BIOLOGY, CLINICAL MANIFESTATIONS, AND TREATMENT OF CANCER
CANCER CHARACTERISTICS AND TERMINOLOGY
Cancer-Causing Mutations in Genes
Types of Genes Misregulated in Cancer
Oncogenes and Tumor-Suppressor Genes: Accelerators and Brakes
Inflammation, Immunity, and Cancer
The Immune System Protects Us Against Viral-Associated Cancers



Cancer is a leading disease, cause of death, and source of morbidity of adults in the Western world. The incidence of cancer increases markedly with advancing age and is strongly affected by gender, lifestyle, ethnicity, infection, and genetics. Over the past 35 years intensive research has led to a significantly enhanced understanding of this complex and frightening disease. We now understand that cancer is a collection of many different diseases, caused by an accumulation of genetic and epigenetic alterations. Environment, heredity, and behavior interact to modify the risk of developing cancer and the response to treatment. Improvements in treatment strategies and supportive care, coupled with new, often individualized therapies based on advances in our fundamental understanding of the basic pathophysiology of malignancy, have contributed to an increasing number of effective options for these diverse, often lethal, disorders collectively called cancer.
CANCER CHARACTERISTICS AND TERMINOLOGY
Any discussion of cancer must start with a definition of what it is and what it is not. Although most readers may have an intuitive understanding of this disorder, composing an exact definition that encompasses this broad category is more challenging. A definition from 1922 may summarize cancer as well as any:
The most generally accepted definition of a tumor is that it is a tissue overgrowth which is independent of the laws governing the remainder of the body. It is usual to add as a qualifying phrase to separate tumors from reparative processes, such as bone callus, that the neoplasm overgrowth serves no useful purpose to the organism.1
The term cancer derives from the Greek word for crab, karkinoma, which the physician Hippocrates used to describe the appendage-like projections extending from tumors. The word tumor originally referred to any swelling that was caused by inflammation, but is now generally reserved for describing a new growth, or neoplasm. Not all tumors or neoplasms, however, are cancer. The term cancer refers to a malignant tumor and is not used to refer to benign growths such as lipomas or hypertrophy of an organ. Yet it is important to recognize that benign neoplasms also can be life threatening if they enlarge in critical locations. For example, a benign meningioma at the base of the skull may cause symptoms by compressing adjacent normal brain tissue. The definitions of benign versus malignant are presented in the following text and in Table 11-1. Box 11-1 contains information on the diagnosis and clinical staging of cancer.
Table 11-1
Characteristics of Benign versus Malignant Tumors
| Benign Tumors | Malignant Tumors |
| Grow slowly | Grow rapidly |
| Have a well-defined capsule | Are not encapsulated |
| Are not invasive | Invade local structures and tissues |
| Well differentiated; looks like the tissue from which it arises | Poorly differentiated; may not be able to tell from what tissue it arose |
| Have a low mitotic index; dividing cells are rare | High mitotic index; many dividing cells |
| Do not metastasize | Can spread distantly, often through blood vessels and lymphatics |
Tumor Classification and Nomenclature
Proper identification of a cancer is important for many reasons. Different lesions will have different causes, different rates and patterns of progression, and different responses to treatment. Cancer classification starts with knowing the site of origin and microscopic appearance of the lesion, but can extend to a detailed description of critical genetic changes in the cancer.
Benign tumors, which are not referred to as cancers, are usually encapsulated and well differentiated. They retain some normal tissue structure and do not invade the capsules surrounding them or spread to regional lymph nodes or distant locations. Benign tumors are generally named according to the tissues from which they arise, and include the suffix “-oma.” For example, a benign tumor of the smooth muscle of the uterus is a leiomyoma, and a benign tumor of fat cells is a lipoma.
Some tumors initially described as benign can progress to cancer and then are referred to as malignant tumors. These tumors are distinguished from benign tumors by their more rapid growth rates and specific microscopic alterations, including loss of differentiation and absence of normal tissue organization. One of the hallmarks of cancer cells, as seen under the microscope, is anaplasia, the loss of cellular differentiation, irregularities of the size and shape of the nucleus, and loss of normal tissue structure. Malignant tumors lack a capsule and grow to invade nearby blood vessels, lymphatics, and surrounding structures. The most important and most deadly characteristic of malignant tumors is their ability to spread far beyond the tissue of origin, a process known as metastasis (Figures 11-1 and 11-2; see Table 11-1).
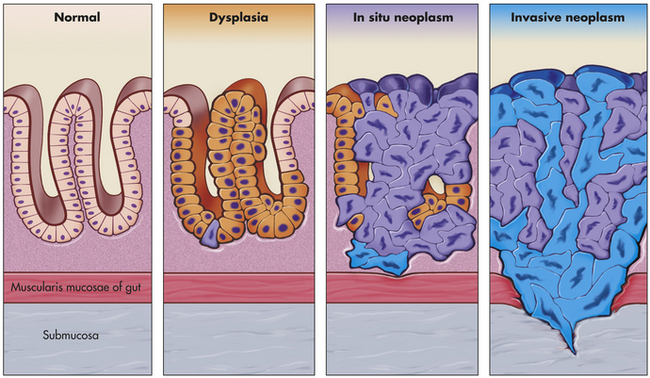
Figure 11-1 Progression from normal to neoplasm. A sequence of cellular and tissue changes progressing from dysplasia to carcinoma in situ and then to invasive cancer is seen often in the development of cancer. In clinical specimens, distinguishing between dysplasia and in situ cancer is difficult. The presence of anaplastic cells and loss of normal tissue architecture signify the development of cancer. This sequence of changes is most easily seen in the squamous epithelium of the uterine cervix, the epidermis of sun-exposed skin, and colonic and gastric mucosa after long-standing inflammation. The high rate of cell division, local mutagens, and inflammatory mediators all contribute to the accumulation of genetic abnormalities that lead to cancer. (Modified from Stevens A, Lowe J: Pathology, ed 2, London, 2000, Mosby.)
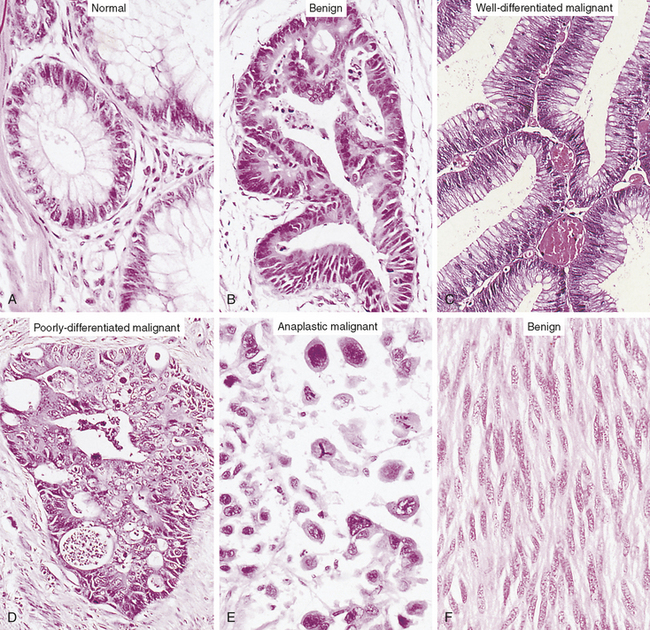
Figure 11-2 Loss of cellular and tissue differentiation during the development of cancer. The cells of a benign neoplasm (B) resemble those of the normal colonic epithelium (A), in that they are columnar and have an orderly arrangement. Loss of some degree of differentiation is evident in that the neoplastic cells do not show much mucin vacuolization. Cells of the well-differentiated malignant neoplasm (C) of the colon have a haphazard arrangement and although gland lumina are formed, they are architecturally abnormal and irregular. Nuclei vary in shape and size, especially when compared with A. Cells in the poorly differentiated malignant neoplasm (D) have an even more haphazard arrangement, with very poor formation of gland lumina. Nuclei show greater variation in shape and size compared with the well-differentiated malignant neoplasm in C. Cells in anaplastic malignant neoplasms (E) bear no relation to the normal epithelium, with no recognizable gland formation. Tremendous variation is found in the size of cells and their nuclei, with very intense staining (hyperchromatic nuclei). Not knowing the site of origin would make it impossible to tell what sort of tumor this is by microscopic appearance alone. Well-differentiated tumors often resemble their cell of origin, as shown in the example of a benign tumor of smooth muscles (F). (From Stevens A, Lowe J: Pathology, ed 2, London, 2000, Mosby.)
In general, cancers are named according to the cell type from which they originate. Cancers arising in epithelial tissue are called carcinomas, and if they arise from or form ductal or glandular structures are named adenocarcinomas. Hence, a malignant tumor arising from breast glandular tissue is a mammary adenocarcinoma. Cancers arising from connective tissue usually have the suffix sarcoma. For example, malignant cancers of skeletal muscle are known as rhabdomyosarcomas. Cancers of lymphatic tissue are called lymphomas, whereas cancers of blood-forming cells are called leukemias. However, many cancers, such as Hodgkin disease and Ewing sarcoma, are named for historical reasons that do not follow this naming convention. Table 11-2 presents the nomenclature and classification of selected tumors.
Table 11-2
Nomenclature and Classification of Benign and Malignant Tumors∗
| Cell or Tissue of Origin | Benign Tumor | Malignant Tumor |
| Tumors of Epithelial Origin | ||
| Squamous cells | Squamous cell papilloma | Squamous cell carcinoma |
| Basal cells | — | Basal cell carcinoma |
| Glandular or ductal epithelium | Adenoma | Adenocarcinoma |
| Cystadenoma | Cystadenocarcinoma | |
| Transitional cells | Transitional cell papilloma | Transitional cell carcinoma |
| Bile duct | Bile duct adenoma | Bile duct carcinoma (cholangiocarcinoma) |
| Liver cells | Hepatocellular adenoma | Hepatocellular carcinoma |
| Melanocytes | Nevus | Malignant melanoma |
| Renal epithelium | Renal tubular adenoma | Renal cell carcinoma |
| Skin adnexal glands | ||
| Sweat glands | Sweat gland adenoma | Sweat gland carcinoma |
| Sebaceous glands | Sebaceous gland adenoma | Sebaceous gland carcinoma |
| Germ cells (testis and ovary) | — | Seminoma (dysgerminoma) |
| Embryonal carcinoma, yolk sac carcinoma | ||
| Tumors of Mesenchymal Origin | ||
| Hematopoietic/lymphoid tissue | ||
| Leukocytes | Leukemias | |
| Granular leukocytes and precursors | Granulocytic leukemia | |
| Myelocytic leukemias | ||
| Myelogenous leukemias | ||
| Plasma cells | Multiple myeloma | |
| Lymphoid | ||
| Nongranular leukocytes and prelymphocytes | Lymphomas | |
| Proliferating lymphocytes and monocytes | Lymphocytic leukemia | |
| Proliferating immature precursor monocytes | Lymphoblastic leukemia | |
| Solid tumors of lymph tissue (thymus, spleen, lymph nodes) | Lymphoma or lymphosarcoma | |
| Neural and retinal tissue | ||
| Nerve sheath | Neurilemoma, neurofibroma | Malignant peripheral nerve sheath tumor |
| Nerve cells | Ganglioneuroma | Neuroblastoma |
| Retinal cells (cones) | — | Retinoblastoma |
| Connective tissue | ||
| Fibrous tissue | Fibromatosis (desmoid) | Fibrosarcoma |
| Fat | Lipoma | Liposarcoma |
| Bone | Osteoma | Osteogenic sarcoma |
| Cartilage | Chondroma | Chrondrosarcoma |
| Muscle | ||
| Smooth muscle | Leiomyoma | Leiomyosarcoma |
| Striated muscle | Rhabdomyoma | Rhabdomyosarcoma |
| Endothelial and related tissues | ||
| Blood vessels | Hemangioma | Angiosarcoma |
| Kaposi sarcoma | ||
| Lymph vessels | Lymphangioma | Lymphangiosarcoma |
| Synovium | — | Synovial sarcoma |
| Mesothelium | — | Malignant mesothelioma |
| Meninges | Meningioma | Malignant meningioma |
| Tumors of Uncertain Origin | — | Ewing tumor |
∗This list is intended to provide only an introduction to tumor nomenclature.
Modified from Murphy GP et al: American Cancer Society’s textbook of clinical oncology, ed 2, New York, 1995, American Cancer Society.
Classification of Tumors—Classical Histology and Modern Genetics
Carcinoma in situ (often abbreviated CIS) refers to preinvasive epithelial malignant tumors of glandular or squamous cell origin. These early stage cancers are localized to the epithelium and have not broken through the local basement membrane or invaded the surrounding stroma (see Figure 11-1). Carcinoma in situ is recognized in a number of sites, including the cervix, skin, oral cavity, esophagus, and bronchus. In glandular epithelium, in situ lesions occur in the stomach, endometrium, breast, and large bowel. In the breast, ductal carcinoma in situ (DCIS) fills the mammary ducts but has not progressed to local tissue invasion. DCIS lesions are readily treatable, although the optimal therapeutic approach is controversial. The time that such preinvasive lesions remain in situ before becoming invasive is unknown.2 Some carcinomas of the cervix appear as preinvasive lesions in situ for several years before they progress to invasive carcinoma and metastatic tumors (see Figure 11-1).
As our knowledge about the molecular alterations that cause cancer increases, it becomes increasingly important for clinicians to gather extensive information about each cancer. The classification of cancers was originally based on gross and light microscopic appearance, but this is now often aided by additional immunohistochemical analysis of protein expression. In selected cases, this is supplemented by extensive molecular analysis of the tumors. Sometimes a single gene is examined (for example, to determine if there is a characteristic translocation diagnostic of chronic myelogenous leukemia [CML]), and sometimes a panel of genes and proteins are examined (e.g., in breast cancer) to determine if the tumor expresses estrogen receptor, progesterone receptor, and the epidermal growth factor (EGF) receptor HER2. In a research setting, panels of gene expression analysis are measured using microarray technology, in which the expression levels of a very large number of genes are measured. This analysis can be used to classify tumors more precisely and may predict what the most effective therapy will be. This detailed analysis of each tumor is a form of personalized medicine offering therapy based on a very detailed knowledge of individuals’ characteristics and their specific cancer.3
The Biology of Cancer Cells
Transformation and Differentiation
Cancer cells behave differently than normal cells in several important ways. Transformation refers to the process by which a normal cell becomes a cancer cell. Autonomy refers to the cancer cell’s independence from normal cellular controls and is part of the transformational process. These differences are most readily seen in specialized laboratory assays, especially those examining the growth patterns of normal and cancerous cells in laboratory incubators. Transformed cells lack many of the normal “social controls” seen in nontransformed cells. Normal cells cease to divide when they fill a Petri (or tissue culture) dish, whereas transformed cells continue to crowd and eventually pile up on each other (Figure 11-3). Normal cells usually will not grow unless they are attached to a firm surface (like a Petri dish). However, cancer cells are often anchorage independent, that is, they continue to divide even when suspended in a soft agar gel. Cancerous cells can be assayed in mice as well; normal human cells when injected into a special type of mouse (genetically engineered to lack an immune system to prevent rejection of human cells) will not grow. However, cancerous cells from humans can continue to grow and even metastasize in these mice. Normal cells have a limited life span in the laboratory; they may divide in a Petri dish 10 or 50 times, but then they cease growing. Cancer cells usually are immortal in that they seem to have an unlimited life span and will continue to divide for years under appropriate laboratory conditions. One of the most commonly used laboratory cell lines, HeLa cells, was derived from a cervical cancer specimen obtained in 1951 that continues to grow and divide in laboratories around the world.4
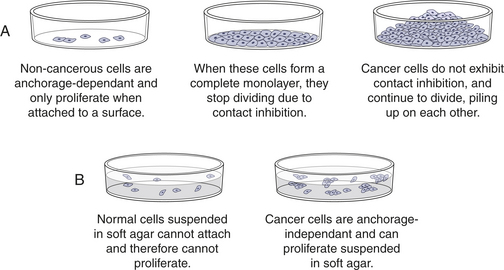
Figure 11-3 Cancerous cells show abnormal growth in the laboratory. Cancer cells, unlike most normal cells, (A) usually continue to grow and pile on top of one another after they have formed a confluent monolayer in culture (loss of contact inhibition) and (B) can grow without being attached to a surface, called anchorage independence.
Cancer cells often show defects in the normal process of differentiation, that is, the process of acquiring a specialized function and organization, such as evolving into a muscle cell (see Chapter 41) or a nerve cell (see Chapter 14). Anaplasia is the absence of differentiation (see Figures 11-2 and 11-4) and means literally “without form.” In clinical specimens, anaplasia is recognized by a loss of organization and a marked increase in nuclear size with evidence of ongoing proliferation. In contrast to normal cells, which are uniform in size and shape, anaplastic cells are of variable size and shape, or pleomorphic. For example, a benign muscle tumor (benign myoma) will retain the ability to make muscle, whereas in a malignant muscle tumor (rhabdomyosarcoma), new muscle formation is seen only rarely, and even then appears highly disorganized. Thus the muscle cancer cells appear undifferentiated (see Figure 11-4). The most malignant tumors tend to have the most anaplasia and be the least differentiated.
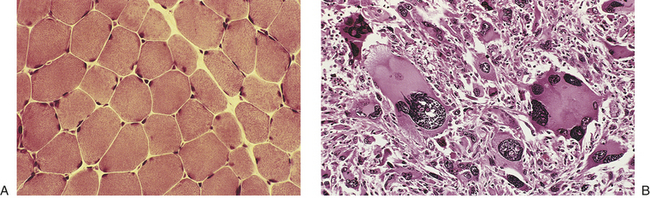
Figure 11-4 Normal and anaplastic skeletal muscle cells. A, Normal skeletal muscle cells. B, Anaplastic tumor of the skeletal muscle (rhabdomyosarcoma). Note the marked cellular and nuclear pleomorphism (cellular and nuclear variation in size and shape), hyperchromatic nuclei, and tumor giant cells. The prominent cell in the center field has an abnormal tripolar spindle. Often the tissue of origin of an anaplastic tumor can be established only by the use of molecular markers, such as immunohistochemical stains and chromosome analysis. (A from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby; B from Kumar V, Abbas AK, Fausto N: Pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders; courtesy of Dr. Trace Worrell, Department of Pathology, University of Texas Southwestern Medical School.)
Cancer Stem Cells
Many tissues, most notably the skin, intestines, and blood-forming cells, continuously renew themselves. The human gut sheds and replaces hundreds of grams of cells each day. This ongoing proliferation of these tissues with a high turnover rate depends on their regeneration from a small fraction of cells known as adult stem cells. Adult stem cells have two essential characteristics: first, they self-renew (that is, some fraction of the cell divisions create new stem cells) and second, they are multipotent, or have the ability to differentiate into multiple different cell types. In the bone marrow, it is estimated that only 0.05% (1 in 20,000) of the blood-forming cells are stem cells, yet this small pool of stem cells can be stimulated to divide and to repopulate all the mature bone marrow–derived cells in approximately 2 weeks after bone marrow transplantation. As few as 10 stem cells are sufficient to entirely repopulate the entire bone marrow of a mouse in bone marrow transplantation experiments. Similarly, the absorptive and goblet cells lining the intestine have a life span of less than 1 week, after which they undergo cell death, or apoptosis, and slough into the intestinal lumen. They too must be replenished by ongoing proliferation of intestinal stem cells. A key feature of stem cells is that they can divide asymmetrically; they can give rise to another stem cell and one daughter cell that ultimately terminally differentiates into diverse cell types, depending on the needs of the tissue (Figures 11-5 and 11-6). Multipotent bone marrow stem cells can self-renew and differentiate into all types of bone marrow–derived cells, such as red cells, lymphocytes, and neutrophils. Certain adult stem cells have a broader range of potential fates. For example, mesenchymal stem cells are able to differentiate into multiple types of cells, such as blood vessels, neurons, and muscle cells (see Figure 11-6). Theoretically, such a stem cell might be able to give rise to all cell types in the body, and as such, could be useful in regeneration of diseased tissues.
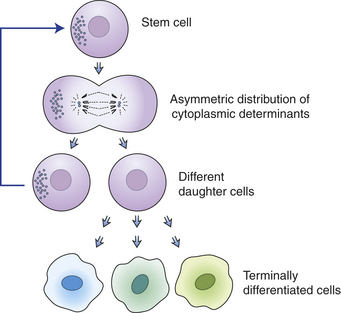
Figure 11-5 Asymmetric division is a requirement for stem cell proliferation. When a stem cell undergoes asymmetric division, cellular contents and cell fates are distributed asymmetrically. One daughter cell remains a stem cell, and the other goes into an amplification pathway that ends in terminal differentiation.
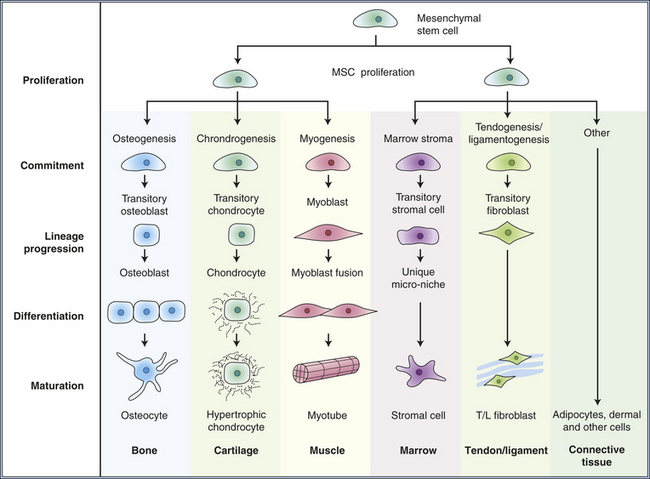
Figure 11-6 Postulated multiple lineage-specific differentiation of mesenchymal stem cells. Differentiation occurs progressively as the offspring of stem cells commit to various lineages. Mesenchymal stem cells can renew themselves and give rise to multiple tissue types. (Modified from Caplan A, Bruder S: Mesenchymal stem cells: Building blocks for molecular medicine in the 21st century, Trends Mol Med 7(6):259-264, 2001.)
Just as normal tissues in adults can arise from a rare tissue stem cell, cancers may arise from cancer stem cells. First shown in acute myeloid leukemias,5 rare cells capable of transmitting the cancer have been demonstrated in many other cancers6 as well (Figure 11-7, A). These studies6,7 show that only a small subset of cancer cells have the ability to divide indefinitely and give rise to full-blown cancer. For example, only a small fraction of cells from a breast cancer are able to divide indefinitely and generate a full-blown breast cancer when transplanted into experimental animals. This emerging concept of the cancer stem cell as the central problem in cancer suggests novel approaches to therapy targeting this specific population.
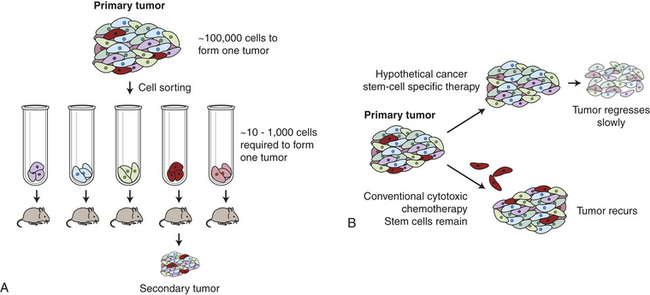
Figure 11-7 The concept of cancer stem cells. A, Only rare cells within a cancer can initiate cancer regrowth. In laboratory experiments it takes as many as 100,000 breast cancer cells injected into a mouse mammary fat pad to form a new cancer. If the breast cancer cells are sorted, one rare subtype (shown here in red) is much more proficient at forming new cancers. B, Conventional chemotherapy can destroy the bulk of a cancer. However, if the cancer stem cells (red cells) are not destroyed, the cancer may regrow. If therapies can be devised that kill the cancer stem cells, then durable long-term responses may be achieved.
In a typical cancer stem cell experiment, up to 100,000 unselected breast cancer cells must be injected into a laboratory mouse to transmit breast cancer, whereas after laboratory procedures (such as cell sorting) to enrich for the cancer stem cells and remove the other cells, it takes only 100 cells or less to transmit the cancer.6 One strong conclusion of this work is that in some but not all tumors,6 greater than 99% of the cells in the cancer are not capable of propagating the cancer, and that the most important cell in the cancer may be a rare “tumor initiating cell” or “cancer stem cell.” An important implication of this work is that current cancer treatments were not designed to kill the rare cancer stem cell. Current treatments that effectively shrink tumors by killing 99% of cancer cells appear relatively ineffective at killing the cancer stem cells.8 Consequently, therapies may shrink cancers but when the cancer stem cell is not killed, the cancer can grow back. This has led to major efforts to find therapies that specifically target the cancer stem cells (see Figure 11-7, B).
Tumor Markers
Tumor markers (biologic markers) are substances produced by cancer cells that are found either in or on the tumor cells or in the blood, spinal fluid, or urine (Table 11-3). Some tumor markers have been known for many decades. For diseases associated with a tumor marker, there is indeed a “blood test for cancer.” Tumor markers include hormones, enzymes, genes, antigens, and antibodies. For example, the adrenal medulla normally secretes the catecholamine epinephrine (adrenaline). Benign tumors of the adrenal medulla can produce catecholamines in vast excess, leading to rapid pulse, high blood pressure, sweats, and tremors. Elevated blood or urine levels of catecholamines in someone with this set of symptoms strongly suggest the presence of an adrenal medullary tumor (pheochromocytoma). Liver and germ cell tumors secrete a protein known as alpha fetoprotein (AFP) into the blood, and prostate tumors secrete prostate specific antigen (PSA) into the blood. These tumor markers can be used in three ways: (1) to screen and identify individuals at high risk for cancer; (2) to help diagnose the specific type of tumor in individuals with clinical manifestations relating to cancer, as in adrenal tumors; and (3) to follow the clinical course of cancer. For example, a falling PSA after therapy for prostate cancer indicates successful treatment, and a later rise in the PSA may indicate a recurrence.
Table 11-3
| Marker Name | Nature | Type of Cancer |
| Alpha fetoprotein (AFP) | 70 kDa protein | Hepatic, germ cell |
| Carcinoembryonic antigen (CEA) | 200 kDa glycoprotein | GI, pancreas, lung, breast, etc. |
| β-Human chorionic gonadotropin (β-HCG) | Glycopeptide hormone | Germ cell |
| Prostate-specific antigen (PSA) | 33 kDa glycoprotein | Prostate |
| Catecholamines | Epinephrine and precursors | Pheochromocytoma (adrenal medulla) |
| Homovanillic acid/vanillylmandelic acid (HVA/VMA) | Catecholamine metabolites | Neuroblastoma |
| Urinary Bence Jones protein | Ig light chain | Multiple myeloma |
| Adrenocorticotropic hormone (ACTH) | Peptide hormone | Pituitary adenomas |
A significant problem in diagnosing cancer using tumor marker assays is that nonmalignant conditions also can produce tumor markers. The presence of an elevated tumor marker therefore may suggest a specific diagnosis, but it is not used alone as a definitive diagnostic test. Identification of ideal sensitive and specific tumor markers that are elevated early in the course of common cancers remains a high priority because the early detection of cancer often improves the treatment outcome.
THE GENETIC BASIS OF CANCER
Cancer-Causing Mutations in Genes
Prior to the advent of modern molecular biology, many different causes of cancer were postulated, based on epidemiologic studies, studies of carcinogens, and studies of viruses. We now understand that changes in the genes of the cancer cell cause the cell to become cancerous. As our knowledge of cancer biology continues to increase so too does our understanding of the many ways that heritable changes in cells can contribute to cancer. These changes include deoxyribonucleic acid (DNA) mutations, but also include changes in DNA and histone chemical modification (epigenetic changes), and, most recently recognized, changes in micro-ribonucleic acid (miRNA) expression (also see Chapters 4 and 12). Although the word mutation is used extensively here, it can also refer to heritable changes in gene expression (epigenetics) that do not involve changes in DNA sequence.
Cancer is predominantly a disease of aging. Perhaps the most telling epidemiologic data are presented in Figure 11-8. The incidence of cancer, that is, the fraction of individuals in each age group who develop cancer, increases dramatically with age. The best explanation for this epidemiologic data is that each individual acquires a number of genetic “hits” or mutations over time. When sufficient mutations have occurred, cancer develops. These epidemiologic data agree well with observation of mutations in early and advanced cancers and from experimental cancers created in the laboratory—four to seven specific hits are required to cause a full-blown cancer.9
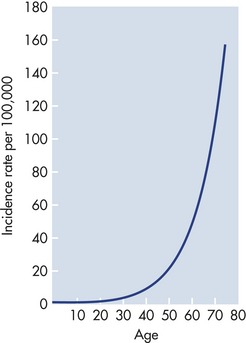
Figure 11-8 Marked increases in cancer with age. The graph depicts the number of cases of colon cancer diagnosed per 100,000 women in England and Wales in 1 year. The incidence of cancer increases dramatically with advancing age. These data suggest that accumulation of genetic and epigenetic alterations over time increases the risk of developing cancer. The slope of the curve suggests that five to seven mutations must occur before full-blown cancer develops. (Modified from Alberts B et al: Molecular biology of the cell, ed 4, New York, 2002, Garland.)
Clonal Selection
As a cell accumulates specific mutations, it can acquire, step by step, the characteristics of a cancer cell, for example, increased growth rate or, alternatively, decreased apoptosis, or death rate. That mutant cell may then have a selective advantage over its neighbors; its progeny can accumulate faster than its nonmutant neighbors. This is referred to as clonal proliferation or clonal expansion (Figure 11-9). As a clone with a mutation proliferates, it may become an early stage tumor, for example, a carcinoma in situ or a benign colonic polyp. Additional heritable changes can occur in these early lesions that permit progression to more advanced tumors. The process of tumor development is a form of darwinian evolution; cells with a genetic change that confers a survival advantage out-compete their neighbors. The progressive accumulation of distinct advantageous (from the point of view of the cancer cell, not the individual!) mutations leads from normal cells to fully malignant cancers.
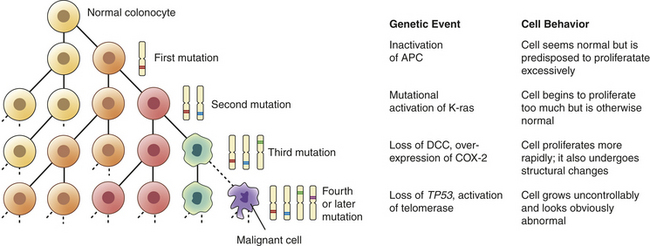
Figure 11-9 Clonal proliferation model of neoplastic progression. During clonal proliferation, progressively altered populations of cells arise over time. As genetic and epigenetic changes occur, different subclones (indicated by different color cells) coexist for a time. Clones that grow the fastest out-compete other clones, producing ever-more malignant, and abnormal-appearing, growths. The sequential accumulation of mutations has been well studied in the progression from a normal colon cell to a benign intestinal polyp to a malignant colon cancer. One of the earliest mutations in colon cancer is loss of the tumor suppressor gene APC. Additional mutations, often in the oncogene ras, activation of COX-2, and loss of the tumor suppressors DCC and TP53 occur as the lesion progresses from a benign polyp to an invasive carcinoma. APC, Adenomatous polyposis coli; DCC, deleted in colon cancer; COX-2, cyclooxygenase-2. (Modified from Mendelsohn I et al: The molecular basis of cancer, ed 2, Philadelphia, 2001, Saunders; Kumar V, Cotran RS, Robbins SL: Basic pathology, ed 6, Philadelphia, 1997, Saunders.)
One organ in which this correlation of genetic and clinical progression has been especially well studied is the colon.10 The colon is accessible to inspection with a colonoscope, and so neoplastic lesions of varying size can readily be detected and removed. Intestinal polyps are benign neoplasms and the first stage in development of colon cancer. Small polyps tend to have only a few detectable mutations. Large polyps have more mutations, whereas frank colon cancers have even more mutations. This type of genetic information provided the framework for the now widely accepted concept that it is the stepwise accumulation of alterations in specific genes that is required for the development of cancer (see Figure 11-9).
Types of Genes Misregulated in Cancer
Alterations in Progrowth and Antigrowth Signals
We now understand that multiple genetic hits are required for the evolution of full-blown cancer. One key question is, what types of genes must be altered to cause a cancer? In 2000, a highly influential paper by Hanahan and Weinberg11 proposed six specific pathways that must be misregulated for cancer to develop (see Figures 11-9 and 11-10). First, cancer cells must have mutations that enable them to proliferate in the absence of external growth signals. To achieve this, some cancers acquire the ability to secrete growth factors that stimulate their own growth, a process known as autocrine stimulation (also see Chapter 1). Other cancers have an increase in growth factor receptors; for example, in breast cancer, the epidermal growth factor (EGF) receptor HER2/neu is up-regulated, and likely sends growth signals into the cell even when growth factors are at very low levels. Inhibitors of HER2 and other EGF receptors that block this pathway are effective in treating selected breast and lung cancers.12 Alternatively, the signal cascade from the cell surface receptor to the nucleus may be mutated in the “on” position. Up to one third of all cancers have an activating mutation in the gene for an intracellular signaling protein called RAS. This mutant RAS stimulates cell growth even when growth factors are missing (Figure 11-11).
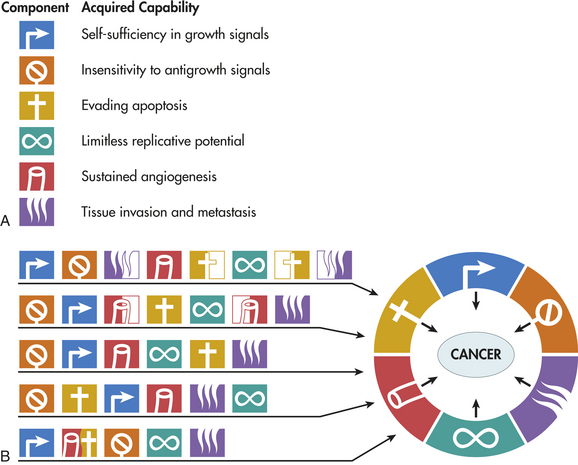
Figure 11-10 Six hallmarks of cancer. A, Most cancers acquire mutations in six distinct areas of cell control during their development. B, Multiple pathways of carcinogenesis. All cancers must acquire mutations in the six areas, but their means of doing so varies mechanistically and chronologically. As shown, the order in which these capabilities are acquired is variable across different cancers. In some tumors, a particular mutation may confer several capabilities simultaneously, decreasing the number of intermediate mutational steps required for full development. Loss of the p53 tumor suppressor gene may facilitate resistance to apoptosis and angiogenesis (e.g., in the five-step pathway shown [bottom pathway]). In other tumors, by comparison, a collaboration of two or more distinct genetic changes may be needed to acquire a given trait. In the eight-step model (top pathway), invasion metastasis and resistance to apoptosis are each acquired in two steps. (Modified from Hanahan D, Weinberg RA: The hallmarks of cancer, Cell 100[1]:57-70, 2000.)
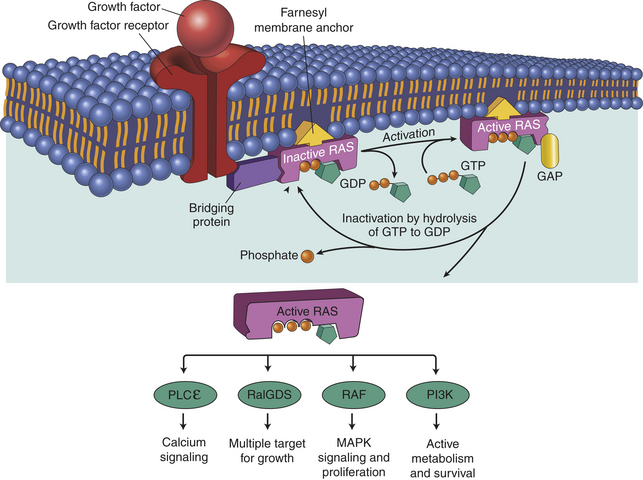
Figure 11-11 Many growth factors signal through the RAS protein. When a normal cell is stimulated through a growth factor binding to its receptor, inactive RAS exchanges GDP for GTP, becoming active. Active (GTP-bound) RAS sends growth signals throughout the cell by interacting with a number of signaling proteins, including RalGDS, RAF, PLCε, and PI3K. RAS is normally inactivated when it hydrolyzes GTP to GDP. Oncogenic Ras has a mutation that blocks hydrolysis of GTP, thereby locking RAS in the active configuration. GDP, guanosine diphosphate; GTP, guanosine triphosphate; MAP, mitogen-activated protein; PI3K, Phosphoinositide-I3 kinase; PLCε, phospholipase C; RalGDS, Ral guanine nucleotide dissociation stimulator; ras, oncogene; ras, protein. (Adapted from Kumar V, Abbas A, Fausto N, Mitchell R: Basic pathology, ed 8, Philadelphia, 2007, Saunders.)
Cells also usually receive diverse “antigrowth” signals from their normal milieu. Contact with other cells, with basement membranes, and with soluble factors all normally signal cells to stop proliferating. These mechanisms can put a halt to unregulated cell growth. In addition, this normal antigrowth signal must be inactivated or ignored. Common mutations that subvert the antigrowth signal include inactivation of the tumor suppressor retinoblastoma or, conversely, activation of the protein kinases that drive the cell cycle, the cyclin-dependent kinases (see Chapter 1). Next, cells have a mechanism that causes them to self-destruct when growth is excessive and cell cycle checkpoints have been ignored. This self-destruct mechanism, called apoptosis, is triggered by diverse stimuli, including normal development and excessive growth (see Chapter 2). The pathway to apoptosis is disabled in advanced cancers. The most common mutations conferring resistance to apoptosis occur in the TP53 gene.
Angiogenesis
If cancers are to grow larger than a millimeter in diameter, they need their own blood supply to deliver oxygen and nutrients. However, in adults new blood vessel growth is normally limited to areas of wound healing and to the uterus during the proliferative phase of the menstrual cycle. Tiny cancers lack the ability to grow new blood vessels and may never grow larger than a grain of sand. More advanced cancers can, however, secrete multiple factors that stimulate new blood vessel growth (called neovascularization or angiogenesis). The angiogenic factors, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and basic fibroblast growth factor (bFGF), by recruiting new vascular endothelial cells and initiating the proliferation of existing blood vessel cells, allow small cancers to become large cancers.13 Therapies directed against new vessel growth are in clinical use; these agents include bevacizumab, a monoclonal antibody that inhibits VEGF; erlotinib, sorafenib, and sunitinib, inhibitors of the VEGF and PDGF receptor tyrosine kinases; and thalidomide, which decreases vascular proliferation (Figure 11-12).14
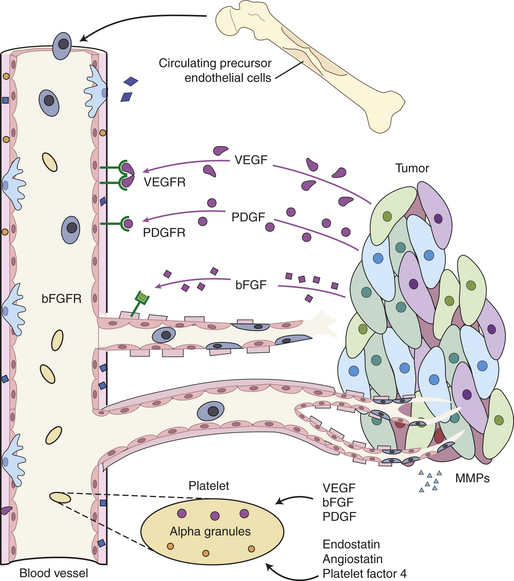
Figure 11-12 Tumor-induced angiogenesis. Malignant tumors secrete angiogenic factors and tissue-remodeling matrix metalloproteinases (MMPs) that actively induce formation of new blood vessels. New blood vessels are formed from both local endothelial cells and circulating precursor cells recruited from the bone marrow. Circulating platelets can also release regulatory proteins into the tumor. MMPs, matrix metalloproteases; PDGF and PDGFR, platelet-derived growth factor and its receptor; VEGF and VEGFR, vascular endothelial growth factor and its receptor; bFGF and bFGFR, basic fibroblast growth factor and its receptor. (Adapted from Folkman J: Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov 6[4]:273-826, 2007.)
Telomeres and Immortality
A hallmark of cancer cells is their immortality. Usually the only cells in the body that are “immortal” are germ cells (those that generate sperm and eggs) and stem cells. Other cells in the body are not immortal and can divide only a limited number of times (known as the Hayflick limit) before they cease dividing. One major block to unlimited cell division (i.e., immortality) is the size of a specialized structure called the telomere. Telomeres are protective ends, or caps, on each chromosome and are placed and maintained by a specialized enzyme called telomerase (Figure 11-13). As you might expect, telomerase is usually active only in germ cells (in ovaries and testes) and in stem cells. All other cells of the body lack telomerase. Therefore, when nongerm cells begin to proliferate abnormally, their telomere caps become smaller and smaller with each cell division. Short telomeres normally signal the cell to cease cell division. If the telomeres become critically small, the chromosomes become unstable and fragment, and then the cells die. Cancer cells, when they reach a critical age, activate telomerase somehow in order to restore and maintain their telomeres and thereby make it possible to divide over and over again.15 Because telomerase is specifically activated in cancer cells, and potentially in cancer stem cells, it is an attractive therapeutic target.16
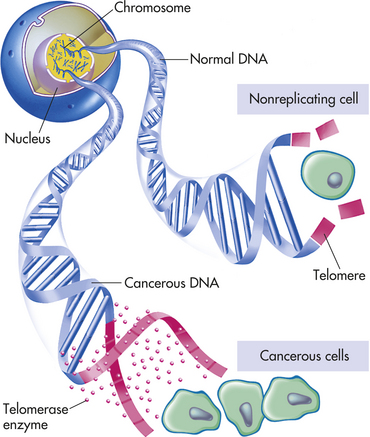
Figure 11-13 Control of immortality: telomeres and telomerase. Normal cells cannot divide indefinitely because the ends of their chromosomes are capped by telomeres. In the absence of the telomerase enzyme, telomeres get shorter and shorter with each division until, when they are critically short, they signal to the cell to stop dividing. In cancer cells the telomerase gene is “switched on,” producing an enzyme that rebuilds the telomeres. Thus the cancer cell becomes immortal and able to divide indefinitely without losing its telomeres.
Finally, it appears genetic differences exist between cells that successfully metastasize and those that do not.17 Specific genes regulate the ability to metastasize. Decreased cell-to-cell adhesion, the secretion of various proteases that digest surrounding barriers, and the ability to grow in new locations, all contribute to successful metastasis18 (discussed later in this chapter).
Oncogenes and Tumor-Suppressor Genes: Accelerators and Brakes
The previous discussion refers to the activating and inactivating of various genes as being key in the development of cancer. Just what types of changes in genes actually occur in cancer? First, it is useful to distinguish between oncogenes and tumor-suppressor genes. Table 11-4 compares the two types of cancer genes. Oncogenes are mutant genes that in their normal nonmutant state direct synthesis of proteins that positively regulate (accelerate) proliferation. Conversely, tumor-suppressor genes encode proteins that in their normal state negatively regulate (halt, or “put the brakes on”) proliferation. Hence, they also have been referred to as anti-oncogenes.
Table 11-4
Comparison of Cancer Gene Types
| Gene Type | Normal Function | Mutation Effect |
| Dominant oncogenes∗ | Encode proteins that promote growth (e.g., growth factors) | Overexpression, amplification, gain of function |
| Tumor suppressors (recessive oncogenes) | Encode proteins that inhibit proliferation and prevent or repair mutations | Loss of function of both alleles |
In its normal, nonmutant state, an oncogene is referred to as a proto-oncogene. An example of a proto-oncogene would be a growth factor (e.g., epidermal growth factor), or a growth factor receptor (e.g., epidermal growth factor receptor). Other positive regulators of proliferation are in the signal transduction pathway that transmits the signal from the growth factor receptor to the cell nucleus. Normally, ras is a proto-oncogene (see Figure 11-11).
Mutation of Normal Genes into Oncogenes
Point Mutations: Several types of genetic events can activate oncogenes (Box 11-1 and Figure 11-14). Perhaps the most common events are small scale changes in DNA such as point mutations, the alteration of one or a few nucleotide base pairs (see Chapter 4). This type of mutation can have profound effects on the activity of proteins. A point mutation in the ras gene converts it from a regulated proto-oncogene to an unregulated oncogene, an accelerator of cellular proliferation. Activating point mutations in ras are found in many cancers, especially pancreatic and colorectal cancer.19 Specialized tests, such as direct DNA sequencing, can detect such point mutations in clinical samples.
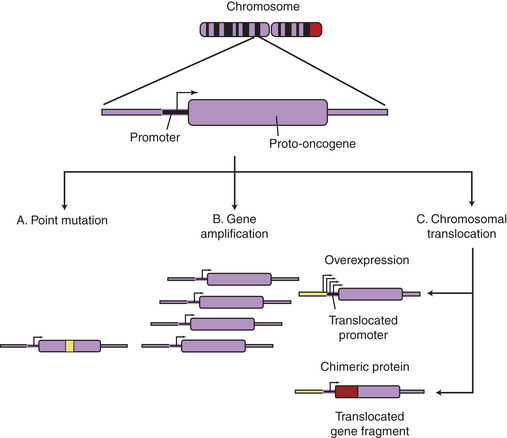
Figure 11-14 Oncogene activation mechanisms. Cellular genes may become cancerous oncogenes as a result of (1) point mutations that alter one or a few nucleotide base pairs, causing the production of a protein that is activated as a result of the altered sequence (e.g., ras); (2) amplification of the cellular gene, resulting in higher levels of protein expression (e.g., N-myc in neuroblastoma); or (3) chromosomal translocations that either (a) lead to the juxtaposition of a strong promoter, causing increased protein expression (c-myc in Burkitt lymphoma), or (b) produce a novel fusion protein that is derived from gene fragments normally present on different chromosomes (Bcr-Abl in chronic myeloid leukemia). (From Haber DA: Molecular genetics of cancer. In ACP Medicine, Danbury, CT, 2004, WebMD.)
Chromosome Translocations: Chromosome translocations, in which a piece of one chromosome is translocated to another chromosome, can activate oncogenes by way of either of two distinct mechanisms. First, a translocation can cause excess and inappropriate production of a proliferation factor. One of the best examples is the t(8;14) translocation found in many Burkitt lymphomas20; t(8;14) designates a chromosome that has a piece of chromosome 8 fused to a piece of chromosome 14 (see Chapter 27). Burkitt lymphoma is an aggressive cancer of B lymphocytes. The myc proto-oncogene found on chromosome 8 is normally turned on at low levels in proliferating lymphocytes and is turned off in mature lymphocytes. The MYC protein is part of the positive signal for cell proliferation. If an accidental formation of the t(8;14) translocation occurs, the myc gene is aberrantly placed under the control of a B cell immunoglobulin (Ig) gene present on chromosome 14. The Ig gene is very active in maturing B lymphocytes. The t(8;14) alters the control of myc; its normal low level is switched to high levels, as directed by an Ig gene promoter. MYC, when inappropriately high, drives proliferation and blocks differentiation. Hence, the t(8;14) translocation causes cancer of maturing B cells (see Figure 11-14, C).
Chromosome translocations also can lead to production of novel proteins with growth-promoting properties. In a different type of leukemia, CML, a specific chromosome translocation is almost always present. This translocation, t(9;22), was first identified in association with CML in Philadelphia in 1960 and so is often referred to as the Philadelphia chromosome.21 This translocation fuses two chromosomes right in the middle of two genes, bcr on chromosome 9 and abl on chromosome 22. The result is production of a BCR-ABL fusion protein containing the first half of BCR and the second half of ABL. BCR-ABL is a misregulated protein tyrosine kinase that promotes growth of myeloid cells. Imatinib, a drug that specifically targets this tyrosine kinase, represents the first successful chemotherapy targeted against the product of a specific oncogenic mutation. Imatinib and related drugs are highly effective in CML and, because of their specificity, lack the toxic side effects noted with nonspecific anticancer drugs.22 These drugs are not effective in those who do not have the t(9;22) translocation or related mutations. In modern personalized cancer therapy, knowing the specific genetic alteration can predict which drugs are best for the individual.
Gene Amplification: Another type of genetic abnormality that turns on oncogenes is gene amplification (see Figures 11-14, B and 11-15). Amplifications are the result of duplication of a small piece of a chromosome over and over again, so that instead of the normal two copies of a gene, tens or even hundreds of copies are present (see Chapter 4). Gene amplification results in increased expression of an oncogene, or in some cases, drug resistance genes. The N-myc oncogene is amplified in 25% of childhood neuroblastoma cases and confers a poor prognosis.23 The epidermal growth factor receptor erbB2 is amplified in 20% of breast cancers.24 Individuals whose cancers have erbB2 amplification respond well to drugs specifically targeted to this oncogene.12
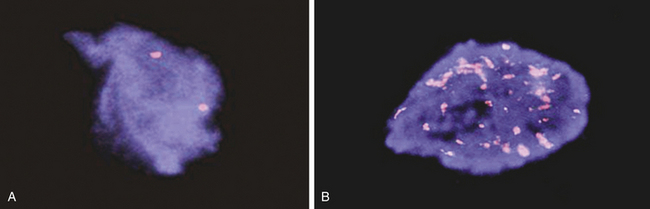
Figure 11-15 N-myc gene amplification in neuroblastoma. The N-myc gene is detected in human neuroblastoma cells using a technique called FISH (fluorescent in situ hybridization) A, A single pair of N-myc genes are detected in normal cells and in low-grade neuroblastoma. B, Multiple, amplified copies of the N-myc gene are detected in some cases of neuroblastoma. Amplification of the N-myc gene strongly associated with a poor prognosis in childhood neuroblastoma. (Courtesy of Arthur R. Brothman, PhD, FACMG, University of Utah School of Medicine.)
Tumor-Suppressor Genes: Tumor suppressor genes are genes whose major function is to negatively regulate cell growth and prevent mutations. Tumor suppressors may normally slow the cell cycle, inhibit proliferation resulting from growth signals, or stop cell division when cells are damaged. Examples of several tumor suppressors are given in Table 11-5. One of the first discovered tumor suppressor genes, the retinoblastoma (Rb) gene, normally strongly inhibits the cell division cycle (see Chapter 1). When it is inactivated, the cell division cycle can proceed unchecked. Rb is mutated in childhood retinoblastoma, and in many lung, breast, and bone cancers as well.
Table 11-5
Some Familial Cancer Syndromes Caused by Tumor-Suppressor Gene Function Loss
| Syndrome | Gene |
| Retinoblastoma | Rb |
| Li-Fraumeni syndrome | TP53 |
| Familial melanoma | p16INK4a |
| Neurofibromatosis | Neurofibromin |
| Familial adenomatous polyps | APC |
| Breast cancer | BRCA1 |
Whereas oncogenes are activated in cancers, tumor suppressors must be inactivated to allow cancer to occur (see Table 11-5 and Figure 11-16). A single genetic event can activate an oncogene because it can act in a dominant manner in the cell. However, we have two copies, or alleles, of each gene, one from each parent. It therefore takes two hits to inactivate the two alleles of a tumor-suppressor gene. The first allele of a tumor suppressor is often inactivated by point mutations. For example, the Rb gene may be inactivated on one chromosome by a point mutation (e.g., the copy inherited from the father). Because the other copy of the retinoblastoma gene (in this example, the one from the mother) is intact, a functional RB protein can still be made and therefore the cell division cycle can be regulated appropriately. If the remaining gene is mutated or silenced, then all RB function is lost and another step toward cancer occurs (see Chapter 5).
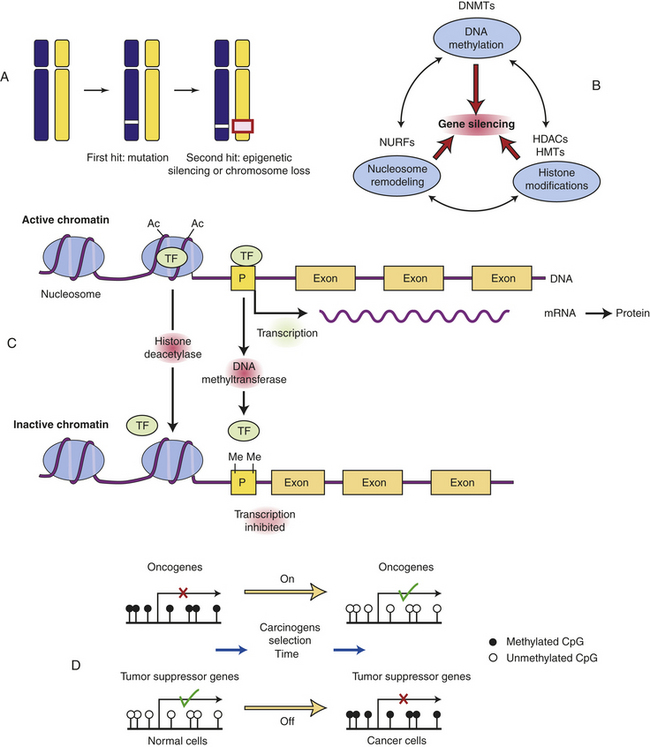
Figure 11-16 Silencing tumor-suppressor genes by epigenetic alterations. Tumor suppressor genes can be turned off by a variety of mechanisms. A, In this example, the first hit is a point mutation in a tumor suppressor gene (white box), followed by either epigenetic silencing or chromosome loss of the second allele (red box). B, Genes can normally be silenced by a variety of interacting processes including DNA methylation, histone modifications, nucleosomal remodeling, and microRNAs (not shown). A number of cellular enzymes contribute to these modifications, including DNA methyltransferases (DNMTs), histone deacetylases (HDACs), histone methyltransferases (HMTs), and complex nucleosomal remodeling factors (NURFs). Gene silencing is essential for normal development and differentiation. C, Histone modification and promoter methylation regulate gene expression. Genes are transcribed when chromatin is modified by addition of acetyl (Ac) groups to specific lysine groups in histones. Gene expression can be turned off when specific acetyl groups are removed (by histone deacetylase (HDACs) or when the CpG-rich promoter regions of genes are modified by direct DNA methylation (by DNA methyltransferase). In addition, small endogenous RNA molecules (microRNAs or miRNA) can bind to mRNA and reduce gene expression. D, Changes in promoter methylation turn cancer genes off and on. Oncogenes can be turned on by promoter hypomethylation, and tumor suppressor genes can be turned off by promoter hypermethylation. Each of these changes can produce selective growth and survival advantage for the cancer cell. (B adapted from Jones PA, Baylin SB: The epigenomics of cancer, Cell 128:683-692, 2007. C from Gluckman PD et al: N Engl J Med 359[1]:66, 2008; D from Shames DS, Minna JF, Gazdar AF: DNA methylation in health, disease, and cancer, Curr Mol Med 7:85-102, 2007.)
Loss of Heterozygosity: For the function of a tumor suppressor to be lost, both chromosomal copies (alleles) of the gene must be inactivated. This is because they act in a recessive manner at the level of the cell. Although it may seem intuitive that simple inactivating mutations might disrupt both alleles, in fact this is not what usually happens.25 Instead, the first allele (in the preceding example, the paternal copy) is inactivated by simple mutation, but the second allele (in this example, the maternal copy) is lost because entire regions of the maternal chromosome are epigenetically silenced or a piece of the chromosome can be simply lost (see Figure 11-16, A). Because you have two chromosomes, one from each parent, you can be heterozygous for nearby genetic markers; loss of a chromosome region in a tumor is referred to as loss of heterozygosity, or LOH. Loss of heterozygosity, like silencing, unmasks inactivating mutations in recessive tumor suppressor genes. For example, the Rb gene resides on chromosome 13, in a region referred to as q14 (13q14). Most individuals with Rb mutations have a subtle mutation in one allele and have lost the other copy of Rb through loss of the 13q14 chromosome region on the other chromosome.
Cancer Epigenetics—Turning Off Genes Without Mutation
Abnormal gene silencing is emerging as a major cause of cancer. Gene expression can be regulated in a heritable manner (i.e., passed from a parent to a child or from a single cell to its progeny) by an “epigenetic” mechanism called silencing that is passed from mother to daughter cells during cell division and does not require mutations or changes in DNA sequence (also see Chapter 4). More simply, the same DNA sequence can produce dramatically different phenotypes depending on chemical modifications that alter the expression of genes. Epigenetic silencing is caused by reversible chemical modification (methylation, addition of methyl group; acetylation, addition of acetyl group) of histones and related chromatin components, as well as methylation of cytosine residues in DNA known as DNA methylation (see Figure 11-16, Figure 4-26 and Chapter 12). Whole regions of chromosomes are normally shut off by silencing, so that the pattern of gene expression is different than in other cells with the same genes. In this way, the progeny of liver cells remain liver cells, and skin cells remain skin cells. Notably, global changes in epigenetic silencing can turn these cells back into stem cells.26
Changes in gene silencing contribute to the development of cancer.27 Many cancers have increased methylation of DNA in the promoter region of tumor suppressor genes (in CpG islands or C-cytosine and G-guanine, p-phosphodiester bond, rich sequences that are often located near promoter regions; see Chapters 4 and 12). They also have associated changes in the modification of histones in the chromatin, including methylation of lysines 9 and 27 in histone H3. In addition, overexpression of chromosome silencing proteins known as the polycomb complex, and loss of expression of histone deacetylases (enzymes that remove acetyl groups from histone proteins) SIRT1 is seen in human cancers. These changes in chromatin-modifying genes alter the promoter regions of genes leading to their silencing. The boundaries of the normally silenced regions can also spread in cancer cells, thereby shutting off previously active genes. In either case, silencing can shut off critical tumor suppressor genes in the absence of mutations in the gene. Early in the development of cancer, these changes in gene expression can lead to a selective advantage for affected cells, perhaps leading to their immortalization and clonal expansion. Silencing of tumor suppressors may be a faster way to create cancer cells than mutational or genetic loss of tumor suppressors.28 Conversely, loss of silencing can contribute to inappropriate expression of oncogenes. Chemotherapeutic drugs that can regulate gene silencing, including histone deacetylase (HDAC) inhibitors and 5 azacytidine (which reverses the effects of DNA methyltransferases (DNMTs), have proven effective in reactivating silenced tumor-suppressor genes in the treatment of selected cancers29,30 (see Figure 11-16).
Guardians of the Genome
The previous discussion of mutations leads naturally to the question of how mutations occur in the first place. The integrity of genetic information can be compromised at several points: during each round of DNA synthesis, during each mitosis when chromosomes are segregated to daughter cells, and when external mutagens (e.g., chemicals and radiation) alter or disrupt DNA. Multiple mechanisms have evolved to protect and repair the genome.31 These repair mechanisms are directed by caretaker genes, genes that are responsible for the maintenance of genomic integrity. Caretaker genes encode proteins that are involved in repairing damaged DNA, such as occurs with errors in DNA replication, mutations caused by ultraviolet or ionizing radiation, and mutations caused by chemicals and drugs. Loss of function of caretaker genes leads to increased mutation rates. If DNA damage is severe, the cell undergoes programmed cell death, or apoptosis, rather than simply dividing with damaged DNA.
Inherited mutations can disrupt the caretaker genes that protect the integrity of the genome. Examples include the disorder xeroderma pigmentosum (XP); affected individuals have defects in the repair of ultraviolet light–induced DNA damage and should avoid direct sunlight exposure. They have a very high incidence of skin cancer. Hereditary nonpolyposis colorectal cancer (HNPCC) results from an inherited defect in repairing DNA base pair mismatches that occur from time to time during DNA replication. Affected individuals have an increased rate of small insertions and deletions in DNA, leading to a high rate of colon and other cancers.32 Finally, there are inherited mutations that threaten the integrity of entire chromosomes. Bloom syndrome and Fanconi aplastic anemia are two autosomal recessive disorders in which affected individuals demonstrate marked chromosomal instability. Chromosome breaks, aberrant fusions, and chromosome loss are common. As a consequence, these individuals have a high risk of developing cancer at an early age.
The rate of individual gene mutation is probably too low to account for the acquisition of many new mutations during the evolution of a malignant cancer clone. In addition to abnormal epigenetic silencing, chromosome instability (often referred to as CIN) also appears to be increased in malignant cells.33 The underlying mechanism of this instability is not clear but may be caused by malfunctions in the cellular machinery that regulates chromosome segregation at mitosis.34 Chromosome instability results in a high rate of chromosome loss, as well as loss of heterozygosity and chromosome amplification. Each of these events can accelerate the loss of tumor-suppressor genes and the overexpression of oncogenes.
Genetics and Cancer-Prone Families
Genetic events are the primary basis of carcinogenesis.35 Most of the genetic and epigenetic alterations that cause cancer occur during the lifetime of the individual within the somatic tissues. As discussed, the frequency of genetic changes can be increased by exposure to mutagens, that is, agents causing mutations, and by defects in DNA repair. Because these genetic events occur in somatic cells as opposed to germ cells, they are not transmitted to future generations. Even though they are genetic events they are not inherited! It is possible, however, for cancer-predisposing mutations to occur in germline cells (cells that produce gametes) (Figure 11-17). Mutations present in germline cells result in the transmission of cancer-causing genes from one generation to the next, producing families with a high incidence of specific cancers. These inherited mutations that predispose to cancer are almost invariably found in tumor-suppressor genes (see Table 11-5).
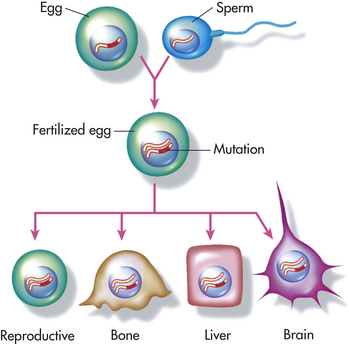
Figure 11-17 Germline mutation. Inherited mutations are carried in the deoxyribonucleic acid (DNA) of reproductive cells. When reproductive cells containing mutations combine to produce offspring, the mutation will be present in all of the offspring’s body cells. (Modified with permission fromLea DN, Jenkins JF, Francomano CA: Genetics in clinical practice, Jones and Bartlett Publishers, 1998. www.jbpub.com.)
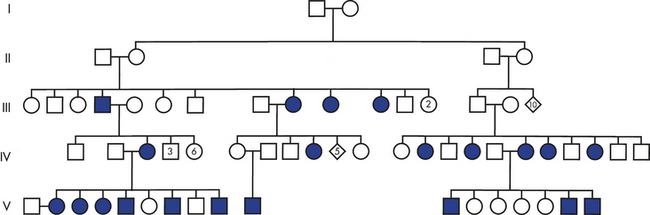
Although rare, such “cancer families” demonstrate that inheritance of a mutated gene can cause cancer (Figure 11-18). In these families, inheritance of one mutant allele predisposes a person to a specific form of cancer: individuals who inherit the germline mutant allele will inevitably suffer loss of the normal allele by loss of heterozygosity (LOH) or epigenetic silencing (see Figure 11-16) in some cells and go on to develop the tumor. Examples of human cancers that can be inherited are retinoblastoma, a childhood cancer of the eye that can be caused by germline mutations in one allele of the Rb gene; Wilms tumor, a childhood cancer of the kidney (Wt1); neurofibromatosis (Nf1); inherited breast cancer (BRCA1); and familial polyposis coli or adenomas of the colon (APC). A specific tumor-suppressor gene has been found in each of these cancers. In many cases, these tumor-suppressor genes also are inactivated in sporadic (as opposed to inherited) cancers. For example, inherited mutations in the APC gene are rare and account for only a few percent of all colon cancers. However, 85% of sporadic colon cancers also have acquired mutations of APC, mutations that occurred over time in the individual. Characterization of cancer-causing genes and other genetic factors helps identify individuals prone to developing cancer (see Figure 11-18) and contributes to our understanding of sporadic cancers. Individuals known to carry mutations in tumor-suppressor genes (for example, women with a germline BRCA1 mutation) are offered targeted cancer screening to facilitate early cancer detection and therapy.36
Inflammation, Immunity, and Cancer
Chronic inflammation has been recognized for close to 150 years as being an important factor in the development of cancer.37,38 Epidemiologic studies strongly support the conclusion that the active immune response in chronic inflammation predisposes to cancer. Individuals who have suffered with ulcerative colitis for 10 years or more have up to a 30-fold increase in the risk of developing colon cancer. Chronic viral hepatitis caused by hepatitis B virus (HBV) or hepatitis C virus (HCV) infection markedly increases the risk of liver cancer. A large study found a 66% increase in risk of lung cancer among women with chronic asthma, an inflammatory disease of the airways.39 Table 11-6 details the various inflammatory conditions and infectious agents associated with cancer.
Table 11-6
Chronic Inflammatory Conditions and Infectious Agents Associated with Neoplasms
| Inflammatory Condition | Associated Neoplasm(s) |
| Asbestosis, silicosis | Mesothelioma, lung carcinoma |
| Bronchitis | Lung carcinoma |
| Cystitis, bladder inflammation | Bladder carcinoma |
| Gingivitis, lichen planus | Oral squamous cell carcinoma |
| Inflammatory bowel disease, Crohn disease, chronic ulcerative colitis | Colorectal carcinoma |
| Lichen sclerosus | Vulvar squamous cell carcinoma |
| Chronic pancreatitis, hereditary pancreatitis | Pancreatic carcinoma |
| Reflux esophagitis, Barrett esophagus | Esophageal carcinoma |
| Sialadenitis | Salivary gland carcinoma |
| Sjögren syndrome, Hashimoto thyroiditis | MALT lymphoma |
| Skin inflammation | Melanoma |
| Infectious Agent | Associated Neoplasm(s) |
| AIDS (HIV, herpesvirus type 8) | Non-Hodgkin lymphoma, squamous cell carcinomas, Kaposi sarcoma |
| Chronic cholecystitis | Gallbladder cancer |
| Chronic cystitis (schistosomiasis) | Bladder, liver, rectal carcinoma; follicular lymphoma of the spleen |
| Gastritis, gastric ulcers (Helicobacter pylori) | Gastric adenocarcinoma, MALT |
| Hepatitis | Liver carcinoma |
| Mononucleosis (Epstein-Barr virus) | B cell non-Hodgkin lymphoma, Burkitt lymphoma |
| Opisthorchis, cholangitis (liver flukes, bile acids) | Cholangiosarcoma, colon carcinoma |
| Osteomyelitis | Skin carcinoma in draining sinuses |
| Pelvic inflammatory disease, chronic cervicitis (HPV, gonorrhea, chlamydia) | Ovarian carcinoma, cervical/anal carcinoma |
HIV, Human immunodeficiency virus; HPV, human papillomavirus; MALT, mucosa-associated lymphoid tissue.
Modified from Coussens LM, Werb Z: Nature 420(6917):860-867, 2002; Dalgleish AG, O'Byrne KJ: Adv Cancer Res 84:231-276, 2002; Shacter E, Weitzman SA: Oncology 16(2):217-226, 229, 2002.
The reasons for the association of inflammation and cancer are complex and differ from site to site. After injury and during infection, inflammatory cells, including neutrophils, lymphocytes, and macrophages release cytokines and growth and survival factors that stimulate local cell proliferation and new blood vessel growth to promote wound healing by tissue remodeling (see Chapter 6). These factors combine in chronic inflammation to promote continued proliferation (Figures 11-19 and 11-20). In addition, inflammatory cells release compounds such as reactive oxygen species (ROS), and other reactive molecules that can promote mutations and block the cellular response to DNA damage. Notably, increased abundance of the enzyme cyclooxygenase-2 (COX-2), which generates prostaglandins during acute inflammation, has been associated with colon and some other cancers. Nonsteroidal anti-inflammatory drugs (NSAIDs), such as aspirin. that inhibit COX-2 can reduce the risk of colon cancer by as much as 20% (see Chapter 5).40
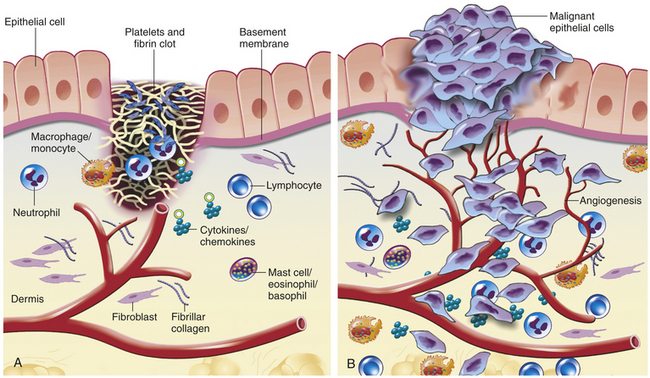
Figure 11-19 Wound healing versus invasive tumor growth. A, Wounds simulate a local inflammatory response. With tissue injury, a blood clot with activated platelets forms, releasing multiple factors that stimulate wound healing. Chemotactic factors such as transforming growth factor-beta and platelet-derived growth factor, derived from activated platelets, initiate granulation tissue formation, activation of fibroblasts, and secretion of proteolytic enzymes (including matrix metalloproteinases) necessary for remodeling of the extracellular matrix. Signaling from many cell types in the wound, including stromal cells, facilitates healing. Once the wound is healed, the signals presumably stop. B, Invasive carcinomas act as disorganized wounds. Neoplastic cells produce angiogenic factors, cytokines and chemokines that are mitogenic and/or chemoattractants for numerous cells. In return, activated fibroblasts and infiltrating inflammatory cells also secrete proteolytic enzymes, cytokines, and chemokines, which are mitogenic for neoplastic cells as well as for endothelial cells involved in neoangiogenesis. These factors, which in the normal situation promote wound healing, now can stimulate tumor growth and angiogenesis, induce fibroblast migration and maturation, and promote metastatic spread through the venous or lymphatic networks. (Adapted from Coussens LM, Werb Z: Nature 420[6917]:860-867, 2002.)
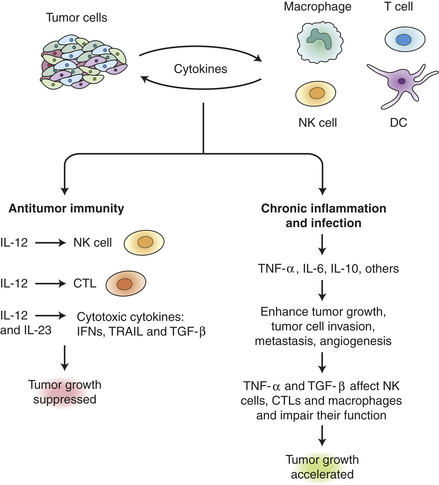
Figure 11-20 Cytokines link innate immunity, inflammation, and cancer. Tumor and immune cells both secrete cytokines that influence tumor growth. Some cytokines (interferons, TRAIL, TGF-β) have antitumor activity, while others stimulate NK and CTL cells to attack the tumor. Balancing the antitumor immunity, however, TNF-α and several interleukins accelerate tumor progression by multiple mechanisms, including growth stimulation, promotion of angiogenesis and by suppressing the activity of infiltrating immune cells. CTL, cytotoxic T lymphocyte; DC, dendritic cell; IFN, interferon; IL, Interleukin; NK, natural killer cells; TGF-β, transforming growth factor beta; TNF, tumor necrosis factor. (Adapted from Lin W, Karin M: J Clin Invest 117[5]:1175-1183.)
The Immune System Protects Us Against Viral-Associated Cancers
It is a popular belief that damage to the immune system may lead to development of common cancers. This idea arose decades ago as our understanding of the immune response against infectious disease developed. Modern data support a more nuanced conclusion. Although the immune system is indeed important in protecting us against cancers caused by specific viral infections (detailed later), it does not effectively protect us against most common cancers. Individuals on chronic powerful immunosuppressive drugs, such as those given for kidney, heart, or liver transplant, have a much higher risk of developing viral-associated cancers, with a 10-fold increased risk of non-Hodgkin lymphoma (caused by Epstein-Barr virus) and up to 1000-fold increased risk of developing Kaposi sarcoma (caused by human herpesvirus 8 [HHV8]) (additional information about viruses and cancer below.) The same immunosuppressed individuals, however, have only a slight increase in the risk of common cancers such as lung and colon cancer (and this could well be because of increased inflammation at those sites) and no increase in the risk of breast or prostate cancer.41,42
In fact, there are many complex interactions between elements of the immune system and tumors (Figure 11-20). Tumors activate surrounding stromal and inflammatory cells, including tumor-infiltrating lymphocytes and macrophages, to secrete multiple cytokines that support tumor growth and spread. In parallel, various cells of the immune system can exert antitumor effects through factors such as TNF-related apoptosis-inducing ligand (TRAIL), interleukin (IL)-10 and IL-12. The double-edged effects of the immune cells, both promoting and inhibiting proliferation, may explain why it has been so difficult to harness the immune system to fight cancer.
Viral Causes of Cancer
As noted, a number of viruses have been associated with human cancer43,44 (Table 11-7). An even broader spectrum of viruses have been associated with cancer in animals. In humans, hepatitis B and C viruses (HBV, HCV), Epstein-Barr virus (EBV), Kaposi sarcoma herpesvirus (KSHV) (also known as HHV8), and human papillomavirus (HPV) are associated with about 15% of all human cancers worldwide. Cancer of the cervix and hepatocellular carcinoma account for about 80% of virus-linked cancer. The initial infection with hepatitis B or C is not associated with cancer; instead, it is acquisition of a chronic viral hepatitis that markedly increases cancer risk (also see Inflammation, Immunity and Cancer). Chronic hepatitis B infections are common in parts of Asia and Sub-Saharan Africa and confer up to a 200-fold increased risk of developing liver cancer. Chronic hepatitis C infections have become increasingly recognized in Western countries. Up to 80% of liver cancer worldwide is associated with chronic hepatitis caused either by HBV or HCV. In both cases, it appears that a lifetime of chronic liver inflammation predisposes to the development of hepatocellular carcinoma. Widespread use of the HBV vaccine is expected to significantly decrease the incidence of chronic hepatitis B and hence hepatocellular carcinoma. Unfortunately, a vaccine for HCV is not yet available.
Table 11-7
Human Viruses Associated with Cancer
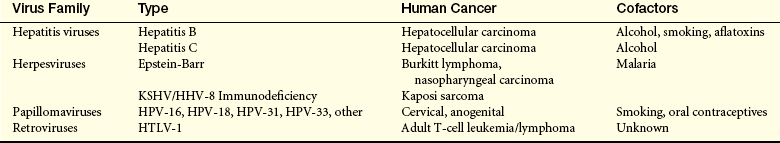
KSHV/HHV-8, Kaposi sarcoma–associated herpesvirus/human herpesvirus-8; HPV, human papillomavirus; HTLV-1, human T-cell leukemia/lymphoma virus-1.
Modified from Mendelsohn J et al, editors: The molecular basis of cancer, ed 2, Philadelphia, 2001, Saunders.
Virtually all cervical cancer is caused by infection with specific subtypes of HPV, which infects basal skin cells and commonly causes warts. There are more than 100 HPV subtypes, but only a few (HPV16, 18, 31, 45, and a few others) are associated with cervical, anogenital, and penile cancer (see Chapters 12 and 23). HPV causes cancer when the viral DNA becomes accidentally integrated into the genomic DNA of the infected basal cell of the cervix and directs the persistent production of viral oncogenes. Early oncogenic HPV infection is readily detected by the Papanicolaou (Pap) test, an examination of cervical epithelia scrapings. Early detection of cellular atypia in a Pap test alerts healthcare providers to the possibility of cervical carcinoma in situ, which can be effectively treated. The Pap test is probably the most effective cancer screening test developed to date. Most recently, vaccines protecting against two or four common oncogenic HPV subtypes have been approved for clinical use; if widely given to young women prior to initial HPV infection, these vaccines can prevent many cases of cervical cancer.45
EBV and HHV8 are members of the herpesviridae family.43 EBV, the cause of infectious mononucleosis, infects B lymphocytes and stimulates their proliferation. In individuals who are immunosuppressed because of HIV infection or because of drugs given for an organ transplant, persistent EBV infection can lead to the development of B-cell lymphomas. This development in those with organ transplants is known as post-transplant lymphoproliferative disorder (PTLD).46 One effective therapy for PTLD is, if possible, to decrease or stop immunosuppressant drugs and allow the immune system to attack the virus. EBV infection also is associated with Burkitt lymphoma in areas of endemic malaria and with nasopharyngeal carcinoma, a cancer endemic in Chinese populations in Southeast Asia.47 HHV8 is linked to the development of Kaposi sarcoma, a cancer that occurs in older adult men and in a markedly more virulent form in immunocompromised individuals, especially those infected with HIV. HHV8 also has been linked to several rare lymphomas.
Human T-cell leukemia-lymphoma virus (HTLV) is an oncogenic retrovirus linked to the development of adult T-cell leukemia and lymphoma (ATLL).44 HTLV is transmitted vertically, that is, inherited by children from infected parents, and horizontally, by breast-feeding, sexual intercourse, blood transfusions, and exposure to infected needles. Infection with HTLV may be asymptomatic, and only a small fraction of infected individuals develop ATLL, often many years after acquiring the virus. It is clear that infection by an oncogenic virus is far from sufficient to cause cancer. For example, in some industrialized regions, Epstein-Barr virus can infect 90% of the adolescent and young adult population, yet only a very small percentage of these individuals develop EBV-related cancer. For each of these infections, important cofactors increase the risk that an infection will develop into cancer.
Bacterial Cause of Cancer
Helicobacter pyloriis a bacterium that infects more than half of the world’s population. Chronic infection with H. pylori is an important cause of peptic ulcer disease and is strongly associated with gastric carcinoma, a leading cause of cancer deaths worldwide. It is also associated with a less common cancer, gastric mucosa–associated lymphoid tissue (MALT) lymphomas.48 H. pylori infection is often acquired in childhood and disproportionately affects lower socioeconomic classes. Although most infections are asymptomatic, prolonged chronic inflammation can lead to atrophic gastritis that can, in a small fraction of individuals, progress to dysplastic changes and finally frank gastric adenocarcinoma. Recent data have shown that H. pylori infection induces methylation of specific genes in the gastric mucosa.49 Eradication of H. pylori from infected individuals prior to the development of dysplasia may prevent the development of cancer.50 However, there is no expert consensus on the value of population screening and treatment strategies.51 The MALT lymphomas associated with chronic H. pylori infections may depend on chronic inflammation and antigenic stimulation associated with infections, and therefore treatment with antibiotics may be useful even in cases of early lymphoma.52
CANCER INVASION AND METASTASIS
Metastasis is the spread of cancer cells from the site of the original tumor to distant tissues and organs through the body. Metastasis is a defining characteristic of cancer, contributes significantly to the pain and suffering from cancer, and is the major cause of death from cancer. When localized, low-stage cancer can often be cured by a combination of surgery, chemotherapy, and radiation. These same therapies are frequently ineffective against cancer that has metastasized. For example, in appropriately treated women with low-stage breast cancer, the 5-year survival rate is often greater than 90%.53 Tragically, less than 30% of women with metastatic breast cancer are alive 5 years after diagnosis.54 A growing body of basic and clinical research is defining the biologic principles of metastasis, with the hope that this improved understanding will lead to novel diagnostic approaches and better therapies to prevent and treat metastatic cancers.55
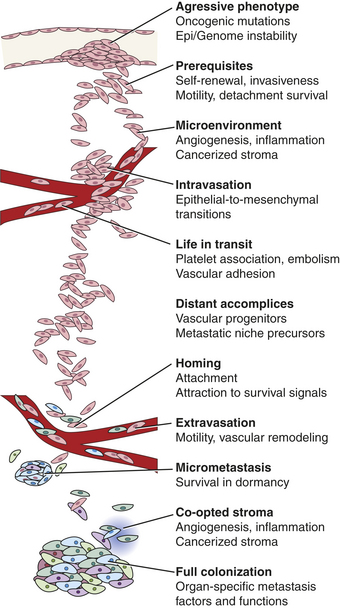
Invasion, or local spread, is a prerequisite for metastasis and is the first step in the metastatic process. In its earliest stages local invasion may occur by direct tumor extension.56 Eventually, however, cells migrate away from the primary tumor and invade the surrounding tissues. Mechanisms important in local invasion include (1) ongoing cancer proliferation; (2) digestion of connective tissue capsules and other structural barriers by secreted proteases; (3) changes in cell-to-cell adhesion, often by changes in expression of cell adhesion molecules, such as cadherins and integrins, making the cancer cells more slippery and mobile; and (4) increased motility of individual tumor cells (Figure 11-22). To transition from local to distant metastasis, the cancer cells must also be able to invade local blood and lymphatic vessels, a task facilitated by stimulation of neoangiogenesis and lymphangiogenesis by factors such as VEGF. Finally, a successful metastatic cell must be able to survive in the circulation, attach in an appropriate new microenvironment, and multiply to produce an entire new tumor, similar to the characteristics of a cancer stem cell. Because metastasis requires successful completion of each and every step, there may be many opportunities to interrupt this potentially lethal pathway.
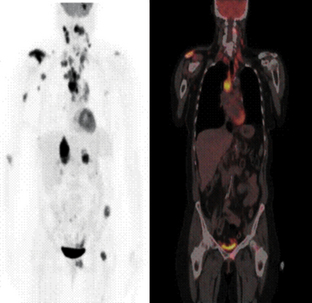
Figure 11-22 Metastatic non-small cell lung cancer. This 54-year-old woman had a non-small cell lung cancer (NSCLC) resected from the left upper lobe. Five years later, these studies were obtained. The positron emission tomography (PET) scan using 18-fluoro-deoxyglucose shows metastatic lesions in the brain, right shoulder, mediastinal and cervical lymph nodes, as well as the liver, left pelvis, and proximal femur. (Left) PET whole-body image. (Right) Representative coronal image from the whole body FDG-PET/CT–fused image of the same patient. The fused image consists of the CT image with the metabolic information superimposed in color. The pattern of spread is most likely from the primary tumor to the large mediastinal lymph nodes, followed by lymphatic spread to cervical nodes. Blood-borne spread produced the bone, brain, and liver metastases. Normally, only the heart, brain, and bladder show strong signal in PET scan. CT, Computed tomography; FDG, fluorodeoxyglucose. (Images courtesy John Hoffman, MD, Huntsman Cancer Institute, Salt Lake City, Utah.)
Only Rare Cells in a Cancer Are Able to Metastasize
Metastasis is a highly inefficient process. A landmark clinical review examined a group of women with advanced ovarian cancer.57 These unfortunate women had accumulated a large amount of peritoneal fluid filled with malignant ovarian cancer cells (malignant ascites). To relieve the pressure caused by the ascites, the fluid was surgically shunted into the venous circulation. This palliative procedure relieved the abdominal pressure but had the side effect of moving millions of ovarian cancer cells an hour directly into the bloodstream. Despite this direct injection of billions of cancer cells into the circulation, these women unexpectedly had no increased number of metastases when they died. The conclusion from this clinical study, that most cancer cells cannot successfully cause metastases, has been supported by many other clinical and laboratory studies as well.56 The reason lies both in the seed and the soil. Cancer cells (the seeds) must surmount multiple physical and physiologic barriers in order to spread, survive, and proliferate in distant locations, and the destination (the soil) must be receptive to the growth of the cancer. It has been suggested that the metastatic cell must, like a decathlon champion, be successful in every event to allow a cancer to spread.56
How do cancer cells develop the ability to metastasize? As cancers grow, they develop increasing heterogeneity. This reflects the genetic instability of cancer and aberrant differentiation of subclones of the malignant cells. The same changes that happen in cells to cause the primary cancer, including gene mutations, deletions, translocations, and epigenetic silencing, all work to provide genetic heterogeneity in the tumor cells as they proliferate. As this diversity increases, this increases the number of cells in the cancer mass with new abilities that can facilitate metastasis.
Detachment and Invasion
In order for cells to move away from their normal niche, they must be able to detach from the stroma and migrate. Cells are normally attached to extracellular matrix (ECM). Epithelial cells are further organized by basement membranes, a meshwork of collagens and other connective proteins. To facilitate cancer spread, many tumors and their associated inflammatory cells secrete proteases and protease activators, such as the matrix metalloproteinases (MMPs) and plasminogen activators. Active proteases digest the extracellular matrix and basement membranes, creating pathways through which cells can move, while releasing bioactive peptides as digestion products that further stimulate tumor growth and mobility. Recognition of the role of proteases led to the development of MMP inhibitors as a weapon against cancer progression. However, it is now recognized that there are also many proteases that block cancer proliferation, perhaps explaining why the first generation of MMP inhibitors showed little or no effect in clinical trials in treating cancer.58
Another key mediator of cell detachment is the down-regulation in the cancer cell of specific adhesion molecules, such as E-cadherin and integrins. E-cadherin is commonly lost in cancers through diverse mechanisms including mutation, epigenetic silencing, proteolysis, and through a tissue remodeling process known as epithelial-mesenchymal transition (EMT) (regulated by the transcription factors with the evocative names Snail, Twist, and Slug). When E-cadherin is lost, cells are able to detach from their extracellular attachments and migrate away more readily.
Survival and Spread in the Circulation
Normal cells, when separated from their ECM, undergo anoikis, a form of apoptosis. In contrast, tumor cells that have already adapted to a hypoxic environment have already been selected for resistance to apoptosis, often by loss of normal cell death pathways. For example, neuroblastomas with loss of the proapoptotic caspase 8 genes are able to avoid apoptosis after loss of integrins, and are more able to metastasize than the same cells with normal levels of caspase 8. Accordingly, individuals whose neuroblastomas have low levels of caspase 8 have a poor prognosis.59 (See Chapter 2.)
Selective Adherence in Favorable Sites
After release from the ECM and digestion of basement membranes, cancer cells gain access to the circulation through new tumor-associated blood vessel growth or angiogenesis (described earlier), also known as neovascularization. Mobile tumor cells are able to enter the circulation, perhaps facilitated by the leaky newly made vessels and attraction of the cells because of chemoattractants coming from these new vessels. Once in the circulation, metastatic cells must be able to withstand the physiologic stresses of travel in the blood and lymphatic circulation, including high shear rates and exposure to immune cells. One mechanism is for tumor cells to bind to blood platelets, giving them a protective coat of nonmalignant blood cells that both shields the tumor cells and creates a small tumor embolus, or cancer clot, that can promote cancer cell survival in distant locations.
The patterns of metastasis are dictated by the interaction between the cancer cells and the microenvironments in which they land. Two distinct mechanisms give rise to patterns of distant spread. First, cancer cells spread through vascular and lymphatic pathways, as well as natural tissue planes. The neovascularization of a cancer offers malignant cells direct access into the venous blood, and draining lymphatics can carry malignant cells to regional lymph nodes. Single cells, clumps, and even tumor fragments can disseminate by these routes. Anatomic patterns of lymphatic and venous blood flow help determine how colon cancers spread to the liver, liver cancers spread through the portal vein to the lungs, lung cancers spread through the systemic circulation to the brain, and breast cancer spreads through lymphatics to axillary lymph nodes (Figure 11-23). There is also a major yet poorly understood selectivity of different cancers for different sites. Thus metastatic breast cancer often spreads through the bloodstream to bones but rarely to kidney or spleen, whereas lymphomas often spread to the spleen but uncommonly spread to bone. In a key study, Schackert and Fidler60 injected different types of cancer cells into the carotid artery of mice. Despite identical blood flow–mediated distribution of the cancer cells, each cancer cell type produced cancers in very different parts of the brain. This tissue selectivity is likely caused by specific interactions between the cancer cells and specific receptors on the small blood vessels in different organs (Table 11-8). Experimental metastasis studies in mice are beginning to reveal additional molecular reasons for this tissue specificity (Figure 11-24). Examples include interaction between α3β1 integrins binding to laminin-5 receptors in the lung, and the chemokine receptor CXCR4 on breast cancer cells promoting homing to lung tissues expressing the ligand CXCL12.55
Table 11-8
| Primary Tumor | Major Anatomic Pathway | Common Site of Distant Metastasis |
| Lung | Pulmonary vein, left ventricle | Multiple organs, including brain |
| Colorectal | Mesenteric lymphatics, portal venous system | Liver |
| Inferior vena cava, right ventricle, pulmonary artery | Lungs | |
| Testicular | Lymphatics to the periaortic area to the subclavian veins to the right ventricle | Lungs, liver, brain |
| Prostate | Regional lymphatics and veins, which drain to Batson plexus of presacral veins | Bones (especially lumbar spine), liver |
| Breast | Axillary, transpectoral, and internal mammary lymphatics | Bone, lung, brain, liver |
| Head and neck | Direct extension | Lymphatics, liver, bones |
| Ovarian | Direct extension, peritoneal seeding, mesenteric veins | Peritoneal surfaces, diaphragm, omentum, liver |
| Sarcoma (extremity) | Inferior vena cava, right ventricle, pulmonary artery | Lungs |
| Melanoma | Regional lymphatics | In transit lymphatics, lung, liver, brain, gastrointestinal tract |
Figure 11-23 Patterns of metastatic spread. A, Sites of hematogenous metastasis. Blood-borne tumor metastasis leads to growth of secondary tumors in several main sites. The macroscopic appearance of bone metastasis are shown in B, where lesions are seen in vertebrae. C, Numerous metastases from a neoplasm of the stomach are seen in the brain in D, The liver is the most common site for metastases from tumors in the gastrointestinal tract which arose from a colonic neoplasm. E, Metastatic tumor has replaced both adrenal glands, as is commonly seen with spread from lung and breast tumors. F, The lung is the most common site for blood-borne metastases from tumors outside the spinal tract, particularly mesenchymal tumors.
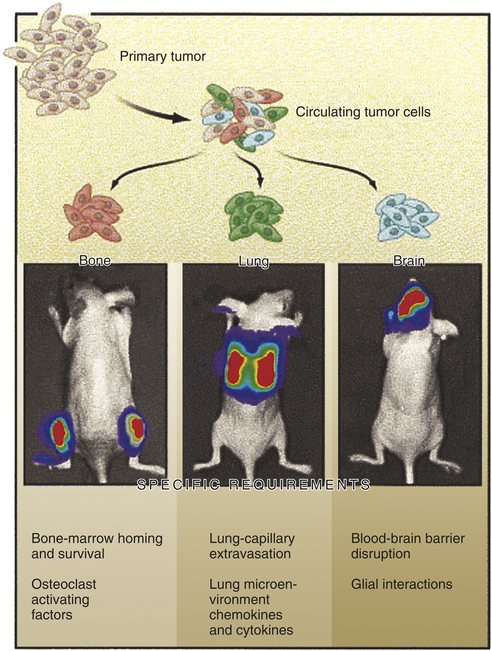
Figure 11-24 Clues as to how the patterns of metastatic spread are determined. Different cancer cells from the same cancer can spread to different locations. This is because tumors are heterogeneous mixtures of cells, with variations in the genes that are turned on and off. Different anatomic locations favor different types of cells. Studies with mice illustrated here show that expression of specific factors such as adhesion molecules, chemokines, and host factors such as blood-brain barrier disruption can favor the spread of specific cancer cells to different locations. In this example, cancer cells were visualized by genetically tagging cells with firefly luciferase for bioluminescent imaging. (From Gupta GP, Massagué J: Cancer metastasis: building a framework, Cell 127[4]:679-695, 2006.)
Escape from the Circulation and Development of a New Microenvironment
As the study with women with ovarian cancer illustrates, pumping millions of cancer cells in the bloodstream does not necessarily cause metastatic disease. Cancer cells may arrive in a new location and survive but not proliferate to form a clinically relevant metastasis. The tumor cells that do not make the transition from simple survival to robust proliferation in a new location are said to be in a state of dormancy. This dormancy may account for the observation that solitary tumor cells can be detected in the blood years after a complete clinical remission in individuals, and that many people with detectable micrometastases will not develop clinically obvious metastases.61,62 One of the factors that allow proliferation of cancer cells in the new environment may be cancer’s recruitment of normal cells from local and circulating bone marrow stem cells. One developing concept is that successful metastatic tumor cells secrete factors that recruit circulating mesenchymal stem cells to the metastatic site (see Figure 11-12). These newly recruited stem cells then differentiate into tumor-supporting stroma and new blood vessels. One area of research is aimed at understanding why some micrometastases remain dormant; new therapies might block their progression to clinically important disease.
CLINICAL MANIFESTATIONS AND TREATMENT OF CANCER
Clinical Manifestations of Cancer
Diagnosis and Staging
Cancer can be discovered in many ways: after screening tests, routine exams, and after investigation of symptoms (see Box 11-2). The symptoms a cancer produces are as diverse as the types of cancer. The location of the cancer can determine symptoms by physical pressure, obstruction, and loss of normal function, or a cancer can cause problems far away from its source by pressing on nerves or secreting bioactive compounds. Whatever the initial complaint, once the diagnosis is suspected and a tumor has been identified, it is essential that tumor tissue be obtained to establish a definitive diagnosis and correctly classify the disease. Various methods of obtaining tissue are described in Table 11-9.
Table 11-9
| Procedure | Purpose | Example |
| Excisional biopsy | Complete removal, usually with a margin of normal tissue | Full resection, e.g., mastectomy, partial colectomy |
| Incisional biopsy | Removal of a portion of a lesion | Lymph node biopsy, muscle mass biopsy |
| Core needle biopsy | Often performed with direct vision, or guided with ultrasound or computed tomography (CT) | Needle biopsy of prostate or liver mass |
| Fine needle aspiration | Obtains dissociated cells for cytology but does not preserve tissue structure | Thyroid, breast mass |
| Exfoliative cytology | Cells shed from the surface, for example, from cervix, sputum (lung) or urine. | Brushings from lung or colon endoscopy |
Once tissue is obtained, it is examined by the pathologist under the microscope for the histologic hallmarks of cancer detailed in the beginning of the chapter. The classification of the cancer can be further facilitated by a variety of clinically available tests, including immunohistochemical stains, flow cytometry, electron microscopy, chromosome analysis, and nucleic acid–based molecular studies.
If the diagnosis of cancer is established, it is critical to establish if the cancer has spread, known as the stage of the cancer. Staging initially involves determining the size of the tumor, the degree to which it has locally invaded, and the extent to which it has spread (metastasized) (Figure 11-25). Specific molecular tests are increasingly used in staging as well. Diverse schemes are used for staging different tumors. In general, a four-stage system is used, with carcinoma in situ regarded as a special case. Cancer confined to the organ of origin is stage 1, cancer that is locally invasive is stage 2, cancer that has spread to regional structures, such as lymph nodes, is stage 3, and cancer that has spread to distant sites, such as a liver cancer spreading to the lung or a prostate cancer spreading to bone, is stage 4. One common scheme for standardizing staging is the World Health Organization’s TNM system: T for tumor spread, N for node involvement, and M indicating the presence of distant metastasis (see Figure 11-25). The prognosis generally worsens with increasing tumor size, lymph node involvement, and metastasis (Table 11-10). Staging also may alter the choice of therapy, with more aggressive therapy being delivered to more invasive disease.

Cancer Treatment
The diagnosis of cancer has a profound effect on individuals and their families. Responses range from depression to resigned fatalism to aggressive no-holds-barred pursuit of therapy. The decision as to what type of therapy to follow should be based on a full consideration by the individual, the family, and the medical team of the individual’s diagnosis, prognosis, and therapeutic options. Many types of cancer can be effectively treated with chemotherapy, radiotherapy, surgery, and combinations of these modalities. Caregivers must recognize that many individuals seek additional nonscience-based explanations and therapies and often use alternative therapies, either concurrently or sequentially. Alternative therapies can be biologically harmless or harmful; rarely is there any evidence they are medically effective and in the worst cases they can be expensive, delay the use of effective therapies, and produce unknown side effects. A challenge for the medical team is to provide the same level of psychosocial comfort and support that alternative therapies can provide, while also providing scientifically rational evidence-based therapies.
Chemotherapy
The era of modern chemotherapy began with the observation in World War II that mustard gas exposure caused suppression of the bone marrow. Related compounds, such as nitrogen mustard and cyclophosphamide, were then tested and produced clinical responses in hematologic malignancies, including lymphomas. Also in the late 1940s, based on the remarkable clinical observation that the vitamin folic acid could increase leukemia growth, antifolate drugs were developed (leading ultimately to methotrexate) that produced remissions in previously untreatable leukemias.72
All chemotherapeutic agents take advantage of specific vulnerabilities in target cancer cells. Antimetabolites, such as methotrexate and L-asparaginase, block normal growth pathways in all cells, but leukemia and other cancer cells are exquisitely sensitive to folic acid and asparagine deprivation, whereas nonmalignant cells are far less sensitive. Similarly, some cancer cells are highly sensitive to DNA-damaging agents, such as cyclophosphamide and anthracyclines, because of the oncogenic mutations that accelerate the cell cycle and DNA synthesis. Cellular checkpoints prevent normal cells treated with microtubule-directed drugs, such as vincristine and the taxanes, from going through mitosis, whereas cancer cells treated with these agents lack normal checkpoints, continue through mitosis, and undergo mitotic catastrophe (see Chapter 1).
Single chemotherapeutic agents often shrink cancers, but these drugs given alone rarely if ever provide a cure. Hence, chemotherapy drugs are usually given in combinations designed to attack a cancer from many different weaknesses at the same time and to limit the dose and therefore the toxicity of any single agent. Cancers contain a very large number of cells, and commonly a small fraction of those cells may be resistant to a particular drug. However, those cells are likely to be sensitive to the second or third drug in a chemotherapy cocktail. Scheduling of drug administration is also very important, with many studies showing cancers are more likely to develop drug resistance if there are significant delays between planned courses of chemotherapy.
The newest highly targeted agents used to treat cancer exploit specific vulnerabilities uncovered by molecular analysis in specific diseases (Table 11-11). These new drugs are still used in combination with conventional chemotherapy and, to be effective, they must be used in diseases in which the molecular target is present. Imatinib is highly effect in treating CML and gastrointestinal stromal tumor (GIST) but ineffective in virtually all other cancers. Fortunately, because these drugs are so tightly targeted they have much less toxicity than conventional chemotherapies that have targets in virtually all cells.
Table 11-11
Examples of Molecular-Era Anticancer Drugs
EGF, Epidermal growth factor; FGF, Fibroblast growth factor; EGF, Vascular endothelial growth factor.
Chemotherapy can be used for several distinct purposes. Induction chemotherapy seeks to cause shrinkage or disappearance of tumors. In Hodgkin disease, chemotherapy alone can be used in some cases to cure the disease. In other settings, chemotherapy may shrink the tumor and improve symptoms without ultimately providing a cure. Adjuvant chemotherapy is given after surgical excision of a cancer with the goal of eliminating micrometastases. Neoadjuvant chemotherapy is given prior to localized (surgical or radiation) treatment of a cancer. As with induction chemotherapy, the effectiveness, or lack thereof, of neoadjuvant therapy can be measured, with follow-up scans. Neoadjuvant therapy can shrink a cancer so that surgery may spare more normal tissue. In the bone cancer osteogenic sarcoma, neoadjuvant therapy often converts a large tumor mass into a much smaller mass, allowing the surgeon to perform a limb-sparing excision rather than an amputation.
Radiation Therapy
Radiation therapy is used to kill cancer cells while minimizing damage to normal structures. Ionizing radiation damages cells by imparting enough energy to cause molecular damage, especially to DNA. The damage may be lethal, in which the cell is killed by radiation; potentially lethal, in which the cell is so severely affected by radiation that modifications in its environment will cause it to die; or sublethal, in which the cell can subsequently repair itself. Cellular compartments with rapidly renewing cells are, in general, more radiosensitive. Effective cell killing by radiation also requires good local delivery of oxygen, something not always present in large cancers. Radiation produces slow changes in most cancers and irreversible changes in normal tissues as well. Because of these irreversible changes, each tissue has a maximum lifetime dose of radiation it can tolerate. Radiation is well suited to treat localized disease in areas that are hard to reach surgically, overused in the brain and pelvis. A number of radiation delivery methods are available, with external beam being the most common. Radiation sources, such as small 125iodine capsules (also called seeds), can also be temporarily placed into body cavities, a delivery method termed brachytherapy. Brachytherapy is useful in the treatment of cervical, prostate, and head and neck cancers.
Surgery
Surgery plays many roles in the care of individuals with cancer. The multiple approaches to obtaining tissue for diagnosis have been discussed. Surgery is often the definitive treatment of cancers that do not spread beyond the limits of surgical excision. It is also indicated for the relief of symptoms, such as those caused by tumor mass obstruction. In selected high-risk diseases, surgery plays a role in the prevention of cancer. Individuals with familial adenomatous polyposis have close to a 100% lifetime risk of colon cancer because of germline mutations of the APC gene, so a prophylactic colectomy is indicated. The role of prophylactic mastectomy in women with BRCA1 mutations is more controversial.
Key principles apply specifically to cancer surgery, including obtaining adequate surgical margins during a resection to prevent local recurrences, placing needle tracks and biopsy incision scars (that may be contaminated with cancer cells) carefully so they can be removed in subsequent incisions, avoidance of the spread of cancer cells during surgical procedures through careful technique, and attention to obtaining adequate tissue specimens during biopsies so that the pathologist can be confident of the diagnosis. Additionally, the surgeon provides critical staging information by inspection, sampling, and removal of local and regional lymph nodes during procedures.
Complications of Cancer and Its Therapy
Paraneoplastic Syndromes: Paraneoplastic syndromes are symptom complexes that are triggered by a cancer but are not caused by direct local effects of the tumor mass. They are most commonly caused by biologic substances released from the tumor (e.g., hormones) or an immune response set off by the tumor. A small fraction of carcinoid tumors release hormones, including serotonin, into the bloodstream that cause flushing, diarrhea, wheezing, and rapid heartbeat. A number of cancers trigger an antibody response that attacks the nervous system, causing a variety of neurologic disorders that can precede other symptoms of cancer by months.73
Although infrequent, paraneoplastic syndromes are significant because they may be the earliest symptom of an unknown cancer and, in affected individuals, can be serious, often irreversible, and sometimes life threatening. Table 11-12 presents the classifications of paraneoplastic syndromes.
Table 11-12
| Clinical Syndromes | Major Forms of Underlying Cancer | Causal Mechanism |
| Endocrinopathies | ||
| Cushing syndrome | Small cell carcinoma of lung | ACTH or ACTH-like substance |
| Pancreatic carcinoma | ||
| Neural tumors | ||
| Syndrome of inappropriate antidiuretic hormone (SIADH) secretion | Small cell carcinoma of lung; intracranial neoplasms | Antidiuretic hormone or atrial natriuretic hormones |
| Hypercalcemia | Squamous cell carcinoma of lung | Parathyroid hormone–related protein (PTHRP), TGF-α, TNF, IL-1 |
| Breast carcinoma | ||
| Renal carcinoma | ||
| Adult T-cell leukemia/lymphoma | ||
| Ovarian carcinoma | ||
| Fibrosarcoma | ||
| Hypoglycemia | Other mesenchymal sarcomas | Insulin or insulin-like substance |
| Hepatocellular carcinoma | ||
| Bronchial adenoma (carcinoid) | ||
| Carcinoid syndrome | Pancreatic carcinoma | Serotonin, bradykinin |
| Gastric carcinoma | ||
| Renal carcinoma | ||
| Polycythemia | Cerebellar hemangioma | Erythropoietin |
| Hepatocellular carcinoma | ||
| Nerve and Muscle Syndromes | ||
| Myasthenia | Bronchogenic carcinoma | Immunologic |
| Disorders of the central and peripheral nervous systems | Breast carcinoma | |
| Dermatologic Disorders | ||
| Acanthosis nigricans | Gastric carcinoma | Immunologic; secretion of epidermal growth factor |
| Lung carcinoma | ||
| Uterine carcinoma | ||
| Bronchogenic, breast carcinoma | ||
| Dermatomyositis | Immunologic | |
| Osseous, Articular, and Soft Tissue Changes | ||
| Hypertrophic osteoarthropathy and clubbing of the fingers | Bronchogenic carcinoma | Unknown |
| Vascular and Hematologic Changes | ||
| Venous thrombosis (Trousseau phenomenon) | Pancreatic carcinoma | Tumor products (mucins that activate clotting) |
| Bronchogenic carcinoma | ||
| Other cancers | ||
| Advanced cancers | ||
| Nonbacterial thrombotic endocarditis | Thymic neoplasms | Hypercoagulability |
| Anemia | Unknown | |
| Others | ||
| Nephrotic syndrome | Various cancers | Tumor antigens, immune complexes |
ACTH, Adrenocorticotropic hormone; IL, interleukin; TGF, transforming growth factor; TNF, tumor necrosis factor.
From Kumar V, Abbas AK, Fausto N: Pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.
Pain: Pain is one of the most feared complications of advanced cancer. Although pain can be one of the presenting symptoms of cancer, most commonly there is little or no pain during the early stages of malignant disease. Significant pain, however, occurs in a large fraction of those individuals who are terminally ill with cancer. Pain is strongly influenced by fear, anxiety, sleep loss, fatigue, and overall physical deterioration. It occurs through an interaction among physiologic, cultural, and psychologic components.30,74 (The neurophysiology of pain is discussed in Chapter 15.)
Cancer-associated pain can arise from a variety of direct and indirect mechanisms. Direct pressure, obstruction, invasion of a sensitive structure, stretching of visceral surfaces, tissue destruction, infection, and inflammation all can cause pain. Pain can occur at the site of the primary tumor or because of a distant metastatic lesion. Furthermore, pain may be referred away from the involved site and manifest, for example, as back pain.
Specific sites are more prone to cancer-associated pain. Bone metastases, common in advanced breast and prostate cancer, can cause significant pain because of periosteal irritation, medullary pressure, vertebral collapse, and pathologic fractures. Brain tumors (primary or metastatic) can, depending on the location, cause headache, seizures, or neurologic deficits. Pain in the abdomen may be caused by bowel obstruction, or inflammation and infection. Hepatic malignancies can stretch the liver, resulting in a dull pain or a feeling of fullness over the right upper abdominal quadrant. Mucosal surfaces can develop painful ulcerative lesions from the cancer, chemotherapy, and radiation or leukopenia, or both.
The diagnosis and treatment of pain are the primary responsibilities of the health care team. The individual’s perception and, hence, reporting of pain can vary widely and be affected by such factors as age and cultural background. The first priority of treatment is to control pain rapidly and completely as judged by the individual.30 The second priority is to prevent recurrence of pain. Objective measurements of pain are increasingly being included along with the reporting of more traditional vital signs. Many institutions are using specialized pain management teams that are trained to recognize different types of acute and chronic pain, as well as the individual’s response to that pain. Many modalities are available to treat pain, ranging from combinations of NSAIDs and narcotics to palliative surgery and radiation therapy. Individual-controlled analgesia provides many benefits, not the least of which is regaining some control over one’s own body. Although cancer pain is a complex problem arising from multiple sources, individuals should be assured that suffering is not inevitable and that relief is attainable.75
Fatigue: Fatigue is the most frequently reported symptom of cancer and cancer treatment. The exact mechanisms that produce fatigue are poorly understood.76 Suggested causes include sleep disturbances, various biochemical changes secondary to disease and treatment, numerous psychosocial factors, level of activity, nutritional status, and other environmental and physical factors.77
The physiologic understanding of fatigue probably includes mechanisms for decreased muscle contractility. Overall, studies of muscle function suggest that some individuals with cancer may lose portions of muscle function needed to perform normal physical activities. Other areas of research include muscle function consequences from metabolic products of cancer treatment and associated muscle loss from circulating cytokines (e.g., tumor necrosis factor [TNF] and IL-1) (Figure 11-26). Similar to pain, fatigue is a subjective clinical manifestation. Fatigue is described by individuals with cancer as tiredness, weakness, lack of energy, exhaustion, lethargy, inability to concentrate, depression, sleepiness, boredom, lack of motivation, and decreased mental status. Some of these symptoms have been termed “chemo brain,” or mild cognitive impairment. The changes in cognitive function can be caused by the cancer itself or the stress associated with the diagnosis of cancer, however, because symptoms similar to “chemo brain” also occur in individuals who have not received chemotherapy.78
Figure 11-26 Theoretic framework for cytokine-induced cancer symptoms. Proinflammatory cytokines and chemokines (IL-1, TNF-α, IL-6, IFN) (solid blue lines) are released by immune cells. They exert their effects on peripheral nerves and the brain. Neurotransmitter responses by the brain are affected. The hypothalamic-pituitary-adrenal axis is activated with increased release of corticosteroids, which provide feedback (dotted red lines) to decrease cytokine production. IL, Interleukin; IFN, interferon; TNF, tumor necrosis factor. (Adapted from Cleeland CS et al: Cancer 97[11]:2919-2925, 2003.)
Cachexia: The syndrome of cachexia includes a constellation of symptoms including anorexia, early satiety (filling), weight loss, anemia, asthenia (marked weakness), taste alterations, and altered protein, lipid, and carbohydrate metabolism. Cachexia is the most severe form of malnutrition associated with cancer and results in wasting, emaciation, and decreased quality of life (Figure 11-27).79 Cachexia occurs in the face of seemingly adequate calorie intake because metabolic disturbances, including insulin resistance, hypertriglyceridemia, muscle wasting, and general increased derangement of metabolism, lead to significant inefficiency of energy usage. Anorexia, or loss of appetite, frequently worsens the weight loss associated with this abnormal metabolic state. Anorexia, itself, can be caused by pain, depression, chemotherapy, and/or radiotherapy. Alterations in taste by the same causes also can account for the anorexia in people with cancer by making foods seem bland or distasteful.
Figure 11-27 Cachexia. This severe form of malnutrition results in wasting and extensive loss of adipose tissue. (From Kamal A, Brockelhurst JC: Color atlas of geriatric medicine, ed 2, St Louis, 1991, Mosby.)
Altered carbohydrate metabolism causes a syndrome resembling diabetes mellitus. Individuals show hyperinsulinemia, insulin resistance, hyperglycemia, and abnormal glucose tolerance test results. These disturbances cause increased gluconeogenesis, which produces glucose from amino acids. In starvation, protein usually is spared to protect vital structures, but in cancer, protein and fatty acids are used to meet energy needs.
An unusual and frustrating component of cancer care is the person’s early satiety, or a sense of being full after only a few mouthfuls of food. Cytokines, including TNF-α, IL-6, and IFN-γ, appear to cause the metabolic alterations associated with tissue loss in cancer wasting. Tumor metabolites may also contribute to a cachectic state. For example, a factor found in the urine of some people with cancer induces the catabolism of muscle. This factor, originally called proteolysis-inducing factor, is a partial fragment of an antimicrobial peptide, dermcidin, normally expressed in the skin.80 (Cytokines are discussed in detail in Chapter 6.)
Anemia: Anemia is commonly associated with malignancy, with 20% of patients having hemoglobin concentrations below 9 g/dl (normal value = 15 g/dl). Mechanisms that cause anemia in people with cancer include chronic bleeding resulting in iron deficiency, severe malnutrition, cytotoxic chemotherapy, and malignancy in blood-forming organs. Chronic bleeding and iron deficiency can accompany colorectal or genitourinary malignancy. Iron also is malabsorbed in individuals with gastric, pancreatic, or upper intestinal cancer. Often there is a defect in the reutilization of iron because of lack of transfer of iron from the storage pool to blood cell precursors. This defect may be caused by increased secretion of IL-6 and hepcidin.81 Defects in erythropoietin production and shortened red cell survival have also been documented. In addition, anorexia can cause iron and folate deficiency. Megaloblastic (large red cell) anemias also may develop after methotrexate treatment.
Administration of erythropoietin, which stimulates production of erythrocytes, has been effective in correcting anemia in people with cancer; fewer red blood cell transfusions were required in most of the studied subjects. In addition, anemias occurring after chemotherapy or radiotherapy have been treated successfully with erythropoietin. However, studies have shown that aggressive use of erythropoietin increases the risk of blood clots and may worsen cancer survival, suggesting it should be reserved only for significant cancer-related anemia.82
Leukopenia and Thrombocytopenia: Direct tumor invasion of the bone marrow causes both leukopenia (decreased total white blood cell count) and thrombocytopenia (decreased number of platelets). More commonly, many chemotherapeutic drugs are toxic to the bone marrow, often causing granulocytopenia and thrombocytopenia. Granulocytopenia also can result from radiation therapy if it encompasses significant areas of the bone marrow. The duration of granulocytopenia and hence the risk of serious infection can be lessened by treatment with recombinant human granulocyte colony-stimulating factor (rhG-CSF, filgrastim).83 rhG-CSF stimulates white blood cell precursors in the marrow to proliferate and differentiate rapidly. Thrombocytopenia is a major cause of hemorrhage in people with cancer and is often treated with platelet transfusions. Thrombocytopenia also is an accompanying disorder of disseminated intravascular coagulation that occurs in individuals with acute promyelocytic leukemia (see Chapter 20) and severe infections (see Chapter 25).
Infection: Infection is the most significant cause of complications and death in patients with malignant disease. When the absolute granulocyte count falls below 500 cells per μL, the risk of serious microbial (bacterial and fungal) infection increases. People with cancer also have debility with advanced disease, and immunosuppression from the underlying cancer and the radiotherapy and chemotherapy used to treat it. (Factors that predispose individuals with cancer to infection are summarized in Table 11-13.) Surgery also can lower resistance to infection because removal of large quantities of tissue, together with hemorrhage, dead spaces, and poor tissue perfusion can create favorable sites for infection. Hospital-acquired (nosocomial) infections increase because of indwelling medical devices, inadequate wound care, and the introduction of microorganisms from visitors and other individuals.

Gastrointestinal Tract: The entire gastrointestinal (GI) tract relies on rapidly growing cells to produce an effective barrier to trauma and infection and to provide an absorptive surface for nutrients. Chemotherapy and radiation therapy may cause a decreased cell turnover, thereby leading to oral ulcers (stomatitis), malabsorption, and diarrhea. The disruption of barrier defenses also increases the risk for infection, especially invasion by a person’s own GI flora. Therapy-induced nausea, thought to be caused by an agent’s direct action on the central nervous system’s vomiting centers, historically has been a major obstacle in therapy.
Aggressive antinausea (antiemetic) therapy, including the centrally acting serotonin 5-HT3 antagonists such as ondansetron or dolasetron, have allowed better tolerance of highly emetogenic protocols. Other popular antiemetics include steroids and phenothiazines. Synthetic cannabinoids, the active ingredients in marijuana, increase appetite in addition to having antinausea properties. Analgesia often includes opiate agents, vital in treating severe cases of mucosal lesions. Supplemental nutrition through enteral or parenteral routes may be needed to combat malnutrition. Good oral care and close attention to hygiene may help prevent complications arising from mucosal membrane breakdown.
Hair and Skin: Alopecia (hair loss) results from chemotherapy effects on hair follicles. Alopecia is usually temporary, although hair may grow back with a different texture initially. Not all chemotherapeutic agents cause alopecia. Decreased renewal rates of the epidermal layers in the skin may lead to skin breakdown and dryness, altering the normal barrier protection against infection. Radiation therapy may cause skin erythema (redness) and contribute to breakdown.
Reproductive Tract: Radiation therapy and chemotherapy may affect the gametes, leading to varying degrees of decreased fertility and premature menopause. These effects are dose and age dependent, with the prepubertal gonad thought to be more resistant to damage. The potential for harm also is dependent on the agent used, with the alkylating category of chemotherapies carrying the greatest risk. Craniospinal irradiation for central nervous system tumors also may affect the hypothalamus or pituitary gland, with subsequent secondary gonadal failure because of lack of production of gonadotropin-releasing hormone, luteinizing hormone, and follicle-stimulating hormone. The potential for reproductive harm should be addressed before therapy, if possible, with provisions made for sperm or embryo banking.
Adenocarcinoma 361
Adjuvant chemotherapy 383
Adult stem cell 364
Anchorage independent 362
Angiogenesis 369
Angiogenic factor 369
Apoptosis 369
Autocrine stimulation 368
Autonomy 362
Benign tumor 361
Brachytherapy 388
Cachexia 390
Cancer 361
Carcinoma 361
Carcinoma in situ (CIS) 361
Caretaker gene 375
Chromosome instability 375
Chromosome translocation 371
Clonal expansion 368
Clonal proliferation 368
DNA methylation 373
Epigenetic change 367
Epigenetic silencing 375
Epigenetics 367
Epithelial-mesenchymal transition (EMT) 382
Gene amplification 372
Human T-cell leukemia-lymphoma virus (HTLV) 380
Immortal 362
Induction chemotherapy 388
Leukemia 361
Loss of heterozygosity (LOH) 373
Lymphoma 361
Malignant tumor 361
Metastasis 381
Multipotent 364
Mutagen 375
MYC protein 370
Neoadjuvant chemotherapy 383
Neoplasm 361
Neovascularization 383
Oncogene 370
Overdiagnosis 386
Overtreatment 386
Paraneoplastic syndrome 388
Personalized medicine 362
Pleomorphic 362
Point mutation 370
Post-transplant lymphoproliferative disorder, (PTLD) 379
Proto-oncogene 370
ras, 370
RAS 368
Retinoblastoma (Rb) gene 372
Sarcoma 361
Silencing 373
Stage 384
Telomerase 369
Telomere 369
Transformation 362
Tumor 361
Tumor marker 367
Tumor-suppressor gene 370
REFERENCES
1. Kern, S.E. Progressive genetic abnormalities in human neoplasia. In: Mendelsohn J., et al, eds. The molecular basis of cancer. Philadelphia: Saunders, 2001.
2. Morrow, M., O’Sullivan, M.J. The dilemma of DCIS. Breast. 2007;16(Suppl 2):S59–S62.
3. van’t Veer, L.J., Bernards, R. Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature. 2008;452(7187):564–570.
4. Skloot R: Henrietta’s dance, Johns Hopkins Magazine, April 2000. Available at www.jhu.edu/~jhumag/0400web/01.html.
5. Reya, T., et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111.
6. Al-Hajj, M., et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988.
7. Lapidot, T., et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice,. Nature. 1994;367(6464):645–648.
8. Bao, S., et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response,. Nature. 2006;444:756–760.
9. Armitage, P., Doll, R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 2004;91(12):1983–1989.
10. Kinzler, K.W., Vogelstein, B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–170.
11. Hanahan, D., Weinberg, R.A. The hallmarks of cancer,. Cell. 2000;100(1):57–70.
12. Ciardiello, F., Tortora, G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–1174.
13. Folkman, J. Angiogenesis. Annu Rev Med. 2006;57:1–18.
14. Folkman, J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6(4):273–286.
15. Mathon, N.F., Lloyd, A.C. Cell senescence and cancer. Nat Rev Cancer. 2001;1(3):203–213.
16. Shay, et al. Targeting telomerase for cancer therapeutics. Br J Cancer. 2008;98(4):677–683.
17. van’t Veer, L.J., et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536.
18. Kang, Y., et al. A multigenic program mediating breast cancer metastasis to bone,. Cancer Cell. 2003;3(6):537–549.
19. Bos, J.L. ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–4689.
20. Goldsby, R.E., Carroll, W.L. The molecular biology of pediatric lymphomas,. J Pediatr Hematol Oncol. 1998;20(4):282–296.
21. Nowell, P., Hungerford, D. A minute chromosome in human granulocytic leukemia,. Science. 1960;132:1497.
22. Druker, B.J., et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037.
23. Brodeur, G.M., et al. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224(4653):1121–1124.
24. Berns, E.M., et al. Prevalence of amplification of the oncogenes c-myc, HER2/neu, and int-2 in one thousand human breast tumours: correlation with steroid receptors. Eur J Cancer. 1992;28(2-3):697–700.
25. Cavenee, W.K., et al. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature. 1983;305(5937):779–784.
26. Takahashi, K., Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676.
27. Jones, P.A., Baylin, S.B. The epigenomics of cancer,. Cell. 2007;128(4):683–692.
28. Baylin, S.B., Ohm, J.E. Epigenetic gene silencing in cancer—a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6(2):107–116.
29. Dokmanovic, M., Clarke, C., Marks, P.A. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5(10):981–989.
30. Karpf, A.R., Jones, D.A. Reactivating the expression of methylation silenced genes in human cancer. Oncogene. 2002;21(35):5496–5503.
31. Rouse, J., Jackson, S.P. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297(5581):547–551.
32. Liu, B., et al. Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients. Nat Med. 1996;2(2):169–174.
33. Lengauer, C., Kinzler, K.W., Vogelstein, B. Genetic instability in colorectal cancers. Nature. 1997;386(6625):623–627.
34. Weaver, B.A., et al. Aneuploidy: instigator and inhibitor of tumororigenesis instability. Cancer Res. 2007;67(21):10103–10105.
35. Jorde, L.B., et al. Medical genetics, ed 3. St. Louis: Mosby; 2005.
36. Schneider, K. Counseling about cancer: strategies for genetic counseling, ed 2. Hoboken, NJ: Wiley; 2001.
37. Coussens, L.M., Werb, Z. Inflammation and cancer. Nature. 2002;420(6917):860–867.
38. Fitzpatrick, F.A. Inflammation, carcinogenesis and cancer. Int Immunopharmacol. 2001;1(9-10):1651–1667.
39. Vesterinen, E., et al. Cancer incidence among 78,000 asthmatic patients. Int J Epidemiol. 1993;22(6):976–982.
40. Dubé, C., et al. The use of aspirin for primary prevention of colorectal cancer: a systematic review prepared for the U.S. Preventive Services Task Force,. Ann Int Med. 2007;146(5):365–375.
41. Stewart, T., et al. Incidence of de-novo breast cancer in women chronically immunosuppressed after organ transplantation. Lancet. 1995;346(8978):796–798.
42. Vajdic, C.M., et al. Cancer incidence before and after kidney transplantation. JAMA. 2006;296(23):2823–2831.
43. Howley, P.M., Ganem, D., Kieff, E. Etiology of cancer: DNA viruses. In DeVita V.T., et al, eds.: Cancer: principles and practice of oncology, ed 8, Philadelphia: Lippincott Williams & Wilkins, 2008.
44. Poeschla, E.M., et al. Etiology of cancer: RNA viruses. In DeVita V.T., et al, eds.: Cancer: principles and practice of oncology, ed 8, Philadelphia: Lippincott Williams & Wilkins, 2008.
45. Paavonen, J., Lehtinen, M. Introducing human papillomavirus vaccines—questions remain. Ann Med. 2008;40(3):162–166.
46. Buell, J.F., et al. Malignancy in pediatric transplant recipients. Semin Pediatr Surg. 2006;15(3):179–187.
47. Yang, X.R., et al. Evaluation of risk factors for nasopharyngeal carcinoma in high-risk nasopharyngeal carcinoma families in Taiwan. Cancer Epidemiol Biomarkers Prev. 2005;14(4):900–905.
48. Helicobacter and Cancer Collaborative Group. Gastric cancer and Helicobacter pylori: a combined analysis of 12 case control studies nested within prospective cohorts. Gut. 2001;49(3):347–353.
49. Ushijima, T. Epigenetic field for cancerization. J Biochem Mol Biol. 2007;40(2):142–150.
50. Wong, B.C., et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291(2):187–194.
51. Correa, P., Houghton, J. Carcinogenesis of Helicobacter pylori. Gastroenterol. 2007;133(2):659–672.
52. Isaacson, P.G., Du, M.Q. MALT lymphoma: from morphology to molecules. Nat Rev Cancer. 2004;4(8):644–653.
53. Fyles, A.W., et al. Tamoxifen with or without breast irradiation in women 50 years of age or older with early breast cancer. N Engl J Med. 2004;351:963–970.
54. Carrick, S., et al. Single agent versus combination chemotherapy for metastatic breast cancer. Cochrane Database Syst Rev. (2):2006. [CD003372].
55. Gupta, G.P., Massagué, J. Cancer metastasis: building a framework. Cell. 2006;127(4):697–708.
56. Fidler, I.J. The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited,. Nat Rev Cancer. 2003;3:453–458.
57. Tarin, D., et al. Clinicopathological observations on metastasis in man studied in patients treated with peritoneovenous shunts. BMJ. 1984;288(6419):749–751.
58. López-Otín, C., Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7(10):800–808.
59. Dwayne, G., et al. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006;439(7072):95–99.
60. Schackert, G., Fidler, I.J. Site-specific metastasis of mouse melanomas and a fibrosarcoma in the brain or meninges of syngeneic animals. Cancer Res. 1998;48:3478–3484.
61. Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7(11):834–846.
62. Meng, S., et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10(24):8152–8162.
63. Armstrong, K., et al, Screening mammography in women 40 to 49 years of age: a systematic review for the American College of Physicians. Ann Intern Med. 2007(146):516–526.
64. Fletcher, S.W., Elmore, J.G., Clinical practice: Mammographic screening for breast cancer. N Engl J Med. 2003(348):1672–1680.
65. Gøtzsche, P.C., Nielsen, M. Screening for breast cancer with mammography. Cochrane database of systematic reviews (Online). 2006. [CD001877,].
66. Qaseem, et al, Screening mammography for women 40 to 49 years of age: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2007(146):511–515.
67. Miller, A.B., et al, Canadian National Breast Screening Study-2: 13-year results of a randomized trial in women aged 50-59 years. J Natl Cancer Inst. 2000(92):1490–1499.
68. Miller, A.B., et al, The Canadian National Breast Screening Study-1: breast cancer mortality after 11 to 16 years of follow-up. A randomized screening trial of mammography in women age 40 to 49 years. Ann Intern Med. 2002(137):305–312.
69. Baines, et al, Impact of menstrual phase on false-negative mammograms in the Canadian National Breast Screening Study. Cancer. 1997(80):720–724.
70. Elmore JG et al: International Variation in Screening Mammography Interpretations in Community-Based Programs, J Natl Cancer Inst 95(18):1384-1395
71. Berrington de González, A., Reeves, G., Mammographic screening before age 50 years in the UK: comparison of the radiation risks with the mortality benefits. Br J Cancer. 2005(93):590–596.
72. Chu, E., DeVita, V.T., Jr. DeVita, V.T., et al, eds. Cancer: Principles and practice of oncologyed 8,. Philadelphia: Lippincott Williams & Wilkins; 2008.
73. Darnell, R.B., Posner, J.B. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349:1543–1554.
74. Esteller, M., et al. A gene hypermethylation profile of human cancer,. Cancer Res. 2001;61(8):3225–3229.
75. Dy, S.M., et al. Evidence-based standards for cancer pain management. J Clin Oncol. 2008;26(23):3879–3885.
76. Ryan, J.L., et al. Mechanisms of cancer-related fatigue. Oncologist. 2007;12(Suppl 1):22–34.
77. Miller, A.H., et al. Neuroendocrine-immune mechanisms of behavioral comorbidities in patients with cancer. J Clin Oncol. 2008;26(6):971–982.
78. Hess, L.M., Insel, K.C. Chemotherapy-related change in cognitive function: a conceptual model. Oncol Nurs Forum. 2007;34(5):981–994.
79. Baracos, V.E. Cancer-associated cachexia and underlying biological mechanisms. Annu Rev Nutr. 2006;26:435–461.
80. Porter, D., et al. A neural survival factor is a candidate oncogene in breast cancer,. Proc Natl Acad Sci. 2003;100(10):10931–10936.
81. Nemeth, E., et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113(9):1271–1276.
82. Kelly, A.M., Khuri, F.R. Unraveling the mystery of erythropoietin-stimulating agents in cancer promotion. Can Res. 2008;68:4013–4017.
83. Bhana, N. Granulocyte colony-stimulating factors in the management of chemotherapy-induced neutropenia: evidence based review. Curr Opin Oncol. 2007;19(4):328–335.