Chapter 2 Inflammation and Repair
See Targeted Therapy available online at studentconsult.com
Overview of Inflammation and Tissue Repair
The survival of all organisms requires that they eliminate foreign invaders, such as infectious agents, and damaged tissues. These functions are mediated by a complex host response called inflammation. Inflammation is a protective response involving host cells, blood vessels, and proteins and other mediators that is intended to eliminate the initial cause of cell injury, as well as the necrotic cells and tissues resulting from the original insult, and to initiate the process of repair. Inflammation accomplishes its protective mission by first diluting, destroying, or otherwise neutralizing harmful agents (e.g., microbes, toxins). It then sets into motion the events that eventually heal and repair the sites of injury. Without inflammation, infections would go unchecked and wounds would never heal. In the context of infections, inflammation is one component of a protective response that immunologists refer to as innate immunity (Chapter 4).
Although inflammation helps clear infections and other noxious stimuli and initiates repair, the inflammatory reaction and the subsequent repair process can themselves cause considerable harm. The components of the inflammatory reaction that destroy and eliminate microbes and dead tissues are also capable of injuring normal tissues. Therefore, injury may accompany entirely normal, beneficial inflammatory reactions, and the damage may even become the dominant feature if the reaction is very strong (e.g., when the infection is severe), prolonged (e.g., when the eliciting agent resists eradication), or inappropriate (e.g., when it is directed against self-antigens in autoimmune diseases, or against usually harmless environmental antigens (e.g., in allergic disorders). Some of the most vexing diseases of humans are disorders that result from inappropriate, often chronic, inflammation. Thus, the process of inflammation is fundamental to virtually all of clinical medicine.
The cells and molecules of host defense, including leukocytes and plasma proteins, normally circulate in the blood, and the goal of the inflammatory reaction is to bring them to the site of infection or tissue damage. In addition, resident cells of vascular walls and the cells and proteins of the extracellular matrix (ECM) are also involved in inflammation and repair (Fig. 2–1). Before we describe the process of inflammation in detail, some of the basic features will be highlighted.
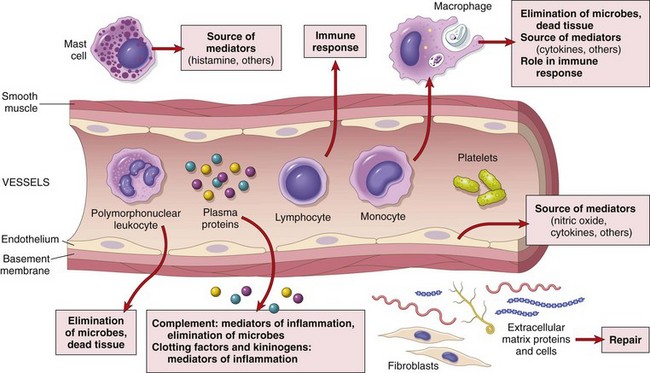
Figure 2–1 The components of acute and chronic inflammatory responses and their principal functions. The roles of these cells and molecules in inflammation are described in this chapter.
Inflammation can be acute or chronic (Table 2–1). Acute inflammation is rapid in onset and of short duration, lasting from a few minutes to as long as a few days, and is characterized by fluid and plasma protein exudation and a predominantly neutrophilic leukocyte accumulation. Chronic inflammation may be more insidious, is of longer duration (days to years), and is typified by influx of lymphocytes and macrophages with associated vascular proliferation and fibrosis (scarring). As we shall see later, however, these two basic forms of inflammation may coexist, and many variables modify their course and histologic appearance.
Table 2–1 Features of Acute and Chronic Inflammation
| Feature | Acute | Chronic |
|---|---|---|
| Onset | Fast: minutes or hours | Slow: days |
| Cellular infiltrate | Mainly neutrophils | Monocytes/macrophages and lymphocytes |
| Tissue injury, fibrosis | Usually mild and self-limited | Often severe and progressive |
| Local and systemic signs | Prominent | Less prominent; may be subtle |
Inflammation is induced by chemical mediators that are produced by host cells in response to injurious stimuli. When a microbe enters a tissue or the tissue is injured, the presence of the infection or damage is sensed by resident cells, mainly macrophages, but also dendritic cells, mast cells, and other cell types. These cells secrete molecules (cytokines and other mediators) that induce and regulate the subsequent inflammatory response. Inflammatory mediators are also produced from plasma proteins that react with the microbes or to injured tissues. Some of these mediators promote the efflux of plasma and the recruitment of circulating leukocytes to the site where the offending agent is located. The recruited leukocytes are activated and they try to remove the offending agent by phagocytosis. An unfortunate side effect of the activation of leukocytes may be damage to normal host tissues.
The external manifestations of inflammation, often called its cardinal signs, are heat (calor), redness (rubor), swelling (tumor), pain (dolor), and loss of function (functio laesa). The first four of these were described more than 2000 years ago by a Roman encyclopedist named Celsus, who wrote the then-famous text De Medicina, and the fifth was added in the late 19th century by Rudolf Virchow, known as the “father of modern pathology.” These manifestations occur as consequences of the vascular changes and leukocyte recruitment and activation, as will be evident from the discussion that follows.
Inflammation is normally controlled and self-limited. The mediators and cells are activated only in response to the injurious stimulus and are short-lived, and they are degraded or become inactive as the injurious agent is eliminated. In addition, various anti-inflammatory mechanisms become active. If the injurious agent cannot be quickly eliminated, the result may be chronic inflammation, which can have serious pathologic consequences.
 Summary
Summary
General Features of Inflammation
• Inflammation is a defensive host response to foreign invaders and necrotic tissue, but it is itself capable of causing tissue damage.
• The main components of inflammation are a vascular reaction and a cellular response; both are activated by mediators derived from plasma proteins and various cells.
• The steps of the inflammatory response can be remembered as the five Rs: (1) recognition of the injurious agent, (2) recruitment of leukocytes, (3) removal of the agent, (4) regulation (control) of the response, and (5) resolution (repair).
• The outcome of acute inflammation is either elimination of the noxious stimulus, followed by decline of the reaction and repair of the damaged tissue, or persistent injury resulting in chronic inflammation.
Acute Inflammation
The acute inflammatory response rapidly delivers leukocytes and plasma proteins to sites of injury. Once there, leukocytes clear the invaders and begin the process of digesting and getting rid of necrotic tissues.
Acute inflammation has two major components (Fig. 2–2):
• Vascular changes: alterations in vessel caliber resulting in increased blood flow (vasodilation) and changes in the vessel wall that permit plasma proteins to leave the circulation (increased vascular permeability). In addition, endothelial cells are activated, resulting in increased adhesion of leukocytes and migration of the leukocytes through the vessel wall.
• Cellular events: emigration of the leukocytes from the circulation and accumulation in the focus of injury (cellular recruitment), followed by activation of the leukocytes, enabling them to eliminate the offending agent. The principal leukocytes in acute inflammation are neutrophils (polymorphonuclear leukocytes).
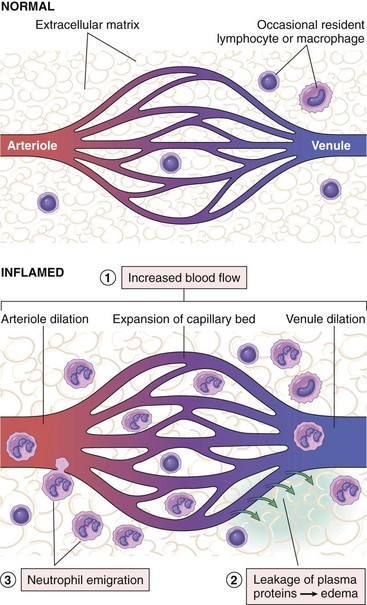
Figure 2–2 Vascular and cellular reactions of acute inflammation. The major local manifestations of acute inflammation, compared with normal, are (1) vascular dilation and increased blood flow (causing erythema and warmth), (2) extravasation of plasma fluid and proteins (edema), and (3) leukocyte (mainly neutrophil) emigration and accumulation.
Stimuli for Acute Inflammation
Acute inflammatory reactions may be triggered by a variety of stimuli:
• Infections (bacterial, viral, fungal, parasitic) are among the most common and medically important causes of inflammation.
• Trauma (blunt and penetrating) and various physical and chemical agents (e.g., thermal injury, such as burns or frostbite; irradiation; toxicity from certain environmental chemicals) injure host cells and elicit inflammatory reactions.
• Tissue necrosis (from any cause), including ischemia (as in a myocardial infarct) and physical and chemical injury
• Foreign bodies (splinters, dirt, sutures, crystal deposits)
• Immune reactions (also called hypersensitivity reactions) against environmental substances or against “self” tissues. Because the stimuli for these inflammatory responses often cannot be eliminated or avoided, such reactions tend to persist, with features of chronic inflammation. The term “immune-mediated inflammatory disease” is sometimes used to refer to this group of disorders.
Although each of these stimuli may induce reactions with some distinctive characteristics, in general, all inflammatory reactions have the same basic features.
In this section, we describe first how inflammatory stimuli are recognized by the host, then the typical reactions of acute inflammation and its morphologic features, and finally the chemical mediators responsible for these reactions.
Recognition of Microbes, Necrotic Cells, and Foreign Substances
A fundamental question relating to activation of the host response is how cells recognize the presence of potentially harmful agents such as microbes in the tissues. It was postulated that microbes and dead cells must elicit some sort of “danger signals” that distinguish them from normal tissues and mobilize the host response. It is now established that phagocytes, dendritic cells (cells in connective tissue and organs that capture microbes and initiate responses to them), and many other cells, such as epithelial cells, express receptors that are designed to sense the presence of infectious pathogens and substances released from dead cells. These receptors have been called “pattern recognition receptors” because they recognize structures (i.e., molecular patterns) that are common to many microbes or to dead cells. The two most important families of these receptors are the following:
• Toll-like receptors (TLRs) are microbial sensors that are named for the founding member called Toll, which was discovered in Drosophila. There are ten mammalian TLRs, which recognize products of bacteria (such as endotoxin and bacterial DNA), viruses (such as double-stranded RNA), and other pathogens (Fig. 2–3, A). TLRs are located in plasma membranes and endosomes, so they are able to detect extracellular and ingested microbes. They are complemented by cytoplasmic and membrane molecules, from several other families, that also recognize microbial products. TLRs and the other receptors recognize products of different types of microbes and thus provide defense against essentially all classes of infectious pathogens. Recognition of microbes by these receptors activates transcription factors that stimulate the production of a number of secreted and membrane proteins. These proteins include mediators of inflammation, antiviral cytokines (interferons), and proteins that promote lymphocyte activation and even more potent immune responses. We return to TLRs in Chapter 4, when we discuss innate immunity, the early defense against infections.
• The inflammasome is a multi-protein cytoplasmic complex that recognizes products of dead cells, such as uric acid and extracellular ATP, as well as crystals and some microbial products. Triggering of the inflammasome results in activation of an enzyme called caspase-1, which cleaves precursor forms of the inflammatory cytokine interleukin-1β (IL-1β) into its biologically active form (Fig. 2–3, B). As discussed later, IL-1 is an important mediator of leukocyte recruitment in the acute inflammatory response, and the leukocytes phagocytose and destroy dead cells. The joint disease, gout, is caused by deposition of urate crystals, which are ingested by phagocytes and activate the inflammasome, resulting in IL-1 production and acute inflammation. IL-1 antagonists are effective treatments in cases of gout that are resistant to conventional anti-inflammatory therapy. Recent studies have shown that cholesterol crystals and free fatty acids also activate the inflammasome, suggesting that IL-1 plays a role in common diseases such as atherosclerosis (associated with deposition of cholesterol crystals in vessel walls) and obesity-associated type 2 diabetes. This finding raises the possibility of treating these diseases by blocking IL-1.
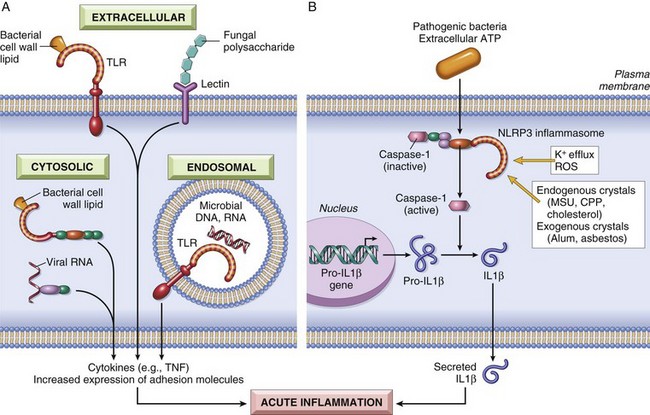
Figure 2–3 Sensors of microbes and dead cells: Phagocytes, dendritic cells, and many types of epithelial cells express different classes of receptors that sense the presence of microbes and dead cells. A, Toll-like receptors (TLRs) located in the plasma membrane and endosomes and other cytoplasmic and plasma membrane receptors (members of families other than TLRs) recognize products of different classes of microbes. The proteins produced by TLR activation have numerous functions; only their role in inflammation is shown. B, The inflammasome is a protein complex that recognizes products of dead cells and some microbes and induces the secretion of biologically active interleukin-1 (IL-1). The inflammasome consists of a sensor protein (a leucine-rich protein called NLRP3), an adaptor, and the enzyme caspase-1, which is converted from an inactive to an active form. (Note that the inflammasome is distinct from phagolysosomes, which also are present in the cytoplasm but are vesicles that serve different functions in inflammation, as discussed later in the chapter.) CPP, calcium pyrophosphate; MSU, monosodium urate.
The functions of these sensors are referred to throughout the chapter. We now proceed with a discussion of the principal reactions of acute inflammation.
Vascular Changes
The main vascular reactions of acute inflammation are increased blood flow secondary to vasodilation and increased vascular permeability, both designed to bring blood cells and proteins to sites of infection or injury. While the initial encounter of an injurious stimulus, such as a microbe, is with macrophages and other cells in the connective tissue, the vascular reactions triggered by these interactions soon follow and dominate the early phase of the response.
Changes in Vascular Caliber and Flow
Changes in blood vessels are initiated rapidly after infection or injury but evolve at variable rates, depending on the nature and severity of the original inflammatory stimulus.
• After transient vasoconstriction (lasting only for seconds), arteriolar vasodilation occurs, resulting in locally increased blood flow and engorgement of the down-stream capillary beds (Fig. 2–2). This vascular expansion is the cause of the redness (erythema) and warmth characteristic of acute inflammation, and mentioned previously as two of the cardinal signs of inflammation.
• The microvasculature becomes more permeable, and protein-rich fluid moves into the extravascular tissues. This causes the red cells in the flowing blood to become more concentrated, thereby increasing blood viscosity and slowing the circulation. These changes are reflected microscopically by numerous dilated small vessels packed with red blood cells, called stasis.
• As stasis develops, leukocytes (principally neutrophils) begin to accumulate along the vascular endothelial surface—a process called margination. This is the first step in the journey of the leukocytes through the vascular wall into the interstitial tissue (described later).
Increased Vascular Permeability
Increasing vascular permeability leads to the movement of protein-rich fluid and even blood cells into the extravascular tissues (Fig. 2–4). This in turn increases the osmotic pressure of the interstitial fluid, leading to more outflow of water from the blood into the tissues. The resulting protein-rich fluid accumulation is called an exudate. Exudates must be distinguished from transudates, which are interstitial fluid accumulations caused by increased hydrostatic pressure, usually a consequence of reduced venous return. Transudates typically contain low concentrations of protein and few or no blood cells. Fluid accumulation in extravascular spaces, whether from an exudate or a transudate, produces tissue edema. Whereas exudates are typical of inflammation, transudates accumulate in various noninflammatory conditions, which are mentioned in Figure 2–4 and described in more detail in Chapter 3.
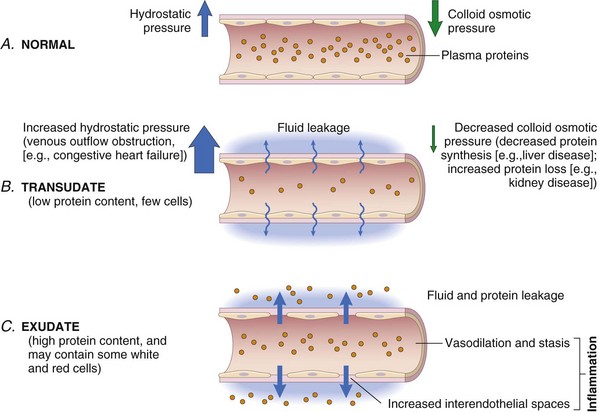
Figure 2–4 Formation of transudates and exudates. A, Normal hydrostatic pressure (blue arrows) is approximately 32 mm Hg at the arterial end of a capillary bed and 12 mm Hg at the venous end; the mean colloid osmotic pressure of tissues is approximately 25 mm Hg (green arrows), which is nearly equal to the mean capillary pressure. Therefore, the net flow of fluid across the vascular bed is almost nil. B, A transudate is formed when fluid leaks out because of increased hydrostatic pressure or decreased osmotic pressure. C, An exudate is formed in inflammation because vascular permeability increases as a result of the increase in interendothelial spaces.
Several mechanisms may contribute to increased vascular permeability in acute inflammatory reactions:
• Endothelial cell contraction leading to intercellular gaps in postcapillary venules is the most common cause of increased vascular permeability. Endothelial cell contraction occurs rapidly after binding of histamine, bradykinin, leukotrienes, and many other mediators to specific receptors, and is usually short-lived (15 to 30 minutes). A slower and more prolonged retraction of endothelial cells, resulting from changes in the cytoskeleton, may be induced by cytokines such as tumor necrosis factor (TNF) and interleukin-1 (IL-1). This reaction may take 4 to 6 hours to develop after the initial trigger and persist for 24 hours or more.
• Endothelial injury results in vascular leakage by causing endothelial cell necrosis and detachment. Endothelial cells are damaged after severe injury such as with burns and some infections. In most cases, leakage begins immediately after the injury and persists for several hours (or days) until the damaged vessels are thrombosed or repaired. Venules, capillaries, and arterioles can all be affected, depending on the site of the injury. Direct injury to endothelial cells may also induce a delayed prolonged leakage that begins after a delay of 2 to 12 hours, lasts for several hours or even days, and involves venules and capillaries. Examples are mild to moderate thermal injury, certain bacterial toxins, and x- or ultraviolet irradiation (i.e., the sunburn that has spoiled many an evening after a day in the sun). Endothelial cells may also be damaged as a consequence of leukocyte accumulation along the vessel wall. Activated leukocytes release many toxic mediators, discussed later, that may cause endothelial injury or detachment.
• Increased transcytosis of proteins by way of an intracellular vesicular pathway augments venular permeability, especially after exposure to certain mediators such as vascular endothelial growth factor (VEGF). Transcytosis occurs through channels formed by fusion of intracellular vesicles.
• Leakage from new blood vessels. As described later, tissue repair involves new blood vessel formation (angiogenesis). These vessel sprouts remain leaky until proliferating endothelial cells mature sufficiently to form intercellular junctions. New endothelial cells also have increased expression of receptors for vasoactive mediators, and some of the factors that stimulate angiogenesis (e.g., VEGF) also directly induce increased vascular permeability.
Although these mechanisms of vascular permeability are separable, all of them may participate in the response to a particular stimulus. For example, in a thermal burn, leakage results from chemically mediated endothelial contraction, as well as from direct injury and leukocyte-mediated endothelial damage.
Responses of Lymphatic Vessels
In addition to blood vessels, lymphatic vessels also participate in the inflammatory response. In inflammation, lymph flow is increased and helps drain edema fluid, leukocytes, and cell debris from the extravascular space. In severe inflammatory reactions, especially to microbes, the lymphatics may transport the offending agent, contributing to its dissemination. The lymphatics may become secondarily inflamed (lymphangitis), as may the draining lymph nodes (lymphadenitis). Inflamed lymph nodes are often enlarged because of hyperplasia of the lymphoid follicles and increased numbers of lymphocytes and phagocytic cells lining the sinuses of the lymph nodes. This constellation of pathologic changes is termed reactive, or inflammatory, lymphadenitis (Chapter 11). For clinicians, the presence of red streaks near a skin wound is a telltale sign of an infection in the wound. This streaking follows the course of the lymphatic channels and is diagnostic of lymphangitis; it may be accompanied by painful enlargement of the draining lymph nodes, indicating lymphadenitis.
Summary
Vascular Reactions in Acute Inflammation
• Vasodilation is induced by chemical mediators such as histamine (described later) and is the cause of erythema and stasis of blood flow.
• Increased vascular permeability is induced by histamine, kinins, and other mediators that produce gaps between endothelial cells; by direct or leukocyte-induced endothelial injury; and by increased passage of fluids through the endothelium. This increased permeability allows plasma proteins and leukocytes to enter sites of infection or tissue damage; fluid leak through blood vessels results in edema.
Cellular Events: Leukocyte Recruitment and Activation
As mentioned earlier, an important function of the inflammatory response is to deliver leukocytes to the site of injury and to activate them. Leukocytes ingest offending agents, kill bacteria and other microbes, and eliminate necrotic tissue and foreign substances. A price that is paid for the defensive potency of leukocytes is that once activated, they may induce tissue damage and prolong inflammation, since the leukocyte products that destroy microbes can also injure normal host tissues. Therefore, host defense mechanisms include checks and balances that ensure that leukocytes are recruited and activated only when and where they are needed (i.e., in response to foreign invaders and dead tissues). Systemic activation of leukocytes can, in fact, have detrimental consequences, as in septic shock (Chapter 3).
Leukocyte Recruitment
Leukocytes normally flow rapidly in the blood, and in inflammation, they have to be stopped and brought to the offending agent or the site of tissue damage, which are typically outside the vessels. The sequence of events in the recruitment of leukocytes from the vascular lumen to the extravascular space consists of (1) margination and rolling along the vessel wall; (2) firm adhesion to the endothelium; (3) transmigration between endothelial cells; and (4) migration in interstitial tissues toward a chemotactic stimulus (Fig. 2–5). Rolling, adhesion, and transmigration are mediated by the interactions of adhesion molecules on leukocytes and endothelial surfaces (see later on). Chemical mediators—chemoattractants and certain cytokines—affect these processes by modulating the surface expression and binding affinity of the adhesion molecules and by stimulating directional movement of the leukocytes.
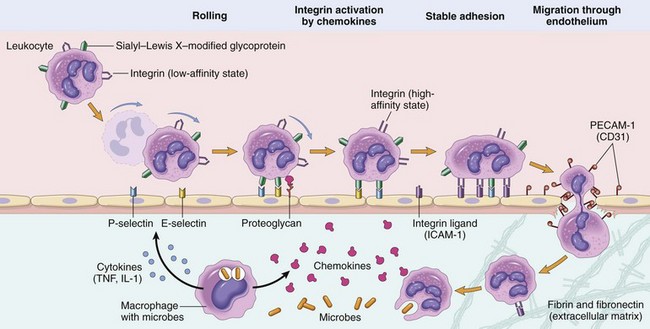
Figure 2–5 Mechanisms of leukocyte migration through blood vessels. The leukocytes (neutrophils shown here) first roll, then become activated and adhere to endothelium, then transmigrate across the endothelium, pierce the basement membrane, and migrate toward chemoattractants emanating from the source of injury. Different molecules play predominant roles in different steps of this process: selectins in rolling; chemokines (usually displayed bound to proteoglycans) in activating the neutrophils to increase avidity of integrins; integrins in firm adhesion; and CD31 (PECAM-1) in transmigration. ICAM-1, intercellular adhesion molecule-1; IL-1, interleukin-1; PECAM-1, platelet endothelial cell adhesion molecule-1; TNF, tumor necrosis factor.
Margination and Rolling
As blood flows from capillaries into postcapillary venules, circulating cells are swept by laminar flow against the vessel wall. Because the smaller red cells tend to move faster than the larger white cells, leukocytes are pushed out of the central axial column and thus have a better opportunity to interact with lining endothelial cells, especially as stasis sets in. This process of leukocyte accumulation at the periphery of vessels is called margination. If the endothelial cells are activated by cytokines and other mediators produced locally, they express adhesion molecules to which the leukocytes attach loosely. These cells bind and detach and thus begin to tumble on the endothelial surface, a process called rolling.
The weak and transient interactions involved in rolling are mediated by the selectin family of adhesion molecules (Table 2–2). Selectins are receptors expressed on leukocytes and endothelium that contain an extracellular domain that binds sugars (hence the lectin part of the name). The three members of this family are E-selectin (also called CD62E), expressed on endothelial cells; P-selectin (CD62P), present on platelets and endothelium; and L-selectin (CD62L), on the surface of most leukocytes. Selectins bind sialylated oligosaccharides (e.g., sialyl–Lewis X on leukocytes) that are attached to mucin-like glycoproteins on various cells. The endothelial selectins are typically expressed at low levels or are not present at all on unactivated endothelium, and are up-regulated after stimulation by cytokines and other mediators. Therefore, binding of leukocytes is largely restricted to endothelium at sites of infection or tissue injury (where the mediators are produced). For example, in unactivated endothelial cells, P-selectin is found primarily in intracellular Weibel-Palade bodies; however, within minutes of exposure to mediators such as histamine or thrombin, P-selectin is distributed to the cell surface, where it can facilitate leukocyte binding. Similarly, E-selectin and the ligand for L-selectin, which are not expressed on normal endothelium, are induced after stimulation by the cytokines IL-1 and TNF.
Table 2–2 Endothelial and Leukocyte Adhesion Molecules
| Endothelial Molecule | Leukocyte Molecule | Major Role(s) |
|---|---|---|
| Selectins and Selectin Ligands | ||
| P-selectin | Sialyl–Lewis X–modified proteins | Rolling |
| E-selectin | Sialyl–Lewis X–modified proteins | Rolling and adhesion |
| GlyCam-1, CD34 | L-selectin* | Rolling (neutrophils, monocytes) |
| Integrins and Integrin Ligands | ||
| ICAM-1 (immunoglobulin family) | CD11/CD18 integrins (LFA-1, Mac-1) | Firm adhesion, arrest, transmigration |
| VCAM-1 (immunoglobulin family) | VLA-4 integrin | Adhesion |
| Others | ||
| CD31 | CD31 (homotypic interaction) | Transmigration of leukocytes through endothelium |
ICAM-1, intercellular adhesion molecule-1; LFA-1, leukocyte function–associated antigen-1; Mac-1, macrophage-1 antigen; VCAM-1, vascular cell adhesion molecule-1; VLA-4, very late antigen-4.
* L-selectin is also involved in the binding of circulating lymphocytes to the high endothelial venules in lymph nodes and mucosal lymphoid tissues, and subsequent homing of lymphocytes to these tissues.
Adhesion
The rolling leukocytes are able to sense changes in the endothelium that initiate the next step in the reaction of leukocytes, which is firm adhesion to endothelial surfaces. This adhesion is mediated by integrins expressed on leukocyte cell surfaces interacting with their ligands on endothelial cells (Fig. 2–5 and Table 2–2). Integrins are transmembrane heterodimeric glycoproteins that mediate the adhesion of leukocytes to endothelium and of various cells to the extracellular matrix. They are normally expressed on leukocyte plasma membranes in a low-affinity form and do not adhere to their specific ligands until the leukocytes are activated by chemokines.
Chemokines are chemoattractant cytokines that are secreted by many cells at sites of inflammation and are displayed on the endothelial surface. (Cytokines are described later in the chapter.) When the adherent leukocytes encounter the displayed chemokines, the cells are activated, and their integrins undergo conformational changes and cluster together, thus converting to a high-affinity form. At the same time, other cytokines, notably TNF and IL-1 (also secreted at sites of infection and injury), activate endothelial cells to increase their expression of ligands for integrins. These ligands include intercellular adhesion molecule-1 (ICAM-1), which binds to the integrins leukocyte function–associated antigen-1 (LFA-1) (also called CD11a/CD18) and macrophage-1 antigen (Mac-1) (i.e., CD11b/CD18), and vascular cell adhesion molecule-1 (VCAM-1), which binds to the integrin very late antigen-4 (VLA-4) (Table 2–2). Engagement of integrins by their ligands delivers signals to the leukocytes that lead to cytoskeletal changes that mediate firm attachment to the substrate. Thus, the net result of cytokine-stimulated increased integrin affinity and increased expression of integrin ligands is stable attachment of leukocytes to endothelial cells at sites of inflammation.
Transmigration
After being arrested on the endothelial surface, leukocytes migrate through the vessel wall primarily by squeezing between cells at intercellular junctions. This extravasation of leukocytes, called diapedesis, occurs mainly in the venules of the systemic vasculature; it has also been noted in capillaries in the pulmonary circulation. Migration of leukocytes is driven by chemokines produced in extravascular tissues, which stimulate movement of the leukocytes toward their chemical gradient. In addition, platelet endothelial cell adhesion molecule-1 (PECAM-1) (also called CD31), a cellular adhesion molecule expressed on leukocytes and endothelial cells, mediates the binding events needed for leukocytes to traverse the endothelium. After passing through the endothelium, leukocytes secrete collagenases that enable them to pass through the vascular basement membrane.
Chemotaxis
After extravasating from the blood, leukocytes move toward sites of infection or injury along a chemical gradient by a process called chemotaxis. Both exogenous and endogenous substances can be chemotactic for leukocytes, including the following:
• Bacterial products, particularly peptides with N-formylmethionine termini
• Cytokines, especially those of the chemokine family
• Components of the complement system, particularly C5
• Products of the lipoxygenase pathway of arachidonic acid (AA) metabolism, particularly leukotriene B4 (LTB4)
These mediators, which are described in more detail later, are produced in response to infections and tissue damage and during immunologic reactions. Leukocyte infiltration in all of these situations results from the actions of various combinations of mediators.
Chemotactic molecules bind to specific cell surface receptors, which triggers the assembly of cytoskeletal contractile elements necessary for movement. Leukocytes move by extending pseudopods that anchor to the ECM and then pull the cell in the direction of the extension. The direction of such movement is specified by a higher density of chemokine receptors at the leading edge of the cell. Thus, leukocytes move to and are retained at the site where they are needed.
The type of emigrating leukocyte varies with the age of the inflammatory response and with the type of stimulus. In most forms of acute inflammation, neutrophils predominate in the inflammatory infiltrate during the first 6 to 24 hours and are replaced by monocytes in 24 to 48 hours (Fig. 2–6). Several factors account for this early abundance of neutrophils: These cells are the most numerous leukocytes in the blood, they respond more rapidly to chemokines, and they may attach more firmly to the adhesion molecules that are rapidly induced on endothelial cells, such as P- and E-selectins. In addition, after entering tissues, neutrophils are short-lived—they die by apoptosis and disappear within 24 to 48 hours—while monocytes survive longer. There are exceptions to this pattern of cellular infiltration, however. In certain infections (e.g., those caused by Pseudomonas organisms), the cellular infiltrate is dominated by continuously recruited neutrophils for several days; in viral infections, lymphocytes may be the first cells to arrive; and in some hypersensitivity reactions, eosinophils may be the main cell type.
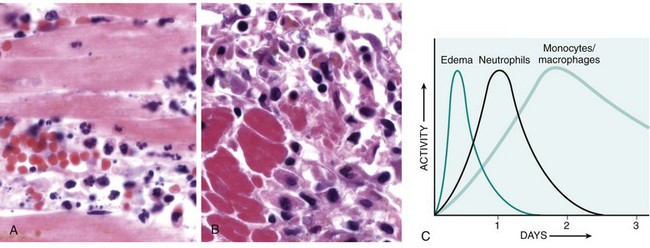
Figure 2–6 Nature of leukocyte infiltrates in inflammatory reactions. The photomicrographs show an inflammatory reaction in the myocardium after ischemic necrosis (infarction). A, Early (neutrophilic) infiltrates and congested blood vessels. B, Later (mononuclear) cellular infiltrates. C, The approximate kinetics of edema and cellular infiltration. For sake of simplicity, edema is shown as an acute transient response, although secondary waves of delayed edema and neutrophil infiltration also can occur.
Summary
Leukocyte Recruitment to Sites of Inflammation
• Leukocytes are recruited from the blood into the extravascular tissue, where infectious pathogens or damaged tissues may be located, and are activated to perform their functions.
• Leukocyte recruitment is a multi-step process consisting of loose attachment to and rolling on endothelium (mediated by selectins); firm attachment to endothelium (mediated by integrins); and migration through interendothelial spaces.
• Various cytokines promote expression of selectins and integrin ligands on endothelium (TNF, IL-1), increase the avidity of integrins for their ligands (chemokines), and promote directional migration of leukocytes (also chemokines); many of these cytokines are produced by tissue macrophages and other cells responding to pathogens or damaged tissues.
• Neutrophils predominate in the early inflammatory infiltrate and are later replaced by macrophages.
Leukocyte Activation
Once leukocytes have been recruited to the site of infection or tissue necrosis, they must be activated to perform their functions. Stimuli for activation include microbes, products of necrotic cells, and several mediators that are described later. As described earlier, leukocytes use various receptors to sense the presence of microbes, dead cells, and foreign substances. Engagement of these cellular receptors induces a number of responses in leukocytes that are part of their normal defensive functions and are grouped under the term leukocyte activation (Fig. 2–7). Leukocyte activation results in the enhancement of the following functions:
• Intracellular destruction of phagocytosed microbes and dead cells by substances produced in phagosomes, including reactive oxygen and nitrogen species and lysosomal enzymes
• Liberation of substances that destroy extracellular microbes and dead tissues, which are largely the same as the substances produced within phagocytic vesicles. A recently discovered mechanism by which neutrophils destroy extracellular microbes is the formation of extracellular “traps.”
• Production of mediators, including arachidonic acid metabolites and cytokines, that amplify the inflammatory reaction, by recruiting and activating more leukocytes
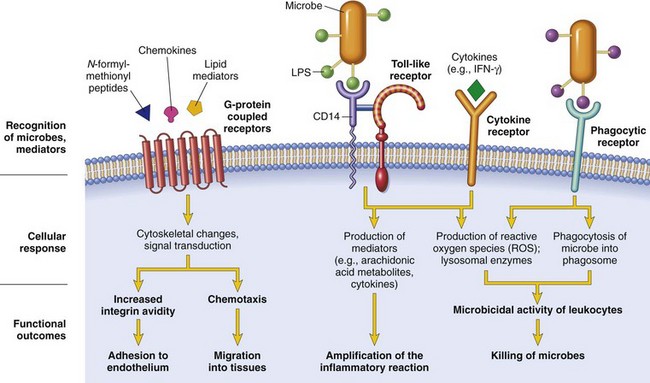
Figure 2–7 Leukocyte activation. Different classes of cell surface receptors of leukocytes recognize different stimuli. The receptors initiate responses that mediate the functions of the leukocytes. Only some receptors are depicted (see text for details). Lipopolysaccharide (LPS) first binds to a circulating LPS-binding protein (not shown). IFN-γ, interferon-γ.
Phagocytosis
Phagocytosis consists of three steps (Fig. 2–8): (1) recognition and attachment of the particle to the ingesting leukocyte; (2) engulfment, with subsequent formation of a phagocytic vacuole; and (3) killing and degradation of the ingested material.
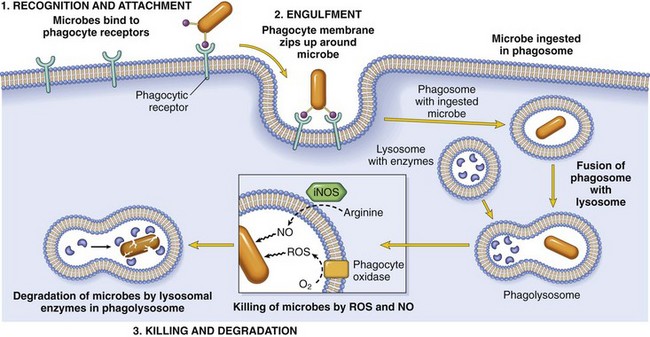
Figure 2–8 Phagocytosis. Phagocytosis of a particle (e.g., a bacterium) involves (1) attachment and binding of the particle to receptors on the leukocyte surface, (2) engulfment and fusion of the phagocytic vacuole with granules (lysosomes), and (3) destruction of the ingested particle. iNOS, inducible nitric oxide synthase; NO, nitric oxide; ROS, reactive oxygen species.
Leukocytes bind and ingest most microorganisms and dead cells by means of specific surface receptors. Some of these receptors recognize components of the microbes and dead cells and other receptors recognize host proteins, called opsonins, that coat microbes and target them for phagocytosis (the process called opsonization). The most important opsonins are antibodies of the immunoglobulin G (IgG) class that bind to microbial surface antigens, breakdown products of the complement protein C3 (described later), and plasma carbohydrate-binding lectins called collectins, which bind to microbial cell wall sugar groups. These opsonins either are present in the blood ready to coat microbes or are produced in response to the microbes. Leukocytes express receptors for opsonins that facilitate rapid phagocytosis of the coated microbes. These receptors include the Fc receptor for IgG (called FcγRI), complement receptors 1 and 3 (CR1 and CR3) for complement fragments, and C1q for the collectins.
Binding of opsonized particles to these receptors triggers engulfment and induces cellular activation that enhances degradation of ingested microbes. In engulfment, pseudopods are extended around the object, eventually forming a phagocytic vacuole. The membrane of the vacuole then fuses with the membrane of a lysosomal granule, resulting in discharge of the granule’s contents into the phagolysosome.
Killing and Degradation of Phagocytosed Microbes
The culmination of the phagocytosis of microbes is killing and degradation of the ingested particles. The key steps in this reaction are the production of microbicidal substances within lysosomes and fusion of the lysosomes with phagosomes, thus exposing the ingested particles to the destructive mechanisms of the leukocytes (Fig. 2–8). The most important microbicidal substances are reactive oxygen species (ROS) and lysosomal enzymes. The production of ROS involves the following steps:
• Phagocytosis and the engagement of various cellular receptors stimulate an oxidative burst, also called the respiratory burst, which is characterized by a rapid increase in oxygen consumption, glycogen catabolism (glycogenolysis), increased glucose oxidation, and production of ROS. The generation of the oxygen metabolites is due to rapid activation of a leukocyte NADPH oxidase, called the phagocyte oxidase, which oxidizes NADPH (reduced nicotinamide adenine dinucleotide phosphate) and, in the process, converts oxygen to superoxide ion (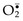 ) (see Fig. 1–18, B, Chapter 1).
) (see Fig. 1–18, B, Chapter 1).
• Superoxide is then converted by spontaneous dismutation into hydrogen peroxide (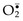 + 2H+ → H2O2). These ROS act as free radicals and destroy microbes by mechanisms that were described in Chapter 1.
+ 2H+ → H2O2). These ROS act as free radicals and destroy microbes by mechanisms that were described in Chapter 1.
• The quantities of H2O2 produced generally are insufficient to kill most bacteria (although superoxide and hydroxyl radical formation may be sufficient to do so). However, the lysosomes of neutrophils (called azurophilic granules) contain the enzyme myeloperoxidase (MPO), and in the presence of a halide such as Cl−, MPO converts H2O2 to HOCl• (hypochlorous radical). HOCl• is a powerful oxidant and antimicrobial agent (NaOCl is the active ingredient in chlorine bleach) that kills bacteria by halogenation, or by protein and lipid peroxidation.
Fortunately, the phagocyte oxidase is active only after its cytosolic subunit translocates to the membrane of the phagolysosome; thus, the reactive end products are generated mainly within the vesicles, and the phagocyte itself is not damaged. H2O2 is eventually broken down to water and O2 by the actions of catalase, and the other ROS also are degraded (Chapter 1). Reactive nitrogen species, particularly nitric oxide (NO), act in the same way as that described for ROS.
The dead microorganisms are then degraded by the action of lysosomal acid hydrolases. Perhaps the most important lysosomal enzyme involved in bacterial killing is elastase.
Of note, in addition to ROS and enzymes, several other constituents of leukocyte granules are capable of killing infectious pathogens. These include bactericidal permeability-increasing protein (causing phospholipase activation and membrane phospholipid degradation), lysozyme (causing degradation of bacterial coat oligosaccharides), major basic protein (an important eosinophil granule constituent that is cytotoxic for parasites), and defensins (peptides that kill microbes by creating holes in their membranes).
Secretion of Microbicidal Substances
The microbicidal mechanisms of phagocytes are largely sequestered within phagolysosomes in order to protect the leukocytes from damaging themselves. Leukocytes also actively secrete granule components including enzymes such as elastase, which destroy and digest extracellular microbes and dead tissues, as well as antimicrobial peptides. The contents of lysosomal granules are secreted by leukocytes into the extracellular milieu by several mechanisms:
• The phagocytic vacuole may remain transiently open to the outside before complete closure of the phagolysosome (regurgitation during feeding).
• If cells encounter materials that cannot be easily ingested, such as immune complexes deposited on immovable surfaces (e.g., glomerular basement membrane), the attempt to phagocytose these substances (frustrated phagocytosis) triggers strong leukocyte activation, and lysosomal enzymes are released into the surrounding tissue or lumen.
• The membrane of the phagolysosome may be damaged if potentially injurious substances, such as silica particles, are phagocytosed.
Neutrophil Extracellular Traps (NETs)
These “traps” are extracellular fibrillar networks that are produced by neutrophils in response to infectious pathogens (mainly bacteria and fungi) and inflammatory mediators (such as chemokines, cytokines, complement proteins, and ROS). NETs contain a framework of nuclear chromatin with embedded granule proteins, such as antimicrobial peptides and enzymes (Fig. 2–9). The traps provide a high concentration of antimicrobial substances at sites of infection, and prevent the spread of the microbes by trapping them in the fibrils. In the process, the nuclei of the neutrophils are lost, leading to death of the cells. NETs also have been detected in blood neutrophils during sepsis. The nuclear chromatin in the NETs, which includes histones and associated DNA, has been postulated to be a source of nuclear antigens in systemic autoimmune diseases, particularly lupus, in which affected persons react against their own DNA and nucleoproteins (Chapter 4).
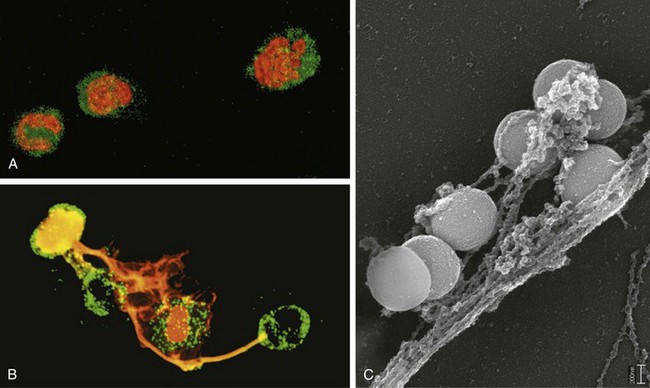
Figure 2–9 Neutrophil extracellular traps (NETs). A, Healthy neutrophils with nuclei stained red and cytoplasm green. B, Release of nuclear material from neutrophils (note that two have lost their nuclei), forming extracellular traps. C, An electron micrograph of bacteria (staphylococci) trapped in NETs.
(From Brinkmann V, Zychlinsky A: Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol 5:577, 2007, with the permission of the authors and publisher.)
Leukocyte-Induced Tissue Injury
Because leukocytes are capable of secreting potentially harmful substances such as enzymes and ROS, they are important causes of injury to normal cells and tissues under several circumstances:
• As part of a normal defense reaction against infectious microbes, when “bystander” tissues are injured. In certain infections that are difficult to eradicate, such as tuberculosis and some viral diseases, the host response contributes more to the pathologic process than does the microbe itself.
• As a normal attempt to clear damaged and dead tissues (e.g., after a myocardial infarction). In an infarct, inflammation may prolong and exacerbate the injurious consequences of the ischemia, especially upon reperfusion (Chapter 1).
• When the inflammatory response is inappropriately directed against host tissues, as in certain autoimmune diseases, or when the host reacts excessively against nontoxic environmental substances, such as allergic diseases including asthma (discussed in Chapter 4)
In all of these situations, the mechanisms by which leukocytes damage normal tissues are the same as the mechanisms involved in the clearance of microbes and dead tissues, because once the leukocytes are activated, their effector mechanisms do not distinguish between offender and host. In fact, if unchecked or inappropriately directed against host tissues, leukocytes themselves become the main offenders. Leukocyte-dependent tissue injury underlies many acute and chronic human diseases (Table 2–3), as is evident in discussions of specific disorders throughout this book.
Table 2–3 Clinical Examples of Leukocyte-Induced Injury
| Disorder* | Cells and Molecules Involved in Injury |
|---|---|
| Acute | |
| Acute respiratory distress syndrome | Neutrophils |
| Acute transplant rejection | Lymphocytes; antibodies and complement |
| Asthma | Eosinophils; IgE antibodies |
| Glomerulonephritis | Antibodies and complement; neutrophils, monocytes |
| Septic shock | Cytokines |
| Chronic | |
| Rheumatoid arthritis | Lymphocytes, macrophages; antibodies? |
| Asthma | Eosinophils; IgE antibodies |
| Atherosclerosis | Macrophages; lymphocytes? |
| Chronic transplant rejection | Lymphocytes, macrophages; cytokines |
| Pulmonary fibrosis | Macrophages; fibroblasts |
IgE, immunoglobulin E.
* Listed are selected examples of diseases in which the host response plays a significant role in tissue injury. Some, such as asthma, can manifest with acute inflammation or a chronic illness with repeated bouts of acute exacerbation. These diseases and their pathogenesis are discussed in much more detail in relevant chapters.
Activated leukocytes, especially macrophages, also secrete many cytokines, which stimulate further inflammation and have important systemic effects, to be discussed later.
Summary
Leukocyte Effector Mechanisms
• Leukocytes can eliminate microbes and dead cells by phagocytosis, followed by their destruction in phagolysosomes.
• Destruction is caused by free radicals (ROS, NO) generated in activated leukocytes and lysosomal enzymes.
• Enzymes and ROS may be released into the extracellular environment.
• The mechanisms that function to eliminate microbes and dead cells (the physiologic role of inflammation) are also capable of damaging normal tissues (the pathologic consequences of inflammation).
Defects in Leukocyte Function
Since leukocytes play a central role in host defense, it is not surprising that defects in leukocyte function, both acquired and inherited, lead to increased susceptibility to infections, which may be recurrent and life-threatening (Table 2–4). The most common causes of defective inflammation are bone marrow suppression caused by tumors or treatment with chemotherapy or radiation (resulting in decreased leukocyte numbers) and metabolic diseases such as diabetes (causing abnormal leukocyte functions). These are described elsewhere in the book.
Table 2–4 Defects in Leukocyte Functions
| Disease | Defect |
|---|---|
| Acquired | |
| Bone marrow suppression: tumors (including leukemias), radiation, and chemotherapy | Production of leukocytes |
| Diabetes, malignancy, sepsis, chronic dialysis | Adhesion and chemotaxis |
| Anemia, sepsis, diabetes, malnutrition | Phagocytosis and microbicidal activity |
| Genetic | |
| Leukocyte adhesion deficiency 1 | Defective leukocyte adhesion because of mutations in β chain of CD11/CD18 integrins |
| Leukocyte adhesion deficiency 2 | Defective leukocyte adhesion because of mutations in fucosyl transferase required for synthesis of sialylated oligosaccharide (receptor for selectins) |
| Chronic granulomatous disease | Decreased oxidative burst |
| X-linked | Phagocyte oxidase (membrane component) |
| Autosomal recessive | Phagocyte oxidase (cytoplasmic components) |
| Myeloperoxidase deficiency | Decreased microbial killing because of defective MPO–H2O2 system |
| Chédiak-Higashi syndrome | Decreased leukocyte functions because of mutations affecting protein involved in lysosomal membrane traffic |
H2O2, hydrogen peroxide; MPO, myeloperoxidase.
Modified from Gallin JI: Disorders of phagocytic cells. In Gallin JI, et al (eds): Inflammation: Basic Principles and Clinical Correlates, 2nd ed. New York, Raven Press, 1992, pp 860, 861.
The genetic disorders, although individually rare, illustrate the importance of particular molecular pathways in the complex inflammatory response. Some of the better understood inherited diseases are the following:
• Defects in leukocyte adhesion. In leukocyte adhesion deficiency type 1 (LAD-1), defective synthesis of the CD18 β subunit of the leukocyte integrins LFA-1 and Mac-1 leads to impaired leukocyte adhesion to and migration through endothelium, and defective phagocytosis and generation of an oxidative burst. Leukocyte adhesion deficiency type 2 (LAD-2) is caused by a defect in fucose metabolism resulting in the absence of sialyl–Lewis X, the oligosaccharide on leukocytes that binds to selectins on activated endothelium. Its clinical manifestations are similar to but milder than those of LAD-1.
• Defects in microbicidal activity. An example is chronic granulomatous disease, a genetic deficiency in one of the several components of the phagocyte oxidase enzyme that is responsible for generating ROS. In these patients, engulfment of bacteria does not result in activation of oxygen-dependent killing mechanisms. In an attempt to control these infections, the microbes are surrounded by activated macrophages, forming the “granulomas” (see later) that give the disease its distinctive pathologic features and its somewhat misleading name.
• Defects in phagolysosome formation. One such disorder, Chédiak-Higashi syndrome, is an autosomal recessive disease that results from disordered intracellular trafficking of organelles, ultimately impairing the fusion of lysosomes with phagosomes. The secretion of lytic secretory granules by cytotoxic T lymphocytes is also affected, explaining the severe immunodeficiency typical of the disorder.
• Rare patients with defective host defenses have been shown to carry mutations in TLR signaling pathways. Inherited defects in components of adaptive immune responses also result in increased susceptibility to infections. These are described in Chapter 4.
• Gain-of-function mutations in genes encoding some components of the inflammasome, one of which is called cryopyrin, are responsible for rare but serious diseases called cryopyrin-associated periodic fever syndromes (CAPSs), which manifest with unrelenting fevers and other signs of inflammation and respond well to treatment with IL-1 antagonists.
Outcomes of Acute Inflammation
Although the consequences of acute inflammation are modified by the nature and intensity of the injury, the site and tissue affected, and the ability of the host to mount a response, acute inflammation generally has one of three outcomes (Fig. 2–10):
• Resolution: Regeneration and repair. When the injury is limited or short-lived, when there has been no or minimal tissue damage, and when the injured tissue is capable of regenerating, the usual outcome is restoration to structural and functional normalcy. Before the process of resolution can start, the acute inflammatory response has to be terminated. This involves neutralization, decay, or enzymatic degradation of the various chemical mediators; normalization of vascular permeability; and cessation of leukocyte emigration, with subsequent death (by apoptosis) of extravasated neutrophils. Furthermore, leukocytes begin to produce mediators that inhibit inflammation, thereby limiting the reaction. The necrotic debris, edema fluid, and inflammatory cells are cleared by phagocytes and lymphatic drainage, eliminating the detritus from the battlefield. Leukocytes secrete cytokines that initiate the subsequent repair process, in which new blood vessels grow into the injured tissue to provide nutrients, growth factors stimulate the proliferation of fibroblasts and laying down of collagen to fill defects, and residual tissue cells proliferate to restore structural integrity. This process is described later in the chapter.
• Chronic inflammation may follow acute inflammation if the offending agent is not removed, or it may be present from the onset of injury (e.g., in viral infections or immune responses to self-antigens). Depending on the extent of the initial and continuing tissue injury, as well as the capacity of the affected tissues to regrow, chronic inflammation may be followed by restoration of normal structure and function or may lead to scarring.
• Scarring is a type of repair after substantial tissue destruction (as in abscess formation, discussed later) or when inflammation occurs in tissues that do not regenerate, in which the injured tissue is filled in by connective tissue. In organs in which extensive connective tissue deposition occurs in attempts to heal the damage or as a consequence of chronic inflammation, the outcome is fibrosis, a process that can significantly compromise function.
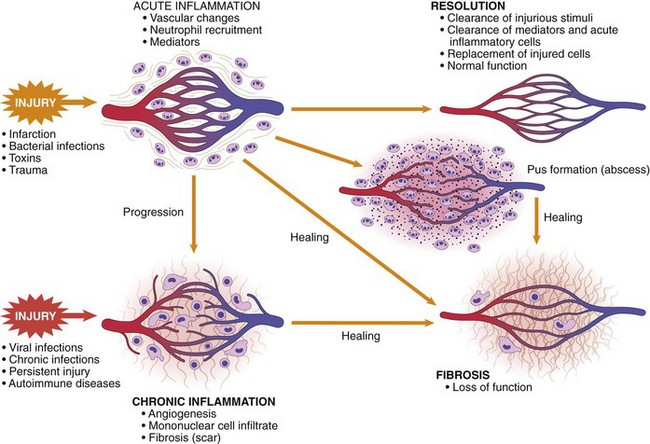
Figure 2–10 Outcomes of acute inflammation: resolution, healing by scarring (fibrosis), or chronic inflammation (see text).
Summary
Sequence of Events in Acute Inflammation
• The vascular changes in acute inflammation are characterized by increased blood flow secondary to arteriolar and capillary bed dilation (erythema and warmth).
• Increased vascular permeability, as a consequence of either widening of interendothelial cell junctions of the venules or direct endothelial cell injury, results in an exudate of protein-rich extravascular fluid (tissue edema).
• The leukocytes, initially predominantly neutrophils, adhere to the endothelium via adhesion molecules and then leave the microvasculature and migrate to the site of injury under the influence of chemotactic agents.
• Phagocytosis, killing, and degradation of the offending agent follow.
• Genetic or acquired defects in leukocyte functions give rise to recurrent infections.
• The outcome of acute inflammation may be removal of the exudate with restoration of normal tissue architecture (resolution); transition to chronic inflammation; or extensive destruction of the tissue resulting in scarring.
Morphologic Patterns of Acute Inflammation
The vascular and cellular reactions that characterize acute inflammation are reflected in the morphologic appearance of the reaction. The severity of the inflammatory response, its specific cause, and the particular tissue involved all can modify the basic morphology of acute inflammation, producing distinctive appearances. The importance of recognizing these morphologic patterns is that they are often associated with different etiology and clinical situations.
 Morphology
Morphology
• Serous inflammation is characterized by the outpouring of a watery, relatively protein-poor fluid that, depending on the site of injury, derives either from the plasma or from the secretions of mesothelial cells lining the peritoneal, pleural, and pericardial cavities. The skin blister resulting from a burn or viral infection is a good example of the accumulation of a serous effusion either within or immediately beneath the epidermis of the skin (Fig. 2–11). Fluid in a serous cavity is called an effusion.
• Fibrinous inflammation occurs as a consequence of more severe injuries, resulting in greater vascular permeability that allows large molecules (such as fibrinogen) to pass the endothelial barrier. Histologically, the accumulated extravascular fibrin appears as an eosinophilic meshwork of threads or sometimes as an amorphous coagulum (Fig. 2–12). A fibrinous exudate is characteristic of inflammation in the lining of body cavities, such as the meninges, pericardium, and pleura. Such exudates may be degraded by fibrinolysis, and the accumulated debris may be removed by macrophages, resulting in restoration of the normal tissue structure (resolution). However, extensive fibrin-rich exudates may not be completely removed, and are replaced by an ingrowth of fibroblasts and blood vessels (organization), leading ultimately to scarring that may have significant clinical consequences. For example, organization of a fibrinous pericardial exudate forms dense fibrous scar tissue that bridges or obliterates the pericardial space and restricts myocardial function.
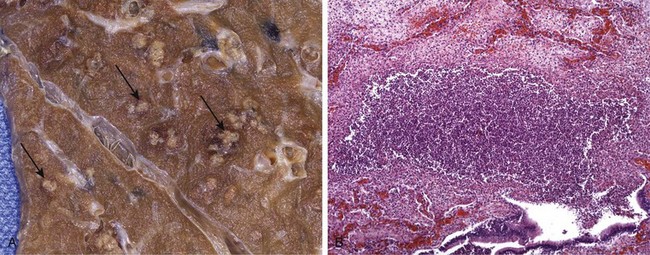
• Suppurative (purulent) inflammation and abscess formation. These are manifested by the collection of large amounts of purulent exudate (pus) consisting of neutrophils, necrotic cells, and edema fluid. Certain organisms (e.g., staphylococci) are more likely to induce such localized suppuration and are therefore referred to as pyogenic (pus-forming). Abscesses are focal collections of pus that may be caused by seeding of pyogenic organisms into a tissue or by secondary infections of necrotic foci. Abscesses typically have a central, largely necrotic region rimmed by a layer of preserved neutrophils (Fig. 2–13), with a surrounding zone of dilated vessels and fibroblast proliferation indicative of attempted repair. As time passes, the abscess may become completely walled off and eventually be replaced by connective tissue. Because of the underlying tissue destruction, the usual outcome with abscess formation is scarring.
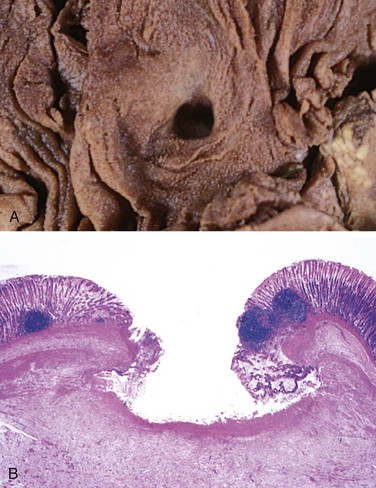
• An ulcer is a local defect, or excavation, of the surface of an organ or tissue that is produced by necrosis of cells and sloughing (shedding) of necrotic and inflammatory tissue (Fig. 2–14). Ulceration can occur only when tissue necrosis and resultant inflammation exist on or near a surface. Ulcers are most commonly encountered in (1) the mucosa of the mouth, stomach, intestines, or genitourinary tract and (2) in the subcutaneous tissues of the lower extremities in older persons who have circulatory disturbances predisposing affected tissue to extensive necrosis. Ulcerations are best exemplified by peptic ulcer of the stomach or duodenum, in which acute and chronic inflammation coexist. During the acute stage, there is intense polymorphonuclear infiltration and vascular dilation in the margins of the defect. With chronicity, the margins and base of the ulcer develop scarring with accumulation of lymphocytes, macrophages, and plasma cells.
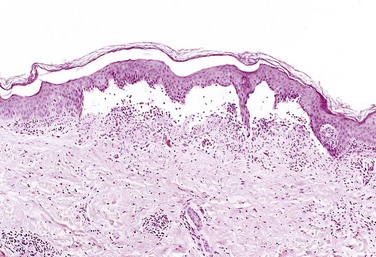
Figure 2–11 Serous inflammation. Low-power view of a cross-section of a skin blister showing the epidermis separated from the dermis by a focal collection of serous effusion.
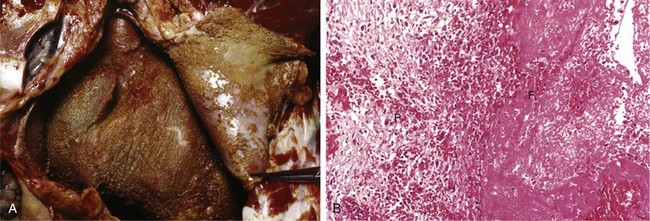
Figure 2–12 Fibrinous pericarditis. A, Deposits of fibrin on the pericardium. B, A pink meshwork of fibrin exudate (F) overlies the pericardial surface (P).
Chemical Mediators and Regulators of Inflammation
Having described the vascular and cellular events in acute inflammation, and the accompanying morphologic alterations, we next discuss the chemical mediators that are responsible for these events. While the harried student may find this list daunting (as do the professors!), it is worthy of note that this knowledge has been used to design a large armamentarium of anti-inflammatory drugs, which are used every day by large numbers of people and include familiar drugs like aspirin and acetaminophen. In this section, we emphasize general properties of the mediators of inflammation and highlight only some of the more important molecules. We also touch upon some of the mechanisms that limit and terminate inflammatory reactions.
• Mediators may be produced locally by cells at the site of inflammation, or may be derived from circulating inactive precursors (typically synthesized by the liver) that are activated at the site of inflammation (Fig. 2–15 and Table 2–5). Cell-derived mediators are normally sequestered in intracellular granules and are rapidly secreted upon cellular activation (e.g., histamine in mast cells) or are synthesized de novo in response to a stimulus (e.g., prostaglandins and cytokines produced by leukocytes and other cells). Plasma protein–derived mediators (complement proteins, kinins) circulate in an inactive form and typically undergo proteolytic cleavage to acquire their biologic activities.
• Most mediators act by binding to specific receptors on different target cells. Such mediators may act on only one or a very few cell types, or they may have diverse actions, with differing outcomes depending on which cell type they affect. Other mediators (e.g., lysosomal proteases, ROS) have direct enzymatic and/or toxic activities that do not require binding to specific receptors.
• The actions of most mediators are tightly regulated and short-lived. Once activated and released from the cell, mediators quickly decay (e.g., arachidonic acid metabolites), are inactivated by enzymes (e.g., kininase inactivates bradykinin), are eliminated (e.g., antioxidants scavenge toxic oxygen metabolites), or are inhibited (e.g., complement regulatory proteins block complement activation).
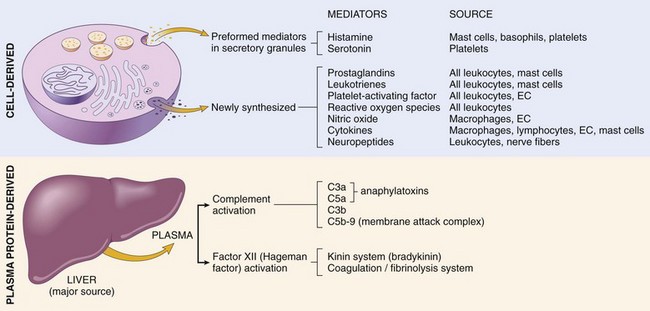
Figure 2–15 Mediators of inflammation. The principal cell-derived and plasma protein mediators are shown. EC, endothelial cells.
Table 2–5 Actions of the Principal Mediators of Inflammation
| Mediator | Source(s) | Actions |
|---|---|---|
| Cell-Derived | ||
| Histamine | Mast cells, basophils, platelets | Vasodilation, increased vascular permeability, endothelial activation |
| Serotonin | Platelets | Vasoconstriction |
| Prostaglandins | Mast cells, leukocytes | Vasodilation, pain, fever |
| Leukotrienes | Mast cells, leukocytes | Increased vascular permeability, chemotaxis, leukocyte adhesion and activation |
| Platelet-activating factor | Leukocytes, mast cells | Vasodilation, increased vascular permeability, leukocyte adhesion, chemotaxis, degranulation, oxidative burst |
| Reactive oxygen species | Leukocytes | Killing of microbes, tissue damage |
| Nitric oxide | Endothelium, macrophages | Vascular smooth muscle relaxation; killing of microbes |
| Cytokines (TNF, IL-1, IL-6) | Macrophages, endothelial cells, mast cells | Local: endothelial activation (expression of adhesion molecules). Systemic: fever, metabolic abnormalities, hypotension (shock) |
| Chemokines | Leukocytes, activated macrophages | Chemotaxis, leukocyte activation |
| Plasma Protein–Derived | ||
| Complement | Plasma (produced in liver) | Leukocyte chemotaxis and activation, direct target killing (MAC), vasodilation (mast cell stimulation) |
| Kinins | Plasma (produced in liver) | Increased vascular permeability, smooth muscle contraction, vasodilation, pain |
| Proteases activated during coagulation | Plasma (produced in liver) | Endothelial activation, leukocyte recruitment |
IL-1, IL-6, interleukin-1 and -6; MAC, membrane attack complex; TNF, tumor necrosis factor.
Cell-Derived Mediators
Tissue macrophages, mast cells, and endothelial cells at the site of inflammation, as well as leukocytes that are recruited to the site from the blood, are all capable of producing different mediators of inflammation.
Vasoactive Amines
The two vasoactive amines, histamine and serotonin, are stored as preformed molecules in mast cells and other cells and are among the first mediators to be released in acute inflammatory reactions.
• Histamine is produced by many cell types, particularly mast cells adjacent to vessels, as well as circulating basophils and platelets. Preformed histamine is released from mast cell granules in response to a variety of stimuli: (1) physical injury such as trauma or heat; (2) immune reactions involving binding of IgE antibodies to Fc receptors on mast cells (Chapter 4); (3) C3a and C5a fragments of complement, the so-called anaphylatoxins (see later); (4) leukocyte-derived histamine-releasing proteins; (5) neuropeptides (e.g., substance P); and (6) certain cytokines (e.g., IL-1, IL-8). In humans, histamine causes arteriolar dilation and rapidly increases vascular permeability by inducing venular endothelial contraction and formation of interendothelial gaps. Soon after its release, histamine is inactivated by histaminase.
• Serotonin (5-hydroxytryptamine) is a preformed vasoactive mediator found within platelet granules that is released during platelet aggregation (Chapter 3). It induces vasoconstriction during clotting. It is produced mainly in some neurons and enterochromaffin cells, and is a neurotransmitter and regulates intestinal motility.
Arachidonic Acid Metabolites: Prostaglandins, Leukotrienes, and Lipoxins
Products derived from the metabolism of AA affect a variety of biologic processes, including inflammation and hemostasis. AA metabolites, also called eicosanoids (because they are derived from 20-carbon fatty acids—Greek eicosa, “twenty”), can mediate virtually every step of inflammation (Table 2–6); their synthesis is increased at sites of inflammatory response, and agents that inhibit their synthesis also diminish inflammation. Leukocytes, mast cells, endothelial cells, and platelets are the major sources of AA metabolites in inflammation. These AA-derived mediators act locally at the site of generation and then decay spontaneously or are enzymatically destroyed.
Table 2–6 Principal Inflammatory Actions of Arachidonic Acid Metabolites (Eicosanoids)
| Action | Eicosanoid |
|---|---|
| Vasodilation | Prostaglandins PGI2 (prostacyclin), PGE1, PGE2, PGD2 |
| Vasoconstriction | Thromboxane A2, leukotrienes C4, D4, E4 |
| Increased vascular permeability | Leukotrienes C4, D4, E4 |
| Chemotaxis, leukocyte adhesion | Leukotriene B4, HETE |
HETE, hydroxyeicosatetraenoic acid.
AA is a 20-carbon polyunsaturated fatty acid (with four double bonds) produced primarily from dietary linoleic acid and present in the body mainly in its esterified form as a component of cell membrane phospholipids. It is released from these phospholipids through the action of cellular phospholipases that have been activated by mechanical, chemical, or physical stimuli, or by inflammatory mediators such as C5a. AA metabolism proceeds along one of two major enzymatic pathways: Cyclooxygenase stimulates the synthesis of prostaglandins and thromboxanes, and lipoxygenase is responsible for production of leukotrienes and lipoxins (Fig. 2–16).
• Prostaglandins and thromboxanes. Products of the cyclooxygenase pathway include prostaglandin E2 (PGE2), PGD2, PGF2α, PGI2 (prostacyclin), and thromboxane A2 (TXA2), each derived by the action of a specific enzyme on an intermediate. Some of these enzymes have a restricted tissue distribution. For example, platelets contain the enzyme thromboxane synthase, and hence TXA2, a potent platelet-aggregating agent and vasoconstrictor, is the major prostaglandin produced in these cells. Endothelial cells, on the other hand, lack thromboxane synthase but contain prostacyclin synthase, which is responsible for the formation of PGI2, a vasodilator and a potent inhibitor of platelet aggregation. The opposing roles of TXA2 and PGI2 in hemostasis are discussed further in Chapter 3. PGD2 is the major metabolite of the cyclooxygenase pathway in mast cells; along with PGE2 and PGF2α (which are more widely distributed), it causes vasodilation and potentiates edema formation. The prostaglandins also contribute to the pain and fever that accompany inflammation; PGE2 augments pain sensitivity to a variety of other stimuli and interacts with cytokines to cause fever.
• Leukotrienes. Leukotrienes are produced by the action of 5-lipoxygenase, the major AA-metabolizing enzyme in neutrophils. The synthesis of leukotrienes involves multiple steps (Fig. 2–16). The first step generates leukotriene A4 (LTA4), which in turn gives rise to LTB4 or LTC4. LTB4 is produced by neutrophils and some macrophages and is a potent chemotactic agent for neutrophils. LTC4 and its subsequent metabolites, LTD4 and LTE4, are produced mainly in mast cells and cause bronchoconstriction and increased vascular permeability.
• Lipoxins. Once leukocytes enter tissues, they gradually change their major lipoxygenase-derived AA products from leukotrienes to anti-inflammatory mediators called lipoxins, which inhibit neutrophil chemotaxis and adhesion to endothelium and thus serve as endogenous antagonists of leukotrienes. Platelets that are activated and adherent to leukocytes also are important sources of lipoxins. Platelets alone cannot synthesize lipoxins A4 and B4 (LXA4 and LXB4), but they can form these mediators from an intermediate derived from adjacent neutrophils, by a transcellular biosynthetic pathway. By this mechanism, AA products can pass from one cell type to another.
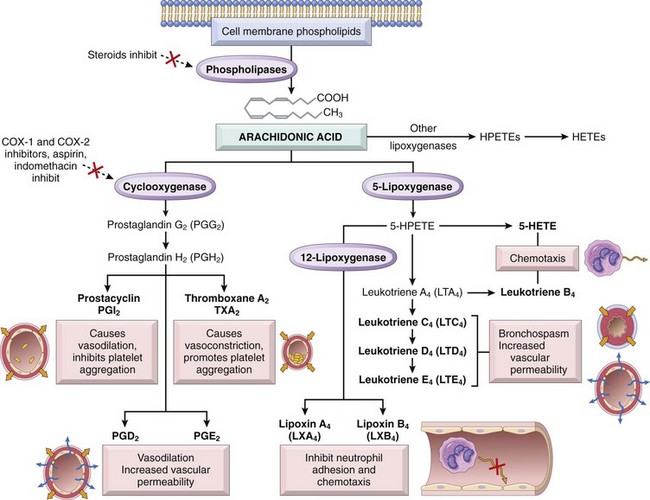
Figure 2–16 Production of arachidonic acid metabolites and their roles in inflammation. Note the enzymatic activities whose inhibition through pharmacologic intervention blocks major pathways (denoted with a red X). COX-1, COX-2, cyclooxygenases 1 and 2; HETE, hydroxyeicosatetraenoic acid; HPETE, hydroperoxyeicosatetraenoic acid.
Anti-inflammatory Drugs That Block Prostaglandin Production
The central role of eicosanoids in inflammatory processes is emphasized by the clinical utility of agents that block eicosanoid synthesis. Nonsteroidal anti-inflammatory drugs (NSAIDs), such as aspirin and ibuprofen, inhibit cyclooxygenase activity, thereby blocking all prostaglandin synthesis (hence their efficacy in treating pain and fever). There are two forms of the cyclooxygenase enzyme, COX-1 and COX-2. COX-1 is produced in response to inflammatory stimuli and also is constitutively expressed in most tissues, where it stimulates the production of prostaglandins that serve a homeostatic function (e.g., fluid and electrolyte balance in the kidneys, cytoprotection in the gastrointestinal tract). By contrast, COX-2 is induced by inflammatory stimuli but it is absent from most normal tissues. Therefore, COX-2 inhibitors have been developed with the expectation that they will inhibit harmful inflammation but will not block the protective effects of constitutively produced prostaglandins. These distinctions between the roles of the two cyclooxygenases are not absolute, however. Furthermore, COX-2 inhibitors may increase the risk for cardiovascular and cerebrovascular events, possibly because they impair endothelial cell production of prostacyclin (PGI2), an inhibitor of platelet aggregation, but leave intact the COX-1–mediated production by platelets of TXA2, a mediator of platelet aggregation. Glucocorticoids, which are powerful anti-inflammatory agents, act in part by inhibiting the activity of phospholipase A2 and thus the release of AA from membrane lipids.
Platelet-Activating Factor
Originally named for its ability to aggregate platelets and cause their degranulation, platelet-activating factor (PAF) is another phospholipid-derived mediator with a broad spectrum of inflammatory effects. PAF is acetyl glycerol ether phosphocholine; it is generated from the membrane phospholipids of neutrophils, monocytes, basophils, endothelial cells, and platelets (and other cells) by the action of phospholipase A2. PAF acts directly on target cells through the effects of a specific G protein–coupled receptor. In addition to stimulating platelets, PAF causes bronchoconstriction and is 100 to 1000 times more potent than histamine in inducing vasodilation and increased vascular permeability. It also stimulates the synthesis of other mediators, such as eicosanoids and cytokines, from platelets and other cells. Thus, PAF can elicit many of the reactions of inflammation, including enhanced leukocyte adhesion, chemotaxis, leukocyte degranulation, and the respiratory burst.
Cytokines
Cytokines are polypeptide products of many cell types that function as mediators of inflammation and immune responses (Chapter 4). Different cytokines are involved in the earliest immune and inflammatory reactions to noxious stimuli and in the later adaptive (specific) immune responses to microbes. Some cytokines stimulate bone marrow precursors to produce more leukocytes, thus replacing the ones that are consumed during inflammation and immune responses. Molecularly characterized cytokines are called interleukins (abbreviated IL and numbered), referring to their ability to mediate communications between leukocytes. However, the nomenclature is imperfect—many interleukins act on cells other than leukocytes, and many cytokines that do act on leukocytes are not called interleukins, for historical reasons.
The major cytokines in acute inflammation are TNF, IL-1, IL-6, and a group of chemoattractant cytokines called chemokines. Other cytokines that are more important in chronic inflammation include interferon-γ (IFN-γ) and IL-12. A cytokine called IL-17, produced by T lymphocytes and other cells, plays an important role in recruiting neutrophils and is involved in host defense against infections and in inflammatory diseases.
Tumor Necrosis Factor and Interleukin-1
TNF and IL-1 are produced by activated macrophages, as well as mast cells, endothelial cells, and some other cell types (Fig. 2–17). Their secretion is stimulated by microbial products, such as bacterial endotoxin, immune complexes, and products of T lymphocytes generated during adaptive immune responses. As mentioned earlier, IL-1 is also the cytokine induced by activation of the inflammasome. The principal role of these cytokines in inflammation is in endothelial activation. Both TNF and IL-1 stimulate the expression of adhesion molecules on endothelial cells, resulting in increased leukocyte binding and recruitment, and enhance the production of additional cytokines (notably chemokines) and eicosanoids. TNF also increases the thrombogenicity of endothelium. IL-1 activates tissue fibroblasts, resulting in increased proliferation and production of ECM.
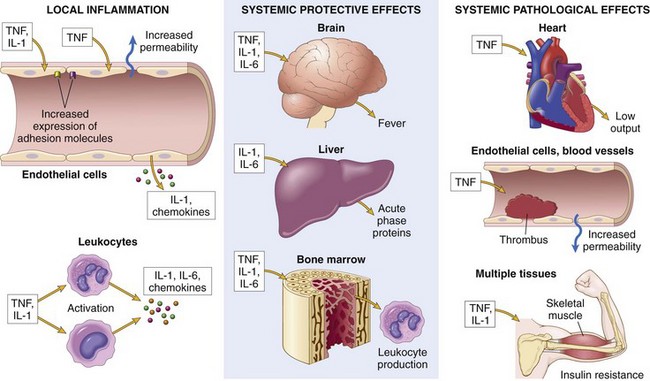
Figure 2–17 The roles of cytokines in acute inflammation. The cytokines TNF, IL-1, and IL-6 are key mediators of leukocyte recruitment in local inflammatory responses and also play important roles in the systemic reactions of inflammation.
Although TNF and IL-1 are secreted by macrophages and other cells at sites of inflammation, they may enter the circulation and act at distant sites to induce the systemic acute-phase reaction that is often associated with infection and inflammatory diseases. Components of this reaction include fever, lethargy, hepatic synthesis of various acute-phase proteins (also stimulated by IL-6), metabolic wasting (cachexia), neutrophil release into the circulation, and fall in blood pressure. These systemic manifestations of inflammation are described later in the chapter.
Chemokines
The chemokines are a family of small (8 to 10 kDa), structurally related proteins that act primarily as chemoattractants for different subsets of leukocytes. The two main functions of chemokines are to recruit leukocytes to the site of inflammation and to control the normal anatomic organization of cells in lymphoid and other tissues. Combinations of chemokines that are produced transiently in response to inflammatory stimuli recruit particular cell populations (e.g., neutrophils, lymphocytes or eosinophils) to sites of inflammation. Chemokines also activate leukocytes; one consequence of such activation, as mentioned earlier, is increased affinity of leukocyte integrins for their ligands on endothelial cells. Some chemokines are produced constitutively in tissues and are responsible for the anatomic segregation of different cell populations in tissues (e.g., the segregation of T and B lymphocytes in different areas of lymph nodes and spleen). Chemokines mediate their activities by binding to specific G protein–coupled receptors on target cells; two of these chemokine receptors (called CXCR4 and CCR5) are important coreceptors for the binding and entry of the human immunodeficiency virus into lymphocytes (Chapter 4).
Chemokines are classified into four groups based on the arrangement of conserved cysteine residues. The two major groups are the CXC and CC chemokines:
• CXC chemokines have one amino acid separating the conserved cysteines and act primarily on neutrophils. IL-8 is typical of this group; it is produced by activated macrophages, endothelial cells, mast cells, and fibroblasts, mainly in response to microbial products and other cytokines such as IL-1 and TNF.
• CC chemokines have adjacent cysteine residues and include monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1α (MIP-1α) (both chemotactic predominantly for monocytes), RANTES (regulated on activation, normal T cell–expressed and secreted) (chemotactic for memory CD4+ T cells and monocytes), and eotaxin (chemotactic for eosinophils).
Reactive Oxygen Species
ROS are synthesized via the NADPH oxidase (phagocyte oxidase) pathway and are released from neutrophils and macrophages that are activated by microbes, immune complexes, cytokines, and a variety of other inflammatory stimuli. The synthesis and regulation of these oxygen-derived free radicals have been described in Chapter 1, in the context of cell injury, and earlier in this chapter in the discussion of leukocyte activation. When the ROS are produced within lysosomes they function to destroy phagocytosed microbes and necrotic cells. When secreted at low levels, ROS can increase chemokine, cytokine, and adhesion molecule expression, thus amplifying the cascade of inflammatory mediators. At higher levels, these mediators are responsible for tissue injury by several mechanisms, including (1) endothelial damage, with thrombosis and increased permeability; (2) protease activation and antiprotease inactivation, with a net increase in breakdown of the ECM; and (3) direct injury to other cell types (e.g., tumor cells, red cells, parenchymal cells). Fortunately, various antioxidant protective mechanisms (e.g., mediated by catalase, superoxide dismutase, and glutathione) present in tissues and blood minimize the toxicity of the oxygen metabolites (Chapter 1).
Nitric Oxide
NO is a short-lived, soluble, free radical gas produced by many cell types and capable of mediating a variety of functions. In the central nervous system it regulates neurotransmitter release as well as blood flow. Macrophages use it as a cytotoxic agent for killing microbes and tumor cells. When produced by endothelial cells it relaxes vascular smooth muscle and causes vasodilation.
NO is synthesized de novo from l-arginine, molecular oxygen, and NADPH by the enzyme nitric oxide synthase (NOS). There are three isoforms of NOS, with different tissue distributions.
• Type I, neuronal NOS (nNOS), is constitutively expressed in neurons, and does not play a significant role in inflammation.
• Type II, inducible NOS (iNOS), is induced in macrophages and endothelial cells by a number of inflammatory cytokines and mediators, most notably by IL-1, TNF, and IFN-γ, and by bacterial endotoxin, and is responsible for production of NO in inflammatory reactions. This inducible form is also present in many other cell types, including hepatocytes, cardiac myocytes, and respiratory epithelial cells.
• Type III, endothelial NOS, (eNOS), is constitutively synthesized primarily (but not exclusively) in endothelium.
An important function of NO is as a microbicidal (cytotoxic) agent in activated macrophages. NO plays other roles in inflammation, including vasodilation, antagonism of all stages of platelet activation (adhesion, aggregation, and degranulation), and reduction of leukocyte recruitment at inflammatory sites.
Lysosomal Enzymes of Leukocytes
The lysosomal granules of neutrophils and monocytes contain many enzymes that destroy phagocytosed substances and are capable of causing tissue damage. Lysosomal granule contents also may be released from activated leukocytes, as described earlier. Acid proteases generally are active only in the low-pH environment of phagolysosomes, whereas neutral proteases, including elastase, collagenase, and cathepsin, are active in extracellular locations and cause tissue injury by degrading elastin, collagen, basement membrane, and other matrix proteins. Neutral proteases also can cleave the complement proteins C3 and C5 directly to generate the vasoactive mediators C3a and C5a and can generate bradykinin-like peptides from kininogen.
The potentially damaging effects of lysosomal enzymes are limited by antiproteases present in the plasma and tissue fluids. These include α1-antitrypsin, the major inhibitor of neutrophil elastase, and α2-macroglobulin. Deficiencies of these inhibitors may result in sustained activation of leukocyte proteases, resulting in tissue destruction at sites of leukocyte accumulation. For instance, α1-antitrypsin deficiency in the lung can cause a severe panacinar emphysema (Chapter 12).
Neuropeptides
Like the vasoactive amines, neuropeptides can initiate inflammatory responses; these are small proteins, such as substance P, that transmit pain signals, regulate vessel tone, and modulate vascular permeability. Nerve fibers that secrete neuropeptides are especially prominent in the lung and gastrointestinal tract.
Summary
Major Cell-Derived Mediators of Inflammation
• Vasoactive amines—histamine, serotonin: Their main effects are vasodilation and increased vascular permeability.
• Arachidonic acid metabolites—prostaglandins and leukotrienes: Several forms exist and are involved in vascular reactions, leukocyte chemotaxis, and other reactions of inflammation; they are antagonized by lipoxins.
• Cytokines: These proteins, produced by many cell types, usually act at short range; they mediate multiple effects, mainly in leukocyte recruitment and migration; principal ones in acute inflammation are TNF, IL-1, IL-6, and chemokines.
• ROS: Roles include microbial killing and tissue injury.
• NO: Effects are vasodilation and microbial killing.
• Lysosomal enzymes: Roles include microbial killing and tissue injury.
Plasma Protein–Derived Mediators
Circulating proteins of three interrelated systems—the complement, kinin, and coagulation systems—are involved in several aspects of the inflammatory reaction.
Complement
The complement system consists of plasma proteins that play an important role in host defense (immunity) and inflammation. Upon activation, different complement proteins coat (opsonize) particles, such as microbes, for phagocytosis and destruction, and contribute to the inflammatory response by increasing vascular permeability and leukocyte chemotaxis. Complement activation ultimately generates a porelike membrane attack complex (MAC) that punches holes in the membranes of invading microbes. Here we summarize the role of the complement system in inflammation.
• Complement components, numbered C1 to C9, are present in plasma in inactive forms, and many of them are activated by proteolysis to acquire their own proteolytic activity, thus setting up an enzymatic cascade.
• The critical step in the generation of biologically active complement products is the activation of the third component, C3 (Fig. 2–18). C3 cleavage occurs by three pathways: (1) the classical pathway, triggered by fixation of the first complement component C1 to antigen-antibody complexes; (2) the alternative pathway, triggered by bacterial polysaccharides (e.g., endotoxin) and other microbial cell wall components, and involving a distinct set of plasma proteins including properdin and factors B and D; and (3) the lectin pathway, in which a plasma lectin binds to mannose residues on microbes and activates an early component of the classical pathway (but in the absence of antibodies).
• All three pathways lead to the formation of a C3 convertase that cleaves C3 to C3a and C3b. C3b deposits on the cell or microbial surface where complement was activated and then binds to the C3 convertase complex to form C5 convertase; this complex cleaves C5 to generate C5a and C5b and initiate the final stages of assembly of C6 to C9.
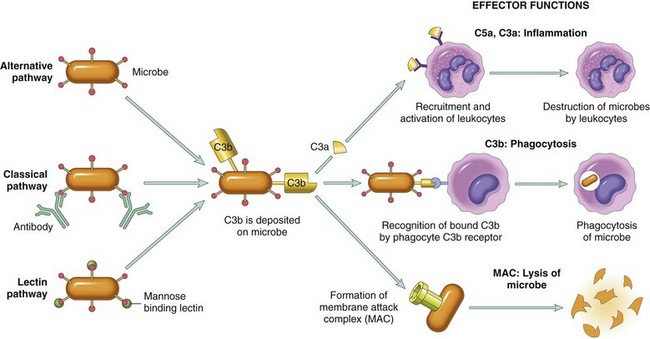
Figure 2–18 The activation and functions of the complement system. Activation of complement by different pathways leads to cleavage of C3. The functions of the complement system are mediated by breakdown products of C3 and other complement proteins, and by the membrane attack complex (MAC).
The complement-derived factors that are produced along the way contribute to a variety of phenomena in acute inflammation:
• Vascular effects. C3a and C5a increase vascular permeability and cause vasodilation by inducing mast cells to release histamine. These complement products are also called anaphylatoxins because their actions mimic those of mast cells, which are the main cellular effectors of the severe allergic reaction called anaphylaxis (Chapter 4). C5a also activates the lipoxygenase pathway of AA metabolism in neutrophils and macrophages, causing release of more inflammatory mediators.
• Leukocyte activation, adhesion, and chemotaxis. C5a, and to lesser extent, C3a and C4a, activate leukocytes, increasing their adhesion to endothelium, and is a potent chemotactic agent for neutrophils, monocytes, eosinophils, and basophils.
• Phagocytosis. When fixed to a microbial surface, C3b and its inactive proteolytic product iC3b act as opsonins, augmenting phagocytosis by neutrophils and macrophages, which express receptors for these complement products.
• The MAC, which is made up of multiple copies of the final component C9, kills some bacteria (especially thin-walled Neisseria) by creating pores that disrupt osmotic balance.
The activation of complement is tightly controlled by cell-associated and circulating regulatory proteins. The presence of these inhibitors in host cell membranes protects normal cells from inappropriate damage during protective reactions against microbes. Inherited deficiencies of these regulatory proteins lead to spontaneous complement activation:
• A protein called C1 inhibitor blocks activation of C1, and its inherited deficiency causes a disease called hereditary angioedema, in which excessive production of kinins secondary to complement activation results in edema in multiple tissues, including the larynx.
• Another protein called decay-accelerating factor (DAF) normally limits the formation of C3 and C5 convertases. In a disease called paroxysmal nocturnal hemoglobinuria, there is an acquired deficiency of DAF that results in complement-mediated lysis of red cells (which are more sensitive to lysis than most nucleated cells) (Chapter 11).
• Factor H is a plasma protein that also limits convertase formation; its deficiency is associated with a kidney disease called the hemolytic uremic syndrome (Chapter 13), as well as spontaneous vascular permeability in macular degeneration of the eye.
Even in the presence of regulatory proteins, inappropriate or excessive complement activation (e.g., in antibody-mediated diseases) can overwhelm the regulatory mechanisms; this is why complement activation is responsible for serious tissue injury in a variety of immunologic disorders (Chapter 4).
Coagulation and Kinin Systems
Some of the molecules activated during blood clotting are capable of triggering multiple aspects of the inflammatory response. Hageman factor (also known as factor XII of the intrinsic coagulation cascade) (Fig. 2–19) is a protein synthesized by the liver that circulates in an inactive form until it encounters collagen, basement membrane, or activated platelets (e.g., at a site of endothelial injury). Activated Hageman factor (factor XIIa) initiates four systems that may contribute to the inflammatory response: (1) the kinin system, producing vasoactive kinins; (2) the clotting system, inducing the activation of thrombin, fibrinopeptides, and factor X, all with inflammatory properties; (3) the fibrinolytic system, producing plasmin and inactivating thrombin; and (4) the complement system, producing the anaphylatoxins C3a and C5a. These are described below.
• Kinin system activation leads ultimately to the formation of bradykinin from its circulating precursor, high-molecular-weight kininogen (HMWK) (Fig. 2–19). Like histamine, bradykinin causes increased vascular permeability, arteriolar dilation, and bronchial smooth muscle contraction. It also causes pain when injected into the skin. The actions of bradykinin are short-lived because it is rapidly degraded by kininases present in plasma and tissues. Of note, kallikrein, an intermediate in the kinin cascade with chemotactic activity, also is a potent activator of Hageman factor and thus constitutes another link between the kinin and clotting systems.
• In the clotting system (Chapter 3), the proteolytic cascade leads to activation of thrombin, which then cleaves circulating soluble fibrinogen to generate an insoluble fibrin clot. Factor Xa, an intermediate in the clotting cascade, causes increased vascular permeability and leukocyte emigration. Thrombin participates in inflammation by binding to protease-activated receptors that are expressed on platelets, endothelial cells, and many other cell types. Binding of thrombin to these receptors on endothelial cells leads to their activation and enhanced leukocyte adhesion. In addition, thrombin generates fibrinopeptides (during fibrinogen cleavage) that increase vascular permeability and are chemotactic for leukocytes. Thrombin also cleaves C5 to generate C5a, thus linking coagulation with complement activation.
• As a rule, whenever clotting is initiated (e.g., by activated Hageman factor), the fibrinolytic system is also activated concurrently. This mechanism serves to limit clotting by cleaving fibrin, thereby solubilizing the fibrin clot (Chapter 3). Plasminogen activator (released from endothelium, leukocytes, and other tissues) and kallikrein cleave plasminogen, a plasma protein bound up in the evolving fibrin clot. The resulting product, plasmin, is a multifunctional protease that cleaves fibrin and is therefore important in lysing clots. However, fibrinolysis also participates in multiple steps in the vascular phenomena of inflammation. For example, fibrin degradation products increase vascular permeability, and plasmin cleaves the C3 complement protein, resulting in production of C3a and vasodilation and increased vascular permeability. Plasmin can also activate Hageman factor, thereby amplifying the entire set of responses.
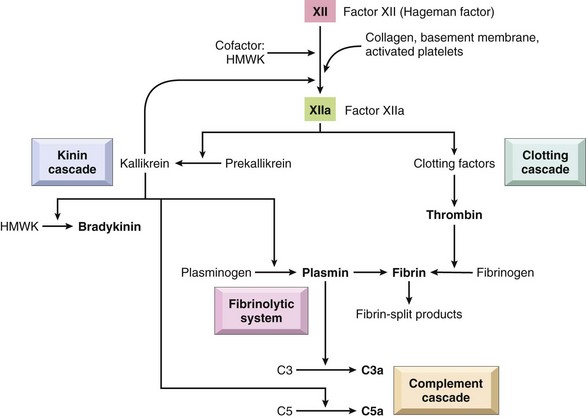
Figure 2–19 Interrelationships among the four plasma mediator systems triggered by activation of factor XII (Hageman factor). See text for details.
As is evident from the preceding discussion, many molecules are involved in different aspects of the inflammatory reaction, and these molecules often interact with, amplify, and antagonize one another. From this almost bewildering potpourri of chemical mediators, it is possible to identify the major contributors to various components of acute inflammation (Table 2–7). The relative contributions of individual mediators to inflammatory reactions to different stimuli have yet to be fully elucidated. Such knowledge would have obvious therapeutic implications since it might allow one to “custom design” antagonists for various inflammatory diseases.
Table 2–7 Role of Mediators in Different Reactions of Inflammation
| Inflammatory Component | Mediators |
|---|---|
| Vasodilation | |
| Increased vascular permeability | |
| Chemotaxis, leukocyte recruitment and activation | |
| Fever | |
| Pain | |
| Tissue damage |
IL-1, interleukin-1; PAF, platelet-activating factor; TNF, tumor necrosis factor.
Summary
Plasma Protein–Derived Mediators of Inflammation
• Complement proteins: Activation of the complement system by microbes or antibodies leads to the generation of multiple breakdown products, which are responsible for leukocyte chemotaxis, opsonization and phagocytosis of microbes and other particles, and cell killing.
• Coagulation proteins: Activated factor XII triggers the clotting, kinin, and complement cascades and activates the fibrinolytic system.
• Kinins: Produced by proteolytic cleavage of precursors, this group mediates vascular reaction and pain.
Anti-inflammatory Mechanisms
Inflammatory reactions subside because many of the mediators are short-lived and are destroyed by degradative enzymes. In addition, there are several mechanisms that counteract inflammatory mediators and function to limit or terminate the inflammatory response. Some of these, such as lipoxins, and complement regulatory proteins, have been mentioned earlier. Activated macrophages and other cells secrete a cytokine, IL-10, whose major function is to down-regulate the responses of activated macrophages, thus providing a negative feedback loop. In a rare inherited disease in which IL-10 receptors are mutated, affected patients develop severe colitis in infancy. Other anti-inflammatory cytokines include TGF-β, which is also a mediator of fibrosis in tissue repair after inflammation. Cells also express a number of intracellular proteins, such as tyrosine phosphatases, that inhibit pro-inflammatory signals triggered by receptors that recognize microbes and cytokines.
Chronic Inflammation
Chronic inflammation is inflammation of prolonged duration (weeks to years) in which continuing inflammation, tissue injury, and healing, often by fibrosis, proceed simultaneously. In contrast with acute inflammation, which is distinguished by vascular changes, edema, and a predominantly neutrophilic infiltrate, chronic inflammation is characterized by a different set of reactions (Fig. 2–20; see also Table 2–1):
• Infiltration with mononuclear cells, including macrophages, lymphocytes, and plasma cells
• Tissue destruction, largely induced by the products of the inflammatory cells
• Repair, involving new vessel proliferation (angiogenesis) and fibrosis
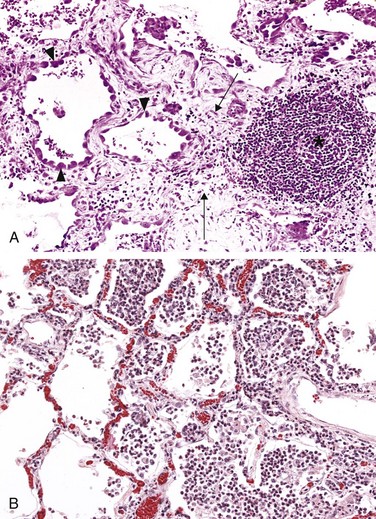
Figure 2–20 A, Chronic inflammation in the lung, showing the characteristic histologic features: collection of chronic inflammatory cells (asterisk); destruction of parenchyma, in which normal alveoli are replaced by spaces lined by cuboidal epithelium (arrowheads); and replacement by connective tissue, resulting in fibrosis (arrows). B, By contrast, in acute inflammation of the lung (acute bronchopneumonia), neutrophils fill the alveolar spaces and blood vessels are congested.
Acute inflammation may progress to chronic inflammation if the acute response cannot be resolved, either because of the persistence of the injurious agent or because of interference with the normal process of healing. For example, a peptic ulcer of the duodenum initially shows acute inflammation followed by the beginning stages of resolution. However, recurrent bouts of duodenal epithelial injury interrupt this process, resulting in a lesion characterized by both acute and chronic inflammation (Chapter 14). Alternatively, some forms of injury (e.g., immunologic reactions, some viral infections) engender a chronic inflammatory response from the outset.
Chronic inflammation may arise in the following settings:
• Persistent infections by microbes that are difficult to eradicate. These include Mycobacterium tuberculosis, Treponema pallidum (the causative organism of syphilis), and certain viruses and fungi, all of which tend to establish persistent infections and elicit a T lymphocyte–mediated immune response called delayed-type hypersensitivity (Chapter 4).
• Immune-mediated inflammatory diseases (hypersensitivity diseases). Diseases that are caused by excessive and inappropriate activation of the immune system are increasingly recognized as being important health problems (Chapter 4). Under certain conditions, immune reactions develop against the affected person’s own tissues, leading to autoimmune diseases. In such diseases, autoantigens evoke a self-perpetuating immune reaction that results in tissue damage and persistent inflammation. Autoimmunity plays an important role in several common and debilitating chronic inflammatory diseases, such as rheumatoid arthritis, inflammatory bowel disease, and psoriasis. Immune responses against common environmental substances are the cause of allergic diseases, such as bronchial asthma. Immune-mediated diseases may show morphologic patterns of mixed acute and chronic inflammation because they are characterized by repeated bouts of inflammation. Since, in most cases, the eliciting antigens cannot be eliminated, these disorders tend to be chronic and intractable.
• Prolonged exposure to potentially toxic agents. Examples are nondegradable exogenous materials such as inhaled particulate silica, which can induce a chronic inflammatory response in the lungs (silicosis, Chapter 12), and endogenous agents such as cholesterol crystals, which may contribute to atherosclerosis (Chapter 9).
• Mild forms of chronic inflammation may be important in the pathogenesis of many diseases that are not conventionally thought of as inflammatory disorders. Such diseases include neurodegenerative disorders such as Alzheimer disease, atherosclerosis, metabolic syndrome and the associated type 2 diabetes, and some forms of cancer in which inflammatory reactions promote tumor development. As mentioned earlier in the chapter, in many of these conditions the inflammation may be triggered by recognition of the initiating stimuli by the inflammasome. The role of inflammation in these conditions is discussed in the relevant chapters.
Chronic Inflammatory Cells and Mediators
The combination of prolonged and repeated inflammation, tissue destruction and fibrosis that characterizes chronic inflammation involves complex interactions between several cell populations and their secreted mediators. Understanding the pathogenesis of chronic inflammatory reactions requires an appreciation of these cells and their biologic responses and functions.
Macrophages
Macrophages, the dominant cells of chronic inflammation, are tissue cells derived from circulating blood monocytes after their emigration from the bloodstream. Macrophages are normally diffusely scattered in most connective tissues and are also found in organs such as the liver (where they are called Kupffer cells), spleen and lymph nodes (where they are called sinus histiocytes), central nervous system (microglial cells), and lungs (alveolar macrophages). Together these cells constitute the so-called mononuclear phagocyte system, also known by the older name of reticuloendothelial system. In all tissues, macrophages act as filters for particulate matter, microbes, and senescent cells, as well as the effector cells that eliminate microbes in cellular and humoral immune responses (Chapter 4).
Monocytes arise from precursors in the bone marrow and circulate in the blood for only about a day. Under the influence of adhesion molecules and chemokines, they migrate to a site of injury within 24 to 48 hours after the onset of acute inflammation, as described earlier. When monocytes reach the extravascular tissue, they undergo transformation into macrophages, which are somewhat larger and have a longer lifespan and a greater capacity for phagocytosis than do blood monocytes.
Tissue macrophages are activated by diverse stimuli to perform a range of functions. Two major pathways of macrophage activation, classical and alternative, have been described (Fig. 2–21):
• Classical macrophage activation is induced by microbial products such as endotoxin, by T cell–derived signals, importantly the cytokine IFN-γ, and by foreign substances including crystals and particulate matter. Classically activated macrophages produce lysosomal enzymes, NO, and ROS, all of which enhance their ability to kill ingested organisms, and secrete cytokines that stimulate inflammation. These macrophages are important in host defense against ingested microbes and in many chronic inflammatory reactions.
• Alternative macrophage activation is induced by cytokines other than IFN-γ, such as IL-4 and IL-13, produced by T lymphocytes and other cells, including mast cells and eosinophils. Alternatively activated macrophages are not actively microbicidal; instead, their principal role is in tissue repair. They secrete growth factors that promote angiogenesis, activate fibroblasts and stimulate collagen synthesis. It may be that in response to most injurious stimuli, macrophages are initially activated by the classical pathway, designed to destroy the offending agents, and this is followed by alternative activation, which initiates tissue repair. However, such a precise sequence is not well documented in most inflammatory reactions.
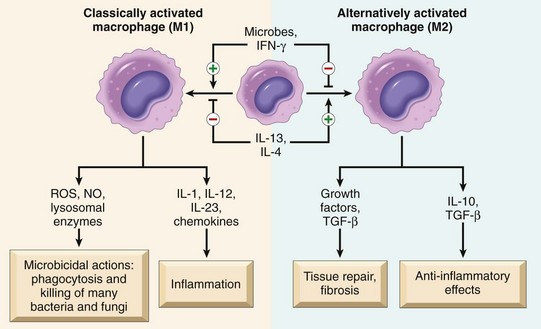
Figure 2–21 Pathways of macrophage activation. Different stimuli activate monocytes/macrophages to develop into functionally distinct populations. Classically activated macrophages are induced by microbial products and cytokines, particularly IFN-γ, and are microbicidal and involved in potentially harmful inflammation. Alternatively activated macrophages are induced by IL-4 and IL-13, produced by TH2 cells (a helper T cell subset) and other leukocytes, and are important in tissue repair and fibrosis. IFN-γ, interferon-γ; IL-4, IL-13, interkeukin-4, -13.
Macrophages have several critical roles in host defense and the inflammatory response.
• Macrophages, like the other type of phagocyte, the neutrophils, ingest and eliminate microbes and dead tissues. Because macrophages respond to activating signals from T lymphocytes, they are the most important phagocytes in the cell-mediated arm of adaptive immune responses (Chapter 4).
• Macrophages initiate the process of tissue repair and are involved in scar formation and fibrosis.
• Macrophages secrete mediators of inflammation, such as cytokines (TNF, IL-1, chemokines, and others) and eicosanoids. These cells are therefore central to the initiation and propagation of all inflammatory reactions.
• Macrophages display antigens to T lymphocytes and respond to signals from T cells, thus setting up a feedback loop that is essential for defense against many microbes by cell-mediated immune responses. The same bidirectional interactions are central to the development of chronic inflammatory diseases. The roles of cytokines in these interactions are discussed later.
After the initiating stimulus is eliminated and the inflammatory reaction abates, macrophages eventually die or wander off into lymphatics. In chronic inflammatory sites, however, macrophage accumulation persists, because of continued recruitment from the blood and local proliferation. IFN-γ can also induce macrophages to fuse into large, multinucleate giant cells.
Lymphocytes
Lymphocytes are mobilized in the setting of any specific immune stimulus (i.e., infections) as well as non–immune-mediated inflammation (e.g., due to ischemic necrosis or trauma), and are the major drivers of inflammation in many autoimmune and other chronic inflammatory diseases. The activation of T and B lymphocytes is part of the adaptive immune response in infections and immunologic diseases (Chapter 4). Both classes of lymphocytes migrate into inflammatory sites using some of the same adhesion molecule pairs and chemokines that recruit other leukocytes. In the tissues, B lymphocytes may develop into plasma cells, which secrete antibodies, and CD4+ T lymphocytes are activated to secrete cytokines.
By virtue of cytokine secretion, CD4+ T lymphocytes promote inflammation and influence the nature of the inflammatory reaction. There are three subsets of CD4+ helper T cells that secrete different sets of cytokines and elicit different types of inflammation:
• TH1 cells produce the cytokine IFN-γ, which activates macrophages in the classical pathway.
• TH2 cells secrete IL-4, IL-5, and IL-13, which recruit and activate eosinophils and are responsible for the alternative pathway of macrophage activation.
• TH17 cells secrete IL-17 and other cytokines that induce the secretion of chemokines responsible for recruiting neutrophils and monocytes into the reaction.
Both TH1 and TH17 cells are involved in defense against many types of bacteria and viruses and in autoimmune diseases. TH2 cells are important in defense against helminthic parasites and in allergic inflammation. These T cell subsets and their functions are described in more detail in Chapter 4.
Lymphocytes and macrophages interact in a bidirectional way, and these interactions play an important role in propagating chronic inflammation (Fig. 2–22). Macrophages display antigens to T cells, express membrane molecules (called costimulators), and produce cytokines (IL-12 and others) that stimulate T cell responses (Chapter 4). Activated T lymphocytes, in turn, produce cytokines, described earlier, which recruit and activate macrophages and thus promote more antigen presentation and cytokine secretion. The result is a cycle of cellular reactions that fuel and sustain chronic inflammation. In some strong and prolonged inflammatory reactions, the accumulation of lymphocytes, antigen-presenting cells, and plasma cells may assume the morphologic features of lymphoid organs, and akin to lymph nodes, may even contain well-formed germinal centers. This pattern of lymphoid organogenesis is often seen in the synovium of patients with long-standing rheumatoid arthritis and the thyroid of patients with autoimmune thyroiditis.
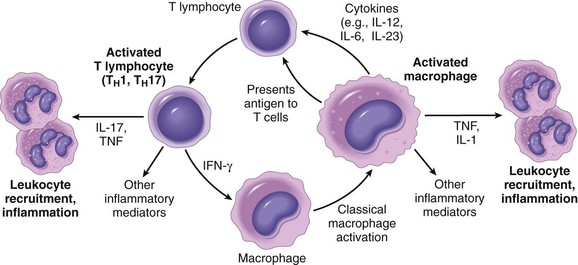
Other Cells
Eosinophils are characteristically found in inflammatory sites around parasitic infections and as part of immune reactions mediated by IgE, typically associated with allergies. Their recruitment is driven by adhesion molecules similar to those used by neutrophils, and by specific chemokines (e.g., eotaxin) derived from leukocytes and epithelial cells. Eosinophil granules contain major basic protein, a highly charged cationic protein that is toxic to parasites but also causes epithelial cell necrosis.
Mast cells are sentinel cells widely distributed in connective tissues throughout the body, and they can participate in both acute and chronic inflammatory responses. In atopic persons (those prone to allergic reactions), mast cells are “armed” with IgE antibody specific for certain environmental antigens. When these antigens are subsequently encountered, the IgE-coated mast cells are triggered to release histamines and AA metabolites that elicit the early vascular changes of acute inflammation. IgE-armed mast cells are central players in allergic reactions, including anaphylactic shock (Chapter 4). Mast cells can also elaborate cytokines such as TNF and chemokines and may play a beneficial role in combating some infections.
An important final point: Although the presence of neutrophils is the hallmark of acute inflammation, many forms of chronic inflammation may continue to show extensive neutrophilic infiltrates, as a result of either persistent microbes or necrotic cells, or mediators elaborated by macrophages. Such inflammatory lesions are sometimes called “acute on chronic”—for example, in inflammation of bones (osteomyelitis).
Granulomatous Inflammation
Granulomatous inflammation is a distinctive pattern of chronic inflammation characterized by aggregates of activated macrophages with scattered lymphocytes. Granulomas are characteristic of certain specific pathologic states; consequently, recognition of the granulomatous pattern is important because of the limited number of conditions (some life-threatening) that cause it (Table 2–8). Granulomas can form under three settings:
• With persistent T-cell responses to certain microbes (such as Mycobacterium tuberculosis, T. pallidum, or fungi), in which T cell–derived cytokines are responsible for chronic macrophage activation. Tuberculosis is the prototype of a granulomatous disease caused by infection and should always be excluded as the cause when granulomas are identified.
• Granulomas may also develop in some immune-mediated inflammatory diseases, notably Crohn disease, which is one type of inflammatory bowel disease and an important cause of granulomatous inflammation in the United States.
• They are also seen in a disease of unknown etiology called sarcoidosis, and they develop in response to relatively inert foreign bodies (e.g., suture or splinter), forming so-called foreign body granulomas.
Table 2–8 Examples of Diseases with Granulomatous Inflammation
| Disease | Cause | Tissue Reaction |
|---|---|---|
| Tuberculosis | Mycobacterium tuberculosis | Caseating granuloma (tubercle): focus of activated macrophages (epithelioid cells), rimmed by fibroblasts, lymphocytes, histiocytes, occasional Langhans giant cells; central necrosis with amorphous granular debris; acid-fast bacilli |
| Leprosy | Mycobacterium leprae | Acid-fast bacilli in macrophages; noncaseating granulomas |
| Syphilis | Treponema pallidum | Gumma: microscopic to grossly visible lesion, enclosing wall of histiocytes; plasma cell infiltrate; central cells are necrotic without loss of cellular outline |
| Cat-scratch disease | Gram-negative bacillus | Rounded or stellate granuloma containing central granular debris and neutrophils; giant cells uncommon |
| Sarcoidosis | Unknown etiology | Noncaseating granulomas with abundant activated macrophages |
| Crohn disease | Immune reaction against intestinal bacteria, self antigens | Occasional noncaseating granulomas in the wall of the intestine, with dense chronic inflammatory infiltrate |
The formation of a granuloma effectively “walls off” the offending agent and is therefore a useful defense mechanism. However, granuloma formation does not always lead to eradication of the causal agent, which is frequently resistant to killing or degradation, and granulomatous inflammation with subsequent fibrosis may even be the major cause of organ dysfunction in some diseases, such as tuberculosis.
Morphology
In the usual H&E preparations (Fig. 2–23), some of the activated macrophages in granulomas have pink, granular cytoplasm with indistinct cell boundaries; these are called epithelioid cells because of their resemblance to epithelia. Typically, the aggregates of epithelioid macrophages are surrounded by a collar of lymphocytes. Older granulomas may have a rim of fibroblasts and connective tissue. Frequently, but not invariably, multinucleate giant cells 40 to 50 μm in diameter are found in granulomas. Such cells consist of a large mass of cytoplasm and many nuclei, and they derive from the fusion of multiple activated macrophages. In granulomas associated with certain infectious organisms (most classically the tubercle bacillus), a combination of hypoxia and free radical injury leads to a central zone of necrosis. On gross examination, this has a granular, cheesy appearance and is therefore called caseous necrosis (Chapters 1 and 13). On microscopic examination, this necrotic material appears as eosinophilic amorphous, structureless, granular debris, with complete loss of cellular details. The granulomas associated with Crohn disease, sarcoidosis, and foreign body reactions tend to not have necrotic centers and are said to be “noncaseating.” Healing of granulomas is accompanied by fibrosis that may be quite extensive.
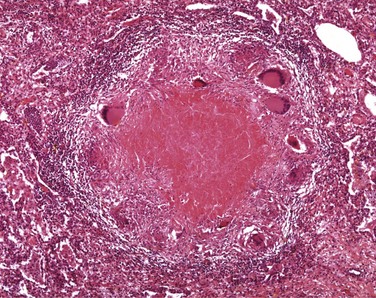
Summary
Features of Chronic Inflammation
• Prolonged host response to persistent stimulus
• Caused by microbes that resist elimination, immune responses against self and environmental antigens, and some toxic substances (e.g., silica); underlies many important diseases
• Characterized by persistent inflammation, tissue injury, attempted repair by scarring, and immune response
• Cellular infiltrate consisting of activated macrophages, lymphocytes, and plasma cells, often with prominent fibrosis
• Mediated by cytokines produced by macrophages and lymphocytes (notably T lymphocytes), with a tendency to an amplified and prolonged inflammatory response owing to bidirectional interactions between these cells
Systemic Effects of Inflammation
Anyone who has suffered a severe bout of viral illness (such as influenza) has experienced the systemic effects of inflammation, collectively called the acute-phase reaction, or the systemic inflammatory response syndrome. The cytokines TNF, IL-1, and IL-6 are the most important mediators of the acute-phase reaction. These cytokines are produced by leukocytes (and other cell types) in response to infection or in immune reactions and are released systemically. TNF and IL-1 have similar biologic actions, although these may differ in subtle ways (Fig. 2–17). IL-6 stimulates the hepatic synthesis of a number of plasma proteins, described further on.
The acute-phase response consists of several clinical and pathologic changes.
• Fever, characterized by an elevation of body temperature, is one of the most prominent manifestations of the acute-phase response. Fever is produced in response to substances called pyrogens that act by stimulating prostaglandin synthesis in the vascular and perivascular cells of the hypothalamus. Bacterial products, such as lipopolysaccharide (LPS) (called exogenous pyrogens), stimulate leukocytes to release cytokines such as IL-1 and TNF (called endogenous pyrogens), which increase the levels of cyclooxygenases that convert AA into prostaglandins. In the hypothalamus the prostaglandins, especially PGE2, stimulate the production of neurotransmitters, which function to reset the temperature set point at a higher level. NSAIDs, including aspirin, reduce fever by inhibiting cyclooxygenase and thus blocking prostaglandin synthesis. Although fever was recognized as a sign of infection hundreds of years ago, it is still not clear what the purpose of this reaction may be. An elevated body temperature has been shown to help amphibians ward off microbial infections, and it is assumed that fever does the same for mammals, although the mechanism is unknown.
• Elevated plasma levels of acute-phase proteins. These plasma proteins are mostly synthesized in the liver, and in the setting of acute inflammation, their concentrations may increase several hundred-fold. Three of the best known of these proteins are C-reactive protein (CRP), fibrinogen, and serum amyloid A (SAA) protein. Synthesis of these molecules by hepatocytes is stimulated by cytokines, especially IL-6. Many acute-phase proteins, such as CRP and SAA, bind to microbial cell walls, and they may act as opsonins and fix complement, thus promoting the elimination of the microbes. Fibrinogen binds to erythrocytes and causes them to form stacks (rouleaux) that sediment more rapidly at unit gravity than individual erythrocytes. This is the basis for measuring the erythrocyte sedimentation rate (ESR) as a simple test for the systemic inflammatory response, caused by any number of stimuli, including LPS. Serial measurements of ESR and CRP are used to assess therapeutic responses in patients with inflammatory disorders such as rheumatoid arthritis. Elevated serum levels of CRP are now used as a marker for increased risk of myocardial infarction or stroke in patients with atherosclerotic vascular disease. It is believed that inflammation is involved in the development of atherosclerosis (Chapter 9), and increased CRP is a measure of inflammation.
• Leukocytosis is a common feature of inflammatory reactions, especially those induced by bacterial infection (see Table 11–6, Chapter 11). The leukocyte count usually climbs to 15,000 to 20,000 cells/mL, but in some extraordinary cases it may reach 40,000 to 100,000 cells/mL. These extreme elevations are referred to as leukemoid reactions because they are similar to those seen in leukemia. The leukocytosis occurs initially because of accelerated release of cells (under the influence of cytokines, including TNF and IL-1) from the bone marrow postmitotic reserve pool. Both mature and immature neutrophils may be seen in the blood; the presence of circulating immature cells is referred to as a “shift to the left.” Prolonged infection also stimulates production of colony-stimulating factors (CSFs), which increase the bone marrow output of leukocytes, thus compensating for the consumption of these cells in the inflammatory reaction. Most bacterial infections induce an increase in the blood neutrophil count, called neutrophilia. Viral infections, such as infectious mononucleosis, mumps, and German measles, are associated with increased numbers of lymphocytes (lymphocytosis). Bronchial asthma, hay fever, and parasite infestations all involve an increase in the absolute number of eosinophils, creating an eosinophilia. Certain infections (typhoid fever and infections caused by some viruses, rickettsiae, and certain protozoa) are paradoxically associated with a decreased number of circulating white cells (leukopenia), likely because of cytokine-induced sequestration of lymphocytes in lymph nodes.
• Other manifestations of the acute-phase response include increased heart rate and blood pressure; decreased sweating, mainly as a result of redirection of blood flow from cutaneous to deep vascular beds, to minimize heat loss through the skin; and rigors (shivering), chills (perception of being cold as the hypothalamus resets the body temperature), anorexia, somnolence, and malaise, probably secondary to the actions of cytokines on brain cells.
• In severe bacterial infections (sepsis), the large amounts of bacterial products in the blood or extravascular tissue stimulate the production of several cytokines, notably TNF, as well as IL-12 and IL-1. TNF can cause disseminated intravascular coagulation (DIC), metabolic disturbances including acidosis, and hypotensive shock. This clinical triad is described as septic shock; it is discussed in more detail in Chapter 3.
Summary
Systemic Effects of Inflammation
• Fever: cytokines (TNF, IL-1) stimulate production of prostaglandins in hypothalamus
• Production of acute-phase proteins: C-reactive protein, others; synthesis stimulated by cytokines (IL-6, others) acting on liver cells
• Leukocytosis: cytokines (CSFs) stimulate production of leukocytes from precursors in the bone marrow
• In some severe infections, septic shock: fall in blood pressure, disseminated intravascular coagulation, metabolic abnormalities; induced by high levels of TNF
Even before the inflammatory reaction ends, the body begins the process of healing the damage and restoring normal structure and function. This process is called repair, and it involves the proliferation and differentiation of several cell types and the deposition of connective tissue. Defects in tissue repair have serious consequences. Conversely, excessive connective tissue deposition (fibrosis) is also a cause of significant abnormalities. Therefore, the mechanisms and regulation of the repair process are of great physiologic and pathologic importance.
Overview of Tissue Repair
Critical to the survival of an organism is the ability to repair the damage caused by toxic insults and inflammation. The inflammatory response to microbes and injured tissues not only serves to eliminate these dangers but also sets into motion the process of repair. Repair, sometimes called healing, refers to the restoration of tissue architecture and function after an injury. It occurs by two types of reactions: regeneration of the injured tissue and scar formation by the deposition of connective tissue (Fig. 2–24).
• Regeneration. Some tissues are able to replace the damaged cells and essentially return to a normal state; this process is called regeneration. Regeneration occurs by proliferation of residual (uninjured) cells that retain the capacity to divide, and by replacement from tissue stem cells. It is the typical response to injury in the rapidly dividing epithelia of the skin and intestines, and some parenchymal organs, notably the liver.
• Scar formation. If the injured tissues are incapable of regeneration, or if the supporting structures of the tissue are severely damaged, repair occurs by the laying down of connective (fibrous) tissue, a process that results in scar formation. Although the fibrous scar cannot perform the function of lost parenchymal cells, it provides enough structural stability that the injured tissue is usually able to function. The term fibrosis is most often used to describe the extensive deposition of collagen that occurs in the lungs, liver, kidney, and other organs as a consequence of chronic inflammation, or in the myocardium after extensive ischemic necrosis (infarction). If fibrosis develops in a tissue space occupied by an inflammatory exudate, it is called organization (as in organizing pneumonia affecting the lung).
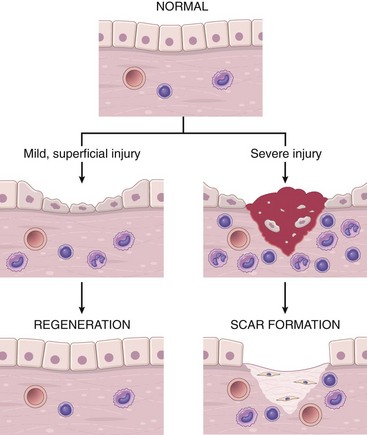
Figure 2–24 Mechanisms of tissue repair: regeneration and scar formation. After mild injury, which damages the epithelium but not the underlying tissue, resolution occurs by regeneration, but after more severe injury with damage to the connective tissue, repair is by scar formation.
After many common types of injury, both regeneration and scar formation contribute in varying degrees to the ultimate repair. Both processes involve the proliferation of various cells and close interactions between cells and the ECM. The next section discusses the principles of cellular proliferation, the roles of growth factors in the proliferation of different cell types involved in repair, and the roles of stem cells in tissue homeostasis. This is followed by a summary of some important properties of the ECM and how it is involved in repair. These sections lay the foundation for a consideration of the salient features of regeneration and healing by scar formation, concluding with a description of cutaneous wound healing and fibrosis (scarring) in parenchymal organs as illustrations of the repair process.
Cell and Tissue Regeneration
The regeneration of injured cells and tissues involves cell proliferation, which is driven by growth factors and is critically dependent on the integrity of the extracellular matrix. Before describing examples of repair by regeneration, we discuss the general principles of cell proliferation and the functions of the ECM in this process.
The Control of Cell Proliferation
Several cell types proliferate during tissue repair. These include the remnants of the injured tissue (which attempt to restore normal structure), vascular endothelial cells (to create new vessels that provide the nutrients needed for the repair process), and fibroblasts (the source of the fibrous tissue that forms the scar to fill defects that cannot be corrected by regeneration). The proliferation of these cell types is driven by proteins called growth factors. The production of polypeptide growth factors and the ability of cells to divide in response to these factors are important determinants of the adequacy of the repair process.
The normal size of cell populations is determined by a balance among cell proliferation, cell death by apoptosis, and emergence of new differentiated cells from stem cells (Fig. 2–25). The key processes in the proliferation of cells are DNA replication and mitosis. The sequence of events that control these two processes is known as the cell cycle, described in detail in Chapter 5 in the context of cancer. At this stage, it is sufficient to note that nondividing cells are in cell cycle arrest in the G1 phase or have exited the cycle and are in the G0 phase. Growth factors stimulate cells to transition from G0 into the G1 phase and beyond into DNA synthesis (S), G2, and mitosis (M) phases. Progression is regulated by cyclins, whose activity is controlled by cyclin-dependent kinases. Once cells enter the S phase, their DNA is replicated and they progress through G2 and mitosis.
Figure 2–25 Mechanisms regulating cell populations. Cell numbers can be altered by increased or decreased rates of stem cell input, cell death by apoptosis, or changes in the rates of proliferation or differentiation.
(Modified from McCarthy NJ, et al: Apoptosis in the development of the immune system: growth factors, clonal selection and bcl-2. Cancer Metastasis Rev 11:157, 1992.)
Proliferative Capacities of Tissues
The ability of tissues to repair themselves is critically influenced by their intrinsic proliferative capacity. On the basis of this criterion, the tissues of the body are divided into three groups.
• Labile (continuously dividing) tissues. Cells of these tissues are continuously being lost and replaced by maturation from stem cells and by proliferation of mature cells. Labile cells include hematopoietic cells in the bone marrow and the majority of surface epithelia, such as the stratified squamous surfaces of the skin, oral cavity, vagina, and cervix; the cuboidal epithelia of the ducts draining exocrine organs (e.g., salivary glands, pancreas, biliary tract); the columnar epithelium of the gastrointestinal tract, uterus, and fallopian tubes; and the transitional epithelium of the urinary tract. These tissues can readily regenerate after injury as long as the pool of stem cells is preserved.
• Stable tissues. Cells of these tissues are quiescent and have only minimal replicative activity in their normal state. However, these cells are capable of proliferating in response to injury or loss of tissue mass. Stable cells constitute the parenchyma of most solid tissues, such as liver, kidney, and pancreas. They also include endothelial cells, fibroblasts, and smooth muscle cells; the proliferation of these cells is particularly important in wound healing. With the exception of liver, stable tissues have a limited capacity to regenerate after injury.
• Permanent tissues. The cells of these tissues are considered to be terminally differentiated and nonproliferative in postnatal life. Most neurons and cardiac muscle cells belong to this category. Thus, injury to brain or heart is irreversible and results in a scar, because neurons and cardiac myocytes cannot regenerate. Limited stem cell replication and differentiation occur in some areas of the adult brain, and there is some evidence that cardiac stem cells may proliferate after myocardial necrosis. Nevertheless, whatever proliferative capacity may exist in these tissues, it is insufficient to produce tissue regeneration after injury. Skeletal muscle is usually classified as a permanent tissue, but satellite cells attached to the endomysial sheath provide some regenerative capacity for this tissue. In permanent tissues, repair is typically dominated by scar formation.
With the exception of tissues composed primarily of nondividing permanent cells (e.g., cardiac muscle, nerve), most mature tissues contain variable proportions of three cell types: continuously dividing cells, quiescent cells that can return to the cell cycle, and cells that have lost replicative ability.
Stem Cells
In most dividing tissues the mature cells are terminally differentiated and short-lived. As mature cells die, the tissue is replenished by the differentiation of cells generated from stem cells. Thus, in these tissues there is a homeostatic equilibrium between the replication, self-renewal, and differentiation of stem cells and the death of the mature, fully differentiated cells. Such relationships are particularly evident in the continuously dividing epithelium of the skin and the gastrointestinal tract, in which stem cells live near the basal layer of the epithelium, and cells differentiate as they migrate to the upper layers of the epithelium before they die and are shed from the surface.
Stem cells are characterized by two important properties: self-renewal capacity and asymmetric replication. Asymmetric replication means that when a stem cell divides, one daughter cell enters a differentiation pathway and gives rise to mature cells, while the other remains an undifferentiated stem cell that retains its self-renewal capacity. Self-renewal enables stem cells to maintain a functional population of precursors for long periods of time. Although the scientific literature is replete with descriptions of various types of stem cells, fundamentally there are two kinds:
• Embryonic stem cells (ES cells) are the most undifferentiated stem cells. They are present in the inner cell mass of the blastocyst and have extensive cell renewal capacity. Hence they can be maintained in culture for over a year without differentiating. Under appropriate culture conditions, ES cells can be induced to form specialized cells of all three germ cell layers, including neurons, cardiac muscle, liver cells, and pancreatic islet cells.
• Adult stem cells, also called tissue stem cells, are less undifferentiated than ES cells and are found among differentiated cells within an organ or tissue. Although, like ES cells, they also have self-renewal capacity, this property is much more limited. In addition, their lineage potential (ability to give rise to specialized cells) is restricted to some or all of the differentiated cells of the tissue or organ in which they are found.
Whereas the normal function of ES cells is to give rise to all cells of the body, adult stem cells are involved in tissue homeostasis. They maintain the compartment size both in tissues with high turnover, such as skin, bone marrow, and gut epithelium, and in those with low cell turnover, such as heart and blood vessels. Although there is much interest in isolation and infusion of tissue stem cells for replenishment of specialized cells in organs such as the heart (after a myocardial infarct) and brain (after a stroke), tissue stem cells are rare and very difficult to isolate to purity. Furthermore, they occur in specialized microenvironments within the organ called stem cell niches. Apparently, signals from other cells in such niches keep the stem cells quiescent and undifferentiated. Stem cell niches have been identified in many organs. In the brain, neural stem cells occur in the subventricular zone and dentate gyrus; in the skin, tissue stem cells are found in the bulge region of the hair follicle; and in the cornea, they are found at the limbus.
Perhaps the most extensively studied tissue stem cells are hematopoietic stem cells found in the bone marrow. Although rare, they can be purified to virtual purity based on cell surface markers. Hematopoietic stem cells can be isolated from bone marrow as well as from the peripheral blood after mobilization by administration of certain cytokines such as granulocyte colony-stimulating factor (G-CSF). As is well known, they can give rise to all blood cell lineages and continuously replenish the formed elements of the blood as these are consumed in the periphery. In clinical practice, marrow stem cells are used for treatment of diseases such as leukemia and lymphomas (Chapter 11). In addition to hematopoietic stem cells, the bone marrow also contains a somewhat distinctive population of tissue stem cells, often called mesenchymal stem cells. These cells can give rise to a variety of mesenchymal cells, such as chondroblasts, osteoblasts, and myoblasts. Hence, there is great interest in their therapeutic potential.
The ability to identify and isolate stem cells has given rise to the new field of regenerative medicine, which has as its main goal the repopulation of damaged organs by using differentiated progeny of ES cells or adult stem cells. Since ES cells have extensive self-renewal capacity and can give rise to all cell lineages, they often are considered ideal for developing specialized cells for therapeutic purposes. However, since ES cells are derived from blastocysts (typically produced from in vitro fertilization), their progeny carry histocompatibility molecules (human leukocyte antigen [HLA] in people) (Chapter 4) of the donors of the egg and sperm. Thus, they are likely to evoke immunologically mediated rejection by the host, just as organs transplanted from genetically disparate hosts do. Hence, much effort has gone into producing cells with the potential of ES cells from patient tissues. To accomplish this goal, the expressed genes in ES cells and differentiated cells have been compared and a handful of genes that are critical for the “stem-cell-ness” of ES cells have been identified. Introduction of such genes into fully differentiated cells, such as fibroblasts or skin epithelial cells, leads, quite remarkably, to reprogramming of the somatic cell nucleus, such that the cells acquire many of the properties of ES cells. These cells are called induced pluripotent stem cells (iPS cells) (Fig. 2–26). Since iPS cells can be derived from each patient, their differentiated progeny should engraft successfully and restore or replace damaged or deficient cells in the patient—for example, insulin-secreting β cells in a patient with diabetes. Although iPS cells hold considerable promise, their clinical usefulness remains to be proved.
Figure 2–26 The production of induced pluripotent stem cells (iPS cells). Genes that confer stem cell properties are introduced into a patient’s differentiated cells, giving rise to stem cells, which can be induced to differentiate into various lineages.
Summary
Cell Proliferation, the Cell Cycle, and Stem Cells
• Regeneration of tissues is driven by proliferation of uninjured (residual) cells and replacement from stem cells.
• Cell proliferation occurs when quiescent cells enter the cell cycle. The cell cycle is tightly regulated by stimulators and inhibitors and contains intrinsic checkpoint controls to prevent replication of abnormal cells.
• Tissues are divided into labile, stable, and permanent, according to the proliferative capacity of their cells.
• Continuously dividing tissues (labile tissues) contain mature cells that are capable of dividing and stem cells that differentiate to replenish lost cells.
• Stem cells from embryos (ES cells) are pluripotent; adult tissues, particularly the bone marrow, contain adult stem cells capable of generating multiple cell lineages.
• Induced pluripotent stem cells (iPS cells) are derived by introducing into mature cells genes that are characteristic of ES cells. iPS cells acquire many characteristics of stem cells.
Growth Factors
Most growth factors are proteins that stimulate the survival and proliferation of particular cells, and may also promote migration, differentiation, and other cellular responses. They induce cell proliferation by binding to specific receptors and affecting the expression of genes whose products typically have several functions: They promote entry of cells into the cell cycle, they relieve blocks on cell cycle progression (thus promoting replication), they prevent apoptosis, and they enhance the synthesis of cellular proteins in preparation for mitosis. A major activity of growth factors is to stimulate the function of growth control genes, many of which are called proto-oncogenes because mutations in them lead to unrestrained cell proliferation characteristic of cancer (oncogenesis) (Chapter 5).
There is a huge (and ever-increasing) list of known growth factors. In the following discussion, rather than attempting an exhaustive cataloguing, we highlight only selected molecules that contribute to tissue repair (Table 2–9). Many of the growth factors that are involved in repair are produced by macrophages and lymphocytes that are recruited to the site of injury or are activated at this site, as part of the inflammatory process. Other growth factors are produced by parenchymal cells or stromal (connective tissue) cells in response to cell injury. We start the discussion by describing general principles of growth factor actions. We return to the roles of individual growth factors in the repair process later in the chapter.
Table 2–9 Growth Factors Involved in Regeneration and Repair
| Growth Factor | Sources | Functions |
|---|---|---|
| Epidermal growth factor (EGF) | Activated macrophages, salivary glands, keratinocytes, and many other cells | Mitogenic for keratinocytes and fibroblasts; stimulates keratinocyte migration; stimulates formation of granulation tissue |
| Transforming growth factor-α (TGF-α) | Activated macrophages, keratinocytes, many other cell types | Stimulates proliferation of hepatocytes and many other epithelial cells |
| Hepatocyte growth factor (HGF) (scatter factor) | Fibroblasts, stromal cells in the liver, endothelial cells | Enhances proliferation of hepatocytes and other epithelial cells; increases cell motility |
| Vascular endothelial growth factor (VEGF) | Mesenchymal cells | Stimulates proliferation of endothelial cells; increases vascular permeability |
| Platelet-derived growth factor (PDGF) | Platelets, macrophages, endothelial cells, smooth muscle cells, keratinocytes | Chemotactic for neutrophils, macrophages, fibroblasts, and smooth muscle cells; activates and stimulates proliferation of fibroblasts, endothelial, and other cells; stimulates ECM protein synthesis |
| Fibroblast growth factors (FGFs), including acidic (FGF-1) and basic (FGF-2) | Macrophages, mast cells, endothelial cells, many other cell types | Chemotactic and mitogenic for fibroblasts; stimulates angiogenesis and ECM protein synthesis |
| Transforming growth factor-β (TGF-β) | Platelets, T lymphocytes, macrophages, endothelial cells, keratinocytes, smooth muscle cells, fibroblasts | Chemotactic for leukocytes and fibroblasts; stimulates ECM protein synthesis; suppresses acute inflammation |
| Keratinocyte growth factor (KGF) (i.e., FGF-7) | Fibroblasts | Stimulates keratinocyte migration, proliferation, and differentiation |
ECM, extracellular membrane.
Signaling Mechanisms of Growth Factor Receptors
Most growth factors function by binding to specific cell-surface receptors and triggering biochemical signals in cells. The major intracellular signaling pathways induced by growth factor receptors are similar to those of many other cellular receptors that recognize extracellular ligands. In general, these signals lead to the stimulation or repression of gene expression. Signaling may occur directly in the same cell that produces the factor (autocrine signaling), between adjacent cells (paracrine signaling), or over greater distances (endocrine signaling).
Receptor proteins are generally located on the cell surface, but they may be intracellular; in the latter case, the ligands must be sufficiently hydrophobic to enter the cell (e.g., vitamin D, or steroid and thyroid hormones). On the basis of their major signaling transduction pathways, plasma membrane receptors fall into three main types, listed in Table 2–10.
• Receptors with intrinsic kinase activity. Binding of ligand to the extracellular portion of the receptor causes dimerization and subsequent phosphorylation of the receptor subunits. Once phosphorylated, the receptors can bind and activate other intracellular proteins (e.g., RAS, phosphatidylinositol 3[PI3]-kinase, phospholipase Cγ [PLC-γ]) and stimulate downstream signals that lead to cell proliferation, or induction of various transcriptional programs.
• G protein–coupled receptors. These receptors contain seven-transmembrane α-helix segments and are also known as seven-transmembrane receptors. After ligand binding, the receptors associate with intracellular guanosine triphosphate (GTP)-binding proteins (G proteins) that contain guanosine diphosphate (GDP). Binding of the G proteins causes the exchange of GDP with GTP, resulting in activation of the proteins. Among the several signaling pathways activated through G protein–coupled receptors are those involving cyclic AMP (cAMP), and the generation of inositol 1,4,5-triphosphate (IP3), which releases calcium from the endoplasmic reticulum. Receptors in this category constitute the largest family of plasma membrane receptors (more than 1500 members have been identified).
• Receptors without intrinsic enzymatic activity. These are usually monomeric transmembrane molecules with an extracellular ligand-binding domain; ligand interaction induces an intracellular conformational change that allows association with intracellular protein kinases called Janus kinases (JAKs). Phosphorylation of JAKs activates cytoplasmic transcription factors called STATs (signal transducers and activators of transcription), which shuttle into the nucleus and induce transcription of target genes.
Table 2–10 Principal Signaling Pathways Used by Cell Surface Receptors
| Receptor Class | Ligands | Signaling Mechanism(s) |
|---|---|---|
| Receptors with intrinsic tyrosine kinase activity | EGF, VEGF, FGF, HGF | Ligand binding to one chain of the receptor activates tyrosine kinase on the other chain, resulting in activation of multiple downstream signaling pathways (RAS-MAP kinase, PI-3 kinase, PLC-γ) and activation of various transcription factors. |
| G protein–coupled seven-transmembrane receptors (GPCRs) | Multiple inflammatory mediators, hormones, all chemokines | Ligand binding induces switch from GDP-bound inactive form of associated G protein to GTP-bound active form; activates cAMP; Ca2+ influx leading to increased cell motility; multiple other effects. |
| Receptors without intrinsic enzymatic activity | Many cytokines including interferons, growth hormone, CSFs, EPO | Ligand binding recruits kinases (e.g., Janus kinases [JAKs]) that phosphorylate and activate transcription factors (e.g., signal transducers and activators of transcription [STATs]). |
cAMP, cyclic adenosine monophosphate; CSFs, colony-stimulating factors; EGF, epidermal growth factor; EPO, epopoietin; FGF, fibroblast growth factor; GDP, guanosine diphosphate; GTP, guanosine triphosphate; HGF, hepatocyte growth factor; PI3, phosphatidylinositol-3; PLC-γ, phospholipase Cγ; MAP, microtubule-associated protein; VEGF, vascular endothelial growth factor.
Summary
Growth Factors, Receptors, and Signal Transduction
• Polypeptide growth factors act in autocrine, paracrine, or endocrine manner.
• Growth factors are produced transiently in response to an external stimulus and act by binding to cellular receptors. Different classes of growth factor receptors include receptors with intrinsic kinase activity, G protein–coupled receptors and receptors without intrinsic kinase activity.
• Growth factors such as epidermal growth factor (EGF) and hepatocyte growth factor (HGF) bind to receptors with intrinsic kinase activity, triggering a cascade of phosphorylating events through MAP kinases, which culminate in transcription factor activation and DNA replication.
• G protein–coupled receptors produce multiple effects via the cAMP and Ca2+ pathways. Chemokines utilize such receptors.
• Cytokines generally bind to receptors without kinase activity; such receptors interact with cytoplasmic transcription factors that move into the nucleus.
• Most growth factors have multiple effects, such as cell migration, differentiation, stimulation of angiogenesis, and fibrogenesis, in addition to cell proliferation.
Role of the Extracellular Matrix in Tissue Repair
Tissue repair depends not only on growth factor activity but also on interactions between cells and ECM components. The ECM is a complex of several proteins that assembles into a network that surrounds cells and constitutes a significant proportion of any tissue. ECM sequesters water, providing turgor to soft tissues, and minerals, giving rigidity to bone. It also regulates the proliferation, movement, and differentiation of the cells living within it, by supplying a substrate for cell adhesion and migration and serving as a reservoir for growth factors. The ECM is constantly being remodeled; its synthesis and degradation accompany morphogenesis, wound healing, chronic fibrosis, and tumor invasion and metastasis.
ECM occurs in two basic forms: interstitial matrix and basement membrane (Fig. 2–27).
• Interstitial matrix: This form of ECM is present in the spaces between cells in connective tissue, and between epithelium and supportive vascular and smooth muscle structures. It is synthesized by mesenchymal cells (e.g., fibroblasts) and tends to form a three-dimensional, amorphous gel. Its major constituents are fibrillar and nonfibrillar collagens, as well as fibronectin, elastin, proteoglycans, hyaluronate, and other elements (described later).
• Basement membrane: The seemingly random array of interstitial matrix in connective tissues becomes highly organized around epithelial cells, endothelial cells, and smooth muscle cells, forming the specialized basement membrane. The basement membrane lies beneath the epithelium and is synthesized by overlying epithelium and underlying mesenchymal cells; it tends to form a platelike “chicken wire” mesh. Its major constituents are amorphous nonfibrillar type IV collagen and laminin (see later).
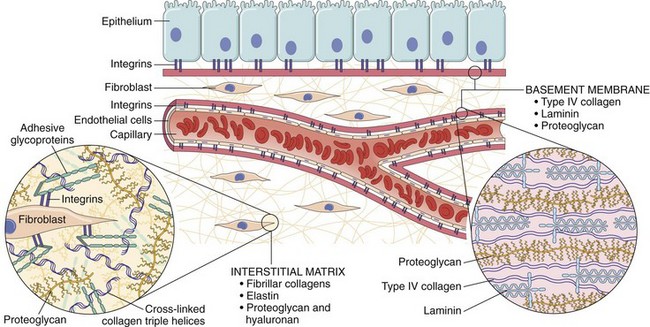
Figure 2–27 The major components of the extracellular matrix (ECM), including collagens, proteoglycans, and adhesive glycoproteins. Note that although there is some overlap in their constituents, basement membrane and interstitial ECM differ in general composition and architecture. Both epithelial and mesenchymal cells (e.g., fibroblasts) interact with ECM through integrins. For simplification, many ECM components have been left out (e.g., elastin, fibrillin, hyaluronan, syndecan).
Components of the Extracellular Matrix
There are three basic components of ECM: (1) fibrous structural proteins such as collagens and elastins, which confer tensile strength and recoil; (2) water-hydrated gels such as proteoglycans and hyaluronan, which permit resilience and lubrication; and (3) adhesive glycoproteins that connect the matrix elements to one another and to cells (Fig. 2–27).
Collagen
The collagens are composed of three separate polypeptide chains braided into a ropelike triple helix. Approximately 30 collagen types have been identified, some of which are unique to specific cells and tissues. Some collagen types (e.g., types I, II, III, and V) form fibrils by virtue of lateral cross-linking of the triple helices. The fibrillar collagens form a major proportion of the connective tissue in healing wounds and particularly in scars. The tensile strength of the fibrillar collagens derives from their cross-linking, which is the result of covalent bonds catalyzed by the enzyme lysyl-oxidase. This process is dependent on vitamin C; therefore, individuals with vitamin C deficiency have skeletal deformities, bleed easily because of weak vascular wall basement membrane, and suffer from poor wound healing. Genetic defects in these collagens cause diseases such as osteogenesis imperfecta and Ehlers-Danlos syndrome. Other collagens are nonfibrillar and may form basement membrane (type IV) or be components of other structures such as intervertebral disks (type IX) or dermal–epidermal junctions (type VII).
Elastin
The ability of tissues to recoil and return to a baseline structure after physical stress is conferred by elastic tissue. This is especially important in the walls of large vessels (which must accommodate recurrent pulsatile flow), as well as in the uterus, skin, and ligaments. Morphologically, elastic fibers consist of a central core of elastin surrounded by a meshlike network of fibrillin glycoprotein. Defects in fibrillin synthesis lead to skeletal abnormalities and weakened aortic walls (as in Marfan syndrome, discussed in Chapter 6).
Proteoglycans and Hyaluronan
Proteoglycans form highly hydrated compressible gels conferring resilience and lubrication (such as in the cartilage in joints). They consist of long polysaccharides, called glycosaminoglycans or mucopolysaccharides (examples are dermatan sulfate and heparan sulfate), linked to a protein backbone. Hyaluronan (also called hyaluronic acid), a huge mucopolysaccharide without a protein core, is also an important constituent of the ECM that binds water, and forms a viscous, gelatin-like matrix. Besides providing compressibility to tissues, proteoglycans also serve as reservoirs for growth factors secreted into the ECM (e.g., fibroblast growth factor [FGF], HGF). Some proteoglycans are integral cell membrane proteins that have roles in cell proliferation, migration, and adhesion—for example, by binding growth factors and chemokines and providing high local concentrations of these mediators.
Adhesive Glycoproteins and Adhesion Receptors
Adhesive glycoproteins and adhesion receptors are structurally diverse molecules involved in cell-to-cell adhesion, the linkage of cells to the ECM, and binding between ECM components. The adhesive glycoproteins include fibronectin (a major component of the interstitial ECM) and laminin (a major constituent of basement membrane); they are described here as prototypical of the overall group. The adhesion receptors, also known as cell adhesion molecules (CAMs), are grouped into four families—immunoglobulins, cadherins, selectins, and integrins—of which only the integrins are discussed here.
• Fibronectin is a large (450-kDa) disulfide-linked heterodimer synthesized by a variety of cells, including fibroblasts, monocytes, and endothelium that exists in tissue and plasma forms. Fibronectins have specific domains that bind to a wide spectrum of ECM components (e.g., collagen, fibrin, heparin, proteoglycans) and can also attach to cell integrins via a tripeptide arginine–glycine–aspartic acid (abbreviated RGD) motif. Tissue fibronectin forms fibrillar aggregates at wound healing sites; plasma fibronectin binds to fibrin within the blood clot that forms in a wound, providing the substratum for ECM deposition and re-epithelialization.
• Laminin is the most abundant glycoprotein in basement membrane. It is an 820-kDa cross-shaped heterotrimer that connects cells to underlying ECM components such as type IV collagen and heparan sulfate. Besides mediating attachment to basement membrane, laminin can also modulate cell proliferation, differentiation, and motility.
• Integrins are a family of transmembrane heterodimeric glycoprotein chains that were introduced in the context of leukocyte adhesion to endothelium. They are also the main cellular receptors for ECM components, such as fibronectins and laminins. We have already discussed some of the integrins as leukocyte surface molecules that mediate firm adhesion and transmigration across endothelium at sites of inflammation, and we shall meet them again when we discuss platelet aggregation in Chapter 3. Integrins are present in the plasma membrane of most cells, with the exception of red blood cells. They bind to many ECM components through RGD motifs, initiating signaling cascades that can affect cell locomotion, proliferation, and differentiation. Their intracellular domains link to actin filaments, thereby affecting cell shape and mobility.
Functions of the Extracellular Matrix
The ECM is much more than a space filler around cells. Its various functions include
• Mechanical support for cell anchorage and cell migration, and maintenance of cell polarity
• Control of cell proliferation by binding and displaying growth factors and by signaling through cellular receptors of the integrin family. The type of ECM proteins can affect the degree of differentiation of the cells in the tissue, again acting largely through cell surface integrins.
• Scaffolding for tissue renewal. Because maintenance of normal tissue structure requires a basement membrane or stromal scaffold, the integrity of the basement membrane or the stroma of parenchymal cells is critical for the organized regeneration of tissues. Thus, although labile and stable cells are capable of regeneration, disruption of the ECM results in a failure of the tissues to regenerate and repair by scar formation (Fig. 2–24).
• Establishment of tissue microenvironments. Basement membrane acts as a boundary between epithelium and underlying connective tissue and also forms part of the filtration apparatus in the kidney.
Summary
Extracellular Matrix and Tissue Repair
• The ECM consists of the interstitial matrix between cells, made up of collagens and several glycoproteins, and basement membranes underlying epithelia and surrounding vessels, made up of nonfibrillar collagen and laminin.
• The ECM serves several important functions:
 It regulates cell proliferation and differentiation; proteoglycans bind growth factors and display them at high concentration, and fibronectin and laminin stimulate cells through cellular integrin receptors.
It regulates cell proliferation and differentiation; proteoglycans bind growth factors and display them at high concentration, and fibronectin and laminin stimulate cells through cellular integrin receptors.• An intact ECM is required for tissue regeneration, and if the ECM is damaged, repair can be accomplished only by scar formation.
Having described the basic components of tissue repair, we now proceed to a discussion of repair by regeneration and by scar formation.
Role of Regeneration in Tissue Repair
The importance of regeneration in the replacement of injured tissues varies in different types of tissues and with the severity of injury.
• In labile tissues, such as the epithelia of the intestinal tract and skin, injured cells are rapidly replaced by proliferation of residual cells and differentiation of tissue stem cells provided the underlying basement membrane is intact. The growth factors involved in these processes are not defined. Loss of blood cells is corrected by proliferation of hematopoietic progenitors in the bone marrow and other tissues, driven by CSFs, which are produced in response to the reduced numbers of blood cells.
• Tissue regeneration can occur in parenchymal organs with stable cell populations, but with the exception of the liver, this is usually a limited process. Pancreas, adrenal, thyroid, and lung have some regenerative capacity. The surgical removal of a kidney elicits in the contralateral kidney a compensatory response that consists of both hypertrophy and hyperplasia of proximal duct cells. The mechanisms underlying this response are not understood.
• The regenerative response of the liver that occurs after surgical removal of hepatic tissue is remarkable and unique among all organs. As much as 40% to 60% of the liver may be removed in a procedure called living-donor transplantation, in which a portion of the liver is resected from a normal person and transplanted into a recipient with end-stage liver disease (Fig. 2–28), or after partial hepatectomy performed for tumor removal. In both situations, the removal of tissue triggers a proliferative response of the remaining hepatocytes (which are normally quiescent), and the subsequent replication of hepatic nonparenchymal cells. In experimental systems, hepatocyte replication after partial hepatectomy is initiated by cytokines (e.g., TNF, IL-6) that prepare the cells for replication by stimulating the transition from G0 to G1 in the cell cycle. Progression through the cell cycle is dependent on the activity of growth factors such as HGF (produced by fibroblasts, endothelial cells, and liver nonparenchymal cells) and the EGF family of factors, which includes transforming growth factor-α (TGF-α) (produced by many cell types).
Figure 2–28 Regeneration of the liver. Computed tomography scans show a donor liver in living-donor liver transplantation. A, The donor liver before the operation. Note the right lobe (outline), which will be resected and used as a transplant. B, Scan of the same liver 1 week after resection of the right lobe; note the enlargement of the left lobe (outline) without regrowth of the right lobe.
(Courtesy of R. Troisi, MD, Ghent University, Flanders, Belgium.)
A point worthy of emphasis is that extensive regeneration or compensatory hyperplasia can occur only if the residual connective tissue framework is structurally intact, as after partial surgical resection. By contrast, if the entire tissue is damaged by infection or inflammation, regeneration is incomplete and is accompanied by scarring. For example, extensive destruction of the liver with collapse of the reticulin framework, as occurs in a liver abscess, leads to scar formation even though the remaining liver cells have the capacity to regenerate.
Scar Formation
As discussed earlier, if tissue injury is severe or chronic and results in damage to parenchymal cells and epithelia as well as the connective tissue, or if nondividing cells are injured, repair cannot be accomplished by regeneration alone. Under these conditions, repair occurs by replacement of the nonregenerated cells with connective tissue, leading to the formation of a scar, or by a combination of regeneration of some cells and scar formation.
Steps in Scar Formation
Repair by connective tissue deposition consists of sequential processes that follow the inflammatory response (Fig. 2–29):
• Formation of new blood vessels (angiogenesis)
• Migration and proliferation of fibroblasts and deposition of connective tissue, which, together with abundant vessels and interspersed leukocytes, has a pink, granular appearance and hence is called granulation tissue
• Maturation and reorganization of the fibrous tissue (remodeling) to produce the stable fibrous scar
Figure 2–29 Steps in repair by scar formation. Injury to a tissue that has limited regenerative capacity first induces inflammation, which clears dead cells and microbes, if any. This is followed by formation of vascularized granulation tissue and then deposition of ECM to form the scar. ECM, extracellular matrix.
Repair begins within 24 hours of injury by the emigration of fibroblasts and the induction of fibroblast and endothelial cell proliferation. By 3 to 5 days, the specialized granulation tissue that is characteristic of healing is apparent. The term granulation tissue derives from the gross appearance, such as that beneath the scab of a skin wound. Its histologic appearance is characterized by proliferation of fibroblasts and new thin-walled, delicate capillaries (angiogenesis) in a loose ECM, often with admixed inflammatory cells, mainly macrophages (Fig. 2–30, A). Granulation tissue progressively accumulates more fibroblasts, which lay down collagen, eventually resulting in the formation of a scar (Fig. 2–30, B). Scars remodel over time. We next describe each of the steps in this process.
Figure 2–30 A, Granulation tissue showing numerous blood vessels, edema, and a loose ECM containing occasional inflammatory cells. Collagen is stained blue by the trichrome stain; minimal mature collagen can be seen at this point. B, Trichrome stain of mature scar, showing dense collagen with only scattered vascular channels. ECM, extracellular matrix.
Angiogenesis
Angiogenesis is the process of new blood vessel development from existing vessels, primarily venules. It is critical in healing at sites of injury, in the development of collateral circulations at sites of ischemia, and in allowing tumors to increase in size beyond the constraints of their original blood supply. Much work has been done to understand the mechanisms underlying angiogenesis, and therapies to either augment the process (e.g., to improve blood flow to a heart ravaged by coronary atherosclerosis) or inhibit it (e.g., to frustrate tumor growth or block pathologic vessel growth such as in diabetic retinopathy) are being developed.
Angiogenesis involves sprouting of new vessels from existing ones and consists of the following steps (Fig. 2–31):
• Vasodilation occurring in response to NO and increased permeability induced by VEGF
• Separation of pericytes from the abluminal surface
• Migration of endothelial cells toward the area of tissue injury
• Proliferation of endothelial cells just behind the leading front of migrating cells
• Remodeling into capillary tubes
• Recruitment of periendothelial cells (pericytes for small capillaries and smooth muscle cells for larger vessels) to form the mature vessel
• Suppression of endothelial proliferation and migration and deposition of the basement membrane
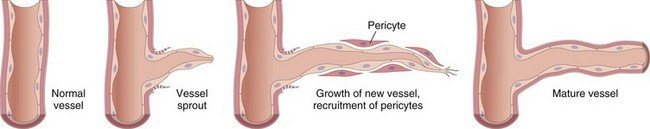
Figure 2–31 Mechanism of angiogenesis. In tissue repair, angiogenesis occurs mainly by growth factor–driven outgrowth of residual endothelium, sprouting of new vessels, and recruitment of pericytes to form new vessels.
The process of angiogenesis involves a variety of growth factors, cell–cell interactions, interactions with ECM proteins, and tissue enzymes.
Growth Factors Involved in Angiogenesis
Several growth factors contribute to angiogenesis; the most important are VEGF and basic fibroblast growth factor (FGF-2).
• The VEGF family of growth factors includes VEGF-A, -B, -C, -D, and -E and placental growth factor (PlGF). VEGF-A is generally referred to as VEGF and is the major inducer of angiogenesis after injury and in tumors; VEGF-B and PlGF are involved in vessel development in the embryo; and VEGF-C and -D stimulate both lymphangiogenesis and angiogenesis. VEGFs are expressed in most adult tissues, with the highest expression in epithelial cells adjacent to fenestrated epithelium (e.g., podocytes in the kidney, pigment epithelium in the retina). They bind to a family of tyrosine kinase receptors (VEGFR-1, -2, and -3). The most important of these receptors for angiogenesis is VEGFR-2, which is expressed by VEGF target cells, especially endothelial cells. Of the many inducers of VEGF, hypoxia is the most important; others are platelet-derived growth factor (PDGF), TGF-α, and TGF-β.
VEGF stimulates both migration and proliferation of endothelial cells, thus initiating the process of capillary sprouting in angiogenesis. It promotes vasodilation by stimulating the production of NO, and contributes to the formation of the vascular lumen. Antibodies against VEGF are approved for the treatment of some tumors that depend on angiogenesis for their spread and growth. These antibodies are also used in the treatment of “wet” (neovascular) age-related macular degeneration, a major cause of visual impairment in adults older than 50 years of age, and is in clinical trials for the treatment of the angiogenesis associated with retinopathy of prematurity and the leaky vessels that lead to diabetic macular edema.
• The FGF family of growth factors has more than 20 members; the best characterized are FGF-1 (acidic FGF) and FGF-2 (basic FGF). These growth factors are produced by many cell types and bind to a family of plasma membrane receptors that have tyrosine kinase activity. Released FGF can bind to heparan sulfate and be stored in the ECM. FGF-2 participates in angiogenesis mostly by stimulating the proliferation of endothelial cells. It also promotes the migration of macrophages and fibroblasts to the damaged area, and stimulates epithelial cell migration to cover epidermal wounds.
• Angiopoietins Ang1 and Ang2 are growth factors that play a role in angiogenesis and the structural maturation of new vessels. Newly formed vessels need to be stabilized by the recruitment of pericytes and smooth muscle cells and by the deposition of connective tissue. Ang1 interacts with a tyrosine kinase receptor on endothelial cells called Tie2. The growth factors PDGF and TGF-β also participate in the stabilization process—PDGF recruits smooth muscle cells and TGF-β suppresses endothelial proliferation and migration, and enhances the production of ECM proteins.
The growth of blood vessels during embryonic development is called vasculogenesis. In vasculogenesis, vessels are formed de novo by the coalescence of endothelial precursors called angioblasts. Angioblasts are derived from hemangioblasts, which also provide the precursors of the hematopoietic system. In addition, there are endothelial progenitors in the adult that are derived from bone marrow stem cells and circulate. The contribution of these cells to angiogenesis in adults is not definitely established.
ECM proteins participate in the process of vessel sprouting in angiogenesis, largely through interactions with integrin receptors in endothelial cells and by providing the scaffold for vessel growth. Enzymes in the ECM, notably the matrix metalloproteinases (MMPs), degrade the ECM to permit remodeling and extension of the vascular tube. Newly formed vessels are leaky because of incomplete interendothelial junctions and because VEGF increases vascular permeability. This leakiness explains why granulation tissue is often edematous and accounts in part for the edema that may persist in healing wounds long after the acute inflammatory response has resolved. Furthermore, it leads to high intratumoral pressure and is the basis for the edema that is so problematic in ocular angiogenesis in pathologic processes such as wet macular degeneration.
Activation of Fibroblasts and Deposition of Connective Tissue
The laying down of connective tissue in the scar occurs in two steps: (1) migration and proliferation of fibroblasts into the site of injury and (2) deposition of ECM proteins produced by these cells. The recruitment and activation of fibroblasts to synthesize connective tissue proteins are driven by many growth factors, including PDGF, FGF-2 (described earlier), and TGF-β. The major source of these factors is inflammatory cells, particularly macrophages, which are present at sites of injury and in granulation tissue. Sites of inflammation are also rich in mast cells, and in the appropriate chemotactic milieu, lymphocytes may be present as well. Each of these cell types can secrete cytokines and growth factors that contribute to fibroblast proliferation and activation.
As healing progresses, the number of proliferating fibroblasts and new vessels decreases; however, the fibroblasts progressively assume a more synthetic phenotype, so there is increased deposition of ECM. Collagen synthesis, in particular, is critical to the development of strength in a healing wound site. As described later, collagen synthesis by fibroblasts begins early in wound healing (days 3 to 5) and continues for several weeks, depending on the size of the wound. Net collagen accumulation, however, depends not only on increased synthesis but also on diminished collagen degradation (discussed later). Ultimately, the granulation tissue evolves into a scar composed of largely inactive, spindle-shaped fibroblasts, dense collagen, fragments of elastic tissue, and other ECM components (Fig. 2–30, B). As the scar matures, there is progressive vascular regression, which eventually transforms the highly vascularized granulation tissue into a pale, largely avascular scar.
Growth Factors Involved in ECM Deposition and Scar Formation
Many growth factors are involved in these processes, including TGF-β, PDGF, and FGF. Because FGF also is involved in angiogenesis, it was described earlier. Here we briefly describe the major properties of TGF-β and PDGF.
• Transforming growth factor-β (TGF-β) belongs to a family of homologous polypeptides (TGF-β1, -β2, and -β3) that includes other cytokines such as bone morphogenetic proteins. The TGF-β1 isoform is widely distributed and is usually referred to as TGF-β. The active factor binds to two cell surface receptors with serine-threonine kinase activity, triggering the phosphorylation of transcription factors called Smads. TGF-β has many and often opposite effects, depending on the cell type and the metabolic state of the tissue. In the context of inflammation and repair, TGF-β has two main functions:
TGF-β stimulates the production of collagen, fibronectin, and proteoglycans, and it inhibits collagen degradation by both decreasing proteinase activity and increasing the activity of tissue inhibitors of proteinases known as TIMPs (discussed later on). TGF-β is involved not only in scar formation after injury but also in the development of fibrosis in lung, liver, and kidneys that follows chronic inflammation. TGF-β is an anti-inflammatory cytokine that serves to limit and terminate inflammatory responses. It does so by inhibiting lymphocyte proliferation and the activity of other leukocytes. Mice lacking TGF-β exhibit widespread inflammation and abundant lymphocyte proliferation.• Platelet-derived growth factor (PDGF) belongs to a family of closely related proteins, each consisting of two chains, designated A and B. There are five main PDGF isoforms, of which the BB isoform is the prototype; it is often referred to simply as PDGF. PDGFs bind to receptors designated as PDGFRα and PDGFRβ. PDGF is stored in platelets and released on platelet activation and is also produced by endothelial cells, activated macrophages, smooth muscle cells, and many tumor cells. PDGF causes migration and proliferation of fibroblasts and smooth muscle cells and may contribute to the migration of macrophages.
• Cytokines (discussed earlier as mediators of inflammation, and in Chapter 4 in the context of immune responses) may also function as growth factors and participate in ECM deposition and scar formation. IL-1 and IL-13, for example, act on fibroblasts to stimulate collagen synthesis, and can also enhance the proliferation and migration of fibroblasts.
Remodeling of Connective Tissue
After its synthesis and deposition, the connective tissue in the scar continues to be modified and remodeled. Thus, the outcome of the repair process is a balance between synthesis and degradation of ECM proteins. We have already discussed the cells and factors that regulate ECM synthesis. The degradation of collagens and other ECM components is accomplished by a family of matrix metalloproteinases (MMPs), which are dependent on zinc ions for their activity. MMPs should be distinguished from neutrophil elastase, cathepsin G, plasmin, and other serine proteinases that can also degrade ECM but are not metalloenzymes. MMPs include interstitial collagenases, which cleave fibrillar collagen (MMP-1, -2, and -3); gelatinases (MMP-2 and -9), which degrade amorphous collagen and fibronectin; and stromelysins (MMP-3, -10, and -11), which degrade a variety of ECM constituents, including proteoglycans, laminin, fibronectin, and amorphous collagen.
MMPs are produced by a variety of cell types (fibroblasts, macrophages, neutrophils, synovial cells, and some epithelial cells), and their synthesis and secretion are regulated by growth factors, cytokines, and other agents. The activity of the MMPs is tightly controlled. They are produced as inactive precursors (zymogens) that must be first activated; this is accomplished by proteases (e.g., plasmin) likely to be present only at sites of injury. In addition, activated MMPs can be rapidly inhibited by specific tissue inhibitors of metalloproteinases (TIMPs), produced by most mesenchymal cells. Thus, during scarring, MMPs are activated to remodel the deposited ECM, and then their activity is shut down by the TIMPs.
Summary
Repair by Scar Formation
• Tissues can be repaired by regeneration with complete restoration of form and function, or by replacement with connective tissue and scar formation.
• Repair by connective tissue deposition involves angiogenesis, migration and proliferation of fibroblasts, collagen synthesis, and connective tissue remodeling.
• Repair by connective tissue starts with the formation of granulation tissue and culminates in the laying down of fibrous tissue.
• Multiple growth factors stimulate the proliferation of the cell types involved in repair.
• TGF-β is a potent fibrogenic agent; ECM deposition depends on the balance among fibrogenic agents, the metalloproteinases (MMPs) that digest ECM, and the TIMPs.
Factors That Influence Tissue Repair
Tissue repair may be altered by a variety of influences, frequently reducing the quality or adequacy of the reparative process. Variables that modify healing may be extrinsic (e.g., infection) or intrinsic to the injured tissue. Particularly important are infections and diabetes.
• Infection is clinically the most important cause of delay in healing; it prolongs inflammation and potentially increases the local tissue injury.
• Nutrition has profound effects on repair; protein deficiency, for example, and especially vitamin C deficiency inhibit collagen synthesis and retard healing.
• Glucocorticoids (steroids) have well-documented anti-inflammatory effects, and their administration may result in weakness of the scar because of inhibition of TGF-β production and diminished fibrosis. In some instances, however, the anti-inflammatory effects of glucocorticoids are desirable. For example, in corneal infections, glucocorticoids are sometimes prescribed (along with antibiotics) to reduce the likelihood of opacity that may result from collagen deposition.
• Mechanical variables such as increased local pressure or torsion may cause wounds to pull apart, or dehisce.
• Poor perfusion, due either to arteriosclerosis and diabetes or to obstructed venous drainage (e.g., in varicose veins), also impairs healing.
• Foreign bodies such as fragments of steel, glass, or even bone impede healing.
• The type and extent of tissue injury affects the subsequent repair. Complete restoration can occur only in tissues composed of stable and labile cells; injury to tissues composed of permanent cells must inevitably result in scarring, as in healing of a myocardial infarct.
• The location of the injury and the character of the tissue in which the injury occurs are also important. For example, inflammation arising in tissue spaces (e.g., pleural, peritoneal, or synovial cavities) develops extensive exudates. Subsequent repair may occur by digestion of the exudate, initiated by the proteolytic enzymes of leukocytes and resorption of the liquefied exudate. This is called resolution, and generally, in the absence of cellular necrosis, normal tissue architecture is restored. In the setting of larger accumulations, however, the exudate undergoes organization: Granulation tissue grows into the exudate, and a fibrous scar ultimately forms.
• Aberrations of cell growth and ECM production may occur even in what begins as normal wound healing. For example, the accumulation of exuberant amounts of collagen can give rise to prominent, raised scars known as keloids (Fig. 2–32). There appears to be a heritable predisposition to keloid formation, and the condition is more common in African-Americans. Healing wounds may also generate exuberant granulation tissue that protrudes above the level of the surrounding skin and hinders re-epithelialization. Such tissue is called “proud flesh” in old medical parlance, and restoration of epithelial continuity requires cautery or surgical resection of the granulation tissue.
Figure 2–32 Keloid. A, Excess collagen deposition in the skin forming a raised scar known as a keloid. B, Thick connective tissue deposition in the dermis.
(A, From Murphy GF, Herzberg AJ: Atlas of Dermatology. Philadelphia, WB Saunders, 1996. B, Courtesy of Z. Argenyi, MD, University of Washington, Seattle, Washington.)
Selected Clinical Examples of Tissue Repair and Fibrosis
Thus far we have discussed the general principles and mechanisms of repair by regeneration and scarring. In this section we describe two clinically significant types of repair—the healing of skin wounds (cutaneous wound healing) and fibrosis in injured parenchymal organs.
Healing of Skin Wounds
Cutaneous wound healing is a process that involves both epithelial regeneration and the formation of connective tissue scar and is thus illustrative of the general principles that apply to healing in all tissues.
Depending on the nature and size of the wound, the healing of skin wounds is said to occur by first or second intention.
Healing by First Intention
One of the simplest examples of wound repair is the healing of a clean, uninfected surgical incision approximated by surgical sutures (Fig. 2–33). This is referred to as primary union, or healing by first intention. The incision causes only focal disruption of epithelial basement membrane continuity and death of relatively few epithelial and connective tissue cells. As a result, epithelial regeneration is the principal mechanism of repair. A small scar is formed, but there is minimal wound contraction. The narrow incisional space first fills with fibrin-clotted blood, which then is rapidly invaded by granulation tissue and covered by new epithelium. The steps in the process are well defined:
• Within 24 hours, neutrophils are seen at the incision margin, migrating toward the fibrin clot. Basal cells at the cut edge of the epidermis begin to show increased mitotic activity. Within 24 to 48 hours, epithelial cells from both edges have begun to migrate and proliferate along the dermis, depositing basement membrane components as they progress. The cells meet in the midline beneath the surface scab, yielding a thin but continuous epithelial layer.
• By day 3, neutrophils have been largely replaced by macrophages, and granulation tissue progressively invades the incision space. Collagen fibers are now evident at the incision margins, but these are vertically oriented and do not bridge the incision. Epithelial cell proliferation continues, yielding a thickened epidermal covering layer.
• By day 5, neovascularization reaches its peak as granulation tissue fills the incisional space. Collagen fibrils become more abundant and begin to bridge the incision. The epidermis recovers its normal thickness as differentiation of surface cells yields a mature epidermal architecture with surface keratinization.
• During the second week, there is continued collagen accumulation and fibroblast proliferation. The leukocyte infiltrate, edema, and increased vascularity are substantially diminished. The long process of “blanching” begins, accomplished by increasing collagen deposition within the incisional scar and the regression of vascular channels.
• By the end of the first month, the scar consists of a cellular connective tissue, largely devoid of inflammatory cells, covered by an essentially normal epidermis. However, the dermal appendages destroyed in the line of the incision are permanently lost. The tensile strength of the wound increases with time, as described later.
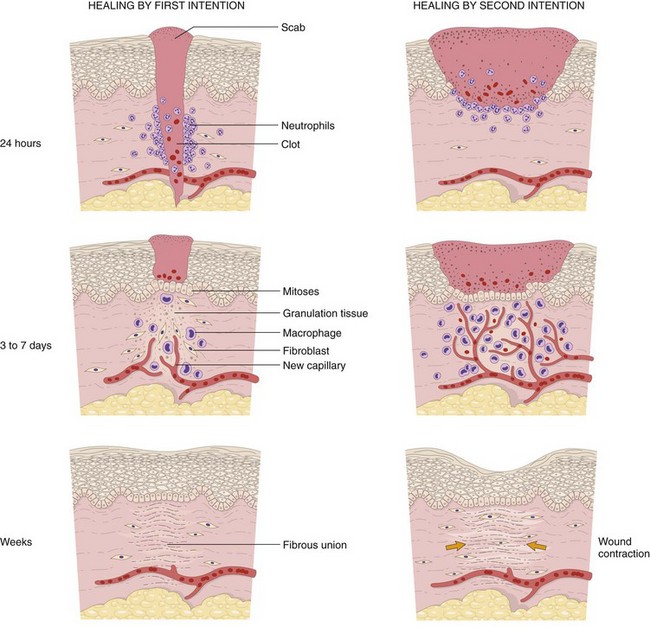
Healing by Second Intention
When cell or tissue loss is more extensive, such as in large wounds, at sites of abscess formation, ulceration, and ischemic necrosis (infarction) in parenchymal organs, the repair process is more complex and involves a combination of regeneration and scarring. In second intention healing of skin wounds, also known as healing by secondary union (Fig. 2–34; see also Fig. 2–33), the inflammatory reaction is more intense, and there is development of abundant granulation tissue, with accumulation of ECM and formation of a large scar, followed by wound contraction mediated by the action of myofibroblasts.
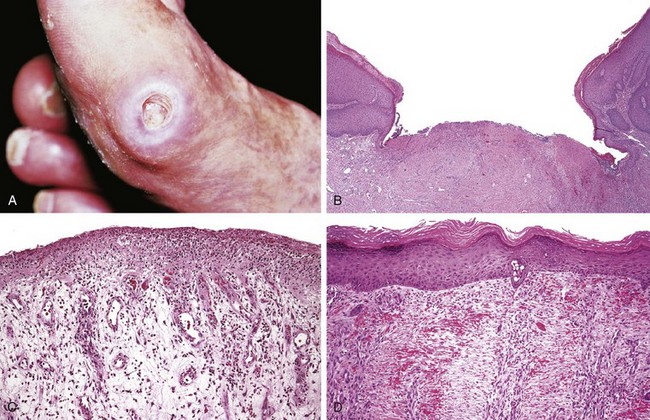
Figure 2–34 Healing of skin ulcers. A, Pressure ulcer of the skin, commonly found in diabetic patients. B, A skin ulcer with a large gap between the edges of the lesion. C, A thin layer of epidermal re-epithelialization, and extensive granulation tissue formation in the dermis. D, Continuing re-epithelialization of the epidermis and wound contraction.
(Courtesy of Z. Argenyi, MD, University of Washington, Seattle, Wash.)
Secondary healing differs from primary healing in several respects:
• A larger clot or scab rich in fibrin and fibronectin forms at the surface of the wound.
• Inflammation is more intense because large tissue defects have a greater volume of necrotic debris, exudate, and fibrin that must be removed. Consequently, large defects have a greater potential for secondary, inflammation-mediated, injury.
• Larger defects require a greater volume of granulation tissue to fill in the gaps and provide the underlying framework for the regrowth of tissue epithelium. A greater volume of granulation tissue generally results in a greater mass of scar tissue.
• Secondary healing involves wound contraction. Within 6 weeks, for example, large skin defects may be reduced to 5% to 10% of their original size, largely by contraction. This process has been ascribed to the presence of myofibroblasts, which are modified fibroblasts exhibiting many of the ultrastructural and functional features of contractile smooth muscle cells.
Wound Strength
Carefully sutured wounds have approximately 70% of the strength of normal skin, largely because of the placement of sutures. When sutures are removed, usually at 1 week, wound strength is approximately 10% of that of unwounded skin, but this increases rapidly over the next 4 weeks. The recovery of tensile strength results from collagen synthesis exceeding degradation during the first 2 months, and from structural modifications of collagen (e.g., cross-linking, increased fiber size) when synthesis declines at later times. Wound strength reaches approximately 70% to 80% of normal by 3 months and usually does not improve substantially beyond that point.
Fibrosis in Parenchymal Organs
Deposition of collagen is part of normal wound healing. The term fibrosis is used to denote the excessive deposition of collagen and other ECM components in a tissue. As already mentioned, the terms scar and fibrosis are used interchangeably, but fibrosis most often refers to the deposition of collagen in chronic diseases.
The basic mechanisms of fibrosis are the same as those of scar formation during tissue repair. However, tissue repair typically occurs after a short-lived injurious stimulus and follows an orderly sequence of steps, whereas fibrosis is induced by persistent injurious stimuli such as infections, immunologic reactions, and other types of tissue injury. The fibrosis seen in chronic diseases such as pulmonary fibrosis is often responsible for organ dysfunction and even organ failure.
Summary
Cutaneous Wound Healing and Pathologic Aspects of Repair
• Cutaneous wounds can heal by primary union (first intention) or secondary union (second intention); secondary healing involves more extensive scarring and wound contraction.
• Wound healing can be altered by many conditions, particularly infection and diabetes; the type, volume, and location of the injury are also important factors in healing.
• Excessive production of ECM can cause keloids in the skin.
• Persistent stimulation of collagen synthesis in chronic inflammatory diseases leads to fibrosis of the tissue.
Bradley JR. TNF-mediated inflammatory disease. J Pathol. 2008;214:149. [An overview of the biology of TNF and the clinical utility of TNF antagonists.]
Carlson BM. Some principles of regeneration in mammalian systems. Anat Rec. 2005;287:4. [A thoughtful review of the evolutionary aspects and general mechanisms of limb and organ regeneration.]
Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932. [A review of the main aspects of normal and abnormal angiogenesis.]
Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610. [An overview of the functions of chemokines in inflammation.]
Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells and stem cells. Hepatology. 2004;39:1477. [A review of the cellular and molecular mechanisms of liver regeneration.]
Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6:232. [An excellent review of the biology of IL-1 and the therapeutic targeting of this cytokine in inflammatory diseases.]
Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314. [An excellent review of the principles of tissue regeneration and repair.]
Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673. [An excellent review of the molecular mechanisms of integrin signaling, linking ECM components to intracellular signal transduction pathways.]
Jiang D, Liang J, Noble PW. Hyaluronans in tissue injury and repair. Annu Rev Cell Dev Biol. 2007;23:435. [A discussion of the role of a major family of ECM proteins in tissue repair.]
Khanapure SP, Garvey DS, Janero DR, et al. Eicosanoids in inflammation: biosynthesis, pharmacology, and therapeutic frontiers. Curr Top Med Chem. 2007;7:311. [A summary of the properties of this important class of inflammatory mediators.]
Lentsch AB, Ward PA. Regulation of inflammatory vascular damage. J Pathol. 2000;190:343. [Discussion of the mechanisms of endothelial damage and increased vascular permeability.]
Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678. [A modern discussion of leukocyte recruitment to sites of inflammation.]
Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad, and the ugly. Trends Cell Biol. 2005;15:599. [Good review on the multiple roles of inflammatory cells in repair.]
Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27:621. [An excellent discussion of autoinflammatory syndromes caused by gain-of-function mutations in components of the inflammasome.]
McAnully RJ. Fibroblasts and myofibroblasts: their source, function, and role in disease. Int J Biochem Cell Biol. 2007;39:666. [A discussion of the two major types of stroma cells and their roles on tissue repair and fibrosis.]
Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011;6:323. [A thoughtful review of the mechanisms by which leukocytes traverse the endothelium.]
Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007;2:251. [A review of the VEGF family of growth factors and their role in angiogenesis in cancer, inflammation, and various disease states.]
Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871. [A discussion of the abnormalities that lead to chronic inflammation.]
Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221. [A review of the function of matrix modifying enzymes in tissue repair.]
Papayannapoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30:513. [A review of a newly discovered mechanism by which neutrophils destroy microbes.]
Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785. [A current overview of the activation and functions of the complement system and its role in disease.]
Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008;3:99. [An excellent discussion of how the immune system recognizes necrotic cells.]
Schultz GS, Wysocki A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009;17:153. [A discussion of the regulation of growth factors by the ECM.]
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821. [An excellent review of the cellular machinery that recognizes products of dead cells, many foreign and abnormal substances, and some microbes.]
Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197. [An excellent discussion of the microbicidal mechanisms of neutrophils.]
Stappenbeck TS, Miyoshi H. The role of stromal stem cells in tissue regeneration and wound repair. Science. 2009;324:1666. [An excellent review of the role of tissue stem cells in repair.]
Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, et al. The pathogenesis of sepsis. Annu Rev Pathol. 2011;6:19. [A discussion of the current concepts of pathogenic mechanisms in sepsis and septic shock.]
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805. [An excellent overview of Toll-like receptors and other pattern recognition receptor families, and their roles in host defense and inflammation.]
Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199. [An overview of the cellular mechanisms of fibrosis, with an emphasis on the role of the immune system in fibrotic reactions to chronic infections.]
Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010;465:704. [A review of the exciting technology for generating iPS cells for regenerative medicine.]