be sustained at or above 7.5 to dissolve or prevent the formation of cystine uroliths. Although sodium bicarbonate can be used to maintain an alkaline urine pH, dietary sodium may enhance cystinuria in humans. Further, alkalinization was not a successful treatment in six cysteinuric dogs also receiving tioproinin.24 Thus potassium citrate may be the preferred urinary alkalinizer in patients with cystine uroliths.
Calcium Oxalate Urolithiasis
Close to 50% of urinary calculi in cats are calcium oxylate rather than struvite, with cats older than 7 years of age more commonly afflicted.25 Uroliths composed of calcium are difficult to dissolve with diet or medications.26‑30 Medications that alter urine concentrations of calcium are likely to detrimentally alter normal calcium homeostasis. Thiazide diuretics decrease renal excretion of calcium by virtue of their effect on the Na-Cl transporter: decreased intracellular sodium results in increases calcium reasorption via the Na+/Ca2+ countertransporter. Volume contraction results in both sodium and calcium reabsorption. Potentiation of the effects of parathyroid hormone on tubular reabsorption of calcium may also be beneficial. Although human patients suffering from normocalcemic hypercalciuria benefit from thiazide therapy, efficacy in comparable small animal patients has not been established. Thiazides may prove useful for patients whose hypercalciuria is associated with normocalcemia but are contraindicated for hypercalcemic patients. Hypokalemia is a potential undesirable side effect of thiazide therapy and can be prevented by oral administration of potassium citrate.
Potassium citrate therapy may also benefit the patient with calcium oxalate urolithiasis. Citrates complex with calcium to form salts that are more soluble than oxalate salts. Citrate also is metabolized to bicarbonate, which may alkalinize the urine, and in dogs, may increase citrate excretion in the urine. Low concentrations of citrate occur in up to 63% of patients with calcium oxalate urolithiasis, but a similar role has not been identified in dogs or cats. Potassium citrate is available in a wax matrix, slow-release preparation. Delayed absorption results in prolonged maintenance of urine citrate concentrations. Potassium citrate is more likely to be effective in dogs with calcium oxalate urolithiasis if hypocitraturia can be documented. Dosing regimens may be guided by measuring urinary pH, which should be maintained at or above 7. Vitamin B6 (2 to 10 mg/kg every 24 hours) and B12 (1 mg/kg every 24 hours) has been recommended to decrease oxalate urolithiasis although clinical trials supporting this use are controversial. Supplementation may be indicated in cases of potential vitamin B deficiency (e,g., senior cats with bowel disease). Dietary management and encouragement of fluid intake (including slightly salting the diet) also are recommended.
Anaerobic microbial oxalate degradation in the gastrointestinal tract appears to be an important mechanism by which the incidence of hyperoxaluria occurs. Deficiency of Oxalobacter formigenes may be associated with calcium oxalate. Supplementation of gastrointestinal flora with the organism in human volunteers was associated with a reduction in urinary oxylate excretion, which suggests that this may be an alternative therapy.31
Urate Urolithiasis
In addition to low-protein diets, dissolution of urate stones can be facilitated by urine alkalinization and administration of allopurinol.26‑30 Urinary pH should be maintained at 7 to 7.5 with oral sodium bicarbonate or potassium citrate. Allopurinol competitively inhibits xanthine oxidase, the enzyme that forms uric acid from xanthines. Plasma and thus urinary concentrations of uric acid are decreased. Because serum concentrations of xanthine increase with allopurinol therapy, urinary alkalinization is recommended with allopurinol therapy in human patients suffering from gout to prevent the formation of xanthine stones. Although allopurinol is recommended for treatment of Dalmatian urate urolithiasis, its use for patients with urate stones associated with portosystemic shunting should be implemented cautiously because the efficacy of allopurinol depends, in part, on hepatic metabolism. Alkalinization also is not recommended for these patients because of the potential for hepatic encephalopathy. The disposition of allopurinol has been described in Dalmations.32 After oral administration of 10 mg/kg, peak plasma concentrations (Cmax) was 6.43 ± 0.18 μg/mL at 3 hours, and the elimination half-life approximated 2.5 to 3 hours. A dosing regimen was not recommended by the authors because of the variability of hyperuricosuria in dogs and the lack of determination of a dose–response relationship to identify a target therapeutic concentration.
Although generally safe, allopurinol is associated with several adverse effects in humans. Included are cutaneous reactions; fever and malaise; and sequelae of drug interactions. Sequential urine urate: creatinine ratios have been advocated for monitoring response to dietary and medical management of urate urolithiasis. A controlled study in healthy dogs, however, found no relationship between random urine samples and 24-hour quantitation of uric acid excretion.33 Thus 24‑hour uric acid measurements probably provide the best means for evaluating response to therapy. Note, however, that urates are easily crystallized and may be difficult to measure in urine. Medications should be continued at least 4 weeks after radiographic evidence of stone dissolution. If dissolution is not evident by 6 to 8 weeks, it is possible that the stones are composed of materials other than urate.
1. Jackson E.K. Diuretics. In: Hardman J.G., Limbird L.E., editors. Goodman and Gilman’s the pharmacological basis of therapeutics. ed 9. New York: McGraw-Hill; 2001:757-787.
2. Okusa M.D. Diuretic drugs. In: Wecker L., editor. Brody’s human pharmacology: molecular to clinical. St Louis: Mosby, 2010.
3. Gross D.R. Diuretics. In: Adams R., editor. Veterinary pharmacology and therapeutics. Ames, Iowa: Iowa State University Press; 1995:523-530.
4. Brater D.C. Diuretic therapy. N Engl J Med. 1998;339(6):387-395.
5. Wakai A., Roberts I., Schierhout G. Mannitol for acute traumatic brain injury. Cochrane Database of Systemic Reviews. 2007. Issue 1. Art. No.: CD001049. doi: 10.1002/14651858.CD001049.pub4
6. Choi H., Won S., Chung W., et al. Effect of intravenous mannitol upon the resistive index in complete unilateral renal obstruction in dogs. J Vet Intern Med. 2003;17:158-162.
7. Skorobohach B.J., Ward D.A., Hendrix D.V. Effects of oral administration of methazolamide on intraocular pressure and aqueous humor flow rate in clinically normal dogs. Am J Vet Res. 2003;64(2):183-187.
8. Hayashi M., Tsutamoto T., Wada A., et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents post-infarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation. 2003;107:2559-2565.
9. Kasma S., Toyama T., Kumakura H., et al. Effect of spironolactone on cardiac sympathetic nerve activity and left ventricular remodeling in patients with dilated cardiomyopathy. J Am Coll Cardio. 2003;41:574-581.
10. Soberman J.E., Weber K.T. Spironolactone in congestive heart failure. Curr Hypertens Rep. 2000;2(5):451-456.
11. Soberman J., Chafin C.C., Weber K.T. Aldosterone antagonists in congestive heart failure. Curr Opin Investig Drugs. 2002;3(7):1024-1028.
12. Abbott L.M., Kovacic J. The pharmacologic spectrum of furosemide. J Vet Emerg Crit Care. 2008;18(1):26-39.
13. Adin D.B., Hill R.C., Scott K.C. Short-term compatibility of furosemide with crystalloid solutions. J Vet Intern Med. 2003;17(5):724-726.
14. Adin D.B., Taylor A.W., Hill R.C., et al. Intermittent bolus injecdtion versus continuous infusion of furosemine in normal adult greyhound dogs. J Vet Intern Med. 2003;17:632-636.
15. Sigrist N.E. Use of dopamine in acute renal failure. J Vet Emerg Crit Care. 2007;17(2):117-126.
16. Flournoy W.S., Wohl J.S., Alnrecht-Schmitt T.J., et al. Pharmacological identification of putative D1 dopamine receptors in feline kidneys. J Vet Pharmacol Therap. 2003;26:283-290.
17. Wohl J.S., Schwartz D.D., Flournoy W.S., et al. Renal hemodynamic and diuretic effects of low-dosage dopamine in anesthetized cats. J Vet Emer Crit Care. 2007;17:45-52.
18. Murphy M.B. Dopamine: a role in the pathogenesis and treatment of hypertension. J Hum Hypertens. 2000;14:S47-S50.
19. Halpenny M., Markos F., Snow H.M., et al. The effects of fenoldopam on renal blood flow and tubular function during aortic cross-clamping in anaesthetized dogs. Eur J Anaesthesiol. 2000;17(8):491-498.
20. Simmons J.P., Wohl J.S., Schwartz D.D., et al. Diuretic effects of fenoldopam in healthy cats. J Vet Emerg Crit Care. 2006;16(2):96-103.
21. Halpenny M., Markos F., Snow H.M., et al. Effects of prophylactic fenoldopam infusion on renal blood flow and renal tubular function during acute hypovolemia in anesthetized dogs. Crit Care Med. 2001;29(4):855-860.
22. Zimmerman-Pope N., Waldron D.R., Barber D.L., et al. Effect of fenoldopam on renal function after nephrotomy in normal dogs. Vet Surg. 2003;32(6):566-573.
23. Wohl J.S., Schwartz D.D., Flournoy W.S., et al. Renal hymodynamic and diuretic effects of low-dosage dopamine in anesthetized cats. J Vet Emerg Crit Care. 2007;17(1):45-52.
24. Hoppe A., Denneberg T. Cystinuria in the dog: clinical studies during 14 years of medical treatment. J Vet Intern Med. 2001;15:361-367.
25. Lekcharoensuk C., Lulich J.P., Osborne C.A., et al. Association between patient-related factors and risk of calcium oxalate and magnesium ammonium phosphate urolithiasis in cats. J Am Vet Med Assoc. 2000;217:520-525.
26. Osborne C.A., Hoppe A., O’Brien T.O. Medical dissolution and prevention of cystine urolithiasis. In: Kirk R.W., Bonagura J.D., editors. Current veterinary therapy X: small animal practice. Philadelphia: Saunders; 1989:1189-1193.
27. Senior D.F. Medical management of urate urolithiasis. In: Kirk R.W., Bonagura J.D., editors. Current veterinary therapy X: small animal practice. Philadelphia: Saunders; 1989:1178-1181.
28. Dieringer T.M., Lees G.E. Nephroliths: approach to therapy. In: Kirk R.W., Bonagura J.D., editors. Current veterinary therapy XI: small animal practice. Philadelphia: Saunders; 1992:889-891.
29. Buffington C.A., Sokolowski J.H. Proceedings of the 16th Annual Waltham/OSU Symposium: Nephrology and Urology. Vernon, Calif: Kal Kan Foods, Inc; 1992.
30. Lulich J.P., Osborne C.A. Canine calcium oxalate urolithiasis: risk factor management. In: Kirk R.W., Bonagura J.D., editors. Current veterinary therapy X: small animal practice. Philadelphia: Saunders; 1992:893-899.
31. Duncan S.H., Richardson A.J., Kaul P., et al. Oxalobacter formigenes and its potential role in human health. Appl Environ Microbiol. 2002;68(8):3841-3847.
32. Ling G.V., Case L.C., Nelson H., et al. Pharmacokinetics of allopurinol in Dalmatian dogs. J Vet Pharmacol Ther. 1997;20(2):134-138.
33. Osborne C., Bartges J.W. Personal communication. University of Minnesota; December 1993.