ALTERED CELLULAR AND TISSUE BIOLOGY



Knowledge of the structural and functional reactions of cells and tissues to injurious agents, including genetic defects, is key to understanding disease processes. Diseases are now defined and interpreted in molecular terms and not just in general descriptions of altered structure. Altered cellular and tissue biology can be the result of adaptation, injury, neoplasia, aging, or death. (Neoplasia is discussed in Chapters 11 through 13.) Adaptation occurs in response to both normal, or physiologic, conditions and adverse, or pathologic, conditions. For example, the uterus adapts to pregnancy—a normal physiologic state—by enlarging. Enlargement occurs because of an increase in the size and number of uterine cells. In response to physiologic stressors or pathologic adaptations, such as high blood pressure, myocardial cells are stimulated to enlarge by the increased work of pumping. Like most of the body’s adaptive mechanisms, however, cellular adaptations to adverse conditions are usually only temporarily successful. Severe or long-term stressors overwhelm adaptive processes, and cellular injury or death ensues.
Cellular injury can be caused by any factor that disrupts cellular structures or deprives the cell of oxygen and nutrients required for survival. Injury may be reversible (sublethal) or irreversible (lethal) and is classified broadly as chemical, hypoxic (lack of sufficient oxygen), free radical, unintentional or intentional, and immunologic or inflammatory. Cellular injuries from various causes have different clinical and pathophysiologic manifestations.
Cellular death is confirmed by structural changes seen when cells are stained and examined with a microscope. The most important changes are nuclear changes; clearly, without a healthy nucleus, the cell cannot survive.
Cellular aging causes structural and functional changes that eventually lead to cellular death or a decreased capacity to recover from injury. Mechanisms explaining how and why cells age are not known, and distinguishing between pathologic changes and physiologic changes that occur with aging is often difficult. Aging clearly causes alterations in cellular structure and function, yet senescence—growing old is both inevitable and normal.
CELLULAR ADAPTATION
Cells adapt to their environment to escape and protect themselves from injury. An adapted cell is neither normal nor injured—its condition lies somewhere between these two states. Cellular adaptations, however, are a common and central part of many disease states. In the early stages of a successful adaptive response, cells may have enhanced function; thus it is hard to know what is a pathologic response vs. an extreme adaptation to an excessive functional demand. The most significant adaptive changes in cells include atrophy (decrease in cell size), hypertrophy (increase in cell size), hyperplasia (increase in cell number), and metaplasia (reversible replacement of one mature cell type by another less mature cell type). Dysplasia (deranged cellular growth) is not considered a true cellular adaptation but rather an atypical hyperplasia. These changes are shown in Figure 2-1.
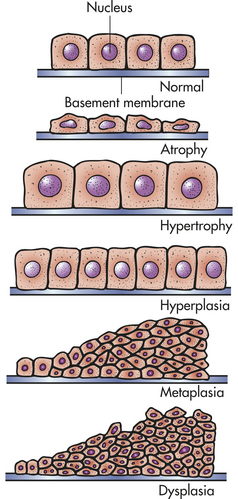
Figure 2-1 Adaptive alterations in simple cuboidal epithelial cells. (From Lewis SM, Heitkemper MM, Dirksen SR: Medical-surgical nursing: assessment and management of clinical problems, ed 6, St Louis, 2004, Mosby.)
Atrophy
Atrophy is a decrease or shrinkage in cellular size. If atrophy occurs in a sufficient number of an organ’s cells, the entire organ shrinks or becomes atrophic. Atrophy can affect any organ, but it is most common in skeletal muscle, the heart, secondary sex organs, and the brain (Figure 2-2). Atrophy can be classified as physiologic or pathologicPhysiologic atrophy occurs with early development. For example, the thymus gland undergoes physiologic atrophy during childhood. Pathologic atrophy occurs as a result of decreases in workload, use, pressure, blood supply, nutrition, hormonal stimulation, and nervous stimulation. Individuals immobilized in bed for a prolonged time exhibit a type of skeletal muscle atrophy called disuse atrophy. Aging causes brain cells to become atrophic and endocrine-dependent organs, such as the gonads, to shrink as hormonal stimulation decreases. Whether atrophy is caused by normal physiologic conditions or by pathologic conditions, atrophic cells exhibit the same basic changes.
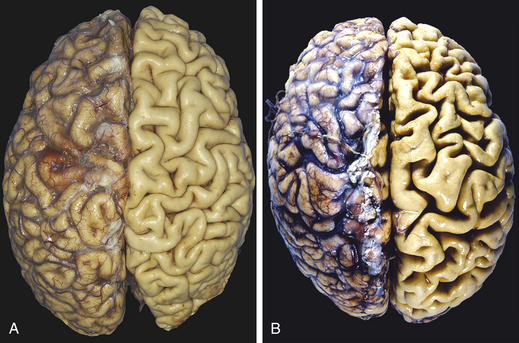
Figure 2-2 Atrophy. A, Normal brain of a young adult. B, Atrophy of the brain in an 82-year-old male with atherosclerotic disease. Atrophy of the brain is because of aging and reduced blood supply. Note that loss of brain substance narrows the gyri and widens the sulci. The meninges have been stripped from the right half of each specimen to reveal the surface of the brain. (From Kumar V, Abbas A, Fausto N: Robbins and Cotran pathologic basis of disease, ed 8, Philadelphia, 2007, Saunders.)
The atrophic muscle cell contains less endoplasmic reticulum and fewer mitochondria and myofilaments (part of the muscle fiber that controls contraction) than does the normal cell. In muscular atrophy caused by nerve loss, oxygen consumption and amino acid uptake are rapidly reduced. The biochemical changes of atrophy are just beginning to be understood. The mechanisms probably include decreased protein synthesis, increased protein catabolism, or both. The primary pathway of protein catabolism is the ubiquitin-proteasome pathway, and up-regulation of proteasome (protein degrading complex) activity is characteristic of atrophic muscle changes.1 Proteins degraded in this pathway are first conjugated to ubiquitin (another small protein) and then degraded by proteaosomes.
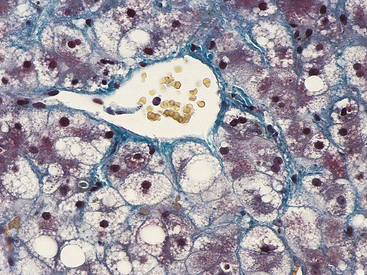
Atrophy as a result of chronic malnutrition is often accompanied by a “self-eating” process called autophagy creating autophagic vacuoles. These vacuoles are membrane-bound vesicles within the cell that contain cellular debris—small fragments of mitochondria and endoplasmic reticulum—and hydrolytic enzymes. Atrophic change causes a rapid increase in hydrolytic enzymes, which are isolated in autophagic vacuoles to prevent uncontrolled cellular destruction. Thus the vacuoles proliferate as needed to protect the uninjured organelles from the injured organelles and are eventually taken up and destroyed by lysosomes (see p. 5). Certain contents of the autophagic vacuole may resist destruction by lysosomal enzymes and persist in membrane-bound residual bodies. An example of this is granules that contain lipofuscin, the yellow-brown age pigment. Lipofuscin accumulates primarily in liver cells, myocardial cells, and atrophic cells.
Hypertrophy
Hypertrophy is an increase in the size of cells and consequently in the size of the affected organ. The cells of the heart and kidneys are particularly responsive to enlargement. The increase in cellular size is associated with an increased accumulation of protein in the cellular components (plasma membrane, endoplasmic reticulum, myofilaments, mitochondria) and not with an increase in cellular fluid. Hypertrophy can be physiologic or pathologic and is caused by specific hormone stimulation or by increased functional demand. For example, physiologic hypertrophy during pregnancy is hormone induced and involves both hypertrophy and hyperplasia. Hypertrophy as an adaptive response—muscular enlargement—occurs in the striated muscle cells of both the heart and skeletal muscles. These cells cannot adapt to increased metabolic demands by mitotic division and production of new cells to share the work. Thus they enlarge and the stimulus appears to be an increased workload. In the heart, pathologic hypertrophy is secondary to hypertension or problem valves. In skeletal muscle, physiologic hypertrophy occurs in response to heavy work. Muscular hypertrophy tends to diminish if the excessive workload diminishes.
In myocardial hypertrophy, initial enlargement is caused by dilation of the cardiac chambers, but this is short lived and is followed by increased synthesis of cardiac muscle proteins, allowing muscle fibers to do more work. The nucleus is also hypertrophic and exhibits increased synthesis of deoxyribonucleic acid (DNA).2 Although fully matured (e.g., terminally differentiated) muscle cells are unable to undergo further mitosis, they are capable of increased DNA synthesis. Why cardiac muscle cells are unable to progress through the cell cycle to mitosis is unclear3 (see Chapters 1 and 11). Eventually, however, advanced hypertrophy can lead to myocardial failure (Figure 2-3) (see Chapter 30).
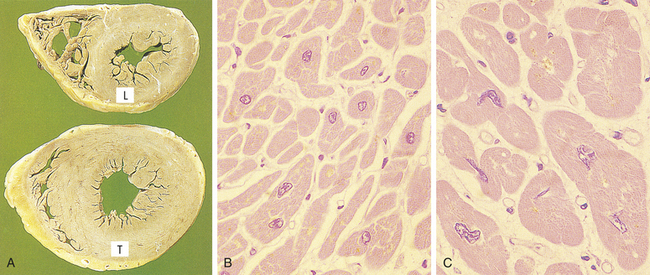
Figure 2-3 Hypertrophy of cardiac muscle in response to valve disease. A, Transverse slices of a normal heart and a heart with hypertrophy of the left ventricle. (L, Normal thickness of left ventricular wall; T, thickened wall from heart in which severe narrowing of aortic valve caused resistance to systolic ventricular emptying.) B, Histology of cardiac muscle from a normal heart. C, Histology of cardiac muscle from a hypertrophied heart. (From Stevens A, Lowe J: Pathology, London, 1995, Mosby.)
A number of genes are activated during hypertrophy, including the genes for atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) also called B-type natriuretic peptide. The ANP gene is usually expressed only during early development; however, with cardiac hypertrophy, it is reinduced and the ANP hormone causes salt secretion by the kidney, decreasing blood volume and pressure and reducing hemodynamic load. BNP is secreted by the heart ventricles and enhances sodium and water excretion, vasodilation, and inhibition of renin and aldosterone. Other genes activated include regulatory factors (e.g., c-fos, c-jun), growth factors, vasoactive agents, certain components involved in receptor-mediated signaling pathways, and kinases. The triggers for hypertrophy include two types of signals: mechanical signals, such as stretch, and trophic signals, such as growth factors and vasoactive agents.
After removal of one kidney, the other kidney adapts to an increased demand for work with an increase in both the size and the number of cells. The major contribution to renal enlargement is hypertrophy.
Hyperplasia
Hyperplasia is an increase in the number of cells resulting from an increased rate of cellular division. Hyperplasia as a response to injury occurs when the injury has been severe and prolonged enough to have caused cell death.2 Loss of epithelial cells and cells of the liver and kidney triggers DNA synthesis and mitotic division. Increased cell growth is a multistep process involving the production of growth factors, which stimulate the remaining cells to synthesize new cell components and, ultimately, to divide. Hyperplasia and hypertrophy often occur together, although the specific mechanism is unknown. Hyperplasia and hypertrophy both take place if the cells are capable of synthesizing DNA; however, in nondividing cells (e.g., myocardial fibers) only hypertrophy occurs.
Two types of normal, or physiologic, hyperplasia are compensatory hyperplasia and hormonal hyperplasia. Compensatory hyperplasia is an adaptive mechanism that enables certain organs to regenerate. For example, removal of part of the liver leads to hyperplasia of the remaining liver cells (hepatocytes) to compensate for the loss. Even with removal of 70% of the liver, regeneration is complete in about 2 weeks. The remarkable regenerating capacity of the liver was even noted by the ancient Greeks. According to one story, Prometheus was chained to a mountain and his liver was eaten daily by a vulture, only to regenerate every night. A protein, hepatocyte growth factor (HGF), is thought to be a mediator in vitro of liver regeneration.4 In addition, other in vitro growth factors and cytokines (cell-signaling proteins) that increase hepatic cell regeneration include transforming growth factor-α (TGF-α), epidermal growth factor (EGF), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α).
Not all types of mature cells have the same capacity for compensatory hyperplastic growth. Some cells, such as nerve, skeletal muscle, and myocardial cells and the lens cells of the eye, do not regenerate. Skeletal muscle cells, however, can be made by the fusion of myoblasts.5 Much research also is being done with the peripheral nervous system (PNS). PNS nerve regeneration enables severed limbs to be reattached and continue growing. Significant compensatory hyperplasia occurs in epidermal and intestinal epithelia, hepatocytes, bone marrow cells, and fibroblasts, and some hyperplasia is noted in bone, cartilage, and smooth muscle cells. An example of compensatory hyperplasia is a callus, or thickening, of the skin as a result of hyperplasia of epidermal cells in response to a mechanical stimulus. Another example is the response to wound healing as part of the inflammation process (see Chapter 6).
Hormonal hyperplasia occurs chiefly in estrogen-dependent organs, such as the uterus and breast. After ovulation, for example, estrogen stimulates the endometrium to grow and thicken for reception of the fertilized ovum. If pregnancy occurs, hormonal hyperplasia, as well as hypertrophy, enables the uterus to enlarge. (Hormone function is described in Chapters 20 and 21.)
Pathologic hyperplasia is the abnormal proliferation of normal cells and can occur as a response to excessive hormonal stimulation or the effects of growth factors on target cells (Figure 2-4). Hyperplastic cells are identified by pronounced nuclear enlargement, clumping of chromatin, and one or more enlarged nucleoli. The most common example is pathologic hyperplasia of the endometrium (which is caused by an imbalance between estrogen and progesterone secretion, with oversecretion of estrogen) (see Chapter 23). Pathologic endometrial hyperplasia, which causes excessive menstrual bleeding, is under the influence of regular growth-inhibition controls. If these controls fail, hyperplastic endometrial cells can undergo malignant transformation. (Malignant cell transformation is discussed in Chapter 11.)
Dysplasia: Not a True Adaptive Change
Dysplasia refers to abnormal changes in the size, shape, and organization of mature cells. Dysplasia is not considered a true adaptive process but is related to hyperplasia and is often called atypical hyperplasia. Dysplastic changes frequently are encountered in epithelial tissue of the cervix and respiratory tract, where they are strongly associated with common neoplastic growths and often are found adjacent to cancerous cells. Importantly, the term dysplasia does not indicate cancer and may not progress to cancer.
Dysplasia is often classified as mild, moderate, or severe; however, this subjective scheme has prompted recommendations to use either “low grade” or “high grade.” Grading of dysplasia, for example, of the female reproductive tract (i.e., Papanicolaou [Pap] test) is discussed in Chapter 23 (Figure 2-5). Data indicate that atypical hyperplasia is a strong predictor of breast cancer development.6,7 If the inciting stimulus is removed, dysplastic changes often are reversible.
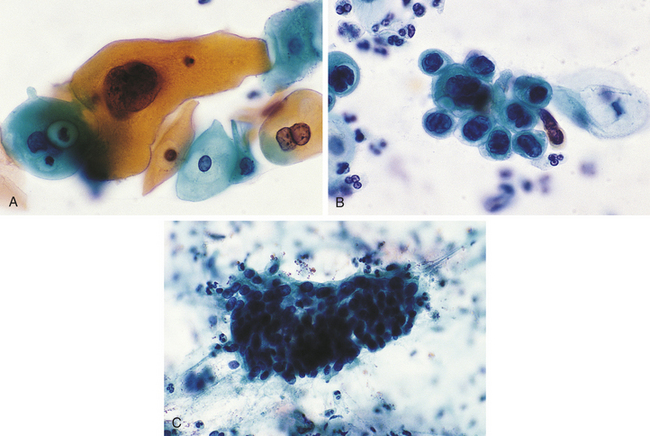
Figure 2-5 Dysplasia of uterine cervix. A, Mild dysplasia. B, Severe dysplasia. C, Carcinoma in situ (see Chapter 11). (From Damjanov I, Linder J: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Metaplasia
Metaplasia is the reversible replacement of one mature cell by another, sometimes less differentiated, cell type. The best example of metaplasia is replacement of normal columnar ciliated epithelial cells of the bronchial (airway) lining by stratified squamous epithelial cells (Figure 2-6). The newly formed squamous epithelial cells do not secrete mucus or have cilia, causing loss of a vital protective mechanism.
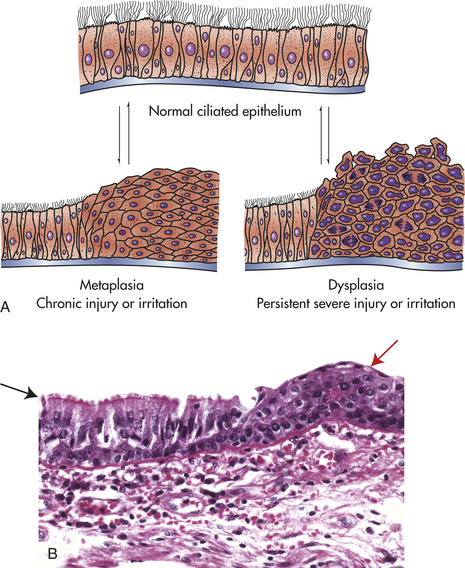
Figure 2-6 Reversible changes in cells lining the bronchi. A, Normal ciliated epithelium, metaplasia and dysplasia. B, Histological slide with upper left (black arrow) normal columnar epithelium and basement membrane, and upper right (red arrow) squamous metaplasia. (B from Kumar V, Abbas A, Fausto N: Robbins and Cotran pathologic basis of disease, ed 8, Philadelphia, 2007, Saunders.)
Metaplasia is thought to develop from a reprogramming of stem cells existing in most epithelia or of undifferentiated mesenchymal (tissue from embryonic mesoderm) cells present in connective tissue. These precursor cells mature along a new pathway because of signals generated by cytokines and growth factors in the cell’s environment.
Bronchial metaplasia can be reversed if the inducing stimulus, usually cigarette smoking, is removed. With prolonged exposure to the inducing stimulus, however, cancerous transformation can occur.
CELLULAR INJURY
Most diseases begin with cell injury, and all forms of loss of function derive from cell injury and cell death. Cellular injury occurs if the cell is unable to maintain homeostasis—a normal or adaptive steady state—in the face of injurious stimuli. Injured cells may recover (reversible injury) or die (irreversible injury). Injurious stimuli include chemical agents, lack of sufficient oxygen (hypoxia), free radicals, infectious agents, physical and mechanical factors, immunologic reactions, genetic factors, and nutritional imbalances. Types of cellular injury and their responses are summarized in Table 2-1 and Figure 2-7.
Table 2-1
Progressive Types of Cell Injury and Responses
| Type | Responses |
| Adaptation | Atrophy, hypertrophy, hyperplasia, metaplasia |
| Active cell injury | Immediate response of “entire” cell |
| Reversible | Loss of adenosine triphosphate (ATP), cellular swelling, detachment of ribosomes, autophagy of lysosomes |
| Irreversible | “Point of no return” structurally when severe vacuolization occurs of the mitochondria and Ca++ moves into the cell including the mitochondria membrane damage |
| Necrosis | Common type of cell death with severe cell swelling and breakdown of organelles |
| Apoptosis, a type of programmed cell death | Cellular self-destruction for elimination of unwanted cell populations |
| Chronic cell injury (subcellular alterations) | Persistent stimuli response may involve only specific organelles or cytoskeleton (e.g., phagocytosis of bacteria) |
| Accumulations or infiltrations | Water, pigments, lipids, glycogen, proteins |
| Pathologic calcification | Dystrophic and metastatic calcification |
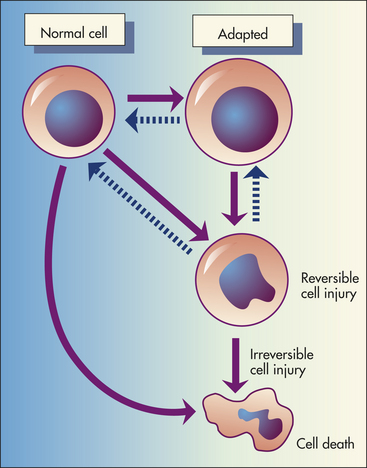
Figure 2-7 Cellular injury and responses. Depicted here is the relationship among normal, adapted (hypertrophy), and reversibly injured cells and cell death of myocardial cells.
Cell injury and cell death often result from exposure to toxic chemicals, infections, and hypoxia. The mechanisms causing chemical and hypoxic injury are perhaps the best understood. (Infections are discussed in Chapter 9.) Both of these mechanisms can lead to disruption of selective permeability (i.e., transport mechanisms) of the plasma membrane; reduction or cessation of cellular metabolism; lack of protein synthesis; damage to lysosomal membranes, with leakage of destructive enzymes into the cytoplasm; enzymatic destruction of cellular organelles; cellular death (exhibited by nuclear changes); and phagocytosis of the dead cell by cellular components of the acute inflammatory response (see Chapter 6). The extent of cellular injury depends on the type, state (including level of cell differentiation and increased susceptibility to fully differentiated cells), and adaptive processes of the cell, as well as the type, severity, and duration of the injurious stimulus. Two individuals exposed to an identical stimulus may incur varying degrees of cellular injury. Modifying factors, such as nutritional status, can profoundly influence the extent of injury. The precise “point of no return” that leads to cellular death is a biochemical puzzle, and the exact mechanisms responsible for the transition from reversible to irreversible cellular damage are being debated.
General Mechanisms of Cell Injury
Cells are complex units, and therefore the mechanisms responsible for cell injury leading to necrotic cell death are numerous and interrelated and depend on a delicate balance between intracellular and extracellular events. There are, however, four common biochemical themes important to cell injury and cell death regardless of the injuring agent (Table 2-2).

The three common forms of cell injury are (1) hypoxic injury, (2) reactive oxygen species and free radical–induced injury, and (3) chemical injury.
Hypoxic Injury
Hypoxia, or lack of sufficient oxygen, is the single most common cause of cellular injury (Figure 2-8). Hypoxia can result from a decreased amount of oxygen in the air, loss of hemoglobin or hemoglobin function, decreased production of red blood cells, diseases of the respiratory and cardiovascular systems, and poisoning of the oxidative enzymes (cytochromes) within the cells. The most common cause of hypoxia is ischemia (reduced blood supply).
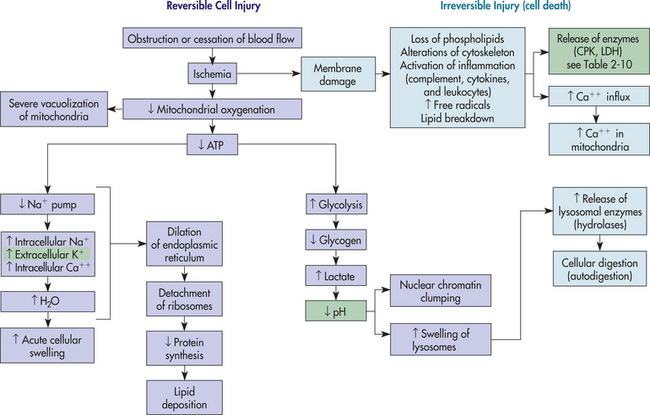
Figure 2-8 Hypoxic injury induced by ischemia. Purple boxes involve reversible cell injury, and light blue boxes involve irreversible cell death. Green boxes are clinical manifestations.
Ischemic injury is often caused by gradual narrowing of arteries (arteriosclerosis) and complete blockage by blood clots (thrombosis). Progressive hypoxia caused by gradual arterial obstruction is better tolerated than the sudden acute anoxia (total lack of oxygen) caused by a sudden obstruction, such as can occur with an embolus (a blood clot or other plug in the circulation). An acute obstruction in a coronary artery can cause myocardial cell death (infarction) within minutes if the blood supply is not restored, whereas the gradual onset of ischemia usually results in myocardial adaptation. Myocardial infarction and stroke, which are common causes of death in the United States, generally result from atherosclerosis (a type of arteriosclerosis) and consequent ischemic injury. (Vascular obstruction is discussed in Chapter 30.)
Cellular responses to hypoxic injury have been extensively studied in heart muscle. Within 1 minute after blood supply to the myocardium is interrupted, the heart becomes pale and has difficulty contracting normally. Within 3 to 5 minutes the ischemic portion of the myocardium ceases to contract. The abrupt lack of contraction is caused by a rapid decrease in mitochondrial phosphorylation, which results in insufficient adenosine triphosphate (ATP) production. Lack of ATP leads to an increase in anaerobic metabolism, which generates ATP from glycogen when there is insufficient oxygen. When glycogen stores are depleted, even anaerobic metabolism ceases.
A reduction in ATP levels causes the plasma membrane’s sodium-potassium (Na+, K+) pump and sodium-calcium exchange to fail, which leads to an intracellular accumulation of sodium and calcium and diffusion of potassium out of the cell. (The Na+, K+ pump is discussed in Chapter 1.) Sodium and water then can enter the cell freely, and cellular swelling results. Because all cells are bathed in a fluid rich in calcium ions, cell membrane damage allows rapid movement of calcium intracellularly. The movement of water and ions into the cell causes early dilation of the endoplasmic reticulum. Dilation causes the ribosomes to detach from the rough endoplasmic reticulum, resulting in reduced protein synthesis. With continued hypoxia, the entire cell becomes markedly swollen, with increased concentrations of sodium, water, and chloride and decreased concentrations of potassium. These disruptions are reversible if oxygen is restored. If oxygen is not restored, however, there is vacuolation (formation of vacuoles or cytoplasmic small cavity) within the cytoplasm, swelling of lysosomes, and marked swelling of the mitochondria resulting from mitochondrial membrane damage. Continued hypoxic injury with accumulation of calcium subsequently activates multiple enzyme systems, including proteases, nitric oxide synthase, phospholipases, and endonuclease, resulting in cytoskeleton disruption, membrane damage, activation of inflammation, DNA degradation, and eventual cell death (see Figure 2-27). Structurally, with plasma membrane damage, extracellular calcium readily moves into the cell and intracellular calcium stores are released. Intracellular calcium results in the activation of enzymes that can further damage membranes, proteins, ATP, and nucleic acids.8 The increased permeability of the membrane causes continued loss of proteins, essential coenzymes, and ribonucleic acids. In addition, the substrates necessary to reconstitute ATP are lost. Irreversible damage is characterized by two events: (1) lack of ATP generation because of mitochondrial dysfunction, and (2) major disturbances and damage in membrane function. Acid hydrolases from leaking lysosomes are activated in the reduced pH of the injured cell and they digest cytoplasmic and nuclear components. Leakage of intracellular enzymes into the peripheral circulation provides a diagnostic tool for detecting tissue-specific cellular injury and death using blood samples; for example, the contractile protein troponin from cardiac muscle is found after myocardial injury and liver transaminases are found after hepatic injury.
Restoration of oxygen, however, can cause additional injury called reperfusion (reoxygenation) injury. Reperfusion is a serious complication and an important mechanism of injury in instances of tissue transplantation and in myocardial, hepatic, intestinal, cerebral, renal, and other ischemic syndromes, including stroke.9,10 Xanthine dehydrogenase, an enzyme that normally uses oxidized nicotinamide adenine dinucleotide (NAD+) as an electron acceptor, is converted during reperfusion with oxygen to xanthine oxidase. During the ischemic period, excessive ATP consumption leads to the accumulation of the purine catabolites hypoxanthine and xanthine, which upon subsequent reperfusion and influx of oxygen are metabolized by xanthine oxidase to make massive amounts of superoxide and hydrogen peroxide. These radicals can all cause membrane damage and mitochondrial calcium overload.11 Cardiac ischemia and reperfusion injury cause excessive reactive oxygen species (ROS) and calcium overload of the mitochondria. These changes lead to the opening of pores on the mitochondrial membrane with massive escape of ATP leading to cell death activation (apoptosis). Interestingly, release of low levels of nitric oxide can acutely protect myocardial mitochondria against reperfusion injury12 (see What’s New? ROS and Proliferation, Apoptosis, and Necrosis). Neutrophils are especially affected with reperfusion injury, and neutrophil adhesion to the endothelium enhances the process. Antioxidant treatment reverses both neutrophil adhesion (leukocyte adhesion) and neutrophil (leukocyte) mediated heart injury in the post-ischemic period.9 Other potential and current treatments include blockage of inflammatory mediators and inhibition of apoptotic pathways.
Free Radicals and Reactive Oxygen Species
An important mechanism of membrane damage is injury induced by free radicals, especially by excess ROS called oxidative stress. Oxidative stress occurs when excess ROS overwhelms endogenous antioxidant systems. A free radical is an electrically uncharged atom or group of atoms having an unpaired electron. Having one unpaired electron makes the molecule unstable; thus to stabilize, it gives up an electron to another molecule or steals one. Therefore, it is capable of injurious chemical bond formation with proteins, lipids, carbohydrates—key molecules in membranes and nucleic acids. Free radicals are difficult to control and initiate chain reactions. Emerging data indicate that ROS play major roles in the initiation and progression of cardiovascular alterations
WHAT’S NEW?
ROS and Proliferation, Apoptosis, and Necrosis
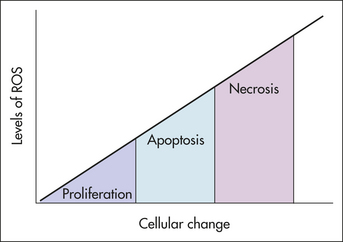
Cellular effect of reactive oxygen species (ROS) may depend on concentration levels. At low concentrations, ROS appear to exert a beneficial growth-stimulatory effect on a wide variety of cells and microorganisms. For example, bacteria such as Escherichia coli and Salmonella typhimurium need 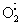 for growth. Certain human cell lines also have shown this dependency in vitro. Yet when ROS levels increase, other signaling pathways may be activated that lead to apoptosis. When ROS levels rise even higher, a cell may die a sudden necrotic death. The apoptosis and necrosis modes are thought to be caused by oxidative stress. Thus the signaling functions of ROS are now appreciated.
for growth. Certain human cell lines also have shown this dependency in vitro. Yet when ROS levels increase, other signaling pathways may be activated that lead to apoptosis. When ROS levels rise even higher, a cell may die a sudden necrotic death. The apoptosis and necrosis modes are thought to be caused by oxidative stress. Thus the signaling functions of ROS are now appreciated.
Data from Buetler TM, Krauskopf A, Ruegg UT: News Physiol Sci 19:120-123, 2004; Valko M et al: Int J Biochem Cell Biol 39(1):44-84, 2007.
associated with hyperlipidemia, diabetes mellitus, hypertension, ischemic heart disease, and chronic heart failure. ROS produced by migrating inflammatory cells (e.g., neutrophils), as well as vascular cells (endothelial cells, vascular smooth muscle cells, and adventitial fibroblasts) have distinct effects on each cell type.13 These cell effects are shown in Figure 2-9.
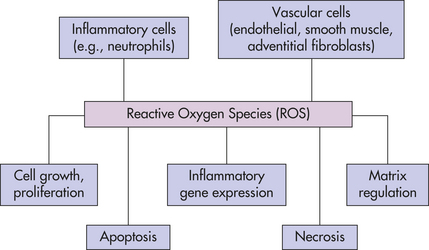
Figure 2-9 ROS can cause distinct functional effects depending on cell type. All cells are capable of making reactive oxygen species (ROS). Emphasis has been on inflammatory and vascular cells because of their widespread disease-causing effect. Some examples include angiotensin II, which can induce vascular smooth muscle cells (VSMC) to hypertrophy; NAD(P)H oxidase-derived ROS has been implicated in the growth response; H2O2 has been shown to induce proliferation and migration of endothelial cells; ROS act as mediators of vascular endothelial growth factor, thus modulating angiogenesis; endothelial injury or exposure to and H2O2 induces apoptosis of endothelial cells. Activity of the extracellular matrix by matrix metalloproteinases (MMPs) can be modulated by ROS. Cytokines play a significant role in the progression of vascular lesions. An important mechanism by which cytokine gene expression is increased is the activation of nuclear factor-κβ (NF-κβ). NF-κβ is a ROS-sensitive transcription factor and has a role in the expression of proinflammatory genes. (Data from Buetler TM, Krauskopf A, Ruegg UT: News Physiol Sci 19:120-123, 2004; Dröge W: Physiol Rev 82:47-95, 2002; Valko M et al: Int J Biochem Cell Biol, 39(1):44-84, 2007.) (Also see What’s New? ROS and Proliferation, Apoptosis, and Necrosis.)
Free radicals may be initiated within cells by (1) the absorption of extreme energy sources (e.g., ultraviolet light, x-rays); (2) endogenous, usually oxidative, reactions that occur during normal metabolic processes (Figure 2-10); or (3) enzymatic metabolism of exogenous chemicals or drugs (e.g., chloromethyl [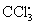 ], a product of carbon tetrachloride [CCl4]). Table 2-3 describes the most significant free radicals.
], a product of carbon tetrachloride [CCl4]). Table 2-3 describes the most significant free radicals.
Table 2-3
Biologically Relevant Free Radicals

Data from Kumar V, Abbas A, Fausto N: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders; Buetler TM, Krauskopf A, Ruegg UT: News Physiol Sci 19:120-123, 2004.
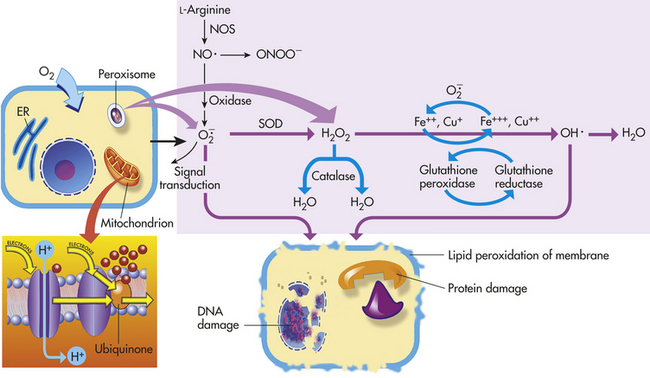
Figure 2-10 Generation of reactive oxygen species (ROS) and antioxidant mechanisms in biologic systems. Mitochondria have four sites of entry for electrons coming into the electron transport system: one for reduced nicotinamide adenine dinucleotide (NADH) and three for reduced form of flavin adenine dinucleotide (FADH2). These pathways meet at the small, lipophilic molecule, ubiquinone (coenzyme Q), at the beginning of the common electron transport pathway. Ubiquinone transfers electrons in the inner membrane, ultimately enabling their interaction with O2 and H2 to yield H2O. In so doing, the transport allows free energy change and the synthesis of one mole of adenosine triphosphate (ATP). With the transport of electrons, free radicals are generated within the mitochondria. ROS (H2O2, OH·, and and nitric oxide [NO]) act as physiologic modulators of some mitochondrial functions but also may cause cell damage. O2 is converted to superoxide () by oxidative enzymes in the mitochondria, endoplasmic reticulum (ER), plasma membrane, peroxisomes, and cytosol. O2 is converted to H2O2 by superoxide dismutase (SOD) and further to OH· by the Cu++/Fe++ Fenton reaction. Superoxide catalyzes the reduction of Fe++ to Fe+++, thus increasing OH· formation by the Fenton reaction. H2O2 is also derived from oxidases in peroxisomes. The NO· (radical) is produced by the oxidation of one of the terminal guanido-nitrogen atoms of L-arginine. Depending on the microenvironment, NO can be converted to other reactive nitrogen species including the highly reactive peroxynitrate (ONOO−). Both OH· and ONOO− are very reactive and can modify cellular macromolecules and cause toxicity. The less reactive molecules and H2O2 can serve as cellular signaling molecules. The major antioxidant enzymes include SOD, catalase, and glutathione peroxidase. (Data from Dröge W: Physiol Rev 82:47-95, 2002; Buetler TM, Krauskopf A, Ruegg UT: News Physiol Sci 19:120-123, 2004.)
Although wide-ranging effects can occur from these reactive species, three are particularly important in regard to cell injury: (1) lipid peroxidation; (2) alterations of proteins causing fragmentation of polypeptide chains; and (3) alterations of DNA, including breakage of single strands. Lipid peroxidation is the destruction of unsaturated fatty acids. Fatty acids of lipids in membranes possess double bonds between some of the carbon atoms. Such bonds are vulnerable to attack by oxygen-derived free radicals, especially OH•. The lipid-radical interactions themselves yield peroxides. The peroxides set off a chain reaction resulting in membrane, organelle, and cellular destruction. Because of our understanding of free radicals, a growing number of diseases and disorders have been linked either directly or indirectly to these reactive species (Table 2-4).
Table 2-4
Diseases and Disorders Linked to Oxygen-Derived Free Radicals
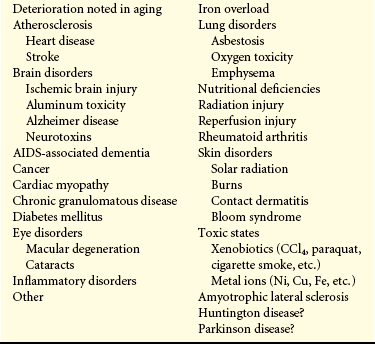
AIDS, Acquired immunodeficiency syndrome.
Data from Knight JA: Ann Clin Lab Sci 25(2):111, 1995; Bergendi L et al: Life Sci 65(18-19): 1865, 1999; Maccarrone M, Ullrich V: Cell Death Differ 11:949-952, 2004.
It is fortunate that the body can sometimes rid itself of free radicals. Superoxide may spontaneously decay into oxygen and hydrogen peroxide. Table 2-5 summarizes other methods that contribute to inactivation or termination of free radicals. The toxicity of certain drugs and chemicals can be attributed to either conversion of these chemicals to free radicals or the formation of oxygen-derived metabolites.9 This process is discussed in Chemical Injury, which follows.
Table 2-5
Methods Contributing to Inactivation or Termination of Free Radicals
| Method | Process |
| Antioxidants | Endogenous or exogenous; either blocks synthesis or inactivates (e.g., scavenges) free radicals; includes vitamin E, vitamin C, cysteine, glutathione, albumin, ceruloplasmin, transferrin |
| Enzymes | Superoxide dismutase,∗ which converts superoxide to H2O2; catalase∗ (in peroxisomes) decomposes H2O2; glutathione peroxidase∗ decomposes OH• and H2O2 |
∗These enzymes are important in modulating the cellular destructive effects of free radicals, also released in inflammation.
Chemical Injury
Mechanisms: Chemical injury begins with a biochemical interaction between a toxic substance and the cell’s plasma membrane, which is ultimately damaged, leading to increased permeability. Not all the mechanisms causing chemically induced membrane destruction are known; however, the two general mechanisms include (1) direct toxicity by combining with a molecular component of the cell membrane or organelles and (2) reactive free radicals and lipid peroxidation.
Because it has been investigated extensively, carbon tetrachloride (CCl4) injury is a useful example of chemical injury. Carbon tetrachloride, an agent formerly used in dry cleaning, harms cells because an enzyme system (P-450) in the smooth endoplasmic reticulum of liver cells converts it into chloromethyl (), a highly toxic free radical.
In CCl4 injury, newly formed rapidly destroys the endoplasmic reticulum of the liver cell by way of lipid peroxidation breaking down the reticulum’s lipid component. The lipid molecules accumulate within the cytoplasm, starting within cisternae of the endoplasmic reticulum (Figure 2-11). Fatty liver develops because CCl4 poisoning blocks the synthesis of lipid-acceptor proteins (apoproteins) that normally bind with triglycerides to form lipoproteins, which are transported out of the cell. Blockage of triglyceride (lipoprotein) secretion begins 10 to 15 minutes after CCl4 exposure. Fat droplets that accumulate in cisternae of the endoplasmic reticulum combine to form larger droplets and fill vacuoles, which in turn fill the entire cytoplasm. Approximately 10 to 12 hours later the liver appears grossly enlarged and pale because of the accumulation of fat. (Accumulation of fat is discussed further on p. 76.)
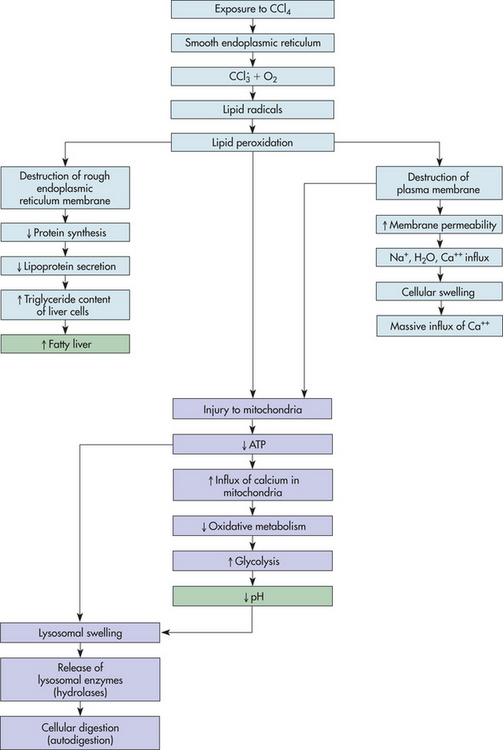
Figure 2-11 Chemical injury of liver cells induced by carbon tetrachloride (CCl4) poisoning. Light blue boxes are mechanisms unique to chemical injury; purple boxes involve hypoxic injury. Green boxes are clinical manifestations.
In the meantime, cellular swelling progresses because of alterations in the selective permeability of the plasma membrane. Cellular swelling becomes severe when the plasma membrane loses its ability to prevent the passive inward diffusion of sodium ions, water, and calcium. The most serious consequence of plasma membrane damage is, as in hypoxic injury, to the mitochondria. An influx of calcium ions from the extracellular compartment activates multiple enzyme systems resulting in cytoskeleton disruption, membrane damage, activation of inflammation, and eventually DNA degradation. Calcium ion accumulation in the mitochondria cause the mitochondria to swell, an occurrence that is associated with irreversible cellular injury. The injured mitochondria can no longer generate ATP, but they do continue to accumulate calcium ions. The influx of calcium into the mitochondria interferes with oxidative metabolism (by uncoupling oxidative phosphorylation).
Decreasing cellular pH (caused by the loss of oxidative phosphorylation and ATP-stimulating glycolysis), together with fluid and electrolyte imbalances (increased sodium, calcium, and water and decreased potassium), leads to lysosomal membrane injury, causing a leakage of lysosomal enzymes into the cytoplasm. Enzymatic digestion of cellular organelles, including the nucleus and nucleolus, ensues, halting synthesis of DNA and ribonucleic acid (RNA). The leakage of lysosomal enzymes apparently occurs late in chemical injury, well after irreversible lipid accumulation, mitochondrial swelling, and ATP loss.
Chemical Agents: Many chemical agents cause cellular injury. Minute amounts of some, such as arsenic and cyanide, can rapidly destroy enough cells to cause death of the individual. Long-term exposure to air pollutants, insecticides, and herbicides can cause cellular injury. Carbon monoxide, carbon tetrachloride, and social drugs, such as alcohol, can significantly alter cellular function and injure cellular structures. Over-the-counter and prescribed drugs also may cause cellular injury, sometimes leading to death. Acetaminophen (outside the United States is known as paracetamol), commonly used as an analgesic, is one of the most common causes of poisoning worldwide.14 In 2005, acetaminophen poisoning was responsible for more than 70,000 visits to healthcare clinics and about 300 deaths.15,16 Drug-induced acute liver failure accounts for about 20% of liver failure in children and a higher percentage in adults17 (see What’s New?, Chapter 39). Accidental or suicidal poisonings by chemical agents cause numerous deaths. The injurious effects of some of these agents—lead, carbon monoxide, ethyl alcohol, and mercury—exemplify common cellular injuries.
Lead: Lead is a heavy metal ubiquitous in the environment. Despite efforts to reduce exposure through government regulation, phasing out production of leaded gasoline, and banning use of lead paint, excessive lead exposure still persists in the environment for many people and lead toxicity is still a primary hazard to children.18 Particularly worrisome is lead exposure to the fetus during pregnancy because the developing nervous system is especially vulnerable. Developing fetuses and young children absorb lead more easily than adults18; however, the exact transport mechanisms have not yet been elucidated. Exposure to lead during neurologic development has significant effects on neurobehavioral and intellectual performance, resulting in learning disorders, hyperactivity, and attention problems.18
Lead-based paint, which has a sweet taste, is often ingested by children when they have access to surfaces painted with it. Other sources of lead in daily life include the dust and soil found in inner-city urban and possibly rural areas, debris from household renovations, baby formula mixed with lead-contaminated tap water, newsprint, water that flows through lead pipes, hair dyes, food stored in soldered tin cans or eaten off of pottery made with lead-based glazes, and contamination from leaded gasoline.19 If nutrition is compromised, especially if dietary intake of iron, calcium, zinc, and vitamin D is insufficient, lead’s toxic effects are enhanced.
The organ systems primarily affected by lead include the nervous system, the hematopoietic system (tissues that produce blood cells), and the kidneys. Lead affects many different biologic activities at the cellular and molecular levels, many of which may be related to its ability to interfere with the functions of calcium.18 Lead is able to increase intracellular calcium concentrations and become a calcium substitute, and some calcium-binding proteins are capable of binding to lead.18 Very tiny concentrations (subnanomolar) of lead activate protein kinase C (PKC) in a process that is partially dependent on calcium.13,20 The PKC-mediated lead-induced rise in intracellular free calcium may be the cause of cellular disruption. Lead appears to have its greatest effects during the later stages of brain development, possibly by altering development of synaptic connections (i.e., trimming/pruning) and neuronal death (apoptosis).18 Alterations in calcium may play a crucial role in the interference with neurotransmitters, which may cause hyperactive behavior and proliferation of capillaries of the white matter and intercerebral arteries.2,18 Lead inhibits several enzymes involved in hemoglobin synthesis. A significant manifestation of lead toxicity is anemia caused by lysis of red blood cells (hemolysis). Other manifestations of brain involvement include convulsions and delirium and, with peripheral nerve involvement, wrist, finger, and sometimes foot paralysis. Renal lesions can cause tubular dysfunction resulting in glycosuria (glucose in the urine), aminoaciduria (amino acids in the urine), and hyperphosphaturia (excess phosphate in the urine). Gastrointestinal symptoms are less severe and include nausea, loss of appetite, weight loss, and abdominal cramping.
Carbon Monoxide: Gaseous substances can be classified according to their ability to asphyxiate (interrupt respiration) or irritate. Toxic asphyxiants, such as carbon monoxide, hydrogen cyanide, and hydrogen sulfide, directly interfere with cellular respiration. Carbon monoxide is widely available.
Carbon monoxide (CO), a gas, is odorless, colorless, and undetectable unless it is mixed with a visible or odorous pollutant. It is produced by the incomplete combustion of such fuels as gasoline. In dense urban environments, CO produced by incomplete combustion from motor vehicles increases air pollution. Although CO is a chemical agent, the ultimate injury it produces is a hypoxic injury, namely, oxygen deprivation. Normally, oxygen molecules are carried to tissues bound to hemoglobin in red blood cells (see Chapter 29). Because CO’s affinity for hemoglobin is 300 times greater than that of oxygen, it quickly binds with the hemoglobin, preventing oxygen molecules from doing so. Minute amounts of CO can produce significant percentages of carboxyhemoglobin (carbon monoxide bound with hemoglobin).
Symptoms related to CO poisoning include headache, giddiness, tinnitus (ringing in the ears), nausea, weakness, and vomiting. At risk for CO exposure are those who: (1) breathe air polluted by gasoline engines or defective furnaces; (2) work in occupations such as coal mining, fire fighting, welding,21 or engine repair; and (3) smoke cigarettes, cigars, or pipes. The fetus is especially at risk from the effects of carbon monoxide because fetal carboxyhemoglobin levels are likely to be 10% to 15% greater than maternal levels.22
Ethanol: Alcohol (ethanol) is the number one mood-altering drug used in the United States. Because alcohol is not only a psychoactive drug but also a food, it is considered part of the basic food supply in many societies.
A large intake of alcohol has enormous effects on nutritional status. Major nutritional deficiencies include magnesium, vitamin B6, thiamine, and phosphorus. Liver and nutritional disorders are the most serious consequences of alcohol abuse. New understandings of the mechanisms of ethanol-induced liver injury have emerged through the clarification of a pathway for ethanol oxidation, the microsomal P-450 oxidase pathway (see following).
The major effects of acute alcoholism involve the central nervous system (CNS). After ingestion, alcohol is absorbed, unaltered, into the stomach and small intestine. Fatty foods and milk slow absorption.23 Alcohol then is distributed to all tissues and fluids of the body in direct proportion to the blood concentration.
Most of the alcohol in the blood is metabolized in the liver through one major and two accessory pathways. The major pathway involves hepatic alcohol dehydrogenase (ADH), an enzyme of the cytosol that catalyzes the conversion of ethanol to acetaldehyde (Figure 2-12).
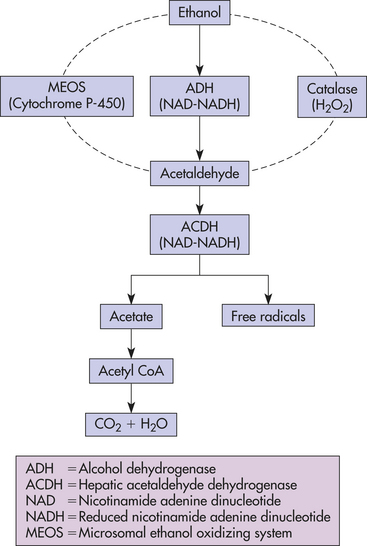
Figure 2-12 Major pathway of metabolism of alcohol in the liver through alcohol dehydrogenase (ADH).
The microsomal ethanol oxidizing system (MEOS) depends on cytochrome P-450, an enzyme necessary for cellular oxidation.24 Activation of the MEOS requires a high ethanol concentration and thus is thought to be important in the accelerated ethanol metabolism (i.e., tolerance) noted in people with chronic alcoholism.24
Individuals differ in their capability to metabolize alcohol. Genetic differences in metabolism of liver alcohol, including aldehyde dehydrogenases, have been identified.25 People with chronic alcoholism develop certain levels of tolerance because of enzyme induction, leading to an increased rate of metabolism (e.g., P-450).
Studies conducted since 1997 have contributed to our understanding of the association between alcohol consumption and cardiovascular disease. Consistent results validate the so-called J-shaped inverse association between alcohol and cardiovascular disease mortality and morbidity. That is, moderate drinkers exhibiting a decreased risk compared with both heavy drinkers and nondrinkers. Surprisingly, consistent epidemiologic studies show that daily light to moderate alcohol intake reduces the risk of coronary heart disease (CHD) as compared with those who do not drink alcoholic beverages at all. Alcohol possibly reduces the risk of CHD through increases in fibrinolysis26 and increases in plasma high density lipoprotein-cholesterol (HDL-C) levels.27 Alcohol also may increase insulin sensitivity.28 Limited data suggest that the level for optimal benefit may be slightly lower for women. Thus the American Heart Association recommends no more than two drinks per day for men and one drink per day for women.27
Acute alcoholism mainly affects the CNS but may induce reversible hepatic and gastric changes. The hepatic changes, initiated from acetaldehyde, include deposition in fat, enlargement of the liver, interruption of microtubular transport of proteins and their secretion, increase in intracellular water, depression of fatty acid oxidation in the mitochondria, increased membrane rigidity, and acute liver cell necrosis (see Chapter 39). In the CNS, alcohol is itself a depressant, initially affecting subcortical structures (probably the brainstem reticular formation).29 Consequently, motor and intellectual activities become disoriented. Acute alcoholism contributes significantly to motor vehicle fatalities. At higher blood levels, medullary centers become depressed, affecting respiration. Much investigation is under way to determine the extent of the relationship between alcohol and snoring and obstructive sleep apnea (cessation of breathing).30,31
Chronic alcoholism causes structural alterations in practically all organs and tissues in the body, especially the liver and stomach. Much progress has been made in understanding the pathogenesis of alcoholic liver disease, which should increase the likelihood of prevention and successful therapy.32 Cellular damage is increased by ROS and oxidative stress (see p. 54). New data on the cellular and molecular mechanisms of liver fibrosis are presented in What’s New? Cellular Mechanisms of Fibrosis and Reversal. In addition, the activation of methionine, an essential amino acid, to S-adenosyl-L-methionine (SAMe) is decreased in those with alcoholism.32 Replacement of SAMe in baboons decreased liver mitochondrial lesions, replenished the antioxidant glutathione, and reduced mortality from cirrhosis.33 Oxidative stress is associated with phospholipid depletion. In baboons, replacement of polyenylphosphatidylcholine (PPC) corrected the phospholipid depletion.33 Clinical trials with PPC involving individuals with alcoholic liver disease are ongoing. Chronic alcoholism is related to several disorders, including
an increased tendency for hypertension, a higher incidence of acute and chronic pancreatitis, and regressive changes in skeletal muscle (see Chapter 39). Ethanol is implicated in the onset of a variety of immune defects, including effects on the production of cytokines involved in inflammatory responses. The deleterious effects of prenatal alcohol exposure (e.g., fetal alcohol syndrome [FAS]) also have been noted. FAS can lead to growth retardation, cognitive impairment, facial anomalies, and ocular disturbances.34,35 In some cases, full-blown FAS may not be indicated but CNS defects may still be present and are classified as alcohol-related birth defects (ARBDs) and alcohol-related neurodevelopmental disorders (ARNDs).36
Autopsies of children with FAS have revealed widespread severe damage, including failure of certain brain regions to develop, malformations of brain tissue, and failure of certain cells to migrate to their necessary location during development.37 Imaging studies reveal that in addition to an overall reduction in brain size, the corpus callosum is reduced in size or missing, the cerebellum is significantly reduced, and the basal ganglia and caudate nucleus are significantly reduced.19,38
Animal studies have shown that ethanol at moderate concentrations inhibits epidermal growth factor–dependent replication of hepatocytes. This finding may account for the growth or development impairment associated with FAS and decreased liver regeneration in those with alcoholic liver disease.39,40 The wide variety of cellular or biochemical effects of ethanol on fetal tissue is itself a puzzle, reflecting a multifactorial problem. These effects are conceptually connected to membrane structure and function involving transport systems, membrane fluidity, Na+, K+ pump expression, and EGF receptor expression.40 Recent evidence points to oxidative stress as being potentially causative of these membrane-related events.41 Additionally, ethanol has been shown to increase apoptotic cell death.42
Whatever the cause, people with chronic alcoholism have a significantly shortened life span related mainly to damage to the liver, stomach, brain, and heart. Alcohol is a well-known cause of hepatic injury, terminating in cirrhosis (see Chapter 39) (Figure 2-13).
Mercury: Mercury has been used medically and commercially for centuries.43 In the past it was a common component in medications. Mercury is still present in some thermometers and blood pressure cuffs and in batteries, switches, and fluorescent light bulbs. Large amounts of mercury exist as part of the electrodes formed in the electrolytic production of chlorine and sodium hydroxide from saline. Today people are exposed to mercury from three major sources: fish consumption, dental amalgams, and vaccines. All of these uses give rise to possible accidental and occupational exposures.43
Dental Amalgams: Dental amalgams have been used for more than 150 years. They are believed to be more durable and easier to use than other types of fillings, as well as being relatively inexpensive. Amalgams consist of about 50% mercury amalgamated or combined with other metals, such as silver and copper. The controversies and heated debates concerning amalgams peaked in the 1970s with the discovery that amalgams can release mercury vapors into the mouth in concentrations that are higher than those deemed safe by occupational health guidelines.
Since then it was realized that the actual inhaled dose was small because of the small volume of the oral cavity.43 Yet, brain, blood, and urinary concentrations correlate with the number of amalgam surfaces present in a person. Removal of amalgam fillings also can cause temporary elevations in blood concentration because the removal transiently increases the amount of mercury vapor inhaled.
Current health risk concerns arise from claims that long-term exposure to low concentrations of mercury vapor either causes or worsens degenerative diseases, such as amyotrophic lateral sclerosis, Alzheimer disease, multiple sclerosis, and Parkinson disease. Concern about the effect of mercury vapor in relation to Alzheimer disease was intensified for a time after a report that the brains of individuals with Alzheimer disease had elevated mercury concentrations. Several epidemiologic investigations, however, failed to provide evidence of a role of dental amalgams in these degenerative diseases; these include a long-term Swedish study,44 an ongoing Swedish study,45 and a study of 129 nuns 75 to 102 years of age.46 Recently, a randomized prospective trial of 507 children failed to show exposure to mercury from amalgams is linked to neurobehavioral or neurologic effects.47 A difficult problem is that mercury can inhibit various biochemical processes in vitro without having the same effects in vivo. Thus, at present it is unknown whether removal of amalgams reduces risk of certain diseases, especially because removal itself affects blood concentrations of mercury vapor, which will rise before they eventually decline, thereby adding to the controversy.
Fish Consumption: The major source of exposure to methyl mercury is the consumption of fish and sea mammals. Clinical reports of mercury poisoning from fish consumption are those from Japan in the 1950s and 1960s. Environmental Protection Agency (EPA) guidelines are derived from reports of neuropsychologic changes noted in the Faeroe Islands study, in which subjects had been inadvertently exposed to methyl mercury mainly from whale consumption.48 A similar study in the United States shows methyl mercury levels to be slightly higher than the EPA guideline for safe consumption.43 The health risk posed by exposure to mercury from fish consumption is being debated. The U.S. Food and Drug Administration (FDA) has, however, recommended that pregnant women, nursing mothers, and young children avoid eating fish with a high mercury content (>1 part per million [ppm]), such as shark, swordfish, tile fish, king mackerel, and whale meat.43 Other advocates, however, have published more extensive lists including fish with the lowest levels, e.g., blue crab, croaker, fish sticks, flounder, haddock, trout, salmon (wild), and shrimp.49
Vaccines: Thimerosal has been used as a preservative in many vaccines since the 1930s.43 It contains the ethyl mercury radical (CH3CH2Hg+). Earlier toxicology studies and a 2007 study50 found either no adverse effects or no support for a causal relationship between thimerosal and deficits in neuropyschologic functioning. An earlier reevaluation of thimerosal, however, performed by applying the revised EPA guideline for methyl mercury to ethyl mercury found the usual U.S. program of recommended vaccines caused patients to receive more ethyl mercury than the EPA guidelines (i.e., >1 mcg of mercury per kilogram per day) deemed safe.51,52 Steps were rapidly taken to remove thimerosal from vaccines by switching to single-dose vials that did not require a preservative. Since 2003 no vaccines contain thimerosal. Recent findings indicate that the half-life of ethyl mercury compared with methyl mercury is shorter.53 The half-life of methyl mercury in blood, which is used to indicate the total body burden, is assumed to be about 50 days.54 For children receiving thimerosal in vaccines, however, the half-life of ethyl mercury in blood was 7 to 10 days, or 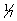 to
to  as long as that of methyl mercury.52,53 Thus in the 2-month periods between vaccinations (at birth and at 2, 4, and 6 months), all of the mercury should be excreted with no accumulation.43
as long as that of methyl mercury.52,53 Thus in the 2-month periods between vaccinations (at birth and at 2, 4, and 6 months), all of the mercury should be excreted with no accumulation.43
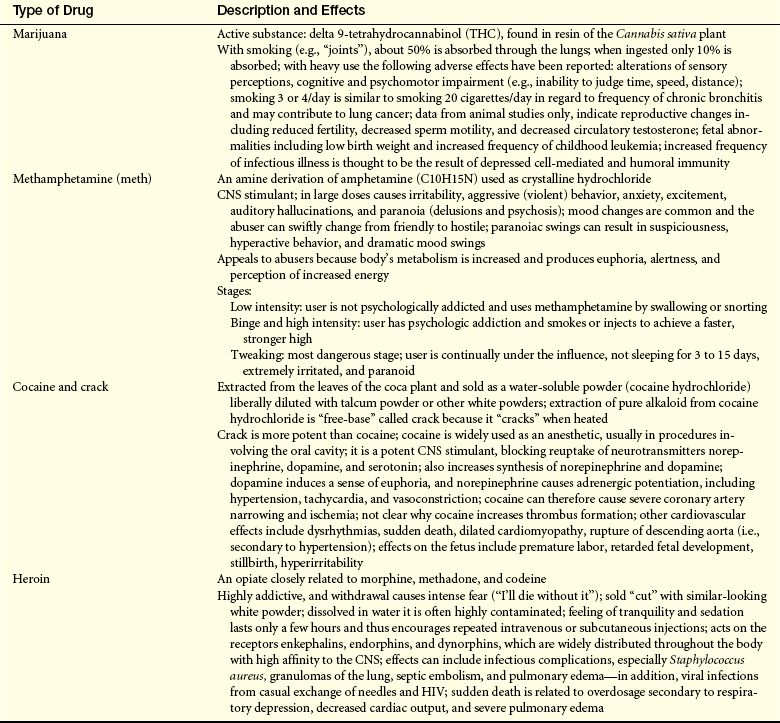
Social or Street Drugs: The social or “recreational” use of psychoactive drugs is widespread in many parts of the world. Most popular and dangerous are the drugs methamphetamine (“meth”), marijuana, cocaine, and heroin. Although the prevalence of cocaine use in the general population decreased in 1986, morbidity and mortality related to cocaine increased sharply in the 1990s. Illicit use of drugs is a prevalent risk behavior among adolescents.55 Table 2-6 summarizes the effects of these drugs.
Table 2-6
Social or Street Drugs and Their Effects
CNS, Central nervous system; HIV, human immunodeficiency virus.
Data from Cotran RS, Kumar V, Colllins T: Robbins pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders; Nahas G, Sutin K, Bennett WM: Review of marijuana and medicine, N Engl J Med 343(7):514, 2000.
Unintentional and Intentional Injuries
Unintentional and intentional injuries are an important health problem in the United States. In 2005 there were 173,573 deaths in this category, an injury death rate of 57.76/100,000.56 Death due to injury is significantly more common for men than women; the overall rate for men is 84.16/100,000 vs. 33.12/100,000 for women. Significant racial differences exist in the death rate, too: whites at 57.42./100,000, blacks at 67.31/100,000, and other racial groups at a combined rate of 35.76/100,000. A bimodal age distribution for injury-related deaths also has been noted, with peaks in the young adult and older adult groups. Unintentional injury is the leading cause of death for people between the ages of 1 and 34 years, with intentional injury (suicide, homicide) ranking between the second and fourth leading causes of death in this age group. A 1999 report published by the Institute of Medicine (IOM) indicated that between 44,000 and 98,000 unnecessary deaths per year occurred in hospitals alone as a result of errors by health care professionals. An accurate account is a tremendous challenge because of disagreements over reported statistics.57 Statistics on nonfatal injuries are harder to document accurately, but they are known to be a significant cause of morbidity and disability and to cost society billions of dollars annually. The more common terms used to describe and classify unintentional and intentional injuries and brief descriptions of important features of these are discussed here.
Blunt force injuries are the result of the application of mechanical energy to the body resulting in the tearing, shearing, or crushing of tissues. They are the most common type of injuries seen in most healthcare settings. Blunt force injury may be caused by blows (a moving object strikes the body), impacts (the moving body strikes a fixed object), or a combination of both. Motor vehicle accidents and falls are the most common causes of these injuries, accounting for 45,520 and 20,426 deaths, respectively, in 2005.
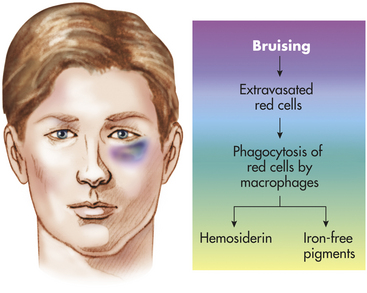
Contusion: A contusion (bruise) is bleeding into the skin or underlying tissues as a consequence of a blow that squeezes or crushes the soft tissues and consequently ruptures blood vessels without breaking the skin. It may take several hours after injury before any change in skin color is seen. A bruise will be red-purple initially, eventually becoming blue-black, and then gradually changing to yellow-brown or green before fully disappearing (Figure 2-14). These color changes reflect the progression of tissue damage and healing that develops in the area of underlying injury. The length of time depends on such factors as the extent and location of the injury and the degree of vascularization in the area. Small contusions may resolve in a matter of days, whereas larger ones can take weeks to completely heal. Bruising of soft tissues may sometimes be confined to deeper structures; thus no injury is visible externally. Blood in deeper structures may dissect along fascial planes, so discoloration of the skin may be seen in areas not directly injured by the initiating blow or impact, such as bruising of the thigh occurring with a hip or pelvis fracture or “black eyes” with orbital plate fractures. Contusions also may be seen in internal organs in cases of severe injury.
A collection of blood in soft tissues or an enclosed space also may be referred to as a hematoma (see Figures 17-3 and 17-6). A subdural hematoma is a collection of blood between the inner surface of the dura mater and the surface of the brain, resulting from the shearing of small veins that bridge the subdural space. Subdural hematomas can result from blows, falls, or sudden acceleration/deceleration of the head, as occurs in shaken baby syndrome. An epidural hematoma is a collection of blood between the inner surface of the skull and the dura. It is caused by a torn artery and is almost always associated with a skull fracture.
Contusions of the brain may result from (1) a blow, or (2) a fall or impact. In blows, when a moving object strikes the stationary head, a cerebral contusion grouped in the portions of the brain underlying the area of scalp and skull injury is known as a coup pattern of injury. In falls or impacts, in which the moving head strikes a fixed object, a cerebral contusion seen in the area of the brain opposite the external injury is known as a contrecoup pattern of injury (see Figure 17-1). Contrecoup injury results when the head accelerates and the brain lags behind and presses into the areas of the skull directly opposite the direction of motion. When the head suddenly stops, the areas of the brain pressing into the skull are injured. For example, a person who falls directly backward striking the occiput (back of the head) will have cerebral contusions of the frontal and temporal tips (these injuries are discussed further in Chapter 17).
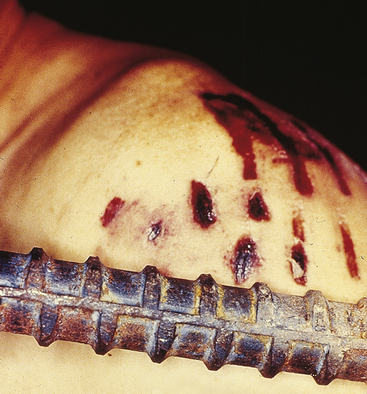
Abrasion: An abrasion (scrape) results from removal of the superficial layers of the skin caused by friction between the skin and injuring object. Abrasions vary in size and severity from fine, thin scratches to large denuded areas (road rash). In cases in which force is applied in a tangential, nonperpendicular direction to the skin surface, tags of tissue may be heaped up at the trailing or downstream edge of the abrasion. An abrasion will have a pale, moist, yellow-brown appearance at first. The color darkens to brown or even black as the injury dries. The injury may ooze fluid for 1 or 2 days until it is completely covered by a crust, or scab, which eventually flakes off of the underlying regenerated skin.
Abrasions and contusions may have a patterned appearance that mirrors the shape and features of an injuring object (Figure 2-15). Patterning of injuries can be of crucial importance in cases of automobile accidents, assaults, or homicides by documenting the connection between the victim’s injuries and a suspect vehicle or weapon. Bite marks (usually a combination of abrasion and contusion) are another example of a patterned injury that can demonstrate a link between an assailant and victim.
Laceration: A laceration is a tear or rip resulting when the tensile strength of the skin or tissue is exceeded. Unlike an incision, in which the tissue is cleanly divided by a sharp edge, a laceration is much more jagged and irregular, and the edges are abraded. The depths of the laceration are irregular, and often tissue “bridges” of small vessels or nerves that have been stretched but not broken are present, crossing from one side of the wound to the other. If the injuring force is applied perpendicularly to the skin, crushing of the surrounding tissue with associated abrasion and contusion will be noted. If force is applied tangentially, undermining of the wound also will occur, with tissues at the trailing edge of the wound being lifted away from the underlying structures, creating a pocket in the direction opposite the blow. An extreme example is an avulsion (Figure 2-16), in which a wide area of tissue may be pulled away, creating a large flap. Usually, the shallower the angle of incidence of the blow, the more extensive the undermining.
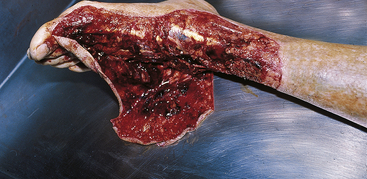
Figure 2-16 Avulsed laceration in motor vehicle accident victim. The victim was the driver and this injury most likely was caused by the brake pedal.
Lacerations of internal organs are not uncommon in blunt impact injuries. Lacerations of the liver, spleen, kidneys, and bowel may occur in cases of blows to the abdomen, often with no externally visible injury to the abdominal wall. The thoracic aorta may be lacerated in sudden deceleration accidents. This results from the arch of the aorta being freely mobile, whereas the descending portion is attached to the spinal column. Rapid deceleration causes horizontal shearing with either partial or complete transection just below the takeoff of the left subclavian artery. Severe blows or impacts to the chest also may cause rupturing of the heart with lacerations of the atria or ventricles.
Fractures: Blunt force blows or impacts also can cause bone to break or shatter. Fractures are extensively covered in Chapter 42 and are not discussed here.
Sharp Force Injuries
Cutting and piercing injuries accounted for 2795 deaths in 2005. As with all injuries, men have a higher rate (1.43/100,000) than women (0.46/100,000). Here, too, are greater differences among races, with whites at 0.73/100,000, blacks at 2.29/100,000, and other racial groups at 0.80/100,000.
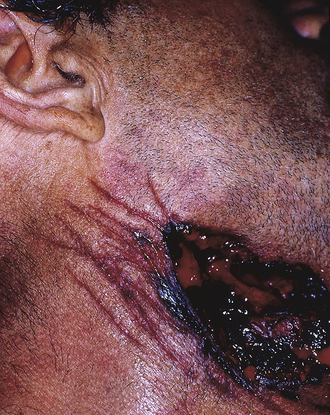
Incised Wounds: An incised wound is a cut that is longer than it is deep. The wound may be straight or jagged, depending on the object used and how the injury occurred; sharp, distinct edges without abrasion. Because the wound is caused by a sharp edge, the tissues are cleanly divided and no tissue bridging or undermining occurs. An incised wound may be thin and narrow or more elliptic and gaping in appearance because of varying lines of tension in the skin, depending on the location and orientation of the wound. Incised wounds tend to produce significant external bleeding with minimal internal hemorrhage. These wounds are often seen in sharp force injury suicides. In most cases, in addition to a deep, lethal cut, multiple superficial incisions are grouped in the surrounding area; these are known as hesitation marks (Figure 2-17).
Stab Wounds: A stab wound is a penetrating sharp force injury that is deeper than it is long. Because a sharp instrument is used, the depths of the wound are clean and distinct with no underlying or associated crushing injury. The edges are usually clean but may be abraded if the object is inserted deeply with enough force so that a wider, blunter portion of the instrument (e.g., the hilt of a knife) impacts the skin. Figure 2-18 illustrates this type of wound.
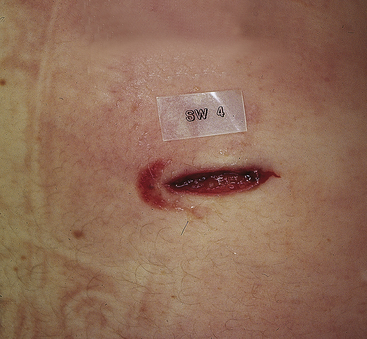
Figure 2-18 Stab wound with associated hilt mark. Note the sharp margin away from the hilt mark with the blunt margin toward it. This wound was caused by a single-edged knife.
A number of the offending blade’s characteristics may be determined from careful examination of the stab wound. If a single-edge blade is used, one margin of the wound will be sharp and the other blunt; if a double-edge blade causes the wound, both margins will have a sharp appearance. Stab wounds produced by a serrated-edge blade are often indistinguishable from those made by a smooth-edge blade. If any hesitation marks or scraping of the skin edges by the blade occurs, an interrupted pattern of abrasion may be seen, but this is uncommon. As with incised wounds, skin tension may cause the wound to gape, giving it an elliptic appearance. The edges must be brought into opposition so there is no distortion before trying to determine whether the margins are sharp or blunt. The length of the stab wound may or may not correlate with the width of the blade, depending on whether there was any cutting or twisting when the blade was inserted or withdrawn. Once the edges are in opposition, the thickness of the blade may be estimated from the width of the wound. Depth of the wound may not correlate with the length of the blade because the blade may not have been inserted fully, or as a consequence of compression of tissues caused by a forceful thrust, the wound may be deeper than the length of the blade.
Depending on size and location of the stab wound, the amount of external bleeding may be surprisingly small. After an initial spurt, even if a major vessel or the heart is struck, the wound track may be almost completely closed by tissue pressure, allowing only a trickle of visible blood externally despite copious internal bleeding.
Puncture Wounds: Instruments or objects with sharp points but without sharp edges may produce penetrating puncture wounds. A classic example is a wound of the foot caused by stepping on a nail. These injuries often will have abrasion of the edges of the wound, are prone to infection, and can be quite deep despite a sometimes innocuous external appearance.
Gunshot Wounds
Injuries caused by gunfire accounted for 30,694 deaths in the United States in 2005. Of these, 17,002 were suicides, 12,682 homicides, 789 unintentional, and 221 classified as undetermined. Men are much more likely to die from gunshot injury than women. The male death rate in 2005 was 18.30/100,000 vs. 2.67/100,000 for women. Black men between the ages of 15 and 24 years have the greatest gunfire injury death rate: 87.07/100,000. To put this statistic into perspective, if this were the rate for the United States as a whole, there would be more than 257,000 gunshot wound deaths per year.
Gunshot wounds may be either penetrating (bullet retained in the body) or perforating (bullet exits the body). In some cases, the bullet may fragment, so pieces of the missile are retained even though there is an exit wound. The most important factors determining the appearance of a gunshot injury are whether it is an entrance or an exit wound and the range of fire.
Entrance Wounds: Although all entrance wounds share some common features, the overall appearance is most affected by the range of fire.
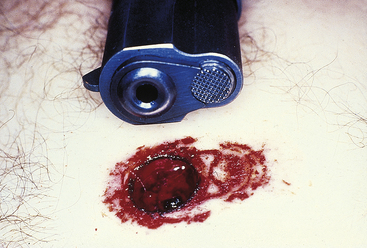
Contact range entrance wounds occur when the gun is held so the muzzle rests on or presses into the skin surface, causing a distinctive type of wound. In addition to the hole made by the bullet, there will be searing of the edges of the wound from the flame and hot gases exiting the barrel and soot or smoke deposited on the edges of and in the depths of the wound. In hard contact wounds, where the barrel is firmly pressed into the skin, there may be minimal soot and searing on the outside of the wound but deep penetration of smoke, burning gunpowder fragments, and hot gases into the depths of the injury. In hard contact wounds of the head, where there is only a thin layer of skin and muscle overlying bone, the large amount of gas and explosive energy sent into the wound may cause severe tearing and disruption of the tissues, giving the wound a large, gaping, and jagged appearance—a phenomenon known as blow back. In areas of the body with thicker layers of soft tissue, the blow back may not cause tearing but will forcefully drive the skin back onto the end of the barrel, producing a patterned abrasion that mirrors the features of the weapon, known as a muzzle imprint (Figure 2-19).
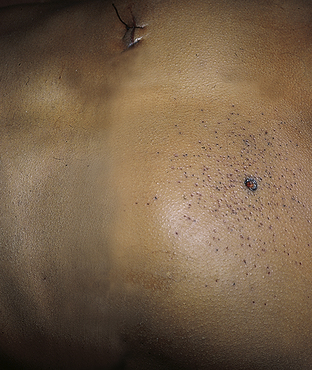
Intermediate-range entrance wounds are surrounded by gunpowder tattooing or stippling (Figure 2-20). Tattooing results from fragments of burning or unburned pieces of gunpowder exiting the barrel and striking the skin surface with enough force to be driven into the epidermis or superficial dermis. Stippling results when fragments of powder strike with enough force to abrade the skin but not actually penetrate the surface. This phenomenon can be seen when the muzzle-to-target range is less than 48 inches of most handguns. Beyond this distance, pieces of gunpowder disperse and slow down so much that tattooing or stippling cannot occur. The closer the muzzle is to the skin, the tighter the distribution and greater the density of powder fragments will be around the actual entrance hole. Soot also may be deposited.
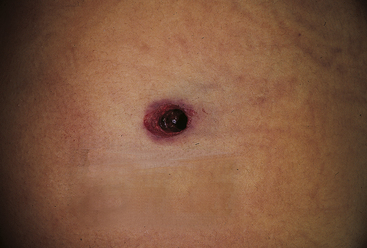
An indeterminate-range (distant) entrance wound occurs when flame, soot, or gunpowder does not reach the skin surface and the only thing striking the body is the bullet. The term indeterminate is used rather than distant because it does not imply that one can actually determine the range of fire from the appearance of the wound. For example, if an individual is shot through multiple layers of clothing, the entrance wound may have no sooting, searing, or stippling even though the actual range of fire is only a matter of inches; the wound would look the same as if the shot came from a range of 6 meters (20 feet) or more. Indeterminate wounds are characterized by a hole surrounded by a rim of abrasion. The size of the hole can vary according to a number of factors. It is important to remember that one cannot say what caliber of weapon inflicted the wound based solely on the size of the entrance wound. The collar of abrasion results from the fact that the bullet first causes stretching and scraping of the skin before it actually perforates. If the bullet strikes perpendicular to the skin, the margin of the abrasion collar is concentrically disturbed about the defect; if it strikes at an angle, the collar is eccentric, with the wider margin pointing in the direction from which the bullet came (Figure 2-21). If the bullet has struck an intermediary target before hitting the skin, it can be turning and tumbling, producing an irregular abrasion collar.
Exit Wounds: Exit wounds, or where the bullet comes out, have the same general appearance no matter what the range of fire. Their shape can vary from round to slitlike to completely irregular. As with entrance wounds, the size does not correlate very well with the caliber of the projectile making the wound. The most important factors affecting exit wounds are the speed of the projectile and the degree of deformation. A smaller, highly deformed bullet exiting at high speed can produce a large, irregular wound, whereas a larger, intact, slower-moving bullet may only make a small hole. Size cannot be used to determine whether the hole is an exit or entrance wound. In most cases, the margins of an exit wound will not have an abrasion collar. An exit wound will have clean edges that can often be reapproximated to cover the defect. The exception is when something is pressing against the skin surface at the exit site, such as tight clothing or the back of a chair. In this situation, the bullet will push the skin against the supporting surface causing rubbing and scraping around the exit defect as it comes out, a defect known as a shored exit wound.
It is important to remember that because the skin is so elastic and deformable, it is one of the toughest structures for a bullet to go through. It is not uncommon for a bullet to pass entirely through the body and be stopped just beneath the skin on the opposing side of the body. Often no visible injury of the overlying skin is seen; however, careful palpation of the area may allow one to locate the bullet.
Wounding Potential of Firearms: The amount of damage done by a bullet is a function of a number of variables. For the most part, the damage caused is a result of the amount of energy transferred to the tissues impacted. The energy a bullet has is determined by the following formula:
where KE is the energy, M is the mass, and V is the speed.
Clearly, increasing the speed of a bullet has a much greater effect on its potential to cause damage than increasing its size. As the bullet passes through tissue and slows down, its energy is dissipated into the surrounding structures. This energy transfer causes tissue destruction in a zone that can be much larger than the actual size of the bullet; the zone of destruction may be several inches in diameter with very high-powered bullets. This transfer of energy in head wounds may lead to orbital plate fractures and palpebral ecchymosis (black eyes) or blood draining from the ears even though the path of the bullet does not come near the base of the skull. The amount of damage caused may be exacerbated by the generation of secondary missiles of bone fragments when portions of the skeleton are struck. Some bullets are designed to expand or fragment when they strike an object, thereby increasing the cross-sectional area of the projectile, increasing drag, and enhancing the transfer of energy into the tissues. Hollow-point ammunition is an example of this kind of bullet.
Obviously the lethality of a gunshot injury depends on what structures are damaged. Depending on the extent of damage, even gunshot wounds of the brain may not be lethal; however, they are usually immediately incapacitating and lead to significant long-term disability. It is important to remember that a victim with a “lethal” injury (wound of the heart or aorta) may not be immediately incapacitated and may engage in varying degrees of physical activity after being injured. Just because the victim is active or even combative when first evaluated does not mean the individual may not have experienced a potentially lethal injury.
Asphyxial Injuries
Asphyxial injuries are caused by a failure of cells to receive or use oxygen. Deprivation of oxygen may be partial (hypoxia) or total (anoxia). Asphyxial injuries can be grouped into four general categories: suffocation, strangulation, chemical, and drowning.
Suffocation: Suffocation, or oxygen failing to reach the blood, can result from a lack of oxygen in the environment (entrapment in an enclosed space or filling the environment with a suffocating gas) or blockage of the external airways. Classic examples of these types of asphyxial injuries are a child who is trapped in an abandoned refrigerator or a person who commits suicide by putting a plastic bag over the head. A reduction in the ambient oxygen level to 16% (normal is 21%) is immediately dangerous. If the level is below 5%, death can ensue within a matter of minutes. The diagnosis of these types of asphyxial injuries depends on the history of what happened, because there will be no specific physical findings.
Diagnosis and treatment in choking asphyxiation (obstruction of the internal airways) depend on locating and removing the obstructing material. Injury or disease also may cause swelling of the soft tissues of the airway, leading to partial or complete obstruction and subsequent asphyxiation. Suffocation also may result from compression of the chest or abdomen (mechanical or compressional asphyxia), preventing normal respiratory movements. Usual signs and symptoms include florid facial congestion and petechiae (pinpoint hemorrhages) of the eyes and face.
Strangulation: Strangulation is caused by compression and closure of the blood vessels and air passages resulting from external pressure on the neck. This causes cerebral hypoxia or anoxia secondary to the alteration or cessation of blood flow to and from the brain. It is important to remember that the amount of force needed to close the jugular veins (2 kg [4.5 lb]) or carotid arteries (5 kg [11 lb]) is significantly less than that required to crush the trachea (15 kg [33 lb]). It is the alteration of cerebral blood flow in most types of strangulation that causes injury or death—not the lack of airflow. With complete blockage of the carotid arteries, unconsciousness can occur within 10 to 15 seconds.
A noose is placed around the neck, and the weight of the body is used to cause constriction of the noose and compression of the neck in hanging strangulations. The body does not need to be completely suspended to produce severe injury or death. Depending on the type of ligature used, there usually is a distinct mark on the neck, an inverted V with the base of the V pointing toward the point of suspension. Internal injuries of the neck are actually quite rare in hangings, and only in judicial hangings, in which the body is weighted and dropped, is significant soft tissue or cervical spinal trauma seen. Petechiae of the eyes or face may be seen, but they are rare.
In ligature strangulation, the mark on the neck is horizontal, without the inverted V pattern seen in hangings. Petechiae may be more common because intermittent opening and closure of the blood vessels may occur as a result of the victim’s struggles. Internal injuries of the neck are rare.
Variable amounts of external trauma on the neck with contusions and abrasions are noted in manual strangulation caused either by the assailant or by the victim clawing at one’s own neck in an attempt to remove the assailant’s hands. Internal damage can be quite severe, with bruising of deep structures and even fractures of the hyoid bone and tracheal and cricoid cartilages. Petechiae are common.
Chemical Asphyxiants: Chemical asphyxiants either prevent the delivery of oxygen to the tissues or block its use. Carbon monoxide is the most common chemical asphyxiant (see p. 59). Cyanide acts as an asphyxiant by combining with the ferric iron atom in cytochrome oxidase, thereby blocking the intracellular use of oxygen. A victim of cyanide poisoning has the same cherry-red appearance as a carbon monoxide intoxication victim because cyanide blocks the use of circulating oxyhemoglobin. An odor of bitter almonds also may be detected. (The ability to smell cyanide is a genetic trait that is absent in a significant portion of the general population.) Hydrogen sulfide (sewer gas) is a chemical asphyxiant in which victims of hydrogen cyanide poisoning may have brown-tinged blood in addition to the nonspecific signs of asphyxiation.
Drowning: Drowning is an alteration of oxygen delivery to tissues resulting from the breathing in of fluid, usually water. In 2005 there were 4248 drowning deaths in the United States. Although research done in the 1940s and 1950s indicated that changes in blood electrolyte levels and volume as a result of absorption of fluid from the lungs may be an important factor in some drownings, the major mechanism of injury is hypoxemia (low blood oxygen levels). Even in freshwater drownings, where large amounts of water can pass through the alveolar-capillary interface, there is no evidence that increases in blood volume cause significant electrolyte disturbances or hemolysis, or that the amount of fluid loading is beyond the compensatory capabilities of the kidneys and heart. Airway obstruction is the more important pathologic abnormality, underscored by the fact that in up to 15% of drownings, little or no water enters the lungs because of vagal nerve–mediated laryngospasms. This phenomenon is called dry-lung drowning.
No matter what mechanism is involved, cerebral hypoxia leads to unconsciousness in a matter of minutes. Whether this progresses to death depends on a number of factors, including age and health of the individual. One of the most important factors is the temperature of the water. Irreversible injury develops much more rapidly in warm water than it does in cold water. Submersion times of up to 1 hour with subsequent survival have been reported in children retrieved from very cold water. Complete submersion is not necessary for a person to drown. An incapacitated or helpless individual (such as a person with epilepsy or alcoholism or an infant) may drown in only a few inches of water.
It is important to remember that there are no specific or diagnostic findings to prove that a person recovered from the water is actually a drowning victim. In cases in which water has entered the lung, there may be large amounts of foam coming from the nose and mouth, although this also can be seen in certain types of drug overdoses. A body recovered from water with signs of prolonged immersion could just as easily be a victim of some other type of injury who has been put in the water to obscure the actual cause of death. When working with a living victim recovered from water, it is essential to keep in mind that an underlying condition may have led to the person’s becoming incapacitated and submersed—a condition that also may need to be treated or corrected while correcting hypoxemia and dealing with its sequelae.
Infectious Injury
The pathogenicity (virulence) of microorganisms lies in their ability to survive and proliferate in the human body, where they injure cells and tissues. The disease-producing potential of a microorganism depends on its ability to (1) invade and destroy cells, (2) produce toxins, and (3) produce damaging hypersensitivity reactions (see Chapter 8 for further discussion).
Immunologic and Inflammatory Injury
Cellular membranes are injured by direct contact with cellular and chemical components of the immune and inflammatory responses, such as phagocytic cells (lymphocytes, macrophages) and substances such as histamine, antibodies, lymphokines, complement, and proteases (see Chapter 6). Complement is responsible for many of the membrane alterations that occur during immunologic injury.
Membrane alterations are associated with rapid leakage of potassium (K+) out of the cell and rapid influx of water. Antibodies can interfere with membrane function by binding to and occupying receptor molecules on the plasma membrane. This type of injury is found in certain forms of diabetes mellitus and in myasthenia gravis. Antibodies also can block or destroy cellular junctions, interfering with intercellular communication (see Chapters 7 and 8).
Injurious Genetic Factors
Genetic disorders may be the result of genetic factors that alter the cell’s nucleus and the plasma membrane’s structure, shape, receptors, or transport mechanisms. For example, enzymatic genetic defects can lead to abnormalities in membrane transport. Genetic disorders can cause structural alterations of the red blood cell, for example, sickle cell anemia. (Mechanisms causing genetic abnormalities are discussed in Unit II.)
Injurious Nutritional Imbalances
Essential nutrients—proteins, carbohydrates, lipids (fats), vitamins, and minerals—are required for cells to function normally. If these nutrients are not consumed in the diet and transported to the body’s cells or if excessive amounts of nutrients are consumed and transported, pathophysiologic cellular effects develop.
Proteins, which consist of chains of amino acids, are the major structural units of the cell and participate in many enzymatic and hormonal functions. Protein deficiency causes a decrease in the intestinal mucosal mass, decreasing the absorptive function. The integrity of the pancreas is also affected, resulting in diminished exocrine secretion. With starvation or malnutrition, the lowered plasma proteins, particularly albumin, cause fluid to move into the interstitium (edema). Protein-calorie malnutrition (PCM) is the predominant worldwide type of malnutrition. Malnourished children are very susceptible to disease and often die of infectious diseases. Even with adequate protein intake, cellular injury can occur if amino acid transport mechanisms fail or are defective. In Fanconi syndrome, for example, renal tubular cells may contain accumulated protein droplets that have been absorbed but cannot be transported.
Glucose is the major carbohydrate obtained from the breakdown of starch (see Chapter 1). Hyperglycemia (excessive glucose in the blood) caused by excessive carbohydrate intake may lead to obesity. Deficiencies of glucose result from starvation or from lack of use, as in diabetes. In both conditions the body compensates by metabolizing fat (lipids). (For details on diabetes, see Chapter 21.)
In lipid deficiency, or hypolipidemia, the body compensates by mobilizing fatty acids from adipose tissue. This causes an increase in the production and circulation of ketone bodies, which are acidic by-products of lipid metabolism. The excretion of ketone bodies results in loss of water and electrolytes and causes dehydration and thirst. Severe increases in ketone bodies cause ketoacidosis, coma, and death. Hyperlipidemia, or an increase in lipoproteins in the blood, results in deposits of fat in the heart, liver, and muscle.
Vitamins are not sources of energy but are necessary for maintaining normal cellular functions. Adequate vitamin intake is necessary because most vitamins are not synthesized by the body. Research from the 1990s resulted in the identification of 13 vitamins as being essential for humans. These include 8 B vitamins (thiamine, niacin, riboflavin, folate, vitamin B6, vitamin B12, biotin, and pantothenic acid), vitamin C or ascorbic acid, and the fat-soluble vitamins A, D, E, and K. Minerals are discussed in Chapter 3. Vitamins are involved in numerous reactions, including metabolism of visual pigments (vitamin A), calcium and phosphate metabolism (vitamin D), prothrombin synthesis (vitamin K), and antioxidation reactions (vitamins E and C). Pyridoxal (vitamin B6) affects amino acid transfer reactions; flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and NAD help the reaction transfer of electrons (see Chapter 1). Table 2-7 presents vitamins and their association with deficiency-related diseases/disorders. New are the many diseases related to vitamin D deficiency, which may include many common cancers, type 1 diabetes, cardiovascular disease, osteoporosis, and fibromyalgia (Figure 2-22 and see Nutrition & Disease, Vitamin D: Importance for Health Promotion).58
Table 2-7
Vitamin Deficiencies and Associated Disorders and Diseases
| Vitamin | Associated Deficiency |
| Niacin | Rough skin (pellagra) symptoms include lassitude, anorexia, dermatitis, diarrhea, inflammation of the mouth and other mucous membranes |
| Riboflavin (vitamin B2) | Decreased growth; skin lesions; soreness and burning of the lips, mouth, and tongue; burning and itching of the eyes; stomatitis; photophobia; vascularization of the cornea; glossitis; anemia; neuropathy |
| Thiamine (vitamin B1) | Beriberi; chronic alcoholism contributes to deficiency; megaloblastic anemia; lactate acidosis; subacute necrotizing encephalomyelopathy; individuals at risk include those undergoing long-term dialysis or intravenous feedings and those with chronic febrile infection |
| Folate | Defects in DNA synthesis (fast-growing tissue, embryo); megaloblastic anemia; vascular disease (e.g., hyperhomocysteinemia); cancers including colon cancer (see Chapter 12); malabsorption syndromes (tropical and nontropical sprue) |
| Vitamin B12 | Pernicious anemia; neurologic (demyelination and peripheral neuropathy); memory loss and dementia; hyperhomocysteinemia and vascular disease |
| Vitamin B6 | Seborrheic dermatitis; microcytic anemia; convulsions; depression; confusion |
| Pantothenic acid (vitamin B5) | Listlessness, fatigue, and weakness; headaches; personality changes; sleep disturbances; impaired motor coordination; gastrointestinal disturbances |
| Biotin | Severe ketoacidosis, seizures, ataxia, lethargy, coma at birth; hair loss; skin rashes; hearing loss; optic atrophy |
| Vitamin C | Scurvy (bleeding under the skin, gums, joint pain, joint effusions, shortness of breath); fatigue; increased risk to infection (decreased immune function) |
| Vitamin K | Hemorrhagic disease of the newborn; depression of vitamin K–dependent coagulation factors; individuals at risk are those on antibiotic therapy (interferes with synthesis) and those who have osteoporosis |
| Vitamin E | Status may depend on selenium and sulfur-containing amino acids; necrotizing myopathy (skeletal, heart, smooth muscle); decreased life span of red blood cells and increased risk of hemolysis; neurologic abnormalities; increased susceptibility to effects of oxidizing agents in the environment; decreased immune function, possibly vascular disease and coronary heart disease; possibly certain cancers (head, neck, lung, colorectal) |
| Vitamin A | Possibly several types of cancer; decreased immune function; fetal malformations; vision abnormalities (including night blindness, cornea changes, drying) |
| Vitamin D | Rickets; type 1 diabetes; cardiovascular disease, some common cancers, osteoporosis; fibromyalgia; multiple sclerosis; parathyroid disorders, autoimmune disorders |
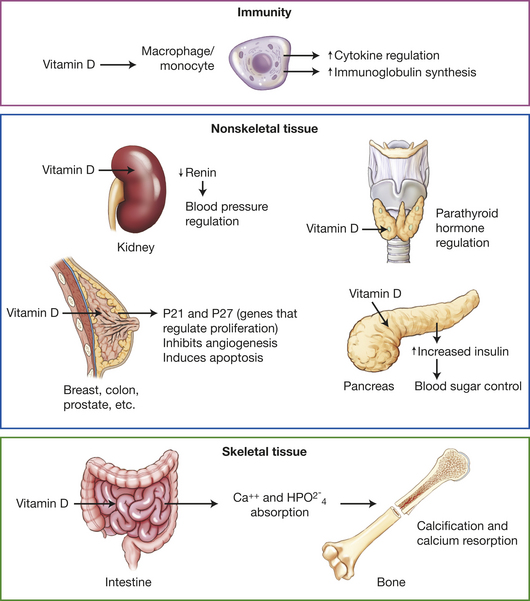
Figure 2-22 The many functions of vitamin D. Most tissues have a vitamin D receptor, thus vitamin D has enormous effects. Vitamin D deficiency is implicated in many chronic diseases including common cancers, autoimmune diseases, infectious diseases, and cardiovascular disease (also see Nutrition & Disease: Vitamin D Importance for Health Promotion, p. 70).
Injurious Physical Agents
Injurious physical agents include temperature extremes, changes in atmospheric pressure, radiation, illumination, mechanical factors, noise, and prolonged vibration. Physical injury can result from excessive exposure to many environmental agents, as well as to agents used for the diagnosis and treatment of illness.
Temperature Extremes
Chilling or freezing of cells causes hypothermic injury. Hypothermia has proved to be strongly injurious to a variety of cells. Prolonged exposure to low ambient temperature is a common risk factor found in homeless persons. Hypothermic injury has long been attributed to disturbances of cellular ion balance or homeostasis, especially of sodium balance (i.e., increased intracellular sodium levels). Hypothermia increases intracellular Ca++ by slowing the Na+, K+-ATPase pump activity, leading to Na+ accumulation intracellularly.59 In recent years, however, a role for ROS has gained importance.60 In animal studies, hypothermia resulted in cell damage caused by formation of ROS.61–63 Hypothermic perfusion of the heart increased (superoxide, see Table 2-3); in turn reacted with nitric oxide (NO) to form another radical peroxynitrate anion (ONOO−).59
Therapeutically, hypothermia is widely used to protect cells and tissues against injurious processes. In some cell types, however, such as hepatocytes and liver endothelial cells, hypothermia can cause pronounced cell injury mediated by ROS.63 During the body’s exposure to cold, injury is inhibited by hypoxia and by a number of antioxidants, especially iron chelators.63
Indirect forms of injury occur because of changes in small blood vessels (the microcirculation). Slow chilling can cause vasoconstriction followed by paralysis of vasomotor control, resulting in vasodilation and increased membrane permeability causing cellular and tissue swelling. With an abrupt drop in temperature, vasoconstriction and increased viscosity of the blood cause ischemic injury—infarction and necrosis (cellular death) in affected tissues. With continued exposure to freezing temperatures, vasodilation produces severe swelling that causes degenerative changes in the myelin sheath that surrounds peripheral nerves, resulting in sensory and motor disturbances. Thrombosis also can occur and may lead to gangrene of the affected part. (Gangrene is discussed on p. 83.) These conditions often are called frostbite.
Hyperthermic injury (injury caused by excessive heat) is common and varies depending on the nature, intensity, and extent of the injury. Three types of hyperthermic injury include heat cramps, heat exhaustion (illness), and heat stroke. (For more detail see Chapter 15.)
Heat cramps are cramping of voluntary muscles, usually as a result of vigorous exercise. Cramps are the result of salt and water loss as a consequence of sweat. Treatment is salt replacement.
Heat exhaustion occurs when sufficient salt and water loss results in hemoconcentration. Hypotension occurs secondary to fluid loss (hypovolemia), and the individual feels weak, nauseated, and can suddenly collapse. Collapsing results from a failure of the cardiovascular system to compensate for hypovolemia. Heat exhaustion is probably the most common heat-related injury.
Heat stroke is a life-threatening condition associated with high environmental temperatures and humidity. Core body temperature rises as a result of thermoregulatory failures. Clinically, a rectal temperature of 106° F is considered a life-threatening sign. Generalized peripheral vasodilation and decreased circulating blood volume are significant. At risk are older adults, athletes, military recruits, and people with cardiovascular disorders.
Burns are caused by local heat injury. A full-thickness burn is an open wound involving skin layers—epidermis, dermis, and subcutaneous layers—and causing extensive loss of fluids and plasma proteins. Cellular regeneration is not possible; therefore, skin from a donor or from the host must be grafted to the site. Partial-thickness burns result in reddening of the area as a result of dilation of small blood vessels and increased permeability of cellular membranes, with loss of protein-rich fluid, resulting in the typical “burn blister.” In surface epithelial cells, membrane permeability increases, causing both cytoplasmic and nuclear swelling. Temperature-sensitive enzymes within certain cells respond to heat by increasing cellular metabolism, with detrimental effects. Intense heat also damages the vascular endothelium and causes coagulation of the blood vessels. (Burns are discussed further in Chapter 46.)
Epidemiologic investigators have reported a relationship between overheating in infants; that is overdressing infants in the winter, and sudden infant deaths. Studies suggest interactions between body temperature and respiratory responses to hypoxia or increased carbon dioxide (hypercapnia). The hypoxia/hypothermia interaction depresses breathing, reducing the ventilatory response to hypercapnia. The effects of hyperthermia seem to be a significant problem only when it accompanies an infection and fever and alters or depresses the breathing responses. (Temperature changes are also discussed in Chapter 15.)
Changes in Atmospheric Pressure
Sudden increases or decreases in atmospheric pressure cause blast injury, which can be transmitted by either air (air blast) or water (immersion blast). With sudden increases in pressure, such as in air blast or explosive injuries, tissue injury is caused by compressive waves of air impinging on the body, followed by a sudden wave of decreased pressure. The pressure changes may collapse the thorax, rupture internal solid organs, and cause widespread hemorrhage. In increased pressure caused by immersion blast, water pressure is applied suddenly to all sides of the body, forcing the body up out of water. The positive pressure compresses the abdomen and ruptures hollow internal organs, such as the spleen, kidneys, and liver.
With sudden decreases in pressure, carbon dioxide and nitrogen that are normally dissolved in the blood come out of solution and form tiny bubbles called gas emboli. At low atmospheric pressure, such as occurs at altitudes above 15,000 feet, there is a significant decrease in available oxygen. This causes hypoxic injury, and compensatory vasoconstriction shunts blood from the peripheral circulation (in the extremities) to the visceral organs, including the lungs. The combination of increases in pulmonary blood flow and systemic hypoxia causes “high-altitude pulmonary edema”64 (see Chapter 33).
Deep-sea divers and underwater construction workers who return to the surface too quickly develop a form of gas embolism called decompression sickness or caisson disease (“the bends”). If water pressure is reduced too rapidly, the gases dissolved in blood bubble out of solution, forming emboli. Oxygen is quickly redissolved, but nitrogen bubbles may persist and obstruct blood vessels. Ischemia resulting from gas emboli causes cellular hypoxia, particularly in the muscles, joints, and tendons, which are especially susceptible to changes in oxygen supply. Emboli and interstitial gas accumulate around the joints and skeletal muscles, causing the individual to double up in pain. Tissues of the heart and brain also may be affected by emboli, causing necrosis. The gases can be promptly redissolved in blood by raising the atmospheric pressure. This is accomplished by placing the individual in a decompression chamber. First, pressure is increased until it approximates pressure at the depth to which the diver had descended. This redissolves the gas bubbles in the blood. Then the pressure in the chamber is decreased gradually until it equals pressure at the surface of the water. The slow decrease in pressure slows the release of gas bubbles out of solution.
Ionizing Radiation
Ionizing radiation is any form of radiation capable of removing orbital electrons from atoms, resulting in the production of negatively charged free electrons and positively charged ionized atoms. Ionizing radiation is emitted by x-rays, gamma rays, and alpha and beta particles (which are emitted from atomic nuclei in the process of radioactive decay) and from neutrons, deuterons, protons, and pions (all of which are emitted from cobalt or linear accelerators). Ionizing radiation of three types (x-radiation, gamma radiation, and neutrons) was classified as a carcinogen in 2004.65
The most abundant source of exposure to ionizing radiation is the environment. This source includes emission from radioactive material inside the body, cosmic rays from outer space, and radiation emitted from such substances as soil and building materials. Environmental radioactivity is emitted primarily by uranium, thorium, and potassium. Other sources are from medical uses (e.g., x-rays, computed tomography [CT] scans, etc.) used for medical diagnosis and treatment, uranium and thorium mines, nuclear weapons, and nuclear reactors that generate electricity. Table 2-8 includes types of ionizing radiation and their magnitude of tissue penetration.
Table 2-8
Types of Ionizing Radiation and Their Tissue Penetration
| Type | Tissue Penetration |
| X-rays | High |
| Gamma (γ) rays | High |
| Beta (β) particles | Low |
| Alpha (α) particles | Very low |
| Protons | Intermediate between α and β |
| Neutrons | High |
Data from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.
Ionizing radiation (x-radiation and gamma rays) causes a large spectrum of genetic changes including gene mutations, mini-satellite mutations (altered numbers of tandem repeats of DNA sequences), micronucleus formation (sign of chromosome damage or loss), chromosomal aberrations (structural or number), ploidy changes (number of sets of chromosomes), DNA strand breaks, and chromosomal instability.65 All phases of the cell cycle can be affected by ionizing radiation. Sensitivity of the cell appears to be greatest in G2, that gap of the cell just before mitosis; irradiation during this phase retards the onset of cell division. Irradiation during mitosis induces chromosomal aberrations. Membrane molecules and enzymes also are damaged by radiation (see Chapter 12). The intensity, duration, and cumulative effects of exposure to ionizing radiation determine the extent of injury. X-radiation and gamma radiation induce genetic alterations in somatic cells and heritable mutations in germ cells (sperm and egg and their precursor cells). DNA may be damaged directly or indirectly by interaction with reactive products (i.e., free electrons, hydroxyl radicals, hydrogen free radicals) from the degradation of water (Figure 2-23). The observed genetic damage is thought to be the result of DNA repair but also may arise from errors in replication.65 Epigenetic mechanisms (a change in gene expression and not in the DNA sequence) that damage genes may be involved in radiation-induced tumor formation. These mechanisms include genomic instability, mutations by irradiation of the cytoplasm (includes ROS, inflammatory cell signaling pathways, extracellular matrix alterations), “bystander effects” induction of genetic damage in innocent or cells not directly irradiated, possibly through cellular gap junctions and signaling pathways (see Chapter 12).
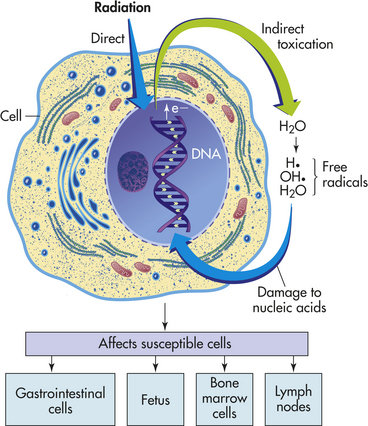
Figure 2-23 Cellular damage caused by ionizing radiation. Radiation can damage macromolecules in two ways: (1) directly, in which the micromolecules are ionized; and (2) indirectly, in which water is ionized and produces free radicals that in turn damage macromolecules. Cells that are particularly susceptible to damage are those of the gastrointestinal tract, bone marrow, lymph nodes, a fetus, and ovarian follicles. (Also see Chapter 18.)
Not all cells and tissues have the same sensitivity to radiation, although all cells can be affected. Radiosensitivity depends on the rate of mitosis and cellular maturity. Because fetal cells are both immature and undergoing rapid cycling, the fetus is at great risk for injury caused by ionizing radiation. Particularly vulnerable are embryonic germ cells, which are precursors of ova and sperm. Throughout life, cells of the bone marrow, intestinal mucosa, testicular seminiferous epithelium, and ovarian follicles are susceptible to injury because they are always undergoing mitosis, which ensures the presence of vulnerable, immature daughter cells. Ionizing radiation is a known human carcinogen. Exposure to x-radiation and gamma radiation is most strongly correlated with leukemia and cancers of the thyroid, breast, and lung; these correlates have been reported at absorbed low levels, less than 0.2 gray (Gy). The risk of developing these cancers may to some extent depend on age at exposure.65 Two studies suggest that radiation could cause liver cancer.66,67 Radiation exposure to children may increase the incidence of lymphomas, leukemias, melanomas, breast cancers, and others. At high-treatment radiation doses, the first study68 of sarcomas added angiosarcoma to the list of radiation-induced cancers.
Studies of A-bomb survivors have found increased mortality from noncancer diseases.69 The majority of radiation-induced noncancer deaths are cardiovascular problems (i.e., myocardial infarction and stroke). Individuals exposed to radiation at less than 40 years of age revealed an excess relative risk of myocardial infarction of 0.25.70 In addition, radiation exposure was significantly related to hypertension and elevated total cholesterol.70 Risk estimates for heart disease and stroke indicated higher susceptibility in women.70 Studies of the late effects of radiation in A-bomb survivors reveal elevated levels of mediators of inflammation (interleukin (IL)-6 and C-reactive protein [CRP]). Persistent elevations were documented of leukocyte counts, erythrocyte sedimentation rates, immunoglobulins, and sialic acid with radiation exposure. Also elevated were other inflammatory markers (TNF-α, IL-10, immunoglobulin (Ig), IgM, IgA, and interferon (IFN-γ). The elevations of autoantibodies and T cell–immune alterations is important because they may activate prolonged inflammation, which is a key contributor to cardiovascular effects.70 The bone marrow and thymus gland are both radiosensitive and documented immunologic effects measured from 40 to 50 years after radiation exposure suggests permanent genetic damage to lymphocyte progenitor cells.70 Thus good evidence exists that at doses of 0.5 sievert (Sv) ionizing radiation is a risk factor for cardiovascular disease.
The effects of ionizing radiation may be acute or delayed. Acute effects of high doses, such as skin redness, skin damage, or chromosomal aberrations, occur within hours, days, or months. The delayed effects of low doses may not be evident for years! Effects are usually (1) somatic, involving the exposed individual’s entire body (e.g., leukemia, other cancers); (2) genetic, involving offspring of the exposed individual; or (3) fetal, involving fetuses that are exposed in utero (see Chapter 12). According to the FDA uncertainty exists regarding the risk estimates for low levels of radiation exposure as commonly experienced in diagnostic radiology procedures71 (see What’s New? Focus on the Department of Energy Low Dose Radiation Research Program). (The carcinogenic effects of radiation are discussed in Chapter 12.)
Illumination
The retina is a highly specialized sense organ that is constantly subjected to environmental stresses, including illumination. Focused light rays can increase oxidative stress and is prevented by a wide array of retinal antioxidant mechanisms.72
Antioxidant mechanisms can, however, be overwhelmed by excessive light exposure, particularly of short wavelength, high-frequency blue light, and ultraviolet light. Since fluorescent lighting was introduced to the workplace, complaints of headaches, eyestrain, and eye discomfort have increased.73 The rapid modulation of light from fluorescent lamps is responsible for eyestrain and headaches.74 The modulation can be reduced by wearing tinted glasses.74 The shorter wavelengths in radiant energy in environmental lighting influence the absorption, scattering, and fluorescence, thus obscuring vision.74
Vision is obstructed at night by decreased illumination and by disabling glare from oncoming vehicle headlights. High-intensity discharge (HID) headlamps project light farther down roads, thus improving the driver’s safety. However, oncoming glare, which is proportional to headlamp brightness, is not good for any drivers of oncoming vehicles and even worse for older drivers.75 Older drivers experience more intraocular light scattering, glare sensitivity, and longer recovery time in reaction to photo stress.76
Studies have demonstrated the in vitro toxicity of halogen lamps.77,78 A pilot study of 12 mice illuminated with varying intensities and durations of halogen exposure resulted in benign forms of skin cancer (papillomas) as well as malignant tumor growth. Fortunately, prevention is simple if commercial models are available with glass or plastic covers.
Mechanical Stresses
Mechanical stimulation of body tissues and cells is constant. For example, gravity as an external force and the pumping of the heart as an internal force are continual. Acutely these forces elicit adaptive responses (to rapidly alter function) chronically; however, the responses may induce tissue remodeling to accommodate load-bearing capabilities.79 When the mechanical forces exceed unknown thresholds, injury results.79 Injury can initiate more reparative responses, transient or continuous dysfunction, or progressive degenerative changes that incorporate nearby and surrounding tissue. Cellularly, the structural responses to deformation and strain (e.g., biomechanical) are causing investigators to focus on the cell membrane. Disruption of cell membranes, or mechanoporation, is central to the biologic progression. Mechanical injury can progress to cell death involving both cell necrosis and delayed apoptosis.79 The heterogeneous distribution of atherosclerosis in the vasculature is possibly related to biomechanical factors, that is, certain arteries (e.g., coronary and carotid arteries) are more susceptible to plaque formation than others.80 Biomechanical forces probably are not systemic and vary with location. Mechanical stimuli include shear forces because of blood flow, strain from pressure distension of the vessel walls, and strain from tethering to a surrounding tissue area (e.g., the heart).80
Recent interest in mechanical injury and blood vessel damage has led investigators to study balloon catheterization or coronary angioplasty.80 Coronary angioplasty involves a catheter that is advanced along a blood vessel until it reaches the blocked region of the vessel. The balloon section of the catheter is then inflated, pushing the walls of the blocked vessel outward. Sometimes metal tubes, called stents, are inserted to keep the vessel open. Without the stents, however, vessel narrowing, or restenosis, often occurs at the balloon site. However, the injury causes an adaptive response of hypertrophy of the smooth muscle cells and an increase in macrophage activity. The macrophages release cytokines and growth factors, causing intimal cell proliferation and subsequent renarrowing.
The major focus of occupational biomechanics is the response of tissue to mechanical stress, especially the prevention of overexertion disorders of the lower back and upper extremities. Many mechanical stresses can cause overt injuries (e.g., a head injury when a worker is struck in the head with a dropped object). Most stresses, however, are subtle and can cause accumulative injuries and disorders.81 Table 2-9 summarizes common types of occupational mechanical stresses and associated types of injury.
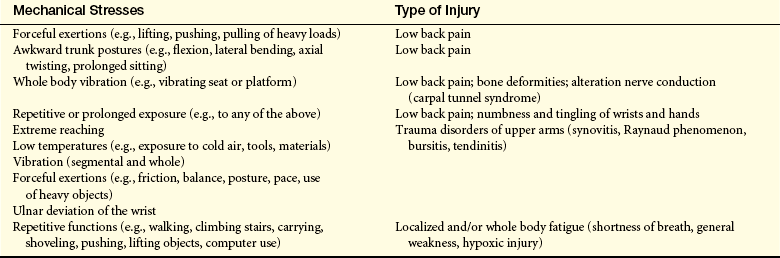
Noise
Noise is sound that has the potential for inflicting bodily harm. The most common pathophysiologic effect of noise is hearing impairment. Noise trauma can be caused by acute loud noise, as well as by the cumulative effects of various intensities, frequencies, and durations of noise. Common irritating noise is caused by numerous sources, including lawn care machinery; high-decibel, low-frequency speakers; loud movies; roaring highways; and so on. According to the National Institutes of Health, more than 10 million Americans suffer some permanent noise-associated hearing loss.82 The largest increase in hearing loss from noise occurs in people 45 to 64 years old. Noise pollution is now considered a public health threat.
Two types of hearing loss are associated with noise: (1) acoustic trauma, or instantaneous damage caused by a single sharply rising wave of sound (e.g., gunfire), and (2) noise-induced hearing loss, the more common type, which is the result of prolonged exposure to intense sound (e.g., noise associated with the workplace and leisure-time activities). Hearing loss can be a serious complication of critical illness. Individuals in intensive care units can experience hearing loss from mechanical or accidental trauma, ototoxic medications, infections, vascular disorders, autoimmune diseases, and environmental noise.83 Acoustic trauma can rupture the eardrum, displace the ossicles of the middle ear, and damage the organ of Corti in the inner ear.
If the offending noise has not been too loud or the exposure to it too long, hearing will return to its original level, a type of hearing loss called a temporary threshold shift (TTS). If the noise is louder than a certain value or the exposure time is long, the hearing threshold never returns to its original value, causing a permanent threshold shift (PTS). Structural changes associated with TTS, although not fully established, include intracellular changes in the sensory cells (hair cells) and swelling of the auditory nerve endings.29 With PTS, cochlear blood flow may be impaired and hair cells are damaged with each exposure. Noise-induced hearing loss is gradual and painless. Symptoms of noise-induced hearing loss include loudness recruitment and tinnitus. In loudness recruitment, soft sounds are not heard but loud sounds are heard normally. Tinnitus is a constant high-pitched ringing that annoys the individual and contributes to loss of sleep.