Chapter 21 Peripheral Nerves and Muscles
See Targeted Therapy available online at studentconsult.com
The major components of the neuromuscular system, the peripheral nerves and skeletal muscles, act as both effectors and sensors for the central nervous system, and in doing so allow thought and sensation to give rise to physical actions and cognitive responses. The principal component of the motor system is the motor unit, which is composed of one lower motor neuron and its associated peripheral axon, neuromuscular junctions, and innervated skeletal muscle fibers. Both the anatomic distribution of lesions and specific signs and symptoms are helpful in classifying neuromuscular diseases and in distinguishing them from diseases of the central nervous system. Accordingly, the discussion of neuromuscular disorders is organized along anatomic lines, highlighting the clinical features that are most useful in their diagnosis.
Disorders of Peripheral Nerves
The two major functional elements of peripheral nerves are axonal processes and their myelin sheaths, which are made by Schwann cells. Axonal diameter and myelin thickness are correlated with each other and with conduction velocity; they can be used to distinguish among different types of axons, which mediate distinct sensory modalities and motor function. Light touch, for example, is transmitted by thickly myelinated large-diameter axons with fast conduction velocities, while temperature sensation is transmitted by slow, unmyelinated thin axons. In the case of myelinated axons, one Schwann cell makes and maintains exactly one myelin segment, or internode, along a single axon (Fig. 21–1, A). Adjacent internodes are separated by nodes of Ranvier. Peripheral nerves contain a mixture of different types of axons. These and the intervening endoneurial connective tissue are arranged into fascicles that are ensheathed by a layer of perineurial cells. The perineurial cells are similar to meningeal cells and help to maintain the blood-nerve barrier in the individual fascicles.
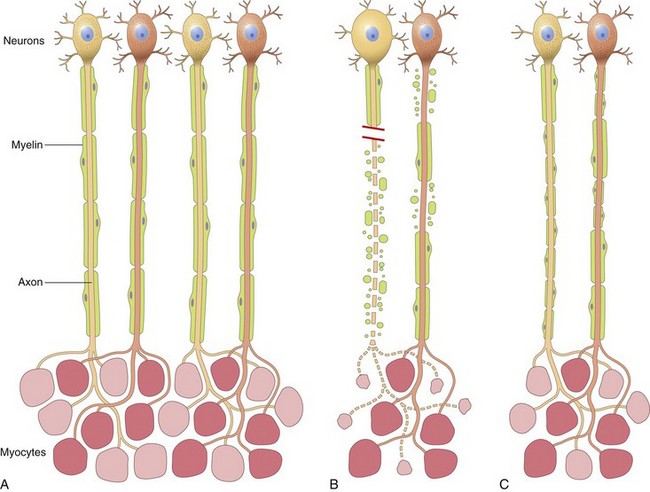
Figure 21–1 Patterns of peripheral nerve damage. A, In normal motor units, type I and type II myofibers are arranged in a “checkerboard” distribution, and the internodes along the motor axons are uniform in thickness and length. B, Acute axonal injury (left axon) results in degeneration of the distal axon and its associated myelin sheath, with atrophy of denervated myofibers. By contrast, acute demyelinating disease (right axon) produces random segmental degeneration of individual myelin internodes, while sparing the axon. C, Regeneration of axons after injury (left axon) allows connections with myofibers to re-form. The regenerated axon is myelinated by proliferating Schwann cells, but the new internodes are shorter and the myelin sheaths are thinner than the original ones. Remission of demyelinating disease (right axon) allows remyelination to take place, but the new internodes also are shorter and have thinner myelin sheaths than flanking normal undamaged internodes.
Patterns of Peripheral Nerve Injury
Most peripheral neuropathies can be subclassified as either axonal or demyelinating, even though some diseases exhibit mixed features. Axonal neuropathies are caused by insults that directly injure the axon. The entire distal portion of an affected axon degenerates. Axonal degeneration is associated with secondary myelin loss (Fig. 21–1, B), a process sometimes referred to as Wallerian degeneration. Regeneration takes place through axonal regrowth and subsequent remyelination of the distal axon (Fig. 21–1, C). The morphologic hallmark of axonal neuropathies is a decrease in the density of axons, which in electrophysiologic studies correlates with a decrease in the strength of amplitude of nerve impulses.
Demyelinating neuropathies are characterized by damage to Schwann cells or myelin with relative axonal sparing, resulting in abnormally slow nerve conduction velocities. Demyelination typically occurs in individual myelin internodes randomly; this process is termed segmental demyelination (Fig. 21–1, B). Morphologically, demyelinating neuropathies show a relatively normal density of axons and features of segmental demyelination and repair. This is recognized by the presence of axons with abnormally thin myelin sheaths and short internodes (Fig. 21–1, C). These latter changes are best demonstrated on teased fiber preparations, which allow the examination of several adjacent myelin internodes along a segment of an individual axon (described later).
Peripheral neuropathies fall into several anatomic patterns and may cause selective sensory or motor axon damage, or a mixture of both.
• Polyneuropathies usually affect peripheral nerves in a symmetric, length-dependent fashion. Axonal loss is typically diffuse and more pronounced in the distal segments of the longest nerves. Patients commonly present with loss of sensation and paresthesias that start in the toes and spread upward to the knees and then involve the hands in a “stocking-and-glove” distribution.
• Polyneuritis multiplex, in which the damage randomly affects portions of individual nerves, resulting (for example) in a right radial nerve palsy and wrist drop together with loss of sensation in the left foot
• A simple mononeuropathy involving only a single nerve most commonly is the result of traumatic injury or entrapment (e.g., carpal tunnel syndrome).
Disorders Associated with Peripheral Nerve Injury
Many different diseases may be associated with peripheral neuropathy (Table 21–1). We discuss next in some detail selected entities that are prototypical for a specific type of polyneuropathy (e.g., Guillain-Barré syndrome) or are common (e.g., diabetic neuropathy).
Table 21–1 Peripheral Neuropathies
| Etiologic Category | Causative Disorders/Agents |
|---|---|
| Nutritional and metabolic | Diabetes mellitus |
| Uremia | |
| Vitamin deficiencies—thiamine, vitamin B6, vitamin B12 | |
| Toxic | Drugs, including vinblastine, vincristine, paclitaxel, colchicine, and isoniazid |
| Other toxins—alcohol, lead, aluminum, arsenic, mercury, acrylamide | |
| Vasculopathic | Vasculitis |
| Amyloidosis | |
| Inflammatory | Autoimmune diseases: systemic lupus erythematosus, rheumatoid arthritis, sarcoidosis, Sjögren syndrome |
| Guillain-Barré syndrome | |
| Chronic inflammatory demyelinating polyneuropathy (CIDP) | |
| Infections | Herpes zoster—most often ganglionitis |
| Leprosy | |
| HIV infection | |
| Lyme disease—often facial nerve palsy | |
| Inherited | Charcot-Marie-Tooth neuropathy, type 1: autosomal dominant (many cases with tandem duplications in PMP22) |
| Charcot-Marie-Tooth neuropathy, type 3: autosomal dominant or recessive (some with point mutations in PMP22) | |
| Charcot-Marie-Tooth neuropathy, X-linked (connexin 32 gene mutations) | |
| Hereditary neuropathy with liability to pressure palsy: autosomal dominant deletions of PMP22 | |
| Others | Paraneoplastic, some leukodystrophies |
Guillain-Barré Syndrome
Guillain-Barré syndrome is one of the most common life-threatening diseases of the peripheral nervous system. It is a rapidly progressive acute demyelinating disorder affecting motor axons that results in ascending weakness that may lead to death from failure of respiratory muscles over a period of only several days. It appears to be triggered by an infection or a vaccine that breaks down self-tolerance, thereby leading to an autoimmune response. Associated infectious agents include Campylobacter jejuni, Epstein-Barr virus, cytomegalovirus, and human immunodeficiency virus. The injury is most extensive in the nerve roots and proximal nerve segments and is associated with mononuclear cell infiltrates rich in macrophages. Both humoral and cellular immune responses are believed to play a role in the disease process. Treatments include plasmapheresis (to remove offending antibodies), intravenous immunoglobulin infusions (which suppress immune responses through unclear mechanisms) and supportive care, such as ventilatory support. Patients who survive the initial acute phase of the disease usually recover over time.
Chronic Inflammatory Demyelinating Polyneuropathy
Chronic inflammatory demyelinating polyneuropathy (CIDP) typically manifests as a symmetric demyelinating disease. Both motor and sensory abnormalities are common, such as difficulty in walking, weakness, numbness, and pain or tingling sensations. Like Guillain-Barré syndrome, CIDP is immune-mediated and occurs at increased frequency in patients with other immune disorders, such as systemic lupus erythematosus and HIV infection. In contrast with Guillain-Barré syndrome, however, CIDP follows a chronic, relapsing-remitting or progressive course. The peripheral nerves show segments of demyelination and remyelination (Fig. 21-2, A). In long-standing cases, chronically regenerating Schwann cells may concentrically wrap around axons in multiple layers in an onion-skin pattern. Treatment includes plasmapheresis and administration of immunosuppressive agents. Some patients recover completely, but more often recurrent bouts of symptomatic disease lead to permanent loss of nerve function.
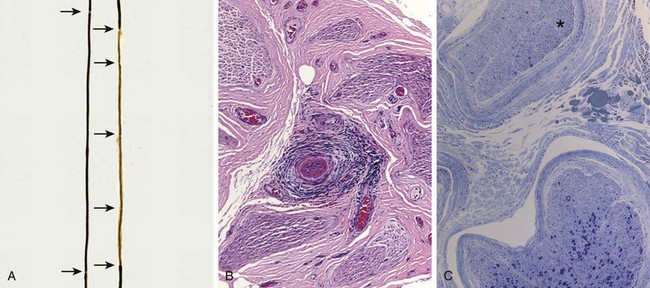
Figure 21–2 Pathologic changes in peripheral neuropathies. A, Regeneration after segmental demyelination. Teased fiber preparations allow for examination of individual axons of peripheral nerves. A normal axon (left) has a myelin sheath of uniform thickness that is interrupted at the nodes of Ranvier (arrows). By contrast, the right axon contains a poorly myelinated segment with unevenly distributed nodes of Ranvier. The area of remyelination is segmental and therefore flanked by internodes with normal myelination. B and C, Vasculitic neuropathy. In B, the perineurial connective tissue contains a vasculocentric inflammatory infiltrate that has obliterated a small vessel. In C, a special stain that colors myelinated axons dark blue reveals that the nerve fascicle in the upper portion of this field (asterisk) has lost almost all of its large myelinated axons, in contrast with the other fascicle shown. Such interfascicular variation in axonal density often is seen in neuropathies resulting from vascular injury.
Diabetic Peripheral Neuropathy
Diabetes is the most common cause of peripheral neuropathy (Chapter 19). Neuropathies usually arise in diabetics with long-standing disease. They include autonomic neuropathy, lumbosacral radiculopathies, and distal symmetric sensorimotor polyneuropathy; these may occur singly or together. Autonomic neuropathy is characterized by changes in bowel, bladder, cardiac, or sexual function. Lumbosacral radiculopathy usually manifests with asymmetric pain that can progress to lower extremity weakness and muscle atrophy. Distal symmetric sensorimotor polyneuropathy is the most common form of diabetic neuropathy. Sensory axons are more severely affected than motor axons, so the clinical presentation usually is dominated by paresthesias and numbness. This form of diabetic polyneuropathy results from the length-dependent degeneration of peripheral nerves and does not neatly fit into the axonal or demyelinating category but instead often exhibits features of both. The pathogenesis of diabetic neuropathy is complex; accumulation of advanced glycosylation end products, hyperglycemia, increased levels of reactive oxygen species, microvascular changes, changes in axonal metabolism, abnormal protein C levels, and neurotrophic factors all have been implicated. Strict glycemic control is the best form of therapy.
Toxic, Vasculitic, and Inherited Forms of Peripheral Neuropathy
• Drugs and environmental toxins that interfere with axonal transport or cytoskeletal function often produce peripheral neuropathies. The longest axons are most susceptible, so the resulting clinical presentation is often most pronounced in the distal extremities.
• Peripheral nerves are often damaged in many different forms of systemic vasculitis (Fig. 21–2, B) (Chapter 9), including polyarteritis nodosa, Churg-Strauss syndrome, and Wegener granulomatosis. Overall, peripheral nerve damage is seen in about a third of all patients with vasculitis at the time of presentation. The most common clinical picture is that of mononeuritis multiplex with a painful asymmetric mixed sensory and motor peripheral neuropathy. Patchy involvement also is apparent at the microscopic level, as single nerves may show considerable interfascicular variation in the degree of axonal damage (Fig. 21–2, C).
• Inherited diseases of peripheral nerves are a heterogeneous but relatively common group of disorders, with a prevalence of 1 to 4 in 10,000. They can be demyelinating or axonal. Most such disorders manifest in adulthood and follow a slowly progressive course that may mimic that of acquired polyneuropathies. The most common cause is mutations in the PMP22 gene, which encodes a protein that is a component of the myelin sheath.
 Summary
Summary
Peripheral Neuropathies
• Peripheral neuropathies may result in weakness and/or sensory deficits and may be symmetric or consist of random involvement of individual nerves.
• Axonal and demyelinating peripheral neuropathies can be distinguished on the basis of clinical and pathologic features. Some disorders are associated with a mixed pattern of injury.
• Diabetes is the most common cause of peripheral neuropathy.
• Guillain-Barré syndrome and chronic idiopathic demyelinating polyneuropathy are immune-mediated demyelinating diseases that follow acute and chronic courses, respectively.
• Metabolic diseases, drugs, toxins, connective tissue diseases, vasculitides, and infections all can result in peripheral neuropathy.
• A number of mutations cause peripheral neuropathy. Many of these are late-onset diseases that may mimic acquired ones.
Disorders of Neuromuscular Junction
The neuromuscular junction is a specialized interface between synaptic nerve endings and muscle fibers. Nerve impulses depolarize the presynaptic membrane, stimulating calcium influx and the release of acetylcholine into the synaptic cleft. Acetylcholine diffuses across the synaptic cleft to bind its receptor on the postsynaptic membrane, leading to depolarization of the myofiber and contraction through electromechanical coupling. Often disorders of the neuromuscular junction produce functional abnormalities in the absence of any significant alterations in morphology beyond ultrastructural changes. Considered in this section are some of the more common or pathogenically interesting disorders that disrupt the transmission of signals across the neuromuscular junction.
Myasthenia Gravis
Myasthenia gravis is caused by autoantibodies that block the function of postsynaptic acetylcholine receptors at motor end plates, which results in the degradation and depletion of the receptors. The disease has an incidence of roughly 3 in 100,000 persons, can manifest at any age, and (like many autoimmune disorders) is more common in females. The disease can be transferred to animals with serum from affected patients, demonstrating the causal role of circulating anti-acetylcholine receptor antibodies. Some 60% of cases are associated with a peculiar reactive hyperplasia of intrathymic B cells (often referred to as thymic hyperplasia), and another 20% are associated with thymoma, a tumor of thymic epithelial cells (Chapter 11). These thymic lesions may perturb tolerance to self antigens, thereby setting the stage for the generation of autoreactive T and B cells.
Clinically, myasthenia gravis frequently manifests with ptosis (drooping eyelids) or diplopia (double vision) due to weakness in the extraocular muscles. This pattern of weakness is distinctly different from that in most primary myopathic processes, in which there is relative sparing of facial and extraocular muscles. The severity of the weakness often fluctuates dramatically, sometimes over periods of only a few minutes. Characteristically, repetitive use or electrophysiologic stimulation of muscles makes the weakness more severe, whereas administration of cholinesterase inhibitors improves strength markedly; both of these features are diagnostically useful. Effective treatments include cholinesterase inhibitory drugs, immunosuppression, plasmapheresis, and (in patients with thymic lesions) thymectomy. These interventions have improved the 5-year survival rate to greater than 95%.
Lambert-Eaton Syndrome
Lambert-Eaton syndrome is caused by autoantibodies that inhibit the function of presynaptic calcium channels, which reduces the release of acetylcholine into the synaptic cleft. In contrast with those suffering from myasthenia gravis, patients with Lambert-Eaton syndrome experience improvement in weakness with repetitive stimulation. This serves to build up sufficient intracellular calcium to facilitate acetylcholine release.
Like myasthenia gravis, however, the disorder can be transferred to animals through the serum of affected patients. It often arises as a paraneoplastic disorder, particularly in patients with small cell lung carcinoma. Cholinesterase inhibitors are not effective, and therapy is therefore directed toward reducing the titer of causative antibodies, through either plasmapheresis or immunosuppression. Owing to the strong link to lung cancer, the overall prognosis for patients with Lambert-Eaton syndrome is substantially worse than for those affected by myasthenia gravis.
Miscellaneous Neuromuscular Junction Disorders
Several other neuromuscular junction disorders merit brief mention.
• Congenital myasthenic syndromes comprise a heterogeneous group of genetic diseases that result from mutations that disrupt the function of various neuromuscular junction proteins. Depending on the affected protein, the defects can occur at the level of acetylcholine release (presynaptic), the transport of acetylcholine across the synaptic cleft (synaptic), or the responsiveness of skeletal muscle (postsynaptic), and may produce symptoms suggestive of Lambert-Eaton syndrome or myasthenia gravis. Some forms respond to treatment with acetylcholinesterase inhibitors.
• Infections may be associated with defects in neural transmission and muscle contraction. Clostridium tetani and Clostridium botulinum (Chapter 8) both release extremely potent neurotoxins that interfere with neuromuscular transmission. Tetanus toxin (known as tetanospasmin) blocks the action of inhibitory neurons, leading to increased release of acetylcholine and sustained muscle contraction and spasm (tetanus). Botulinum toxin, by contrast, inhibits acetylcholine release, producing a flaccid paralysis. The purified toxin (Botox) is remarkably stable after injection, an attribute that has led to its widespread use as an antidote for wrinkles and a variety of other conditions associated with unwanted muscular activity (e.g., blepharospasm and strabismus).
Summary
Neuromuscular Junction Disorders
• Disorders of neuromuscular junctions manifest with weakness that often affects facial and extraocular muscles and may show marked fluctuation in severity.
• Both myasthenia gravis and Lambert-Eaton syndrome, the most common forms, are immune-mediated, being caused by antibodies to postsynaptic acetylcholine receptors and presynaptic calcium channels, respectively.
• Myasthenia gravis often is associated with thymic hyperplasia or thymoma. Lambert-Eaton syndrome in a majority of cases is a paraneoplastic disorder; the strongest association is with small cell lung cancer.
• Genetic defects in neuromuscular junction proteins and bacterial toxins also can cause symptomatic disturbances in neuromuscular transmission.
Disorders of Skeletal Muscle
Patterns of Skeletal Muscle Injury
Skeletal muscle consists of different fiber types broadly classified as slow twitch “aerobic” type I and fast twitch “anaerobic” type II fibers. They are normally distributed in checkerboard pattern (Fig. 21–1, A). Function of both types of fibers depends on the unique protein complexes that make up the sarcomeres and the dystrophin-glycoprotein complex (Fig. 21–3), as well as enzymes that meet the special metabolic requirements of muscle.
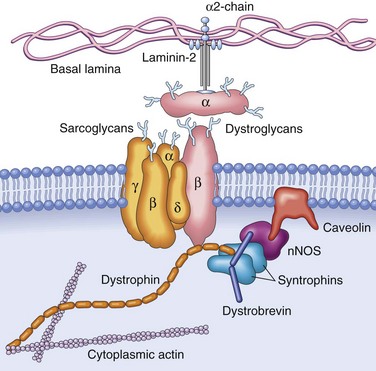
Figure 21–3 The dystrophin-glycoprotein complex (DGC). This complex of glycoproteins serves to couple the cell membrane (the sarcolemma) to the extracellular matrix proteins such as laminin-2 and the intracellular cytoskeleton. One key set of connections is made by dystrophin, a scaffolding protein that tethers the myofibrillar cytoskeleton to the transmembrane dystroglycans and sarcoglycans, and also binds complexes containing dystrobrevin, syntrophin, neuronal nitric oxide synthetase (nNOS), and caveolin, which participate in intracellular signaling pathways. Mutations in dystrophin are associated with X-linked Duchenne and Becker muscular dystrophies; mutations in caveolin and the sarcoglycan proteins, with autosomal limb-girdle muscular dystrophies; and mutations in α2-laminin (merosin), with a form of congenital muscular dystrophy.
Primary muscle diseases or myopathies have to be distinguished from secondary neuropathic changes caused by disorders that disrupt muscle innervation. Both are associated with altered muscle function and morphology, but each has distinctive features (Fig. 21–4). Myopathic conditions are often associated with segmental necrosis and regeneration of individual muscle fibers (Fig. 21–4, B). As discussed later on, specific types of myopathies have additional morphologic features, such as inflammatory infiltrates or intracellular inclusions. Disruption of muscle by endomysial fibrosis and fatty replacement is a feature of disease chronicity associated with myopathic or neuropathic conditions.
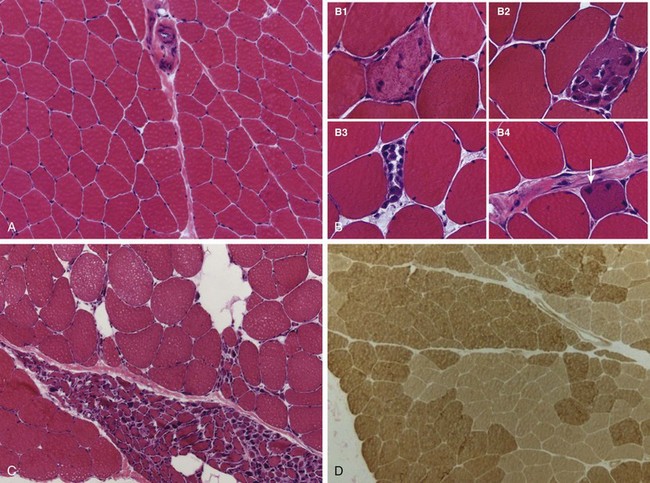
Figure 21–4 Patterns of skeletal muscle injury. A, Normal skeletal muscle has relatively uniform polygonal myofibers with peripherally placed nuclei that are tightly packed together into fascicles separated by scant connective tissue. A perimysial interfascicular septum containing a blood vessel is present (top center). B, Myopathic conditions often are associated with segmental necrosis and regeneration of individual myofibers. Necrotic cells (B1-B3) are infiltrated by variable numbers of inflammatory cells. Regenerative myofibers (B4, arrow) are characterized by cytoplasmic basophilia and enlarged nucleoli (not visible at this power). C and D, Clusters of both atrophic myofibers (C) (grouped atrophy) and fiber-type grouping (D), patchy areas in which myofibers share the same fiber type, are features of neurogenic remodeling. The ATPase reaction shown in D is one method of distinguishing between fiber types, as type I fibers stain more lightly than type II fibers. Note loss of the “checkerboard” pattern (Fig 21–1, A).
Muscle fiber atrophy is shared by both neuropathic and myopathic processes. However, certain disorders are associated with particular patterns of atrophy, as follows:
• Neuropathic changes are characterized by fiber type grouping and grouped atrophy. Changes in muscle innervation result in larger groups of fibers that share the same fiber type with the resultant replacement of the normal checkerboard distribution by groups of fibers that are type I or II (Fig. 21–4, D). The presence of fewer but larger motor units and the segregation of innervated fibers results in groups of atrophic fibers (Fig. 21–4, C). Remarkably, the fiber type of myofibers is not an inherent feature, but is dictated by the innervating motor neuron. Thus, if injury and regeneration of peripheral nerves alters muscle innervation, it will change the distribution of type I and type II myofibers. Degeneration and regeneration of individual fibers and inflammatory infiltrates usually are absent in skeletal muscle disorders caused by abnormal innervation.
• Prolonged disuse of muscles due to any cause (e.g., prolonged bed rest in the sick, casting of a broken bone) can cause focal or generalized muscle atrophy, which tends to affect type II fibers more than type I fibers.
• Glucocorticoid exposure, whether exogenous or endogenous (e.g., in Cushing syndrome), also can cause muscle atrophy. Proximal muscles and type II myofibers are affected preferentially by these agents.
Inherited Disorders of Skeletal Muscle
Genetic disorders affecting skeletal muscle include muscular dystrophies, congenital muscular dystrophies, and congenital myopathies. Muscular dystrophies are inherited diseases that result in progressive muscle injury in patients who usually appear normal at birth. Congenital muscular dystrophies are progressive, early-onset diseases. Some are also associated with central nervous system manifestations. Congenital myopathies are a heterogeneous group of inherited diseases that often have a perinatal or early childhood presentation and result in relatively static deficits.
The following discussion of the muscular dystrophies follows a long-standing classification that is based on inheritance patterns and clinical features. Of note, however, the classification of the muscular dystrophies is evolving based on new insights into the molecular pathogenesis of these disorders and genotype-phenotype relationships. For example, mutations in several different genes present as autosomal recessive limb-girdle muscular dystrophy, whereas different mutations in a single gene (such as dystrophin) can lead to two very different clinical phenotypes, the Duchenne and Becker types of muscular dystrophy.
Dystrophinopathies: Duchenne and Becker Muscular Dystrophy
Dystrophinopathies are the most common form of muscular dystrophy. Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are the two most important disease manifestations linked to mutations in the dystrophin gene. Duchenne muscular dystrophy has an incidence of about 1 per 3500 live male births and follows an inexorable fatal course. DMD becomes clinically evident by the age of 5 years; most patients are wheelchair-bound by the time they are teenagers and dead of their disease by early adulthood. The Becker type of muscular dystrophy is less common and much less severe.
 Morphology
Morphology
The histologic alterations in skeletal muscles affected by DMD and BMD are similar, except that the changes are milder in BMD (Fig. 21–5). The hallmarks of these as well as other muscular dystrophies are ongoing myofiber necrosis and regeneration. Progressive replacement of muscle tissue by fibrosis and fat is the result of degeneration outpacing repair. As a result of ongoing repair muscles typically show marked variation in myofiber size and abnormal internally placed nuclei. Both DMD and BMD also affect cardiac muscles, which show variable degrees of myofiber hypertrophy and interstitial fibrosis.
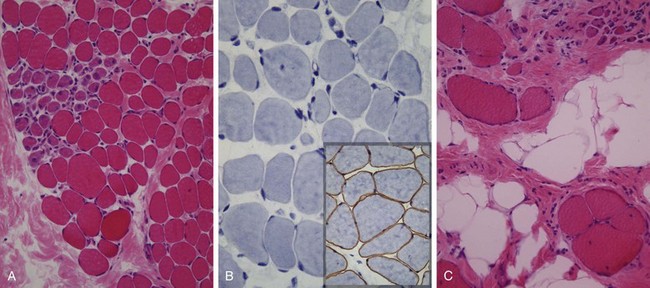
Figure 21–5 Duchenne muscular dystrophy. Histologic images of muscle biopsy specimens from two brothers. A and B, Specimens from a 3-year-old boy. C, Specimen from his brother, 9 years of age. As seen in A, at a younger age fascicular muscle architecture is maintained, but myofibers show variation in size. Additionally, there is a cluster of basophilic regenerating myofibers (left side) and slight endomysial fibrosis, seen as focal pink-staining connective tissue between myofibers. In B, immunohistochemical staining shows a complete absence of membrane-associated dystrophin, seen as a brown stain in normal muscle (inset). In C, the biopsy from the older brother illustrates disease progression, which is marked by extensive variation in myofiber size, fatty replacement, and endomysial fibrosis.
 Pathogenesis
Pathogenesis
Both DMD and BMD are caused by loss-of-function mutations in the dystrophin gene located on the short arm of the X chromosome (Xp21). Dystrophin is a very large protein (427 kD in molecular weight) found in skeletal and cardiac muscle, brain, and peripheral nerves; it is part of the dystrophin-glycoprotein complex (Fig. 21–3). This complex stabilizes the muscle cell during contraction and may be involved in cell signaling through interaction with other proteins. Dystrophin-glycoprotein complex defects are thought to make muscle cells vulnerable to transient membrane tears during contraction that lead to calcium influx, and may also disrupt intracellular signaling. The result is myofiber degeneration that with time outpaces the capacity for repair. The dystrophin-glycoprotein complex also is important for cardiac muscle function; this explains why cardiomyopathy eventually develops in many patients.
The dystrophin gene spans roughly 2.4 megabases (about 1% of the X chromosome), making it one of the largest human genes. Its enormous size may explain in part its vulnerability to sporadic mutations that disrupt dystrophin production. The most common mutations are deletions, followed by frameshift and point mutations. Muscle biopsy specimens from patients with DMD show a complete absence of dystrophin, whereas patients with BMD have mutations that permit some dystrophin (albeit often a defective form) to be made; thus, the severity of the disease correlates with the degree of the dystrophin deficiency.
Clinical Features
Often the first symptoms of DMD are clumsiness and an inability to keep up with peers due to muscle weakness. The weakness typically begins in the pelvic girdle and next involves the shoulder girdle. Enlargement of the calf muscles, termed pseudohypertrophy, is an important early physical finding. The increased muscle bulk initially stems from myofiber hypertrophy, but as myofibers progressively degenerate, an increasing part of the muscle is replaced by adipose tissue and endomysial fibrosis. Cardiac muscle damage and fibrosis can lead to heart failure and arrhythmias, which may prove fatal. Although no structural abnormalities in the central nervous system have been described, cognitive impairment is also sometimes seen and may be severe enough to manifest as mental retardation. Owing to ongoing muscle degeneration, high serum creatine kinase levels are present at birth and persist through the first decade of life but fall as muscle mass is lost during disease progression. Death results from respiratory insufficiency, pneumonia, and cardiac decompensation.
BMD becomes symptomatic later in childhood or adolescence and progresses at a slower and more variable rate. Many patients live well into adulthood and have a nearly normal life span. Cardiac involvement can be the dominant clinical feature and may result in death in the absence of significant skeletal muscle weakness.
Other X-Linked and Autosomal Muscular Dystrophies
Other forms of muscular dystrophy share some features with DMD and BMD but have distinct clinical, genetic, and pathologic features.
• Myotonic dystrophy. Myotonia, the sustained involuntary contraction of a group of muscles, is the cardinal neuromuscular symptom in myotonic dystrophy. Patients often complain of stiffness and difficulty releasing their grip, for instance, after a handshake. Myotonic dystrophy is inherited as an autosomal dominant trait. More than 95% of patients with myotonic dystrophy have mutations in the gene that encodes the dystrophia myotonica protein kinase (DMPK). In normal subjects, this gene contains fewer than 30 repeats of the sequence CTG, whereas in severely affected persons, several thousand repeats may be present. Myotonic dystrophy thus falls into the group of disorders associated with trinucleotide repeat expansions (Chapter 6). As is the case with other disorders with similar mutations, myotonic dystrophy exhibits the phenomenon of anticipation, characterized by worsening of the disease manifestations with each passing generation due to further trinucleotide repeat expansion. The CTG repeat expansion is located in the 3′ untranslated region of the DMPK mRNA, and the manner in which it produces disease is unclear. The disease often manifests in late childhood with gait abnormalities due to weakness of foot dorsiflexors, with subsequent progression to weakness of the intrinsic muscles of the hands and wrist extensors, atrophy of the facial muscles, and ptosis. Other tissues may also be affected, presenting as cardiac arrhythmias, cataracts, early frontal balding, endocrinopathies, and testicular atrophy.
• Limb-girdle muscular dystrophies. These autosomal muscular dystrophies preferentially affect the proximal musculature of the trunk and limbs. The genetic basis for these is heterogeneous. The growing list includes at least 6 dominant subtypes and 12 autosomal recessive subtypes. Some of the responsible mutations affect components of the dystrophin-glycoprotein complex other than dystrophin. Others affect proteins involved in vesicle transport and repair of cell membrane after injury (caveolin-3 and dysferlin), cytoskeletal proteins, or posttranslational modification of dystroglycan, a component of the dystrophin-glycoprotein complex.
• Emery-Dreifuss muscular dystrophy (EMD) is a rare but fascinating disorder caused by mutations affecting structural proteins found in the nucleus. An X-linked form results from mutations in the gene encoding the protein emerin, while an autosomal dominant form is caused by mutations in the gene encoding lamin A/C. It is hypothesized that defects in these proteins compromise the structural integrity of the nucleus in cells that are subjected to repetitive mechanical stress (e.g., cardiac and skeletal muscle). These proteins also may regulate chromatin structure. The clinical picture is characterized by progressive muscle weakness and wasting, contractures of the elbows and ankles, and cardiac disease. The cardiac involvement is severe, being associated with cardiomyopathy and arrhythmias that lead to sudden death in up to 40% of patients.
• Fascioscapulohumeral dystrophy is an autosomal dominant form of muscular dystrophy that is usually associated with deletions in chromosomal region 4q35. The pathophysiologic relationship between this chromosomal defect and the disease phenotype is not known. Most patients become symptomatic by the age of 20 years, usually owing to weakness in the facial muscles and the shoulder. Patients also exhibit weakness in the lower trunk and the dorsiflexors of the foot. Most affected persons have a normal life expectancy.
Channelopathies, Metabolic Myopathies, and Mitochondrial Myopathies
Other important inherited disorders of skeletal muscle are the result of defects in ion channels (channelopathies), metabolism, and mitochondrial function.
• Ion channel myopathies are a group of familial disorders characterized by myotonia, relapsing episodes of hypotonic paralysis associated with abnormal serum potassium levels, or both. As implied by their name, these diseases stem from inherited defects in genes encoding ion channels. Hyperkalemic periodic paralysis results from mutations in the gene encoding the skeletal muscle sodium channel protein SCN4A, which regulates sodium entry during contraction. Malignant hyperthermia is a rare syndrome characterized by a dramatic hypermetabolic state (tachycardia, tachypnea, muscle spasms, and finally hyperpyrexia). It is triggered when patients carrying mutations in the ryanodine receptor, a calcium release channel protein, are given halogenated anesthetic agents or succinylcholine during surgery. Some of these patients also show features of a congenital myopathy referred to as central core disease, so called because the center of the myofiber contains a collection of disorganized myofibrils.
• Myopathies due to inborn errors of metabolism include disorders of glycogen synthesis and degradation (Chapter 6), and abnormalities in lipid handling. The latter include disorders of the carnitine transport system or deficiencies of the mitochondrial dehydrogenase enzyme system, both of which can lead to significant accumulation of lipid in myocytes (lipid myopathies). These storage disorders can manifest as systemic disease or result in a muscle-specific phenotype. Some are associated with ongoing muscle damage and weakness. Others manifest with recurring episodes of massive exercise- or fasting-induced muscle damage, sometimes associated with acute renal failure and myoglobulinuria (rhabdomyolysis).
• Mitochondrial myopathies can stem from mutations in either the mitochondrial or nuclear genomes, the latter because some mitochondrial enzymes are encoded in nuclear DNA. The forms caused by mitochondrial mutations show maternal inheritance (Chapter 6). Mitochondrial myopathies usually manifest in early adulthood with proximal muscle weakness and sometimes with severe involvement of the ocular musculature (external ophthalmoplegia). There can also be neurologic signs and symptoms, lactic acidosis, and cardiomyopathy. Some mitochondrial diseases are associated with normal muscle morphology, whereas others show aggregates of abnormal mitochondria; the latter impart a blotchy red appearance in special stains—hence the term ragged red fibers. On ultrastructural examination, these correspond to abnormal aggregates of mitochondria with abnormal shape and size, some containing paracrystalline parking lot inclusions.
Acquired Disorders of Skeletal Muscle
A diverse group of acquired disorders can manifest with muscle weakness, muscle cramping, or muscle pain. These include inflammatory myopathies, toxic muscle injuries, postinfectious rhabdomyolysis, and muscle infarction in the setting of diabetes. In most instances these are disorders of adults with acute or subacute onsets.
Inflammatory Myopathies
Polymyositis, dermatomyositis, and inclusion body myositis are the most important primary inflammatory myopathies. Other immune disorders (e.g., SLE, sarcoidosis) also can involve skeletal muscle.
• Polymyositis is an autoimmune disorder associated with increased expression of MHC class I molecules on myofibers and predominantly endomysial inflammatory infiltrates containing CD8+ cytotoxic T cells. The autoimmune attack leads to myofiber necrosis and subsequent regeneration (Fig. 21–6, A). Patients with polymyositis are often successfully treated with corticosteroids or other immunosuppressive agents.
• Dermatomyositis is the most common inflammatory myopathy in children, in whom it appears as an isolated entity. In adults, it can manifest as a paraneoplastic disorder. In both contexts, it is believed to have an autoimmune basis. On microscopic examination, it is associated with perivascular mononuclear cell infiltrates, “dropout” of capillaries, the presence of so-called tubuloreticular inclusions in endothelial cells, and myofiber damage in a paraseptal or perifascicular pattern (Fig. 21–6, B). Type 1 interferon-induced gene products are strongly upregulated in affected muscles. Some patients have autoantibodies that are relatively specific for dermatomyositis; these include antibodies against Mi-2 (a nuclear helicase) and p155 and p140, proteins with uncertain functions.
• Inclusion body myositis is the most common inflammatory myopathy in patients older than 60 years of age. It is lumped in with other forms of myositis, but it has yet to be determined whether inflammation is cause or effect in this disorder. The morphologic hallmark of inclusion body myositis is the presence of rimmed vacuoles (Fig. 21–6, C) that contain aggregates of the same proteins that accumulate in the brains of patients with neurodegenerative diseases—hyperphosphorylated tau, amyloid derived from β-amyloid precursor protein and TDP-43 (Chapter 20)—leading some to speculate that this is a degenerative disorder of aging. Other features typical of chronic inflammatory myopathies, including myopathic changes, mononuclear cell infiltrates, endomysial fibrosis, and fatty replacement, also are evident. The disease follows a chronic, progressive course and generally does not respond well to immunosuppressive agents, another feature suggesting that inflammation is a secondary event.
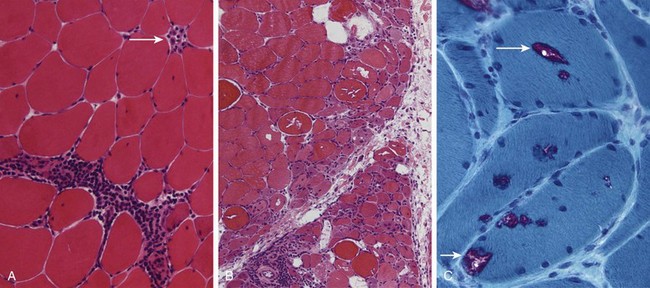
Figure 21–6 Inflammatory myopathies. A, Polymyositis is characterized by endomysial inflammatory infiltrates and myofiber necrosis (arrow). B, Dermatomyositis often shows prominent perifascicular and paraseptal atrophy. C, Inclusion body myositis, showing myofibers containing rimmed vacuoles (arrows).
Modified Gomori trichrome stain.
Toxic Myopathies
A number of insults can cause toxic muscle injury, including intrinsic factors (e.g., thyroxine) and extrinsic factors (e.g., acute alcohol intoxication, various drugs).
• Thyrotoxic myopathy may take the form of either acute or chronic proximal muscle weakness, and can be the first indication of thyrotoxicosis. Histologic findings include myofiber necrosis and regeneration.
• Ethanol myopathy occurs after an episode of binge drinking. The degree of rhabdomyolysis may be severe, sometimes leading to acute renal failure secondary to myoglobinuria. Patients usually complain of acute muscle pain, which may be generalized or confined to a single muscle group. Microscopically, there is myocyte swelling, necrosis, and regeneration.
• Drug myopathy can be produced by a variety of agents. Currently the most commonly implicated drugs are those belonging to the statin family. The affected muscles show evidence of myopathic injury, usually without an inflammatory component.
Summary
Disorders of Skeletal Muscle
• Skeletal muscle function can be impaired secondarily because of problems with muscle innervation or by a primary myopathy that can be inherited or acquired.
• The genetic forms of myopathy fall into several fairly distinct clinical phenotypes, including muscular dystrophy, congenital myopathy, and congenital muscular dystrophy.
• Dystrophinopathies are X-linked disorders caused by mutations in the dystrophin gene and disruption of the dystrophin-glycoprotein complex. Depending on the type of mutation the disease may be severe, such as Duchenne muscular dystrophy, or mild (e.g., Becker dystrophy).
• Acquired myopathies have diverse causes, including inflammation and toxic exposures.
Peripheral Nerve Sheath Tumors
A number of different tumors arise from peripheral nerves. Such tumors may manifest as soft tissue masses or with pain or loss of function related to impingement on nerves or other surrounding structures. In most peripheral nerve tumors, the neoplastic cells show evidence of Schwann cell differentiation. These tumors usually occur in adults and include both benign and malignant variants. An important feature is their frequent association with the familial tumor syndromes neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2). Tumors with skeletal muscle differentiation also occur; these are discussed in Chapter 20, along with other tumors of soft tissues.
Schwannomas and Neurofibromatosis Type 2
Schwannomas are benign encapsulated tumors that may occur in soft tissues, internal organs, or spinal nerve roots. The most commonly affected cranial nerve is the vestibular portion of the eighth nerve. Tumors arising in a nerve root or the vestibular nerve may be associated with symptoms related to nerve root compression, which includes hearing loss in the case of vestibular schwannomas.
Most schwannomas are sporadic, but about 10% are associated with familial neurofibromatosis type 2. NF2 patients are at risk of developing multiple schwannomas, meningiomas, and ependymomas (the latter are described in Chapter 22). The presence of bilateral vestibular schwannomas is a hallmark of NF2. Affected patients carry a dominant loss of function mutation of the merlin gene on chromosome 22. Merlin is a cytoskeletal protein that functions as a tumor suppressor by facilitating E-cadherin–mediated contact inhibition (Chapter 5). Of note, merlin expression is also disrupted in sporadic schwannomas. Despite the name of the syndrome, neurofibromas are not a feature of NF2. Schwannomatosis is a familial condition associated with multiple schwannomas in which vestibular schwannomas are absent. Some cases have recently been linked to loss-of-function mutations in a tumor suppressor gene on chromosome 22 that encodes a protein that regulates chromatin structure.
Morphology
On gross inspection, most schwannomas appear as circumscribed masses abutting an adjacent nerve. On microscopic examination, these tumors often show an admixture of dense and loose areas referred to as Antoni A and B, respectively (Fig. 21–7, A and B). They are comprised of a uniform proliferation of neoplastic Schwann cells. In the dense Antoni A areas, bland spindle cells with buckled nuclei are arranged into intersecting fascicles. These cells often align to produce nuclear palisading, resulting in alternating bands of nuclear and anuclear areas called Verocay bodies. Axons are largely excluded from the tumor. Thick-walled hyalinized vessels often are present. Hemorrhage or cystic change are also seen sometimes.
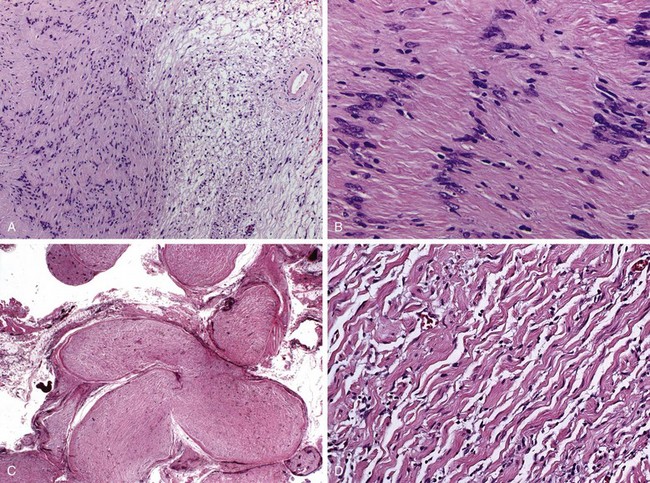
Figure 21–7 Schwannoma and plexiform neurofibroma. A and B, Schwannoma. As seen in A, schwannomas often contain dense pink Antoni A areas (left) and loose, pale Antoni B areas (right), as well as hyalinized blood vessels (right). B, Antoni A area with the nuclei of tumor cells aligned in palisading rows. C and D, Plexiform neurofibroma. Multiple nerve fascicles are expanded by infiltrating tumor cells (C), which at higher power (D) are seen to consist of bland spindle cells admixed with wavy collagen bundles likened to carrot shavings.
Neurofibromas
Neurofibromas are benign peripheral nerve sheath tumors. Three important subtypes are recognized:
• Localized cutaneous neurofibromas arise as superficial nodular or polypoid tumors. These occur either as solitary sporadic lesions or as often multiple lesions in the context of neurofibromatosis type 1 (NF1).
• Plexiform neurofibromas grow diffusely within the confines of a nerve or nerve plexus. Surgical enucleation of such lesions is therefore difficult and is often associated with lasting neurologic deficits. Plexiform neurofibromas are virtually pathognomonic for NF1 (discussed later on). Unlike other benign nerve sheath tumors, these tumors are associated with a small but real risk of malignant transformation.
• Diffuse neurofibromas are infiltrative proliferations that can take the form of large, disfiguring subcutaneous masses. These also are often associated with NF1.
Morphology
Unlike schwannomas, neurofibromas are not encapsulated. They may appear circumscribed, as in localized cutaneous neurofibromas, or exhibit a diffuse infiltrative growth pattern. Also in contrast with schwannomas, the neoplastic Schwann cells in neurofibroma are admixed with other cell types, including mast cells, fibroblast-like cells and perineurial-like cells. As a result, the cellular growth pattern of neurofibromas is more haphazard than that of schwannomas. The background stroma often contains loose wavy collagen bundles but also can be myxoid or contain dense collagen (Fig. 21–7, D). Plexiform neurofibromas involve multiple fascicles of individual affected nerves (Fig. 21–7, C). Residual axons are found embedded within the diffuse neoplastic Schwann cell proliferation, which expand the fascicles while leaving the perineurium intact. Diffuse neurofibromas show an extensive infiltrative pattern of growth within the dermis and subcutis of the skin.
Malignant Peripheral Nerve Sheath Tumors
Malignant peripheral nerve sheath tumors are neoplasms seen in adults that typically show evidence of Schwann cell derivation and sometimes a clear origin from a peripheral nerve. They may arise from transformation of a neurofibroma, usually of the plexiform type. About one half of such tumors arise in patients with NF1, and 3% to 10% of all patients with NF1 develop a malignant peripheral nerve sheath tumor during their lifetime.
Morphology
Malignant peripheral nerve sheath tumors manifest as large, poorly defined soft tissue masses. On histologic examination, these tumors are highly cellular and exhibit features of overt malignancy, including anaplasia, necrosis, infiltrative growth pattern, pleomorphism, and high proliferative activity. The low-power view often shows alternating areas of high and low cellularity that result in an appearance described as “marble-like.” Also frequently seen are perivascular areas of increased cellular density.
Neurofibromatosis Type 1
NF1 is an autosomal dominant disorder caused by mutations in the tumor suppressor neurofibromin, encoded on the long arm of chromosome 17 (17q). Neurofibromin is a negative regulator of the potent oncoprotein Ras (Chapter 5). Disruption of neurofibromin function and Ras hyperactivity appear to be a cardinal feature of NF1-associated tumors. As would be anticipated for a tumor suppressor gene, the sole normal neurofibromin allele is mutated or silenced in tumors arising in the setting of NF1, which include neurofibromas of all three main types, malignant peripheral nerve sheath tumors, optic gliomas, and other glial tumors. In addition, patients with NF1 exhibit learning disabilities, seizures, skeletal abnormalities, vascular abnormalities with arterial stenoses, pigmented nodules of the iris (lisch nodules), and pigmented skin lesions (axillary freckling and café au lait spots) in various degrees.
Traumatic Neuroma
Traumatic neuroma is a non-neoplastic proliferation associated with previous injury of a peripheral nerve. Injuries that lead to the transection of axons activate a regenerative program (see Fig. 21–1) characterized by sprouting and elongation of processes from the proximal axonal stump. With severe injuries that disrupt the perineurial sheath, these new processes may “miss” their target, the distal end of the transected nerve. The misguided elongating axonal processes can induce a reactive proliferation of Schwann cells, leading to the formation of a painful localized nodule that consists of a haphazard mixture of axons, Schwann cells, and connective tissue.
Summary
Peripheral Nerve Sheath Tumors
• In most peripheral nerve sheath tumors, the neoplastic cells show evidence of Schwann cell differentiation.
• Peripheral nerve sheath tumors are important features of the familial tumor syndromes neurofibromatosis type 1 (NF1) and type 2 (NF2).
• Schwannomas and neurofibromas are benign nerve sheath tumors.
• Schwannomas are circumscribed, usually encapsulated tumors that abut the nerve of origin and are a feature of NF2.
• Neurofibromas may manifest as a sporadic subcutaneous nodule, as a large, poorly defined soft tissue lesion, or as a growth within a nerve. Neurofibromas are associated with NF1.
• About 50% of malignant peripheral nerve sheath tumors occur de novo in otherwise normal persons, while the remainder arise from the malignant transformation of a preexisting NF1-associated neurofibroma.
Amato AA, Barohn RJ. Evaluation and treatment of inflammatory myopathies. J Neurol Neurosurg Psychiatry. 2009;80:1060. [Review of idiopathic inflammatory myopathies focused especially on clinical features and therapy.]
Briemberg HR. Peripheral nerve complications of medical disease. Semin Neurol. 2009;29:124. [Review of the ways medical diseases including diabetes, connective tissue diseases, cancer, and infections affect peripheral nerves.]
Dalakas MC. Inflammatory muscle diseases: a critical review on pathogenesis and therapies. Curr Opin Pharmacol. 2010;10:346. [Discussion of current concepts on the pathophysiology of idiopathic inflammatory myopathies.]
Finsterer J, Stollberger C. Primary myopathies and the heart. Scand Cardiovasc J. 2008;42:9. [Review of inherited myopathies with focus on associated cardiac involvement.]
Gorson KC. Vasculitic neuropathies: an update. Neurologist. 2007;13:12. [A good review of peripheral nerve disease with vasculitis.]
Greenberg SA. Inflammatory myopathies: disease mechanisms. Curr Opin Neurol. 2009;22:516. [Discussion of current concepts on the pathophysiology of idiopathic inflammatory myopathies.]
Habib AA, Brannagan THIII. Therapeutic strategies for diabetic neuropathy. Curr Neurol Neurosci Rep. 2010;10:92. [Review focused especially on clinical features and therapy of diabetic neuropathy.]
Hewer E, Goebel HH. Myopathology of non-infectious inflammatory myopathies—the current status. Pathol Res Pract. 2008;204:609. [Review focused on the pathologic features of inflammatory myopathies.]
Klopstock T. Drug-induced myopathies. Curr Opin Neurol. 2008;21:590. [Review focused especially on the effects of statins and nucleoside analogue reverse transcriptase inhibitors for HIV infection/AIDS.]
Mahadeva B, Phillips LH, Juel VC. Autoimmune disorders of neuromuscular transmission. Semin Neurol. 2008;28:212. [Review of myasthenia gravis and Lambert-Eaton syndrome.]
McClatchey AI. Neurofibromatosis. Annu Rev Pathol. 2007;2:191. [Review of features that distinguish neurofibromatosis type 1, neurofibromatosis type 2, and schwannomatosis, with a focus on the genetics.]
Nelson SF, Crosbie RH, Miceli MC, et al. Emerging genetic therapies to treat Duchenne muscular dystrophy. Curr Opin Neurol. 2009;22:532. [Good review of recent developments in the search for new therapies.]
North K. What’s new in congenital myopathies? Neuromuscul Disord. 2008;18:433. [Review on new developments in congenital myopathies.]
Obrosova IG. Diabetes and the peripheral nerve. Biochim Biophys Acta. 2009;1792:931. [Detailed discussion of the pathophysiology of diabetic neuropathy.]
Silberman J, Lonial S. Review of peripheral neuropathy in plasma cell disorders. Hematol Oncol. 2008;26:55. [Review of the ways in which peripheral nerve diseases are related to plasma cell disorders and the chemotherapies used in their treatment.]
van Adel BA, Tarnopolsky MA. Metabolic myopathies: update 2009. J Clin Neuromuscul Dis. 2009;10:97. [Review of metabolic myopathies.]