STRUCTURE, FUNCTION, AND DISORDERS OF THE INTEGUMENT


The skin covers the entire body and is the body’s largest organ accounting for approximately 20% of the body’s weight. Combined with the accessory structures of hair, nails, and glands, it forms the integumentary system. The primary function of the skin is to protect the body from the environment by serving as a barrier against microorganisms, ultraviolet (UV) radiation, loss of body fluids, and the stress of mechanical forces. The skin also regulates body temperature within a very narrow range and is involved in the production of vitamin D. Touch and pressure receptors provide important protective functions and pleasurable sensations.
STRUCTURE AND FUNCTION OF THE SKIN
The skin consists of three layers: the outer layer of epidermis, a deeper layer of dermis, and the subcutaneous layer (Figure 44-1 and Table 44-1). This underlying subcutaneous layer of connective tissue contains macrophages, fibroblasts, and fat cells.
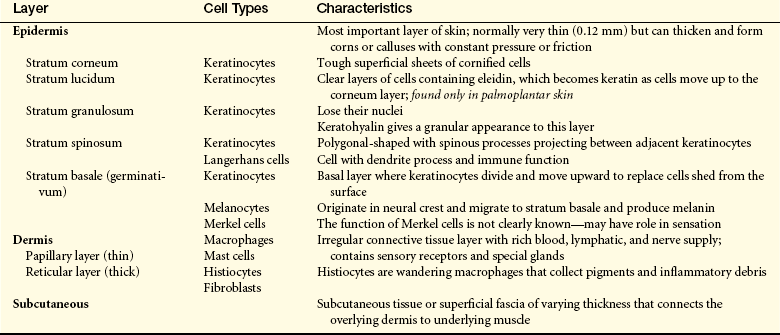
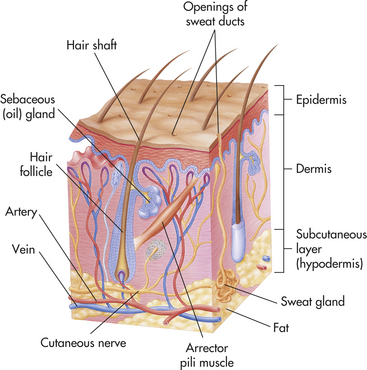
Figure 44-1 Structure of the skin. (From Thibodeau GA, Patton KT: Anatomy and physiology, ed 5, St Louis, 2003, Mosby.)
Epidermis
The epidermis grows continually by shedding the superficial layer of stratum corneum, which is formed primarily of keratinocytes and melanocytes. These cells are named for the substances they produce. Keratinocytes produce keratin, a scleroprotein that provides protection from mechanical stress. Keratin is the main constituent of skin, hair, and nail cells. The thickness of the epidermis varies from 0.3 mm on the eyelids to 1.5 mm on the palms of the hands and soles of the feet. New cells (keratinocytes) formed in the basal layer (stratum basale) move upward and differentiate, forming the spinous layer (stratum spinosum). Together they form the germinative layer (stratum germinativum). The cells enlarge and then become flattened, stacked, and cornified (stratum corneum) as they ascend to the skin surface. Cornification, or keratinization, prevents dehydration of deeper skin layers. The average turnover of the epidermis is about 30 days.
The epidermis has three additional types of cells that facilitate its functional characteristics: melanocytes, Langerhans cells, and Merkel cells. The melanocytes are usually located near the base of the epidermis. They synthesize and secrete the pigment melanin with exposure to UV light in response to melanocyte-stimulating hormone (MSH). Melanin in the epidermis provides a shield against UV radiation and determines skin color. Langerhans cells migrate to the epidermis from the bone marrow. Langerhans cells (a type of dendritic cell) and dermal dendritic cells initiate an immune response by presenting processed antigen to T cells, thus providing a defense against environmental antigens.1 Merkel cells are associated with touch receptors and function as slowly adapting mechanoreceptors when stimulated by deformation of the epidermis.
Dermis
The dermis is 1 to 4 mm thick and is composed of three types of connective tissue: (1) collagen, (2) elastin and reticulin, and (3) a gel-like ground substance. The haphazard arrangement of connective tissue allows the skin to be mobile and to stretch and contract with body movement. Hair follicles, sebaceous glands, sweat glands, blood vessels, lymphatic vessels, and nerves are contained in the dermis. The conelike projections of the papillary dermis interface with the epidermis. The papillae provide texture to the surface of the skin by forming rete pegs.
The cells of the dermis include fibroblasts, mast cells, and macrophages. Fibroblasts secrete the connective tissue matrix and collagen. Mast cells release histamine and play a role in hypersensitivity reactions in the skin. Macrophages are phagocytic and participate in immune responses. Histiocytes are macrophages that reside in loose connective tissue and phagocytize pigments and the debris of inflammation.
Subcutaneous Layer
The third layer of the skin is subcutaneous tissue and consists of fat cells or adipocytes. The lobules are separated by fibrous walls (septa) of collagen and large blood vessels. Dermal collagen is continuous with the collagen found in the subcutaneous tissue.2
Dermal Appendages
The dermal appendages include the nails, hair, sebaceous glands, and the eccrine and apocrine sweat glands. The nails are protective keratinized plates that appear at the ends of fingers and toes. Each nail is composed of four structural units: (1) the proximal nail fold, (2) the matrix from which the nail grows, (3) the hyponychium (nail bed), and (4) the nail plate (Figure 44-2). Nail growth is continuous throughout life at a rate of 1 mm or less per day.
Figure 44-2 Structures of the nail. (Redrawn from Thompson JM et al: Mosby’s clinical nursing, ed 5, St Louis, 2002, Mosby.)
Hair follicles and sebaceous glands are integrated units (see Figure 44-1). Hair color, density, grain, and pattern of distribution have considerable variability and depend on age, gender, and race. Hair follicles arise from the matrix (or bulb) located deep in the dermis. They extend from the dermis at an angle and have an erector pili muscle attached near the mid-dermis that straightens the follicle when contracted, causing the hair to stand up. Hair growth begins in the bulb, with cellular differentiation of stem cells occurring as the hair progresses up the follicle.3,4 Hair is fully hardened, or cornified, by the time it emerges at the skin surface. Hair growth is cyclic, with periods of growth and rest that vary over different body surfaces.
The sebaceous glands open onto the surface of the skin through a canal. They are found in greatest numbers on the face, chest, and back; modified glands are found on the eyelids, lips, nipple, glans penis, and prepuce. Sebaceous glands secrete sebum that is composed primarily of lipids; sebum oils the skin and hair and prevents drying. Growth of sebaceous glands is stimulated by androgens, and their enlargement is one of the early signs of puberty.
The eccrine sweat glands are distributed over the body, with the greatest numbers in the palms of the hands, soles of the feet, and forehead. These secretions are important in thermoregulation and cooling of the body through evaporation. The apocrine sweat glands are fewer in number and are located in the axillae, scalp, face, abdomen, and genital area and have very limited proven function.
Blood Supply and Innervation
The blood supply to the skin is limited to the papillary capillaries, or plexus, of the dermis. These capillary loops arise from a subpapillary plexus that is supplied by a deeper horizontal cutaneous arterial plexus. Branches from the deep plexus supply hair follicles and sweat glands. A subpapillary network of veins drains the capillary loops. Arteriovenous anastomoses in the dermis facilitate the regulation of body temperature. Heat loss can be regulated by varying blood flow through the skin by opening and closing the arteriovenous anastomoses in conjunction with evaporative heat loss of sweat. The sympathetic nervous system regulates vasoconstriction and vasodilation through α-adrenergic receptors. The lymphatic vessels arise in the papillary dermis and drain into larger subcutaneous trunks, removing cells, proteins, and immunologic mediators.
Aging and Skin Integrity
Many age-associated changes in the skin are readily observable and appear over the body surface. Environmental and genetic factors, particularly UV radiation from sun exposure (photoaging) and inflammatory responses, contribute to cutaneous changes with aging.4,5 Structurally the skin becomes thinner, drier, and wrinkled with a change in pigmentation.6,7 The cellular alterations contributing to the changes include a flattening of the dermoepidermal junction with a shortening and decrease in the number of capillary loops. There are fewer melanocytes, resulting in decreased protection against UV radiation. A significant decrease in the number of Langerhans cells decreases the skin’s immune response with aging. The thickness of the dermis also decreases and accounts for the translucent, paper-thin quality of the skin. Loss of the rete pegs gives the skin a smooth, shiny appearance.8
The decreased vasculature and lymphatic drainage contribute to loss of barrier protection and the atrophy of eccrine, apocrine, and sebaceous glands that causes dry skin.9 Loss of elastin fibers is associated with wrinkling. Collagen fibers become fragmented, and fibroblasts decrease in number, resulting in a decreased ability of the skin to stretch and regain shape. Decreased cell proliferation, decreased blood supply, and depressed immune responses also delay wound healing in aging skin.10 Changes in hair color and distribution also occur. Graying is caused by loss of melanocytes from hair bulbs, and thinning occurs from a gradual decline in the number of hair follicles and growth of finer hair.
Epidermal cells change shape, and the barrier function of the stratum corneum is reduced. There is increased permeability and decreased clearance of substances from the dermis. The accumulation of such substances is related to decreased vascularity and can cause skin irritation. Temperature regulation is compromised in older adults, with an increased risk for heat stroke and hypothermia. Loss of cutaneous vasomotion and subcutaneous fat, decreased vascularity, and decreased eccrine sweat production are contributing factors. The pressure and touch receptors and free nerve endings decrease in number and reduce sensory perception. With aging many of the protective functions of the skin decrease, whereas infection and delay in wound healing increase.11
Tests of Skin Function
Diagnostic evaluations of skin disorders often can be completed by gathering historical information, performing a physical examination, and observing the distribution and characteristics of the presenting lesions. Additional diagnostic studies are summarized in Table 44-2.
Table 44-2
Summary of Skin Diagnostic Procedures
| Test | Purpose |
| Skin biopsy | Histologic examination of tissue to determine differential diagnosis of cellular structure (i.e., benign growths vs. carcinoma, chronic infections, blistering diseases, and vasculitis) |
| Microscopic immunofluorescence | Identification of antibodies, immunoglobulins, and complement components for diseases such as pemphigus, vasculitis, and discoid lupus erythematosus using fluorescent light on slide-mounted biopsy specimens |
| Gram stain | Differentiation of gram-positive from gram-negative bacteria according to stain absorption |
| Culture | Identification of chronic bacterial and fungal infections by incubating skin specimens in culture media |
| Wood lamp examination | Examination of skin or hair to identify fungus that fluoresces bright yellow-green under ultraviolet light |
| Patch and scratch tests | Application of suspected allergens to skin by patch or scratch for evaluation of immune system responses to known allergens and evaluation of cell-mediated immune function (Candida albicans, skin fungus, chemicals, aeroallergens, and foods) |
| Skin scrapings | Application of potassium hydroxide and low heat to skin scrapings on a glass slide to identify dermatophytes and C. albicans |
| Side lighting | Indirect lighting of the skin using light to the side of the lesions to evaluate patterns of depression and elevation of skin lesions |
| Diascopy | Use of glass or clear plastic pressed on the skin to differentiate erythema caused by dilated capillaries (blanching) from extravasation of blood (no blanching) |
| Tzanck smear | A microscopic examination of cellular material from skin lesions to help diagnose vesicular diseases, including herpes simplex virus and varicella zoster |
Clinical Manifestations of Skin Dysfunction
Lesions of the skin are readily observable and easily assessed for distribution and structure. Identification of the morphologic structure and appearance of the skin in combination with a health history is necessary to identify the underlying pathophysiology. Table 44-3 describes and illustrates the basic lesions of the skin. Special skin lesions are described in Table 44-4.
Table 44-3
Primary and Secondary Skin Lesions
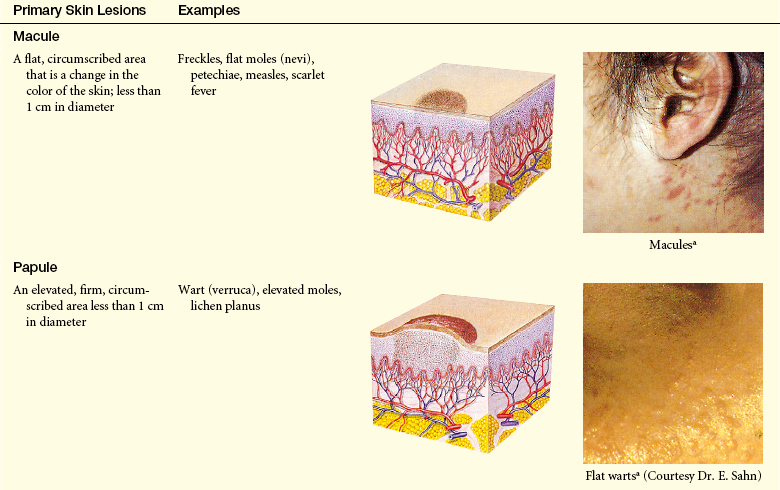
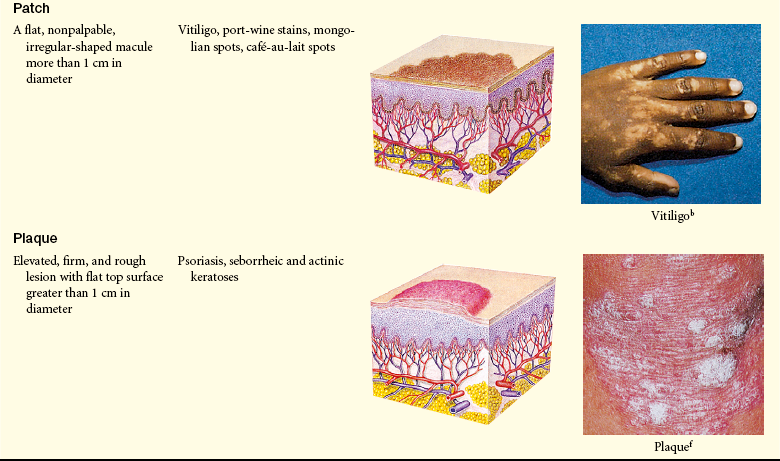
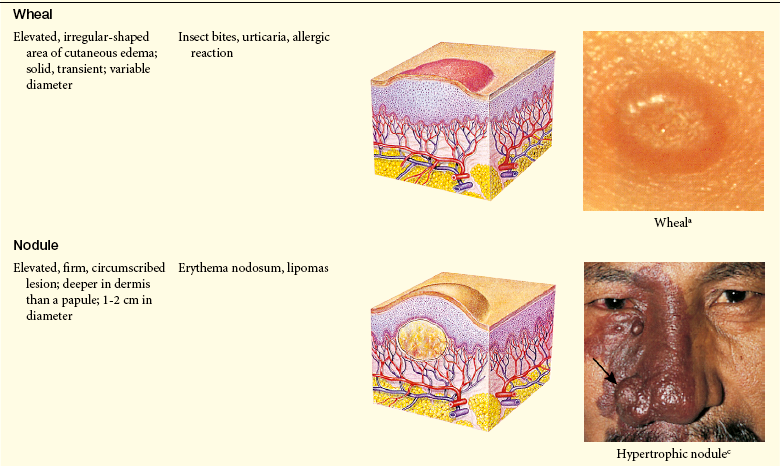
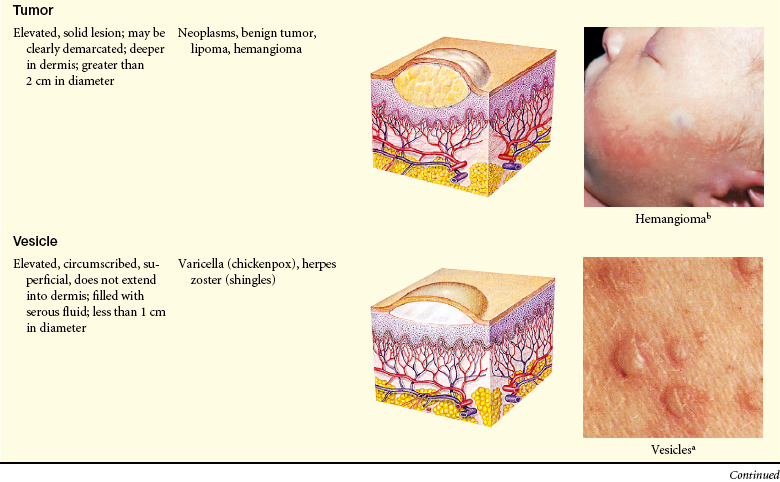
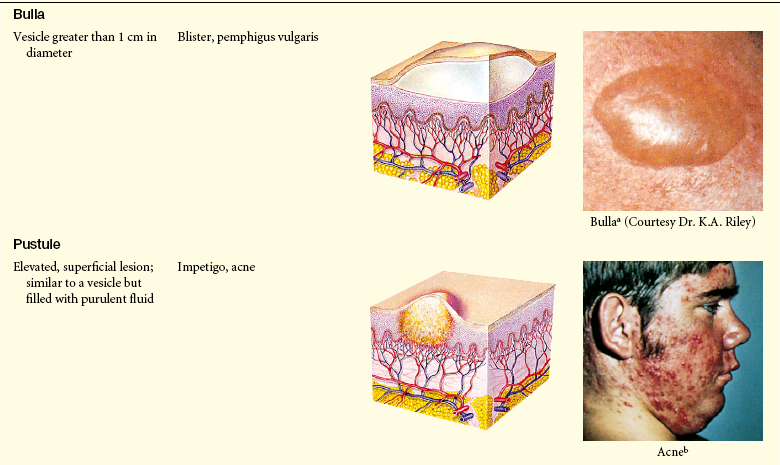
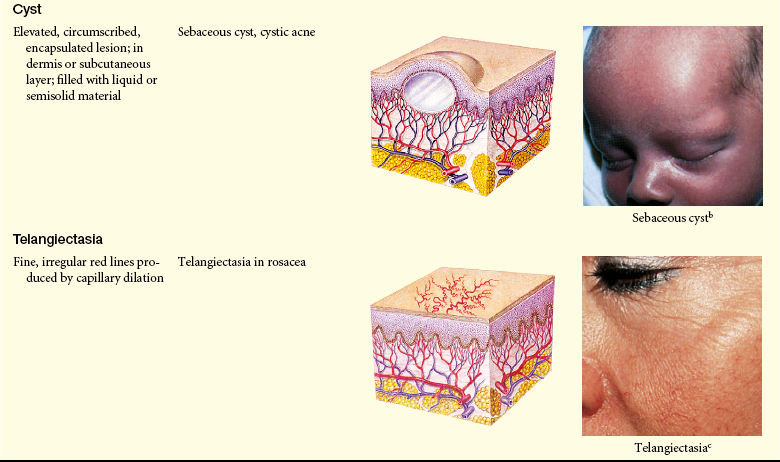
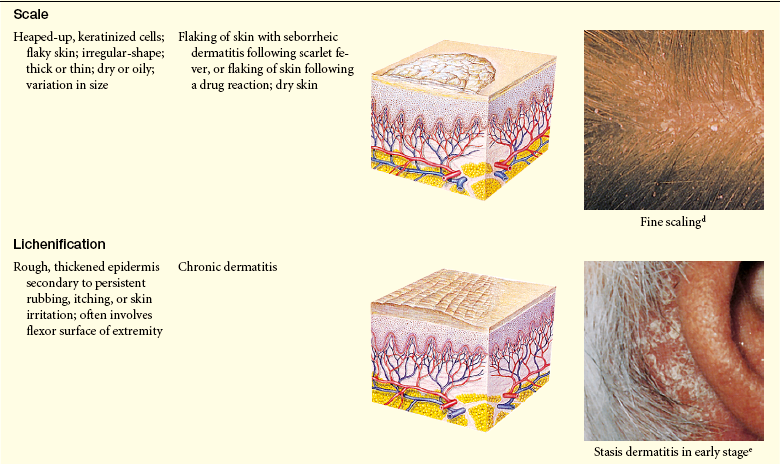
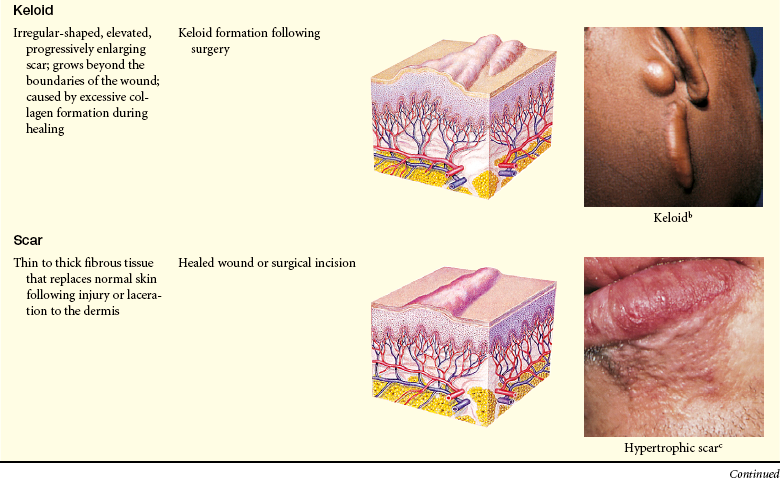
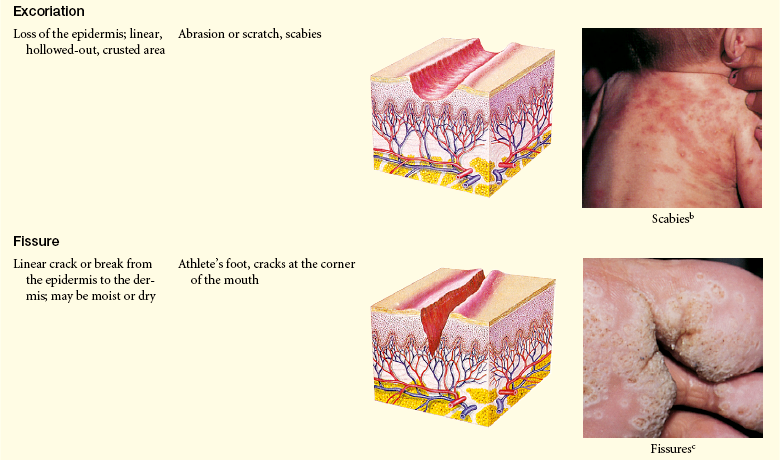
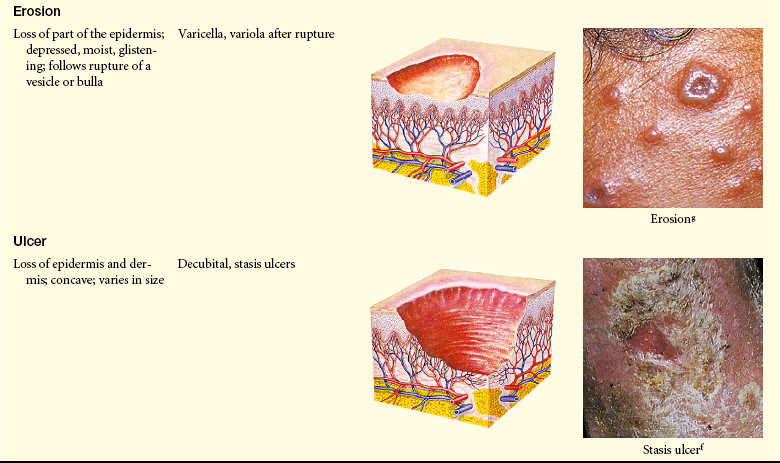
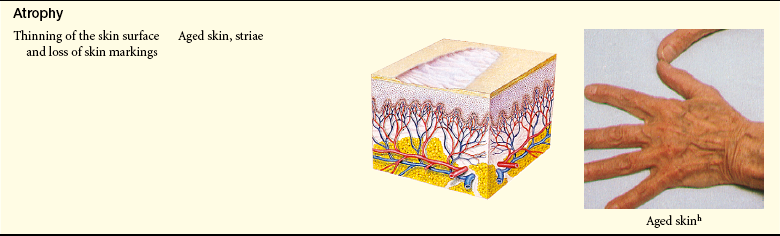
Photo credits on p. 1653.
aFarrar WE et al: Infectious diseases, ed 2, London, 1992, Gower.
bWeston WL, Lane AT: Color textbook of pediatric dermatology, ed 3, St Louis, 2002, Mosby.
cGoldman MP, Fitzpatrick RE: Cutaneous laser surgery: the art and science of selective photo thermolysis, ed 2, St Louis, 1998, Mosby.
dBaran R, Dawber RR, Levene GM: Color atlas of the hair, scalp, and nails, St Louis, 1991, Mosby.
eMarks JG Jr, DeLeo VA: Contact and occupational dermatitis, St Louis, 1991, Mosby.
fHabif TP: Clinical dermatology, ed 4, St Louis, 2004, Mosby.
gCohen BA: Pediatric dermatology, London, 1993, Mosby-Wolfe.
hSeidel HM et al: Mosby’s guide to physical examination, ed 5, St Louis, 2003, Mosby.
From Thompson JM, Wilson SF: Health assessment for nursing practice, St Louis, 2002, Mosby.
Table 44-4
| Type | Clinical Manifestations |
| Comedone | A plug of sebaceous and keratin material lodged in the opening of a hair follicle; an open comedone has a dilated orifice (blackhead), and a closed comedone has a narrow opening (whitehead) |
| Burrow | A narrow, raised, irregular channel caused by a parasite |
| Petechiae | A circumscribed area of blood less than 0.5 cm in diameter |
| Purpura | A circumscribed area of blood greater than 0.5 cm in diameter |
Pressure Ulcers: Pressure ulcers are lesions caused by unrelieved pressure resulting in damage of underlying tissue. Four factors contribute to the development of pressure ulcers: pressure, shearing forces, friction, and moisture. Pressure that consistently interrupts arterial and venous blood flow to and from the skin or deeper tissue is the most significant cause.12,13 The term decubitus ulcer refers to an ulcer or pressure sore that results when an individual lies in the recumbent position for a long time. The more general terms of pressure sore or ulcer are used here. Factors associated with greatest risk are as follows14:
1. Older adults in hospitals and nursing homes
2. Neurologic disorders that result in loss of mobility and/or sensation (spinal cord injuries, dementia, or cerebrovascular disease)
6. Lying in bed without changing position or relieving pressure over an extended period
7. Lying for hours on hard imaging and operating tables
8. Chronic diseases accompanied by anemia, edema, renal failure, malnutrition, sepsis, and urinary or fecal incontinence
9. Coarse bed sheets used for turning by dragging, which produces a shearing force
Additional risk factors for the critically ill include the following15,16:
Most individuals with darkly pigmented skin are at greater risk for developing pressure ulcers because early signs of skin damage may not be clearly visible.17,18 Pressure sores usually develop over bony prominences: the sacrum, heels, ischia, and greater trochanters are the most common sites. Continuous pressure on tissue between the bony prominence and a resistant outside surface distorts capillaries and occludes the blood flow and oxygen supply. If the pressure is relieved within a few hours, a brief period of reactive hyperemia (redness) occurs with no lasting tissue damage. If the pressure continues unrelieved, the endothelial cells lining the capillaries become disrupted with platelet aggregation, forming microthrombi that block blood flow and cause anoxic necrosis of surrounding tissues. Pressure ulcers can be classified by stages19:
Suspected deep tissue injury: localized area of purple or maroon discolored intact skin or blood-filled blister caused by underlying soft tissue damage from pressure and/or shear.
I Nonblanchable erythema of intact skin usually over bony prominence; darkly pigmented skin may not have visible blanching
II Partial-thickness skin loss involving epidermis or dermis presenting as a shallow open ulcer with a red-pink wound bed, without slough; May also present as an intact or open/ruptured serum-filled blister
III Full-thickness skin loss involving damage or necrosis of subcutaneous tissue that may extend to, but not through, underlying fascia; may include undermining and tunneling
IV Full-thickness skin loss with extensive destruction, tissue necrosis, or damage to muscle, bone, or supporting structures
Unstageable: full-thickness tissue loss with base of ulcer covered by slough and/or eschar in the wound bed
A layer of dead tissue forms that appears as a blister when there is superficial damage or as a reddish blue discoloration when there is deeper tissue damage. Superficial sores are more common on the sacrum as a result of shearing or friction forces (forces parallel to the skin). Deep sores develop closer to the bone as a result of tissue distortion and vascular occlusion from pressure that is perpendicular to the tissue (over the heels, trochanter, and ischia) (Figure 44-3).
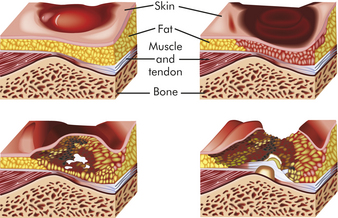
Figure 44-3 Progression of decubitus ulcer. Sustained pressure over a bony prominence compresses the tissue and reduces blood flow resulting in progressive ischemia and necrosis of tissue.
The necrotic tissue initiates an inflammatory response, with pain, fever, and leukocytosis. Although bacteria colonize the dead tissue, the infection is usually localized and self-limiting. Proteolytic enzymes from bacteria and macrophages dissolve necrotic tissues and cause a foul-smelling discharge that resembles, but is not, pus.
Pressure sores are often painful in individuals who do not have loss of sensation from spinal cord trauma or neuropathy. The presence of necrotic tissue produces an inflammatory response with hyperemia, fever, and increased white blood cell count. If the ulceration is large, toxicity and pain lead to loss of appetite, debility, and renal insufficiency. Individuals who are immunosuppressed or have diabetes mellitus may develop infection and inflammation of adjacent tissues (cellulitis) or septicemia.
The primary goal for those at risk for pressure ulcers is prevention. Several scales are available for predicting pressure sore risk, and the Braden Scale is one of those most frequently used. 20,21 Pressure sores are not prevented by topical agents because they do not relieve the pressure. Frequent skin assessment with repositioning and turning; use of pressure reduction surfaces; elimination of incontinence, moisture, and drainage; and maintenance of fluid, protein, and caloric intake are effective preventive techniques.22,23 Nutrition, oxygenation, and fluid balance must be maintained.
Superficial ulcers should be covered with flat, nonbulky dressings that cannot wrinkle and cause increased pressure or friction. Spontaneous healing will occur more quickly when the ulcer is kept moist with an occlusive dressing.24 Antibiotics are seldom required. Antiseptics, such as hydrogen peroxide or iodine, are damaging to granulation tissue and should not be used.25 Successful healing requires continued adequate relief of pressure and débridement of dead tissue.26
Large, deep pressure ulcers may require surgical débridement of necrotic tissue, opening of deep pockets for drainage, and skin grafting for wound closure and successful healing. The myocutaneous flap, a single unit of skin with its underlying muscle and vasculature, has been an effective treatment in large avascular areas over bony prominences.27 Application of wound tension by range of motion may also promote healing.28 Tools are available to evaluate and document progress of healing and further research is needed to advance clinical assessment and interdisciplinary communication regarding the management of pressure ulcers.
Keloids and Hypertrophic Scars: Keloids are elevated, rounded, and firm with irregular clawlike margins that extend beyond the original site of injury. In contrast hypertrophic scars are elevated erythematous fibrous lesions that do not expand beyond the border of injury. Both lesions are caused by abnormal wound healing with excessive fibroblast activity and collagen formation during dermal connective tissue repair.29 Many genes are involved.30 Keloids are most common in darkly pigmented skin types and burn scars (see Chapter 46). Excessive or poorly aligned tension on a wound, introduction of foreign material into the skin, and certain types of trauma (e.g., burns) are provocative factors. At risk are the shoulders, back, chin, ears, and lower legs. Most keloids appear within 1 year of trauma. Individuals 10 to 30 years of age develop lesions much more commonly than do children before puberty or older adults. Hypertrophic scars usually regress within a year.
Type III collagen is increased with keloids. The increased synthesis of collagen is associated with interleukin-6 (IL-6) signaling and dermal fibroblasts that have high metabolic and mitotic rates and aberrant expression of various growth factors.31 Myofibroblasts, cells with characteristics of fibroblasts and smooth muscle cells, are the principal cells in keloids. Collagenase activity in keloids is normal or increased, but the collagen may be protected from degradation by proteoglycan, a glycoprotein present in connective tissue that serves as a binding (cementing) material, and by specific inhibitors of proteolytic enzymes. Genes regulating fibroblasts may be up-or down-regulated in keloid tissue.32 A familial tendency for keloid formation has been found, with autosomal recessive and autosomal dominant inheritance patterns reported.33
Keloids start as pink or red, firm, well-defined rubbery plaques that persist for several months after trauma. Later, uncontrolled overgrowth causes extension beyond the site of the original wound and the tumor becomes smoother, irregularly shaped, hyperpigmented, harder, and more symptomatic. Keloids typically send out clawlike prolongations (Figure 44-4).
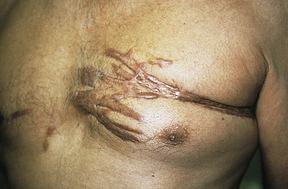
Keloids are the most extreme example of cutaneous scarring and the most difficult to treat. Preventive measures such as avoiding unnecessary, elective surgeries are of paramount importance. Various treatments are available for the management of keloids and hypertrophic scars, and there is a need for research to improve treatment outcome.34,35
Pruritus
Pruritus, or itching, is the most common symptom associated with many primary skin disorders, such as eczema, psoriasis, or lice infestations, or it can be a manifestation of systemic disease (e.g., xerosis, chronic renal failure, cholestatic liver disease, thyroid disorders, iron deficiency) or drug reactions. Pruritus may be localized or generalized and may move from one location to another.36 Central and peripheral nerve pathways are activated.37
Significant progress has been made in understanding the pathophysiology of itch. Studies show that peripheral itch mediators include neuropeptides, serotonin, prostaglandins, bradykinin, histamine, substance P, and acetylcholine and that the itch sensation is carried by specific unmyelinated C-nerve fibers.38,39 These nerve fibers may also interact with dermal mast cells.40
Itching also has been linked to pain because many stimuli that induce pain produce itching at lower intensities. Central nervous system mechanisms also can modulate itching, which is less perceptible when the mind is concentrating on other things. How the central nervous system influences the itch sensation is unclear. Neuropathic itch is related to pathology along an afferent pathway (i.e., postherpetic neuropathy). Psychogenic itch is associated with psychologic disorders (i.e., depression and obsessive-compulsive disorder).41
Chronic itching is an unpleasant sensation relieved by scratching—often done so intensely that trauma to the skin occurs, resulting in infection and scarring. Some individuals become so distraught with the constant irritation that they apply heat with enough intensity and duration to produce burns.
Management of localized itching depends on the cause, and the primary condition must be treated. Symptomatic relief may be obtained from antihistamines, which also have a sedative effect. Minor tranquilizers, such as promethazine, may be effective for some causes of pruritus. Itching related to dry, rough skin (xerosis) can be managed with applications of emollients and increased environmental humidity. Topical steroids are immediately effective with some occurrences of pruritus; however, in some instances, pruritus is resistant to any type of therapy. New topical treatment therapies are being developed, as are the use of phototherapy treatment with narrow band UVB and vagal nerve stimulation42 and cutaneous field stimulation.43 Analgesics used to treat neuropathic pain also can be effective for treating pruritus.44
DISORDERS OF THE SKIN
Disruptions in skin integrity may be precipitated by trauma, abnormal cellular function, infection and inflammation, and systemic diseases. Many skin disorders are benign and self-limiting, whereas others are severe and life threatening.
Inflammatory Disorders
The most common inflammatory disorder of the skin is eczema, or dermatitis. Eczema and dermatitis are general terms that describe a particular type of inflammatory response in the skin—the terms can be used interchangeably. Diseases considered eczematous are generally characterized by pruritus, lesions with indistinct borders, and epidermal changes. These lesions can appear as either erythema, papules, or scales, and they can present in an acute, subacute, or chronic phase. Atopic dermatitis, contact dermatitis (whether nonallergic [irritant] or allergic), lichen simplex chronicus, nummular eczema, and seborrheic dermatitis are examples of specific types of eczema or dermatitis.45 Edema, serous discharge, and crusting occur with continued irritation and scratching. In chronic eczema the skin becomes thickened, leathery, and hyperpigmented from recurrent irritation and scratching. The location of eczema is related to the underlying cause. Eczematous inflammations need to be differentiated from other rashes and dermatoses, particularly psoriasis.
Allergic Contact Dermatitis
Allergic contact dermatitis is a common form of T-cell mediated or delayed hypersensitivity (type IV).46 (See Chapter 8 for various types of allergic responses.) Allergens (e.g., microorganisms, chemicals, foreign proteins, drugs, metals, latex) can form the sensitizing antigen; contact with poison ivy is a common example (Figure 44-5). The response is a reaction to irritants with release of cytokines, chemokines, and cytotoxins from keratinocytes, dendritic cells (Langerhans cells), and natural killer cells. When the allergen comes into contact with the skin the allergen is bound to a carrier protein, forming a hapten-specific sensitizing antigen. Langerhans cells process the antigen and carry it to T cells that then become sensitized to the antigen releasing cytokines and chemokines leading to leukocyte infiltration and inflammation.47 Latex allergy can be either a type IV hypersensitivity to chemicals used in latex rubber processing or a type I immediate hypersensitivity with IgE antibodies formed in response to latex rubber protein.48
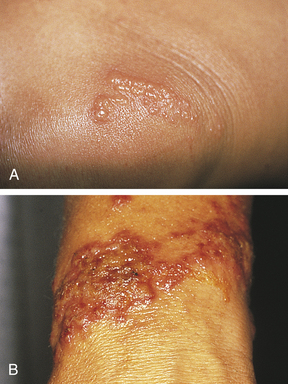
Figure 44-5 Poison ivy. A, Poison ivy on knee. B, Poison ivy dermatitis. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
In delayed hypersensitivity, several hours pass before an immunologic response is apparent. The T cells play an important role because they differentiate and secrete lymphokines that affect macrophage movement and aggregation, coagulation, and other inflammatory responses (see Chapter 8). Sensitization usually develops with first exposure to the antigen, and symptoms of dermatitis occur with reexposure.
The manifestations of allergic contact dermatitis include erythema and swelling with pruritic (itching) vesicular lesions in the areas of allergen contact. The pattern of distribution provides clues to the source of the antigen (e.g., hands exposed to chemical solutions or boundaries from rings and bracelets). Patch tests with specific antigens may assist with diagnosis. Removal of the allergen is necessary for resolution of the inflammatory response and tissue repair. Topical or systemic steroids, as well as other symptomatic treatment, may be required depending on the severity of the lesion.47
Atopic Dermatitis
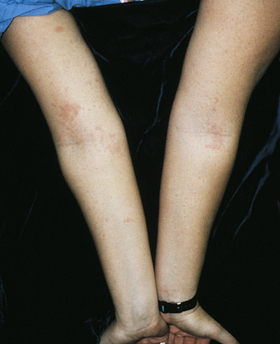
Atopic dermatitis (allergic dermatitis) is more common in infancy and childhood; however, some individuals are affected throughout life. A family history of asthma, allergic rhinitis, dry skin, food allergy, and eczema often accompanies this disorder. During adolescence and childhood the lesions are usually localized to the hands and feet or flexor surfaces (i.e., antecubital fossa, popliteal space) of the arms and legs (Figure 44-6). The erythema, scaling, and lichenification (thickened and leather-like skin) are exacerbated by scratching because the lesions manifest by itching. The scratching increases susceptibility to infections from Staphylococcus aureus and predisposition to cutaneous dissemination of viruses, particular herpes simplex. The pathogenesis and treatment of atopic dermatitis in adults are similar to that in children49 and are discussed in detail in Chapter 45.
Stasis Dermatitis
Stasis dermatitis usually occurs on the legs as a result of venous stasis and edema. The disorder is associated with varicosities (incompetent venous valves), phlebitis, and vascular trauma. Pooling of venous blood traps leukocytes that may release proteolytic enzymes. Increased venous pressure widens interendothelial pores with deposition of fibrin and other macromolecules making them unavailable for repair.50 Edema evolves to erythema and pruritus and progression to scaling, petechiae, and hyperpigmentation. Progressive lesions become ulcerated (stasis ulcers), particularly around the ankles and tibia (Figure 44-7).
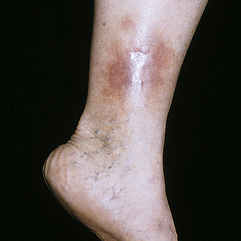
Figure 44-7 Stasis ulcer. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Treatment includes elevating the legs as often as possible, not wearing tight clothes around the legs, and not standing for long periods. Defined infections are treated with antibiotics. Chronic lesions with ulceration are treated with moist dressings, external compression, and vein ablation surgery.51,52
Irritant Contact Dermatitis
Irritant contact dermatitis is a common nonimmunologically mediated inflammation of the skin. The intensity of the inflammation is related to the concentration of the irritant, exposure time, and disruption of the skin barrier.53 Irritation can occur from almost anything, especially if the epidermal barrier is compromised in any way (Box 44-1). The skin lesions are similar in appearance to allergic contact dermatitis. Removing the source of irritation and use of topical agents (i.e., corticosteroids and petroleum-based emollients) and non-irritating soaps constitute effective treatment.
Seborrheic Dermatitis
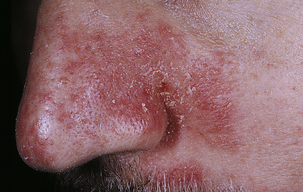
Seborrheic dermatitis is a common chronic inflammation of the skin involving the scalp, eyebrows, eyelids, ear canals, nasolabial folds, axillae, chest, and back (Figure 44-8). In infants it is known as cradle cap. The cause is unknown, but genetic predisposition an immunologic response to yeasts from the genus Malassezia have been implicated.54 The lesions appear from infancy to old age, with periods of remission and exacerbation. The lesions appear as greasy, scaly, white, or yellowish inflammatory plaques in sebaceous areas with mild pruritus. Mild cases are treated with shampoos containing sulfur, salicylic acid, or tar. Ketoconazole has antifungal and anti-inflammatory effects and has been used with success.55 Topical calcineurin inhibitors have also been effective and corticosteroid applications are useful for suppression of severe symptoms but should not be used for maintenance therapy.54a
Papulosquamous Disorders
Psoriasis, pityriasis rosea, and lichen planus are disorders characterized by inflammatory processes associated with papules, scales, plaques, and erythema. Collectively they are described as papulosquamous disorders.
Psoriasis
Psoriasis is a chronic, relapsing, proliferative skin disorder that involves the skin, scalp and nails and can occur at any age. The disease affects about 2% of the population.56 Psoriasis is a T-cell mediated autoimmune disease. Inflammatory cytokines (i.e., tumor necrosis factor [TNF], interferon-gamma [IFN-γ], IL-6, IL-12, IL-15, IL-17, IL-22, IL-23) from activated T cells, B cells, and macrophages cause the skin changes and comorbidities occurring in psoriasis.57 The onset is generally established by 20 years of age. A family history of psoriasis is common. The genetic mechanisms are complex and the human leukocyte antigen (HLA)-Cw6 allele (PSORS1) is a major susceptibility gene.58
The dermis and epidermis are thickened, with cellular hyperproliferation, altered keratinocyte differentiation, expanded dermal vasculature, infiltration of neutrophils and lymphocytes, and inflammation.59 The turnover time for shedding the epidermis is decreased from the normal 26 to 30 days to 3 to 4 days. There are increased numbers of germinative cells and an increase in transit time of cells through the dermis. The rapid cellular proliferation does not allow time for cell maturation and keratinization to occur, resulting in a thickened epidermis and plaque formation. The loosely cohesive keratin gives the lesion a silvery, scaly appearance. There is often capillary dilation and increased vascularization to accommodate the increased cell metabolism. The increased vascularity causes erythema.
The types of psoriasis include plaque (psoriasis vulgaris), inverse, guttate, pustular, and erythrodermic. Plaque psoriasis is the most common and affects 80% to 90% of individuals with psoriasis. The disease can be mild, moderate, or severe, depending on the size, distribution, and inflammation of the lesions. Early onset psoriasis is an inflammatory lesion with epidermal hyperproliferation and the presence of activated T lymphocytes.57 The typical lesion of plaque psoriasis is a well-demarcated, thick, silvery, scaly, erythematous plaque surrounded by normal skin (Figure 44-9). Initial lesions usually develop insidiously as small erythematous papules that enlarge and coalesce into larger inflammatory lesions. The lesions are commonly located on the scalp, elbows, and knees and at sites of trauma. The scales are usually loosely adherent and may cause small bleeding points when removed. Inverse psoriasis involves lesions that develop in skinfolds (i.e., axilla or groin). They are large, smooth, dry, and deep red. The progress of psoriasis is characterized by remissions and exacerbations. Antimalarial drugs, lithium, nonsteroidal anti-inflammatory drugs, and beta-blockers, tend to exacerbate existing psoriasis.
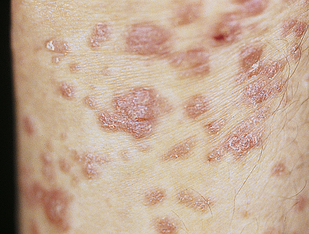
Figure 44-9 Psoriasis. Typical oval plaques with well-defined borders and silvery scale. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
In guttate psoriasis, small papules (1 to 10 mm) appear suddenly on the trunk and extremities (Figure 44-10). The lesions may appear a few weeks after a streptococcal respiratory infection and are more common in children. Guttate psoriasis may resolve spontaneously in weeks or months. Pustular psoriasis appears as blisters of noninfectious pus (collections of neutrophils) that develop over areas of plaque psoriasis. Erythrodermic (exfoliative) psoriasis is often accompanied by itching or pain with widespread red, scaling lesions that cover a large area of the body.
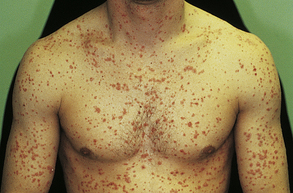
Figure 44-10 Guttate psoriasis after streptococcal infection. Numerous uniformly small lesions may abruptly occur after streptococcal pharyngitis. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Psoriatic arthritis and ankylosing spondylitis are associated with the proinflammatory cytokines that cause psoriatic skin lesions. Joints of the hands, feet, knees, and ankles are involved, and 5% to 50% of individuals with psoriasis have seronegative joint involvemment.60 Psoriatic nail disease can occur in all psoriasis subtypes with pitting, onycholysis, subungual hyperkeratosis, and nail plate dystrophy. Psoriasis is also a risk factor for a number of comorbidities including inflammatory bowel disease, metabolic syndrome, including hypertension, insulin resistance, dyslipidemias, and abdominal obesity, and increased risk for atherosclerosis and myocardial infarction that is independent of traditional risk factors for these diseases.
Treatment is individualized and related to reducing epidermal cell turnover. Mild lesions are usually treated with emollients, keratolytic agents, and corticosteroids. Moderate lesions may respond to UV light, tar preparations, or a combination of both, and to methotrexate, cyclosporine A, and acitretin. Vitamin D3 (calcitriol) is used to reduce epidermal proliferation. Moderate to severe disease is the indication for biologic treatment, including drugs that inhibit the activation and number of T lymphocytes (alefacept), drugs that block T-cell adhesion (efalizumab), or TNF-α inhibitors (adalimumab, etanercept, infliximab).56
Pityriasis Rosea
Pityriasis rosea is a benign self-limiting inflammatory disorder that occurs more often in young adults, usually during winter months. The cause is unknown but is thought to be associated with a virus because of the timing and clustering of the outbreaks.61 Pityriasis rosea begins as a single lesion known as a herald patch (Figure 44-11) that is circular, demarcated, salmon-pink, approximately 3 to 4 cm in diameter, and usually located on the trunk. Early lesions are macular and papular; secondary lesions develop within 14 to 21 days and extend over the trunk and upper part of the extremities. Lesions are rarely located on the face. They emerge as small erythematous papules that expand into characteristic oval lesions. There may be few or hundreds of lesions. The pattern of distribution follows the skin lines around the trunk and resembles a drooping pine tree. As scales flake off from the margin of the lesions, a collarette pattern is formed. Itching is the most common symptom. Headache, fatigue, or sore throat may precede the development of the lesions.62
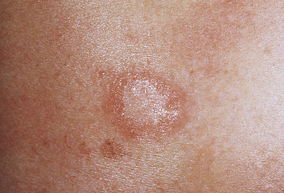
Figure 44-11 Pityriasis rosea herald patch. A collarette pattern has formed around the margins. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
The diagnosis of pityriasis rosea is made by the clinical appearance of the lesion. It can be confused with secondary syphilis, psoriasis, or seborrheic dermatitis. The disorder is usually self-limiting and resolves in a few months with symptomatic treatment for pruritus. UV light, antihistamines, or topical corticosteroids may be used to control itching, and erythromycin may control the rash.63 Sun exposure facilitates resolution of the lesions.
Lichen Planus
Lichen planus is a benign, autoimmune inflammatory disorder of the skin and mucous membranes. The cause is unknown, but T cells, adhesion molecules, inflammatory cytokines, perforin, and antigen-presenting cells are involved.64 The infiltrate of T cells mediates immunoreactivity against basal layer keratinocytes, which have altered surface antigens and adhesion molecules.65 Lichen planus is also linked to hepatitis C virus.66 Some individuals develop lichenoid lesions after exposure to drugs or film-processing chemicals. The age of onset is usually between 30 and 70 years. The disorder begins with nonscaling, violet-colored pruritic papules, 2 to 4 mm in size, usually located on the wrists, ankles, lower legs, and genitalia (Figure 44-12). The papules are flat topped and have a polygonal shape. New lesions are pale pink and evolve into a dark violet. Persistent lesions may be thickened and red, forming hypertrophic lichen planus. The lesions often involve the oral mucous membranes, appearing as lacy white rings that must be differentiated from leukoplakia or oral candidiasis.67 Fine white lines, known as Wickham striae, can be seen throughout the oral lesions on magnification. These lesions also can develop on the penis and vulvovaginal area. More commonly, oral lesions do not ulcerate, but localized or extensive painful ulcerations do, and frequently occur. Chronic ulcerated lesions become malignant in 1% of individuals with the disease. Thinning and splitting of nails are common, and part or all of the nail may be shed.
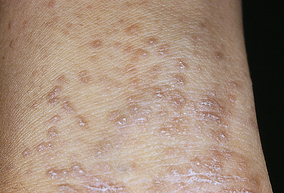
Figure 44-12 Hypertrophic lichen planus on arms. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Pruritus is the most distressing symptom. The lesions are self-limiting and may last for months or years, with an average duration of 6 to 18 months. Postinflammatory hyperpigmentation is a common consequence of the lesion. Approximately 20% of individuals have a recurrence. Diagnosis is commonly made by the clinical appearance of the lesion. Treatment is individualized. Antihistamines are given for itching, and topical or systemic corticosteroids may be used to control inflammation. Mucous membrane lesions are treated with topical steroids, topical tacrolimus, or pimecrolimus.68
Acne Vulgaris
Acne vulgaris is an inflammatory disorder of the pilosebaceous follicle (the sebaceous gland contiguous with a hair follicle). It occurs most commonly during adolescence. Details of this disorder are presented in Chapter 45.
Acne Rosacea
Acne rosacea is an inflammation of the skin that develops in middle-age adults. The disease is chronic with episodes of exacerbation. The most common lesion types are erythematotelangiectatic, papulopustular, phymatous, and ocular.69 They occur in the middle third of the face, including the forehead, nose, cheeks, and chin (Figure 44-13). The cause is unknown, but immune-mediated inflammation may be a factor.70 The lesions are associated with chronic, inappropriate vasodilation resulting in flushing and sensitivity to the sun. Sebaceous hypertrophy, fibrosis, and telangiectasia may be severe enough to produce an irreversible bulbous appearance of the nose, known as rhinophyma. Disorders of the eye often accompany rosacea, particularly conjunctivitis and, more rarely, keratitis, which can result in visual impairment.71 Facial application of fluorinated topical steroids may precipitate rosacea-like lesions that are difficult to treat. There is controversy regarding the association between Demodex folliculorum (mites), Helicobacter pylori infection, and rosacea.72,73
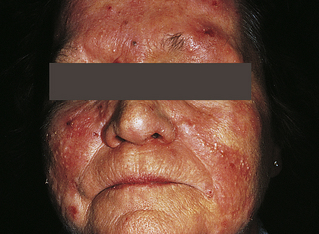
Figure 44-13 Granulomatous rosacea. Pustules and erythema occur on the forehead, cheeks, and nose. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Hot drinks or alcohol should be taken cautiously because the heat and vasodilation accentuate erythema. Tetracycline, though photosensitizing, continues to be the drug of choice for treatment, and a low-maintenance dose may be required after the most severe lesions are controlled. Daily use of photoprotection, including sunscreens, is recommended; 1% topical metronidazole and 15% azelaic acid gel may be effective.74,75 Surgical excision of excessive tissue may be required for rhinophyma.
Lupus Erythematosus
Lupus erythematosus is an inflammatory, autoimmune, systemic disease with cutaneous manifestations. Discoid (or cutaneous) lupus erythematosus (DLE) is limited to the skin and can lead to systemic lupus erythematosus (SLE) in approximately 5% of individuals. DLE may be described as a subset of SLE, with cutaneous manifestations as the only symptom76 (Figure 44-14). (SLE, a diffuse, multisystem disease, is discussed in Chapter 8.)
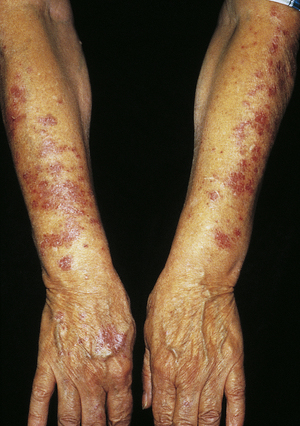
Figure 44-14 Subacute cutaneous lupus (discoid lupus erythematosus). (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Discoid (Cutaneous) Lupus Erythematosus: Discoid (cutaneous) lupus erythematosus (DLE) usually occurs in genetically susceptible adults, particularly in women in their late 30s or early 40s. There are three forms of the disease: acute, subacute, and chronic. The lesions may be single or multiple and of various sizes. Often the lesions are located on light-exposed areas of the skin, and photosensitivity is common. The face is the most common site of lesion involvement; a butterfly pattern of distribution is found over the nose and cheeks.77
The cause is thought to be an altered immune response to an unknown antigen or response to UV wavelengths with the development of self-reactive T and B cells, decreased number of regulatory T cells, and increased proinflammatory cytokines.78 Autoantibodies and immune complexes cause tissue damage.79
The lesions of DLE usually begin as red macules or papules with an adherent scale that resolve with residual atrophy, scarring, or pigment changes. The characteristic manifestations of the different categories of DLE are summarized in Table 44-5.
Table 44-5
Categories and Manifestations of Discoid Lupus Erythematosus (DLE)

Data from: Kuhn A, Biji M: Lupus 17(5):389-393, 2008; Rothfield N, Sontheimer RD, Bernstein M: Clin Dermatol 24(5):348-362, 2006.
Diagnosis of DLE is made from the presenting symptoms, biopsy of skin lesions with direct immunofluorescence, as well as histology. Skin biopsy with immunofluorescent observation reveals lumpy deposits of immunoglobulins, especially IgM, in some individuals. Individuals with DLE must use sunscreen and limit direct exposure to the sun because this initiates or exacerbates lesions. Initial treatment with potent topical steroids relieves symptoms. Calcineurin inhibitors have been used with some success.80 Antimalarial drugs (e.g., hydroxychloroquine sulfate) provide first-line systemic therapy and usually lead to clinical improvement within 1 to 3 months.81 These medications must be used with caution to prevent serious side effects.
Vesiculobullous Disorders
Vesiculobullous skin disorders represent a group of diseases that have different causes and clinical courses but share a common characteristic of vesicle, or blister, formation. Two such diseases are pemphigus and erythema multiforme.
Pemphigus
Pemphigus (meaning to blister or bubble) is a rare autoimmune blistering disease of the skin and oral mucous membranes caused by circulating autoantibodies directed against the cell surface adhesion molecule, desmoglein, at the desmosomal cell junction in the suprabasal layer of the epidermis. Immunoglobulin G (IgG) autoantibodies and C3 complement bind to the desmoglein adhesion molecules resulting in the destruction of cell-to-cell adhesion (acantholysis) in the epidermis with fluid accumulation and the resulting symptom of blister formation. A subset of autoantibodies may block keratinocyte acetylcholine receptors, disrupting keratinocyte cohesion.82 IgA autoantibodies have been found in some individuals.83 Pemphigus can occur in all age groups but is more prevalent between 40 and 50 years of age. There is a genetic predisposition as well as environmental triggers.84
Pemphigus presents in varying forms. Pemphigus vulgaris is the most common form with acantholysis at the suprabasal level. Oral lesions precede the onset of skin blistering, which is more prominent on the face, scalp, and axilla. The blisters rupture easily because of the thin, fragile overlying portion of epidermis. Pemphigus vegetans is a variant of pemphigus vulgaris with large blisters occurring in tissue folds of the axilla and groin. Pemphigus foliaceus is a milder form of the disease and involves acantholysis at the subcorneal level with blistering, erosions, scaling, crusting, and erythema usually of the face and chest. Oral mucous membranes are rarely involved. Pemphigus erythematosus is a subset of pemphigus foliaceus often associated with systemic lupus erythematosus with positive antinuclear antibodies. The lesions are generally less widely distributed.
The diagnosis of pemphigus is made from the clinical manifestations and histologic examination of the skin. Immunofluorescence demonstrates the presence of antibodies at the site of blister formation. The clinical course of the disease may range from rapidly fatal to relatively benign. The primary treatment for pemphigus is systemic corticosteroids, usually in high doses during acute episodes or when there is widespread involvement. Adjuvant immunosuppressive therapy also may be used and decreases the steroid dosage requirement. Newer methods of treatment and a clearer understanding of the pathogenesis have improved the prognosis and decreased mortality.85
Bullous Pemphigoid
Bullous pemphigoid (BP) is a more benign disease than pemphigus vulgaris, with the presence of serum and bound IgG and blistering of the subepidermal skin layer.86 Autoantibodies have been found to hemidesmosomal proteins designated BP 180 and BP 230. Loss of dermal-epidermal adhesion is caused by proteinases released by granulocytes.87 The lesions of pemphigoid begin with localized erythema or as pruritic plaques that extend and become edematous. The plaques turn reddish-purple by 2 to 3 weeks, with vesicles and bullae emerging on the surface (Figure 44-15). The bullae do not extend with pressure. The blisters rupture within 1 week and heal rapidly. BP occurs more commonly after 60 years of age.
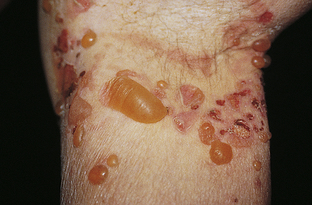
Figure 44-15 Bullous pemphigoid. Generalized eruption with blisters arising from an edematous, erythematous annular base. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Diagnosis is by skin biopsy and immunofluorescent examination. The presence of subepidermal blistering and eosinophils distinguishes pemphigoid from pemphigus. Treatment usually includes hydroxyzine (Atarax) for itching and prednisone with an immunosuppressive drug to control blistering. Individuals who respond to treatment with sulfapyridine or dapsone do not require prednisone.
Erythema Multiforme
Erythema multiforme is not a single disease but rather a syndrome characterized by inflammation of skin and mucous membranes. It often is associated with a T-cell mediated immunologic reaction to microorganisms (e.g., Mycoplasma pneumoniae, herpes simplex virus) or a toxic reaction to drugs in which TNF-α causes tissue damage.88 Overall, it is relatively rare and can occur at any age but is more common between 20 and 40 years of age. Immune complex formation and deposition of complement (C3), IgM, and fibrinogen around the superficial dermal blood vessels, basement membrane, and keratinocytes are found in most individuals with erythema multiforme. Edema develops in the superficial dermis, leading to the formation of vesicles and bullae. The lesions vary in clinical presentation and may involve the skin or mucous membranes or both. The characteristic “bull’s-eye” or “target” lesions occur on the skin surface with a central dusky region surrounded by concentric rings or alternating edema and inflammation.89 The lesions usually occur suddenly in groups over 2 to 3 weeks. Urticarial plaques, 1 to 2 cm in diameter, can develop without the target lesion. A vesiculobullous form is characterized by mucous membrane lesions and erythematous plaques over elbows and knees. Single or multiple vesicles or bullae may arise on a part of the plaque, accompanied by pruritus and burning. In the minor form there may be ten to hundreds of lesions.90 The lesions heal within 3 to 4 weeks (Figure 44-16).
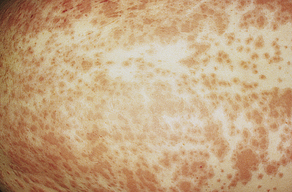
Figure 44-16 Erythema multiforme caused by doxepin. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
The most common severe forms in children and young adults are Stevens-Johnson syndrome (severe bullous form) and toxic epidermal necrolysis, in which there are numerous erythematous bullous lesions on the skin and mucous membranes. These diseases may have a different etiology than erythema multiforme.91 The cause is unknown, but an immune mechanism related to drug administration is involved.92 Bursts of nitric oxide formation have been proposed as the cause of epidermal apoptosis and necrosis.93 There is destruction of the epidermis in toxic epidermal necrolysis. Cytotoxic lymphocytes (early) and monocytes-macrophages (late) are involved in this severe blistering disease.94
Prodromal symptoms of fever, headache, malaise, sore throat, and cough develop in approximately one third of cases. The bullous lesions form erosions and crusts when they rupture. The mouth, air passages, esophagus, urethra, and conjunctiva may be involved. Blindness can result from corneal ulcerations. Difficulty with eating, breathing, and urinating may develop with severe manifestations. The disease can involve the kidneys and extend from the upper respiratory passages into the lungs. Severe forms of the disease can be fatal.
Diagnosis is made by medication history, recognition of the target lesion, or by skin biopsy if the target lesion is absent. Mild acute forms of the disease last 10 to 14 days. Mild forms of the disease, usually self-limiting, require no treatment. Any ongoing drug therapy should be withdrawn or reevaluated and underlying infections treated. Fluid and electrolyte balance should be monitored in severe forms of the disease, and mucous membranes must be carefully managed with a bland diet, warm saline eyewashes, topical anesthetics, or corticosteroids to maintain comfort and prevent infection. Use of systemic steroids is controversial.95 Cutaneous blisters can be treated with wet compresses of nanocrystalline silver. Ophthalmic, kidney, and lung involvement requires special care. Resolution occurs in 8 to 10 days, usually without scarring. Mucosal lesions may take 6 weeks to heal.
Infections
Cutaneous infections are common forms of skin disease. They generally remain localized; however, serious complications can develop with systemic involvement. The types of skin infection include bacterial, viral, and fungal. Most infections occur superficially; however, systemic signs and symptoms occasionally develop and rarely become life threatening. Aerobes, yeast, and anaerobes comprise the normal flora of the skin and often provide protection against pathogens that cause skin infections, including Staphylococcus and Streptococcus.96
Bacterial Infections
Most bacterial infections of the skin are caused by local invasion of pathogens. Coagulase-positive S. aureus and, less often, beta-hemolytic streptococci are the common causative microorganisms.97 Community-acquired methicillin-resistant Staphylococcus aureus (C-MRSA) is also a cause of serious skin infection (Box 44-2).
Folliculitis: Folliculitis is usually caused by a bacterial infection of the hair follicle. S. aureus is a common causative organism. The infection develops from proliferation of the organism around the opening of the follicle and then spreads into the follicle. Inflammation is caused by the release of chemotactic factors and enzymes from the bacteria. The lesions appear as pustules with a surrounding area of erythema. They are most prominent on the scalp and extremities and rarely cause systemic symptoms. Prolonged skin moisture, skin trauma, and poor hygiene are associated contributing factors to the development of folliculitis. Cleaning with soap and water and topical application of antibiotics are effective forms of treatment.
Furuncles and Carbuncles: A furuncle, or “boil,” is an inflammation of the hair follicles that may develop from a preceding folliculitis and spread through the follicular wall into the surrounding dermis. The invading organism is usually S. aureus. The infecting strain may spread to the skin from the anterior nares. Any skin area with hair can be infected, and one or several lesions may be present. The precipitating events are similar to folliculitis. The initial lesion is a deep, firm, red, painful nodule 1 to 5 cm in diameter (Figure 44-17). Within a few days the initial erythematous nodule changes to a large fluctuant and tender cystic nodule that may be accompanied by cellulitis. No systemic symptoms are present, and the lesion may drain large amounts of pus and necrotic tissue.
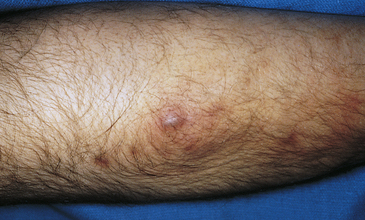
Figure 44-17 Furuncle on the forearm. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
A carbuncle is a collection of infected hair follicles occurring most often on the back of the neck, the upper back, and the lateral thighs. The lesion begins in the subcutaneous tissue and lower dermis as a firm mass that evolves into an erythematous, painful, swollen mass that drains through many openings. Abscesses may develop. Chills, fever, and malaise are systemic symptoms that can occur during the early stages of lesion development.
Furuncles and carbuncles are treated with warm compresses to provide comfort and promote localization and spontaneous drainage. Abscess formation requires incision and drainage, and recurrent infections are treated with systemic antibiotics.
Cellulitis: Cellulitis is an infection of the dermis and subcutaneous tissue usually caused by Staphylococcus or group B streptococci.98 Cellulitis can occur as an extension of a skin wound, an ulcer, or from furuncles or carbuncles. Risk factors include diabetes mellitus, edema, peripheral vascular disease, tinea pedis, insect bites, and immune suppression.99 The infected area is erythematous, warm, swollen, and painful and can extend to lymph nodes and the blood. The infection responds to systemic antibiotics, and Burow soaks can be used to relieve pain.
Erysipelas: Erysipelas is an acute superficial infection of the upper dermis (a superficial form of cellulitis) most often caused by group A streptococci. The face, ears, and lower legs are common sites of involvement, and the site of initial infection may not be identified. Chills, fever, and malaise precede the onset of lesions by 4 hours to 20 days. The initial lesions appear as firm, red spots that enlarge and coalesce to form a clearly circumscribed, advancing, bright red, hot lesion with a raised border. Vesicles may appear over the lesion and at the border, producing a bullous form of the disease. Itching, burning, and tenderness accompany the development of the lesion. Cold compresses provide symptomatic relief, and systemic antibiotics are required to arrest the infection.100
Impetigo: Impetigo is a superficial lesion of the skin caused by coagulase-positive Staphylococcus or alpha-hemolytic streptococci. It may complicate atopic dermatitis.101 The disease occurs in adults but is more common in children (see Chapter 45).
Viral Infections
Herpes Simplex Virus: There are eight types of herpes simplex virus (HSV), a group of deoxyribonucleic acid (DNA) viruses: HSV-1 (type 1), HSV-2 (type 2), cytomegalovirus (CMV), varicella-zoster virus (VZV; type 3), Epstein-Barr virus, and human herpesviruses 6, 7, and 8 (Kaposi sarcoma–associated) cause substantial neurologic morbidity among infants and children.102 A “cold sore” or “fever blister” is a type of HSV-1 infection and is the most common manifestation of HSV.103 HSV-1 usually causes infection of the cornea (herpes keratitis), mouth (gingivostomatitis), and labia (labialis). Individuals receiving cytotoxic therapy for cancer are at risk.
The lesions of HSV-1 appear as a rash or clusters of inflamed and painful vesicles within the mouth, over the tongue, or on the lips and around the nose (Figure 44-18). Increased sensitivity, paresthesias, and mild burning may occur before onset of the lesion. The vesicles rupture, forming a crust. Lesions may last 2 to 6 weeks. Occasionally there is associated upper respiratory infection. HSV-1 is transmitted by contact with infected saliva. Treatment is symptomatic, and the lesions usually resolve within 2 weeks.
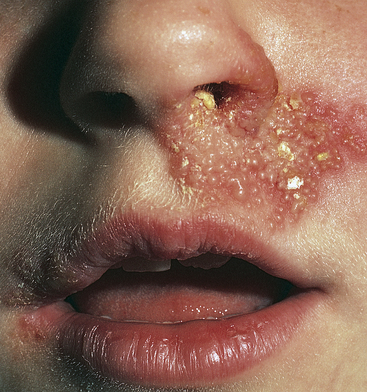
Figure 44-18 Herpes simplex labialis. Typical presentation with tense vesicles appearing on the lips and extending onto the skin. (From Habif TP: Clinical dermatology: a color guide to diagnosis and therapy, ed 4, St Louis, 2004, Mosby.)
Genital infections are more commonly caused by HSV-2. The virus is spread by skin-to-skin mucous membrane contact during viral shedding. Risk of infection is high after sexual contact with infected individuals and when there is immunosuppression. Vertical transmission from mother to neonate is associated with significant neonatal morbidity and mortality.104 After penetrating the skin, HSV is established in the sensory nerve ganglion innervating the primary site. Infection in one area does not protect other areas from subsequent infection. The primary infection is asymptomatic and can be determined only by a rising antibody titer.105
The incubation period ranges from 2 to 14 days, and clinical symptoms last 1 to 3 weeks. An individual then continues to shed the virus for 2 to 6 weeks. The virus remains dormant within sensory or autonomic nerve ganglia and can lead to recurrence of the disease. A number of factors stimulate recurrence, including sun exposure, fever, or stress, and lesions are usually located at or near the primary site. Because anti-HSV antibodies develop in response to infection, recurrence is also related to the titer or amount of antibodies present.
Genital herpes (HSV-2) also may occur in primary or recurrent forms, and a large number of infections are sexually transmitted, usually within 3 to 14 days after exposure (see Chapter 24). The lesions begin as small vesicles that progress to ulceration within 3 to 4 days with pain, itching, and weeping. Treatment includes oral or topical administration of an antiviral drug that decreases new lesion formation and promotes healing. Progress is being made with development of both therapeutic and prophylactic vaccines.106
Herpes Zoster and Varicella: Herpes zoster (shingles) and varicella (chickenpox) are caused by the same herpesvirus—VZV. Varicella is a primary infection followed years later by herpes zoster, particularly among those who are immunosuppressed.107 Chickenpox usually occurs in children (see Chapter 45).
Herpes zoster, or shingles, has initial symptoms of pain and paresthesia localized to the affected dermatome (the cutaneous area innervated by a single spinal nerve; see Chapter 14), followed by vesicular eruptions along a facial, cervical, or thoracic lumbar dermatome (Figure 44-19). Some individuals have vesicles scattered outside the area of the dermatome but lesions do not usually cross the midline. Local symptoms are alleviated with compresses, calamine lotion, or baking soda. Persistent pain is a debilitating complication, particularly in older adults and requires treatment.108 Approximately 20% of individuals experience postherpetic neuralgias.109 Antiviral drugs are useful if used within the first 72 hours.110 Treatment includes a topical lidocaine patch, anti-convulsant medication, controlled-release narcotics, and tricyclic antidepressants.108 Topical capsaicin may be used to relieve post-herpetic neuralgia. The varicella vaccine is safe and effective and may boost humoral and cellular immunity in older adults.111
Warts: Warts (verrucae) are benign lesions of the skin caused by the human papillomavirus (HPV). There are many different types of HPV, and specific viruses are associated with specific kinds and locations of lesions. An oncoprotein expressed by HPV is thought to inactivate growth controls regulated by p53 tumor-suppressor protein.112 The lesions are round and elevated with a rough, grayish surface; can occur anywhere on the skin113; and are transmitted by touch. Common warts (verrucae vulgaris) occur most often in children and are usually on the fingers, although they may be located on any skin surface or mucous membrane. Warts vary in shape, size (flat, round, or fusiform), and location (Figure 44-20). Plantar warts are usually located at pressure points on the bottom of the feet.
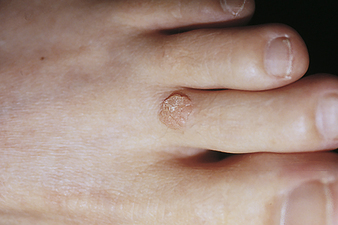
Figure 44-20 Verruca vulgaris. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Diagnosis of warts is by visualization. Treatment considers age of the individual and size and location of the lesion. Warts can be removed by freezing with liquid nitrogen, electrocautery, vaporization with lasers, application of keratolytics, or application of irritants and corrosives such as salicylic acid, formaldehyde, interferons, or podophyllum.114,115 Many warts resolve spontaneously but often recur.
Condylomata acuminata (venereal warts) are highly contagious and sexually transmitted. The cauliflower-like lesions occur in moist areas, along the glans of the penis, vulva, and anus (see Chapter 24). Oncogenic HPV is a primary cause of cervical cancer116 (see Chapter 23).
Fungal Infections
The fungi causing superficial skin infections are called dermatophytes, and they thrive on keratin (stratum corneum, hair, nails). Fungal disorders are known as mycoses; when caused by dermatophytes, the mycoses are termed tinea (dermatophytosis or ringworm).
Tinea Infections: Tinea infections are fungal infections of the skin and are classified according to their location on the body.117 The most common sites are summarized in Table 44-6. These infections are common in children (see Chapter 45). Tinea pedis is a chronic, superficial fungal infection of the skin of the foot common in adults (Figure 44-21). In prepubertal children, most scaling disorders of the toes and feet are eczema. Tinea corporis (ringworm) and tinea capitis (a fungal infection of the scalp) are much more common in children than adults. (See Chapter 45 for a discussion of fungal infections in children.)
Table 44-6
Common Sites of Tinea Infections
| Site | Clinical Manifestations |
| Tinea capitis (scalp) | Scaly, pruritic scalp with bald areas; hair breaks easily |
| Tinea corporis (skin areas, excluding scalp, face, hands, feet, groin) | Circular, clearly circumscribed, mildly erythematous scaly patches with a slightly elevated ringlike border; some forms are dry and macular, and other forms are moist and vesicular |
| Tinea cruris (groin, also known as “jock itch”) | Small erythematous and scaling vesicular patches with a well-defined border that spreads over the inner and upper surfaces of the thighs; occurs with heat and high humidity |
| Tinea pedis (foot, also known as “athlete’s foot”) | Occurs between the toes and may spread to the soles of the feet, nails, and skin of toes; slight scaling, macerated painful skin, occasionally with fissures and vesiculation |
| Tinea manus (hand) | Dry, scaly, erythematous lesions, or moist vesicular lesions that begin with clusters of intensely itching, clear vesicles; often associated with fungal infection of the feet |
| Tinea unguium or onychomycosis (nails) | A superficial or deep inflammation of the nail that develops yellow-brown accumulations of brittle keratin over all or portions of the nail |
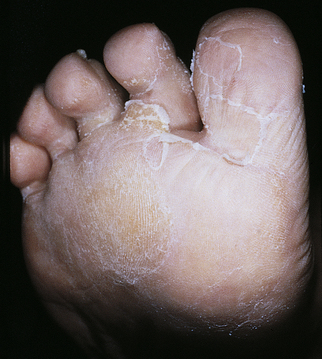
Figure 44-21 Tinea pedis. Inflammation has extended from the web area onto the dorsum of the foot. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Tinea is diagnosed by culture, microscopic examination of skin scrapings prepared with potassium hydroxide wet mount, or observation of the skin with a UV light (Wood lamp). Cultures establish the particular type of fungus and are necessary for hair and nail infections. Fungi have characteristic spores and filaments known as hyphae that are more prominent when prepared in potassium hydroxide. The spores fluoresce blue-green when exposed to UV light. Treatment is related to the type of fungi and includes both topical and systemic antifungal medication.
Candidiasis: Candidiasis is caused by the yeastlike fungus Candida albicans and normally can be found on mucous membranes, on the skin, in the gastrointestinal tract, and in the vagina. C. albicans can, under certain circumstances, change from a commensal organism to a pathogen, particularly in the critically ill and those who are immunosuppressed.118 Factors that predispose to infection include (1) a local environment of moisture, warmth, maceration, or occlusion; (2) the systemic administration of antibiotics; (3) pregnancy; (4) diabetes mellitus; (5) Cushing disease; (6) debilitated states; (7) age younger than 6 months (more likely to get an infection because of decreased immune reactivity); (8) immunosuppression; and (9) certain neoplastic diseases of the blood and monocyte-macrophage system. The resident bacteria on the skin, mainly cocci, inhibit proliferation of C. albicans. Cell-mediated immunity plays a major role in the defense against monilial infections. C. albicans can activate the complement system by the alternative pathway and can include small abscesses. Candidiasis affects only the outer layers of mucous membranes and skin and occurs in the mouth, vagina, uncircumcised penis, and large skinfolds (under breast, in arm folds and abdominal creases). Table 44-7 lists the different sites of candidiasis. Innate and adaptive immune responses are required for elimination of C. albicans.119,120
The initial lesion is a thin-walled pustule that extends under the stratum corneum with an inflammatory base that may burn or itch. The accumulation of inflammatory cells and scale produces a whitish yellow curdlike substance over the infected area. The lesion ceases to spread when it reaches dry skin.121 Antifungal medication is used for treatment.
Vascular Disorders
Vascular abnormalities are commonly associated with skin diseases, or they may be present as congenital vascular malformations (see Chapter 45) or as vascular responses to local or systemic vasoactive substances. Blood vessels may increase in number, dilate, constrict, or become obliterated by disease processes.
Cutaneous Vasculitis
Vasculitis (angiitis) is an inflammation of the blood vessels of the skin. The vasculitis is often idiopathic or can be triggered by infection, decreased blood flow (i.e., venous stasis), drugs, or autoimmune disorders. The initiating site of inflammation may be the blood, the vessel wall, or the adjacent tissue. Small vessels are usually affected. Immune complexes, which initiate an uncontrolled inflammatory response, are often the cause of damage, and the lesions are often polymorphic.
Cutaneous vasculitis develops from the deposit of immune complexes in small blood vessels as a toxic response to drugs (phenothiazines, barbiturates, sulfonamides) or allergens as a response to streptococcal or viral infection or as a component of systemic vasculitic syndromes. The precise mechanism is not known, but the deposit of immune complex activates complement, which is chemotactic for polymorphonuclear leukocytes and other mediators of inflammation which disrupt adhesion molecules and the vessel wall. The cutaneous form usually resolves in a few weeks and is treated with steroids.
The disorder is also known as allergic vasculitis and occurs primarily in adults. A systemic form (cutaneous systemic vasculitis) can involve other organs, including the kidneys, lungs, and gastrointestinal tract.122 The extremities are the chief sites, primarily the lower legs and feet. The lesions appear as palpable purpuras (from the leakage of blood from damaged vessels) and progress to hemorrhagic bullae with necrosis and ulceration from occlusion of the vessel (Figure 44-22). Lesions appear in clusters and remain from 1 to 4 weeks. Recurrences are common. Biopsy may disclose the presence of complement or immunoglobulins in the vessel walls.
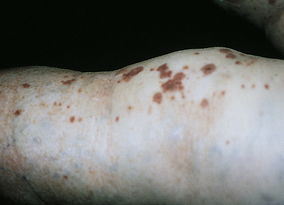
Figure 44-22 Vasculitis of the leg. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Identifying and removing the antigen (chemical, drug, or source of infection) is the first step of treatment. Corticosteroids and other drugs may be used when symptoms are severe.123
Urticaria
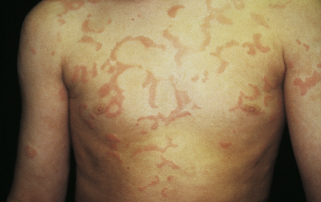
Urticaria (hives) is a circumscribed area of raised erythema and edema of the superficial dermis. Urticarial lesions are most commonly associated with type I hypersensitivity reactions to drugs (e.g., penicillin, aspirin), certain foods (e.g., strawberries, shellfish), systemic diseases (e.g., intestinal parasites, lupus erythematosus), physical agents (e.g., heat, cold), or complement-mediated reactions (see Chapter 8). The lesions are mediated by IgE-stimulated release of histamine, bradykinin, kallikrein from mast cells or basophils, or both, which causes the endothelial cells of skin blood vessels to contract.124 Other inflammatory mediators such as serotonin, leukotrienes, prostaglandins and kinins may also be mediators of urticaria. The leakage of fluid from the vessels appear as wheals, welts, or hives and may be a few leaks or many leaks distributed over the entire body (Figure 44-23). Most lesions resolve spontaneously within 24 hours, but new lesions may appear. All possible causes should be removed. Antihistamines (H1 antagonists) usually reduce hives and provide relief of itching. Epinephrine or corticosteroids and α-adrenergic agonists may be required for treatment of severe attacks (i.e., angioderma). Chronic urticaria (recurrent wheals for more than 6 weeks) is either an autoimmune or idiopathic disease.125
Scleroderma
Scleroderma means sclerosis of the skin, and the disease is associated with immune dysregulation and several autoantibodies.126 The disease is more prominent in women. Genetic predisposition, autoimmunity, and an immune reaction to a toxic substance are possible initiating mechanisms of the disease. Impaired regulation of growth factors, collagen gene expression by fibroblasts, probably underlies the persistent fibrosis.127 Systemic scleroderma (sclerosis) involves the connective tissues of many organs, including the kidney, heart, peripheral nervous system, gastrointestinal tract, and lungs.128 Only a few organs are involved in some individuals. The cutaneous lesions are most often on the face and hands, neck, and upper chest. The entire skin can be involved, however.
There are massive deposits of collagen with fibrosis, accompanied by inflammatory reactions, vascular changes in the capillary network with a decrease in the number of capillary loops, dilation of the remaining capillaries, enhanced expression of adhesion molecules, endothelial injury and dysfunction, perivascular infiltrates, and ischemia.129 Fibrosis occurs in the papillary and reticular dermis and in the subcutaneous tissue and deep fascia. The skin is hard, hypopigmented, taut, shiny, and tightly connected to the underlying tissue. The tightness of the facial skin projects an immobile masklike appearance, and the mouth may not open completely. The nose may assume a beaklike appearance. The hands are shiny and sometimes red and edematous (Figure 44-24). The fingers become tapered and flexed, often with depressed scars and loss of fingertips from atrophy. Raynaud phenomenon with episodic arteriolar vasoconstriction of the fingers contributes to ulcer formation and gangrene.130 The nails may be shed. Calcium deposits develop in the subcutaneous tissue and erupt through the skin. Progression to body organs may occur, and death is caused by subsequent respiratory failure, renal failure, cardiac dysrhythmias, or esophageal or intestinal obstruction or perforation.
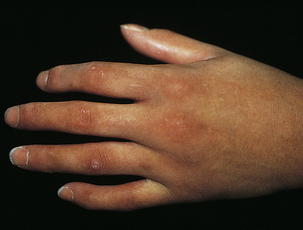
Figure 44-24 Scleroderma (acrosclerosis). Note inflammation and shiny skin. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Suitable clothing and a warm environment are essential to protecting the hands. Trauma and smoking should be avoided. Vasodilator drugs (i.e., angiotensin-converting enzyme inhibitors) or sympathectomy rarely has lasting effects. Symptomatic treatment is required for involved organs (e.g., intestinal resection for obstruction, antibiotics for pneumonitis, and regulation of hypertension).131 There is no specific treatment, and progression of the disease is variable. Broad-spectrum immunosuppression and hematopoietic bone marrow or stem cell transplantation may be used early in the disease.128,132 Fifty percent of individuals die within 5 years of the onset of scleroderma.
Insect Bites
Ticks are significant vectors of transmitted diseases, including Rocky Mountain spotted fever and other rickettsial diseases, tularemia, and Lyme disease.133 Ticks vary from 1 cm to about the size of a comma on this printed page. They embed their heads in the skin to obtain blood. As they gorge themselves on blood, they enlarge to many times their normal size and may release toxins or transmit microorganisms during feeding. In most instances, there is no consequence from a tick bite, with the exception of papular urticaria at the site of the bite. If mouthparts remain in the skin when the tick is removed, a persistent nodule remains that may require excision; ideally the tick should be removed completely intact. Irritant substances, such as camphor, soft wax, or heat from a match, may stimulate the tick to withdraw its head. Wearing protective clothing and applying tick repellant, such as diethyltoluamide (DEET), butopyronoxyl (Indalone), or benzylbenzoate, helps prevent tick bites.
Lyme disease is a multisystem inflammatory disease caused by the spirochete Borrelia burgdorferi transmitted by tick bites and is the most frequently reported vector-borne illness.134 The highest incidence is among children (50% of infected individuals are symptom free). An immune response to B. burgdorferi may contribute to the pathogenesis of the disease.135 The microorganism is difficult to culture and it escapes immunodefenses through antigenetic diversity, blocks complement-mediated killing, and hides in tissue.136,137
Symptoms of the disease occur in three stages.134 Localized infection occurs soon after the bite with erythema migrans (rash), fever, fatigue, malaise, myalgias, and arthralgias. Within days to weeks after the onset of the illness, there is disseminated infection with secondary erythema migrans, arthralgias, meningitis, neuritis, or cardiovascular symptoms. Late persistent infection can continue for years with arthritis, encephalopathy, or polyneuropathy (see Chapter 16). The diagnosis of Lyme disease is based on the clinical presentation and history of tick bite, if known. Serologic tests often are used to confirm the diagnosis.138 Antibiotics (i.e., doxycycline [not used in children younger than 8 years] or amoxicillin) are used for treatment.134 Reinfection can occur. Vaccines are in development, and personal protection is important to disease prevention.139
Mosquitoes and Flies
There are thousands of species of mosquitoes throughout the world. Species from the Culicidae family are responsible for malaria, yellow fever, dengue fever, filariasis, and St. Louis encephalitis. Several different species of mosquitoes can carry the West Nile virus which causes encephalitis (see Chapters 9 and 17). Mosquitoes can bite through thin, loose clothing and are attracted by warmth and sweat. The edema, pruritus, and papular lesions of the mosquito bite are caused by the disruption of the skin from the insertion of a blood tube by a female mosquito. Irritating salivary secretions also contain anticoagulants. Reactions vary depending on the sensitivity of the victim.
Several species of flies are blood suckers. The black fly (Simuliidae) is usually found in swarms—near moving bodies of water in the late spring and early summer—and is a vicious biter. The initial bite is painless because the fly injects an anesthetic with the bite. Subsequent lesions are painful and accompanied by significant swelling of surrounding tissues. Systemic reactions, such as fever, headache, and nausea, are common.
Very small flies of the Ceratopogonidae family, also known as “no-see-ums,” “midges,” “punkies,” or sand fleas, are also blood suckers. The bite of the female is particularly miserable and produces immediate pain, erythema, and vesicles. Itching and vesicular reactions may persist for weeks.
The fiercest blood-sucking flies are the Tabanidae, or horseflies, deerflies, gadflies, greenheads, and clegs. These flies vary in size from 1 to 5 cm and produce painful, bleeding bites because of their large mouthparts. The bites produce urticaria that may be accompanied by weakness, dizziness, and wheezing.
Wounds produced by biting insects should be cleansed with soap and water, and a local antiseptic should be applied. Local applications of steroid creams or antihistamine will reduce symptoms. Systemic reactions may require more specific medical care.
Benign Tumors
Most benign tumors of the skin are associated with aging. Benign tumors include seborrheic keratosis, keratoacanthoma, actinic keratosis, and moles.
Seborrheic Keratosis
Seborrheic keratosis is a benign proliferation of cutaneous basal cells that produces smooth or warty elevated lesions. The pathogenesis is unknown. They are usually seen in older people and occur as multiple lesions on the chest, back, and face. The color varies from tan to waxy yellow, flesh colored, or dark brown-black. Lesion size varies from a few millimeters to several centimeters, and they are often oval and greasy appearing with a hyperkeratotic stuck-on scaly appearance (Figure 44-25). Cryotherapy with liquid nitrogen or electrocautery are effective treatments, and the lesions usually slough 2 to 3 weeks after treatment.
Keratoacanthoma
A keratoacanthoma is a benign self-limiting tumor of squamous cell differentiation arising from hair follicles. It usually occurs on sun-damaged skin of older adults and smokers; the incidence is high in males. The most commonly affected sites are the face, back of the hands, forearms, neck, and legs (Figure 44-26). The lesion develops over a period of 1 to 2 months and has a histology resembling a well-differentiated squamous cell carcinoma as follows140:
Figure 44-26 Keratoacanthoma. Classic presentation of a fully developed tumor. Round, smooth, dome-shaped mass with a central keratin-filled crater. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Proliferative stage: lesion develops as a rapidly growing, dome-shaped nodule with central crust.
Mature stage: Lesion fills with whitish keratin and requires differentiation from squamous cell carcinoma.141
Involution stage: Occurs over a 3- to 4-month period with regression of the lesion.
Although the lesions resolve spontaneously, they can be removed by curettage or excision to improve cosmetic appearance. Intralesional methotrexate also has been effective.142
Actinic Keratosis
Actinic keratosis is a premalignant lesion found on skin surfaces exposed to the UV radiation of the sun. The prevalence is highest in individuals with unprotected light-colored skin. Actinic keratosis is rare in black skin. The lesions appear as rough or scaly, poorly defined pink to reddish or reddish- brown papules that are felt more than seen (Figure 44-27) and are considered an early in situ squamous cell carcinoma.143 Surrounding areas may have telangiectasia. Treatment options include ablative and topical therapies. The lesions should continue to be evaluated for progressive squamous cell carcinoma.144 Protection from the sun with clothing or a sunblock to prevent lesions from developing elsewhere is advised.
Nevi
Nevi (moles) are pigmented or nonpigmented lesions that form from melanocytes beginning at ages 3 to 5 years. During the early stages of development, the cells accumulate at the junction of the dermis and epidermis and are macular lesions. Over time the cells move down into the dermis and the nevi become nodular and palpable. Nevi may appear on any part of the skin, and vary in size. They occur singly or in groups and are not considered disfiguring. Nevi may undergo transition to malignant melanomas (see p. 1670). Nevi irritated by clothing can be excised.
Cancer
Skin cancers account for about 5% of all cancers, and their incidence is increasing.145 Basal cell carcinoma and squamous cell carcinoma are the most common. Incidence of these carcinomas is greater in men than in women, and increases steadily with age. Malignant melanoma is the most serious: an estimated 11,590 people die of skin cancer each year, 8650 of which are from malignant melanoma.145 Important trends related to skin cancer are presented in Box 44-3.
UV solar radiation causes most skin cancers by inducing mutations in the TP53 tumor-suppressor gene.146 Protection from the sun during the childhood years of life significantly reduces the risk of skin cancer in later years.147 Areas widely exposed to the sun’s rays—face, neck, and hands—are highly vulnerable for such lesions. Outdoor workers (farmers, sailors, fishermen) are high-risk populations. Like other cancers, skin cancers progress through stages of initiation, progression, and metastasis.
Basal Cell Carcinoma
Basal cell carcinoma is a surface epithelial tumor of the skin originating from undifferentiated basal or germinative cells. The tumors grow upward and laterally or downward to the dermal epidermal junction (Figure 44-28). They usually have depressed centers and rolled borders. Early tumors are so small that they are not clinically apparent.
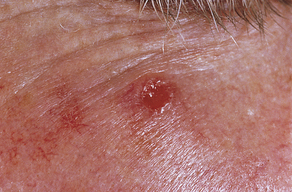
Figure 44-28 Basal cell carcinoma. Center has ulcerated. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Basal cell carcinoma is the most common type of skin cancer in whites and is thought to be caused by UV radiation exposure.148 Lesions are seen most often on people who live in regions with intense sunlight and on those areas of the skin most exposed—namely, the face and neck. Dark-skinned persons and those avoiding sunlight are significantly less likely to develop these malignant tumors. In dark-skinned persons, basal cells contain the pigment melanin, a protective factor against sun exposure. Basal cell carcinoma arises as a consequence of UV-associated mutation in the TP53 tumor-suppressor gene, leading to loss of keratinocyte repair functions and apoptosis resistance of DNA-damaged cells.149 Other factors also are implicated: arsenic from groundwater wells150 (alters DNA repair and causes mutation in TP53 tumor-suppressor gene), autosomal dominant nevoid basal cell carcinoma syndrome with mutation in the PTCH1 gene that has tumor-suppressor activity, and alteration in the Sonic Hedgehog signaling pathway genes (important for cell growth and differentiation).151,152
The lesion starts as a nodule (greater than 5 mm across) that is pearly or ivory in appearance and slightly elevated above the skin surface; it has small blood vessels on the surface. As the lesion grows, it often ulcerates, develops crusting, and becomes firm to the touch. If left untreated basal cell lesions invade surrounding tissues and, over months or years, can destroy a nose, an eyelid, or an ear (for treatment, see Box 44-3). Metastatic spread is rare because these tumors do not invade blood or lymph vessels.
Squamous Cell Carcinoma
Squamous cell carcinoma is a tumor of the epidermis characterized by two types: in situ (Bowen disease) and invasive. Areas affected are the head and neck (75%) and the hands (15%), with 10% of squamous cell carcinomas occurring elsewhere on the body. These tumors are more predominant in countries where arsenic is found in higher rates in drinking water. Gamma rays and x-rays are also associated with squamous cell carcinoma. In addition, patients who are immunosuppressed experience a greater occurrence of this carcinoma.
UV exposure causes squamous cell carcinoma, particularly with mutation of the TP53 gene.153 It is unclear how UV light produces alterations in DNA, and DNA repair.154 Invasive squamous cell carcinoma can arise from premalignant lesions of the skin. It rarely arises from normal-appearing skin or de novo. The premalignant lesions include sun-damaged skin or dysplasias (actinic dermatitis); leukoplakia, or whitish, discolored areas; scars; radiation-induced keratosis; tar and oil keratosis; and chronic ulcers and sinuses. The invasive type grows more rapidly than basal cell carcinomas and can spread to regional lymph nodes with metastasis. These tumors are firm and increase in elevation and diameter. The surface may be granular and bleed easily (Figure 44-29).
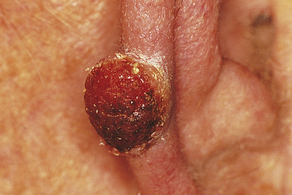
Figure 44-29 Squamous cell carcinoma. The sun-exposed ear is a common site for squamous cell carcinoma. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
In situ squamous cell carcinoma is usually confined to the epidermis (intraepidermal) but may extend into the dermis. Common premalignant skin lesions associated with in situ squamous cell carcinomas are actinic (solar) keratosis and Bowen disease. Actinic keratosis is a white, scaly, keratotic (horny) lesion on the exposed areas of the body (see p. 1668). Bowen disease is a dysplasia of the basal layer of the dermis or carcinoma in situ. It often is found on unexposed areas of the body and is demonstrated by flat, reddish, scaly patches. These lesions may enlarge to more than 1 cm in diameter, rarely invading surrounding tissue and almost never metastasizing. Other cellular components in the skin (sweat glands, hair follicles, etc.) can give rise to skin cancer, but these cancers are relatively uncommon.
Malignant Melanoma
Melanoma is a malignant tumor of the skin originating from melanocytes, or cells that synthesize the pigment melanin. The incidence of melanoma is increasing, and young to middle-age adults are at highest risk. Risk factors implicated in melanoma induction include genetic predisposition, exposure to ultraviolet light (solar and artificial), steroid hormone activity, fair hair, light skin with a propensity to sunburn, moles, and susceptibity genes155,156 (see What’s New? Melanoma, UVA Exposures and Cutaneous Vitamin D3).
Melanomas arise as a result of malignant degeneration of melanocytes located either along the basal layer of the epidermis or in a benign melanocytic nevus. The pathogenesis of malignant melanoma is complex and a number of proto-oncogenes have been identified.157
A nevus, or mole, is an aggregation of melanocytes (Figure 44-30). These clusters of cells may not be apparent until puberty, when the pigmentation process is initiated by steroid
WHAT’S NEW?
Melanoma, UVA Exposures and Cutaneous Vitamin D3
The incidence of cutaneous malignant melanoma (CMM) has been increasing over the past decades among those with white skin, primarily among indoor workers and those living in Australia, North America, and Europe; however, the mortality rate is stable or decreasing. Although intermittent, intense overexposures to the sun with sunburns can initiate CMM, sun exposure, particularly exposure to ultraviolet B (UVB) wavelengths (290-320 nm) may also have a protective action or an action that improves survival. One theory proposes that indoor workers are overexposed to ultraviolet A (UVA) wavelengths (321-400 nm) because UVA passes through glass windows whereas UVB does not. UVB promotes formation of vitamin D3 in the skin which is then converted by the liver and kidney to its active form calcitriol which kills melanoma cells in-vitro and reduces tumor growth in-vivo. Melanoma cells can also convert skin vitamin D3 to the active form. The action of UVA causes mutations in melanocytes and breaks down vitamin D3. Inadequately maintained vitamin D3 decreases its protective effects of reducing the promotion of melanoma. Thus, although intermittent overexposure to UVB can cause melanoma, overexposure to UVA and inadequate calcitriol may promote melanoma. Continuing research is needed to determine the actual UV exposures and levels of calcitriol needed to prevent melanoma.
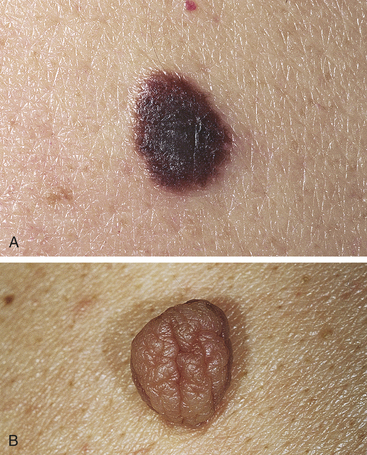
Figure 44-30 Nevi. A, Junction nevus: slightly raised, dark, and uniform. B, Dermal nevus: pedunculated with a soft, flabby, wrinkled surface. (From Habif TP: Clinical dermatology: a color guide to diagnosis and therapy, ed 4, St Louis, 2004, Mosby.)
Data from: Godar DE, Landry RJ, Lucas AD: Med Hypotheses 72(4):434-443, 2009; Moan J, Porojnicu AC, Dahlback A: Adv Exp Med Biol 624:104-116, 2008; Leiter U, Garbe C: Adv Exp Med Biol 624:89-103, 2008; Bennett DC: Pigment Cell Melanoma Res 21(5):520-524, 2008.
hormones. The relationship between nevi and melanoma makes it important for the clinician to understand the various neval forms (Table 44-8). Most nevi never become suspicious; however, suspicious pigmented nevi should be removed.158 Indications for biopsy include color change, size change, irregular notched margin, itching, bleeding or oozing, nodularity, scab formation, ulceration, or an unusual pattern of presentation. The ABCDE rule is used as a guide: Asymmetry, Border irregularity, Color variation, Diameter larger than 6 mm, and Elevation which includes raised appearance or rapid enlargement.158 Clinical characteristics are summarized in Table 44-9.
Table 44-8
| Nevi | Common Characteristics |
| Junctional nevus | Flat, well circumscribed, vary in size up to 2 cm, dark color, hairs may be present; originate in basal layer of epidermis and can eventually reach the cutaneous surface; rarely develop into a melanoma |
| Compound nevus | Most common in adolescents; the majority of pigmented lesions are in children; rarely develops into melanoma; usually 1 cm in size; hairs may be present; surface is elevated and smooth |
| Intradermal nevus | Small (less than 1 cm) with regular edges and bristle-like hairs; color ranges from skin tone to light brown; has a slight likelihood of developing into a melanoma |

Staging of melanoma is determined from tissue biopsy, by assessing the thickness of the lesion using the Breslow microstage, and the Clark levels of tumor invasion159 (Figure 44-31). The clinical classifications of cutaneous melanoma include lentigo malignant melanoma (LMM) (Figure 44-32), superficial spreading melanoma (SSM) (Figure 44-33), primary nodular melanoma (PNM), and acral lentiginous melanoma. A melanotic melanoma is the rarest subtype and is a nonpigmented melanoma.160 Efforts are in progress to refine the morphologic classifications by adding subgroups that include molecular markers for proto-oncogene mutations and specific signaling pathways.161,162 Early recognition of cutaneous melanomas can have a major effect on surgically curing this disease.
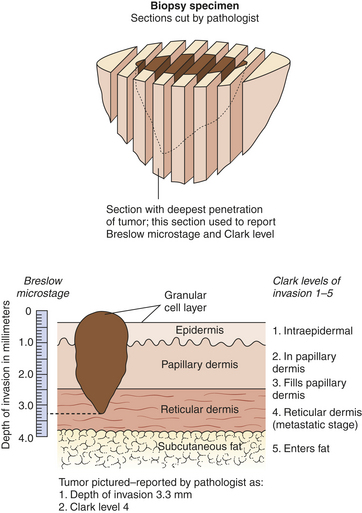
Figure 44-31 Melanoma staging using Breslow and Clark levels of invasion. (From Habif TP: Clinical dermatology: a color guide to diagnosis and therapy, ed 4, St Louis, 2004, Mosby.)

Figure 44-32 Lentigo malignant melanoma. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
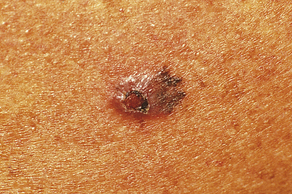
Figure 44-33 Level IV melanoma. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Treatment of melanoma with no evidence of metastatic disease involves surgical excision to the primary site and regional lymph nodes. The extent of surgery is determined by the staging of disease. Lesions of the extremities have the best prognosis; head and neck lesions and trunk lesions have the poorest prognosis. Survival rate for advanced disease is very low. Immunotherapy is advancing and vaccines and gene therapy are under investigation.163,164
Kaposi Sarcoma
Kaposi sarcoma (KS) is a vascular malignancy with four different presentations:
1. In association with drug-induced immunosuppressions, for example, after kidney transplantation
2. An endemic form in equatorial Africa
3. A classic form presenting on the lower legs of older men
4. In association with acquired immunodeficiency syndrome (AIDS)
Kaposi-associated herpesvirus 8 is found in all four forms of KS and may be a common etiology for all types of KS through the development of an angiogenic-inflammatory state.165 Immunosuppression allows for opportunistic infections and malignancy. Proliferation of the tumor depends on the presence of platelet-derived and other growth factors.166
The human immunodeficiency virus (HIV) and CMV have been proposed as cofactors in the development of KS.167 The endothelial cell is thought to be the progenitor of KS but the specific origin is elusive. The lesions emerge as purplish- brown macules and develop into plaques and nodules. They tend to be multifocal rather than spreading by metastasis. The lesions initially appear over the lower extremities in the classic form (Figure 44-34). The rapidly progressive form associated with AIDS tends to spread symmetrically over the upper body, particularly the face and oral mucosa. The lesions are often pruritic and painful. About 75% of individuals with epidemic KS have involvement of lymph nodes, particularly in the gastrointestinal tract and lungs. Organ involvement is much less common in the classic form. The rapidly progressive form has a poor prognosis and shorter survival rates than the classic form. (See Chapter 9 for a further discussion on AIDS.)
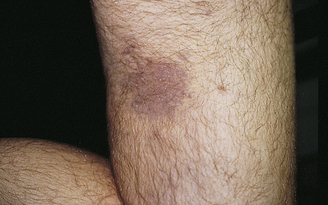
Figure 44-34 Kaposi sarcoma. The purple lesion commonly seen on the skin. (Courtesy Department of Dermatology, School of Medicine, University of Utah.)
Diagnosis is by skin biopsy, with a high index of suspicion for those with immunodeficiency. The disease is incurable. Local lesions can be excised. Multiple disseminated lesions may be treated with a combination of immunomodulator, cytotoxic, and antiviral drugs. The new, highly active antiretroviral therapy (HAART) for AIDS treatment is decreasing the incidence of KS, but new treatments are needed.168
Frostbite
Frostbite is injury to the skin caused by exposure to extreme cold. The areas most commonly affected are fingers, toes, ears, nose, and cheeks. Initially the body responds with alternating cycles of vasoconstriction and vasodilation—“the burning reaction.” The mechanism of injury is complex but appears to be related to direct cold injury to cells, indirect injury and cell death from extracellular and intracellular ice crystal formation, and impaired circulation with anoxia because of thrombosis in the exposed area.169 The inflammatory mediators of frostbite are similar to burns and include prostaglandins, thromboxanes, bradykinin, and histamine. Reperfusion injury is part of the pathophysiology.170 Frozen skin becomes white or yellowish and is waxy. There is numbness and no sensation of pain.
Skin damage can range from mild to severe. With mild frostbite, redness and discomfort occur during rewarming, followed by a return to normal in a few hours. In more severe cases, cyanosis and mottling develop, followed by redness, swelling, and burning pain on rewarming. Within 24 to 48 hours, vesicles and bullae appear and resolve into crusts that eventually slough off, leaving thin, newly formed skin. The most severe cases result in gangrene with loss of the affected part. Frostbite may be classified by depth of injury after rewarming as follows171:
First degree: Superficial, characterized by a numb central white area surrounded by erythema and edema and including partial skin freezing without blistering
Second degree:. Full-thickness skin freezing with blistering surrounded by edema and hyperemia
Third degree: Deep, characterized by full-thickness skin and subcutaneous freezing with tissue necrosis and hemorrhagic vesicles
Fourth degree: Deep tissue freezing with full-thickness necrosis and gangrene
Immediate treatment of frostbite is to cover affected areas with other body surfaces and warm clothing. The area should not be rubbed or massaged. Local, dry heat should be avoided. Immersion in a warm water bath (40° to 42° C 104° to 107.6° F]) until frozen tissue is thawed is the best treatment. Aspirin is used to inhibit prostaglandins, and aloe vera is a topical inhibitor of thromboxane.172 Thrombolytic therapy reduces the incidence of amputation when administered with 24 hours.173 Pain during the thawing period is severe and should be treated with potent analgesics. Gentle cleansing and no pressure on the skin should be maintained during healing. Amputation of necrotic tissue is delayed until a clear line of demarcation is established.
DISORDERS OF THE HAIR
Male-Pattern Alopecia (Androcentric Alopecia)
Alopecia means loss of hair. Localized hair loss in men is not a disease but rather a genetically predisposed response to androgens. The mechanism of inheritance is unknown. Within the distribution of hair over the scalp, androgen-sensitive hair follicles are on top and androgen-insensitive follicles are on the sides and back. In genetically predisposed men, the androgen-sensitive follicles are transformed into vellus follicles. The normal hair is shed and replaced by fine, light, short hair. Male-pattern baldness begins with frontotemporal recession and progresses to loss of hair over the top of the scalp. Minoxidil may be used to stimulate hair growth and finasteride may decrease the effect of androgens on hair follicles.174 Affected men may choose to wear wigs or have hair transplants.
Female-Pattern Alopecia
Some women in their 20s and 30s experience progressive thinning and loss of hair over the central part of the scalp.175 Contrary to male-pattern baldness, no loss of hair occurs along the frontal hairline. Many of these women have elevated levels of serum adrenal androgen dehydroepiandrosterone sulfate (DHEAS), and treatment with antiandrogens can be effective.176,177 In rare instances a male-pattern baldness develops. Laboratory evaluation of serum androgenic hormones shows elevations, and some women have decreased hair loss when treated with daily doses of spironolactone.
Alopecia Areata
Alopecia areata is an autoimmune T-cell–mediated chronic inflammatory disease directed at hair follicles that results in baldness.178 Hair loss occurs in multiple areas of the scalp, usually in round patches.179 The eyebrows, eyelashes, beard, and other areas of the body are rarely involved. The cause is unknown, but stressful events, cell-mediated immune factors, and genetic susceptibility are linked to hair loss. Metabolic disorders, such as Addison disease, thyroid disease, and lupus erythematosus, also are associated with alopecia areata.180
The affected areas of skin are smooth or may have short shafts of hair. The hair shaft is poorly developed and breaks at the surface. Regrowth occurs within 1 to 3 months, but hair loss may recur at the same site. Permanent regrowth of hair usually occurs. Total loss of hair (alopecia totalis) occurs in some young people; the long-term prognosis for total hair regrowth is poor.
Diagnosis is made by observation of the pattern of hair loss. Biopsy may show a lymphocytic infiltrate around the follicle. There are several treatments for alopecia areata including corticosteroids and topical immunotherapy.181
Hirsutism
Hirsutism is the abnormal growth and distribution of hair on the face, body, and pubic area in a male pattern that occurs in women. There is also frontotemporal hair recession. These areas of hair growth are androgen sensitive.182 Variations of hair growth in women are great, and a male pattern can be normal. Women who develop hirsutism may be secreting hormones associated with ovarian or adrenal disease, and such women should be evaluated for polycystic ovaries, adrenal hyperplasia, or adrenal tumors.183If no hormonal pathologic conditions exist undesirable hair can be mechanically removed.184 Medical treatments are available by prescription.185
DISORDERS OF THE NAIL
Paronychia is an acute or chronic infection of the cuticle.186 Acute paronychia is manifest by the rapid onset of painful inflammation of the cuticle, usually after minor trauma. An abscess may develop, requiring incision and drainage for relief of pain. The most common causative organisms are staphylococci, streptococci, and occasionally Candida.
Chronic paronychia develops slowly, with tenderness and swelling around the proximal or lateral nail folds. One or more fingers or toes may be involved. Individuals whose hands are frequently exposed to moisture are at greatest risk. Manipulation of the cuticle can be predisposing because it opens the space between the proximal nail fold and nail plate, leaving a moist, warm medium for pathogenic organisms to incubate. The skin around the nail becomes more edematous and painful with progressive infection. Pus may be expressed from the proximal nail fold. The nail plate is usually not affected, although it can become discolored and develop ridges.
Treatment includes keeping the hands dry. Oral antifungals are not very effective because they do not penetrate the affected tissues. Topical application of steroid creams can be effective.
Onychomycosis
Onychomycosis is a fungal or dermatophyte infection of the nail plate that occurs in 2% to 18% of the population.187 The most common pattern is a nail plate that turns yellow or white (infection develops from the dorsal surface) and becomes elevated as a result of the accumulation of hyperkeratotic debris within the plate. Fungal infections of the nail may require culture and microscopy. In psoriasis, pitting often is found on the nail surface.188 Treatment includes débridement and systemic antifungal therapy.189 Surgical excision of the nail may be required. New systemic and topical antifungals are being investigated.190
Acne rosacea 1659
Acne vulgaris 1659
Actinic keratosis 1668
Allergic contact dermatitis 1655
Alopecia 1673
Alopecia areata 1674
Apocrine sweat gland 1646
Atopic dermatitis 1656
Basal cell carcinoma 1669
Basal layer (stratum basale) 1644
Bullous pemphigoid 1661
Candidiasis 1665
Carbuncle 1663
Cellulitis 1663
Clawlike prolongation 1654
Condylomata acuminata 1664
Cutaneous vasculitis 1666
Dermal appendage 1646
Dermis 1645
Discoid (cutaneous) lupus erythematosus (DLE) 1659
Eccrine sweat gland 1646
Eczema 1655
Epidermis 1644
Erysipelas 1663
Erythema multiforme 1661
Erythrodermic (exfoliative) psoriasis 1658
Flies 1668
Folliculitis 1662
Frostbite 1673
Furuncle 1662
Germinative layer (stratum germinativum) 1644
Guttate psoriasis 1658
Hair follicle 1646
Herald patch 1658
Herpes simplex virus (HSV) 1663
Herpes zoster 1664
Hirsutism 1674
Hypertropic scar 1654
Hyphae 1665
Impetigo 1663
Inverse psoriasis 1657
Irritant contact dermatitis 1656
Kaposi sarcoma (KS) 1672
Keloid 1654
Keratin 1644
Keratinocytes 1644
Keratoacanthoma 1668
Langerhans cell 1645
Lichen planus 1658
Lupus erythematosus 1659
Lyme disease 1667
Melanocyte 1645
Melanoma 1670
Merkel cell 1645
Mosquito 1668
Myofibroblast 1654
Nail 1646
Nevi (sing., nevus; also known as a mole) 1669
Onychomycosis 1674
Papillary capillary 1646
Papulosquamous disorder 1657
Paronychia 1674
Pemphigus 1660
Pityriasis rosea 1658
Plaque psoriasis 1657
Proteoglycan 1654
Pruritus 1654
Psoriasis 1657
Psoriatic arthritis 1658
Psoriatic nail disease 1658
Pustular psoriasis 1658
Scleroderma 1667
Sebaceous gland 1646
Seborrheic dermatitis 1656
Seborrheic keratosis 1668
Spinous layer (stratum spinosum) 1644
Squamous cell carcinoma 1670
Stasis dermatitis 1656
Stevens-Johnson syndrome 1661
Stratum corneum 1644
Systemic scleroderma (sclerosis) 1667
Tinea capitis 1664
Tinea corporis (ringworm) 1664
Tinea infection 1664
Tinea pedis 1664
Toxic epidermal necrolysis 1661
Urticaria 1666
Urticarial lesion 1666
Varicella 1664
Wart 1664
REFERENCES
1. Loser, K., Beissert, S. Dendritic cells and T cells in the regulation of cutaneous immunity. Adv Dermatol. 2007;23:307–333.
2. Nicol, N.H. Anatomy and physiology of the skin. In Hill M.J., ed.: Dermatologic nursing essentials: a core curriculum, ed 2, Pitman, NJ: Anthony J. Jannetti, Inc, 2003.
3. Blanpain, C., Fuchs, E. Epidermal stem cells of the skin. Ann Rev Cell Dev Biol. 2006;22:339–373.
4. Landau, M. Exogenous factors in skin aging. Curr Probl Dermatol. 2007;35:1–13.
5. Thornfeldt, C.R. Chronic inflammation is etiology of extrinsic aging. J Cosmet Dermatol. 2008;7(1):78–82.
6. Baumann, L. Skin aging and its treatment. J Pathol. 2007;211(2):241–251.
7. Imokawa, G. Recent advances in characterizing biological mechanisms underlying UV-induced wrinkles: a pivotal role of fibroblast derived elastase. Arch Dermatol Res. 2008;300(Suppl 1):s7–s20.
8. Gilchrist, B.A. Skin and aging processes. Boca Raton, FL: CRC Press; 1984.
9. Ryan, T. The ageing of the blood supply and the lymphatic drainage of the skin. Micron. 2004;35(3):161–171.
10. Bennett, M.F., et al. Skin immune systems and inflammation: protector of the skin or promoter of aging? J Investig Dermatol Symp Proc. 2008;13(1):15–19.
11. Farage, M.A., et al. Clinical implications of aging skin: cutaneous disorders in the elderly. Am J Clin Dermatol. 2009;10(2):73–86. [doi: 10.2165/00128071-200910020-00001.].
12. Cannon, B.C., Cannon, J.P. Management of pressure ulcers. Am J Health Syst Pharm. 2004;61(18):1895–1905.
13. Stekelenburg, A., et al. Deep tissue injury: how deep is our understanding. Arch Phys Med Rehabil. 2008;89(7):141–143.
14. Berlowitz, D.R., et al. Predictors of pressure ulcer healing among long-term care residents. J Am Geriatric Soc. 1997;45(1):30–34.
15. Theaker, C., et al. Risk factors for pressure sores in the critically ill. Anesthesia. 2000;55(3):221.
16. Frankel, H., Sperry, J., Kaplan, L. Risk factors for pressure ulcer development in a best practice surgical intensive care unit. Am Surg. 2007;73(12):1215–1217.
17. Baumgarten, M., et al. Black/white differences in pressure ulcer incidence in nursing home residents. J Am Geriatr Soc. 2004;19(6):339–341.
18. Scanlon, E., Stubbs, N. Pressure ulcer risk assessment in patients with darkly pigmented skin. Prof Nurse. 2004;19(5):339–341.
19. National Pressure Ulcer Advisory Panel: Pressure ulcer stages revised by NPUAP, 2007. Available at www.npuap.org/pr2.htm.
20. Ayello EA: How and why to do pressure ulcer risk assessment, Advances in Skin & Wound Care. FindArticles.com. 28 May, 2009. Available at: http://findarticles.com/p/articles/mi_qa3977/is_200205/ai_n9033549/
21. Royal College of Nursing, The management of pressure ulcers in primary and secondary care. London: Royal College of Nursing; 2005. Available at www.nice.org.uk/nicemedia/pdf/cg029fullguideline_appendices.pdf
22. Butler, F. Essence of care and the pressure ulcer benchmark-an evaluation. J Tissue Viabil. 2008;17(2):44–59.
23. Reddy, M., Gill, S.S., Rochon, P.A. Preventing pressure ulcers: a systematic review. J Am Med Assoc. 2006;296(8):974–984.
24. Smith, D.M. Pressure ulcers in the nursing home. Ann Intern Med. 1995;123(6):433.
25. Fine, N.A., Mustoe, T.A. Wound healing. In Greenfield L.J., et al, eds.: Surgery: scientific principles and practice, ed 2, Philadelphia: Lippincott-Raven, 1997.
26. Bauer, J., Phillips, L.G. MOC-PSSM CME article: pressure sores. Plast Reconstr Surg. 2008;121(1Suppl):1–10.
27. Lee, J.T., et al. A new technique of transferring island pedicled anterolateral thigh and vastus lateralis myocutaneous flaps for reconstruction of recurrent ischial pressure sores. J Plast Reconstr Aesthet Surg. 2007;60(9):1060–1066.
28. Sorenson, J.L., Jorgensen, B., Gottrup, F. Surgical treatment of pressure ulcers. Am J Surg. 2004;188(Suppl 1A):42–51.
28a. George-Saintilus, E., et al. Pressure ulcer PUSH score and traditional nursing assessment in nursing home residents: do they correlate? J Am Med Dir Assoc. 2009;10(2):141–144.
29. Köse, O., Waseem, A. Keloids and hypertrophic scars: are they two different sides of the same coin? Dermatol Surg. 2008;34(3):336–346.
30. Seifert, O., et al. Identification of unique gene expression patterns within different lesional sites of keloids. Wound Repair Regen. 2008;16(2):254–265.
31. Ghazizadeh, M., et al. Functional implications of the IL-6 signaling pathway in keloid pathogenesis. J Invest Dermatol. 2007;127(1):98–105.
32. Satish, L., et al. Gene expression patterns in isolated keloid fibroblasts. Wound Repair Regen. 2006;14(4):463–467.
33. Bayat, A., et al. Genetic susceptibility to keloid disease: transforming growth factor beta receptor gene polymorphisms are not associated with keloid disease. Exp Dermatol. 2004;13(2):120–124.
34. Durani, P., Bayat, A. Levels of evidence for the treatment of keloid disease. J Plast Recontr Aesthet Surg. 2008;61(1):4–17.
35. Robles, D.T., et al. Keloids: pathophysiology and management. Dermatol Online J. 2007;13(3):9.
36. Greaves, M.W., Wall, P.D. Pathophysiology of itching. Lancet. 1996;348(9032):938.
37. Pogatzki-Zahn, F., et al. Chronic pruritus: targets, mechanisms and future therapies. Drug News Perspect. 2008;21(10):541–551.
38. Ständer, S., Weisshaar, E., Luger, T.A. Neurophysiological and neurochemical basis of modern pruritus treatment. Exp Dermatol. 2008;17(3):161–169.
39. Weldon, D. What lies beneath the surface of the itch in adults. Allergy Asthma Proc. 2007;28(2):153–162.
40. Greaves, M.W. Recent advances in pathophysiology and current management of itch. Ann Acad Med Singapore. 2007;36(9):788–792.
41. Yosipovitch, G., Samuel, L.S. Neuropathic and psychogenic itch. Dermatol Ther. 2008;21(1):32–41.
42. Langner, M.D., Maibach, H., I. Pruritus measurement and treatment. Clin Exp Dermatol. 2009;34(3):285–288.
43. Nilsson, H.J., et al. Profound inhibition of chronic itch induced by stimulation of thin cutaneous nerve fibres. J Eur Acad Dermatol Venereol. 2004;18(1):37–43.
44. Ständer, S., Weisshaar, E., Luger, T.A. Neurophysiological and neurochemical basis of modern pruritus treatment. Exp Dermatol. 2008;17(3):161–169.
45. Nicol, N.H. Dermatitis/eczema. In Hill M.J., ed.: Dermatologic nursing essentials: a core curriculum, ed 2, Pitman, NJ: Anthony J. Jannetti, Inc, 2003.
46. Gober, M.D., Gaspari, A.A. Allergic contact dermatitis. Curr Dir Autoimmun. 2008;10:1–26.
47. Fyhrquist-Vanni, N., Alenius, H., Lauerma, A. Contact dermatitis. Dermatol Clin. 2007;25(4):613–623.
48. Rolland, J.M., O’Hehir, R.E. Latex allergy: a model for therapy. Clin Exp Allergy. 2008;38(6):898–912.
49. Chan, L.S. Atopic dermatitis in 2008: Curr Dir Autoimmun. 2008;10:76–118.
50. Trent, J.T., et al. Venous ulcers: pathophysiology and treatment options. Ostomy Wound Manage. 2005;51(5):38–54.
51. Howard, D.P., et al. The role of superficial venous surgery in the management of venous ulcers: a systematic review. Eur J Vasc Endovasc Surg. 2008;36(4):458–465.
52. Palfreyman, S.J., et al. Dressings for healing venous leg ulcers. Cochrane Database Syst Rev. 3, 2006. [CD001103].
53. Fluhr, J.W., et al. Skin irritation and sensitization: mechanisms and new approaches for risk assessment. 1. Skin irritation. Skin Pharmacol Physiol. 2008;21(3):124–135.
54. Dawson, T.L., Jr. Malassezia globosa and restricta: breakthrough understanding of the etiology and treatment of dandruff and seborrheic dermatitis through whole-genome analysis. J Investig Dermatol Symp Proc. 2007;12(2):15–19.
54a. Cook, B.A., Warshaw, E.M. Role of topical calcineurin inhibitors in the treatment of seborrheic dermatitis: a review of pathophysiology, safety and efficacy. Am J Clin Dermatol. 2009;10(2):103–118.
55. Borgers, M., Degreef, H. The role of ketoconazole in seborrheic dermatitis. Cutis. 2007;80(4):359–363.
56. Menter, A., et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care of the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58(5):826–850.
57. Hueber, A.J., McInnes, I.B. Immune regulation in psoriasis and psoriatic arthritis—recent developments. Immunol Lett. 2007;114(2):59–65.
58. Duffin, K.C., Krueger, G.G. Genetic variations in cytokines and cytokine receptors associated with psoriasis found by genome-wide association. J Invest Dermatol. 2009;129(4):827–833.
59. Murphy, M., Kerr, P., Grant-Kels, J.M. The histopathologic spectrum of psoriasis. Clin Dermatol. 2007;25(6):624–628.
60. Kleinert, S., et al. Psoriatic arthritis: clinical spectrum and diagnostic procedures. Clin Dermatol. 2007;25(6):519–523.
61. Chuh, A., Chan, H., Zawar, V. Pityriasis rosea—evidence for and against an infectious aetiology. Epidemiol Infect. 2004;132(32):381–390.
62. Stulberg, D.L., Wolfrey, J. Pityriasis rosea. Am Fam Physician. 2004;69(1):87–91.
63. Chuh, A.A., et al. Interventions for pityriasis rosea. Cochrane Database Syst Rev. (2):2007. [CD005068].
64. Prpic Massari, L., et al. Perforin expression in peripheral blood lymphocytes and skin-infiltrating cells in patients with lichen planus. Br J Dermatol. 2005;151(2):433–439.
65. Shiohara, T., et al. Pathomechanisms of lichen planus autoimmunity elicited by cross-reactive T cells. Curr Dir Autoimmun. 2008;10:206–226.
66. Carrozzo, M. Oral diseases associated with hepatitis C virus infection. Part 2: lichen planus and other diseases. Oral Dis. 2008;14(3):217–228.
67. DeRossi, S.S., Ciarrocca, K.N. Lichen planus, lichenoid drug reactions, and lichenoid mucositis. Dent Clin North Am. 2005;49(1):77–89.
68. Gorruhi, F., et al. Randomized trial of pimecrolimus cream versus triamcinolone acetonide paste in the treatment of oral lichen planus. J Am Acad Dermatol. 2007;57(5):806–813.
69. Marks, R. The enigma of rosacea. J Dermatol Treat. 2007;18(6):326–328.
70. Millikan, L.E. Rosacea as an inflammatory disorder: a unifying theory? Cutis. 2004;73(1 Suppl):5–8.
71. Stone, D.U., Chodosh, J. Ocular rosacea: an update on pathogenesis and therapy. Curr Opin Ophthalmol. 2004;15(6):499–502.
72. Crawford, G.H., Pelle, M.T., James, W.D. Rosacea: I, etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51(3):327–341.
73. Lacey, N., et al. Mite-related bacterial antigens stimulate inflammatory cells in rosacea. Br J Dermatol. 2007;157(3):474–481.
74. Conde, J.F., et al. Managing rosacea: a review of the use of metronidazole alone and in combination with oral antibiotics. J Drugs Dermatol. 2007;6(5):495–498.
75. Gooderham, M. Rosacea and its topical management. Skin Therapy Lett. 2009;14(2):1–3.
76. Panjwani, S. Early diagnosis and treatment of discoid lupus erythematosus. J Am Board Fam Med. 2009;22(2):206–213.
77. Pramatarov, K.D. Chronic cutaneous lupus erythematosus—clinical spectrum. Clin Dermatol. 2004;22(2):113–120.
78. Kuhn, A., Biji, M. Pathogenesis of cutaneous lupus erythematosus. Lupus. 2008;17(5):389–393.
79. Rothfield, N., Sontheimer, R.D., Bernstein, M. Lupus erythematosus: systemic and cutaneous manifestations. Clin Dermatol. 2006;1495:348–362.
80. Sárdy, M., Ruzicka, T., Kuhn, A. Topical calcineurin inhibitors in cutaneous lupus erythematosus. Arch Dermatol Res. 2009;301(1):93–98.
81. Fabbri, P., et al. Cutaneous lupus erythematosus: diagnosis and management. Am J Clin Dermatol. 2003;4(7):449–465.
82. Baroni, A., et al. Vesicular and bullous disorders: pemphigus. Dermatol Clin. 2007;25(4):597–603.
83. Hashimoto, T. Recent advances in the study of the pathophysiology of pemphigus. Arch Dermatol Res. 2003;395(Suppl 1):S2–11.
84. Tron, F., et al. Immunogenetics of pemphigus: an update. Autoimmunity. 2006;39(7):531–539.
85. Prajapati, V., Mydlarski, P.R. Advances in pemphigus therapy. Skin Therapy Lett. 2008;13(3):4–7.
86. Olasz, E.F., Yancey, K.B. Bullous pemphigoid and related subepidermal autoimmune blistering diseases, Curr Dir Autoimmun. 2008;10:141–166.
87. Kasperkiewicz, M., Zillikens, D. The pathophysiology of bullous pemphigoid. Clin Rev Allergy Immunol. 2007;33(1-2):67–77.
88. Scully, C., Bagan, J. Oral mucosal diseases: erythema multiforme. Br J Oral Maxillofac Surg. 2008;46(20):90–95.
89. Katta, R. Taking aim at erythema multiforme. How to spot target lesions and less typical presentations. Postgrad Med. 2000;107(1):87.
90. Provost, T.T., Weston, W.L. Bullous diseases. St Louis: Mosby; 1993.
91. Parrillo, S.J. Stevens-Johnson syndrome and toxic epidermal necrolysis. Curr Allergy Asthma Rep. 2007;7(4):243–247.
92. Roujeau, J.C. Stevens-Johnson syndrome and toxic epidermal necrolysis are severity variants of the same disease which differs from erythema multiforme. J Dermatol. 1997;24(11):726.
93. Paquet, P., Pierard, G.E. Toxic epidermal necrolysis: revisiting the tentative link between early apoptosis and late necrosis. Int J Mol Med. 2007;19(1):3–10.
94. Pereira, F.A., Mudgil, A.V., Rosmarin, D.M. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56(2):181–200.
95. Mukasa, Y., Craven, N. Management of toxic epidermal necrolysis and related syndromes. Postgrad Med J. 2008;84(988):60–65.
96. Singh, G., Marples, R.R., Klingman, A.M. Staphylococcus infections in humans. J Invest Dermatol. 1971;57(3):149–162.
97. Hirschmann, J.V. Antimicrobial therapy for skin infections. Cutis. 2007;79(Suppl):26–36.
98. Bernard, P. Management of common bacterial infections of the skin. Curr Opin Infect Dis. 2008;21(2):122–128.
99. Koutkia, P., Mylonakis, E., Boyce, J. Cellulitis: evaluation of possible predisposing factors in hospitalized patients. Diagn Microbiol Infect Dis. 1999;34(4):325.
100. Gabillot-Carré, M., Roujeau, J.C. Acute bacterial skin infections and cellulites. Curr Opin Infect Dis. 2007;20(2):118–123.
101. Kiken, D.A., Silverberg, N.B. Atopic dermatitis in children, part 1: epidemiology, clinical features, and complications. Cutis. 2006;78(4):241–247.
102. Baringer, J.R. Herpes simplex infections of the nervous system. Neurol Clin. 2008;26(3):657–674. [viii].
103. Hobbs, M.R., et al. Identification of a herpes simplex labialis susceptibility region on human chromosome 21. J Infect Dis. 2008;197(3):340–346.
104. Brown, Z. Preventing herpes simplex virus transmission to the neonate. Herpes. 2004;11(Suppl 3):175A–186A.
105. Gupta, R., Warren, T., Wald, A. Genital herpes. Lancet. 2007;370(9605):2127–2137.
106. Koelle, D.M., Corey, L. Herpes simplex: insights on pathogenesis and possible vaccines. Annu Rev Med. 2008;59:381–395.
107. Ahmed, A.M., et al. Managing herpes zoster in immunocompromised patients. Herpes. 2007;14(2):32–36.
108. Johnson, R.W., et al. Postherpetic neuralgia: epidemiology, pathophysiology and management. Exp Rev Neurother. 2007;7(11):1581–1595.
109. Ragozzino, M.W., et al. Population based study of herpes zoster and its sequelae. Medicine (Baltimore). 1982;5(61):310.
110. Dworkin, R.H., et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(Suppl 1):S1–S26.
111. Oxman, M.N., Levin, M.J. Shingles Prevention Study Group: vaccination against herpes zoster and postherpectic neuralgia. J Infect Dis. 2008;197(Suppl 2):S228–S236.
112. Lassus, J., Ranki, A. Simultaneously detected aberrant p53 tumor-suppressor protein and HPV-DNA localized mostly in separate keratinocytes in anogenital and common warts. Exp Dermatol. 1996;5(2):72.
113. Prasad, C.J. Pathobiology of human papillomavirus. Clin Lab Med. 1995;15(3):685.
114. Gibbs, S., Harvey, I. Topical treatments for cutaneous warts. Cochrane Database Syst Rev. 2006;3:CD001781.
115. Lipke, M.M. An armamentarium of wart treatments. Clin Med Res. 2006;4(4):273–293.
116. Steben, M., Duarte-Franco, E. Human papillomavirus infection: epidemiology and pathophysiology. Gynecol Oncol. 2007;107(2 Suppl 1):A2–A5.
117. Woodfolk, J.A. Allergy and dermatophytes. Clin Microbiol Rev. 2005;18(1):30–43.
118. Armstron-James, D. Invasive Candida species infection: the importance of adequate empirical antifungal therapy. J Antimicrob Chemother. 2007;60(3):459–460.
119. Ashman, R.B. Protective and pathologic immune responses against Candida albicans infection. Front Biosci. 2008;13:3334–3351.
120. Netea, M.G., et al. An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol. 2008;6(1):67–78.
121. Levitz, S.M. Overview of host defenses in fungal infections. Clin Infect Dis. 1992;14(Suppl 1):537.
122. Lotti, T.M., Comacchi, C., Ghersetich, I. Cutaneous necrotizing vasculitis. Relation to systemic disease. Adv Exp Med Biol. 1999;455:115.
123. Chen, K.R., Carlson, J.A. Clinical approach to cutaneous vasculitis. Am J Clin Dermatol. 2008;9(2):71–92.
124. Kapp, A., Wedi, B. Chronic urticaria: clinical aspects and focus on a new antihistamine, levocetirizine. Drugs Dermatol. 2004;3(6):632–639.
125. Brodell, L.A., Beck, L.A., Saini, S.S. Pathophysiology of chronic urticaria. Ann Allergy Asthma Immunol. 2008;100(4):291–297.
126. Steen, V.D. The many faces of scleroderma. Rheum Dis Clin North Am. 2008;34(1):1–15. [v].
127. Varga, J.A., Trojanowska, M. Fibrosis in systemic sclerosis. Rheum Dis Clin North Am. 2008;34(1):115–143. [cii].
128. Gilliam, A.C. Scleroderma. Curr Dir Autoimmun. 2008;10:258–279.
129. Kahaleh, B. Vascular disease in scleroderma: mechanisms of vascular injury. Rheum Dis Clin North Am. 2008;34(1):57–71.
130. Herrick, A. Diagnosis and management of scleroderma peripheral vascular disease. Rheum Dis Clin North Am. 2008;34(1):89–114. [vii].
131. Shah, A.A., Wigley, F.M. Often forgotten manifestations of systemic sclerosis. Rheum Dis Clin North Am. 2008;34(1):221–238. [ix].
132. Nihtyanova, S.I., Denton, C.P. Current approaches to the management of early active diffuse scleroderma skin disease. Rheum Dis Clin North Am. 2008;34(1):161–179.
133. Amsden, J.R., Warmack, S., Gubbins, P.O. Tick-borne bacterial, rickettsial, spirochetal, and protozoal infectious disease in the United States: a comprehensive review. Pharmacotherapy. 2005;25(2):191–210.
134. Bratton, R.L., et al. Diagnosis and treatment of Lyme disease. Mayo Clin Proc. 2008;83(5):566–571.
135. Cruz, A.R., et al. Phagocytosis of Borrelia burgdorferi, the Lyme disease spirochete, potentiates innate immune activation and induces apoptosis in human monocytes. Infect Immun. 2008;76(1):56–70.
136. Rupprechi, T.A., et al. The pathogenesis of lyme neuroborreliosis: from infection to inflammation. Mol Med. 2008;14(3-4):205–212.
137. Hovius, J.W., van Dan, A.P., Fikrig, E. Tick-host-pathogen interactions in Lyme disease. Trends Parasitol. 2007;23(9):434–438.
138. Aguero-Rosenfeld, M.E. Lyme disease: laboratory issues. Infect Dis Clin North Am. 2008;22(2):301–313. [vii].
139. Vázquez, M., et al. Effectiveness of personal protective measures to prevent Lyme disease. Emerg Infect Dis. 2008;14(2):210–216.
140. Magalhães, R.F., et al. Diagnosis and follow-up of keratoacanthoma-like lesions: clinical-histologic study of 43 cases. J Cutan Med Surg. 2008;12(4):163–173.
141. Kossard, S., Tan, K.B., Choy, C. Keratoacanthoma and infundibulocystic squamous cell carcinoma. Am J Dermatopathol. 2008;30(2):127–134.
142. Annest, N.M., et al. Intralesional methotrexate treatment for keratoacanthoma tumors: a retrospective study and review of the literature. J Am Acad Dermatol. 2007;56(6):989–993.
143. Roewert-Huberj, J., Stockfleth, E., Kerl, H. Pathology and pathobiology of actinic (solar) keratosis—an update. Br J Dermatol. 2007;157(Suppl 2):18–20.
144. McIntyre, W.J., Downs, M.R., Bedwell, S.A. Treatment options for actinic keratoses. Am Fam Physician. 2007;76(5):667–671.
145. American Cancer Society. Cancer facts and figures 2009. Atlanta: The Society; 2009.
146. Benjamin, C.L., Meinikova, V.O., Ananthaswamy, H.N. P53 protein and pathogenesis of melanoma and nonmelanoma skin cancer. Adv Exp Med Biol. 2008;624:265–282.
147. Kyle, J.W., et al. Economic evaluation of the US Environmental Protection Agency’s SunWise program: sun protection education for young children. Pediatrics. 2008;121(5):e1074–e1084.
148. Lear, J.T., et al. Basal cell carcinoma: from host response and polymorphic variants to tumour suppressor genes. Clin Exp Dermatol. 2005;30(1):49–55.
149. Erb, P., et al. Apoptosis and pathogenesis of melanoma and nonmelanoma skin cancer. Adv Exp Med Biol. 2008;624:283–295.
150. Zakl-Prelich, M., Narbutt, J., Sysa-Jedrzejowska, A. Environmental risk factors predisposing to the development of basal cell carcinoma. Dermatol Surg. 2004;30(2 Pt 2):248–252.
151. Lindström, E., et al. PTCH mutations: distribution and analyses. Hum Mutat. 2006;27(3):215–219.
152. Lupi, O. Correlations between the Sonic Hedgehog pathway and basal cell carcinoma. Int J Dermatol. 2007;46(11):1113–1117.
153. Rigel, D.S. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J Am Acad Dermatol. 2008;58(5 Suppl 2):S129–S132.
154. Rass, K., Reichrath, J. UV damage and DNA repair in malignant melanoma and nonmelanoma skin cancer. Adv Exp Med Biol. 2008;624:162–178.
155. Berwick, M., Erdei, E., Hay, J. Melanoma epidemiology and public health. Dermatol Clin. 2009;27(2):205–214. [viii].
156. Rodenas, J.M. sun exposure, pigmentary traits, and risk of cutaneous maligant melanoma: a case-control study in a Mediterranean population,. Cancer Control. 1996;7(2):275.
157. Dahl, C., Guldberg, P. The genome and epigenome of malignant melanoma. APMIS. 2007;115(10):1161–1176.
158. O’Conner, K.M., Chien, A.J. Management of melanocytic lesions in the primary care setting. Mayo Clin Proc. 2008;83(2):208–213.
159. Habif, T.B. Clinical dermatology: a color guide to diagnosis and therapy. St Louis: Mosby; 2004.
160. Bishop, J.A. Melanoma. Hosp Med. 2000;61(2):103.
161. Fetcher, L.A., et al. Toward a molecular classification of melanoma. J Clin Oncol. 2007;25(12):1606–1620.
162. Viros, A., et al. Improving melanoma classification by integrating genetic and morphologic features. PLoS Med. 5(6), 2008. [e120].
163. Kirkwood, J.M., et al. Next generation of immunotherapy for melanoma. J Clin Oncol. 2008;26(20):3445–3455.
164. Lejeune, F.J., Rimoldi, D., Speiser, D. New approaches in metastatic melanoma: biological and molecular targeted therapies. Expert Rev Anticancer Ther. 2007;7(5):701–713.
165. Szajerka, T., Jablecki, J. Kaposi’s sarcoma revisited. AIDS Rev. 2007;9(4):230–236.
166. Dezube, B.J., Sullivan, R., Koon, H.B. Emerging targets and novel strategies in the treatment of AIDS-related Kaposi’s sarcoma: bidirectional translations science. J Cell Physiol. 2006;209(3):659–662.
167. Goopman, J. Neoplasms in the acquired immune deficiency syndrome: the multidisciplinary approach. Semin Oncol. 1987;14(2 Suppl 3):1.
168. Nguyen, H.Q., et al. Persistent Kaposi sarcoma in the era of highly active antiretroviral therapy: characterizing the predictors of clinical response. AIDS. 2008;22(8):937–945.
169. Grandberg, P.O. Freezing cold injury. Arctic Med Res. 1991;50(Suppl 6):76.
170. Murphy, J.V., et al. Frostbite: pathogenesis and treatment. J Trauma. 2000;48(1):171.
171. Greenfield, L.J., et al. Surgery: scientific principles and practice, ed 2. Philadelphia: Lippincott-Raven; 1997.
172. Raine, T.J., London, M.D., Goluch, L. Antiprostaglandins and antithromboxanes for treatment of frostbite. Surg Forum. 1980;31:557.
173. Bruen, K.J., et al. Reduction of the incidence of amputation in frostbite injury with thrombolytic therapy. Arch Surg. 2007;142(6):546–551.
174. Savin, R.C., Atton, A.V. Minoxidil: update on its clinical role. Dermatol Clin. 1993;11(1):55.
175. Norwood, O.T., Lehr, B. Female androgenetic alopecia: a separate entity. Dermatol Surg. 2000;26(7):679.
176. Dinh, Q.Q., Sinclair, R. Female pattern hair loss: current treatment concepts. Clin Interven Aging. 2007;2(2):189–199.
177. Rushton, D.H. Management of hair loss in women. Dermatol Clin. 1993;11(1):47.
178. King, L.E., Jr., McElwee, K.J., Sundberg, J.P. Alopecia areata. Curr Dir Autoimmun. 2008;10:280–312.
179. Callen, J.P., et al. Color atlas of dermatology. Philadelphia: Saunders; 2000.
180. Gilhar, A., Paus, R., Kalish, R.S. Lymphocytes, neuropeptides and genes involved in alopecia areata. J Clin Invest. 2007;117(8):2019–2027.
181. Avgerinou, G., et al. Alopecia areata: topical immunotherapy treatment with diphencyprone. J Eur Acad Dermatol Venereal. 2008;22(3):320–323.
182. Somani, N., Harrison, S., Bergfeld, W.F. The clinical evaluation of hirsutism. Dermatol Ther. 2008;21(5):376–391.
183. Archer, J.S., Chang, R.J. Hirsutism and acne in polycystic ovary syndrome. Best Pract Res Clin Obstet Gynaecol. 2004;18(5):737–754.
184. Elghblawi, E. idiopathic hirsutism: excessive bodily and facial hair in women. Br J Nurs. 2008;17(3):192–197.
185. Martin, K.A., et al. Evaluation and treatment of hirsutism in premenopausal women: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93(4):1105–1120.
186. Rigopoulos, D., et al. Acute and chronic paronychia. Am Fam Physician. 2008;77(3):339–346.
187. Scher, R.K. Onychomycosis: therapeutic update. J Am Acad Dermatol. 1999;40(6 Pt 2):S21.
188. Elewski, B.E. Diagnostic techniques for confirming onychomycosis. J Am Acad Dermatol. 1996;35(3 Pt 2):S6.
189. Baran, R., Hay, R.J., Garduno, J.I. Review of antifungal therapy and the severity index for assessing onychomycosis: part 1. J Dermatolog Treat. 2008;19(2):72–81.
190. Nunley, K.S., Cornelius, L. Current management of onychomycosis. J Hand Surg Am. 2008;33(7):1211–1214.