CANCER EPIDEMIOLOGY


Although cancer arises from a complicated and an interacting web of multiple causes, preventing exposures to individual carcinogens, or cancer-causing substances, may prevent many cancers. Research has shown that environmental-lifestyle factors and occupational exposure fuels the number of cancer cases and deaths.1–3 Widespread general exposure to pollutants from water; air; the work environment; personal lifestyle choices (such as smoking, excessive alcohol, and poor diet) and involuntary or unknowing exposures to carcinogens in the air, water, and occupational environments are major contributors to cancer. The National Cancer Institute (NCI) and the National Institute for Environmental Health Sciences (NIEHS) notes in the document titled “Cancer and the Environment” that two thirds of all cancers are caused by environmental-lifestyle factors.3
GENES, ENVIRONMENTAL-LIFESTYLE FACTORS, AND RISK FACTORS
Cancers are caused by environmental-lifestyle and genetic factors. At the level of the cell, cancer is genetic. Although complex, investigators are struggling to connect the complex web between genotype, phenotype, and the environment to understand a person’s chances of developing cancer. Environmental-lifestyle factors include cigarette smoking, excessive alcohol consumption, poor diet, lack of exercise, excessive sunlight exposure, and sexual behavior that increases exposure to certain viruses. Additional factors include exposure to radiation, hormones, medical drugs, viruses, bacteria, pesticides, and other environmental chemicals present in air, water, food, soil, and the workplace. Investigations of occupational groups with high exposure to chemicals have identified numerous chemicals as carcinogens (Table 12-1).
Table 12-1
Summary of Environmental and Occupational Links with Cancer
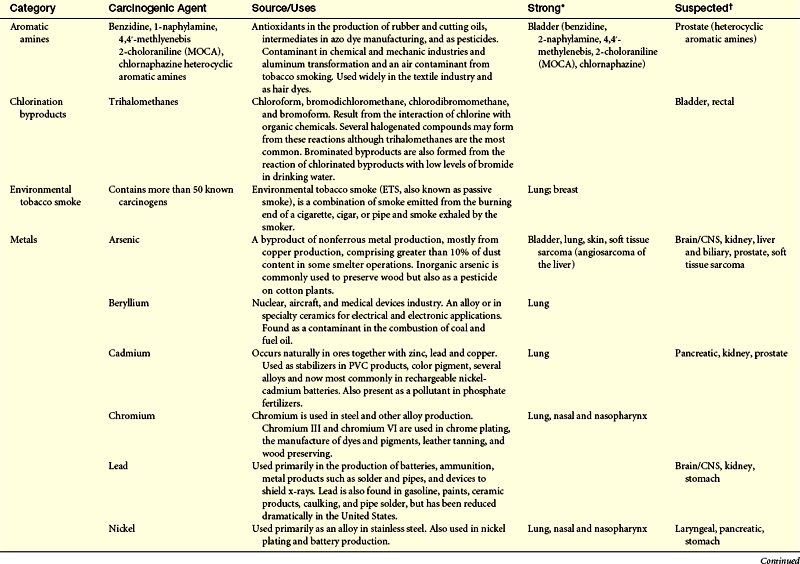

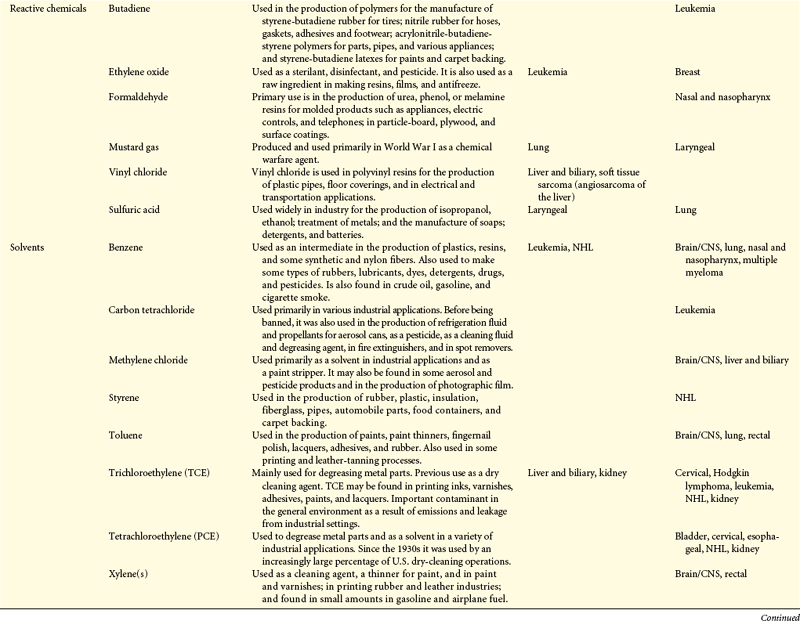

CNS, Central nervous system; DES, diethylstilbestrol; EPA, Environmental Protection Agency; NHL, non-Hodgkin lymphoma.
∗Strong causal evidence of a causal link is based primarily on a Group 1 designation by the International Agency for Research on Cancer.
†Suspected evidence of a causal link is based on our assessment that results of epidemiologic studies is mixed, yet positive findings from well-designed and conducted studies warrant precautionary action and additional scientific investigation.
From Clapp RW, Jacobs MM, Loechler EL: Environmental and occupational causes of cancer: new evidence, 2005-2007. Lowell Center for Sustainable Production, Lowell, MA. Cancer working Group of the Collaborative on Health and the Environment.
Studies of gene-environmental interactions whereby individuals with particular genetic predispositions may be more susceptible to the biologic effects of environmental exposures cannot explain the increased cancer risk in studies of exposed groups. More simply, it appears that the majority of cancers are caused by carcinogen exposure, rather than by rare genetic conditions.2 For example, for women who have mutated cancer susceptibility genes, BRCA1 or BRCA2, the risk of having breast cancer at age 50 is 24% for those born before 1940 but 67% for those born later.4 The implication here is related to lifestyle factors that changed from 1940 (i.e., hormone therapy, later age at first pregnancy, increased nulliparity, etc.). Investigations of more complex gene-gene environment interactions and proteomics may or may not alter these conclusions.
Strong evidence suggests that certain environmental-lifestyle and occupational exposures are associated with bladder cancer, bone, brain and central nervous system (CNS) cancer, breast cancer, Ewing sarcoma, liver cancer, laryngeal cancer, melanoma, mesothelioma, non-Hodgkin lymphoma (NHL), renal cancer, scrotal cancer, skin cancer, soft tissue sarcoma, and thyroid cancer (see Table 12-1).
Although tobacco smoke remains the single most preventable cause of cancer, it is not linked to the majority of cancers that have increased substantially in the United States. These cancers include melanoma, non-Hodgkin lymphoma, testicular, brain, and thyroid.2 For example, testicular cancer affects men mostly in their 20s and 30s. Incidence rates in these age groups in the United States increased about 75% from the 1970s to the 1980s and remain about 11 to 12/100,000. This increase is not attributed to improved diagnostic methods and cancer registration rates.5 Among all population groups, brain and nervous system cancers increased from 10 in 1973 to 20.9 in 1992—an astounding 109% increase.2,6
Lung cancer rates have risen and fallen paralleling the prevalence of smoking. Meanwhile, stomach cancer in the United States dropped dramatically over the past century.2 The decrease may be caused by lifestyle factors or decreasing or controlling bacterial infection from Helicobacter pylori, or both.
Compelling is the role of environmental-lifestyle contributions to cancer from studies comparing different populations around the world. Breast cancer, for example, is prevalent among northern Europeans and Americans but is relatively rare among women in developing countries. If ethnicity played a major role, then immigrants should retain the cancer incidence rates of their country of origin. Instead, immigrants acquire the same cancer rates of where they move within one or two generations.7,8 A particularly instructive group of studies has been the Multiethnic Cohort (MEC) study. These studies are focused on ethnic and migrant populations in Hawaii, which has large populations of ethnic groups that assist with interethnic comparisons and disentangling the effects of genes, the environment, and lifestyle on cancer rates. These results and others support the claim that cancer risk is substantially influenced by environmental factors (Box 12-1).
Elevated cancer rates are more common in cities, in farming locations, near hazardous waste sites, downwind of industrial and radiation activities, and near contaminated water wells. In addition, cancers are associated with areas of high pesticide use, toxic work exposures, waste incinerators, and other sources of pollution.9,10
Farmers have increased death rates from brain, multiple myeloma, prostate cancer, Hodgkin lymphoma, leukemia, non-Hodgkin lymphoma, and lip and stomach cancers.11 Migrant farmers also experience elevated rates of some of these cancers.2
A new paradigm shift suggests that susceptibility to disease is set in utero or neonatally as the result of nutrition and exposures to environmental toxins or stressors, or both.12 Children also may be affected by prenatal exposures, parental exposures prior to conception, and breast milk. Epidemiologic studies have linked higher risks of childhood leukemia and brain and CNS cancers with parental and childhood exposure to particular solvents, pesticides, petrochemicals, dioxins, and polycyclic aromatic hydrocarbons.13
Environmental-lifestyle factors play important roles in cancer development, but there are major gaps in our knowledge of how these factors affect our individual resistances to cancer-causing agents. Research needed for shaping public policy concerning environmental exposures should focus on (1) the relationship between the timing of exposures (periods of vulnerability), multiple exposures, and chronic exposures; (2) risks among racial groups and gender; (3) human contamination (biomonitoring); and (4) scrutinization of unexplained patterns of risk. In addition, public health advocates have argued for mandating that producers of environmental hazards (chemicals, tobacco, drugs, radiologic products, etc.) assess health, safety, and environmental effects before introducing them to the marketplace and making that information publicly available. Table 12-2 summarizes the estimated new cases and deaths caused by cancer, by gender, for specified sites.
Table 12-2
Estimated New Cancer Cases and Deaths by Sex, U.S., 2009∗
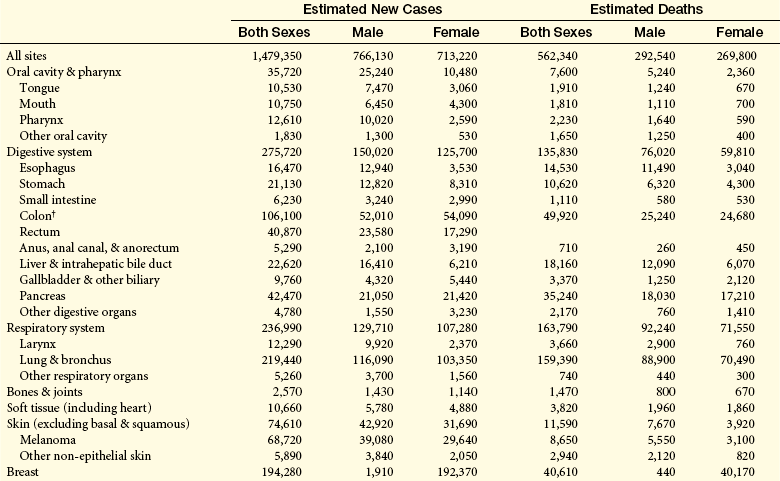
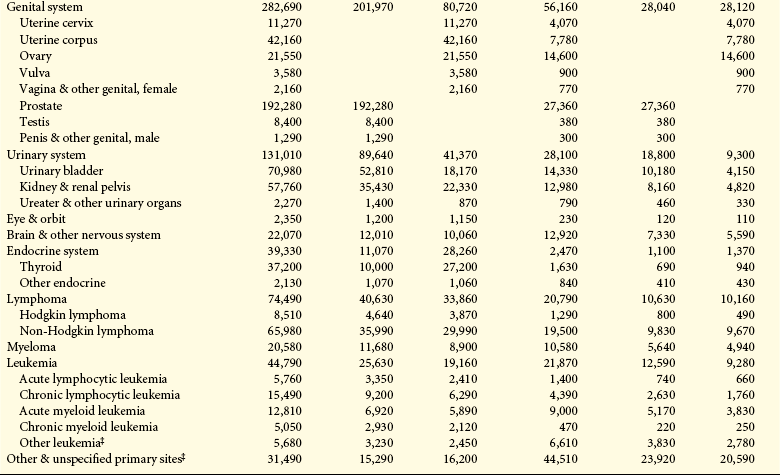
∗Rounded to the nearest 10; estimated new cases exclude basal and squamous cell skin cancers and in situ carcinomas except urinary bladder. About 62,280 female carcinoma in situ of the breast and 53,120 melanoma in situ will be newly diagnosed in 2009.
†Estimated deaths for colon and rectum cancers are combined.
‡More deaths than cases suggests lack of specificity in recording underlying causes of death on death certificates.
Source: Estimated new cases are based on 1995-2005 incidence rates from 41 states and the District of Columbia as reported by the North American Association of Central Cancer Registries (NAACCR), representing about 85% of the US population. Estimated deaths are based on data from US Mortality Data, 1969-2006, National Center for Health Statistics, Centers for Disease Control and Prevention, 2009. © 2009, American Cancer Society.
Epigenetics and Genetics
The idea that increased risk of disease originates from interactions among genes and environmental-lifestyle factors, infact, may not be driven by the genetic code. A hot debate has been the relative importance of genetic versus epigenetic processes. Although it is clear that inherited variation in deoxyribonucleic acid (DNA) sequence influences individual risk of cancer, this occurs in only a small percentage of the population.14 In addition, how structural variation of the genome increases cancer risk is unknown. An explosion of data now indicates the importance of epigenetic processes, especially those with resultant gene silencing of key regulatory genes (see Chapter 11). Epigenetic changes collaborate with genetic changes and environmental-lifestyle factors to cause the development of cancer. These changes are mitotically and meiotically heritable.15,16
The three major areas of epigenetics are (1) methylation (the addition of methyl group [CH3] to cytosine ring) (see Figure 11-16); aberrant methylation, which can lead to silencing of tumor-suppressor genes; (2) histone modifications (histone acetylation, alterations in chromatin); and (3) micro-ribonucleic acids (miRNAs), small RNA molecules that can target gene expression post-transcriptionally. The expression of miRNAs has been linked to carcinogenesis because they can act as either oncogenes or tumor-suppressor genes.17 An important feature of epigenetic mechanisms and their role in development and disease is that epigenetic processes can be modified by lifestyle, particularly diet and the environment, pharmacologic interventions, or both.18 Data on aging also have shown to affect DNA methylation in many cell types in various organisms.19–21 Significant evidence for environment-lifestyle as the major contributor to these age-related effects on the “epigenome” involves a recent study of monozygotic (MZ) twins. Cell types and patterns of DNA methylation across the genome were similar in young MZ twins, but the patterns diverged in older twins.22 These data suggest that environmental-lifestyle factors act on individuals throughout life, changing gene expression through epigenetic mechanisms with subsequent implications for health.
Nutrition has become a major focus because it influences DNA methylation in several ways (see p. 405). Biologically active food components modify DNA methylation directly (see p. 406). Nutrition influences metabolic effects associated with energy balance. Because adipose tissue is endocrine tissue, obese individuals accumulate macrophages that secrete various proinflammatory signaling molecules and cytokines (see p. 414). Inflammation is strongly associated with cancer development, and inflammatory bowel disease is related to methylation in the colon.23 The role of nutrition and diet is discussed on p. 405.
In Utero and Early Life Conditions
From studies of the etiology of certain cancers, it is widely accepted that a long latency period precedes the onset of adult cancers. Accumulating data suggest early life events influence later susceptibility to certain chronic diseases.24 Developmental plasticity is the degree to which an organism’s development is contingent on its environment. It requires stable gene expression that in part appears to be modulated by epigenetic processes such as DNA methylation and histone modification (Figure 12-1).24 Sensitivity to environmental-lifestyle factors influences the mature phenotype and is dependent on the interactions of both the genome and epigenome.
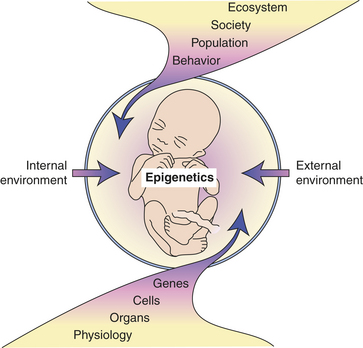
Figure 12-1 Fetal vulnerability to external and internal environments. The fetus is particularly vulnerable to changes in the external and internal environments, which can have immediate and lifelong consequences. Such environmentally induced changes can occur at multiple levels, including molecular and behavioral. Ultimately these alterations may be epigenetic, inducing mitotically heritable alterations in gene expression without changing the DNA. (Adapted from Crews E, McLachlan JA: Endocrinology 147[6 Suppl]: S4-S10, 2006.)
Perhaps one of the best examples of early life events and future cancer is the chemical exposure to diethylstilbestrol (DES), a synthetic estrogen. This medication was prescribed between 1938 and 1971 to attempt to prevent multiple pregnancy-related problems, such as miscarriage, premature birth, and abnormal bleeding.25 In 1953, a clinical study found that DES did not reduce the risk of miscarriage, and by the 1950s it became clear that DES interfered with the development of the reproductive system in the fetus. Recent data suggest that DES-associated increase in clear cell adenocarcinoma is elevated throughout a woman’s reproductive years.26 More recent studies have revealed that daughters of women who took DES during pregnancy may have a slight increased risk of breast cancer before age 40 (i.e., 1.9 times the risk compared with unexposed women at age 40).27 For every 1000 DES-exposed women ages 45 to 49 it is estimated that 4 will be diagnosed with breast cancer.
Research from animal studies has demonstrated a relationship between DES exposure and an increased rate of a rare type of testicular cancer (rete testis) and prostate cancer.28,29 In terms of in utero exposures, testicular cancer has been linked to exposure to abnormal levels of estrogen,30 and testicular cancer is a risk factor for men with undescended testicles, a factor in some studies correlated with DES exposure. However, studies in humans for risk of testicular or prostate cancer and DES exposure are unclear and continuing.31
In summary, epidemiologic and animal studies reveal that small changes in the developmental environment can alter phenotypic changes, resulting in individual responses in adulthood. Continuing evidence indicates that epigenetic mechanisms are responsible for tissue-specific gene expression during cellular differentiation and that these mechanisms modulate developmental phenotypic changes.24 The phenotypic effects of epigenetic modifications during development may need long latency periods, such as in cancer, thus manifesting later in life. In addition, epigenetic effects may help explain transgeneration effects. For example, Newbold and colleagues29 demonstrated in mice the occurrence of DES-related reproductive cancers also occurred in the grandsons and granddaughters of mothers treated with DES!
In addition to altering some cancer risks, investigators are studying diet during pregnancy (metabolic alterations, including obesity, diabetes, leptin and insulin resistance; hypertension; and vascular endothelial dysfunction). Recently, a striking experiment in mice demonstrated how extra vitamin doses during pregnancy in the mother’s diet changed the fur color of pups.32 This was the first study to show maternal nutrition and subsequent phenotype and disease. The nutrients (B12, folic acid, choline, and betaine) silenced the gene that rendered mice fat and yellow but did not alter its DNA sequence. Silencing, or switching the gene off, linked prenatal diet to such diseases as diabetes, obesity, and cancer. These concepts are defining the hypothesis of disease onset called the developmental basis of health and disease. Subsequently the focus of disease prevention and intervention needs to include the decades prior to onset—that is, in utero and neonatal periods.
Tobacco Use
Cigarette smoking is carcinogenic and remains the most important cause of cancer. The risk is greatest in those who begin to smoke when young and continue throughout life.33 Globally, tobacco use is greatest in developing countries, where 84% of 1.3 billion current smokers live.34 The World Health Organization (WHO) World Cancer Report35 reports that in the twentieth century approximately 100 million people died worldwide from tobacco-associated diseases (cancer, chronic lung disease, cardiovascular disease, and stroke). About 50% of regular smokers are killed by the habit. About 25% of smokers will die prematurely during middle age (35 to 69 years).35
Cigarette smoking accounts for 1 of every 5 deaths each year in the United States.36 About 21% of all U.S. adults smoke cigarettes. Estimates of cigarette smoking by age are 23.6% ages 18 to 24, 23.8% ages 25 to 44, 22.4% ages 45 to 64, and 8.8% ages 65 and older.37 Cigarette smoking is more common among men (23.9%) than women (18.5%), and the prevalence of cigarette smoking is highest among Native Americans/Native Alaskans (32.4%) than whites (22.2%), blacks (20.2%), Hispanics (15%), and Asians (10.4%).37 It is more common among adults living below the poverty level (30.6%) than those at or above the poverty level (20.4%).37
Overall, cigarette smoking in developed countries is responsible for 30% of all cancer deaths, and an epidemic of cancer deaths is expected in developing countries.35 Tobacco use is associated primarily with squamous and small cell adenocarcinomas. In addition, smoking causes even more deaths from vascular, respiratory, and other diseases than from cancer. Smoking tobacco is linked to cancers of the lower urinary tract (renal, penis, and bladder), upper aerodigestive tract (oral cavity, pharynx, larynx, nasal cavity, paranasal sinuses, esophagus, and stomach), liver, kidney, pancreas, cervix, and uterus, as well as myeloid leukemia.38 Evidence is lacking that smoking causes breast, prostate, or endometrial cancer of the uterus.35 However, tobacco smoke is known to cause mammary tumors in animals. In 2005, Japanese researchers reported that both active and passive smoking increased the risk of breast cancer in pre-menopausal women.38a
Secondhand smoke, also called environmental tobacco smoke (ETS), is the combination of sidestream smoke (burning end of a cigarette, cigar, or pipe) and mainstream smoke (exhaled by the smoker). More than 4000 chemicals have been identified in mainstream tobacco smoke (250 chemicals as toxic)39 of which 60 are considered carcinogenic.40 Measuring secondhand smoke is difficult. Nonsmokers who live with smokers are at greatest risk for lung cancer as well as numerous noncancerous conditions.40
Cigar or pipe smoking, or both, is strongly and causally related to cancers of the oral cavity, oropharynx, hypopharynx, larynx, esophagus, and lung. Cigar smokers who inhale deeply may be at increased risk for developing coronary heart disease and chronic obstructive pulmonary disease.41 Pipe smokers have a lower risk of dying from tobacco than cigarette smokers, but it is as harmful as and perhaps more harmful than cigar smoking.42 Bidi smoking, a small amount of tobacco wrapped in the leaf of another plant (used in South Asia), delivers higher amounts of nicotine per gram of tobacco and comparable or greater amounts of tar compared with cigarettes.38 Case-controlled studies indicate bidi smoking can cause cancers of the respiratory and digestive sites. Epidemiologic data from the United States and Asia reveal an increased risk of oral cancer with smokeless tobacco products.43 These data, however, are not confirmed in northern European studies.43
Measures that prevent young adults from starting smoking would substantially avoid future disease burden. A public health approach is therefore needed that prevents young people from starting smoking and helps others stop smoking.
Diet
Understanding dietary factors that increase the risk for cancer can be difficult. The ways in which diet affects one’s likelihood of developing cancer are complicated by the variety of foods consumed, the many constituents of foods, the metabolic consequences of eating, and the temporal changes in the patterns of food use. Cancer risks in older adults may depend as much on diet in early life as on current eating practices.33,44 In addition, studies in humans targeting diet and disease associations face a variety of challenges including measurements of specific nutrients, food types, and dietary patterns.
Humans are constantly exposed to a variety of compounds termed xenobiotics (Greek xenos, “foreign”; bios, “life”) that include toxic, mutagenic, and carcinogenic chemicals. Many of these chemicals are found in the human diet. Most xenobiotics are transported in the blood by lipoproteins and penetrate lipid membranes. These chemicals can react with cellular macromolecules, such as proteins and DNA, or can react directly with cell structures to cause cell damage.45 The body has two defense systems for counteracting these effects: (1) detoxification enzymes and (2) antioxidant systems (see Chapter 2). Enzymes that activate xenobiotics are called phase I activation enzymes and are represented by the multigene cytochrome P-450 family, aldehyde oxidase, xanthine oxidases, and peroxidases. Phase II detoxification enzymes then protect further against a large array of reactive intermediates and nonactivated xenobiotics.45 These enzymes are located predominantly in the liver and provide clearance of compounds through the portal circulation, thereby preventing the potentially carcinogenic agent(s) from entering the body through the gastrointestinal tract and portal circulation. These enzymes also occur in the skin epithelia and can be induced in other extrahepatic tissue, such as the lung.
Dietary sources of carcinogenic substances include compounds produced in the cooking of fat, meat, or protein, and naturally occurring carcinogens associated with plant food substances, such as alkaloids or mold byproducts.45 The most studied and most relevant carcinogens produced by cooking are the polycyclic aromatic hydrocarbons benzo[a]pyrene and heterocyclic aromatic amines generated by meat protein. The greatest levels are found in well-done charbroiled beef. People, likewise, ingest xenobiotics that are found in environmental or industrial contaminants (e.g., particulate matter of diesel exhaust, contaminating pesticides in food and water supplies) and in certain prescribed and over-the-counter medicines.
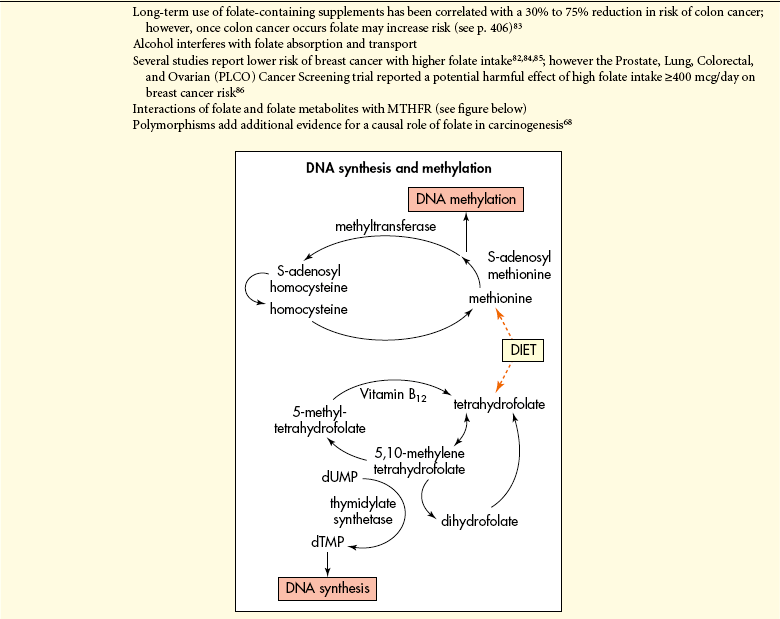
Nutrition may directly influence epigenetic factors that silence genes that should be active or activate genes that should be silent.46 Dietary components can act directly as mutagens or interfere with mutagen elimination. Nutritional factors may alter cellular environments by modulating hormonal axes or influencing cellular proliferation, or both.46 Importantly, specific nutrients may directly affect the phenotype or expression of key genes, for example, epigenetically through abnormalities of methylation of the promoter regions of genes or histones. These alterations can affect DNA structure and mRNA for transcription.46 Clearly, epigenetic events are susceptible to change, thus offering potential explanations of how environmental factors (e.g., diet) may modify cancer risk and tumor behavior. DNA methylation—the attachment of a methyl group to the 5-position of cytosine within cytosine guanine dinucleotides (CpGs)—is one of several epigenetic changes important in gene regulation and expression (Figure 12-2). CpGs are distributed evenly throughout the genome and remain in short stretches, or clusters, called CpG islands. These islands are located in the promoter region of genes found in half of all human genes.47 DNA is also susceptible to hypomethylation, which can cause overexpression of transcription of proto-oncogenes, increased recombination and mutation (i.e., oncogenes), and failure to imprint. These alterations can all promote cancer.48 Aberrant DNA methylation patterns occur in several cancers (colon, lung, prostate, and breast). Dietary factors may be related to DNA methylation in four ways: (1) they may influence the supply of methyl groups for the formation of S-adenosylhomocysteine (SAM) (see p. 411); (2) they may modify the use of methyl groups, e.g., by shifts in an important enzyme, DNA methyltransferase (DNMT) (see p. 411); (3) they may reduce methyl groups called demethylation; and (4) DNA methylation patterns may influence the response to a dietary factor.49
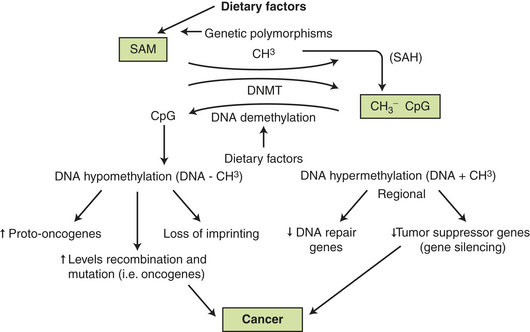
Figure 12-2 Dietary factors, DNA methylation, and cancer. Certain dietary factors (see Table 12-5) may supply methyl groups (+CH3) that can be donated through S-adenosylmethonine (SAM) to many acceptors in the cell (DNA, proteins, lipids, and metabolites). Donation and removal (demethylation) are affected by numerous enzymes, including DNA methyltransferase (DNMT). Increased DNMT activity is known to occur in many tumor cells. Hypermethylation can inhibit or silence tumor-suppressor genes (see Chapter 11) and DNA methylation inhibitors as anticancer agents can block DNMT and, thus, reactivate tumor-suppressor genes. DNA hypomethylation can reactivate and mutate genes, including cancer-causing oncogenes. SAH, S-adenolyhomocysteine.
Specific nutritional factors seem to influence susceptibility to cancer. Continuing studies are determining whether certain deficiencies, such as vitamin D and compounds found in fruits and vegetables, can increase cancer incidence.48 Excess intake of alcohol can promote cancer (see p. 415). Aflatoxin (produced by mold) can contaminate corn, peanuts, and rice stored in hot, humid environments, and Chinese-style salted fish are known to cause cancer. With emphasis on epigenetic and aberrant methylation (Figure 12-3) the role of folic acid in cancer is prominent. The methyl group of 5-methylenetetrahydrofolate is the precursor of methyl group of methionine and eventually SAM (see Table 12-5, p. 410). Cancer cells reveal both genome overall (global) hypomethylation and regional hypermethylation (see Figure 12-2). Global hypomethylation in cancer is widely observed and can lead to genomic instability.50 Regional hypermethylation has been observed in various cancers and when it involves tumor methylation can cause their inactivation or silencing (see Figure 12-2). Low intake of folate combined with high intake of alcohol is associated with global hypomethylation and colorectal cancer. Aberrant folate metabolism leading to DNA methylation is prevalent in the highly aggressive HER2/neu-positive breast cancers.51 Regional hypermethylation and gene silencing are found in the majority of invasive breast lobular carcinomas.52 Aberrant methylation is progressively acquired in the early stages of colorectal cancer.53 The role of folic acid in cancer is complicated. For example, low intake of folic acid is correlated with an increased risk of colorectal cancer. Uracil is misincorporated into DNA as a result of folate deficiency.48 However, it is important to note that once a cancerous lesion is present folate intake may increase tumor growth.54
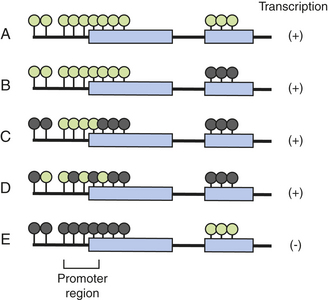
Figure 12-3 Methylation of a gene region and its effect on gene transcription. Green circles, unmethylated cytosine guanine dinuceotide (CpG) sites; (A) and dark circles, methylated CpG sites. Methylation of exons (B, C) does not block gene transcription. Mosaic methylation of promoter CpG island also does not block transcription (D). However, dense methylation of promoter CpG islands completely blocks transcription, and is often associated with hypomethylation of downstream regions (E). (From Ushijima T: J Biochem Mol Biol 40[2]:143, 2007.)
Selective dietary agents that inhibit histone deacetylase (HDAC) and, therefore, abnormal patterns of histone modification include sulforaphane (SFN). SFN is an isothiocyanate found in cruciferous vegetables, such as broccoli and broccoli sprouts. A growing body of evidence suggests that SFN acts through epigenetic mechanisms to inhibit HDAC activity in human colon and prostate cancer lines.55 In human subjects, a single ingestion of 68 g (1 cup) of broccoli sprouts inhibited HDAC activity in circulating white blood cells 3 to 6 hours after eating.56
In summary, epidemiologic and laboratory evidence suggests that diet is a significant factor in the cause, progression, and prevention of cancer (Table 12-3). Diet affects many pathways to cancer including cell cycle control, differentiation, DNA repair, gene silencing, inflammation, apoptosis, and carcinogen metabolism. Many of these processes are likely influenced, if not regulated, by DNA methylation, an epigenetic mechanism that affects gene function. Imbalances of nutrients can lead to global hypomethylation, and there is reason to believe that diet can affect gene-specific hypomethylation or hypermethylation, or both.49 Much more research is needed to identify how specific nutrients can alter DNA methylation and restore gene function as well as other pathways (i.e., apoptosis, etc.) that can prevent tumor development. Studies on diet and cancer need testing in a variety of settings and among different population groups.
Table 12-3
Relationship of Dietary Factors with Risk of Major Cancersa
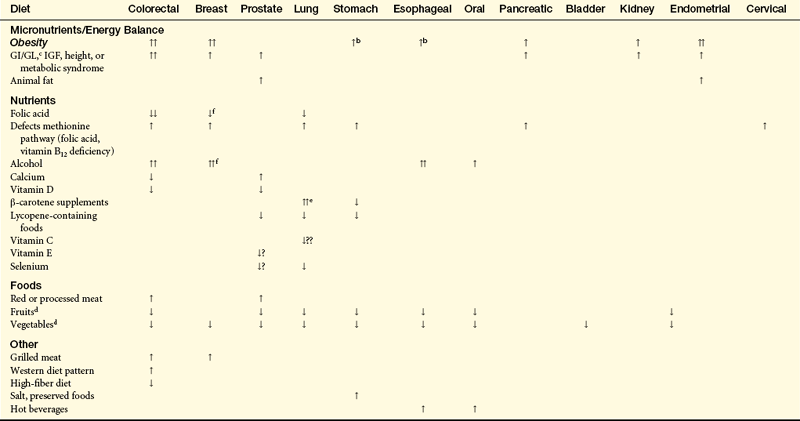
IGF, Insulin-like growth factor.
aTwo arrows indicate more consistent evidence.
bCancers of the gastric cardia.
cGI/GL, glycemic index/glycemic load.
dEvidence for a potential benefit from some components of fruits and vegetables (not necessarily blanket effect).
eIncreased risk limited to smokers.
fData support hypothesis that higher folate intake reduces estrogen receptor–breast cancer, particularly important with alcohol consumption.
From Koushik A et al: J Natl Cancer Inst 99(19):1471–1483, 2007; McCullough ML, Giovannucci EL: Oncogene 23(38):6349–6364, 2004; Zhang SM et al: Cancer Epidemiol Biomarkers Prev 14(8):2004–2008, 2005.
Obesity
Obesity in most developed countries (and in urban areas of many developing countries) has been increasing rapidly over the past 20 years (Figure 12-4). The only globally accepted criteria for overweight-ness and obesity are based on body-mass index (BMI). Widely accepted standards based on BMI criteria for overweight-ness and obesity are recommended by the WHO54 and supported by other panels and federal agencies (WHO classifications are shown in Table 12-4).
Table 12-4
WHO Classification of Body Mass Index (BMI)
| BMI (kg/m2)∗ | WHO Classification | Other Descriptions |
| <18.5 | Underweight | Thin |
| 18.5-24.9 | Normal range | “Healthy,” “normal,” or “acceptable” weight |
| 25-29.9 | Grade 1 overweight | Overweight |
| 30-39.9 | Grade 2 overweight | Obesity |
| ≥40 | Grade 3 overweight | Morbidly overweight |
WHO, World Health Organization.
∗The cutoffs are somewhat arbitrary, although they are derived from epidemiologic studies of BMI and overall mortality. It is important to understand that within each category of BMI there can be substantial individual variation in total and visceral adiposity and in related metabolic factors. These variations are also true for the normal range BMI.
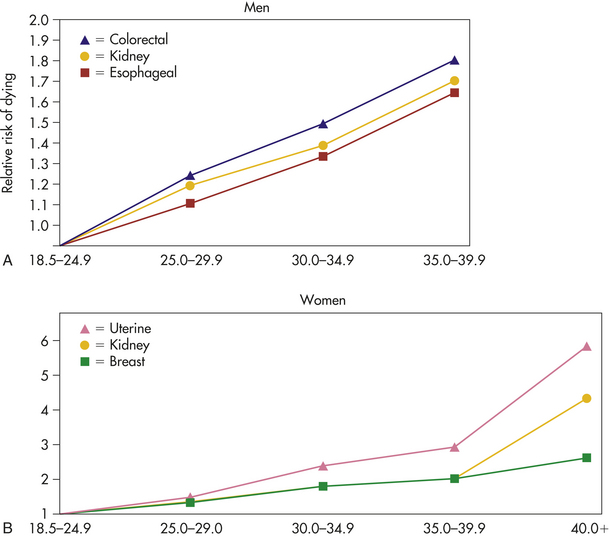
Figure 12-4 Weight and risk of dying from cancer. A, As a man’s body mass index (BMI) rises above the normal range (18.5 to 24.9), his risk of dying of colorectal, esophageal, kidney, and other cancers also rises. For example, the risk of dying of colorectal cancer is 10% higher for men who are overweight (BMI 25 to 29.9) than for men of normal or lower BMI. For the most obese men (BMI 35 or higher) the risk is almost double (84%). B, As a woman’s BMI rises above the normal range (18.5 to 24.9), her risk of dying of breast, kidney, uterine, and other cancers rises. For example, the risk of dying of breast cancer is 34% higher for women who are overweight. For the most obese women (BMI >40), the risk of dying of breast cancer is double. The risk of kidney disease is almost five times higher, and the risk of uterine cancer is six times higher. (Data from Calle EE et al: N Engl J Med 348:1625-1638, 2003.)
Studies in the United States have suggested that obesity associated with some cancers can increase mortality rates.57 Because obesity is associated with other chronic diseases such as cardiovascular and diabetes, it can increase overall mortality, though it may not be a causal factor involved in cancer mortality—yet obesity was found to be related to increased incidence of several cancer types. A recent hypothesis states that the observed increased incidence of such cancers as breast, endometrium, colon, liver, kidney, and the esophagus may be associated with obesity.58,59
A large prospective study of 900,000 American adults showed obesity is linked to cancer. Starting with a mean age of 57 years, individuals were followed for 16 years and cancer mortality data were collected during that interval.57 Compared with men whose BMI was in the normal range (18.5 to 24.9), men with substantial obesity (BMI ≥40) showed significant increases in cancer mortality. Women had a similar risk. Although significant, people with lesser degrees of obesity had slighter increases in cancer mortality. In men with higher BMI, there were higher rates of death from esophageal, stomach, colorectal, liver, gallbladder, pancreatic, prostate, and kidney cancers and non-Hodgkin lymphoma, multiple myeloma, and leukemia.57 Among women, high BMI correlated to greater morbidity from colorectal, liver, gallbladder, pancreatic, breast, uterine, cervical, ovarian, and kidney cancers and from non-Hodgkin lymphoma and multiple myeloma.
The mechanisms of obesity-associated cancer risks are unclear and may vary by type of tumor and distribution of body fat. Abdominal obesity, as defined by waist circumference or waist/hip ratio, has been shown to be more strongly related to some tumor types than obesity as defined by BMI.58 Possible associated mechanisms include insulin resistance and resultant chronic hyperinsulinemia, increased insulin-like growth factors (IGFs), or increased steroid hormones, increased tissue-derived hormones, and cytokines (adipokines) or inflammatory mediators.58
Endogenous Hormones: Three mechanisms involve how adiposity influences the synthesis and bioavailability of endogenous sex steroids, the estrogens, progesterone, and androgens (see Figure 12-6):
1. Adipose tissue expresses various sex-steroid metabolizing enzymes that promote the formation of estrogens from androgenic precursors (secreted by the gonads and adrenal glands).
2. Adipose cells increase the circulating levels of insulin and increase IGF-1 biologic activity. This results in reduced liver synthesis and blood levels of sex hormone–binding globulin (SHBG), a binding hormone with affinity for estradiol and testosterone. The adiposity-related decrease in SHBG increases bioavailable estradiol in men and women. In women, decreased SHBG also leads to increased levels of testosterone; in men, contrarily, decreases in SHBG generally lead to reduction in total testicular testosterone production and no increase in bioavailable testosterone.
3. High insulin levels can increase ovarian and, possibly, adrenal androgen synthesis and in some genetically susceptible premenopausal women cause the development of polycystic ovary syndrome (PCOS).60 PCOS is characterized by ovarian hyperandrogenism, chronic anovulation, and progesterone deficiency. PCOS is relatively common with an estimated prevalence of 4% to 6%.61
Epidemiologic evidence shows that adiposity-induced alterations in blood levels of sex steroids could explain the correlation noted between indices of excess weight and risks of breast cancer (postmenopausal only) and endometrial cancer (both pre- and postmenopausal).61
For breast and endometrial cancers, estrogen and progesterone play a central role, as established by a large body of experimental and clinical evidence. These sex steroids are important regulators of cellular proliferation, differentiation, and apoptosis. Further evidence that increased endogenous estrogen levels might drive the association between BMI and breast cancer comes from studies of hormone replacement therapy (HRT). BMI is more strongly related to breast cancer incidence among postmenopausal women who have never received HRT compared with women who have.62–64 Possibly, only in women whose levels of estrogen are low (after menopause with no HRT) does increased adiposity and, therefore, an increase in peripheral aromatase activity by androgenous activity and androgenous estrogen production, lead to an increase in breast cancer risk. In addition, mortality is higher among heavier women than among leaner women.61,65
Several case-control and prospective studies have reported increased risks among pre- and postmenopausal women who have lower blood levels of SHBG and thus high levels of androgens and testosterone. Among postmenopausal women, risk is also correlated to levels of estrone and total bioavailable (free) estradiol.66
Among men, prostate carcinogenesis is thought to be related to endogenous hormone metabolism, including androgen production and, possibly, estrogens. Yet excess weight does not appear to be a prominent risk factor except with advanced disease.67 Other dietary factors and the outcome of studies for cancer risk are presented in Table 12-5. Hormones and cancer are discussed in Chapter 23.






Biologic Mechanisms: Although hypothetical, the role of obesity and cancer may involve alterations in the hormonal milieu. Adipose tissue is active endocrine and metabolic tissue and can have greater effects on the physiology of other tissue (see Chapter 39). In response to endocrine and metabolic signals from other organs, adipose tissue responds by increasing or decreasing the release of free fatty acids—fuel for skeletal muscle and other tissues. When triglycerides, the main storage lipid, are metabolically hydrolyzed, they release free fatty acids into the blood. Abdominal visceral adipocytes are more metabolically active than abdominal subcutaneous adipocytes and because visceral adipocytes have high lipolytic activity and release large amounts of free fatty acids, accurate measurements of adiposity must consider the amount and the site of deposition of the adipose tissue. Adipose tissue is very important in the regulation of energy balance and lipid metabolism through the release of peptide hormones, including leptin, adiponectin, resistin, and tumor necrosis factor-gamma (TNF-γ) (Figure 12-5). Increased release of free fatty acids, resistin, and TNF-γ by adipose tissue and reduced release of adiponectin give rise to insulin resistance—a state characterized by the reduced metabolic response of tissues (muscle, liver, adipose tissue) to insulin and to compensatory hyperinsulinemia61 (Figure 12-5). Involvement of signaling pathways (i.e., AKT, JAK/STAT) and other inflammatory cascades (i.e., nuclear factor kappa beta [NF-кβ]) has been linked with obesity and cancer.116 Adipose tissue cells also produce various steroid-hormone-metabolizing enzymes and are an important source of circulating estrogens in postmenopausal women (see Figure 12-6).
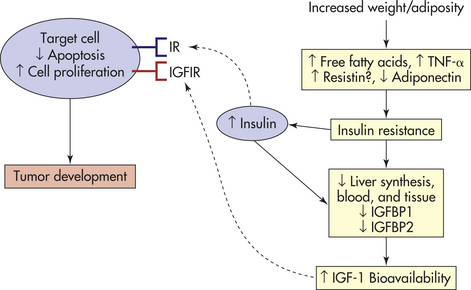
Figure 12-5 Energy balance, lipid metabolism, and insulin sensitivity and tumor development. In obesity, increased release from adipose tissue of free fatty acids (FFAs), tumor necrosis factor-alpha (TNF-α) and resistin, and reduced release of adiponectin lead to insulin resistance and compensatory chronic hyperinsulinemia. Increased insulin levels ultimately lead to decreased liver synthesis and blood levels of insulin-like growth factor–binding protein-1 (IGFBP1) and, theoretically, also decrease IGFBP1 synthesis locally in other tissues. Increased fasting levels of insulin in plasma are also correlated with decreased levels of IGFBP2 in the blood, leading to increased levels of bioavailable IGF-1. Insulin and IGF-1 signal through the insulin receptors (IRs) and IGF-1 receptor (IGF1R) to stimulate cellular proliferation and inhibit apoptosis in many tissue types. These effects could promote tumor development. (Adapted from Calle EE, Kaaks R: Nat Rev Cancer 4[8]:579-591, 2004.)
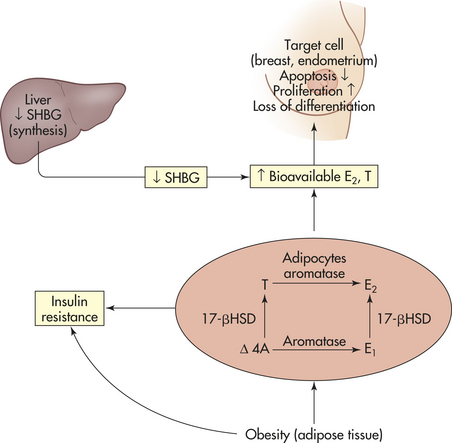
Figure 12-6 Effects of obesity on hormone alterations. Adipose tissue produces the enzymes aromatase and 17-beta hydroxysteroid dehydrogenase (17-βHSD). In obese persons, therefore, there is an increased conversion of the androgens Δ4-androstenedione (Δ4A) and testosterone (T) into the estrogens estrone (E1) and estradiol (E2), respectively, by the enzyme aromatase. The important enzyme 17-βHSD converts the less biologically active hormones Δ4A and E1 into the more active hormones T and E2, respectively. In parallel, obesity leads to hyperinsulinemia, which causes a decrease in the liver synthesis and blood circulating levels of sex hormone-binding globulin (SHBG). The combined effect of increased synthesis of estrone and testosterone, along with reduced levels of SHBG (their transporter), leads to an increase in the bioavailablity (or free fractions) of E2 and T that can diffuse to target tissue, where they bind to estrogen and androgen receptors. Binding to their respective receptors in some tissue (e.g., breast and endometrium) promotes cellular proliferation and inhibits apoptosis. Thus they can increase tumor development. In both men and women, adiposity-related decreases in SHBG generally increase the fraction of bioavailable or free estradiol. In contrast, decreases of SHBG in men only generally lead to reductions in testicular production of testosterone and no increase in bioavailable testosterone. (Adapted from Calle EE, Kaaks R: Nat Rev Cancer 4[8]:579-591, 2004.)
Excess weight, increased plasma triglyceride levels, low levels of physical activity, and certain dietary factors can contribute to chronic hyperinsulinemia. Chronically increased insulin levels have been correlated with the development of colon, breast, pancreatic, and endometrial cancers67,117,118 (see Figure 12-6). These pathogenic effects of insulin might be mediated by insulin receptors in the preneoplastic or neoplastic target cells or could be due to alterations in endogenous hormone metabolism secondary to hyperinsulinemia. For example, insulin promotes the synthesis and biologic activity of the growth factor insulin-like growth factor-1 (IGF-1). IGF-1 is a peptide hormone with a molecular
structure similar to insulin that regulates cellular proliferation in response to available energy and nutrients from diet and body constituents (see Figure 12-6).119 Insulin also can promote the synthesis and biologic availability of the male and female sex hormones, including estrogens, progesterone, and androgens61 (see Figure 12-6).
In vitro studies have established that insulin and IGF-1 act as growth factors that promote cell proliferation and inhibit apoptosis.120–123 Epidemiologic evidence supports the hypothesis that chronic hyperinsulinemia increases cancer risk. Type 2 diabetes mellitus associated with insulin resistance and increased pancreatic insulin secretion for long periods before and after disease onset is associated with increased risks of cancer of the colon, endometrium, kidney, and pancreas.67,118,124,125 Prospective cohort studies have shown increased risk of cancers of the colon or colorectum among individuals with increased prediagnostic blood levels of C-peptide (a marker for pancreatic insulin secretion), fasting glucose levels, or insulin measured 2 hours after absorption of a standard oral dose of glucose.126 Similar data have found a direct relationship between cancer risk and prediagnostic C-peptide levels for endometrial cancer.66 The study also found inverse relationships between cancer risk and blood levels of IGF-binding protein 1 (IGFBP1) and IGF-binding protein 2 (IGFBP2),66 which reduce the amount of bioavailable IGF-1. However, a case-control study nested within the European Prospective Investigation found the C-peptide risk association was substantially decreased after adjustment for free estradiol in postmenopausal women.127 The Physicians Health Study reported the first association between plasma insulin levels or C-peptide before prostate cancer diagnosis (prediagnostic) and the risk of prostate cancer mortality.128 This finding suggests excess body weight and a high plasma level of C-peptide predispose men to the development of prostate cancer and to an increased likelihood of dying from their disease. More studies are needed to confirm these findings.
The IGFBPs regulate the availability of IGF-1 because they stabilize the large pool of IGF-1 in the circulation, the efflux of IGF-1 from this circulation pool toward target tissues and binding of IGF-1 to its receptor.61 IGFBP3 increases tissue apoptosis, so decreased amounts may contribute to carcinogenesis.
More than 80% of IGF-1 is provided by growth hormone (GH). In overnourished states and in individuals with type 2 diabetes mellitus, endogenous insulin levels and liver GH-receptor levels are high and large amounts of IGF-1 are produced.61 Contrarily, however, obese individuals have lower blood levels of IGF-1 than normal weight individuals.129 A compelling explanation for the lower levels of IGF-1 in obese individuals, despite increased GH sensitivity of liver and other tissues, is that reduction in IGFBP1 and IGFBP2 levels leads to increased negative feedback by free IGF-1 (unbound to IGFBPs) on pituitary gland secretion of GH. Overall, this feedback results in reduced synthesis of IGF-1 and reduced plasma IGF-1 concentrations.
IGF-1 has been shown to promote proliferation of normal epithelial breast cells.119 The IGF-signaling pathway has been linked to breast carcinogenesis in animal studies,130 and previous epidemiologic studies have reported increased blood levels of IGF-1 as directly related to different forms of cancer. These studies reported that high levels of IGF-1 and low levels of IGFBP3 are correlated with increased risk of premenopausal breast cancer.131,132 Similar risks were reported for cancers of the prostate and colorectum.133–136 An analysis for the Breast and Prostate Cancer Cohort Consortium (BPC3) from six large cohorts found no association between common genetic variations in the IGF-1, IGFBP1, and IGFBP3 genes in relation to circulating levels of IGF-1 and IGFBP3 and breast cancer risk (also no effect by menopausal status).
From the epidemiologic studies, breast and prostate cancer showed no clear relationship with BMI or other indices of adiposity. Colon cancer, however, did show positive relationships with BMI and other indices of adiposity. In addition, there was no clear linear relationship between circulating levels of IGF-1 and the degree of adiposity.61
Alcohol Consumption
Chronic alcohol consumption is a strong risk factor for cancer of the oral cavity, pharynx, hypopharynx, larynx, esophagus, and liver.137 Although evidence is inconsistent, alcohol consumption is less strongly related to breast cancer and colorectal cancer; however, it is known to increase cell growth of human breast cancer cells in vitro.138 In addition, although the risk is lower, breast carcinogenesis can be enhanced with relatively low daily amounts of alcohol.137 A meta-analysis showed no consistent relationship between alcohol and cancers of the pancreas, lung, prostate, or bladder.139 Alcohol interacts with smoke, increasing the risk of malignant tumors, possibly by acting as a solvent for the carcinogenic chemicals in smoke products. In individuals who have never smoked, substantial alcohol consumption (i.e., three or more drinks per day) has been associated with head and neck cancers.140 Inherited factors also put some individuals at increased risk in the ability to repair DNA, carcinogen metabolism, and cell cycle control.141 The strongest genetic associations to alcoholism are those with alcohol dehydrogenase (ADH) and mitochondrial aldehyde dehydrogenase (ALDH2). Specifically, individuals having the genes encoding 32-ADH or the dominant negative allele for ALDH2 are at reduced risk of alcoholism, despite being at much higher risk for oropharyngeal cancer.142
Mechanisms involved in alcohol-related carcinogenesis include the effect of acetaldehyde, the first metabolite of ethanol oxidation; the induction of cytochrome P-450 2E1 (genetic variant CYP2E1) leading to the generation of reactive oxygen species (ROS); increased procarcinogen activation (e.g., nitrosamines) and modulation of cellular regeneration (cell cycle); and nutritional deficiencies (retinol, retinyl esters, folic acid, other vitamins). Nutritional deficiencies may give rise to altered mucosal integrity, enzyme and metabolic dysfunction, and other structural abnormalities. The median age for diagnosis is the early 60s, with a male predominance, especially in laryngeal cancer.140
Ionizing Radiation
Much of the knowledge of the effects of ionizing radiation on human cancer has stemmed from observations of the Hiroshima and Nagasaki atomic bomb exposures, particularly the Life Span Study. These data provide the best estimate of human cancer risk over the dose range from 20 to 250 cGy for low linear energy transfer (LET) radiation, such as x-rays or γ-rays. Other data are derived from groups exposed for medical reasons (Table 12-6), underground miners exposed to radon gas, and workers exposed to high doses while in the nuclear weapons program of the former USSR. The atomic bomb exposures in Japan caused acute leukemias in adults and children and increased frequencies of thyroid and breast carcinomas. Lung, stomach, colon, esophageal, and urinary tract cancers and multiple myeloma have been added to the list. At Nagasaki and Hiroshima, leukemia incidence in individuals 15 years or younger reached its peak 6 to 7 years after the explosions and has steadily declined since 1952. People 45 years and older at the time of exposure had a latent period of 20 years before developing acute leukemia.
Table 12-6
Estimated Doses of Ionizing Radiation from a Single Exposure During Certain Diagnostic Procedures
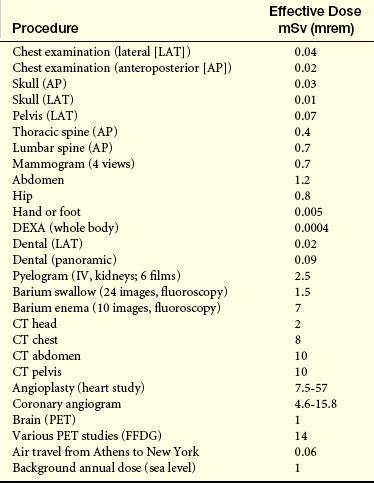
CT, Computed tomography (CAT scan); FFDG, nuclear medicine with F-fluorodeoxyglucose; IV, intravenous; PET, positron-emission tomography.
Data from Health Physics Society: Diagnostic imaging procedures. Available at www.hps.org/documents/mediaimaging.pdf. Accessed June 2008.
Human exposure to ionizing radiation includes emissions from x-rays, radioisotopes, and other radioactive sources. Health risks involve not only neoplastic diseases but also cardiovascular disease and stroke following high doses in therapeutic medicine and lower doses in A-bomb survivors (BEIR VII).143,144 Late effects of radiation in A-bomb survivors show persistent elevations of inflammatory markers (e.g., interleukin [IL]-6, CRP, TNF-α, etc.) implying immunologic damage may be the cause of later cardiovascular effects.145 Other risks include somatic mutations that may contribute to other diseases (e.g., birth defects and eye maladies) and from animal studies, inherited mutations that may affect the incidence of diseases in future generations (see What’s New? Radiation and Vulnerable Populations). Heritable mutations are of particular concern for women because the number of oocytes are presumably fixed at birth and mutations, if not repaired, are cumulative (see p. 421).146 An important summary point in BEIR VII143 is the concern from high-dose medical exposure, for example computed tomography (CT) (see What’s New? Increasing Use of Computed Tomography Scans and Risks). In 2009, the National Council on Radiation Protection and Measurements147 reported Americans were exposed to more than seven times as much ionizing radiation from medical procedures compared to the 1980s (Figure 12-7).
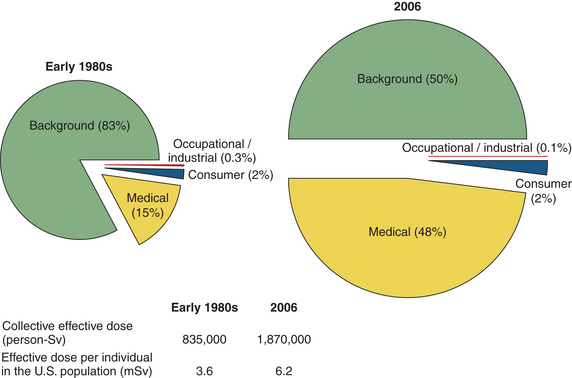
Figure 12-7 Ionizing radiation exposure of the population of the United States. (NCRP report no. 160.)
The risks of low-dose radiation are being debated among radiobiologists, geneticists, physicists, and others because of the potential effect on the health of current and future generations.146 Two opposing hypotheses have emerged: (1) there is no dose of radiation considered safe and the use of radiation must always be considered on the basis of risk versus benefit and (2) the health risks of diagnostic doses less than 10 cGy are not now measurable and may be nonexistent. Limiting is that general findings on the health risks of low-dose radiation are made by analyses of data on the risk of cancer alone.146 The expression of radiation-induced damage depends not only on dose, fractionation, and protraction but also on repair mechanisms, bystander effects, radioprotective substances such as antioxidants, and how it is delivered.146
Radiation-Induced Cancer
Ionizing radiation (IR) is a mutagen and carcinogen and can penetrate cells and tissues and deposit energy in tissues at random in the form of ionizations (e.g., excitation or removal of an electron from the target atom). These ionizations can lead to irreversible or indirect damage from formation and
attack by water-based free radicals (radiolysis).148 IR affects many cell processes, including gene expression, disruption of mitochondrial function, cell cycle arrest, and cell death. IR is a potent DNA damaging agent causing cross-linking, nucleotide base damage, and single- and double-strand breaks (Figure 12-8).149 Damage to DNA and disrupted cellular regulation processes can lead to carcinogenesis.149–151 The double-strand break (DSB) (see Figure 12-8) is considered the characteristic lesion observed for the effects of IR. In certain experimental systems, a single DSB may lead to cell cycle arrest and possible further repair. Yet many DSBs appear to result from clustered damage, a consequence of the pattern of distribution of ionizations with DNA. These patterns of clustered damage may be more difficult to accurately repair.148 Importantly, DSBs are mostly repaired by the nonhomologous end joining (NHEJ) pathway. This pathway is efficient for joining the DNA broken ends; however, errors can occur. Irradiated human cells unable to execute the NHEJ are supersensitive to the introduction of large-scale mutations and chromosomal aberrations.148
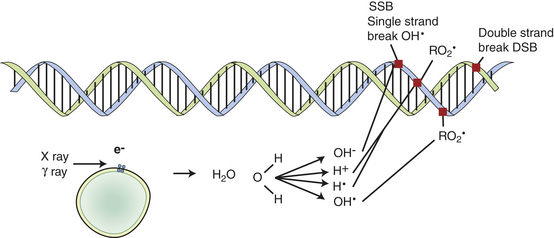
Figure 12-8 Free radicals. Free radicals formed by water nearby and around DNA cause indirect effects. These effects have a short life of single free radicals. Oxygen can modify reaction, enabling longer lifetimes of oxidative free radicals.
A long-held assumption is that cellular alterations—mutations and malignant transformation—occur only in cells directly radiated. It is now known that radiation may induce a type of genomic instability to the progeny of the directly irradiated cells over many generations of cell radiation, leading to an increased rate at which the genetic effects (i.e., mutations/chromosomal aberrations) arise in these distant progeny called transgeneration effects. The directly irradiated cells also can lead to genetic effects in so-called bystander cells or innocent cells (called bystander effects) even though they themselves received no direct radiation exposure.148 For example, using an in vivo mouse model, investigators found that localized radiation to the head led to induced bystander effects in the lead-shielded distant spleen tissue.152 The vast majority of bystander effects have been described in cell-culture systems. In vivo (i.e., in an organism) was reported in lead shielded mouse heads after radiation exposure of the remainder of the body (Mancuso et al 2008).153 These mice showed unexpected enhancement of medulloblastoma in cerebellum of radiosensitive Patched-1 (Ptch1) heterozygous mice. Both double-strand DNA breaks and apoptotic cell death were induced by bystander effects supporting the role of gap-junctional intercellular communication (GJIC, see p. 423). The bystander and genomic instability effects also have been termed “nontargeted” effects (see pp. 420-422). Although bystander and transgeneration IR effects are associated with induced genomic instability leading to chromosome aberrations, gene mutations, late cell death, and aneuploidy, all of these effects may be epigenetically mediated (see p. 401). The epigenetic changes include DNA methylation, histone modification, and RNA-associated silencing (see p. 403 and Chapter 11).152
Radiation-induced cancer in humans seems to have long latent periods; 10 years for leukemia and more than 30 years for solid tumors.154 This implies that radiation-induced gene mutations or chromosomal alterations that can be detected early (within 24 hours of radiation exposure) are not solely responsible for tumor development in normal human cells. Such mutations, however, provide a critical hit or induce genetic instability that makes cells more susceptible to accumulation of genetic alterations caused by other spontaneous or induced mutations. The accumulation of mutations leads to full transformation and cancer.148
The majority of evidence on radiation-induced human cancer risk is from epidemiologic studies of exposed populations. For example, an increase of total cancers after the Chernobyl radioactive fallout was reported in Sweden.155 Most direct data, however, are available only at relatively high doses (greater than 0.1 Gy) from mainly low-LET radiation (x- and γ-rays). Data, however, from cell-culture systems and in vivo mouse studies are emerging for low dose radiation. Radiation-induced cancers include some leukemias and lymphomas, thyroid cancers, some sarcomas, skin cancers, and some lung and breast carcinomas.
Constant debate involves risk estimates for human exposure at low-dose, low-LET ionizing radiation (0 to 100 mSv or less than 0.1 Gy). The problem is complicated because setting regulation levels at which levels of interest are so low that data endpoints—mutations and cancer—become difficult to measure with statistical significance144; thus theoretic models are used to estimate response curves. Several models include the linear no-threshold (LNT) relationship, in which any dose, including very low doses, has the potential to cause mutations (Figure 12-9). The threshold model proposes a threshold dose below which radiation may not cause cancer in humans. Proponents of this model argue that such thresholds derive, for example, from the ability to repair damage caused by lower doses of radiation. Another model, the linear-quadratic relationship, proposes there is a risk mathematical term that is directly proportional to the dose (linear term) and another proportional to the square of the dose (quadratic term) (see Figure 12-9). There is some evidence that low doses may actually produce a higher level of risk per unit of dose called the supra-linear hypothesis (see Figure 12-9).154 Currently, the shape of the response curve for the low-dose region is really unknown.144 In 1990, the Fifth Biological Effects of Ionizing Radiation Panel (BEIR V) estimated that the risk of radiation was considerably higher than prior official studies.154 It supported the LNT relationship for solid cancers and included estimates of cancer risk.
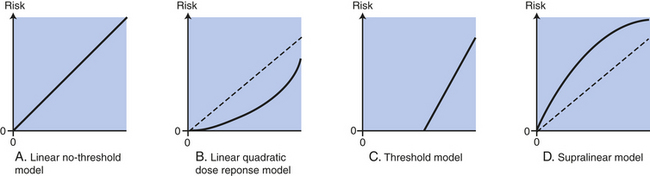
Figure 12-9 Theoretic models for estimating risk of low-dose ionizing radiation. Collective population dose is expressed as a person-rem (roentgen equivalent man) (see Table 12-7). Estimating a collective dose then enables an application of a “constant risk factor” to get a statistical estimate of the number of additional cancers (above background radiation) resulting from that exposure. These computations apply to low doses—low-dose rates only (A). Many propose the best fit is the Linear No-Threshold (LNT) model (B). The most common alternative to the LNT Model is the Linear-Quadratic Model. The quadratic term is the square of the dose. The linear term is equal to zero (C). The threshold model is a threshold below which there is no increase in cancer risk. Proponents of this model argue that some toxic chemicals/materials exhibit such thresholds and that radiation must, too. Their arguments are related to repair of the radiation damage caused by lower doses of radiation (D). Some evidence exists that low levels of radiation produce a higher level of risk per unit dose called the Supra-linear Model. (From Makhijani A, Smith B, Thorne MC: Science for the vulnerable: setting radiation and multiple exposure environmental health standards to protect those at most risk, IEER, Takoma Park, Maryland, 2006.)
Recent data, however, show that the LNT model underestimates the risk from low radiation.156 The researchers used a precision microbeam device to fire alpha particles into nuclei of human-hamster hybrid cells in Petri dishes. When the researchers irradiated the nuclei with just one alpha particle each, 98 mutations of a particular gene occurred per 100,000 surviving cells. Zapping only 5% of the nuclei produced 57 such mutations per 100,000 cells, rather than the 5 mutations that a linear model predicts. These data suggest that the relevant target for radiation-induced mutagenesis is larger than an individual cell and thus supports the need to reconsider the validity of the linear extrapolation model.156
An important conclusion from BEIR VII was hormesis, or proposed adaptive/stimulatory (beneficial) effects of low doses of ionizing radiation, that exceeds detrimental effects; it is unwarranted at this time and not addressed further.144 A summary of the BEIR VII estimates of cancer risk from low-level radiation changed from BEIR V and is included in Table 12-7. The risk to women is now estimated to be considerably higher.157 BEIR VII also has provided both estimates of risk of cancer incidence in addition to fatal cancer risk. The risks also have estimated the cancer risk by age.
Table 12-7
Cancer Incidence and Fatality (BEIR VII) Estimates of Low-Level Radiation per Gender

Estimated number of cancer cases and deaths expected to result in 100,000 persons.
∗Estimates correspond to 95% confidence interval in parenthesis.
Carcinogenesis: Genomic Instability
Genomic instability is an increased tendency of the genome to acquire mutations when various processes involved in maintaining and replicating the genome are dysfunctional. Biologic consequences of exposure to ionizing radiation include cell death, gene mutations, and chromosome aberrations. Conventional dogma attributes these effects to alterations resulting from the deposition of energy to the DNA of an irradiated cell. Eventually the cell is presumed repaired by DNA enzymatic mechanisms that also can occur during DNA replication. It was widely accepted that most of these changes took place immediately after exposure. Thus if the damage were faithfully repaired, the descendants of an irradiated cell would be normal (Figure 12-10, A). If misrepaired, however, the descendants would be expected to pass on radiation-induced genetic change and all cells derived from such a cell would have the identical genetic alteration, or, more simply, the effect would be clonal (Figure 12-10, B). Yet many in vitro studies have demonstrated nonclonal chromosome aberrations and mutations in the clonal progeny of irradiated cells.158 Furthermore, it has been known for many years that radiation-induced cellular alterations (cytotoxicity), identified as a loss of reproductive potential, might be delayed for several generations of cell replication, with death occurring randomly among the progeny cells.158
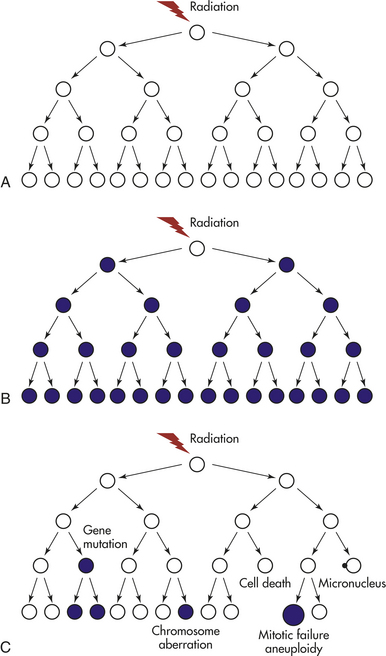
Figure 12-10 Models of the responses of clonogenic cells to ionizing radiation. Mutations and/or chromosomal aberrations are shown as filled circles and apparently normal cells as open circles. A, If a cell faithfully repairs DNA damage, then its clonal descendents will appear normal. B, If a cell is directly mutated by radiation, then all of its descendants will express the same mutation. C, Radiation- induced genomic instability is characterized by nonclonal effects in descendant cells. (From Lorimore SA, Coates PJ, Wright EG: Oncogene 22[45]:7058-7069, 2003.)
Now it is known that the progeny of irradiated cells can exhibit an increased death rate and loss of reproductive potential that continues for several generations—perhaps indefinitely. This delayed cell death phenotype is known as “lethal mutation” and “delayed reproductive death.” Significant is that various genetic alterations are demonstrated in cells that are not themselves irradiated, but in so-called innocent cells that are referred to as bystander effects and are considered manifestations of a radiation-induced genomic instability (see following and Figure 12-10, C). This instability is similar to inherited chromosome instability syndromes with spontaneously high levels of chromosomal alterations and mutations.159 Why is radiation-induced genomic instability relevant to low-dose exposure? Little160 provides a hypothetical model for multistep carcinogenesis incorporating radiation induced genomic instability (RIGI). As part of the multistep model, it is proposed that a number of mutations in individual genes must accumulate in a given group of cells to give rise to an invasive tumor. Thus hypothetically, radiation might act at an initial stage of carcinogenesis but could act at any time in later stages after one or more initiating mutations have occurred.160 Radiation may induce or increase genomic instability by facilitating new mutations in later generations. This effect may also be mediated by a nontargeted bystander mechanism. Little160 includes these important summary characteristics of RIGI relevant to low doses:
• RIGI is a “high-frequency” event occurring in 10% or more cells in an irradiated population.
• The biologic effects are manifested later after many rounds of cell division, making it possible that RIGI induced in germinal cells could be passed on to the offspring possibly conferring genetic effects including cancer in these children.
Induction of genetic changes in bystander cells, as well as those directly radiated, could lead to a “hyperlinearity” response; that is, a higher level of risk per unit dose, also called the supralinear model of the dose response curve for low dose (low particle fluences) (see Figure 12-9, D, p. 419) when only a small number of the cells are irradiated.160 Direct evidence of the dose-response relationship of a supra-linear relationship was found with an increase in double strand breaks (DSB) (see Figure 12-8) in the dose range of 1.2-5mGy was largely from bystander effects.161 In addition, Little160 speculates that individuals with a decreased ability to repair their DNA might be more susceptible to bystander effects.
The genome is constantly challenged by destabilizing factors, including normal DNA replication and cell division; intracellular and extracellular environmental stresses, such as oxidative metabolism; exposure to genotoxic chemical agents; and background radiation. Cells have complex mechanisms for trying to maintain genomic stability. These processes include proofreading of DNA replication, enzymatic repair of DNA damage, and checkpoints monitoring progression through the cell cycle. Failure of any of these processes can result in destabilization of the genome, deleterious mutations, and alterations in cell proliferation.
Although similar to alterations to the chromosome instability syndromes, the radiation-induced genomic instability seems to reflect epigenetic phenomena rather than mutation of genome genes.149 Experiments have shown that irradiation can induce growth factors and extracellular matrix (microenvironment) remodeling. A major function of the microenvironment is to control cell differentiation and proliferation, and its disruption is required for the establishment of cancer.162 These data suggest that such epigenetic events after radiation, including alterations in pathways affecting cell adhesion, extracellular matrix interactions, and cell-to-cell communication, may override the positive or repairing influence of tissue signaling and architecture that inhibits neoplastic progression.162 The chromosomal instability noted in mouse hematopoietic tissue163 and mouse mammary epithelium164 is, however, strongly influenced by genetic factors, with some genotypes being more susceptible than others.
Bystander Effects: In vivo and in vitro culture experiments performed over the past two decades indicate that low-dose ionizing radiation causes significantly divergent biologic responses from high-dose radiation. Two important findings concerning the biologic effects of a low dose (or low fluences) of radiation occur in the irradiated cells and in cells that are not themselves radiated—the so-called bystander effect: (1) radiation-induced genomic instability occurs in the descendant cells of the irradiated cell after several generations of cell division, and (2) radiation-induced bystander effects are caused as a consequence of damage signals transmitted from neighboring irradiated cells whereby transmission may be mediated by either direct or intercellular communication through gap junctions or by factors released into the surrounding medium.165 Bystander effects occur in a wide variety of cell types.166 The biologic effects may be associated with oxidative stress and the generation of ROS (e.g., superoxide, hydrogen peroxide, COX-2) (Figure 12-11). Nitric oxide, however, may initiate intercellular signaling pathways that influence the bystander effect.165,167 The first “proof-of-principle” in vivo experiment suggested that COX-2 (e.g., inflammatory/oxidative stress) did not influence the bystander damage and investigators speculated the role of connexin (gap junction) proteins.153 Numerous intercellular and intracellular signaling pathways are implicated in the bystander response, and effects have been shown to be transmitted to their descendants (see Figure 12-11). Bystander effects include mutations, sister chromatid exchanges, chromosomal aberrations, neoplastic transformation, and cell death, proliferation, and differentiation.166
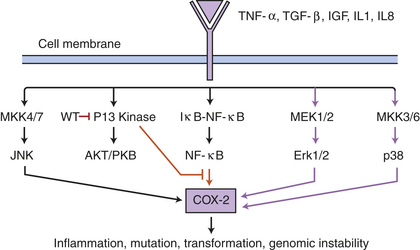
Figure 12-11 Bystander effect and signaling pathways: a working model. The signaling pathways involved in the radiation-induced bystander effect include binding of tumor necrosis factor-alpha (TNF-α), transforming growth factor-beta (TGF-β), insulin-like growth factor (IGF), interleukin-1 (IL-1), and IL-8 ligands to their receptors, thus activating the signaling pathways including MEK1/2, mitogen-activated protein kinase (MKK3/6), and p38 kinase (purple arrows) leading to COX-2 gene expression. Specific inhibitors of these pathways, such as Wortmannin (wt), can help investigators understand the specific routes in the bystander effect. (Data from Zhou H, et al: Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway, Proc Natl Acad Sci U S A 102[41]:14641-14646, 2005.)
In vivo experiments also have shown that inflammatory-type responses occur after exposure to ionizing radiation.168,169 Theses studies revealed that activation of macrophages and neutrophil accumulations were not direct effects of irradiation but were instead a consequence of the recognition and phagocytosis of radiation-induced apoptotic cells. The phagocytotic mechanism is suggested for the interactions between irradiated and nonirradiated (i.e., bystander effects) hematopoietic cells, both in vivo and in vitro.169 Persistent activation of these inflammatory-type responses is implicated as a contributory bystander mechanism for causing delayed DNA damage (see Figure 12-12).168 Oxidative-stress mediators also have been implicated in the cytotoxic effects observed in solid tumors located at distinct sites away from those receiving radiation.170 Genes responsible for oxidative stress have been identified.171 Direct evidence of how these oxidative events occur is, however, lacking; also unclear is the source of the oxidants—are they strictly derived from cytoplasmic membranes or from the mitochondria?
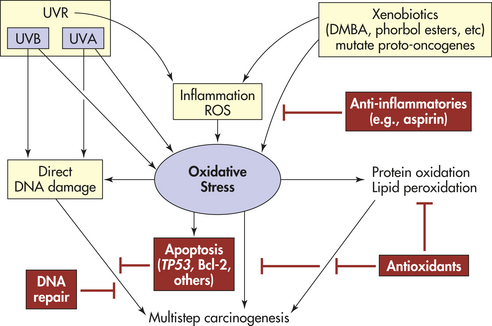
Figure 12-12 Theoretic scheme of multistep skin carcinogenesis. Ultraviolet radiation (UVR), inflammation, and xenobiotics (see p. 405) lead to oxidative stress, resulting in direct DNA damage, protein oxidation, lipid peroxidation, and apoptosis. The protective mechanisms shown in red include apoptosis, DNA repair, and antioxidants. DMBA, Dimethylbenzene anthracene; ROS, reactive oxygen species; UVA, ultraviolet A; UVB, ultraviolet B. (Adapted from Sander CD et al: Intl J Dermatol 43[5]:326-340, 2004.)
Gap Junction Intercellular Communication: Confluent cell cultures respond as an integrated whole rather than as separate individual cells that have been irradiated, indicating a critical role for cell-to-cell communication in mediating the bystander effect. This mediation could be controlled by gap junctions. Gap junctions consist of a cell-to-cell channel spanning two plasma membranes; they result from the bridging of two half channels, or connexons, contributed separately by each of the two participating cells.172 Exposure of cells to low levels of radiation (≤0.16 cGy) has been shown to induce the expression of connexin 43, suggesting that oxidizing mediators increase the expression of proteins involved in gap junction intercellular communication (GJIC).171
In conclusion, some data support a role for oxidative stress and GJIC in the radiation-induced bystander effects, and studies of the molecular mechanisms underlying bystander cells should increase our understanding of the overall risk of ionizing radiation.
Ultraviolet Radiation
Ultraviolet sunlight causes basal cell carcinoma and squamous cell carcinoma (i.e., photocarcinogenesis), two common skin cancers found in white individuals. Exposure to ultraviolet radiation (UVR) can emanate from natural and artificial sources; however, the principal source of exposure for most people is sunlight. With further depletion of the stratospheric ozone layer, people and the environment will be exposed to higher intensities of UVR. The degree of damage in skin depends on the intensity and wavelength content (i.e., ultraviolet A [UVA] or ultraviolet B [UVB]) and the depth of penetration. UV radiation is known to cause specific gene mutations; for example, squamous cell carcinoma involves mutation in the TP53 gene, basal cell carcinoma in the patched gene, and melanoma in the p16 gene.173 In addition, UV light induces the release of TNF-α in the epidermis, which may reduce immune surveillance against skin cancer.174
Skin exposure to UVR and ionizing radiation, as well as chemical (xenobiotic) agents/drugs, produces ROS in large quantities that can overwhelm tissue antioxidants and other oxygen-degrading pathways.175 Uncontrolled release of ROS is an important contributor to skin carcinogenesis.175 Imbalances in ROS and antioxidants can lead to oxidative stress, tissue injury, and direct DNA damage (see Figure 12-12). ROS can induce a number of transcription factors (e.g., activator protein-1 [AP-1] and NF-κβ).176 In addition, UVA radiation of skin fibroblast releases iron, which is involved in activation of the transcription factor NF-κβ and other free radicals important in regulating genes that induce inflammation.175,177 Inflammation is a critical component of tumor progression.
Healthy genes are needed to coordinate the levels of antioxidants and decrease harmful ROS. Antioxidants decrease ROS and oxidative stress and other protective mechanisms including DNA repair and apoptosis (see Figure 12-12). The genetic alterations in proto-oncogenes and tumor-suppressor genes may make epidermal cells resistant to signals for terminal differentiation.177 With oxidative stress, DNA damage occurs and calcium-dependent enzymes (endonucleases) are activated that produce DNA strand breaks.177 In addition, ROS are involved in the activation of procarcinogens, such as polycyclic aromatic hydrocarbons including 7,12-dimethylbenzene(a)anthracene (DMBA). DMBA is capable of initiating a point mutation in the ras proto-oncogene, which is evidenced to be the triggering event in mouse carcinogenesis.178 The essential step appears to be the activation of DMBA by nicotinamide adenine dinucleotide dehydrogenase (NADH) and cytochrome P-450–dependent oxidases.179 The pathophysiology of skin carcinogenesis is discussed further in Chapter 44.
Basal cell carcinoma commonly occurs on the head and neck. Individuals with these tumors generally have light complexions, light eyes, and fair hair. They tend to sunburn rather than tan and live in areas of high sunlight exposure. Usually these cancers arise on areas of the body that receive the greatest sun exposure, although they are not necessarily restricted to these skin sites. Squamous cell carcinoma is found more commonly in men who work outdoors. These tumors are distributed over the head, neck, and exposed areas of the upper extremities (see Chapter 44).
The incidence of melanoma has been increasing annually at rates of 2% to 7% for white populations.180 The steady rise in incidence has resulted in an alarming 165% increase in mortality rate.181 Sun exposure and the risk of melanoma, a malignant pigmented mole, remain complex. Melanomas can appear suddenly without warning but can arise from or near a mole (melanocytic nevus). When detected in the early stages, melanoma is highly curable.181 About 20% of melanomas, however, are diagnosed at nonlocalized and advanced stages.181 The pathogenesis of melanoma is complex, involving genetic and environmental factors. The genetic factors can be inherited, for example, in high-susceptibility genes (i.e., cyclin-dependent kinase inhibitor 2A [CDKN2A]) or in low-susceptibility genes (i.e., melanocortin-1). Epidemiologic and case-control studies suggest that UVR exposure is the most significant factor for the development of melanoma. Other evidence, however, reports rates of melanoma are uncommon in persons with outdoor occupations.182 Although nonmelanoma skin cancers are related to cumulative exposure to UV radiation, melanoma is related to episodes of intense, intermittent exposure (measured as history of sunburn).183 Melanomas more commonly occur in areas less continually exposed to sunlight, like the trunks in men and backs of the legs in women. Family history (i.e., genetic factors), skin type, and the density of moles are important in determining the risk of developing melanoma. For example, the incidence of melanoma in white populations is 10 times greater than in black, Asian, or Hispanic populations residing in the same area.182 Most importantly, the risk of melanoma from sunlight is certainly modified by risk factors.182 Traits associated with a high risk of melanoma are light-colored hair, eyes, and skin; an inability to tan; and a tendency to freckle, sunburn, and develop nevi.184
Until 1998 a direct causal relationship between UVB light and melanoma in humans had not been established. Grafted newborn human foreskin (xenograft) onto RAG-1 immunodeficient mice showed such a relationship.185 Melanocytic hyperplasia occurred in 73% of UVB-treated xenografts. One graft treated with DMBA and UVB light developed a human malignant melanoma. This was the first experiment to show that UVB light and an exogenous carcinogen could result in a new malignant melanoma.
A similar study using UVB light and overexpression of an endogenous growth factor, basic fibroblast growth factor (bFGF), also induced human melanoma.186 Numerous local factors may result in increased cytokine production, including trauma, infection, diet, obesity, hormones, and other causes of inflammation. Sunburn reflects an overdose of UV light, triggering inflammation with an increase in cytokine production.
A very significant finding in 2002 jolted the melanoma field. Davies and colleagues179 detected an activating point mutation in the B-raf (BRAF) proto-oncogene (i.e., somatic, not germline, mutation) in 60% to 70% of malignant melanomas. This mutation results in a marked increase in BRAF kinase activity, leading to activation of the mitogen-activated protein kinase (MAPK) pathway (see Table 1-4). This BRAF mutation and others have recently been linked to sun exposure.187 Activation of MAPK in melanoma can occur through several mechanisms: (1) mutation in the B-raf gene, (2) stimulation of fibroblast growth factor (FGF) and hepatocyte growth factor (HGF), (3) exogenous stimulation by IGF-1, and (4) by adhesion molecule receptor signaling. The development of melanoma is associated with the loss of E-cadherin and the appearance of N-cadherin adhesion molecules. Cadherins are cell surface glycoproteins that promote calcium-dependent cell-to-cell adhesion. The major adhesion molecule between keratinocytes and normal melanocytes is E-cadherin, which disappears during melanoma progression,188 and is expressed in melanoma cells, allowing them to adhere to fibroblasts and endothelial cells. Thus N-cadherin allows the melanoma cells to survive as they migrate through the dermis.189 (For further discussion, see Chapter 44.)
Increased knowledge of the intricate cellular interactions in melanoma will increase knowledge of melanoma etiology and pathogenesis. This knowledge is essential for early detection and treatment.
Electromagnetic Radiation
Health risks associated with electromagnetic radiation (EMR) are very controversial. Exposure to electric and magnetic fields is widespread. EMRs are a type of nonionizing, low-frequency radiation without enough energy to break off electrons from their orbits around atoms and ionize (charge) the atoms. Microwaves, radar, and power frequency radiation associated with electricity and radio waves, fluorescent lights, computers, and other electric equipment create EMRs of varying strength. The major debate for more than four decades has focused on the association of exposure to EMR and resultant health consequences, including cancer. Scientific evidence has been accumulating slowly because it is hampered by methods to accurately measure exposure, the lack of a clear dose-response relationship, and the difficulty in reproducing effects. In addition, with competing priorities like convenience, financial interest, health necessity, a consensus of the risk/benefit ratio of EMR exposure may be difficult to achieve.190 A recent case-control study in Japan linked high EMR exposure with a significantly higher risk of childhood leukemia.191 Other recent studies report the apparent link between cordless and cellular phone use with lymphoma and benign and malignant brain tumors.192–195 Another case-control study reported a link between childhood leukemia and prenatal proximity to high-voltage powerlines.196 Data from Sweden show adverse EMR has the potential to induce certain skin abnormalities and is a positive factor in the development of melanoma.197,198 Yet another recent study denied an association between EMR exposure and female breast cancer.199 A recent population-based study (N = 5400 women) linked residential EMR exposure from high voltage power lines to a 60% increased risk of breast cancer in Norwegian women of all ages.200 In 1998, however, a National Institute of Environmental Health Sciences Electric and Magnetic Fields Working Group recommended that low-frequency electromagnetic fields (EMFs) be classified as possible carcinogens.201 Sweden has officially categorized electrohypersensitivity as a functional impairment.202 The United Kingdom Childhood Cancer Study published in 1999 and updated in 2000203,204 did not support a link between EMR exposure and childhood cancer. A pooled analysis from Europe showed no risk of childhood cancers with average exposures (less than 0.1 micro T), no increased risk at intermediate exposures, but at the highest average exposures (greater than 0.3 micro T American studies or 0.4 micro T European study) increased risk affecting few children (1.4%) with a significant relative risk of 2.205 In response to public concern, the WHO requested further studies in high-exposure areas like Japan. Thus a case-control study from Japan (N = 312 children) found acute lymphocytic leukemia (ALL) cases in the highest exposure category above 0.4 micro T.191
The controversy about potential health hazards associated with the exposure to EMFs has been stimulated by the increased use of mobile telecommunication devices and emissions from cell towers. Cell phones emit electromagnetic radiation in the range of 800 to 2000 MHz, which is in the microwave range (300 MHz to 300 GHz).
EMR from a cell phone can penetrate the skull and deposit energy 4 to 6 cm into the brain.206 This energy can result in thermal heating of the tissue. The debate therefore has been whether these thermal effects could induce carcinogenesis. One thermal mechanism proposed is change in protein phosphorylation.207,208 Exposure of human peripheral blood lymphocytes to EMR associated with cell phones found a linear increase in chromosome 17 aneuploidy.209 Control experiments (without EMR) involving temperature changes from 24.5° to 38.5° C (76° to 101.3° F) showed that elevated temperature is not associated with genetic or epigenetic alterations. Thus these findings indicated a genotoxic effect of the EMRs is not elicited by a thermal pathway.209 Increasing evidence indicates that the mechanism of harm from EMR is induction of cell stress and damage of intracellular components (e.g., free radical formation and altered protein conformation).210 Adverse EMR has been reported to affect DNA synthesis, alter cell division, alter electrical charge of ions, and alter molecules within cells.211,212 Interference with cellular electrical charges may modify ionic structures, disturbing movement of ions across the membrane, including calcium ions.188,213
In 2007, a meta-analysis of cell phones and brain tumors concluded that studies of individuals using cell phones for more than 10 years “give a consistent pattern of an increased risk for acoustic neuroma and glioma.”193 The risk of a tumor is highest on the same side of the head that the phone is used. The studies reported are international including Sweden, Finland, the United Kingdom, Germany, Japan, and the United States. There are no studies of adults who have used cell phones as children or adolescents. Concern is for children in whom the effects may be compounded because of increased vulnerability to radiation and their longer use of cell phones into adulthood. Ongoing unbiased research is desperately needed. Absolute proof of causation may be hindered because of the ethical questions of exposing individuals to potentially harmful interventions.189
Sexual and Reproductive Behavior: Human Papillomaviruses
The past decade has demonstrated that sexually transmitted infection with carcinogenic types of human papillomavirus (HPV), referred to as high-risk types of HPV, is required for the development of most cervical cancers. In addition, HPV is a newly identified causal factor for squamous cell carcinoma of the head and neck (SCCHN).140 HPV infections, however, are very common in sexually active women, and the majority of these infections will resolve or only cause transient, minor problems.214 Eighty HPV types have been sequenced—30 of these types infect the female and male genital tract and two thirds of these are classified as high-risk types. HPV-16, in most countries, accounts for 50% to 60% of cervical cancer cases, followed by HPV-18 (10% to 12%) and HPV-31 and HPV-45 (4% to 5% each).215,216 HPV types correlated with genital warts, HPV-6 and HPV-11, are called low risk because they are rarely associated with cancer.216 HPV-16 is directly mutagenic by inducing the viral genes E6 and E7. Persistence of infection with high-risk HPV is a prerequisite for the development of cervical intraepithelial neoplasia (CIN) (see Figure 23-17) lesions and invasive cervical cancers.215 Biologic factors that determine persistence are not understood; controversial risk factors include long-term use of oral contraceptives and smoking.217 Newer risk factors being studied include drug addiction and reproductive factors (i.e., age at menarche and menopause). In fact, it has been shown that a second peak of high-risk HPV prevalence occurs in postmenopausal women.217 Smoking has been implicated in acquiring high-risk HPV (HR-HPV) but not as an independent risk factor for high-grade CIN.218 Earlier reported risk factors, for example, number of sexual partners, are probably indicators of HPV exposure rather than independent risk factors. HPV can be transmitted by genital contact (oral, touching, or sexual intercourse); therefore, condoms are not necessarily protective (see Chapter 23).
About 500,000 cases of invasive cervical cancer are diagnosed each year worldwide—the majority in developing countries. Although HPV is the most prevalent sexually transmitted infection in the United States, less than one third of women and men in the general population have heard of it, and awareness is low among women in high school and college settings.219 Cervical cancer mortality has decreased over the past five decades in the United States by more than 70%, probably due to screening with the Papanicolaou (Pap) test.214 HPV vaccination programs have made it possible to eliminate about 70% of all invasive cervical cancers worldwide (see Chapter 23).219 Human immunodeficiency virus (HIV)–infected individuals have demonstrated an increase in oral and anogenital pathologic conditions because of HPV infection.220
The incidence of invasive cervical cancer is substantially higher in women of low socioeconomic standing and is more common in Central and South America, eastern Africa, and the Caribbean.221 Consensus is that newborn babies can be exposed to cervical HPV infection of the mother. The possible modes of transmission in children, however, are controversial.222
Other Viruses and Microorganisms
A discussion on viruses and bacteria and cancer is contained in Chapter 11. Other microorganisms include particular parasites, such as Opisthorchis viverrini and Schistosoma haematobium. Their specific roles in carcinogenesis are thought to be related to cofactors or carcinogens, or both.
Physical Activity
Physical activity reduces the risk of breast and colon cancers and may reduce the risk of other cancers. Several biologic mechanisms causing this effect have been proposed and include decreasing insulin and IGF levels; decreasing obesity; increasing free radical scavenger systems; altering inflammatory mediators; decreasing circulating sex hormones and metabolic hormones; improving immune function; enhancing cytochrome P-450, thus modifying carcinogen activation; and increasing gut motility.222–226 For colon cancer, physical activity increases gut motility, which reduces the length of time (transit time) that the bowel lining is exposed to potential mutagens.227 For breast cancer, vigorous physical activity may decrease exposure of breast tissue to ovarian hormones, insulin, and IGF. A randomized trial found that after 12 months of moderate-intensity exercise, postmenopausal women had significantly decreased serum estrogens.228 Physical activity also helps prevent type 2 diabetes, which has been associated with risk of cancer of the colon and pancreas.228,229
Many questions are unanswered regarding frequency, intensity, and duration of exercise. Much of the literature suggests that between 3.5 and 4 hours of vigorous activity per week are necessary to optimize protection for colon cancer.226 There is likely a dose-response relationship for colon cancer and breast cancer, and 30 to 60 minutes per day of moderate to vigorous intensity is proposed to decrease breast cancer risk.230 A randomized controlled trial (12 months) recently supported the Institute of Medicine and Department of Agriculture guidelines of 60 minutes per day of moderate to vigorous physical activity for decreasing weight, BMI, body fat, and intra-abdominal fat.231
Chemicals and Occupational Hazards as Carcinogens
An estimated 80,000 synthetic chemicals are used in the United States. Of those, only about 7% have been tested for their health effects.232 Disturbing is that another 1000 are added each year. Exposure to chemicals occurs everyday — they are present in air, soil, food, water, household products, toys, personal care products, workplaces, and homes. Table 12-1 provides a summary of the chemicals according to strong and suspected links to various types of cancer. Box 12-2 identifies known occupational carcinogenic agents classified by the International Agency for Research on Cancer (IARC), and Box 12-3 identifies probable occupational carcinogenic agents classified by the IARC. A substantial percentage of cancers of the upper respiratory passages, lung, bladder, and peritoneum are attributed to occupational factors; however, fewer studies of nonsmokers exist.233 One notable occupational factor is asbestos, which increases the risk of mesothelioma and lung cancer. Asbestos was used in homes and buildings built before the 1970s to insulate ceiling tiles, flooring, and pipe covers. In western Europe, the epidemic of mesothelioma in building workers and other workers born after 1940 did not become apparent until the 1990s because of long latency. Carcinoma of the bladder has been linked with the manufacture of dyes, rubber, paint, and aromatic amines, especially β-naphthylamine and benzidine. Benzol inhalation is linked to leukemia in shoemakers and in workers in the rubber cement, explosives, and dyeing industries. Other notable occupational hazards include heavy metals (e.g., high-nickel alloy, chromium VI compounds, inorganic arsenic), silica, polycyclic aromatic hydrocarbons, sulfuric acid, and chloromethyl ether. Studies of occupational exposure to diesel exhaust indicate an increased risk of lung cancer.234 Disentangling data related to lung cancer, air pollution, and occupational risks is complex, especially in combination with active and passive smoking and the interplay of environmental factors and genetic polymorphisms at multiple loci.
Air Pollution
A person inhales about 20,000 L of air every day; thus even modest contamination of the atmosphere can result in inhalation of appreciable doses of pollutants. Contaminants include outdoor and indoor air pollutants. Concerns include industrial emissions, including arsenicals, benzene, chloroform, formaldehyde, sulfuric acid, mustard gas, vinyl chloride, and acrylonitrile.235 Living close to certain industries is a recognized cancer risk factor, although it is difficult to determine cancer risk from outdoor pollution alone because investigators must accurately control for smoking and radon. Studies that controlled or stratified for smoking demonstrated associations between excess lung cancer rates and heavy metal and aromatic hydrocarbon emissions in polluted air. Evidence for cancers, other than lung cancer and childhood cancer, is inconsistent.236
Indoor pollution generally is considered worse than outdoor pollution, partly because of cigarette smoke. Environmental tobacco smoke (ETS; passive smoking) can cause the formation of reactive oxygen free radicals and thus DNA damage. The IARC has classified ETS as a human carcinogen. Another significant indoor air pollutant is radon gas. Radon is a natural radioactive gas derived from the radioactive decay of uranium that is ubiquitous in rock and soil; it can become trapped in houses and gives rise to radioactive decay products known to be carcinogenic to humans. The most hazardous houses can be identified by testing and then by being modified to prevent further radon contamination. Exposure levels are greater from underground mines than from houses. Most of the lung cancers associated with radon are bronchogenic; however, small cell carcinoma does occur with greater frequency in underground miners. Radon increases the risk of lung cancer in underground miners whether they smoke or not.
In China, some regions report very high levels of lung cancer in women who spend much of their time indoors. Exposures from heating and cooking combustion sources (e.g., oil vapors) are identified as risk factors for lung cancer.237 In addition, domestic coal use and ETS increase the risk of lung cancer in women and men.238
Inorganic arsenic (known as a carcinogen since the late 1960s), found principally in underground water (from 1000 to 4000 mcg/L), is found in many regions of the world. According to the IARC, strong evidence indicates an increased risk of bladder, skin, and lung cancers following consumption of water with high levels of arsenic (generally greater than 200 mcg/L).239 Evidence for cancers of the liver, colon, and kidney is weaker. Other sources of inorganic arsenic are related to occupational exposures (see Box 12-2).
The central hypothesis, based on rat studies, for the mechanisms related to particle-induced lung carcinogenesis is that insoluble particles cause pulmonary inflammation (e.g., cytokine release, ROS), which leads to genotoxic stress, proliferative response, and tissue remodeling progressing toward fibrosis and tumor development. Additional research is needed to understand the surface chemistry and lung tissue remodeling in relation to insoluble particles, lung carcinogenesis, and other respiratory problems.240,241
Asbestos 426
Bystander effect 421
Cadherin 424
C-peptide 414
Connexon 422
Developmental plasticity 403
Environmental tobacco smoke (ETS) 404
Genomic instability 420
Hormesis 419
Individual carcinogen 396
Insulin-like growth factor-1 (IGF-1) 409
Linear no-threshold (LNT) relationship 419
Linear-quadratic relationship 419
Nontargeted effect 418
Phase I activation enzyme 405
Phase II detoxification enzyme 405
Radon 426
Supra-linear hypothesis 419
Threshold model 419
Transgeneration effect 418
Xenobiotics 404
REFERENCES
1. Clapp, R.W., et al. Environmental and occupational causes of cancer; new evidence 2005-2007. Rev Environ Health. 2008;23(1):1–37.
2. Clapp, R.W., Howe, G.K., Jacobs, M.M. Environmental and occupational causes of cancer: a call to act on what we know. Biomed Pharmacother. 2007;61(10):631–639.
3. National Cancer Institute: Cancer and the environment: what you need to know, what you can do, U.S. Department of Health and Human Services, 2004. Available at www.nci.nih.gov/newscenter/benchmarks-vol4-issue3.
4. King, M.C., Marks, J.H., Mandell, J.B. New York breast cancer study group: breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2002;302:643–646.
5. Bergstrom, H.O., et al. Increase in testicular cancer incidence in six European countries: a birth cohort phenomenon. J Natl Cancer Inst. 1996;88(11):727.
6. National Cancer Institute-DCCPS-Surveillance Research Program-Cancer Statistics Branch: Surveillance, epidemiology, and end results (SEER) program. Available at http://.seer.cancer.gov SEER∗ Stat Database: Incidence-SEER 9 Regs Public-Use, Nov 2005 Sub (1973-2003), SEER Cancer Query Systems, released April 2006. http://seer.cancer.gov/canques. Accessed July 2008.
7. Flood, D.M., et al. Colorectal cancer incidence in Asian migrants to the United States and their descendants. Cancer Causes Control. 2000;11(5):403.
8. Liao, K.A., et al. Endometrial cancer in Asian migrants to the United States and their descendants. Cancer Causes Control. 2003;11(5):357.
9. Knox, E.G. Childhood cancers, birthplaces, incinerators, and landfill sites. Int J Epidemiol. 2000;29:391.
10. Litt, J.S., Tran, N.L., Burke, T.A. Examining urban brownfields through the public health “macroscope,”. Environ Health Perspect. 2002;110(Suppl 2):183.
11. Davis, D.L., Blair, A., Hoel, D.G. Agricultural exposures and cancer trends in developed countries. Environ Health Perspect. 1992;100:39.
12. Heindel, J.J., et al. Animal models for probing the developmental basis of disease and dysfunction paradigm. Basic Clin Pharmacol Toxicol. 2008;102(2):76–81.
13. Gouveia-Vigeant T, Tickner J: Toxic chemicals and childhood cancer: a review of the evidence, Lowell Center for Sustainable Production, 2003, University of Massachusetts at Lowell. Available at www.sustainableproduction.org.
14. Kolonel, L.N., Altshuler, D., Henderson, B.E. The multiethnic cohort study: exploring genes, lifestyle and cancer risk. Nat Rev Cancer. 2004;4(7):519–527.
15. Chuang, J.C., Jones, P.A. Epigenetics and microRNAs. Pediatr Res. 2007;61(5 Pt 2):24R–29R.
16. Sasaki, H., Matsui, Y. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat Rev Genet. 2008;9(2):129–140.
17. Lu, J., et al. Micro RNA expression of profiles classify human cancers. Nature. 2005;435:834–838.
18. Johnson, I.T., Belshaw, N.J. Environment, diet and CpG methylation: epigenetic signals in gastrointestinal neoplasia. Food Chem Toxicol. 2007;46(4):1346–1359.
19. Issa, J.P. CpG-island methylation in aging and cancer. Curr Top Microbiol Immunol. 2000;249:101–118.
20. Liu, L., et al. Aging, cancer and nutrition: the DNA methylation connection. Mech Ageing Dev. 2003;124:989–998.
21. Richardson, B. Impact of aging on DNA methylation. Ageing Res Rev. 2003;2:243–261.
22. Fraga, M.F., et al. Epigenetic differences arise during the lifetime of monzygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609.
23. Issa, J.P., et al. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573–3577.
24. Gluckman, P.D., et al. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359(1):61–73.
25. Rubin, M.M. Antenatal exposure to DES: lesions learned…future concerns. Obstet Gynecol Surv. 62(8), 2007. [548–444].
26. Triosi, R., Potischman, N., Hoover, R.N. Exploring the underlying hormonal mechanisms of prenatal risk factors for breast cancer: a review and commentary. Cancer Epidemiol Biomarkers Prev. 2007;16(9):1700–1712.
27. Palmer, J.R., et al. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(8):1509–1514.
28. McLachlan, J.A. Commentary: prenatal exposure to diethylstilbestrol (DES): a continuing story. Int J Epidemiol. 2006;35(4):868–870.
29. Newbold, R.R., et al. Proliferative lesions and reproductive tract tumors in male descendents of mice exposed developmentally to diethylstilbestrol. Carcinogenesis. 2000;21:1355–1363.
30. Petridou, E., et al. Baldness and other correlates of sex hormones in relation to testicular cancer. Int J Cancer. 1997;71(6):982–985.
31. Strohsnitter, W.C., et al. Cancer risk in men exposed in utero to diethylstilbestrol. J Natl Cancer Inst. 2001;93(7):545–551.
32. Waterland, R.A., Jirtle, R.L. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300.
33. Peto, J. Cancer epidemiology in the last century and the next decade. Nature. 2001;411(6835):390–395.
34. Centers for Disease Control and Prevention: Global youth tobacco surveillance, 2000-2007. MMWR Morb Mortal Wkly Rep. 2008;57(5501):1–21.
35. WHO World Cancer Report: Global cancer rates could increase by 50% to 15 million by 2020. Available at www.WHO.int/mediacentre/news/release/2003/pr27/en/. Accessed July 2008.
36. Centers for Disease Control and Prevention: Cigarette smoking among adults—United States. MMWR Morb Mortal Wkly Rep. 2005;54(44):1121–1124.
37. Centers for Disease Control and Prevention: Cigarette smoking among adults—United States. MMWR Morb Mortal Wkly Rep. 2006;56(44):1157–1161. [2007].
38. Vineis, P., et al. Tobacco and cancer: recent epidemiological evidence. J Natl Cancer Inst. 2004;96(2):99–106.
38a. Hanaoka, T., et al. Active and passive smoking and breast cancer risk in middle-aged Japanese women. Int J Cancer. 2005;114:317–322.
39. Centers for Disease Control and Prevention: Smoking and tobacco use, fact sheet September 2006. Available at www.CDC.gov/tobacco/data_statistics/factsheets/secondhandsmoke.htm.
40. National Cancer Institute: Secondhand smoke: questions and answers. Available at www.cancer.gov/cancertopics/factsheet/tobacco/ETS, 2005. Accessed July 2008.
41. Centers for Disease Control and Prevention: Cigars, fact sheet March 2007. Available at www.CDC.gov/tobacco/data_statistics/factsheets/secondhandsmoke.htm. Accessed July 2008.
42. Henley, S.J., et al. Association between exclusive pipe smoking and mortality from cancer and other diseases. J Natl Cancer Inst. 2004;96(11):853–861.
43. Boffetta, P., et al. Smokeless tobacco and cancer. Lancet Oncol. 2008;9(7):667–675.
44. Working Group on Diet and Cancer of the Committee on Medical Aspects of Food and Nutrition Policy: Nutritional aspects of the development of cancer. London: Department of Health Rep Health Social Subjects 48, The Stationery Office; 1998.
45. Jones, D.P., Delong, M.J. Detoxification and protective functions of nutrients. In: Stipanuk M., ed. Biochemical and physiological aspects of nutrition. Philadelphia: Saunders, 2000.
46. Wiseman, M. The second World Cancer Research Fund/American Institute for Cancer Research expert report. Food, nutrition, physical activity, and the prevention of cancer: a global perspective. Proc Nutr Soc. 2008 May 1;67(3):253–256. [Epub].
47. Baylin, S.B. DNA methylation and gene silencing in cancer, Nat Clin Pract Oncol. 2005;2(Suppl 1):S4–S11. [review].
48. Agrawal, A., Murphy, R.F., Agrawal, D.K. DNA methylation in breast and colorectal cancers. Mod Pathol. 2007;20(7):711–721. [review].
49. Ross, S.A. Diet and DNA methylation interactions in cancer prevention. Ann N Y Acad Sci. 2003;983:197–207. [review].
50. Ushijima, T. Epigenetic field for cancerization. J Biochem Mol Biol. 2007;40(2):142–150.
51. Ross, J.S., et al. Breast cancer biomarkers and molecular medicine. Expert Rev Mol Diagn. 2003;3:573–585.
52. Droufakou, S., et al. Multiple ways of silencing E-cadherin gene expression in lobular carcinoma of the breast. Int J Cancer. 2001;92:404–408.
53. Shames, D.S., et al. DNA methylation in health, disease, cancer. Curr Mol Med. 2007;7:85–102.
54. World Health Organization (WHO): Global strategy on diet, physical activity, and health (online), 2004. Available at www.who.int/dietphysicalactivity/strategy/eb11344/en/.
55. Dashwood, R.H., Ho, E. Dietary histone deacetylase inhibitors: from cells to mice to man. Sem Cancer Biol. 2007;17:363–369.
56. Myzak, M.C., et al. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med. 2007;232:227–234.
57. Calle, E.E., et al. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348(17):1625–1638.
58. Pischon, T., Nöthlings, U., Boeing, H. Obesity and cancer. Proc Nutr Soc. 2008;67(2):128–145.
59. World Cancer Research Fund. Food, nutrition, and the prevention of cancer: a global perspective, pp 371–373. Washington, DC: American institute for Cancer Research; 1997.
60. Dunaif, A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. 1997;18(6):774–800.
61. Calle, E.E., Kaaks, R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4(8):579–591.
62. Collaborative Group on Hormonal Factors in Breast Cancer: Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet. 1997;350(9084):1047–1059.
63. Feigelson, H.S., et al. Weight gain, body mass index, hormone replacement therapy, and postmenopausal breast cancer in a large prospective study. Cancer Epidemiol Biomarkers Prev. 2004;13(2):220–224.
64. Schairer, C., et al. Menopausal estrogen and estrogen-progestin replacement therapy and breast cancer risk. JAMA. 2000;283(4):485–491.
65. Coates, R.J. Race, nutritional status, and survival from breast cancer. J Natl Cancer Inst. 1990;82(21):1684–1692.
66. Lukanova, A., et al. Prediagnostic levels of C-peptide, IGF-I, IGFBP-1, -2, and -3, and risk of endometrial cancer. Int J Cancer. 2004;108(2):262–268.
67. Giovannucci, E. Insulin and colon cancer. Cancer Causes Control. 1995;6(2):164–179.
68. McCullough, M.L., Giovannacci, E.L. Diet and cancer prevention. Oncogene. 2004;23(38):6349–6364.
69. Sandhu, M.S., White, I.R., McPherson, K. Systematic review of the prospective cohort studies on meat consumption and colorectal cancer risk: a meta-analytical approach. Cancer Epidemiol Biomarkers Prev. 2001;10(5):439–446.
70. Cho, E., et al. Premenopausal dietary carbohydrate, glycemic index, glycemic load, and fiber in relation to risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2003;12(11 Pt 1):1153–1158.
71. Cramer, D.W., et al. A case-control study of galactose consumption and metabolism in relation to ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2000;9(1):95–101.
72. Baron, J.A., et al. Calcium supplements for the prevention of colorectal adenomas, Calcium Polyp Prevention Study Group. N Engl J Med. 1999;340(2):101–107.
73. Martinez, M.E., Marshall, J.R., Giovannucci, E. Diet and cancer prevention: the roles of observation and experimentation. Nat Rev Cancer. 2008;8(9):694–703.
74. Rodriguez, C., et al. Calcium, dairy products, and risk of prostate cancer in a prospective cohort of United States men. Cancer Epidemiol Biomarkers Prev. 2003;12(7):597–603.
75. McMichael, A.J., Potter, J.D. Dietary influences upon colon carcinogenesis. Princess Takamatsu Symp. 1985;16:275–290.
76. Lamprecht, S.A., Lipkin, M. Cellular mechanisms of calcium and vitamin D in the inhibition of colorectal carcinogenesis. Ann N Y Acad Sci. 2001;952:73–87.
77. Duthie, S.J. Folic acid deficiency and cancer: mechanisms of DNA instability. Br Med Bull. 1999;55(3):578–592.
78. Blount, B.C., et al. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci U S A. 1997;94(7):3290–3295.
79. Wickramasinghe, S.N., Fida, S. Bone marrow cells form vitamin B12- and folate-deficient patients misincorporate uracil into DNA. Blood. 1994;83(6):1656–1661.
80. Duthie, S.J., et al. Folate deficiency in vitro induces uracil misincorporation and DNA hypomethylation and inhibits DNA excision repair in immortalized normal human colon epithelial cells. Nutr Cancer. 2000;37(2):245–251.
81. Giovannucci, E. Epidemiologic studies of folate and colorectal neoplasia: a review. J Nutr. 2002;132(Suppl):2350S–2355S.
82. Eichholzer, M., et al. Folate and the risk of colorectal, breast, and cervix cancer: the epidemiological evidence. Swiss Med Wkly. 2001;131(37-38):539–549.
82a. Figuerido, J., et al. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 101(432), 2009.
83. Jacobs, E.J., et al. Multivitamin use and colon cancer mortality in the Cancer Prevention Study II cohort (United States). Cancer Causes Control. 2001;12(10):927–934.
84. Feigelson, H.S., et al. Alcohol, folate, methionine, and risk of incident breast cancer in the American Cancer Society Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol Biomarkers Prev. 2003;12(2):161–164.
85. Zhang, S.M., et al. Plasma folate, vitamin B6, vitamin B12, homocysteine, and risk of breast cancer. J Natl Cancer Inst. 2003;95(5):373–380.
86. Kim, Y.I. Does high folate intake increase the risk of breast cancer? Nutr Rev. 2006;64(10 Pt 1):468–475.
87. Arab, L., Steck-Scott, S., Bowen, P. Participation of lycopene and beta-carotene in carcinogenesis: defenders, aggressors, or passive bystanders? Epidemiol Rev. 2001;23(2):211–230.
88. Toniolo, P., et al. Serum carotenoids and breast cancer. Am J Epidemiol. 2001;153(12):1142–1147.
89. Blumberg, J., Block, G. The Alpha-Tocopherol Beta-Carotene Cancer Prevention Study in Finland. Nutr Rev. 1994;52(7):242–245.
90. Albanes, D., et al. Alpha-tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst. 1996;88(21):1560–1570.
91. Liu, C., Russel, R.M., Wang, X.D. Alpha-tocopherol and ascorbic acid decrease the production of beta-apo-carotenals and increase the formation of retinoids from beta-carotene in the lung tissues of cigarette smoke-exposed ferrets in vitro. J Nutr. 2004;134(2):426–430.
92. Baron, J.A., et al. Neoplastic and antineoplastic effects of beta-carotene on colorectal adenoma recurrence: results of a randomized trial. J Natl Cancer Inst. 2003;95(10):717–722.
93. Mares-Perlman, J.A., et al. The body of evidence to support a protective role for lutein and zeaxanthin in delaying chronic disease. Overview. J Nutr. 2002;132(3):518S–524S.
94. Jacob, R.A. The role of micronutrients in DNA synthesis and maintenance. Adv Exp Med Biol. 1999;472:101–113.
95. Correa, P., et al. Chemoprevention of gastric dysplasia: randomized trial of antioxidant supplements and anti-. Helicobacter pylori therapy, J Natl Cancer Inst. 2000;92(23):1881–1888.
96. Helzlsouer, K.J., et al. Association between alpha-tocopherol, gamma-tocopherol, selenium, and subsequent prostate cancer. J Natl Cancer Inst. 2000;92(24):2018–2023.
97. Lippman, S.M. Effect of selenium and Vitamin E on risk of prostate cancer and other cancers: the selenium and Vitamin E cancer prevention trial (SELECT). JAMA. 2008;301(1):39–51.
98. Pryor, W.A. Vitamin E and heart disease: basic science to clinical intervention. Free Radic Biol Med. 2000;28(1):141–164.
99. Nomura, A.M., et al. Serum selenium and subsequent risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 2000;9(9):883–887.
100. Yoshizawa, K., et al. Study of prediagnostic selenium level in toenails and the risk of advanced prostate cancer. J Natl Cancer Inst. 1998;90(16):1219–1224.
101. Reagan-Shaw, S., et al. Combination of vitamin E and selenium causes an induction of apoptosis of human prostate cancer cells by enhancing Bax/Bcl-2 ratio. Prostate. 2008;68(15):1624–1634.
102. Bingham, S.A., et al. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003;361(9368):1496–1501.
103. Alberts, D.S., et al. Lack of effect of a high-fiber cereal supplement on the recurrence of colorectal adenomas. Phoenix Colon Cancer Prevention Physicians’ Network. N Engl J Med. 2000;342(16):1156–1162.
104. Bonithon-Kopp, C., et al. Calcium and fibre supplementation in prevention of colorectal adenoma recurrence: a randomised intervention trial. European Cancer Prevention Organisation Study Group. Lancet. 2000;356(9238):1300–1306.
105. Schatzkin, A. Going against the grain? Current status of the dietary fiber-colorectal cancer hypothesis. Biofactors. 2000;12(1-4):305–311.
106. Burkitt, D.P. Epidemiology of cancer of the colon and rectum. Cancer. 1971;28(1):3–13.
107. International Agency for Research on Cancer (IARC): Cancer epidemiology. Lyon, France: IARC Press; 2004.
108. Smith-Warner, S.A., et al. Intake of fruits and vegetables and risk of breast cancer: a pooled analysis of cohort studies. JAMA. 2001;285(6):769–776.
109. Terry, P., et al. Prospective study of major dietary patterns and colorectal cancer risk in women. Am J Epidemiol. 2001;154(12):1143–1149. [2001].
110. Flood, A., et al. Dietary patterns as identified by factor analysis and colorectal cancer among middle-aged Americans. Am J Clin Nutr. 2008;88(1):176–184.
111. Ames, B.N., Gold, L.S., Willett, W.C. The causes and prevention of cancer. Proc Natl Acad Sci U S A. 1995;92(12):5258–5265.
112. Lampe, J.W., et al. Modulation of human glutathione S-transferases by botanically defined vegetable diets. Cancer Epidemiol Biomarkers Prev. 2000;9(8):787–793.
113. Aldercreutz, H. Phyto-oestrogens and cancer. Lancet Oncol. 2002;3(6):364–373.
114. Steinmetz, K.A., Potter, J.D. Vegetables, fruit, and cancer. I. Epidemiology. Cancer Causes Control. 1991;2(5):325–357.
115. Doll, R., Peto, R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66(6):1191–1308.
116. Bond, V.P., Benary, V., Sondhaus, C.A. A different perception of the linear, nonthreshold hypothesis for low-dose irradiation. Proc Natl Acad Sci U S A. 1991;88(19):8666–8670.
117. Kaaks, R., Lukanova, A., Kurzer, M.S. Obesity, endogenous hormones, and endometrial cancer risk: a synthetic review. Cancer Epidemiol Biomarks Prev. 2002;11(12):1531–1542.
118. Weiderpass, E., et al. Occurrence, trends, and environment etiology of pancreatic cancer. Scand J Work Environ Health. 1998;24(3):165–174.
119. Patel, A.V., et al. IGF-I, IGFBP-1, and IGFBP-3 polymorphisms predict circulating IGF levels but not breast cancer risk: findings from the Breast and Prostate Cancer Cohort Consortium (BPC3). PLos ONE. 2008;3(7):e2578.
120. Khandwala, H.M., et al. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr Rev. 2000;21(3):215–244.
121. Lawlor, M.A., Alessi, D.R. PKB/Akt: a key mediator of cell proliferation, survival, and insulin responses? J Cell Sci. 2001;114(Pt 16):2903–2910.
122. Le Roith, D. Regulation of proliferation and apoptosis by the insulin-like growth factor I receptor. Growth Horm IGF Res. 2000;10(Suppl A):S12–S13.
123. Prisco, M., et al. Insulin and IGF-I receptors signaling in protection from apoptosis. Horm Metab Res. 1999;31(2-3):80–89.
124. Everhart, J., Wright, D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA. 1995;273(20):1605–1609.
125. Lindblad, P., et al. The role of diabetes mellitus in the aetiology of renal cell cancer. Diabetologia. 1999;42(1):107–112.
126. Schoen, R.E., et al. Increased blood glucose and insulin, body size, and incident colorectal cancer. J Natl Cancer Inst. 1999;9(13):1147–1154. [1].
127. Cust, A.E., et al. Serum levels of C-peptide, IGFBP-1 and IGFBP-2 and endometrial cancer risk; results from the European prospective investigation into cancer and nutrition. Int J Cancer. 2007;120(12):2656–2664.
128. Ma, J., et al. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncol. 2008 Oct 3. [[Epub ahead of print]].
129. Kaaks, R., Lukanova, A. Energy balance and cancer: the role of insulin and insulin-like growth factor I. Proc Nutr Soc. 2001;60(1):91–106.
130. Hadsell, D.L., Bonnette, S.G. IGF and insulin action in the mammary gland and in breast cancer. J Mammary Gland Biol Neoplasia. 2000;5(1):19–30.
131. Ne, Allen, et al. A prospective study of serum insulin-like growth factor-I (IGF-I), IGF-II, IGF-binding protein-3 and breast cancer risk. Br J Cancer. 2005;92(7):1283–1287.
132. Fletcher, O., et al. Polymorphisms and circulating levels in the insulin-like growth factor system and risk of breast cancer: a systematic review. Cancer Epidemiol Biomarkers Prev. 2005;14(1):2–19.
133. Chan, J.M., et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. 1998;279(5350):563–566.
134. Kaaks, R., et al. Serum C-peptide, insulin-like growth factor IGF-I, IGF binding proteins, and colorectal cancer risk in women. J Natl Cancer Inst. 2000;92(19):1592–1600.
135. Palmqvist, R., et al. Plasma insulin-like growth factor 1, insulin-like growth factor binding protein 3, and risk of colorectal cancer: a prospective study in northern Sweden. Gut. 2002;50(5):642–646.
136. Stattin, P., et al. Plasma insulin-like growth factor-I, insulin-like growth factor-binding proteins, and prostate cancer risk: a prospective study. J Natl Cancer Inst. 2000;92(2):1910–1917.
137. Poschl, G., Seitz, H.K. Alcohol and cancer. Alcohol Alcohol. 2004;39(3):155–165.
138. Izevbigie, E.B., et al. Ethanol modulates the growth of human breast cancer cells in vitro. Exp Biol Med. 2002;227(4):260–265.
139. Bagnardi, V., et al. A meta-analysis of alcohol drinking and cancer risk. Br J Cancer. 2001;85(11):1700–1705.
140. Argiris, A., et al. Head and neck cancer. Lancet. 2008;9625(371):1695–1709.
141. Sturgis, E.M., Wei, Q. Genetic susceptibility—molecular epidemiology of head and neck cancer. Curr Opin Oncol. 2002;14(3):310–317.
142. Crabb, D.W., et al. Overview of the role of alcohol dehydrogenese and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc Nutr Soc. 2004;63(1):49–63.
143. Committee on the Biological Effects of Ionizing Radiation: Health risks from exposure to low levels of ionizing radiation, BEIR VII Phase 2. Washington DC: National Academies Press; 2006.
144. Preston, D.L., et al. Studies of mortality of atomic bomb survivors. Report 13: solid cancer and noncancer disease mortality: 1950-1997. Rad Res. 2003;160:381–407.
145. Hoel, D.G. Ionizing radiation and cardiovascular disease. Ann N Y Acad Sci. 2006;1076:309–317.
146. Prasad, K.N., Cole, W.C., Hasse, G.M. Health risks of low dose ionizing radiation in humans: a review. Exp Biol Med. 2004;229(5):378–382.
147. NCRP 1929-2009 Medical Radiation Exposure of the U.S. Population Greatly Increased Since the Early 1980s. March, 2009 http://NCRPonline,org.
148. Little, J.B. Cellular radiation effects and the bystander response. Mutat Res. 2006;597:113–118.
149. Kovalchuk, O., Baulch, J.E. Epigenetic changes and nontargeted radiation effects—is there a link? Environ Mol Mutagen. 2008;49(1):16–25.
150. Barcellos-Hoff, M.H. Integrative radiation carcinogenesis: interactions between cell and tissue responses to DNA damage. Semin Cancer Biol. 2005;15:138–148.
151. Sowa, M., et al. Effects of ionizing radiation on cellular structures, induced instability and carcinogenesis. EXS. 2006;96:293–301.
152. Koturbash, I., et al. In vivo bystander effects: cranial x-irradiation leads to elevated DNA damage, altered cellular proliferation and apoptosis, and increased p53 levels in shielded spleen. Int J Radiat Oncol Bio Phys. 2008;70(2):554–562.
153. Mancuso, M., et al. Oncogenic bystander radiation effects in. Patched heterozygous mouse cerebellum, PNAS. 2008;105(34):12445–12450.
154. Committee on the Biological Effects of Ionizing Radiation: Biological effects of ionizing radiation BEIR V. Washington, DC: National Academy Press; 1990.
155. Tondel, M., et al. Increased incidence of malignancies in Sweden after the Chernobyl accident—a promoting effect? Am J Ind Med. 2006;49(3):159–168.
156. Zhou, H., et al. Induction of a bystander mutagenic effect of alpha particles in mammalian cells. Proc Natl Acad Sci U S A. 2000;97(5):2099–2104.
157. Makhijani, A., Smith, B., Thorne, M.C. Science for the vulnerable setting radiation and multiple exposure environmental health standards to protect those at most risk. Maryland: IEER, Takoma Park; 2006.
158. Lorimore, S.A., Coates, P.J., Wright, E.G. Radiation-induced genomic instability and bystander effects: inter-related nontargeted effects of exposure to ionizing radiation. Oncogene. 2003;22(45):7058–7069.
159. Futaki, M., Liu, J.M. Chromosomal breakage syndromes and the BRCA1 genome surveillance complex. Trends Mol Med. 2001;7(12):560–565.
160. Little, J.B. Lauriston S. Taylor lecture: nontargeted effects of radiation: implications for low dose exposures. Health Phys. 2006;91(5):416–426.
161. Ojima, M., Ban, N., Kai, M. DNA double-strand breaks induced by very low x-ray doses are largely due to bystander effects. Radiat Res. 2008;170(3):365–371.
162. Park, C.C., et al. Ionizing radiation induces heritable disruption of epithelial cell interactions. Proc Natl Acad Sci USA. 2003;100(19):10728–10733.
163. Watson, G.E., et al. In vivo chromosomal instability and transmissible aberrations in the progeny of haemopoietic stem cells induced by high- and low-LET radiations. Int J Radiat Biol. 2001;77(4):409–417.
164. Ponnaiya, B., Cornforth, M.N., Ullrich, R.L. Radiation-induced chromosomal instability in BALB/c and C57BL/6 mice: the difference is as clear as black and white. Radiat Res. 1997;147(2):121–125.
165. Little, J.B. Genomic instability and bystander effects: a historical perspective. Oncogene. 2003;22(45):6978–6987.
166. Hamada, N., et al. Intercellular and intracellular signaling pathways mediating ionizing radiation-induced bystander effects. J Radiat Res (Tokyo). 2007;48(2):87–95. [review].
167. Shao, C., et al. Targeted cytoplasmic irradiation induces bystander responses. Proc Natl Acad Sci U S A. 2004;101(37):13495–13500.
168. Coates, P.J., et al. Ongoing activation of p53 pathway is a long-term consequence of radiation exposure in vivo and associates with altered macrophage activities. J Pathol. 2008;214(5):610–616.
169. Lorimore, S.A., et al. Inflammatory-type responses after exposure to ionizing radiation in vivo: a mechanism for radiation-induced bystander effects? Oncogene. 2001;20(48):7085–7095.
170. Ohba, K., et al. Primary biliary cirrhosis among atomic bomb survivors in Nagasaki, Japan. J Clin Epidemiol. 2001;54(8):845–850.
171. Azzam, E.I., de Toldeo, S.M., Little, J.B. Oxidative metabolism, gap junctions, and the ionizing radiation-induced bystander effect. Oncogene. 2003;22(45):7050–7057.
172. Spitz, D.R., et al. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23(3-4):311–322.
173. Cleaver, J.E., Crowley, E. UV damage, DNA repair, and skin carcinogenesis. Front Biosci. 2002;7:d1024–d1043.
174. Streilein, J.W., et al. Immune surveillance and sunlight-induced skin cancer. Immunol Today. 1994;15(4):174–179.
175. Bickers, D.R., Ather, M. Oxidative stress in the pathogenesis of skin disease. J Invest Dermatol. 2006;126:2562–2575.
176. Dhar, A., Young, M.R., Colburn, N.H. The role of AP-1, NF-kappaB, and ROS/NOS in skin carcinogenesis: The JB6 model is predictive. Mol Cell Biochem. 2002;234-125(1-2):185–193.
177. Sander, C.D., et al. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Intl J Dermatol. 2004;43(5):326–335.
178. Wei, S., et al. Incidence of p53 and ras gene mutations in DMBA-induced rat leukemias. , J Exp Clin Cancer Res. 2002;21(3):389–396.
179. Davies, H., et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954.
180. Lin, J., et al. Genetics of melanoma. Br J Dermatol. 2008;159(2):286–291.
181. Heymann, W.R. Screening for melanoma. J Am Acad Dermatol. 2007;56(1):144–145.
182. Polsky, D., et al. Molecular biology of melanoma. In Mendelsohn J., et al, eds.: The molecular basis of cancer, ed 2, Philadelphia: Saunders, 2001.
183. Perlis, C., Herlyn, M. Recent advances in melanoma biology. Oncologist. 2004;9(2):182–187.
184. Rees, J.L. The melanocortin 1 receptor (MC1R): more than just red hair. Pigment Cell Res. 2000;13(3):135–140.
185. Berking, C., et al. Basic fibroblast growth factor and ultraviolet B transform melanocytes in human skin. Am J Pathol. 1998;158(3):943–953.
186. Berking, C., et al. Basic fibroblast growth factor and ultraviolet B transform melanocytes in human skin. Am J Pathol. 2002;158:943–954.
187. Besaratinia, A., Pfeifer, G.P. Sunlight ultraviolet irradiation and BRAF V600 mutagenesis in human melanoma. Hum Mutat. 2008;29(8):983–1991.
188. Tang, A., et al. E-cadherin is the major mediator of human melanocyte adhesion to keratinocytes in vitro. J Cell Sci. 1994;107(pt 4):983–992.
189. Li, G., Satayamoorthy, K., Herlyn, M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001;61(9):3819–3825.
190. Genuis, S.J. Fielding a current idea: exploring the public health impact of electromagnetic radiation. Public Health. 2008;122(2):113–124.
191. Kabuto, M., et al. Childhood leukemia and magnetic fields in Japan: a case-control study of childhood leukemia and residential power-frequency magnetic fields in Japan. Int J Cancer. 2006;119:643–650.
192. Hardell, L., Carlberg, M., Hansson Mild, K. Use of cellular telephones and brain tumour risk in urban and rural areas. Occup Environ Health. 2005;62(6):390–394.
193. Hardell, L., et al. Long-term use of cellular phones and brain tumours: increased risk associated with use for >or =10 years. Occup Environ Med. 2007;64(9):626–632. [Review].
194. Hardell, L., Sage, C. Biological effects from electromagnetic field exposure and public exposure standards. Biomed Pharmacother. 2008;62(2):104–109. [review].
195. Hardell, L., et al. Meta-analysis of long-term phone use and the association with brain tumours. Int J Oncol. 2008;32(5):1097–1103.
196. Draper, G., et al. Childhood cancer in relation to distance from high voltage power lines in England and Wales: a case-control study. BMJ. 2005;330:1290.
197. Hallberg, O., Johansson, O. Melanoma incidence and frequency modulation (FM) broadcasting. Arch Environ Health. 2002;57:32–40.
198. Hallberg, O., Johansson, O. Maligant melanomas of the skin—not a sunshine story!. Med Sci Monit. 2004;10:CR336–CR340.
199. Feychting, M., Forssén, U. Electromagnetic fields and female breast cancer. Cancer Causes Control. 2006;17(4):553–558. [review].
200. Kliukiene, J., Tynes, T., Andersen, A. Residential and occupational exposure to 50-Hz magnetic fields and breast cancer in women: a population-based study. Am J Epidemiol. 2004;159(9):852–861.
201. National Institute of Environmental Health Sciences (NIEHS) Working Group Report. Assessment of health effects from exposure to power-line frequency electric and magnetic fields. Washington, DC: US Government Printing Office; 1998.
202. Johansson, O. Electrohypersensitivity: state-of-art of a functional impairment. Electroman Biol Med. 2006;25(4):245–258. [review].
203. UK Childhood Cancer Study Investigators. Exposure to power-frequency magnetic fields and the risk of childhood cancer. Lancet. 1999;354(9194):1925–1931.
204. UK Childhood Cancer Study Investigators. Childhood cancer and residential proximity to power lines. Br J Cancer. 2000;83(11):1573–1580.
205. Ahlbom, A., et al. A pooled analysis of magnetic fields and childhood leukaemia. Br J Cancer. 2000;83(5):692–698.
206. Christensen, H.C., et al. Cellular telephone use and risk of acoustic neuroma. Am J Epidemiol. 2004;159(3):277–283.
207. Independent Expert Group on Mobile Phones: Mobile phones and health (the Stewart report). Didcot, Oxon, United Kingdom, 2000, National Radiological Protection Board. Available at www.iegmp.org.uk/report/text.htm.
208. Repacholi, M.H. Radiofrequency field exposure and cancer: what do the laboratory studies suggest? Environ Health Perspect. 1997;105(Suppl 6):1565–1568.
209. Mashevich, M., et al. Exposure of human peripheral blood lymphocytes to electromagnetic fields associated with cellular phones leads to chromosomal instability. Bioelectromagnetics. 2003;24(2):82–90.
210. World Health Organization (WHO): Sensitivity of children to EMF exposure. Proceedings of a symposium sponsored by the WHO International EMF Project, June 9-10, 2004, Istanbul, Turkey, Bioelectromagnetics (Suppl 7):S1-S160 Erratum 27(5):430, 2006.
211. Cherry N: World conference on breast cancer—Ottawa, Canada, July 26-31, 1999. Lincoln: New Zealand Lincoln University; 2002. Available at: www.neilcherry.com/cart/specific+health+effect+review?mode=show_category.
212. Havas, M. Biological effects of non-ionizing electromagnetic energy: a critical review of the reports by the US National Research Council and the US National Institute of Environment Health Sciences as they relate to the broad realm of EMF bioeffects. Environ Rev. 2000;89:173–253.
213. Blackman, C.F., Benane, S.G., House, D.E. The influence of temperature during electric- and magnetic-field-induced alteration of calcium-ion release from in vitro brain tissue. Bioelectromagnetics. 1991;12(3):173–182.
214. Anhang, R., Goodman, A., Goldie, S.J. HPV communication: review of existing research and recommendation for patient education. CA Cancer J Clin. 2004;54(5):248–259.
215. Bosch, F.X., de Sanjose, S. Human papillomavirus and cervical cancer—burden and assessment of causality. J Natl Cancer Inst Monogr. 2003;31:3–13.
216. Stanley, M. Immunobiology of HPV and HPV vaccines. Gynecol Oncol. 2008;109(2 Supp):S15–S21. [review].
217. Syrjänen, K., et al. New concepts on risk factors of HPV and novel screening strategies for cervical cancer precursors. Eur J Gynaecol Oncol. 2008;29(3):205–221.
218. Syrjänen, K., et al. Smoking is an independent risk factor for oncogenic human papillomavirus (HPV) infections but not for high-grade CIN. Eur J Epidemiol. 2007;22(10):723–735.
219. Herzog, T.J., et al. Initial lessons learned in HPV vaccination. Gynecol Oncol. 2008;109(2 Suppl):S4–S11.
220. Hagensee, M.E., et al. Human papillomavirus infection and disease in HIV-infected individuals. Am J Med Sci. 2004;328(1):57–63.
221. Stuver, S. Adami H-O: Cervical cancer. In: Adami H., Hunter D., Trichopoulos D., eds. Textbook of cancer epidemiology. Oxford: Oxford University Press, 2002.
222. Syrjanen, S., Puranen, M. Human papillomavirus infections in children: the potential role of maternal transmission. Crit Rev Oral Biol Med. 2000;11(2):259–274.
223. Eyre, H., et al. Preventing cancer, cardiovascular disease, and diabetes: a common agenda for the American Cancer Society, the American Diabetes Association, and the American Heart Association. Stroke. 2004;35(8):1999–2010.
224. McTiernan, A. Mechanisms linking physical activity with cancer. Nat Rev Cancer. 2008;8(3):205–211.
225. Rogers, C.J., et al. Physical activity and cancer prevention: pathways and targets for intervention. Sports Med. 2008;38(4):271–296.
226. Slattery, M.L. Physical activity and colorectal cancer. Sports Med. 2004;34(4):239–252.
227. McTiernan, A., et al. Physical activity and cancer etiology: associations and mechanisms. Cancer Causes Control. 1998;9(5):487–509. [review].
228. McTiernan, A., et al. Effect of exercise on serum estrogens in postmenopausal women: a 12-month randomized clinical trial. Cancer Res. 2004;64(8):2923–2928.
229. Calle, E.E., et al. Diabetes mellitus and pancreatic cancer mortality in a prospective cohort of United States Adults. Cancer Causes Control. 1998;9(4):403–410.
230. Lee, I.M. Physical activity and cancer prevention…data from epidemiologic studies. Med Sci Sports Exerc. 2003;35(11):1823–1827.
231. McTiernan, A., et al. Exercise effect on weight and body fat in men and women. Obesity. 2007;15(6):1496–1512.
232. Gray, J. State of the Evidence. In The connection between breast cancer and the environment. San Francisco: Breast Cancer Fund; 2008.
233. Neuberger, J.S., Field, R.W. Occupation and lung cancer in nonsmokers. Rev Environ Health. 2003;18(4):251–267. [review].
234. Vineis, P., et al. Outdoor air pollution and lung cancer: recent epidemiologic evidence. Int J Cancer. 2004;111(5):647–652.
235. Blair, A., Kazerouni, N. Reactive chemicals and cancer. Cancer Causes Control. 1997;8(3):473–490.
236. Boffetta, P., et al. Mortality among workers employed in the titanium dioxide production industry in Europe. Cancer Causes Control. 2004;15(7):697–706.
237. Boffetta, P. Involuntary smoking and lung cancer. Scand J Work Environ Health. 2002;28(Suppl 2):30–40.
238. Zhao, Y., et al. Air pollution and lung cancer risks in China—meta-analysis. Sci Total Environ. 2006;366(2-3):500–513.
239. Borm, P.J., Schins, R.P., Albrecht, C. Inhaled particles and lung cancer, part B: paradigms and risk assessment. Int J Cancer. 2004;110(1):3–14.
240. Holguin, F. Traffic, outdoor air pollution, and asthma. Immunol Allergy Clin North Am. 2008;28:577–588.