Chapter 1 Cell Injury, Cell Death, and Adaptations
See Targeted Therapy available online at studentconsult.com
Introduction to Pathology
Literally translated, pathology is the study (logos) of disease (pathos, suffering). It involves the investigation of the causes of disease and the associated changes at the levels of cells, tissues, and organs, which in turn give rise to the presenting signs and symptoms of the patient. There are two important terms that students will encounter throughout their study of pathology and medicine:
• Etiology is the origin of a disease, including the underlying causes and modifying factors. It is now clear that most common diseases, such as hypertension, diabetes, and cancer, are caused by a combination of inherited genetic susceptibility and various environmental triggers. Understanding the genetic and environmental factors underlying diseases is a major theme of modern medicine.
• Pathogenesis refers to the steps in the development of disease. It describes how etiologic factors trigger cellular and molecular changes that give rise to the specific functional and structural abnormalities that characterize the disease. Whereas etiology refers to why a disease arises, pathogenesis describes how a disease develops.
Defining the etiology and pathogenesis of disease not only is essential for understanding a disease but is also the basis for developing rational treatments. Thus, by explaining the causes and development of disease pathology provides the scientific foundation for the practice of medicine.
To render diagnoses and guide therapy in clinical practice, pathologists identify changes in the gross or microscopic appearance (morphology) of cells and tissues, and biochemical alterations in body fluids (such as blood and urine). Pathologists also use a variety of morphologic, molecular, microbiologic, and immunologic techniques to define the biochemical, structural, and functional changes that occur in cells, tissues, and organs in response to injury. Traditionally, the discipline is divided into general pathology and systemic pathology; the former focuses on the cellular and tissue alterations caused by pathologic stimuli in most tissues, while the latter examines the reactions and abnormalities of different specialized organs. In this book we first cover the broad principles of general pathology and then progress to specific disease processes in individual organs.
Overview of Cellular Responses to Stress and Noxious Stimuli
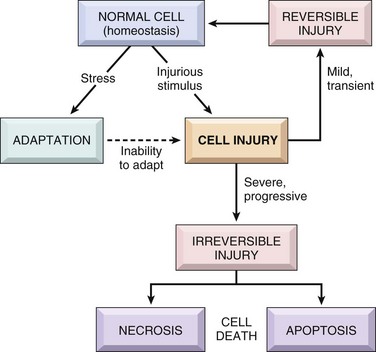
Cells are active participants in their environment, constantly adjusting their structure and function to accommodate changing demands and extracellular stresses. Cells normally maintain a steady state called homeostasis in which the intracellular milieu is kept within a fairly narrow range of physiologic parameters. As cells encounter physiologic stresses or pathologic stimuli, they can undergo adaptation, achieving a new steady state and preserving viability and function. The principal adaptive responses are hypertrophy, hyperplasia, atrophy, and metaplasia. If the adaptive capability is exceeded or if the external stress is inherently harmful, cell injury develops (Fig. 1–1). Within certain limits, injury is reversible, and cells return to a stable baseline; however, if the stress is severe, persistent and rapid in onset, it results in irreversible injury and death of the affected cells. Cell death is one of the most crucial events in the evolution of disease in any tissue or organ. It results from diverse causes, including ischemia (lack of blood flow), infections, toxins, and immune reactions. Cell death also is a normal and essential process in embryogenesis, the development of organs, and the maintenance of homeostasis.
The relationships among normal, adapted, and reversibly and irreversibly injured cells are well illustrated by the responses of the heart to different types of stress (Fig. 1–2). Myocardium subjected to persistent increased load, as in hypertension or with a narrowed (stenotic) valve, adapts by undergoing hypertrophy—an increase in the size of the individual cells and ultimately the entire heart—to generate the required higher contractile force. If the increased demand is not relieved, or if the myocardium is subjected to reduced blood flow (ischemia) from an occluded coronary artery, the muscle cells may undergo injury. Myocardium may be reversibly injured if the stress is mild or the arterial occlusion is incomplete or sufficiently brief, or it may undergo irreversible injury and cell death (infarction) after complete or prolonged occlusion. Also of note, stresses and injury affect not only the morphology but also the functional status of cells and tissues. Thus, reversibly injured myocytes are not dead and may resemble normal myocytes morphologically; however, they are transiently noncontractile, so even mild injury can have a significant clinical impact. Whether a specific form of stress induces adaptation or causes reversible or irreversible injury depends not only on the nature and severity of the stress but also on several other variables, including basal cellular metabolism and blood and nutrient supply.
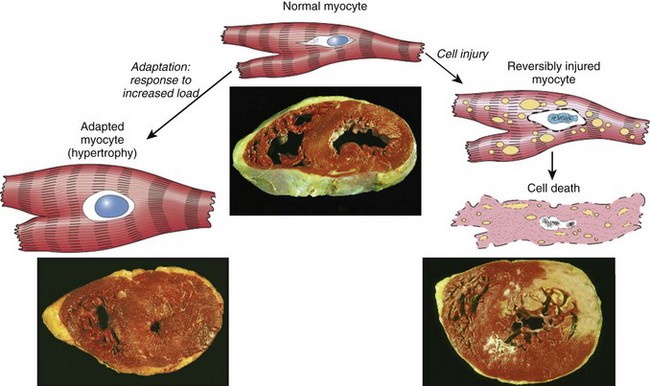
Figure 1–2 The relationship among normal, adapted, reversibly injured, and dead myocardial cells. The cellular adaptation depicted here is hypertrophy, the type of reversible injury is ischemia, and the irreversible injury is ischemic coagulative necrosis. In the example of myocardial hypertrophy (lower left), the left ventricular wall is thicker than 2 cm (normal, 1–1.5 cm). Reversibly injured myocardium shows functional effects without any gross or light microscopic changes, or reversible changes like cellular swelling and fatty change (shown here). In the specimen showing necrosis (lower right) the transmural light area in the posterolateral left ventricle represents an acute myocardial infarction. All three transverse sections of myocardium have been stained with triphenyltetrazolium chloride, an enzyme substrate that colors viable myocardium magenta. Failure to stain is due to enzyme loss after cell death.
In this chapter we discuss first how cells adapt to stresses and then the causes, mechanisms, and consequences of the various forms of acute cell damage, including reversible cell injury, subcellular alterations, and cell death. We conclude with three other processes that affect cells and tissues: intracellular accumulations, pathologic calcification, and cell aging.
Cellular Adaptations to Stress
Adaptations are reversible changes in the number, size, phenotype, metabolic activity, or functions of cells in response to changes in their environment. Physiologic adaptations usually represent responses of cells to normal stimulation by hormones or endogenous chemical mediators (e.g., the hormone-induced enlargement of the breast and uterus during pregnancy). Pathologic adaptations are responses to stress that allow cells to modulate their structure and function and thus escape injury. Such adaptations can take several distinct forms.
Hypertrophy
Hypertrophy is an increase in the size of cells resulting in increase in the size of the organ. In contrast, hyperplasia (discussed next) is characterized by an increase in cell number because of proliferation of differentiated cells and replacement by tissue stem cells. Stated another way, in pure hypertrophy there are no new cells, just bigger cells containing increased amounts of structural proteins and organelles. Hyperplasia is an adaptive response in cells capable of replication, whereas hypertrophy occurs when cells have a limited capacity to divide. Hypertrophy and hyperplasia also can occur together, and obviously both result in an enlarged (hypertrophic) organ.
Hypertrophy can be physiologic or pathologic and is caused either by increased functional demand or by growth factor or hormonal stimulation.
• The massive physiologic enlargement of the uterus during pregnancy occurs as a consequence of estrogen-stimulated smooth muscle hypertrophy and smooth muscle hyperplasia (Fig. 1–3). In contrast, in response to increased demand the striated muscle cells in both the skeletal muscle and the heart can undergo only hypertrophy because adult muscle cells have a limited capacity to divide. Therefore, the chiseled physique of the avid weightlifter stems solely from the hypertrophy of individual skeletal muscles.
• An example of pathologic cellular hypertrophy is the cardiac enlargement that occurs with hypertension or aortic valve disease (Fig. 1–2).
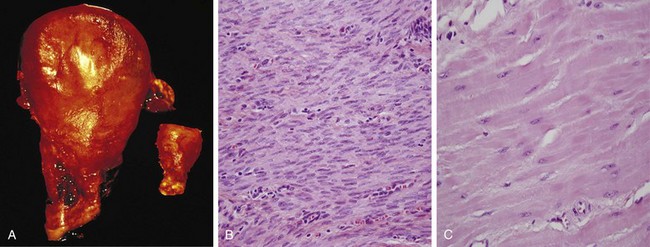
Figure 1–3 Physiologic hypertrophy of the uterus during pregnancy. A, Gross appearance of a normal uterus (right) and a gravid uterus (left) that was removed for postpartum bleeding. B, Small spindle-shaped uterine smooth muscle cells from a normal uterus. C, Large, plump hypertrophied smooth muscle cells from a gravid uterus; compare with B. (B and C, Same magnification.)
The mechanisms driving cardiac hypertrophy involve at least two types of signals: mechanical triggers, such as stretch, and trophic triggers, which typically are soluble mediators that stimulate cell growth, such as growth factors and adrenergic hormones. These stimuli turn on signal transduction pathways that lead to the induction of a number of genes, which in turn stimulate synthesis of many cellular proteins, including growth factors and structural proteins. The result is the synthesis of more proteins and myofilaments per cell, which increases the force generated with each contraction, enabling the cell to meet increased work demands. There may also be a switch of contractile proteins from adult to fetal or neonatal forms. For example, during muscle hypertrophy, the α-myosin heavy chain is replaced by the β form of the myosin heavy chain, which produces slower, more energetically economical contraction.
Whatever the exact mechanisms of hypertrophy, a limit is reached beyond which the enlargement of muscle mass can no longer compensate for the increased burden. When this happens in the heart, several “degenerative” changes occur in the myocardial fibers, of which the most important are fragmentation and loss of myofibrillar contractile elements. The variables that limit continued hypertrophy and cause the regressive changes are incompletely understood. There may be finite limits of the vasculature to adequately supply the enlarged fibers, of the mitochondria to supply adenosine triphosphate (ATP), or of the biosynthetic machinery to provide the contractile proteins or other cytoskeletal elements. The net result of these changes is ventricular dilation and ultimately cardiac failure, a sequence of events that illustrates how an adaptation to stress can progress to functionally significant cell injury if the stress is not relieved.
Hyperplasia
As discussed earlier, hyperplasia takes place if the tissue contains cell populations capable of replication; it may occur concurrently with hypertrophy and often in response to the same stimuli.
Hyperplasia can be physiologic or pathologic. In both situations, cellular proliferation is stimulated by growth factors that are produced by a variety of cell types.
• The two types of physiologic hyperplasia are (1) hormonal hyperplasia, exemplified by the proliferation of the glandular epithelium of the female breast at puberty and during pregnancy, and (2) compensatory hyperplasia, in which residual tissue grows after removal or loss of part of an organ. For example, when part of a liver is resected, mitotic activity in the remaining cells begins as early as 12 hours later, eventually restoring the liver to its normal weight. The stimuli for hyperplasia in this setting are polypeptide growth factors produced by uninjured hepatocytes as well as nonparenchymal cells in the liver (Chapter 2). After restoration of the liver mass, cell proliferation is “turned off” by various growth inhibitors.
• Most forms of pathologic hyperplasia are caused by excessive hormonal or growth factor stimulation. For example, after a normal menstrual period there is a burst of uterine epithelial proliferation that is normally tightly regulated by stimulation through pituitary hormones and ovarian estrogen and by inhibition through progesterone. However, a disturbed balance between estrogen and progesterone causes endometrial hyperplasia, which is a common cause of abnormal menstrual bleeding. Hyperplasia also is an important response of connective tissue cells in wound healing, in which proliferating fibroblasts and blood vessels aid in repair (Chapter 2). In this process, growth factors are produced by white blood cells (leukocytes) responding to the injury and by cells in the extracellular matrix. Stimulation by growth factors also is involved in the hyperplasia that is associated with certain viral infections; for example, papillomaviruses cause skin warts and mucosal lesions composed of masses of hyperplastic epithelium. Here the growth factors may be encoded by viral genes or by the genes of the infected host cells.
An important point is that in all of these situations, the hyperplastic process remains controlled; if the signals that initiate it abate, the hyperplasia disappears. It is this responsiveness to normal regulatory control mechanisms that distinguishes pathologic hyperplasias from cancer, in which the growth control mechanisms become dysregulated or ineffective (Chapter 5). Nevertheless, in many cases, pathologic hyperplasia constitutes a fertile soil in which cancers may eventually arise. For example, patients with hyperplasia of the endometrium are at increased risk of developing endometrial cancer (Chapter 18).
Atrophy
Shrinkage in the size of the cell by the loss of cell substance is known as atrophy. When a sufficient number of cells are involved, the entire tissue or organ diminishes in size, becoming atrophic (Fig. 1–4). Although atrophic cells may have diminished function, they are not dead.
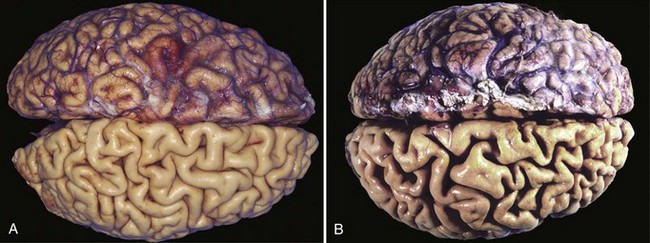
Figure 1–4 Atrophy as seen in the brain. A, Normal brain of a young adult. B, Atrophy of the brain in an 82-year-old man with atherosclerotic disease. Atrophy of the brain is due to aging and reduced blood supply. Note that loss of brain substance narrows the gyri and widens the sulci. The meninges have been stripped from the bottom half of each specimen to reveal the surface of the brain.
Causes of atrophy include a decreased workload (e.g., immobilization of a limb to permit healing of a fracture), loss of innervation, diminished blood supply, inadequate nutrition, loss of endocrine stimulation, and aging (senile atrophy). Although some of these stimuli are physiologic (e.g., the loss of hormone stimulation in menopause) and others pathologic (e.g., denervation), the fundamental cellular changes are identical. They represent a retreat by the cell to a smaller size at which survival is still possible; a new equilibrium is achieved between cell size and diminished blood supply, nutrition, or trophic stimulation.
The mechanisms of atrophy consist of a combination of decreased protein synthesis and increased protein degradation in cells.
• Protein synthesis decreases because of reduced metabolic activity.
• The degradation of cellular proteins occurs mainly by the ubiquitin-proteasome pathway. Nutrient deficiency and disuse may activate ubiquitin ligases, which attach multiple copies of the small peptide ubiquitin to cellular proteins and target them for degradation in proteasomes. This pathway is also thought to be responsible for the accelerated proteolysis seen in a variety of catabolic conditions, including the cachexia associated with cancer.
• In many situations, atrophy is also accompanied by increased autophagy, with resulting increases in the number of autophagic vacuoles. Autophagy (“self-eating”) is the process in which the starved cell eats its own components in an attempt to survive. We describe this process later in the chapter.
Metaplasia
Metaplasia is a reversible change in which one adult cell type (epithelial or mesenchymal) is replaced by another adult cell type. In this type of cellular adaptation, a cell type sensitive to a particular stress is replaced by another cell type better able to withstand the adverse environment. Metaplasia is thought to arise by reprogramming of stem cells to differentiate along a new pathway rather than a phenotypic change (transdifferentiation) of already differentiated cells.
Epithelial metaplasia is exemplified by the squamous change that occurs in the respiratory epithelium of habitual cigarette smokers (Fig. 1–5). The normal ciliated columnar epithelial cells of the trachea and bronchi are focally or widely replaced by stratified squamous epithelial cells. The rugged stratified squamous epithelium may be able to survive the noxious chemicals in cigarette smoke that the more fragile specialized epithelium would not tolerate. Although the metaplastic squamous epithelium has survival advantages, important protective mechanisms are lost, such as mucus secretion and ciliary clearance of particulate matter. Epithelial metaplasia is therefore a double-edged sword. Moreover, the influences that induce metaplastic change, if persistent, may predispose to malignant transformation of the epithelium. In fact, squamous metaplasia of the respiratory epithelium often coexists with lung cancers composed of malignant squamous cells. It is thought that cigarette smoking initially causes squamous metaplasia, and cancers arise later in some of these altered foci. Since vitamin A is essential for normal epithelial differentiation, its deficiency may also induce squamous metaplasia in the respiratory epithelium. Metaplasia need not always occur in the direction of columnar to squamous epithelium; in chronic gastric reflux, the normal stratified squamous epithelium of the lower esophagus may undergo metaplastic transformation to gastric or intestinal-type columnar epithelium. Metaplasia may also occur in mesenchymal cells but in these situations it is generally a reaction to some pathologic alteration and not an adaptive response to stress. For example, bone is occasionally formed in soft tissues, particularly in foci of injury.
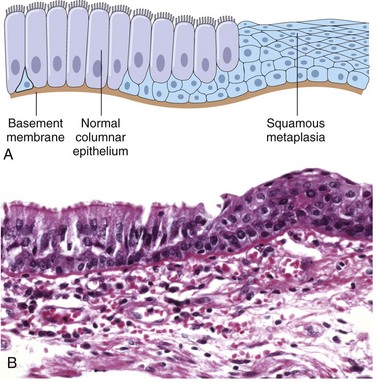
Figure 1–5 Metaplasia of normal columnar (left) to squamous epithelium (right) in a bronchus, shown schematically (A) and histologically (B).
 Summary
Summary
Cellular Adaptations to Stress
• Hypertrophy: increased cell and organ size, often in response to increased workload; induced by growth factors produced in response to mechanical stress or other stimuli; occurs in tissues incapable of cell division
• Hyperplasia: increased cell numbers in response to hormones and other growth factors; occurs in tissues whose cells are able to divide or contain abundant tissue stem cells
• Atrophy: decreased cell and organ size, as a result of decreased nutrient supply or disuse; associated with decreased synthesis of cellular building blocks and increased breakdown of cellular organelles
• Metaplasia: change in phenotype of differentiated cells, often in response to chronic irritation, that makes cells better able to withstand the stress; usually induced by altered differentiation pathway of tissue stem cells; may result in reduced functions or increased propensity for malignant transformation
Overview of Cell Injury and Cell Death
As stated at the beginning of the chapter, cell injury results when cells are stressed so severely that they are no longer able to adapt or when cells are exposed to inherently damaging agents or suffer from intrinsic abnormalities (e.g., in DNA or proteins). Different injurious stimuli affect many metabolic pathways and cellular organelles. Injury may progress through a reversible stage and culminate in cell death (Fig. 1–1).
• Reversible cell injury. In early stages or mild forms of injury the functional and morphologic changes are reversible if the damaging stimulus is removed. At this stage, although there may be significant structural and functional abnormalities, the injury has typically not progressed to severe membrane damage and nuclear dissolution.
• Cell death. With continuing damage, the injury becomes irreversible, at which time the cell cannot recover and it dies. There are two types of cell death—necrosis and apoptosis—which differ in their mechanisms, morphology, and roles in disease and physiology (Fig. 1–6 and Table 1–1). When damage to membranes is severe, enzymes leak out of lysosomes, enter the cytoplasm, and digest the cell, resulting in necrosis. Cellular contents also leak through the damaged plasma membrane into the extracellular space, where they elicit a host reaction (inflammation). Necrosis is the major pathway of cell death in many commonly encountered injuries, such as those resulting from ischemia, exposure to toxins, various infections, and trauma. When a cell is deprived of growth factors, or the cell’s DNA or proteins are damaged beyond repair, typically the cell kills itself by another type of death, called apoptosis, which is characterized by nuclear dissolution without complete loss of membrane integrity. Whereas necrosis is always a pathologic process, apoptosis serves many normal functions and is not necessarily associated with pathologic cell injury. Furthermore, in keeping with its role in certain physiologic processes, apoptosis does not elicit an inflammatory response. The morphologic features, mechanisms, and significance of these two death pathways are discussed in more detail later in the chapter.
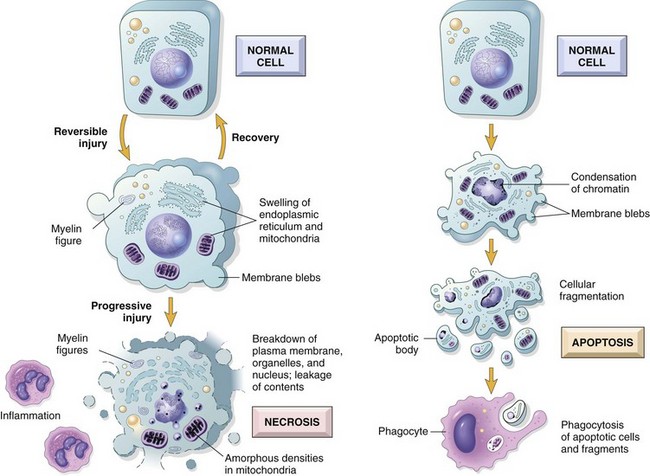
Table 1–1 Features of Necrosis and Apoptosis
| Feature | Necrosis | Apoptosis |
|---|---|---|
| Cell size | Enlarged (swelling) | Reduced (shrinkage) |
| Nucleus | Pyknosis → karyorrhexis → karyolysis | Fragmentation into nucleosome size fragments |
| Plasma membrane | Disrupted | Intact; altered structure, especially orientation of lipids |
| Cellular contents | Enzymatic digestion; may leak out of cell | Intact; may be released in apoptotic bodies |
| Adjacent inflammation | Frequent | No |
| Physiologic or pathologic role | Invariably pathologic (culmination of irreversible cell injury) | Often physiologic; means of eliminating unwanted cells; may be pathologic after some forms of cell injury, especially DNA and protein damage |
DNA, deoxyribonucleic acid.
Causes of Cell Injury
The causes of cell injury range from the gross physical trauma of a motor vehicle accident to the single gene defect that results in a nonfunctional enzyme underlying a specific metabolic disease. Most injurious stimuli can be grouped into the following categories.
Oxygen Deprivation
Hypoxia, or oxygen deficiency, interferes with aerobic oxidative respiration and is an extremely important and common cause of cell injury and death. Hypoxia should be distinguished from ischemia, which is a loss of blood supply in a tissue due to impeded arterial flow or reduced venous drainage. While ischemia is the most common cause of hypoxia, oxygen deficiency can also result from inadequate oxygenation of the blood, as in pneumonia, or from reduction in the oxygen-carrying capacity of the blood, as in blood loss anemia or carbon monoxide (CO) poisoning. (CO forms a stable complex with hemoglobin that prevents oxygen binding.)
Chemical Agents
An increasing number of chemical substances that can injure cells are being recognized; even innocuous substances such as glucose, salt, or even water, if absorbed or administered in excess, can so derange the osmotic environment that cell injury or death results. Agents commonly known as poisons cause severe damage at the cellular level by altering membrane permeability, osmotic homeostasis, or the integrity of an enzyme or cofactor, and exposure to such poisons can culminate in the death of the whole organism. Other potentially toxic agents are encountered daily in the environment; these include air pollutants, insecticides, CO, asbestos, and “social stimuli” such as ethanol. Many therapeutic drugs can cause cell or tissue injury in a susceptible patient or if used excessively or inappropriately (Chapter 7). Even oxygen at sufficiently high partial pressures is toxic.
Infectious Agents
Agents of infection range from submicroscopic viruses to meter-long tapeworms; in between are the rickettsiae, bacteria, fungi, and protozoans. The diverse ways in which infectious pathogens cause injury are discussed in Chapter 8.
Immunologic Reactions
Although the immune system defends the body against pathogenic microbes, immune reactions can also result in cell and tissue injury. Examples are autoimmune reactions against one’s own tissues and allergic reactions against environmental substances in genetically susceptible individuals (Chapter 4).
Genetic Factors
Genetic aberrations can result in pathologic changes as conspicuous as the congenital malformations associated with Down syndrome or as subtle as the single amino acid substitution in hemoglobin S giving rise to sickle cell anemia (Chapter 6). Genetic defects may cause cell injury as a consequence of deficiency of functional proteins, such as enzymes in inborn errors of metabolism, or accumulation of damaged DNA or misfolded proteins, both of which trigger cell death when they are beyond repair. Genetic variations (polymorphisms) contribute to the development of many complex diseases and can influence the susceptibility of cells to injury by chemicals and other environmental insults.
Nutritional Imbalances
Even in the current era of burgeoning global affluence, nutritional deficiencies remain a major cause of cell injury. Protein–calorie insufficiency among underprivileged populations is only the most obvious example; specific vitamin deficiencies are not uncommon even in developed countries with high standards of living (Chapter 7). Ironically, disorders of nutrition rather than lack of nutrients are also important causes of morbidity and mortality; for example, obesity markedly increases the risk for type 2 diabetes mellitus. Moreover, diets rich in animal fat are strongly implicated in the development of atherosclerosis as well as in increased vulnerability to many disorders, including cancer.
Physical Agents
Trauma, extremes of temperature, radiation, electric shock, and sudden changes in atmospheric pressure all have wide-ranging effects on cells (Chapter 7).
Aging
Cellular senescence leads to alterations in replicative and repair abilities of individual cells and tissues. All of these changes result in a diminished ability to respond to damage and, eventually, the death of cells and of the organism. The mechanisms underlying cellular aging are discussed separately at the end of the chapter.
The Morphology of Cell and Tissue Injury
It is useful to describe the structural alterations that occur in damaged cells before we discuss the biochemical mechanisms that bring about these changes. All stresses and noxious influences exert their effects first at the molecular or biochemical level. Cellular function may be lost long before cell death occurs, and the morphologic changes of cell injury (or death) lag far behind both (Fig. 1–7). For example, myocardial cells become noncontractile after 1 to 2 minutes of ischemia, although they do not die until 20 to 30 minutes of ischemia have elapsed. These myocytes may not appear dead by electron microscopy for 2 to 3 hours, or by light microscopy for 6 to 12 hours.
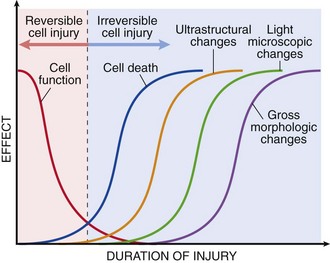
Figure 1–7 The relationship among cellular function, cell death, and the morphologic changes of cell injury. Note that cells may rapidly become nonfunctional after the onset of injury, although they are still viable, with potentially reversible damage; with a longer duration of injury, irreversible injury and cell death may result. Note also that cell death typically precedes ultrastructural, light microscopic, and grossly visible morphologic changes.
The cellular derangements of reversible injury can be corrected, and if the injurious stimulus abates, the cell can return to normalcy. Persistent or excessive injury, however, causes cells to pass the nebulous “point of no return” into irreversible injury and cell death. The events that determine when reversible injury becomes irreversible and progresses to cell death remain poorly understood. The clinical relevance of this question is obvious; if the biochemical and molecular changes that predict cell death can be identified with precision, it may be possible to devise strategies for preventing the transition from reversible to irreversible cell injury. Although there are no definitive morphologic or biochemical correlates of irreversibility, two phenomena consistently characterize irreversibility: the inability to correct mitochondrial dysfunction (lack of oxidative phosphorylation and ATP generation) even after resolution of the original injury, and profound disturbances in membrane function. As mentioned earlier, injury to lysosomal membranes results in the enzymatic dissolution of the injured cell, which is the culmination of injury progressing to necrosis.
As mentioned earlier, different injurious stimuli may induce death by necrosis or apoptosis (Fig. 1–6 and Table 1–1). Below we describe the morphology of reversible cell injury and necrosis; the sequence of morphologic alterations in these processes is illustrated in Figure 1–6, left. Apoptosis has many unique features, and we describe it separately later in the chapter.
Reversible Injury
The two main morphologic correlates of reversible cell injury are cellular swelling and fatty change. Cellular swelling is the result of failure of energy-dependent ion pumps in the plasma membrane, leading to an inability to maintain ionic and fluid homeostasis. Fatty change occurs in hypoxic injury and in various forms of toxic or metabolic injury and is manifested by the appearance of small or large lipid vacuoles in the cytoplasm. The mechanisms of fatty change are discussed in Chapter 15.
In some situations, potentially injurious insults induce specific alterations in cellular organelles, like the ER. The smooth ER is involved in the metabolism of various chemicals, and cells exposed to these chemicals show hypertrophy of the ER as an adaptive response that may have important functional consequences. For instance, barbiturates are metabolized in the liver by the cytochrome P-450 mixed-function oxidase system found in the smooth ER. Protracted use of barbiturates leads to a state of tolerance, with a decrease in the effects of the drug and the need to use increasing doses. This adaptation is due to increased volume (hypertrophy) of the smooth ER of hepatocytes and consequent increased P-450 enzymatic activity. Although P-450–mediated modification is often thought of as “detoxification,” many compounds are rendered more injurious by this process; one example is carbon tetrachloride (CCl4), discussed later. In addition, the products formed by this oxidative metabolism include reactive oxygen species (ROS), which can injure the cell. Cells adapted to one drug have increased capacity to metabolize other compounds handled by the same system. Thus, if patients taking phenobarbital for epilepsy increase their alcohol intake, they may experience a drop in blood concentration of the antiseizure medication to subtherapeutic levels because of induction of smooth ER in response to the alcohol.
 Morphology
Morphology
Cellular swelling (Fig. 1–8, B), the first manifestation of almost all forms of injury to cells, is a reversible alteration that may be difficult to appreciate with the light microscope, but it may be more apparent at the level of the whole organ. When it affects many cells in an organ, it causes some pallor (as a result of compression of capillaries), increased turgor, and increase in weight of the organ. Microscopic examination may reveal small, clear vacuoles within the cytoplasm; these represent distended and pinched-off segments of the endoplasmic reticulum (ER). This pattern of nonlethal injury is sometimes called hydropic change or vacuolar degeneration. Fatty change is manifested by the appearance of lipid vacuoles in the cytoplasm. It is principally encountered in cells participating in fat metabolism (e.g., hepatocytes, myocardial cells) and is also reversible. Injured cells may also show increased eosinophilic staining, which becomes much more pronounced with progression to necrosis (described further on).
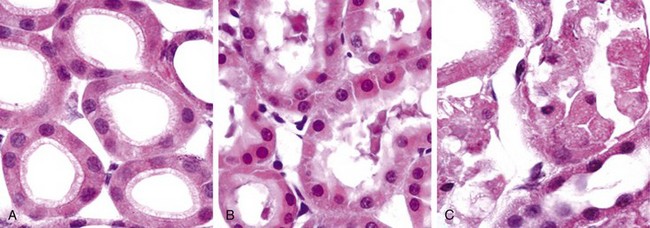
Figure 1–8 Morphologic changes in reversible and irreversible cell injury (necrosis). A, Normal kidney tubules with viable epithelial cells. B, Early (reversible) ischemic injury showing surface blebs, increased eosinophilia of cytoplasm, and swelling of occasional cells. C, Necrotic (irreversible) injury of epithelial cells, with loss of nuclei and fragmentation of cells and leakage of contents.
(Courtesy of Drs. Neal Pinckard and M.A. Venkatachalam, University of Texas Health Sciences Center, San Antonio, Tex.)
The intracellular changes associated with reversible injury (Fig. 1–6) include (1) plasma membrane alterations such as blebbing, blunting, or distortion of microvilli, and loosening of intercellular attachments; (2) mitochondrial changes such as swelling and the appearance of phospholipid-rich amorphous densities; (3) dilation of the ER with detachment of ribosomes and dissociation of polysomes; and (4) nuclear alterations, with clumping of chromatin. The cytoplasm may contain phospholipid masses, called myelin figures, which are derived from damaged cellular membranes.
Necrosis
Necrosis is the type of cell death that is associated with loss of membrane integrity and leakage of cellular contents culminating in dissolution of cells, largely resulting from the degradative action of enzymes on lethally injured cells. The leaked cellular contents often elicit a local host reaction, called inflammation, that attempts to eliminate the dead cells and start the subsequent repair process (Chapter 2). The enzymes responsible for digestion of the cell may be derived from the lysosomes of the dying cells themselves and from the lysosomes of leukocytes that are recruited as part of the inflammatory reaction to the dead cells.
Morphology
Necrosis is characterized by changes in the cytoplasm and nuclei of the injured cells (Figs. 1–6, left, and 1–8, C).
• Cytoplasmic changes. Necrotic cells show increased eosinophilia (i.e., pink staining from the eosin dye—the E in the hematoxylin and eosin [H&E] stain), attributable in part to increased binding of eosin to denatured cytoplasmic proteins and in part to loss of the basophilia that is normally imparted by the ribonucleic acid (RNA) in the cytoplasm (basophilia is the blue staining from the hematoxylin dye—the H in “H&E”). Compared with viable cells, the cell may have a more glassy, homogeneous appearance, mostly because of the loss of glycogen particles. Myelin figures are more prominent in necrotic cells than during reversible injury. When enzymes have digested cytoplasmic organelles, the cytoplasm becomes vacuolated and appears “moth-eaten.” By electron microscopy, necrotic cells are characterized by discontinuities in plasma and organelle membranes, marked dilation of mitochondria with the appearance of large amorphous densities, disruption of lysosomes, and intracytoplasmic myelin figures.
• Nuclear changes. Nuclear changes assume one of three patterns, all due to breakdown of DNA and chromatin. The basophilia of the chromatin may fade (karyolysis), presumably secondary to deoxyribonuclease (DNase) activity. A second pattern is pyknosis, characterized by nuclear shrinkage and increased basophilia; the DNA condenses into a solid shrunken mass. In the third pattern, karyorrhexis, the pyknotic nucleus undergoes fragmentation. In 1 to 2 days, the nucleus in a dead cell may completely disappear. Electron microscopy reveals profound nuclear changes culminating in nuclear dissolution.
• Fates of necrotic cells. Necrotic cells may persist for some time or may be digested by enzymes and disappear. Dead cells may be replaced by myelin figures, which are either phagocytosed by other cells or further degraded into fatty acids. These fatty acids bind calcium salts, which may result in the dead cells ultimately becoming calcified.
Patterns of Tissue Necrosis
There are several morphologically distinct patterns of tissue necrosis, which may provide clues about the underlying cause. Although the terms that describe these patterns do not reflect underlying mechanisms, such terms are in common use, and their implications are understood by both pathologists and clinicians. Most of these types of necrosis have distinct gross appearance; fibrinoid necrosis is detected only by histologic examination.
Morphology
• Coagulative necrosis is a form of necrosis in which the underlying tissue architecture is preserved for at least several days (Fig. 1–9). The affected tissues take on a firm texture. Presumably the injury denatures not only structural proteins but also enzymes, thereby blocking the proteolysis of the dead cells; as a result, eosinophilic, anucleate cells may persist for days or weeks. Leukocytes are recruited to the site of necrosis, and the dead cells are digested by the action of lysosomal enzymes of the leukocytes. The cellular debris is then removed by phagocytosis. Coagulative necrosis is characteristic of infarcts (areas of ischemic necrosis) in all of the solid organs except the brain.
• Liquefactive necrosis is seen in focal bacterial or, occasionally, fungal infections, because microbes stimulate the accumulation of inflammatory cells and the enzymes of leukocytes digest (“liquefy”) the tissue. For obscure reasons, hypoxic death of cells within the central nervous system often evokes liquefactive necrosis (Fig. 1–10). Whatever the pathogenesis, the dead cells are completely digested, transforming the tissue into a liquid viscous mass. Eventually, the digested tissue is removed by phagocytes. If the process was initiated by acute inflammation, as in a bacterial infection, the material is frequently creamy yellow and is called pus (Chapter 2).
• Although gangrenous necrosis is not a distinctive pattern of cell death, the term is still commonly used in clinical practice. It usually refers to the condition of a limb, generally the lower leg, that has lost its blood supply and has undergone coagulative necrosis involving multiple tissue layers. When bacterial infection is superimposed, coagulative necrosis is modified by the liquefactive action of the bacteria and the attracted leukocytes (resulting in so-called wet gangrene).
• Caseous necrosis is encountered most often in foci of tuberculous infection. Caseous means “cheese-like,” referring to the friable yellow-white appearance of the area of necrosis (Fig. 1–11). On microscopic examination, the necrotic focus appears as a collection of fragmented or lysed cells with an amorphous granular pink appearance in the usual H&E-stained tissue. Unlike with coagulative necrosis, the tissue architecture is completely obliterated and cellular outlines cannot be discerned. The area of caseous necrosis is often enclosed within a distinctive inflammatory border; this appearance is characteristic of a focus of inflammation known as a granuloma (Chapter 2).
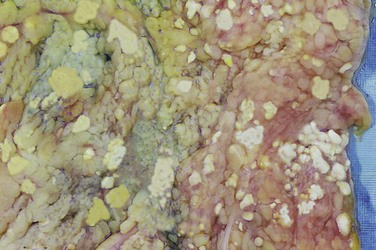
• Fat necrosis refers to focal areas of fat destruction, typically resulting from release of activated pancreatic lipases into the substance of the pancreas and the peritoneal cavity. This occurs in the calamitous abdominal emergency known as acute pancreatitis (Chapter 16). In this disorder, pancreatic enzymes that have leaked out of acinar cells and ducts liquefy the membranes of fat cells in the peritoneum, and lipases split the triglyceride esters contained within fat cells. The released fatty acids combine with calcium to produce grossly visible chalky white areas (fat saponification), which enable the surgeon and the pathologist to identify the lesions (Fig. 1–12). On histologic examination, the foci of necrosis contain shadowy outlines of necrotic fat cells with basophilic calcium deposits, surrounded by an inflammatory reaction.
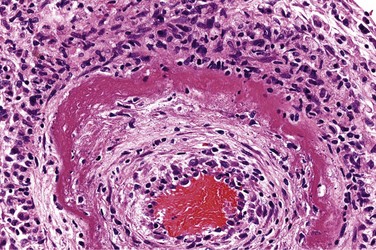
• Fibrinoid necrosis is a special form of necrosis, visible by light microscopy, usually in immune reactions in which complexes of antigens and antibodies are deposited in the walls of arteries. The deposited immune complexes, together with fibrin that has leaked out of vessels, produce a bright pink and amorphous appearance on H&E preparations called fibrinoid (fibrin-like) by pathologists (Fig. 1–13). The immunologically mediated diseases (e.g., polyarteritis nodosa) in which this type of necrosis is seen are described in Chapter 4.
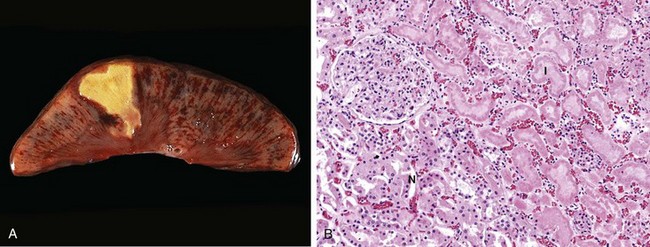
Figure 1–9 Coagulative necrosis. A, A wedge-shaped kidney infarct (yellow) with preservation of the outlines. B, Microscopic view of the edge of the infarct, with normal kidney (N) and necrotic cells in the infarct (I). The necrotic cells show preserved outlines with loss of nuclei, and an inflammatory infiltrate is present (difficult to discern at this magnification).
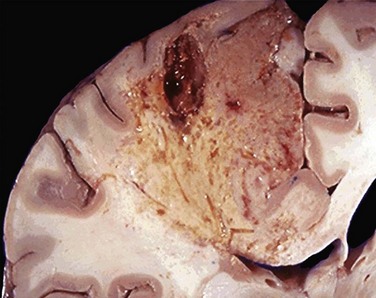
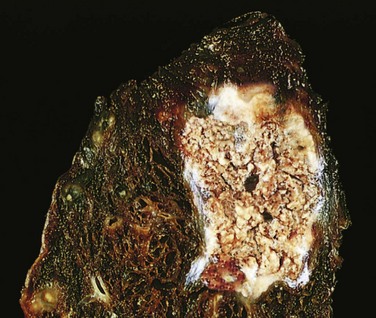
Figure 1–11 Caseous necrosis. Tuberculosis of the lung, with a large area of caseous necrosis containing yellow-white (cheesy) debris.
Leakage of intracellular proteins through the damaged cell membrane and ultimately into the circulation provides a means of detecting tissue-specific necrosis using blood or serum samples. Cardiac muscle, for example, contains a unique isoform of the enzyme creatine kinase and of the contractile protein troponin, whereas hepatic bile duct epithelium contains a temperature-resistant isoform of the enzyme alkaline phosphatase, and hepatocytes contain transaminases. Irreversible injury and cell death in these tissues result in increased serum levels of such proteins, and measurement of serum levels is used clinically to assess damage to these tissues.
Summary
Morphologic Alterations in Injured Cells and Tissues
• Reversible cell injury: cell swelling, fatty change, plasma membrane blebbing and loss of microvilli, mitochondrial swelling, dilation of the ER, eosinophilia (due to decreased cytoplasmic RNA)
• Necrosis: increased eosinophilia; nuclear shrinkage, fragmentation, and dissolution; breakdown of plasma membrane and organellar membranes; abundant myelin figures; leakage and enzymatic digestion of cellular contents
• Patterns of tissue necrosis: Under different conditions, necrosis in tissues may assume specific patterns: coagulative, liquefactive, gangrenous, caseous, fat, and fibrinoid.
Mechanisms of Cell Injury
Now that we have discussed the causes of cell injury and the morphologic changes in necrosis, we next consider in more detail the molecular basis of cell injury, and then illustrate the important principles with a few selected examples of common types of injury.
The biochemical mechanisms linking any given injury with the resulting cellular and tissue manifestations are complex, interconnected, and tightly interwoven with many intracellular metabolic pathways. Nevertheless, several general principles are relevant to most forms of cell injury:
• The cellular response to injurious stimuli depends on the type of injury, its duration, and its severity. Thus, low doses of toxins or a brief duration of ischemia may lead to reversible cell injury, whereas larger toxin doses or longer ischemic intervals may result in irreversible injury and cell death.
• The consequences of an injurious stimulus depend on the type, status, adaptability, and genetic makeup of the injured cell. The same injury has vastly different outcomes depending on the cell type; thus, striated skeletal muscle in the leg accommodates complete ischemia for 2 to 3 hours without irreversible injury, whereas cardiac muscle dies after only 20 to 30 minutes. The nutritional (or hormonal) status can also be important; clearly, a glycogen-replete hepatocyte will tolerate ischemia much better than one that has just burned its last glucose molecule. Genetically determined diversity in metabolic pathways can contribute to differences in responses to injurious stimuli. For instance, when exposed to the same dose of a toxin, individuals who inherit variants in genes encoding cytochrome P-450 may catabolize the toxin at different rates, leading to different outcomes. Much effort is now directed toward understanding the role of genetic polymorphisms in responses to drugs and toxins. The study of such interactions is called pharmacogenomics. In fact, genetic variations influence susceptibility to many complex diseases as well as responsiveness to various therapeutic agents. Using the genetic makeup of the individual patient to guide therapy is one example of “personalized medicine.”
• Cell injury results from functional and biochemical abnormalities in one or more of several essential cellular components (Fig. 1–14). The principal targets and biochemical mechanisms of cell injury are: (1) mitochondria and their ability to generate ATP and ROS under pathologic conditions; (2) disturbance in calcium homeostasis; (3) damage to cellular (plasma and lysosomal) membranes; and (4) damage to DNA and misfolding of proteins.
• Multiple biochemical alterations may be triggered by any one injurious insult. It is therefore difficult to assign any one mechanism to a particular insult or clinical situation in which cell injury is prominent. For this reason, therapies that target individual mechanisms of cell injury may not be effective.
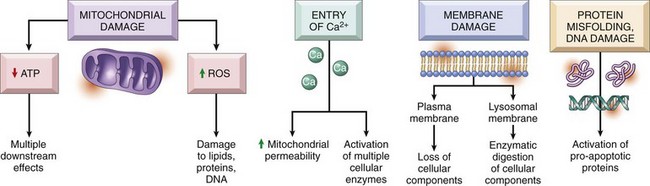
Figure 1–14 The principal biochemical mechanisms and sites of damage in cell injury. ATP, adenosine triphospate; ROS, reactive oxygen species.
With this background, we can briefly discuss the major biochemical mechanisms of cell injury.
Depletion of ATP
ATP, the energy store of cells, is produced mainly by oxidative phosphorylation of adenosine diphosphate (ADP) during reduction of oxygen in the electron transport system of mitochondria. In addition, the glycolytic pathway can generate ATP in the absence of oxygen using glucose derived either from the circulation or from the hydrolysis of intracellular glycogen. The major causes of ATP depletion are reduced supply of oxygen and nutrients, mitochondrial damage, and the actions of some toxins (e.g., cyanide). Tissues with a greater glycolytic capacity (e.g., the liver) are able to survive loss of oxygen and decreased oxidative phosphorylation better than are tissues with limited capacity for glycolysis (e.g., the brain). High-energy phosphate in the form of ATP is required for virtually all synthetic and degradative processes within the cell, including membrane transport, protein synthesis, lipogenesis, and the deacylation-reacylation reactions necessary for phospholipid turnover. It is estimated that in total, the cells of a healthy human burn 50 to 75 kg of ATP every day!
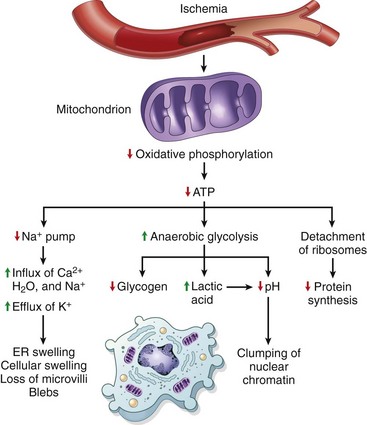
Significant depletion of ATP has widespread effects on many critical cellular systems (Fig. 1–15):
• The activity of plasma membrane ATP-dependent sodium pumps is reduced, resulting in intracellular accumulation of sodium and efflux of potassium. The net gain of solute is accompanied by iso-osmotic gain of water, causing cell swelling and dilation of the ER.
• There is a compensatory increase in anaerobic glycolysis in an attempt to maintain the cell’s energy sources. As a consequence, intracellular glycogen stores are rapidly depleted, and lactic acid accumulates, leading to decreased intracellular pH and decreased activity of many cellular enzymes.
• Failure of ATP-dependent Ca2+ pumps leads to influx of Ca2+, with damaging effects on numerous cellular components, described later.
• Prolonged or worsening depletion of ATP causes structural disruption of the protein synthetic apparatus, manifested as detachment of ribosomes from the rough ER (RER) and dissociation of polysomes into monosomes, with a consequent reduction in protein synthesis. Ultimately, there is irreversible damage to mitochondrial and lysosomal membranes, and the cell undergoes necrosis.
Mitochondrial Damage and Dysfunction
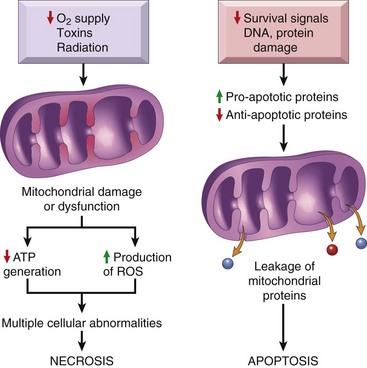
Mitochondria may be viewed as “mini-factories” that produce life-sustaining energy in the form of ATP. Not surprisingly, therefore, they are also critical players in cell injury and death (Fig. 1–16). Mitochondria are sensitive to many types of injurious stimuli, including hypoxia, chemical toxins, and radiation. Mitochondrial damage may result in several biochemical abnormalities:
• Failure of oxidative phosphorylation leads to progressive depletion of ATP, culminating in necrosis of the cell, as described earlier.
• Abnormal oxidative phosphorylation also leads to the formation of reactive oxygen species, which have many deleterious effects, described below.
• Damage to mitochondria is often associated with the formation of a high-conductance channel in the mitochondrial membrane, called the mitochondrial permeability transition pore. The opening of this channel leads to the loss of mitochondrial membrane potential and pH changes, further compromising oxidative phosphorylation.
• The mitochondria also contain several proteins that, when released into the cytoplasm, tell the cell there is internal injury and activate a pathway of apoptosis, discussed later.
Influx of Calcium
The importance of Ca2+ in cell injury was established by the experimental finding that depleting extracellular Ca2+ delays cell death after hypoxia and exposure to some toxins. Cytosolic free calcium is normally maintained by ATP-dependent calcium transporters at concentrations as much as 10,000 times lower than the concentration of extracellular calcium or of sequestered intracellular mitochondrial and ER calcium. Ischemia and certain toxins cause an increase in cytosolic calcium concentration, initially because of release of Ca2+ from the intracellular stores, and later resulting from increased influx across the plasma membrane. Increased cytosolic Ca2+ activates a number of enzymes, with potentially deleterious cellular effects (Fig. 1–17). These enzymes include phospholipases (which cause membrane damage), proteases (which break down both membrane and cytoskeletal proteins), endonucleases (which are responsible for DNA and chromatin fragmentation), and adenosine triphosphatases (ATPases) (thereby hastening ATP depletion). Increased intracellular Ca2+ levels may also induce apoptosis, by direct activation of caspases and by increasing mitochondrial permeability.
Accumulation of Oxygen-Derived Free Radicals (Oxidative Stress)
Free radicals are chemical species with a single unpaired electron in an outer orbital. Such chemical states are extremely unstable, and free radicals readily react with inorganic and organic chemicals; when generated in cells, they avidly attack nucleic acids as well as a variety of cellular proteins and lipids. In addition, free radicals initiate reactions in which molecules that react with free radicals are themselves converted into other types of free radicals, thereby propagating the chain of damage.
Reactive oxygen species (ROS) are a type of oxygen-derived free radical whose role in cell injury is well established. Cell injury in many circumstances involves damage by free radicals; these situations include ischemia-reperfusion (discussed later on), chemical and radiation injury, toxicity from oxygen and other gases, cellular aging, microbial killing by phagocytic cells, and tissue injury caused by inflammatory cells.
There are different types of ROS, and they are produced by two major pathways (Fig. 1–18).
• ROS are produced normally in small amounts in all cells during the reduction-oxidation (redox) reactions that occur during mitochondrial respiration and energy generation. In this process, molecular oxygen is sequentially reduced in mitochondria by the addition of four electrons to generate water. This reaction is imperfect, however, and small amounts of highly reactive but short-lived toxic intermediates are generated when oxygen is only partially reduced. These intermediates include superoxide ( ), which is converted to hydrogen peroxide (H2O2) spontaneously and by the action of the enzyme superoxide dismutase. H2O2 is more stable than
), which is converted to hydrogen peroxide (H2O2) spontaneously and by the action of the enzyme superoxide dismutase. H2O2 is more stable than  and can cross biologic membranes. In the presence of metals, such as Fe2+, H2O2 is converted to the highly reactive hydroxyl radical •OH by the Fenton reaction.
and can cross biologic membranes. In the presence of metals, such as Fe2+, H2O2 is converted to the highly reactive hydroxyl radical •OH by the Fenton reaction.
• ROS are produced in phagocytic leukocytes, mainly neutrophils and macrophages, as a weapon for destroying ingested microbes and other substances during inflammation and host defense (Chapter 2). The ROS are generated in the phagosomes and phagolysosomes of leukocytes by a process that is similar to mitochondrial respiration and is called the respiratory burst (or oxidative burst). In this process, a phagosome membrane enzyme catalyzes the generation of superoxide, which is converted to H2O2. H2O2 is in turn converted to a highly reactive compound hypochlorite (the major component of household bleach) by the enzyme myeloperoxidase, which is present in leukocytes. The role of ROS in inflammation is described in Chapter 2.
• Nitric oxide (NO) is another reactive free radical produced in leukocytes and other cells. It can react with  to form a highly reactive compound, peroxynitrite, which also participates in cell injury.
to form a highly reactive compound, peroxynitrite, which also participates in cell injury.
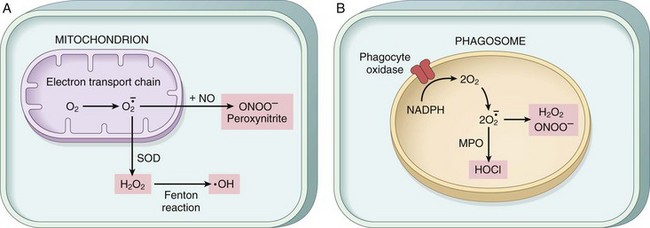
Figure 1–18 Pathways of production of reactive oxygen species. A, In all cells, superoxide (O2•) is generated during mitochondrial respiration by the electron transport chain and may be converted to H2O2 and the hydroxyl (•OH) free radical or to peroxynitrite (ONOO−). B, In leukocytes (mainly neutrophils and macrophages), the phagocyte oxidase enzyme in the phagosome membrane generates superoxide, which can be converted to other free radicals. Myeloperoxidase (MPO) in phagosomes also generates hypochlorite from reactive oxygen species (ROS). NO, nitric oxide; SOD, superoxide dismutase.
The damage caused by free radicals is determined by their rates of production and removal (Fig. 1–19). When the production of ROS increases or the scavenging systems are ineffective, the result is an excess of these free radicals, leading to a condition called oxidative stress.
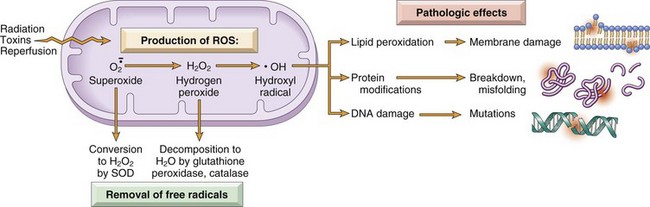
Figure 1–19 The generation, removal, and role of reactive oxygen species (ROS) in cell injury. The production of ROS is increased by many injurious stimuli. These free radicals are removed by spontaneous decay and by specialized enzymatic systems. Excessive production or inadequate removal leads to accumulation of free radicals in cells, which may damage lipids (by peroxidation), proteins, and deoxyribonucleic acid (DNA), resulting in cell injury.
The generation of free radicals is increased under several circumstances:
• The absorption of radiant energy (e.g., ultraviolet light, x-rays). Ionizing radiation can hydrolyze water into hydroxyl (•OH) and hydrogen (H•) free radicals.
• The enzymatic metabolism of exogenous chemicals (e.g., carbon tetrachloride—see later)
• Inflammation, in which free radicals are produced by leukocytes (Chapter 2)
Cells have developed many mechanisms to remove free radicals and thereby minimize injury. Free radicals are inherently unstable and decay spontaneously. There are also nonenzymatic and enzymatic systems that contribute to inactivation of free radicals (Fig. 1–19).
• The rate of decay of superoxide is significantly increased by the action of superoxide dismutases (SODs) found in many cell types.
• Glutathione (GSH) peroxidases are a family of enzymes whose major function is to protect cells from oxidative damage. The most abundant member of this family, glutathione peroxidase 1, is found in the cytoplasm of all cells. It catalyzes the breakdown of H2O2 by the reaction 2 GSH (glutathione) + H2O2 → GS-SG + 2 H2O. The intracellular ratio of oxidized glutathione (GSSG) to reduced glutathione (GSH) is a reflection of this enzyme’s activity and thus of the cell’s ability to catabolize free radicals.
• Catalase, present in peroxisomes, catalyzes the decomposition of hydrogen peroxide (2H2O2 → O2 + 2H2O). It is one of the most active enzymes known, capable of degrading millions of molecules of H2O2 per second.
• Endogenous or exogenous antioxidants (e.g., vitamins E, A, and C and β-carotene) may either block the formation of free radicals or scavenge them once they have formed.
Reactive oxygen species cause cell injury by three main reactions (Fig. 1–19):
• Lipid peroxidation of membranes. Double bonds in membrane polyunsaturated lipids are vulnerable to attack by oxygen-derived free radicals. The lipid–radical interactions yield peroxides, which are themselves unstable and reactive, and an autocatalytic chain reaction ensues.
• Cross-linking and other changes in proteins. Free radicals promote sulfhydryl-mediated protein cross-linking, resulting in enhanced degradation or loss of enzymatic activity. Free radical reactions may also directly cause polypeptide fragmentation.
• DNA damage. Free radical reactions with thymine in nuclear and mitochondrial DNA produce single-strand breaks. Such DNA damage has been implicated in cell death, aging, and malignant transformation of cells.
In addition to the role of ROS in cell injury and killing of microbes, low concentrations of ROS are involved in numerous signaling pathways in cells and thus in many physiologic reactions. Therefore, these molecules are produced normally but, to avoid their harmful effects, their intracellular concentrations are tightly regulated in healthy cells.
Defects in Membrane Permeability
Increased membrane permeability leading ultimately to overt membrane damage is a consistent feature of most forms of cell injury that culminate in necrosis. The plasma membrane can be damaged by ischemia, various microbial toxins, lytic complement components, and a variety of physical and chemical agents. Several biochemical mechanisms may contribute to membrane damage (Fig. 1–20):
• Decreased phospholipid synthesis. The production of phospholipids in cells may be reduced whenever there is a fall in ATP levels, leading to decreased energy-dependent enzymatic activities. The reduced phospholipid synthesis may affect all cellular membranes, including the membranes of mitochondria, thus exacerbating the loss of ATP.
• Increased phospholipid breakdown. Severe cell injury is associated with increased degradation of membrane phospholipids, probably owing to activation of endogenous phospholipases by increased levels of cytosolic Ca2+.
• ROS. Oxygen free radicals cause injury to cell membranes by lipid peroxidation, discussed earlier.
• Cytoskeletal abnormalities. Cytoskeletal filaments act as anchors connecting the plasma membrane to the cell interior, and serve many functions in maintaining normal cellular architecture, motility, and signaling. Activation of proteases by increased cytosolic Ca2+ may cause damage to elements of the cytoskeleton, leading to membrane damage.
• Lipid breakdown products. These include unesterified free fatty acids, acyl carnitine, and lysophospholipids, all of which accumulate in injured cells as a result of phospholipid degradation. These catabolic products have a detergent effect on membranes. They may also either insert into the lipid bilayer of the membrane or exchange with membrane phospholipids, causing changes in permeability and electrophysiologic alterations.
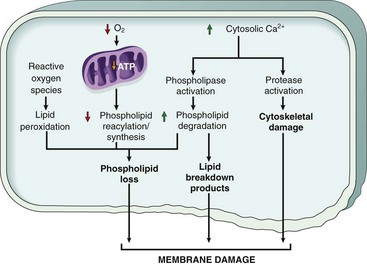
Figure 1–20 Mechanisms of membrane damage in cell injury. Decreased O2 and increased cytosolic Ca2+ typically are seen in ischemia but may accompany other forms of cell injury. Reactive oxygen species, which often are produced on reperfusion of ischemic tissues, also cause membrane damage (not shown).
The most important sites of membrane damage during cell injury are the mitochondrial membrane, the plasma membrane, and membranes of lysosomes.
• Mitochondrial membrane damage. As discussed earlier, damage to mitochondrial membranes results in decreased production of ATP, with many deleterious effects culminating in necrosis.
• Plasma membrane damage. Plasma membrane damage leads to loss of osmotic balance and influx of fluids and ions, as well as loss of cellular contents. The cells may also leak metabolites that are vital for the reconstitution of ATP, thus further depleting energy stores.
• Injury to lysosomal membranes results in leakage of their enzymes into the cytoplasm and activation of the acid hydrolases in the acidic intracellular pH of the injured (e.g., ischemic) cell. Lysosomes contain ribonucleases (RNases), DNases, proteases, glucosidases, and other enzymes. Activation of these enzymes leads to enzymatic digestion of cell components, and the cells die by necrosis.
Damage to DNA and Proteins
Cells have mechanisms that repair damage to DNA, but if this damage is too severe to be corrected (e.g., after radiation injury or oxidative stress), the cell initiates its suicide program and dies by apoptosis. A similar reaction is triggered by the accumulation of improperly folded proteins, which may result from inherited mutations or external triggers such as free radicals. Since these mechanisms of cell injury typically cause apoptosis, they are discussed later in the chapter.
Summary
Mechanisms of Cell Injury
• ATP depletion: failure of energy-dependent functions → reversible injury → necrosis
• Mitochondrial damage: ATP depletion → failure of energy-dependent cellular functions → ultimately, necrosis; under some conditions, leakage of mitochondrial proteins that cause apoptosis
• Influx of calcium: activation of enzymes that damage cellular components and may also trigger apoptosis
• Accumulation of reactive oxygen species: covalent modification of cellular proteins, lipids, nucleic acids
• Increased permeability of cellular membranes: may affect plasma membrane, lysosomal membranes, mitochondrial membranes; typically culminates in necrosis
• Accumulation of damaged DNA and misfolded proteins: triggers apoptosis
Clinicopathologic Correlations: Examples of Cell Injury and Necrosis
To illustrate the evolution and biochemical mechanisms of cell injury, we conclude this section by discussing some commonly encountered examples of reversible cell injury and necrosis.
Ischemic and Hypoxic Injury
Ischemia, or diminished blood flow to a tissue, is a common cause of acute cell injury underlying human disease. In contrast with hypoxia, in which energy generation by anaerobic glycolysis can continue (albeit less efficiently than by oxidative pathways), ischemia, because of reduced blood supply, also compromises the delivery of substrates for glycolysis. Consequently, anaerobic energy generation also ceases in ischemic tissues after potential substrates are exhausted or when glycolysis is inhibited by the accumulation of metabolites that would normally be removed by blood flow. Therefore, ischemia injures tissues faster and usually more severely than does hypoxia. The major cellular abnormalities in oxygen-deprived cells are decreased ATP generation, mitochondrial damage, and accumulation of ROS, with its downstream consequences.
The most important biochemical abnormality in hypoxic cells that leads to cell injury is reduced intracellular generation of ATP, as a consequence of reduced supply of oxygen. As described above, loss of ATP leads to the failure of many energy-dependent cellular systems, including (1) ion pumps (leading to cell swelling, and influx of Ca2+, with its deleterious consequences); (2) depletion of glycogen stores and accumulation of lactic acid, thus lowering the intracellular pH; and (3) reduction in protein synthesis.
The functional consequences may be severe at this stage. For instance, heart muscle ceases to contract within 60 seconds of coronary occlusion. If hypoxia continues, worsening ATP depletion causes further deterioration, with loss of microvilli and the formation of “blebs” (Fig. 1–6). At this time, the entire cell and its organelles (mitochondria, ER) are markedly swollen, with increased concentrations of water, sodium, and chloride and a decreased concentration of potassium. If oxygen is restored, all of these disturbances are reversible, and in the case of myocardium, contractility returns.
If ischemia persists, irreversible injury and necrosis ensue. Irreversible injury is associated with severe swelling of mitochondria, extensive damage to plasma membranes, and swelling of lysosomes. ROS accumulate in cells, and massive influx of calcium may occur. Death is mainly by necrosis, but apoptosis also contributes; the apoptotic pathway is activated by release of pro-apoptotic molecules from mitochondria. The cell’s components are progressively degraded, and there is widespread leakage of cellular enzymes into the extracellular space. Finally, the dead cells may become replaced by large masses composed of phospholipids in the form of myelin figures. These are then either phagocytosed by leukocytes or degraded further into fatty acids that may become calcified.
Ischemia-Reperfusion Injury
If cells are reversibly injured, the restoration of blood flow can result in cell recovery. However, under certain circumstances, the restoration of blood flow to ischemic but viable tissues results, paradoxically, in the death of cells that are not otherwise irreversibly injured. This so-called ischemia-reperfusion injury is a clinically important process that may contribute significantly to tissue damage in myocardial and cerebral ischemia.
Several mechanisms may account for the exacerbation of cell injury resulting from reperfusion into ischemic tissues:
• New damage may be initiated during reoxygenation by increased generation of ROS from parenchymal and endothelial cells and from infiltrating leukocytes. When the supply of oxygen is increased, there may be a corresponding increase in the production of ROS, especially because mitochondrial damage leads to incomplete reduction of oxygen, and because of the action of oxidases in leukocytes, endothelial cells, or parenchymal cells. Cellular antioxidant defense mechanisms may also be compromised by ischemia, favoring the accumulation of free radicals.
• The inflammation that is induced by ischemic injury may increase with reperfusion because of increased influx of leukocytes and plasma proteins. The products of activated leukocytes may cause additional tissue injury (Chapter 2). Activation of the complement system may also contribute to ischemia-reperfusion injury. Complement proteins may bind in the injured tissues, or to antibodies that are deposited in the tissues, and subsequent complement activation generates by-products that exacerbate the cell injury and inflammation.
Chemical (Toxic) Injury
Chemicals induce cell injury by one of two general mechanisms:
• Some chemicals act directly by combining with a critical molecular component or cellular organelle. For example, in mercuric chloride poisoning (as may occur from ingestion of contaminated seafood) (Chapter 7), mercury binds to the sulfhydryl groups of various cell membrane proteins, causing inhibition of ATP-dependent transport and increased membrane permeability. Many antineoplastic chemotherapeutic agents also induce cell damage by direct cytotoxic effects. In such instances, the greatest damage is sustained by the cells that use, absorb, excrete, or concentrate the compounds.
• Many other chemicals are not intrinsically biologically active but must be first converted to reactive toxic metabolites, which then act on target cells. This modification is usually accomplished by the cytochrome P-450 in the smooth ER of the liver and other organs. Although the metabolites might cause membrane damage and cell injury by direct covalent binding to protein and lipids, the most important mechanism of cell injury involves the formation of free radicals. Carbon tetrachloride (CCl4)—once widely used in the dry cleaning industry but now banned—and the analgesic acetaminophen belong in this category. The effect of CCl4 is still instructive as an example of chemical injury. CCl4 is converted to the toxic free radical 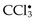 , principally in the liver, and this free radical is the cause of cell injury, mainly by membrane phospholipid peroxidation. In less than 30 minutes after exposure to CCl4, there is breakdown of ER membranes with a decline in hepatic protein synthesis of enzymes and plasma proteins; within 2 hours, swelling of the smooth ER and dissociation of ribosomes from the smooth ER have occurred. There is reduced lipid export from the hepatocytes, as a result of their inability to synthesize apoprotein to form complexes with triglycerides and thereby facilitate lipoprotein secretion; the result is the “fatty liver” of CCl4 poisoning. Mitochondrial injury follows, and subsequently diminished ATP stores result in defective ion transport and progressive cell swelling; the plasma membranes are further damaged by fatty aldehydes produced by lipid peroxidation in the ER. The end result can be calcium influx and eventually cell death.
, principally in the liver, and this free radical is the cause of cell injury, mainly by membrane phospholipid peroxidation. In less than 30 minutes after exposure to CCl4, there is breakdown of ER membranes with a decline in hepatic protein synthesis of enzymes and plasma proteins; within 2 hours, swelling of the smooth ER and dissociation of ribosomes from the smooth ER have occurred. There is reduced lipid export from the hepatocytes, as a result of their inability to synthesize apoprotein to form complexes with triglycerides and thereby facilitate lipoprotein secretion; the result is the “fatty liver” of CCl4 poisoning. Mitochondrial injury follows, and subsequently diminished ATP stores result in defective ion transport and progressive cell swelling; the plasma membranes are further damaged by fatty aldehydes produced by lipid peroxidation in the ER. The end result can be calcium influx and eventually cell death.
Apoptosis
Apoptosis is a pathway of cell death in which cells activate enzymes that degrade the cells’ own nuclear DNA and nuclear and cytoplasmic proteins. Fragments of the apoptotic cells then break off, giving the appearance that is responsible for the name (apoptosis, “falling off”). The plasma membrane of the apoptotic cell remains intact, but the membrane is altered in such a way that the cell and its fragments become avid targets for phagocytes. The dead cell and its fragments are rapidly cleared before cellular contents have leaked out, so apoptotic cell death does not elicit an inflammatory reaction in the host. Apoptosis differs in this respect from necrosis, which is characterized by loss of membrane integrity, enzymatic digestion of cells, leakage of cellular contents, and frequently a host reaction (Fig. 1–6 and Table 1–1). However, apoptosis and necrosis sometimes coexist, and apoptosis induced by some pathologic stimuli may progress to necrosis.
Causes of Apoptosis
Apoptosis occurs in many normal situations and serves to eliminate potentially harmful cells and cells that have outlived their usefulness. It also occurs as a pathologic event when cells are damaged beyond repair, especially when the damage affects the cell’s DNA or proteins; in these situations, the irreparably damaged cell is eliminated.
Apoptosis in Physiologic Situations
Death by apoptosis is a normal phenomenon that serves to eliminate cells that are no longer needed and to maintain a constant number of cells of various types in tissues. It is important in the following physiologic situations:
• The programmed destruction of cells during embryogenesis. Normal development is associated with the death of some cells and the appearance of new cells and tissues. The term programmed cell death was originally coined to denote this death of specific cell types at defined times during the development of an organism. Apoptosis is a generic term for this pattern of cell death, regardless of the context, but it is often used interchangeably with programmed cell death.
• Involution of hormone-dependent tissues upon hormone deprivation, such as endometrial cell breakdown during the menstrual cycle, and regression of the lactating breast after weaning
• Cell loss in proliferating cell populations, such as intestinal crypt epithelia, in order to maintain a constant number
• Elimination of cells that have served their useful purpose, such as neutrophils in an acute inflammatory response and lymphocytes at the end of an immune response. In these situations, cells undergo apoptosis because they are deprived of necessary survival signals, such as growth factors.
• Elimination of potentially harmful self-reactive lymphocytes, either before or after they have completed their maturation, in order to prevent reactions against the body’s own tissues (Chapter 4)
• Cell death induced by cytotoxic T lymphocytes, a defense mechanism against viruses and tumors that serves to kill virus-infected and neoplastic cells (Chapter 4)
Apoptosis in Pathologic Conditions
Apoptosis eliminates cells that are genetically altered or injured beyond repair and does so without eliciting a severe host reaction, thereby keeping the extent of tissue damage to a minimum. Death by apoptosis is responsible for loss of cells in a variety of pathologic states:
• DNA damage. Radiation, cytotoxic anticancer drugs, extremes of temperature, and even hypoxia can damage DNA, either directly or through production of free radicals. If repair mechanisms cannot cope with the injury, the cell triggers intrinsic mechanisms that induce apoptosis. In these situations, elimination of the cell may be a better alternative than risking mutations in the damaged DNA, which may progress to malignant transformation. These injurious stimuli cause apoptosis if the insult is mild, but larger doses of the same stimuli result in necrotic cell death. Inducing apoptosis of cancer cells is a desired effect of chemotherapeutic agents, many of which work by damaging DNA.
• Accumulation of misfolded proteins. Improperly folded proteins may arise because of mutations in the genes encoding these proteins or because of extrinsic factors, such as damage caused by free radicals. Excessive accumulation of these proteins in the ER leads to a condition called ER stress, which culminates in apoptotic death of cells.
• Cell injury in certain infections, particularly viral infections, in which loss of infected cells is largely due to apoptotic death that may be induced by the virus (as in adenovirus and human immunodeficiency virus infections) or by the host immune response (as in viral hepatitis).
• Pathologic atrophy in parenchymal organs after duct obstruction, such as occurs in the pancreas, parotid gland, and kidney
Morphology
In H&E-stained tissue sections, the nuclei of apoptotic cells show various stages of chromatin condensation and aggregation and, ultimately, karyorrhexis (Fig. 1–21); at the molecular level this is reflected in fragmentation of DNA into nucleosome-sized pieces. The cells rapidly shrink, form cytoplasmic buds, and fragment into apoptotic bodies composed of membrane-bound vesicles of cytosol and organelles (Fig. 1–6). Because these fragments are quickly extruded and phagocytosed without eliciting an inflammatory response, even substantial apoptosis may be histologically undetectable.
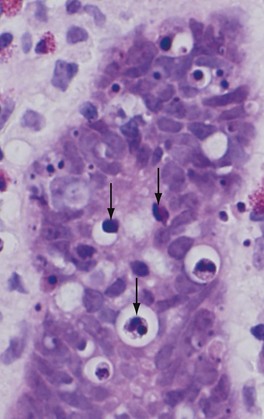
Figure 1–21 Morphologic appearance of apoptotic cells. Apoptotic cells (some indicated by arrows) in a normal crypt in the colonic epithelium are shown. (The preparative regimen for colonoscopy frequently induces apoptosis in epithelial cells, which explains the abundance of dead cells in this normal tissue.) Note the fragmented nuclei with condensed chromatin and the shrunken cell bodies, some with pieces falling off.
(Courtesy of Dr. Sanjay Kakar, Department of Pathology, University of California San Francisco, San Francisco, Calif.)
Mechanisms of Apoptosis
Apoptosis results from the activation of enzymes called caspases (so named because they are cysteine proteases that cleave proteins after aspartic residues). The activation of caspases depends on a finely tuned balance between production of pro- and anti-apoptotic proteins. Two distinct pathways converge on caspase activation: the mitochondrial pathway and the death receptor pathway (Fig. 1–22). Although these pathways can intersect, they are generally induced under different conditions, involve different molecules, and serve distinct roles in physiology and disease.
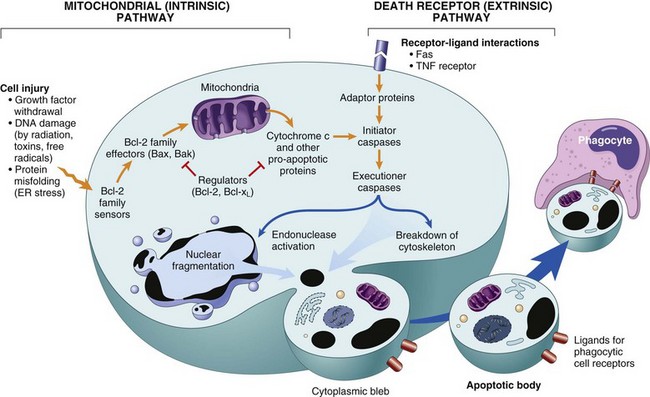
Figure 1–22 Mechanisms of apoptosis. The two pathways of apoptosis differ in their induction and regulation, and both culminate in the activation of caspases. In the mitochondrial pathway, proteins of the Bcl-2 family, which regulate mitochondrial permeability, become imbalanced and leakage of various substances from mitochondria leads to caspase activation. In the death receptor pathway, signals from plasma membrane receptors lead to the assembly of adaptor proteins into a “death-inducing signaling complex,” which activates caspases, and the end result is the same.
The Mitochondrial (Intrinsic) Pathway of Apoptosis
Mitochondria contain several proteins that are capable of inducing apoptosis; these proteins include cytochrome c and other proteins that neutralize endogenous inhibitors of apoptosis. The choice between cell survival and death is determined by the permeability of mitochondria, which is controlled by a family of more than 20 proteins, the prototype of which is Bcl-2 (Fig. 1–23). When cells are deprived of growth factors and other survival signals, or are exposed to agents that damage DNA, or accumulate unacceptable amounts of misfolded proteins, a number of sensors are activated. These sensors are members of the Bcl-2 family called “BH3 proteins” (because they contain only the third of multiple conserved domains of the Bcl-2 family). They in turn activate two pro-apoptotic members of the family called Bax and Bak, which dimerize, insert into the mitochondrial membrane, and form channels through which cytochrome c and other mitochondrial proteins escape into the cytosol. These sensors also inhibit the anti-apoptotic molecules Bcl-2 and Bcl-xL (see further on), enhancing the leakage of mitochondrial proteins. Cytochrome c, together with some cofactors, activates caspase-9. Other proteins that leak out of mitochondria block the activities of caspase antagonists that function as physiologic inhibitors of apoptosis. The net result is the activation of the caspase cascade, ultimately leading to nuclear fragmentation. Conversely, if cells are exposed to growth factors and other survival signals, they synthesize anti-apoptotic members of the Bcl-2 family, the two main ones of which are Bcl-2 itself and Bcl-xL. These proteins antagonize Bax and Bak, and thus limit the escape of the mitochondrial pro-apoptotic proteins. Cells deprived of growth factors not only activate the pro-apoptotic Bax and Bak but also show reduced levels of Bcl-2 and Bcl-xL, thus further tilting the balance toward death. The mitochondrial pathway seems to be the pathway that is responsible for apoptosis in most situations, as we discuss later.
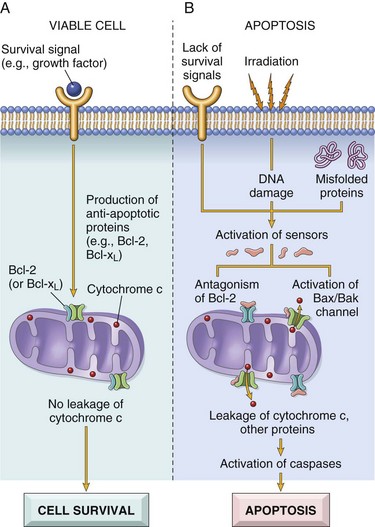
Figure 1–23 The mitochondrial pathway of apoptosis.The induction of apoptosis by the mitochondrial pathway is dependent on a balance between pro- and anti-apoptotic proteins of the Bcl family. The pro-apoptotic proteins include some (sensors) that sense DNA and protein damage and trigger apoptosis and others (effectors) that insert in the mitochondrial membrane and promote leakage of mitochondrial proteins. A, In a viable cell, anti-apoptotic members of the Bcl-2 family prevent leakage of mitochondrial proteins. B, Various injurious stimuli activate cytoplasmic sensors and lead to reduced production of these anti-apoptotic proteins and increased amounts of pro-apoptotic proteins, resulting in leakage of proteins that are normally sequestered within mitochondria. The mitochondrial proteins that leak out activate a series of caspases, first the initiators and then the executioners, and these enzymes cause fragmentation of the nucleus and ultimately the cell.
The Death Receptor (Extrinsic) Pathway of Apoptosis
Many cells express surface molecules, called death receptors, that trigger apoptosis. Most of these are members of the tumor necrosis factor (TNF) receptor family, which contain in their cytoplasmic regions a conserved “death domain,” so named because it mediates interaction with other proteins involved in cell death. The prototypic death receptors are the type I TNF receptor and Fas (CD95). Fas ligand (FasL) is a membrane protein expressed mainly on activated T lymphocytes. When these T cells recognize Fas-expressing targets, Fas molecules are cross-linked by FasL and bind adaptor proteins via the death domain. These in turn recruit and activate caspase-8. In many cell types caspase-8 may cleave and activate a pro-apoptotic member of the Bcl-2 family called Bid, thus feeding into the mitochondrial pathway. The combined activation of both pathways delivers a lethal blow to the cell. Cellular proteins, notably a caspase antagonist called FLIP, block activation of caspases downstream of death receptors. Interestingly, some viruses produce homologues of FLIP, and it is suggested that this is a mechanism that viruses use to keep infected cells alive. The death receptor pathway is involved in elimination of self-reactive lymphocytes and in killing of target cells by some cytotoxic T lymphocytes.
Activation and Function of Caspases
The mitochondrial and death receptor pathways lead to the activation of the initiator caspases, caspase-9 and -8, respectively. Active forms of these enzymes are produced, and these cleave and thereby activate another series of caspases that are called the executioner caspases. These activated caspases cleave numerous targets, culminating in activation of nucleases that degrade DNA and nucleoproteins. Caspases also degrade components of the nuclear matrix and cytoskeleton, leading to fragmentation of cells.
Clearance of Apoptotic Cells
Apoptotic cells entice phagocytes by producing “eat-me” signals. In normal cells phosphatidylserine is present on the inner leaflet of the plasma membrane, but in apoptotic cells this phospholipid “flips” to the outer leaflet, where it is recognized by tissue macrophages and leads to phagocytosis of the apoptotic cells. Cells that are dying by apoptosis also secrete soluble factors that recruit phagocytes. This facilitates prompt clearance of the dead cells before they undergo secondary membrane damage and release their cellular contents (which can induce inflammation). Some apoptotic bodies express adhesive glycoproteins that are recognized by phagocytes, and macrophages themselves may produce proteins that bind to apoptotic cells (but not to live cells) and target the dead cells for engulfment. Numerous macrophage receptors have been shown to be involved in the binding and engulfment of apoptotic cells. This process of phagocytosis of apoptotic cells is so efficient that dead cells disappear without leaving a trace, and inflammation is virtually absent.
Although we have emphasized the distinctions between necrosis and apoptosis, these two forms of cell death may coexist and be related mechanistically. For instance, DNA damage (seen in apoptosis) activates an enzyme called poly-ADP(ribose) polymerase, which depletes cellular supplies of nicotinamide adenine dinucleotide, leading to a fall in ATP levels and ultimately necrosis. In fact, even in common situations such as ischemia, it has been suggested that early cell death can be partly attributed to apoptosis, with necrosis supervening later as ischemia worsens.
Examples of Apoptosis
Cell death in many situations is caused by apoptosis. The examples listed next illustrate the role of the two pathways of apoptosis in normal physiology and in disease.
Growth Factor Deprivation
Hormone-sensitive cells deprived of the relevant hormone, lymphocytes that are not stimulated by antigens and cytokines, and neurons deprived of nerve growth factor die by apoptosis. In all these situations, apoptosis is triggered by the mitochondrial pathway and is attributable to activation of pro-apoptotic members of the Bcl-2 family and decreased synthesis of Bcl-2 and Bcl-xL.
DNA Damage
Exposure of cells to radiation or chemotherapeutic agents induces DNA damage, which if severe may trigger apoptotic death. When DNA is damaged, the p53 protein accumulates in cells. It first arrests the cell cycle (at the G1 phase) to allow the DNA to be repaired before it is replicated (Chapter 5). However, if the damage is too great to be repaired successfully, p53 triggers apoptosis, mainly by stimulating sensors that ultimately activate Bax and Bak, and by increasing the synthesis of pro-apoptotic members of the Bcl-2 family. When p53 is mutated or absent (as it is in certain cancers), cells with damaged DNA that would otherwise undergo apoptosis survive. In such cells, the DNA damage may result in mutations or DNA rearrangements (e.g., translocations) that lead to neoplastic transformation (Chapter 5).
Accumulation of Misfolded Proteins: ER Stress
During normal protein synthesis, chaperones in the ER control the proper folding of newly synthesized proteins, and misfolded polypeptides are ubiquitinated and targeted for proteolysis. If, however, unfolded or misfolded proteins accumulate in the ER because of inherited mutations or environmental perturbations, they induce a protective cellular response that is called the unfolded protein response (Fig. 1–24). This response activates signaling pathways that increase the production of chaperones and retard protein translation, thus reducing the levels of misfolded proteins in the cell. In circumstances in which the accumulation of misfolded proteins overwhelms these adaptations, the result is ER stress, which leads to the activation of caspases and ultimately apoptosis. Intracellular accumulation of abnormally folded proteins, caused by mutations, aging, or unknown environmental factors, may cause diseases by reducing the availability of the normal protein or by inducing cell injury (Table 1–2). Cell death as a result of protein misfolding is now recognized as a feature of a number of neurodegenerative diseases, including Alzheimer, Huntington, and Parkinson diseases, and possibly type 2 diabetes. Deprivation of glucose and oxygen and stresses such as infections also result in protein misfolding, culminating in cell injury and death.
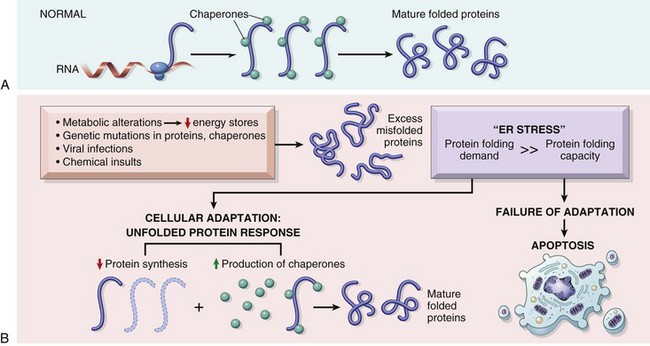
Figure 1–24 The unfolded protein response and ER stress. A, In healthy cells, newly synthesized proteins are folded with the help of chaperones and are then incorporated into the cell or secreted. B, Various external stresses or mutations induce a state called ER stress, in which the cell is unable to cope with the load of misfolded proteins. Accumulation of these proteins in the ER triggers the unfolded protein response, which tries to restore protein homeostasis; if this response is inadequate, the cell dies by apoptosis.
Table 1–2 Diseases Caused by Misfolding of Proteins
| Disease | Affected Protein | Pathogenesis |
|---|---|---|
| Cystic fibrosis | Cystic fibrosis transmembrane conductance regulator (CFTR) | Loss of CFTR leads to defects in chloride transport |
| Familial hypercholesterolemia | LDL receptor | Loss of LDL receptor leading to hypercholesterolemia |
| Tay-Sachs disease | Hexosaminidase β subunit | Lack of the lysosomal enzyme leads to storage of GM2 gangliosides in neurons |
| Alpha-1-antitrypsin deficiency | α-1 antitrypsin | Storage of nonfunctional protein in hepatocytes causes apoptosis; absence of enzymatic activity in lungs causes destruction of elastic tissue giving rise to emphysema |
| Creutzfeld-Jacob disease | Prions | Abnormal folding of PrPsc causes neuronal cell death |
| Alzheimer disease | Aβ peptide | Abnormal folding of Aβ peptides causes aggregation within neurons and apoptosis |
Shown are selected illustrative examples of diseases in which protein misfolding is thought to be the major mechanism of functional derangement or cell or tissue injury.
Apoptosis of Self-Reactive Lymphocytes
Lymphocytes capable of recognizing self antigens are normally produced in all individuals. If these lymphocytes encounter self antigens, the cells die by apoptosis. Both the mitochondrial pathway and the Fas death receptor pathway have been implicated in this process (Chapter 4). Failure of apoptosis of self-reactive lymphocytes is one of the causes of autoimmune diseases.
Cytotoxic T Lymphocyte–Mediated Apoptosis
Cytotoxic T lymphocytes (CTLs) recognize foreign antigens presented on the surface of infected host cells and tumor cells (Chapter 4). On activation, CTL granule proteases called granzymes enter the target cells. Granzymes cleave proteins at aspartate residues and are able to activate cellular caspases. In this way, the CTL kills target cells by directly inducing the effector phase of apoptosis, without engaging mitochondria or death receptors. CTLs also express FasL on their surface and may kill target cells by ligation of Fas receptors.
Summary
Apoptosis
• Regulated mechanism of cell death that serves to eliminate unwanted and irreparably damaged cells, with the least possible host reaction
• Characterized by enzymatic degradation of proteins and DNA, initiated by caspases; and by recognition and removal of dead cells by phagocytes
• Initiated by two major pathways:
 Mitochondrial (intrinsic) pathway is triggered by loss of survival signals, DNA damage and accumulation of misfolded proteins (ER stress); associated with leakage of pro-apoptotic proteins from mitochondrial membrane into the cytoplasm, where they trigger caspase activation; inhibited by anti-apoptotic members of the Bcl family, which are induced by survival signals including growth factors.
Mitochondrial (intrinsic) pathway is triggered by loss of survival signals, DNA damage and accumulation of misfolded proteins (ER stress); associated with leakage of pro-apoptotic proteins from mitochondrial membrane into the cytoplasm, where they trigger caspase activation; inhibited by anti-apoptotic members of the Bcl family, which are induced by survival signals including growth factors.Autophagy
Autophagy (“self-eating”) refers to lysosomal digestion of the cell’s own components. It is a survival mechanism in times of nutrient deprivation, such that the starved cell subsists by eating its own contents and recycling these contents to provide nutrients and energy. In this process, intracellular organelles and portions of cytosol are first sequestered within an autophagic vacuole, which is postulated to be formed from ribosome-free regions of the ER (Fig. 1–25). The vacuole fuses with lysosomes to form an autophagolysosome, in which lysosomal enzymes digest the cellular components. Autophagy is initiated by multi-protein complexes that sense nutrient deprivation and stimulate formation of the autophagic vacuole. With time, the starved cell eventually can no longer cope by devouring itself; at this stage, autophagy may also signal cell death by apoptosis.
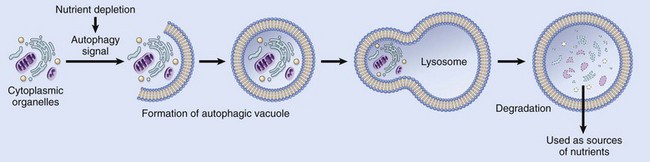
Figure 1–25 Autophagy. Cellular stresses, such as nutrient deprivation, activate autophagy genes (Atg genes), which initiate the formation of membrane-bound vesicles in which cellular organelles are sequestered. These vesicles fuse with lysosomes, in which the organelles are digested, and the products are used to provide nutrients for the cell. The same process can trigger apoptosis, by mechanisms that are not well defined.
Autophagy is also involved in the clearance of misfolded proteins, for instance, in neurons and hepatocytes. Therefore, defective autophagy may be a cause of neuronal death induced by accumulation of these proteins and, subsequently, neurodegenerative diseases. Conversely, pharmacologic activation of autophagy limits the build-up of misfolded proteins in liver cells in animal models, thereby reducing liver fibrosis. Polymorphisms in a gene involved in autophagy have been associated with inflammatory bowel disease, but the mechanistic link between autophagy and intestinal inflammation is not known. The role of autophagy in cancer is discussed in Chapter 5. Thus, a once little-appreciated survival pathway in cells may prove to have wide-ranging roles in human disease.
We have now concluded the discussion of cell injury and cell death. As we have seen, these processes are the root cause of many common diseases. We end this chapter with brief considerations of three other processes: intracellular accumulations of various substances and extracellular deposition of calcium, both of which are often associated with cell injury, and aging.
Intracellular Accumulations
Under some circumstances cells may accumulate abnormal amounts of various substances, which may be harmless or associated with varying degrees of injury. The substance may be located in the cytoplasm, within organelles (typically lysosomes), or in the nucleus, and it may be synthesized by the affected cells or may be produced elsewhere.
There are four main pathways of abnormal intracellular accumulations (Fig. 1–26):
• Inadequate removal of a normal substance secondary to defects in mechanisms of packaging and transport, as in fatty change in the liver
• Accumulation of an abnormal endogenous substance as a result of genetic or acquired defects in its folding, packaging, transport, or secretion, as with certain mutated forms of α1-antitrypsin
• Failure to degrade a metabolite due to inherited enzyme deficiencies. The resulting disorders are called storage diseases (Chapter 6).
• Deposition and accumulation of an abnormal exogenous substance when the cell has neither the enzymatic machinery to degrade the substance nor the ability to transport it to other sites. Accumulation of carbon or silica particles is an example of this type of alteration.
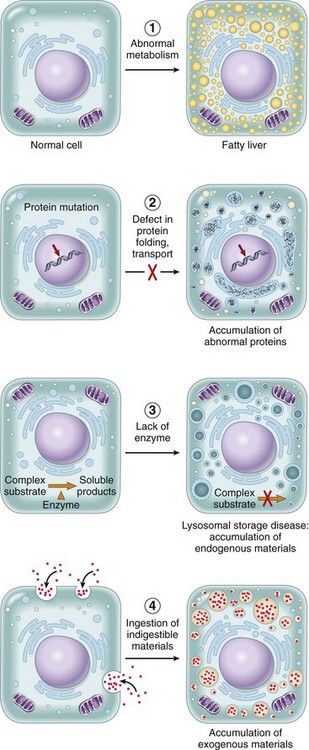
Figure 1–26 Mechanisms of intracellular accumulation: (1) Abnormal metabolism, as in fatty change in the liver. (2) Mutations causing alterations in protein folding and transport, so that defective molecules accumulate intracellularly. (3) A deficiency of critical enzymes responsible for breaking down certain compounds, causing substrates to accumulate in lysosomes, as in lysosomal storage diseases. (4) An inability to degrade phagocytosed particles, as in carbon pigment accumulation.
Fatty Change (Steatosis)
Fatty change refers to any abnormal accumulation of triglycerides within parenchymal cells. It is most often seen in the liver, since this is the major organ involved in fat metabolism, but it may also occur in heart, skeletal muscle, kidney, and other organs. Steatosis may be caused by toxins, protein malnutrition, diabetes mellitus, obesity, or anoxia. Alcohol abuse and diabetes associated with obesity are the most common causes of fatty change in the liver (fatty liver) in industrialized nations. This process is discussed in more detail in Chapter 15.
Cholesterol and Cholesteryl Esters
Cellular cholesterol metabolism is tightly regulated to ensure normal cell membrane synthesis without significant intracellular accumulation. However, phagocytic cells may become overloaded with lipid (triglycerides, cholesterol, and cholesteryl esters) in several different pathologic processes. Of these, atherosclerosis is the most important. The role of lipid and cholesterol deposition in the pathogenesis of atherosclerosis is discussed in Chapter 9.
Proteins
Morphologically visible protein accumulations are much less common than lipid accumulations; they may occur when excesses are presented to the cells or if the cells synthesize excessive amounts. In the kidney, for example, trace amounts of albumin filtered through the glomerulus are normally reabsorbed by pinocytosis in the proximal convoluted tubules. However, in disorders with heavy protein leakage across the glomerular filter (e.g., nephrotic syndrome), there is a much larger reabsorption of the protein, and vesicles containing this protein accumulate, giving the histologic appearance of pink, hyaline cytoplasmic droplets. The process is reversible: If the proteinuria abates, the protein droplets are metabolized and disappear. Another example is the marked accumulation of newly synthesized immunoglobulins that may occur in the RER of some plasma cells, forming rounded, eosinophilic Russell bodies. Other examples of protein aggregation are discussed elsewhere in this book (e.g., “alcoholic hyaline” in the liver in Chapter 15; neurofibrillary tangles in neurons in Chapter 22).
Glycogen
Excessive intracellular deposits of glycogen are associated with abnormalities in the metabolism of either glucose or glycogen. In poorly controlled diabetes mellitus, the prime example of abnormal glucose metabolism, glycogen accumulates in renal tubular epithelium, cardiac myocytes, and β cells of the islets of Langerhans. Glycogen also accumulates within cells in a group of closely related genetic disorders collectively referred to as glycogen storage diseases, or glycogenoses (Chapter 6).
Pigments
Pigments are colored substances that are either exogenous, coming from outside the body, such as carbon, or endogenous, synthesized within the body itself, such as lipofuscin, melanin, and certain derivatives of hemoglobin.
• The most common exogenous pigment is carbon (an example is coal dust), a ubiquitous air pollutant of urban life. When inhaled, it is phagocytosed by alveolar macrophages and transported through lymphatic channels to the regional tracheobronchial lymph nodes. Aggregates of the pigment blacken the draining lymph nodes and pulmonary parenchyma (anthracosis) (Chapter 12).
• Lipofuscin, or “wear-and-tear pigment,” is an insoluble brownish-yellow granular intracellular material that accumulates in a variety of tissues (particularly the heart, liver, and brain) as a function of age or atrophy. Lipofuscin represents complexes of lipid and protein that derive from the free radical–catalyzed peroxidation of polyunsaturated lipids of subcellular membranes. It is not injurious to the cell but is a marker of past free radical injury. The brown pigment (Fig. 1–27), when present in large amounts, imparts an appearance to the tissue that is called brown atrophy. By electron microscopy, the pigment appears as perinuclear electron-dense granules (Fig. 1–27, B).
• Melanin is an endogenous, brown-black pigment that is synthesized by melanocytes located in the epidermis and acts as a screen against harmful ultraviolet radiation. Although melanocytes are the only source of melanin, adjacent basal keratinocytes in the skin can accumulate the pigment (e.g., in freckles), as can dermal macrophages.
• Hemosiderin is a hemoglobin-derived granular pigment that is golden yellow to brown and accumulates in tissues when there is a local or systemic excess of iron. Iron is normally stored within cells in association with the protein apoferritin, forming ferritin micelles. Hemosiderin pigment represents large aggregates of these ferritin micelles, readily visualized by light and electron microscopy; the iron can be unambiguously identified by the Prussian blue histochemical reaction (Fig. 1–28). Although hemosiderin accumulation is usually pathologic, small amounts of this pigment are normal in the mononuclear phagocytes of the bone marrow, spleen, and liver, where aging red cells are normally degraded. Excessive deposition of hemosiderin, called hemosiderosis, and more extensive accumulations of iron seen in hereditary hemochromatosis, are described in Chapter 15.
Pathologic Calcification
Pathologic calcification is a common process in a wide variety of disease states; it implies the abnormal deposition of calcium salts, together with smaller amounts of iron, magnesium, and other minerals. When the deposition occurs in dead or dying tissues, it is called dystrophic calcification; it occurs in the absence of derangements in calcium metabolism (i.e., with normal serum levels of calcium). In contrast, the deposition of calcium salts in normal tissues is known as metastatic calcification and is almost always secondary to some derangement in calcium metabolism (hypercalcemia). Of note, while hypercalcemia is not a prerequisite for dystrophic calcification, it can exacerbate it.
Dystrophic Calcification
Dystrophic calcification is encountered in areas of necrosis of any type. It is virtually inevitable in the atheromas of advanced atherosclerosis, associated with intimal injury in the aorta and large arteries and characterized by accumulation of lipids (Chapter 9). Although dystrophic calcification may be an incidental finding indicating insignificant past cell injury, it may also be a cause of organ dysfunction. For example, calcification can develop in aging or damaged heart valves, resulting in severely compromised valve motion. Dystrophic calcification of the aortic valves is an important cause of aortic stenosis in elderly persons (Fig. 10-17, Chapter 10).
The pathogenesis of dystrophic calcification involves initiation (or nucleation) and propagation, both of which may be either intracellular or extracellular; the ultimate end product is the formation of crystalline calcium phosphate. Initiation in extracellular sites occurs in membrane-bound vesicles about 200 nm in diameter; in normal cartilage and bone they are known as matrix vesicles, and in pathologic calcification they derive from degenerating cells. It is thought that calcium is initially concentrated in these vesicles by its affinity for membrane phospholipids, while phosphates accumulate as a result of the action of membrane-bound phosphatases. Initiation of intracellular calcification occurs in the mitochondria of dead or dying cells that have lost their ability to regulate intracellular calcium. After initiation in either location, propagation of crystal formation occurs. This is dependent on the concentration of Ca2+ and PO4−, the presence of mineral inhibitors, and the degree of collagenization, which enhances the rate of crystal growth.
Metastatic Calcification
Metastatic calcification can occur in normal tissues whenever there is hypercalcemia. The major causes of hypercalcemia are (1) increased secretion of parathyroid hormone, due to either primary parathyroid tumors or production of parathyroid hormone–related protein by other malignant tumors; (2) destruction of bone due to the effects of accelerated turnover (e.g., Paget disease), immobilization, or tumors (increased bone catabolism associated with multiple myeloma, leukemia, or diffuse skeletal metastases); (3) vitamin D–related disorders including vitamin D intoxication and sarcoidosis (in which macrophages activate a vitamin D precursor); and (4) renal failure, in which phosphate retention leads to secondary hyperparathyroidism.
Morphology
Regardless of the site, calcium salts are seen on gross examination as fine white granules or clumps, often felt as gritty deposits. Dystrophic calcification is common in areas of caseous necrosis in tuberculosis. Sometimes a tuberculous lymph node is essentially converted to radiopaque stone. On histologic examination, calcification appears as intracellular and/or extracellular basophilic deposits. Over time, heterotopic bone may be formed in the focus of calcification.
Metastatic calcification can occur widely throughout the body but principally affects the interstitial tissues of the vasculature, kidneys, lungs, and gastric mucosa. The calcium deposits morphologically resemble those described in dystrophic calcification. Although they generally do not cause clinical dysfunction, extensive calcifications in the lungs may be evident on radiographs and may produce respiratory deficits, and massive deposits in the kidney (nephrocalcinosis) can lead to renal damage.
Summary
Abnormal Intracellular Depositions and Calcifications
Abnormal deposits of materials in cells and tissues are the result of excessive intake or defective transport or catabolism.
Fatty change: accumulation of free triglycerides in cells, resulting from excessive intake or defective transport (often because of defects in synthesis of transport proteins); manifestation of reversible cell injury Cholesterol deposition: result of defective catabolism and excessive intake; in macrophages and smooth muscle cells of vessel walls in atherosclerosis• Deposition of proteins: reabsorbed proteins in kidney tubules; immunoglobulins in plasma cells
• Deposition of glycogen: in macrophages of patients with defects in lysosomal enzymes that break down glycogen (glycogen storage diseases)
• Deposition of pigments: typically indigestible pigments, such as carbon, lipofuscin (breakdown product of lipid peroxidation), or iron (usually due to overload, as in hemosiderosis)
Cellular Aging
Individuals age because their cells age. Although public attention on the aging process has traditionally focused on its cosmetic manifestations, aging has important health consequences, because age is one of the strongest independent risk factors for many chronic diseases, such as cancer, Alzheimer disease, and ischemic heart disease. Perhaps one of the most striking discoveries about cellular aging is that it is not simply a consequence of cells’ “running out of steam,” but in fact is regulated by a limited number of genes and signaling pathways that are evolutionarily conserved from yeast to mammals.
Cellular aging is the result of a progressive decline in the life span and functional capacity of cells. Several mechanisms are thought to be responsible for cellular aging (Fig. 1–29):
• DNA damage. A variety of metabolic insults that accumulate over time may result in damage to nuclear and mitochondrial DNA. Although most DNA damage is repaired by DNA repair enzymes, some persists and accumulates as cells age. Some aging syndromes are associated with defects in DNA repair mechanisms, and the life span of experimental animals in some models can be increased if responses to DNA damage are enhanced or proteins that stabilize DNA are introduced. A role of free radicals in DNA damage leading to aging has been postulated but remains controversial.
• Decreased cellular replication. All normal cells have a limited capacity for replication, and after a fixed number of divisions cells become arrested in a terminally nondividing state, known as replicative senescence. Aging is associated with progressive replicative senescence of cells. Cells from children have the capacity to undergo more rounds of replication than do cells from older people. In contrast, cells from patients with Werner syndrome, a rare disease characterized by premature aging, have a markedly reduced in vitro life span. In human cells, the mechanism of replicative senescence involves progressive shortening of telomeres, which ultimately results in cell cycle arrest. Telomeres are short repeated sequences of DNA present at the ends of linear chromosomes that are important for ensuring the complete replication of chromosome ends and for protecting the ends from fusion and degradation. When somatic cells replicate, a small section of the telomere is not duplicated, and telomeres become progressively shortened. As the telomeres become shorter, the ends of chromosomes cannot be protected and are seen as broken DNA, which signals cell cycle arrest. Telomere length is maintained by nucleotide addition mediated by an enzyme called telomerase. Telomerase is a specialized RNA-protein complex that uses its own RNA as a template for adding nucleotides to the ends of chromosomes. Telomerase activity is expressed in germ cells and is present at low levels in stem cells, but it is absent in most somatic tissues (Fig. 1–30). Therefore, as most somatic cells age their telomeres become shorter and they exit the cell cycle, resulting in an inability to generate new cells to replace damaged ones. Conversely, in immortalized cancer cells, telomerase is usually reactivated and telomere length is stabilized, allowing the cells to proliferate indefinitely. This is discussed more fully in Chapter 5. Telomere shortening may also decrease the regenerative capacity of stem cells, further contributing to cellular aging. Despite such alluring observations, however, the relationship of telomerase activity and telomere length to aging has yet to be fully established.
• Defective protein homeostasis. Over time, cells are unable to maintain normal protein homeostasis, because of increased turnover and decreased synthesis caused by reduced translation of proteins and defective activity of chaperones (which promote normal protein folding), proteasomes (which destroy misfolded proteins) and repair enzymes. Abnormal protein homeostasis can have many effects on cell survival, replication, and functions. In addition, it may lead to accumulation of misfolded proteins, which can trigger pathways of apoptosis.
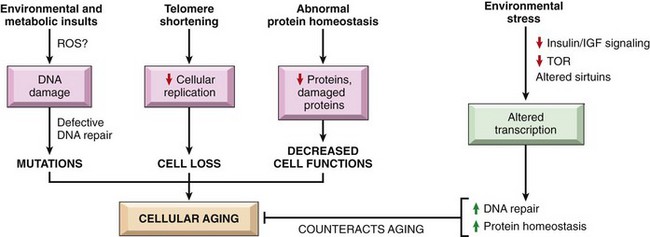
Figure 1–29 Mechanisms that cause and counteract cellular aging. DNA damage, replicative senescence, and decreased and misfolded proteins are among the best described mechanisms of cellular aging. Some environmental stresses, such as calorie restriction, counteract aging by activating various signaling pathways and transcription factors. IGF, insulin-like growth factor; TOR, target of rapamycin.
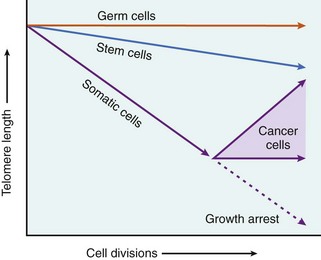
Figure 1–30 The role of telomeres and telomerase in replicative senescence of cells. Telomere length is plotted against the number of cell divisions. In most normal somatic cells there is no telomerase activity, and telomeres progressively shorten with increasing cell divisions until growth arrest, or senescence, occurs. Germ cells and stem cells both contain active telomerase, but only the germ cells have sufficient levels of the enzyme to stabilize telomere length completely. In cancer cells, telomerase is often reactivated.
(Data from Macmillan Publishers Ltd, from Holt SE, et al: Refining the telomer-telomerase hypothesis of aging and cancer. Nat Biotechnol 14:836, 1996.)
There has been great interest in defining signaling pathways that counteract the aging process, not only because of their obvious therapeutic potential (the search for the “elixir of youth”) but also because elucidating these pathways might tell us about the mechanisms that cause aging. It is now thought that certain environmental stresses, such as calorie restriction, alter signaling pathways that influence aging (Fig. 1–29). Among the biochemical alterations that have been described as playing a role in counteracting the aging process are reduced signaling by insulin-like growth factor receptors, reduced activation of kinases (notably the “target of rapamycin,” [TOR], and the AKT kinase), and altered transcriptional activity. Ultimately these changes lead to improved DNA repair and protein homeostasis and enhanced immunity, all of which inhibit aging. Environmental stresses may also activate proteins of the Sirtuin family, such as Sir2, which function as protein deacetylases. These proteins may deacetylate and thereby activate DNA repair enzymes, thus stabilizing the DNA; in the absence of these proteins, DNA is more prone to damage. Although the role of sirtuins has received a great deal of attention recently, their importance in the aging process is not yet established.
Summary
Cellular Aging
• Results from combination of accumulating cellular damage (e.g., by free radicals), reduced capacity to divide (replicative senescence), and reduced ability to repair damaged DNA
• Accumulation of DNA damage: defective DNA repair mechanisms; conversely DNA repair may be activated by calorie restriction, which is known to prolong aging in model organisms
• Replicative senescence: reduced capacity of cells to divide secondary to progressive shortening of chromosomal ends (telomeres)
• Other factors: progressive accumulation of metabolic damage; possible roles of growth factors that promote aging in simple model organisms
It should be apparent that the various forms of cellular derangements and adaptations described in this chapter cover a wide spectrum, ranging from adaptations in cell size, growth, and function, to the reversible and irreversible forms of acute cell injury, to the regulated type of cell death represented by apoptosis. Reference is made to these many different alterations throughout this book, because all instances of organ injury and ultimately all cases of clinical disease arise from derangements in cell structure and function.
Auten RL, Davis JM. Oxygen toxicity and reactive oxygen species: the devil is in the details. Pediatr Res. 2009;66:121. [A review of the production and degradation of reactive oxygen species, and their roles in cell injury.]
Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483. [A good review of the role of free radicals in aging.]
Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361:2353. [An excellent review of the basic biology of telomeres, and how their abnormalities may contribute to cancer, aging, and other diseases.]
Chipuk JE, Moldoveanu T, Llambl F, et al. The BCL-2 family reunion. Mol Cell. 2010;37:299. [A review of the biochemistry and biology of the BCL-2 family of apoptosis-regulating proteins.]
de Groot H, Rauen U. Ischemia-reperfusion injury: processes in pathogenetic networks: a review. Transplant Proc. 2007;39:481. [A review of the roles of intrinsic cell injury and the inflammatory response in ischemia-reperfusion injury.]
Dong Z, Saikumar P, Weinberg JM, Venkatachalam MA. Calcium in cell injury and death. Annu Rev Pathol. 2006;1:405. [A review of the links between calcium and cell injury.]
Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059. [An excellent review of the mechanisms by which apoptotic cells are cleared, and how abnormalities in these clearance pathways may result in disease.]
Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45. [Excellent discussion of the mechanisms of muscle hypertrophy, using the heart as the paradigm.]
Galluzzi L, Aaronson SA, Abrams J, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093. [A practical summary of the morphologic and other techniques for detecting and quantifying dead cells.]
Haigis MC, Yankner BA. The aging stress response. Mol Cell. 2010;40:333. [A review of the role of cellular stresses in controlling the aging process.]
Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570. [Excellent review of the major pathways of cell death (necrosis, apoptosis, and autophagy-associated death), and their clinical implications and therapeutic targeting.]
Kenyon CJ. The genetics of ageing. Nature. 2010;464:504. [An excellent review of the genes that influence aging, based on human genetic syndromes and studies with mutant model organisms.]
Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280. [An excellent discussion of the biology, biochemical pathways, and physiologic roles of autophagy.]
Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427. [A discussion of the biology of autophagy and its potential contribution to a variety of disease states.]
Lin JH, Walter P, Yen TSB. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399. [A review of the biology and disease relevance of the unfolded protein response and ER stress induced by unfolded proteins.]
Lombard DB, Chua KF, Mostoslavsky R, et al. DNA repair, genome stability, and aging. Cell. 2005;120:497. [The role of DNA damage in cellular aging.]
McKinnell IW, Rudnicki MA. Molecular mechanisms of muscle atrophy. Cell. 2004;119:907. [Discussion of the mechanisms of cellular atrophy.]
Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481. [Excellent review of the many functions of mitochondria, with an emphasis on their role in cell death.]
Sahin E, DePinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520. [An excellent review of stem cell abnormalities that contribute to aging.]
Tosh D, Slack JM. How cells change their phenotype. Nat Rev Mol Cell Biol. 2002;3:187. [Review of metaplasia and the roles of stem cells and genetic reprogramming.]
Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44. [An interesting discussion of the biochemistry of reactive oxygen and nitrogen-derived free radicals, their roles in cell injury, and their physiologic functions as signaling molecules.]