CHAPTER 71 Disorders of Peripheral Nerves and the Neuromuscular Junction
GENERAL CONSIDERATIONS
The clinically important peripheral nerves are the peripheral nerves arising from the spinal nerves in the cervical and lumbar intumescences to innervate the muscles of the limbs and the 12 pairs of cranial nerves originating in the brainstem. Spinal nerve or peripheral nerve lesions result in lower motor neuron (LMN) motor signs of weakness, decreased tone, and decreased reflexes in affected muscles and limbs. When sensory components of the peripheral nerves are involved, there may also be decreased, absent, or altered sensation in the skin supplied by that nerve.
At the neuromuscular junction (NMJ) a nerve impulse reaching the nerve terminal initiates the release of acetylcholine (ACh) into the synaptic cleft. ACh binds to ACh receptors on the postsynaptic (muscle) membrane, inducing a conformational change and ion flux that results in muscular contraction. Presynaptic NMJ disorders that interfere with the release of ACh from the nerve terminal result in generalized LMN signs of weakness and hyporeflexia similar to disorders affecting peripheral nerves. Myasthenia gravis is a postsynaptic disorder that causes partial failure of neuromuscular transmission, resulting in weakness with normal spinal reflexes, similar to the muscle disorders discussed in Chapter 72.
FOCAL NEUROPATHIES
TRAUMATIC NEUROPATHIES
Traumatic neuropathies are common. They result from mechanical blows, fractures, pressure, stretching, laceration, and the injection of agents into or adjacent to the nerve. Diagnosis is usually straightforward and is based on the history and clinical findings. Individual nerves or a group of adjacent nerves may be damaged. Traumatic radial nerve paralysis, complete avulsion of the entire brachial plexus, and sciatic nerve injury are most common in the dog and cat (Table 71-1; Fig. 71-1).
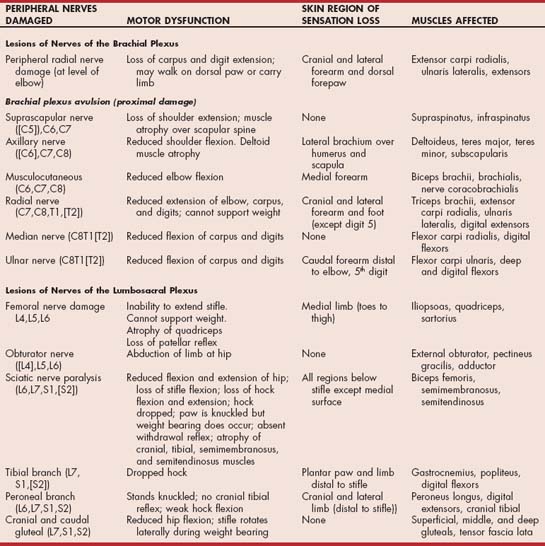

FIG 71-1 A, Traumatic brachial plexus avulsion in a Chesapeake Bay Retriever. B, Horner’s syndrome in the same dog.
Electrodiagnostic testing, when available, can be used to evaluate the extent of nerve damage. In 5 to 7 days after denervation of a muscle, electromyography detects denervation action potentials (i.e., increased insertional activity and spontaneous action potentials) in the muscles normally supplied by the damaged nerve (see Table 71-1). Nerve conduction studies proximal and distal to the site of injury are also useful in assessing nerve integrity.
When an animal is presented with a peripheral nerve injury, careful mapping and assessment of cutaneous sensation and motor function help determine the precise location of the injury, and sequential mapping can be used to monitor progress (Fig. 71-2). The regenerative ability of a nerve is proportional to the continuity of connective tissue structures remaining around the damaged portion of the nerve. If adequate connective tissue scaffolding is left, axonal regeneration can occur at a rate of 1 to 4 mm/day. Severed nerve ends should be surgically brought into apposition and anastomosed to increase the likelihood of regeneration. The closer a nerve injury is to the innervated muscle, the better the chances are of recovery.
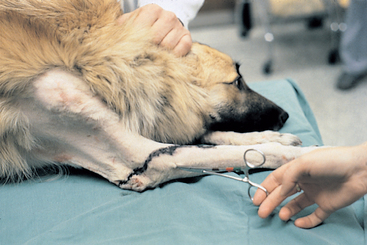
FIG 71-2 Mapping the region of sensory loss is important in localizing lesions and monitoring improvement. This dog has a caudal brachial plexus avulsion, so he has lost superficial sensation on the limb distal to the elbow.
Physical therapy such as swimming, limb manipulation, heat therapy, and massage help delay muscle atrophy and tendon contracture and speed return of function in animals with incomplete lesions. Self-mutilation may become a problem 2 to 3 weeks after injury because regeneration of sensory nerves can result in abnormal sensation lasting 7 to 10 days. Lack of improvement in motor function after 1 month warrants consideration of amputation of the affected limb or, when feasible, arthrodesis for limb salvage.
PERIPHERAL NERVE SHEATH TUMORS
Tumors of nerve sheath origin arise from cells surrounding the axons in peripheral nerves or nerve roots. Most of these tumors are anaplastic with a high mitotic index and aggressive biological behavior and are therefore classified as malignant peripheral nerve sheath tumors (PNSTs) regardless of their cell of origin. These tumors are a relatively common cause of lameness and neuropathy when they involve the nerves of the brachial plexus. Lymphoma may also involve the nerve roots or peripheral nerves of dogs and cats (Fig. 71-3).
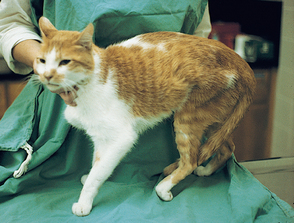
FIG 71-3 Dramatic muscle atrophy and sensory loss in a cat with lymphoma involving the L6-S1 nerve roots.
Clinical Features
Clinical signs depend on tumor location and the nerves involved. Trigeminal nerve sheath tumors cause ipsilateral atrophy of the temporalis and masseter muscles. Malignant PNSTs in dogs most commonly affect the caudal cervical (C6-C8) or cranial thoracic (T1-T2) nerve roots of the brachial plexus, resulting in lameness, muscle atrophy, pain, and lifting of the affected leg (root signature). The insidious onset of these tumors may make it difficult to differentiate them from lameness caused by a vague musculoskeletal injury or nerve root compression caused by intervertebral disk disease. With progression of the tumor, atrophy, weakness, and loss of reflexes may occur as the affected peripheral nerve is destroyed. Tumors involving the T1-T3 nerve roots commonly interrupt the sympathetic pathway and result in ipsilateral Horner’s syndrome. Similarly, the ipsilateral cutaneous trunci reflex will be absent if the C8-T1 ventral nerve roots are damaged. Tumors originating in the spinal canal and extending peripherally and tumors originating in the brachial plexus and extending proximally into the vertebral canal will often cause upper motor neuron (UMN) deficits in the ipsilateral hindlimb as the tumor expands, but this may not be clinically apparent until significant spinal invasion has taken place.
Diagnosis
Radiographs of the spine are indicated if a neoplasm involving a spinal nerve root is suspected. Nerve sheath tumors rarely cause bony changes, although expanding tumors that pass through an intervertebral foramen may cause widening of the foramen as a result of pressure necrosis. Myelography can be useful to identify spinal cord compression. Electromyography and nerve conduction velocity determinations may confirm the presence of a peripheral nerve lesion and aid in localization. Deep palpation of the axilla under general anesthesia and ultrasound examination may reveal a mass. Advanced diagnostic imaging (i.e., computed tomography [CT], magnetic resonance imaging [MRI]), when used with contrast enhancement, is the best way to delineate tumor masses and detect vertebral canal invasion (Fig. 71-4).
Treatment
The treatment of choice for a PNST is surgical removal. Aggressive removal of distally located tumors can result in a cure. Extensive neurologic damage by the tumor, damage affecting several spinal nerves or nerve roots, or severely atrophied muscles usually require amputation of the limb. Nerve root tumors that have progressed to cause spinal cord compression usually involve multiple nerve roots, are rarely completely resectable, and are associated with a poor prognosis. Postoperative irradiation may be indicated in an attempt to slow tumor recurrence.
FACIAL NERVE PARALYSIS
Facial nerve (CN7) paralysis is recognized frequently in dogs and cats. In 75% of dogs and 25% of cats with acute facial nerve paralysis, there are no associated neurologic or physical abnormalities and no underlying cause can be found, prompting a diagnosis of idiopathic facial nerve paralysis. Canine hypothyroidism is occasionally associated with a mononeuropathy involving the facial nerve, but the causality is uncertain. Traumatic injury to the facial nerve can also occur at the level of the brainstem or peripherally as the nerve courses through the petrous temporal bone.
The most common identifiable cause of facial nerve paralysis, however, is damage to branches of the facial nerve within the middle ear secondary to inflammation, infection, or neoplasia. Otitis media and otitis interna usually result from extension of bacterial otitis externa, particularly in breeds predisposed to chronic bacterial otitis externa (e.g., Cocker Spaniels, German Shepherd Dogs, and Setters. Foreign bodies (e.g., grass awns), malignant tumors (in both dogs and cats), and benign nasopharyngeal polyps (in cats) involving the middle ear can also cause facial nerve paralysis.
Clinical Features
Clinical manifestations of facial nerve paralysis include an inability to close the eyelid, move the lip, or move the ear. Affected animals are unable to blink spontaneously or in response to visual or palpebral sensory stimulation. Corneal ulceration may occur because of an inability to distribute the tear film by blinking (neuroparalytic keratitis) and loss of facial nerve (parasympathetic)–stimulated lacrimal gland secretion (neurogenic keratitis). Drooping of the ear and lip as a result of loss of muscle tone on the affected side is common (Fig. 71-5). Many dogs and cats with facial nerve paralysis caused by middle ear disease also develop peripheral vestibular signs and/or Horner’s syndrome because of the close proximity of the nerves in the area of the middle and inner ear. Rarely, a painful syndrome of hemifacial spasm with facial muscle contracture and lip retraction may occur as a result of facial nerve irritation. This should be differentiated from nonpainful muscle atrophy and contracture, which occur relatively commonly in animals with long-standing facial nerve paralysis (Fig. 71-6).

Diagnosis
Idiopathic facial nerve paralysis can be diagnosed only after excluding all other causes. A complete neurologic examination should be performed to ensure that there are no other cranial nerve deficits, ataxia, or proprioceptive deficits suggesting a brainstem lesion. Clinicopathologic testing (i.e., complete blood count [CBC], serum biochemistry profile, urinalysis) is required to evaluate for systemic or metabolic disease. A suspicion of hypothyroidism warrants evaluation of thyroid function (see Chapter 51).
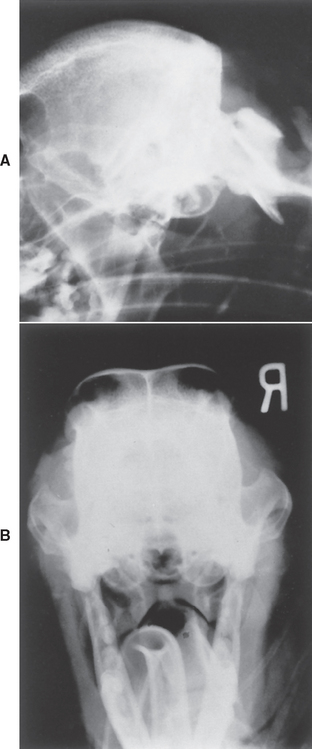
All dogs and cats with facial nerve paralysis should be evaluated carefully for disease of the middle and inner ear. Careful otoscopic examination is important, even if general anesthesia is required. Most animals with otitis media or otitis interna have obvious otitis externa and a tympanic membrane that appears abnormal or ruptured, but occasionally the otoscopic examination is normal. If the suspicion for middle and inner ear disease is high, general anesthesia for radiographs, advanced imaging, and myringotomy to collect a sample from the middle ear are warranted (Fig. 71-7).
Treatment
No treatment exists for idiopathic facial nerve paralysis. If keratoconjunctivitis sicca is present, the eye should be medicated as needed. The paralysis may be permanent, or spontaneous recovery may occur in 2 to 6 weeks.
If evaluation of the middle and inner ear reveals bony lysis or extensive soft tissue proliferation, this suggests that neoplasia could be the cause of facial nerve paralysis. A biopsy should be performed, and surgery to debulk or remove the tumor should be considered. The prognosis for cure with feline benign inflammatory nasopharyngeal polyps in this location is excellent. Tumors of the bulla, bony labyrinth, ear canal, or peripheral nerve are less likely to be treated effectively using surgery alone. Radiotherapy or chemotherapy may be beneficial in some cases.
Medical treatment of dogs and cats with bacterial otitis media and otitis interna is discussed in Chapter 68.
TRIGEMINAL NERVE PARALYSIS
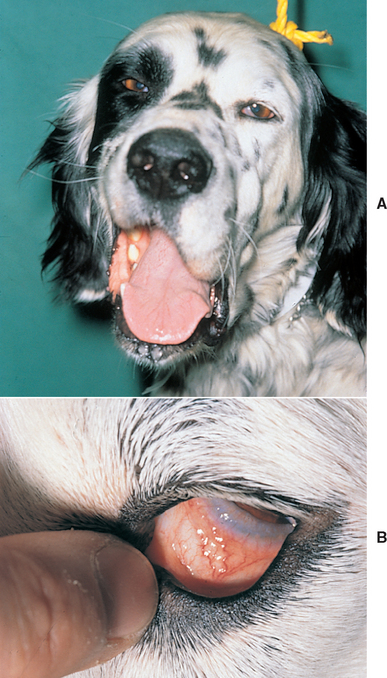
Bilateral motor paralysis of the trigeminal nerves results in the sudden onset of an inability to close the jaw or prehend food. The mouth hangs open, but it can be physically closed and manipulated without resistance (Fig. 71-8). Swallowing is usually normal. Severe rapid atrophy of the muscles of mastication may occur, and a few animals display concurrent Horner’s syndrome. Sensory loss (trigeminal distribution) is variable, but if hyposensitization of the corneal surface occurs, there will be decreased reflex tear formation and loss of trophic factors, leading to corneal ulceration without significant discomfort (neurotrophic keratitis).
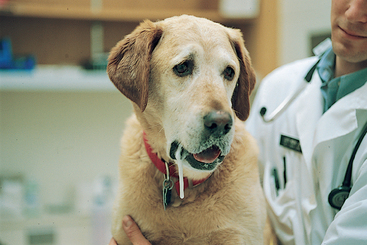
FIG 71-8 Idiopathic trigeminal nerve motor paralysis resulting in a dropped jaw and excessive drooling in a 9-year-old Labrador Retriever. The paralysis resolved in 14 days without therapy.
Idiopathic trigeminal paralysis is seen in middle-aged and older dogs and rarely in cats. The diagnosis relies on clinical signs and on ruling out other possible causes. Rabies and other inflammatory central nervous system (CNS) diseases are unlikely in the absence of other clinical signs. Neoplastic and traumatic disorders are not usually bilateral, although bilateral motor trigeminal nerve infiltration has been reported in a dog with multicentric lymphoma.
The etiology of this idiopathic disorder is unknown. If biopsy of the nerve is performed, it reveals bilateral nonsuppurative neuritis of all motor branches of cranial nerve 5 associated with demyelination. Treatment consists of supportive care. Most dogs can drink and maintain adequate hydration if they are given water in a deep container, such as a bucket. Hand-feeding may be necessary. Holding the mouth partially closed in a sling may facilitate eating and drinking during recovery (Fig. 71-9). Lubricating eye ointments may help prevent corneal ulceration. The prognosis is excellent, with most animals recovering completely within 2 to 4 weeks.

HYPERCHYLOMICRONEMIA
Peripheral neuropathies have been observed in cats of all ages with a mutation in the gene encoding lipoprotein lipase. Affected cats have delayed clearance of chylomicrons from the circulation, resulting in the formation of lipid granulomas (xanthomas) in the skin and other tissues. These xanthomas may compress a nerve against bone, resulting in neuropathology. Horner’s syndrome and tibial and radial nerve paralysis are most often seen. Clinicopathologic testing reveals fasting hyperchylomicronemia and blood that looks like cream-of-tomato soup. Diagnosis is by biopsy of the xanthomas or measurement of lipoprotein lipase concentration. The neurologic signs are reversible if hyperchylomicronemia can be controlled by feeding affected cats a low-fat diet (see Chapter 54).
ISCHEMIC NEUROMYOPATHY
Caudal aortic thromboembolism causes paralysis from ischemic damage to affected muscles and peripheral nerves. Ischemia is caused by vasoconstriction of the collateral circulation to the limbs as a result of release of thromboxane A2 and serotonin from platelets in a clot lodged in the aortic trifurcation. Caudal aortic thromboembolism is common in cats and rare in dogs. An acute onset of LMN pelvic limb paralysis or paresis is seen. Femoral pulses are weak or absent. The legs and feet are cool, and the pads are no longer pink (Fig. 71-10). Hemorrhage does not occur when a toenail is cut short on an affected foot. The affected muscles are swollen and painful. LMN paralysis with complete areflexia of the rear limbs are common, although occasionally the patellar reflex is maintained. Within hours, rigid extension of the legs may occur as a result of contracture of ischemic muscle. In cats cardiomyopathy is most common. In dogs a disorder associated with hypercoagulability can usually be identified (see Chapter 12), and the dog should be evaluated for nephrotic syndrome, hyperadrenocorticism, heartworm disease, neoplasia, and endocarditis. Treatment of feline aortic thromboembolism is discussed in Chapter 12.
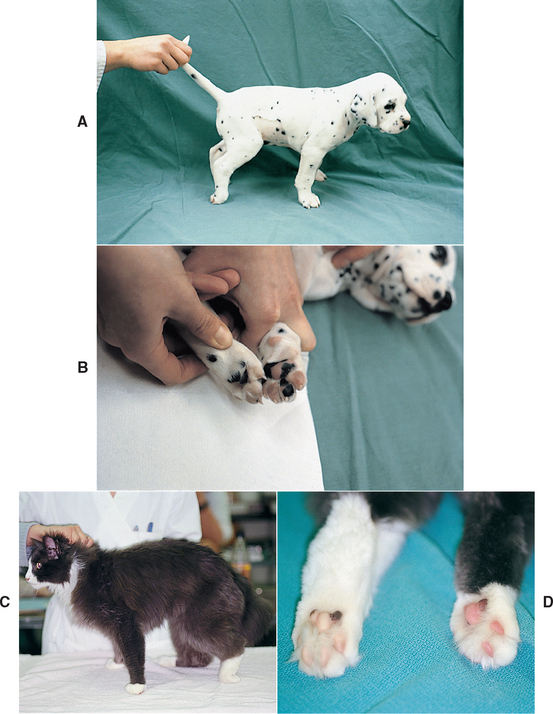
FIG 71-10 A, Acute, severe lower motor neuron paralysis of the rear limbs occurred in this 6-week-old Dalmatian puppy. The limbs were cool, and no femoral pulses were palpable. B, The footpads on the front feet were warm and pink, whereas those on the rear feet were cool and pale. Ultrasound examination revealed a caudal aortic thrombus. C, Acute lower motor neuron paralysis in the left hindlimb of a 9-year-old cat caused by an iliac artery thrombus. D, The left hindlimb was cool, had no palpable femoral arterial pulse, and had pale footpads.
POLYNEUROPATHIES
CONGENITAL/INHERITED POLYNEUROPATHIES
A number of breed-associated degenerative peripheral neuropathies occur. They usually affect young animals and are presumed to have a hereditary basis. Most of these disorders cause progressive generalized LMN dysfunction with severe tetraparesis, muscle atrophy, and hyporeflexia. Pathologic lesions vary with each disorder but may involve the motor neurons in the ventral horn of the spinal cord, ventral nerve roots, or peripheral nerves. In Rottweilers, Dalmatians, and Great Pyrenees, concurrent laryngeal paralysis is common. Certain inherited storage diseases will cause CNS signs as well as diffuse LMN paresis. Familial sensory neuropathies causing diminished sensation /nociception and self-mutilation (English Pointers) and sometimes mild ataxia and loss of proprioception (Longhaired Dachshunds) can also occur. All of these conditions are extremely rare and are reviewed in detail in Suggested Readings. Presumptive diagnosis is by recognition of typical breed and age of onset and presentation and ruling out of other disorders. Definitive diagnosis requires electrophysiological evaluation of nerve function and nerve biopsy.
ACQUIRED CHRONIC POLYNEUROPATHIES
Polyneuropathies affect more than one group of peripheral nerves, resulting in generalized LMN signs that include flaccid muscle weakness or paralysis, marked muscle atrophy, decreased muscle tone, and reduced or absent reflexes. Proprioceptive deficits may be evident if the sensory portions of the nerves are severely affected. Electromyography, when available, reveals evidence of denervation, and nerve conduction velocity is decreased in affected nerves. Nerve biopsies typically reveal axonal degeneration and demyelination regardless of the underlying cause, so a thorough systemic investigation of possible etiologies is required to reach a diagnosis and recommend appropriate treatment (Box 71-1).
 BOX 71-1 Generalized Disorders of the Peripheral Nerves and the Neuromuscular Junction
BOX 71-1 Generalized Disorders of the Peripheral Nerves and the Neuromuscular Junction
Chronic Lower Motor Neuron Paresis
Breed-associated degenerative neuropathies
Diabetic Polyneuropathy
Clinical signs of diabetic polyneuropathy are usually subtle or inapparent in the dog but may be dramatic in the cat. Weakness of the rear limbs, reluctance to jump, a plantigrade pelvic limb stance, and weakness of the tail are characteristic (Fig. 71-11). Physical examination findings may include rear limb hyporeflexia and marked muscle atrophy. (See Chapter 52 for more information.) If diabetic polyneuropathy is recognized early, establishing improved glucose regulation can result in stabilization or improvement of neurologic signs in some cats.

Hypothyroid Polyneuropathy
Hypothyroidism has been associated with a variety of peripheral nerve abnormalities, including diffuse LMN paralysis, unilateral peripheral vestibular disease, facial nerve paralysis, laryngeal paralysis, and megaesophagus in dogs. Nerve and muscle biopsies in affected dogs may show neuronal degeneration and regeneration, as well as muscle fiber type grouping that is most indicative of neurogenic atrophy. In some hypothyroid dogs neurologic signs resolve once supplementation with thyroid hormone is initiated (Fig. 71-12). (See Chapter 51 for more information).
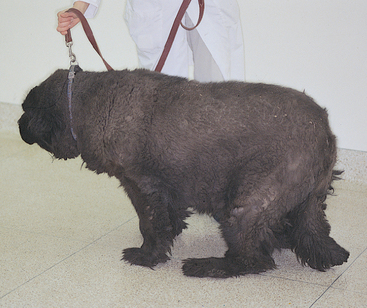
Insulinoma Polyneuropathy
Insulin-secreting tumors have been associated with a paraneoplastic polyneuropathy in dogs. Affected dogs may initially have a stiff rear limb gait, but this progresses to generalized weakness, muscle atrophy, and hyporeflexia. Treatment of the insulinoma may result in resolution of the polyneuropathy. (See Chapter 52 for more information.)
Paraneoplastic Polyneuropathy
Although clinically significant paraneoplastic neuropathies are infrequently recognized in dogs and cats, histologic lesions of polyneuropathy are evident in most dogs with cancer. LMN paresis caused by paraneoplastic polyneuropathy has been reported in dogs with bronchogenic carcinoma, hemangiosarcoma, mammary carcinoma, pancreatic carcinoma, prostatic carcinoma, lymphoma, and multiple myeloma. Complete systemic evaluation and cancer search (thorough physical examination, thoracic and abdominal radiographs, abdominal ultrasound, lymph node aspirates) are warranted in all animals presented for chronic progressive LMN dysfunction. In some cases treatment or removal of the offending neoplasm is associated with resolution of the clinical signs of polyneuropathy.
Immune-Mediated Polyneuritis
Polyneuritis can occur as a primary immune-mediated disorder or as a component of a systemic immune-mediated disease such as systemic lupus erythematosus (SLE; Fig. 71-13). Weakness and exercise intolerance may be the initial manifestations, followed by progressive muscle atrophy and hyporeflexia. Some animals have concurrent cranial nerve dysfunction. Tests should be performed to eliminate endocrine and neoplastic causes of neuropathy and to investigate other systemic manifestations of immune-mediated disease. Screening tests should include evaluation of a CBC, measurement of protein in the urine (i.e., protein : creatinine ratio), and analysis of synovial fluid. Skin lesions, if present, should be biopsied, and blood should be submitted for an antinuclear antibody (ANA) titer. Immunosuppressive therapy should be initiated using prednisone and azathioprine. The short-term prognosis for recovery may be good, but there is a tendency for these disorders to relapse and progress over time.
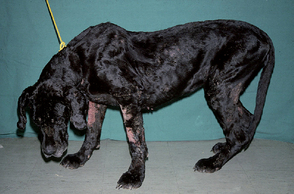
FIG 71-13 A 4-year-old Great Dane with severe weakness, hyporeflexia, and muscle atrophy caused by polyneuritis resulting from systemic lupus erythematosus. The dog also had dermatitis, polyarthritis, glomerulonephritis, and a positive antinuclear antibody test. Polyneuritis was confirmed on postmortem examination.
Idiopathic Polyneuropathy
Demyelinating polyneuropathies with no known etiology have been observed in dogs and cats. Systemic evaluation does not reveal an underlying cause. Often, they are treated as an immune-mediated disorder, with no response.
Ehrlichiosis
Rarely, dogs are identified with a mononeuropathy or polyneuropathy and concurrent positive serologic or polymerase chain reaction (PCR) testing for Ehrlichia canis, but they have no other clinical signs indicating ehrlichiosis. In some dogs the neuropathy resolves after appropriate treatment with doxycycline (5 mg/kg, administered orally q12h) or imidocarb dipropionate (5 mg/kg, administered twice intramuscularly, 14 days apart). Definitive evidence for a causative relationship is lacking.
Delayed Organophosphate Intoxication
Some toxins, including organophosphates, heavy metals, and industrial chemicals, can cause peripheral nerve damage. Organophosphates, in particular, can have a delayed neurotoxic effect that may be related to their inhibition of neurotoxic esterase, an enzyme necessary for nutrient transport within neurons. Exposure to the toxin may have been a single severe exposure with clinical signs of acute intoxication or chronic mild to moderate exposure repeated over weeks or months without acute signs. Between 1 and 6 weeks after exposure a neuropathy develops. Affected animals are weak but do not have classic autonomic signs of organophosphate intoxication, such as salivation, vomiting, diarrhea, or miosis. With chronic exposure, hair, blood, fat, or liver samples may contain the toxin. Plasma acetylcholinesterase activity is usually low. Toxic neuropathy may be suspected on the basis of nerve biopsy results. Spontaneous improvement should occur in 3 to 12 weeks, provided that the toxic substance is removed and reexposure is prevented.
ACQUIRED ACUTE POLYNEUROPATHIES
Acute Polyradiculoneuritis
Acute canine polyradiculoneuritis (ACP) is the only acute polyneuropathy commonly diagnosed in dogs. The disease affects dogs of any breed and gender, and most affected dogs are adults. A similar disease occurs rarely in cats.
The popular name Coonhound paralysis originates in the fact that in many of the early cases the syndrome developed 7 to 10 days after hunting dogs were bitten by a raccoon. Although raccoon saliva injection does not reliably produce the disorder, it has been shown that in a few susceptible dogs the disorder results from an immune response against some component of raccoon saliva (Fig. 71-14).
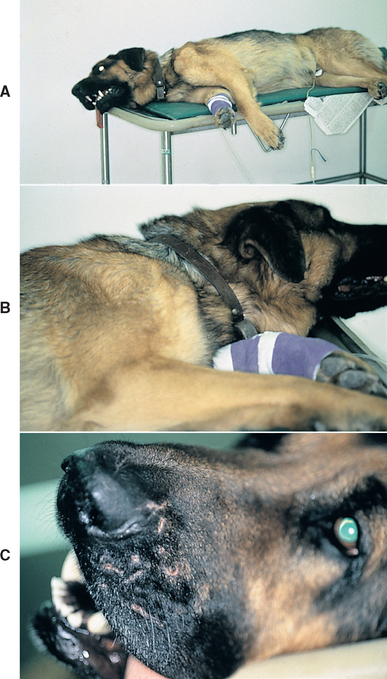
FIG 71-14 A 4-year-old German Shepherd Dog with (A) rapidly progressive ascending lower motor neuron paralysis, (B) severe appendicular muscle atrophy, and (C) healing facial wounds presumed to be from an encounter with a raccoon. The tentative diagnosis in this dog was acute polyradiculoneuritis. Supportive care was initiated, and the dog returned to normal after a prolonged recovery lasting 3 months.
Acute polyradiculoneuritis also occurs in many dogs with no possible exposure to raccoons. Previous systemic illness or vaccination (particularly rabies vaccination) has been implicated as an initiating factor in some of these cases, but in most cases no initiating factor can be identified. The pathologic manifestations include extensive demyelination, inflammatory cell infiltration, and disruption of the ventral root components of peripheral nerves. The disease is very similar to allergic neuritis and Guillain-Barré syndrome in humans, making an immunologic pathogenesis suspect.
Clinical Features
The first clinical sign may be a change in character of the bark; affected dogs sound hoarse. They also develop a stilted, short-strided gait with severe weakness in the rear limbs that ascends rapidly to involve all limbs. Some dogs retain voluntary movement, but most progress to complete paralysis within 5 to 10 days of onset. Neurologic examination reveals remarkably decreased muscle tone, severely diminished or absent reflexes, and rapid muscle atrophy. Some dogs seem to be hyperesthetic, reacting vigorously to mild stimulation such as palpation of the muscles or pinching of the toes. This hyperesthesia is a feature of polyradiculoneuritis that does not occur in association with tick paralysis or botulism, the two major differential diagnoses. Despite the severe paresis or paralysis, dogs remain bright and alert, continue to eat and drink when supported, and can wag their tail vigorously. Bladder and rectum functions remain normal. As a rule, cranial nerves are not involved; no problems with chewing or swallowing exist; neither do any pupillary abnormalities. A small percentage of very severely affected dogs have concurrent bilateral facial nerve paralysis. In a few dogs respiratory paralysis can lead to death.
Diagnosis
The diagnosis is suspected on the basis of clinical and neurologic findings. The most important and challenging aspect of diagnosis is differentiating this disorder from NMJ disorders causing acute LMN tetraparesis (tick paralysis, botulism, and acute fulminating myasthenia gravis) using clinical features and (when available) electrodiagnostic testing (Table 71-2). Owners should be questioned regarding any possible inciting event or exposure 7 to 14 days earlier. Normal cranial nerve and esophageal function and the presence of hyperesthesia make ACP most likely. Cerebrospinal fluid (CSF) is usually normal, although a mild increase in protein concentration may occur. Electromyography reveals diffuse denervation (spontaneous activity) after 6 or more days of paralysis, a finding not expected with the NMJ disorders. Definitive diagnosis can also be established by nerve biopsy, but this is rarely necessary.

Treatment
No specific treatment exists for this disorder. During the initial progressive phase dogs must be monitored for respiratory compromise. Signs typically stabilize after 5 to 10 days, after which the patients can usually be managed with supportive care at home. They may require assistance in sitting up to eat and drink. If possible, they should be kept on an air mattress, waterbed, lounge chair, or bed of straw and turned periodically to prevent lung atelectasis and pressure sores. Corticosteroid treatment is not beneficial.
Prognosis
The prognosis for recovery is good. Most dogs begin to improve after the first week and are fully recovered within 2 to 4 weeks. Recovery may take 4 to 6 months in severely affected dogs. Some dogs never recover completely, and the prognosis for complete recovery in the cat is poor. Affected animals that have recovered may be prone to recurrences, particularly if exposed again to the initiating antigen.
Neospora Polyradiculoneuritis
Adult dogs
Neosporosis can cause a wide range of signs in adult dogs, depending on the site of infection within the nervous system. Paraparesis, tetraparesis, cerebellar signs, muscle pain, seizures, and cranial nerve abnormalities are all reported. Rarely, a rapidly progressive LMN paralysis similar to acute idiopathic polyradiculoneuritis has been reported. Definitive diagnosis is based on a positive serum test for anti-Neospora caninum antibodies, and occasionally the organism can be demonstrated within muscle or nerve biopsies by immunohistochemistry (see Table 69-1). Treatment with clindamycin hydrochloride (10 mg/kg PO q8h for at least 4 weeks) is most effective.
Puppies
Young puppies infected transplacentally by N. caninum begin showing signs of LMN paraparesis between 6 weeks and 6 months of age. Inflammation of ventral nerve roots and peripheral nerves in the rear limbs results in progressive rear limb weakness, muscle atrophy, and hyporeflexia. Over a period of weeks these LMN signs progress to severe pelvic limb extension as muscle atrophy and fibrosis lock the pelvic limbs in extensor rigidity (see Chapter 69, p. 1062; see also Figs. 69-4 and 69-5). Diagnosis should be suspected on the basis of typical history, clinical, and neurologic findings in a young puppy. Litter mates are often affected. There may be mild to moderate elevations in serum creatine kinase (CK) and aspartate aminotransferase (AST) if muscles are involved. Diagnosis and treatment are as described for adult dogs, with most affected puppies having positive serology and organisms identified in muscle biopsies. Multifocal signs, rapid progression of signs, pelvic limb rigid hyperextension, and delayed treatment are all associated with a poor prognosis for recovery.
DISORDERS OF THE NEUROMUSCULAR JUNCTION
TICK PARALYSIS
A flaccid, rapidly ascending motor paralysis has been recognized in dogs infested with certain species of ticks. Most of the reported cases in North America are associated with selected strains of Dermacentor andersoni, Dermacentor variabilis, or Amblyomma americanum ticks. The feeding of a female tick results in the elaboration and circulation of a salivary neurotoxin that interferes with acetylcholine release at the neuromuscular junction. Signs occur within 4 to 9 days after tick attachment.
Clinical Features
Dogs with tick paralysis exhibit a rapid progression from pelvic limb weakness to recumbency, usually resulting in complete LMN paralysis. Muscles are flaccid, and spinal reflexes are decreased or absent. Muscle atrophy does not occur. Pain is perceived normally, with no evidence of hyperesthesia. In most cases the cranial nerves are not significantly affected, but facial weakness, an altered voice, dysphagia, or decreased jaw tone may be recognized. Muscles of respiration may become paralyzed in severely affected patients.
Diagnosis
This disease can be confused with other causes of acute tetraparesis such as acute polyradiculoneuritis, botulism, and myasthenia gravis (see Table 71-2). Tick paralysis is diagnosed on the basis of the history, clinical signs, and knowledge of the geographic region. Sometimes a tick can be found on the animal, and diagnosis is confirmed by documenting rapid improvement after tick removal. When electromyography is available, there is no evidence of denervation. Diminished amplitude of the muscle action potential occurs in response to a single supramaximal stimulus, as would be expected with a defect in neuromuscular transmission.
BOTULISM
Botulism is rarely recognized in dogs and has not been clinically seen in cats. It results from the ingestion of spoiled food or carrion containing a preformed type C neurotoxin produced by the bacterium Clostridium botulinum. Only very small amounts of toxin are needed to cause clinical signs. This toxin blocks the release of acetylcholine from the neuromuscular junction, resulting in complete LMN paralysis. Clinical signs occur hours to days after ingestion of the toxin.
Clinical Features
Affected dogs are weak and develop a short-strided, shuffling gait that rapidly progresses to recumbency. Muscle tone is poor and spinal reflexes are absent, but the tail wag is preserved. Proprioception and pain perception are normal, without hyperesthesia. Affected dogs often have cranial nerve dysfunction resulting in dilated pupils, loss of the pupillary light reflex, weak voice/bark, facial weakness, decreased jaw tone, and dysphagia. Regurgitation resulting from megaesophagus is common. The amount of ingested toxin determines severity of signs. Clinical signs can last for weeks, and death can occur if respiratory muscles are impaired.
Diagnosis
The diagnosis is based on clinical findings and/or a history of ingestion of spoiled food. Botulism is especially likely if an outbreak of LMN paralysis is seen in a group of dogs. Rabies must be considered as a differential diagnosis in severely affected dogs, but it is usually associated with abnormal mentation. Weakness of the muscles of the face, jaw, and pharynx is much more pronounced with botulism than would be expected with acute polyradiculoneuritis or tick paralysis. When electromyography is available, it reveals the absence of spontaneous activity (no denervation potentials) and diminished amplitude of the muscle action potential in response to a supramaximal stimulus, similar to tick paralysis. Botulinum toxin (type C) may be demonstrated in the blood, vomitus, feces, or stomach contents of affected dogs.
Treatment
No specific treatment for botulism exists. The administration of laxatives and enemas may help remove unabsorbed toxin from the gastrointestinal tract if ingestion was recent. Administration of commercially available human trivalent antitoxin (types A, B, and E) will not be effective. If Type C antitoxin is available, administration of 10,000 units intramuscularly twice, 4 hours apart, is recommended, but this will simply bind and inactivate circulating toxin that has not yet penetrated nerve endings. Most dogs recover in 1 to 3 weeks with supportive care, although aspiration pneumonia is a common complication during recovery.
MYASTHENIA GRAVIS
Myasthenia gravis (MG) is a neuromuscular disorder characterized by weakness that is exacerbated by exercise and alleviated by rest. Two forms have been described: congenital and acquired. The congenital form of MG results from an inherited deficiency of acetylcholine receptors (AChRs) at the postsynaptic membranes in skeletal muscle. Signs of impaired neuromuscular transmission first become evident in puppies or kittens 6 to 9 weeks old. The disorder has been recognized in English Springer Spaniels, Smooth Fox Terriers, and Jack Russell Terriers, with rare reports in other breeds and a few cats. An unusual, poorly classified transient congenital myasthenic syndrome has also been identified in Miniature Dachshunds; the signs in these dogs resolve with maturation.
The acquired form of MG is a common immunemediated disorder in which antibodies are directed against a portion of the nicotinic AChRs of skeletal muscle. Antibodies bind to the receptors, reducing the sensitivity of the postsynaptic membrane to the transmitter acetylcholine.
The acquired form of MG affects dogs of all breeds and both genders. German Shepherd Dogs, Golden Retrievers, Labrador Retrievers, and Dachshunds are most commonly affected, but this may merely reflect the popularity of these breeds. Breeds that seem to be at increased risk for acquired MG relative to their popularity include the Akita, some terrier breeds, German Shorthaired Pointers, and Chihuahuas. Young adult dogs (mean age, 2 to 3 years) and old dogs (mean age, 9 to 10 years), make up most of the affected population. Cats are rarely affected, but breed predispositions include the Abyssinian and Somali.
Clinical Features
The characteristic clinical abnormality in most animals with MG is appendicular muscle weakness that worsens with exercise and improves with rest. Mentation, postural reactions, and reflexes are normal, although reflexes may be fatiguable with repeated stimulation. Excessive salivation and regurgitation is common, caused by megaesophagus (seen in 90% of dogs with acquired MG). Megaesophagus is less common in cats with MG and in congenital MG. Dysphagia, hoarse character of the bark or meow, persistently dilated pupils, and facial muscle weakness are sometimes seen.
A focal form of MG occurs in approximately 40% of affected dogs and 14% of affected cats, causing megaesophagus with no detectable appendicular weakness. Affected dogs exhibit weakness of the pharyngeal, laryngeal, and/or facial muscles and may have a fatiguable palpebral reflex. Approximately 25% to 40% of all dogs with adult-onset megaesophagus suffer from acquired focal MG, so this disorder should always be considered as a differential diagnosis early in the course of evaluation of dogs with megaesophagus.
An acute, fulminating form of acquired MG has also been recognized, causing a rapid onset of severe appendicular muscle weakness. Affected animals are often unable to stand and cannot even raise their head. This form of MG is usually associated with severe megaesophagus and aspiration pneumonia. Profound muscle weakness and severe pneumonia commonly lead to respiratory failure and death.
Diagnosis
MG should be considered as a differential diagnosis in any dog with generalized muscular weakness and in all dogs with acquired megaesophagus. Definitive diagnosis is made by demonstrating circulating antibodies against AChRs by immunoprecipitation radioimmunoassay. This test is readily available (Comparative Neuromuscular Laboratory, University of California, San Diego) and is positive in 85% of all dogs and cats with acquired disease and in 98% of those with generalized acquired disease. False-positive results have not been documented. Although the serum anti-AChR antibody titer does not correlate directly with the severity of clinical signs, dogs with focal MG tend to have lower titers and dogs with acute fulminating MG have the highest titers. Rarely, dogs with acquired MG are negative for circulating AChR antibodies, but immune complexes can be demonstrated in muscle biopsies at the NMJ using immunocytochemical methods. These dogs may have very-high-affinity antibody that remains bound to AChRs and does not circulate or antibodies directed against junctional antigens other than AChRs.
When results of the serum test for antibodies are not yet available, or in animals with suspected congenital disease, support for the diagnosis of MG can be gained by demonstrating a positive response to administration of the ultra-short-acting anticholinesterase edrophonium chloride (Tensilon; Box 71-2). This drug inhibits enzymatic hydrolysis of ACh at the NMJ, increasing the effective concentration of ACh and the duration of its effect in the synaptic cleft, optimizing the opportunities for successful interactions between ACh and the AChRs. Most animals with MG exhibit obvious improvement in clinical signs (e.g., resolution of weakness) within 30 to 60 seconds after administration of edrophonium chloride, with the effect lasting approximately 5 minutes. Some dogs with other myopathic and neuropathic disorders may also show some minor improvement, but a dramatic unequivocal response is very suggestive of MG. A failure to respond does not rule out MG. The response can be difficult to assess in dogs and cats with focal MG, and approximately 50% of dogs with acute fulminating MG will have no response because there has been marked antibody-mediated destruction of AChRs.
Electrodiagnostic testing (showing a decremental response of muscle action potentials to repetitive nerve stimulation) can be performed as an aid to reaching a definitive diagnosis of MG. However, whenever possible, anesthesia should be avoided in animals with megaesophagus because of the risk of aspiration during recovery.
Thoracic radiographs should be assessed for megaesophagus, aspiration pneumonia, or thymoma, and the animal should be evaluated systemically for underlying or associated immune-mediated and neoplastic disorders. If a cranial mediastinal mass is identified, fine-needle aspiration cytology should be used to confirm the suspicion that it is a thymoma—a tumor that has been identified in fewer than 5% of dogs with acquired MG and in more than 25% of cats. Concurrent immune-mediated disorders are common in dogs with MG, including hypothyroidism, immunemediated thrombocytopenia, immune-mediated hemolytic anemia, hypoadrenocorticism, polymyositis, and SLE. MG may also develop as a paraneoplastic disorder in association with a wide variety of tumors, including hepatic carcinoma, anal sac adenocarcinoma, osteosarcoma, cutaneous lymphoma, and primary lung tumors. Acquired drug-induced MG has also been documented in hyperthyroid cats being treated with methimazole.
Treatment
Treatment of acquired MG includes supportive care and the administration of anticholinesterase drugs and occasionally immunosuppressive agents. Surgical removal of thymoma should be considered if identified because many animals with MG will have a decrease in AChR antibody titer and dramatic resolution of their signs after thymectomy. Animals with megaesophagus and regurgitation should be maintained in an upright position during feeding and for 10 to 15 minutes after feeding to facilitate the movement of esophageal contents into the stomach, decreasing the chance of aspiration (Fig. 71-15). If severe regurgitation remains a problem, a gastrostomy tube can be placed to assist in the delivery of nutrients, fluids, and medications (see Chapter 30). Whenever aspiration pneumonia is present, a transtracheal wash (see Chapter 20) should be performed for culture and then aggressive treatment for the pneumonia should be initiated using antibiotics, fluids, nebulization, and coupage. Administration of antibiotics that impair neuromuscular transmission (ampicillin, aminoglycosides) should be avoided.

FIG 71-15 Upright feeding in animals with megaesophagus facilitates emptying of esophageal contents into the stomach. Animals should be maintained in this position for 10 to 15 minutes after eating.
Anticholinesterase drugs are commonly administered in an attempt to improve muscular strength. Pyridostigmine bromide (Mestinon, 1 to 3 mg/kg, administered orally q8h) has been used in dogs. In cats pyridostigmine bromide syrup (0.25 to 1.0 mg/kg, administered orally q12h, diluted 1 : 1 with water to decrease gastric irritation) has been recommended. For both dogs and cats the dose must be individualized on the basis of clinical response. Ideally, feeding should be timed to coincide with peak drug effect (2 hours). In dogs initially unable to tolerate oral medication because of severe megaesophagus, neostigmine methylsulfate (Prostigmin 0.04 mg/kg, administered intramuscularly q6-8h) can be used.
If an animal appears to be responding to anticholinesterase treatment but then suddenly gets worse, it is important to determine whether the deterioration is due to underdosage of the anticholinesterase drug (myasthenic crisis) or overdosage (cholinergic crisis). Clinically, these are indistinguishable, but the administration of one dose of edrophonium (Tensilon) allows the clinician to distinguish between them. The animal in a myasthenic crisis improves after edrophonium administration, whereas the condition of an animal in a cholinergic crisis becomes transiently worse or does not change. Dosing adjustments can then be made on the basis of the observed response.
Acquired MG is an immune-mediated disease, and administration of corticosteroids and other immunosuppressive drugs may be associated with a more rapid clinical response, a decrease in AChR antibody, and an improved outcome in some dogs. Ideally, immunosuppressive drugs should be administered only to stable patients without aspiration pneumonia. Because corticosteroids at standard immunosuppressive doses commonly cause transient worsening of muscular weakness in dogs with MG, treatment should be initiated with a low-dose (oral prednisone, 0.5 mg/kg/day) and the dosage gradually increased over 2 to 4 weeks. The oral administration of azathioprine (Imuran, 2 mg/kg/day) or mycophenolate mofetil (CellCept, 10 to 20 mg/kg q12h) alone or in combination with prednisone has been associated with a positive clinical response in some dogs.
Prognosis
Response to medical management of MG can be good if aspiration pneumonia is not severe and the complications of aspiration and anticholinesterase overdosage are avoided. Severe aspiration pneumonia, persistent megaesophagus, acute fulminating MG, and the presence of a thymoma or another underlying neoplasm are all associated with a poor prognosis for recovery. Many affected dogs die of either acute fatal aspiration or euthanasia within 12 months of diagnosis. Anticholinesterase drugs effectively control appendicular muscle weakness in most animals, but their effect on esophageal function is variable. Response to various immunosuppressive protocols is difficult to determine because most dogs with acquired MG will go into a spontaneous permanent clinical and immunologic remission within 18 months after diagnosis (average, 6.4 months), regardless of the treatment used. Remission is unlikely in animals with thymoma or other neoplastic disease. Sequential antibody determinations in an individual animal are correlated with disease progression or remission; thus it is recommended that AChR antibody concentrations be measured and monitored every 4 to 8 weeks in animals being treated for MG.
DYSAUTONOMIA
Dysautonomia is a polyneuropathy affecting both sympathetic and parasympathetic nerves of the autonomic nervous system. Historically, it was recognized as a problem of cats in the United Kingdom, but since the late 1980s it has become a common problem affecting dogs in the Midwest United States, particularly in rural Kansas, Missouri, Oklahoma, and Wyoming. The etiology is unknown, although toxic and autoimmune mechanisms have been proposed. Clinical signs reflect failure of autonomic function in multiple organ systems.
Clinical Features
The disease affects primarily young adult dogs with a median age of 18 months. Cats are occasionally affected. Affected animals have a rapid onset of clinical signs, which progress over days to weeks. Common presenting complaints are vomiting or regurgitation, diarrhea (constipation in cats), straining to urinate, dribbling urine, photophobia, dyspnea, coughing, depression, and anorexia. Physical examination findings include decreased or absent anal tone, dilated pupils that do not respond to light, dry nose and mucous membranes, and prolapse of the nictitating membrane. The bladder may be distended and easy to express.
Diagnosis
Diagnosis is suspected on the basis of the observed clinical signs. Thoracic and abdominal radiographs may reveal megaesophagus, aspiration pneumonia, generalized ileus, and a large distended urinary bladder. The bladder is easily expressed, suggesting diminished urethral sphincter tone. Anal tone is usually decreased. Pharmacologic testing can be used to support the diagnosis. When very dilute (0.05% to 0.1%) pilocarpine (Isoptocarpine 1%, Alcon Laboratories, diluted with saline) is applied to the eye of a dog with dysautonomia, pupillary constriction and nictitating membrane retraction will occur within 60 minutes or less, documenting denervation hypersensitivity. Administration of bethanechol (0.04 mg/kg SQ) may also enable an affected dog with a distended bladder and urine dribbling to void normally. The subcutaneous administration of atropine (0.04 mg/kg) does not produce any change in heart rate in affected dogs. These findings suggest the diagnosis of dysautonomia, but definitive diagnosis requires the demonstration of lesions within the autonomic nervous system at postmortem examination. A loss of nerve cell bodies results in decreased neuron density in all autonomic ganglia, especially the pelvic, mesenteric, and ciliary ganglia.
Treatment
Treatment is largely supportive and includes the administration of fluids, total parenteral nutrition or percutaneous gastrostomy tube feeding, bladder and colon emptying, lubricating eye ointments, and physical therapy. Pilocarpine (1%, one drop q6-12h) may improve lacrimation and decrease photophobia. Subcutaneous bethanechol (0.05 mg/kg q 8-12h) may improve urinary function, and prokinetic drugs (metoclopramide, cisapride) may improve gastrointestinal tract motility. The prognosis is generally poor, with a mortality rate of about 70% to 90%.
Braund KG. Degenerative disorders of the central nervous system. Braund KG, editor. Clinical neurology in small animals : localization, diagnosis and treatment. Ithaca, NY: International Veterinary Information Service. 2003. www.ivis.org.
Bruchim Y, et al. Toxicological, bacteriological and serological diagnosis of botulism in a dog. Vet Rec. 2006;158:768.
Coates JR, et al. Congenital and inherited neurologic disorders of dogs and cats. In: Bonagura JD, editor. Current veterinary therapy XIII. Philadelphia: WB Saunders, 2001.
Cuddon PA. Acquired canine peripheral neuropathies. Vet Clin N Am. 2000;32(1):207.
Dewey CW, et al. Treatment of a myasthenic dog with mycophenolate mofetil. J Vet Emerg Crit Care. 2000;10:177.
Harkin KR, Andrews GA, Nietfeld JC. Dysautonomia in dogs: 65 cases (1993-2000). J Am Vet Med Assoc. 2002;220(5):633.
Jones BR. Hyperchylomicronemia in the cat. In: Bonagura JD, et al, editors. Kirk’s current veterinary therapy XII. Philadelphia: WB Saunders, 1995.
Kern TJ, et al. Facial neuropathy in dogs and cats: 95 cases. J Am Vet Med Assoc. 1987;191(12):1604.
Kocan AA. Tick paralysis. J Am Vet Med Assoc. 1988;192(11):1498.
Malik R, Farrow BRH. Tick paralysis in North America and Australia. Vet Clin N Am Small Anim Pract. 1991;21(1):157.
Mayhew PD, Bush WW, Glass EN. Trigeminal neuropathy in dogs: a retrospective study of 29 cases (1991-2000). J Am Anim Hospl Assoc. 2002;38:262.
Shelton GD. Myasthenia gravis and other disorders of neuromuscular transmission. Vet Clin N Am Small Anim Pract. 2002;32(1):188.
Shelton GD, et al. Acquired myasthenia gravis: selective involvement of esophageal, pharyngeal and facial muscles. J Vet Intern Med. 1990;4(6):181.
Vitale CL, Olby NJ. Neurologic dysfunction in hypothyroid, hyperlipidemic Labrador Retrievers. J Vet Intern Med. 2007;21:1316.