MECHANISMS OF HORMONAL REGULATION


The endocrine system is composed of various glands located throughout the body (Figure 20-1). These glands are capable of synthesizing and releasing special chemical messengers called hormones. The endocrine system has five general functions:
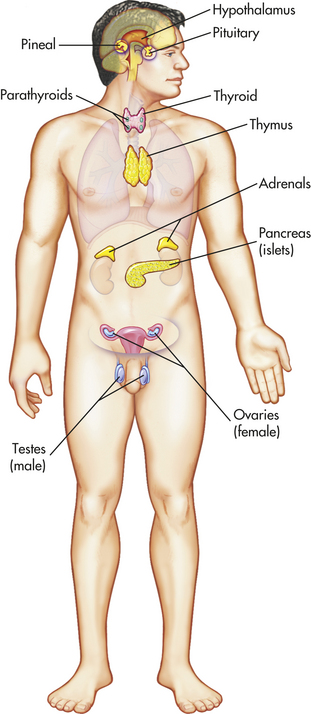
Figure 20-1 Principal endocrine glands. (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
1. Differentiation of the reproductive and central nervous systems in the developing fetus
2. Stimulation of sequential growth and development during childhood and adolescence
3. Coordination of the male and female reproductive systems, which makes sexual reproduction possible
4. Maintenance of an optimal internal environment throughout the life span
5. Initiation of corrective and adaptive responses when emergency demands occur
Hormones convey specific regulatory information among cells and organs and are integrated with the nervous system to maintain communication and control. The mechanisms of communication include autocrine (within cell), paracrine (between local cells), and endocrine (between remote cells).1
MECHANISMS OF HORMONAL REGULATION
The endocrine glands respond to specific signals by synthesizing and releasing hormones into the circulation. Although a wide variety of hormones function within the body, they share certain general characteristics:
1. Hormones have specific rates and rhythms of secretion. Three basic secretion patterns are (1) circadian or diurnal patterns, (2) pulsatile and cyclic patterns, and (3) patterns that depend on levels of circulating substrates (e.g., calcium, sodium, potassium, or the hormones themselves).
2. Hormones operate within feedback systems, either positive or negative, to maintain an optimal internal environment.
3. Hormones affect only cells with appropriate receptors and then act on those cells to initiate specific cell functions or activities.
4. Hormones are either excreted directly by the kidneys or metabolized by the liver, which inactivates them and renders the hormone more water soluble for renal excretion.
Hormones may be classified according to their structure, gland of origin, effects, or chemical composition. (Table 20-1 categorizes hormones based on structure.) The secretion and mechanisms of action of hormones represent an extremely complex system of integrated responses. Although much has been learned about these complex systems, many of the specific mechanisms of action are not yet understood. The endocrine and nervous systems work together to regulate responses to the internal and external environments.

Regulation of Hormone Release
The release of hormones occurs either in response to an alteration in the cellular environment or in the process of maintaining a regulated level of certain hormones or certain substances.2 Hormone release is regulated by one or more of the following mechanisms: (1) chemical factors (such as blood sugar or calcium levels); (2) endocrine factors (a hormone from one endocrine gland controlling another endocrine gland); and (3) neural control. Of these regulatory mechanisms, endocrine regulation by way of feedback circuits (systems) is perhaps the most important way in which hormonal secretion is maintained within a physiologic range.1,2
Negative feedback is the most common type of feedback system. In a negative-feedback system, plasma levels of one type of hormone influence the level of other types of hormones. An example of hormone negative feedback is shown in Figure 20-2, A. Increased anterior pituitary release of thyroid-stimulating hormone (TSH) stimulates the synthesis and secretion of thyroid hormones. TSH is inhibited by thyroxine (T4) and to a lesser extent by triiodothyronine (T3). TSH secretion is regulated by thyrotropin-releasing hormone primarily in the hypothalamus and by negative-feedback inhibition from thyroid hormones.
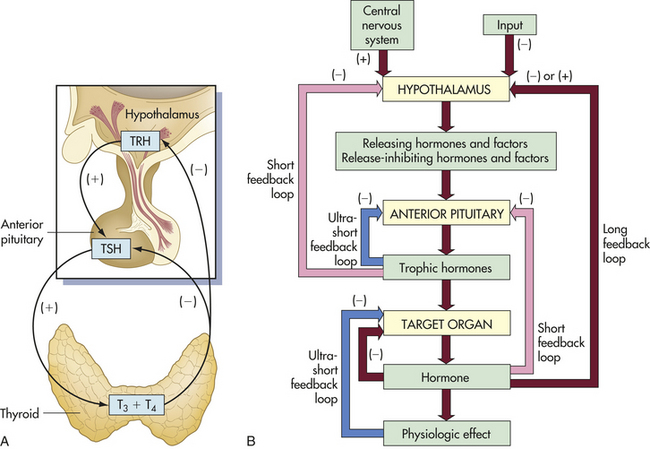
Figure 20-2 Feedback loops. A, Endocrine feedback loops involving the hypothalamus-pituitary gland and end organs; in this example, the feedback loops for the thyroid gland (endocrine regulation). B, General model for control and negative feedback to hypothalamic-pituitary target organ systems. Negative-feedback regulation is possible at three levels: target organ (ultrashort feedback), anterior pituitary (short feedback), and hypothalamus (long feedback). TRH, Thyroid-releasing hormone; TSH, thyroid-stimulating hormone; T3, triiodothyronine; T4, tetraiodothyronine-thyroxine.
Negative-feedback systems are important in maintaining hormones within physiologic ranges. The lack of negative-feedback inhibition on hormonal release often results in pathologic conditions. As discussed in Chapter 21, various hormonal imbalances and related conditions are caused by excessive hormone production, which is the result of failure to “turn off” the system. These negative-feedback regulatory systems are diagrammed in Figure 20-2, B.
An example of neural regulation is the release of epinephrine from the adrenal medulla as a result of activation of the sympathetic division of the autonomic nervous system in response to stress. When the stress is removed, the nervous stimulation decreases and less epinephrine is released. Neural regulation is often integrated with temporal (e.g., circadian) and environmental stimuli. For example, the quantity and pattern of growth hormone secretion is a dynamic one affected by hypothalamic responses to stress, nutrients, adrenergic pathways, and other osmoreceptors.2
Hormone Transport
Once hormones are released into the circulatory system by the endocrine glands, they are circulated throughout the body. Peptide or protein hormones (insulin, pituitary, hypothalamic, parathyroid) are water soluble and circulate in free (unbound) forms. Water-soluble hormones generally have a short half-life because they are catabolized by circulating enzymes.2 For example, insulin has a half-life of 3 to 5 minutes and is catabolized by insulinases. Lipid-soluble hormones, such as cortisol and adrenal androgens, are primarily circulated bound to a carrier or binding protein (Table 20-2). A small percentage of the lipid-soluble hormone circulates in a free or active form. For example, approximately 10% of the circulating cortisol is free, whereas 75% is bound to corticosteroid-binding globulin. Only free hormones can signal a target cell. A large change in the concentration of binding protein can affect the concentration of free hormones and therefore hormone effects (see Table 20-2). As is discussed later in this chapter, water-soluble hormones bind to one of four classes of cell surface receptors, whereas lipid-soluble hormones bind to plasma membrane receptors or diffuse through the plasma membrane and bind to cytosolic or nuclear receptors.

Cellular Mechanisms of Hormone Action
When a hormone is released into the circulatory system, it is distributed throughout the body, but only those cells with appropriate receptors for that hormone are affected. The target cell hormone receptors have two main functions: (1) to recognize and bind with high affinity to their particular hormones and (2) to initiate a signal to appropriate intracellular effectors. See Chapter 1 for cell signaling pathways, particularly Figures 20-16 and 20-17 on pages 18-19. The binding of hormones with their receptors stimulates three general types of responses by:
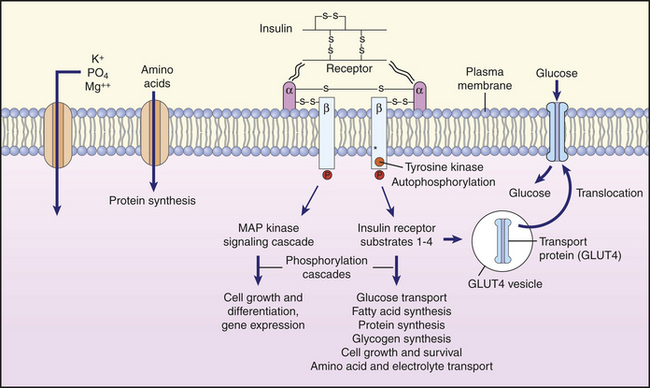
Figure 20-16 Insulin action on cells. Binding of insulin to its receptor causes autophosphorylation of the receptor, which then itself acts as a tyrosine kinase that phosphorylates insulin receptor substrate 1 (IRS-1). Numerous target enzymes, such as protein kinase B and MAP kinase are activated, and these enzymes have a multitude of effects on cell function. The glucose transporter, GLUT4, is recruited to the plasma membrane, where it facilitates glucose entry into the cell. The transport of amino acids, potassium, magnesium, and phosphate into the cell is also facilitated. The synthesis of various enzymes is induced or suppressed, and cell growth is regulated by signal molecules that modulate gene expression. MAP, mitogen-activated protein. (Redrawn from Berne RM, Levy MN: Principles of physiology, ed 3, St Louis, 2000, Mosby.)
1. Acting on preexisting channel-forming proteins to alter membrane channel permeability
2. Activating preexisting proteins through a second messenger system
The sensitivity of the target cell to a particular hormone is related to the total number of receptors per cell. Low concentrations of hormone increase the number of receptors per cell; this is called up-regulation (Figure 20-3, A). High concentrations of hormone decrease the number of receptors; this is called down-regulation (Figure 20-3, B). Thus the cell can adjust its sensitivity to the concentration of the signaling hormone.
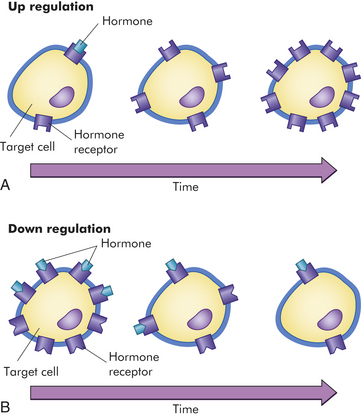
Figure 20-3 Regulation of target cell sensitivity. A, Low hormone level and up-regulation, or an increase in the number of receptors. B, High hormone level and down-regulation, or a decrease in the number of receptors. (From Thibodeau GA, Patton KT: Anatomy & physiology, ed 6, St Louis, 2007, Mosby.)
Hormones have two general types of effects on target cells: direct and permissive. Direct effects are the obvious changes in cell function that specifically result from stimulation by a particular hormone. Permissive effects are less obvious hormone-induced changes that facilitate the maximal response or functioning of a cell. For example, insulin has a direct effect on skeletal muscle cells with insulin receptors, causing increased glucose transport into these cells. Insulin also has a permissive effect on mammary cells, facilitating the response of these cells to the direct effects of prolactin.
Some hormones have biphasic pharmacologic effects that are dependent on the concentration of the hormone. For example, low or physiologic levels of antidiuretic hormone (ADH, or arginine vasopressin) stimulate renal tubular reabsorption of sodium and water. However, at supraphysiologic levels (i.e., those that can be achieved by exogenous administration), ADH acts as a vasoconstrictor.
Hormone Receptors
Hormone receptors may be located in or on the plasma membrane or in the intracellular compartment of the target cell (Figure 20-4). Water-soluble hormones (see Table 20-1) have a high molecular weight and cannot diffuse across the cell membrane. They interact or bind with receptors in or on the cell membrane.3 Steroid hormones are lipid soluble. These hormones easily diffuse across the plasma membrane and bind to either cytosolic or nuclear receptors. The hormone-receptor complex binds to a specific region in the deoxyribonucleic acid (DNA) and alters the expression of a specific gene. The recent discovery of nongenomic rapid actions of thyroid and steroid hormones indicates plasma membrane receptors that activate second messenger systems are also present.4–6 (Types of hormones, their corresponding receptors, and the mechanisms by which they affect the cell are summarized in Table 20-3.)
Table 20-3
Types of Hormones, Their Receptors, and Their Mechanisms of Action
| Hormone | Type of Receptor | Mechanism of Action |
| Water-Soluble Hormones | ||
| Glycoproteins, amines, small peptides | Plasma membrane receptors | Second messengers; cAMP, cGMP, Ca++, IP3, DAG and proteins (except insulin) |
| Insulin | Plasma membrane receptors | Involves receptor autophosphorylation and activation of the receptor protein tyrosine kinase |
| Growth hormone, prolactin, leptin | Plasma membrane receptors | Involves intracellular JAK and activation of STAT pathway |
| Lipid-Soluble Hormones | ||
| Steroid hormones | Plasma membrane receptors | Rapid nongenomic action |
| Nuclear receptors | Nuclear translocation and altered genome transcription | |
| Thyroid hormones (iodothyronines) | Nuclear receptor | Altered genome transcription |
| Cytosolic receptors | Promote cytosolic signal transduction |
cAMP, Cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; DAG, diacylglycerol; IP3, inositol triphosphate; JAK, Janus family of tyrosine kinases; STAT, signal transducers and activators of transcription.
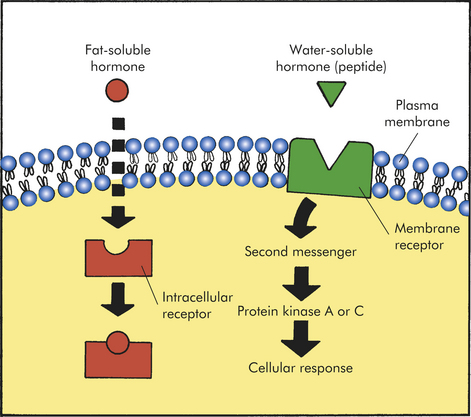
Plasma Membrane Receptors and Signal Transduction
First Messenger: Receptors for most water-soluble hormones are located on the plasma membrane of a target cell. Sometimes a hormone or ligand that binds to a receptor is referred to as a first messenger. This is because a hormone binding to its specific receptor represents the first signal within an elaborate signal transduction cascade. Signal transduction is the process by which extracellular signals (e.g., hormones) are communicated into a cell. In general, signal transduction involves a series of steps that includes receptor activation or binding of a hormone to its receptor, activation of a G protein (transducer) and membrane-associated enzyme (effector enzyme), and production of a second messenger (See Figure 1-20, page 22 and Figure 20-5). The final event is activation of an intracellular enzyme, such as protein kinase A or C, usually leading to alterations in gene transcription and the target cell response to the hormone.7
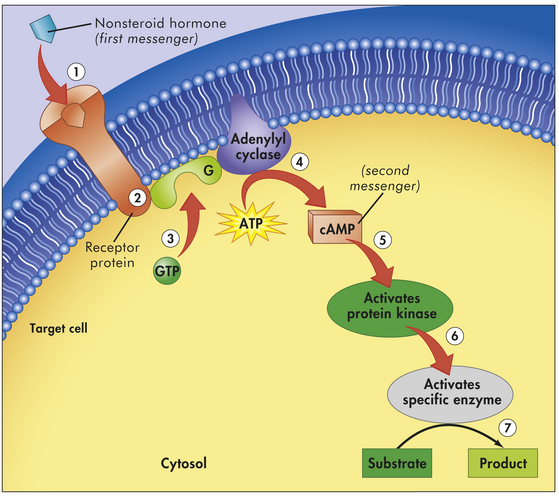
Figure 20-5 Example of first- and second-messenger signaling. A nonsteroid hormone (first messenger) binds to a fixed receptor of the target cell (1). The hormone-receptor complex activates the G protein (2). The activated G protein (G) reacts with guanosine triphosphate (GTP), which in turn activates the membrane-bound enzyme adenylyl cyclase (3). Adenylyl cyclase catalyzes the conversion of adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP; second messenger) (4). cAMP activates protein kinase (5). Protein kinases activate specific intracellular enzymes (6). These activated enzymes then influence specific cellular reactions and metabolic pathways, thus producing the target cell’s response to the hormone (7). (From Thibodeau GA, Patton KT: Anatomy & physiology, ed 6, St Louis, 2007, Mosby).
The signal transduction process begins at the receptor, which is a protein. Receptors on the plasma membrane are continuously synthesized and degraded, so the receptor number can vary from one cell type to another. Various physiochemical conditions can affect both the receptor number and the affinity at which the hormone binds to its receptor. Some of these physiochemical conditions are the fluidity and structure of the plasma membrane, pH, temperature, ion concentration, diet, and the presence of other chemicals (e.g., drugs). Mutations in receptor structure also can affect target cell activation such that normal cellular responses are increased or decreased.2
Cell surface receptors usually are classified according to their function: (1) G protein–linked receptors, (2) ion-channel receptors, and (3) enzyme-linked receptors (including tyrosine-kinase, serine kinase, and the cytokine-receptor superfamily with intrinsic enzyme activity—such as the Janus family of tyrosine kinases [JAK] and signal tranducers and activators of transcription [STAT] molecules).1,3,7,8 With the exception of insulin, growth hormone, and prolactin, most water-soluble hormones—such as adrenocorticotropic hormone (ACTH), glucagon, norepinephrine, and epinephrine—activate G protein–linked receptors.3,8,9 Other hormones, such as angiotensin II, activate G protein–linked and ion-channel receptors. Insulin activates a tyrosine-kinase receptor. Growth hormones, prolactin, and cytokines—such as interleukins—activate the JAK/STAT receptors.10
Second-Messenger Molecules: cAMP, Ca++, and cGMP:
Cyclic Adenosine Monophosphate (cAMP): Second-messenger molecules are the initial link between the first signal (hormone) and the inside of the cell (Table 20-4). For example, binding of epinephrine to a β-adrenergic receptor subtype activates (through a stimulatory G protein [Gs]) the enzyme adenylyl cyclase. Adenylyl cyclase catalyzes the conversion of adenosine triphosphate (ATP) to the second messenger, 3′,5′-cAMP. Elevation of cAMP activates the enzyme cAMP-dependent protein kinase A (PKA). Kinase enzymes, by adding a phosphate moiety to cellular proteins, either activate or deactivate intracellular proteins or enzymes. PKA phosphorylates and activates nuclear transcription factors (cAMP response element-binding [CREB] proteins) that influence numerous cellular functions.11 Alterations in CREB activity have been implicated in many disease states including cancer and stroke.12,13 In cardiac muscle, cAMP-dependent protein kinase phosphorylation of cellular membrane proteins associated with the L-type channel increase the influx of calcium into the cell. Increased intracellular calcium levels increase myocardial contractility. The actions of cAMP are terminated by the enzyme phosphodiesterase (PDE) III, which hydrolyzes cAMP into inactive adenosine monophosphate (AMP).
Table 20-4
Second Messengers Identified for Specific Hormones
| Second Messenger | Associated Hormones |
| Cyclic AMP | Adrenocorticotropic hormone (ACTH) |
| Luteinizing hormone (LH) | |
| Human chorionic gonadotropin (hCG) | |
| Follicle-stimulating hormone (FSH) | |
| Thyroid-stimulating hormone (TSH) | |
| Antidiuretic hormone (ADH) | |
| Thyrotropin-releasing hormone (TRH) | |
| Parathyroid hormone (PTH) | |
| Glucagon | |
| Cyclic GMP | Atrial natriuretic peptide |
| Calcium | Angiotensin II |
| Gonadotropin-releasing hormone (GnRH) | |
| Antidiuretic hormone (ADH) | |
| IP3 and DAG | Angiotensin II |
| Antidiuretic hormone (ADH) | |
| Luteinizing hormone–releasing hormone (LHRH) | |
| Tyrosine phosphorylation | |
| Tyrosine kinase | Insulin |
| JAK-STAT | Growth hormone |
| Leptin | |
| Prolactin |
AMP, Adenosine monophosphate; DAG, diacylglycerol; GMP, guanosine monophosphate; IP3, inositol triphosphate; JAK, Janus family of tyrosine kinases; STAT, signal transducers and activators of transcription.
Calcium (Ca++): In addition to being an important ion that participates in a multitude of cellular actions, Ca++ is considered an important second messenger.14,15 The binding of a hormone (such as norepinephrine or angiotensin II) to a surface receptor activates the enzyme phospholipase C through a G protein inside the plasma membrane. This enzyme breaks down membrane phospholipid phosphatidylinositol biphosphate (PIP2) into second messengers inositol triphosphate (IP3) and diacylglycerol (DAG) (See Figure 1-21, page 22). IP3 mobilizes Ca++ from intracellular stores (endoplasmic reticulum). In several cell types an increase in intracellular Ca++ activates specific physiologic effects. For example, when Ca++ binds with intracellular calmodulin, the Ca++-calmodulin complex activates specific proteins.
DAG, together with Ca++, activates protein kinase C (PKC). Similar to other kinase enzymes, PKC either activates (by phosphorylation) or deactivates (dephosphorylates) other proteins or enzymes. PKC initiates a variety of cellular responses that are linked to cell metabolism and growth. For example, PKC activates glycogen synthase in liver cells to convert glucose to glycogen. Calcium signaling systems are crucial to healthy functioning of virtually every tissue system in the body including heart, brain, bone, smooth muscle, and many others.14–18
Cyclic Guanosine Monophosphate (cGMP): The production of the second messenger 3′,5′-cGMP is associated with the activation of the intracellular enzyme guanylyl cyclase. cGMP activates cGMP-dependent kinase, which in turn activates a number of physiologic processes. The effects of various ligands, such as atrial natriuretic factor and nitric oxide, are mediated by the second messenger cGMP. Drugs that target the actions of cGMP are being explored for the treatment of vascular and pulmonary disorders.19,20 A summary of second messenger systems is presented in Figure 20-5.
Steroid (Lipid-Soluble) Hormones
The lipid-soluble hormones are classified as steroid hormones and include androgens, estrogens, progestins, glucocorticoids, mineralocorticoids, and thyroid hormones. Steroid hormones are relatively small hydrophobic molecules (synthesized from cholesterol) and therefore cross the plasma membrane by simple diffusion (see Chapter 1). Some steroid hormones bind to receptor molecules in the cytoplasm and then diffuse into the nucleus, whereas others bind to receptors in the nucleus. The resulting hormone-receptor complex binds to a specific site on the promoter region of DNA. This binding activates ribonucleic acid (RNA) polymerase, which stimulates DNA transcription and increased synthesis of specific proteins (increased gene expression) (Figure 20-6). Modulation of gene expression can take hours to days.
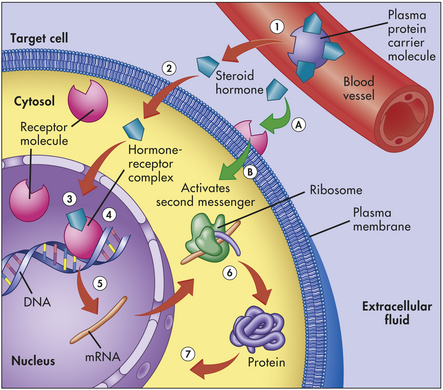
Figure 20-6 Steroid hormone mechanism. Lipid-soluble steroid hormone molecules detach from the carrier protein (1) and pass through the plasma membrane (2). Hormone molecules then diffuse into the nucleus, where they bind to a receptor to form a hormone-receptor complex (3). This complex then binds to a specific site on a DNA molecule (4), triggering transcription of the genetic information encoded there (5). The resulting messenger ribonucleic acid (mRNA) molecule moves to the cytosol, where it associates with a ribosome, initiating synthesis of a new protein (6). This new protein—usually an enzyme or channel protein—produces specific effects on the target cell (7). The classical genomic action is typically slow (red arrows). Steroids also may exact rapid effects by binding to receptors on the plasma membrane (A) and activating an intercellular second messenger (B). (Modified from Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
Recent studies indicate there are steroid hormone receptors in the plasma membrane associated with rapid response (seconds or minutes) that have nongenomic and genomic effects.4,21–24 These receptors are still being explored, and it has been found that the nongenomic actions of steroid hormones involve many second messengers. Crosstalk between gene transcription and nongenomic responses modulates each other, allowing cells to adapt rapidly to environmental changes.2 Thyroid hormone, a nonsteroid lipid-soluble hormone, also has been recently found to use a cell surface receptor for its nongenomic actions.5,6 It first binds to an integrin receptor on the plasma membrane, then uses a specific transport mechanism to gain access to its nuclear receptors.5
STRUCTURE AND FUNCTION OF THE ENDOCRINE GLANDS
The hypothalamic-pituitary axis (HPA) forms the structural and functional basis for central integration of the neurologic and endocrine systems, creating what is called the neuroendocrine system.25 The HPA produces a number of releasing/inhibitory hormones and tropic hormones that affect a number of diverse body functions (Figure 20-7). For example, the functions of the thyroid gland, adrenal gland, and male and female reproductive glands, as well as somatic growth and lactation, are regulated by hormones originating from the HPA.
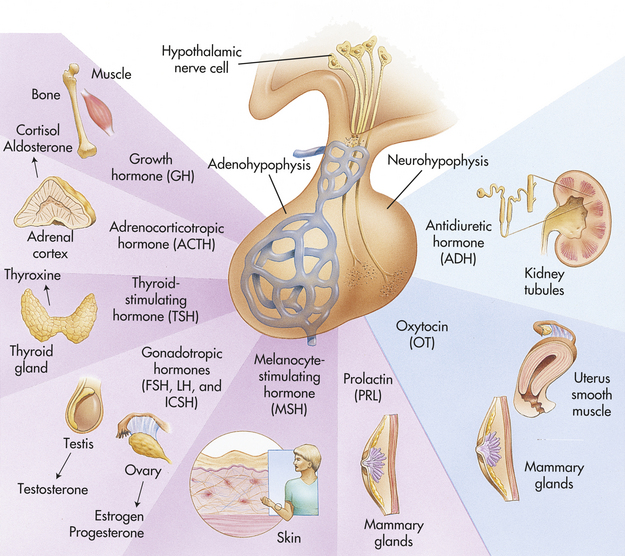
Figure 20-7 Pituitary hormones and their target organs. FSH, Follicle-stimulating hormone; ICSH, male analog of LH (interstitial cell–stimulating hormone) LH, luteinizing hormone. (Modified from Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
Hypothalamus
The hypothalamus is divided into several nuclei and nuclear areas and is located at the base of the brain (Figures 20-8 and 20-9). The pituitary gland is located at the sella turcica (a saddle-shaped depression on the superior surface of the sphenoid bone). The communication or anatomic connection (blood vessels and neural tract) between the hypothalamus and anterior and posterior pituitary is quite elaborate and well described. However, simply described, the hypothalamus is connected to the anterior pituitary by way of portal osmoreceptor blood vessels (Figure 20-10), whereas the hypothalamus is connected to the posterior pituitary by way of a nerve tract referred to as the supraopticohypophsial tract (see Figure 20-9). These connections are vital to the functioning of the hypothalamus-pituitary system.25
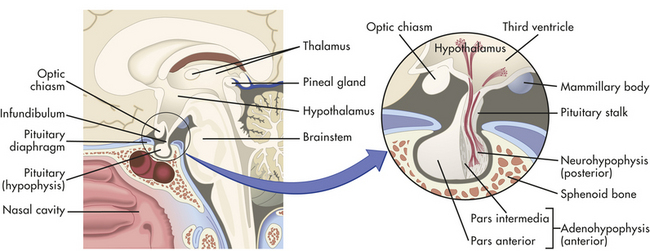
Figure 20-8 Location and structure of the pituitary gland (hypophysis). The pituitary gland is located within the sella turcica of the skull’s sphenoid bone and is connected to the hypothalamus by a stalklike infundibulum. The infundibulum passes through a gap in the portion of the dura mater that covers the pituitary (the pituitary diaphragm). The inset shows that the pituitary is divided into an anterior portion, the adenohypophysis, and a posterior portion, the neurohypophysis. The adenohypophysis is further subdivided into the pars anterior and pars intermedia. The pars intermedia is almost absent in the adult pituitary. (Modified from Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
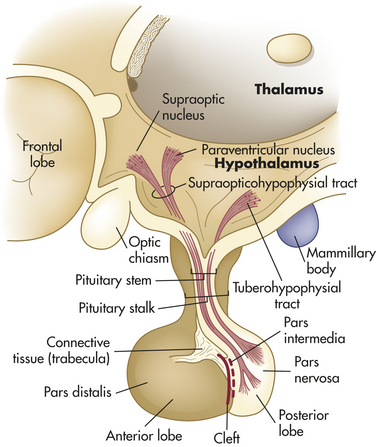
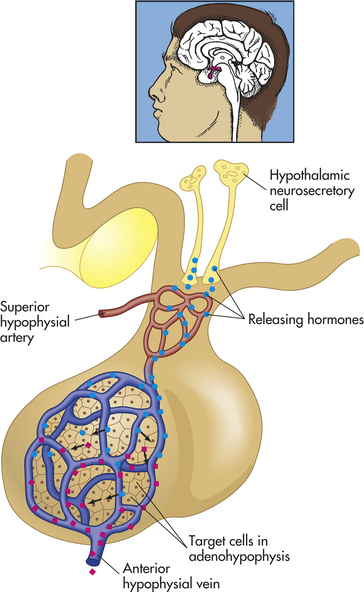
Figure 20-10 Hypophysial portal system. Neurons in the hypothalamus secrete releasing hormones into veins that carry the releasing hormones directly to the vessels of the adenohypophysis, thus bypassing the normal circulatory route. (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
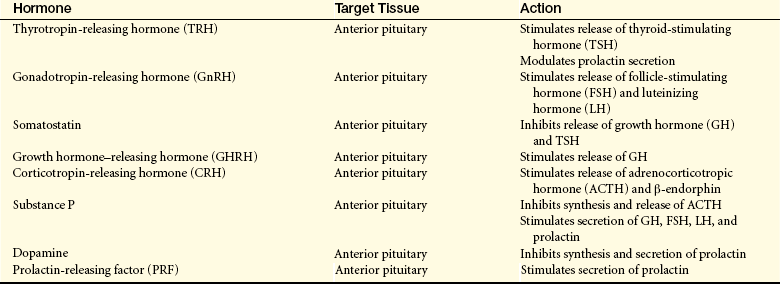
The special cells of the hypothalamus are like other neurons in that they have similar electrical properties, organelles, membranes, and synapses. Neurosecretory cells, however, can synthesize and secrete the hypothalamic-releasing hormones and synthesize the hormones of the posterior portion of the pituitary gland.25 For example, antidiuretic hormone (ADH) and oxytocin are synthesized in hypothalamic neurons but are stored and secreted by the posterior pituitary. ADH and oxytocin travel to the posterior pituitary by way of the hypothalamohypophysial nerve tract. Releasing/inhibitory hormones (see Figure 20-17, p. 716) are also synthesized in the hypothalamus and are secreted into the portal osmoreceptor blood vessels through which they travel to the anterior pituitary and control the release of tropic hormones. These releasing/inhibitory hormones from the hypothalamus include prolactin-inhibiting factor (PIF), thyrotropin-releasing hormone (TRH), gonadotropin-releasing hormone (GnRH), somatostatin, growth hormone–releasing factor (GRF), corticotropin-releasing hormone (CRH), and substance P. These hormones are summarized in Table 20-5.
Pineal Gland
The pineal gland is located within the brain itself (see Figure 20-1) and is composed of photoreceptive cells. It is innervated by noradrenergic sympathetic nerve terminals controlled by pathways within the hypothalamus.25 The primary role of the pineal gland is to secrete the hormone melatonin. Melatonin release is stimulated by exposure to dark and inhibited by light exposure. It is synthesized from tryptophan. Tryptophan is first synthesized to serotonin and then to melatonin. It regulates circadian rhythms and reproductive systems, including the secretion of GnRH and the onset of puberty.25 It also plays an important role in immune regulation and is postulated to affect the aging process.26,27 Further effects of melatonin include increasing nitric oxide release from blood vessels, removing toxic oxygen radicals, and decreasing insulin secretion.28–30 Melatonin has been used therapeutically in humans to help with sleep disturbances, jet lag, and psychologic disorders, and its utility for numerous other disorders is being explored.31
Pituitary Gland
The anterior pituitary (adenohypophysis) accounts for 75% of the total weight of the pituitary gland. It is composed of three regions: (1) the pars distalis, (2) the pars tuberalis, and (3) the pars intermedia. The pars distalis is the major component of the anterior pituitary and the source of the anterior pituitary hormones. The pars tuberalis is a thin layer of cells on the anterior and lateral portions of the pituitary stalk. The pars intermedia lies between the two lobes of the pituitary gland. In the adult the distinct intermediate lobe disappears, and the individual cells are distributed diffusely throughout the pars distalis and pars nervosa (neural lobe).
The posterior pituitary (neurohypophysis) arises embryologically from an outpouching of the floor of the third ventricle within the brain. The posterior pituitary consists of three parts: (1) the median eminence located at the base of the hypothalamus, (2) the pituitary stalk, and (3) the infundibular process, also known as the pars nervosa or neural lobe. The median eminence is composed largely of the nerve endings of axons that arise primarily in the ventral hypothalamus. The median eminence often is designated as part of the posterior pituitary but contains at least 10 biologically active hypothalamic-releasing hormones, as well as the neurotransmitters dopamine, norepinephrine, serotonin, acetylcholine, and histamine. The median eminence therefore might be more appropriately considered part of the hypothalamus. The pituitary stalk contains the axons of neurons that originate in the supraoptic and paraventricular nuclei of the hypothalamus. The pituitary stalk thus connects the pituitary gland to the brain. Axons originating in the hypothalamus terminate in the pars nervosa, which secretes the hormones of the posterior pituitary.
Because of the anatomic location and connection of the pituitary gland to the brain, several neurotransmitters as well as physical and emotional stressors influence the release of specific hypothalamic releasing–inhibitory hormones and their respective tropic hormones. For example, the neurotransmitter norepinephrine stimulates CRH, TRH, and GnRH secretion, whereas the neurotransmitter gamma-aminobutyric acid (GABA) inhibits CRH, TRH, and GnRH secretion.25 In terms of tropic hormone release from the anterior pituitary, norepinephrine stimulates the secretion of TSH, growth hormone (GH), luteinizing hormone (LH), and follicle-stimulating hormone (FSH), whereas the secretion of ACTH is inhibited. Physical (trauma) and emotional (pain) stress, starvation, and inflammation, as well as hypoglycemia, can influence the release of stimulating hormones, such as TRH and CRH, therefore ultimately affecting the amount of TSH and ACTH (the tropic hormones) released by the anterior pituitary.25 This example emphasizes the integrated and coordinated function of the hypothalamic-pituitary axis.
Interestingly, hypothalamic hormones also are synthesized outside the HPA. For example, CRH is synthesized in cells of the immune system, female and male reproductive organs, and the placenta.32 These peripherally synthesized neuropeptides are thought to play a role in the reproductive and immune responses to stress.32,33
Hormones of the Posterior Pituitary
The posterior pituitary secretes two polypeptide hormones: (1) ADH, also called arginine vasopressin; and (2) oxytocin. These peptide hormones are similar in structure, differing by only two amino acids. They are synthesized, along with their binding proteins, the neurophysins, in the supraoptic and paraventricular nuclei of the hypothalamus (see Figure 20-9). Once synthesized, these hormones and their carrier proteins are packaged in secretory vesicles. They are moved down the axons of the pituitary stalk to the pars nervosa for storage. The posterior pituitary thus can be seen as a storage and releasing site for hormones synthesized in the hypothalamus.
Similar to the anterior pituitary hormones, the release of ADH and oxytocin is influenced by neurotransmitter release. The major stimulus to ADH and oxytocin release is glutamate, whereas the major inhibitory input is through GABA.34
Antidiuretic Hormone: The major homeostatic function of the posterior pituitary is the control of plasma osmolality, as regulated by ADH (see Chapter 3). At physiologic levels, ADH acts on the vasopressin 2 (V2) receptors of the renal tubular cells to increase their permeability (see Chapter 35). This increased permeability leads to an increase in water reabsorption into the blood and the production of more concentrated urine. These effects may be inhibited by hypercalcemia, prostaglandin E, and hypokalemia. ADH has no direct effect on electrolyte levels, but by increasing water reabsorption, serum electrolyte concentrations may decrease because of a dilutional effect.34
The secretion of ADH is regulated primarily by the osmoreceptors of the hypothalamus, located near or in the supraoptic nuclei (osmoreceptors are stimulated by increased osmolality). These osmoreceptors also control thirst. The plasma osmolality is maintained at the mean set point of approximately 280 mOsm/kg. As plasma osmolality increases, the rate of ADH secretion increases, more water is reabsorbed from the kidney, and the plasma is diluted back to its set-point osmolarity.
Other mechanisms also affect ADH secretion. ADH secretion is increased by changes in intravascular volume. Intravascular volume changes are monitored by mechanoreceptors in the left atrium and in the carotid and aortic arches. A volume loss of 7% to 25% acts through these receptors to stimulate ADH secretion. This mechanism for regulating ADH secretion is much less sensitive than that of the osmoreceptors. Stress, trauma, pain, exercise, nausea, nicotine, exposure to heat, and drugs such as chloroform and morphine also increase ADH secretion, apparently by activating cholinergic neurotransmitters in the hypothalamus. ADH secretion decreases with a decrease in plasma osmolality, an increase in intravascular volume, hypertension, estrogen, progesterone, angiotensin II, and alcohol ingestion.
ADH originally was named vasopressin because in extremely high doses it does cause vasoconstriction and a resulting increase in arterial blood pressure. This baroreceptor-mediated response is much less sensitive than the ADH response to changes in osmolarity. Therefore, physiologic levels of ADH do not significantly affect vessel tone. However, significant vasoconstriction may be achieved pharmacologically. For example, high doses of ADH (given as the drug vasopressin) may be administered to achieve hemostasis during hemorrhage and to raise blood pressure in shock states.35,36
Oxytocin: Oxytocin is primarily responsible for contraction of the uterus and milk ejection in lactating women and may have a role in sperm motility in men, although this effect has not yet been clearly elucidated. In both sexes, oxytocin has an antidiuretic effect similar to that of ADH. The mechanisms by which this effect is achieved appear to be similar to those of ADH, but the physiologic significance is not clear. (The function of this hormone is discussed in Chapter 22.)
The release of oxytocin has been studied more extensively in women than in men. In a woman, oxytocin is secreted in response to suckling and mechanical distention of the female reproductive tract.34 Oxytocin is required for the milk “let-down” reflex. Stimulated by sucking, oxytocin binds to its receptors on myoepithelial cells in the mammary tissues and causes contraction of those cells. This results in increased intramammary pressure and milk expression.
Oxytocin also acts on the uterus to stimulate contractions. Its role in initiating labor has been debated because levels of oxytocin do not increase until near the end of labor. However, it is used clinically to induce uterine contraction. It is hypothesized that near the end of labor oxytocin functions to enhance effectiveness of contractions, to promote delivery of the placenta, and to stimulate postpartum contractions to prevent excessive bleeding.37
Oxytocin has been implicated in behavior responses, especially in women. It has been suggested that oxytocin reduces the brain’s responsiveness to stressful stimuli, especially in the pregnant and postpartum states.38,39 Its potential role in the treatment of a variety of anxiety disorders and autism is being explored.38,40
Hormones of the Anterior Pituitary
The anterior pituitary is composed of two main cell types: (1) the chromophobes, which appear to be nonsecretory, and (2) the chromophils, which are considered the secretory cells of the adenohypophysis. The chromophils are subdivided into six secretory cell types, each type secreting one or more specific hormones (Table 20-6). In general, the regulation of the anterior pituitary hormones is achieved by (1) feedback of hypothalamic releasing–inhibitory hormones and factors, (2) feedback from target gland hormones (i.e., cortisol, estrogen), and (3) direct effects of neurotransmitters.
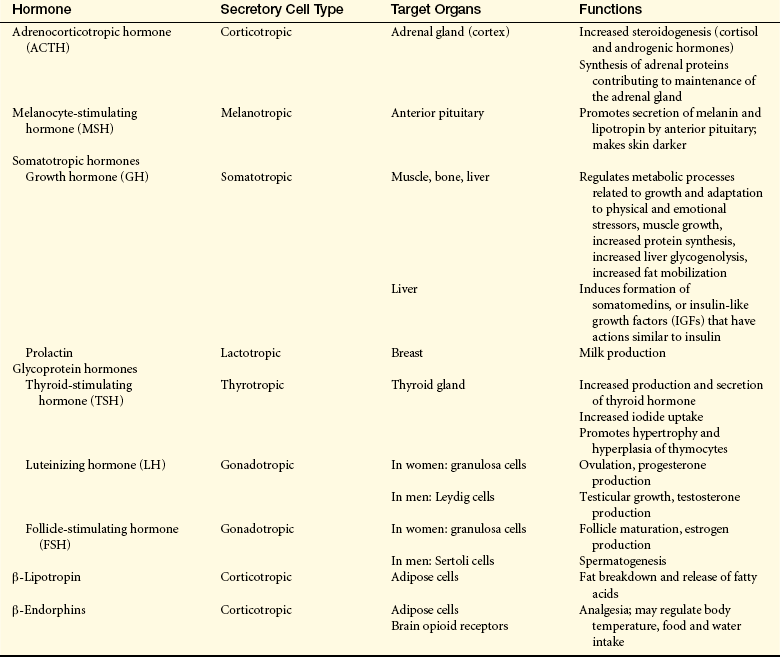
The tropic hormones secreted by the anterior pituitary include ACTH, melanocyte-stimulating hormone (MSH), LH, GH, prolactin, FSH, and TSH. The actions of these anterior pituitary tropic hormones are summarized in Table 20-6. Even though six major stimulatory hormones are released by the anterior pituitary, they can be grouped into three categories: corticotropin-related hormones (ACTH, β-lipoprotein, MSH, and related endorphins), glycoproteins (LH, FSH, and TSH), and somatomammotropins (GH and prolactin). The corticotropin-related hormones are all derived from the precursor pro-opiomelanocortin (POMC). POMC is the precursor for ACTH and β-lipotropin; MSH exists within the ACTH amino acid sequence. β-endorphin and met-enkephalin are derived from β-lipotropin. (The role of ACTH is discussed later in this chapter. The glycoprotein gonadotropins FSH and LH control reproductive processes in men and women and are discussed in Chapter 22. TSH and its effects on the thyroid gland are discussed later in this chapter.) The somatomammotropins have diverse and important effects on body tissues.
Growth Hormone (GH): GH secretion is controlled by two hormones from the hypothalamus: GnRH, which increases GH secretion, and somatostatin, which inhibits it.41 GH is released from the pituitary in a pulsatile fashion, and overall secretion peaks during adolescence. This hormone is essential to normal tissue growth and maturation, and affects aging, sleep, nutritional status stress, and reproductive hormones.42 In the bone, GH stimulates epiphyseal growth and increases osteoclast and osteoblast activity, resulting in increased bone mass.41 GH also increases amino acid transport in muscles. Other functions of GH include lipolysis and enhancement of hepatic protein synthesis.
Many of the anabolic functions of GH are mediated, at least in part, by the insulin-like growth factors (IGFs), also known as the somatomedins.41 There are two primary forms of IGF: IGF-1 and IGF-2, of which IGF-1 is the most biologically active. They both circulate bound to a group of binding proteins (IGFBP). IGF-1 binds to both insulin receptors, providing an insulin-like effect on skeletal muscle, and with the IGF-1 receptor, which mediates the anabolic effects of GH. The IGF-2 receptor causes a negative effect on tissue growth, thus balancing the activity of the IGF-1 receptor.43 IGF and IGFBPs have been linked to many forms of cancer because of their growth-stimulating effects.43,44
Prolactin: Prolactin primarily functions to induce milk production during pregnancy and lactation. It also has effects on ovulation and immune function. Its synthesis is stimulated by vasoactive intestinal polypeptide, serotonin, and growth factors. Its release is inhibited by dopamine. Hyperprolactinemia is a complication of tumors of the pituitary gland and some medications.41
Thyroid and Parathyroid Glands
The thyroid gland, located in the neck just below the larynx, produces hormones that control the rates of metabolic processes throughout the body. The four parathyroid glands are located near the posterior side of the thyroid and function to control serum calcium levels.
Thyroid Gland
The thyroid gland is composed of two lobes that lie on either side of the trachea, inferior to the thyroid cartilage (Figure 20-11). The lobes are joined by a small band of tissue, the isthmus, which crosses the anterior surface of the trachea and larynx at the cricoid cartilage. The normal thyroid gland is not visible on inspection, but it may be palpated on swallowing, which causes upward displacement of the gland.
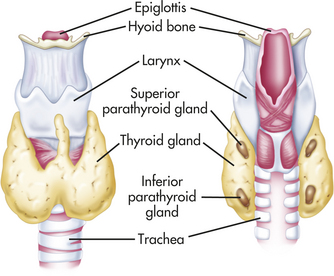
Figure 20-11 Thyroid and parathyroid glands. Note the relationship of the thyroid and parathyroid glands to each other, to the larynx (voice box), and to the trachea. (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
The thyroid gland is composed of follicles (Figure 20-12). The follicles are composed of follicular cells that surround a viscous substance called colloid. The follicular cells synthesize and secrete some of the thyroid hormones. Neurons terminate on blood vessels within the thyroid gland and on the follicular cells themselves. Acetylcholine, catecholamines, and other peptides directly affect secretory activity of the follicular cells and thyroid blood flow.
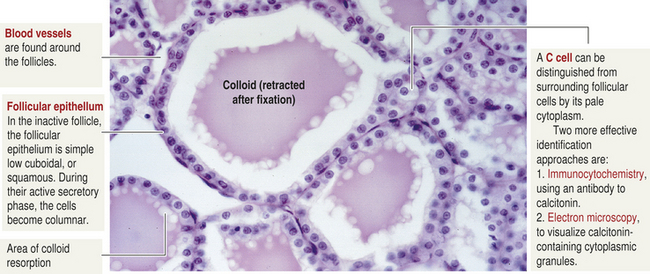
Figure 20-12 Thyroid follicle cells. (From Kierszenbaum A: Histology and cell biology: an introduction to pathology, ed 2, St Louis, 2007, Mosby.)
Also found in the tissue of the thyroid are parafollicular cells, or C cells (see Figure 20-12). The C cells secrete various polypeptides, including calcitonin and somatostatin. Calcitonin, also called thyrocalcitonin, acts to lower serum calcium levels by inhibition of bone-resorbing osteoclasts. High levels of calcitonin are required for these effects, and deficiencies of calcitonin do not lead to hypocalcemia. (Bone resorption is explained in Chapter 41.) Consequently, the metabolic consequences of calcitonin deficiency or excess do not appear to be significant in humans (Table 20-7). However, calcitonin is used for treatment of osteoporosis, osteoarthritis, Paget bone disease, hypercalcemia, osteogenesis imperfecta, and metastatic cancer of the bone.45–47 The precursor molecule to calcitonin, called procalcitonin, is a stress hormone that is elevated in significant infections and systemic inflammatory disorders.48 Its measurement can aid in the diagnosis of these serious diseases.48,49
Table 20-7
Thyroid Gland Hormones and Their Regulation and Functions
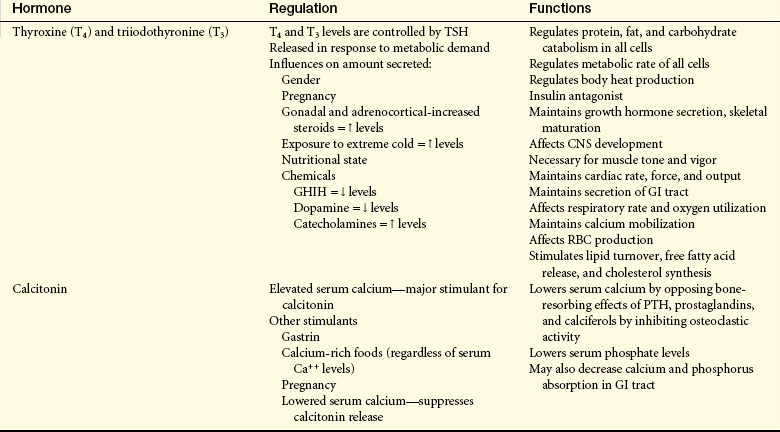
CNS, Central nervous system; GHIH, growth hormone–inhibiting hormone; GI, gastrointestinal; PTH, parathyroid hormone; RBC, red blood cell; TSH, thyroid-stimulating hormone.
From Monahan FD, et al: Phipps’ medical-surgical nursing: health and illness perspective, ed 8, St Louis, 2007, Mosby.
Regulation of Thyroid Hormone Secretion: Thyroid hormone (TH) is regulated through a negative-feedback loop involving the hypothalamus, the anterior pituitary, and the thyroid gland (see Figure 20-2). (Figure 20-12 illustrates the thyroid and parathyroid glands.) The initiating hormone is termed thyrotropin-releasing hormone (TRH), and it is synthesized and stored within the hypothalamus. TRH is released into the hypothalamic-pituitary portal system and circulates to the anterior pituitary, where it stimulates the release of TSH. TRH is increased with exposure to cold, stress, and decreased levels of thyroxine (T4).
Thyroid-stimulating hormone (TSH) is a glycoprotein hormone synthesized and stored within the anterior pituitary. Once TSH is secreted by the anterior pituitary, it circulates to bind with TSH receptor sites located on the outer side of the thyroid cell’s plasma membrane. The effects of TSH on the thyroid include (1) an immediate increase in the release of stored thyroid hormones, (2) an increase in iodide uptake and oxidation, (3) an increase in thyroid hormone synthesis, and (4) an increase in the synthesis and secretion of prostaglandins by the thyroid. Thyroid gland hormones and their regulation and function are summarized in Table 20-7. TSH is also important in stimulating the growth and maintenance of the thyroid gland by stimulating thymocyte hypertophy and hyperplasia and decreasing apoptosis.50
When TH is secreted by the thyroid gland, it acts on the thyroid gland, the anterior pituitary, and the median eminence to regulate further TH production. Thyroid hormones have a negative-feedback effect and inhibit TRH and TSH, which decreases TH synthesis and secretion. Recent studies suggest that thyroid hormone synthesis is also controlled by circulating enzymes called deiodinases that inactivate the precursor molecule thyroxine.51
Synthesis of Thyroid Hormone: The thyroid gland is stimulated to produce thyroid hormone by pituitary TSH, by low serum iodide levels, or by drugs interfering with the thyroid gland’s uptake of iodide from the blood. (Iodide is the inorganic or ionic form of iodine and is the form in which iodine enters the thyroid gland.) Iodide is oxidized to iodine in the presence of thyroidal peroxidase. The major naturally occurring source of iodine is seafood; in the United States iodine is added to salt and flour. Approximately 25% of ingested iodine is trapped by the thyroid gland.
Thyroid hormone synthesis is summarized in the following steps:
1. Uniodinated thyroglobulin (a large glycoprotein) is produced by the endoplasmic reticulum of the follicular cells.
2. Tyrosine is incorporated into the thyroglobulin as it is synthesized.
3. Iodide is actively transferred (pumped) from the blood into the colloid by carrier proteins located in the outer membrane of the follicular cells. This active transport system is called the iodide trap and is very efficient at accumulating the trace amounts of iodide from the blood.
4. Iodide quickly attaches to tyrosine within the thyroglobulin molecule.
5. Coupling of iodinated tyrosine forms thyroid hormones. Triiodothyronine (T3) is formed from coupling of monoiodotyrosine (one iodine atom and tyrosine) and diiodotyrosine (two iodine atoms and tyrosine). Tetraiodothyronine (T4), commonly known as thyroxine, is formed from coupling of two diiodotyrosines.
6. Thyroid hormones are stored attached to thyroglobulin within the colloid until it is released into the circulation.
The thyroid gland normally produces 90% T4 and 10% T3. In the body tissues, however, T4 is converted to T3, and T3 has the greatest metabolic effect. Once released into the circulation, T3 and T4 are primarily transported bound to one of three carrier proteins: thyroxine-binding globulin, thyroxine-binding prealbumin (transthyretin), or albumin. A small amount of thyroid hormone is also carried by lipoproteins.52
Thyroid hormones affect many body tissues, primarily by affecting growth and maturation of tissues. Similar to some steroid hormones, thyroid hormones bind to intracellular receptor complexes and then influence the genetic expression of specific proteins. Thyroid hormones also affect cell metabolism by altering protein, fat, and glucose metabolism and, as a result, heat production and oxygen consumption are increased. Thus these hormones are essential for maintaining healthy metabolic processes, and their use is being explored for the therapy of many metabolic disorders.53
It is important to note that thyroid hormones exert a number of permissive effects on many organs, which are rather modest at physiologic thyroid hormone levels. However, these effects can become very pronounced when there is either high or low levels of circulating thyroid hormones. For example, in the heart, T3 stimulates the synthesis of specific contractile proteins (e.g., α-myosin heavy chain), sarcolemmal ion pumps (Na+/K+-ATPase pump, Ca++--ATPase pump) and membrane receptors (β-adrenergic receptors). Therefore, in hyperthyroidism, which is associated with elevated levels of thyroid hormones, cardiac effects include increased heart rate and cardiac output, as well as the development of a cardiomyopathy. Thyroid hormones also affect the respiratory center, contributing to the normal hypoxic and hypercapnic drive. In severe hypothyroidism, ventilation can become very depressed. Thyroid hormone also stimulates bone resorption, and hyperthyroidism is associated with osteopenia, hypercalcemia, and hypercalciuria.54 Thyroid hormone is essential for normal neurologic development in the fetus and infant and affects neurologic functioning in adults.55 Other manifestations of thyroid hormone alteration are explained in Chapter 21.
Parathyroid Glands
Two pairs of parathyroid glands normally are present, but the number may range from two to six. They are small and located behind the upper pole of the thyroid gland and behind the lower pole (see Figure 20-11).
The parathyroid glands produce parathyroid hormone (PTH), a regulator of serum calcium. PTH is regulated primarily by the level of ionized plasma calcium, although how these regulatory mechanisms work is not precisely clear. Calcium also increases intraparathyroid destruction of PTH but apparently does not affect the rate of PTH synthesis.
Magnesium and phosphate levels also affect PTH secretion. Hypomagnesemia in persons with normal calcium levels acts as a mild stimulant to PTH secretion. In hypocalcemic individuals, hypomagnesemia decreases PTH secretion. Hyperphosphatemia leads to hypocalcemia because of calcium-phosphate precipitation in soft tissue and bone. Alterations in serum phosphate levels therefore may indirectly influence PTH secretion by affecting serum calcium levels (Figure 20-13). The overall effect of PTH is to increase serum calcium and to decrease serum phosphate concentration.
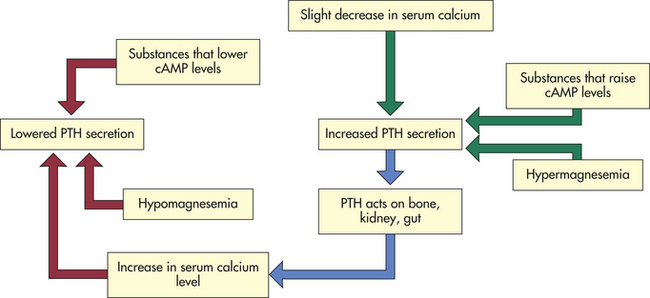
Figure 20-13 Variables affecting parathyroid hormone (PTH) secretion. cAMP, Cyclic adenosine monophosphate.
Once the parathyroid gland is stimulated, PTH is secreted. On release, PTH enters the circulation in unbound form. The hormone attaches to plasma membrane receptors in target tissues, where the biologic effects of PTH are mediated primarily by activation of the adenylyl cyclase system (see Chapter 1).
PTH is the single most important factor in the regulation of serum calcium levels (Figure 20-14). To achieve regulation of serum calcium, PTH acts directly on bone and the kidneys. In bone, PTH has at least two effects. In acute hypocalcemia, PTH secretion stimulates osteoblasts to release receptor activator for nuclear factor-kappa beta (NF-kβ) ligand (RANKL) and macrophage-colony stimulating factor (M-CSF) which results in osteoclast proliferation, maturation, and release of acidic enzymes, such as cathepsin. These enzymes mobilize calcium from bone which increases the serum calcium. Chronic stimulation by PTH results in bone remodeling, a process in which bone is broken down and reformed. Chronic stimulation can occur because of parathyroid tumors (primary hyperparathyroidism) or because of chronic hypocalcemia like that seen in end-stage renal disease (secondary hyperparathyroidism). This process can weaken the bone, leading to an increased risk for fracture (e.g., renal osteodystrophy).56 Paradoxically, intermittent therapeutic bursts of PTH can actually strengthen bone and are used in individuals with osteoporotic fractures.57,58
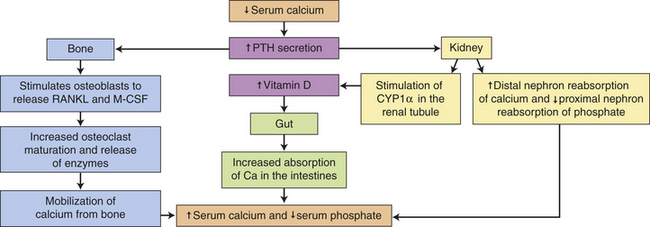
Figure 20-14 Normal calcium metabolism regulated by PTH and vitamin D. CYP1α-hydrolase, cytochrome P4501a-hydrolase; M-CSF, macrophage-colony stimulating factor; PTH, Parathyroid hormone; RANKL, Receptor activator of NF-kβ ligand.
In the kidneys, PTH acts on its plasma membrane receptor in the distal and proximal tubules of the nephron to increase reabsorption of calcium and to decrease reabsorption of phosphorus, respectively. PTH also decreases proximal tubule reabsorption of bicarbonate. In the kidney, PTH stimulates the synthesis of a biologically active form of vitamin D (1,25-dihydroxy-vitamin D3). Vitamin D3 serves as a cofactor with PTH for osteoblast stimulation and, as a potent stimulator of calcium and phosphate absorption in the intestine.59 In this way PTH increases gastrointestinal absorption of calcium.
Endocrine Pancreas
The pancreas is both an endocrine gland that produces hormones and an exocrine gland that produces digestive enzymes. (The exocrine pancreas is discussed in Chapter 38.) The pancreas therefore has important metabolic functions within the body. A major disorder of the endocrine pancreas is diabetes mellitus.
The pancreas is located behind the stomach, between the spleen and the duodenum. It houses the islets of Langerhans, which secrete glucagon and insulin, hormones that help to regulate much of the carbohydrate metabolism within the body. The islets of Langerhans have three types of hormone-secreting cells: (1) alpha cells, which secrete glucagon; (2) beta cells, which secrete insulin; and (3) delta cells, which secrete somatostatin and gastrin. The alpha cells and delta cells are located at the periphery of the islet, and beta cells are located in the middle. F cells, a fourth type of pancreatic cell, secrete pancreatic polypeptide. (The pancreas is illustrated in Figure 20-15.) Nerves from both divisions of the autonomic nervous system innervate the pancreatic islets.
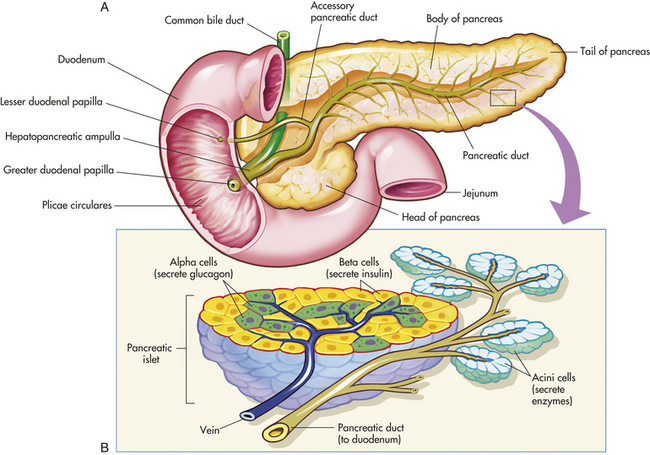
Figure 20-15 The pancreas. A, Pancreas dissected to show main and accessory ducts. The main duct may join the common bile duct, as shown here, to enter the duodenum by a single opening at the major duodenal papilla, or the two ducts may have separate openings. The accessory pancreatic duct is usually present and has a separate opening into the duodenum. B, Exocrine glandular cells (around small pancreatic ducts) and endocrine glandular cells of the pancreatic islets (adjacent to blood capillaries). Exocrine pancreatic cells secrete pancreatic juice, alpha endocrine cells secrete glucagon, and beta cells secrete insulin. (From Thibodeau GA, Patton KT: Anatomy & physiology, ed 5, St Louis, 2003, Mosby.)
The parasympathetic nervous system stimulates hormonal secretion and the sympathetic nervous system inhibits secretion. The perfusion of the anterior lobe of the pancreas where alpha, beta, and delta cells are most numerous comes from branches of the superior mesenteric artery. The posterior lobe is perfused by branches of the celiac artery. The pancreatic islets receive 10% of the pancreatic blood flow but represent only 1% of pancreatic mass. This is necessary for oxygenation and delivery of islet hormones to target cells.
Insulin
The beta cells of the pancreas synthesize insulin from the precursor, proinsulin. Proinsulin is formed from a larger and earlier precursor molecule, preproinsulin. Proinsulin is composed of an A peptide and a B peptide connected by a C peptide and two disulfide bonds. C peptide is cleaved by proteolytic enzymes, leaving the A and B peptide chains connected by the disulfide bonds. The bonded A and B chains become insulin. Insulin circulates freely in the plasma and is not bound to a carrier. C peptide can be measured in the blood as an indirect measure of serum insulin synthesis. Recent studies have shown that C peptide has biologic activity and binds to cells through a G protein receptor resulting in increased intracellular calcium levels.60 Its role in health and disease is being explored.60–62
Secretion of insulin is regulated by chemical, hormonal, and neural control. Insulin secretion is promoted by increased blood levels of glucose, amino acids (arginine and lysine), serum free fatty acids, and gastrointestinal hormones, and by parasympathetic stimulation of the beta cells. Insulin secretion diminishes in response to low blood levels of glucose (hypoglycemia), high levels of insulin (through negative feedback to the beta cells), and sympathetic stimulation of the alpha cells in the islets. Prostaglandin (PGE2) also inhibits insulin secretion.
Insulin facilitates the rate of glucose uptake into many cells within the body. Binding of insulin to its tyrosine-kinase receptor subtype initiates a series of events that involves autophosphorylation of the insulin receptor substrate 1 (IRS-1) and the activation (phosphorylation) of other proteins, including GRB2, a PI-3 kinase, and a tyrosine phosphatase. The net effect is that glucose transporters (GLUT) migrate from the cytosol to the cell surface. Translocation of the GLUT4 transporter is associated with a 10- to 20-fold increase in glucose diffusion into the cell, particularly in skeletal and cardiac muscle, liver, and adipose cells (Figure 20-16). The sensitivity of the insulin receptor is a key component in maintaining normal cellular function, and insulin resistance has been implicated in numerous cardiovascular diseases, including hypertension and diabetes. Adipocytes release a number of hormones that are altered in obesity and have an important effect on insulin sensitivity63 (see What’s New? Hormones from Adipose Tissue—The Adipokines).
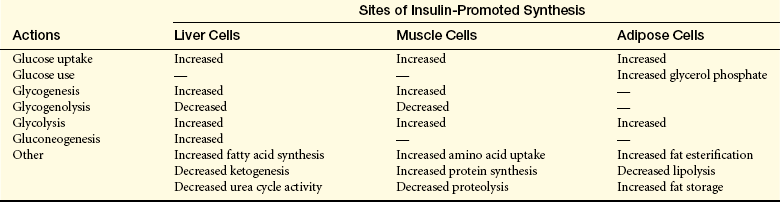
Insulin is an anabolic hormone that promotes the synthesis of proteins, lipids, and nucleic acids. The major sites of insulin-promoted synthesis are the liver, muscle, and adipose tissue (Table 20-8). The net effect of insulin in these tissues is to stimulate cellular metabolism. Overall, however, the major consequence of insulin release is to decrease blood glucose. Insulin also facilitates the intracellular transport of potassium. The brain and red blood cells do not require insulin for glucose transport.
Insulin is metabolized in the liver and kidney by enzymes that split disulfide bonds. Very little insulin is excreted unchanged in the urine.
Amylin
Amylin is a peptide hormone co-secreted with insulin in response to nutrient stimuli. It regulates blood glucose by delaying nutrient uptake and suppressing glucagon secretion
after meals. Amylin also has a satiety effect. Through these mechanisms, amylin has an antihyperglycemic effect.64
Glucagon
Glucagon is produced by the alpha cells of the pancreas and by a number of cells lining the gastrointestinal tract. High glucose levels cause glucagon release to be inhibited; low glucose levels and sympathetic stimulation promote glucagon release, particularly in the liver. Amino acids, such as alanine, glycine, and asparagine, also stimulate glucagon secretion. A protein-rich meal has the same effect.
Glucagon acts primarily in the liver and increases blood glucose by stimulating glycogenolysis and gluconeogenesis. Glucagon acts as an antagonist to insulin. Much controversy exists regarding the role of glucagon in carbohydrate regulation, both normally and in diabetes mellitus.64 Glucagon also stimulates lipolysis, which has a ketogenic effect caused by the metabolism of free fatty acids in the liver.
Somatostatin
Pancreatic somatostatin is produced by delta cells of the pancreas and is a hormone essential in carbohydrate, fat, and protein metabolism (i.e., homeostasis of ingested nutrients). It differs from hypothalamic somatostatin, which inhibits release of growth hormone and TSH. Little is known about pancreatic somatostatin, but in animal studies it has been found to be involved in the regulation of alpha cell and beta cell function within the islets.65 Presumably, somatostatin inhibits glucagon and insulin secretion, and it may prevent excess secretion of insulin.
Gastrin, Grehlin, and Pancreatic Polypeptide
The function of pancreatic gastrin has not been established; however, it likely controls the secretion of glucagon.64 Grehlin stimulates GH secretion, controls appetite, and plays a role in the regulation of insulin sensitivity. Pancreatic polypeptide is released by F cells in response to hypoglycemia and protein-rich meals and signals satiety.66 It also inhibits gallbladder contraction and exocrine pancreas secretion and increases gastric acid secretion. It is frequently increased in pancreatic tumors and in diabetes.
Adrenal Glands
The adrenal glands are paired pyramid-shaped organs located behind the peritoneum and close to the upper pole of each kidney. Each gland is surrounded by a capsule embedded in fat and well supplied with blood from the phrenic and renal arteries and the aorta. Venous return on the left is to the renal vein and on the right is to the inferior vena cava.
Each adrenal gland consists of two separate portions: an inner medulla and an outer cortex. These two portions have different embryonic origins, different structures, and different hormonal functions. In effect, each adrenal gland functions like two separate glands, although there are interrelationships between functions of each portion (Figure 20-17).
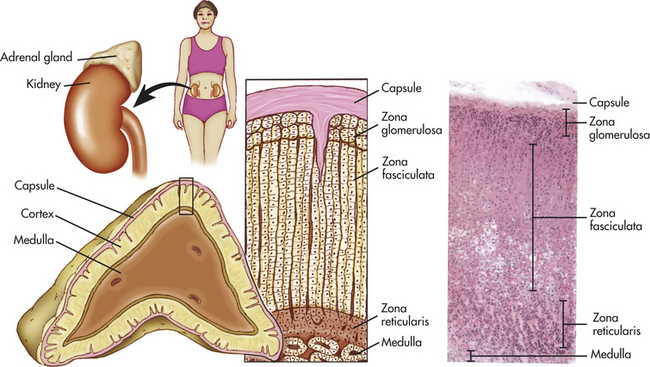
Figure 20-17 Structure of the adrenal gland showing cell layers (zones) of the cortex. Zona glomerulosa secretes aldosterone. Zona fasciculata secretes abundant amounts of glucocorticoids, chiefly cortisol. Zona reticularis secretes minute amounts of sex hormones and glucocorticoids. A portion of the medulla is visible at lower right in the photomicrograph (× 35) and at the bottom of the drawing. (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
The adrenal cortex, or outer region of the gland, accounts for 80% of the weight of the adult gland. The cortex is histologically subdivided into three zones. The outer layer, the zona glomerulosa, constitutes approximately 15% of the cortex and primarily produces the mineralocorticoid aldosterone. The middle layer, the zona fasciculata (78% of the cortex), and the inner layer, the zona reticularis (7% of the cortex), secrete other mineralocorticoids, the adrenal androgens and estrogens, and the glucocorticoids.67 The adrenal medulla, accounting for 20% of the gland’s total weight, secretes the catecholamines epinephrine (adrenaline), and norepinephrine (noradrenaline). Sympathetic and parasympathetic cholinergic fibers innervate the adrenal medulla; the adrenal cortex does not appear to be directly innervated.
Adrenal Cortex
The cells of the adrenal cortex are stimulated by the anterior pituitary hormone adrenocorticotropic hormone (ACTH). The adrenal cortex secretes several steroid hormones, including the glucocorticoids (mainly cortisol), the mineralocorticoids (mainly aldosterone), and the adrenal androgens and estrogens. These hormones are all synthesized from cholesterol. The best-known pathway of steroidogenesis involves the conversion of cholesterol to pregnenolone, which is then converted to the major corticosteroids.67
Glucocorticoids: The glucocorticoids have metabolic, neurologic, anti-inflammatory, and growth-suppressing effects. They act through nuclear and nongenomic pathways in the cell.21,68 The term glucocorticoid refers to those steroid hormones that have direct effects on carbohydrate metabolism. These hormones increase blood glucose concentration by promoting gluconeogenesis in the liver and by decreasing uptake of glucose into muscle cells, adipose cells, and lymphatic cells.67 In extrahepatic tissues the glucocorticoids stimulate protein catabolism and inhibit amino acid uptake and protein synthesis. In hepatic tissue, however, glucocorticoids act primarily to stimulate glucose formation and synthesis of enzymes that mediate glucocorticoid effects. The ultimate effect on the body is protein breakdown (catabolism).
The glucocorticoids act at several sites to influence immune and inflammatory reactions (described in Chapters 6 and 10). They affect innate immunity through several pathways, including decreasing the activity of pattern receptors on the surface of macrophages (see Chapter 6).69 Another major immune suppressant effect is a glucocorticoid-mediated decrease in the proliferation of T lymphocytes, primarily T-helper lymphocytes. There is a greater effect on T-helper 1 cytokine production (including antiviral interferons) than there is T-helper 2 cytokine production and therefore greater depression of cellular immunity than humoral immunity (see Chapter 7). Glucocorticoids also decrease immune and inflammatory responses by decreasing natural killer cell activity, promoting microphage phagocytosis of apoptotic granulocytes, and suppressing the synthesis, secretion, and actions of chemical mediators involved in inflammatory and immune responses. Glucocorticoids suppress the inflammatory response by blocking phospholipase A and the synthesis of prostaglandins, thromboxanes, and leukotrienes; and by inhibiting inflammatory gene expression.67 In addition, glucocorticoids stimulate anti-inflammatory cytokines (e.g., IL-10 and transforming growth factor-beta). Lysosomal membranes are also stabilized, decreasing the release of proteolytic enzymes. This suppression of innate and adaptive immunity by glucocorticoids means that infection and poor wound healing are some of the most problematic complications of the use of glucocorticoids in the treatment of disease. Similarly, psychologic and physiologic stress increases glucocorticoid production, which provides a pathway for the well-described decrease in immunity seen in both acute and chronic stress conditions (see Chapter 10).
Pathologically high levels of glucocorticoids include increasing circulating erythrocytes, leading to polycythemia; increasing the appetite; promoting fat deposits in the face and cervical areas; increasing uric acid excretion; decreasing serum calcium levels, possibly by inhibiting gastrointestinal absorption of calcium; and suppressing GH secretion so that somatic growth is inhibited. The glucocorticoids also have important “permissive” effects, sensitizing arterioles to the vasoconstrictive effects of norepinephrine. Glucocorticoids appear to potentiate the effects of catecholamines, thyroid hormone, and GH on adipose tissue. It also has been speculated that a metabolite of cortisol may act like a barbiturate and depress nerve cell function in the brain. This may account for the noted effects on mood associated with steroid fluctuation in disease or stress.
The most potent of the naturally occurring glucocorticoids is cortisol. It is the main secretory product of the adrenal cortex and is necessary for the maintenance of life and for protection from stress (see Chapter 10, particularly Figure 10-2). Cortisol has a biologic half-life of approximately 90 minutes, with the liver primarily responsible for its deactivation.
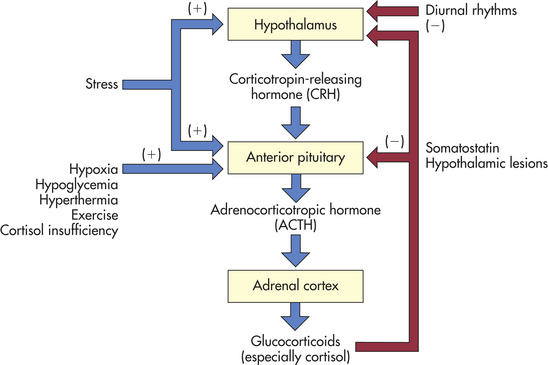
The secretion of cortisol is regulated primarily by the hypothalamus and the anterior pituitary gland (Figure 20-18). In the hypothalamus, CRH is produced in several nuclei and stored in the median eminence. Once released, CRH travels through the portal vessels to stimulate the production of ACTH from POMC, β-lipotropin, γ-lipotropin, endorphins, and enkephalins by the anterior pituitary. ACTH is the main regulator of cortisol secretion and adrenocortical growth.
Three factors appear to be primarily involved in regulating the secretion of ACTH: (1) high circulating levels of cortisol and synthetic glucocorticoids suppress CRH and ACTH, whereas low cortisol levels stimulate their secretion; (2) diurnal rhythms affect ACTH and cortisol levels (in persons with regular sleep-wake patterns, ACTH peaks 3 to 5 hours after sleep begins and declines throughout the day; and cortisol levels follow a similar pattern, peaking right before awakening); and (3) stress has been shown to increase ACTH secretion, leading to increased cortisol levels. (Neuroendocrine mechanisms regulating sleep are discussed in Chapter 15.) ACTH secretion also is controlled by hypothalamic arginine vasopressin.67 A form of ACTH (i.e., irACTH) also is produced by the cells of the immune system. It is detectable through laboratory techniques, and physiologically it appears to exert the usual feedback effects (see Chapter 10). This mechanism may account in part for integration of the immune and endocrine systems.
Once ACTH is secreted, it binds to specific plasma membrane receptors on the cells of the adrenal cortex and on other extra-adrenal tissues. Because both adrenal and extra-adrenal tissues have ACTH receptors, a number of effects result from stimulation by ACTH. (These are summarized in Box 20-1.) Both adrenal and extra-adrenal effects appear to be mediated through the activation of the adenylyl cyclase system. Melanocyte-stimulating hormone is also synthesized from the precursor POMC and increases skin pigmentation.
Once ACTH stimulates the cells of the adrenal cortex, cortisol synthesis and secretion immediately occur. In the normal person the secretory patterns of ACTH and cortisol are nearly identical. After secretion, most cortisol circulates in bound form: 15% to 30% is bound to albumin, and 55% to 75% is tightly but reversibly bound to a plasma glycoprotein called transcortin, or corticosteroid-binding globulin. The levels of transcortin play a role in the HPA feedback system controlling cortisol secretion.70 Transcortin levels are significantly elevated by increased estrogen levels that occur with pregnancy and hormone therapy—10% to 15% of the cortisol secreted circulates unbound. The unbound portion is free to diffuse into cells, but only those cells with specific intracellular glucocorticoid receptors respond to cortisol stimulation. ACTH is rapidly inactivated in the circulation, and the liver and kidneys remove the deactivated hormone.
Mineralocorticoids: Aldosterone: Mineralocorticoid steroids directly affect ion transport by epithelial cells, causing sodium retention and potassium and hydrogen loss. Aldosterone is the most potent of the naturally occurring mineralocorticoids and acts to conserve sodium by increasing the activity of the sodium pump of the epithelial cells. (The sodium pump is described in Chapter 1.)
The initial stages of aldosterone synthesis occur in the zona fasciculata and zona reticularis. The final conversion of corticosterone to aldosterone, however, apparently is confined to the zona glomerulosa. Aldosterone synthesis and secretion are regulated primarily by the renin-angiotensin-aldosterone system (described in Chapter 3 and Chapter 35; see Figure 35-10), although other factors also may be involved. Sodium and potassium levels may directly affect aldosterone secretion; however, the mechanisms involved are not understood. ACTH may transiently stimulate aldosterone synthesis but does not appear to be a major regulator of aldosterone secretion.
Aldosterone synthesis and secretion are stimulated by angiotensin II. The conversion of angiotensin I to angiotensin II is stimulated by the enzyme angiotensin I–converting enzyme (Figure 20-19). The conversion of angiotensinogen to angiotensin I is stimulated by renin. Renin secretion is stimulated primarily by decreased renal blood flow because of sodium and water depletion and a diminished effective blood volume. Renin secretion also is stimulated by increased serum potassium.
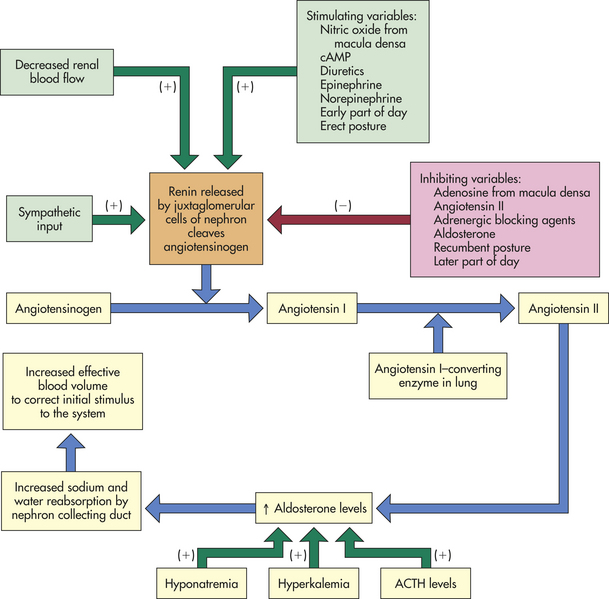
Figure 20-19 The feedback mechanisms regulating aldosterone secretion. ACTH, Adrenocorticotropic hormone; cAMP, cyclic adenosine monophosphate.
When sodium and potassium levels are within normal limits, normal serum levels of aldosterone are 5-30 mg/d; 50% to 75% of the secreted aldosterone bind to plasma proteins, including albumin, transcortin, and an α1-acid glycoprotein (AAG). The relatively large proportion of unbound aldosterone contributes to its rapid metabolic turnover in the liver, its low plasma concentration, and its short half-life of approximately 15 minutes. The main site of aldosterone degradation is the liver, with the metabolic end products being excreted by the kidney.
In the kidney aldosterone primarily acts on the epithelial cells of the nephron collecting duct to increase sodium ion reabsorption (thus promoting water reabsorption) and increase potassium and hydrogen ion excretion. High levels of aldosterone may result in alkalosis and hypokalemia (see Chapter 3). (Kidney function is discussed in Chapter 35.) This renal effect takes 1½ to 6 hours to occur after stimulation by aldosterone. Aldosterone also reduces sodium in sweat, saliva, and gastric juice.
Aldosterone affects many other tissues in the body, especially the cardiovascular system. Pathologically elevated levels of aldosterone have been implicated in hypertension, atherosclerosis, and heart failure.71
Adrenal Estrogens and Androgens: Estrogen secretion by the normal adrenal cortex is so minimal as to be considered physiologically unimportant. The adrenal cortex also secretes androgens. Adrenal androgen secretion is regulated by ACTH rather than the gonadotropins.67 Some of the weak androgenic substances secreted by the cortex are then converted by peripheral tissues to stronger androgens, such as testosterone, thus accounting for some androgenic effect initiated by the adrenal cortex. An increased capacity for peripheral conversion of adrenal androgens to estrogens occurs in particular cases, however, including aging, obesity, liver disease, and hyperthyroidism. The biologic effects and metabolism of the adrenal sex steroids do not vary from those produced by the gonads (see Chapter 22).
Adrenal Medulla
The adrenal medulla, together with the sympathetic divisions of the autonomic nervous system, is embryonically derived from neural crest cells. Chromaffin cells (pheochromocytes) are the cells of the adrenal medulla. The major products secreted by the chromaffin cells are the catecholamines epinephrine (adrenaline) and norepinephrine, although the medulla is only a minor source of norepinephrine. The adrenal medulla functions as a sympathetic ganglion without postganglionic processes. Sympathetic cholinergic preganglion fibers terminate on the chromaffin cells and secrete catecholamines directly into the bloodstream.72 The catecholamines are therefore hormones and not neurotransmitters.
Only 30% of circulating epinephrine comes from the adrenal medulla. The other 70% is released from nerve terminals. Catecholamine production in the adrenal medulla consists of approximately 75% to 85% epinephrine and approximately 15% to 25% norepinephrine. Epinephrine is about 10 times more potent than norepinephrine in producing direct metabolic effects. The adrenal medulla synthesizes the catecholamines from the amino acid phenylalanine (Figure 20-20).
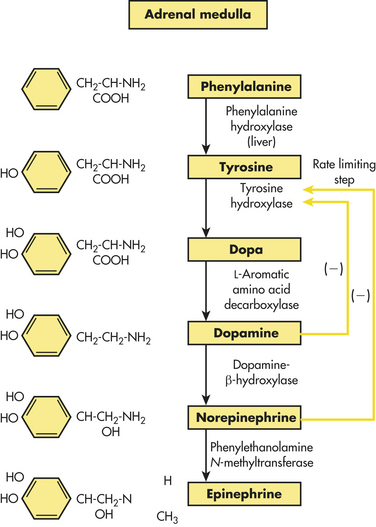
Adrenal catecholamines are stored in secretory granules within the chromaffin cells. Physiologic stress to the body (e.g., traumatic injury, hypoxia, hypoglycemia, and many others) triggers release of adrenal catecholamines through acetylcholine (from the preganglionic sympathetic fibers), which depolarizes the chromaffin cells.72 Depolarization causes exocytosis of the storage granules from the chromaffin cells with release of epinephrine and norepinephrine into the bloodstream. The control of exocytosis probably involves calcium, although this mechanism is not fully understood.72 Secretion of adrenal catecholamines is also increased by ACTH and the glucocorticoids.
Once released, the catecholamines remain in the plasma for only seconds to minutes. The catecholamines exert their biologic effects after binding to a plasma membrane receptors (α1, α2, β1, β2, β3) in target cells (see Table 14-7). This binding activates the adenylyl cyclase system. Catecholamines are rapidly removed from the plasma by being taken up by neurons for storage in new cytoplasmic granules, or they may be metabolically inactivated and excreted in the urine.
Catecholamines have diverse effects on the entire body. Their release and the body’s response have been characterized as the “fight or flight” response (see Chapter 10). In general, the metabolic effects of catecholamines promote hyperglycemia through a variety of mechanisms and through interfering with usual glucose regulatory feedback mechanisms.
Neuroendocrine Response to Stressors
The endocrine system acts together with the nervous system to respond to stressors. The integrated response to stressors also includes the immune system. Hormones of the neuroendocrine system affect components of the immune system, and mediators produced by immune components regulate the neuroendocrine response.
Perception that an event is stressful may be essential to the emotional arousal and initiation of the stress response (discussed in Chapter 10). Some events, such as bacterial invasion, can activate the stress response without emotional arousal. The hypothalamus receives input from a variety of areas within the brain and ultimately directs the neuroendocrine response to stress through the actions of CRH, the locus ceruleus–norepinephrine autonomic (sympathetic) nervous system, and the pituitary-adrenal axis. In addition to the neuroendocrine components of the stress response, the gamma motor neuron system is activated to increase skeletal motor tone. Enhanced availability of vital substrates occurs, and growth and reproduction are inhibited to preserve energy for protective responses. Details of the stress response are presented in Chapter 10.
Tests of Endocrine Function
Tests of the endocrine system involve several general types of clinical evaluation.29 Measurement of hormone level is accomplished by radioimmunoassay, by enzyme-linked immunosorbent assay, and less commonly by bioassay. Radioimmunoassay (RIA) is a technique for measuring the minute quantities of hormones in the blood. Antibody that is specific for the hormone is mixed with plasma containing the hormone to be measured. Standard radiolabeled hormone is also added to the mixture. The amount of antibody present in the body is not enough to bind with both the tagged hormone and the hormone to be measured. The competition between the radiolabeled hormone and the unlabeled hormone in an antigen-antibody reaction determines the concentration of the unlabeled hormone. A quantitative value is established by use of standard reference curves.
Enzyme-linked immunosorbent assay (ELISA) also is used to determine circulating hormone levels. This method is similar to that of RIA but is less expensive and easier to conduct. Instead of radiolabeled hormones, an enzyme-labeled hormone is used. The enzyme activity in either the bound or unbound fraction is determined and related to the concentration of the unlabeled hormone.
A bioassay involves the use of graded doses of hormone in a reference preparation and then comparison of the results with an unknown sample. Bioassays are used more commonly in investigative endocrinology than in clinical laboratories.
The concentration of hormones in serum can be measured to assess endocrine function in health and disease. If the serum level is greater or less than the reference values, more definitive tests are required to determine the source of the problem. Measurement of individual hormones does not always permit differentiation between normal and abnormal values when hormone levels are changing over time. For an accurate interpretation, the broad normal range of some hormones requires a knowledge of previous hormonal levels and timed sampling.
The major problems in evaluating the endocrine system include (1) the complexity of the clinical presentation because of multiple organ system involvement, (2) the nonspecific nature of complaints frequently associated with endocrine dysfunction, and (3) the inappropriate use of laboratory test interpretations.
Aging and the Endocrine System
The precise relationship between aging and the endocrine system is not clear. Perhaps most important, the question of whether changes in endocrine function are a consequence or a cause of aging has yet to be resolved. These relationships have been difficult to identify, in part because of a number of age-related variables that may coexist, such as acute and chronic nonendocrine disease; use of medications; alterations in diet, body composition, and weight; and changes in sleep-wake cycles. However, the endocrine system is so integral to health that changes in endocrine function have been used as “biomarkers” for unhealthy aging.73
Theories About the Effects of Aging
Investigation into the role of the endocrine glands and their interactions in the aging process has generated much data, although the evidence is contradictory. There are complex changes within the HPA, and altered biologic activity of hormones, altered circulating levels of hormones, altered secretory response of the endocrine glands, altered metabolism of hormones, and loss of circadian control of hormone secretion are among the findings. The most studied changes in hormone function with aging affect the levels of reproductive hormones and gonadotropins (menopause and andropause) (discussed in Chapter 22). Two of the most important endocrine changes associated with aging affect the pancreas and the thyroid gland.74 Other important hormone changes associated with aging include a decline in serum levels of growth hormone, IGF-1, and dehydroepiandrosterone.75
Theories of cellular damage deal with adverse cellular conditions that produce the biologic effects associated with aging. (These theories are discussed in Chapter 2.) The endocrine system has not been specifically implicated in any of these theories, particularly as a causative variable. The cellular changes or consequences described by these theories, however, do affect specific endocrine glands and might contribute to endocrine gland dysfunction or alterations in responsiveness of target organs.
Theories of stress and adaptation suggest that body structures wear out from overuse or are no longer able to adapt to the cumulative effects of physiologic stress. One such endocrine function that may be affected is the sympathoadrenal axis. Exhaustion of this axis may be associated with an inability of the body to respond effectively to stressors.
Theories of programmed change are concerned with genetic control of cell function. Certain secretory cells may be programmed genetically to secrete hormones for a prescribed length of time. Changes seen with female reproductive function may represent the phenomenon of programmed change. Reduced signaling of insulin and insulin-like peptides is associated with increased life spans.
All changes in cellular activity—as a result of damage, programmed change, or wear and tear—may affect neuroendocrine regulation. Changes in secretion of hypothalamic regulatory factors and hormones or changes in hypothalamic feedback sensitivity may contribute to alterations in control of an optimal internal environment. The dynamic equilibrium of the endocrine system also may be affected by altered secretion of neurotransmitters within certain areas of the brain, affecting hypothalamic and pituitary function. Such alterations may include an excess or deficit in secretion of pituitary hormones and loss of appropriate secretory pattern of those hormones. Loss of endocrine steady states may be associated with or contribute to aging.
Effects of Aging on Specific Glands and Hormones
Thyroid Gland: Changes in thyroid structure and function occur with aging. Structurally, some glandular atrophy and fibrosis occur with nodularity and increasing inflammatory infiltrates. These infiltrative changes may reflect age-related autoimmune damage. Overall, it is estimated that there is some evidence of thyroid dysfunction in 5% to 10% of older adult women.74 The presence of thyroid nodules increases after the age of 70 years.76
Changes relative to thyroid hormone and its function are more difficult to assess. One difficulty is finding older adults who are free of all systemic and thyroid-related illness, so that the resulting changes can be attributed to aging. Much of the available data are contradictory. Most evidence, however, supports the following age-related changes74:
1. Overall TSH secretion is diminished.
2. Responsiveness of plasma TSH concentration to TRH administration is reduced, especially in men.
In addition, the average dose for TH replacement appears to be lower in older adults because the peripheral metabolism of TH decreases with age. TH must be replaced slowly in elderly individuals with coronary artery disease to prevent angina and myocardial infarction. Clinical signs of thyroid disease are more difficult to detect in older adults.76
Pancreas: It is estimated that 40% to 50% of individuals older than age 65 have impaired glucose tolerance or diabetes.74 With aging, the pancreatic cells are increasingly replaced with fat tissue. Dysfunction of the pancreas with decreased insulin secretion of the beta cells, insulin receptors, and cellular responses to insulin are all noted. These changes have significant implications for many target organs, particularly the cardiovascular system, which is increasingly at risk for both vascular (hypertension, atherosclerosis, glomerulosclerosis) and cardiac (infarction, failure) disorders. Exciting research is exploring the relationship between insulin and activation of genes for the “sirtuins,” which are a group of proteins that have been linked with the aging process.77
Growth Hormone and Insulin-like Growth Factors: The amounts of GH and IGF decline with aging, a process that has been called the “somatopause.” This decline in anabolic stimuli is linked to decreases in muscle size and function, fat and bone mass, and changes in reproductive and cognitive function.42,74 Clinical findings related to somatotropic hormone changes with aging include increases in visceral fat, decreased lean body mass, and decreased bone density. Despite the initial enthusiasm for the use of therapeutic doses of GH as a way to slow the aging process, studies have not been consistently positive and the risk for GH- and IGF-mediated oncogenesis is being explored.42
Parathyroid Glands: An age-related alteration in PTH secretion has been proposed to explain alterations in calcium homeostasis that have been noted in older adults.78 Such an alteration, however, has not been documented consistently. Calcium intake, especially in women, tends to decrease with aging and may contribute to osteoporosis (see Chapter 42). The average daily intake of 450 to 500 mg/day causes a negative calcium balance greater than 40 mg/day and may be related to the absolute bone loss of approximately 1.5% per year. Older adults show decreased intestinal adaptation to variations in calcium intake. Hyperparathyroidism may occur secondary to calcium malabsorption and hypocalcemia with increased bone remodeling that results in cortical bone thinning and porosity. Elevated levels of parathyroid hormone have been linked with an increase in mortality in older adults,79 many of whom also have a mild, persistent hypercalciuria, which indicates a defective renal mechanism for responding to decreased calcium intake. Decreased circulating levels of vitamin D are common in older adults, especially those in long-term care institutions.78 Vitamin D deficiency has been linked to not only osteoporosis but also cancer, autoimmune diseases, diabetes, cardiovascular disease, and mental health disorders.80
The decrease in calcium intake, an age-related decrease in circulating vitamin D, and a blunted response in older adults to PTH may explain these changes seen in aging. Additional investigation into mechanisms of altered calcium metabolism is required before the age-associated alterations can be explained.
Adrenal Glands: The adrenal cortex loses some weight and has more fibrous tissue after the age of 50 years. Age does not appear to affect the feedback mechanisms involved in maintaining glucocorticoid levels, but the decrease in the metabolic clearance rate of the glucocorticoids is age related.
The metabolic clearance of cortisol decreases with an age-related decline in liver and kidney function. Further, less cortisol appears to be used by the body when aging is accompanied by a loss of lean body mass. Decreased clearance and reduced use of cortisol contribute to higher circulating cortisol levels, but diurnal variation is maintained. Because feedback mechanisms are intact, the higher cortisol levels cause a decrease in cortisol secretion. Circadian patterns of ACTH and cortisol secretion may change with aging.
Plasma levels of the adrenal androgens, as well as urinary excretion of the metabolic end products, decrease gradually but dramatically with age, to as much as 50% to 70% of the young adult level. This change in adrenal function has been called the “adrenopause” and is correlated with decreased synthesis activity of dehydroepiandrosterone (DHEA).74,75 This change appears to reflect a decline in the function of the zona reticularis. In postmenopausal women, this decline in adrenal androgen secretion is especially important because nearly all sex steroids after menopause come from adrenal and ovarian production of androgen precursors converted to estrogens in the periphery. In older adult men, adrenal androgen production accounts for more than half of circulating testosterone levels.
Antidiuretic Hormone: Although hyponatremia is a common finding in older adults, it appears related to changes in renal function rather than to ADH-related mechanisms. Morphologic studies have not shown significant age-related degenerative changes in the neuroendocrine pathways that regulate the synthesis and secretion of ADH. It appears that ADH secretion is augmented when stimulated by changes in osmotic concentration, whereas baroreceptor-mediated ADH secretion is reduced.
Adrenal cortex 716
Adrenal gland 715
Adrenal medulla 715
Adrenocorticotropic hormone (ACTH) 716
Aldosterone 717
Amylin 714
Anterior pituitary 704
Antidiuretic hormone (ADH) 703
Beta cell 713
Bioassay 720
C cell 709
Calcitonin 709
Chromaffin cell (pheochromocyte) 719
Chromophil 707
Chromophobe 707
Corticotropin-releasing hormone (CRH) 704
Cortisol 717
Diacylglycerol (DAG) 702
Direct effect 699
Down-regulation 699
Enzyme-linked immunosorbent assay (ELISA) 720
First messenger 700
Follicle 709
Gastrin 715
Gonadotropin-releasing hormone (GnRH) 704
Grehlin 715
Growth hormone–releasing factor (GRF) 704
Glucagon 714
Glucocorticoid 716
Hormone 696
Hormone receptor 699
Inositol triphosphate (IP3) 702
Insulin 713
Islets of Langerhans 712
Isthmus 709
Median eminence 705
Melatonin 704
Mineralocorticoid 717
Neuroendocrine system 703
Oxytocin 703
Pancreas 712
Pancreatic polypeptide 715
Parathyroid hormone (PTH) 711
Pars distalis 704
Pars intermedia 704
Pars nervosa (neural lobe) 705
Pars tuberalis 704
Permissive effect 699
Pituitary stalk 705
Posterior pituitary 705
Prolactin-inhibiting factor (PIF) 704
Radioimmunoassay (RIA) 720
Signal transduction 701
Somatostatin 714
Substance P 704
Target cell 699
Thyroid gland 709
Thyroid hormone (TH) 709
Thyroid-stimulating hormone (TSH) 710
Thyrotropin-releasing hormone (TRH) 704, 709
Up-regulation 699
Zona fasciculata 716
Zona glomerulosa 716
Zona reticularis 716
REFERENCES
1. Jameson, L.J., Degroot, L.J. Endocrinology: impact on science and medicine. In DeGroot L.J., Jamerson J.L., eds.: Endocrinology, ed 5, St Louis: Saunders, 2006.
2. Kronenberg, H.M., et al. Principles of endocrinology. In Kronenberg H.M., et al, eds.: Williams textbook of endocrinology, ed 11, Philadelphia: Saunders, 2008.
3. Spiegel, A., Carter-Su, C., Taylor, S.I. Mechanisms of action of hormones that act at the cell surface. In Kronenberg H.M., et al, eds.: Williams textbook of endocrinology, ed 11, Philadelphia: Saunders, 2008.
4. Losel, R., Wehling, M. Nongenomic actions of steroid hormones. Nat Rev Mol Cell Biol. 2003;4(1):46–56.
5. Davis, P.J., Leonard, J.L., Davis, F.B. Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol. 2008;29(2):211–218.
6. Oetting, A., Yen, P.M. New insights into thyroid hormone action. Best Pract Res Clin Endocrinol Metab. 2007;21(2):193–208.
7. Avruch, J. Hormone signaling via tyrosine kinase receptors. In DeGroot L.J., Jamerson J.L., eds.: Endocrinology, ed 5, St Louis: Saunders, 2006.
8. Gonzales-Maeso, J., Sealfon, S.C. Hormone signaling via G protein-coupled receptors. In DeGroot L.J., Jamerson J.L., eds.: Endocrinology, ed 5, St Louis: Saunders, 2006.
9. Lohse, M.J., et al. Kinetics of G-protein-coupled receptor signals in intact cells. Br J Pharmacol. 2008;153(Suppl 1):S125–S132.
10. Debrincat, M.A., Hilton, D.J. Hormone signaling via cytokine receptors. In DeGroot L.J., Jamerson J.L., eds.: Endocrinology, ed 5, St Louis: Saunders, 2006.
11. Takemori, H. Transcription factor cAMP response element-binding protein CREB. FEBS J. 2007;274(13):3201.
12. Kitagawa, K. CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J. 2007;274(13):3210–3217.
13. Siu, Y.T., Jin, D.Y. CREB—a real culprit in oncogenesis. FEBS J. 2007;274(13):3224–3232.
14. Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49.
15. Colomer, J., Means, A.R. Physiological roles of the Ca2+/CaM-dependent protein kinase cascade in health and disease. Subcell Biochem. 2007;45:169–214.
16. Blair, H.C., et al. Calcium signalling and calcium transport in bone disease. Subcell Biochem. 2007;45:539–562.
17. Friel, D.D., Chiel, H.J. Calcium dynamics: analyzing the Ca2+ regulatory network in intact cells. Trends Neurosci. 2008;31(1):8–19.
18. Jude, J.A., et al. Calcium signaling in airway smooth muscle. Proc Am Thorac Soc. 2008;5(1):15–22.
19. Omori, K., Kotera, J. Overview of PDEs and their regulation. Circ Res. 2007;100(3):309–327.
20. Sallustio, F., et al. Saving the ischemic penumbra: potential role for statins and phosphodiesterase inhibitors. Curr Vasc Pharmacol. 2007;5(4):259–265.
21. Haller, J., Mikics, E., Makara, G.B. The effects of non-genomic glucocorticoid mechanisms on bodily functions and the central neural system. A critical evaluation of findings. Front Neuroendocrinol. 2008;29(2):273–291.
22. Losel, R.M., Wehling, M. Classic versus non-classic receptors for nongenomic mineralocorticoid responses: emerging evidence. Front Neuroendocrinol. 2008;29(2):258–267.
23. Michels, G., Hoppe, U.C. Rapid actions of androgens. Front Neuroendocrinol. 2008;29(2):182–198.
24. Vasudevan, N., Pfaff, D.W. Non-genomic actions of estrogens and their interaction with genomic actions in the brain. Front Neuroendocrinol. 2008;29(2):238–257.
25. Low, M.J., et al. Neuroendocrinology. In Kronenberg H.M., ed.: Williams textbook of endocrinology, ed 11, Philadelphia: Saunders, 2008.
26. Karasek, M. Does melatonin play a role in aging processes? J Physiol Pharmacol. 2007;6(58 Suppl):105–113.
27. Szczepanik, M. Melatonin and its influence on the immune system. J Physiol Pharmacol. 2007;6(58 Suppl):115–124.
28. Paulis, L., Simko, F. Blood pressure modulation and cardiovascular protection by melatonin: potential mechanisms behind. Physiol Res. 2007;56(6):671–684.
29. Peschke, E. Melatonin, endocrine pancreas and diabetes. J Pineal Res. 2008;44(1):26–40.
30. Reiter, R.J., et al. Melatonin defeats neurally derived free radicals and reduces the associated neuromorphological and neurobehavioral damage. J Physiol Pharmacol. 2007;58(Suppl 6):5–22.
31. Maharaj, D.S., Glass, B.D., Daya, S. Melatonin: new places in therapy. Biosci Rep. 2007;27(6):299–320.
32. Kalantaridou, S., et al. Peripheral corticotropin-releasing hormone is produced in the immune and reproductive systems: actions, potential roles and clinical implications. Front Biosci. 2007;12:572–580.
33. Smith, E.M. Neuropeptides as signal molecules in common with leukocytes and the hypothalamic-pituitary-adrenal axis. Brain Behav Immun. 2008;22(1):3–14.
34. Robinson, A.G., Verbalis, J. Posterior pituitary. In Kronenberg H.M., et al, eds.: Williams textbook of endocrinology, ed 11, Philadelphia: Saunders, 2008.
35. Dunser, M.W., Westphal, M. Arginine vasopressin in vasodilatory shock: effects on metabolism and beyond. Curr Opin Anaesthesiol. 2008;21(2):122–127.
36. Franchini, M. The use of desmopressin as a hemostatic agent: a concise review. Am J Hematol. 2007;82(8):731–735.
37. Su, L.L., Chong, Y.S., Samuel, M. Oxytocin agonists for preventing postpartum haemorrhage. Cochrane Database Syst Rev. (3):2007. [CD005457].
38. Neumann, I.D. Stimuli and consequences of dendritic release of oxytocin within the brain. Biochem Soc Trans. 2007;35(Pt 5):1252–1257.
39. Slattery, D.A., Neumann, I.D. No stress please! Mechanisms of stress hyporesponsiveness of the maternal brain. J Physiol. 2008;586(2):377–385.
40. Carter, C.S. Sex differences in oxytocin and vasopressin: implications for autism spectrum disorders? Behav Brain Res. 2007;176(1):170–186.
41. Reiter, E.O., Rosenfield, R.G. Normal and aberrant growth. In Kronenberg H.M., ed.: Williams textbook of endocrinology, ed 11, Philadelphia: Saunders, 2008.
42. Sherlock, M., Toogood, A.A. Aging and the growth hormone/insulin like growth factor-I axis. Pituitary. 2007;10(2):189–203.
43. Samani, A.A., et al. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28(1):20–47.
44. Ryan, P.D., Goss, P.E. The emerging role of the insulin-like growth factor pathway as a therapeutic target in cancer. Oncologist. 2008;13(1):16–24.
45. Karsdal, M.A., et al. Calcitonin affects both bone and cartilage: a dual action treatment for osteoarthritis? Ann N Y Acad Sci. 2007;1117:181–195.
46. Mercadante, S., Fulfaro, F. Management of painful bone metastases. Curr Opin Oncol. 2007;19(4):308–314.
47. Simon, L.S. Osteoporosis. Rheum Dis Clin North Am. 2007;33(1):149–176.
48. Müller, B. Endocrine aspects of critical illness. Ann Endocrinol (Paris). 2007;68(4):290–298.
49. Becker, K.L., Snider, R., Nylen, E.S. Procalcitonin assay in systemic inflammation, infection, and sepsis: clinical utility and limitations. Crit Care Med. 2008;36(3):941–952.
50. Szkudlinski, M.S., Kazlauskaite, R., Weintraub, B.D. Thyroid stimulating hormone and regulation of the thyroid axis. In DeGroot L.J., Jamerson J.L., eds.: Endocrinology, ed 5, St Louis: Saunders, 2006.
51. Gereben, B., et al. Activation and inactivation of thyroid hormone by deiodinases: local action with general consequences. Cell Moll Life Sci. 2008;65(4):570–590.
52. Larsen, P.R., et al. Thyroid physiology and diagnostic evaluation of patients with thyroid disorders. In Kronenberg H.M., ed.: Williams textbook of endocrinology, ed 11, Philadelphia: Saunders, 2008.
53. Grover, G.J., Mellstrom, K., Malm, J. Therapeutic potential for thyroid hormone receptor-beta selective agonists for treating obesity, hyperlipidemia and diabetes. Curr Vasc Pharmacol. 2007;5(2):141–154.
54. Wexler, J.A., Sharretts, J. Thyroid and bone. Endocrinol Metab Clin North Am. 2007;36(3):673–705. [vi].
55. Rivas, M., Naranjo, J.R. Thyroid hormones, learning and memory. Genes Brain Behav. 2007;6(Suppl 1):40–44.
56. Schumock, G.T., Sprague, S.M. Clinical and economic burden of fractures in patients with renal osteodystrophy. Clin Nephrol. 2007;67(4):201–208.
57. Bergenstock, M.K., Partridge, N.C. Parathyroid hormone stimulation of noncanonical Wnt signaling in bone. Ann N Y Acad Sci. 2007;1116:354–359.
58. Khosla, S., Westendorf, J.J., Oursler, M.J. Building bone to reverse osteoporosis and repair fractures. J Clin Invest. 2008;118(2):421–428.
59. Heaney, R.P. Vitamin D endocrine physiology. J Bone Miner Res. 2007;22(Suppl 2):V25–V27.
60. Hills, C.E., Brunskill, N.J. Intracellular signalling by C-peptide. Exp Diabetes Res. 2008;51(8):1534–1543.
61. Ekberg, K., Johansson, B.L. Effect of C-peptide on diabetic neuropathy in patients with type 1 diabetes. Exp Diabetes Res. 2008. [2008:457912].
62. Wilhelm, B., Kann, P., Pfutzner, A. Influence of C-peptide on glucose utilization. Exp Diabetes Res. 2008. [2008:769483].
63. Smith, S.R., Ravussin, E. Role of the adipocyte in metabolism and endocrine function. In DeGroot L.J., Jamerson J.L., eds.: Endocrinology, ed 5, St Louis: Saunders, 2006.
64. Drucker, D.J. The role of gut hormones in glucose homeostasis. J Clin Invest. 2007;117(1):24–32.
65. Ehrman, M.M., et al. Regulation of pancreatic somatostatin gene expression by insulin and glucagon. Mol Cell Endocrinol. 2005;235(1-2):31–37.
66. Wren, A.M. Gut and hormones and obesity. Front Horm Res. 2008;36:165–181.
67. Stewart, P.M. The adrenal cortex. In Kronenberg H.M., ed.: Williams textbook of endocrinology, ed 11, Philadelphia: Saunders, 2008.
68. Heitzer, M.D., et al. Glucocorticoid receptor physiology. Rev Endocr Metab Disord. 2007;8(4):321–330.
69. Chinenov, Y., Rogatsky, I. Glucocorticoids and the innate immune system: crosstalk with the toll-like receptor signaling network. Mol Cell Endocrinol. 2007;275(1-2):30–42.
70. Kumsta, R., et al. Cortisol and ACTH responses to psychosocial stress are modulated by corticosteroid binding globulin levels. Psychoneuroendocrinol. 2007;32(8-10):1153–1157.
71. Marney, A.M., Brown, N.J. Aldosterone and end-organ damage. Clin Sci. 2007;113(6):267–278.
72. Young, W.F. Endocrine hypertension. In Kronenberg H.M., ed.: Williams textbook of endocrinology, ed 11, Philadelphia: Saunders, 2008.
73. Simm, A., et al. Potential biomarkers of ageing. Biol Chem. 2008;389(3):257–265.
74. Lambers, S.W. Endocrinology and aging. In Kronenberg H.M., ed.: Williams textbook of endocrinology, ed 11, Philadelphia: Saunders, 2008.
75. Chahal, H.S., Drake, W.M. The endocrine system and ageing. J Pathol. 2007;211(2):173–180.
76. Peeters, R.P. Thyroid hormones and aging. Hormones. 2008;7(1):28–35.
77. Engel, N., Mahlknecht, U. Aging and anti-aging: unexpected side effects of everyday medication through sirtuin1 modulation. Int J Mol Med. 2008;21(2):223–232.
78. Adami, S., et al. Relationship between serum parathyroid hormone, vitamin D sufficiency, age, and calcium intake. Bone. 2008;42(2):267–270.
79. Bjorkman, M.P., Sorva, A.J., Tilvis, R.S. Elevated serum parathyroid hormone predicts impaired survival prognosis in a general aged population. Eur J Endocrinol. 2008;158(5):749–753.
80. Holick, M.F. Vitamin D deficiency. N Engl J Med. 2007;357:266–281.