CHAPTER 3 Management of Heart Failure
OVERVIEW OF HEART FAILURE
Heart failure entails abnormalities of cardiac systolic or diastolic function, or both. These can occur without evidence of abnormal fluid accumulation (congestion), especially in the initial stages of disease. Congestive heart failure (CHF) is characterized by high cardiac filling pressure, which leads to venous congestion and tissue fluid accumulation. It is a complex clinical syndrome rather than a specific etiologic diagnosis. The pathophysiology of heart failure is complex. It involves structural and functional changes within the heart and vasculature as well as other organs. The process of progressive cardiac remodeling inherent to heart failure can develop secondary to cardiac injury or stress from valvular disease, genetic mutations, acute inflammation, ischemia, increased systolic pressure load, and other causes.
CARDIAC RESPONSES
Cardiac remodeling refers to the changes in myocardial size, shape, and stiffness that occur in response to various mechanical, biochemical, and molecular signals induced by the underlying injury or stress. These changes include myocardial cell hypertrophy, cardiac cell drop-out or self-destruction (apoptosis), excessive interstitial matrix formation, fibrosis, and destruction of normal collagen binding between individual myocytes. The latter, resulting from effects of myocardial collagenases or matrix metalloproteinases, can cause dilation or distortion of the ventricle from myocyte slippage. Stimuli for remodeling include mechanical forces (e.g., increased wall stress from volume or pressure overload) and also various neurohormones (e.g., angiotensin II, norepinephrine, endothelin, aldosterone) and cytokines (e.g., tumor necrosis factor [TNF]-alpha). Contributing biochemical abnormalities related to cellular energy production, calcium fluxes, protein synthesis, and catecholamine metabolism have been variably identified in different models of heart failure and in clinical patients. Myocyte hypertrophy and reactive fibrosis increase total cardiac mass by eccentric and, in some cases, concentric patterns of hypertrophy. Ventricular hypertrophy can increase chamber stiffness, impair relaxation, and increase filling pressures; these abnormalities of diastolic function can also contribute to systolic failure. Ventricular remodeling also promotes the development of arrhythmias. The initiating stimulus underlying chronic cardiac remodeling may occur years before clinical evidence of heart failure appears.
Acute increases in ventricular filling (preload) induce greater contraction force and blood ejection. This response, known as the Frank-Starling mechanism, allows beat-to-beat adjustments that balance the output of the two ventricles and increase overall cardiac output in response to acute increases in hemodynamic load. The Frank-Starling effect helps normalize cardiac output under conditions of increased pressure and/or volume loading, but these conditions also increase ventricular wall stress and oxygen consumption.
Ventricular wall stress is directly related to ventricular pressure and internal dimensions and inversely related to wall thickness (Laplace’s law). Myocardial hypertrophy can reduce wall stress. The pattern of hypertrophy that develops depends on the underlying disease. A ventricular systolic pressure load induces “concentric” hypertrophy; myocardial fibers and ventricular walls thicken as contractile units are added in parallel. A volume load causes “eccentric” hypertrophy; myocardial fiber elongation and chamber dilation occur as new sarcomeres are laid down in series. Compensatory hypertrophy lessens the importance of the Frank-Starling mechanism in stable, chronic heart failure. Although volume loads are better tolerated because myocardial oxygen demand is not as severe, both abnormal pressure and volume loading impair cardiac performance over time. Eventually, decompensation and myocardial failure develop. In patients with primary myocardial diseases, initial cardiac pressure and volume loads are normal, but intrinsic defects of the heart muscle lead to the hypertrophy and dilation observed.
Cardiac hypertrophy and other remodeling begin long before heart failure becomes manifest. In addition to myocyte hypertrophy, cardiac remodeling can include cell loss or self-destruction (apoptosis), excessive interstitial matrix formation, and loss of normal collagen binding. Myocyte hypertrophy and reactive fibrosis increase total cardiac mass as well as ventricular stiffness. This promotes elevated filling pressures and predisposes the patient to ischemia. Increased chamber size increases wall stress and myocardial O2 demand. Biochemical abnormalities involving cell energy production, calcium fluxes, and contractile protein function can develop. Clinical heart failure can be considered a state of decompensated hypertrophy; ventricular function progressively deteriorates as contractility and relaxation become more deranged.
Continued exposure to increased sympathetic stimulation reduces cardiac sensitivity to catecholamines. Down-regulation (reduced number) of myocardial β1-receptors and other changes in cellular signaling may help protect the myocardium against the cardiotoxic and arrhythmogenic effects of catecholamines. Beta-blocking agents can reverse β1-receptor down-regulation but may worsen heart failure. Cardiac β2- and α1-receptors are also present but are not down-regulated; these are thought to contribute to myocardial remodeling and arrhythmogenesis. Another cardiac receptor subtype (β3-receptors) may promote myocardial function deterioration through a negative inotropic effect.
SYSTEMIC RESPONSES
Neurohormonal Mechanisms
Neurohormonal (NH) responses contribute to cardiac remodeling and also have more far-reaching effects. Over time, excessive activation of neurohormonal “compensatory” mechanisms leads to the clinical syndrome of CHF. Although these mechanisms support circulation in the face of acute hypotension and hypovolemia, their chronic activation accelerates further deterioration of cardiac function. Major neurohormonal changes in heart failure include increases in sympathetic nervous tone, attenuated vagal tone, activation of the renin-angiotensin-aldosterone system, and release of antidiuretic hormone (ADH-vasopressin). These neurohormonal systems work independently and together to increase vascular volume (by sodium and water retention and increased thirst) and vascular tone (Fig. 3-1). Excessive volume retention results in edema and effusions. Prolonged systemic vasoconstriction increases the workload on the heart, can reduce forward cardiac output, and may exacerbate valvular regurgitation. The extent to which these mechanisms are activated varies with the severity and etiology of heart failure. In general, as failure worsens, neurohormonal activation increases. Increased production of endothelins and proinflammatory cytokines, as well as altered expression of vasodilatory and natriuretic factors, also contribute to the complex interplay among these NH mechanisms and their consequences.
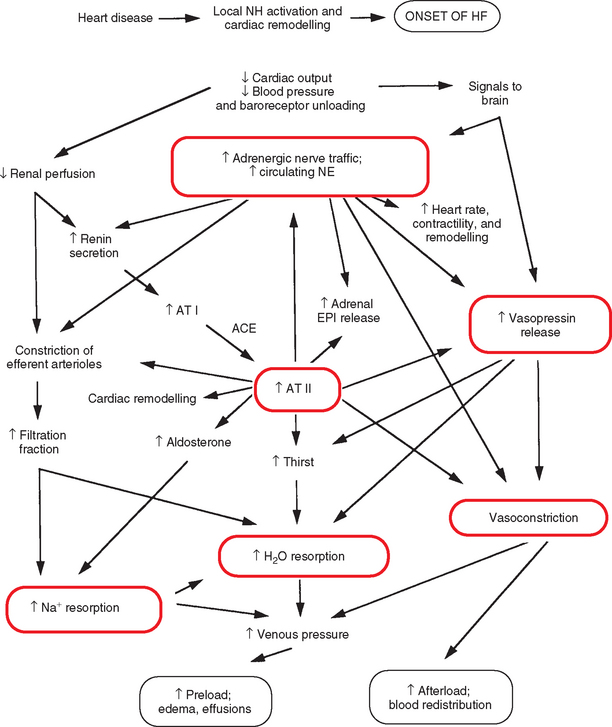
FIG 3-1 Major neurohormonal mechanisms leading to volume retention and increased afterload in congestive heart failure. ACE, Angiotensin-converting enzyme; AT, angiotensin; EPI, epinephrine; HF, heart failure; NE, norepinephrine.
The effects of sympathetic stimulation (e.g., increased contractility, heart rate, and venous return) can increase cardiac output initially, but over time these effects become detrimental by increasing afterload stress and myocardial oxygen requirements, contributing to cellular damage and myocardial fibrosis, and enhancing the potential for cardiac arrhythmias. Normal feedback regulation of sympathetic nervous and hormonal systems depends on arterial and atrial baroreceptor function. Baroreceptor responsiveness becomes attenuated in chronic heart failure, which contributes to sustained sympathetic and hormonal activation and reduced inhibitory vagal effects. Baroreceptor function can improve with reversal of heart failure, increased myocardial contractility, decreased cardiac loading conditions, or inhibition of angiotensin II (which directly attenuates baroreceptor sensitivity). Digoxin has a positive effect on baroreceptor sensitivity.
The renin-angiotensin system has far-reaching effects. Whether systemic renin-angiotensin-aldosterone activation always occurs before overt congestive failure is unclear, and may depend on the underlying etiology. Renin release from the renal juxtaglomerular apparatus occurs secondary to low renal artery perfusion pressure, renal β-adrenergic receptor stimulation, and reduced Na+ delivery to the macula densa of the distal renal tubule. Stringent dietary salt restriction and diuretic or vasodilator therapy can promote renin release. Renin facilitates conversion of the precursor peptide angiotensinogen to angiotensin I (an inactive form). Angiotensin-converting enzyme (ACE), found in the lung and elsewhere, converts angiotensin I to the active angiotensin II and is involved in the degradation of certain vasodilator kinins. There are also other pathways that generate angiotensin II.
Angiotensin II has several important effects, including potent vasoconstriction and stimulation of aldosterone release from the adrenal cortex. Additional effects of angiotensin II include increased thirst and salt appetite, facilitation of neuronal norepinephrine synthesis and release, blockade of neuronal norepinephrine reuptake, stimulation of antidiuretic hormone (vasopressin) release, and increased adrenal epinephrine secretion. Inhibition of ACE can reduce NH activation and promote vasodilation and diuresis. Local production of angiotensin II also occurs in the heart, vasculature, adrenal glands, and other tissues. Local activity affects cardiovascular structure and function by enhancing sympathetic effects and promoting tissue remodeling that can include hypertrophy, inflammation, and fibrosis.
Aldosterone promotes sodium and chloride reabsorption as well as potassium and hydrogen ion secretion in the renal collecting tubules; the concurrent water reabsorption augments vascular volume. Increased aldosterone concentration can promote hypokalemia, hypomagnesemia, and impaired baroreceptor function. Aldosterone is also produced locally in the cardiovascular system and mediates inflammation and fibrosis. Chronic exposure can be detrimental to ventricular function and contribute to pathologic remodeling and myocardial fibrosis.
Antidiuretic hormone is released from the posterior pituitary gland. This hormone directly causes vasoconstriction and also promotes free water reabsorption in the distal nephrons. Although increased plasma osmolality or reduced blood volume are the normal stimuli for ADH release, reduced effective circulating volume and other nonosmotic stimuli cause continued release of ADH in patients with heart failure. The continued release of ADH contributes to the dilutional hyponatremia sometimes found in patients with heart failure.
Increased circulating concentrations of other substances that play a role in abnormal myocardial hypertrophy and/or fibrosis, including cytokines (e.g., TNFα) and endothelins, have also been detected in animals with severe heart failure. Endothelin production is stimulated by hypoxia and vascular mechanical factors but also by angiotensin II, ADH, norepinephrine, cytokines (including TNFα and interleukin-I), and other factors.
Endogenous mechanisms that oppose the vasoconstrictor responses also are activated. These include natriuretic peptides, nitric oxide, and vasodilator prostaglandins. Normally, a balance between vasodilator and vasoconstrictor effects maintains circulatory homeostasis as well as renal solute excretion. As heart failure progresses, the influence of the vasoconstrictor mechanisms predominates despite increased activation of vasodilator mechanisms.
Natriuretic peptides are synthesized in the heart and play an important role in regulation of blood volume and pressure. Atrial natriuretic peptide (ANP) is synthesized by atrial myocytes as a prohormone, which is then cleaved to the active peptide after release stimulated by mechanical stretch of the atrial wall. Brain natriuretic peptide (BNP) is also synthesized in the heart, mainly by the ventricles in response to myocardial dysfunction or ischemia. Natriuretic peptides cause diuresis, natriuresis, and peripheral vasodilation. They act to antagonize the effects of the renin-angiotensin system and can also alter vascular permeability and inhibit growth of smooth muscle cells. Natriuretic peptides are degraded by neutral endopeptidases. Circulating concentrations of ANP and BNP increase in patients with heart failure. This increase has been correlated with pulmonary capillary wedge pressure and severity of heart failure in both dogs and people.
Nitric oxide (NO), produced in vascular endothelium in response to endothelial-nitric oxide synthetase (NOS), is a functional antagonist of endothelin and angiotensin II. This response is impaired in patients with heart failure. At the same time, myocardial inducible–NOS expression is enhanced; myocardial NO release has negative effects on myocyte function. Intrarenal vasodilator prostaglandins oppose the action of angiotensin II on the renal vasculature. The use of prostaglandin synthesis inhibitors in dogs or cats with severe heart failure could potentially reduce glomerular filtration (by increasing afferent arteriolar resistance) and enhance sodium retention.
Renal Effects
Renal efferent glomerular arteriolar constriction, mediated by sympathetic stimulation and angiotensin II, helps maintain glomerular filtration in the face of reduced cardiac output and renal blood flow. Higher oncotic and lower hydrostatic pressures develop in the peritubular capillaries, enhancing the reabsorption of tubular fluid and sodium. Angiotensin II–mediated aldosterone release further promotes sodium and water retention. Continued activation of these mechanisms leads to clinical edema and effusions.
Afferent arteriolar vasodilation mediated by endogenous prostaglandins and natriuretic peptides can partially offset the effects of efferent vasoconstriction, but progressive impairment of renal blood flow leads to renal insufficiency. Diuretics not only can magnify azotemia and electrolyte loss but can further reduce cardiac output and activate the neurohormonal mechanisms.
Other Effects
Reduced exercise capacity, along with skeletal muscle atrophy, occurs in patients with heart failure. Poor diastolic filling, inadequate forward output, and pulmonary edema or pleural effusion can interfere with exercise ability. Furthermore, impaired peripheral vasodilation during exercise contributes to inadequate skeletal muscle perfusion and fatigue. Excessive peripheral sympathetic tone, angiotensin II (both circulating and locally produced), and vasopressin can contribute to impaired skeletal muscle vasodilatory capacity in patients with CHF. Increased vascular wall sodium content and interstitial fluid pressure stiffen and compress vessels. Other mechanisms can include impaired endothelium-dependent relaxation, increased endothelin concentration, and vascular wall changes induced by the growth factor effects of various neurohormonal vasoconstrictors. ACE inhibitor therapy, with or without spironolactone, may improve endothelial vasomotor function and exercise capacity. Pulmonary endothelial function is improved by ACE inhibitors in dogs with CHF.
GENERAL CAUSES OF HEART FAILURE
The causes of heart failure are quite diverse; it can be useful to think of them in terms of underlying pathophysiology. In most cases of heart failure, the major initiating abnormality is myocardial (systolic pump) failure, systolic pressure overload, volume overload, or reduced ventricular compliance (impaired filling). Nevertheless, several pathophysiologic abnormalities often coexist; both systolic and diastolic function abnormalities are common in patients with advanced failure.
Myocardial failure is characterized by poor ventricular contractile function, and it is most commonly secondary to idiopathic dilated cardiomyopathy; valvular insufficiency may or may not be present initially but usually develops as the affected ventricle dilates. Persistent tachyarrhythmias, some nutritional deficiencies, and other cardiac insults also can lead to myocardial failure (see Chapters 7 and 8). Diseases that cause a volume or flow overload to the heart usually involve a primary “plumbing” problem (e.g., a leaky valve or abnormal systemic-to-pulmonary connection). Cardiac pump function is often maintained at a near-normal level for a prolonged time, but myocardial contractility does eventually deteriorate (see Chapters 5 and 6). Pressure overload results when the ventricle must generate higher-than-normal systolic pressure to eject blood. Concentric hypertrophy increases ventricular wall thickness and stiff ness and predisposes the patient to ischemia. Excessive pressure loads eventually lead to a decline in myocardial contractility. Myocardial pressure overload results from congenital ventricular outflow obstruction and systemic or pulmonary hypertension (see Chapters 5, 10, and 11). Diseases that restrict ventricular filling impair diastolic function. These include hypertrophic and restrictive myocardial disease and pericardial disease (see Chapters 8 and 9). Contractile ability is usually normal initially, but high filling pressure leads to congestion behind the ventricle(s) and may diminish cardiac output. Examples of common diseases are listed in Table 3-1 according to their main initiating pathophysiology and typical clinical manifestation of CHF signs.
 TABLE 3-1 Common Causes of Congestive Heart Failure (CHF)
TABLE 3-1 Common Causes of Congestive Heart Failure (CHF)
| MAJOR PATHOPHYSIOLOGY | TYPICAL CHF MANIFESTATION* |
|---|---|
| Myocardial Failure | |
| Idiopathic dilated cardiomyopathy | Either L- or R-CHF |
| Myocardial ischemia/infarction | L-CHF |
| Drug toxicities (e.g., doxorubicin) | L-CHF |
| Infective myocarditis | Either L- or R-CHF |
| Volume-Flow Overload | |
| Mitral valve regurgitation (degenerative, congenital, infective) | L-CHF |
| Aortic regurgitation (infective endocarditis, congenital) | L-CHF |
| Ventricular septal defect | L-CHF |
| Patent ductus arteriosus | L-CHF |
| Tricuspid valve regurgitation (degenerative, congenital, infective) | R-CHF |
| Tricuspid endocarditis | R-CHF |
| Chronic anemia | Either L- or R-CHF |
| Thyrotoxicosis | Either L- or R-CHF |
| Pressure Overload | |
| (Sub)aortic stenosis | L-CHF |
| Systemic hypertension | L-CHF (rare) |
| Pulmonic stenosis | R-CHF |
| Heartworm disease | R-CHF |
| Pulmonary hypertension | R-CHF |
| Impaired Ventricular Filling | |
| Hypertrophic cardiomyopathy | L-(+/- R-) CHF |
| Restrictive cardiomyopathy | L-(+/- R-) CHF |
| Cardiac tamponade | R-CHF |
| Constrictive pericardial disease | R-CHF |
* L-CHF, Left-sided congestive heart failure (pulmonary edema as main congestive sign); R-CHF, right-sided congestive heart failure (pleural effusion and/or ascites as main congestive sign). Weakness and other low-output signs can occur with any of these diseases, especially those associated with arrhythmias.
APPROACH TO TREATING HEART FAILURE
Most current treatment strategies are aimed at modifying either the results of NH activation (i.e., sodium and water retention) or the activation process itself (e.g., ACE inhibition). In most cases, therapy centers on controlling edema and effusions, improving cardiac output, reducing cardiac workload, supporting myocardial function, and managing concurrent arrhythmias. The approach to these goals varies somewhat with different diseases, most notably those causing restriction to ventricular filling.
The evolving perspective on CHF management is based on blocking excessive NH activation and preventing progression of myocardial remodeling and dysfunction, with diuretics being used to control signs of congestion. Future strategies may also involve drugs that block cytokines, antagonize endothelins, and enhance atrial peptides, as well as other strategies to block the effects of NH activation.
Classification of Severity
Guidelines for clinical staging of heart failure (based on the American Heart Association and American College of Cardiology [AHA/ACC] system) are being increasingly applied to veterinary patients (Table 3-2). These describe disease progression through four stages over time. This staging system emphasizes the importance of early diagnosis and evidence-based management of heart dysfunction. It also deemphasizes the term “congestive” in congestive heart failure because volume overload is not consistently present at all stages. Nevertheless, attention to the patient’s fluid status is highly important.
 TABLE 3-2 Classification Systems for Heart Failure Severity
TABLE 3-2 Classification Systems for Heart Failure Severity
| CLASSIFICATION | DEGREE OF SEVERITY |
|---|---|
| Modified AHA/ACC Heart Failure Staging System | |
| A | Patient “at risk” for the development of heart failure, but apparent cardiac structural abnormality not yet identified |
| B | Structural cardiac abnormality is evident, but no clinical signs of heart failure |
| C | Structural cardiac abnormality, with past or present clinical signs of heart failure |
| D | Persistent or end-stage heart failure signs, refractory to standard therapy |
| Modified NYHA Functional Classification | |
| I | Heart disease is present but no evidence of heart failure or exercise intolerance; cardiomegaly is minimal to absent |
| II | Signs of heart disease with evidence of exercise intolerance; radiographic cardiomegaly is present |
| III | Signs of heart failure with normal activity or at night (e.g., cough, orthopnea); radiographic signs of significant cardiomegaly and pulmonary edema or pleural/abdominal effusion |
| IV | Severe heart failure with clinical signs at rest or with minimal activity; marked radiographic signs of CHF and cardiomegaly |
| International Small Animal Cardiac Health Council Functional Classification | |
| I | Asymptomatic patient |
| Ia | Signs of heart disease without cardiomegaly |
| Ib | Signs of heart disease and evidence of compensation (cardiomegaly) |
| II | Mild to moderate heart failure. Clinical signs of failure evident at rest or with mild exercise, and adversely affect quality of life |
| III | Advanced heart failure. Clinical signs of CHF are immediately obvious |
| IIIa | Home care is possible |
| IIIb | Hospitalization recommended (cardiogenic shock, life-threatening edema, large pleural effusion, refractory ascites) |
| Forrester’s Classification (Group) | |
| I | Normal cardiac output and pulmonary venous pressures |
| II | Pulmonary congestion but normal cardiac output |
| III | Low cardiac output and peripheral hypoperfusion with no pulmonary congestion |
| IV | Low cardiac output with pulmonary congestion |
AHA/ACC, American Heart Association and American College of Cardiology; CHF, congestive heart failure.
The clinical severity of heart failure is also sometimes described according to a modified New York Heart Association (NYHA) classification scheme or the International Small Animal Cardiac Health Council (ISACHC) criteria. These systems group patients into functional categories on the basis of clinical observations rather than underlying cardiac disease or myocardial function. Such classification can be helpful conceptually and for categorizing study patients. Forrester’s classification is another method of grouping heart failure patients. Dogs with chronic mitral regurgitation often fall into group II; severe dilated cardiomyopathy is the most common diagnosis in group IV. Diseases causing group III heart failure are rare in dogs and cats. Regardless of the clinical classification scheme, identifying the underlying etiology and pathophysiology, as well as the clinical severity, is important for individualized therapy.
TREATMENT FOR ACUTE CONGESTIVE HEART FAILURE
GENERAL CONSIDERATIONS
Fulminant CHF is characterized by severe cardiogenic pulmonary edema, with or without pleural and/or abdominal effusions or poor cardiac output. Therapy is aimed at rapidly clearing pulmonary edema, improving oxygenation, and optimizing cardiac output (Box 3-1). Thoracocentesis should be performed expediently if marked pleural effusion exists. Likewise, large-volume ascites should be drained to improve ventilation. Animals with severe CHF are greatly stressed. Physical activity must be maximally curtailed to reduce total oxygen consumption; cage confinement is preferred. Environmental stresses such as excess heat and humidity or extreme cold should be avoided. When transported, the animal should be placed on a cart or carried. Unnecessary handling of the patient and administration of oral medications should be avoided, when possible.
BOX 3-1 Acute Treatment of Decompensated Congestive Heart Failure
ACE, Angiotensin-converting enzyme; CRI, constant rate infusion; D5W, 5% dextrose in water.
* Dilution of 250 mg dobutamine into 500 ml of D5W or lactated Ringer’s solution yields a solution of 500 μg/ml; CRI of 0.6 ml/kg/hr provides 5 μg/kg/min.
† Dilution of 40 mg of dopamine into 500 ml of D5W or lactated Ringer’s solution yields a solution of 80 μg/ml; a volume of 0.75 ml/kg/hr provides 1 μg/kg/min.
SUPPLEMENTAL OXYGEN
Oxygen administered by face mask or improvised hood, nasal catheter, endotracheal tube, or oxygen cage is beneficial as long as the method chosen does not increase the patient’s distress. An oxygen cage with temperature and humidity controls is preferred, and a setting of 65° F is recommended for normothermic animals. Oxygen flow of 6 to 10 L/min is usually adequate. Concentrations of 50% to 100% oxygen may be needed initially, but this should be reduced within a few hours to 40% to avoid lung injury. When a nasal tube is used, humidified O2 is delivered at a rate of 50 to 100 ml/kg/min. Extremely severe pulmonary edema with respiratory failure may respond to endotracheal or tracheotomy tube placement, airway suctioning, and mechanical ventilation. Positive end-expiratory pressure helps clear small airways and expand alveoli. Positive airway pressures can adversely affect hemodynamics, however, and chronic high oxygen concentrations (>70%) can injure lung tissue (see Suggested Readings for more information). Continuous monitoring is essential for intubated animals.
DRUG THERAPY
Diuresis
Rapid diuresis can be achieved with IV furosemide; effects begin within 5 minutes, peak by 30 minutes, and last about 2 hours. This route also provides a mild venodilating effect. Some patients require aggressive initial doses or cumulative doses administered at frequent intervals (see Box 3-1). Furosemide can be given by constant rate infusion (CRI), which may provide greater diuresis than bolus injection. The veterinary formulation (50 mg/ml) can be diluted to 10 mg/ml for CRI using 5% dextrose in water (D5W), lactated Ringer’s solution (LRS), or sterile water. Dilution to 5 mg/ml in D5W or sterile water is also described. The patient’s respiratory rate, as well as other parameters (discussed in more detail later), guide the intensity of continued furosemide therapy. Once diuresis has begun and respiration improves, the dosage is reduced to prevent excessive volume contraction or electrolyte depletion. An ancillary approach that has been described for patients with fulminant cardiogenic edema is phlebotomy (up to 25% of total blood volume), but this is not generally done.
Vasodilation
Vasodilator drugs can reduce pulmonary edema by increasing systemic venous capacitance, lowering pulmonary venous pressure, and reducing systemic arterial resistance. Although ACE inhibitors are a mainstay of CHF management, more immediate afterload reduction is desirable for animals with acute pulmonary edema. Arteriolar vasodilation is not recommended for heart failure caused by diastolic dysfunction or ventricular outflow obstruction.
Sodium nitroprusside is a potent arteriolar and venous dilator, with direct action on vascular smooth muscle. It is given by IV infusion because of its short duration of action. Blood pressure must be closely monitored when using this drug. The dose is titrated to maintain mean arterial pressure at about 80 mm Hg (at least >70 mm Hg) or systolic blood pressure between 90 and 110 mm Hg. Nitroprusside CRI is usually continued for 12 to 24 hours. Dosage adjustments may be needed because drug tolerance develops rapidly. Profound hypotension is the major adverse effect. Cyanide toxicity can result from excessive or prolonged use (e.g., longer than 48 hours). Nitroprusside should not be infused with other drugs, and should be protected from light.
Hydralazine, a pure arteriolar dilator, is an alternative to nitroprusside. It is useful for refractory pulmonary edema caused by mitral regurgitation (and sometimes dilated cardiomyopathy) because it can reduce regurgitant flow and lower left atrial pressure. An initial dose of 0.75 to 1 mg/kg is given orally, followed by repeated doses every 2 to 3 hours until the systolic blood pressure is between 90 and 110 mm Hg or clinical improvement is obvious. If blood pressure cannot be monitored, an initial dose of 1 mg/kg is repeated in 2 to 4 hours if sufficient clinical improvement has not been observed. The addition of 2% nitroglycerin ointment may provide beneficial venodilating effects.
An ACE inhibitor or amlodipine, with or without nitroglycerin ointment, is an alternative to hydralazine/nitroglycerine. The onset of action is slower and the effects are less pronounced, but this regimen can still be helpful.
Nitroglycerin (and other orally or transcutaneously administered nitrates) act mainly on venous smooth muscle to increase venous capacitance and reduce cardiac filling pressure. The major indication for nitroglycerin is acute cardiogenic pulmonary edema. Nitroglycerin ointment (2%) is usually applied to the skin of the groin, axillary area, or ear pinna, although the efficacy of this in heart failure is unclear. An application paper or glove is used to avoid skin contact by the person applying the drug.
Other Acute Therapy
Some dogs with severe pulmonary edema and bronchoconstriction benefit from bronchodilator therapy. Aminophylline, given by slow IV administration or intramuscular (IM) injection, has mild diuretic and positive inotropic actions as well as a bronchodilating effect; it also decreases fatigue of respiratory muscles. Adverse effects include increased sympathomimetic activity and arrhythmias. The oral route can be used when respiration improves because gastrointestinal (GI) absorption is rapid.
Mild sedation (butorphanol or morphine for dogs, butorphanol with acepromazine for cats) can reduce anxiety. Because morphine can induce vomiting, butorphanol may be a better choice in dogs. Nevertheless, other beneficial effects of morphine include slower, deeper breathing from respiratory center depression and redistribution of blood away from the lungs via dilation of capacitance vessels. Morphine is contraindicated in dogs with neurogenic edema because it can raise intracranial pressure. Morphine is not used in cats.
Inotropic Support
Positive inotropic therapy is indicated when heart failure is caused by poor myocardial contractility. Oral therapy with pimobendan or digoxin can be started as soon as practical for animals needing chronic inotropic support (see Table 3-3 and p. 65). Treatment for one to three days with an IV sympathomimetic (catecholamine) or phosphodiesterase (PDE) inhibitor drug can help support arterial pressure, forward cardiac output, and organ perfusion when myocardial failure or hypotension is severe.
 TABLE 3-3 Drugs for Managing Chronic Heart Failure
TABLE 3-3 Drugs for Managing Chronic Heart Failure
| DRUG | DOGS | CATS |
|---|---|---|
| Diuretics | ||
| Furosemide | 1-3 mg/kg PO q8-24h (long term); use smallest effective dose | 1-2 mg/kg PO q8-12h; use smallest effective dose |
| Spironolactone | 0.5-2 mg/kg PO q(12-)24h | 0.5-1 mg/kg PO q(12-)24h |
| Chlorothiazide | 20-40 mg/kg PO q12h | 20-40 mg/kg PO q12h |
| Hydrochlorothiazide | 2-4 mg/kg PO q12h | 1-2 mg/kg PO q12h |
| ACE Inhibitors | ||
| Enalapril | 0.5 mg/kg PO q(12-)24h | 0.25-0.5 mg/kg PO q24(−12)h |
| Benazepril | 0.25-0.5 mg/kg PO q(12-)24h | 0.25-0.5 mg/kg PO q24(−12)h |
| Captopril | 0.5-2.0 mg/kg PO q8-12h (low initial dose) | 0.5-1.25 mg/kg PO q12-24h |
| Lisinopril | 0.25-0.5 mg/kg PO q(12-)24h | 0.25-0.5 mg/kg PO q24h |
| Fosinopril | 0.25-0.5 mg/kg PO q24h | — |
| Ramipril | 0.125-0.25 mg/kg PO q24h | — |
| Imidapril | 0.25 mg/kg PO q24h | — |
| Other Vasodilators | ||
| Hydralazine | 0.5-2 mg/kg PO q12h (to 1 mg/kg initial) | 2.5 (up to 10) mg/cat PO q12h |
| Amlodipine | 0.05 (initial) to 0.3(−0.5) mg/kg PO q(12-)24h | 0.3125-0.625 mg/cat PO q24(−12)h |
| Prazosin | Medium dogs: 1 mg PO q8-12hr; large dogs: 2 mg PO q8h | — |
| Nitroglycerin 2% ointment | ½-1½ inch cutaneously q4-6h | ¼-½ inch cutaneously q4-6h |
| Isosorbide dinitrate | 0.5-2 mg/kg PO q(8-)12h | — |
| Isosorbide mononitrate | 0.25-2 mg/kg PO q12h | — |
| Positive Inotropes | ||
| Pimobendan | 0.1-0.3 mg/kg PO q12h, start low; give at least 1 hour before feeding | |
| Digoxin | 0.007 mg/kg (or ¼ of 0.125 mg tab) PO q48h. See Box 3-1 for IV dose. | |
CRI, Constant rate infusion.
Catecholamines enhance contractility via a cAMP-mediated increase in intracellular Ca++. They can provoke arrhythmias and increase pulmonary and systemic vascular resistance (potentially exacerbating edema formation). Their short half-life (<2 minutes) and extensive hepatic metabolism necessitate constant IV infusion. Dobutamine (a synthetic analog of dopamine) has lesser effect on heart rate and afterload and is preferred over dopamine. Dobutamine stimulates β1-receptors, with only weak action on β2- and α-receptors. Lower doses (e.g., 3 to 7 μg/kg/min) have minimal effects on heart rate and blood pressure. The initial infusion rate should be low; this can be gradually increased over hours to achieve greater inotropic effect and maintain systolic arterial pressure between 90 and 120 mm Hg. Heart rate, rhythm, and blood pressure must be monitored closely. Although dobutamine is less arrhythmogenic than other catecholamines, higher infusion rates (e.g., 10 to 20 μg/kg/min) can precipitate supraventricular and ventricular arrhythmias. Adverse effects are more likely in cats; these include seizures at relatively low doses.
Dopamine at low doses (<2-5 μg/kg/minute) also stimulates vasodilator dopaminergic receptors in some regional circulations. Low-to-moderate doses enhance contractility and cardiac output, but high doses (10-15 μg/kg/minute) cause peripheral vasoconstriction and increase heart rate, O2 consumption, and the risk of ventricular arrhythmias. An initial IV infusion of 1 μg/kg/min can be titrated upward to desired clinical effect. The infusion rate should be decreased if sinus tachycardia or other tachyarrhythmias develop.
Bipyridine PDE inhibitors such as amrinone and milrinone increase intracellular Ca++ by inhibiting PDE III, an intracellular enzyme that degrades cAMP. These drugs also cause vasodilation, as increased cAMP promotes vascular smooth muscle relaxation. Hypotension, tachycardia, and GI signs can occur when giving high doses. These drugs can exacerbate ventricular arrhythmias. The effects of amrinone are short-lived (<30 minutes) after IV injection in normal dogs, so CRI is required for sustained effect. Peak effects occur after 45 minutes of CRI in dogs. Amrinone is sometimes used as an initial slow IV bolus followed by CRI; half the original bolus dose can be repeated after 20 or 30 minutes. Milrinone has a much greater potency than amrinone, but there is little veterinary data with the IV form. A PDE inhibitor can be used concurrently with digoxin and a catecholamine.
Digoxin is generally not used intravenously except for some supraventricular tachyarrhythmias when other acute therapy is unavailable or ineffective (see Chapter 4). Acidosis and hypoxemia associated with severe pulmonary edema can increase myocardial sensitivity to digitalis-induced arrhythmias. If digoxin is used intravenously, it must be given slowly (over at least 15 minutes); rapid injection causes peripheral vasoconstriction. The calculated dose is usually divided, and boluses of one fourth the dose are given slowly over several hours.
If arrhythmias develop during IV inotropic therapy, the infusion rate is reduced or the drug is discontinued. In animals with atrial fibrillation, catecholamine infusion is likely to increase the ventricular response rate by enhancing atrioventricular (AV) conduction. If dobutamine or dopamine is deemed necessary for such a case, diltiazem (administered rapidly by mouth or cautiously by IV) is used to reduce the heart rate. Digoxin, administered either by mouth (loading) or cautiously by IV, is an alternative.
HEART FAILURE CAUSED BY DIASTOLIC DYSFUNCTION
When acute CHF is caused by hypertrophic cardiomyopathy, thoracocentesis (if needed), diuretics, and oxygen therapy are given as outlined previously. Cutaneous nitroglycerin can also be used. Diltiazem or a β1-blocker such as atenolol can be given to slow heart rate and increase ventricular filling time once severe dyspnea has abated; alternatively, IV administration of diltiazem or esmolol could be used. Propranolol (or other nonselective β-blockers) is generally avoided in patients with fulminant pulmonary edema because β2-blockade could induce bronchoconstriction.
Arteriolar vasodilators can be detrimental if dynamic left ventricular (LV) outflow obstruction coexists, because afterload reduction provokes greater systolic obstruction (see Chapter 8). ACE inhibitors at standard doses do not appear to worsen the LV outflow gradient. Addition of an ACE inhibitor is recommended as soon as oral therapy is possible.
MONITORING AND FOLLOW-UP
Repeated assessment is important to monitor the effectiveness of therapy and to prevent hypotension or severe azotemia caused by excessive diuresis. Mild azotemia commonly occurs. Hypokalemia and metabolic alkalosis can occur after aggressive diuresis. A serum potassium concentration within the mid- to high-normal range is especially important for animals with arrhythmias. Serum biochemical testing every 24 to 48 hours is advised until the patient is eating and drinking well.
Arterial blood pressure can be monitored indirectly or directly, but gaining arterial access can increase patient stress. Indirect measures of organ perfusion such as capillary refill time, mucous membrane color, urine output, toe-web temperature, and mentation can also be useful. Body weight should be monitored, especially with aggressive diuretic therapy.
Central venous pressure (CVP) does not adequately reflect left heart filling pressures. It should not be used to guide diuretic or fluid therapy in patients with cardiogenic pulmonary edema. Although pulmonary capillary wedge pressure can reliably guide therapy, the placement and care of an indwelling pulmonary artery catheter require meticulous attention to asepsis and close monitoring.
Pulse oximetry is a helpful noninvasive means of monitoring oxygen saturation (SpO2). Supplemental O2 should be given if SpO2 is <90%; mechanical ventilation is indicated if SpO2 is <80% despite O2 therapy. Arterial sampling for blood gas analysis is more accurate but is stressful for the patient. Resolution of radiographic evidence for pulmonary edema usually lags behind clinical improvement by a day or two.
After respiratory signs begin to abate and diuresis is evident, low-sodium water is offered. Fluid administration (either subcutaneously or intravenously) is generally not advised in patients with fulminant CHF. In most cases, gradual rehydration by free choice (low sodium) water intake is preferred even after aggressive diuretic therapy. However, fluid therapy may be necessary for patients with heart failure and renal failure, marked hypokalemia, hypotension, digoxin toxicity, persistent anorexia, or other serious systemic disease. Some animals require relatively high cardiac filling pressure to maintain cardiac output, especially those with myocardial failure or markedly reduced ventricular compliance (e.g., from hypertrophic cardiomyopathy or pericardial disease). Diuresis and vasodilation in such cases can cause inadequate cardiac output and hypotension.
In most patients with decompensated CHF, the smallest fluid volume possible should be used to deliver drugs by CRI. Careful monitoring and continued diuretic use is important to prevent recurrent pulmonary edema. When additional fluid therapy is necessary, D5W or a reduced sodium fluid (e.g., 0.45% NaCl with 2.5% dextrose) with added KCl is administered at a conservative rate (e.g., 15 to 30 ml/kg/day IV). Alternatively, 0.45% NaCl with 2.5% dextrose or lactated Ringer’s solution can be administered subcutaneously.
Potassium supplementation at a maintenance rate is provided by 0.05 to 0.1 mEq/kg/hour (or more conservatively, 0.5 to 2.0 mEq/kg/day). For animals with hypokalemia, higher rates are used: 0.15 to 0.2 mEq/kg/hour for mild K+ deficiency; 0.25 to 0.3 mEq/kg/hour for moderate deficiency; and 0.4 to 0.5 mEq/kg/hour for severe deficiency. Measuring serum K+ concentration in 4 to 6 hours is advised when supplementing for moderate to severe deficiency. Hyponatremia and worsened fluid retention can develop after using low-sodium IV solutions in some patients. These require a more balanced crystalloid solution. Other supportive therapies for CHF and any underlying disease(s) depend on individual patient needs. Parenteral fluid administration is tapered off as the animal is able to resume oral food and water intake.
MANAGEMENT OF CHRONIC HEART FAILURE
GENERAL CONSIDERATIONS
A general approach to chronic heart failure therapy is presented in this section. Additional information is found in the chapters describing different diseases. Therapy is tailored to the individual animal’s needs by adjusting dosages, adding or substituting drugs, and modifying lifestyle or diet. Pleural effusion and large-volume ascites that accumulate despite medical therapy should be drained to facilitate respiration. Likewise, pericardial effusion that compromises cardiac filling must be drained. As heart disease progresses, more aggressive therapy is usually needed. Support of cardiac function with digoxin or pimobendan is often indicated in dogs and sometimes in cats.
Exercise restriction helps reduce cardiac workload regardless of heart failure etiology. Strenuous exercise can provoke dyspnea and potentially serious cardiac arrhythmias even in animals with compensated CHF. Chronic heart failure is associated with skeletal muscle changes that lead to fatigue and dyspnea. Physical training can improve cardiopulmonary function and quality of life in patients with chronic heart failure. This is partly mediated by improvement in vascular endothelial function and restoration of flow-dependent vasodilation. Although it is difficult to know how much exercise is best, regular (not sporadic) mild to moderate activity is encouraged, as long as excessive respiratory effort is not induced. Bursts of strenuous activity should be avoided. Dietary salt restriction and other nutritional issues are also important in the management of patients with chronic heart failure.
DIURETICS
Diuretic therapy is indicated for controlling cardiogenic pulmonary edema and effusions. Diuretics remain fundamental to the management of CHF because of their ability to decrease venous congestion and fluid accumulation (see Table 3-3). Agents that interfere with ion transport in the loop of Henle (e.g., furosemide) have potent ability to promote both salt and water loss. Diuretics of other classes, such as thiazides and potassium-sparing agents, are sometimes combined with furosemide for chronic heart failure therapy. Given to excess, diuretics promote excessive volume contraction and activate the renin-angiotensin-aldosterone cascade. Diuretics also can exacerbate preexisting dehydration or azotemia. Therefore the indication for their use in such animals should be clearly established, and the lowest effective dose should be used.
Furosemide
Furosemide is the loop diuretic used most widely for cats and dogs with heart failure. It acts on the ascending limb of the loop of Henle to inhibit active Cl−, K+, and Na+ cotransport, thereby promoting excretion of these electrolytes; Ca++ and Mg++ are also lost in the urine. Loop diuretics can increase systemic venous capacitance, possibly by mediating renal prostaglandin release. Furosemide may also promote salt loss by increasing total renal blood flow and by preferentially enhancing renal cortical flow. The loop diuretics are well absorbed when given orally. After oral administration, diuresis occurs within 1 hour, peaks between 1 to 2 hours, and may last for 6 hours. Furosemide is highly protein bound; about 80% is actively secreted unchanged in the proximal renal tubules, with the remainder excreted as glucuronide.
Although aggressive furosemide treatment is indicated for acute, fulminant pulmonary edema, the smallest effective doses should be used for chronic heart failure therapy. The dosage varies, depending on the clinical situation. Respiratory pattern, hydration, body weight, exercise tolerance, renal function, and serum electrolyte concentrations are used to monitor response to therapy. Furosemide (or other diuretic) alone is not recommended as the sole treatment for chronic heart failure because it can exacerbate NH activation and reduce renal function.
Adverse effects are usually related to excessive fluid and/or electrolyte losses. Because they are more sensitive than dogs, lower doses are used in cats. Although hypokalemia is the most common electrolyte disturbance, it is unusual in dogs that are not anorexic. Hyponatremia develops in some patients with severe CHF and results from an inability to excrete free water (dilutional hyponatremia) rather than from a total body sodium deficit.
Spironolactone
Spironolactone may be a useful adjunct therapy in patients with chronic refractory heart failure. Its anti-aldosterone effects are also thought to be important locally within the heart. Spironolactone is a competitive antagonist of aldosterone. It promotes Na+ loss and K+ retention in the distal renal tubule and can reduce the renal potassium wasting of furosemide and other diuretics, especially when circulating aldosterone concentration is high. But its diuretic effect in normal dogs is questionable. Spironolactone’s onset of action is slow; peak effect occurs within 2 to 3 days.
Aldosterone release can occur despite the use of an ACE inhibitor (so-called aldosterone escape); this may involve reduced hepatic clearance, increased release stimulated by K+ elevation or Na+ depletion, and local tissue aldosterone production. Spironolactone’s anti-aldosterone effect is thought to mitigate aldosterone-induced cardiovascular remodeling in some cases. The drug has improved survival in people with moderate to severe CHF, but it is not yet clear whether similar survival benefit occurs clinically in dogs and cats.
A potassium-sparing diuretic must be used cautiously in patients receiving an ACE inhibitor or potassium supplement and is absolutely contraindicated in hyperkalemic patients. Adverse effects relate to excess K+ retention and GI disturbances. Spironolactone may decrease digoxin clearance.
Thiazide Diuretics
Thiazide diuretics decrease Na+ and Cl− absorption and increase Ca++ absorption in the distal convoluted tubules. Mild to moderate diuresis with excretion of Na+, Cl−, K+, and Mg++ results. The thiazides decrease renal blood flow and should not be used in azotemic animals. Adverse effects are uncommon in the absence of azotemia, but hypokalemia or other electrolyte disturbance and dehydration can occur with excessive use or in anorectic patients. Thiazides can cause hyperglycemia in diabetic or prediabetic animals by inhibiting conversion of proinsulin to insulin. Chlorothiazide’s effects begin within 1 hour, peak at 4 hours, and last 6 to 12 hours. Hydrochlorothiazide produces diuresis within 2 hours, with peak effect at 4 hours, and duration of about 12 hours.
ANGIOTENSIN-CONVERTING ENZYME INHIBITORS
ACE inhibitors (ACEIs) are indicated for most causes of chronic heart failure, especially dilated cardiomyopathy and chronic valvular insufficiency (see Table 3-3). Their use has led to clinical improvement and lowered mortality rates in people with heart failure; similar benefits seem to occur in dogs with myocardial failure or volume overload and in cats with diastolic dysfunction. They moderate excess NH responses in several ways; therefore ACEIs have considerable advantages over hydralazine and other arteriolar dilators. ACEIs have only modest diuretic and vasodilatory effects; their main benefits arise from opposing the effects of NH activation and abnormal cardiovascular remodeling changes. By blocking the formation of angiotensin II, ACEIs allow arteriolar and venous vasodilation. The secondary inhibition of aldosterone release helps reduce Na+ and water retention and therefore edema/effusions, as well as the adverse effects of aldosterone directly on the heart. ACEIs reduce ventricular arrhythmias and the rate of sudden death in people (and probably animals) with heart failure, likely because angiotensin II–induced facilitation of norepinephrine and epinephrine release is inhibited. Their vasodilating effects may be enhanced by vasodilator kinins normally degraded by ACE. A local vasodilating effect may occur through inhibition of ACE found within vascular walls, even in the absence of high circulating renin concentrations. Local ACE inhibition may be beneficial by modulating vascular smooth muscle and myocardial remodeling. However, it is unclear whether ACE inhibitors prevent ventricular remodeling and dilation in dogs with heart disease. ACE inhibitors have been variably effective in treating dogs with hypertension.
Most ACEIs (except captopril and lisinopril) are prodrugs that are converted to their active form in the liver; therefore severe liver dysfunction can interfere with this conversion. Adverse effects of ACEIs include hypotension, GI upset, deterioration of renal function, and hyperkalemia (especially when used with a potassium-sparing diuretic or potassium supplement). Angiotensin II is important in mediating renal efferent arteriolar constriction, which maintains glomerular filtration when renal blood flow decreases. As long as cardiac output and renal perfusion improve with therapy, renal function is usually maintained. Poor glomerular filtration is more likely to result with overdiuresis, excess vasodilation, or severe myocardial dysfunction. Azotemia is first addressed by decreasing the diuretic dosage. If necessary, the ACEI dosage is decreased or discontinued. Hypotension can usually be avoided by starting with low initial doses. Other adverse effects reported in people include rash, pruritus, impairment of taste, proteinuria, cough, and neutropenia. The mechanism of ACEI-induced cough in people is unclear but may involve inhibition of endogenous bradykinin degradation or may be associated with increased NO generation. NO has an inflammatory effect on bronchial epithelial cells.
Enalapril
Enalapril maleate is absorbed well when taken orally; administration with food does not decrease its bioavailability. It is hydrolyzed in the liver to enalaprilat, its most active form. Peak ACE-inhibiting activity occurs within 4 to 6 hours in dogs. Duration of action is 12 to 14 hours, and effects are minimal by 24 hours at the recommended once-daily dose. Enalapril is generally administered once daily, although some dogs respond better when dosed every 12 hours. In cats maximal activity occurs within 2 to 4 hours after an oral dose of either 0.25 or 0.5 mg/kg; some ACE inhibition (50% of control) persists for two to three days. Enalapril and its active metabolite are excreted in the urine. Renal failure and severe CHF prolong its half-life, so reduced doses or benazepril are used in such patients. Severe liver dysfunction will interfere with the conversion of the prodrug to the active enalaprilat; lisinopril or captopril should be considered in such patients instead. Injectable enalaprilat is also available, but little veterinary data exist on its use; this form is not well absorbed orally.
Benazepril
Benazepril is metabolized to its active form, benazeprilat. Only about 40% is absorbed when administered orally, but feeding does not affect absorption. After oral administration, peak ACE inhibition occurs within 2 hours in dogs and cats; its effect can last over 24 hours. In cats doses of 0.25 to 0.5 mg/kg result in 100% inhibition of ACE that is maintained at >90% for 24 hours, and tapers off to about 80% by 36 hours. Benazapril has an initial half-life of 2.4 hours and terminal half-life of about 29 hours in cats. Repeated doses produce moderate increases in drug plasma concentration. Benazepril is a preferred ACEI for animals with renal disease. This drug is eliminated equally in urine and bile in dogs. In cats about 85% of the drug is excreted in the feces and only 15% in urine. The drug is generally well tolerated. It may also slow renal function deterioration in cats with kidney disease.
Captopril
Captopril was the first ACEI used clinically. Captopril contains a sulfhydryl group, in contrast to enalapril and others. Disulfide metabolites can act as free radical scavengers. This might have beneficial effects for the treatment of some heart diseases, although the clinical significance is presently unclear. Captopril appears to be less effective than several other agents in reducing ACE activity in normal dogs. Captopril is well absorbed when taken orally (75% bioavailable); however, food decreases its bioavailability by 30% to 40%. In dogs hemodynamic effects appear within 1 hour, peak in 1 to 2 hours, and last less than 4 hours. Captopril is excreted in the urine.
Lisinopril
Lisinopril is a lysine analog of enalaprilat with direct ACE-inhibiting effects. It is 25% to 50% bioavailable, and absorption is not affected by feeding. The time to peak effect is 6 to 8 hours. The duration of ACE inhibition appears long, but more specific information in animals is lacking. Once-daily administration has been tried with apparent effectiveness.
Fosinopril
Fosinopril is structurally different in that it contains a phosphinic acid radical (rather than sulfhydryl or carboxyl), and it may be retained longer in myocytes. Fosinopril is also a prodrug that is converted to the active fosinoprilat in the GI mucosa and liver. Elimination occurs equally between kidney and liver; compensatory increases in one pathway occur with impairment of the other. Its duration of action is well over 24 hours in people. Fosinopril may cause falsely low serum digoxin measurements using certain RIA assays.
POSITIVE INOTROPIC AGENTS
A positive inotropic agent is indicated for patients with dilated cardiomyopathy or other causes of myocardial failure, including dogs with advanced mitral regurgitation (see Table 3-3). Pimobendan, now approved in the United States, and digoxin are the inotropic agents available for chronic oral therapy. Pimobendan improves cardiac pump function both by enhancing contractility as well as by vasodilation. Digoxin is still used in some cases and can be combined with pimobendan. Digoxin also is indicated for treating some supraventricular tachyarrhythmias (see Chapter 4), except in cats with hypertrophic cardiomyopathy.
Pimobendan
Pimobendan (Vetmedin) is a benzimidazole-derivative phosphodiesterase III inhibitor. It slows cAMP breakdown and enhances adrenergic effects on Ca++ fluxes and myocardial contractility. Pimobendan also has a calcium-sensitizing effect on the contractile proteins, which promotes increased contractility without an increase in myocardial O2 requirement. Pimobendan is known as an inodilator because it increases contractility while also causing systemic and pulmonary vasodilation. The drug may have other beneficial effects by modulating NH and proinflammatory cytokine activation. Peak plasma concentrations occur within an hour of oral dosing. Bioavailability is about 60% in dogs, but this decreases in the presence of food. Pimobendan is highly protein bound. Elimination is mainly via hepatic metabolism and biliary excretion. Concurrent Ca++ or β-blocker therapy may diminish the drug’s positive inotropic effect.
Clinical improvement has occurred in many dogs when this agent was added to conventional CHF therapy (e.g., furosemide, an ACE inhibitor, and digoxin). Pimobendan appears to improve clinical status and increase survival time in dogs with dilated cardiomyopathy (DCM) or chronic mitral valve disease. Pimobendan does not appear to increase the frequency of ventricular arrhythmias and sudden death, as has occurred with other phosphodiesterase inhibitors. There are limited anecdotal reports of pimobendan use in cats.
Digoxin
The benefits of digoxin arise from its modest positive inotropic effect as well as its supraventricular antiarrhythmic activity. Its ability to sensitize baroreceptors and thereby modulate neurohormonal activation is probably its most important attribute in patients with heart failure. Because digoxin is potentially toxic, low doses are used and serum concentrations should be monitored. Serum concentrations in the low to mid therapeutic range are desired (discussed in more detail later).
Digoxin is indicated in patients with heart failure caused by myocardial dysfunction, chronic mitral insufficiency, and other chronic volume or pressure overloads. Digoxin is usually contraindicated in patients with hypertrophic cardiomyopathy, especially those with ventricular outflow obstruction; it is not useful in dogs or cats with pericardial diseases. Digoxin is only moderately effective in slowing the ventricular response rate to atrial fibrillation and does not cause conversion to sinus rhythm. It is usually contraindicated when sinus or AV node disease is present. Digoxin is relatively contraindicated in most patients with serious ventricular arrhythmias because it can exacerbate such arrhythmias.
Digoxin, in addition to other digitalis glycosides, increases the Ca++ available to contractile proteins by competitively binding and inhibiting the Na+, K+-ATPase pump at the myocardial cell membrane. Intracellular Na+ accumulation then promotes Ca++ entry via the sodium-calcium exchange. In diseased myocardial cells in which diastolic sequestration and systolic release of Ca++ is impaired, digitalis glycosides may be ineffective as inotropic agents and could predispose the patient to cellular Ca++ overload and electrical instability.
The antiarrhythmic effects of digoxin are mediated primarily via increased parasympathetic tone to the sinus and AV nodes and the atria. Some direct effects further prolong conduction time and refractory period of the AV node. Sinus rate slowing, reduced ventricular response rate to atrial fibrillation and flutter, and suppression of atrial premature depolarizations are resulting effects. Although some ventricular arrhythmias might be suppressed (probably via enhanced vagal tone), the digitalis glycosides have potential arrhythmogenic effects, especially in patients with heart failure.
Oral maintenance doses of digoxin are used to initiate therapy in most cases because loading doses can result in toxic serum concentrations. When more rapid achievement of therapeutic serum concentrations is important (e.g., for supraventricular tachyarrhythmia), the drug can be given at twice the oral maintenance dose for 1 to 2 doses or intravenously with caution (see Table 3-3). But alternate IV therapy for supraventricular tachycardia is usually more effective (see Chapter 4). Other IV-positive inotropic drugs (see p. 60 and Box 3-1) are safer and more effective than digoxin for immediate support of myocardial contractility.
Digoxin is well absorbed orally and undergoes minimal hepatic metabolism. Absorption is approximately 60% for the tablet form and 75% for the elixir. Bioavailability is decreased by kaolin-pectin compounds, antacids, the presence of food, and malabsorption syndromes. About 27% of the drug in serum is protein bound. The serum half-life in dogs ranges from 23 to 39 hours; therapeutic serum concentrations are achieved within 2 to 41/2 days with dosing every 12 hours. In cats the reported serum half-life ranges widely, from about 25 to over 78 hours; chronic oral administration increases the half-life. The alcohol-based elixir, which is poorly palatable, results in serum concentrations approximately 50% higher than the tablet form of digoxin. Administration of the tablets with food has resulted in serum concentrations about 50% lower than in the fasted state in cats. The pharmacokinetics in cats with heart failure are similar to those in control cats receiving aspirin, furosemide, and a low-salt diet, although much interpatient variation is present. Digoxin treatment every 48 hours in cats produces effective serum concentrations, with steady state achieved in about 10 days. Because approximately 50% of cats become toxic at a dose of 0.01 mg/kg every 48 hours, a dose of 0.007 mg/kg every 48 hours has been recommended. Serum concentrations can be measured 8 hours postdosing once steady state is reached (after about 10 days). Digoxin elimination is primarily by glomerular filtration and renal secretion in dogs, although approximately 15% is metabolized by the liver. Renal and hepatic elimination appear equally important in cats.
Serum digoxin concentration (and risk of toxicity) increases with renal failure because of reduced clearance and volume of distribution. There appears to be no correlation between the degree of azotemia and the serum digoxin concentration in dogs, making extrapolations from human formulas for calculating drug dosage in renal failure unusable in this species. Lower doses and close monitoring of serum digoxin concentration are recommended in animals with renal disease.
There is only a weak correlation between digoxin dose and serum concentration in dogs with heart failure, indicating that other factors influence the serum concentrations of this drug. Because much of the drug is bound to skeletal muscle, animals with reduced muscle mass or cachexia and those with compromised renal function can easily become toxic at the usual calculated doses. The dose should be based on the patient’s calculated lean body weight because digoxin has poor lipid solubility. This consideration is especially important in obese animals. Management of digoxin toxicity is outlined later in this section. Conservative dosing and measurement of serum digoxin concentrations help to prevent toxicity.
Measurement of serum concentration is recommended 7 to 10 days after initiation of therapy (or dosage change). Samples should be drawn 8 to 10 hours postdose. Many veterinary and most human hospital laboratories can provide this service. The therapeutic serum concentration range is 1 to 2 (or 2.4) ng/ml. If the serum concentration is less than 0.8 ng/ml, the digoxin dose can be increased by 25% to 30% and the serum concentration measured the following week. But a serum concentration in the mid to low therapeutic range is probably safer. People with high-normal serum digoxin concentrations have greater risk for sudden death. If serum concentrations cannot be measured and toxicity is suspected, the drug should be discontinued for one to two days and then reinstituted at half of the original dose.
Certain drugs affect serum digoxin concentrations when administered concurrently. Quinidine increases serum digoxin concentrations by displacing the drug from skeletal muscle binding sites and reducing its renal clearance. This drug combination is therefore not recommended, but, if both must be used, the digoxin dose is reduced by 50% initially and guided by serum concentration measurement. Other drugs known to increase serum digoxin concentration include verapamil and amiodarone. Diltiazem, prazosin, spironolactone, and triamterene possibly increase serum digoxin concentration. Hypokalemia especially, as well as other electrolyte and thyroid disturbances, can potentiate digoxin toxicity. Drugs affecting hepatic microsomal enzymes may also have effects on digoxin metabolism.
Digoxin Toxicity
As discussed previously, azotemia, hypokalemia, or concurrent use of certain drugs predispose the patient to digoxin toxicity. Therefore it is important to monitor renal function and serum electrolytes during digoxin therapy. Hypokalemia predisposes the patient to myocardial toxicity by leaving more available binding sites on membrane Na+, K+-ATPase for digitalis; conversely, hyperkalemia displaces digitalis from those binding sites. Hypercalcemia and hypernatremia potentiate both the inotropic and the toxic effects of the drug. Abnormal thyroid hormone concentrations can also influence the response to digoxin. Hyperthyroidism may potentiate the myocardial effects of the drug, whereas hypothyroidism prolongs the half-life of digoxin in people but has no pharmacokinetic effect in dogs. Hypoxia sensitizes the myocardium to the toxic effects of digitalis. Quinidine increases serum digoxin concentration by reducing renal clearance and competing for Na/K binding sites in skeletal muscle. Verapamil and amiodarone also increase serum digoxin concentration; other drugs that may do so include diltiazem, prazosin, and spironolactone. In addition, alteration of hepatic and renal function may affect the clearance of these drugs.
Digoxin toxicity causes GI, myocardial, or sometimes central nervous system (CNS) signs. GI toxicity may develop before signs of myocardial toxicity. Signs include anorexia, depression, vomiting, borborygmi, and diarrhea. Some of these GI signs result from the direct effects of digitalis on chemoreceptors in the area postrema of the medulla. CNS signs include depression and disorientation.
Myocardial toxicity from digitalis glycosides can cause almost any cardiac rhythm disturbance, including ventricular tachyarrhythmias, supraventricular premature complexes and tachycardia, sinus arrest, Mobitz type I second-degree AV block, and junctional rhythms. Myocardial toxicity can occur before any other signs and can lead to collapse and death, especially in animals with myocardial failure. Therefore the appearance of PR interval prolongation or signs of GI toxicity should not be used to guide progressive dosing of digoxin. Digitalis glycosides can aggravate cellular calcium overloading and electrical instability common in failing myocardial cells. Digitalis can stimulate spontaneous automaticity of myocardial cells by inducing and potentiating late afterdepolarizations; cellular stretch, calcium overloading, and hypokalemia enhance this effect. Toxic concentrations of digitalis also enhance automaticity by increasing sympathetic tone to the heart. Furthermore, the parasympa thetic effects of slowed conduction and altered refractory period facilitate development of reentrant arrhythmias. Digitalis intoxication should be suspected in patients taking the drug when ventricular arrhythmias and/or tachyarrhythmias with impaired conduction appear.
Therapy for digitalis toxicity depends on its manifestations. GI signs usually respond to drug withdrawal and correction of fluid or electrolyte abnormalities. AV conduction disturbances resolve after drug withdrawal, although sometimes anticholinergic therapy is needed. Digitalis-induced ventricular tachycardia and frequent ventricular premature complexes are generally treated with lidocaine. This drug reduces sympathetic nervous tone and can suppress arrhythmias caused by reentry and late afterdepolarizations; it has little effect on sinus rate or AV nodal conduction. If lidocaine is ineffective, phenytoin (diphenylhydantoin) is the second drug of choice in dogs; its effects are similar to those of lidocaine. IV administration of phenytoin must be slow to prevent hypotension and myocardial depression caused by the propylene glycol vehicle. Phenytoin has occasionally been used orally to treat or prevent ventricular tachyarrhythmias caused by digitalis.
Other measures are also helpful for digoxin toxicity, including IV potassium supplementation if the serum potassium concentration is <4 mEq/L (see p. 62). Magnesium supplementation may also be effective in suppressing arrhythmias; MgSO4 has been used at 25 to 40 mg/kg via slow intravenous bolus, followed by infusion of the same dose over 12 to 24 hours. Fluid therapy is indicated to correct dehydration and maximize renal function. A β-blocker may help control ventricular tachyarrhythmias, but this is not used if AV conduction block is present. Quinidine should not be used because it increases the serum concentration of digitalis. Oral administration of the steroid-binding resin cholestyramine is useful only very soon after accidental overdose of digoxin because this drug undergoes minimal enterohepatic circulation. A preparation of digoxin-specific antigen-binding fragments (digoxin-immune Fab) derived from ovine antidigoxin antibodies has occasionally been used for digoxin and digitoxin overdose. The Fab fragment binds with antigenic determinants on the digoxin molecule, preventing and reversing the pharmacologic and toxic effects of digoxin. The Fab fragment-digoxin complex is subsequently excreted by the kidney. Each 38 mg vial will bind about 0.6 mg digoxin. The recommended human dose is: # vials needed = (serum digoxin concentration [ng/ml] × body weight [kg])/100. A modified formula (Senior et al, 1991) taking the volume of distribution of digoxin in the dog into account is: # vials needed = body load of digoxin (mg)/0.6 mg of digoxin. The body load of digoxin = (serum digoxin concentration [ng/ml] /1000) × 14 L/kg × body weight [kg].
OTHER VASODILATORS
Vasodilators can affect arterioles, venous capacitance vessels, or both (“balanced” vasodilators). Arteriolar dilators relax arteriolar smooth muscle and thereby decrease systemic vascular resistance and afterload on the heart. This facilitates ejection of blood and also can be useful in treating animals with hypertension. In patients with mitral regurgitation, arteriolar dilators decrease the systolic pressure gradient across the mitral valve, reduce regurgitant flow, and enhance forward flow into the aorta. Reduced regurgitant flow can diminish left atrial pressure, pulmonary congestion, and possibly left atrial size.
Arteriolar or mixed vasodilator therapy is generally begun with low doses to avoid hypotension and reflex tachycardia. Reduction in concurrent diuretic dosage may be advisable. Monitoring for signs of hypotension is especially important. Sequential arterial blood pressure measurement for several hours after dosage increase is preferred. A mean arterial pressure of 70 to 80 mm Hg or a venous pO2 of >30 mm Hg (from a free-flowing jugular vein) is the suggested therapeutic goal for dosage titration. Systolic pressures of less than 90 to 100 mm Hg should be avoided. Clinical signs of drug-induced hypotension include weakness, lethargy, tachycardia, and poor peripheral perfusion. The vasodilator dose can be titrated upward, if necessary, while monitoring for hypotension with each increase in dose.
Venodilators relax systemic veins, increase venous capacitance, decrease cardiac filling pressures (preload), and reduce pulmonary congestion. Goals of venodilator therapy are to maintain central venous pressure at 5 to 10 cm H2O and pulmonary capillary wedge pressure at 12 to 18 mm Hg.
Hydralazine
Hydralazine directly relaxes arteriolar smooth muscle when the vascular endothelium is intact, but it has little effect on the venous system. The drug reduces arterial blood pressure, improves pulmonary edema, and increases jugular venous oxygen tension (presumably from increased cardiac output) in dogs with mitral insufficiency and heart failure. The most common indication for hydralazine is acute, severe CHF from mitral regurgitation. Hydralazine has been associated with significant reflex tachycardia in some animals; the dosage should be reduced if this occurs. Hydralazine can contribute to the enhanced NH response in patients with heart failure, which makes it less desirable than ACEIs for chronic use. However, it can be useful for animals that cannot tolerate an ACEI.
Hydralazine has a faster onset of action than amlodipine. Its effect peaks within 3 hours and lasts up to 12 hours. Administration of hydralazine with food decreases bioavailability by over 60%. There is also extensive first-pass hepatic metabolism of this drug. However, in dogs increased doses saturate this mechanism and increase bioavailability. General precautions for initiating and titrating therapy are outlined in the preceding section.
Hypotension is the most common adverse effect of hydralazine therapy. GI upset also can occur, which may require drug discontinuation. High dosages have been associated with a lupuslike syndrome in people, although this has not been reported in animals.
Amlodipine
This dihydropyridine L-type Ca++ channel blocker causes peripheral vasodilation as its major action, which tends to offsets any negative inotropic effect. Amlodipine has little effect on AV conduction. Besides being used to treat hypertension in cats and sometimes dogs (see Chapter 11), it is an adjunctive therapy for refractory CHF. In dogs that cannot tolerate ACEIs, amlodipine could be used in combination with a nitrate.
Amlodipine’s oral bioavailability is good. It has a long duration of action (at least 24 hours in dogs). Plasma concentration peaks in 3 to 8 hours; half-life is about 30 hours. Plasma concentrations increase with long-term therapy. Maximal effect develops over 4 to 7 days after therapy is begun in dogs. The drug is metabolized in the liver. Elimination is through the urine and feces. Because of the delay in achieving maximum effect, low initial doses and weekly blood pressure monitoring during slow up-titration are recommended.
Prazosin
Prazosin selectively blocks α1-receptors in both arterial and venous walls. It is not often used for chronic CHF management because drug tolerance can develop over time and the capsule dose-size is inconvenient in small animals. In addition, controlled clinical studies in dogs are lacking. Hypotension is the most common adverse effect, especially after the first dose. Tachycardia should occur less frequently than with hydralazine because presynaptic α2-receptors, important in the feedback control of norepinephrine release, are not blocked.
Nitrates
Nitrates act as venodilators. They are metabolized in vascular smooth muscle to produce NO, which indirectly mediates vasodilation. Nitroglycerin ointment or isosorbide dinitrate are used occasionally in the management of chronic CHF, either combined with standard therapy for refractory CHF or with hydralazine or amlodipine in animals that cannot tolerate ACEIs. Nitrates effect blood redistribution in people, but there are few studies involving dogs, especially using the oral route for CHF management. There is extensive first-pass hepatic metabolism, and the efficacy of oral nitrates is questionable. Nitroglycerine ointment (2%) is usually applied cutaneously (see p. 60). Self-adhesive, sustained-release preparations may be useful, but they have not been systematically evaluated in small animals. Transdermal patches, 5 mg, applied for 12 hours per day, have been used with anecdotal success in large dogs. Large doses, frequent application, or long-acting formulations are most likely to be associated with drug tolerance. Whether intermittent treatment (with drug-free intervals) will prevent nitrate tolerance from developing in dogs and cats is unknown.
DIETARY CONSIDERATIONS
Heart failure can interfere with the kidney’s ability to excrete sodium and water loads. Therefore dietary sodium restriction is recommended to help control fluid accumulation and reduce necessary drug therapy. Chloride restriction also appears important. However, very low salt intake can increase rennin-angiotension system activation. It is unclear whether a reduced-salt diet is necessary before overt CHF develops, but refraining from feeding the patient high-salt table scraps or treats would appear prudent. High-salt foods include processed meats; liver and kidney; canned fish; cheese, margarine, or butter; canned vegetables; breads; potato chips, pretzels, and other processed snack foods; and dog treats such as rawhide and biscuits.
Moderate salt restriction is advised when clinical heart failure develops. This represents a sodium intake of about 30 mg/kg/day (about 0.06% sodium for canned food or 210 to 240 mg/100 g of dry food). Diets for senior animals or those with renal disease usually provide this level of salt. Prescription cardiac diets usually have greater sodium restriction (e.g., 13 mg sodium/kg/day, or about 90 to 100 mg sodium/100 g of dry food, or 0.025% sodium in a canned food) and can be helpful in patients with advanced heart failure. Severe sodium restriction (e.g., 7 mg/kg/day) can exacerbate NH activation and contribute to hyponatremia. A well-balanced diet and adequate caloric and protein intake are important. Recipes for homemade low-salt diets are available, but providing balanced vitamin and mineral content may be difficult. Drinking water in some areas can contain high sodium concentrations. Nonsoftened water or (where water from the public water supply contains more than 150 ppm of sodium), distilled water can be recommended to further decrease salt intake. Supplementation of specific nutrients is important in some cases (discussed in more detail later in this section).
Inappetence is common in dogs and cats with advanced heart failure. However, more calories may be needed because of increased cardiopulmonary energy consumption or stress. Malaise, increased respiratory effort, azotemia, digoxin toxicity, and adverse effects of other medications all can contribute to poor appetite. Meanwhile, poor splanchnic perfusion, bowel and pancreatic edema, and secondary intestinal lymphangiectasia may reduce nutrient absorption and promote protein loss. Hypoalbuminemia and reduced immune function may develop. Such factors, as well as renal or hepatic dysfunction, also can alter the pharmacokinetics of certain drugs.
Strategies that sometimes help improve appetite include warming the food to enhance its flavor, adding small amounts of very palatable human foods (e.g., nonsalted meats or gravy, low-sodium soup), using a salt substitute (KCl) or garlic powder, handfeeding, and providing small quantities of the diet several times a day. If a change in diet is indi-cated, a gradual switch improves acceptance (e.g., mixing the new with the old diet in a 1 : 3 ratio for several days, then 1 : 1 for several days, then 3 : 1, and finally the new diet alone).
Cardiac cachexia is the syndrome of muscle wasting and fat loss associated with some cases of chronic CHF. Loss of muscle over the spine and gluteal region is usually noted first. Weakness and fatigue are seen with loss of lean body mass; cardiac mass also can be affected. Cardiac cachexia is thought to be a predictor of poor survival, and it is associated with reduced immune function in people. The pathogenesis of cardiac cachexia involves multiple factors, including proinflammatory cytokines, TNFα, and interleukin-1. These substances suppress appetite and promote hypercatabolism. Dietary supplementation with fish oils, which are high in omega-3 fatty acids (eicosapentaenoic [EPA] and docosahexaenoic [DHA] acids) can reduce cytokine production and may improve endothelial function, among other beneficial effects. Approximate doses of 27 mg/kg/day EPA and 18 mg/kg/day DHA produced improvement in cachexia and lower interleukin-1 levels in dogs with dilated cardiomyopathy, although there was no effect of fish oil on overall mortality (Freeman, 1998). Whether higher EPA and DHA doses would provide added benefit is not known; 30 to 40 mg/kg/day EPA and 20 to 25 mg/kg/day DHA orally have been recommended.
Grossly overweight pets with heart disease may benefit from a weight-reducing diet. Obesity increases metabolic demands on the heart and expands blood volume. This could increase cardiac filling and stimulate hypertrophy, increase venous pressure, and predispose the patient to arrhythmias as well as alter cardiac metabolism. Mechanical interference with respiration promotes hypoventilation; this can contribute to cor pulmonale and complicate preexisting heart disease.
Taurine
Taurine is an essential nutrient for cats. Prolonged deficiency causes myocardial failure as well as other abnormalities (see p. 151). Most commercial and prescription cat foods are well supplemented with taurine, which has markedly reduced the prevalence of taurine-responsive dilated cardiomyopathy in cats. But taurine concentrations should be measured in cats diagnosed with dilated cardiomyopathy, because the diet of some cats may still be deficient. Taurine-deficient cats are given oral supplements of taurine (250 to 500 mg) twice daily.
Some dogs with dilated cardiomyopathy appear deficient in taurine and/or l-carnitine, most notably American Cocker Spaniels but also others (see p. 136). Dogs fed protein-restricted diets can become taurine deficient, and some develop evidence of dilated cardiomyopathy. Taurine supplementation for dogs <25 kg is 500 to 1000 mg every 8 hours; for dogs 25 to 40 kg the dose is 1 to 2 g every 8 to 12 hours. Although not all taurine-deficient American Cocker Spaniels need both taurine and l-carnitine, most appear to.
l-carnitine
Although l-carnitine deficiency has been identified in Boxers and Doberman Pinschers with dilated cardiomyopathy, its prevalence is thought to be low and the number of affected dogs responsive to l-carnitine supplementation even lower. Nevertheless, a trial period of supplementation (at a higher dosage) may be worthwhile. After at least 4 months, reevaluation by echocardiogram is done to assess LV functional improvement. Dogs treated with carnitine supplementation may give off a peculiar odor.
The minimum effective dose of l-carnitine is not known; it may vary with the type of deficiency, if present at all. Several dose ranges have been suggested, including 50 to 100 mg/kg every 8 to 12 hours for systemic deficiency or 200 mg/kg every 8 hours for myopathic deficiency. Others use 1 g of oral l-carnitine every 8 hours for dogs <25 kg and a dose of 2 g every 12 hours for dogs between 25 and 40 kg. About ½ teaspoonful of pure l-carnitine powder is the equivalent of 1 g. Both taurine and l-carnitine can be mixed with food for easier administration.
Other Supplements
The role of other dietary supplements is unclear. Oxidative stress and free-radical damage probably play a role in the pathogenesis of myocardial dysfunction. Cytokines such as TNF, shown to increase in the circulation in heart failure, can promote oxidative stress. In people vitamin C supplementation has a beneficial effect on endothelial function, cardiac morbidity, and mortality. But the role of supplemental antioxidant vitamins in CHF is unclear, especially in animals. Whether coenzyme Q-10 provides any measurable benefit is controversial.
Beta-Blockers in Patients With Heart Failure
β-blockers must be used cautiously, especially in animals with myocardial failure, because of their negative inotropic effects. An important role is in the management of certain arrhythmias, such as atrial fibrillation, and some ventricular tachyarrhythmias (see Chapter 4). Another potential role for some β-blockers is in modulating the processes that lead to pathologic cardiac remodeling in patients with heart failure. It is well known that certain agents, in people, can improve cardiac function, reverse pathologic ventricular remodeling, and reduce mortality with chronic therapy. Carvedilol (a third-generation β-blocker) appears to be most effective in this regard, but some second-generation β-blockers (e.g., metoprolol) also show a survival benefit. It is possible that carvedilol or metoprolol might play a similar beneficial role in dogs, especially those with dilated cardiomyopathy, but the clinical efficacy of this in dogs (and cats) is presently not known.
Carvedilol blocks β1-, β2-, and α1-adrenergic receptors but is without intrinsic sympathomimetic activity. It has antioxidant effects, reduces endothelin release, has some Ca++ blocking effect, and also is thought to promote vasodilation by affecting either NO or prostaglandin mechanisms. Peak plasma concentrations appear to be quite variable after oral administration. The drug is eliminated mainly through hepatic metabolism. The half-life is short (<2 hours) in dogs; an active metabolite is thought to account for the nonselective β-blocking effect, which lasts for 12 to 24 hours. Some experimental evidence suggests that metoprolol also may produce beneficial effects on myocardial function in dogs, but ability to improve function and survival in clinical cases is unknown.
Dogs with occult myocardial dysfunction, stable compensated CHF (e.g., no evidence of congestion for at least a week or more) caused by cardiomyopathy, or chronic mitral regurgitation that show signs of myocardial dysfunction (and compensated CHF) are likely good candidates for carvedilol (or metoprolol) therapy. There are presently no definitive guidelines. Initially, very low doses are used, along with conventional CHF therapy as indicated. β-blocker up-titration is done slowly. The dosage is increased every 1 to 2 weeks, if possible, over a 2-month period, to a target dose or as tolerated. Anecdotal experience suggests a starting dose of 0.05 to 0.1 mg/kg every 24 hours for carvedilol, with an eventual target of 0.2 to 0.3 mg/kg every 12 hours (or higher) if tolerated. An initial metoprolol dose might be 0.1 to 0.2 mg/kg/day, with an eventual target of 1 mg/kg (if tolerated). Careful monitoring is important because CHF decompensation, bradycardia, and hypotension can occur.
CHRONIC DIASTOLIC DYSFUNCTION
Furosemide is continued orally in patients that have developed CHF from hypertrophic cardiomyopathy and other causes of diastolic dysfunction. Gradual reduction to the lowest dosage level and frequency that are effective for controlling edema is the aim. A β-blocker or diltiazem has traditionally also been used, but the efficacy of this in cats with chronic CHF from hypertrophic cardiomyopathy is unclear. An ACEI in such cases is thought to be beneficial, unless it provokes hypotension, particularly in cats with dynamic LV outflow obstruction (see Chapter 8). Spironolactone can also be useful as an adjunct therapy, especially for cases with recurrent pleural effusion.
REEVALUATION AND MONITORING
Client education is important when managing dogs and cats with chronic heart failure. A good understanding of the pet’s underlying disease, the signs of heart failure, and the purpose and potential adverse effects of each medication make early identification of complications more likely. Frequent reevaluation is important in patients with chronic heart failure because underlying diseases progress and complications often develop. The time frame for recheck visits may vary from weekly to every 6 months or so, depending on the severity of heart disease and the clinical stability of the patient.
Medications and dosage schedules should be reviewed at each visit, and problems with drug administration or signs of adverse effects ascertained. How well the animal has responded to medications, the diet and appetite level, activity level, and any other concerns should also be discussed. It is helpful to have owners monitor their pet’s respiratory (and, if possible, heart) rate when the animal is asleep or resting at home. Resting respiratory rates for normal animals in the home environment are usually ≤30 breaths/minute. A persistent increase (of ≥20%) in resting respiratory rate is often an early sign of worsening heart failure. This is because pulmonary edema increases lung stiffness, which induces faster, more shallow respiration. Likewise, a persistent increase in resting heart rate accompanies the heightened sympathetic tone of decompensating failure.
A thorough physical examination, with emphasis on the cardiovascular system (see Chapter 1), is important at each evaluation. Depending on the patient’s status, clinical tests might include a resting electrocardiogram (ECG) or ambulatory monitoring, thoracic radiographs, serum biochemistry analyses, an echocardiogram, serum digoxin concentration, or others. Periodic measurement of serum electrolyte and creatinine or BUN concentrations is recommended. Electrolyte imbalance (especially hypokalemia or hyperkalemia, hypomagnesemia, and sometimes hyponatremia) can occur from the use of diuretics, ACEIs, and salt restriction. Prolonged anorexia can contribute to hypokalemia, but potassium supplementation should not be used without documenting hypokalemia, especially when ACEIs and spironolactone are prescribed. Serum magnesium concentration does not accurately reflect total body stores; however, supplementation may be especially beneficial in animals that develop ventricular arrhythmias while receiving furosemide and digoxin.
Many factors can exacerbate the signs of heart failure, including physical exertion, infection, anemia, exogenous fluid administration (excess volume or sodium load), high-salt diet or dietary indiscretion, erratic administration of medication, inappropriate medication dosage for the level of disease, development of cardiac arrhythmias, environmental stress (e.g., heat, humidity, cold, smoke), development or worsening of concurrent extracardiac disease, and progression of underlying heart disease (e.g., ruptured chordae tendineae, left atrial tear, secondary right heart failure). Repeated episodes of acute, decompensated congestive failure that may require hospitalization and intensive diuresis are relatively common in patients with chronic progressive heart failure.
STRATEGIES FOR REFRACTORY CONGESTIVE HEART FAILURE
Recurrent CHF while on initial therapy with furosemide and an ACEI is usually first handled by increasing the dose of furosemide and/or maximizing the ACEI dose. Pimobendan or digoxin, if not previously used, is added if inotropic support is indicated. Other ancillary therapy could include an additional diuretic or vasodilator. Spironolactone is recommended because of its action as an aldosterone antagonist and its likely cardioprotective effects. Because its benefits are thought to extend beyond additional diuresis, use of spironolactone earlier in the course of therapy may be advantageous. Some animals benefit from the addition of a thiazide diuretic as failure becomes more refractory.
Low doses of an arteriolar vasodilator to further reduce afterload (e.g., amlodipine or hydralazine) can further intensify therapy for dogs with CHF caused by mitral regurgitation or dilated cardiomyopathy. Blood pressure should be monitored. An arteriolar vasodilator is not recommended for cats with hypertrophic cardiomyopathy or dogs with fixed ventricular outflow obstruction (e.g., subaortic stenosis).
Pathophysiology of Heart Failure
Asano K, et al. Plasma atrial and brain naturietic peptide levels in dogs with congestive heart failure. J Vet Med Sci. 1999;61:523.
Biondo AW, et al. Genomic sequence and cardiac expression of atrial natriuretic peptide in cats. Am J Vet Res. 2002;63:236.
Burger AJ, Aronson D. Activity of the neurohormonal system and its relationship to autonomic abnormalities in decompensated heart failure. J Card Fail. 2001;7:122.
Constable P, Muir W, Sisson D. Clinical assessment of left ventricular relaxation. J Vet Intern Med. 1999;13:5.
deMorais HA, Schwartz DS. Pathophysiology of heart failure. In: Ettinger SJ, Feldman EC, editors. Textbook of veterinary internal medicine. ed 6. Philadelphia: WB Saunders; 2005:914-940.
Francis GS. Pathophysiology of chronic heart failure. Am J Med. 2005;110:37S.
Freeman LM, et al. Antioxidant status and biomarkers of oxidative stress in dogs with congestive heart failure. J Vet Intern Med. 2005;19:537.
Haggstrom J, et al. Effects of naturally acquired decompensated mitral valve regurgitation on the renin-angiotensin-aldosterone system and atrial natriuretic peptide concentration in dogs. Am J Vet Res. 1997;58:77.
Kramer GA, et al. Plasma taurine concentrations in normal dogs and in dogs with heart disease. J Vet Intern Med. 1995;9:253.
Meredith IT, et al. Cardiac sympathetic nervous activity in congestive heart failure. Circulation. 1993;88:136.
Meurs KM, et al. Plasma concentrations of tumor necrosis factor-alpha in cats with congestive heart failure. Am J Vet Res. 2002;63:640.
Oyama MA, Sisson DD. Cardiac troponin-I concentration in dogs with cardiac disease. J Vet Intern Med. 2004;18:831.
Pedersen HD, et al. Activation of the renin-angiotensin system in dogs with asymptomatic and mildly symptomatic mitral valvular insufficiency. J Vet Intern Med. 1995;9:328.
Sanderson SL, et al. Effects of dietary fat and l-carnitine on plasma and whole blood taurine concentrations and cardiac function in healthy dogs fed protein-restricted diets. Am J Vet Res. 2001;62:1616.
Sisson DD. Neuroendocrine evaluation of cardiac disease. Vet Clin North Am: Small Anim Pract. 2004;34:1105.
Spratt DP, et al. Cardiac troponin I: evaluation of a biomarker for the diagnosis of heart disease in the dog. J Small Anim Pract. 2005;46:139.
Tidholm A, Haggstrom J, Hansson K. Vasopressin, cortisol, and catecholamine concentrations in dogs with dilated cardiomyopathy. Am J Vet Res. 2005;66:1709.
Turk JR. Physiologic and pathophysiologic effects of natriuretic peptides and their implication in cardiopulmonary disease. J Am Vet Med Assoc. 2000;216:1970.
Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345:1689.
Abbott JA. Beta-blockade in the management of systolic dysfunction. Vet Clin North Am: Small Anim Pract. 2004;34:1157.
Abbott JA, et al. Hemodynamic effects of orally administered carvedilol in healthy conscious dogs. Am J Vet Res. 2005;66:637.
Adin DB, Hill RC, Scott KC. Short-term compatibility of furosemide with crystalloid solutions. J Vet Intern Med. 2003;17:724.
Adin DB, et al. Intermittent bolus injection versus continuous infusion of furosemide in normal adult greyhound dogs. J Vet Intern Med. 2003;17:632.
Adin DB, et al. Efficacy of a single oral dose of isosorbide 5-mononitrate in normal dogs and in dogs with congestive heart failure. J Vet Intern Med. 2001;15:105.
Atkins CE, Snyder PS, Keene BW. Effect of aspirin, furosemide, and commercial low-salt diet on digoxin pharmacokinetic properties in clinically normal cats. J Am Vet Med Assoc. 1988;193:1264.
Atkins CE, et al. Effects of compensated heart failure on digoxin pharmacokinetics in cats. J Am Vet Med Assoc. 1989;195:945.
Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation. 2000;101:558.
Clare M, Hopper K. Mechanical ventilation: indications, goals and prognosis. Compend Cont Educ Pract Vet. 2005;27:195.
Clare M, Hopper K. Mechanical ventilation: ventilator settings, patient management, and nursing care. Compend Cont Educ Pract Vet. 2005;27:256.
COVE Study Group. Controlled clinical evaluation of enalapril in dogs with heart failure: results of the Cooperative Veterinary Study Group. J Vet Intern Med. 1995;9:243.
Davidson G. Enalapril maleate. Compend Contin Educ Small Anim Pract. 1999;21:1118.
Ettinger SJ, et al. Effects of enalapril maleate on survival of dogs with naturally acquired heart failure. J Am Vet Med Assoc. 1998;213:1573.
Freeman LM, et al. Nutritional alterations and the effect of fish oil supplementation in dogs with heart failure. J Vet Intern Med. 1998;12:440.
Garg R, et al. The effects of digoxin on mortality and morbidity in patients with heart failure. New Engl J Med. 1997;336:525.
Gordon SG, et al. Pharmacodynamics of carvedilol in conscious, healthy dogs. J Vet Intern Med. 2006;20:297.
Hamlin RL, et al. Effects of enalapril on exercise tolerance and longevity in dogs with heart failure produced by iatrogenic mitral regurgitation. J Vet Intern Med. 1996;10:85.
Hamlin RL, Nakayama T. Comparison of some pharmacokinetic parameters of 5 angiotensin-converting enzyme inhibitors in normal Beagles. J Vet Intern Med. 1998;12:93.
Hoffman RL, et al. Vitamin C inhibits endothelial cell apoptosis in congestive heart failure. Circulation. 2001;104:2182.
Hornig B, Maier V, Drexler H. Physical training improves endothelial function in patients with chronic heart failure. Circulation. 1996;93:210.
IMPROVE Study Group. Acute and short-term hemodynamic, echocardiographic, and clinical effects of enalapril maleate in dogs with naturally acquired heart failure: results of the Invasive Multicenter Prospective Veterinary Evaluation of Enalapril study. J Vet Intern Med. 1995;9:234.
Keister DM, et al. Milrinone: a clinical trial in 29 dogs with moderate-to-severe congestive heart failure. J Vet Intern Med. 1990;4:79.
Khatta M, Alexander BS, Krichten CM, et al. The effect of coenzyme Q10 in patients with congestive heart failure. Ann Intern Med. 2000;132:636.
King JN, Humbert-Droz E, Maurer M. Pharmacokinetics of benazepril and inhibition of plasma ACE activity in cats. Abstr. J Vet Intern Med. 1996;10:163.
King JN, Mauron C, Kaiser G. Pharmacokinetics of the active metabolite of benazepril, benazeprilat, and inhibition of plasma angiotensin-converting enzyme activity after single and repeated administration to dogs. Am J Vet Res. 1995;56:1620.
Kittleson MD, Johnson LE, Pion PD. The acute hemodynamic effects of milrinone in dogs with severe idiopathic myocardial failure. J Vet Intern Med. 1987;1:121.
Kvart C, et al. Efficacy of enalapril for prevention of congestive heart failure in dogs with myxomatous valve disease and asymptomatic mitral regurgitation. J Vet Intern Med. 2002;16:80.
Lee SC, et al. Iron supplementation inhibits cough associated with ACE inhibitors. Hypertension. 2001;38:166.
Lefebvre HP, et al. Effects of renal impairment on the disposition of orally administered enalapril, benazepril and their active metabolites. J Vet Intern Med. 1999;13:21.
Lombarde CW, Jöns O, Bussadori CM. Clinical efficacy of pimobendan versus benazepril for the treatment of acquired atrioventricular valvular disease in dogs. J Am Anim Hosp Assoc. 2006;42:249.
Lovern CS, et al. Additive effects of a sodium chloride restricted diet and furosemide administration in healthy dogs. Am J Vet Res. 2001;62:1793.
Luis Fuentes V. Use of pimobendan in the management of heart failure. Vet Clin North Am: Small Anim Pract. 2004;34:1145.
Luis Fuentes V, et al. A double-blind, randomized, placebo-controlled study of pimobendan in dogs with cardiomyopathy. J Vet Intern Med. 2002;16:255.
Morita H, et al. Effects of long-term monotherapy with metoprolol CR/XL the progression of left ventricular dysfunction and remodeling in dogs with chronic heart failure. Cardiovasc Drugs Ther. 2002;16:443.
Packer M. Current role of beta-adrenergic blockers in the management of chronic heart failure. Am J Med. 2001;110:81S.
Parameswaran N, et al. Increased splenic capacity in response to transdermal application of nitroglycerine in the dog. J Vet Intern Med. 1999;13:44.
Pion PD, Sanderson SL, Kittelson MD. The effectiveness of taurine and levo-carnitine in dogs with heart disease. Vet Clin North Am: Small Anim Pract. 1998;28:1495.
Pouchelon JL, et al. Long-term tolerability of benazepril in dogs with congestive heart failure. J Vet Cardiol. 2004;6:7.
Roudebush P, et al. The effect of combined therapy with captopril, furosemide, and a sodium-restricted diet on serum electrolyte levels and renal function in normal dogs and dogs with congestive heart failure. J Vet Intern Med. 1994;8:337.
Rush JE, et al. Clinical, echocardiographic and neurohormonal effects of a sodium-restricted diet in dogs with heart failure. J Vet Intern Med. 2000;14:512-520.
Rush JE, et al. Use of metoprolol in dogs with acquired cardiac disease. J Vet Cardiol. 2002;4:23.
Senior DF, et al. Treatment of acute digoxin toxicosis with digoxin immune Fab (ovine). J Vet Intern Med. 1991;5:302.
Smith PJ, et al. Efficacy and safety of pimobendan in canine heart failure caused by myxomatous mitral valve disease. J Small Anim Pract. 2005;46:121.
Straeter-Knowlen IM, et al. ACE inhibitors in HF restore canine pulmonary endothelial function and ANGII vasoconstriction. Am Physiol Soc. 1999;277:H1924.
Uechi M, et al. Cardiovascular and renal effects of carvedilol in dogs with heart failure. J Vet Med Sci. 2002;64:469.
Ward DM, et al. Treatment of severe chronic digoxin toxicosis in a dog with cardiac disease, using ovine digoxin-specific immunoglobulin G Fab fragments. J Am Vet Med Assoc. 1999;215:1808.