Chapter 12 Lung
See Targeted Therapy available online at studentconsult.com
The major function of the lung is to replenish oxygen and excrete carbon dioxide from blood. Developmentally, the respiratory system is an outgrowth from the ventral wall of the foregut. The midline trachea develops two lateral outpocketings, the lung buds. The right lung bud eventually divides into three main bronchi, and the left into two main bronchi, thus giving rise to three lobes on the right and two on the left. The main bronchi branch dichotomously, giving rise to progressively smaller airways, termed bronchioles, which are distinguished from bronchi by the lack of cartilage and submucosal glands within their walls. Additional branching of bronchioles leads to terminal bronchioles; the part of the lung distal to the terminal bronchiole is called an acinus. Pulmonary acini are composed of respiratory bronchioles (emanating from the terminal bronchiole) that proceed into alveolar ducts, which immediately branch into alveolar sacs, the blind ends of the respiratory passages, whose walls are formed entirely of alveoli, the ultimate site of gas exchange. The microscopic structure of the alveolar walls (or alveolar septa) consists of the following components, proceeding from blood to air (Fig. 12–1):
• The capillary endothelium and basement membrane.
• The pulmonary interstitium is composed of fine elastic fibers, small bundles of collagen, a few fibroblast-like cells, smooth muscle cells, mast cells, and rare mononuclear cells. It is most prominent in thicker portions of the alveolar septum.
• Alveolar epithelium contains a continuous layer of two principal cell types: flattened, platelike type I pneumocytes covering 95% of the alveolar surface and rounded type II pneumocytes. The latter synthesize pulmonary surfactant and are the main cell type involved in repair of alveolar epithelium after damage to type I pneumocytes. The alveolar walls are not solid but are perforated by numerous pores of Kohn, which permit passage of air, bacteria, and exudates between adjacent alveoli.
• A few alveolar macrophages usually lie free within the alveolar space. In the adult, these macrophages often contain phagocytosed carbon particles.
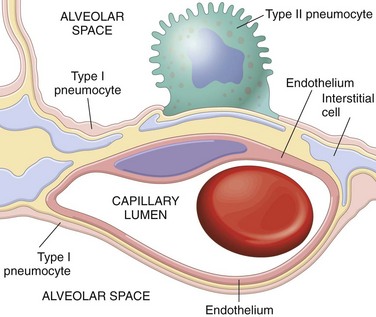
Figure 12–1 Microscopic structure of the alveolar wall. Note that the basement membrane (yellow) is thin on one side and widened where it is continuous with the interstitial space. Portions of interstitial cells are shown.
There are multiple primary lung diseases that can broadly be divided into those primarily affecting (1) the airways, (2) the interstitium, and (3) the pulmonary vascular system. This division into discrete compartments is, of course, deceptively neat. In reality, disease in one compartment often causes secondary alterations of morphology and function in other areas.
Atelectasis (Collapse)
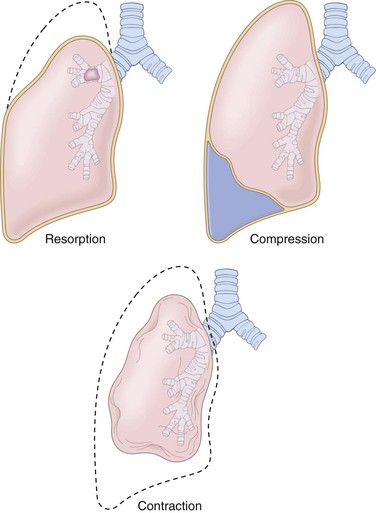
Atelectasis, also known as collapse, is loss of lung volume caused by inadequate expansion of air spaces. It results in shunting of inadequately oxygenated blood from pulmonary arteries into veins, thus giving rise to a ventilation-perfusion imbalance and hypoxia. On the basis of the underlying mechanism or the distribution of alveolar collapse, atelectasis is classified into three forms (Fig. 12–2).
• Resorption atelectasis. Resorption atelectasis occurs when an obstruction prevents air from reaching distal airways. The air already present gradually becomes absorbed, and alveolar collapse follows. Depending on the level of airway obstruction, an entire lung, a complete lobe, or one or more segments may be involved. The most common cause of resorption collapse is obstruction of a bronchus by a mucous or mucopurulent plug. This frequently occurs postoperatively but also may complicate bronchial asthma, bronchiectasis, chronic bronchitis, tumor, or foreign body aspiration, particularly in children.
• Compression atelectasis. Compression atelectasis (sometimes called passive or relaxation atelectasis) is usually associated with accumulation of fluid, blood, or air within the pleural cavity, which mechanically collapses the adjacent lung. This is a frequent occurrence with pleural effusion, caused most commonly by congestive heart failure (CHF). Leakage of air into the pleural cavity (pneumothorax) also leads to compression atelectasis. Basal atelectasis resulting from the elevated position of the diaphragm commonly occurs in bedridden patients, in patients with ascites, and during and after surgery.
• Contraction atelectasis. Contraction (or cicatrization) atelectasis occurs when either local or generalized fibrotic changes in the lung or pleura hamper expansion and increase elastic recoil during expiration.
Atelectasis (except when caused by contraction) is potentially reversible and should be treated promptly to prevent hypoxemia and superimposed infection of the collapsed lung.
Acute Lung Injury
The term acute lung injury encompasses a spectrum of bilateral pulmonary damage (endothelial and epithelial), which can be initiated by numerous conditions. Clinically, acute lung injury manifests as (1) acute onset of dyspnea, (2) decreased arterial oxygen pressure (hypoxemia), and (3) development of bilateral pulmonary infiltrates on the chest radiograph, all in the absence of clinical evidence of primary left-sided heart failure. Since the pulmonary infiltrates in acute lung injury are usually caused by damage to the alveolar capillary membrane, rather than by left-sided heart failure (Chapter 10), such accumulations constitute an example of noncardiogenic pulmonary edema. Acute lung injury can progress to the more severe acute respiratory distress syndrome, described next.
Acute Respiratory Distress Syndrome
Acute respiratory distress syndrome (ARDS) is a clinical syndrome caused by diffuse alveolar capillary and epithelial damage. The usual course is characterized by rapid onset of life-threatening respiratory insufficiency, cyanosis, and severe arterial hypoxemia that is refractory to oxygen therapy and may progress to multisystem organ failure. The histologic manifestation of ARDS in the lungs is known as diffuse alveolar damage (DAD). ARDS can occur in a multitude of clinical settings and is associated with either direct injury to the lung or indirect injury in the setting of a systemic process (Table 12–1). It should be recalled that respiratory distress syndrome of the newborn is pathogenetically distinct; it is caused by a primary deficiency of surfactant.
Table 12–1 Clinical Disorders Associated with the Development of Acute Lung Injury/Acute Respiratory Distress Syndrome
| Direct Lung Injury | Indirect Lung Injury |
|---|---|
| Common Causes | |
| Pneumonia | Sepsis |
| Aspiration of gastric contents | Severe trauma with shock |
| Uncommon Causes | |
| Pulmonary contusion | Cardiopulmonary bypass |
| Fat embolism | Acute pancreatitis |
| Near-drowning | Drug overdose |
| Inhalational injury | Transfusion of blood products |
| Reperfusion injury after lung transplantation | Uremia |
Modified from Ware LB, Matthay MA: The acute respiratory distress syndrome. N Engl J Med 342:1334, 2000.
 Pathogenesis
Pathogenesis
The alveolar-capillary membrane is formed by two separate barriers: the microvascular endothelium and the alveolar epithelium. In ARDS, the integrity of this barrier is compromised by either endothelial or epithelial injury, or, more commonly, both. The acute consequences of damage to the alveolar capillary membrane include increased vascular permeability and alveolar flooding, loss of diffusion capacity, and widespread surfactant abnormalities caused by damage to type II pneumocytes (Fig. 12–3). Although the cellular and molecular basis of acute lung injury and ARDS remains an area of active investigation, recent work suggests that in ARDS, lung injury is caused by an imbalance of pro-inflammatory and anti-inflammatory mediators. As early as 30 minutes after an acute insult, there is increased synthesis of interleukin 8 (IL-8), a potent neutrophil chemotactic and activating agent, by pulmonary macrophages. Release of this and similar mediators, such as IL-1 and tumor necrosis factor (TNF), leads to endothelial activation as well as sequestration and activation of neutrophils in pulmonary capillaries. Neutrophils are thought to have an important role in the pathogenesis of ARDS. Histologic examination of lungs early in the disease process shows increased numbers of neutrophils within the vascular space, the interstitium, and the alveoli. Activated neutrophils release a variety of products (e.g., oxidants, proteases, platelet-activating factor, leukotrienes) that cause damage to the alveolar epithelium and endothelium. Combined assault on the endothelium and epithelium perpetuates vascular leakiness and loss of surfactant that render the alveolar unit unable to expand. Of note, the destructive forces unleashed by neutrophils can be counteracted by an array of endogenous antiproteases, antioxidants, and anti-inflammatory cytokines (e.g., IL-10) that are upregulated by pro-inflammatory cytokines. In the end, it is the balance between the destructive and protective factors that determines the degree of tissue injury and clinical severity of ARDS.
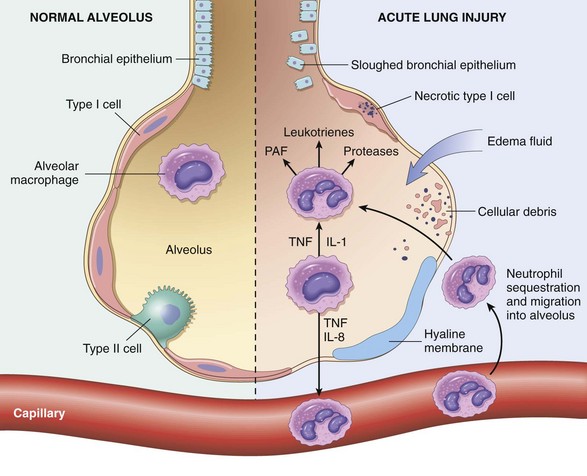
Figure 12–3 The normal alveolus (left), compared with the injured alveolus in the early phase of acute lung injury and the acute respiratory distress syndrome. Under the influence of proinflammatory cytokines such as interleukins IL-8 and IL-1 and tumor necrosis factor (TNF) (released by macrophages), neutrophils initially undergo sequestration in the pulmonary microvasculature, followed by margination and egress into the alveolar space, where they undergo activation. Activated neutrophils release a variety of factors such as leukotrienes, oxidants, proteases, and platelet-activating factor (PAF), which contribute to local tissue damage, accumulation of edema fluid in the air spaces, surfactant inactivation, and hyaline membrane formation. Subsequently, the release of macrophage-derived fibrogenic cytokines such as transforming growth factor-β (TGF-β) and platelet-derived growth factor (PGDF) stimulate fibroblast growth and collagen deposition associated with the healing phase of injury.
(Modified from Ware LB: Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med 27:337, 2006.)
 Morphology
Morphology
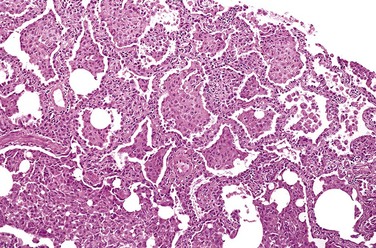
In the acute phase of ARDS, the lungs are dark red, firm, airless, and heavy. Microscopic examination reveals capillary congestion, necrosis of alveolar epithelial cells, interstitial and intra-alveolar edema and hemorrhage, and (particularly with sepsis) collections of neutrophils in capillaries. The most characteristic finding is the presence of hyaline membranes, particularly lining the distended alveolar ducts (Fig. 12–4). Such membranes consist of fibrin-rich edema fluid admixed with remnants of necrotic epithelial cells. Overall, the picture is remarkably similar to that seen in respiratory distress syndrome in the newborn (Chapter 6). In the organizing stage, vigorous proliferation of type II pneumocytes occurs in an attempt to regenerate the alveolar lining. Resolution is unusual; more commonly, there is organization of the fibrin exudates, with resultant intra-alveolar fibrosis. Marked thickening of the alveolar septa ensues, caused by proliferation of interstitial cells and deposition of collagen.
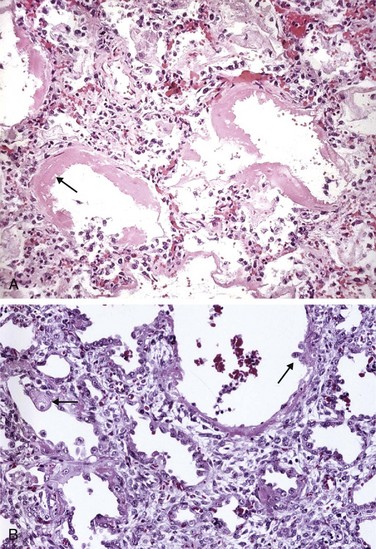
Figure 12–4 A, Diffuse alveolar damage in acute lung injury and acute respiratory distress syndrome. Some alveoli are collapsed; others are distended. Many are lined by bright pink hyaline membranes (arrow). B, The healing stage is marked by resorption of hyaline membranes with thickening of alveolar septa containing inflammatory cells, fibroblasts, and collagen. Numerous reactive type II pneumocytes also are seen at this stage (arrows), associated with regeneration and repair.
Clinical Features
Approximately 85% of patients develop the clinical syndrome of acute lung injury or ARDS within 72 hours of the initiating insult. With improvements in supportive therapy, the mortality rate for the 190,000 ARDS cases occurring yearly has decreased from 60% to 40% in the last decade. Predictors of poor prognosis include advanced age, underlying bacteremia (sepsis), and the development of multisystem (especially cardiac, renal, or hepatic) failure. Should the patient survive the acute stage, diffuse interstitial fibrosis may occur, with continued compromise of respiratory function. However, in most patients who survive the acute insult and are spared the chronic sequelae, normal respiratory function returns within 6 to 12 months.
 Summary
Summary
Acute Respiratory Distress Syndrome
• ARDS is a clinical syndrome of progressive respiratory insufficiency caused by diffuse alveolar damage in the setting of sepsis, severe trauma, or diffuse pulmonary infection.
• Neutrophils and their products have a crucial role in the pathogenesis of ARDS by causing endothelial and epithelial injury.
• The characteristic histologic picture is that of alveolar edema, epithelial necrosis, accumulation of neutrophils, and presence of hyaline membranes lining the alveolar ducts.
Obstructive Versus Restrictive Pulmonary Diseases
Diffuse pulmonary diseases can be classified into two categories: (1) obstructive (airway) disease, characterized by limitation of airflow, usually resulting from an increase in resistance caused by partial or complete obstruction at any level, and (2) restrictive disease, characterized by reduced expansion of lung parenchyma accompanied by decreased total lung capacity.
The major diffuse obstructive disorders are emphysema, chronic bronchitis, bronchiectasis, and asthma. In patients with these diseases, forced vital capacity (FVC) is either normal or slightly decreased, while the expiratory flow rate, usually measured as the forced expiratory volume at 1 second (FEV1), is significantly decreased. Thus, the ratio of FEV to FVC is characteristically decreased. Expiratory obstruction may result either from anatomic airway narrowing, classically observed in asthma, or from loss of elastic recoil, characteristic of emphysema.
By contrast, in diffuse restrictive diseases, FVC is reduced and the expiratory flow rate is normal or reduced proportionately. Hence, the ratio of FEV to FVC is near normal. The restrictive defect occurs in two general conditions: (1) chest wall disorders in the presence of normal lungs (e.g., with severe obesity, diseases of the pleura, and neuromuscular disorders, such as the Guillain-Barré syndrome [Chapter 21], that affect the respiratory muscles) and (2) acute or chronic interstitial lung diseases. The classic acute restrictive disease is ARDS, discussed earlier. Chronic restrictive diseases (discussed later) include the pneumoconioses, interstitial fibrosis of unknown etiology, and most of the infiltrative conditions (e.g., sarcoidosis).
Obstructive Lung (Airway) Diseases
In their prototypical forms, the four disorders in this group—emphysema, chronic bronchitis, asthma, and bronchiectasis—have distinct clinical and anatomic characteristics (Table 12–2), but overlaps between emphysema, bronchitis, and asthma are common.
Table 12–2 Disorders Associated with Airflow Obstruction: The Spectrum of Chronic Obstructive Pulmonary Disease
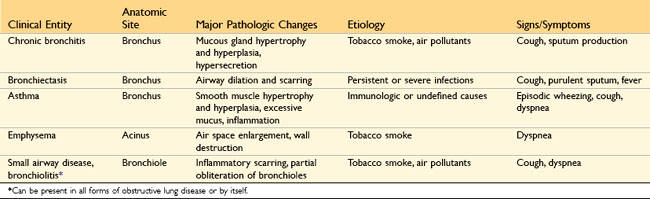
At the outset, it should be recognized that the definition of emphysema is morphologic, whereas chronic bronchitis is defined on the basis of clinical features such as the presence of chronic and recurrent cough with excessive mucus secretion. Second, the anatomic distribution is partially different; chronic bronchitis initially involves the large airways, whereas emphysema affects the acinus. In severe or advanced cases of both, small airway disease (chronic bronchiolitis) is characteristic. Although chronic bronchitis may exist without demonstrable emphysema, and almost pure emphysema may occur (particularly in patients with inherited α1-antitrypsin deficiency) (discussed later), the two diseases usually coexist. This is almost certainly because the major cause—cigarette smoking, especially long-term, heavy tobacco exposure—is common to both disorders. In view of their propensity to coexist, emphysema and chronic bronchitis often are clinically grouped together under the rubric of chronic obstructive pulmonary disease (COPD). COPD affects more than 10% of the U.S. adult population and is the fourth leading cause of death in this country. The primarily irreversible airflow obstruction of COPD distinguishes it from asthma, which, as described later, is characterized largely by reversible airflow obstruction; however, patients with COPD commonly have some degree of reversible obstruction as well (Fig. 12–5).
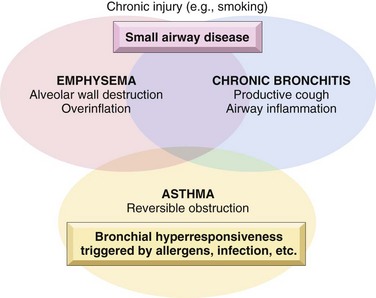
Emphysema
Emphysema is characterized by abnormal permanent enlargement of the air spaces distal to the terminal bronchioles, accompanied by destruction of their walls without significant fibrosis.
Types of Emphysema
Emphysema is classified according to its anatomic distribution within the lobule; as described earlier, the acinus is the structure distal to terminal bronchioles, and a cluster of three to five acini is called a lobule (Fig. 12–6, A). There are four major types of emphysema: (1) centriacinar, (2) panacinar, (3) distal acinar, and (4) irregular. Only the first two types cause clinically significant airway obstruction, with centriacinar emphysema being about 20 times more common than panacinar disease.
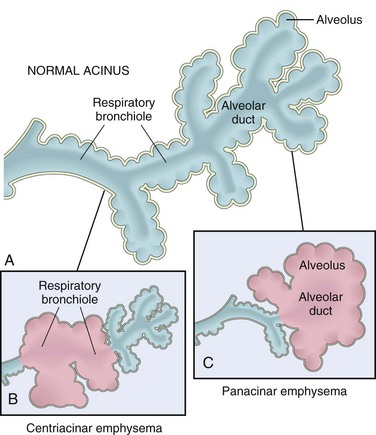
Figure 12–6 Major patterns of emphysema. A, Diagram of normal structure of the acinus, the fundamental unit of the lung. B, Centriacinar emphysema with dilation that initially affects the respiratory bronchioles. C, Panacinar emphysema with initial distention of all the peripheral structures (i.e., the alveolus and alveolar duct); the disease later extends to affect the respiratory bronchioles.
Centriacinar (Centrilobular) Emphysema
The distinctive feature of centriacinar (centrilobular) emphysema is the pattern of involvement of the lobules: The central or proximal parts of the acini, formed by respiratory bronchioles, are affected, while distal alveoli are spared. Thus, both emphysematous and normal air spaces exist within the same acinus and lobule (Fig. 12–6, B). The lesions are more common and severe in the upper lobes, particularly in the apical segments. In severe centriacinar emphysema the distal acinus also becomes involved, and thus, the differentiation from panacinar emphysema becomes difficult. This type of emphysema is most commonly seen as a consequence of cigarette smoking in people who do not have congenital deficiency of α1-antitrypsin.
Panacinar (Panlobular) Emphysema
In panacinar (panlobular) emphysema, the acini are uniformly enlarged, from the level of the respiratory bronchiole to the terminal blind alveoli (Fig. 12–6, C). In contrast with centriacinar emphysema, panacinar emphysema tends to occur more commonly in the lower lung zones and is the type of emphysema that occurs in α1-antitrypsin deficiency.
Distal Acinar (Paraseptal) Emphysema
In distal acinar (paraseptal) emphysema, the proximal portion of the acinus is normal but the distal part is primarily involved. The emphysema is more striking adjacent to the pleura, along the lobular connective tissue septa, and at the margins of the lobules. It occurs adjacent to areas of fibrosis, scarring, or atelectasis and is usually more severe in the upper half of the lungs. The characteristic finding is the presence of multiple, contiguous, enlarged air spaces ranging in diameter from less than 0.5 mm to more than 2.0 cm, sometimes forming cystic structures that, with progressive enlargement, are referred to as bullae. The cause of this type of emphysema is unknown; it is seen most often in cases of spontaneous pneumothorax in young adults.
Irregular Emphysema
Irregular emphysema, so named because the acinus is irregularly involved, is almost invariably associated with scarring, such as that resulting from healed inflammatory diseases. Although clinically asymptomatic, this may be the most common form of emphysema.
Pathogenesis
Exposure to toxic substances such as tobacco smoke and inhaled pollutants induces ongoing inflammation with accumulation of neutrophils, macrophages and lymphocytes in the lung. Elastases, cytokines (including IL-8) and oxidants are released causing epithelial injury and proteolysis of the extracellular matrix (ECM). Elastin degradation products further increase the inflammation. Unless checked by antielastases (e.g., α1-antitrypsin) and antioxidants, the cycle of inflammation and ECM proteolysis continues. Indeed, more than 80% of patients with congenital α1-antitrypsin deficiency develop symptomatic panacinar emphysema, which occurs at an earlier age and with greater severity if the affected person smokes.
There is marked individual variation in susceptibility to the development of emphysema/COPD. Multiple genetic factors control the response to injury after smoking. For example, the TGFB gene exhibits polymorphisms that influence susceptibility to the development of COPD by regulating the response of mesenchymal cells to injury. For example, with certain polymorphisms, mesenchymal cell response to TGF-β signaling is reduced, which in turn results in inadequate repair of elastin injury caused by inhaled toxins. Matrix metalloproteinases (MMPs), especially MMP-9 and MMP-12, have also been shown to have a pathogenic role in emphysema. MMP-9 gene polymorphisms and higher levels of both MMP-9 and MMP-12 have been found in some emphysema patients. Moreover, MMP-12–deficient mice are protected from cigarette smoke–induced emphysema. Although much remains to be studied, the current understanding of emphysema pathogenesis is summarized in Figure 12–7.
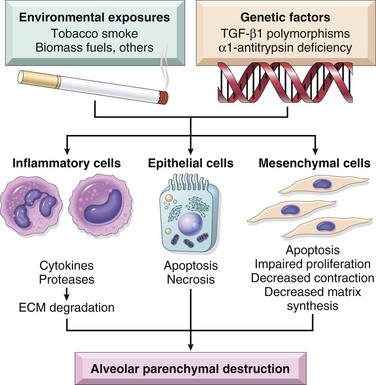
Figure 12–7 Loss of cellular homeostasis in emphysema pathogenesis. Exposure to inhaled toxins (such as cigarette smoke) leads to epithelial cell death, inflammation, and extracellular matrix proteolysis. In susceptible persons, mesenchymal cell survival and reparative functions are impaired by direct effects of inhaled toxic substances and inflammatory mediators and by the loss of the peri- and extracellular matrix. The result is loss of structural cells of the alveolar wall and the associated matrix components.
(Reproduced with permission from Horowitz JC, Martinez FJ, Thannickal VJ: Mesenchymal cell fate and phenotypes in the pathogenesis of emphysema. COPD 6:201, 2009.)
Complex interactions between inflammatory mediators, cell signaling and inappropriate activation of repair mechanisms may result in very different diseases: tissue destruction without fibrosis (emphysema) or interstitial fibrosis (discussed later). Recent data indicate that mesenchymal cell response may be a key factor in determining which of these two processes ensues. In emphysema there is loss of not only epithelial and endothelial cells but also mesenchymal cells, leading to lack of extracellular matrix, the scaffolding upon which epithelial cells would have grown. Thus, emphysema can be thought of as resulting from insufficient wound repair. By contrast, patients with fibrosing lung diseases have excessive myofibroblastic or fibroblastic response to injury, leading to unchecked scarring.
Morphology
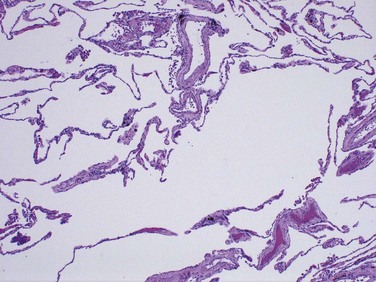
The diagnosis and classification of emphysema depend largely on the macroscopic appearance of the lung. Panacinar emphysema, when the pathologic process is well developed, produces pale, voluminous lungs that often obscure the heart when the anterior chest wall is removed at autopsy. The macroscopic features of centriacinar emphysema are less impressive. The lungs are a deeper pink than in panacinar emphysema and less voluminous, unless the disease is well advanced. Generally, in centriacinar emphysema the upper two thirds of the lungs are more severely affected than the lower lungs. Histologic examination reveals destruction of alveolar walls without fibrosis, leading to enlarged air spaces (Fig. 12–8). In addition to alveolar loss, the number of alveolar capillaries is diminished. Terminal and respiratory bronchioles may be deformed because of the loss of septa that help tether these structures in the parenchyma. With the loss of elastic tissue in the surrounding alveolar septa, radial traction on the small airways is reduced. As a result, they tend to collapse during expiration—an important cause of chronic airflow obstruction in severe emphysema. Bronchiolar inflammation and submucosal fibrosis are consistently present in advanced disease.
Clinical Features
Dyspnea usually is the first symptom; it begins insidiously but is steadily progressive. In patients with underlying chronic bronchitis or chronic asthmatic bronchitis, cough and wheezing may be the initial complaints. Weight loss is common and may be so severe as to suggest a hidden malignant tumor. Pulmonary function tests reveal reduced FEV1 with normal or near-normal FVC. Hence, the ratio of FEV1 to FVC is reduced.
The classic presentation in emphysema with no “bronchitic” component is one in which the patient is barrel-chested and dyspneic, with obviously prolonged expiration, sitting forward in a hunched-over position, attempting to squeeze the air out of the lungs with each expiratory effort. In these patients, air space enlargement is severe and diffusing capacity is low. Dyspnea and hyperventilation are prominent, so that until very late in the disease, gas exchange is adequate and blood gas values are relatively normal. Because of prominent dyspnea and adequate oxygenation of hemoglobin, these patients sometimes are called “pink puffers.”
At the other extreme of the clinical presentation in emphysema is a patient who also has pronounced chronic bronchitis and a history of recurrent infections with purulent sputum. Dyspnea usually is less prominent, with diminished respiratory drive, so the patient retains carbon dioxide, becomes hypoxic, and often is cyanotic. For reasons not entirely clear, such patients tend to be obese—hence the designation “blue bloaters.” Often they seek medical help after the onset of CHF (cor pulmonale) (Chapter 10) and associated edema.
Most patients with emphysema and COPD, however, fall somewhere between these two classic extremes. In all cases, secondary pulmonary hypertension develops gradually, arising from both hypoxia-induced pulmonary vascular spasm and loss of pulmonary capillary surface area from alveolar destruction. Death from emphysema is related to either pulmonary failure, with respiratory acidosis, hypoxia, and coma, or right-sided heart failure (cor pulmonale).
Summary
Emphysema
• Emphysema is a chronic obstructive airway disease characterized by permanent enlargement of air spaces distal to terminal bronchioles.
• Subtypes include centriacinar (most common; smoking-related), panacinar (seen in α1-antitrypsin deficiency), distal acinar, and irregular.
• Smoking and inhaled pollutants cause ongoing accumulation of inflammatory cells, releasing elastases and oxidants, which destroy the alveolar walls without adequate mesenchymal repair response.
• Most patients with emphysema demonstrate elements of chronic bronchitis concurrently, since cigarette smoking is an underlying risk factor for both; patients with pure emphysema are characterized as “pink puffers.”
Conditions Related to Emphysema
Several conditions resemble emphysema only superficially but nevertheless are (inappropriately) referred to as such:
• Compensatory emphysema is a term used to designate the compensatory dilation of alveoli in response to loss of lung substance elsewhere, such as occurs in residual lung parenchyma after surgical removal of a diseased lung or lobe.
• Obstructive overinflation refers to the condition in which the lung expands because air is trapped within it. A common cause is subtotal obstruction by a tumor or foreign object. Obstructive overinflation can be a life-threatening emergency if the affected portion extends sufficiently to compress the remaining normal lung.
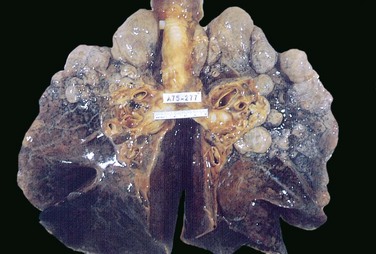
• Bullous emphysema refers merely to any form of emphysema that produces large subpleural blebs or bullae (spaces greater than 1 cm in diameter in the distended state) (Fig. 12–9). Such blebs represent localized accentuations of one of the four forms of emphysema; most often the blebs are subpleural, and on occasion they may rupture, leading to pneumothorax.
• Mediastinal (interstitial) emphysema is the condition resulting when air enters the connective tissue stroma of the lung, mediastinum, and subcutaneous tissue. This may occur spontaneously with a sudden increase in intra-alveolar pressure (as with vomiting or violent coughing) resulting in a tear, with dissection of air into the interstitium. Sometimes it develops in children with whooping cough. It is particularly likely to occur in patients on respirators who have partial bronchiolar obstruction or in persons who suffer a perforating injury (e.g., a fractured rib). When the interstitial air enters the subcutaneous tissue, the patient may literally blow up like a balloon, with marked swelling of the head and neck and crackling crepitation all over the chest. In most instances, the air is resorbed spontaneously after the site of entry is sealed.
Chronic Bronchitis
Chronic bronchitis is common among cigarette smokers and urban dwellers in smog-ridden cities; some studies indicate that 20% to 25% of men in the 40- to 65-year-old age group have the disease. The diagnosis of chronic bronchitis is made on clinical grounds: it is defined by the presence of a persistent productive cough for at least 3 consecutive months in at least 2 consecutive years. In early stages of the disease, the productive cough raises mucoid sputum, but airflow is not obstructed. Some patients with chronic bronchitis may demonstrate hyperresponsive airways with intermittent bronchospasm and wheezing. A subset of bronchitic patients, especially heavy smokers, develop chronic outflow obstruction, usually with associated emphysema.
Pathogenesis
The distinctive feature of chronic bronchitis is hypersecretion of mucus, beginning in the large airways. Although the single most important cause is cigarette smoking, other air pollutants, such as sulfur dioxide and nitrogen dioxide, may contribute. These environmental irritants induce hypertrophy of mucous glands in the trachea and main bronchi, leading to a marked increase in mucin-secreting goblet cells in the surface epithelium of smaller bronchi and bronchioles. In addition, these irritants cause inflammation with infiltration of CD8+ lymphocytes, macrophages, and neutrophils. In contrast with asthma, there are no eosinophils in chronic bronchitis. Whereas the defining feature of chronic bronchitis (mucus hypersecretion) is primarily a reflection of large bronchial involvement, the morphologic basis of airflow obstruction in chronic bronchitis is more peripheral and results from (1) small airway disease, induced by goblet cell metaplasia with mucous plugging of the bronchiolar lumen, inflammation, and bronchiolar wall fibrosis, and (2) coexistent emphysema. In general, while small airway disease (also known as chronic bronchiolitis) is an important component of early and relatively mild airflow obstruction, chronic bronchitis with significant airflow obstruction is almost always complicated by emphysema.
It is postulated that many of the respiratory epithelial effects of environmental irritants (e.g., mucus hypersecretion) are mediated by local release of T cell cytokines such as IL-13. The transcription of the mucin gene MUC5AC in bronchial epithelium and the production of neutrophil elastase are increased as a consequence of exposure to tobacco smoke. Microbial infection often is present but has a secondary role, chiefly by maintaining the inflammation and exacerbating symptoms.
Morphology
As seen in gross specimens, the mucosal lining of the larger airways usually is hyperemic and swollen by edema fluid. It often is covered by a layer of mucinous or mucopurulent secretions. The smaller bronchi and bronchioles also may be filled with similar secretions. On histologic examination, the diagnostic feature of chronic bronchitis in the trachea and larger bronchi is enlargement of the mucus-secreting glands (Fig. 12–10). The magnitude of the increase in size is assessed by the ratio of the thickness of the submucosal gland layer to that of the bronchial wall (the Reid index—normally 0.4). Inflammatory cells, largely mononuclear but sometimes admixed with neutrophils, are frequently present in variable density in the bronchial mucosa. Chronic bronchiolitis (small airway disease), characterized by goblet cell metaplasia, mucous plugging, inflammation, and fibrosis, is also present. In the most severe cases, there may be complete obliteration of the lumen as a consequence of fibrosis (bronchiolitis obliterans). It is the submucosal fibrosis that leads to luminal narrowing and airway obstruction. Changes of emphysema often co-exist.
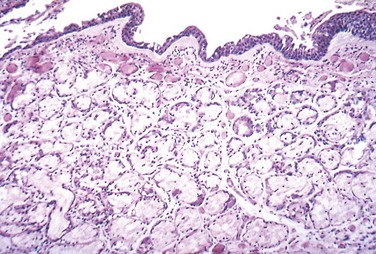
Figure 12–10 Chronic bronchitis. The lumen of the bronchus is above. Note the marked thickening of the mucous gland layer (approximately twice-normal) and squamous metaplasia of lung epithelium.
(From the Teaching Collection of the Department of Pathology, University of Texas, Southwestern Medical School, Dallas, Texas.)
Clinical Features
In patients with chronic bronchitis, a prominent cough and the production of sputum may persist indefinitely without ventilatory dysfunction. As alluded to earlier, however, some patients develop significant COPD with outflow obstruction. This clinical syndrome is accompanied by hypercapnia, hypoxemia, and (in severe cases) cyanosis (hence the term “blue bloaters”). Differentiation of this form of COPD from that caused by emphysema can be made in the classic case, but many such patients have both conditions. With progression, chronic bronchitis is complicated by pulmonary hypertension and cardiac failure (Chapter 10). Recurrent infections and respiratory failure are constant threats.
Summary
Chronic Bronchitis
• Chronic bronchitis is defined as persistent productive cough for at least 3 consecutive months in at least 2 consecutive years.
• Cigarette smoking is the most important underlying risk factor; air pollutants also contribute.
• Chronic obstructive component largely results from small airway disease (chronic bronchiolitis) and coexistent emphysema.
• Histologic examination demonstrates enlargement of mucus-secreting glands, goblet cell metaplasia, and bronchiolar wall fibrosis.
Asthma
Asthma is a chronic inflammatory disorder of the airways that causes recurrent episodes of wheezing, breathlessness, chest tightness, and cough, particularly at night and/or early in the morning. The hallmarks of the disease are intermittent and reversible airway obstruction, chronic bronchial inflammation with eosinophils, bronchial smooth muscle cell hypertrophy and hyperreactivity, and increased mucus secretion. Some of the stimuli that trigger attacks in patients would have little or no effect in persons with normal airways. Many cells play a role in the inflammatory response, in particular eosinophils, mast cells, macrophages, lymphocytes, neutrophils, and epithelial cells. Of note, there has been a significant increase in the incidence of asthma in the Western world over the past four decades. This epidemiologic observation has led to the “hygiene hypothesis,” according to which the eradication of infections may alter immune homeostasis and promote allergic and other harmful immune responses.
Asthma may be categorized into atopic (evidence of allergen sensitization, often in a patient with a history of allergic rhinitis, eczema) and nonatopic. In either type, episodes of bronchospasm can be triggered by diverse mechanisms, such as respiratory infections (especially viral), environmental exposure to irritants (e.g., smoke, fumes), cold air, stress, and exercise. There is emerging evidence for differing patterns of inflammation: eosinophilic, neutrophilic, mixed inflammatory, and pauci-granulocytic. These subgroups may differ in etiology, immunopathology, and response to treatment. Asthma also may be classified according to the agents or events that trigger bronchoconstriction.
Pathogenesis
The major etiologic factors of asthma are genetic predisposition to type I hypersensitivity (atopy), acute and chronic airway inflammation, and bronchial hyperresponsiveness to a variety of stimuli. The inflammation involves many cell types and numerous inflammatory mediators, but the role of type 2 helper T (TH2) cells may be critical to the pathogenesis of asthma. The classic atopic form of asthma is associated with an excessive TH2 reaction against environmental antigens. Cytokines produced by TH2 cells account for most of the features of asthma—IL-4 stimulates IgE production, IL-5 activates eosinophils, and IL-13 stimulates mucus production and also promotes IgE production by B cells. IgE coats submucosal mast cells, which, on exposure to allergen, release granule contents. This induces two waves of reaction: an early (immediate) phase and a late phase (Fig. 12-11). The early reaction is dominated by bronchoconstriction, increased mucus production and variable vasodilation. Bronchoconstriction is triggered by direct stimulation of subepithelial vagal receptors. The late-phase reaction consists of inflammation, with activation of eosinophils, neutrophils, and T cells. In addition, epithelial cells are activated to produce chemokines that promote recruitment of more TH2 cells and eosinophils (including eotaxin, a potent chemoattractant and activator of eosinophils), as well as other leukocytes, thus amplifying the inflammatory reaction. Repeated bouts of inflammation lead to structural changes in the bronchial wall, collectively referred to as airway remodeling. These changes include hypertrophy of bronchial smooth muscle and mucus glands, and increased vascularity and deposition of subepithelial collagen, which may occur as early as several years before initiation of symptoms.
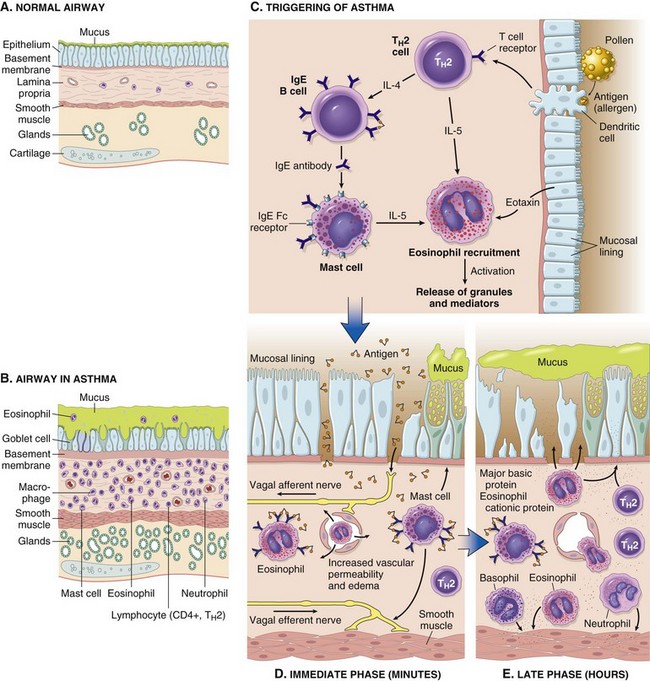
Figure 12–11 A and B, Comparison of a normal bronchus with that in a patient with asthma. Note the accumulation of mucus in the bronchial lumen resulting from an increase in the number of mucus-secreting goblet cells in the mucosa and hypertrophy of submucosal glands. In addition, there is intense chronic inflammation due to recruitment of eosinophils, macrophages, and other inflammatory cells. Basement membrane underlying the mucosal epithelium is thickened, and smooth muscle cells exhibit hypertrophy and hyperplasia. C, Inhaled allergens (antigens) elicit a TH2-dominated response favoring IgE production and eosinophil recruitment (priming or sensitization). D, On reexposure to antigen (Ag), the immediate reaction is triggered by antigen-induced cross-linking of IgE bound to IgE receptors on mast cells in the airways. These cells release preformed mediators. Collectively, either directly or through neuronal reflexes, the mediators induce bronchospasm, increase vascular permeability and mucus production, and recruit additional mediator-releasing cells from the blood. E, The arrival of recruited leukocytes (neutrophils, eosinophils, basophils, lymphocytes, and monocytes) signals the initiation of the late phase of asthma and a fresh round of mediator release from leukocytes, endothelium, and epithelial cells. Factors, particularly from eosinophils (e.g., major basic protein, eosinophil cationic protein), also cause damage to the epithelium. IgE, immunoglobulin E.
Asthma is a complex genetic disorder in which multiple susceptibility genes interact with environmental factors to initiate the pathologic reaction. There is significant variation in the expression of these genes and in the combinations of polymorphisms that effect the immune response or tissue remodeling. One of the susceptibility loci is on the long arm of chromosome 5 (5q), where several genes involved in regulation of IgE synthesis and mast cell and eosinophil growth and differentiation map. The genes at this locus include IL13 (genetic polymorphisms linked with susceptibility to the development of atopic asthma), CD14 (single-nucleotide polymorphisms associated with occupational asthma), class II HLA alleles (tendency to produce IgE antibodies), β2-adrenergic receptor gene, and IL-4 receptor gene (atopy, total serum IgE level, and asthma). Another important locus is on 20q where ADAM-33 that regulates proliferation of bronchial smooth muscle and fibroblasts is located; this controls airway remodeling. Upregulation of various chitinase enzymes has been shown to be important in TH2 inflammation and severity of asthma; high serum YKL-40 levels (a chitinase family member with no enzymatic activity) correlate with the severity of asthma.
Types of Asthma
Atopic Asthma
This is the most common type of asthma, usually beginning in childhood, and is a classic example of type I IgE–mediated hypersensitivity reaction (Chapter 4). A positive family history of atopy and/or asthma is common, and asthmatic attacks are often preceded by allergic rhinitis, urticaria, or eczema. The disease is triggered by environmental antigens, such as dusts, pollen, animal dander, and foods. Infections can also trigger atopic asthma. A skin test with the offending antigen results in an immediate wheal-and-flare reaction. Atopic asthma also can be diagnosed based on serum radioallergosorbent tests (RASTs) that identify the presence of IgE specific for a panel of allergens.
Non-Atopic Asthma
Patients with nonatopic forms of asthma do not have evidence of allergen sensitization, and skin test results usually are negative. A positive family history of asthma is less common. Respiratory infections due to viruses (e.g., rhinovirus, parainfluenza virus) and inhaled air pollutants (e.g., sulfur dioxide, ozone, nitrogen dioxide) are common triggers. It is thought that virus-induced inflammation of the respiratory mucosa lowers the threshold of the subepithelial vagal receptors to irritants. Although the connections are not well understood, the ultimate humoral and cellular mediators of airway obstruction (e.g., eosinophils) are common to both atopic and nonatopic variants of asthma, so they are treated in a similar way.
Drug-Induced Asthma
Several pharmacologic agents provoke asthma, aspirin being the most striking example. Patients with aspirin sensitivity present with recurrent rhinitis and nasal polyps, urticaria, and bronchospasm. The precise mechanism remains unknown, but it is presumed that aspirin inhibits the cyclooxygenase pathway of arachidonic acid metabolism without affecting the lipoxygenase route, thereby shifting the balance of production toward leukotrienes that cause bronchial spasm.
Occupational Asthma
This form of asthma is stimulated by fumes (epoxy resins, plastics), organic and chemical dusts (wood, cotton, platinum), gases (toluene), and other chemicals. Asthma attacks usually develop after repeated exposure to the inciting antigen(s).
Morphology
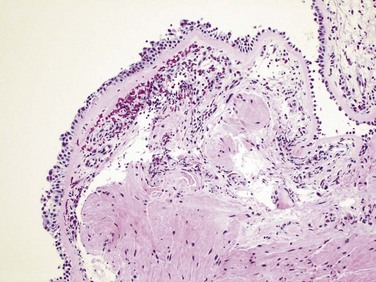
The morphologic changes in asthma have been described in persons who die of prolonged severe attacks (status asthmaticus) and in mucosal biopsy specimens of persons challenged with allergens. In gross specimens obtained in fatal cases, the lungs are overdistended because of overinflation, and there may be small areas of atelectasis. The most striking macroscopic finding is occlusion of bronchi and bronchioles by thick, tenacious mucous plugs. Histologically, the mucous plugs contain whorls of shed epithelium (Curschmann spirals). Numerous eosinophils and Charcot-Leyden crystals (collections of crystalloids made up of eosinophil proteins) also are present. Other characteristic morphologic changes in asthma, collectively called “airway remodeling,” include (Fig. 12–11, B):
• Sub-basement membrane fibrosis (Fig. 12–12)
• Increased vascularity in submucosa
• An increase in size of the submucosal glands and goblet cell metaplasia of the airway epithelium
• Hypertrophy and/or hyperplasia of the bronchial muscle (this is the basis for the novel therapy of bronchial thermoplasty, which involves controlled delivery of thermal energy during bronchoscopy; this reduces the mass of smooth muscles which in turn reduces airway hyperresponsiveness)
Clinical Features
An attack of asthma is characterized by severe dyspnea with wheezing; the chief difficulty lies in expiration. The victim labors to get air into the lungs and then cannot get it out, so that there is progressive hyperinflation of the lungs with air trapped distal to the bronchi, which are constricted and filled with mucus and debris. In the usual case, attacks last from 1 to several hours and subside either spontaneously or with therapy, usually bronchodilators and corticosteroids. Intervals between attacks are characteristically free from overt respiratory difficulties, but persistent, subtle deficits can be detected by spirometry. Occasionally a severe paroxysm occurs that does not respond to therapy and persists for days and even weeks (status asthmaticus). The associated hypercapnia, acidosis, and severe hypoxia may be fatal, although in most cases the condition is more disabling than lethal.
Summary
Asthma
• Asthma is characterized by reversible bronchoconstriction caused by airway hyperresponsiveness to a variety of stimuli.
• Atopic asthma is caused by a TH2 and IgE-mediated immunologic reaction to environmental allergens and is characterized by acute-phase (immediate) and late-phase reactions. The TH2 cytokines IL-4, IL-5, and IL-13 are important mediators.
• Triggers for nonatopic asthma are less clear but include viral infections and inhaled air pollutants, which can also trigger atopic asthma.
• Eosinophils are key inflammatory cells found in almost all subtypes of asthma; eosinophil products such as major basic protein are responsible for airway damage.
• Airway remodeling (sub-basement membrane thickening and hypertrophy of bronchial glands and smooth muscle) adds an irreversible component to the obstructive disease.
Bronchiectasis
Bronchiectasis is the permanent dilation of bronchi and bronchioles caused by destruction of the muscle and the supporting elastic tissue, resulting from or associated with chronic necrotizing infections. It is not a primary disease but rather secondary to persisting infection or obstruction caused by a variety of conditions. Once developed, it gives rise to a characteristic symptom complex dominated by cough and expectoration of copious amounts of purulent sputum. Diagnosis depends on an appropriate history along with radiographic demonstration of bronchial dilation. The conditions that most commonly predispose to bronchiectasis include:
• Bronchial obstruction. Common causes are tumors, foreign bodies, and occasionally impaction of mucus. With these conditions, the bronchiectasis is localized to the obstructed lung segment. Bronchiectasis can also complicate atopic asthma and chronic bronchitis.
• Congenital or hereditary conditions—for example:
 In cystic fibrosis, widespread severe bronchiectasis results from obstruction caused by the secretion of abnormally viscid mucus thus predisposing to infections of the bronchial tree. This is an important and serious complication (Chapter 6). In immunodeficiency states, particularly immunoglobulin deficiencies, localized or diffuse bronchiectasis is likely to develop because of an increased susceptibility to repeated bacterial infections. Kartagener syndrome is a rare autosomal recessive disorder that is frequently associated with bronchiectasis and with sterility in males. In this condition, structural abnormalities of the cilia impair mucociliary clearance in the airways, leading to persistent infections, and reduce the mobility of spermatozoa.
In cystic fibrosis, widespread severe bronchiectasis results from obstruction caused by the secretion of abnormally viscid mucus thus predisposing to infections of the bronchial tree. This is an important and serious complication (Chapter 6). In immunodeficiency states, particularly immunoglobulin deficiencies, localized or diffuse bronchiectasis is likely to develop because of an increased susceptibility to repeated bacterial infections. Kartagener syndrome is a rare autosomal recessive disorder that is frequently associated with bronchiectasis and with sterility in males. In this condition, structural abnormalities of the cilia impair mucociliary clearance in the airways, leading to persistent infections, and reduce the mobility of spermatozoa.• Necrotizing, or suppurative, pneumonia, particularly with virulent organisms such as Staphylococcus aureus or Klebsiella spp., may predispose affected patients to development of bronchiectasis. Posttuberculosis bronchiectasis continues to be a significant cause of morbidity in endemic areas.
Pathogenesis
Two processes are crucial and intertwined in the pathogenesis of bronchiectasis: obstruction and chronic persistent infection. Either of these may come first. Normal clearance mechanisms are hampered by obstruction, so secondary infection soon follows; conversely, chronic infection over time causes damage to bronchial walls, leading to weakening and dilation. For example, obstruction caused by a primary lung cancer or a foreign body impairs clearance of secretions, providing a favorable substrate for superimposed infection. The resultant inflammatory damage to the bronchial wall and the accumulating exudate further distend the airways, leading to irreversible dilation. Conversely, a persistent necrotizing inflammation in the bronchi or bronchioles may cause obstructive secretions, inflammation throughout the wall (with peribronchial fibrosis and traction on the walls), and eventually the train of events already described.
Morphology
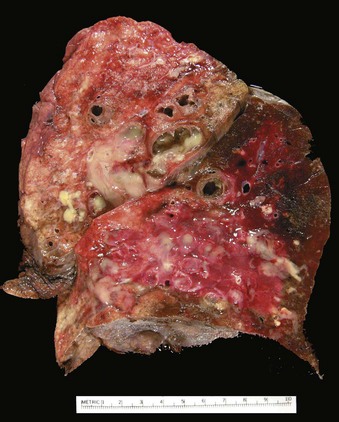
Bronchiectasis usually affects the lower lobes bilaterally, particularly those air passages that are most vertical. When caused by tumors or aspiration of foreign bodies the involvement may be sharply localized to a single segment of the lungs. Usually, the most severe involvement is found in the more distal bronchi and bronchioles. The airways may be dilated to as much as four times their usual diameter and on gross examination of the lung can be followed almost to the pleural surfaces (Fig. 12–13). By contrast, in normal lungs, the bronchioles cannot be followed by ordinary gross examination beyond a point 2 to 3 cm from the pleural surfaces. The histologic findings vary with the activity and chronicity of the disease. In the full-blown active case, an intense acute and chronic inflammatory exudate within the walls of the bronchi and bronchioles and the desquamation of lining epithelium cause extensive areas of ulceration. In the usual case, a mixed flora can be cultured from the involved bronchi, including staphylococci, streptococci, pneumococci, enteric organisms, anaerobic and microaerophilic bacteria, and (particularly in children) Haemophilus influenzae and Pseudomonas aeruginosa. When healing occurs, the lining epithelium may regenerate completely; however, usually so much injury has occurred that abnormal dilation and scarring persist. Fibrosis of the bronchial and bronchiolar walls and peribronchiolar fibrosis develop in more chronic cases. In some instances, the necrosis destroys the bronchial or bronchiolar walls resulting in the formation of an abscess cavity within which a fungus ball may develop.
Clinical Features
The clinical manifestations consist of severe, persistent cough with expectoration of mucopurulent, sometimes fetid, sputum. The sputum may contain flecks of blood; frank hemoptysis can occur. Symptoms often are episodic and are precipitated by upper respiratory tract infections or the introduction of new pathogenic agents. Clubbing of the fingers may develop. In cases of severe, widespread bronchiectasis, significant obstructive ventilatory defects are usual, with hypoxemia, hypercapnia, pulmonary hypertension, and (rarely) cor pulmonale. Metastatic brain abscesses and reactive amyloidosis (Chapter 4) are other, less frequent complications of bronchiectasis.
Chronic Interstitial (Restrictive, Infiltrative) Lung Diseases
Chronic interstitial diseases are a heterogeneous group of disorders characterized predominantly by bilateral, often patchy, and usually chronic involvement of the pulmonary connective tissue, principally the most peripheral and delicate interstitium in the alveolar walls. The pulmonary interstitium is composed of the basement membrane of the endothelial and epithelial cells (fused in the thinnest portions), collagen fibers, elastic tissue, fibroblasts, a few mast cells, and occasional mononuclear cells (Fig. 12–1). Many of the entities in this group are of unknown cause and pathogenesis; some have an intra-alveolar as well as an interstitial component, and there is frequent overlap in histologic features among the different conditions. Nevertheless, the similarity in clinical signs, symptoms, radiographic alterations, and pathophysiologic changes justifies their consideration as a group. The hallmark feature of these disorders is reduced compliance (i.e., more pressure is required to expand the lungs because they are stiff), which in turn necessitates increased effort of breathing (dyspnea). Furthermore, damage to the alveolar epithelium and interstitial vasculature produces abnormalities in the ventilation–perfusion ratio, leading to hypoxia. Chest radiographs show diffuse infiltration by small nodules, irregular lines, or “ground-glass shadows.” With progression, patients can develop respiratory failure, often in association with pulmonary hypertension and cor pulmonale (Chapter 10). Advanced forms of these diseases may be difficult to differentiate because they result in scarring and gross destruction of the lung, referred to as end-stage or “honeycomb” lung. Chronic interstitial lung diseases are categorized based on clinicopathologic features and characteristic histology (Table 12–3).
Table 12–3 Major Categories of Chronic Interstitial Lung Disease
| Fibrosing |
| Granulomatous |
| Eosinophilic |
| Smoking-Related |
Fibrosing Diseases
Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF), also known as cryptogenic fibrosing alveolitis, refers to a pulmonary disorder of unknown etiology. It is characterized by patchy but progressive bilateral interstitial fibrosis, which in advanced cases results in severe hypoxemia and cyanosis. Males are affected more often than females, and approximately two thirds of patients are older than 60 years of age at presentation. The radiologic and histologic pattern of fibrosis is referred to as usual interstitial pneumonia (UIP), which is required for the diagnosis of IPF. Of note, however, similar pathologic changes in the lung may be present in well-defined entities such as asbestosis, the collagen vascular diseases, and a number of other conditions. Therefore, known causes must be ruled out before the appellation of idiopathic is used.
Pathogenesis
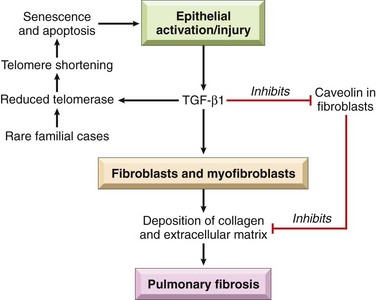
The current concept is that IPF is caused by “repeated cycles” of epithelial activation/injury by some unidentified agent (Fig. 12–14). Histopathologic features include inflammation and induction of TH2 type T cell response with eosinophils, mast cells, IL-4, and IL-13 in the lesions. There has been considerable interest in the idea that “alternatively activated macrophages” are dominant in patients with lung fibrosis and may be important in its pathogenesis (Chapter 2). Abnormal epithelial repair at the sites of damage and inflammation gives rise to exuberant fibroblastic or myofibroblastic proliferation, leading to the characteristic fibroblastic foci. Although the mechanisms of abnormal repair are incompletely understood, recent data point to TGF-β1, which is released from injured type I pneumocytes and induces transformation of fibroblasts into myofibroblasts leading to excessive and continuing deposition of collagen and ECM. Some patients with familial IPF have mutations that shorten telomeres (Chapter 1) leading to rapid senescence and apoptosis of pneumocytes. TGF-β1 also downregulates fibroblast caveolin-1, which acts as an endogenous inhibitor of pulmonary fibrosis.
Morphology
Grossly, the pleural surfaces of the lung have the appearance of cobblestones because of the retraction of scars along the interlobular septa. The cut surface shows fibrosis (firm, rubbery white areas), with lower lobe predominance and a distinctive distribution in the subpleural regions and along the interlobular septa. The pattern of fibrosis in IPF is referred to as usual interstitial pneumonia (UIP). The histologic hallmark of UIP is patchy interstitial fibrosis, which varies in intensity (Fig. 12–15) and worsens with time. The earliest lesions demonstrate exuberant fibroblastic proliferation and appear as fibroblastic foci (Fig. 12–16). Over time these areas become more collagenous and less cellular. Quite typical is the existence of both early and late lesions (temporal heterogeneity). The dense fibrosis causes collapse of alveolar walls and formation of cystic spaces lined by hyperplastic type II pneumocytes or bronchiolar epithelium (honeycomb fibrosis). The interstitial inflammation usually is patchy and consists of an alveolar septal infiltrate of mostly lymphocytes and occasional plasma cells, mast cells, and eosinophils. Secondary pulmonary hypertensive changes (intimal fibrosis and medial thickening of pulmonary arteries) are often present.
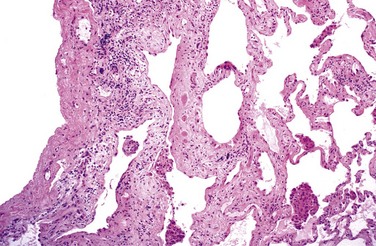
Clinical Features
IPF usually manifests insidiously, with the gradual onset of a nonproductive cough and progressive dyspnea. On physical examination, most patients with IPF have characteristic “dry” or “Velcro”-like crackles during inspiration. Cyanosis, cor pulmonale, and peripheral edema may develop in later stages of the disease. The clinical and radiologic findings often are diagnostic; surgical lung biopsy is needed for diagnosis in selected cases. Unfortunately, progression of IPF is relentless despite medical therapy, and the mean survival is 3 years or less. Lung transplantation is the only definitive therapy available.
Nonspecific Interstitial Pneumonia
Nonspecific interstitial pneumonia (NSIP) is a chronic bilateral interstitial lung disease of unknown etiology, which despite its nonspecific name, has distinct clinical, radiologic, and histologic features. It is important to recognize this disease, since it carries a much better prognosis than that for IPF. On the basis of the histologic appearance, NSIP is divided into cellular and fibrosing patterns. The cellular pattern features mild-to-moderate chronic interstitial inflammation (lymphocytes and a few plasma cells) in a uniform or patchy distribution. The fibrosing pattern consists of diffuse or patchy interstitial fibrosis, without the temporal heterogeneity characteristic of UIP. Fibroblastic foci and honeycombing are typically absent in both variants. Patients present with dyspnea and cough of several months’ duration. Patients with the cellular pattern have a better outcome than those with the fibrosing pattern and UIP.
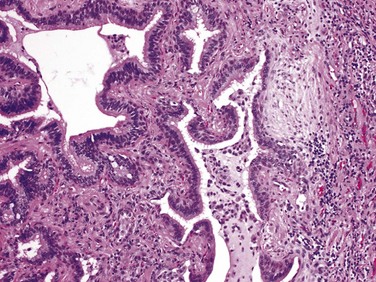
Cryptogenic Organizing Pneumonia
Cryptogenic organizing pneumonia is synonymous with the previously popular designation bronchiolitis obliterans organizing pneumonia (“BOOP”); the former term is now preferred, however, because it emphasizes the unknown etiology of this clinicopathologic entity. Patients present with cough and dyspnea, and chest radiographs demonstrate subpleural or peribronchial patchy areas of air space consolidation. On histologic examination, cryptogenic organizing pneumonia is characterized by the presence of polypoid plugs of loose organizing connective tissue within alveolar ducts, alveoli, and often bronchioles (Fig. 12–17). The connective tissue is all of the same age, and the underlying lung architecture is normal. Some patients recover spontaneously, but most require treatment with oral steroids for 6 months or longer. Of note, organizing pneumonia with intra-alveolar fibrosis also can be seen as a response to infection (e.g., pneumonia) or inflammatory injury (e.g., collagen vascular disease, transplantation injury) to the lung; in such cases, the etiology obviously is not “cryptogenic,” and the outcome is determined by the underlying disease.
Pulmonary Involvement in Collagen Vascular Diseases
Many collagen vascular diseases (e.g., systemic lupus erythematosus, rheumatoid arthritis, systemic sclerosis, dermatomyositis-polymyositis) are associated with pulmonary manifestations. Several histologic variants can be seen, depending on the underlying disorder, with NSIP, UIP pattern (similar to that seen in IPF), vascular sclerosis, organizing pneumonia, and bronchiolitis (small airway disease, with or without fibrosis) being the most common. Pleural involvement (pleuritis, pleural nodules, and pleural effusion) may also be present. Pulmonary involvement in these diseases is usually associated with a poor prognosis, although it is still better than that with IPF.
Summary
Chronic Interstitial Lung Diseases
• Diffuse interstitial fibrosis of the lung gives rise to restrictive lung diseases characterized by reduced lung compliance and reduced forced vital capacity (FVC). The ratio of FEV to FVC is normal.
• The diseases that cause diffuse interstitial fibrosis are heterogeneous. The unifying pathogenetic factor is injury to the alveoli with activation of macrophages and release of fibrogenic cytokines such as TGF-β.
• Idiopathic pulmonary fibrosis is prototypic of restrictive lung diseases. It is characterized by patchy interstitial fibrosis, fibroblastic foci, and formation of cystic spaces (honeycomb lung). This histologic pattern is known as usual interstitial pneumonia (UIP).
Pneumoconioses
Pneumoconiosis is a term originally coined to describe the non-neoplastic lung reaction to inhalation of mineral dusts. The term has been broadened to include diseases induced by organic as well as inorganic particulates, and some experts also regard chemical fume- and vapor-induced non-neoplastic lung diseases as pneumoconioses. The mineral dust pneumoconioses—the three most common of which result from exposure to coal dust, silica, and asbestos—nearly always result from exposure in the workplace. However, the increased risk of cancer as a result of asbestos exposure extends to family members of asbestos workers and to other persons exposed to asbestos outside of the workplace. Table 12–4 indicates the pathologic conditions associated with each mineral dust and the major industries in which the dust exposure is sufficient to produce disease.
Table 12–4 Mineral Dust–Induced Lung Disease
| Agent | Disease | Exposure |
|---|---|---|
| Coal dust | Simple coal worker’s pneumoconiosis: macules and nodules | Coal mining |
| Complicated coal worker’s pneumoconiosis: PMF | ||
| Silica | Silicosis | Sandblasting, quarrying, mining, stone cutting, foundry work, ceramics |
| Asbestos | Asbestosis, pleural effusions, pleural plaques, or diffuse fibrosis; mesothelioma; carcinoma of the lung and larynx | Mining, milling, and fabrication of ores and materials; installation and removal of insulation |
PMF, progressive massive fibrosis.
Pathogenesis
The reaction of the lung to mineral dusts depends on many variables, including size, shape, solubility, and reactivity of the particles. For example, particles greater than 5 to 10 µm are unlikely to reach distal airways, whereas particles smaller than 0.5 µm move into and out of alveoli, often without substantial deposition and injury. Particles that are 1 to 5 µm in diameter are the most dangerous, because they get lodged at the bifurcation of the distal airways. Coal dust is relatively inert, and large amounts must be deposited in the lungs before lung disease is clinically detectable. Silica, asbestos, and beryllium are more reactive than coal dust, resulting in fibrotic reactions at lower concentrations. Most inhaled dust is entrapped in the mucus blanket and rapidly removed from the lung by ciliary movement. However, some of the particles become impacted at alveolar duct bifurcations, where macrophages accumulate and engulf the trapped particulates. The pulmonary alveolar macrophage is a key cellular element in the initiation and perpetuation of lung injury and fibrosis. Many particles activate the inflammasome and induce IL-1 production. The more reactive particles trigger the macrophages to release a number of products that mediate an inflammatory response and initiate fibroblast proliferation and collagen deposition. Some of the inhaled particles may reach the lymphatics either by direct drainage or within migrating macrophages and thereby initiate an immune response to components of the particulates and/or to self-proteins that are modified by the particles. This then leads to an amplification and extension of the local reaction. Tobacco smoking worsens the effects of all inhaled mineral dusts, more so with asbestos than with any other particle.
Coal Worker’s Pneumoconiosis
Worldwide dust reduction in coal mines has greatly reduced the incidence of coal dust–induced disease. The spectrum of lung findings in coal workers is wide, ranging from asymptomatic anthracosis, in which pigment accumulates without a perceptible cellular reaction, to simple coal worker’s pneumoconiosis (CWP), in which accumulations of macrophages occur with little to no pulmonary dysfunction, to complicated CWP or progressive massive fibrosis (PMF), in which fibrosis is extensive and lung function is compromised (Table 12–4). Although statistics vary, it seems that less than 10% of cases of simple CWP progress to PMF. Of note PMF is a generic term that applies to a confluent fibrosing reaction in the lung; this can be a complication of any one of the pneumoconioses discussed here.
Although coal is mainly carbon, coal mine dust contains a variety of trace metals, inorganic minerals, and crystalline silica. The ratio of carbon to contaminating chemicals and minerals (“coal rank”) increases from bituminous to anthracite coal; in general, anthracite mining has been associated with a higher risk of CWP.
Morphology
Pulmonary anthracosis is the most innocuous coal-induced pulmonary lesion in coal miners and also is commonly seen in all urban dwellers and tobacco smokers. Inhaled carbon pigment is engulfed by alveolar or interstitial macrophages, which then accumulate in the connective tissue along the lymphatics, including the pleural lymphatics, or in lymph nodes.
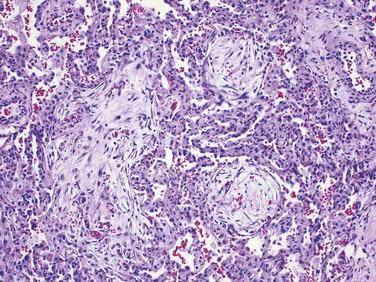
Simple CWP is characterized by coal macules and the somewhat larger coal nodule. The coal macule consists of dust-laden macrophages; in addition, the nodule contains small amounts of collagen fibers arrayed in a delicate network. Although these lesions are scattered throughout the lung, the upper lobes and upper zones of the lower lobes are more heavily involved. In due course, centrilobular emphysema can occur. Functionally significant emphysema is more common in the United Kingdom and Europe, probably because the coal rank is higher than in the United States.
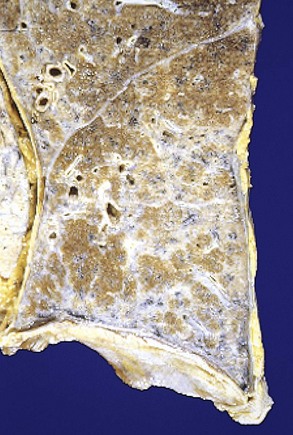
Complicated CWP (PMF) occurs on a background of simple CWP by coalescence of coal nodules and generally requires many years to develop. It is characterized by usually multiple, intensely blackened scars larger than 2 cm, sometimes up to 10 cm in greatest diameter. On microscopic examination the lesions are seen to consist of dense collagen and pigment (Fig. 12–18).
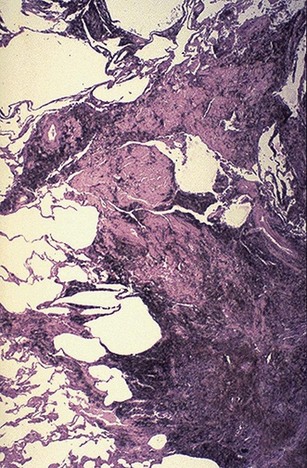
Clinical Features
CWP is usually a benign disease that produces little decrement in lung function. In those in whom PMF develops, there is increasing pulmonary dysfunction, pulmonary hypertension, and cor pulmonale. Progression from CWP to PMF has been linked to a variety of conditions including coal dust exposure level and total dust burden. Unfortunately, PMF has a tendency to progress even in the absence of further exposure. Once smoking-related risk has been taken into account, there is no increased frequency of lung carcinoma in coal miners, a feature that distinguishes CWP from both silica and asbestos exposures (discussed next).
Silicosis
Silicosis is currently the most prevalent chronic occupational disease in the world. It is caused by inhalation of crystalline silica, mostly in occupational settings. Workers in several occupations but especially those involved in sandblasting and hard-rock mining are at particular risk. Silica occurs in both crystalline and amorphous forms, but crystalline forms (including quartz, cristobalite, and tridymite) are by far the most toxic and fibrogenic. Of these, quartz is most commonly implicated in silicosis. After inhalation the particles interact with epithelial cells and macrophages. Ingested silica particles cause activation and release of mediators by pulmonary macrophages, including IL-1, TNF, fibronectin, lipid mediators, oxygen-derived free radicals, and fibrogenic cytokines. Especially compelling is the evidence incriminating TNF, since anti-TNF monoclonal antibodies can block lung fibrosis in mice that are given silica intratracheally. When mixed with other minerals, quartz has been observed to have a reduced fibrogenic effect. This phenomenon is of practical importance, because quartz in the workplace is rarely pure. Thus, miners of the iron-containing ore hematite may have more quartz in their lungs than some quartz-exposed workers and yet have relatively mild lung disease, because the hematite provides a protective effect.
Morphology
Silicotic nodules are characterized grossly in their early stages by tiny, barely palpable, discrete, pale-to-blackened (if coal dust is also present) nodules in the upper zones of the lungs (Fig. 12–19). Microscopically, the silicotic nodule demonstrates concentrically arranged hyalinized collagen fibers surrounding an amorphous center. The “whorled” appearance of the collagen fibers is quite distinctive for silicosis (Fig. 12–20). Examination of the nodules by polarized microscopy reveals weakly birefringent silica particles, primarily in the center of the nodules. As the disease progresses, the individual nodules may coalesce into hard, collagenous scars, with eventual progression to PMF. The intervening lung parenchyma may be compressed or overexpanded, and a honeycomb pattern may develop. Fibrotic lesions may also occur in the hilar lymph nodes and pleura. Sometimes, thin sheets of calcification occur in the lymph nodes and are appreciated radiographically as “eggshell” calcification (e.g., calcium surrounding a zone lacking calcification).
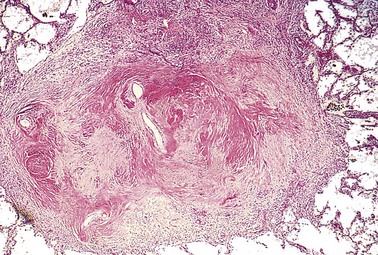
Clinical Features
Silicosis usually is detected on routine chest radiographs obtained in asymptomatic workers. The radiographs typically show a fine nodularity in the upper zones of the lung, but pulmonary function is either normal or only moderately affected. Most patients do not develop shortness of breath until late in the course, after PMF is present. At this time, the disease may be progressive, even if the person is no longer exposed. Many patients with PMF develop pulmonary hypertension and cor pulmonale, as a result of chronic hypoxia-induced vasoconstriction and parenchymal destruction. The disease is slow to kill, but impaired pulmonary function may severely limit activity. Silicosis is associated with an increased susceptibility to tuberculosis. It is postulated that silicosis results in a depression of cell-mediated immunity, and crystalline silica may inhibit the ability of pulmonary macrophages to kill phagocytosed mycobacteria. Nodules of silicotuberculosis often contain a central zone of caseation. The relationship between silica and lung cancer has been a contentious issue. In 1997, based on evidence from several epidemiologic studies, the International Agency for Research on Cancer concluded that crystalline silica from occupational sources is carcinogenic in humans. However, this subject continues to be controversial.
Asbestosis and Asbestos-Related Diseases
Asbestos is a family of crystalline hydrated silicates with a fibrous geometry. On the basis of epidemiologic studies, occupational exposure to asbestos is linked to (1) parenchymal interstitial fibrosis (asbestosis); (2) localized fibrous plaques or, rarely, diffuse fibrosis in the pleura; (3) pleural effusions; (4) lung carcinomas; (5) malignant pleural and peritoneal mesotheliomas; and (6) laryngeal carcinoma. An increased incidence of asbestos-related cancers in family members of asbestos workers has alerted the general public to the potential hazards of asbestos in the environment.
Pathogenesis
Concentration, size, shape, and solubility of the different forms of asbestos dictate whether inhalation of the material will cause disease. There are two distinct forms of asbestos: serpentine, in which the fiber is curly and flexible, and amphibole, in which the fiber is straight, stiff, and brittle. Several subtypes of curly and straight asbestos fibers are recognized. The serpentine chrysotile accounts for most of the asbestos used in industry. Amphiboles, even though less prevalent, are more pathogenic than the serpentine chrysotile, but both types can produce asbestosis, lung cancer, and mesothelioma. The greater pathogenicity of straight and stiff amphiboles is apparently related to their structure. The serpentine chrysotiles, with their more flexible, curled structure, are likely to become impacted in the upper respiratory passages and removed by the mucociliary elevator. Those that are trapped in the lungs are gradually leached from the tissues, because they are more soluble than amphiboles. The straight, stiff amphiboles, in contrast, align themselves in the airstream and are hence delivered deeper into the lungs, where they may penetrate epithelial cells to reach the interstitium. Despite these differences, both asbestos forms are fibrogenic, and increasing exposure to either is associated with a higher incidence of all asbestos-related diseases. Asbestosis, like other pneumoconioses, causes fibrosis by a process involving interaction of particulates with lung macrophages.
In addition to cellular and fibrotic lung reactions, asbestos probably also functions as both a tumor initiator and a promoter. Some of the oncogenic effects of asbestos on the mesothelium are mediated by reactive free radicals generated by asbestos fibers, which preferentially localize in the distal lung close to the mesothelial layer. However, potentially toxic chemicals adsorbed onto the asbestos fibers undoubtedly contribute to the pathogenicity of the fibers. For example, the adsorption of carcinogens in tobacco smoke onto asbestos fibers may well be important to the remarkable synergy between tobacco smoking and the development of lung carcinoma in asbestos workers.
Morphology
Asbestosis is marked by diffuse pulmonary interstitial fibrosis. These changes are indistinguishable from UIP, except for the presence of asbestos bodies, which are seen as golden brown, fusiform or beaded rods with a translucent center. They consist of asbestos fibers coated with an iron-containing proteinaceous material (Fig. 12–21). Asbestos bodies apparently are formed when macrophages attempt to phagocytose asbestos fibers; the iron is derived from phagocyte ferritin. Asbestos bodies sometimes can be found in the lungs of normal persons, but usually in much lower concentrations and without an accompanying interstitial fibrosis.
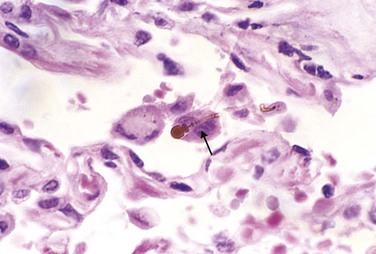
Figure 12–21 High-power detail of an asbestos body, revealing the typical beading and knobbed ends (arrow).
In contrast with CWP and silicosis, asbestosis begins in the lower lobes and subpleurally, but the middle and upper lobes of the lungs become affected as fibrosis progresses. Contraction of the fibrous tissue distorts the normal architecture, creating enlarged air spaces enclosed within thick fibrous walls. In this way the affected regions become honeycombed. Simultaneously, fibrosis develops in the visceral pleura, causing adhesions between the lungs and the chest wall. The scarring may trap and narrow pulmonary arteries and arterioles, causing pulmonary hypertension and cor pulmonale.
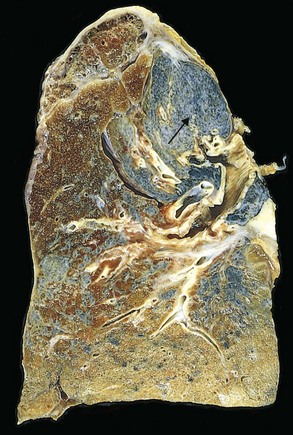
Pleural plaques are the most common manifestation of asbestos exposure and are well-circumscribed plaques of dense collagen (Fig. 12–22), often containing calcium. They develop most frequently on the anterior and posterolateral aspects of the parietal pleura and over the domes of the diaphragm. They do not contain asbestos bodies, and only rarely do they occur in persons with no history or evidence of asbestos exposure. Uncommonly, asbestos exposure induces pleural effusion or diffuse pleural fibrosis.
Clinical Features
The clinical findings in asbestosis are indistinguishable from those of any other chronic interstitial lung disease. Typically, progressively worsening dyspnea appears 10 to 20 years after exposure. The dyspnea is usually accompanied by a cough associated with production of sputum. The disease may remain static or progress to congestive heart failure, cor pulmonale, and death. Pleural plaques are usually asymptomatic and are detected on radiographs as circumscribed densities. Both lung carcinoma and malignant mesothelioma develop in workers exposed to asbestos. The risk of lung carcinoma is increased about five-fold for asbestos workers; the relative risk for mesothelioma, normally a very rare tumor (2 to 17 cases per 1 million persons), is more than 1000 times greater. Concomitant cigarette smoking greatly increases the risk of lung carcinoma but not that of mesothelioma. Lung or pleural cancer associated with asbestos exposure carries a particularly grim prognosis.
Summary
Pneumoconioses
• Pneumoconioses encompass a group of chronic fibrosing diseases of the lung resulting from exposure to organic and inorganic particulates, most commonly mineral dust.
• Pulmonary alveolar macrophages play a central role in the pathogenesis of lung injury by promoting inflammation and producing reactive oxygen species and fibrogenic cytokines.
• Coal dust–induced disease varies from asymptomatic anthracosis, to simple coal worker’s pneumoconiosis (coal macules or nodules, and centrilobular emphysema), to progressive massive fibrosis (PMF), manifested by increasing pulmonary dysfunction, pulmonary hypertension, and cor pulmonale.
• Silicosis is the most common pneumoconiosis in the world, and crystalline silica (e.g., quartz) is the usual culprit.
• The manifestations of silicosis can range from asymptomatic silicotic nodules to PMF; persons with silicosis also have an increased susceptibility to tuberculosis. The relationship between silica exposure and subsequent lung cancer is controversial.
• Asbestos fibers come in two forms; the stiff amphiboles have a greater fibrogenic and carcinogenic potential than the serpentine chrysotiles.
• Asbestos exposure is linked with six disease processes: (1) parenchymal interstitial fibrosis (asbestosis); (2) localized fibrous plaques or, rarely, diffuse pleural fibrosis; (3) pleural effusions; (4) lung cancer; (5) malignant pleural and peritoneal mesotheliomas; and (6) laryngeal cancer.
• Cigarette smoking increases the risk of lung cancer in the setting of asbestos exposure; moreover, even family members of workers exposed to asbestos are at increased risk for cancer.
Drug- and Radiation-Induced Pulmonary Diseases
Drugs can cause a variety of both acute and chronic alterations in respiratory structure and function. For example, bleomycin, an anticancer agent, causes pneumonitis and interstitial fibrosis, as a result of direct toxicity of the drug and by stimulating the influx of inflammatory cells into the alveoli. Similarly, amiodarone, an antiarrhythmic agent, also is associated with risk for pneumonitis and fibrosis. Radiation pneumonitis is a well-known complication of therapeutic irradiation of pulmonary and other thoracic tumors. Acute radiation pneumonitis, which typically occurs 1 to 6 months after therapy in as many as 20% of the patients, is manifested by fever, dyspnea out of proportion to the volume of irradiated lung, pleural effusion, and development of pulmonary infiltrates corresponding to the area of radiation. These signs and symptoms may resolve with corticosteroid therapy or progress to chronic radiation pneumonitis, associated with pulmonary fibrosis.
Granulomatous Diseases
Sarcoidosis
Although sarcoidosis is considered here as an example of a restrictive lung disease, it is important to note that sarcoidosis is a multisystem disease of unknown etiology characterized by noncaseating granulomas in many tissues and organs. Other diseases, including mycobacterial or fungal infections and berylliosis, sometimes also produce noncaseating granulomas; therefore, the histologic diagnosis of sarcoidosis is one of exclusion. Although the multisystem involvement of sarcoidosis can manifest in many clinical guises, bilateral hilar lymphadenopathy or lung involvement (or both), visible on chest radiographs, is the major presenting manifestation in most cases. Eye and skin involvement each occurs in about 25% of cases, and either may occasionally be the presenting feature of the disease.
Epidemiology
Sarcoidosis occurs throughout the world, affecting both genders and all races and age groups. There are, however, certain interesting epidemiologic trends, including:
• There is a consistent predilection for adults younger than 40 years of age.
• A high incidence has been noted in Danish and Swedish populations, and in the United States among African Americans (in whom the frequency of involvement is 10 times greater than in whites).
• Sarcoidosis is one of the few pulmonary diseases with a higher prevalence among nonsmokers.
Etiology and Pathogenesis
Although the etiology of sarcoidosis remains unknown, several lines of evidence suggest that it is a disease of disordered immune regulation in genetically predisposed persons exposed to certain environmental agents. The role of each of these contributory influences is summarized in the following discussion.
Several immunologic abnormalities in sarcoidosis suggest the development of a cell-mediated response to an unidentified antigen. The process is driven by CD4+ helper T cells. These abnormalities include:
• Intra-alveolar and interstitial accumulation of CD4+ TH1 cells
• Oligoclonal expansion of T cell subsets as determined by analysis of T cell receptor rearrangement
• Increases in T cell–derived TH1 cytokines such as IL-2 and IFN-γ, resulting in T cell expansion and macrophage activation, respectively
• Increases in several cytokines in the local environment (IL-8, TNF, macrophage inflammatory protein-1α) that favor recruitment of additional T cells and monocytes and contribute to the formation of granulomas
• Anergy to common skin test antigens such as Candida or purified protein derivative (PPD), that may result from pulmonary recruitment of CD4+ T cells and consequent peripheral depletion
• Polyclonal hypergammaglobulinemia, another manifestation of TH cell dysregulation
• The role of genetic factors is suggested by familial and racial clustering of cases and association with certain human leukocyte antigen (HLA) genotypes (e.g., class I HLA-A1 and HLA-B8)
After lung transplantation, sarcoidosis recurs in the new lungs in 75% of patients. Finally, several putative “antigens” have been proposed as the inciting agent for sarcoidosis (e.g., viruses, mycobacteria, Borrelia, pollen), but thus far there is no unequivocal evidence to suggest that sarcoidosis is caused by an infectious agent.
Morphology
The diagnostic histopathologic feature of sarcoidosis is the noncaseating epithelioid granuloma, irrespective of the organ involved (Fig. 12–23). This is a discrete, compact collection of epithelioid cells rimmed by an outer zone of largely CD4+ T cells. The epithelioid cells are derived from macrophages and are characterized by abundant eosinophilic cytoplasm and vesicular nuclei. It is not uncommon to see intermixed multinucleate giant cells formed by fusion of macrophages. A thin layer of laminated fibroblasts is present peripheral to the granuloma; over time, these proliferate and lay down collagen that replaces the entire granuloma with a hyalinized scar. Two other microscopic features are sometimes seen in the granulomas: (1) Schaumann bodies, laminated concretions composed of calcium and proteins; and (2) asteroid bodies, stellate inclusions enclosed within giant cells. Their presence is not required for diagnosis of sarcoidosis—they also may occur in granulomas of other origins. Rarely, foci of central necrosis may be present in sarcoid granulomas, suggesting an infectious process. Caseation necrosis typical of tuberculosis is absent.
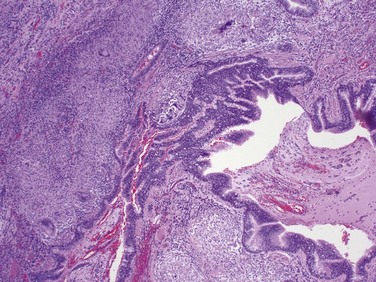
The lungs are involved at some stage of the disease in 90% of patients. The granulomas predominantly involve the interstitium rather than air spaces, with some tendency to localize in the connective tissue around bronchioles and pulmonary venules and in the pleura (“lymphangitic” distribution). The bronchoalveolar lavage fluid contains abundant CD4+ T cells. In 5% to 15% of patients, the granulomas eventually are replaced by diffuse interstitial fibrosis, resulting in a so-called honeycomb lung.
Intrathoracic hilar and paratracheal lymph nodes are enlarged in 75% to 90% of patients, while a third present with peripheral lymphadenopathy. The nodes are characteristically painless and have a firm, rubbery texture. Unlike in tuberculosis, lymph nodes in sarcoidosis are “nonmatted” (nonadherent) and do not ulcerate.
Skin lesions are encountered in approximately 25% of patients. Erythema nodosum, the hallmark of acute sarcoidosis, consists of raised, red, tender nodules on the anterior aspects of the legs. Sarcoidal granulomas are uncommon in these lesions. By contrast, discrete painless subcutaneous nodules can also occur in sarcoidosis, and these usually reveal abundant noncaseating granulomas.
Involvement of the eye and lacrimal glands occurs in about one fifth to one half of patients. The ocular involvement takes the form of iritis or iridocyclitis and may be unilateral or bilateral. As a consequence, corneal opacities, glaucoma, and (less commonly) total loss of vision may develop. The posterior uveal tract also is affected, with resultant choroiditis, retinitis, and optic nerve involvement. These ocular lesions are frequently accompanied by inflammation in the lacrimal glands, with suppression of lacrimation (sicca syndrome). Unilateral or bilateral parotitis with painful enlargement of the parotid glands occurs in less than 10% of patients with sarcoidosis; some go on to develop xerostomia (dry mouth). Combined uveoparotid involvement is designated Mikulicz syndrome.
The spleen may appear unaffected grossly, but in about three fourths of cases, it contains granulomas. In approximately 10%, it becomes clinically enlarged. The liver demonstrates microscopic granulomatous lesions, usually in the portal triads, about as often as the spleen, but only about one third of the patients demonstrate hepatomegaly or abnormal liver function. Sarcoid involvement of bone marrow is reported in as many as 40% of patients, although it rarely causes severe manifestations. Other findings may include hypercalcemia and hypercalciuria. These changes are not related to bone destruction but rather are caused by increased calcium absorption secondary to production of active vitamin D by the mononuclear phagocytes in the granulomas.
Clinical Features
In many affected persons the disease is entirely asymptomatic, discovered on routine chest films as bilateral hilar adenopathy or as an incidental finding at autopsy. In others, peripheral lymphadenopathy, cutaneous lesions, eye involvement, splenomegaly, or hepatomegaly may be presenting manifestations. In about two thirds of symptomatic cases, there is gradual appearance of respiratory symptoms (shortness of breath, dry cough, or vague substernal discomfort) or constitutional signs and symptoms (fever, fatigue, weight loss, anorexia, night sweats). Because of the variable and nondiagnostic clinical features, resort is frequently made to lung or lymph node biopsies. The presence of noncaseating granulomas is suggestive of sarcoidosis, but other identifiable causes of granulomatous inflammation must be excluded.
Sarcoidosis follows an unpredictable course characterized by either progressive chronicity or periods of activity interspersed with remissions. The remissions may be spontaneous or initiated by steroid therapy and often are permanent. Overall, 65% to 70% of affected persons recover with minimal or no residual manifestations. Another 20% develop permanent lung dysfunction or visual impairment. Of the remaining 10% to 15%, most succumb to progressive pulmonary fibrosis and cor pulmonale.
Summary
Sarcoidosis
• Sarcoidosis is a multisystem disease of unknown etiology; the diagnostic histopathologic feature is the presence of noncaseating granulomas in various tissues.
• Immunologic abnormalities include high levels of CD4+ T cells in the lung that secrete TH1-dependent cytokines such as IFN-γ and IL-2 locally.
• Clinical manifestations include lymph node enlargement, eye involvement (sicca syndrome [dry eyes], iritis, or iridocyclitis), skin lesions (erythema nodosum, painless subcutaneous nodules), and visceral (liver, skin, marrow) involvement. Lung involvement occurs in 90% of cases, with formation of granulomas and interstitial fibrosis.
Hypersensitivity Pneumonitis
Hypersensitivity pneumonitis is an immunologically mediated inflammatory lung disease that primarily affects the alveoli and is therefore often called allergic alveolitis. Most often it is an occupational disease that results from heightened sensitivity to inhaled antigens such as in moldy hay (Table 12–5). Unlike bronchial asthma, in which bronchi are the focus of immunologically mediated injury, the damage in hypersensitivity pneumonitis occurs at the level of alveoli. Hence, it manifests as a predominantly restrictive lung disease with decreased diffusion capacity, lung compliance, and total lung volume. The occupational exposures are diverse, but the syndromes share common clinical and pathologic findings and probably have a very similar pathophysiologic basis.
Table 12–5 Selected Causes of Hypersensitivity Pneumonitis
| Syndrome | Exposure | Antigens |
|---|---|---|
| Fungal and Bacterial Antigens | ||
| Farmer’s lung | Moldy hay | Micropolyspora faeni |
| Bagassosis | Moldy pressed sugar cane (bagasse) | Thermophilic actinomycetes |
| Maple bark disease | Moldy maple bark | Cryptostroma corticale |
| Humidifier lung | Cool-mist humidifier | Thermophilic actinomycetes, Aureobasidium pullulans |
| Malt worker’s lung | Moldy barley | Aspergillus clavatus |
| Cheese washer’s lung | Moldy cheese | Penicillium casei |
| Insect Products | ||
| Miller’s lung | Dust-contaminated grain | Sitophilus granarius (wheat weevil) |
| Animal Products | ||
| Pigeon breeder’s lung | Pigeon droppings | Pigeon serum proteins in droppings |
| Chemicals | ||
| Chemical worker’s lung | Chemical industry | Trimellitic anhydride, isocyanates |
Several lines of evidence suggest that hypersensitivity pneumonitis is an immunologically mediated disease:
• Bronchoalveolar lavage specimens consistently demonstrate increased numbers of T lymphocytes of both CD4+ and CD8+ phenotype.
• Most patients with hypersensitivity pneumonitis have specific precipitating antibodies in their serum, and complement and immunoglobulins have been demonstrated within vessel walls by immunofluorescence, indicating type III hypersensitivity. The presence of noncaseating granulomas in two thirds of patients with this disorder suggests a role for type IV hypersensitivity as well.
In summary, hypersensitivity pneumonitis is an immunologically mediated response to an extrinsic antigen that involves both immune complex and delayed-type hypersensitivity reactions.
Morphology
The histopathologic picture in both acute and chronic forms of hypersensitivity pneumonitis includes patchy mononuclear cell infiltrates in the pulmonary interstitium, with a characteristic peribronchiolar accentuation. Lymphocytes predominate, but plasma cells and epithelioid cells also are present. In acute forms of the disease, variable numbers of neutrophils may also be seen. Interstitial noncaseating granulomas are present in more than two thirds of cases, usually in a peribronchiolar location (Fig. 12–24). In advanced chronic cases, diffuse interstitial fibrosis occurs.
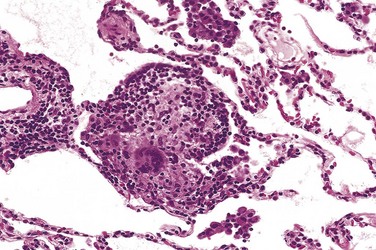
Clinical Features
Hypersensitivity pneumonitis may manifest either as an acute reaction, with fever, cough, dyspnea, and constitutional signs and symptoms arising 4 to 8 hours after exposure, or as a chronic disease characterized by insidious onset of cough, dyspnea, malaise, and weight loss. With the acute form of this disease, the diagnosis is usually obvious because of the temporal relationship of symptom onset to exposure to the incriminating antigen. If antigenic exposure is terminated after acute attacks of the disease, complete resolution of pulmonary symptoms occurs within days. Failure to remove the inciting agent from the environment eventually results in an irreversible chronic interstitial pulmonary disease.
Pulmonary Eosinophilia
A number of clinical and pathologic pulmonary entities are characterized by an infiltration and activation of eosinophils, the latter by elevated levels of alveolar IL-5. These diverse diseases generally are of immunologic origin, but the etiology is not understood. Pulmonary eosinophilia is divided into the following categories:
• Acute eosinophilic pneumonia with respiratory failure, characterized by rapid onset of fever, dyspnea, hypoxia, and diffuse pulmonary infiltrates on chest radiographs. The bronchoalveolar lavage fluid typically contains more than 25% eosinophils. There is prompt response to corticosteroids.
• Simple pulmonary eosinophilia (Loeffler syndrome), characterized by transient pulmonary lesions, eosinophilia in the blood, and a benign clinical course. The alveolar septa are thickened by an infiltrate containing eosinophils and occasional giant cells.
• Tropical eosinophilia, caused by infection with microfilariae and helminthic parasites
• Secondary eosinophilia, seen, for example, in association with asthma, drug allergies, and certain forms of vasculitis
• Idiopathic chronic eosinophilic pneumonia, characterized by aggregates of lymphocytes and eosinophils within the septal walls and the alveolar spaces, typically in the periphery of the lung fields, and accompanied by high fever, night sweats, and dyspnea. This is a disease of exclusion, once other causes of pulmonary eosinophilia have been ruled out.
Smoking-Related Interstitial Diseases
The role of cigarette smoking in causing obstructive pulmonary disease (emphysema and chronic bronchitis) has been discussed. Smoking also is associated with restrictive or interstitial lung diseases. Desquamative interstitial pneumonia (DIP) and respiratory bronchiolitis are the two related examples of smoking-associated interstitial lung disease. The most striking histologic feature of DIP is the accumulation of large numbers of macrophages with abundant cytoplasm containing dusty-brown pigment (smoker’s macrophages) in the air spaces (Fig. 12–25). The alveolar septa are thickened by a sparse inflammatory infiltrate (usually lymphocytes), and interstitial fibrosis, when present, is mild. Pulmonary functions usually show a mild restrictive abnormality, and patients with DIP typically have a good prognosis with excellent response to steroid therapy and smoking cessation. Respiratory bronchiolitis is a common histologic lesion found in smokers, characterized by the presence of pigmented intraluminal macrophages akin to those in DIP, but in a “bronchiolocentric” distribution (first- and second-order respiratory bronchioles). Mild peribronchiolar fibrosis also is seen. As with DIP, affected patients present with gradual onset of dyspnea and dry cough, and the symptoms recede with cessation of smoking.