Chapter 7 Antimicrobial Drugs
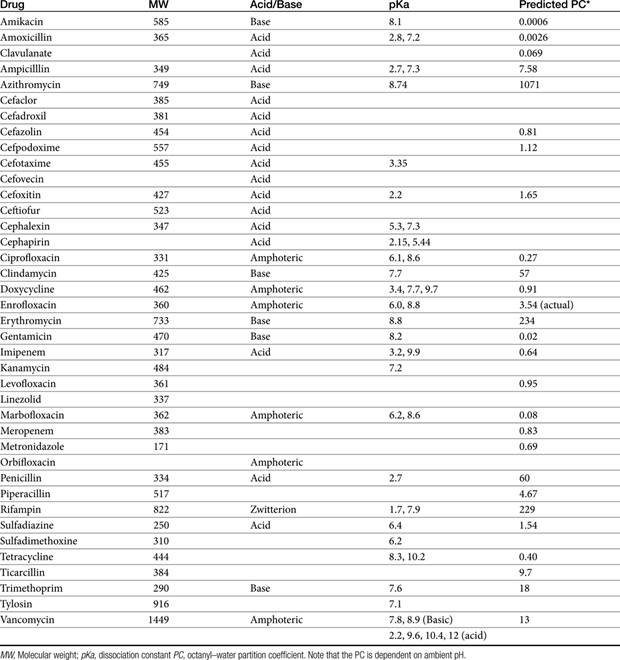
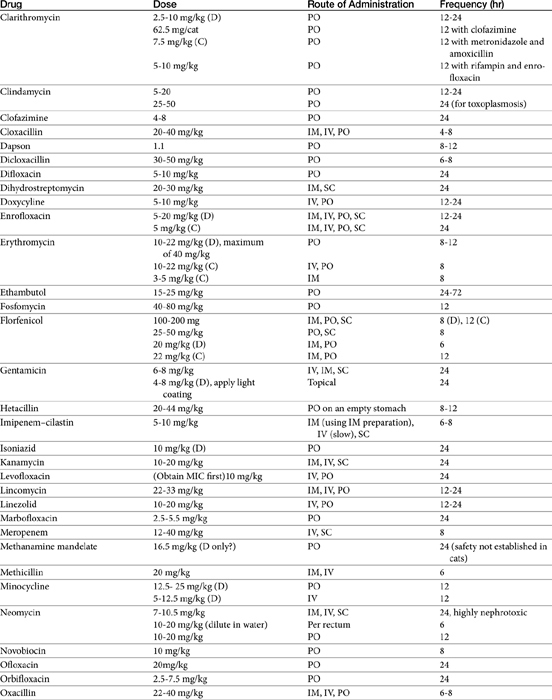
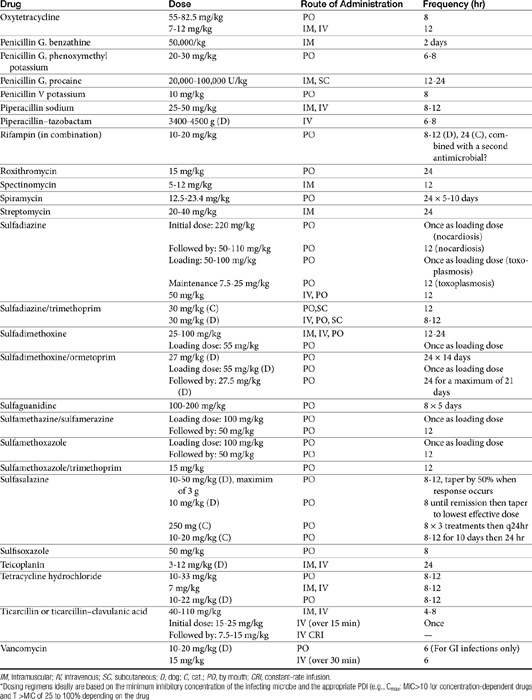
The principles that guide proper antimicrobial selection are discussed in Chapter 6. This chapter focuses on the individual drugs or drug classes and their use to successfully treat bacterial infections. This includes not only resolution of clinical signs but avoidance of resistance. Characteristics discussed for each drug class include structure–activity releationship; the mechanism of antimicrobial action, including whether the drug is time- or concentration- dependent (Table 7-1); the spectrum of antimicrobial activity (Table 7-2), including pharmacodynamics (minimum inhibitory concentrations [MIC] (Tables 7-3 and 7-4) for selected organisms; mechanisms of antimicrobial resistance; clinically relevant aspects of the drug; the disposition of the drug in the patient (as it relates to both safety and efficacy); adverse drug effects; and drug interactions. The breakpoint MICs (the concentration at which an infecting isolate is considered susceptible or resistant to a drug of interest) are delineated in Chapter 6, Table 6-2). Pharmacokinetics were drawn from individual manuscripts, and the Antimicrobial’s Monograph issue of the Journal of Veterinary Pharmacology and Therapeutics2 In addition, Albarellos1 also has provided a review of disposition of selected antimicrobials; these have been included, when appropriate, in Table 7-1. Tissue distribution of antimicrobials is addressed when available; Table 7-5 provides information regarding the relative proportion of tissue versus serum concentrations of drugs, with a focus on body fluids and phagocytic cells. As a reminder (see Chapter 6), drug concentrations measured in tissue homogenates are minimally relevant to concentrations to which microbes are exposed. Data collected by ultrafiltration probes is preferred. However, interstitial fluid is not free of factors that might preclude drug activity (i.e., proteins or ionization; see discussion of cefovecin in cats); as such, dosing errors should be on the side that increase concentrations in tissues. Therapeutic indications are offered when relevant. The dissociation constant of a drug (pKa) and selected information regarding the chemical characteristics of selected drugs or preparation stability are provided for selected drugs in Table 7-6. Doses are indicated in Table 7-7; however, doses ideally should be designed on the basis of intergration of pharmacokinetic (PK) and pharmacodynamic (PD) data (see Chapter 6). Treatment of specific infection is addressed by system in Chapter 8.
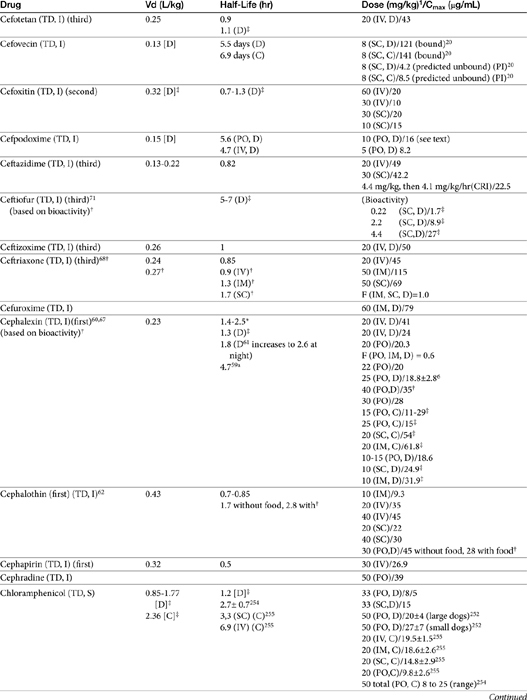
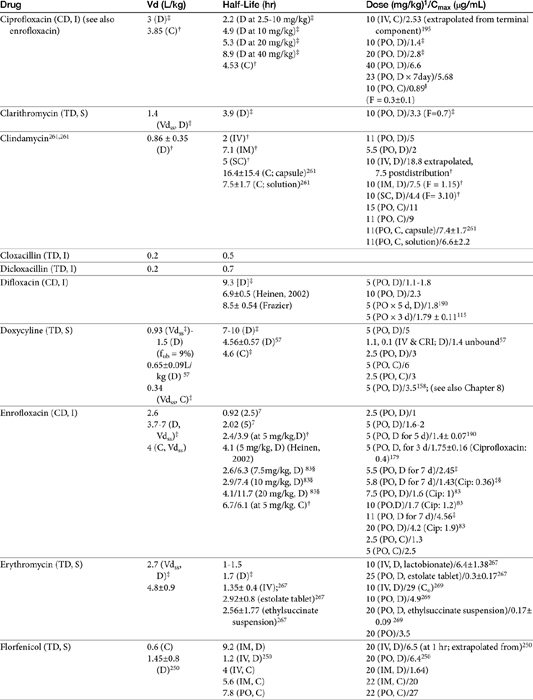
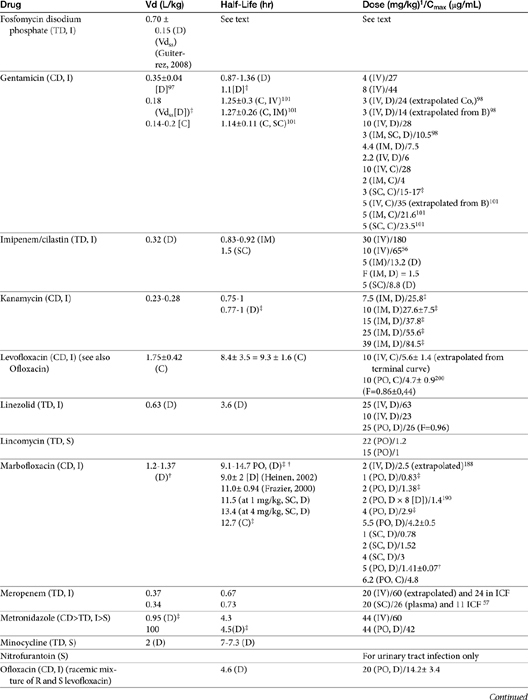
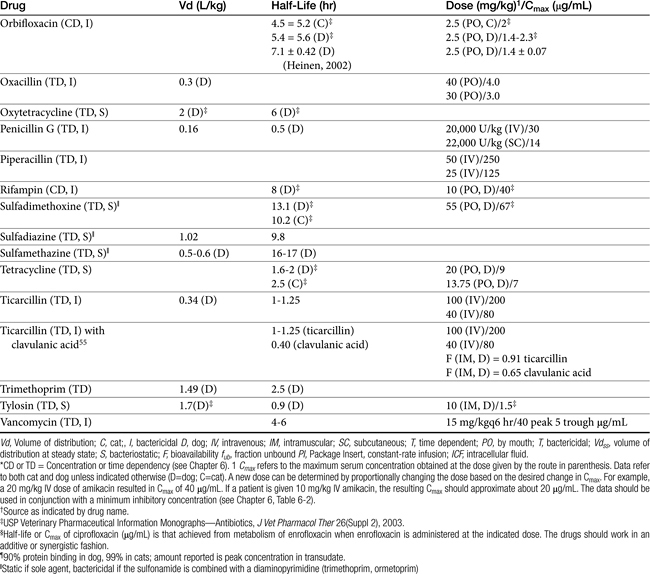
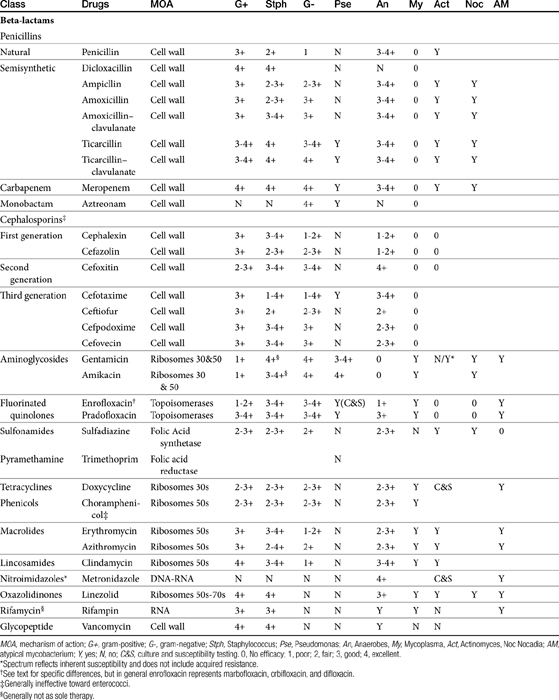
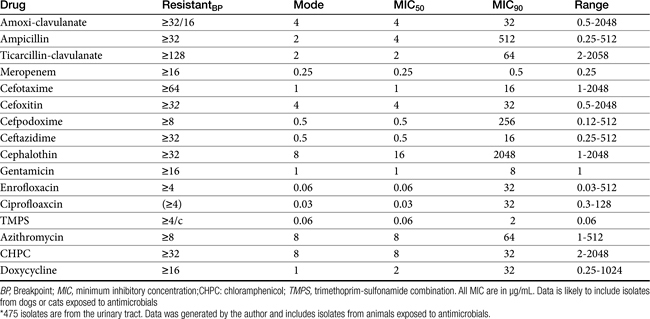
Table 7-4 Susceptibility Data for Selected Drugs and Selected Human Pathogens Associated with Skin and Soft Tissue Infections106
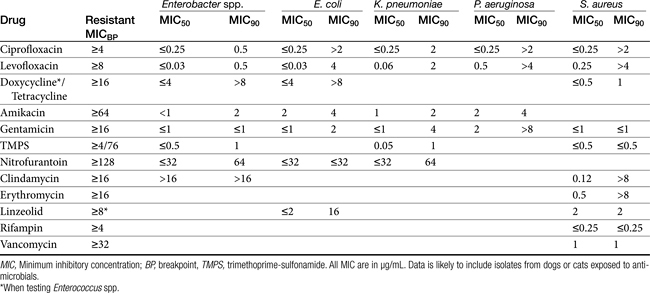
Table 7-5 Serum Concentration of Drugs Achieved in (Human) Tissues∗292
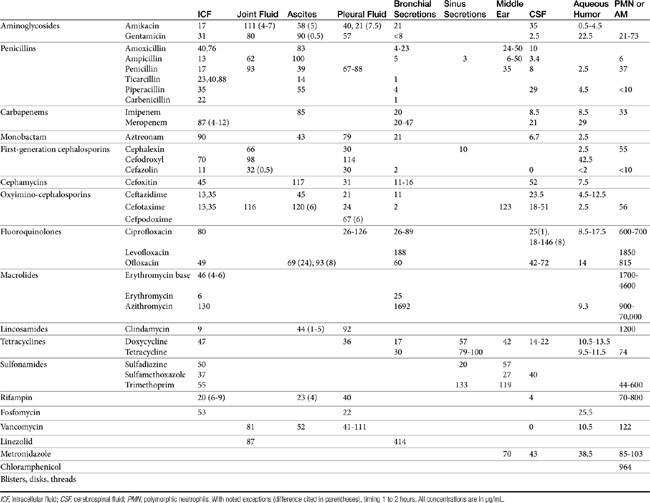
Chapter 6 addressed the importance of integrating PK and PD MIC data when designing a dosing regimen. The PK parameters on which integration is most commonly based are the maximum drug concentration (for both time-dependent and concentration-dependent drugs) and elimination half-life. The latter is particularly important for time-dependent drugs but will also increase area under the curve (AUC), which predicts the efficacy of selected concentration-dependent drugs (e.g., fluoroquinolones; see Table 7-1). Among the sources of PK data to be consulted beyond this chapter are the Antimicrobial Monographs published by the United States Pharmacopiea2 in conjunction with the Journal of Veterinary Pharmacology and Therapeutics. The PD data on which integration is based ideally is the MIC of the isolate cultured from the site of infection in the patient. If not available, the high range of the MIC or the MIC90 might be a reasonable population statistic surrogate indicator of “what is needed” (see Tables 7-3 and 7-4). When available, PD information for canine and feline pathogens (e.g., see Table 7-3) is offered for selected drugs; in addition, relevant information from the human-medicine literature is provided (see Table 7-4). Care should be taken when extrapolating information regarding human pathogens to dogs and cats, although a growing amount of evidence suggests that relative susceptibility of isolates is similar for many drugs (indeed, isolates are likely to be shared), and the data are likely to include both patients that have previously received and not been exposed to antimicrobials. For time-dependent drugs, the relevant PD index (PDI) to be targeted is T > MIC, with a target of at least 50% to 75% of the dosing interval necessary to enhance efficacy, and longer to avoid resistance. An exception can be made for the carbapenems, for which T > MIC of 25% of the dosing interval is sufficient. For concentration-dependent drugs, the relevant PDI is a Cmax/MIC ≥10.3 This ratio should be reached at the site of infection. Alternatively, the AUC/MIC should target 125 to 250. Although as low as 30 has been supported for selected gram-positive drugs, this is particularly true for Streptotoccus pneumoniae, which is an organism that is particularly problematic in humans. This low AUC/MIC may not be relevant to other gram-positive organisms, including other streptococci. Because availability of AUC data is limited, this chapter will focus on Cmax/MIC as the target for concentration-dependent drugs. For PDI for both time- and concentration-dependent drugs, doses should be modified as indicated by drug, host, and microbial factors.
The discussion of antimicrobial drugs is based on their classification by mechanism of action (Figure 7-1; see Table 7-1). The mechanism of action of each drug determines drug efficacy (i.e., bactericidal versus bacteriostatic) and mechanisms of resistance4; influences time- versus concentration-dependence and duration of postantibioitic effect; and, for some drugs, affects safety. Mechanisms of action also influence the selection of combination antimicrobial therapy. For drugs that are approved for use in humans but not animals and for which information regarding use in dogs and cats is not available, PK information in humans will be summarized.
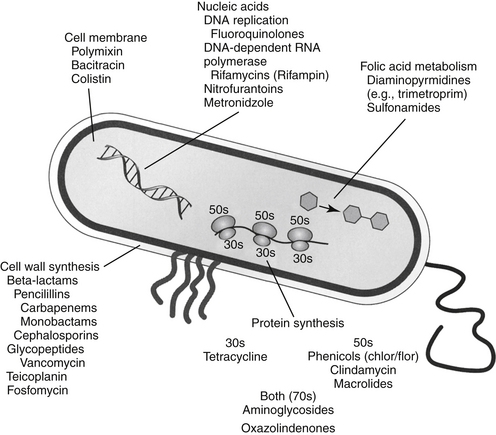
Figure 7-1 Drug mechanisms of action determine drug efficacy, bactericidal or bacteriostatic effects, mechanisms of bacterial resistance, and appropriateness of combination therapy. Occasionally, the mechanism of drug action predicts the mechanism of host toxicity.
Drugs that Target the Cell Wall
Beta-Lactam Antimicrobials
The broad spectrum, low toxicity, and reasonable cost of beta-lactam antibiotics contribute to their frequent use for treatment of infections. In addition, their effects on cell wall synthesis result in their frequent selection for combination antimicrobial therapy. The beta-lactam antibiotics include the cephalosporins, penicillins (including combination penicillin/beta–lactamase inhibitors), carbepenems, and monobactams (see Table 7-1).
Structure–Activity Relationship
Beta-lactam antibiotics contain a four-member beta-lactam ring as the active site. A second member ring establishes the drug as either a cephalosporin—one carbon larger—or a penicillin (Figure 7-2).5-9 Chemically, the beta-lactams are classified as weak acids (see Table 7-6). They include natural, and semisynthetic drugs (see Table 7-2). Penicillin is a natural drug derived from the molds of the genus Penicillium. Penicillin serves as a base for the semisynthetic aminopenicillins (ampicillin, amoxicillin), the extended-spectrum penicillins (carbenicillin, ticarcillin, piperacillin), the carpabenems (imipenem, meropenem), and the monobactams (aztreonam). Penicillin G is the basis for the definition of the international unit (IU) of penicillin, which is equivalent to 0.6 mg of the international pure crystalline sodium penicillin (1.6 IU/mg). The conversion of USP units varies with the salt, with 1 mg of penicillin G equivelant to the following units: sodium (1500-1750); potassium (1440-1680), and procaine (900-1050).As a group the natural penicillins are unstable and subject to hydrolysis at the beta-lactam ring. Degradation can occur when combined with other solutions. Degradation also occurs for most penicillins exposed to gastric acidity, precluding oral absorption.9
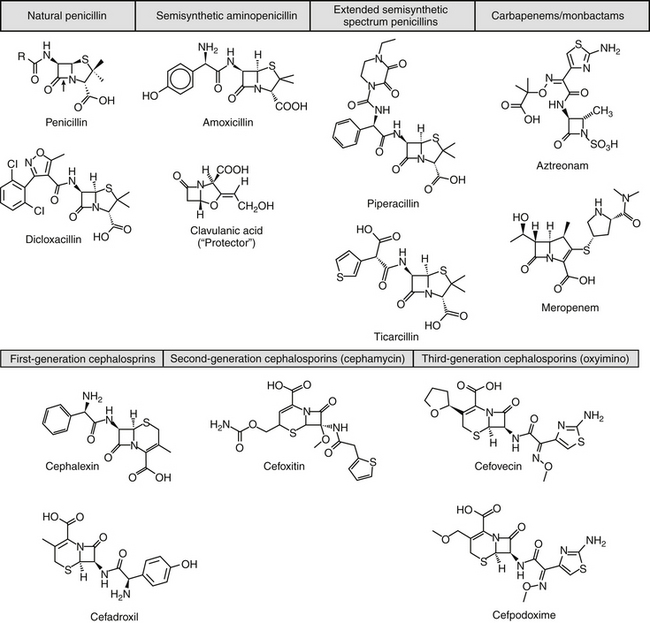
Figure 7-2 Beta-lactam antibiotics include the penicillins and cephalosporins. The four membered beta-lactam ring of each drug mimics the substrate of the transpeptidase enzyme (a penicillin-binding protein), and specifically the terminal portion of p-D-Ala-Asp-D-Ala (boxed inset). This ring structure also is the target of beta-lactamase enzyme destruction. Penicillins have an adjacent five-member ring, cephalosporins a seven-member ring. The addition of larger structures to the basic ring structure may help reduce emerging resistance by beta-lactamase rings but will not avoid methicillin resistance.
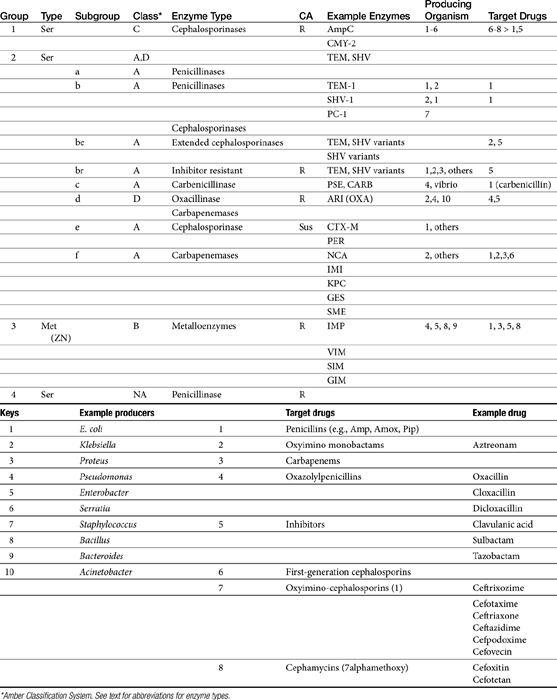
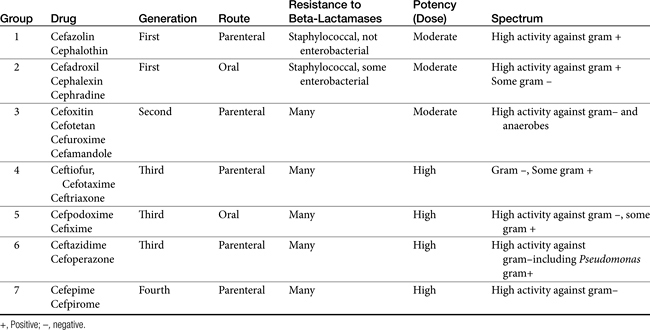
The cephalosporins are derived from a chemical produced by the fungus Cephalosporium acremonium. The six-member ring of the cephalosporins renders them more stable; this increased stability also causes them to be less susceptible to resistance. More than 22 cephalosporins are approved for use in the United States, including the cephamycins (e.g., cefoxitin, cefotetan) and oxyimino-cephalosporins (e.g., ceftazidime, cefotoxime, ceftiofur, cefpodoxime, cefovecin) (see Figure 7-2). The cephalosporins have been variably categorized, with the original “generation” designation being the most widely accepted (Table 7-8).8,10,11 The designations began as an indicator of chronologic approval but have evolved such that each indicates relative resistance to beta-lactamase destruction; the first generation is most and the later generations least susceptible to destruction.10 The advent of extended-spectrum beta-lactamases renders the classification less clear in that these beta-lactams specifically target later-generation drugs. Spectrum and pharmacologic properties of drugs within the generations vary, particularly in the third or later generations. Reclassifying the cephalosporins into groups according to the route of administration, and spectrum has been proposed (see Table 7-8).
Mechanism of Action
The mechanism of action of beta-lactams reflects interference with bacterial cell wall synthesis (Figure 7-3). The bacterial cell wall comprises several layers of a peptidoglycan matrix. The peptidoglycan strands are composed of five repeating disaccharide units of N-acetylglucosamine and N-acetylmuramate; these units are formed by the bacteria in stages. A pentapeptide, which ends with a D-Ala-D-Ala terminus, is attached to each of the repeating units of these disaccharides. The units are joined to form a chain or peptidoglycan strand. The resulting chains are then cross-linked to provide cell wall rigidity. Cross-linking between the D-Ala-D-Ala terminals is catalyzed by transpeptidase enzymes, one of several types of proteins that bind penicillin (referred to as penicillin-binding proteins [PBPs]) located in the cell wall (see Figure 7-3).12 The bacterial substrate for the transpeptidase enzyme is the pentapeptide of the peptidoglycan and, specifically, the terminal amino acids D-Ala-D-Ala. The beta-lactam ring is the functional (active) group of all drugs in this class. It is structurally similar to the D-Ala-D-Ala terminus of the pentapeptide, acting as a substrate and subsequently inhibiting the D-D transpeptidase enzyme (see Figure 7-3). In an actively growing cell, as peptidoglycan precursors increase in response to inhibition of synthesis, autolysins, particularly in gram-positive organisms, contribute to cell wall degradation. Degradation coupled with impaired cell wall synthesis causes the bacterial cell wall to lose rigidity. The cell becomes permeable to the surrounding environment, which, although isotonic to the host, is hypotonic to the organism. Influx of surrounding fluid into the hypertonic bacterial cell results in cytolysis, or osmotic lysis, particularly in gram-negative organisms. Cell wall instability induces the secretion of autolysins, particularly in gram-positive organisms. Because organisms continually break down and rebuild cell walls, the efficacy of the beta-lactam antibiotic ideally is constantly present and, as such, this class of drugs is considered time-dependent (see Chapter 6). However, the duration that the plasma drug concentration (PDC) should be above the MIC varies with the drug, with the desired duration being 50% to 75% of the dosing interval for most drugs. However, T > MIC may be as little as 25% to 50% for carbapenems because they are characterized by more rapid bacterial killing.13 A longer T > MIC is indicated to decrease the risk of resistance.
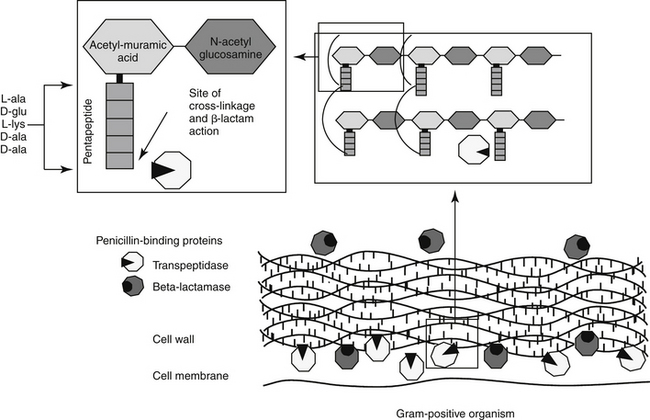
Figure 7-3 The antibacterial mechanism of action of the beta-lactams. The pentapeptide containing the D-Ala-D-Ala terminus (the structure mimicked and thus inhibited by beta-lactam antibiotics) provides the cross-linking of the strands of the cell wall, which are critical to rigidity. Two types of penicillin-binding proteins (PB) are located in the cell wall of bacteria. Transpeptidase enzymes are responsible for catalyzing the cross-bridging between the pentapeptides thus providing rigidity; changes in the structure of these proteins confers resistance to methicillin (PB-2) or other drugs. Beta-lactamase penicillin-binding proteins destroy susceptible beta-lactam antibiotics.
Although all PBPs are able to covalently bind beta-lactam antibiotics, the numbers bound and subsequent activity vary among organisms. Up to nine PBPs are encoded by the genome of Escherichia coli; each PBP generally has subgroups. The diversity of PBPs is responsible, in part, for differences in the spectrum of activity of the beta-lactams. High-molecular-weight PBPs (1, 2, and 3) are essential for microbial growth and survival in Staphylocccocus spp., whereas only PBPs 1 and 2 are critical for Streptococcus spp.; as such, these PBPs are the critical targets of antimicrobial therapy in these organisms.14 In E. coli PBP-2 is essential for cell elongation and PBP-3 for cell division. Because PB-3 appears to complex with PBP-1, -4, and -7 as well as with other proteins,12 effective antimicrobial binding to PBP-3 might have a greater impact than binding to other PBPs in E. coli. The PBP targeted is known for some drugs (e.g, cefpodoxime targets PBP-1a and 1b and PBP-3 (see package insert).
KEY POINT 7-1
The mechanism of action of the beta-lactams necessitates the presence of the drug throughout most of the dosing interval.
KEY POINT 7-2
The spectrum of the penicillins becomes broader as the class “extends,” whereas the spectrum of the later-generation cephalosporins varies with the drug.
Although beta-lactams are very effective antimicrobials, their unique mechanism of action increases the risk of therapeutic failure in certain conditions, independent of bacterial resistance. Efficacy, particularly toward gram-negatives, is reduced in a hypertonic environment (e.g., the renal intersitium of the normally functioning kidney, an abcess) because osmotic lysis may not occur. Slow growth impairs autolysin activity, which may result in the loss of the bactericidal effect of the beta-lactam antibiotic. Examples might include the combined use of a beta-lactam with a drug that slows growth of the organisms (i.e., a ribosomal inhibitor [see Figure 7-1]), or in a hypoxic environment (e.g., abscess).
Spectrum of Activity
The spectrum of activity of beta-lactam antibiotics varies (see Table 7-2). PD data are available for both the dog and cat for limited drugs (Table 7-9), with selected information provided on human pathogens associated with skin or soft tissue infections (Table 7-10). Penicillin G, a natural antibiotic, is effective against selected gram-positive cocci and both gram-negative and gram-positive anaerobes, but it is beta-lactamase sensitive.5 Selected enterococci are not susceptible to penicillin, and most staphylococci produce beta-lactamases. The gram-negative spectrum of penicillin G is limited but includes Pasteurella multocida. Penicillin V is an orally bioavailable natural penicillin, but its antimicrobial efficacy is reduced.9 Beta-lactamase–resistant isoxazolyl-derivative penicillins include dicloxacillin, cloxacillin, methicillin, and oxacillin. These drugs are effective against gram-positive organisms, including Staphylococcus spp., and gram-negative and anaerobic organisms.
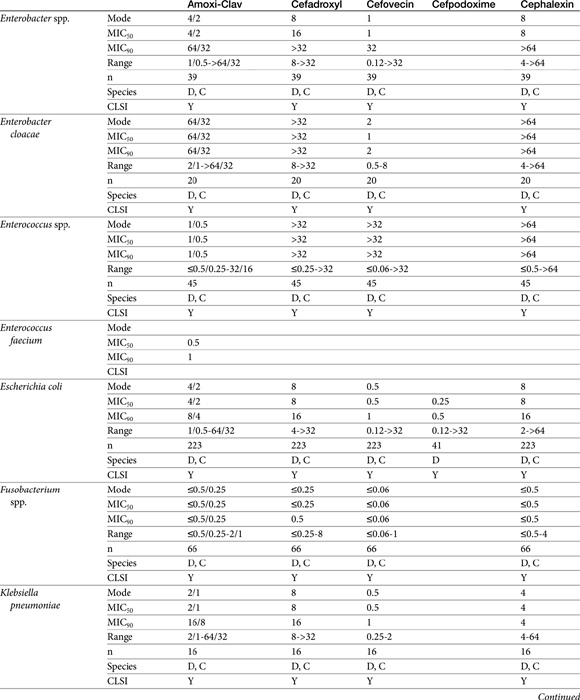
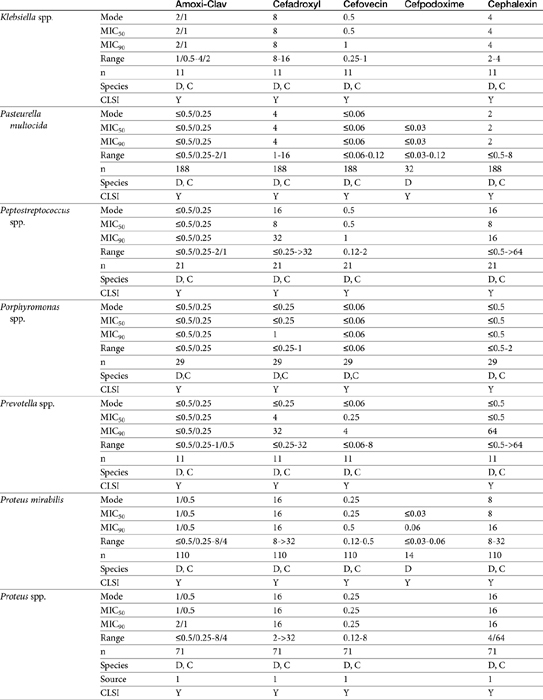
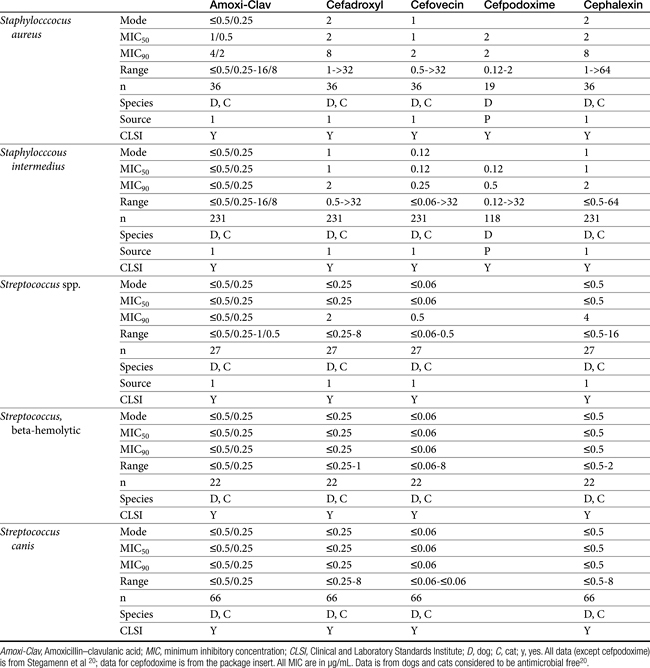
Table 7-10 Susceptibility Data for Selected Human Pathogens Associated with Skin and Soft Tissue Infections160
The spectrum of the beta-lactams was expanded with the production of the semisynthetic aminopenicillins. Amoxicillin and ampicillin (aminopenicillins) are considered broad-spectrum drugs; however, this classification has largely been muted by acquired resistance unless combined with clavulanic acid or sulbactam. They target PBP-1a. The anaerobic and gram-positive spectrum of penicillin G is maintained (although the aminopenicillins are slightly less efficacious against anaerobes). The aminopenicillins are generally effective against enterococci, although Enterococcus faecium often expresses resistance. In addition, many gram-negative organisms are added to the spectrum, including E. coli, Pasteurella, some Proteus species, Klebsiella, and selected others (e.g., Salmonella, Shigella). Serratia, Enterobacter, and Pseudomonas are not, however, included in the spectrum of the aminopenicillins. The spectrum of ampicillin is generally similar to that of amoxicillin, and it serves as the model drug for amoxicillin on culture and susceptibility (C&S) testing whereas amoxicillin–clavulanic acid indicates data for ampicillin–sulbactam. However, the potency of ampicillin generally is less than that of amoxicillin against enterococci and Salmonella but greater against Shigella and Enterobacter. The aminopenicillins are less effective compared with the penicillins against Bacteroides fragilis, although efficacy remains good to excellent.9 Like penicillin, the aminopenicillins are beta-lactamase sensitive. Combination with a beta-lactamase protector (e.g., clavulanic acid or sulbactam) improves efficacy and thus broadens the spectrum against susceptible organisms that have acquired resistance through beta-lactamase production. This includes Staphylococcus, E. coli, Klebsiella spp., and some Proteus spp.15 Pseudomonas spp. and other gram-negative organisms remain resistant.7,9 Further modifications led to the extended-spectrum penicillins characterized by a markedly enhanced spectrum, particularly against gram-negative organisms, including Pseudomonas aeruginosa, Serratia, Proteus spp., some Klebsiella spp. Shigella spp., and Enterobacter spp. Examples include the carboxypenicillins carbenicillin and ticarcillin, with ticarcillin having two to four times higher activity toward Pseudomonas spp. than carbenicillin, and the ampicillin-derived ureidopenicillin piperacillin, which has the highest antipseudomonal activity.5,7,16,17 The extended-spectrum penicillins are effective against anaerobic organisms, although they may be less effective than the natural penicillins. They maintain, however, good to excellent activity against B. fragilis.9 The extended-spectrum penicillins are beta-lactamase sensitive; however, a ticarcillin/clavulanic acid combination product is available.
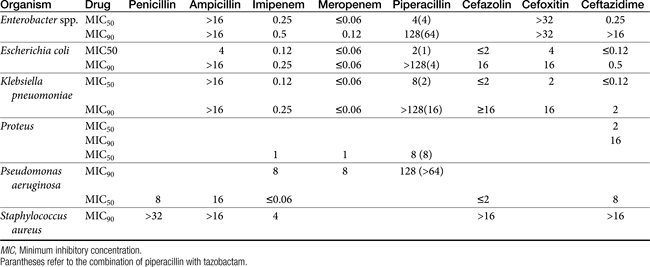
The carbapenems (imipenem and meropenem) and monobactams (aztreonam) represent the most recent members of the beta-lactam penicillins.18 Imipenem targets PBP-1a, -1b, and -2, with its efficacy based on binding to PBP-2 and -1b. It is prepared in combination with cilastatin, which inhibits renal tubular degradation (metabolism by dehydropeptidase-1) of imipenem. As a result, drug half-life may be prolonged (although the clinical relevance of this effect in animals is questionable), and the formation of potentially nephrotoxic metabolites is reduced. Imipenem and meropenem have the broadest antimicrobial spectrums available against bacterial organisms with cell walls, including Pseudomonas spp. Imipenem and meropenem are relatively resistant to beta-lactamase destruction. However, an extended beta-lactamase enzyme has recently been reported, particularly in Klebsiella pneumoniae,emerging as a nosocomial pathogen.19 An advantage of the carbopenems has been their very low MICs (0.05 to 2 μg/mL) for most susceptible organisms. Meropenem is generally similar to imipenem for empirical treatment of serious infections.
Aztreonam is a monobactam (see Figure 7-2), with a high affinity for PBP-3 and lesser affinity for PBP-1a. It is particularly effective against gram-negative aerobes, including Pseudomonas spp. but is ineffective against gram-positive organisms and anaerobes.
The spectrum of the cephalosporins is more diverse than that of the penicillins and is not as easily categorized. Although generalizations regarding the spectrum of activity of each successive generation might be made, variability in efficacy among the drugs within and certainly among generations may result in therapeutic failure if attention is not paid to differences.9 Thus either the package insert or C&S data should be consulted before selecting a cephalosporin, particularly beyond the first generation. In general, cephalosporins are ineffective against enterococci. With each successive generation, the cephalosporins become increasingly more resistant to beta-lactamase destruction, and all generations are generally more resistant as a class than are the penicillins. As such, they are often chosen as empirical first-choice treatment of Staphylococcus spp. Cephalothin (no longer commercially available) has been the drug designated by the Clinical Laboratory Standards Institute (CLSI; previously National Committee for Clinical Laboratory Standards [NCCLS]) as the model indicator for susceptibility for the first-generation cephalosporins. However, it does not represent the class equally. The aerobic spectrum of the first-generation cephalosporins is similar to that of the aminopenicillins,9 although efficacy is more similar to amoxicillin–clavulanic acid combinations. First-generation cephalosporins such as cefazolin, cephalothin, and cephalexin are active (although not equally so) against gram-positive and gram-negative organisms such as E. coli, K. pneumoniae, and Proteus mirabilis. Among the first-generation drugs, cefazolin has better efficacy than cephalexin against gram-negative organisms (e.g., E. coli) but poorer efficacy against Staphylococcus spp.5,9 Efficacy of cephalexin against E. coli is fair to poor. The anaerobic spectrum of the first-generation cephalosporins is fair but less than that of the aminopenicillins.
The second-generation cephalosporins, cefamandole, cefaclor, cefoxitin, and others, are characterized by enhanced activity toward Enterobacter spp., some Proteus spp., E. coli, and Klebsiella spp.5 Cefoxitin has an excellent anaerobic spectrum, particularly against Bacteroides spp.,5,8 although it is less effective than first-generation drugs against gram-positive organisms. Third- (cefotaxime, ceftazidime, cefpodoxime, cefoperazone, cefovecin, and the oxa-beta lactam moxalactam) and fourth-generation (cefepime; not approved in the United States) cephalosporins are generally reserved for serious gram-positive or gram-negative infections (e.g., P. aeruginosa, Enterobacter spp., and Serratia spp.). However, although the efficacy of most of the second-plus generation cephalosporins against E. coli tends to be good to excellent, efficacy against P. aeruginosa9 is variable, and cross-efficacy among members of these generations to any organism should not be assumed. For example, cefoperazone and ceftazidime are among the most effective drugs against P. aeruginosa, although efficacy is less than that of the newer extended-spectrum penicillins. Cefpodoxime and cefovecin are generally effective against E. coli but not effective against Pseudomonas spp. Selected third-generation cephalosporins (e.g., cefotaxime) are effective against anaerobic organisms, whereas others (e.g., ceftazidime, ceftriaxone, and cefpodoxime) are not. Ceftiofur is a third-generation cephalosporin approved for use for canine urinary tract infections. The antimicrobial spectrum of ceftiofur includes gram-positive (Streptococcus spp. and Corynebacterium spp.), gram-negative (Pasteurella, E. coli, and Salmonella spp. but not Pseudomonas spp.), and anaerobic organisms. Ceftiofur is effective against many staphylococcal organisms; however, selection against Staphylococcus spp. should be based on C&S data.9 The first-generation drug cefazolin has been inappropriatly promoted as a generic version of ceftiofur.19a The spectrum of the third-generation drugs cefpodoxime and cefovecin (the former approved in dogs and cats, the latter approved in dogs but used in cats) includes Staphylococcus spp.; cefovecin is also approved for use in the treatment of Streptococcus spp. Both drugs are effective against a variety of gram-negative organisms, including E. coli and Klebsiella, but are not generally effective toward Pseudomonas spp. Stegemann20 has provided PD statistics for a large number of organisms for cefovecin, as well as selected other beta-lactams, some of which are provided in Table 7-9.
Resistance
Bacteria develop resistance to beta-lactams through four major mechanisms: altered or different PBPs such that antibiotic binding does not occur (e.g., staphylococcal organisms and penicillins; enterococcal organisms and cephalosporins); efflux through specific pumps; loss of or changes in porins (especially P. aeruginosa); and inactivation by beta-lactamases. Inactivation by beta-lactamases is most common. Staphylococcus resistance to penicillin appeared as early as 1942; by the late 1960s, more than 80% of medically relevant isolates were resistant to penicillin as a result of beta-lactamase production. Today more than 90% of isolates (human) produce penicillinase.21 The approval of “protected” drugs (i.e., improved the efficacy of selected penicillins), but along with the cephalsoporins, is likely to have contributed to the emergence of altered PBP. This most notorious mechanism of resistance has yielded methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE).
Beta-lactamases
Beta-lactamases are structurally and mechanistically similar to PBPs; indeed, certain PBPs are capable of beta-lactamase activity. Destruction of the beta-lactam (amide) ring reflects its hydrolysis (see Figure 7-2).22 Currently, more than 400 distinct beta-lactamase enzymes are produced by gram-negative, gram-positive, and anaerobic organisms.23,24 Selected examples are listed in Table 7-11. Altlhough clearly a major mechanism of resistance in gram-positive organisms, beta-lactamase production is also the major mechanism by which gram-negative organisms develop resistance.22 Beta-lactamase production occurs as a result of either chromosomal mutations, particularly in gram-positive organisms, or plasmid-mediated resistance in both gram-positive and gram-negative organisms. Beta-lactamases are either constitutive, already present in the cell wall (particularly in gram-negative organisms), or induced by the presence of the antimicrobial drug (in both gram-negative and gram-positive organisms).25 Gram-negative bacteria have the added advantage of secreting beta-lactamases into the periplasmic space such that they are strategically placed before the antibiotic can penetrate the cell wall.9 The beta-lactams are variably susceptible to destruction by beta–lactamases; microbes vary in which enzyme they produce and whether the enzyme is constitutive or inducible (see Table 7-11).
Two major types of beta-lactamases exist: serine-based enzymes and the metallo-beta lactamases. The latter contain a zinc atom that activates water as the destructive site (see Table 7-11).22 Several schemes have been proposed to classify beta-lactamases.The most common scheme is based on the molecular structure (Amber Classification); however, classification according to the target substrate (Bush–Jacoby Classification) may be easier to follow (see Table 7-11). According to the Amber system, Class B enzymes contain the metallo-beta lactamases, but the other three classes are serine-based enzymes. These include classes A (TEM, SHV), C (ampC, targeting cephamycins [cefotetan, cefoxitin]), and D (OXA; targeting protected drugs, such as dicloxacillin, but also protectors such as clavulanic acid). The most prevalent beta-lactamases are class A penicillinases and cephalosporinases, including clinically relevant TEM-1 and 2 or SHV-1 enzymes found in E. coli and K. pneumoniae, and PC-1 enzymes produced by S. aureus.22
KEY POINT 7-3
Microbes have been able to adapt to each pharmaceutical manipulation intended to decrease beta-lactamase activity.
TEM-1 and SHV-1 confer high-level resistance to penicillins and first generation cephalosporines but generally do not target the extended-spectrum (selected second- and third- or fourth-generation) cephalosporins or carbapenems. As such, the cephalosporins (cephalosporin C) are generally less impacted by beta-lactamases, particularly those produced by Staphylococcus species.10 However, only a few cephalosporins are stable against anaerobic beta-lactamases. Selected semi-synthetic beta-lactams also are less impacted by beta-lactamases, including most third-generation cephalosporins and imipenem. The semisynthetic dicloxacillin (and oxacillin) is beta-lactamase resistant, with the exception of class D (group 2d). The combination of beta-lactam antibiotics with drugs that inhibit beta-lactamase activity (e.g., clavulanic acid, sulbactam, and tazobactam) increases the potency of the beta-lactam antibiotic, (but not the spectrum) toward susceptible organisms (see Tables 7-2, 7-9 and 7-10). Clavulanic acid irreversibly binds to some but not all beta-lactamases (see Table 7-11).26 Combinations of beta-lactams with beta-lactamase inhibitors are particularly useful against mixed infections and have shown efficacy against selected multiresistant pathogens such as Acinetobacter spp. Aztreonam is generally resistant to beta-lactamase destruction but is susceptible to extended-spectrum beta-lactamases (ESBLs). The presence and diversity of beta-lactamases in canine and feline staphylococcal organisms has been described. As in other species, production is encoded by the blaZ gene, with all four classes of enzymes (A to D) represented genes for classes A, C, and D being plasmid mediated and class B chromosomally mediated.27
Microbes have adapted to each pharmaceutical manipulation intended to combat emergent resistance resulting from beta-lactamase destruction. Third-generation cephalosporins such as cefotaxime and ceftazidime initially were considered indestructible by beta-lactamases.25 However, high-level use has been accompanied by induction and selection for ESBLs in multiple-resistant coliforms,28 particularly in those organisms that produce TEM and SHV enzymes. The genes encoding ESBLs are carried by large plasmids and are able to confer information between bacterial species and strains. The ESBLs are most commonly found in Klebsiella spp. (incidence in North America, 4.4%), E. coli (3.3% to 4.7%), or P. mirabilis (3.1-9.5%), but they also have been detected in other members of the family Enterobacteriaceae and in P. aeruginosa isolates.29-32 The resistant gene codes for mutations in one or more amino acid (serine) substitutions in class A enzymes (TEM or SHV). The resultant change in configuration allows the enzyme to gain access to the drug despite the large oxyimino side chain of these newer-generation drugs.24 Drugs amenable to destruction by ESBL include third-generation cefotaxime, ceftazidime and ceftriaxone, cefpodoxime, and (presumably) cefovecin.22,33 Selected fourth-generation drugs are also susceptible, including cefepime (no longer marketed in the United States).28 Cephamycins (e.g., second-generation cephalosporins cefoxitin, cefotetan) do not appear to be destroyed (although they are destroyed by ampC). Monobactams (i.e., aztreonam) are destroyed. Carbapenems are generally not destroyed by ESBL, nor are beta-lactamase protectors such as clavulanic acid. The use of beta-lactamase protectors appears to reduce the clinical emergence of ESBLs and may reduce the emergence of other resistant pathogens such as Clostridium difficile and vancomycin-resistant enterococci.34 However, the effect (e.g., of the beta-lactamase in the presence of ESBLs) is not always predictable. Decreasesd cephalosporin usage also reduces the advent of ESBLs.
Resistance to ESBLs often is incorporated in plasmids simultaneously conferring resistance to aminoglycosides and sulfonamides.22 Further, ESBL resistance may be associated with non–plasmid-mediated resistance mechanisms such as occurs for quinolones.22 An “inoculum effect” of ESBLs has been described for some drugs and may explain discrepancies among studies: the MIC of the organisms toward cephalosporins increases with a larger (107) compared with smaller (105) inoculum. Because susceptibility may depend on the size of the inoculum at the site of infection,22 ESBLs may not be detected on routine C&S testing.31 Lack of detection of ESBLs may also reflect different levels of activity against the different cephalosporins.
Detection of ESBLs has been based on double disk diffusion techniques. The susceptible cephalosporin (e.g., cefpodoxime, ceftazidime) is incubated with the isolate as the sole drug and in the presence of a beta-lactamase inhibitor; a substantial reduction in the MIC (e.g., fourfold to eightfold) with the combination drugs compared with the cephalosporin by itself indicates an ESBL.35,36 Not all clinical microbiology laboratories have incorporated tests for ESBLs in routine testing procedures.22 The presence of an ESBL should be suspected with organisms resistant to or with high MIC to cefotaxime but susceptible to beta-lactam/beta-lactamase combinations.22 The detection of an isolate with ESBL in a patient with a serious gram-negative bacillary infection should lead to the use of a carbapenem. However, a novel carbapenemase also has been described following isolation in Serratia spp., K. pneumoniae and Enterobacter cloacae.19,22,37 Alternatively, combination of the cephalosporin with clavulanic acid should be considered.
Altered pencillin-binding proteins
The advent of MRSA and multidrug-resistant Enterococcus spp. also has been associated with cephalosporins although it is likely that beta-lactamase inhibitors contributed to its emergence.25 The approval of the cephalosporins in the 1980s was followed by the first MRSA epidemics in the mid-1980s in the United Kingdom; the use of second- and third-generation cephalosporins also was associated with an outbreak of MRSA in Japan.25 In humans, mortality associated with S. aureus bacteremia is 20% to 40%; MRSA has become a leading cause of nosocomial infections in human medicine. The term MRSA was coined in the early 1960s, when these penicillinase-resistant drugs were relatively new, and refers to resistance expressed in vitro to methicillin.21 Although this discussion will focus on MRSA, increasingly, methicillin resistance is being recognized in other species and much of this information is relevant to all methicillin resistant staphylococci (MRS). Over the 30 to 40 years since MRSA was identified, MRSA infections have led to increased mortality and morbidity. The sequelae of MRSA are worse than those associated with beta-lactamase resistance because no alternative therapy remains that is predictably effective.21 In contrast to resistance resulting from penicillinase production which is generally considered low level, infection with MRSA is considered high-level resistance. Further, MRSA isolates are essentially multidrug resistant, that is, expressing resistance to classes other than beta-lactams.
MRSA and methicillin-resistant Staphylococcus pseudintermedius (MRSIG)38 are indicated by the presence of the mecA gene. This gene encodes a mutation in penicillin-binding protein 2a, thus reducing its affinity for the beta-lactam ring, rendering the organism resistant to all beta-lactams. The mecA gene is carried on the staphylococcal chromosomal cassette (SCC); currently five SCCmec have been described.39 Protectors such as clavulanic acid are also unable to bind and thus are ineffective.21 Detection of MRSA or MRSIG (or methicillin-resistance in other staphylococci [MRS]) on C&S testing generally is based on resistance to oxacillin, which is more stable than methicillin in disks used for testing. However, variability in testing methods can profoundly alter results; therefore, cefoxitin might be a more appropriate indicator of multidrug resistance in these organisms.40 Alternative procedures such as polymerase chain reaction or latex agglutination have been used to detect the gene responsible for the formation of penicillin-binding protein 2a (mecA) of MRSA, and other techniques such as pulsed-field gel electrophoresis or multilocus sequence typing identify the specific strain of MRSA (e.g., USA100 or USA300). It is likely that this area of diagnostics will be refined in the next decade and will be applied to other MRS.
KEY POINT 7-5
Changes in the penicillin-binding protein (PBP2) by Staphylococcus spp. renders the microbe resistant to all beta-lactams.
Antimicrobials are associated with induction, selection, and propagation of MRSA. The wide use of cephalosporins, in particular, may have contributed significantly to the advent of MRSA. MRSA in human patients has evolved from a hospital-acquired (HA-MRSA; nosocomial) infection (usually USA100) that occurs most commonly in patients whose immune systems are compromised by a community-acquired infection (CA-MRSA), in which otherwise healthy persons are infected, usually in the skin or soft tissue. Crowded conditions, shared items, and poor hygiene increase the risk of CA-MRSA. It is CA-MRSA strain USA300 that appears to be most commonly associated with increased colonization in dogs and cats. In contrast, it is HA-MRSA (USA-100) that is most commonly associated with infections in dogs and cats.40a According to the Center for Disease Control, the incidence of MRSA doubled in human medicine between 1999 and 2006. The impact of MRSA (or other MRS) in veterinary medicine is increasingly problematic, not only because of its impact on the patient but also because of public health considerations. The mec gene has been detected in MRSA organisms infecting dogs,40-42 and MRSA has been associated with infection in dogs.43 However, MRSA also has been found in up to 4% of healthy dogs, with identification complicated by the need for multiple sampling sites (nasal and rectal or perineal). Risk factors for the presence of MRSA in pets or working dogs (e.g., detection and aid dogs) include contact with human hospitals (particularly if patients fed the dogs treats or were licked by the dogs) and children.42 Infections have been isolated to family members and pets in the same household, but this is likely to reflect original transmission from humans to the pet.40-4244 It is likely that colonization is transient in animals. However, healthy pets have been demonstrated to be potential reservoirs for transmission of MRSA to healthy handlers and a potential health risk to immunocompromised patients (humans and presumably other animals in the household). According to the American Veterinary Medical Association, colonization by MRSA is suggested to be an occupational risk for veterinarians, although the frequency of infection associated with MRSA in veterinarians compared with other health professionals has not been documented.
MRSIG45 has a prevalence of 0.58% to 2% in healthy dogs and up to 4% in healthy cats,42,46 with the mec gene present in each canine MRSIG isolate in one study.47 Human colonization with MRSIG is unusual.42 However, MRSIG has been reported as a cause of infection in human patients,42 and transmission from pets with pyoderma to humans has been confirmed.48,49 Although the true public health significance of MRSA and MRSIG (or other multidrug-resistant organisms) in pets is not clear, the fear of infection may be as important as true risk, necessitating proper hygiene and other proactive measures such that human or animal health (including unnecessary euthanasia) is not risked.
The American Veterinary Medical Association offers a website that includes a discussion of MRSA zoonoses, including sources of guidelines that might decrease the risk presented to susceptible humans.49a Among the more important actions that can be taken is establishment of infection control policies and guidelines in each veterinary practice. In general, common sense approaches should prevail (e.g., minimizing intimate contact, maintaining good personal and environmental hygiene practices; see the three D’s approach described in Chapter 6). This includes cleansing of hands of handlers and the paws (or body) of animals that might be exposed to MRSA, including those visiting human health care facilities. Immunocompromised patients are at most risk for MRSA infection acquired from an animal. In such cases the carrier or infected animal should be removed from the environment until successfully treated for MRSA. For dogs with skin infections, cultures are indicated to detect MRSA, particularly in animals for which infection does not resolve. Successful resolution of colonized or infected animals may require both topical (for skin infections) and systemic therapy. Evidence of successful treatment might be based on skin swabs of the ear, nose, and perianal region. Care must be taken to ensure that the laboratory providing culture procedures is well-versed in the diagnosis of MRSA, including speciation of coagulase-positive organisms.
KEY POINT 7-6
Methicillin-resistant Staphylococcus aureus likely originated with a human, whereas Methicillin-resistant Staphylococcus pseudintermedius likely originated from a dog or cat.
The multidrug resistance associated with MRSA is now evolving toward other (non–beta-lactam) antimicrobials. This reflects, in part, other resistance genes in the gene cassette carrying the mec gene.42 Drugs that are affected include fluorinated quinolones and aminoglycosides. Although newer fluorinated quinolones (e.g., levfloxacin) appear to be more effective than older drugs in vitro, particularly to Staphylococcus, whether this translates to better clinical efficacy is unclear.21 Glycopeptides such as vancomycin are the initial drugs used to treat MRSA in humans, although increasingly vancomycin-resistant Staphylococcus aureus (VRSA) infections have emerged. Linezolid and rifampin are alternative drug choices.
Multidrug resistant Enterococcus spp. also is an emerging issue; its emergence also appears to be correlated to use of cephalosporins. Enterococcus faecalis more so than Enterococcus faecalis is likely to develop resistance, and speciating Enterococcus spp. susceptibility testing might be prudent. Resistance reflects a change in penicillin-binding protein (PB-V), and the risk is increased when drugs effective against Enterococcus spp. are used.
Pharmacokinetics
The beta-lactams are weak acids, which favor oral absorption. Many of the beta-lactam antibiotics, however, are destroyed by the acidity of the gastrointestinal tract and thus cannot be given orally. Penicillin exceptions include penicillin V, dicloxacillin, the aminopenicillins (ampicillin and amoxicillin, including combinations with clavulanic acid), and carbenicillin (indanyl form only; effective concentrations can be achieved only in urine). Lack of stability also may affect the shelf-life of reconstituted products; expiration dates should be adhered to as indicated for the reconstituted product. Orally bioavailable cephalosporins include cephalexin, cefadroxil, and cefpodoxime (third or fourth generation). The oral bioavailability of the cephalosporins also varies among drugs and species.5,9
Many beta-lactams are available as intravenous or parenteral preparations. Absorption from parenteral sites tends to be rapid and complete, with the exception of products that are specifically formulated to allow slow release (e.g., esterified penicillins). Although drug concentrations may persist in circulation longer than non–slow-release preparations (an appealing aspect for time-dependent antimicrobials), older dosing regimens were designed for efficacy against organisms considerably more susceptible to drugs at the time of approval compared with current microorganisms. Thus consideration should be taken to design the dose of these products to compensate for any increase in MIC that may have emerged since the approval of the labeled dose. Selected beta-lactams are highly bound to plasma proteins. Although binding limits distribution into tissues, it also contributes to a long disappearance half-life. Cefpodoxime and, to a greater degree, cefovecin are example of beta-lactams whose long half-life reflects slow release from intravascular protein.20
KEY POINT 7-7
As water-soluble drugs, all beta-lactams distribute to extracellular fluid, do not penetrate sanctuary tissues well, and are renally excreted.
Distribution of beta-lactams is limited to extracellular fluid (volume of distribution [Vd or Vdss] of unbound drug generally ≤0.3 L/kg), but, barring a marked host inflammatory response, adequate concentrations of unbound drug can usually be achieved in the interstitial fluid (the site of most infections) in many tissues (see Table 7-5).5,9 Penicillins and cephalosporins are thus widely distributed throughout most extracellular body fluids, including kidneys, lungs, joints, bone, soft tissues, and bile5,8,11 Interstitial fluid concentrations in normal tissues generally can be predicted by, but are not necessarily equivalent to, the concentration of (unbound) drug in plasma. Comparisons of AUC frequently reveal interstitial fluids to be 30% or less than that in plasma. Among the first-generation cephalosporins, cefodroxil appears to have the better tissue-to-PDC ratio in humans (see Table 7-5). Neither penicillins nor cephalosporins traverse sanctuaries well, including mammary, prostatic, or blood–brain barriers. Imipenem, but generally not antipseudomonal penicillins such as ticarcillin and piperacillin, can reach effective concentrations in the brain. However, first- and second-generation cephalosporins should not be used for central nervous system (CNS) infections because many are destroyed by local enzymes or transported out of the CNS. Beta-lactams in general achieve 25% or less in bronchial secretions compared with PDCs (see Table 7-5).50-52 Inflammation increases the penetration of many beta-lactams. For example, cefuroxime, cefotaxime, ceftriaxone, and ceftazidime can reach therapeutic concentrations when the cerebral spinal fluid (CSF) is inflamed.9 Acute inflammation may also increase beta-lactam penetration of abscesses and pleural, peritoneal, and synovial fluids because of changes in vascular permeability. However, those drugs characterized by high binding to plasma protein will likewise be bound to inflammatory proteins. As response to therapy decreases, resolution of inflammation may decrease distribution. Further, if inflammation does not resolve but progresses, efficacy of beta-lactams is likely to decrease as a result of poor penetrabiltity of lipid tissue. The beta-lactams do not significantly accumulate in phagocytic cells (see Table 7-5). Beta-lactams are concentrated in the urine, enhancing efficacy for cystitis; the clinician must not assume that the high concentration will be achevied in other tissues that also are infected (e.g, nephritis or other urinary tract sites, and even high urinary concentrations may be ineffective in the presence of biofilm (see Chapter 8).
The small Vd that characterizes the unbound beta-lactams contributes to their relatively short half-lives, which often are less than 1 to 4 hours (see Table 7-1). Slow release of highly-protein bound drugs will prolong presence in the plasma. Because beta-lactams in general do not exhibit a long postantibiotic effect, dosing intervals for such drugs may be inconvenient; for critical patients, administering the drug as a constant-rate infusion may be appropriate. The attributes of constant-rate infusion for critical human patients receiving beta-lactams with short half-lives are well recognized and have been demonstrated in animal models.3 The advantages may reflect better steady-state concentrations of drugs in peripheral tissues. Exceptions occur for selected drugs that have a longer half-life, drugs characterized by metabolism to active metabolites, or slowly absorbed or released preparations. The former includes cefpodoxime (4- to 5-hour half-life and 80% to 90% protein bound) and cefovecin (approximate 4- to 5-day half-life and 90% to 99% bound to serum proteins in dogs or cats). Penicillins designed for slow release include slow-release esters (e.g., procaine or benzathine penicillins) or highly protein-bound drugs that may be slowly released from plasma to tissue (e.g., cefovecin). For the latter, generally either absorption or distribution, rather than elimination, half-life is prolonged, resulting in a “flip-flop” model (see Chapter 1). The beta-lactam antibiotics are eliminated, in general, by active tubular secretion in the renal tubules. Clavulanic acid, which is a beta-lactam antibiotic, albeit with poor efficacy by itself, is excreted primarily in the urine of dogs.53 With the exception of hetacillin (no longer available), hepatic metabolism does not play a role in the elimination of the penicillins. Some cephalosporins are eliminated in the urine after deacetylation by the liver, often generate no active metabolites. Examples include cephalothin, cephapirin, cefotaxime, and ceftiofur. Imipenem is degraded to inactive metabolites in the kidney. Reabsorption from the urine is facilitated by an acid urinary pH. Deacetylation of ceftiofur results in an active metabolite; dosing regimens and C&S testing are based on ceftiofur bioactivity.54 Ceftriaxone and cefoperazone are eliminated in the bile in humans and appear to be at least partially eliminated in the bile in dogs.9
Disposition of selected beta-lactam antibiotics
Penicillins
Preparations of penicillin G intended for intramuscular use (e.g., procaine and benzathine) may be prepared as esters, which hydrolyze at variable rates and thus prolong absorption. Procaine penicillin is absorbed for at least 24 hours and benzathine penicillin for approximately 120 hours in some species.9
For the aminopenicillins the oral bioavailability of amoxicillin is greater than that of ampicillin and, unlike ampicillin, is not impaired by the presence of food.5 Clavulanic acid appears to be about 30% to 65% orally bioavailable.15,53,55 The absorption of both amoxicillin and clavulanic acid appears to occur through a saturable process. As with humans, a maximum rate may be reached in dogs at 10 mg/kg and 5 mg/kg, respectively. As the oral dose of amoxicillin reaches 25 mg/kg and clavulanic acid 6.25 mg/kg, amoxicillin may interfere with oral absorption of clavulanic acid. Thus ratios that favor clavulanic acid might be preferred to ensure sufficient absorption.26 Other disposition paramenters of the aminopenicillins are summarized in Table 7-1. The disposition of amoxicillin is such that care should be taken to ensure that underdosing does not occur. This is likely to require administration beyond the label dose (12.5 mg/kg, alone or as clavulanic acid). For treatment of S. pseudintermedius, Stegemann20 has reported an MIC90 of <0.5 μg/mL for amoxicillin–clavulanic acid (see Table 7-9). The MIC50 and MIC90 for amoxicillin–clavulanic acid and E. coli are 2 and 8 μg/mL, respectively. Integration of PK–PD for these organisms indicates that an alternative drug to amoxicillin with or without clavulanic acid might be considered; an exception might occur with UTI because higher drug concentrations will be achieved in the target tissue (urine). However, precaution is also suggested with this approach (see Chapter 8). Note that CLSI has recently re-set breakpoint MIC’s such that many isolates considered susceptible before this change will now be considered resistant.
Carbepenems
Both imipenem and meropenem have been studied in dogs.56,57 Imipenem is minimally protein bound in dogs.56 Peak concentrations (see Table 7-1) occur at 30 minutes for intramuscular and 50 minutes for subcutaneous administration. Extrapolated PDCs after intravenous administration appear to approximate 40 mg/L. The volume of distribution of 0.32 L/kg indicates distribution to extracellular fluid; clearance (CL) is 0.26 L/hr/kg. The elimination half-life varies almost twofold with the route (see Table 7-1). Bioavailability is high after intramuscular or subcutaneous administration.56 In dogs given 5 mg/kg subcutaneously, targeting a 12-hour interval and a T ≥ MIC- (25%) (acceptable for carbapenems), the highest MIC that might be treated is 2 μg/mL. The dose should be increased (approximately 30%) to adjust for ≤70% drug movement from plasma into normal interstitial fluid, particularly if the drug is given subcutaneously.
Meropenem has been studied in dogs after single dose58 and constant-rate infusion.57 As with imipenem, it is minimally (12%) protein bound in dogs. Clearance is 5.6 to 6.5 mL/min/kg. After a dose of 20 mg/kg, mean meropenem (μg/mL) in interstitial fluid (using ultrafiltration techniques) was 24 ± 8 μg/mL. After subcutaneous administration, Cmax (μg/mL) in plasma and interstitial fluid, respectively, were 25 and 11 (ratio = 0.44), and AUCs were 63 and 43 μg ∗ hr/mL, (ratio 0.68) respectively. The better ratio for AUC reflects a longer mean residence time in intracellular fluid (ICF) compared with plasma (2, 4, and 0.9 hours, respectively). Although interstitial fluid concentrations correlated very well with PDC, the doses based on plasma Cmax values might be increased at least 40% when basing dosing on PDC to compensate for differential distribution to extracellular sites of infection. The AUC in interstitial fluid after 20 mg/kg administered intravenously or subcutaneously was 73 μg ∗ hr/mL, and 43 μg ∗ hr/mL, respectively, indicating that intravenous administration might be preferred to subcutaneous administration from a cost standpoint. Note that the time to maximum concentration in interstitial fluid after subcutaneous administration was 3.7 hours (2 hours for intravenous administration), indicating a potential delay in response in the acute situation.58 Based on plasma Cmax after 20 mg/kg administered subcutaneously in dogs, a 12-hour dosing interval, and T > MIC of 25%, the highest MIC that might be treated is 4 μg/mL. If concentrations are used to design the dosing regimen, the highest MIC that could be treated would be 1 μg/mL. Anuric renal failure in humans prolongs the half-life of meropenem fourfold.59
First-generation cephalosporins
Papich et al. described the tissue distribution of cephalexin.59a The ratio of cephalexin Cmax or AUC in plasma versus interstitial fluid were approximately 50% and 57%, respectively. The eliminaton half-life of cephalexin appears to be somewhat route dependent, being almost twice as long as after oral administration (150 minutes) compared with intramuscular or intravenous administration (80 minutes; see Table 7-1). However, Papich et al. reported a much longer half-life of 4.7 + 1 hours in dogs after oral administration of 25 mg/kg.59a Plasma clearance is 2.5 mL/min/kg.60 Bioavailability approximates 60% after either oral or intramuscular administration.60 Oral bioavailability in dogs is affected by the time of day of administration, with Cmax 22% lower in the evening; however, this is more than offset (as a time-dependent drug) by a prolongation of half-life by 50%.61 The oral bioavailability of cephalexin also is affected by pretreatment with metaclopramide, which increases Cmax and AUC, respectively, by 17% and 25%.61 Based on the original half-life reported for cephalexin, targeting T > MIC (50%), the maximum MIC that can be treated using an oral dose of 20 mg/kg is 1 μg/mL. This is equivalent to the MIC50 but less than the MIC90 (2 μg/mL) reported for S. intermedius and cephalexin in dogs.20 A dose of 40 mg/kg is needed for twice-daily dosing, or the interval should be reduced to every 8 hours. Doses would need to be further increased to compensate for differential distribution to tissues or other host or microbial factors. However, if the half-life of 5 hours is used, then twice-daily dosing of cephalexin will result in drug concentration in both plasma and interstitial fluid above the MIC90 for S. intermedius20 for 12 hours or more.59a Note that the MIC50 and MIC90, respectively, for E. coli and cephalexin are 8 and 16,20 indicating that this drug should not be used to treat infections associated with E. coli, including urinary tract infections.
Cephalothin (no longer available in the United States, although it remains the model drug for first-generation cephalosporins at the time of publication) has been studied in dogs after oral administration at 30 mg/kg. Food affects its absorption: Cmax of 45 μg/mL is reduced to 28 μg/mL with food at a Tmax of 1.7 and 2.8 hours, respectively. Elimination half-life is 1.8 and 2.6 hours without and with food, respectively.62
Cefadroxil achieves a Cmax of 35 μg/mL at a Tmax of 20 minutes after an oral dose of 30 mg/kg. Food minimally affects rate or extent of absorption according to one study, but it does increase half-life from 1.7 to 4 hours.
Cefazolin has been studied in two separate groups of canine patients undergoing elective orthopedic procedures. In one study63 clinical canine patients (n = 15) undergoing total hip replacement were administered 22 mg/kg intravenously over 2 minutes at the time of surgical positioning; animals were dosed 2 more times.64 The distribution of the central compartment (Vc; before distribution) was 0.083 ± 0.008 L/kg. The distribution half-life approximated 5 minutes, and the elimination half-life approximated 45 minutes. Tissues from the coxofemoral joint capsule, acetabulum, and femoral cancellous bone were collected from each patient as the site was approached surgically; serum samples were collected at the same general time for each patient. Peak serum concentrations after the first dose were 178 ± μg/mL; tissue (homogenate) concentrations and mean time of collection were as follows: joint capsule, 58 + 5.7 μg/mL at 20 min, acetabulum 157 + 23 at 52 minutes and bone cancellous 227 + 29 at 68 minutes. Peak serum concentrations approximated 178 μg/mL (before distribution) and 119 μg/mL (after distribution). Based on simulations, ideal dosing was suggested to be either 22 mg/kg every 2 hours or 8 mg/kg every hour, to ensure drug concentrations remained above the MIC of Staphylococcus spp. (reported at 2 μg/mL).
Second- and third-generation cephalosporins
Cefuroxime is a second-generation cephalosporin approved for use in humans. Oral administration is in the form of the axetil ester; as a prodrug, desterification occurs before oral absorption. It has been studied both orally and parenterally in Beagles (n=6) as part of a toxicity study.64a Intravenous doses up to 500 mg/kg every 24 hours were well tolerated for 1 week. Jung65 compared cefuroxime in serum to that in cortical tissues in dogs. At approximately 1.25 hours, after 10 and 20 mg/kg administered intravenously, serum concentrations were 12.5 and 28.7 μg/mL, respectively. The elimination half-life was 2.9 hours. Spurling66 reported limited PDCs after oral administration in Beagles. Concentrations (μg/mL) after oral administration of the axetil form at 100 or 400 mg/kg were approximately 28.7 ± 5, and 77 ± 17, respectively.66,67
After a dose of 50 mg/kg ceftriaxone (third generation) was given to apparently healthy dogs, clearance was 3.61 ± 0.8 mL/kg/hr; Tmax occurred at 30 minutes compared with 90 minutes after subcutaneous administration. Pain occurred at the injection site after both intramuscular and subcutaneous administration, whereas intravenous administration was not associated with any adversity.68
Based on studies in dogs after an intravenous dose of 14 mg/kg, cefepime was distributed to a volume of 0.14 l/kg, suggesting that the drug might be protein bound. However, both the elimination half-life and MRT were short at 60 minutes. Clearance was 0.13 ± 0.04 l/kg/hr. The dose necessary to maintain the breakpoint MIC of 8 μg/mL for at least two-thirds of the dosing interval (above 2 μg/mL for the entire interval) (for humans) in dogs was recommended by the author to be 40 mg/kg every 6 hours.28
Ceftazidime is a third-generation drug characterized by an elimination half-life of 0.8 hours in dogs. After subcutaneous injection, Tmax occurs at 1 hour after administration of 30 mg/kg. When given an initial dose of 4.4 mg/kg followed by a constant-rate infusion of 4.1 mg/kg/hr for 36 hours, Cmax at steady state is 22.2 μg/mL. Total body clearance is 0.19 L/kg/hr.69 The MIC90 for clinical isolates (n = 101) of P. aeruginosa was ≤ 4 μg/mL.69 Using 4μg/mL as the basis for a subcutaneous dose of 30 mg/kg, only 3 half-lives can elapse for T = MIC, indicating a 6-hour dosing interval might be appropriate for Pseudomonas spp. Ceftazidime has been studied in cats (n = 5) after intravenous and intramuscular (30 mg/kg) administration.70 After intravenous administration, the Vd was 18 ± 0.04 L/kg; protein binding was not described. Plasma clearance was 0.19 ± 0.08 L/hr/kg, and elimination half-life was 0.77 ± 0.06 hour. After intramuscular administration, bioavailability was 82.47 ± 4.37%, resulting in a Cmax of 89.42 ± 12.15 μg/mL, at a Tmax of approximately 30 minutes. The authors indicated that for an 8- to 12-hour dosing interval, T > MIC would range from 35% to 52% of the dosing interval for intravenous and 48% to 72% for intramuscular administration for isolates with an MIC ≤ 4 μg/mL.
Ceftiofur is a third-generation drug approved for use in dogs for treatment of urinary tract infections. It has been studied at 0.22, 2.2, and 4.4 mg/kg administered subcutaneously in dogs (n = 9).71 PDCs increase proportionately (see Table 7-1). It has a relatively long half-life compared with other cephalosporins, reflecting, in part, its active metabolite. Accordingly, a longer dosing interval is likely to be more reasonable for ceftiofur compared with the first-generation drugs. When administered subcutaneously, peak PDCs (Cmax) were 1.66 ± 0.2, 8.91 ± 6.42, and 27 ± 1 μg/mL at 0.22, 2.2, and 4.4 mg/kg, respectively.71 At the Cmax of approximately 9 μg/mL at a dose of 2.2 mg/kg, targeting T > MIC of 50%, the highest MIC that can be treated at 12-hour intervals is 4 μg/mL. At 4.4 mg/kg administered subcutaneously, the Cmax disproportionately increases to 29 μg/mL, and the highest MIC that could be treated using the same targets is 16 μg/mL, which actually exceeds the MICBP (≥8 μg/mL). Urine concentrations were also reported for ceftiofur bioactivity in the dog. At 24 hours, urine concentrations at 2.2 and 4.4 mg/kg were 8.1 and 29.6 μg/mL, respectively. These concentrations surpassed the the MIC90 for E. coli (4.0 μg/mL) and P. mirabilis (1.0 μg/mL).71
Cefpodoxime is a relatively new third-generation cephalosporin to be approved in dogs for treatment of canine pyoderma. Orally, it is administered as a prodrug, cefpodoxime proxetil, which is desterified in the gastrointestinal tract such that it is absorbed as cefpodoxime. According to the package insert and technical mongraphs, oral bioavailability in dogs is 63% and food does not impair absorption. At 10 mg/kg administered orally, Cmax is variable at 16.4 ± 11 μg/mL, suggesting that dosing should err on the high side for higher MIC; Tmax occurs at 2 to 3 hours. Plasma clearance is 23 mL/hr/kg. Cefpodoxime is excreted largely in the urine with more than 75% excreted as the parent drug. The elimination half-life of 5.6 hours (MRT 9 hours) is longer than that of many beta-lactams; therefore a longer dosing interval is possible (i.e., 12 to 24 hours, depending on the dose and MIC of the infecting microbe). PDCs after 10 mg/kg appear to approximate 1 μg/mL at the end of a 24-hour dosing interval. Thus PDC will stay above the MIC90 for E. coli (0.5), and for S. pseudintermedius (0.5) well beyond the targeted T>MIC of 50% to 75%. (assuming MIC does not change dramatically overtime). However, at 5 mg/kg administered orally in dogs, the highest MIC that can be treated with a 12-hour dosing interval is 4 μg/mL, and with a 24-hour dosing interval, 2 μg/mL, both of which are still above the MIC90 of the approved pathogens. Cefpodoxime is well tolerated in dogs at doses as high as 400 mg/kg/day for 6 months.
Tissue kinetics of cefpodoxime compared with cephalexin have been described in dogs.59a The free and thus diffusible fraction of drug in plasma ranged from 9% to 34%. Maximum drug concentrations after administration of 8.5 mg/kg (single dose) in dogs (n = 6) was (extrapolated from plot) approximately 10 μg/mL free drug (33±7 μg/mL total) in plasma compared with 4.3 +1.9 in interstitial fluid, suggesting less than 50% of the drug in plasma reaches interstitial tissues. Unbound AUC in plasma was not provided, but the disappearance half-life of cefpodoxime from interstitial fluid was twice as long as that from plasma (10 + 3 hours versus 5.6 + 0.9 hours, respectively). The reason for this difference is not clear, although factors that influence diffusibility from tissue into serum might also influence antibacterial activity potentially precluding drug efficacy. Nonetheless, on the basis of these data, interstitial concentrations of cefpodoxime exceeded the MIC90 of S. intermedius and E. coli as reported on the package insert for 24 hours.59a This is in contrast to cephalexin, which is <20% bound to plasma proteins and for which interstitial concentrations exceeded the MIC90 for S. pseudintermedius (as reported by Stegemann20) for 12 hours but did not achieve the MIC90 for E. coli.
Cefovecin (third-generation) is the newest cephalosporin to be approved in dogs at the time of this publication. Its PD and PK have been very well described including either concentrations or bioactivity in interstitial fluid in dogs or cats in part because its disposition is complicated by extensive binding to plasma proteins.20,72,73 Accordingly, care must be taken when designing dosing regimens to base decisions on unbound, rather than total, drug. Based on protein-binding studies (microdialysis) at cefovecin concentrations ranging from 10 to 300 μg/mL in dog plasma, 96% to 98% is bound at concentrations below 100 μg/mL, with the fraction increasing to 72% at 200 μg/mL and 56% at 300 μg/mL. Avid protein-binding results in a slow release and a long elimination half-life of 136 or 133 hours when given intravenously or subcutaneously, respectively. Protein-binding also affects Tmax, which does not occur until 6 hours (based on total drug), and the apparent Vd (0.12 L/kg), which is higher than total blood volume but considerably lower than extraceullar fluid volume. Cmax of unbound, active drug approximates about 5 μg/mL. Predicted unbound concentrations suggest that T > MIC90 of, S. pseudintermedius (0.25 μg/mL) occurs at approximately day 12 after dosing 8 mg/kg subcutaneously; however, this is reduced to day 8 on the basis of the lowest unbound concentration predicted by the 95% confidence interval of 1 μg/mL, which is the more prudent statistic to follow (see package insert). For organisms with MIC ≥ 2 μg/mL (see Table 7-9) (e.g., S. aureus, not an approved indication), T > MIC of mean (predicted) unbound drug at approximately 1 to 2 days; however, if based on the lowest (95% confidence interval) predicted unbound concentrations, 2 μg/mL would not be reached in plasma. In contrast, the MIC90 of Streptococcus canis (an approved indication) is much lower (< 0.06 μg/mL); thus T > MIC exceeds 14 days even when based on the lowest predicted unbound concentration in plasma. The same is true for Pasteurella, the approved indication in cats; the targeted T > MIC90 is not reached until 12 days after treatment.
KEY POINT 7-10
The high fraction of cefovecin binding to plasma proteins prolongs its half-life, but less than 10% of total drug is active.
Studies of unbound cefovecin in tissue have been published using tissue cage models in dogs.72 The studies demonstrate that unbound cefovecin effectively moves from plasma into tissues, as indicated by antibacterial activity against S. pseudintermedius across time). After 8 mg/kg administered subcutaneously in dogs, cefovecin (total) Cmax (total, μg/mL) was 116, 32, and 40 in plasma, transudate, and exudate, respectively, with elimination half-life from transudate similar to that in plasma (147 hours and 136 hours, respectively). Antibacterial activity was detectable in transudate at 4 hours; however, Tmax of cefovecin antibacterial activity did not occur until approximately 2 days. Interestingly, antibacterial activity in transudate actually exceeded antibacterial activity in plasma at all time points after 8 hours and far exceeded it from day 5 forward. Peak antibacterial effects for S. pseudintermedius persisted in transudate until day 10 after injection, with log 2 reduction in CFUs still present at day 18; activity was gone by day 21.
Urine concentrations of cefovecin have been reported in dogs after subcutaneous administration of 8 mg/kg. Peak urine (presumably unbound) concentrations of 66 μg/mL were achieved at 54 hours and approximated 2.9 μg/mL at 18 days.
These data support the use of cefovecin for treatment of susceptible isolates causing urinary tract infections. Cefovecin also is approved for use in cats. Compared with the dog, cevovecin at 8 mg/kg reaches a higher total plasma Cmax; however, it is 99% or more bound to plasma proteins in the cat. Although mean predicted unbound concentrations approximate 10 μg/mL, the predicted variability is great, yielding as little as 0.2 if based on the lower 95% confidence interval (see package insert). The elimination half-life in cats is slightly longer at 166 hours (compared with 136 hours in dogs). The Tmax for plasma is only 2 hours in cats (compared with 6 hours in dog). Peak concentrations of cefovecin in transudate (occurring at 1 day) were approximately 65 μg/mL (compared with approximately 30 μg/mL in dogs). However, 99% of the drug in transudate also was bound, despite the assumption that transudate is protein free. Antibacterial studies were not performed in the transudate of cats and it is not clear what impact, binding has on transudate bioactivity. The concentration of free drug in transudate in cats approximated or exceeded the MIC90 (T > MIC90) P. multocida (0.012 μg/mL; the approved target organism in cats) for 10 days.
The percentage of a radiolabeled dose of cefovecin recovered in urine of dogs (approximately 28%) was only slightly higher than that in feces (24%), indicating that the impact of cefovecin on normal gastrointestinal microbiota may not necessarily be less than that of orally administered drugs. Although urine contamination of feces may have occurred during the collection process, a second peak in PDCs occurs in cats, indicating that enteroheptic circulation may occur.
KEY POINT 7-11
Indiscriminate use of cefovecin must be avoided such that emergence of methicillin-resistant Staphylococcus aureus or methicillin-resistant Staphylococcus intermedius will not be facilitated.
Stegemann20 has reported the PD activity of many anaerobic and aerobic gram-positive and gram-negative (potentially) pathogenic organisms collected from dogs and cats in the United States and Europe. Isolates were tested toward cefovecin, amoxicillin–clavulanic acid, cephalexin, and cefodroxil. The number of isolates in general for each organism exceeded 25, although exceptions exist (e.g, Klebsiella, coryneforms). Acinetobacter and Enterococcus spp. (n ≥ 25) were characaterized by an MIC50 of 16 or higher, well above the Cmax of unbound drug; cefovecin should not be used to treat infections caused by these organisms. For the remaining isolates, integration of PD data with PK data (see Table 7-6) reveals that T > MIC for cefovecin that is superior to the other three drugs studied.
Several considerations should be made when selecting cefovecin as empirical choice for treatment of (presumed) susceptible infections in the dog or cat. First, recognizing the historical relationship between cephalosporins and MRSA might lead to judicious, if not limited, use. Second, not all organisms are equally susceptible to cefovecin. Caution is recommended when using cefovecin for treatment of organisms whose MIC90 ≥ 2 μg/mL. Third, if the decision is made to redose cefovecin, doing so probably should be considered at 2 to 4 days rather than 7 to 14 days for those organisms whose MIC is equal to or greater than 2 μg/mL. The need for redosing might be limited to those patients at risk for persistent and thus resistant infections. A final consideration for cefovecin therapy is the time that must lapse to detectable (4 to 8 hours in plasma or transudate) and peak (2 to 3 days) antibacterial activity of cefovecin in interstitial fluid.72 Cefovecin may not be a wise choice if rapid antibacterial efficacy is needed. This includes the surgical patient. Because of its long time to onset and persistence, cefovecin should not be used for surgical prophylaxis. Fourth, increasingly in human medicine, the duration of antimicrobial therapy is being shortened (e.g., to 5 days or less) for treatment of uncomplicated infections such that emergent resistance might be minimized (see Chapter 6); with cefovecin, “hit hard, get out quick” is not possible.
Drug interactions
The potential synergistic and antagonistic effects of beta-lactams with other antimicrobials was discussed in Chapter 6. Synergisim resulting from enhanced antimicrobial uptake associated with altered cell wall permeability has been demonstrated for a number of antimicrobials. Antagonism should be anticipated with drugs whose impact slows organism growth (i.e., single subunit ribosomal inhibitors); efficacy of beta-lactams may be reduced to bacteriostatic rather than bactericidal effects. An exception may occur for chloramphenicol and selected Enterobacteriaceae (see the discussion of chloramphenicol). As weak acids, the beta-lactams may chemically interact with and inactivate weak bases (see the discussion of aminoglycosides). Inactivation occurs at high concentrations, as might occur with mixing of medications, or potentially, in urine. High protein binding of beta-lactams may result in drug intractions with other highly protein-bound drugs because of competition for protein-binding sites, as is exemplified for cefovecin. Drugs for which higher concentrations have been demonstrated when combined with cefovecin and include carprofen, furosemide, doxycycline, and ketoconazole (PI). It should be anticipated that concurrent use of cefovecin with other highly protein-bound drugs will result in increased free drug concentrations. Beta-lactams will compete for active tubular secretion proteins in the proximal tubule with other organic acids (e.g., penicillins, cephalosporins, nonsteroidal antiinflammatory drugs, sulfonamides, diuretics). The prototypic example drug is probenecid, the combination of which with penicillins was used therapeutically to prolong elimination before implementation of mass production technology. According to the package insert accompanying probenecid, combined use with penicillin results in a twofold to fourfold increase in penicillin drug concentrations.
Adverse Effects
Mammalian cells lack a cell wall; therefore, the beta-lactam antibiotics are very safe. Diarrhea is a common side effect that may reflect altered intestinal microbial flora. Experimentally, co-oral administration with a recombinant beta-lactamase minimally altered fecal microflora but did not negatively influence PDCs.73a Increasing the ratio of amoxicillin to clavulanic acid reduces gastrointestinal upset in humans (but may decrease the absorption of clavulanic acid; see previous discussion), but ratios less than 4:1 can only be accomplished using human-approved drugs, whose equivalent bioavailability has not been established in dogs and cats. The role of probiotics in preventing diarrhea has yet to be established but warrants consideration. Hypersensitivity is an infrequent reaction and occurs less often with cephalosporins. Penicilloic acid (results from breakdown of the beta-lactam ring) is the more likely mediator of hypersensitivity reactions; it is generated from the activity of several beta-lactamase or other enzymes from various sources. Thrombocytopenia has been reported to occur with some members of this class. With the exception of the carbapenems and selected later-generation cephalosporins, the beta-lactams may cause endotoxin release (see Chapter 6), which may prove detrimental to the patient, although relevance to dogs and cats is not clear.74 Penicillins, including imipenem, antagonize gamma-aminobutyric acid type A receptors and may thus lower the seizure threshold.75 The risk may be greater in patients with renal disease.76 Cephalexin can cause false glucosuria.77
Therapeutic Use
The broad spectrum and wide safety margin of the beta-lactam antibiotics lead to their common use. Caution is recommended, however, when they are used to treat complicated infections without the benefit of C&S data. For many drugs, because of the short half-life, Cmax achieved at recommended doses often is not sufficient to allow a convenient dosing interval. Exceptions occur for those cephalosporins with a long half-life or carpabenems for which T > MIC of 25% is acceptable. Resistance develops to beta-lactams relatively rapidly, and the drugs are not characterized by an excellent distribution pattern, with interstitial fluid concentrations of active drug often being 50% to 30% or less of plasma concentrations, depending on the tissue and the drug. Caution should be taken with third- and fourth-generation cephalosporins despite indications of susceptibility on culture data because of inducible ESBLs that require special testing, especially in the presence of a high infecting inoculum. The spectrum of natural penicillins is relatively narrow, particularly when considered in the context of resistance that has emerged through decades of use. Resistance to aminopenicillins also limits their use as empirical drugs of choice. Exceptions might include anaerobic infections. Because the extended penicillins are susceptible to beta-lactamase destruction, combination with a beta-lactamase protector (e.g., ticarcillin and clavulanic acid) or use of imipenem—which is inherently more resistant to beta-lactamase destruction—should be considered. Imipenem or meropenem should be considered before other beta-lactams for treatment of infections associated with endotoxemia because either drug is associated with the least endotoxin release. Constant-rate infusion should be considered for those penicillins with a short half-life to maintain effective concentrations in the critical patient; alternatively, and preferably, carbapenems should be considered in lieu of penicillins. Use of beta-lactamase–protected products should be considered even in uncomplicated infections. Indiscriminate use of beta-lactams, and particularly cephalosporins, should be avoided to minimize the advent of MRSIG.
The first-generation cephalosporins have been excellent first-choice antimicrobials for many infections, including urinary, skin, and respiratory tract infections. Their relative resistance to beta-lactamases produced by Staphylococcus spp. leads to their frequent empirical selection for infections in which Staphylococcus spp. are assumed to be involved. However, their empirical use increasingly is being limited, particularly at dosing regimens currently recommended. Their efficacy against Staphylococcus spp. as well as against many gram-negative organisms leads to their selection for surgical prophylaxis. Cefovecin should not be included in this category because of its long time to antibacterial effect and time to maximum effect and the persistence of drug concentrations well beyond the immediate postoperative period. Of the second-generation cephalosporins, cefoxitin, which is not impacted by ESBL, might be more safely considered for empirical therapy requiring a broad-spectrum antimicrobial and for anaerobic infections. With the exception of P. aeruginosa, cefoxitin is effective against most other organisms. The use of other second-generation and the third-generation cephalosporins is best based on C&S data because the spectra of these drugs are so variable. Caution should accompany use of second- through fourth-generation cephalosporins when based on in vitro data that may not reflect the production of ESBLs. Note also that the (over) use of cephalosporins has been associated with the emergence of multidrug-resistant microorganisms, including MRSA, Enterococcus spp., and P. aeruginosa.25
Beta-lactams should be the first drugs considered for combination antimicrobial therapy (if used at appropriate dosing regimens). Their unique mechanism of action facilitates movement of other drugs into bacteria, which should facilitate efficacy of other antimicrobials. The risk of resistance should also be reduced as antimicrobial movement into the cell is improved. Beta-lactams are combined with drugs effective against gram-negative organisms when broad-spectrum therapy is needed, as in the case of life-threatening infections for which the causative organisms are not known, polymicrobial infections involving anaerobes and aerobes, or gram-positive and gram-negative organisms.
Vancomycin
Vancomycin has had an important role in the treatment of human patients infected with methicillin-resistant staphylococci (see Chapter 6), but the advent of penicillinase-resistant beta-lactams and the incidence of adverse reactions have curtailed its use. Vancomycin is a large glycopeptide with three components, each of which may be responsible for its antimicrobial action on bacterial cell walls (Figure 7-4).78 The D-Ala-D-Alanine precursor of the pentapeptide fits into a pocket formed by the large molecule, sterically interfering with further cell wall elongation. The spectrum of activity of vancomycin is limited to Staphylococcus and Streptococcus spp. and anaerobes (see Table 7-4). Selected Enterococcus, Clostridium, and Corynebacterium spp. are also generally susceptible. With the exception of enterococcal organisms, the effects of vancomycin are generally bactericidal, although they act slowly. As with other cell wall–active antimicrobials, vancomycin exhibits time-dependent killing effects, with efficacy also related to AUC. Resistance has been impeded by the high specificity of the drug. Multiple mutations are required to change the enzymes currently targeted by vancomycin. Resistance that has developed by E. faecalis has resulted from synthesis of a new protein that interferes with vancomycin. More recently, vancomycin-resistant staphylococci have emerged. A strain of vancomycin-intermediate S. aureus (VISA) has been described, the mechanism of which includes thickening of the cell wall, coupled with “clogging” of the cell wall by vancomycin itself. 79
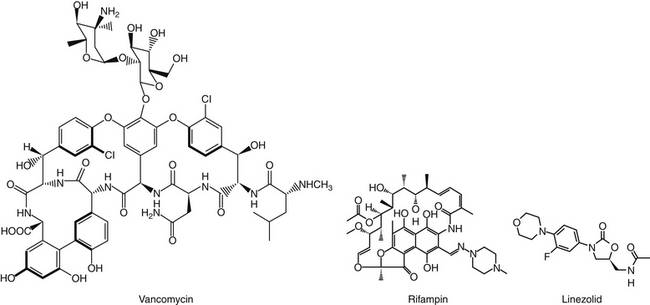
Although vancomycin is available as an oral preparation, this preparation is intended for topical (gastrointestinal) administration, most commonly indicated for pseudomembranous colitis caused by C. difficile. Systemic effects require intravenous administration. Vancomycin is distributed to most body tissues. The exception is the CNS, unless the meninges are inflamed; even then only 30% or less will penetrate. It is renally eliminated; drug concentrations may become toxic if doses are not modified for the patient with renal disease. The risk of nephrotoxicity is increased dramatically if the drug is given in combination with another nephrotoxic drug. Hypersensitivity in human patients warrants slow (60-minute) intravenous infusion of drug diluted in fluid. Ototoxicity has been reported in humans when concentrations reach 60 to 100 μg/mL.80 Its use for veterinary patients should be limited to treatment of organisms resistant to other drugs as based on C&S data.
Teicoplanin
Teicoplanin is a mixture of several molecules (teicoplanins A2 1-5). The molecules compose a fused glycopeptide core ring structure (teicoplanin) to which are attached two carbohydrates (differing from those in vancomycin), mannose and n-acetylglycosamine, and an acyl (fatty acid). It is the latter structure that confers better lipid solubility compared with vancomyin. Its mechanism of action and impact on bacterial killing and spectrum is similar to those of vancomycin. Its use has largely been replaced by vancomycin or daptomycin.
Fosfomycin
Fosfomycin is a phosphonic acid that contains a carbon–phosphorous bond (see Figure 7-12). It is a natural antibiotic produced by Streptomyces fradiae. Its in vitro spectrum is broad, and it expresses potential efficacy against isolates expressing multidrug resistance, including E. coli and gram-positive organisms. As a phosphoenolpyruvate analog, fosfomycin irreversibly inhibits phosphoenol pyruvate transferase, an enzyme that catalyzes the first step of cell wall peptidoglycan synthesis of microbial cell walls.81 As a cell wall inhibitor, fosfomycin is bactericidal when present at the site of infection at therapeutic concentrations. Its irreversible nature contributes to a concentration-dependent effect. Fosfomycin exhibits in vitro activity against a broad range of gram-positive and gram-negative aerobic microorganisms associated with uncomplicated urinary tract infections. The MIC breakpoints reported for humans are 64 (S), 28 Intermediate (I), and 256 (R). Although its mechanism of action is similar to that of the beta-lactams, fosfomycin is not susceptible to destruction by any class of beta-lactamases. Rather, resistance to fosfomycin, which is unusual, reflects the FosX or FosA enzyme, which hydrolyzes the drug in a manner similar to that of glutathione S-transferases. The gene for this protein is chromosomally mediated. Thus when resistance does occur, it is usually only toward fosfomycin (single drug resistance) with cross-resistance not occurring between fosfomycin and other classes of antimicrobial agents. Therefore resistance is not associated with multidrug resistance.81 Further, compared with susceptible strains, fosfomycin-resistant mutants are impaired, exhibiting poorer growth rates and reduced adherence to uroepithelial cells. Fosfomycin appears to reduce bacterial adherence to uroepithelial cells, and decreased adherence is facilitated by N–acetylcystein82 and urinary catheters.83 Studies in humans have demonstrated that fosfomycin distributes well to soft tissues, reaching therapeutic breakpoints.84 Other attributes of fosfomycin that support its use for treatment of E. coli urinary tract infections include renal excretion, synergistic interaction with several other classes of antimicrobials,86 and preparation as a 3-g sachet (granules), which is mixed with water to orally deliver approximately 40 mg/kg (in humans).
The disposition of fosfomycin disodium (pure substrate) has been described in dogs (n = 8)88 after intravenous, intramuscular, subcutaneous, and oral administration at both 40 and 80 mg/kg day for 3 days. Plasma protein binding was negligible; drug concentrations increased in a dose-dependent manner and did not change during the study period, including across each 3-day treatment period. At 40 mg/kg, peak PDCs (µg/mL) were as follows: 51.8 ± 3.4 (extrapolated peak PDC; Co, intravenous) and 5.4 ± 0.04 (oral); and at 80 mg/kg, 113 ± 12 (Co, intravenous) and 10.8 ± 0.5 (oral). Oral bioavailability (F) was 30%. Clearance was 14.9 ± 1.26 mL/kg/hr, elimination half-life was 1.3 ± 0.06 hours, and mean residence time was 1.62 ± 0.4 and 5.2 ± 0.7 (oral).
The PD and PK of fosfomycin have also been studied by the author. The distribution MIC for fosfomycin for clinical E. coli isolates, regardless of the presence of multidrug resistance, appears to be well below the susceptible breakpoint (≤ 64 μg/mL) for fosfomycin. In more than 100 clinical isolates collected from dogs and cats, the MIC range was 0.25 to 4 μg/mL; the MIC50 and MIC90 were, respectively, 1 and 1.5 μg/mL. Fosfomycin tromethamine was administered as a single oral dose of 80 mg/kg. After oral administration, Cmax, elimination half-life and mean residence time were 66 ± 21 (μg/mL), 2.5 ± 1.09 hours and 5.1 ± 1.7, hours, respectively. Drug was detected at concentrations exceeding the MIC90 of fosfomycin for multidrug-resistant E. coli (1.5 μg/mL) until 7 (2.5 μg/mL) and 12 hours (9 μg/mL) after intravenous and oral administration, respectively. Drug was present in urine at concentrations above 10 μcg/mL at 24 hr post dosing. Gastrointestinal upset manifesting as mild to moderate diarrhea was observed in 4 of the 12 dogs. Food decreased oral bioavailability: without food, 109 ± 31% (95% confidence interal CI: 84%-135%) and with food, 66 ± 16% (95% CI: 52%-79%). Gender had no impact on oral bioavailability. Kill studies in our laboratory indicate that for treatment of E. coli, the drug is not concentration dependent, as is suggested by other studies that indicate both time- and concentration-dependent effects.88,89 Further studies are warranted to establish efficacy for treatment of multidrug-resistant–associated urinary tract infections.
Although fosfomycin is appealing for treatment of urinary tract infections and potentially other infections caused by multidrug-resistant isolates, differences in bioavailability (oral) among different fosfomycin salts necessitates that PK be the basis, particularly of oral dosing regimens in the dog. Its efficacy appears to be both time and concentration dependent; if the latter, this should facilitate efficacy despite the short-half-life of the drug.88 The drug appears to interact in an additive to synergistic fashion with a number of other antimicrobials.
Drugs that Target Ribosomes (Bactericidal)
Aminoglycosides
Despite their potential nephrotoxicity, aminoglycosides remain the cornerstone of aerobic gram-negative therapy in many complicated or serious infections. Minor differences in the chemical structures of these drugs lead to differences in efficacy and toxicity. Clinically useful aminoglycosides include neomycin, gentamicin, amikacin, netilimicin, streptomycin (or dihydrostreptomycin), and tobramycin.
Structure–Activity Relationship
Aminoglycoside compounds are composed of an amino sugar linked through glycosidic bonds to an aminocyclitol.5,90 They vary in the amino sugar and the specific number and location of the amine groups (Figure 7-5). The different name endings indicate the microbe of origin for the natural antibiotic: The suffix “icin” (e.g., gentamicin) originates from Micromonospora sp., whereas the “mycin” suffix (e.g., tobramycin) derives from Streptomyces. Amikacin is a semisynthetic derivative of kanamycin, and netilmicin, a semisynthetic derivative of sisomicin. Tobramycin is most similar to gentamicin in both spectrum and toxicity. The aminoglycosides are polycationic, depending on the number of amine groups. Kanamycin and gentamicin have two amino sugars, whereas neomycin has three amino sugars. The amine group of gentamicins is variably methylated, yielding three different gentamicins. Streptomycin has a different aminocyclitol sugar compared with the other drugs, whereas spectinomycin is an aminocyclitol that does not contain any amino sugars.
Mechanism of Action
Aminoglycosides target bacterial ribosomes (Figure 7-6). The drugs enter gram-negative organisms initially through porins in the lipopolysaccharide layer. Subsequent penetration of aerobic bacteria at the level of the cell membrane appears to occur in three binding stages: the negatively charged moieties of phospholipids are first ionically attracted and bound by the positive moieties of the drug, followed by the lipopolysaccharides and finally membrane proteins. Energy-dependent uptake follows binding to lipopolysaccharides. An acidic environment external to the cell membrane has been associated with increased transport, perhaps because of an increase in the membrane potential differential. However, a lower pH more commonly has been associated with increased membrane resistance; the disparity may reflect the different molecules of each aminoglycoside. An alkaline environment consistently appears to facilitate transport as does movement of cations out of the cell membrane. Uptake depends on a membrane-bound respiratory protein that is lacking in anaerobic organisms, leading to inherent resistance. The system also is deficient in facultative anaerobes such as Enterococcus spp. Active transport depends on a high oxygen tension in the environment rendering obligate anaerobes inherently resistant, and facultative anaerobes resistant in an anaerobic environment.91 Cations such as calcium and magnesium in the lipopolysaccharide covering and cell membrane repel the aminoglycosides, impairing transport into bacterial cells (and renal tubular cells). Removal of calcium (e.g., through use of chelating agents such as ethylenediaminetetraacetic acid [EDTA]) or a decrease in serum calcium (i.e., hypocalcemia) facilitates aminoglycoside movement into the cell.5,90 Hyperosmolarity and decreased pH also decrease drug movement into the cell.80
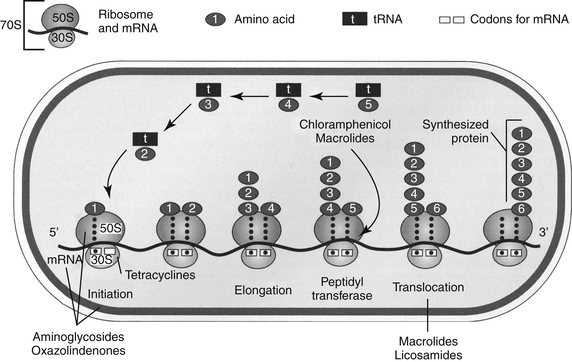
Figure 7-6 The mechanism of action of ribosomal inhibitors. The bacterial ribosome is a complex structure, composed of three RNA molecules (peptidyl and aminoacyl tRNAs and mRNA) and more than 50 proteins. The ribosome is formed as two subunits, 30S (including a 16S portion) and 50S (including a 5S portion; S referring to sedimentation rate), which join when protein synthesis is initiated and separate when completed. The process of initiation begins by the formation of a functional ribosome. The 30S subunit complexes with mRNA (which codes tRNA synthesis) and forms an initiation complex consisting of tRNA, the first amino acid (methionine), and three initiation factors (IF 1-3), one of which is an energy source, GTP. The initiation complex joins the 50S subunit, forming the (mature) 70S ribosome; it is the mature 70S ribosome that initiates protein synthesis. The mature ribosome is composed of an “A” or amino acid site (30S), the “P” or peptidyl site (50S), which contains a peptidyl transferase center; and an E or exit site adjacent to the P site. The aminoacyl tRNA carrying the amino acid binds to the A site, which then complexes to an elongation factor. Release of energy by GTP causes a conformational change or contracting motion, and the the peptide forming at the P site joins the amino acid at the A site. A nucleophilic attack initiated by the aminoacyl tRNA results in bonding of the amino acid to the growing peptide (transpeptidation); the growing peptide is then translocated to the P site. The elongation step is repeated until protein synthesis is completed.278 The aminoglycosides inhibit ribosomal initiation (as the 30S subunit becomes activated to 70S); binding is irreversible, contributing to a bactericidal effect. Tetracyclines bind to the 16S portion of the 30S subunit of ribosomes, preventing the translocation of the amino acid from transfer RNA (tRNA) to the codon of messenger RNA (mRNA). Chloramphenicol and erythromycin prevent the transfer of peptides by binding to the 50S subunit. Erythromycin and clindamycin prevent translocation of the peptide. Drugs that act at the same site should not be used in combination.
KEY POINT 7-12
Effiacy of aminoglycosides is dependent on active transport. Accordingly, efficacy is markedly reduced to absent in an anaerobic environment, and anaerobes are not susceptible.
Once inside the cell, aminoglycosides bind to ribosomes (see Figure 7-6). Although their mechanism of action is not completely understood, aminoglycoside antimicrobials bind to the 30S ribosomal subunit, which, as the initiator of protein synthesis, plays a crucial role in providing high-fidelity translation of genetic material.92 Binding is so effective that polyribosome formation is prevented, and protein synthesis is impaired because of altered synthesis and misreading. Thus, in contrast to most bacteriostatic drugs, which bind to 50S ribosomes, the aminoglycosides are more likely to achieve bactericidal concentrations safely in animals. Although only a small amount of aminoglycoside appears to penetrate the cell membrane, the initial impact on ribosomes is sufficient to alter cell membrane proteins and permeability such that additional drug is able to penetrate the cell. Irreversible saturation of the ribosomes results in cell death and accounts for the concentration-dependent killing effects of the drugs; the irreversible nature of binding contributes to bactericidal effects.92 Aminoglycosides are rapidly bactericidal, with efficacy and the postantibiotic effect of aminoglycosides correlating to peak concentrations, which ideally should be at least 10 times the MIC of the target organism.93-97 Drugs that target the 50S ribosomal unit (e.g., chloramphenhicol, linezolid) may interfere with intracellular movement and thus rapid killing effects of aminoglycosides.94 Because toxicity of aminoglycosides is correlated with trough concentrations (later discussed as adverse effects of aminoglycosides), treatment is implemented with once-daily therapy at high doses. This approach is both clinically85,97,98 and experimentally95,96,99 equal to or more efficacious and safer than the traditional frequency of administration (i.e., two to three times daily). The appropriateness of this dosing method may vary with the organism and the immunocompetence of the patient.
Spectrum of Activity
The spectrum of activity of aminoglycosides (see Tables 7-2 through 7-4, 7-9 and 7-10) includes most aerobic gram-negative bacteria, particularly E. coli, K. pneumoniae, P. aeruginosa, Proteus spp. and Serratia spp.5,16,80,91,101 Newer aminoglycosides such as gentamicin, tobramycin, amikacin, and netilmicin have a wider spectrum compared with older compounds such as streptomycin and kanamycin. These drugs are also effective against selective aerobic gram-positive organisms, most notably Staphylococcus spp. However, they generally should not be used as sole agents against gram-positive organisms. Synergism against gram-positive isolates has been demonstrated when combined with penicillins or vancomycin.80 Aminoglycoside activity against Enterococci spp. is adequate only when used synergistically with a cell wall–active antibiotic, such as beta-lactams or vancomycin.92 Among the aminoglyocsides, based on clinical isolates in humans, netilimicin has the lowest MIC90 toward Enterococcus spp. and, along with tobramycin, Staphylococcus spp. Of the aminoglycosides most commonly used in dogs and cats, gentamicin has a much lower MIC90 than amikacin toward Staphylococcus spp., even accounting for differences in breakpoint MICs. Gentamicin is preferred to amikacin for treatment of Staphylococcus infections, based on a rabbit model of endocarditis.101 Further, a recent comparison of activity of 1000 isolates also found gentamicin to be more effective than amikacin toward Staphylococcus spp.102 In this same report, the authors noted that gentamicin also had lower MIC toward many enterobacteriacea but that amikacin achieved higher serum concentrations (and has a higher breakpoint MIC), thus negating this benefit.102 Gentamicin and tobramycin have a very similar spectrum toward gram-negative aerobes. They and amikacin are effective against P. aeruginosa, Proteus spp. and Serratia spp. Gentamicin is the least effective of the three against P. aeruginosa but most effective against Serratia marcescens.92 Amikacin generally is most effective against P. aeruginosa. With the exception of Pseudomonas species (usually an obligate aerobe, although exceptions have been reported), these organisms are facultative anaerobes and, if cultured aerobically from an anaerobic environment, may fail to respond to aminoglycoside therapy in the patient. The aminoglycosides are also effective against Nocardia and selected atypical mycobacterial organisms.
Resistance
Besides the inherent resistance of anaerobic organisms (owing to decreased active transport), resistance to aminoglycosides is acquired as a result of decreased cell entry; altered porin size in the gram-negative organism is less important.90 Resistance also includes altered ribosomal structure (uncommon except for Enterococcus spp.) and, more commonly, destruction by microbial enzymes inside the cell. Resistance to gentamicin involving altered ribosomal structure by Enterococcus spp. generally affects all aminoglycosides, as well as penicillins and vancomycin. An exception is streptomycin, which is destroyed by a different enzyme and may remain effective toward Enterococcus.90
Enzymatic destruction is the most important mechanism of acquired resistance in clinical isolates, in part because it is acquired through conjugative plasmids. Resistance reflects enzyme modification of the amino or hydroxyl groups of the drugs. The modified drug can no longer bind to ribosomes. Impact on efficacy varies among the different aminoglycosides. For example, target sites of destruction by the enzymes are harder to reach with amikacin. Consequently, amikacin is less vulnerable to resistance than are other aminoglycosides and is frequently effective toward otherwise multidrug-resistant isolates.5,80,90 At least three different enzyme classes exist, classified by phenotypes as to phosphotransferases, acetyltransferases, and nucleotidyltransferases. Among the aminoglycosides used clinically in veterinary medicine, gentamicin and kanamycin more commonly act as substrates for phosphotransferases and acetyltransferases, wheraeas amikacin and tobramycin are more common substrates for the nucleotidyltransferases. Of the three enzymes, the phosphotransferases are more likely to be associated with high-level resistance.
Resistance to aminoglycosides by Staphylococcus spp. reflects chromosomal mutations in transmembrane potentials and thus drug uptake. Mutational resistance caused by changes in ribosome binding sites has been identified primarily against streptomycin, the use of which is limited. However, whereas the four gram-negative organisms most commonly causing (blood) infection in humans (Pseudomonas, Klebsiella, E. coli, and Enterobacter) remain susceptible (>95%) to the greatest number of aminoglycosides, up to 40% of S. aureus organisms are resistant to gentamicin. Current investigations are attempting to identify the mechanism by which enzymatic destruction of aminoglycosides might be inhibited, much the same as beta-lactamases have been used to prevent beta-lactam destruction.92 Low-level resistance caused by multidrug efflux mechanisms has been identified in P. aeruginosa, Burkholderia sp. (previously Pseudomonas), Acinetobacter, spp. and E. coli.92
KEY POINT 7-15
Enzymatic destruction of aminoglycosides is increasingly limiting efficacy, particularly for gentamicin toward Staphylococus spp.
Adaptive resistance has been described for the aminoglycosides (see Chapter 6). In humans up to 40 hours may need to elapse between doses for full bacterial susceptibility to commence.103 This phenomenon supports the once-daily use of the aminoglycosides.
Pharmacokinetics
The aminoglycosides are polar, water-soluble weak bases, and as such they are poorly absorbed from the gastrointestinal tract. An exception might occur in very young animals that are still absorbing colostrum or in the presence of inflammatory gastrointestinal disease.90,91 Kanamycin, which is structurally very similar to amikacin, behaves similarly to amikacin.91 Aminoglycosides are administered topically (including aerosolization and incorporation in beads) or parenterally but can be used orally for local bacterial cleansing of the gastrointestinal tract. However, absorption from body cavities may be sufficiently rapid to cause neuromuscular blockade.90 Absorption will also occur when applied topically to large wounds with subcutaneous exposure; absorption may be sufficient to cause toxicity.104
Although aminoglycosides are distributed to extracellular fluids, their penetration into many tissues is considered poor (see Table 7-5). However, therapeutic concentrations can be attained in synovia and in pleural and peritoneal fluid, particularly if membranes are inflamed. Penetration of bronchial secretions is generally better than that of many beta-lactam antibiotics. However, therapeutic concentrations generally are not attained in CSF, ocular fluids, bile, milk, and prostatic secretions. Further, killing of intracellular (e.g., Enterobacter spp.) spp.) organisms may be limited.94a Intrathecal administration has been indicated for CNS infections, but the advent of third- and fourth-generation cephalsoporins and carbapenems has preempted this need.90 Aminoglycosides are actively accumulated by renal tubular cells, but this may be of more relevance to toxicity rather than efficacy. In addition to anaerobic environments, the efficacy of aminoglycosides is reduced in an acidic environment such as might occur in the urine, ascitic fluid, and abscesses.
KEY POINT 7-16
As water-soluble weak bases, aminoglycosides are not orally absorbed, do not penetrate tissues well, and are excreted in proportion to the glomerular filtration rate.
Drug elimination half-life of the aminoglycosides is generally less than 2 to 4 hours (see Table 7-1). The aminoglycosides are eliminated by glomerular filtration, which is a relatively inefficient process. Drug accumulates in acidic urine, and alkaline urine pH facilitates reabsorption. Urine concentrations have been described for selected aminoglycosides in dogs.105 Dosing of gentamicin (6.6 mg/kg), tobramycin (3 mg/kg), and amikacin (15 mg/kg) subcutaneously in divided doses at 8-hour intervals (not recommended) for five consecutive doses and kanamycin at 11 mg/kg at 12-hour intervals (also not recommended) for 4 doses generated mean interval urine concentrations (μg/mL) of 107 ± 33 for gentamicin: 66 ± 39 for tobramycin, 342 ± 153 for amikacin, and 473 ± 306 for kanamycin.105
The disposition of aminoglycosides varies somewhat among animals, primarily because of differences in glomerular filtration rates. Elimination is slower in larger animals because glomerular filtration rate decreases with body size; this may be offset by differences in Vd. Dosing based on metabolic rate normalizes the rate of elimination and might be considered in patients predisposed to aminoglycoside nephrotoxicity, although estimates of glomerular filtration rate based on extracellular fluid volume may be more accurate.106
A number of investigators have described the disposition of aminoglycosides in dogs or cats (Table 7-1). Gentamicin has been studied in dogs by multiple investigators. Riviere107 described the disposition in 5-month-old Beagles (n = 11). Clearance was 4.1 ± 0.6 mL/min∗kg and Vd (area) was 0.4 ± 0.04L/kg. Elimination half-life was 61 ± 8 minutes. Wilson108 studied gentamicin (3 mg/kg) in dogs (n = 6) after intravenous, intramuscular, and subcutaneous administration. After intravenous administration, clearance was 2.29 ± 0.48 mL/min∗kg and Vdss was 0.172 ± 0.025. Bioavailability approximated 95% for both intramuscular and subcutaneous routes, yielding a Cmax of approximately 10 μg/mL for either route, with time to peak concentration for intramuscular administration being 27 minutes compared with 43 minutes for subcutaneous administration. The elimination half-life was 54 ± 15 minutes. Albarellos204 studied gentamicin after intramuscular administration of 6 mg/kg for 5 days. After day 1, assuming 100% bioavailability, clearance was 1.24 ± 0.6 mL/min∗kg (1.10 ± 0.4 by day 5), and Vd (area) was 0.084 L/kg (0.1 ± 0.05 day 5). Mean residence time was 1.48 ± 0.54 hour (1.77 ± 0.48 by day 5; significantly prolonged) and half-life ranged from 0.55 to 1.46 hours. For IV administration, the Vdss after intravenous administration was 0.23±0.04 L/kg and clearance was 2.64 ± 0.24 mL/min∗kg.
Jernigan and coworkers100 have described the disposition of several aminoglycosides in cats (see Table 7-1). After intravenous administration of gentamicin (3 mg/kg) in cats (n = 6), Vdss was 0.12 ± 0.02 l/kg and clearance was 1.1 ± 0.25 mL/kg∗min. Bioavailability after subcutaneous administration was 83 ± 14.8%. Gentamicin was also studied in cats (n = 6) after intravenous, intramuscular, and subcutaneous administration of 5 mg/kg.110 After intravenous administration, Vdss was 0.14 ± 0.02 L/kg and clearance was 1.38 ± 0.35 mL/min∗kg; mean residence time was 1.8 ± 43 hour. Bioavailability after intramuscular and subcutaneous administration was 67.8 and 76.2%, respectively. Tobramycin was studied in six cats after 5 mg/kg.111 After intravenous administration, Vdss was 0.19 ± 0.03 l/kg and clearance was 2.21 ± 0.6 mL/min∗kg; mean residence time was 90 ± 16 minutes. Bioavailability after intramuscular and subcutaneous administration was 103% and 99% respectively; bioavailability was also measured at greater than 150% for both routes in one set of studies, perhaps indicating decreased clearance owing to nephrotoxicity. Finally, amikacin (5 mg/kg) was studied in cats (n = 6) after intravenous, intramuscular, and subcutaneous administration.112 After intravenous administration, Vdss was 0.17 ± 0.02 L/kg, and clearance was 1.46 ± 0.26 mL/min∗kg; mean residence time was 118 ± 14 minutes. Bioavailability after intramuscular and subcutaneous administration was 95 ± 20% and 12.3 ± 33%, respectively.
Disposition of the aminoglycosides appears to vary among breeds. Kukanich113 has compared the PK of amikacin (10 mg/kg, administered intravenously) in Greyhounds and Beagles (n = 6 each). The volume of distribution (L/kg) was smaller (0.18 versus 0.23), but clearance was less (2.1 versus 3.3 mL∗kg/min) in Greyhounds, thus elimination half-life did not differ (0.8 and 0.9 hour for Greyhounds and Beagles, respectively). The bioavailability of amikacin in Greyhounds after subcutaneous administration was approximately 90%. Although extrapolated time 0 PDC was reported for both species after intravenous administration (86 and 70 μg/mL, respectively, for Greyhounds and Beagles), this is not an appropriate target on which to base Cmax/MIC (i.e, the Cmax should be measured after distribution has occurred). However, compartmental analysis yielded concentrations extrapolated from the terminal curve (presumably reflecting postdistributional concentration; see Table 7-1). On the basis of these data and a target Cmax/MIC of 8 (rather than 10), the respective subcutaneous doses (mg/kg) of amikacin recommended by the authors to target an MIC of 2, 4, and 8 μg/mL, respectively, were for the Greyhound 6, 12, and 24 and for the Beagle, 11.5, 22, and 40.
The influence of endotoxemia on gentamicin disposition has been described in cats.114,115 Elimination half-life was shorter (77 ± 13 minutes before and 65 ± 14 after), but this change is not likely to be significant, in part because neither Vdss nor clearance was significantly different.
The disposition of aminoglycosides also differs among ages. PDCs are less in the neonate and pediatric patient because greater total body water and extracellular fluid compartments increase the Vd of the drugs from 0.25 to 0.35 L/kg (see Table 7-1). Renal clearance of aminoglycosides is less. Thus for young animals the dose of aminoglycosides should be increased; although elimination half-life may be longer, the current use of a 24-hour interval should preclude the need to lengthen it further in the pediatric patient. Disposition is also altered by disease. Dehydration and obesity increase PDCs, which may be of benefit for these concentration-dependent drugs. Intensive fluid therapy or other syndromes associated with accumulation of fluid at a site to which aminoglycosides distribute and endotoxemia decrease plasma aminoglycoside concentrations.91 Ascites also will increase the Vd and half-life of aminoglyocosides.116 Aminoglycosides may accumulate and cause nephrotoxicity in the fetus and should not be used during pregnancy.90 Elimination is impaired in the patient with renal disease; dosing regimens are usually modified by lengthening the interval on the basis of serum creatinine concentration (see the section on therapeutic use).
Adverse Effects
The aminoglycosides induce a glomerular and (principally) tubular nephrotoxicity; however, because of the regenerative capacity of the proximal tubule, toxicity is largely reversible unless allowed to progress to an irreversible state (i.e., destruction of basement membrane). Toxicity results from active uptake into the renal tubular cell and disruption of cellular lysosomes (Figure 7-7). Impaired cellular respiration and synthesis of protective vasodilatory renal prostaglandins by the aminoglycoside may be important in the development of nephrotoxicity.
Figure 7-7 Nephrotoxicity of aminoglycosides occurs primarily in the proximal tubular cells. The cationic charge of the drugs is attracted to the anionic charge of the phosopholipids in the cell membrane. The drug is actively accumulated in the cell by pinocytosis. Inside the cell the drugs accumulate in lysozymes, causing lysosomal disruption and release of myeloid bodies. Intracellular movement into lysozymes also limits intracellular efficacy. Mitochondrial function is also impaired. The effects of prostaglandin on renal blood flow may contribute to the toxicity of aminoglycosides. A number of factors increase or decrease the risk of toxicity (see text).
KEY POINT 7-17
Aminoglycoside nephrotoxicity can be minimized if kidneys are allowed a drug-free period such that drug that has been actively accumulated in the kidney can be eliminated.
Reversible renal impairment occurs in up to 25% to 55% of human patients receiving aminoglycosides for more than 3 days, although the better-designed studies indicate a rate of 10% to 20%.90,117 In humans aminoglycoside-induced nephrotoxicity is defined as an increase in serum creatinine concentration of 0.5 mg/dL in patients for which baseline concentration is < 3 mg/dL, or an increase in 1 mg/dL if the baseline is at or above 3 mg/dL.117
The exact mechanism of aminoglycoside-induced nephrotoxicity is not known. Toxicity begins as the anionic phospholipids of the renal tubular cell membranes attract and bind the cationically charged drugs. The relative nephrotoxicity of the different aminoglycosides reflects differences in their renal accumulation.90 Nephrotoxicity may be related to the number of positively charged amino groups on the drugs; hence, neomycin is expected to be among the most nephrotoxic of the drugs.5 An acidic local pH may enhance uptake by ionizing the aminoglycoside and thus increase the risk of toxicity. Of the clinically used aminoglycosides, neomycin is the most nephrotoxic and dihydrostreptomycin, the least. The nephrotoxicities of the other aminoglycosides are between these two extremes. Several studies have compared tobramycin and gentamicin (the latter is more concentrated), but controlled clinical trials in humans have failed to find a clinical difference in the nephrotoxicity potential between the two.90 Studies comparing the nephrotoxic potential of amikacin with other aminolgycosides (but not gentamicin) also have been inconclusive.90 A number of drugs increase the risk of nephrotoxicity (see the section on drug interactions).
The attraction of aminoglycoside cations to the renal tubular cell membrane can be competitively inhibited by divalent (e.g., magnesium or calcium) cations (e.g., ethylenediaminetetraacetic acid [EDTA]) or decreased in an alkaline urine (un-ionizing amine groups). Hypocalcemia or hypomagnesemia may increase the risk of aminoglycoside toxicity; in contrast, dietary calcium loading may protect against toxicity. Uptake of aminoglycosides also may be related to the amount of phosphatidylinositol in the cell membrane; the amount is disproportionately higher in renal cortex and cochlear tissues.91
Once the renal tubular cells are entered, aminoglycosides are then actively accumulated in the cell by pinocytosis; intracellular accumulation may result in concentrations greater than fiftyfold of that in plasma. Inside renal tubular cells, probably in part because of ion trapping, aminoglycosides are sequestered in lysosomes, which subsequently appear morphologically as myeloid bodies. The drugs are slowly eliminated in the urine as myeloid bodies, which contain drug, RNA, and DNA after the tubular cell dies.
The cause of tubular cell death induced by aminoglycosides remains unclear, although a number of cellular functions (in addition to lysosomal damage) are impaired; examples include phospholipases, sphingomyelinases, and ATPases. Mitochondrial respiration is decreased, impairing energy resources of the cell. Again, this may reflect interaction between the drug and mitochondrial cell membrane. Proximal tubular permeability may be impaired both directly as drugs interact with the cell membrane and indirectly as a result of impaired Na+,K+-ATPase activity. Aminoglycosides also alter glomerular function, perhaps by reducing the number and size of glomerular endothelial cells.91 Finally, phospholipases important for renal prostaglandin synthesis are among the enzymes impaired by aminoglycosides. The initial decrease in glomerular filtration that accompanies aminoglycoside therapy may reflect the inability of the kidney to vasodilate in response to vasoconstrictor actions such as that signaled by angiotensin II.91 This may reflect altered prostaglandin synthesis. As glomerular filtration declines, so may clearance of the aminoglycoside, thus increasing the risk of toxicity.90
The half-life of renal cortical aminoglycosides is approximately 100 hours. This and the fact that a critical aminoglycoside concentration must be reached before nephrotoxicity emerges generally preclude renal cortical nephrotoxicity before the first 3 days of therapy (Box 7-1).117 No study has demonstrated a threshold of dosing or interval that ensures or predicts toxicity. Studies that have focused on aminoglycoside toxicity in dogs and cats have used dosing interval that ranges from 12 hours to constant intravenous infusion. Studies regarding aminoglycoside nephrotoxicity in cats have focused on doses of 35 mg/kg or more at intervals of 12 hours or less.91 A bimodal course of aminoglycoside-induced nephrotoxicity has been described in the dog, with an initial subclinical phase characterized by a concentrating defect and an azotemic phase; different disease states might be predictable based on changes in pharmacokinetics.108 Under experimental conditions, gentamicin at 4 mg/kg every 12 hours in dogs changes urine osmolarity within 7 days and an increase in serum creatinine by 17 days. Urinary prostaglandin E activity decreases before azotemia, which may be responsible for the state of nephrogenic diabetes insipidus. Whereas a single dose of 15 mg/kg gentamicin was associated with subclinical and morphologic changes in the kidney of young Beagles,119 higher doses of 30 mg/kg administered at 8-hour intervals in dogs result in increases in urine gamma-glutamyltransferase within 2 days and serum creatinine within 9 to 12 days. Interestingly, a study that describes the disposition of gentamicins C1, C1a, and C2 in dogs found clearance of C1 to be twice as fast and Vd to be twice as high as for the other two gentamicins.120 The investigators found that the renal binding of C1 is likely to be greater, suggesting that it is more likely to be nephrotoxic compared with C1a and C2. Gentamicin (3 mg/kg) administered intravenously every 8 hours for 5 days to cats (n = 6) was not associated with changes in serum or histologic indicators of renal or vestibular dysfunction.121 Endotoxemia appears to cause more gentamicin renal medullary accumulation in cats but does not appear to be associated with increased renal pathology.115 Tobramycin was associated with increased serum creatinine and/or BUN in 9 of 12 cats dosed twice with tobramycin despite washout periods,111 suggesting that it may be more nephrotoxic than other aminoglycosides, at least in cats.
Box 7-1 Minimization of Aminoglycoside Nephrotoxicity
No indicator of renal damage induced by the aminoglycosides is sufficiently sensitive to prevent damage; indeed, damage will continue beyond detection with current methods. Changes in urine osmolality or sodium fractional clearance typical of the initial subclinical phase may detect a concentrating defect. However, this should be preceded by a release of renal tubular enzymes such as gamma-glutamyltransferase into urine. Measurement of the enzyme has been used experimentally to measure aminoglycoside toxicity. The enzymes increase within several days after damage has begun. However, 24-hour sample collection for these procedures is impractical. Measurement of the urine creatinine to gamma-glutamyltransferase ratio in spot samples of urine have proved useful in experimental models of aminoglycoside toxicity.123 Ratios may not, however, change until several days after toxicity has begun.91 A change in aminoglycoside clearance may be the most sensitive indicator of aminoglycoside toxicity (see Chapter 5.81,114,115 In humans serum creatitine may increase up to 1 week after therapy is discontinued, indicating the potential for continued damage once the drug is discontinued,117 presumably because accumulated drug remains in the tubules. Accordingly, nephrotoxicity is best avoided (see Box 7-1 and the section on therapeutic use).
The presence of renal disease is not a contraindication for aminoglycoside use, although it certainly raises the risk. Normograms have been designed in human medicine to reduce the risk of further damage (see the section on therapeutic use). The risk of nephrotoxicity is greater if any condition of the patient depends on renal prostaglandin formation, such as hypotension, shock, endotoxemia, renal or cardiac disease, or with concurrent drug therapy that impairs prostaglandin synthesis, such as nonsteroidal antiinflammatory drugs.5,126 Metabolic acidosis (or an acidic urine pH) also predisposes the patient to aminoglycoside nephrotoxicity because drugs are ionized and attracted to the anionic changes of cell membranes.127 Consequently, if the source of infection is in the urinary tract, maintaining an alkaline pH will enhance the efficacy of the aminoglycosides by facilitating their diffusion back into infected tissue (and bacteria), while decreasing renal tubular cell uptake of aminoglycosides, presumably because of decreased ionization of the drugs. Aminoglycoside toxicity was demonstrated to be temporal in rats,128 being worse when rats were resting and least when active. Accordingly, dosing in the morning may be prudent for dogs; dosing at night might be considered for cats. Some patients (e.g., pediatric dogs <14 days of age, patients with diabetes mellitus or hypothyroidism) are protected against aminoglycoside- (gentamicin)-induced nephrotoxicity because renal accumulation in the cortical tissues is limited.130,131 Symptomatic hypomagnesemia, hypocalcemia, and hypokalemia associated with inappropriate urinary excretion of potassium despite low serum concentrations has been reported in humans after gentamicin therapy.132 The magnitude correlated with the total cumulative dose of gentamicin. Risk factors included older age and long duration of therapy.123 Note that hypomagnesemia and hypocalcemia may increase the risk of aminoglycoside toxicity by increasing the ease with which drugs enter the renal tubular cell.
KEY POINT 7-18
The presence of renal disease is not a contraindication for aminoglycoside use, although it certainly raises the risk of adverse effects.
Studies have attempted to identify therapies that might treat or prevent aminoglycoside-induced nephrotoxicity. The role of prostaglandin analogs (e.g., misoprostol) in the prevention or treatment of aminoglycoside toxicity has not yet been established. Melatonin administered simultaneously to rats receiving gentamicin was associated with reduced nephrotoxicity.124 Rate receiving l-Carnitine (40 to 200 mg/kg/day, injected) beginning 4 days before receiving doses of gentamicin ranging from 50 to 80 mg/kg had less nephroxicity (based on serum creatinine and histology) compared with untreated rats. Renal gentamicin concentrations were not different, suggesting that decreased aminoglycoside uptake by the renal tubular cell was not the mechanism of prevention. Proposed mechanisms were promotion of fatty-acid oxidation, increased mitochondrial ATP, and decreased formation of oxygen radicals.135 Again, in rats, N–acetylcystein (10 mg/kg intraperitoneally [IP]) protected against gentamicin (100 mg/kg subcutaneously/day × 5 days) induced nephrotoxicity.136 This treatment apparently also has also been demonstrated to be otoprotective in human patients undergoing hemodialysis that are treated with gentamicin.137 A federally funded human clinical trial is currently underway to validate the beneficial effects of N–acetylcysteine in patients with or at risk to develop aminoglycoside nephrotoxicity.
Aminoglycosides can cause an irreversible ototoxicity, although this is not likely to occur at therapeutic doses as long as trough concentrations are lower than 2 to 5 μg/mL (lower should be targeted for gentamicin, higher for amikacin). However, a single dose of tobramycin was associated with ototoxicy in humans.80 Like nephrotoxicity, ototoxicity reflects active uptake of the drug by hair cells of the cochlea. Both auditory and vestibular toxicity may occur. As with nephrotoxicity, the ototoxic potential of each drug varies. The drugs typically should not be given to a patient with a perforated eardrum. Aminoglycosides can cause neuromuscular blockade owing to impaired calcium release at myoneural junctions. The risk appears to be dose dependent and is greater with intravenous administration, in the presence of hypocalcemia, or when combined with other agents active at the myoneural junction (e.g., anesthetics, skeletal muscle relaxants). Neuromuscular blockade can be reversed by cholinesterase inhibitors and (cautiously) calcium.
Drug Interactions
The risk of aminoglycoside ototoxicity and nephrotoxicity is increased when aminoglycosides are used in combination with one another or with nonsteroidal antiinflammatory drugs, diuretics (particularly loop-acting), angiotensin-converting enzyme inhibitors, amphotericin B, and other nephrotoxic (or nephroactive) or ototoxic drugs. The risk of neuromuscular blockade is increased with the combination of aminoglycosides and intravenous calcium, calcium channel blockers, and gas anesthetics and other neuromuscular blocking agents, including atacurium. Edrophonium will reverse the latter, whereas calcium supplementation can reverse any neuromuscular blockade.2
As weak bases, the aminoglycosides may chemically inactivate weak acids; inactivation has been documented in vitro138 and in vivo139 between tobramycin and extended-spectrum penicillins but not carbapenems.140 Tobramycin appears more amenable to inactivation than does amikacin.139 In vivo inactivation is more likely to occur in patients with renal disease for which PDC may be higher than in normal patients. Chemical inactivation might also occur in urine as higher concentrations are achieved. In general, the aminoglycoside is inactivated rather than the penicillin simply because the penicillin is present at much higher concentrations compared with the aminoglycoside.
Synergism between aminoglycosides and cell wall–active antimicrobials has been documented against Enterococcus spp. as well as some strains of Enterobacteriaceae, P. aeruginosa, staphylococci (including MRSA), and other microorganisms. However, these organisms are not always inhibited by the combination of aminoglycoside and cell wall–active compounds. Indeed, antagonism has been described between aminoglycosides and beta-lactams against a MRSA, presumably owing to induction of an aminoglycoside-modifying enzyme.92
Therapeutic Use
Despite their ability to cause nephrotoxicity, the aminoglycosides remain the most effective drugs for the treatment of serious gram-negative infections. They are also effective (combination therapy recommended), against Staphylococcus, Nocardia, Mycoplasma, and selected Mycobacteria spp. Caution is recommended in their use for infections in tissues that are difficult to penetrate and infections that may be located in an anaerobic environment. Combination therapy and topical therapy (in concert with systemic therapy) should be considered whenever possible for serious or complicated infections or in the presence of intracellular infections. Aminoglycoside-impregnated calcium hydroxyapatite or methyl methacrylate beads and methyl methacrylate cement have been used with apparent success in orthopedic procedures (see Chapter 6). 140a Aminoglycosides cannot be given orally with the intent of systemic effects, and their use might be limited to hospitalized patients. However, once-daily therapy increases the convenience and safety of outpatient aminoglycoside therapy.
The pharmacologic rationale for once-daily (also called extended-interval) dosing of aminoglycosides includes their concentration-dependent bacterial killing, minimization of the adaptive resistance, the presence of a postantibiotic effect, and avoidance of renal cortical drug accumulation (i.e., providing a drug-free period to facilitate excretion) such that trough concentrations reach a low target.117 As early as 1984,141 a fixed-dose, prolonged interval was known to be safer than a reduced dose and fixed interval in regard to nephrotoxicity in dogs. Recent studies in dogs, humans, and experimental models have supported a 24–hour dosing interval (administering the total daily dose once a day) for aminoglycoside therapy. The once-daily dose of an aminoglycoside necessary to impair renal function has not been determined, in part because different drugs are studied at different doses and intervals. Because clinical patients are likely to be characterized by changes that predispose to toxicity, studies in normal animals may not be relevant. Once-daily administration of gentamicin was concluded to be safe for 5 days in dogs at a single daily dose of 6 mg/kg.108 Maximum concentration (Cmax (μg/mL), was 9.2 at a Tmax of 0.48 hours. Mean trough gentamicin serum concentrations were 0.1 μg/mL. Although deemed safe, serum creatinine and urea nitrogen were increased and specific urine gravity decreased in one dog and granular casts were evident in two dogs.
Many clinical trials have been performed in humans to assess the safety and efficacy of once-versus multiple-daily dosing of aminoglycosides. Differences in objectives, patients, methodologies, and conclusions have led to confusion. Several meta-analyses have been performed in humans that focuses on clinical efficacy and either nephrotoxicity or ototoxicity in patients treated with aminoglycosides once versus multiple times daily. The number of trials included in each meta-analysis ranged from 21 to 26; the number of persons studied by each meta-analysis was 2100 to more than 3000. Barza’s group142 found that once-daily administration of aminoglycosides in patients without preexisting renal failure was as effective as multiple-daily dosing and was associated with a lower risk of nephrotoxicity and no greater risk of ototoxicity. Further, once-daily dosing was more convenient and less costly. A second (22 studies)143 and third meta-analysis (26 studies)144 found the rates of efficacy and toxicitity were similar and convenience and reduced cost justified the once-daily approach. Another study found that gentamicin (once or multiple times daily) and ticarcillin-clavulanic acid, either alone or combined with gentamicin, was associated with the same efficacy and nephrotoxicity renal function was better preserved with either once-daily gentamicin combined with ticarcillin–clavulanic acid or ticarcillin–clavulanic acid alone.145 However, in humans, experts continue to advise that extended-interval aminoglycoside dosing not be used in patients with endocarditis, mycobacterial infections, or burns. Further, a simple once-daily approach to aminoglycoside therapy should not be used if the patient’s creatinine clearance is less than 20 mL/min or in patients in hemodialysis because of marked alteration of PK in these patients. Rather, monitoring should be the basis of dosing in these patients.146 Further, for obese patients (actual body weight > 20% above ideal body weight [IBW]), the dose should be reduced using the following formula that adjusts weight: obese dosing weight = IBW + 0.4 (actual weight − IBW).147 A number of normograms have been developed for use in humans to support the design of aminoglycoside dosing regimens that will be effective yet safe in patients with renal disease. Generally, the normograms are based on creatinine clearance and other patient factors. However, in general, the normograms underestimate the dose necessary to achieve a therapeutic maximum drug concentration. Methods using probabilistic or deterministic methods are currently being investigated.148 However, therapeutic drug monitoring continues to be the preferred method to allow calculation of individual patient PK.117,146 Indeed, a meta-anlysis that compared once-daily multiple-dosing therapy and dosing based on PK found that basing doses on individual PK was the safest approach to dosing with aminoglycosides.117 AUC based on two time points has enhanced prediction of dosing regimens for aminoglycosides in children with cystic fibrosis.149 However, the distribution phase of aminoglycosides is sufficiently slow that the first sample probably should be collected no earlier than 1 hour after dosing is complete. Monitoring peak (no earlier than 1 hour, to ensure complete distribution) and detectable trough concentrations (no later than 2 to 3 half-lives after the peak to ensure concentrations are still detectable) will allow estimation of half-life, and (if given intravenously) Vd and clearance (see Chapter 5). Pretreatment and posttreatment comparisons may be useful in the early detection of significant changes in renal function, which will also help guide safe therapy. The clinical pharmacologist offering recommendations will be able to determine these parameters regardless of the actual timing (i.e., 1 versus 1.5 hours for peak, 4 versus 8 hours for trough); however, accuracy in reporting the time that samples were collected is critical for proper recommendations when the samples are collected for determination of half-life.
Maintaining hydration is probably the single most important means by which the risk of aminoglycoside-induced nephrotoxicity can be minimized. Ototoxicity also can be minimized by hydration and avoidance of topical administration, particularly in the presence of a perforated tympanum. Although gentamicin is the most economical aminoglycoside, amikacin should be considered for serious infections because of its improved resistance to antimicrobial destruction and better efficacy against some organisms, including P. aeruginosa. The aminoglycosides are often used in combination with other antimicrobials that have a less comprehensive gram-negative spectrum. As with imipenem, the aminoglycosides cause minimal endotoxin release in patients suffering from gram-negative infections associated with a large inoculum.74
Drugs that Target Nucleic Acids
Fluorinated Quinolones
The fluorinated quinolones (FQs) are among the most recent classes of antimicrobials to be developed for treatment of bacterial infections. These synthetic drugs are minimally toxic yet have been effective in the treatment of many aerobic gram-negative organisms and selected gram-positive organisms. The desire to expand their spectrum of activity and the advent of resistance has led to innovated structural changes.
Structure–Activity Relationship
A review of the development of FQs is worthwhile, not only to facilitate understanding of their actions but also to provide insight regarding the advantages of so-called designer drugs. Two decades elapsed between the development of nalidixic acid, the progenitor of the FQs, and norfloxacin, the first of the FQs to be approved for use. Among the FQs currently used for treatment of susceptible infections in dogs and cats, ciprofloxacin was first approved for use in humans in 1986, with its veterinary counterpart, enrofloxacin, rapidly following in 1991. Extensive use of these drugs has exposed the need for improvements and newer clinical indications; pharmaceutical companies have been attentive to addressing these needs.
Nalidixic acid is the progenitor of the FQs (Figure 7-8). Synthetic manipulations, including but not limited to the addition of a fluorine atom, have broadened the antibacterial spectrum; enhanced tissue penetrability; reduced (some) side effects (perhaps while contributing to others); and, most recently, decreased the risk of resistance. Currently marketed FQs generally consist of a quinolone ring nucleus, the target of most initial structural manipulations (Figure 7-9), or a napthyridone ring structure, which replaces the nitrogen at carbon 8 on the quinolone structure (enoxacin, tosufloxacin, trovafloxacin, and gemifloxacin). The quinolone nucleus contains a carboxylic acid group at position 3 and an exocyclic oxygen at position 4 (hence the term “4-quinolones”); these are the active DNA gyrase binding sites, and thus these sites generally are not chemically manipulated. The structures yield two pKas for most FQs, rendering them amphoteric; they can act as weak bases, weak acids, or neutral compounds. For example, the carboxylic acid of enrofloxacin has a pKa of 6 and the amine group a pKa of 8.8. The side chain attached to the nitrogen at position 1 affects potency. The ethyl group at this position on nalidixic acid and the first of the clinically used FQs, norfloxacin, was replaced with a bulkier group (e.g., the cyclopropyl group of ciprofloxacin), which enhanced both gram-negative and -positive spectra. Substitution at position 5 also improved the gram-positive spectrum; however, it was the addition of a fluorine atom at position 6 that profoundly enhanced the gram-positive spectrum. The addition of a piperazyl ring, containing a heterocyclic nitrogen, at position 7 also was a critical improvement. This addition improved bacterial penetration (potency) and added P. aeruginosa to the gram-negative spectrum. The combination of the fluorine atom with a piperanyl ring produced the “breakthrough” class of FQs used today; norfloxacin was the first of these FQs to be approved in the United States.
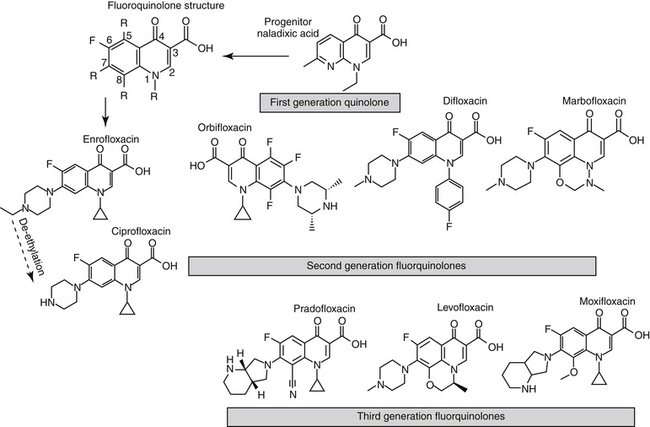
Figure 7-8 Various substitutions of the core chemical structure of the fluorinated quinolones have improved their spectrum, efficacy, and tissue penetration. The efficacy of the fluorinated quinolones depends on the ketone group at position 4 and on the carboxylic acid at position 3 (necessary for inhibition of DNA gyrase). The combination of the fluorine at position 6 (which markedly expanded the gram-positive spectrum) and the substitution of a piperyl ring at position (which enhanced efficacy towards Pseudomonas aeruginosa as well as increased tissue penetrability) represented a “breakthrough” for the fluorinated quinolones (e.g., enrofloxacin and its active metabolite, ciprofloxacin). Substitutions at position 8 increase the anaerobic spectrum (e.g., pradofloxacin). The addition of larger side chains may impair microbial resistance mechanisms.
Figure 7-9 The mechanism of action of fluorinated quinolones. During DNA synthesis, the double strands of circular bacterial DNA are in a tightly (negatively) coiled state (negative referring to the direction of the coils). The DNA strands are “unzipped” to allow either messenger RNA or a new DNA strand to be synthesized. The unzipping induces stress and the subsequent formation of positive supercoils, that ultimately must be removed. DNA gyrase, a topoisomerase, directs double-stranded breaks in the DNA. After DNA synthesis, the daughter chromosomes are unlinked by topoisomerase IV. Both DNA gyrase and topoisomerase IV are essential to bacteria and either or both are targeted by the fluorinated quinolones. Drugs that target both enzymes may require multiple mutations for resistance to emerge.
Chemical manipulations continue to improve the FQs in terms of spectrum, potency, and avoidance of resistance. Substitutions on the piperazyl (e.g., ofloxacin, its L isomer, levofloxacin, and sparfloxacin) enhance the gram-positive penetration, whereas substitutions at position 8 enhance anaerobic activity (e.g., sparfloxacin, pradofloxacin, moxifloxacin). Substitutions at these sites with halogens such as chlorine or fluorine (e.g., 8-chloroquinolones or 8-fluoroquinolones [sparfloxacin]) result in ultraviolet unstable compounds (particularly the chloro substitution), which can cause phototoxicity. In contrast, substitution of a methoxy-group at the 8 position (e.g., moxifloxacin, gatifloxacin) confers good anaerobic activity but without risk of phototoxicity. Recent improvements (in human medicine) focus on increasing the efficacy of FQs toward pneumococci and MRSA, as well as other gram-positive cocci, Enterobacteriaceae, Pseudomonas, and anaerobes, and methods by which resistance might be minimized.
KEY POINT 7-19
Chemical manipulations of fluorinated quinolones improve potency, broaden the spectrum, and decrease resistance.
Four drugs are currently approved for oral use in small animals in the United States: enrofloxacin (the first approved, for both dogs and cats, also approved for injectable [SC] use in dogs), followed rapidly by orbifloxacin (dogs and cats), difloxacin (dogs), and marbofloxacin (dogs and cats) (see Figure 7-8). Pradofloxacin may be undergoing consideration for approval for use in dogs in the United States. Variations in the chemical structures of these drugs may result in subtle differences in potency, efficacy, and tissue distribution. Human-marketed FQs, particularly ciprofloxacin and increasingly levofloxacin, continue to be prescribed by veterinarians. Care should be taken to ensure that differences in disposition between humans and dogs or cats are considered when using these drugs. In their guidance to industry, the FQs have been indicated by the Food and Drug Administration (FDA) as “drugs of interest”; as such, veterinary use of these or newer FQs approved for use in humans is likely to draw scrutiny by allied health professions, including regulatory agencies. Note that use of drugs intended for human use (including cheaper generic drugs) instead of veterinary drugs solely because the former are less expensive is likely to be a disincentive for veterinary manufacturers with regard to future approvals of drugs for animals. Further, Animal Medical Drug Use Clarification Act stipulates that the conditions underwhich extra-label drug use is allowed include the lack of availability of a veterinary approved drug that meets the patient’s needs. Extra precautions should be taken when prescribing human-medicine drugs to ensure judicious use.
Because enrofloxacin was the first of the veterinary FQs to be approved for use in dogs and cats, it often is the gold standard on which subsequent drug approvals are based and upon which clinical trials evaluating FQ efficacy are based. Because it is structurally similar to ciprofloxacin and because it is metabolized (up to 50% of the AUC of bioactivity) to ciprofloxacin in many species, much of the PD information in the human literature regarding efficacy for ciprofloxacin is applicable to enrofloxacin. However, exceptions occur, particularly with regard to PK considerations. Further, some differences exist in regard to pharmacodynamics between ciprofloxacin and enrofloxacin. Although marbofloxacin has been approved for a shorter period in the United States compared with enrofloxacin, it has been used since 1994 in Europe, and a considerable amount of information is available regarding this drug. In contrast, less information is available for orbifloxacin and particularly difloxacin.
Mechanism of Action
The FQs currently are the only veterinary-approved antimicrobials that directly inhibit DNA synthesis. Bacterial DNA, is circular and can be up to 1.3 mm long, necessitating a negatively supercoiled state surrounding the RNA core (see Figure 7-9).150,151 During DNA synthesis, the double strands of DNA must be uncoiled or “unzipped” to allow either messenger RNA to interpret or a new DNA strand to be synthesized. The unzipping of the double strands induces positive supercoiling, which leads to undue stress in the individual strands. Accordingly, DNA gyrase (topoisomerase II), directs double-stranded breaks in the DNA, thus inducing a negative supercoil configuration, balancing the positive supercoils. Once DNA polymerase passes through a break in the strand, the break is repaired. Topoisomerase IV separates the daughter DNA molecules produced by DNA replication.152 Both DNA gyrase and topoisomerase IV are essential to bacteria replication; both are targeted by FQs either individually or sequentially, depending on the drug and organism.151
Bacterial topoisomerases are ATPase-dependent enzymes. Each exists as a tetramer consisting of two A and two B subunits. For DNA gyrase, the subunits are encoded by the genes gyrA (2517 bp) and gyrB (2060 bp), respectively, and for topoisomerase parC and parA, respectively. The primary enzyme responsible for activity varies with the organism and influences the target of the FQ. DNA gyrase is the primary target in E. coli, other gram-negative organisms, and Mycobacterium tuberculosis, whereas topoisomerase IV is the primary target of S. aureus and (probably) other gram-positive organisms.151,153 The efficacy of the FQs against various microbes can be explained, in part, by the presence or absence of the target enzymes, as well as drug preference for different enzymes (which in turn can be related to chemical structure [see above]). For example, unlike most other bacteria, M. tuberculosis lacks topoisomerase IV and might be less susceptible than other microbes that have both targets. Ciprofloxacin prefers topoisomerase IV, whereas moxifloxacin prefers DNA gyrase. Accordingly, bactericidal activity of moxifloxacin might be (and clinically appears to be) better compared with ciprofloxacin against M. tuberculosis. Efficacy of FQs is related to the number of molecules that interfere with the target topoisomerase; interference is irreversible, resulting in concentration-dependent effects.
KEY POINT 7-20
Differences in efficacy of the fluorinated quinolones reflects, in part, the preferred topoisomerase targeted by the drug.
The MICs of the FQ for susceptible organisms tend to be low compared with most other antimicrobial drugs. DNA gyrase actions are inhibited at concentrations of 0.1 to 10 μg/mL. The precise mechanisms by which FQs kill are not fully understood, but strand breakage, autolysis associated with SOS DNA repair systems, and blockade of replication by the gyrase FQ complex may cause bacterial inhibition without bacterial killing.154 However, the concentration of FQs necessary to inhibit the growth of organisms (MIC) is very close to that necessary to kill the organism (MBC). Although mammal DNA replication also depends on a topoisomerase, its function is somewhat different. More important, affinity of host topoisomerases is less than 0.001 of that of bacterial DNA gyrase. Thus the unique mechanism of action of the FQs renders rapid bactericidal activity with minimal effects on the host. The time to effect for FQs is very short (30 minutes); their rapidity of action often is the reason for preference of these drugs compared with other equally but more slowly effective antimicrobials (e.g., amoxicillin–clavulanic acid combinations for the treatment of selected pyodermas). Interestingly, cellular factors such as intracellular magnesium concentration, salt, and ATP may influence the affinity of FQs for their target enzymes; the clinical implications of this observation are not clear.151
KEY POINT 7-21
The irreversible interaction between drug and topoisomerase results in a concentration-dependent effect for the fluorinated quinolone.
The efficacy of the FQs occurs, in part, because of a long postantibiotic effect, which also is concentration dependent. Depending on the organisms, drug, and concentration, the postantibiotic effects can approximate 5 to 8 hours.155-159 The efficacy of the FQs appears to correlate more closely with peak concentrations (i.e., concentration dependent) than with duration of PDC above the MIC.160,161 Consequently, efficacy is more likely when Cmax/MIC exceeds 10 or more. However, duration of time that PDCs are above the MIC (AUC/MIC) also is an effective predictor of efficacy, and may be better than Cmax/MIC for selected organisms.162 Analysis of multiple studies focusing on the best predictor of successful bacterial killing indicated that the area under the inhibitory curve (AUIC), an index that is similar to AUC/MIC (see Chapter 6) was the best predictor of efficacy. If AUIC is greater than 100 but less than 250, bacterial killing is slow (evident by day 7 of therapy), whereas an AUIC greater than 250 produced rapid killing, with eradication occurring within 24 hours. The effect occurred for both gram-negative and gram-positive organisms.153 These data suggest that the most effective use of the FQs is to administer at a dose that will achieve rapid killing. A comparison of Cmax/MIC or AUC/MIC may be helpful in comparing relative efficacy among the FQs used to treat feline or canine pathogens164 (see Table 7-12).
Spectrum of Activity
The (human-medicine) FQs have been categorized into 3 to 4 generations based on their spectrum of activity (see Figure 7-8).165 Athough not often used, the classification is helpful for perspective on the development of the FQs. The spectrum of nalidixic acid, the first-generation drug, is narrow. However, it was improved through pharmaceutical manipulation, yielding the second-generation drugs. This generation is exemplified by the human-marketed drug ciprofloxacin and the current veterinary FQs approved for use in dogs and cats. Their spectrum includes a broad gram-negative and less broad gram-positive spectrum. Third-generation drugs include levofloxacin, the L-isomer of ofloxacin, sparfloxacin, gatifloxacin, and moxifloxacin. This generation is characterized by enhanced potency, improved spectrum (which includes anaerobes), and reduced resistance. The fourth-generation drugs are characterized by the broadest spectrum and are exemplified by trovafloxacin. Each generation has been designed such that drug molecules target specific molecules of the target enzymes, thus increasing efficacy, and for some reducing the emergence of resistance.
The second-generation veterinary FQs have been referred to as broad in spectrum, but this term is appropriate only when referring to the gram-negative spectrum; the term broad is more appropriate for third-generation drugs, for which their currently is no veterinary approved example in the United States. The gram-positive spectrum is more selective, and anaerobes, in general, are not susceptible. However, other microbes are targeted, including cell wall–deficient microbes and mycobacterium. Organisms particularly susceptible to FQs include Pasteurella (among the lowest MICs), E. coli, Klebsiella spp. E. cloacae, P. mirabilis, Citrobacter freundii, and S. marcescens Pseudomonas spp. also is included in the spectrum but generally is characterized by the highest MICs, with efficacy toward Pseudomonas spp. varying with the individual drugs (Table 7-12; see also Tables 7-3 and 7-4).154 Among the drugs used in dogs or cats, ciprofloxacin, enrofloxacin, and marbofloxacin tend to have the lowest MICs. Ciprofloxacin is most potent toward gram-negative isolates, particularly for E. coli and P. aeruginosa.164,166 The gram-positive spectrum includes Staphylococcus spp. and some Corynebacterium. The FQs have exhibited variable efficacy against Streptococcus species and E. faecalis.156,167 Other susceptible organisms generally include Campylobacter, Salmonella, Shigella, and Yersinia. Efficacy of the FQs toward leptospirosis is supported by limited studies. Some rickettsial organisms may be susceptible; in vitro data and limited in vivo data indicate potential efficacy against organisms causing ehrlichiosis and Rocky Mountain spotted fever.168
Table 7-12 Pharmacodynamic Data for Selected Fluoroquinolones and Selected Feline and Canine Pathogens(164,180)
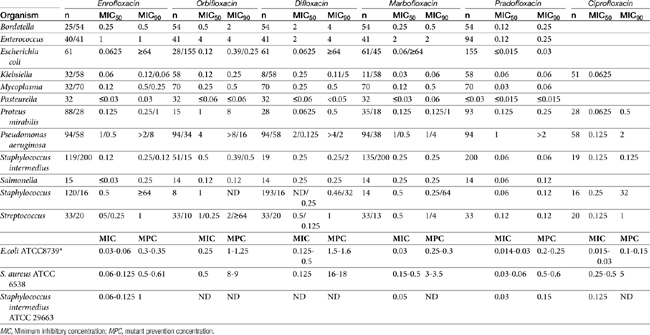
KEY POINT 7-22
The second-generation fluorinated quinolones have a broad gram-negative spectrum and a more limited gram-positive spectrum and are generally not effective toward anaerobes.
Integration of PK and PD of the FQs reveals some differences in predicted efficacy among the FQs used in cats and dogs toward organisms within the spectrum (see Table 7-12). Based on PK reported either in the literature or on the package insert, two PDIs were determined: the Cmax/MIC (target 10) or AUC/MIC (target 125). The PDIs were compared among drugs for the susceptible isolates of each organism at the lowest and highest labeled dose for each drug. In general, at the low dose the only organism for which the target PDIs were reached for all drugs was E. coli. For all other organisms, even at the high dose, targets were reached consistently only for ciprofloxacin, enrofloxacin, and marbofloxacin.154 The authors concluded that the highest dose of the FQ is generally recommended when possible and that enrofloxacin, marbofloxacin, and ciprofloxacin performed in vitro better than difloxacin and marbofloxacin.
Levofloxacin is a human-marketed third-generation FQ that increasingly is being used in dogs and cats. It is twice as potent against gram-positive isolates (topoisomerase IV) and equally potent against gram-negative isolates (DNA gyrase) compared with ciprofloxacin, although more recent data suggest that this is not consistent (see Table 7-4).169 For example, the MIC50/90 (μg/mL) for human organisms isolated from skin or soft tissue infections are as follows: S. aureus (0.25, > 4), E. coli (≤0.03, 4), or P. aeruginosa (0.5, >4).170 The potential efficacy of levofloxacin cannot be assessed for dogs because PK have not been established and neither Cmax nor AUC is available. Kinetics have been reported for levofloxacin in the cat, but at 10 mg/kg, the Cmax does not reach the MIC90 for Staphylococcus or Pseudomonas spp. The Cmax/MIC90 is only 1 (rather than the target ≥10) for E. coli. The target ≥10 would be reached based on the MIC50 for Staphylococcus spp. and E. coli but not for Pseudomonas spp. The safety of levofloxacin in cats at doses that will be necessary to reach the target PDI has not been established. These data suggest that PK and PD studies are needed in the dog before levofloxacin is used and that the organisms against which levofloxacin is used in cats at the dose of 10 mg/kg should be characterized by an MIC of 0.5 μg/mL or less. Once-daily administration was demonstrated to be more effective against Staphyloccus spp., including an MRSA isolate, compared with twice-daily dosing.169
Anaerobic organisms have been considered generally resistant to the FQs. However, the spectrum of the newer drugs, particularly those substituted at position 8, has been expanded to include anaerobes. Levofloxacin, sparfloxacin, grepafloxacin, and pradofloxacin each has greater activity against anaerobes compared with older drugs. This includes the B. fragilis group, as well as Clostridium, Peptostreptococcus, Prevotella, and Fusobacterium spp.171
The FQs are effective against mycobaterial organisms. However, using M. tuberculosis as an example, the MIC (μg/mL) for the newer FQs are lower compared with the second-generation drugs: 1 μg/mL for levofloxacin, 0.1 to 0.5 for sparfloxacin, 0.2 to 0.25 for gatifloxacin, and 0.12 to 0.5 for moxifloxacin, compared with 0.5 to 4 for ciprofloxacin.154 Of the FQs, gatifloxacin and moxifloxacin have been demonstrated to exceed the mutant potential concentration (MPC; see Chapter 8) for M. tuberculosis. Like other organisms, and despite their slow growth, the activity of FQs against Mycobacterium spp. is concentration dependent. However, tubercular organisms are able to enter a dormant, persistant, and antimicrobial-resistant phase, necessitating long-term therapy.
Each of the veterinary FQs has been approved with a “flexible” dosing regimen, indicating low to high doses, with the choice depending on the MIC of the infecting organism. However, as previously discussed, increasing evidence suggests that the highest concentration should be targeted whenever possible. The concept of the MPC emerged in the context of emerging FQ resistance in mycobacteria. Targeting simply the MIC is likely to select for stepwise mutants (see Chapter 6).162 Flexibility also occurs for the interval: for enrofloxacin and orbifloxacin, the label allows once- or twice-daily dosing, whereas for marbofloxacin and difloxacin, the dose is limited to once a day. Because FQs are concentration dependent, administration of the total daily dose as a once-daily dose is generally preferred, as has been demonstrated for ciprofloxacin173 and levofloxacin.169 P. aeruginosa is an example of an organism whose tendency toward resistance suggests the higher, once-daily dose.174 Because efficacy of an FQ is based on AUC/MIC as well as Cmax/MIC, a second dose (not half the dose twice) might be considered, particularly for selected organisms (e.g., S. aureus).
KEY POINT 7-23
Failure to achieve the mutant prevention concentration (MPC) may allow emergence of multistep mutants.
The amphoteric nature of the FQs complicates the impact of pH on efficacy. For example, difloxacin was shown to be most potent (based on MIC differences) at a pH of 7.1 compared with 5.9 or 7.9, with a fourfold increase in the MIC at the alkaline pH occurring for E. coli, K. pneumoniae, P. mirabilis, and S. intermedius.159
Resistance
A major advantage of the FQs promoted during marketing, was the lack of clinically relevant plasmid-mediated quinolone resistance. Rather, the major mechanism of resistance reflects genetic mutations in the target topoisomerase enzymes (e.g., DNA gyrase [topoisomerase II] and topoisomerase IV). However, several observations dampen the importance of the predominance of mutational, rather than plasmid-mediated, resistance. First, history has demonstrated that resistance of any antimicrobial (plasmid or otherwise) may take several decades of intense antimicrobial use, suggesting that, as with other antimicrobials, the use of FQs ultimately was to be limited by resistance. Secondly, resistance to norfloxacin emerged as little as 3 years after its approval, regardless of the mechanism. This rapid development of resistance foretold a similar problem with other second-generation FQs. Thus, as the medical community enters the third decade of ciprofloxacin use in human medicine and the second decade of FQ use in veterinary medicine, increasing resistance, albeit not necessarily plasmid mediated, has emerged and is limiting the widespread effective use of these drugs in both human and veterinary medicine. Finally, plasmid-mediated resistance has appeared and plays a role in horizontal transmission of FQ resistance.165 The development of FQ resistance by human bacterial organisms has influenced the decision to ban extralabel use of FQs in food animals or use as food additives (i.e., growth promotants). Clinically, the increasing pattern of resistance for veterinary FQs and ciprofloxacin has emerged toward several organisms, including S. aureus, P. aeruginosa, E. coli, and other gram-negative organisms (see Chapter 6). In chronic otitis of dogs, 14% of S. pseudintermedius cultured from the middle ear and more than 65% of Pseudomonas spp. cultured from the external and middle ear were resistant to enrofloxacin.175 A prospective study of more than 300 organisms submitted to commercial laboratories found nearly 30% of E. coli resistant to all veterinary FQs, as well as ciprofloxacin.The MIC90 for Pseudomonas surpassed the CLSI MIC breakpoint for all drugs except ciprofloxacin, and for E. coli and Staphylococcus spp. (not including S. intermedius) exceeding it for all drugs by fourfold to eightfold.154 A subsequent prospective study of more than 350 E. coli isolates (collected from all body tissues, with the vast majority associated with urinary tract infections) found that 30% demonstrated an MIC90 greater than 32 μg/mL (MIC breakpoint ≥ 4 μg/mL), with regional geographical differences demonstrated.166 Resistance to FQs is associated with FQ use; in humans a single dose of ciprofloxacin lead to FQ-resistant microorganisms.167 That FQ resistance can be associated with FQ use in dogs was demonstrated by Debavalya et al.:178 Close to 100% of fecal E. coli developed high level resistance to FQs (associated with multi-drug resistance) within 3 to 9 days of therapy of enrofloxacin in dogs (5 mg/kg every 24 hours).
Susceptibility data from laboratories that test both ciprofloxacin and enrofloxacin may report susceptibility to ciprofloxacin but resistance to enrofloxacin. Interpretive standards on culture reports for ciprofloxacin are based on human data and may not take into account differences in oral bioavailability, just as standards for enrofloxacin do not include bioactivity contributed by ciprofloxacin. Although ciprofloxacin is more potent toward E. coli and Pseudomonas aeruginosa compared to enrofloxacin, the difference is usually within 1 tube dilution. A prospective study compared the proportion of resistance and the relative susceptibility (efficacy) among ciprofloxacin, difloxacin, enrofloxacin (alone or with ciprofloxacin), marbofloxacin, and orbifloxacin FQs toward six organisms collected from canine and feline patients.154 The proportion of resistant isolates, which was based on CLSI interpretive criteria, did not differ among drugs, suggesting that expression of resistance by an isolate to one (second-generation) FQ might be prudently interpreted as resistance to all, despite the not uncommon finding of susceptibility to ciprofloxacin and resistance to another FQ (e.g., enrofloxacin).
Three major mechanisms of FQ resistance have been identified,55,167 with the most studied being changes in the structure of the target topoisomerase enzymes. However, mutations, which impart resistance within the FQ class of drugs, are often accompanied by decreased expression of porin membranes and increased activity of efflux pumps, which imparts multidrug resistance.155,169 Thus far, resistance to FQs acquired through changes in DNA gyrase has been documented clinically only after chromosomal point mutations; at least 10 different mutations have been identified so far. Resistance is stepwise, with the first step occurring primarily through mutations that reduce FQ affinity for the preferred topoisomerase target, which varies with the organism. Gram-negative bacteria tend to more commonly target DNA gyrase; changes occur more often in the GyrA subunit compared with GyrB.141 The primary target of gram-positive organisms tends to be changes in topoisomerase IV, targeting parC and parE followed by changes in DNA gyrase. Recent evidence suggests that the drug (and its primary target) select for the mechanism of resistance.141 High-level resistance generally reflects a second step mutation that leads to additional changes in the amino acid sequence of either (the alternate) topoisomerase target, thus further decreasing affinity, or the generation of efflux pump mechanisms. The MIC of the organisms progressively increases with each step. The role of reduced porin membranes and efflux pumps in FQ resistance was more recently discovered. Gram-negative isolates are associated with both mechanisms of reduced drug accumulation (i.e., porins and pumps), as well as decreased lipids in the lipopolysaccharide covering, impeding drug transport; gram-positive isolates (S. aureus) have been associated with increased drug efflux.145,147,167 The efflux pumps affect multiple drugs, contributing to multidrug resistance, including resistance to drugs structurally unrelated to FQs.167,169, 169 bib169a These include tetracyclines, phenicols, and macrolides. Beta-lactams may also be involved; resistance to antiseptics and disinfectants may occur. Expression of the pump is chromosomally mediated. For example, mutations in the mar operon may induce the acrAB proteins of a stress-induced efflux pump, resulting in high-level resistance, even for isolates with no or single mutations in topoisomerase.155 Plasmid-mediated quinolone resistance (PMQR), associated with the qnr gene, has recently been identified in clinical bacterial isolates, generally associated with class I integrons. However, while initially rare, in 2003, several strains of E. coli and Klebsiella spp. were found to transmit qnr resistance, and isolates have since been identified in the United States. The author has reported a high incidence of PMQR in clinical canine and feline E. coli isolates.165a Resistance mediated by PMQR and qnr tends to be low level and thus may be difficult to detect on C&S testing. Mechanisms include production of a protein that prevents quinolone binding to the target, and enzymatic destruction of the drug. Its impact appears to be related to its ability to increase the incidence of spontaneous mutations and facilitation of altered porin or efflux protein activity. Despite its low level, PMQR resistance associated with qnr appears to affect other drug classes, including cephalosporins (including second- and third-generation), aminoglycosides, and potentiated sulfonamides.
The emergence of stepwise resistance is generally indicated by an increase in the MIC of the organism toward the drug. In human medicine, isolates characterized by an MIC greater than 0.125 μg/mL for ciprofloxacin are treated as “reduced susceptibility,” indicating that a first step toward mutation (or resistance) has occurred, whereas isolates greater than 2 μg/mL are considered to have “high-level” resistance.165 These reports are likely, in part, to be the basis of “susceptible” MIC breakpoint promulgated by CLSI. However, it is important to note that despite a susceptible designation for some isolates, reduced susceptibility is an indication that resistance has begun and use of a FQ should be done cautiously and judiciously. Actions such as using a second dose or using the drug in combination with a second, synergistic drug should be strongly considered. Current clinical microbiology laboratories often do not perform susceptibility testing at concentrations below 0.125 to 0.25 μg/mL for FQs, thus precluding the identification of isolates that are characterized by reduced susceptibility. Thus it is important to note that reduced susceptibility to an FQ of intererest may characterize a “susceptible” isolate, and use of FQs should be done judiciously.
The term MPC was coined after substantial evidence emerged that resistance to FQs reflects multistep or stepwise selection of mutants when the FQ is used therapeutically at a dose that targets the MIC of a cultured infecting microble (see Chapter 6).172 At drug concentrations below the MPC, first step mutants will continue to grow in the absence of effective host response, and may replace the wild-type (nonmutant) population.180 Consequently, the MPC, rather than the MIC, ideally is targeted with drug therapy. Predicting the MPC on the basis of MIC is not possible; the relationship between the two appears to be larger for gram-positive than gram-negative isolates, and varies among the FQs (see Table 7-12).180 Among the veterinary FQs, using quality assurance isolates, the ratio of MPC to MIC seems to be similar for gram-negative isolates, being less than 10, and the MPC might be reasonably targeted with doses that are within recommendations based on a Cmax/MIC ratio of 10. However, Pasquali181 demonstrated that the MPC/MIC for E. coli was fourfold to sixteenfold higher for enrofloxacin compared with ciprofloxacin. In this study the authors found that targeting the MPC for P. aeruginosa was not effective, postulating that the reason reflects efflux pump activity rather than point mutation (the basis of the MPC theory) as the major mechanism of resistance. Enrofloxacin and pradofloxacin have the lowest MPC/MIC ratio for gram-positive isolates; concentrations necessary to target the MPC for gram-positive isolates may be achievable with these drugs but may not be achievable at recommended doses, particularly for difloxacin and orbifloxacin. Use of the highest dose of any FQ is recommended because of the risk of resistance. If reduced susceptibility is suspected (e.g., MIC > 0.25 μg/mL), then the addition of a second dose or use as part of combination therapy might be prudent. Combination therapy has been described as a mechanism to reduce emergent resistance to FQs. For example, in an in vitro model, rifampin prevented emergence of resistance to ciprofloxacin.169 The addition of a FQ decreased the advent of resistance to cephalosporins in another study.182
Newer drugs, including gemifloxacin, trovafloxacin, gatifloxacin, and pradofloxacin, may target both DNA gyrase and topoisomerase IV. Thus for these drugs, multistep resistance may be necessary to neutralize their antibacterial effects. Newer FQs appear to avoid resistance because their stereochemistry interferes with altered porin sizes and efflux mechanism. For example, for pradofloxacin the cyclopropyl ring at N1 provides bacterial killing, but the diazabicyclononyl moiety at C7 appears to physically block porins.154 Wetzstein180 compared the MPCs for older and newer FQs. That resistance may be more likely with older compared with newer drugs was suggested by an in vitro study,169 in which resistance could be induced for ciprofloxacin but not levofloxacin. However, surveillance studies in humans infected with Streptoccocus spp. as well as other isolates, report variable findings, including lower, similar, or higher rates of resistance for levofloxacin, compared with ciprofloxacin.169,183,184 Because resistance is likely to emerge even to the newer FQs, use based on C&S testing and design of a dosing regimen that targets the MPC as much as possible is prudent.
FQ resistance by Mycobacterium spp. occurs primarily as part of multidrug-resistant tuberculosis, which develops when an FQ is used as the only active agent in a failing multidrug regimen.154 Thus combination with traditional antitubercular drugs (isoniazid, rifampin) enhances antimicrobial efficacy.154
Pharmacokinetics
The PK of the veterinary FQs are largely comparable among the drugs, particularly if structurally similar, although individual differences may become important for some infections. Maximum drug concentrations of the FQs do not always increase linearly with dose (see Table 7-1) This may reflect, for some drugs, variability in peak concentrations measured among different investigators, including different analytical methods. In particular, attention must be paid to the method of drug detection, with those based on bioactivity (i.e., bioassay) frequently yielding higher concentrations if an active metabolite is present (e.g., enrofloxacin and ciprofloxacin).
KEY POINT 7-24
The fluorinated quinolones are characterized by good to excellent tissue distribution because of their lipid solubility and accumulation in phagocytic white blood cells.
The only injectable preparation approved for dogs is for enrofloxacin, although an injectable preparation is available for human FQs, including ciprofloxacin. All remaining veterinary FQs approved in dogs or cats are available for oral administration. Enrofloxacin is available as a topical combination preparation. Marbofloxacin, enrofloxacin, difloxacin, and orbifloxacin are characterized by close to 100% oral bioavailability in young adult animals. A number of factors, however, influence absorption of FQs in general, and several drugs specifically. Magnesium and aluminum decrease oral absorption, and food may also, which may be undesirable for concentration-dependent drugs. The oral bioavailiabity of FQs may not be predictable, with extrapolation among species not recommended. For example, norfloxacin is characterized by 60% or less oral bioavailability in dog, and ciprofloxacin, generally less than 60%. Extrapolation of levofloxacin between humans and cats appears to be more appropriate than that of ciprofloxacin. Oral absorption also may be impaired in neonates, as has been demonstrated for enrofloxacin.185
As a class, the FQs are well distributed to most body tissues (see Table 7-5). Protein binding of enrofloxacin, ciprofloxacin, and marbofloxacin in dogs is 34 ± 2%, 18.5 ± 2%, and 21 ± 6%, respectively.176 Although the Vd of the drugs ranges from a low of 1.12 (marbofloxacin) to a high of 3.2 (difloxacin), the clinical relevance of these differences is not likely to be a sufficient cause to select one over another. The respective PCs for selected FQs have been variably reported, with enrofloxacin characterized by the highest lipophilicity of the three: 2.4, 0.02, and 0.11;187 and 3.54, 0.07, and 0.08, for enrofloxacin, ciprofloxacin, and marbofloxacin, respectively (Figure 7-10).176 However, as with Vd, predicting tissue distribution based on PC is difficult. This reflects, in part, the common use of homogenate data for solid tissues. Homogenate data include both interstitial fluid and ICF. As such, drugs that penetrate cell membranes and accumulate in cells, not necessarily in active form, may be characterized by higher concentrations compared with drugs that distribute to interstitial fluid only. Intracellular trapping of drugs may limit access to microbes in interstitial fluid, although movement from the cell back into interstitial fluid may prolong the presence of drug in interstitial fluid by slow release from the cell. The relevance of the data is then influenced by the location of the infection (i.e., intracellular versus extracellular) and host (e.g., inflammation) or microbial (e.g., biofilm) factors that might affect efficacy. Fluid tissue concentrations (based on homogenate data) are generally greater in organs of elimination compared with plasma for all FQs. Solid tissue concentrations are often higher (e.g., if drug is trapped in the cells), particularly for the liver and kidney (organs of elimination) but also spleen and lung (perhaps reflecting phagocytic cell accumulation), prostate (perhaps reflecting ion trapping), and muscle161,188 Homogenate tissue data are available on the package inserts of several of the veterinary approved FQs. Interestingly, the concentration of difloxacin in cortical bone (but not bone marrow), exceeds that in plasma by threefold, but did not change across a 24-hour period. This might suggest that FQs (or difloxacin) bind to bone, which may preclude activity. Frazier and coworkers189 compared the disposition and homogenate tissue concentrations of difloxacin (5 mg/kg), enrofloxacin (5 mg/kg; ciprofloxacin also measured), and marbofloxacin (2.75 mg/kg) after multiple dosing (5 days) in the same dogs using a randomized crossover design (21 day washout period); drugs were detected using HPLC. Their studies demonstrate that the FQs accumulate in tissues with multiple dosing. Concentrations increased in the skin to reach a 4-day peak that exceeded the 1-day concentration by at least threefold. The concentrations in skin (μg/mL) at 1 and 4 days were, respectively, as follows: marbofloxacin (1.87 and 4.9), enrofloxacin (1.38 and 5.99), ciprofloxacin (0.2 and 0.5 for a total bioactivity of 1.59 and 6.9), and difloxacin (1 and 3.8). Urine concentrations also were higher at day 4 compared to day 1, with the magnitude varying for each drug. The concentrations in urine (μg/mL) were at 24 and 98 hours, respectively: marbofloxacin (14 and 50), enrofloxacin (0.14 and 1.83) plus ciprofloxacin (5.61 and 33.3 for a total bioactivity of 5.9 and 39), and difloxacin (0.56 and 1.8).
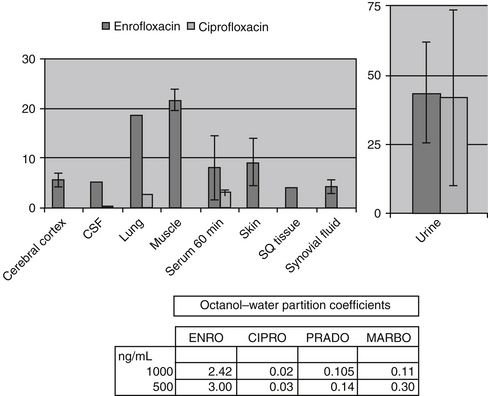
Figure 7-10 Selected tissue homogenate concentrations of enrofloxacin 2 hours after intravenous administration of 20 mg/kg. Concentrations in fluids are most relevant to bacterial exposure. Ciprofloxacin concentrations reflect metabolism of enrofloxacin to ciprofloxacin. Octanol–water partition coefficients suggest that enrofloxacin would distribute best into fluids at physiologic pH.
Homgenate data has been reported for enrofloxacin in anesthestized dogs (n = 4) receiving 20 mg/kg of enroflxoacin IV dogs.188 The 1- and 2-hour serum concentrations were 8.2 and 6.4 μg/mL (ciprofloxacin 3.1 and 2.8 μg/mL), respectively. Homogenate tissue to plasma ratios at 2 hours from lowest to highest were, in order, tracheal cartilage (0.2), aqueous humor (0.3), synovial fluid and subcutaneous tissue (0.4), peritonenal fluid and CSF (0.5), and brain (0.6).178 For fluids located in sanctuaries, at 1 hour aqueous humor (n = 2) achieved 2.5 μg/mL enrofloxacin and 0.5 μg/mL ciprofloxacin; peak CSF concentration of 5.3 μg/mL occurred at 2 hours (one dog). For aqueous humor a second study documented, 0.23 μg/mL of enrofloxacin and 0.064 μg/mL of ciprofloxacin 3 hours after 4 days of oral and 1 day of intravenous dosing at 5 mg/kg.180 The ratio of tissue to plasma concentrations were similar for ciprofloxacin and enrofloxacin (see Table 7-13).Another study documented that marbofloxacin (2 mg/kg, administered intravenously) achieves 0.41 μg/mL in aqueous humor at 3.5 hours in dogs.191 Other ratios of plasma to tissue enrofloxacin after 20 mg/kg administered intravenously192 included ligament (0.6), ear cartilage (0.7), and bone marrow (0.8). Concentrations in the prostate were 2.5-fold higher and urine 4.5-fold higher than in plasma (urine concentration of 45 μg/mL). Interstitial fluid concentrations of enrofloxacin (and formed ciprofloxacin) and marbofloxacin have also been measured using ultrafiltration. After 10 mg/kg enrofloxacin administered intravenously, the ratio of Cmax in interstitial fluid (2.41 μg/mL) compared with plasma (5.54 μg/mL) was 0.47; the ratio for AUC, however, was 1.3, indicating that the drug appears to stay longer in interstitial fluid compared with plasma.177 A second study176 determined plasma to interstitial fluid ratios after 5 mg/kg, administered orally, for marbofloxacin (approximating the highest labeled dose) and enrofloxacin (the lowest once-daily dose). Plasma to interstitial fluid Cmax ratio was 0.75 for marbofloxacin and 0.7 for enrofloxacin plus ciprofloxacin and for AUC was 1.11, for marbofloxacin and 1.3 for enrofloxacin and ciprofloxacin. The higher AUC for marbofloxacin reflected in part the higher Cmax but also a longer elimination half-life (8.5 hours) compared with enrofloxacin (3 hours). All FQs that have been studied thus far (enrofloxacin, marbofloxacin, pradofloxacin, and ciprofloxacin) accumulate in phagocytic WBCs; concentrations may be up to 140-fold higher compared with plasma (see Table 7-5).164,193-196 Drug in phagocytes will be distributed to sites of inflammation, thus increasing concentrations at the site of infection.196 Impact on intracellular killing is controversial. Whereas some studies have demonstrated that FQs retain intracellular killing effects compared with macrolides,197 another in vitro study demonstrated reduced intracellular killing ability for a variety of FQs.184
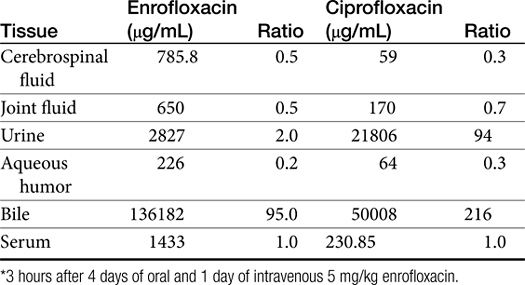
KEY POINT 7-25
The bioactivity of enrofloxacin can be doubled by formation of its more potent metabolite, ciprofloxacin.
The organ of elimination varies among the FQs. Difloxacin is eliminated almost exclusively by hepatic metabolism to inactive metabolites. Orbifloxacin is 40% eliminated unchanged in the urine. Marbofloxacin (clearance of 1.6 L/min) is largely excreted into the urine. However, up to 15% is metabolized in the liver to inactive metabolites,198 with the proportion changing in the presence of renal disease.198 Enrofloxacin also is eliminated in the urine as the unchanged drug, although approximately 25% of the drug is metabolized to ciprofloxacin, which subsequently achieves concentrations severalfold higher than enrofloxacin (Figure 7-11; see also Figure 7-8 and Table 7-13). Therapeutic concentrations of ciprofloxacin can be achieved in other tissues after administration of enrofloxacin, depending on the target organism.161,193,199 The parent and metabolite should act in an additive fashion.160 Because ciprofloxacin is characterized by a longer half-life than enrofloxacin in dogs (see Table 7-1), as a metabolite, ciprofloxacin can double the AUC of enrofloxacin bioactivity (see Table 7-1 and Figure 7-11).193 Longer elimination half-lives also characterize difloxacin and marbofloxacin compared with orbifloxacin and enrofloxacin, contributing to higher AUC for these drugs (see Table 7-1). Elimination half-lives are somewhat dose dependent,193 at least for enrofloxacin and ciprofloxacin (see Table 7-1). Alkaline urine increases the passive reabsorption of FQs from the renal tubules and may also prolong the elimination half-life. The longer half-lives should increase efficacy by increasing the likelihood that the drug will achieve the target AUC/MIC. Heinen200 compared PDIs among the FQs after oral administration, using a bioassay that detects both parent compound and active metabolites (see Table 7-1). Based on MIC90 determined for E. coli and Staphyloccoccus spp. (from isolates before 1999), for no drug was the targeted AUC/MIC achieved for Staphylococcus spp. and only enrofloxacin achieved the Cmax/MIC for Staphylococcus spp. Enrofloxacin (5 mg/kg) had the highest Cmax/MIC toward E. coli, followed by marbofloxacin (2 mg/kg) and orbifloxacin (2.5 mg/kg); difloxacin (5 mg/kg) did not reach the targeted Cmax/MIC or AUC/MIC for either organism. A similar pattern of efficacy was found among the veterinary FQs by Boothe using isolate MIC and reported Cmax,164 with enrofloxacin plus ciprofloxacin > ciprofloxacin > marbofloxacin > orbifloxacin > difloxacin being the general pattern of magnitude in PDI. However, the higher dose was generally needed to reach desired targets for PK/PD indices; isolates had been collected from 1998 to 2000 suggesting the likelihood of achieving targeted PDI with current isolates is less likely.
Figure 7-11 Enrofloxacin is metabolized by de-ethylation to ciprofloxacin. The two compounds will act in an additive fashion. The dotted line in the top graph indicates the predicted amount of bioactivity resulting from both enrofloxacin and its active metabolite, ciprofloxacin, after administration of 10 mg/kg. The longer half-life of ciprofloxacin can contribute to a longer duration. The graph demonstrates the accumulation of both enrofloxacin and ciprofloxacin in white blood cells (top two plots).
The disposition of enrofloxacin in neonatal kittens differs from that in adults, appearing to be age dependent even in the pediatric patient.185 Administration of 5 mg/kg to 2- to 8-week-old kittens185 revealed a shorter half-life at all ages but a Vdss that was less at 2 to 4 weeks and greater at 6 to 8 weeks compared with that of adults. Accordingly, Cmax was lower in the 6- to 8-week-old kittens. Enrofloxacin was generally poorly bioavailable at all ages.
Pradofloxacin
Pradofloxacin is a newer-generation FQ that may be undergoing approval in animals in the United States and the European Union. Structurally, it is characterized by a cyclopropyl ring at N1 (see Figure 7-8) that increases bacterial killing. A diazabicyclononyl moiety at C7 appears to physically block drug efflux through porins and targets both topoisomerases such that mutation must be multistep.154 Its spectrum includes P. aeruginosa. However, many anaerobes also will be effectively targeted. At 3 mg/kg orally for 5 days in dogs, Cmax was 1.7 ± 0.9 μg/mL and 6.2 ± 2.3 μg/mL in dogs (n = 6); half-life was 10 ± 7 hr at 3 mg/kg and 5.9 ± 1.5 hr at 12 mg/kg.190 The long half-life results in an AUC/MIC that is favorable compared with the other FQs. Pradofloxacin also has been studied in anesthetized dogs. It appears to be well distributed among the tissues.190 Aqueous humor concentrations achieved 0.32 μg/mL after 5 days of administration (4 oral followed by 1 IV) at 5 mg/kg of pradofloxacin.
Ciprofloxacin
Although ciprofloxacin has been studied in dogs following intravenous and oral202 administration (Table 7.1), the studies used different animals, and limited information is available on its oral bioavailability in dogs. However, reports provided by the manufacturer indicate that ciprofloxacin is only 33% to 40% bioavailable in dogs203 compared with nearly 80% to 100% in humans. Oral absorption of ciprofloxacin in dogs involves a dose-dependent nonlinear component that may affect its oral absorption.202 Oral and bioavailability of ciprofloxacin in cats (using pure powder in gelatin capsules) appears to be less than that in dogs, being 20% ± 11% following single dosing and 33% ± 12% after multiple dosing.205 Oral absorption was characterized by marked interanimal variability, suggesting that oral absorption may be minimal in some cats.205 This suggests that oral ciprofloxacin should be avoided in cats, and oral dosing in both cats and dogs should err on the side of higher doses to compensate for unpredictable oral bioavailability. More than several human-marketed generic preparations of oral ciprofloxacin are now available at a greatly reduced cost compared with oral enrofloxacin. However, whereas bioequivalence of a generic product must be proved to the pioneer product, this proof is generated only in the species in which the drug is approved. That the PK behavior of an orally administered generic drug will behave the same way in a nonapproved species should not be assumed.
The disposition of ciprofloxacin has been described in cats after intravenous administration of 10 mg/kg (see Table 7-1). In cats Vdss of ciprofloxacin is 3.85 ± 1.34 L/k, and plasma clearance is 0.64 ± 0.28 L/hr/kg, which exceeds the normal feline glomerular filtration rate (0.15-0.25 L/h/kg), suggesting that active tubular secretion occurs.206 AUCs after intravenous and oral administration are 17 ± 5 and 3 ± 1.2 μg∗hr/mL, respectively, in cats. Drug accumulation was not significant after seven oral administrations.205 Ciprofloxacin is metabolized into active (in humans) and inactive metabolites (N-oxide [the primary metabolite in dogs] and N-desmethyl). However, high concentrations of unchanged drug are achieved in urine, as is demonstrated after administration of enrofloxacin (see Figure 7-10).
Levofloxacin
Levofloxacin is the optical S-isomer of the racemic drug substance ofloxacin (see Figure 7-8). Compared with older FQs, its spectrum includes mycoplasma and gram-negative organisms, but the spectrum is broader toward gram-positive organisms and includes anaerobes.207 Ofloxacin is marketed as the levo isomer (i.e., levofloxacin) rather than the racemic mixture because the L-isomer is much more active against bacterial pathogens than the R-isomer. In humans levofloxacin is well absorbed orally, is distributed to a volume of 1.1 L/kg, and is renally excreted. Concentrations in the CSF approximate 16% of that in plasma, suggesting that the drug may not be well distributed into sanctuaries. Excretion is correlated with creatinine clearance, and half-life is prolonged with renal disease, requiring dose adjustments in patients with significant renal dysfunction.208
Because of its spectrum and improved antibacterial activity compared with veterinary FQs, levofloxacin has been used anecdotally in dogs but does not appear to have been studied in dogs. However, ofloxacin (but not its isomers) has been studied after oral administration in young and mature Beagles.209 Peak concentrations (measured by HPLC) at 20 mg/kg were 14.2 ± 0.4 μg/mL. The dispositions of the L-and D-isomer are likely to differ, precluding prediction of the proportion of the Cmax represented by levofloxacin. However, even if 100% of the drug is the L-isomer, concentrations are still well below the MIC90 of levofloxacin. The disposition of levofloxacin has been well described in cats on the basis of a bioassay after intravenous and oral administration,210 and it does not appear to be substantially different from that in humans. In cats the drug is well, albeit slowly, absorbed orally (Tmax 1.6 hours), with bioavailability at 87%. The drug is rapidly distributed, reaching a Vdss of 1.75 L/kg; clearance is 0.14 L∗hr/kg, and mean residence time is 13 hours (see Table 7-1). The Cmax following oral administration was 4.7 μg/mL, indicating that the drug should be used in cats only for organisms with an MIC of 0.5 μg/mL or less.
Drug Interactions
The FQs inhibit selected hepatic drug-metabolizing enzymes and are known to prolong the elimination of selected drugs. Theophylline toxicity has been documented in humans and dogs (see Chapter 2) simultaneously receiving theophylline and ciprofloxacin or enrofloxacin.211 Marbofloxacin also impairs the elimination of theophylline in dogs, but the effect is dose dependent, being absent at 2 mg/kg. However, at 5 mg/kg, theophylline clearance is decreased by 26% (compared with 50% reduction by enrofloxacin at 5 mg/kg IV once a day for 5 days), resulting in a change in theophylline half-life from 3.6 to 5.4 hours and a change in Cmax from 32 (no marbofloxacin) to 44 μg/mL (5 mg/kg marbofloxacin).212 Ciprofloxacin has been associated with increased cyclosporine concentrations, prolonged anticoagulant effects of warfarin, and enhanced hypoglycemic effects of oral hypoglycemics and insulin. Presumably, enrofloxacin and other FQs might have similar effects. Because of chelation by magnesium, calcium, and other cations, drugs such as antacids, sucralfate, and multiple vitamins should not be administered orally at the same time as a FQ. Because FQs competitively inhibit gamma-aminobutyric acid receptor binding, drugs that act similarly (e.g., selected nonsteroidal antiinflammatory drugs) when used in combination may increase the risk of seizural or other CNS activity. Enrofloxacin has been associated with false glucosuria.77
The use of FQs in combination with other antimicrobials may result in synergistic activity (e.g., aminoglycosides for gram-negative organisms; beta-lactams for gram-positive or gram–negative organisms) (see Chapter 6) or antagonistic (e.g., ribosomal inhibitors).
Adverse Effects
Adverse reactions to the FQs do not reflect interaction with mammalian topoisomerases. Most adverse reactions are predictable and can be prevented with proper administration. Gastrointestinal upset manifested by vomiting, nausea, and possibly diarrhea may occur after any route of administration but particularly oral administration. The intramuscular administration of enrofloxacin frequently causes pain on injection. Nausea and vomiting have been reported when the intramuscular solution is given intravenously and may reflect mast cell degranulation and histamine release. The intramuscular solution also is very alkaline (pH 10). Diluting the drug in saline and administering it over a 30-minute period may reduce nausea and clinical signs consistent with an anaphylactoid response. FQs have been associated with allergic reactions; however, the lack of previous exposure in some (human) patients (and in the author’s experience with ciprofloxacin) suggests an anaphylactoid rather than anaphylactic reaction.213Acute cardiovascular toxicity (hypotension, decreased left ventricular function) has been described for levofloxacin (Freedom of Information [FOI]) after an intravenous bolus (≥6 mg/kg) or intravenous infusion (≥20 mg/kg, but not ≤10 mg/kg). Increased circulating histamine concentrations accompanied the high-dose intravenous infusion, indicating a potential anaphylactoid reaction at 10, 15, 30, and 60 mg/kg intravenous bolus. Death occurred in dogs in association with neurologic and cardiac signs at 200 mg/kg, administered intravenously. Enrofloxacin also is available as a more concentrated solution (100 mg/mL) approved for use in cattle. However, it is prepared an an arganine-based vehicle, which is painful on injection and will cause perivascular inflammation if given parenterally by any route other than intravenous. Ulcers may occur if the large animal prepration is given orally.
Cartilage deformities and ligament and tendon repair
The FQs are associated with cartilage damage in dogs (and other species) (see package inserts). Enrofloxacin’s original package insert cited clinical signs indicative of cartilage damage in Beagle puppies within 3 days of treatment at 12.5 mg/kg. Lesions have been documented in dogs treated with other FQs. For levofloxacin, arthropathies occurred in juvenile dogs at ≥10 mg/kg/day for 7 days (FOI). Lesions in adult dogs require much higher concentrations, as was demonstrated for levofloxacin: the no-observed-effect level was 3 mg/kg/day in normal 7- to 8-month-old dogs compared with 30 mg/kg/day in normal 18-month-old dogs. The arthropathic potential of ofloxacin (the racemic mixture of levo and the R-isomer of ofloxacin) also has been studied in dogs.209 At 20 mg/kg for 8 days, eight out of eight 3-month-old animals developed histologic lesions, whereas only two developed clinical signs; the associated serum ofloxacin concentration was 14 μg/mL. The mechanism of cartilage damage is not known, although the most likely mechanism appears to be chelation of magnesium ions leading to dysfunction of integrins. These cell membrane proteins regulate a variety of cellular functions, including chondrocyte adherence to extracellular matrix and proteoglycan synthesis.215 Magnesium-deficient diets in juvenile rats led to cartilage damage similar to that caused by FQs.216 Indeed, magnesium supplementation may reverse the effects of FQs on canine chondrocytes.217 Dogs may be among the most sensitive and the most likely to exhibit clinical lameness caused by FQ-induced cartilage damage.204 Note that cartilage lesions as a result of FQs might be considered when FQs are used in any situation that involves growing or repairing cartilage, such as septic or immune-mediated arthritis and potentially osteoarthritis. Lesions have also been reported in other species, including humans.218 Use of chondroprotectants (i.e., polysulfated glycosaminoglycans) might be considered if FQ therapy must be instituted in growing dogs or other situations involving cartilage growth or repair.
The FQs appear to negatively affect healing in damaged ligaments.219 Connective tissue proteins decreased by up to 73% in dogs treated with as little as 30 to 200 mg/kg ciprofloxacin orally. Lesions were similar to those produced in magnesium-deficient dogs, suggesting that FQs induce tendon or ligament damage by antagonizing magnesium effects in the affected tissues.
The impact of FQs on bone repair also may be of concern. Based on experimental fracture healing in rats receiving placebo, cefazolin, or ciprofloxacin (50 mg/kg every 12 hours subcutaneously for any of the aforementioned drugs), fracture callus healing appeared to be impaired by FQs.218 In vivo studies in dogs of the effects of ciprofloxacin at 30 to 200 mg/kg/day orally (equivalent to approximately 15 to 65 mg/kg bioavailable drug) in dogs on either a normal or magnesium-deficient diet found a number of proteins were decreased in both groups at all doses, including collagen, elastin, and fibronectin.219 Of these effects, the authors concluded that magnesium deficiency increases the risk of impaired healing in the presence of FQs.
Seizures and other central nervous system disorders
Seizures and other CNS disorders have been precipitated in human and veterinary patients220 and animal models receiving FQs;221 predisposing factors include a preepileptic state, high doses, and concurrent use of nonsteroida1 antiinflammatory drugs.220 Newer drugs may be more likely to cause CNS side effects.222 FQs (and imipenem) inhibit GABA release, leading to hyperexcitability;76 inhibition of N-methyl-d-aspartate or adenosine may also be involved.143 FQs also lower seizure threshold and impede neuromuscular transmission. Peripheral neuropathies are a recognized side effect of FQs in humans.13,223 Clinical signs in humans have been described as severe, involving multiple organs. Onset is described as rapid (within 24 hours of onset of therapy; 84% afflicted within 1 week) and long term in duration, with symptoms lasting more than 3 months in 71% of afflicted patients and more than 1 year in 58%. The majority of cases involved levofloxacin (64%), despite ciprofloxacin (21%) being the most commonly prescribed drug. The most frequent complaints included both sensory (tingling, burning, or numbness) and motor (musculoskletal, cardiovascular, skin, gastrointestinal [cramping]) abnormalities; symptoms were described as severe in 80% of the patients.
Dose-dependent retinal degeneration
Dose-dependent retinal degeneration has been associated with use of FQs in cats. The incidence of ocular toxicity is very rare, occurring in 1 of 125,000 cats receiving enrofloxacin. The incidence at high doses is sufficiently low that toxicity was not detected in preapproval toxicity studies. During preapproval in cats, 25 mg/kg/day for 30 days and 125 mg/kg for 5 days were not associated with detectable toxicity. It is not clear whether ocular toxic-specific outcomes were addressed. Doses in clinical reports224 in which ocular toxicity occurred (retrospective study) ranged from 4.6 to 54 mg/kg/day, with duration of dosing ranging fom 4 to 120 days. Clinical signs began with mydriasis, rapidly followed by acute blindness. Age may be a factor, with cats younger than 9 years seemingly requiring a higher (>20 mg/kg) dose. Diseases associated with changes in disposition that might result in high plasma enrofloxacin concentrations (e.g., renal disease, heart disease) may also increase the risk. Intravenous administration may increase the risk, further supporting the concentration dependence of toxicity.
Experimental studies by Bayer Animal Health in young, apparently healthy cats at 5, 20, and 50 mg/kg/day for 21 days found electroretinography changes in one of six cats at 20 mg/kg and severe changes in six of six cats within 1 week at 50 mg/kg. Manufacturers of other veterinary FQs have likewise performed follow-up ocular toxicity studies. Marbofloxacin was not associated with lesions in young cats treated with up to 27 mg/kg/day for 6 weeks or 55 mg/kg/day for 14 days. Orbifloxacin was not associated with lesions at 15 mg/kg/day orally for 30 days, but changes occurred at 45 and 75 mg/kg.225
KEY POINT 7-27
Among the fluorinated quinolones currently approved for use in cats in the United States, marbofloxacin appears to be the least likely to cause retinal degeneration in cats.
The mechanism of ocular toxicity appears to reflect a mutation in four amino acids of an efflux protein in the blood-retina barrier, rendering it ineffective. Effective protein activity is absent in all cats. (personal communication, Dr. Katrina Mealey, Washington State University). The FQs are structurally similar to compounds known to cause accumulation in lysosomes of retinal pigment cells and subsequent ocular toxicity. Additionally, FQs have a predilection for pigmented cells of the eye. The FQs also have been associated with phototoxicity. The combination of FQs with ultraviolet radiation produces both a time- and concentration-dependent ocular toxicity, with a methyl group at position 8 of the quinolone ring reducing the risk.226 Reducing exposure to sunlight (dosing at night, or keeping cats indoors) might be prudent for cats receiving FQs.
Induction of bacteriophage supergenes
Induction of bacteriophage supergenes has been associated with the use of FQs, and in dog bacterial isolates, specifically enrofloxacin. Shortly after approval of enrofloxacin in Canada, seven canine cases of streptococcal toxic shock syndrome (STSS) and/or necrotizing fasciitis (NF) were reported; four of the dogs had been treated with enrofloxacin in the early stages of infection. Treatment was not only ineffective, but the syndrome appeared to be worsened by the antimicrobial therapy.227 Further investigation has provided some insight into the possible relationships between STSS and NF and bacteriophage supergenes in S. canis. Using polymerase chain reaction analysis, 22 of 23 S. canis isolates in one study exhibited a bacteriophage-encoded streptococcal superantigen gene. Under culture conditions, induction of the bacteriophage by enrofloxacin at therapeutic concentrations resulted in a 58-fold enhancement of expression of the gene.228 Apparently, the FQ stimulates autoingestion of a repressor protein that otherwise would prevent the bacteriophage from becoming lytic. FQs apparently also can induce bacteriophage lysis and enhanced Shiga toxin production in E. coli. For example, ciprofloxacin-treated mice experimentally colonized by Shiga-toxigenic E. coli died while their untreated colonized cohorts did not; increased Shiga toxin was demonstrated in their feces. However, induction requires ideal conditions, being dependent in part on stage and rate of growth and ideal drug concentration; conditions favoring bacteriophage induction in clinical patients have not yet been described.
Therapeutic Use
The FQs originated from nalidixic acid, itself a by product of chloroquine.150 Nalidixic acid is characterized by a narrow spectrum, and its use was limited to treatment of urinary tract infections. Modifications of chemical structures increasingly have improved the drugs, yielding drugs that have among the broadest of antibacterial spectrums. However, caution should be exercised with selected drugs because efficacy toward specific organisms (e.g., Pseudomonas, spp. anaerobes) varies. The FQs also are characterized as a class among those with the greatest tissue and antimicrobial distribution patterns. However, differences in tissue distribution (e.g., enrofloxacin versus ciprofloxacin, bone distribution of difloxacin) does indicate prudence when comparing FQ use. The rapid bactericidal effect of FQs is of clinical benefit in life-threatening situations or immune-suppressed patients; concentration dependence allows once-daily dosing that improves owner compliance. Intracellular accumulation of these drugs supports use for recurrent infections caused by intracellular organisms or at sites characterized by marked inflammation. Plasmid-mediated resistance has been slow to develop, although increasingly resistance, particularly that associated with multidrug resistance, is limiting FQ use. Oral bioavailability allows prolonged administration on an outpatient basis. However, bioavailability of the different drugs varies among the species, and good oral bioavailability should not be assumed. Rather, extrapolation of oral doses should be based on scientific studies. The unique mechanism of action of these drugs renders them appealing for combination antimicrobial therapy.
However appealing these numerous attributes of the FQs, common use of these drugs is discouraged. Widespread use—and abuse—of these drugs in the past 2 decades has proved that antimicrobial resistance can and will occur. Resistance, when it does occur, is often associated with multidrug resistance affecting chemically unrelated drugs. The emergence of of MDR with newer FQs needs to be assessed. Confirmation of the need for the drug and attention to MPCs (see Chapter 6) in the design of the dosing regimen should be two hurdles that are consciously addressed each time these drugs are considered. The metabolism of enrofloxacin to ciprofloxacin and the reduced oral bioavailability of ciprofloxacin in dogs and cats coupled with the importance of ciprofloxacin as a human-medicine drug call for extra caution to be taken. Once the decision is made to use an FQ, strict adherence to the principles of antimicrobial therapy, with a special focus on proper dosing regimens, is paramount to protecting this class of antimicrobial drugs, which is so critical to the medical community.
Rifamycins
Rifamycins are macrocylic antibiotics produced by Amycolatopsis mediterranei. Several semisynthetic derivatives) of natural rifamycins (rifamycin SV, Rifampin, rifampicin, rifamiderifamide) have been used as extended-spectrum antibiotics.150 Rifampin is among them. A large molecule (MW 823; see Figure 7-4) as with all rifamycins, it inhibits the B subunit of DNA-dependent RNA polymerase, suppressing RNA synthesis. Because mammalian RNA polymerase does not bind to rifamycins, its inhibition requires much higher concentrations. Rifampin can achieve bactericidal concentrations in some tissues. Effects are concentration-dependent for mycobacterium but unclear for other organisms. However, resistance develops very rapidly, markedly curtailing its use, and in general, rifampin should be used only in combination with other effective antimicrobials. Resistance may develop in as little as 2 days when it is used as the sole antimicrobial; rifampin is used experimentally to study mutation frequencies in some organisms. The use of rifampin as sole agent for treating pyoderma is addressed in Chapter 8. Resistance generally reflects a single mutation that changes the affinity of the target enzyme for the drug. Resistance (and efficacy) can be decreased with combination therapy with a number of drugs, including erythromycin, most beta-lactam antibiotics, chloramphenicol, doxycycline, and selected aminoglycosides. Rifampin has shown some efficacy against fungal microorganisms.
Spectrum
The spectrum of activity of rifampin includes primarily gram-positive (especially Staphylococcus spp.) organisms (see Table 7-4). However, it also is effective against Mycobacterium, Neisseria, and Chlamydia spp. and has been used to treat Clostridium and Bacteroides species. Rifampin has limited activity against gram-negative organisms (including Brucella). Resistant gram-negative organisms include E. coli, Enterobacter spp. K. pneumoniae, Proteus spp. Salmonella spp., and P. aeruginosa. However, an Internet search reveals a number of papers that indicate efficacy toward P. aeruginosa when combined with a number of other drugs. Highly susceptible gram-positive organisms are considered to have an MIC of 0.25 μg/mL or less; MICs are often less than 0.1 μg/mL. In contrast, the MIC of gram-negative organisms is generally 8 to 32 μg/mL; the higher MICs reflect limited penetration of gram-negative organisms. A dose of 10 mg/kg in the dog achieves a Cmax of 40 μg/mL (see Table 7-1); accordingly, its use for gram-negative isolates (and ideally, all isolates) should be based on C&S testing.
Pharmacokinetics
Rifampin may be administered intramuscularly, intravenously, or orally with systemic effects. Oral absorption of rifampin is incomplete in humans (∼40%) with peak plasma concentrations occurring in 2 to 4 hours. Concurrent feeding may reduce or delay absorption. Because it is a substrate for P-glycoprotein,229 oral absorption may be much higher in dogs exhibiting P-glycoprotein deficiency. Approximately 75% to 80% of rifampin is bound to plasma proteins. Rifampin is very lipid soluble, distributing well to most body tissues. It concentrates in white blood cells and is characterized by immunmodulation.230 Because rifamycins penetrate tissues and cells to a substantial degree, they are particularly effective against intracellular organisms. Rifampin is rapidly eliminated after acetylation to a metabolite (desacetyl rifampin) that is equal in efficacy to the parent compound. Whether the dog is a deficient acetylator of rifampin is unclear. Both the parent and metabolite are excreted in the bile (supporting its use for cholangitis in humans); the parent compound and metabolite undergo enterohepatic circulation. The elimination half-life of rifampin is dose dependent, being about 8 hours in dogs.
Adverse Effects
Rifampin is usually well tolerated and produces few side effects. However, gastrointestinal disturbances and abnormalities in liver function (icterus) have been reported in humans and may lead to discontinuation of therapy. Hypersensitivity reactions can also result from rifampin administration, and renal failure is a possible consequence when intermittent dosage schedules are followed. Partial, reversible immunosuppression of lymphocytes occurs. Urine, feces, saliva, sputum, sweat, and tears are often colored red-orange by rifampin and its metabolites; urine may stain. Plasma will also be orange and may be misinterpreted as hemoglobinemia. CNS depression after intravenous administration and temporary inappetence may occur. Interestingly, intermittent administration (less than twice weekly) increases the risk of side effects in humans, resulting in a flulike syndrome that is associated with clinical signs indicative of a drug reaction (eosinophilia, thrombocytopenia, hemolytic anemia [note potential for orange discoloration of plasma] and renal disease).150 In a limited number of dogs, marked increases in serum alkaline phosphatase have been observed by the author. No other liver enzyme or function tests were affected, and dogs did not become clinically ill. The increase may reflect induction of the enzymes (much the same as glucocorticoids or phenobarbital), but monitoring of hepatic function may be prudent in at-risk dogs receiving rifampin.
Drug Interactions
Rifampin is a broad, potent inducer of microsomal enzymes, including CYP1A2, 2C9, 2C19, and 3A4;150 as such, it will shorten the elimination half-life of a number of drugs and may increase the risk of toxicity associated with drug metabolism.229 Therapeutic failure may occur for other drugs metabolized by the liver if modifications in dosing regimens are not made. Rifampin PDCs will decrease after multiple dosing because of induction, with plasma elimination half-life of rifampin progressively shortening by approximately 40% during the first 2 weeks of treatment in humans. Other affected drugs include the imidazoles, cyclosporine, digoxin, and several sodium channel– and beta receptor–blocking cardiac antiarrhythmics. Endogenous substrates of hepatic metabolism also may be affected; several steroids will be more rapidly catabolized.150 Withdrawal syndromes have been reported in humans receiving opioid analgesics.150 Because rifampin is a substrate for P-glycoprotein, dogs with the MDR-1 (ABC) deletion will have an increased risk of adverse reactions; the risk is increased if rifampin is used in combination with other drugs that interact with this protein. Finally, rifampin also has decreased biliary secretion of some compounds, notably contrast imaging media.150 Rifampin has been used in combination with a number of drugs to enhance efficacy (and reduce resistance; see the section on resistance) for treatment of MRSA, VRE, and Mycobacterium spp. and others. Use in combination with doxycycline has been recommended for canine brucellosis, although clinical efficacy has not been demonstrated.2
Two other rifamycins are approved for use in humans. Rifabutin is a derivative of rifampin that is characterized by less induction of drug-metabolizing enzymes. Used for the treatment of Mycobacterium spp., it is characterized by unique side effects, including polymyalgia, anterior uveitis, and others. Rifapentine is used to treat tuberculosis associated with human immunodeficiency virus infections in humans. Its longer half-life allows once-weekly dosing, and its impact on drug-metabolizing enzymes has been described as intermediate.150 Rifaximin is a semisynthetic derivative of rifamycin that is not orally absorbed. It is indicated for treatment of enteric pathogens, including Campylobacter, C. difficile, E. coli, Helicobacter pylori, and Salmonella and Shigella.231 A potential advantage of rifaximin is an apparent minimal long-term effect on the gastrointestinal flora: both E. coli and Enterocococcus spp. were minimally affected after 3 to 14 days of therapy. Resistance to rifaximin seems to emerge only slowly, compared with systemic use of rifampin.231 Indications in humans have been a variety of (nonbloody) diarrheas, including small bowel overgrowth, intestinal gas, and inflammatory bowel disease.
Metronidazole
Metronidazole is deriviative of the antibiotic azomycin (2 nitro-imidazole) secreted by a streptomycete (Figure 7-12).232 A number of other nitroimidazoles were developed from azomycin.232 Among the other closely related imadazoles used outside the United States are tinidazole, and benznidazole, the latter being used to treat acute Chagas disease. Metronidazole impairs microbial RNA and DNA synthesis but must first undergo nitrous reduction in the organism. As such, metronidazole is a prodrug, with efficacy depending on the nitrous group and a low redox potential that can be achieved only in an anaerobic environment.233 Only organisms that live in a low-oxygen environment have developed anaerobic energy or electron-generating pathways (e.g., ferredoxins) capable of generating single electrons. Transfer of the electron to the nitrous group of metronidazole results in a highly reactive nitro radical ion. Although DNA is the primary target, other macromolecular structures may be targeted. Metronidazole will be regenerated on death of the microbe, thus facilitating its efficacy. Efficacy appears to be predominantly bactericidal, although actions may be bacteriostatic toward some organisms (e.g., Eubacterium spp.). Metronidazole acts as a concentration-dependent drug against trichomoniasis, and, although this is not always clear, it also appears to be concentration dependent when treating other microbes. However, time dependence has also been ascribed234 (e.g., Clostridium and its efficacy appear to be similar if administered once or twice daily).235
Spectrum
Metronidazole is rapidly bactericidal against all gram-negative (e.g., B. fragilis) and most gram-positive (e.g., Clostridium spp.) anaerobic bacilli, generally at MIC equal to or less than 8 μg/mL. Microaerophilic microbes such as Helicobacter and Campylobacter spp. are susceptible. Metronidazole is effective toward a number of protozoa, with efficacy dependent on the nitro group at position 5 and enhanced with substitutions at the 2 position.236 Susceptible infections include trichomoniasis (MIC of 0.05 μg/mL if anaerobic conditions), amebiasis, and giardiasis (1 to 50 μg/mL).
Resistance
Aerobes and facultative anaerobic bacteria lack electron transport systems necessary to generate single electrons and thus are resistant to metronidazole. Further, in higher oxygen environments, oxygen will compete for the electrons generated by anaerobic organisms, thus decreasing efficacy of metronidazole. Higher doses are necessary if the infection occurs in an environment of 1% or more oxygen.232 Interestingly, protozoa may develop resistance ot metronidazole in patients with impaired oxygen-radical scavenging abilities.232 Microbes also acquire resistance by decreasing proteins that generate the electrons (e.g., ferredoxin). The mechanism of bacterial resistance is not totally clear, but increased production of interfering enzymes is likely. Resistance by Helicobacter spp. can be rapid.
Pharmacokinetics
Metronidazole is well distributed to all body tissues and can penetrate the blood–brain barrier. It is minimally protein bound (in humans). Elimination is dose dependent and occurs primarily by hepatic metabolism. At least one metabolite has 50% of the activity of the parent compound toward trichomonads. Intestinal microbes can produce a small amount of the reduced (active) metabolites. Peak concentrations in dogs after 44 mg/kg reached 42 μg/mL. Vd is 0.95 ± 0.1 L/kg, and clearance is 2.5 ± 0.54 mL/kg/min.237 Oral bioavailability is variable, ranging from 59% to 100%. Elimination half-life in one study was 4.5 ± 9 hours (see Table 7-1). Metronidazole disposition has been described in the cat after single intraveous (5 mg/kg) administration as the salt-free product and then at 20 mg/kg orally of the benzoate salt (12.4 mg/kg active drug).238 Extrapolated plasma concentration after intravenous administration at time 0 averaged 7.8 ± 2 μg/mL; Vd was 0.7 ± 0.3 L/kg, and plasma clearance was 91 mL/kg/hr. Elimination half-life and mean residence time were 5.3 ± 0.7 and 7.6 ± 1 hours, respectively. The benzoate salt was fairly well absorbed but was characterized by clinically significant variability, with a bioavailability of 65% ± 27% (range 28% to 80%). The Cmax also varied, with a mean of 8.8 + 5.4, reflecting a range of 4.9 to 17.8 μg/mL; Tmax also varied from 1 to 8 hours (mean 3.6 ± 2.9 hours). Elimination half-life and mean residence time after oral administration were 5.2 ± 0.5 and 8.7 ± 1.3 hours, respectively.
Adverse Effects
Metronidazole may discolor urine (red-brown).232 More problematic adverse reactions include gastrointestinal upset (including hepatotoxicity when given at high doses) and CNS adversities, including seizures.237,239 The risk of neurotoxicity is increased with intravenous administration; as such, oral administration is the preferred route whenever possible. The caustic nature of the intravenous solution also necessitates slow intravenous administration. The mechanism of neurotoxicity is not known, but in mice degenerative lesions have been demonstrated in the Purkinje cells, vestibular tracts, and several nuclei associated with equilibrium and fine motor control. These areas are also the site of the majority of gamma-aminobutyric acid–minergic receptors. In humans, characteristic lesions seen on magnetic resonance imaging indicate that the cerebellum may be most sensitive to damage; because interstitial edema was evident, with axonal swelling was suggested as a cause.240
In dogs, seizures are indicative of toxicity. One study in dogs (n = 21) induced seizures at doses of 60 to 110 mg/kg for a total of 10 to 110 days.241 The most common clinical signs were vertical nystagmus, ataxia, inability to walk (≥50% each), and paraparesis (30%); less frequent neurologic signs included tetraparesis, hypermetria, tremors, head tilt, torticollis, and opisthotonus. Treatment with diazepam proved effective based on a shorter response time (resolution of debilitating clinical signs; 13 hours versus 4.5 days) as well as recovery time (return to normalcy; 11 versus 36 hours). The dose of diazepam was approximately 0.5 mg/kg, administered intravenously followed by oral administration every 8 hours for 3 days. Neurologic reaction to metronidazole has also been reported in cats (n = 2). The dose and duration associated with clinical signs were 111 mg/kg body for 9 weeks followed by 222 mg/kg/day for 2 days in one cat and 58 mg/kg for 6 months in the second.242 Clinical signs in cats included ataxia, altered mentation, and progression to seizures. Neurologic signs resolved within days of discontinuation of the drug and supportive therapy. Histologic lesions have also been described in another 14-year-old cat that developed fatal presumed metronidaozle toxicity after treatment for inflammatory bowel disease at 73 to 147 mg/kg/day. Among the neurologic clinical signs was acute tetraparesis; lesions included diffuse, multifocal areas of necrosis throughout the brainstem.243
Metronidazole as either the free form or when administered as the benazoate salt was genotoxic (disruptive of lymphocytic DNA) but not cytotoxic to feline polymorphonuclear cells. Genotoxicity resolved within 7 days after the drug was discontinued.
Preparations
Metronidazole is available as either a hydrochloride (used in the approved product) salt (oral or intravenous) or, in pure drug substrate form (i.e., for compounding), the benzoate salt. It can be administered as a loading dose infused over 30 to 60 minutes in fluids, followed by an intravenous drip. It also can be given intermittently as an 8- to 12-hour maintenance dose as long as the infusion takes place slowly. For intravenous administration the dose should be neutralized with sodium bicarbonate and mixed with lactated Ringer’s solution, saline, bacteriostatic water, or 5% dextrose in water (see package insert). Because intravenous administration of metronidazole is complicated, oral administration is preferred whenever possible.
The benzoate salt of metronidazole, which is not commercially available, is less bitter tasting and more tolerable than the commercially available hydrochloride salt. The oral disposition of the benzoate salt was previously described.238 However, the benzoate moiety is larger than the hydrochloride moiety, representing 38% of the drug product. As such, when dosed on total drug weight, the dose of metronidazole benzoate should be 1.6 times the dose of metronidazole hydrochloride.244 Further, the benzoate must be removed by desterification before its absorption; it is not clear if oral administration of the benzoate form will be as effective against gastrointestinal microbes compared with a nonbenzoate form.
Metronidazole (not studied as a salt) has been demonstrated to be stable in solutions when stored at 40° C for 90 days.245 However, it reacts with the aluminum of needles or other canulas. Metronidazole is subject to drug interactions associated with inhibition (e.g., cimetidine) or induction (e.g., phenobarbital, prednisone, rifampin) of drug-metabolizing enzymes.
Metronidazole is available as a topical gel, which provides wound odor control. Although it can be prepared as a transdermal PLO gel, studies by the author demonstrated minimal absorption when applied to the pinna of the ear for 3 weeks at 15 mg/kg.
Metronidazole is a drug of choice for treating infections caused by obligate anaerobes, particularly those associated with gastrointestinal flora. Increasingly, it is used in lieu of oral vancomycin to treat C. difficile. Frequently, it is cited as a treatment for inflammatory bowel diseases in animals or humans (particularly Crohn’s disease). Its efficacy may reflect, in part, immunomodulatory properties (see Chapter 19) or its ability to target those microbes most likely to produce inflammatory mediators.