CHAPTER 72 Disorders of Muscle
GENERAL CONSIDERATIONS
Skeletal muscle functions to maintain posture and produce movement. Patients with generalized muscle disease generally present with weakness. This may manifest as a stiff and stilted gait, trembling while standing, a low head carriage (ventral neck flexion), and exercise intolerance. When a complete nervous system examination is performed, animals with muscle disease have normal postural reactions, are not ataxic, and usually have normal spinal reflexes. Some muscle disorders cause muscle pain and muscle swelling, whereas others cause muscle atrophy and/or fibrosis.
Myopathies in dogs and cats can be either acquired or inherited. Acquired muscle disorders include infectious and immune-mediated inflammatory disorders as well as metabolic and endocrine disorders. Characteristic clinical findings may suggest a specific diagnosis, but muscle biopsy and systemic evaluation are usually required for definitive diagnosis. Metabolic testing may be required to demonstrate and characterize functional abnormalities.
EXERCISE INTOLERANCE
Reluctance to exercise or inability to exercise for a prolonged period is a common complaint among dog owners. Exercise intolerance can result from orthopedic, cardiovascular, respiratory, hematologic, metabolic/endocrine, neurologic, neuromuscular, and muscular disorders (Box 72-1). When evaluating a dog for exercise intolerance, the veterinarian must perform a careful physical and neurologic examination. Muscle atrophy or pain and weakness at rest with normal postural reactions may suggest a muscle disorder. Joint pain may indicate that the dog has polyarthritis or degenerative joint disease. Abnormalities of cardiac auscultation or arterial pulse character should prompt thorough cardiac evaluation. Routine systemic evaluation with clinicopathologic tests and survey radiographs should be completed. When all examinations and tests are normal at rest, affected dogs should be evaluated while performing the exercise historically associated with their exercise intolerance. Characteristic clinical features during the exercise intolerance (e.g., weakness, stridor, arrhythmia) sometimes provide a clue regarding the etiology. Depending on clinical findings, additional testing may be recommended, including measuring antibodies against acetylcholine receptors (AChRs), continuous electrocardiographic monitoring, thyroid and adrenal function evaluation, arterial blood gas, and measuring preexercise and postexercise parameters (i.e., electrolytes, glucose, creatinine kinase, lactate, and pyruvate). When neurologic examination and ancillary testing suggest a muscular cause for exercise intolerance, muscle biopsies should be performed.
 BOX 72-1 Important Causes of Acquired Exercise Intolerance in Dogs
BOX 72-1 Important Causes of Acquired Exercise Intolerance in Dogs
An inherited syndrome of exercise induced collapse (EIC) has been identified in young adult (7 months to 2 years old) Labrador Retrievers being trained for field work. Affected dogs are normal at rest and with moderate exercise. Strenuous exercise in conjunction with excitement results in ataxia and rear limb weakness, sometimes progressing to collapse (Fig. 72-1). During collapse, affected dogs are hyperthermic and they hyperventilate, but physiologic and clinicopathologic parameters are not dramatically different from those of normal exercise-tolerant Labrador Retrievers taking part in the same exercise. Patellar reflexes are absent during collapse, and many affected dogs experience a profound loss of balance (disequilibrium) during collapse and recovery. A few dogs die during an episode of collapse, but most recover within 10 to 20 minutes, with no residual clinical or clinicopathologic abnormalities. Muscle biopsies are normal. The condition is not progressive, so a normal lifespan is expected if participation in the activities triggering collapse is restricted. Diagnosis is made by eliminating other causes of exercise intolerance and demonstrating that the affected dog is homozygous for the causative mutation.

INFLAMMATORY MYOPATHIES
MASTICATORY MYOSITIS
Masticatory muscle myositis (MMM) is a common immune-mediated disorder involving only the muscles of mastication in dogs. The masticatory muscles are composed primarily of a unique myofiber (type 2M) that is not present in limb muscles, and in dogs with MMM, IgG is directed against the unique myosin component of these fibers. Masticatory myositis can occur in any breed of dog, but the German Shepherd Dog, the retrieving breeds, the Doberman Pinscher, and other large breeds are most commonly affected. Primarily, young or middle-aged dogs are affected, and no apparent gender predilection exists. The disorder has not been documented in cats.
Clinical Features
The acute form of the disease involves recurrent painful swelling of the temporal and masseter muscles. Pyrexia, submandibular and prescapular lymphadenopathy, and tonsillitis are variably present. Dogs are reluctant to eat and are usually presented for anorexia and depression. Palpation of the muscles of the head and attempts to open the mouth are met with resistance because of pain.
As this disorder progresses, there is progressive, severe atrophy of the temporal and masseter muscles, resulting in a skull-like appearance of the head. Opening the mouth is no longer painful but is restricted by atrophy and fibrosis of the masticatory muscles (Fig. 72-2). The globes may sink deep into the orbits because of the dramatic loss of muscle mass (see Fig. 66-9). Many dogs will be presented for evaluation at a stage wherein they have pain on opening the mouth together with muscle atrophy as they progress from the acute to the chronic form of the disease. Occasionally, dogs will present with nonpainful severe atrophy without any history of signs related to previous acute episodes of pain.
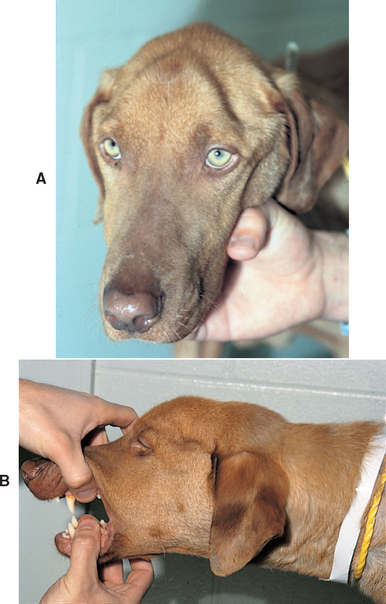
Diagnosis
Diagnosis is suspected on the basis of the clinical findings. In dogs with pain on opening the mouth, differentials must include retrobulbar abscess or mass, dental disease, and abnormalities of the temporomandibular joint or the bullae. The severe, nonpainful atrophy observed in chronically affected dogs must be differentiated from atrophy caused by disorders of the trigeminal nerve, widespread polymyositis (any etiology), or systemic disorders such as hypothyroidism or hyperadrenocorticism.
A hemogram may be normal or may reveal mild anemia and neutrophilic leukocytosis; occasionally, a peripheral eosinophilia is found. Serum creatine kinase (CK), aspartate aminotransferase (AST), and globulin concentrations may be increased. Proteinuria sometimes occurs. Circulating antibodies against type 2M fibers can be detected in the serum of many (80%) dogs with acute MMM, but they may not be present in dogs with chronic disease. Electromyography (EMG), when available, can demonstrate the presence of muscle disease in the masticatory muscles and confirm that other muscle groups are unaffected. Histopathologic evaluation of a biopsy from the affected muscles establishes the diagnosis. Fresh and formalin-fixed muscle should be submitted to permit the use of histochemical and immunohistochemical stains to identify antibody bound to type 2M muscle fibers.
Treatment
The oral administration of corticosteroids (prednisone, 1 to 2 mg/kg q12h) usually results in rapid elimination of pain in acutely affected dogs and an improved ability to open the mouth in chronically affected dogs. After approximately 3 weeks, the dose of corticosteroids can be decreased (to 1 mg/kg q24h) and then gradually tapered over 4 to 6 months to the lowest possible alternate-day dose. Inadequate dosing or treatment for an insufficient period of time is associated with a high rate of relapse. Dogs that do not respond adequately to corticosteroid therapy and dogs that relapse each time the dose is decreased may benefit from the use of other immunosuppressive drugs such as azathioprine (Imuran; Burroughs Wellcome) given 2 mg/kg orally once a day until the patient shows signs of improvement, then every 48 hours. Dogs treated aggressively have a good prognosis for recovery. They should be carefully monitored for relapse (using jaw mobility and discomfort and serum CK), particularly as the corticosteroid dose is tapered. Lifelong treatment may be required.
Historically, it was recommended that dogs with chronic MMM have their jaws opened by force under anesthesia to stretch the fibrous tissue and muscle. This practice is no longer recommended because it does not improve clinical outcome, it increases the inflammation in torn muscle fibers, and it carries an inherent risk of iatrogenic mandibular luxation or fracture.
EXTRAOCULAR MYOSITIS
A unique form of myositis confined to the extraocular muscles resulting in acute exophthalmos has been described in dogs (Fig. 72-3). Affected dogs are usually young, with a median age at presentation of 8 months. Golden Retrievers and other large-breed dogs are especially susceptible. Bilateral exophthalmos and eyelid retraction are common, often with concurrent chemosis. Vision may be impaired. Serum CK concentrations are usually normal. Orbital sonography confirms swollen extraocular muscles and eliminates retrobulbar abscess or mass as differentials. Definitive diagnosis requires biopsy of affected muscles. Treatment is as for MMM, with a good prognosis for recovery.
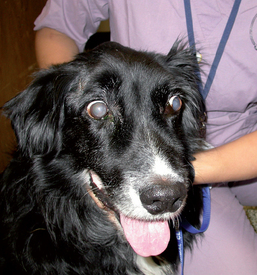
CANINE IDIOPATHIC POLYMYOSITIS
Idiopathic polymyositis (PM) is a diffuse inflammation of skeletal muscle presumed to be an autoimmune process. Large-breed adult dogs are most commonly affected, with many reported cases in German Shepherd Dogs and Boxers. Newfoundlands may also be overrepresented.
Clinical Features
Mild to severe weakness and a stiff, stilted gait that may be exacerbated by exercise are the most common features. Muscles are painful in some dogs, whereas nonpainful, severe atrophy occurs in others. Affected dogs may regurgitate as a result of megaesophagus or exhibit dysphagia, excessive salivation, and a weak bark. Signs may be intermittent in mild cases or early in the course of the disease. Some dogs with acute severe disease are pyrexic and experience generalized pain. Neurologic examination reveals normal proprioception, spinal reflexes, mental status, and cranial nerve exam. Muscle atrophy is usually prominent, especially involving the temporalis and masseter muscles.
Diagnosis
The diagnosis of PM is based on clinical signs, CK determination, EMG, and muscle biopsy. High serum CK (twofold to hundredfold increase) and AST activities are seen in most affected dogs at rest, and even more dramatic increases are common after exercise. Gamma globulins may also be increased. When available, EMG can be performed to document that multiple muscle groups are involved and to select a severely affected muscle for biopsy. A definitive diagnosis of idiopathic PM requires muscle biopsy. Typical histopathologic findings include multifocal necrosis and phagocytosis of type 1 and type 2 myofibers, perivascular lymphocytic and plasmacytic infiltration, and evidence of muscle regeneration and fibrosis. Muscle biopsy results may be normal in some dogs because of the multifocal, patchy nature of the disease. This should not preclude a diagnosis of myositis if the clinical findings, EMG, and serum CK and AST activities suggest the diagnosis.
PM can occur as an idiopathic primary immunemediated disorder, or it can be secondary to systemic immune-mediated disease (e.g., systemic lupus erythematosus), protozoal infection (e.g., Toxoplasma, Neospora myositis), or systemic neoplasia. All dogs with PM should have a complete blood count (CBC), biochemistry profile, synovial fluid analysis, urinalysis, serum antinuclear antibody (ANA) titer, and protozoal serology and/or immunohistochemical staining of muscle biopsies for protozoal antigens. Assessment of thoracic radiographs and abdominal ultrasound should focus on a search for neoplasia and identification of megaesophagus and aspiration pneumonia. Lymph node, spleen, and liver aspirates and bone marrow biopsy are indicated because lymphoma is the most common cancer associated with PM. If all of these tests are normal, a diagnosis of idiopathic PM is made.
Treatment
Prednisone administration (1 to 2 mg/kg q12h for 14 days, then q24h for 14 days, then q48h) results in dramatic clinical improvement and recovery for most dogs. In dogs with megaesophagus upright feeding of small meals (see Fig. 71-15) may be beneficial to prevent aspiration. Aspiration pneumonia, if it occurs, should be treated with antibiotics. Prednisone treatment should continue for at least 4 to 6 weeks at decreasing doses, with long-term treatment for 12 months or longer occasionally required. Azathioprine should be administered if the response to prednisone is inadequate.
FELINE IDIOPATHIC POLYMYOSITIS
An acquired inflammatory disorder of skeletal muscle similar to canine PM has been described in a few cats. Affected cats experience a sudden onset of weakness with pronounced ventral neck flexion, an inability to jump, and a tendency to sit or lie down after walking short distances. Muscle pain may be evident. Neurologic examination reveals normal mentation, cranial nerves, proprioception, and reflexes.
Diagnosis is made on the basis of clinical features, increases of serum CK and AST activities, and multifocal EMG abnormalities. Many affected cats (70%) are slightly hypokalemic, suggesting a possible relationship between this disorder and hypokalemic polymyopathy. Some clinical features of PM also mimic mild thiamine deficiency; thus evaluation of the response to treatment of affected cats with thiamine (10 to 20 mg/day, administered intramuscularly) and correction of hypokalemia are recommended before proceeding with extensive diagnostic testing for PM.
Serum titers against Toxoplasma gondii should be evaluated, as should tests for feline leukemia virus antigen and feline immunodeficiency virus antibody. A complete drug history should be obtained to eliminate the possibility of drug-induced PM. Thoracic and abdominal radiographs and abdominal ultrasound should be considered to look for an underlying neoplastic cause of the PM. PM has been diagnosed in many cats with thymoma. Muscle biopsy reveals myofiber necrosis and phagocytosis, muscle regeneration, variation in muscle fiber size, lymphocytic inflammation, and fibrosis. Empiric treatment for Toxoplasma myositis is sometimes recommended (clindamycin 12.5 to 25 mg/kg, administered orally q12h); if the animal has a dramatic response to clindamycin, the treatment should be continued for at least 6 weeks. It is important to realize, however, that spontaneous recovery or remission is observed in at least one third of all cats with PM. Corticosteroid therapy (prednisone, 4 to 6 mg/kg/day initially, tapered over 2 months) may aid recovery in some cats. Recurrences are common.
DERMATOMYOSITIS
Dermatomyositis is an uncommon disease characterized by dermatitis and polymyositis. Familial canine dermatomyositis has been reported in juvenile rough-coated and smooth-coated Collies and in Shetland Sheepdogs (i.e., Shelties). Sporadic cases have been observed in a few other breeds, including Welsh Corgis, Australian Cattle Dogs, and Border Collies. The disease has not been recognized in cats. Skin lesions include erythema, ulcers, crusts, scales, and alopecia on the inner surfaces of the pinnae and on the head and skin surfaces subjected to trauma (e.g., tail, elbows, hocks, sternum; Fig. 72-4). Mild pruritus may occur. Histopathologic findings include hydropic degeneration of basal cells and separation of the dermoepidermal junction. A perivascular mononuclear infiltrate may be seen. Dermatologic lesions appear during the first 3 months of life and may improve or resolve with time. The course often fluctuates.

FIG 72-4 A Shetland Sheepdog with typical skin lesions of dermatomyositis. This dog also had megaesophagus and generalized muscular weakness.
Dogs severely affected by dermatomyositis may develop signs of concurrent muscle disease, including generalized muscle weakness and atrophy, facial palsy, decreased jaw tone, and a stiff gait. Mentation, proprioception, and reflexes are normal. Dysphagia is common, as is regurgitation as a result of megaesophagus. EMG reveals spontaneous myofiber discharges, including fibrillation potentials, positive sharp waves, and bizarre high-frequency discharges in affected muscles. Nerve conduction velocities are normal. Muscle biopsies reveal myofiber necrosis with mononuclear cell infiltrates, atrophy, regeneration, and fibrosis. Some dogs with relatively severe dermatologic lesions exhibit no evidence of muscle disease.
Biopsies of skin and muscle, as well as EMG, may confirm a diagnosis of dermatomyositis. Breeding should be discouraged. Dogs with muscular manifestations of this disorder are usually treated with immunosuppressive doses of corticosteroids with a variable response. Dermatologic lesions may respond to oral administration of tetracycline and niacinamide (250 mg of each q8h if <10 kg, 500 mg of each q 8h if >10 kg) or pentoxifylline (Trental, 10 to 25 mg/kg q 8-12h).
PROTOZOAL MYOSITIS
Myositis caused by T. gondii can occur alone or in conjunction with myelitis, meningitis, or polyradiculoneuritis in dogs and cats, and similar syndromes caused by Neospora caninum can occur in the dog (see Chapters 69 and 71). Clinical signs referable to protozoal myositis typically include muscle pain, swelling or atrophy, and weakness. Increases in CK and AST activities are common, and serum titers for the offending organism may be positive. EMG may reveal spontaneous activity in affected muscles (definitive diagnosis requires muscle biopsy). A mononuclear inflammatory reaction is present, and organisms are often seen. Immunohistochemical stains can be used to identify the organisms and differentiate between T. gondii and N. caninum in affected dogs. Success has been reported in the treatment of protozoal myositis with oral clindamycin (12.5 to 25 mg/kg q12h) for 14 days, but more prolonged treatment (4 to 6 weeks) is advised.
ACQUIRED METABOLIC MYOPATHIES
In addition to the myopathies associated with infectious and inflammatory disease, myopathies may accompany hyperadrenocorticism (i.e., Cushing’s disease), the administration of exogenous corticosteroids, and perhaps hypothyroidism. In cats a myopathy associated with hypokalemia has been recognized.
GLUCOCORTICOID EXCESS
Glucocorticoid excess causes a degenerative myopathy. Spontaneous hyperadrenocorticism or exogenous administration of glucocorticoids, especially in high doses, can result in the syndrome. Muscle weakness and atrophy are common. Rarely, affected dogs develop limb rigidity, stiff gait, and hyperextension of all four limbs.
Diagnosis is suspected on the basis of a history of exogenous steroid administration or clinical findings consistent with steroid excess (e.g., polyuria, polydipsia, hair loss, pendulous abdomen, thin skin). Muscle biopsy reveals nonspecific changes, including type 2 myofiber atrophy, focal necrosis, and fiber size variation. Diagnostic tests for hyperadrenocorticism may confirm the diagnosis (see Chapter 53). Supplementation with l-carnitine, coenzyme Q10 and riboflavin may improve muscular strength. Control of excess glucocorticoids may result in some clinical improvement; however, in most dogs the prognosis is poor for complete resolution of the myopathy.
HYPOTHYROIDISM
Hypothyroidism may be associated with a mild myopathy in dogs, causing weakness, muscle atrophy, and reduced exercise tolerance. Spinal reflexes are normal unless concurrent polyneuropathy is present. Biopsy reveals mild type 2 myofiber atrophy. Documentation of hypothyroidism and response to thyroid hormone supplementation are required for diagnosis.
HYPOKALEMIC POLYMYOPATHY
A polymyopathy linked to decreased dietary intake or increased urinary excretion of potassium leading to total body potassium depletion has been recognized in cats of all breeds, ages, and genders. Cats with chronic renal failure and those consuming acidifying diets are most commonly affected, but cats with polyuria or polydipsia secondary to hyperthyroidism and cats with anorexia from any etiology may also be at risk. Cats with primary hyperaldosteronism because of functional adrenal neoplasia also present with hypokalemic polymyopathy.
The predominant clinical feature in all of these cats is weakness characterized by persistent ventroflexion of the neck (Fig. 72-5); a stiff, stilted gait; and reluctance to move. Some cats exhibit excessive scapular movement during walking. Muscle pain may be apparent, but the neurologic examination is otherwise unremarkable, with normal postural reactions and spinal reflexes. Clinical signs may have an acute onset and be episodic. Serum CK activity is usually high (10 to 30 times normal), serum potassium concentration is decreased (<3.5 mEq/L), and increased fractional urinary excretion of potassium (normal is 4.7-14.3%) may occur. Because most affected cats have renal dysfunction, serum urea and creatinine concentrations may be increased. Interpretation of these parameters and the urine specific gravity can be difficult because hypokalemia can itself decrease renal blood flow and glomerular filtration rate (GFR), interfering with urine-concentrating mechanisms.
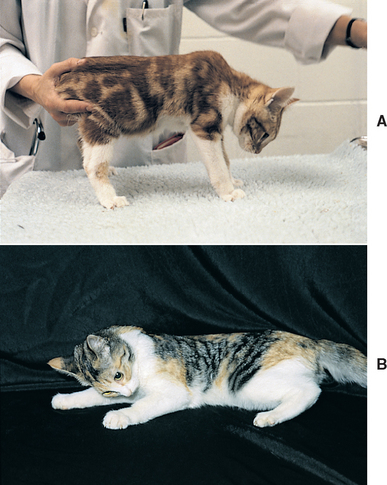
FIG 72-5 Feline hypokalemic myopathy resulting in weakness and cervical ventroflexion in (A) a kitten with congenital renal disease and (B) a hyperthyroid cat. The weakness resolved in both cats after potassium supplementation.
EMG abnormalities are found in multiple muscle groups and include frequent positive sharp waves, fibrillation potentials, and occasional bizarre high-frequency discharges with normal nerve conduction velocities. Muscle histopathology usually is normal.
Signs of hypokalemic polymyopathy usually resolve after parenteral or oral supplementation of potassium. Oral treatment with potassium gluconate is recommended for mildly affected cats (Kaon Elixir; Adria Laboratories, Columbus, Ohio) at a dose of 2.5 to 5.0 mEq/cat twice a day for 2 days, then once a day. The dose administered is adjusted on the basis of serum potassium levels. Cats with more dramatic hypokalemia (less than 2.5 mEq/L) or those with severe muscular weakness causing respiratory compromise require parenteral administration of lactated Ringer’s solution, intravenously or subcutaneously supplemented with at least 80 mEq/L of potassium chloride per liter of fluid. Intravenous (IV) supplementation of potassium should not exceed 0.5 mEq/kg/hour. Long-term oral supplementation with potassium gluconate may be required. Periodic monitoring of serum potassium concentration is recommended.
INHERITED MYOPATHIES
MUSCULAR DYSTROPHY
The muscular dystrophies (MDs) are a heterogeneous group of inherited degenerative noninflammatory muscle disorders. Most of the MDs recognized in dogs are associated with an absence of the cytoskeletal protein dystrophin caused by genetic mutation of the dystrophin gene. This very large dystrophin gene is located on the X-chromosome, so MD is generally inherited as an X-linked recessive trait, clinically apparent in male dogs and transmitted by females that are asymptomatic. Canine X-linked muscular dystrophy (CXMD) has been most completely described in Golden Retrievers but has also been reported in many other breeds of dogs, including the Irish Terrier, Samoyed, Rottweiler, Belgian Shepherd, Miniature Schnauzer, Pembroke Welsh Corgi, Alaskan Malamute, Wire-Haired Fox Terrier, German Shorthaired Pointer, Brittany Spaniel, Labrador Retriever, and Rat Terrier.
Dogs with CXMD typically show clinical signs at birth or very early in life. Golden Retriever muscular dystrophy (GRMD) has been well described, and despite the fact that all affected male dogs have the same genetic lesion, the severity of clinical expression within a litter is variable. Puppies with GRMD are often stunted even before weaning. Abduction of the elbows, a bunny-hopping gait, and difficulty opening the mouth may be noted. With time, affected puppies develop a progressively stilted gait; exercise intolerance; a plantigrade stance; atrophy of the truncal, limb, and temporalis muscles; and muscle contractures. Muscle strength deteriorates until approximately 6 months of age, when the signs tend to stabilize. Proprioceptive positioning and spinal reflexes are normal, but spinal reflexes may be difficult to elicit once muscle fibrosis and joint contractures occur. Severely affected dogs may develop pharyngeal or esophageal dysfunction. Cardiac failure occurs occasionally.
MD should be suspected when typical clinical signs are seen in a young male puppy of a predisposed breed. Serum CK levels are markedly increased as early as 1 week of age and peak at 6 to 8 weeks of age. Very dramatic increases in CK occur after exercise. EMG reveals pseudomyotonic discharges in most muscles by 10 weeks of age. Biopsies reveal marked myofiber size variation, necrosis, and regeneration with multifocal myofiber mineralization. Immunocytochemical studies document the absence of the sarcolemmal protein dystrophin. No effective treatment exists.
An X-linked MD has also been reported in the cat. Clinical signs first appear at 5 to 6 months of age. Affected cats exhibit marked generalized muscular hypertrophy, protrusion of the tongue, excessive salivation, stiff gait, and bunny hopping. Megaesophagus is common. Serum CK is greatly elevated (often >30,000 U/L). Diagnosis requires muscle biopsy and dystrophin immunostaining.
CENTRONUCLEAR MYOPATHY OF LABRADOR RETRIEVERS
Centronuclear myopathy (CNM) is a relatively common disorder in the Labrador Retriever. This disorder has been previously reported as hereditary Labrador Retriever myopathy (HLRM), autosomal recessive muscular dystrophy, and type 2 myofiber deficiency. Affected puppies appear normal at birth. Muscular weakness, an awkward gait, exercise intolerance, and muscle atrophy without myalgia typically become apparent by 3 to 5 months of age, with a few puppies showing signs at 6 to 8 weeks. The age of onset and severity of clinical signs varies dramatically among affected litter mates. Severely affected dogs exhibit a low head carriage and a short-strided, stilted gait (Fig. 72-6). Their back may be arched, and a bunny-hopping gait may develop with exercise. Muscle atrophy may be marked, especially in the proximal limbs and the muscles of mastication. Neurologic examination is normal except for consistent patellar hyporeflexia or areflexia. Megaesophagus causing regurgitation has been seen in a few affected dogs. Clinical signs are worse with stress, exercise, excitement, or cold temperatures. Muscular weakness and atrophy are typically slowly progressive, but a few affected puppies will be recumbent within 1 to 2 months. Clinical signs stabilize after 12 months of age in mildly affected dogs. Serum CK is normal or moderately elevated, and on EMG examination spontaneous electrical activity and bizarre high-frequency discharges are seen. CNM is histologically characterized by mild to marked variation in fiber size, atrophic type 1 and type II myofibers, replacement of type 2 myofibers by type 1 myofibers resulting in a type 2 predominance, and a marked increase in centralization of nuclei within muscle cells. CNM has an autosomal recessive inheritance pattern. Recently, the causative genetic mutation has been identified and a DNA test is commercially available. No treatment is available, but mildly affected dogs can function as pets.

MYOTONIA
Myotonia is a rare disorder of muscle that has been recognized in Chow Chows, Cocker Spaniels, Staffordshire Bull Terriers, Miniature Schnauzers, Labrador Retrievers, Rhodesian Ridgebacks, Samoyeds, West Highland White Terriers, Great Danes, and individual dogs of a number of breeds. Affected kittens have also been identified. Myotonia causes involuntary contraction of muscle that persists after voluntary movement or stimulation. This results from altered chloride conductance, which causes postexcitement depolarization of the muscle membrane and continued contraction. In Miniature Schnauzers the mutant skeletal muscle chloride channel allele has been identified, and a PCR-based test has been developed.
Clinical signs include generalized muscle stiffness and hypertrophy that begin at a young age (i.e., 2 to 6 months). Dogs with myotonia are neurologically normal. No abnormalities of proprioception or mentation exist. Cold weather, excitement, and exercise exacerbate the clinical signs. Affected dogs may remain in rigid recumbency for up to 30 seconds if they are suddenly placed in lateral recumbency. Serum CK and AST activities may be increased, indicating muscle fiber necrosis. Bizarre high-frequency discharges that wax and wane (“dive-bomber sound”) are revealed by EMG and, when present, confirm the diagnosis. Muscle biopsy alone is rarely diagnostic. Membrane-stabilizing agents such as procainamide (10 to 30 mg/kg, administered orally q6h) and phenytoin (20 to 35 mg/kg, administered orally q12h) and the sodium channel blocker mexiletine (Mexitil; Boehringer Ingelheim: 8 mg/kg, administered orally q8h), have been beneficial in the treatment of some cases. The avoidance of cold temperatures is also advised. Most dogs are euthanized because of the severity of their signs.
INHERITED METABOLIC MYOPATHIES
A number of genetically based metabolic myopathies have been described in dogs and cats. In each of these disorders there is a biochemical defect of the skeletal muscle energy system, resulting in inefficient muscle performance. All of these disorders cause signs of muscle dysfunction, including exercise intolerance; muscular weakness; a stiff, stilted gait; muscle pain; muscle tremors; and muscle atrophy. Mitochondrial myopathies, glycogen storage diseases, lipid storage myopathies, and disorders causing nemaline rod accumulation within myofibers have all been reported. Establishing the precise cause of a metabolic myopathy can be difficult because of the wide range of biochemical abnormalities that can arise and the co-dependence of all of the structural proteins making up a muscle fiber. Sometimes metabolic testing can be beneficial; for example, inappropriate lactic acid accumulation with exercise suggests mitochondrial dysfunction. Evaluation of plasma lactate and pyruvate before and after exercise and quantitative analysis of urinary organic acids and plasma, urine, and muscle carnitine will help to document that a metabolic myopathy is present and may help determine the affected biochemical pathway. After metabolic testing, histologic and ultrastructural examination of skeletal muscle should be performed. This metabolic testing and biopsy evaluation should be performed by a laboratory specializing in metabolic disorders of dog and cat muscle. When testing suggests a mitochondrial myopathy or a lipid myopathy, nonspecific treatment with an oral combination of l-carnitine (50 mg/kg q12h), coenzyme Q10 (100 mg/dog q 24h), and riboflavin (100 mg/dog q 24h) may result in improved muscle strength.
INVOLUNTARY ALTERATIONS IN MUSCLE TONE
Tetanus, opisthotonus, myoclonus, and dyskinesias are all involuntary alterations of muscle tone that are not the result of muscle disease. Tetanus is a sustained tonic contraction of the muscles. Opisthotonos is a very severe form of tetanus in which spasm of the limb and neck muscles results in lateral recumbency with dorsiflexion of the neck and extensor rigidity of the limbs. Myoclonus is the rhythmic repetitive contraction of a particular group of muscles. Dykinesias, a group of poorly defined movement disorders, are discussed in Chapter 65.
OPISTHOTONOS AND TETANUS
Loss of consciousness occurring in association with tetanus and opisthotonos (decerebrate rigidity, see Fig. 63-9, A) is seen in dogs and cats with severe brainstem disease caused by infection, trauma, or neoplasia. Brainstem disease in these animals is suspected on the basis of the history, neurologic findings, and results of clinicopathologic tests. Opisthotonos and tetanus with no altered state of consciousness may be seen after trauma to the rostral cerebellum (decerebellate rigidity, Fig. 63-9, B), and in Clostridium tetani infection.
C. tetani is a gram-positive, anaerobic bacillus that produces spores that persist for long periods in the environment. If a deep wound or an area of tissue damage becomes contaminated with these spores, the spores may be anaerobically converted to a vegetative form and a toxin (tetanospasmin) is produced. The toxin ascends peripheral nerves to the spinal cord, where it blocks the release of neurotransmitter from the inhibitory interneurons (Renshaw cells), releasing extensor muscles from inhibition and resulting in tetany. Cats are more resistant to the toxin than dogs.
Clinical signs of tetanus appear 3 to 20 days after wound infection. Animals with mild or early tetanus show a stiff gait, erect ears, an elevated tail, and contraction of the facial muscles (risus sardonicus; Fig. 72-7). The signs may be most severe in the area of the body adjacent to where the toxin is being produced. In severe disease the animal is recumbent and shows extensor rigidity of all four limbs and opisthotonos. The animal may die as a result of an inability to ventilate adequately. Tetanus is diagnosed on the basis of clinical signs and the history of a recent wound.

FIG 72-7 Tetanus in two dogs, with the erect ears and risus sardonicus resulting from contraction of the head and facial muscles. Both dogs had wounds on a forelimb, which were presumed to be the site of entry of the toxin.
Treatment should consist of rest, immediate wound debridement, antibiotics, neutralization of the toxin, and intensive supportive care. Initially, aqueous penicillin G is administered intravenously (40,000 U/kg q8h), after which the procaine salt can be given by intramuscular injection (40,000 U/kg q12h). Alternatively, metronidazole (10 to 15 mg/kg IV q8h) may be administered; it is bactericidal against most anaerobes and achieves a therapeutic concentration even in necrotic tissues. Antibiotics are administered for 2 weeks or until clinical recovery occurs.
A test dose of tetanus antitoxin (equine origin) is injected intradermally 15 to 30 minutes before the administration of a treatment dose. If no wheal develops, the antitoxin is administered intravenously (200 to 1000 U/kg; maximum, 20,000 U). This dose is not repeated because a therapeutic blood concentration persists for 7 to 10 days after a single injection, and repeated administration of antitoxin increases the chance of an anaphylactic reaction. The injection of a small dose of antitoxin (1000 U) just proximal to the wound site may be beneficial in dogs and cats with localized tetanus.
The animal is maintained in a quiet, dark environment. Muscle spasms are controlled with oral or IV diazepam (0.5 to 1 mg/kg, as needed) and IV chlorpromazine (0.5 mg/kg q8h) or intramuscular acepromazine (0.1 to 0.2 mg/kg q6h). Phenobarbital (2 mg/kg q8h, administered intravenously or intramuscularly) or pentobarbital (5 to 15 mg/kg IV to effect) may be administered as needed. IV fluids are administered, and nutritional support is achieved using nasogastric or gastrotomy tube feeding. The animal is hand-fed as soon as it is able to prehend food and swallow. In some animals urinary and fecal retention must be managed by repeated catheterization and enemas. Improvement is usually noticeable within 1 week, but signs may persist for 3 to 4 weeks. The prognosis is poor if the signs progress rapidly, but approximately 50% of affected dogs survive if managed intensively.
MYOCLONUS
Myoclonus is a rhythmic, repetitive contraction of a portion of a muscle, an individual muscle, or a group of muscles occurring as often as 60 times per minute. These rhythmic contractions do not abate during sleep or general anesthesia. Limb and facial muscles are most often involved. Myoclonus is most commonly associated with canine distemper meningoencephalomyelitis, but other focal inflammatory or neoplastic lesions of the spinal cord can also produce myoclonus in rare cases. The prognosis for resolution of the myoclonus is grave.
Familial reflex myoclonus has been recognized in 4- to 6-week-old Labrador Retriever puppies. Clinical signs include intermittent spasms of the axial and appendicular muscles with occasional episodes of opisthotonos. These signs worsen when the animal is stressed or excited. Treatment with diazepam and clonazepam has not been successful. The prognosis for recovery is grave.
Allgoewer I, et al. Extraocular muscle myositis and restrictive strabismus in 10 dogs. Vet Ophthalmol. 2000;3(1):21.
Bandt C, et al. Retropective study of tetanus in 20 dogs: 1988-2004. J Am Anim Hosp Assoc. 2007;43:143.
Braund KG. Myopathic disorders. Braund KG, editor. Clinical neurology in small animals: localization, diagnosis, and treatment. Ithaca, NY: International Veterinary Information Service. 2005. www.ivis.org, 2005.
Evans J, Levesque D, Shelton GD. Canine inflammatory myopathies: A clinicopathologic review of 200 cases. J Vet Intern Med. 2004;18:679.
Gaschen F, Jaggy A, Jones B. Congenital diseases of feline muscle and neuromuscular junction. J Feline Med Surg. 2004;6:355.
Klopp LS, et al. Autosomal recessive muscular dystrophy in Labrador Retrievers. Comp Cont Educ Sm Anim Pract. 2000;22(2):121.
Platt SR, Shelton GD. Exercise intolerance, collapse and paroxysmal disorders. In: Platt SR, Olby NJ, editors. BSAVA manual of canine and feline neurology. Gloucester: BSAVA, 2004.
Shelton GD, Engvall E. Muscular dystrophies and other inherited myopathies. Vet Clin N Am: Sm Anim Pract. 2002;32:103.
Taylor SM. Selected disorders of muscle and the neuromuscular junction. Vet Clin N Am Sm Anim Pract. 2000;30(1):59.
Taylor SM. Exercise-induced weakness/collapse in Labrador Retrievers. In Tilley LP, Smith FW, editors: Blackwell’s five minute veterinary consult: canine and feline, ed 4, Ames, Iowa: Blackwell, 2007.
Vite CH. Myotonia and disorders of altered muscle cell membrane excitability. Vet Clin North Am Small Anim Prac. 2002;32:169.
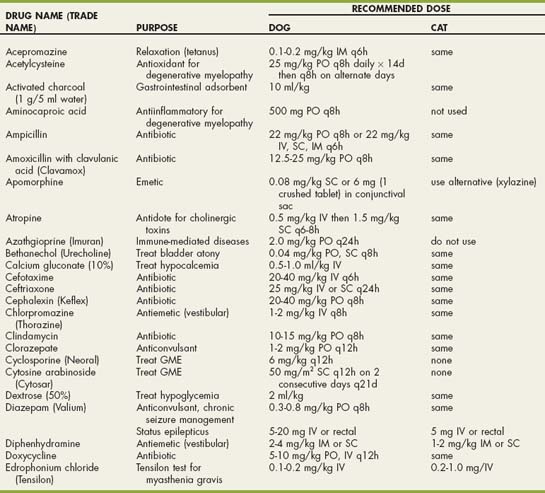
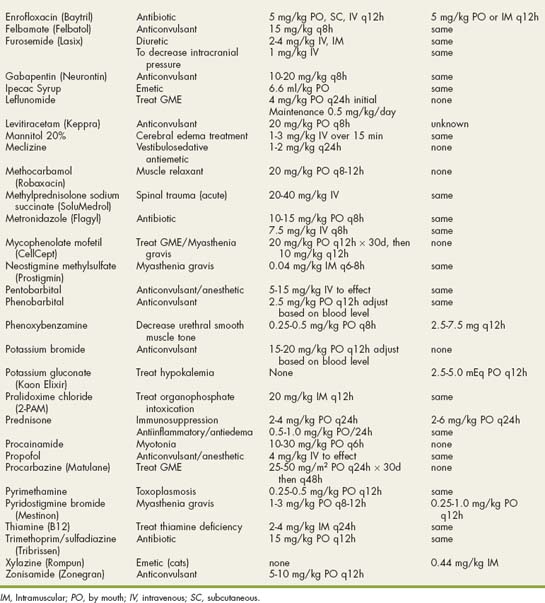
 Drugs Used in Neurologic Disorders
Drugs Used in Neurologic Disorders